Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
1. Physiological roles of...
2. Autophagy in tumor biology
3. Neurological influences on...
4. Tumor-induced remodeling of...
5. Therapeutic strategies for...
6. Concluding remarks and future...
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2026; 16(14):8077-8103. doi:10.7150/thno.134896 This issue Cite
Review
Decoding autophagy in neuro-tumor crosstalk: from underlying mechanisms to translational opportunities
Huifang Cheng1,#, Hongyao Li2,#, Peiyan Liu3,#, Xin Jin1, Yi Qu4, ![]() , Bo Liu2,
, Bo Liu2, ![]() , Aizhuo Li1,
, Aizhuo Li1, ![]()
1. Department of Ultrasound, The First Hospital of China Medical University, Shenyang, 110001, China.
2. Department of Biotherapy, Cancer Center and State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu, 610041, China.
3. Department of Obstetrics, The First Hospital of China Medical University, Shenyang, 110001, China.
4. Department of Hematology, The First Hospital of China Medical University, Shenyang, 110001, China.
#These authors contributed equally to this work.
Received 2026-3-22; Accepted 2026-6-9; Published 2026-7-13
Abstract

Recent advances in cancer neuroscience have established the nervous system as an active regulator of tumor initiation and progression, emphasizing the complex bidirectional crosstalk between neural networks and oncology. In order to deconstruct the relationship between primary central nervous system malignancies and brain metastasis, this review systematically studies the specific mechanisms of neuron tumor synaptic communication, paracrine signaling, and neuroimmune interactions. Innovatively, autophagy is considered as a regulatory bridge connecting neuronal activity and tumor behavior. When functioning in neurons, autophagic flux determines synaptic plasticity and regulates neurotransmitter turnover. Inside tumor cells, autophagy may lead to uncontrolled proliferation and help evade immune surveillance. Integrating these molecular observations establishes the neuro-autophagy-tumor axis, and interconnected nodes in this axis provide emerging therapeutic strategies that target neural signaling and autophagy. These insights highlight the importance of incorporating neurobiological context into cancer research and position autophagy as a promising target for disrupting neural regulation in cancer therapy.
Keywords: autophagy, cancer neuroscience, cancer therapy, neurotransmitter, neurotrophin
Introduction
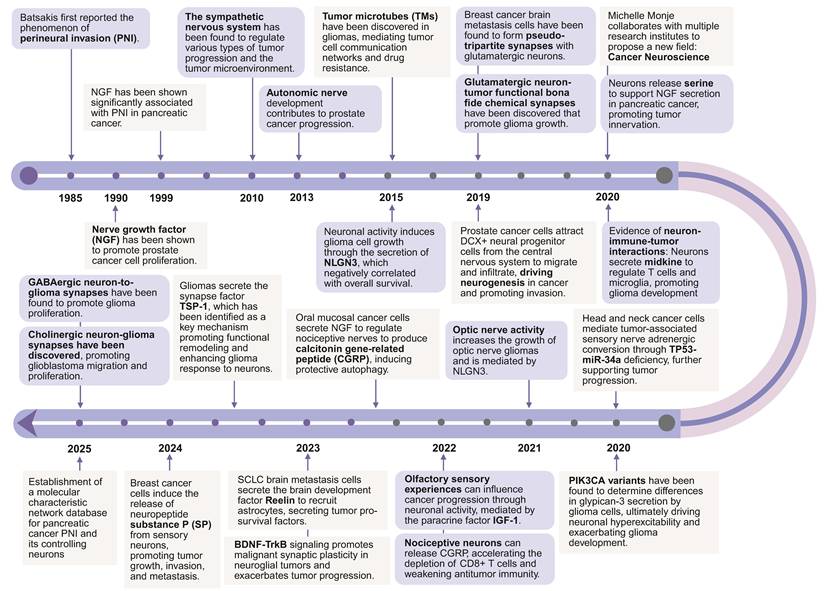
Epidemiological studies show an increase in the occurrence of tumors in the nervous system, particularly primary central nervous system (CNS) tumors such as glioblastoma and brain metastases [1]. These tumors generally lead to a poor prognosis due to their aggressiveness, resistance to treatment, and the complexity of the neuroanatomical environment [2]. Effectively, researchers are increasingly recognizing that chronic psychological stress, depression, and anxiety mediated by neuronal dysfunction may be risk factors that exacerbate tumorigenesis and progression [3]. Similarly, growing evidence indicates the nervous system plays an active role in promoting tumor oncology. The concept of cancer neuroscience was proposed, and related fields have grown rapidly in recent years [4,5]. One of the most striking findings is the discovery of neuron-to-tumor synaptic communication, wherein glioma cells form functional synapses with neurons [5]. These synapses directly transmit excitatory electrical signals that enhance tumor cell proliferation and invasion. Similar phenomena have also been observed in brain metastases (Figure 1).
A brief history of cancer neuroscience. Key discoveries in cancer neuroscience have been summarized.
Neurons influence tumor progression through paracrine signaling. Neurons release neurotransmitters and neurotrophic factors. These factors bind to corresponding receptors expressed on tumor cells and activate oncogenic pathways. The nervous system also regulates immune responses within the tumor microenvironment (TME), contributing to immune escape and creating a niche favorable to tumor persistence [6]. This neuro-immune-tumor axis is now recognized as a key driver of the failure of immunotherapies in neurological and systemic malignancies. Our current approaches to treating cancer might be missing a crucial aspect the role of the nervous system. Traditional therapies like chemotherapy and radiotherapy often cause neurotoxicity, namely chemotherapy-related cognitive impairment and radiation-induced brain injury (RIBI) [7,8]. The nervous system may adapt to these treatments by activating pro-tumorigenic circuits [9,10]. These observations indicate the crucial need to incorporate neural regulation into cancer therapy.
This intracellular degradation machinery strictly governs the complex physical interactions that link neural tissues with oncological lesions. When operating inside healthy neural networks, the autophagic flux dictates how synaptic junctions remodel themselves and actively calibrates the exact pool of available neurotransmitters, ultimately preventing the structural collapse of the neuron [11,12]. Once hijacked by a malignant clone, this precise regulatory node forces the cell to navigate a biological fork: the degradative machinery either actively dismantles the tumor via programmed execution or metabolically shields the neoplasm to guarantee its survival [13].
Because it controls the rate at which presynaptic terminals release neurochemicals and how postsynaptic membranes recycle their receptors, this degradative network directly intercepts the cross-talk between nerves and cancer cells. This hijacked flux simultaneously dictates how invading macrophages polarize and how these immune sentinels present antigens when exposed to local inflammatory stress [14,15]. High-resolution cellular profiling maps this dependency directly to invasive cellular architectures. Autophagic cascades physically assemble tumor microtubes (TMs) and tunneling nanotubes (TNTs), delicate membrane extensions that bridge distant cells and render the interconnected tumor network profoundly resistant to cytotoxic drugs [16]. Utilizing these exact pathways, the expanding lesion actively recruits new neural fibers and physically tracks along existing perineural tracts (PNI), permanently forcing the surrounding neural architecture to supply the electrical inputs required for its own pathogenesis [17].
This review describes how the brain affects tumors. It looks at the main ways the nervous system influences both new brain tumors and those that have spread there. We also discuss the remodeling effects of tumors and their treatments on the nervous system. We summarize current drug strategies targeting the neuro-tumor axis. Importantly, this review extends beyond current cancer neuroscience frameworks by providing an in-depth exploration of autophagy as a critical regulatory bridge within these processes. We propose that autophagy acts as both mediator and effector in neural-tumor crosstalk, influencing tumor progression, immune evasion, and therapeutic outcomes. By integrating recent discoveries in neuroscience, tumor oncology, and autophagy research, this review provides a comprehensive framework for understanding the neuro-autophagy-tumor axis. It further outlines potential therapeutic opportunities, including the repositioning of neural signaling inhibitors, autophagy modulators, and dual-targeting agents to disrupt neuro-tumor interactions.
1. Physiological roles of autophagy in cellular and nervous system homeostasis
Autophagy is a conserved lysosome-dependent degradation pathway essential for cellular homeostasis, with particular importance in the nervous system. It comprises three major forms as macroautophagy, chaperone-mediated autophagy (CMA), and microautophagy, which collectively mediate the clearance of misfolded proteins, damaged organelles, and pathogenic aggregates. Neurons are in a postmitotic state and face a demand for metabolic resources that remains high across a long lifespan. These cells require autophagic flux to maintain the structural integrity of their networks (Figure 2).
Physiological function of autophagy in the nervous system. Autophagy, including macroautophagy, chaperone-mediated autophagy, and microautophagy, plays essential roles in maintaining neuronal homeostasis. It supports organelle quality control, regulates synaptic development and plasticity, coordinates neuron–glia metabolic crosstalk, and buffers proteotoxic stress.
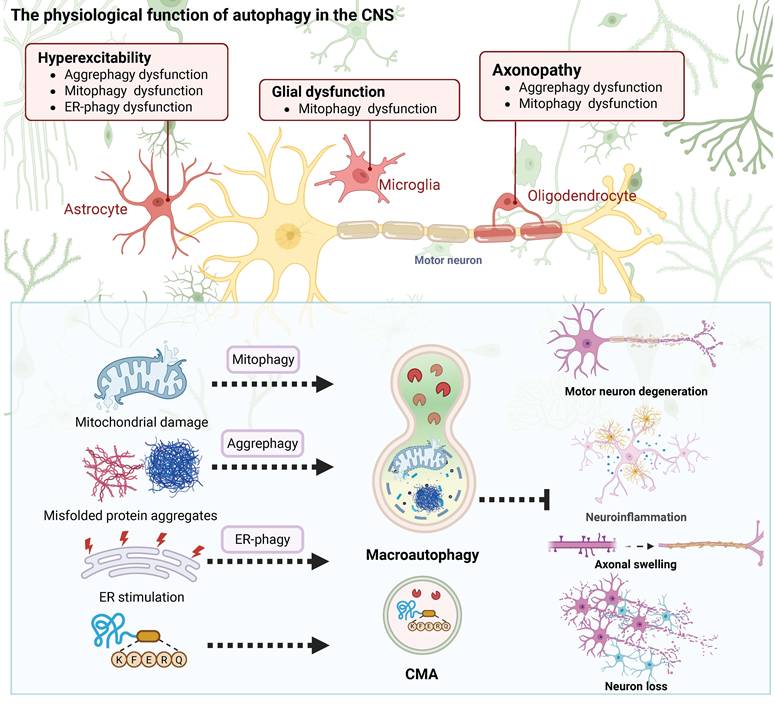
Autophagy works as a recycling system, balancing biosynthesis and degradation in response to nutrients and stress. Macro-autophagy involves autophagosome formation and lysosomal degradation orchestrated by conserved ATG proteins. When specific components need to be cleared, receptors like Parkin and p62/SQSTM1 identify the targets. This process, called selective autophagy, then removes damaged mitochondria, parts of the endoplasmic reticulum, and clumps of proteins [18]. CMA mediates HSC70-dependent lysosomal translocation of KFERQ motif-containing substrates via LAMP2A, whereas microautophagy contributes to basal turnover through direct lysosomal membrane invagination [19-21].
As neural tissue matures, its cleanup system does more than remove waste. It directly controls how neural networks survive and talk to each other. Neurons cannot divide again and need a lot of energy to work. For this reason, they completely depend on a constant, active cleanup process. This essential cleanup happens most importantly inside long axons and complex dendrites. There, it maintains healthy proteins and removes damaged cell parts. During brain development, this same cleanup process shapes the brain's ability to change and adapt. It removes unused synapses and manages the movement of neurotransmitter receptors and synaptic vesicles in established neural pathways. This activity affects learning, memory, and the overall stability of neural networks [22,23]. Glial cells also use autophagy to help maintain balance in the nervous system. For example, astrocytes rely on it to clear away excess proteins and waste from outside cells. Microglia need autophagy to control inflammatory signals and prevent excessive brain inflammation. Oligodendrocytes require this process to maintain the protective myelin sheath and manage their membrane proteins [24]. Autophagy often links to several brain disorders. Problems with this process are seen in conditions that involve poor synaptic function and abnormal myelin, including some neurodevelopmental and psychiatric illnesses [25,26]. Impaired autophagy, which often occurs with aging, is a common feature in major neurodegenerative diseases [27,28]. Its roles in synaptic plasticity, glial-neuronal crosstalk, and circadian coordination underscore the pathway’s complexity and therapeutic potential [29].
2. Autophagy in tumor biology
2.1 Autophagy in tumors of the nervous system
In primary nervous system tumors, autophagy exerts context-dependent dual functions. In glioblastoma and medulloblastoma, enhanced autophagic flux can sustain stemness, tumorigenicity, radio- or chemoresistance, and immune evasion, partly through pathways such as MST4-ATG4B signaling, AMBRA1-STAT3 activation, and autophagy-associated macrophage polarization. Conversely, under specific molecular contexts, autophagy may suppress tumor growth as shown in SPARC-overexpressing medulloblastoma, dopamine receptor D5 (DRD5)-activated pituitary adenoma, and CaMKII–Beclin1-regulated neuroblastoma. Thus, autophagy in primary nervous system tumors should be viewed as a dynamic and tumor-specific regulatory process, providing opportunities either to inhibit cytoprotective autophagy or to exploit pro-death autophagy for therapeutic benefit.
2.1.1 Glioblastoma
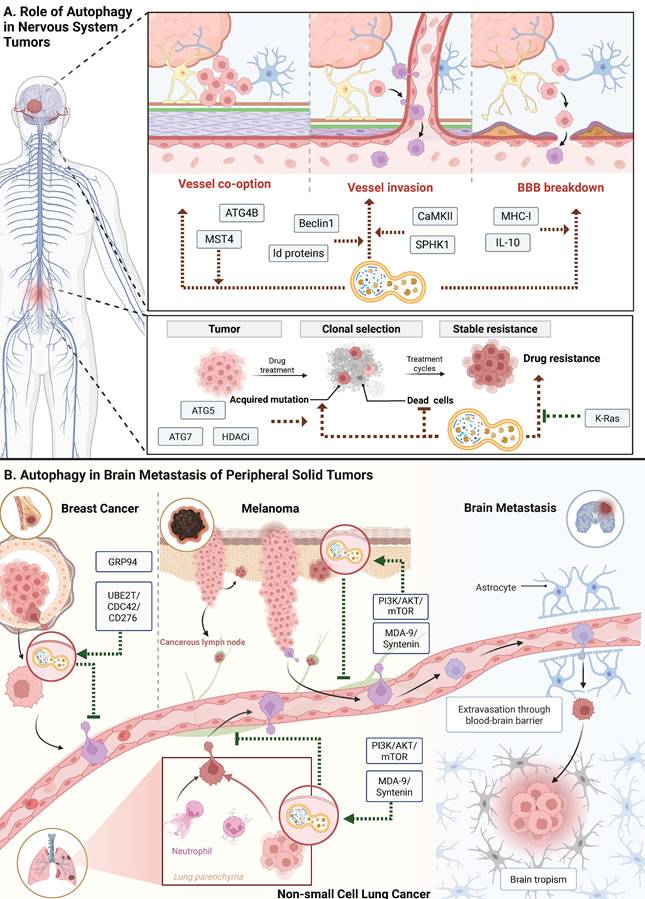
Autophagy plays dual roles in glioblastoma progression and therapy resistance through tumor-intrinsic survival pathways and immune microenvironment remodeling [30,31]. Specifically, the kinase MST4 phosphorylates ATG4B at Ser383, thereby amplifying autophagic flux. This MST4-ATG4B phosphorylation axis sustains the stemness of glioblastoma cells and protects them from radiation [32]. Simultaneously, glioblastoma-derived autophagy promotes an immunosuppressive microenvironment by releasing metabolites and cytokines that drive macrophage M2 polarization. This phenotypic shift, which impairs T-cell infiltration and downregulates the expression of MHC-I, allows the tumor to evade immune surveillance [33] (Figure 3A).
Pathogenic mechanism of autophagy in primary tumors and brain metastases. A. Role of Autophagy in Nervous System Tumors. Autophagy plays a key role in the occurrence and progression of central nervous system tumors, involving processes such as co-option of blood vessels, vessel invasion, and disruption of the blood-brain barrier. In addition, autophagy helps tumor cells acquire drug tolerance by regulating clonal selection and cellular stress response. B. Autophagy in Brain Metastasis of Peripheral Solid Tumors. Autophagy supports brain metastasis of peripheral solid tumors such as breast cancer, melanoma, and lung cancer by promoting tumor cell extravasation across the BBB and survival within the brain microenvironment. It modulates multiple signaling pathways that enable metastatic cells to adapt to the neural niche.
2.1.2 Medulloblastoma
In SPARC-overexpressing medulloblastoma cells, enhanced autophagic flux promotes lysosomal membrane permeabilization and caspase-dependent apoptosis, sensitizing tumor cells to stress-induced death [34]. In contrast, the autophagy regulator AMBRA1 sustains cancer stem cells by coordinating pro-survival autophagy with STAT3 signaling, thereby promoting self-renewal and chemoresistance [35,36]. Subtype-specific strategies that either exploit the apoptosis driven by SPARC or block the survival pathways supported by AMBRA1 may enhance therapeutic efficacy.
2.1.3 Pituitary adenoma
For pituitary adenomas, autophagy exerts context-dependent effects governed by mTOR signaling and dopamine receptor activation. When these tumors lack the protein DEPTOR, mTORC1 hyperactivates and suppresses autophagy. This suppression drives uncontrolled proliferation and causes resistance to dopamine agonists. Restoring DEPTOR expression inhibits mTORC1 and induces cytoprotective autophagy, resulting in cell cycle arrest and enhanced drug sensitivity [37]. When DRD5 is activated, it induces too much autophagy through the AMPK and mTOR pathways, thereby inducing autophagic cell death through lysosomal membrane permeabilization and cathepsin-dependent caspase activation. This destructive form of autophagy seems to target only tumor cells, leaving normal pituitary cells unharmed [38]. These results show that adjusting autophagy levels can be a way to treat cancer. Controlling this process can either stop tumors from growing or trigger their death, which supports using combination therapies that take advantage of these two different effects [39,40].
2.1.4 Neuroblastoma
Problems in autophagy genes like LC3B2 and ATG4A can disrupt the fusion of autophagosomes with lysosomes. This damage may make cells less able to handle stress from cancer-causing viruses. The resulting weakness promotes genetic instability and speeds up tumor growth [41]. The kinase CaMKII adds a phosphate group to the protein Beclin1. This specific change significantly increases the cell's baseline level of autophagy. The boosted autophagy system then captures Id proteins and breaks them down. Removing these Id proteins disrupts the MYCN signaling pathway. As a result, the aggressive neuroblastoma cells stop dividing and change into mature, specialized neurons [42]. In differentiated SH-SY5Y cells, glycolysis sustains autophagy induced by 4-hydroxynonenal, buffering oxidative stress and limiting apoptosis. But if researchers restrict this glucose metabolism, the autophagic defense collapses and shifts cells toward caspase-dependent death [43]. Additionally, sphingolipid signaling exhibits marked cell-type specificity. While sphingosine kinase 1 promotes autophagic clearance of damaged organelles in neurons, it sustains cytoprotective autophagy by suppressing ceramide accumulation and apoptotic signaling in neuroblastoma cells [44].
2.2 Autophagy in brain metastasis of solid tumors
When solid tumors metastasize to the brain, disseminating cells activate autophagy to survive the severe metabolic and oxidative stresses that colonizing this new niche entails. By conferring resistance to anoikis and nutrient deprivation, autophagic flux helps tumor cells cross the blood-brain barrier (BBB). Following brain entry, the metastatic cells rely on autophagy to control mitochondrial quality and maintain metabolic flexibility, thereby promoting long-term adaptation to the nutrient-limited cerebral microenvironment. These conserved mechanisms render autophagy a targetable vulnerability for brain metastatic tumors (Figure 3B).
2.2.1 NSCLC
Autophagy plays a context-dependent role in the formation and progression of brain metastases from non-small cell lung cancer (NSCLC), functioning both as an adaptive survival mechanism and a therapeutic vulnerability. Disruption of mitochondrial metabolism using mitochondria-targeted lonidamine inhibits electron transport chain complex I, leading to ATP depletion and activation of the AMPK/mTOR axis. This metabolic stress induces excessive mitophagy, which paradoxically results in mitochondrial loss and energy collapse, thereby impairing NSCLC cell survival and metastatic colonization in the brain [45]. In contrast, genetic evidence from patients carrying the ATG16L1 Thr300Ala polymorphism indicates that reduced autophagic capacity is associated with a lower incidence of brain metastasis. Impaired autophagy limits the ability of circulating tumor cells to withstand oxidative and metabolic stress during dissemination and reduces secretion of pro-metastatic factors, including matrix metalloproteinases, thereby restricting BBB penetration [46]. Collectively, these studies indicate that autophagy supports metabolic adaptation during NSCLC brain metastasis, and that pharmacological or genetic inhibition of autophagic pathways may provide a potential strategy to limit metastatic progression.
2.2.2 Breast cancer
Activation of lysosomal autophagy through the UBE2T/CDC42/CD276 axis enhances matrix metalloproteinase secretion, facilitating degradation of BBB components and enabling tumor cell extravasation [47]. Additionally, molecular CMA, regulated by proteins such as GRP94, contributes to the stabilization of Snail transcription factors, promoting epithelial-mesenchymal transition and enhancing metastatic potential and resistance to microenvironmental stress conditions, including hypoxia and therapeutic interventions [48]. In parallel, CMA, regulated by GRP94, stabilizes the transcription factor Snail, thereby promoting epithelial-mesenchymal transition and enhancing metastatic potential as well as resistance to hypoxia and therapeutic stress [49]. The resident astrocytes can secrete KISS1 to activate autophagy in metastatic breast cancer cells, supporting metabolic flexibility and survival. Drug-based strategies can block cancer spread. Pharmacologists can use drugs that completely block lysosomes. They can also develop inhibitors that target specific proteins like CD276, GRP94, or the specific CMA recycling pathway [50,51].
2.2.3 Melanoma
To colonize the brain, metastatic melanoma cells activate autophagy. This process helps them resist cell death and cope with the brain’s oxidative environment. The switch is controlled by MDA-9/Syntenin, which stabilizes the pro-survival PI3K/AKT/mTOR pathway [52]. The tumor’s need for continuous autophagy creates a therapeutic opportunity. The compound trifluoperazine exploits this by blocking the fusion of autophagosomes with lysosomes. Autophagosomes accumulate, trapping waste and crippling the cell’s metabolism. This effect is selective, killing the melanoma cells without harming normal brain cells [53].
3. Neurological influences on tumors
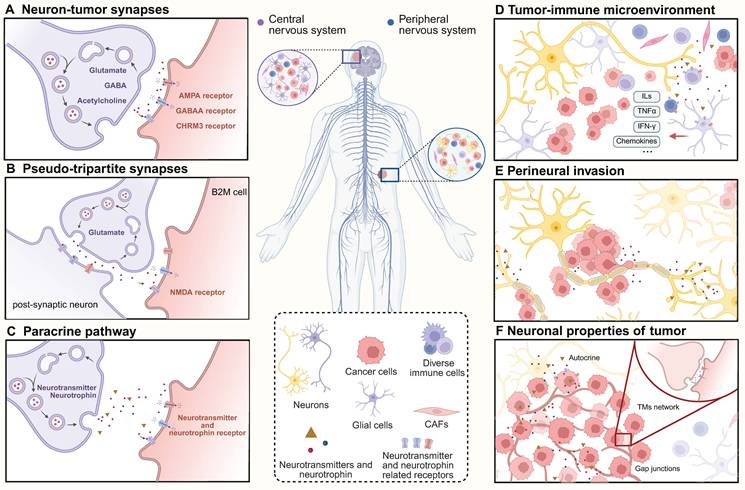
The nervous system is mainly made of neurons and glial cells. In the tumor microenvironment, these glial cells are important. They talk directly to cancer cells. Chronic stress, anxiety, and depression are known to be risk factors for cancer getting worse. Nervous system activity is associated with carcinogenesis and can significantly influence tumor development and metastasis (Figure 4) (Table 1). Autophagy acts as the exact molecular bridge linking neuronal firing to tumor expansion.
Neural inputs drive tumor progression. A. Neurons and tumor cells form real synaptic structures and regulate tumor progression through neurotransmitters such as glutamate, GABA, and acetylcholine. B. Tumor cells recruit at neuronal synaptic structures and form pseudo-tripartite synapses to receive neurotransmitter regulation. C. Neurons secrete factors into the tumor microenvironment, modulating tumor cell activity through paracrine signaling. D. Neuronal activity induces alterations in immune cells and remodels the immune microenvironment, impairing tumor immune responses. E. Peripheral neurons secrete factors that drive tumor cells growing along nerves and provide more accessible metastasis pathways. F. Tumor cells utilize TMs to form communication networks and accelerate progression via autocrine neurotransmitters and neurotrophins.
The key signaling pathways in neuro-tumor interactions
| Pathway/Target | Classification | Mechanism | Disease | Ref. |
|---|---|---|---|---|
| Glutamatergic signaling pathway-AMPA/NMDA | synaptic communication | Neurons form functional bona fide chemical synapses with glioma cells or “pseudo-tripartite” structures with B2BM cell, and release glutamate, glioma/B2BM cells receive excitatory neural signals through AMPA/ NMDA receptors that promote tumor proliferation and invasion | Glioma, Breast cancer | [60,64] |
| Paracrine signaling | 1. Para-neuronal secretion of glutamate into the tumor microenvironment promotes tumor proliferation and invasion 2. Gliomas secrete massive excitatory neurotransmitter glutamate, which over-activates AMPA and NMDA receptors in neuronal cells, causing seizure disorders and leading to neuronal death | Glioma, PDAC | [60] | |
| GABAergic signaling pathways-GABAA | synaptic communication | In neuron-glioma synapses, neurons produce GABA, depolarizing glioma cells, leading to elevated intracellular chloride ion concentrations and increasing glioma proliferation | Glioma | [61] |
| Paracrine signaling | 1. Para-neuronal secretion of GABA into the tumor microenvironment promotes tumor proliferation and invasion 2. Elevated glutamate concentration in the tumor microenvironment decreases KCC2 expression in peritumoral neurons, leading to GABA receptor activation of chloride efflux and functional excitation of neurons | Glioma, Breast cancer, Colorectal cancer | [65] | |
| Cholinergic signaling pathways-CHRMs | synaptic communication | Neurons form cholinergic synapses with glioma, and glioblastoma receives acetylcholine signaling through CHRM3 receptor, promoting tumor proliferation and migration | Glioma | [62] |
| Paracrine signaling | 1. Enhancing Wnt and Notch signaling pathway activity to enhance tumor growth and metastasis through CHRM3 2.Inhibiting downstream MAPK/EGFR and PI3K/AKT pathways in PDAC cells through CHRM1 | Prostate cancer, Gastric cancer, Pancreatic cancer | [87,88] | |
| Immunity modulation | Suppressing CD8+ cell infiltration and decreasing the Th1/Th2 ratio, leading to a suppressive immune microenvironment | PDAC | [125] | |
| Adrenergic signaling pathway | Paracrine signaling | Sympathetic nerves release catecholamines, promoting tumor-cell proliferation, invasion, migration, angiogenesis, and resistance to cell death | Prostate cancer, Gastric cancer, Pancreatic cancer, Breast cancer, NSCLC, Colorectal cancer | [83] |
| Immunity modulation | 1. Agonizing astrocytes β2-AR, regulating TNFα and downstream inflammatory genes 2. Activating T cell β2-AR, leading to its depletion, impairing anti-tumor immune response 3. Activating MDSCβ2-AR, thus activating STATs signaling to regulate the expression of immunosuppressive factors and altering the proliferative capacity of T cells; reprogramming MDSC metabolism, leading to immunosuppression 4. Inhibiting cytotoxicity and cytokine production by NK cells | Colorectal cancer, Melanoma | [111,124] | |
| Dopamine signaling pathway | Paracrine signaling | Activating dopamine receptor D2 on tumor cells to inhibit tumor growth | Glioma, Breast cancer, Lung cancer, Pancreatic cancer | [224] |
| Immunity modulation | 1. Antagonizing dopamine receptor D2, affecting the immune metabolism of TAMs and leading to a pro-inflammatory tumor microenvironment 2. Activating NK cell via D1-like receptors or inhibiting it by D2-like receptors | Glioma | [108] | |
| MENK | Paracrine signaling | MENK binds opioid receptors on tumor cells and relocates NFAT1 to the nucleus, ultimately leading to apoptosis | Glioma, Melanoma, Colorectal cancer, Ovarian cancer | [74] |
| Immunity modulation | Remodeling the tumor immune microenvironment, increasing the infiltration of macrophages, NK cells, CD8+ T cells, CD4+ T cells, and other immune cells, which is conducive to overcoming tumor immune escape and immune resistance | NSCLC, Colon cancer | [130,131] | |
| BDNF-TrkB | synaptic communication | Promotes NGS formation and the transport of AMPA receptors on the surface of glioma cells, thereby amplifying glutamate-induced currents in malignant cells and promoting glioma growth | Glioma | [75] |
| Paracrine signaling | Neurons or tumors secrete BNDF, binding to TrkB receptors to activate downstream signaling pathways and promote tumor proliferation and invasion | Breast cancer, Retinoblastoma, Oral squamous cell carcinoma, Melanoma | [225] | |
| Immunity modulation | 1. Reducing microglia/macrophage infiltration and CD68 immunoreactivity, which in turn reduces glioma migration 2. Stimulating IL-15 production by microglia, shifting myeloid cells to an antitumor state 3. Re-educating TAM, developing an immunosuppressive microenvironment | Glioma, Prostate cancer, Lung cancer | [109,110] | |
| NLGN3 | synaptic communication | Promoting neuron-glioma synapse formation | Glioma | [77] |
| Paracrine signaling | Activating PI3K-mTOR pathway activity in glioma cells and driving their proliferation, and inducing cancer stem cell characterization through autocrine | Glioma, Neuroblastoma | [226] | |
| IGF1 | Paracrine signaling | TuAstrocytes and olfactory receptor neurons indirectly or directly secrete IGF1 to promote tumor proliferation and progression | Glioma | [80] |
| LCN2 | Paracrine signaling | Promoting tumor progression via STAT3 pathway | Medulloblastoma | [81] |
| Reelin | Paracrine signaling | SCLC secretes reelin to recruit reactive astrocytes, the latter promoting SCLC growth by secreting neuronal pro-survival factors | Brain metastases from SCLC | [82] |
| GDNF- GFRα1 | Paracrine signaling | 1. Nerve-released GFRα1 enhances tumor nerve invasion via GDNF-RET signaling 2. Endoneurial macrophages secrete GDNF, activating RET to induce PNI | PDAC | [126] |
| ARTN | Paracrine signaling | ARTN promotes pancreatic cancer invasion and perineural dissemination | Pancreatic cancer | [92] |
| NCAM1 | Paracrine signaling | Direct contact between Schwann cells and cancer cells induces process formation and directed migration, a process that depends on NCAM1 in Schwann cells | Pancreatic cancer | [93] |
| NGF- TrkA | Paracrine signaling | 1. Tumor secretes NGF, recruiting nerves into the tumor microenvironment and hijacking nociceptive nerves to secrete CGRP to support tumor growth 2. Nerves secrete NGF, inducing the tumor to migrate along nerves 3. Binding to TrkA receptors on the tumor, activating the PI3K/AKT and MAPK signaling pathways to promote tumor survival, migration, and invasion | Pancreatic cancer, Gastric cancer, Oral mucosa carcinoma | [95] |
| Immunity modulation | Desensitizing the tumor to IFN-γ signaling, repelling T cells and NK cells, and suppressing T cell function | Melanoma | [127] | |
| substance P | Paracrine signaling | Neurons secrete SP, inducing tumor cell death and ssRNAs release, indirectly activating tumor metastasis expression | Breast cancer | [102] |
| Midkine-CCL4-CCL5 | Immunity modulation | Neurons secrete midkine, activating CD8+ T cells to produce CCL4, which in turn induces microglia/macrophages to produce CCL5, thus regulating LGG stem cell survival | Glioma | [107] |
| IL-6-STAT3 | Immunity modulation | Astrocytes secrete IL-6, which binds to IL-6R and activates STAT3 signaling, thereby promoting tumor-cell proliferation and migration | Glioma | [112] |
| NAA/NAT8L | Immunity modulation | Neurons secrete NAA, inhibiting NK cells and CD8+ T cells, promoting immune escape from brain cancer cells | Breast cancer | [114] |
| CGRP | Immunity modulation | Nociceptor neurons in sensory nerves secrete CGRP, weakening anti-tumor immunity by accelerating CD8+ T cell depletion | Melanoma | [116] |
| TSP-1 | synaptic communication | Glioma cells secrete TSP-1, promoting the synaptic connectivity of neurons and increasing glioblastoma proliferation | Glioma | [63] |
3.1 Direct neuron interaction
3.1.1 Neuron-to brain tumor synaptic communication
Tumors located within the central nervous system are continually bathed in neural activity. This interaction occurs through a process termed neuron-to-brain tumor synaptic communication, which involves both direct and indirect forms of synaptic signaling. Gliomas are the most common malignant primary brain tumors in adults, originating primarily from neural precursor cells (NPCs) or oligodendrocyte precursor cells (OPCs) [54,55]. Healthy OPCs form synapses with neurons. This natural interaction helps control their own development [56].
Glioma cells co-opt this same biological mechanism. They grow very thin, synapse-like structures called TMs. Using these microtubes, the cancer cells spread through healthy brain tissue. The microtubes also link glioma cells into a connected network. This network makes the tumor highly resistant to radiotherapy [57,58]. To maintain these invasive structures, glioma cells must keep their internal skeleton stable. They also need to continuously move mitochondria to the growing tips of the microtubes. Both of these vital processes rely completely on well-functioning autophagy.
Glioma cells express various glutamate receptor subunits and genes associated with postsynaptic components [59]. Approximately 10% of glioma cells form neuro-glioma synapses (NGSs), through which TMs establish functional, chemical synapses with neurons. These synapses generate unidirectional postsynaptic currents mediated by glutamatergic AMPA receptors, resulting in excitatory signals that depolarize glioma cells and enhance their proliferation [60]. Additionally, neuronal activity induces non-synaptic, activity-dependent potassium currents. These currents are amplified via gap junction-mediated communication among glioma cells, forming an electrically coupled network that further supports tumor growth and malignancy [59]. Cellular connectivity via gap junctions can be influenced by autophagy, which regulates the turnover of connexin proteins.
In addition to the well-characterized glutamatergic NGS, recent studies have identified functional GABAergic neuron-glioma synapses mediated by GABA receptors in diffuse midline gliomas (DMG). GABAergic input depolarizes DMG cells and promotes their proliferation, an effect that is further amplified by administration of the GABA-enhancing benzodiazepine lorazepam [61]. Another study demonstrated that cholinergic neurons can also form cholinergic neuron-glioma synapses with glioblastoma cells, thus facilitating their migration and proliferation, and inhibition of CHRM3 receptors reduces tumor progression [62]. Collectively, these findings suggest that glioma cells can hijack neuronal signaling, enhancing their survival and malignancy by increasing synaptic strength and synapse number. Such remodeling of neural circuits also contributes to cognitive impairment in affected individuals [63]. At physiological synapse, autophagy directly controls how neurotransmitter receptors turn over and how vesicles recycle. When experimental models lack functional autophagic networks, the intracellular trafficking of GABA receptors completely stalls.
When cancer spreads from organs like the breast or lung to the brain, the new tumors also use the brain's own wiring to survive. Breast-to-brain metastases (B2BM) own a unique "pseudo-tripartite" architecture. Rather than constructing de novo synapses, the invading breast cancer cells position themselves around existing connections between healthy neurons. When these normal neurons fire, they release the neurotransmitter glutamate. The glutamate then activates NMDA receptors on the surface of the nearby cancer cells. This stolen signal drives the tumor cells to grow and invade [64]. If the genes for these NMDA receptors are turned off, the cancer cells lose their ability to establish themselves in the brain tissue.
These metastatic cancer cells also develop features of GABA-releasing neurons. They make the same receptors and transporters that normal inhibitory brain cells use. This lets the cancer cells pick up extra survival signals from the surrounding brain network [65]. SCLC in the brain uses a similar strategy. Researchers can use light to activate nearby brain neurons. When this happens, the neighboring cancer cells show an immediate electrical change. This shared electrical activity directly fuels the tumor's spread [66].
3.1.2 Neuron-to extracerebral tumor synaptic-like communication
The PNS has also been implicated in cancer oncology. SCLC cells at the primary site look and act like nerve cells very early on and can generate electrical signals [67]. These cells are believed to originate from neuroendocrine cells [68]. These aggressive cancer cells can even grow long, thin extensions that look like axons, a process controlled by genes involved in nerve development. This lets them move more easily and spread to other parts of the body [69]. Surgical interventions provide the ultimate proof of this anatomical dependency. Cutting the vagus nerve can stop the growth and spread of these lung tumors [66].
Besides these structures, cells can also connect through TNTs. These tiny tubes let cells share many things, from proteins and RNA to entire organelles, and are important in both health and disease [70,71]. In the nervous system, TNTs facilitate communication between neurons and between neurons and glial cells, contributing to neurodevelopment and neurological disorders. In tumor cells, TNTs have been strongly associated with enhanced invasiveness and resistance to radiotherapy [72]. Whether TNTs can serve as a communication bridge between neurons and tumor cells remains an important question for future investigation. Interestingly, the formation and function of TNTs are closely linked to autophagy-related processes, as they share molecular machinery for membrane trafficking and can transfer autophagosomes, potentially serving as a conduit for autophagic cargo between cells.
3.2 Paracrine neuro-tumor signaling pathways
3.2.1 Paracrine secretion in the central nervous system
In the CNS, neuronal activity leads to the release of a variety of neurotransmitters and neurotrophins into the TME, which exert diverse regulatory effects on tumor cells. As previously described, tumor cells can directly receive neurotransmitters via synaptic connections, and they also express neurotransmitter receptors that respond to signals present in the surrounding environment. For instance, glutamatergic neurons can promote glioma proliferation and invasion not only through direct synaptic transmission but also via paracrine glutamate signaling [60]. Similarly, cholinergic signaling through CHRM3 receptors and GABAergic signaling via GABA receptors have both been implicated in promoting tumor cell proliferation [62,65]. Autophagic networks govern these exact physiological processes by dictating how neurons synthesize transmitters and recycle their vesicular pools.
The neuropeptide methionine enkephalin (MENK) can stop tumor growth. In gliomas, MENK binds to opioid receptors on the cancer cell surface. This binding causes the transcription factor NFAT1 to move into the nucleus, which then triggers cell death [73]. This tumor-suppressive effect of MENK is also seen in cancers outside the brain, like melanoma and colon cancer [74]. BDNF enhances the excitability and growth of glioma cells by increasing surface AMPA receptors. Genetically silencing or pharmacologically blocking the specific BDNF receptor TRKB inhibits this process [75]. BDNF and GRP78 can also work together to stimulate tumor growth [75,76]. Interestingly, autophagy helps regulate BDNF levels, and BDNF signaling can in turn affect autophagy, creating a two-way relationship.
Another neural factor, neuroligin-3 (NLGN3), also promotes cancer. When released, NLGN3 binds to glioma cells and over-activates the pro-growth PI3K-mTOR pathway [76]. Furthermore, NLGN3 plays a crucial role in the establishment of NGSs [77]. Its release from neurons is mediated by the sheddase ADAM10, and inhibition of ADAM10 or knockdown of NLGN3 significantly reduces glioma growth [78].
Not all paracrine interactions are limited to neuron-tumor communication. Within the TME of medulloblastoma, tumor granule neuron precursors can transdifferentiate into astrocyte-like cells, termed TuAstrocytes, which secrete interleukin-4 (IL-4) to activate microglia.
Once activated by the local microenvironment, these microglial sentinels release insulin-like growth factor 1 (IGF1). Rather than acting as a simple one-way signal, this specific secretion locks the immune cells and the malignant clones into a self-sustaining paracrine feedback loop, which continuously drives the expansion of the tumor [79]. The cellular origin of IGF1 dictates its specific role in oncogenesis. In a different setting, neurons can also release IGF1 to start tumors. For instance, stimulating the sense of smell causes neurons to secrete IGF1, which can trigger glioma formation. This process relies on IGF1. When scientists reduce IGF1 levels in the brain's oligodendrocyte precursor cells, these cells stop multiplying, and tumor growth is blocked [80]. Tumor-associated astrocytes fuel tumor growth. These astrocytes, reprogrammed by the TME and often found at the tumor edge, work by secreting lipocalin-2 (LCN2) and activating the STAT3 signaling pathway [81]. Inhibiting LCN2 expression or blocking STAT3 signaling suppresses tumor growth, particularly in the Sonic Hedgehog subtype of medulloblastoma.
In brain metastases from SCLC, tumor cells secrete Reelin, a developmental cue that recruits astrocytes to the metastatic niche. These recruited astrocytes then secrete neuronal survival factors such as SERPINE1, which enhance SCLC proliferation and survival [82].
3.2.2 Paracrine secretion in the peripheral nervous system
Within the PNS, the autonomic nervous system regulates the function of internal organs through the sympathetic (adrenergic) and parasympathetic (cholinergic) divisions. The sympathetic nervous system utilizes neurotransmitters such as epinephrine, norepinephrine, and dopamine, whereas the parasympathetic nervous system primarily relies on acetylcholine.
Neurotransmitters released from peripheral neurons influence cancer oncology. β-adrenergic signaling, in particular, has been shown to promote tumor cell proliferation, invasion, migration, angiogenesis, and resistance to cell death. These effects are largely mediated by activation of the cAMP/PKA and MAPK signaling pathways [83]. Recent studies have revealed that adrenergic activity also accelerates colorectal cancer (CRC) progression via ADRA2A/Gi-mediated activation of Yes-associated protein [84]. Furthermore, both ablation of adrenergic nerves and knockdown of adrenergic receptors significantly suppress prostate cancer growth and metastasis [85]. Similarly, blockade of β-adrenoceptors (β-AR) has been shown to inhibit metastasis in breast cancer [86].
Cholinergic signaling has also been implicated in cancer progression. In gastric cancer, vagotomy reduces the activity of Wnt and Notch signaling pathways, consistent with findings that cholinergic input enhances Wnt signaling and promotes tumorigenesis [87]. Cholinergic pathways similarly contribute to prostate cancer metastasis. However, these effects are context-dependent; for example, in pancreatic cancer, cholinergic signaling appears to suppress tumor growth, suggesting that neuronal subtypes may exert cancer-specific effects depending on the tumor type [88].
Substantial evidence indicates that N-methyl-D-aspartate receptors (NMDARs) are expressed in cancer cells outside the CNS, responding to glutamate released via autocrine and paracrine signaling [89]. In pancreatic ductal adenocarcinoma (PDAC), glutamate from neurons induces calcium influx via NMDARs, activating the Ca2+-dependent CaMKII/ERK-MAPK pathway and enhancing glycolysis to promote PNI [90]. Additionally, cancer cells adapt to oxidative stress and support rapid growth by upregulating glutamine metabolism through glutamate/glutamine interconversion. Importantly, the abovementioned β-adrenergic, cholinergic, and calcium signaling pathways are directly associated with the induction of autophagy in various types of cancer, positioning autophagy as a key mediator of tumor progression mediated by these pathways.
The PNS also secretes a variety of neurotrophic factors. Beyond their roles in axonogenesis and neural recruitment in the CNS, neurotrophins play critical roles in tumor progression. PNI, the invasion of tumor cells into and along nerve fibers, is associated with increased tumor aggressiveness and poor prognosis. Nerve fibers serve as low-resistance conduits for migration, while neurotrophins and chemokines secreted by neurons act as chemoattractants that facilitate PNI. For instance, glial cell line-derived neurotrophic factor (GDNF), released by nerves, attracts cancer cells via activation of RET receptors. The RET co-receptor GFRα1, expressed by cancer cells and secreted by nerve cells, enhances this signaling. Even in the absence of GFRα1 expression in cancer cells, nerve-derived GFRα1 promotes PNI through GDNF-RET signaling [91]. Similarly, artemin, another neurotrophic factor, contributes to the neural invasion and dissemination of pancreatic cancer cells [92]. Schwann cells, key components of peripheral nerves, can also promote PNI by secreting NCAM1, facilitating tumor cell migration toward and into nerves [93].
NGF, which supports neuronal survival and differentiation, has been shown to induce tumor cells to migrate toward nerves and recruit neuronal elements into the TME [94]. NGF promotes tumor cell survival, migration, and invasion, especially in pancreatic and gastric cancers, via TrkA receptor activation and downstream PI3K/AKT and MAPK signaling [95]. Under nutrient deprivation, oral mucosal cancer cells secrete NGF, which stimulates nociceptors to release calcitonin gene-related peptide (CGRP). This peptide, in turn, enhances tumor cell growth [96]. BDNF, similar to its role in gliomas, promotes tumor invasion and metastasis in breast and ovarian cancers through activation of TrkB-mediated signaling pathways [97]. Interestingly, there is growing recognition of crosstalk between neurotransmitters and neurotrophic factors. β-AR activation enhances NGF secretion, axonogenesis, and tumor proliferation [98]. β2-adrenergic nerves also stimulate cancer-associated fibroblasts (CAFs) to express NGF, thereby promoting sympathetic innervation and driving CRC progression [84]. β-AR activation increases catecholamine secretion, and catecholamines have been shown to induce NGF production in PDAC cells [99,100]. Additionally, neuro-related cholinergic factors can stimulate NGF expression and promote gastric cancer development [87].
Peripheral neurons also release serine, which triggers metabolic crosstalk and increases NGF expression and secretion in PDAC, further enhancing tumor innervation and growth [101]. Furthermore, neurons release substance P (SP), a neuropeptide that indirectly supports breast cancer growth, invasion, and metastasis. SP acts on TACR1 to induce tumor cell death, releasing single-stranded RNAs that activate TLR7 in adjacent tumor cells, thereby driving metastatic gene expression [102].
Finally, nerve fibers contribute to PNI not only by providing structural pathways, but also by secreting matrix metalloproteinases (MMPs), fibronectin, vascular endothelial growth factor (VEGF), and other molecules that remodel the TME. PNI is also driven by tumor cell-derived factors, which will be discussed in the following section.
3.3 Neuro-immune regulation of the tumor microenvironment
In addition to the direct regulation of tumor cells by neuronal signals, immune cells constitute a critical component of the TME and secrete a variety of regulatory molecules that influence both tumor cells and neurons. Notably, there is significant crosstalk between the nervous and immune systems. Immune cells express receptors for various neurotransmitters, enabling them to be modulated by neuronal input. Conversely, immune cells can also synthesize and release neurotransmitters, acting in both paracrine and autocrine manners.
Norepinephrine is released into the tumor microenvironment and binds to specific receptors on immune cells. This interaction does not send a normal physiological signal. Instead, it suppresses the anti-tumor immune response. One key effect is that receptor activation increases the expression of the immune checkpoint protein PD-1. Concurrently, this signaling cascade, hijacking pathways normally reserved for acute stress responses, dictates the suppressive behavior of local macrophages and myeloid-derived suppressor cells (MDSCs). It reprograms T-cell metabolism, impairing their ability to activate effectively and sustain anti-tumor functions [103]. When dopamine engages D1-like receptors, the resulting biochemical cascade activates effector T lineages and aggressively amplifies the cytotoxicity of NK cells. Activation of D2-like receptors produces the opposite effect, suppressing natural killer cell activity and concurrently restraining the expansion of regulatory T cells (Tregs) [104]. The parasympathetic neurotransmitter acetylcholine, in contrast, primarily delivers anti-inflammatory signals that help modulate overall immune activity [105].
3.3.1 Neuro-immune crosstalk in the central nervous system
Within low-grade gliomas (LGGs) driven by neurofibromatosis type 1, local firing neurons actively synthesize and release the growth factor midkine. Intercepting this neural signal, adjacent CD8⁺ T cells undergo immediate activation, an acquired state that forces the lymphocytes to secrete the chemokine CCL4. Notably, CCL5 expression is negatively correlated with patient survival in LGG, providing direct evidence for the existence of a neuron–immune–tumor regulatory axis [106,107].
In gliomas, pharmacological inhibition of dopamine receptor D2 modulates TAM immune metabolism, leading to a pro-inflammatory TME [108]. Additionally, infusion of BDNF significantly reduces microglial/macrophage infiltration and CD68 expression, thereby inhibiting glioma migration [109]. BDNF also stimulates IL-15 production in microglia, which enhances IFN-γ secretion by NK cells and reprograms myeloid cells toward an anti-tumor phenotype [110].
Astrocytes, another key component of the CNS TME, express β₂-ARs. Activation of β₂-ARs modulates TNF-α expression and its downstream inflammatory gene network, influencing inflammatory homeostasis in the CNS [111]. Astrocytes facilitate glioma progression by secreting interleukin-6. This cytokine engages IL-6 receptors on tumor cells, triggering the STAT3 signaling pathway that drives proliferation and invasion [112].
In drug-resistant breast cancer brain metastases, elevated levels of the neuronal compound N-acetylaspartate (NAA) and its transporter NAT8L suppress the activity of key immune cells like NK and CD8⁺ T cells. Tumor cells appear to co-opt this native neuro-immune checkpoint to avoid elimination [113]. NAA-secreting neurons may facilitate immune evasion in both metastatic and primary CNS tumors [114].
3.3.2 Neuro-immune crosstalk in the peripheral nervous system
The immune composition of the PNS TME is diverse, primarily comprising TAMs, T cells, NK cells, MDSCs, and B cells. Previous studies have demonstrated that norepinephrine signaling, released upon activation of intestinal sympathetic neurons, enhances macrophage-mediated immune responses to bacterial infections, confirming neuro-immune communication between intestinal neurons and macrophages [115]. Sensory neurons, excluding autonomic nerves, release the neuropeptide CGRP, which impairs anti-tumor immunity by promoting CD8⁺ T cell depletion [116].
Drug repurposing research has shown that propranolol, a non-selective β1- and β2-adrenergic receptor blocker, inhibits CRC progression, likely through its role in blocking β₂-AR-mediated T cell depletion. Combination therapy with propranolol and the chemotherapeutic agent irinotecan exerts synergistically enhanced anti-tumor effects in CRC models [117]. Inhibition of β2-adrenergic stress signaling also improves the efficacy of radiotherapy [118].
Further studies have demonstrated that activation of the sympathetic nervous system enhances β₂-AR signaling in CD8+ T cells, impairing their anti-tumor activity and promoting tumor immune evasion. Pharmacological inhibition of β2-AR can augment the effectiveness of anti-PD-1 immunotherapy [119]. Moreover, chronic stress-induced adrenergic signaling disrupts T cell metabolism by depleting glycolytic and oxidative phosphorylation capacity, leading to immunosuppression in melanoma models [120].
Sympathetically released catecholamines activate β-adrenergic receptors on MDSCs within the TME, promoting the expression of immunosuppressive molecules such as arginase-1 and PD-L1 via STAT3 signaling, and enhancing their capacity to suppress T cell proliferation [121]. Additionally, β2-AR signaling reprograms MDSC metabolism by reducing glycolysis while increasing oxidative phosphorylation and fatty acid oxidation, thereby reinforcing immunosuppression [122]. Epinephrine and norepinephrine have also been shown to inhibit NK cell cytotoxicity and cytokine production [104].
Psychological stress further modulates tumor immunity. Sympathetic release of norepinephrine induces prostate tumor cells to secrete neuropeptide Y via β2-AR signaling, which promotes MDSC recruitment and IL-6 secretion. IL-6 subsequently activates STAT3 signaling and facilitates tumor progression [123,124]. Excessive activation of cholinergic pathways can inhibit CD8+ T cell infiltration and decrease the Th1/Th2 ratio, contributing to an immunosuppressive microenvironment [125]. The secretion of GDNF by perineurial macrophages is a key step that facilitates pancreatic cancer invasion. This neurotrophic factor acts by activating RET signaling pathways in the tumor cells [126].
Neuronal signaling molecules can influence cancer pathophysiology independently of neuronal structures. In the tumor-immune microenvironment, NGF signaling via TrkA receptors impairs IFN-γ signaling in melanoma cells, reduces T and NK cell infiltration, and suppresses T cell function [127]. Similarly, BDNF contributes to immunosuppressive TAM reprogramming in lung adenocarcinoma [128]. In prostate cancer, activation of leukemia inhibitory factor receptor signaling upregulates BDNF, promoting neuroendocrine differentiation and creating an immunosuppressive TME [129].
Other neurotransmitters also exert immunomodulatory effects. For instance, GABA suppresses T cell responses and infiltration in CRC, promoting a TAM-mediated immunosuppressive phenotype. Serotonin influences CD8+ T cell infiltration and modulates immune function [5]. MENK, a neuropeptide, acts as a crucial immunoregulatory bridge between the nervous and immune systems. In lung and colorectal cancers, MENK remodels the tumor immune microenvironment by increasing the infiltration of macrophages, NK cells, and CD4+ and CD8+ T cells, thereby countering immune escape and resistance [130,131]. Furthermore, nociceptor neurons in sensory nerves, except autonomic nerves, release a neuropeptide called CGRP, which impairs anti-tumor immunity by accelerating the depletion of CD8+ T cells [116]. It is worth noting that many of the immunomodulatory effects of these molecules on the TME are mediated by autophagy, such as increasing tumor immunogenicity through autophagy activation.
The latest research has also discovered that cancer cells under immune pressure stimulate transcription factor 4 and secrete slit guidance ligand 2, which later activate tumor-draining nociceptive neurons. This action not only exacerbates cancer-induced pain but also induces nociceptive neurons to secrete CGRP, contributing to the remodeling of tumor-draining lymph nodes into an immunosuppressive state [132].
Finally, the nervous system exerts systemic control over the immune response. It modulates immune cell trafficking through cholinergic, adrenergic, and sensory peptidergic signaling and possesses "immune memory" capabilities, whereby neurons reinitiate specific immune responses upon reactivation by prior immune stimuli.
3.4 Autophagy-related regulation of the neural-tumor axis
Given the central role of autophagy in neuronal and tumor biology, it has been proposed as a potential regulatory link in the neuro-tumor axis. Here, we discuss the potential mechanistic involvement of autophagy in paracrine neuro-tumor signaling pathways, neuro-immune regulation, and neural-tumor synapse formation (Figure 5). Based on these associations, the evidence supporting a role for autophagy in neuro-tumor interactions can be regarded as a proposed mechanism that deserves future experimental validation.
Autophagy maintains neuronal homeostasis while exerting context-dependent effects on tumor survival, stemness, metastasis, and therapy resistance. It also mediates neuro–tumor paracrine signaling by integrating neurotransmitter, neurotrophic, and stromal cues, and modulates immune responses through antigen presentation, immune-checkpoint regulation, DAMP release, and immunosuppressive mediators. The bottom panel illustrates a proposed mechanistic model in which autophagy may influence tumor microtube formation, synaptic plasticity, and neurotrophic signaling.
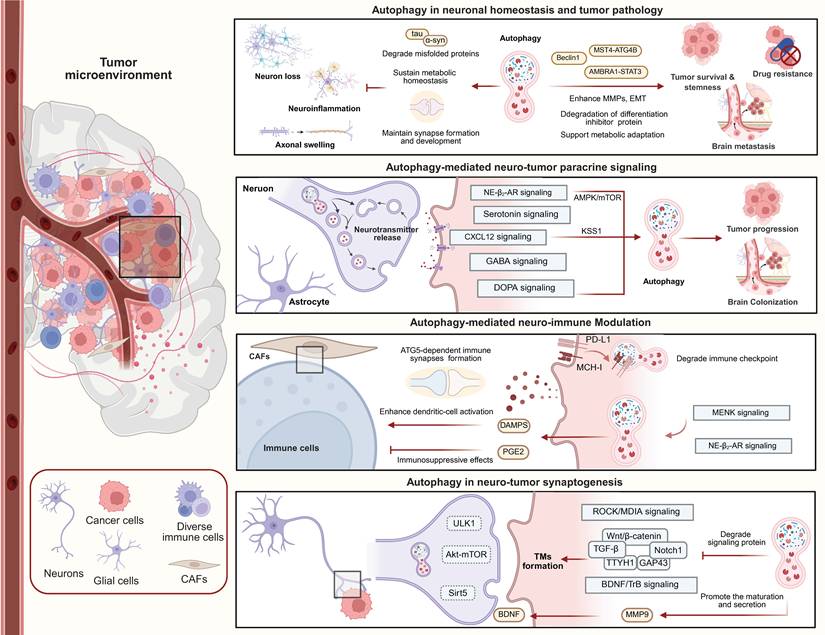
In terms of paracrine signaling and TME remodeling, autophagy plays a crucial role through interactions with neurotransmitters and neurotrophic factors. Autophagy participates in neurotransmitter synthesis and synaptic vesicle turnover. For example, autophagy maintains the homeostasis of tyrosine hydroxylase-positive neurons and supports dopamine synthesis [133], while autophagy induction in hepatocytes upregulates DOPA decarboxylase expression [134]. In neurons, neurotransmitter release depends on synaptic vesicle recycling, and autophagy regulates the degradation and turnover of presynaptic membranes and vesicular proteins [135]. Importantly, presynaptic autophagy is directly coupled to synaptic vesicle cycling through ATG9 trafficking [136]. Activity-dependent exo-endocytosis of ATG9 links neuronal activity with autophagosome biogenesis at presynaptic terminals, providing strong mechanistic evidence that autophagy participates in synaptic communication machinery relevant to neuron-tumor signaling. Additional evidence supports a direct role for autophagy in neuronal secretory signaling. Glutamate-stimulated autophagosomes mediate the autophagic secretion of α-synuclein (α-Syn), and autophagy inducers increase α-Syn secretion [137]. Since α-Syn expression is reduced in gliomas and inversely correlates with malignancy, restoration of α-Syn may exert tumor-suppressive effects [138]. Together, these findings suggest that autophagy-dependent neuronal secretory pathways may influence glioma biology.
Most importantly, direct evidence also supports neurotransmitters and neurotrophic factors acting in autocrine or paracrine manners, regulating tumor cell autophagy. Chronic stress-induced activation of β2-adrenergic receptors promotes autophagy and accelerates gastric cancer progression in gastric cancer cells [139]. Blockade of the DRD4 receptor inhibits the autophagy-lysosome pathway, inducing glioblastoma stem cells to undergo cell-cycle arrest and apoptosis [140]. Serotonin exhibits context-dependent effects on autophagy. It can suppress starvation-induced autophagy in hepatocellular carcinoma and contribute to tumor progression through non-mTOR-dependent activation of mTOR downstream targets [141]. Another study suggested that serotonin may promote hepatocarcinogenesis by inducing hepatic steatosis through activation of autophagy and the Notch pathway [142]. Additionally, autophagy defects resulting from Atg7 deletion impair GABA receptor trafficking, highlighting a critical role for autophagy in neurotransmitter signaling [143]. Furthermore, elevated serotonin signaling in depressive states activates the autophagy/P-STAT3 axis, creating an immunosuppressive microenvironment that promotes lung adenocarcinoma progression [144]. Collectively, these studies directly link neurotransmitter signaling, autophagy regulation, immune modulation, and tumor progression.
Strong evidence additionally exists for autophagy-mediated glial–tumor interactions. Hypoxic gliomas secrete exosomes rich in miR-423-3p, which further enhance astrocytic autophagy and establish an immunosuppressive microenvironment favorable for glioma progression [145]. Similarly, astrocytes secrete the chemokine CXCL12, which downregulates KISS1 expression in breast cancer brain metastasis cells. This KISS1 downregulation triggers the activation of ATG5/ATG7-dependent autophagy pathways, providing the necessary metabolic and structural signaling to drive the metastatic invasion and colonization of breast cancer in the brain parenchyma [49]. This astrocyte-tumor-autophagy signaling loop represents one of the most mechanistically complete examples of autophagy-dependent neural stromal regulation in metastatic brain tumors.
Autophagy also contributes directly to immune synapse regulation and neuroimmune interactions within the TME. The immunosuppressive effects of β2-AR signaling are mediated by increased autophagy and activation of the arachidonic acid cycle, which ultimately enhances the release of the immunosuppressive mediator PGE2 [122]. In cutaneous squamous cell carcinoma, MENK activates autophagy and stimulates DAMP release, thereby enhancing dendritic-cell activation and tumor immunogenicity [146]. Besides, autophagy directly dictates intrinsic immune cell function in the TME. Defective autophagy in microglia accelerates NLRP3 inflammasome activation, which in turn modulates inflammatory responses and immune cell function, significantly influencing glioma progression and brain metastasis [147-149]. Furthermore, immune cells also form immune synapses with target cells. CAFs form immune synapses with Tregs in an ATG5-dependent manner, and ATG5 knockdown reduces Treg infiltration and tumor development [150]. Inhibition of the autophagy protein Beclin-1 restores cytotoxic T lymphocyte-mediated tumor cell killing [151]. These findings strongly support the concept that autophagy bridges neural, immune, and tumor signaling networks.
Evidence linking autophagy to PNI. Several autophagy-associated proteins have been identified as independent risk factors for PNI and poor prognosis in PDAC. Specifically, ubiquitin C has been identified as a key molecular link bridging the metabolic demands of PNI and autophagic flux, providing a foundation for future functional studies [152].
Auxiliary evidence supports the hypothesis that autophagy regulates TM formation, neural synaptic plasticity, and neurotrophic signaling. These conclusions are currently based primarily on inferences drawn from pathway interactions and indirect mechanisms, and warrant further validation. The synaptic ultrastructure of NGS is localized on TMs, and autophagy may be involved in TM formation. TM formation relies on dynamic remodeling of microtubules and actin, regulated by Rho GTPases, including RhoA, Cdc42, and Rac1 [57]. These factors activate ROCK and MDIA signaling to coordinate protrusion formation [153]. Autophagy plays a central role in cytoskeletal remodeling and selectively degrades microtubule-associated and RhoA-associated proteins through autophagic degradation [154].
Furthermore, multiple signaling pathways that coregulate TMs are themselves modulated by autophagy. The TGF-β pathway and its downstream mediator TSP1 promote TMs communication through calcium signaling enhancement [155]. Autophagy has been shown to modulate TGF-β activity in a context-dependent manner, either by promoting p62-dependent degradation of TGF-β or by enhancing TGF-β signaling through Smad7 degradation [156,157]. TMs suppress Wnt signaling to promote neuronal degeneration, while simultaneously exploiting Wnt/β-catenin-driven JNK/MMP signaling to reinforce TM expansion, forming a positive feedback loop [158]. Autophagy has been proven to negatively regulate Wnt/β-catenin signaling through β-catenin degradation and relocalization in glioblastoma cells [159]. Autophagy can also degrade Notch1, a Wnt pathway activator whose downregulation promotes TM formation, whereas Notch1 upregulation is observed in TM-deficient oligodendrogliomas [160].
Several neuronal growth- and synapse-associated proteins further link autophagy to TM-dependent neuron-tumor interactions. Growth-associated protein 43 (GAP43) and tweety-homolog 1 (TTYH1), which mediate axon growth and synaptogenesis in neurons, are enriched in TMs. GAP43 drives TM-dependent tumor invasion, interconnection, proliferation, and radio-resistance, and its expression is regulated by autophagy-associated proteins, with autophagy inhibition reducing GAP43 levels [57]. Moreover, TTYH1 is an endolysosomal membrane protein, promoting autophagy and mitochondrial quality control in glial cells. TTYH1 deficiency or autophagy defects impair mitochondrial turnover, increase oxidative stress, and alter cellular metabolism toward glycolysis, thereby impairing TMs' integrity and glioma progression [161-163]. Neurotrophic signaling provides a functional link between autophagy and malignant synapse plasticity. BDNF levels are regulated by autophagic degradation, while secretory autophagy enhances extracellular BDNF maturation through stress-induced upregulation of MMP9 secretion [164,165]. In glioblastoma, autophagy inhibition activates the NT-3-TrkC signaling and p38 MAPK phosphorylation, promoting tumor cell survival [166]. Additionally, reciprocal involvement between autophagy and TrkB/BDNF signaling has been reported, where inhibition of one pathway activates the other, and dual inhibition produces greater antitumor effects than single-pathway targeting [167].
These findings support a mechanistic link between autophagy and TM-associated neural plasticity programs, which may be linked to functional neuron–tumor synapses and warrants further research to confirm.
Furthermore, to facilitate a broader discussion, some studies originally extrapolated from neuronal autophagy biology, neurodevelopmental systems, or non-neoplastic neurological disorders have also been proposed as hypotheses indicating that autophagy regulates neuro-tumor interactions. These hypotheses may be worth future validation within the context of neuro-tumor communication. For example, cylindromatosis promotes synapse elimination through Akt-mTOR-dependent autophagy activation, while mTOR inhibition rescues synaptic pruning defects [168]. RPM-1 suppresses neuronal autophagy through regulation of UNC-51/ULK signaling, thereby affecting synapse maintenance and axonal development [169]. Similarly, impaired neuronal macroautophagy in the prelimbic cortex contributes to synaptic dysfunction and anxiety-associated neural remodeling in chronic neuropathic pain models [170]. SIRT5 regulates synaptic remodeling pathways and restores autophagic flux dysfunction in neurodegenerative models [171,172], while CHI3L1 influences TM connectivity and promotes autophagy through JNK signaling [173,174]. These findings strongly support a role for autophagy in synaptic biology, but their extensions to neuron–tumor synapses remain speculative.
Overall, current evidence indicates that autophagy likely acts as a regulatory hub linking neuronal signaling, glial remodeling, neurotransmitter secretion, immune modulation, and tumor adaptation. The strongest support exists for autophagy-mediated neuroimmune signaling and glia–tumor communication, whereas the proposed role of autophagy in TM-dependent neuron–tumor synapse formation remains largely mechanistic and requires further experimental validation.
4. Tumor-induced remodeling of the nervous system
The connection between the nervous system and tumors is reciprocal. Tumor cells can directly invade neural tissue, secrete factors that disrupt neural function, and trigger systemic responses that profoundly alter both the structure and function of the nervous system.
4.1 Tumor-mediated neuromodulation and neural remodeling
First of all, primary brain tumors directly infiltrate brain tissue, causing compression or destruction of surrounding neurons and glial cells. The occupying effects of brain metastases can also disrupt the normal function of the nervous system. Beyond physical occupancy, neuron-to-brain tumor synaptic communication critically alters neuronal activity. For example, gliomas increase neuronal excitability, a hallmark characteristic of epilepsy. Seizures are commonly observed in glioma patients, primarily due to glioma cells secreting large amounts of the excitatory neurotransmitter glutamate, which hyperactivates AMPA and NMDA receptors on neuronal cells, leading to cell death [175,176]. Additionally, gliomas can activate their own NMDAR and AMPAR in an autocrine manner, promoting glioma growth and invasiveness [177].
Elevated glutamate concentrations in the TME also strongly downregulate KCC2 levels. Peritumoral neurons experience a chloride imbalance due to deregulated expression of NKCC1 and KCC2, resulting in chloride efflux upon GABA receptor activation and generating a functional incentive [178,179]. Paradoxically, endogenous GABAA receptor activity in glioma cells is associated with a reduction in their proliferation [180]. Recent studies indicate that glioma cells with PIK3CA mutations differentially secrete glypican-3, contributing to neuronal hyperexcitability [181].
Tumor cells also induce neurogenesis and PNI by releasing paracrine factors, thereby creating a "neural microenvironment" that promotes tumor progression. In the PNS, pancreatic cancer cells use GDNF and ARTN to promote peripheral invasion and secrete NGF, which induces aberrant nerve fiber proliferation and extension into the tumor environment. This NGF-mediated axonogenesis is often associated with increased adrenergic or cholinergic signaling [100]. Besides, tumor cells mediate neurogenesis in NPCs and facilitate neural reprogramming. Some studies have shown that peripheral tumors can establish communication with the brain at a distance. For instance, prostate cancer attracts DCX+ neural progenitor cells from the subventricular zone of the brain, prompting them to migrate through the bloodstream after crossing the BBB. These neural progenitor cells infiltrate the tumor, initiating intratumoral neurogenesis and generating new adrenergic neurons [182]. Gliomas localized in functionally connected regions also secrete the synaptogenic factor thrombospondin-1 (TSP-1), which contributes to neuron-glioma interactions, ultimately remodelling neural circuits, promoting glioma progression, and impairing cognitive performance [63]. Another study revealed that in head and neck cancer, loss of TP53 leads to loss of the microRNA miR-34a, contributing to tumor-associated axonogenesis of sensory nerves and adrenergic transformation, thereby supporting tumor progression [183].
Paraneoplastic syndromes are abnormal immune responses or ectopic hormone production induced by tumor products, including hormones secreted by tumors. When tumor cells express antigens similar to those found in neurons (e.g., Hu /Yo /Ri proteins), an autoimmune response is triggered, leading to antibody-mediated attack on the nervous system. This results in neurological disorders known as paraneoplastic neurological syndromes [184]. Examples include opsoclonus-myoclonus ataxia syndrome in neuroblastoma, characterized by cerebellar and brainstem autoimmunity, and paraneoplastic cerebellar degeneration associated with SCLC.
Furthermore, tumor cells cause nerve cell dysfunction through abnormal metabolism, which leads to lactate accumulation and an enrichment of reactive oxygen species along with inflammatory mediators in the neural-TME [185]. For example, CRC may cause sensory neuron dysfunction via cytokine and chemokine signaling, such as CXCL10 [186]. In addition to neuronal cells, breast and lung cancer cells can form a gap junction network with astrocytes by expressing PCDH7. These channels facilitate the transport of cGAMP from brain metastatic cancer cells, activate the STING pathway, and produce inflammatory factors like TNF and IFNα. This, in turn, activates the STAT1 and NF-κB pathways, contributing to tumor progression and chemoresistance [187].
4.2 Tumor therapy-induced neurotoxicity and neural dysfunction
Tumor treatment harms more than just the cancer. The radiation and chemotherapy used to fight the disease also damage the healthy nervous system. Many patients receiving these treatments develop problems with thinking and memory, a side effect often called "chemo-brain." This includes poor concentration, serious memory loss, and slower thinking. These drugs do not only target fast-growing cancer cells. They also attack and harm normal brain structure and function. Chemotherapy can hurt the brain by causing inflammation, increasing oxidative stress, and stopping the growth of new neurons [188,189]. One common drug, paclitaxel, works by blocking cell division. However, this same action disrupts the microtubules that neurons need for their proper shape and function. This disruption leads to dysfunctional neuronal growth, development, and communication. Additionally, chemotherapeutic drugs like methotrexate, which inhibit DNA synthesis, can result in DNA damage accumulation and reduced generation of new neurons. Furthermore, treatments with adriamycin and methotrexate have been shown to reduce BDNF levels, thereby suppressing neurogenesis [190,191].
RIBI is another common complication of radiation therapy for tumors. This condition refers to brain tissue damage caused by radiotherapy, leading to neuronal and glial cell degeneration, necrosis, and CNS disorders. RIBI is primarily driven by direct DNA damage, apoptosis, vascular damage, and inflammation. For example, radiation prompts microglia to secrete pro-inflammatory chemokines, such as CCL2 and CCL8, which mediate CD8+ T-cell infiltration and contribute to chronic neuroinflammation [192]. Conversely, some studies have indicated that radiotherapy may enhance neuronal-tumor connectivity by increasing neuronal activity [62].
4.3 Autophagy-related regulation of tumor-induced neural remodeling
Autophagy not only mediates the neurological impact on tumor progression, but also plays an indispensable role in how tumors affect the nervous system. In terms of tumor progression, autophagy has duality. On the one hand, autophagy supports tumor cell survival by removing defective organelles, reducing oxidative stress, preventing DNA damage, and resisting metabolic stress, thus promoting tumor cell survival. On the other hand, autophagy facilitates neurological invasion by influencing processes related to tumor metastasis. Additionally, autophagy contributes to the creation of a suppressive tumor immune microenvironment, which aids in immune evasion. In contrast, excessive autophagy activation can lead to cytotoxic effects, inducing cell death and inhibiting tumor progression [193]. This dual role is likely dependent on factors such as tumor type, progression stage, and the TME.
Autophagy also mediates the suppressive immune microenvironment within tumors. It not only regulates the transcription and translation of immune checkpoint molecules such as PD-L1 but also facilitates the autophagic degradation of PD-L1 and MHC-I. This promotes immune evasion by tumor cells, influencing their ability to invade the nervous system [194]. Thus, autophagy plays a critical role in both promoting tumor survival and invasiveness, further impacting the nervous system.
Beyond its role in intracellular degradation and recycling, autophagy also regulates extracellular release pathways [195]. Autophagy influences the accumulation and release of inflammatory factors such as IL-6, CXCL8, ROS, and HMGB1, which can have either anti-inflammatory or pro-inflammatory effects [196]. These released inflammatory factors may contribute to neuronal damage in the tumor’s vicinity. Moreover, autophagy is involved in neurotransmitter release in neurons, and tumor cell-derived neurotransmitter crosstalk and neurotrophic factors may also depend on autophagy for secretion. For instance, in pancreatic cancer, the NGF/ATG paracrine pathway activates autophagy in Schwann cells, which not only repairs nerves and promotes nerve fiber extension towards tumor cells but also drives tumor cells towards nerve-directed growth, facilitating neural infiltration [17]. Blocking both NGF and autophagy inhibits PNI. In oral mucosa carcinoma, autophagy-induced neuronal growth inhibition is associated with CGRP-mediated protection, which activates autophagy in cancer cells by disrupting Rap1-mediated mTOR-Raptor interactions [96]. Furthermore, tumor cells themselves exploit autophagic pathways to dictate the composition of the surrounding TME. For instance, glioblastoma utilizes secretory autophagy to release PAI-1 into the extracellular space, supporting an immunosuppressive niche. Pharmacological inhibition of this autophagic release restricts extracellular PAI-1, thereby remodeling the TME into a pro-inflammatory, anti-tumor state and significantly prolonging survival [197].
Neurotoxicity induced by tumor treatments, such as chemotherapy and radiotherapy, is also linked to autophagy. For example, mitoxantrone, a chemotherapeutic agent used to treat various tumors and multiple sclerosis, induces neurotoxicity through excessive autophagy, leading to neuronal death [198]. Similarly, sunitinib, a targeted drug against VEGFR, causes cognitive and memory impairments by impairing autophagy in neurons [199].
RIBI is commonly associated with cerebrovascular dysfunction, which is considered a significant cause of cognitive impairment. Recent studies indicate that autophagy defects in pericytes, which are part of the neurovascular unit, promote BBB dysfunction by inducing senescence. This contributes to cognitive decline and aids in glioma cell growth and invasion [200]. Interestingly, autophagy activation through rapamycin eliminates senescent cells and significantly alleviates radiation-induced cognitive dysfunction.
Furthermore, autophagy plays a crucial role in chemoresistance in gliomas. For example, temozolomide (TMZ) treatment induces autophagy and leads to treatment resistance, which is associated with impaired down-regulation of ATG9B expression by downregulated DAB2IP through the Wnt/β-catenin pathway in drug-resistant cells [201]. Similarly, autophagy flux has been shown to correlate with glioma sensitivity to radiation, and blocking autophagy may enhance treatment efficacy [202]. In conclusion, autophagy mediates resistance to conventional tumor therapies and indirectly contributes to the tumor's detrimental effects on neural tissue.
5. Therapeutic strategies for targeting neural-tumor interactions
With the rapid advancement of cancer neuroscience, an increasing number of neural–tumor interactions are being elucidated. These discoveries hold significant potential for translation into novel cancer therapies. Based on mechanistic insights into the neural–tumor interaction network, key signaling molecules, such as neurotransmitters, their receptors, and neurotrophic factors, have emerged as promising targets for the development of targeted therapeutics. Furthermore, these findings offer a rationale for the repurposing of existing drugs that modulate neural signaling pathways (Table 2).
The promising drugs targeting neuro-tumor interactions
| Classification | Drug | Target | Mechanism | Disease | Phase | Ref. |
|---|---|---|---|---|---|---|
| Neurotransmitter blocker | Talampanel perampanel | AMPA receptor | Blocking AMPAR, inhibiting glioma-induced epilepsy, and suppressing calcium-related invasiveness and growth of gliomas | Glioblastoma | II | [60,206] NCT00064363 |
| Memantine | NMDA receptor | Blocking NMDAR, attenuating brain necrosis from radiotherapy, altering the immunosuppressive activity of TAM, and promoting anti-tumor immunity | Brain tumors, Hepatocellular sarcoma | III | [205] NCT00566852 | |
| Propranolol carvedilol | Adrenergic receptor | Blocking β-adrenergic receptor signaling, inhibiting the tumor from receiving catecholamine stimulation from multiple sources, and inhibiting tumor progression by reducing tumor cell proliferation, metastasis, modulating the immunosuppressive environment, and inducing death | Colorectal cancer, Prostate cancer, Breast cancer, Ovarian cancer, Glioblastoma, PDAC, Melanoma | IV | [204] NCT02962947 | |
| ONC201 | Dopamine receptor | Blocking dopamine receptors, inhibiting proliferation, mediating apoptosis, and remodeling the pro-inflammatory tumor environment | Glioma, Prostate cancer, Endometrial cancer, Neuroendocrine tumors | III | [108,209] NCT05580562 | |
| Atropine | Choline receptor | Blocking cholinergic receptors, inhibiting tumor EMT, deregulating the immunosuppressive environment, and suppressing cancer stem cells | Colorectal cancer, Breast cancer, PDAC | IV | [88,207,208] NCT00575523 | |
| Neurotrophin inhibitor | Tanezumab | NGF | NGF Antibody, relieving pain, inhibiting tumor invasion and migration, and reducing nerve infiltration | Cancer bone metastases, Pancreatic cancer | III | [210] NCT02609828 |
| Larotrectinib Entrectinib | TRK | Inhibiting NTRK1/2/3, suppressing the signaling pathway associated with the aberrant proliferation of tumor cells due to persistent activation of the aberrant conformation of TRK proteins | TRK fusion-positive solid tumors | FDA-approved | [211] | |
| Synapse inhibitor | Meclofenamate | Gap junctions | Inhibiting TM-dependent tumor intercellular communication, increasing susceptibility to chemotherapy | Glioblastoma | II | [212] NCT02429570 |
| INCB7839 | NLGN3-ADAM10 | Inhibiting ADAM10, blocking NLGN3 shedding, and preventing tumor proliferation and progression | Glioma | II | [77] | |
| Neuro-anesthetic | Botulinum toxin | Intratumoral innervation | Inhibiting acetylcholine vesicle release at the neuromuscular junction, suppressing tumor cells' proliferation, and promoting apoptosis | Head Neck Cancer, Gastric cancer, Breast Cancer, Neuroma, Cutaneous Leiomyomas | IV | NCT01374191 |
| Other repurposing drugs | Aprepitant | Substance P | Inhibiting TACR1 and blocking ssRNA release from SP-activated dying cells, suppressing breast cancer cell growth, invasion, and metastasis | Breast cancer | Pre-clinical | [102] |
| Telmisartan | IL-6 | Agonizing PPARγ, inhibiting astrocyte IL-6 paracrine secretion, and inhibiting glial cell proliferation and migration | Glioma | Pre-clinical | [112] | |
| Rimegepant | CGRP | Blocking CGRP, inhibiting cancer cells from utilizing sensory nerves to induce protective autophagy | Oral mucosa carcinomas | Pre-clinical | [96] | |
| Fluoxetine | Glutamatergic synaptic transmission | Blocking NMDA receptors and interfering with glutamatergic synaptic transmission between neurons and glioma cells, exhibiting antiproliferative, pro-apoptotic, and synergistic chemotherapeutic anti-tumor effects | Glioma, Colorectal cancer | I | NCT05634707 | |
| Imipramine | Serotonin | Blocking serotonin reuptake, inhibiting cell migration and invasion, inducing apoptosis, and augmenting autophagy, reprogramming of the immunosuppressive tumor microenvironment through inhibition of histamine receptor signaling | Glioma, breast cancer | I | [33] NCT04863950 | |
| Gabapentin | TSP-1 | Pharmacologically inhibiting TSP-1, reducing glioma proliferation and network synchrony | Glioma | Pre-clinical | [63] |
5.1 Current therapeutic strategies targeting neural-tumor communication pathways
From the perspective of targeting neurotransmitters, inhibition of β-adrenergic receptor signaling has been shown to suppress both neuronal secretion and norepinephrine stimulation originating from various sources within the TME. This inhibition can impede tumor progression by reducing tumor cell proliferation and metastasis, modulating the immunosuppressive environment, and inducing tumor cell death [203]. Representative agents include non-selective β-blockers such as propranolol and carvedilol. Propranolol, a lipophilic compound capable of crossing the blood–brain barrier, has demonstrated antitumor effects in preclinical studies across a range of cancers, including colorectal, prostate, breast, ovarian, and various hematologic malignancies [204]. Both propranolol and carvedilol are currently being evaluated in clinical trials.
Inhibiting glutamate receptors offers a direct way to stop tumors driven by neural activity. AMPA and NMDA receptors help pass excitatory signals from neurons to cancer cells. Blocking them with drugs can halt cancer cell multiplication and also shield healthy neurons from harm. A known NMDA receptor blocker, memantine, is used in the clinic. It is given with radiotherapy to lessen brain injury and cognitive side effects. It has also shown immunomodulatory and antitumor effects in preclinical models [205]. Talampanel, an AMPA receptor inhibitor, demonstrated safety and therapeutic potential in glioma patients when combined with radiotherapy and TMZ, suggesting its utility in glioblastoma treatment [206]. Perampanel, another AMPA receptor antagonist, has been reported to reduce glioma invasion and progression by disrupting glutamatergic signaling between neurons and tumor cells [60]. Additionally, it has shown efficacy in mitigating glioma-associated epilepsy and is currently under investigation in multiple clinical trials as an adjunct therapy.
Other neurotransmitter-targeting agents include the cholinergic receptor antagonist atropine, which has shown preclinical efficacy in breast and colorectal cancers by inhibiting epithelial–mesenchymal transition and alleviating immunosuppression [207,208]. Bethanechol, a muscarinic receptor agonist, has also demonstrated effectiveness in PDAC and is under clinical evaluation [88].
ONC201 has been identified as an antagonist of dopamine receptors DRD2 and DRD3. It blocks AKT/ERK signaling in tumor cells and has antitumor activity in multiple cancer types like glioma, prostate cancer, and endometrial cancer by inhibiting proliferation, mediating apoptosis, and contributing to a pro-inflammatory microenvironment. ONC201 is being evaluated in a clinical trial in patients with advanced solid tumors and hematological malignancies [108,209]. These neurotransmitter receptor antagonists are frequently incorporated into therapeutic strategies targeting nerve–tumor interactions, often through drug repurposing approaches.
Moreover, inhibition of neurotrophic factor signaling represents another crucial approach for disrupting the neural–tumor axis. Notably, targeting the NGF-Trk and BDNF-TrkB signaling pathways has shown therapeutic potential. CAFs, through interactions with sympathetic nerves, can promote tumor progression via NGF secretion within the TME. Inhibition of Trk signaling can disrupt these neuro-mesenchymal interactions and has been proposed as a strategy for CRC treatment [84]. Tanezumab, a monoclonal antibody against NGF, has reached Phase III clinical trials for bone cancer, where it demonstrates efficacy in pain relief and inhibition of nerve infiltration (NCT02609828). Additionally, it has been reported to suppress neural invasion by pancreatic cancer cells and reduce nociceptive transmission [210]. Pan-Trk inhibitors such as entrectinib and larotrectinib have been approved for solid tumors harboring NTRK gene fusions [211]. These agents offer targeted therapeutic options for tumors driven by neurotrophic signaling and characterized by neural–tumor interactions.
Emerging targets such as the NLGN3-ADAM10 signaling axis also show promise. INCB7839, an ADAM10 inhibitor, is currently under clinical investigation for multiple brain and extracranial tumors, including glioma and breast cancer [77]. Similarly, meclofenamate, an inhibitor of gap junctions, has been reported to disrupt TMs formation in gliomas, thereby impairing TM-mediated tumor cell communication [212].
Given the unique properties of the CNS, repurposing drugs capable of crossing the blood–brain barrier may offer an additional strategy to target the neural–tumor axis. Several antipsychotics and antidepressants have demonstrated antitumor activity [213]. For example, aprepitant, a TACR1 antagonist approved for chemotherapy-induced nausea, has been shown to inhibit neuronal hyperactivation induced by SP released from breast cancer cells. This agent targets the neuropeptide/extracellular ssRNA-sensing axis and suppresses breast cancer growth and metastasis in various preclinical models [102].
Telmisartan, an angiotensin II type 1 receptor blocker with PPARγ agonist activity, has been shown to counteract the pro-tumorigenic effects of IL-6 secreted by astrocytes, suggesting its potential use as an adjuvant therapy in glioma patients, particularly those with hypertension [112]. The FDA-approved drug gabapentin, used as an antiepileptic drug, can decrease glioblastoma proliferation through the inhibition of TSP-1 [63]. Likewise, rimegepant, an anti-migraine drug, has demonstrated efficacy in inhibiting oral mucosal carcinoma by blocking neurogenic CGRP signaling [96].
Additionally, the rabies virus, beyond its established use as a neural tracer, has been engineered to selectively induce apoptosis in glioma-connected neurons, thereby halting glioblastoma progression. This represents a novel viral strategy for glioblastoma treatment [62].
5.2 Therapeutic potential of autophagy-directed modulation of neural-tumor interactions
As previously discussed, autophagy serves as a critical mediator in the crosstalk between the nervous system and tumors: (1) plays an essential role in neuronal development, (2) regulates the release of neuron-related factors, (3) exerts a dual regulatory effect on tumor cells, and (4) functions as a downstream effector of neural–tumor interaction signaling. Therefore, targeting autophagy may disrupt neural–tumor interactions through multiple mechanisms, offering a novel avenue for cancer research and therapeutic intervention.
Evidence suggests that autophagy inhibition can impair neural–tumor dynamics. For instance, the autophagy inhibitor chloroquine suppresses autophagic flux in Schwann cells within the pancreatic cancer microenvironment, thereby interrupting NGF signaling derived from pancreatic cancer cell paracrine secretion. Simultaneously, it induces apoptosis in pancreatic cancer cells and prevents neural infiltration [17]. Clinical pathological analysis has further revealed a positive correlation between PNI and elevated autophagy in pancreatic cancer patients [214]. The antitumor effects of many neuro-modulatory drugs are mediated through changes in autophagy. Certain beta-blockers can inhibit autophagic flux, and this inhibition may be central to their anti-neurotropic activity [215]. Conversely, memantine has been identified as an autophagy activator that restores autophagic flux and improves neuronal viability [216].
In addition, some preclinical evidence has shown that autophagy modulators combined with neuro-tumor axis blockers exhibit synergistic effects.
In CRC, BDNF inhibitor K252a, combined with chloroquine, has been shown to significantly reduce tumor cell metabolic activity and enhance apoptosis, demonstrating superior efficacy compared to monotherapies [167]. Induction of autophagic dysfunction has also been reported to enhance the cytotoxic activity of propranolol against hemangioma cells [217]. In addition, Chloroquine was shown to suppress ONC206-induced protective autophagy, thereby potentiating the pro-apoptotic effects of ONC206 and enhancing its antitumor efficacy in hepatocellular carcinoma [218]. Similarly, chloroquine inhibited the protective autophagic response induced by imipramine in esophageal squamous cell carcinoma, further augmenting tumor suppression [219]. Silencing of the autophagy-related gene ATG5 was likewise reported to enhance Melatonin-induced apoptosis in liver cancer cells [220].
Conversely, another study demonstrated that Larotrectinib induced autophagic cell death through activation of autophagic flux, whereas co-treatment with chloroquine disrupted its antitumor activity against CRC, highlighting the context-dependent and dual role of autophagy modulation in cancer therapy [221].
In addition, results from clinical trials of glioblastoma treatment using a combination of chloroquine and TMZ chemotherapy, along with radiation therapy, indicate that this combination regimen can prolong overall survival [222]. However, further expanded clinical trials will require the development of more potent and less toxic autophagy inhibitors.
Interestingly, owing to their intrinsic ability to penetrate the blood–brain barrier, repurposed neuropsychiatric agents with antitumor activity, particularly antidepressants, have emerged as promising therapeutic candidates for brain malignancies. Notably, the antineoplastic effects of many of these agents appear to be closely associated with modulation of autophagy pathways. Among them, imipramine, maprotiline, fluoxetine, and escitalopram have been reported to induce autophagy, whereas nortriptyline, clomipramine, and paroxetine function predominantly as autophagy inhibitors. In contrast, sertraline and desipramine exhibit context-dependent effects on autophagy, acting either as autophagy inducers or inhibitors depending on the specific tumor setting [223].
Given autophagy’s inherently dual role, contextually promoting either tumor suppression or survival, its regulatory state within the neuro–neurosecretory factor-tumor axis is highly dependent on the spatial and temporal context of the TME.
In the context of neurodevelopment and neuronal homeostasis, physiological autophagy is primarily protective and functionally indispensable. Inducing autophagy or restoring autophagic flux is generally associated with maintaining neuronal stability and preventing neurodegeneration. Excessive inhibition of autophagy within the CNS may consequently impair neuronal survival, synaptic stability, and cognitive function. Conversely, during tumor progression and metastasis, autophagy frequently acts as an adaptive survival mechanism. Blocking this protective autophagy can thereby overcome microenvironmental adaptation and therapeutic resistance. In highly aggressive tumors such as glioblastoma and metastatic brain tumors, autophagy primarily functions as a pro-survival mechanism that supports tumor stemness, metabolic adaptation, immune evasion, and therapeutic resistance. For example, the MST4-ATG4B axis enhances autophagic flux to sustain glioblastoma stemness and radioresistance [32], while astrocyte-associated autophagy promotes brain metastasis adaptation to the cerebral microenvironment [49]. Excessive or dysregulated autophagy can also induce tumor cell death and represents another exploitable therapeutic vulnerability.
Most importantly, in the currently strongest-supported neuro–tumor interaction models, autophagy primarily promotes tumor-supportive communication between neurons, glial cells, immune cells, and tumor cells, suggesting that selective inhibition of tumor-associated autophagy may provide therapeutic benefit. For instance, β2-adrenergic signaling promotes tumor progression through autophagy activation [139], while astrocyte-derived CXCL12 induces ATG5/ATG7-dependent autophagy to facilitate breast cancer brain metastasis [49]. Similarly, hypoxic glioma-derived exosomes enhance astrocytic autophagy to establish an immunosuppressive microenvironment [145]. In these contexts, autophagy inhibition may suppress neural-derived trophic support, reverse immunosuppression, and sensitize tumors to therapy. Consequently, in current therapeutic developments, pharmacological blockers of neuro-tumor interactions are increasingly being combined with autophagy inhibitors to achieve robust synergistic effects.
However, excessive systemic autophagy inhibition within the CNS may simultaneously impair neuronal synaptic homeostasis and disrupt normal neuroimmune regulation. Conversely, controlled autophagy activation in neurons or immune cells may preserve neuronal integrity and enhance antitumor immunity in selected contexts. For example, MENK-induced autophagy promotes DAMP release and dendritic-cell activation [146], while autophagy-mediated degradation of oncogenic signaling intermediates may suppress TM-associated plasticity and malignant synapse reinforcement. Thus, within the neural–tumor axis, the most promising future strategy may not involve global activation or inhibition of autophagy, but rather spatially and cell-type selective modulation, in which autophagy is inhibited in tumor-supportive compartments while preserved or restored in neurons and antitumor immune populations.
6. Concluding remarks and future directions
Cancer neuroscience reconstructs the traditional boundaries of oncology. The nerves in our body are closely linked to growing tumors. They do not just grow into TMEs, but also help tumors grow and resist treatment. Nerves talk to tumors through connections and by releasing special chemicals. Neurotransmitters and neurotrophic factors operating through β-adrenergic, glutamatergic, and cholinergic cascades form the molecular basis of these regulations. Several pharmacological agents have exhibited either preclinical or clinical evidence of efficacy in modulating neural inputs to tumors. Furthermore, repurposed CNS medications, such as antidepressants, antipsychotics, and anti-migraine agents, have demonstrated potential in disrupting neurogenic signaling within the TME.
Autophagy has emerged as a potential molecular bridge linking neural activity to tumor behavior. Modulating autophagy provides an additional avenue for therapeutic intervention, as demonstrated by pharmacological agents such as chloroquine and memantine. Translating these mechanisms into real therapies demands more precision. Because both neurotransmitter release and autophagy status between tumor-suppressive and supportive states remain vague. Conditionally activated prodrugs or targeted delivery platforms should be considered and constructed, therefore selectively restricting pathological neuro-tumor communication and keeping the autophagic baseline that normal neurons require. The physical architecture of these circuits, especially the feedback loops that connect the central nervous system to peripheral ganglia, remains largely unexplored. Before oncologists can integrate neural-targeted regimens into standard care protocols, large-scale clinical trials must strictly quantify the off-target neurological damage these agents inflict.
In conclusion, targeting the neural-tumor axis represents a promising therapeutic strategy in the field of oncology. Further elucidation of underlying mechanisms and more precise pharmacological interventions is required in the future. This review outlines the current understanding of neural–tumor interactions, identifies key signaling pathways with therapeutic potential, and suggests autophagy as a pivotal mechanism connecting the neural, tumor, and immune microenvironments. These findings provide valuable directions for future investigations and contribute to the emerging discipline of cancer neuroscience.
Abbreviations
α-Syn: α-synuclein
BBB: blood-brain barrier
B2BM: breast-to-brain metastasis
β-AR: β-adrenoceptors
CAFs: cancer-associated fibroblasts
CGRP: calcitonin gene-related peptide
CNS: central nervous system
CMA: chaperone-mediated autophagy
CRC: colorectal cancer
DMG: diffuse midline gliomas
DRD5: dopamine receptor D5
GDNF: glial cell line-derived neurotrophic factor
GAP43: growth-associated protein 43
IGF1: insulin-like growth factor 1
IL-4: interleukin-4
LCN2: lipocalin-2
LGGs: low-grade gliomas
MMPs: matrix metalloproteinases
MENK: methionine enkephalin
MDSCs: myeloid-derived suppressor cells
NAA: N-acetylaspartate
NGF: nerve growth factor
NPCs: neural precursor cells
NLGN3: neuroligin-3
NGSs: neuro-glioma synapses
NMDARs: N-methyl-D-aspartate receptors
NSCLC: non-small cell lung cancer
OPCs: oligodendrocyte precursor cells
PDAC: pancreatic ductal adenocarcinoma
PNI: perineural invasion
PNS: peripheral nervous system
RIBI: radiation-induced brain injury
Tregs: regulatory T cells
SCLC: small cell lung cancer
SP: substance P
TMZ: temozolomide
TSP-1: thrombospondin-1
TME: tumor microenvironment
TMs: tumor microtubes
TNTs: tunneling nanotubes
TTYH1: tweety-homolog 1
VEGF: vascular endothelial growth factor
Acknowledgements
We acknowledge BioRender.com, which was used to create several figures in this manuscript. Language polishing was supported by an AI language model (Gemini 3) to enhance readability and grammar.
Funding
This work was supported in part by grants from National Natural Science Foundation of China (Grant No. 82373193), and Natural Science Foundation of Sichuan Province (Grant No. 2024NSFSC0627).
Author Contributions
A.Z. Li, B. Liu, and Y. Qu conceived and supervised the project. H.F. Cheng, H.Y. Li, and P.Y. Liu drafted the original manuscript. H.F. Cheng and H.Y. Li prepared the figures and tables. A.Z. Li and Y. Qu proofread the structure and figures. B. Liu polished the manuscript. All the authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Price M, Neff C, Nagarajan N, Kruchko C, Waite KA, Cioffi G. et al. CBTRUS statistical report: American Brain Tumor Association & NCI Neuro-Oncology Branch adolescent and young adult primary brain and other central nervous system tumors diagnosed in the United States in 2016-2020. Neuro Oncol. 2024;26:iii1-53
2. Tan AC, Ashley DM, López GY, Malinzak M, Friedman HS, Khasraw M. Management of glioblastoma: state of the art and future directions. CA Cancer J Clin. 2020;70:299-312
3. Weller M, Le Rhun E, Van den Bent M, Chang SM, Cloughesy TF, Goldbrunner R. et al. Diagnosis and management of complications from the treatment of primary central nervous system tumors in adults. Neuro Oncol. 2023;25:1200-24
4. Hanahan D, Monje M. Cancer hallmarks intersect with neuroscience in the tumor microenvironment. Cancer Cell. 2023;41:573-80
5. Mancusi R, Monje M. The neuroscience of cancer. Nature. 2023;618:467-79
6. Cervantes-Villagrana RD, Albores-García D, Cervantes-Villagrana AR, García-Acevez SJ. Tumor-induced neurogenesis and immune evasion as targets of innovative anti-cancer therapies. Signal Transduct Target Ther. 2020;5:99
7. Lange M, Joly F, Vardy J, Ahles T, Dubois M, Tron L. et al. Cancer-related cognitive impairment: an update on state of the art, detection, and management strategies in cancer survivors. Ann Oncol. 2019;30:1925-40
8. Gibson EM, Monje M. Microglia in cancer therapy-related cognitive impairment. Trends Neurosci. 2021;44:441-51
9. Horowitz TS, Suls J, Treviño M. A call for a neuroscience approach to cancer-related cognitive impairment. Trends Neurosci. 2018;41:493-6
10. Dietrich J, Parsons MW, Santarnecchi E. Exploring novel therapeutic avenues for chemotherapy-related cognitive impairment. Cancer Res. 2024;84:2041-2
11. Palmer JE, Wilson N, Son SM, Obrocki P, Wrobel L, Rob M. et al. Autophagy, aging, and age-related neurodegeneration. Neuron. 2025;113:29-48
12. Fu Y, Zhang J, Qin R, Ren Y, Zhou T, Han B. et al. Activating autophagy to eliminate toxic protein aggregates with small molecules in neurodegenerative diseases. Pharmacol Rev. 2025;77:100053
13. Debnath J, Gammoh N, Ryan KM. Autophagy and autophagy-related pathways in cancer. Nat Rev Mol Cell Biol. 2023;24:560-75
14. Karpova A, Hiesinger PR, Kuijpers M, Albrecht A, Kirstein J, Andres-Alonso M. et al. Neuronal autophagy in the control of synapse function. Neuron. 2025;113:974-90
15. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722-37
16. Sadeghsoltani F, Avci ÇB, Hassanpour P, Haiaty S, Rahmati M, Mota A. et al. Autophagy modulation effect on homotypic transfer of intracellular components via tunneling nanotubes in mesenchymal stem cells. Stem Cell Res Ther. 2024;15:189
17. Zhang W, He R, Yang W, Zhang Y, Yuan Q, Wang J. et al. Autophagic Schwann cells promote perineural invasion mediated by the NGF/ATG7 paracrine pathway in pancreatic cancer. J Exp Clin Cancer Res. 2022;41:48
18. Xiang H, Zhang J, Lin C, Zhang L, Liu B, Ouyang L. Targeting autophagy-related protein kinases for potential therapeutic purpose. Acta Pharm Sin B. 2020;10:569-81
19. Hegdekar N, Sarkar C, Bustos S, Ritzel RM, Hanscom M, Ravishankar P. et al. Inhibition of autophagy in microglia and macrophages exacerbates innate immune responses and worsens brain injury outcomes. Autophagy. 2023;19:2026-44
20. Yang G, Song W, Postoak JL, Chen J, Martinez J, Zhang J. et al. Autophagy-related protein PIK3C3/VPS34 controls T cell metabolism and function: PIK3C3/VPS34 in T cell metabolism and function. Autophagy. 2021;17:1193-204
21. Mizushima N, Levine B. Autophagy in human diseases. N Engl J Med. 2020;383:1564-76
22. Fleming A, Bourdenx M, Fujimaki M, Karabiyik C, Krause GJ, Lopez A. et al. The different autophagy degradation pathways and neurodegeneration. Neuron. 2022;110:935-66
23. Zhang K, Zhu S, Li J, Jiang T, Feng L, Pei J. et al. Targeting autophagy using small-molecule compounds to improve potential therapy of Parkinson’s disease. Acta Pharm Sin B. 2021;11:3015-34
24. Nixon RA, Rubinsztein DC. Mechanisms of autophagy-lysosome dysfunction in neurodegenerative diseases. Nat Rev Mol Cell Biol. 2024;25:926-46
25. Saitoh T, Akira S. Regulation of inflammasomes by autophagy. J Allergy Clin Immunol. 2016;138:28-36
26. Qiao L, Ma J, Zhang Z, Sui W, Zhai C, Xu D. et al. Deficient chaperone-mediated autophagy promotes inflammation and atherosclerosis. Circ Res. 2021;129:1141-57
27. Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M. et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014;112:24-49
28. Zhou T, Zheng Y, Sun L, Badea SR, Jin Y, Liu Y. et al. Microvascular endothelial cells engulf myelin debris and promote macrophage recruitment and fibrosis after neural injury. Nat Neurosci. 2019;22:421-35
29. Martini-Stoica H, Xu Y, Ballabio A, Zheng H. The autophagy-lysosomal pathway in neurodegeneration: a TFEB perspective. Trends Neurosci. 2016;39:221-34
30. Hong B, Yang E, Su D, Ju J, Cui X, Wang Q. et al. EPIC-1042 as a potent PTRF/Cavin1-caveolin-1 interaction inhibitor to induce PARP1 autophagic degradation and suppress temozolomide efflux for glioblastoma. Neuro Oncol. 2024;26:100-14
31. Li H, Chen L, Li JJ, Zhou Q, Huang A, Liu WW. et al. miR-519a enhances chemosensitivity and promotes autophagy in glioblastoma by targeting STAT3/Bcl2 signaling pathway. J Hematol Oncol. 2018;11:70
32. Huang T, Kim CK, Alvarez AA, Pangeni RP, Wan X, Song X. et al. MST4 phosphorylation of ATG4B regulates autophagic activity, tumorigenicity, and radioresistance in glioblastoma. Cancer Cell. 2017;32:840-855.e8
33. Chryplewicz A, Scotton J, Tichet M, Zomer A, Shchors K, Joyce JA. et al. Cancer cell autophagy, reprogrammed macrophages, and remodeled vasculature in glioblastoma triggers tumor immunity. Cancer Cell. 2022;40:1111-1127.e9
34. Bhoopathi P, Chetty C, Gujrati M, Dinh DH, Rao JS, Lakka S. Cathepsin B facilitates autophagy-mediated apoptosis in SPARC overexpressed primitive neuroectodermal tumor cells. Cell Death Differ. 2010;17:1529-39
35. Petersen W, Liu J, Yuan L, Zhang H, Schneiderjan M, Cho YJ. et al. Dasatinib suppression of medulloblastoma survival and migration is markedly enhanced by combining treatment with the aurora kinase inhibitor AT9283. Cancer Lett. 2014;354:68-76
36. Nazio F, Po A, Abballe L, Ballabio C, Diomedi Camassei F, Bordi M. et al. Targeting cancer stem cells in medulloblastoma by inhibiting AMBRA1 dual function in autophagy and STAT3 signalling. Acta Neuropathol. 2021;142:537-64
37. Yao H, Tang H, Zhang Y, Zhang QF, Liu XY, Liu YT. et al. DEPTOR inhibits cell proliferation and confers sensitivity to dopamine agonist in pituitary adenoma. Cancer Lett. 2019;459:135-44
38. Leng ZG, Lin SJ, Wu ZR, Guo YH, Cai L, Shang HB. et al. Activation of DRD5 (dopamine receptor D5) inhibits tumor growth by autophagic cell death. Autophagy. 2017;13:1404-19
39. Yang Y, Chen Y, Wu J, Ren Y, Liu B, Zhang Y. et al. Targeting regulated cell death with plant natural compounds for cancer therapy: a revisited review of apoptosis, autophagy-dependent cell death, and necroptosis. Phytother Res. 2023;37:1488-525
40. Corno C, Perego P. KiSS1 in regulation of metastasis and response to antitumor drugs. Drug Resist Updat. 2019;42:12-21
41. Hait AS, Olagnier D, Sancho-Shimizu V, Skipper KA, Helleberg M, Larsen SM. et al. Defects in LC3B2 and ATG4A underlie HSV2 meningitis and reveal a critical role for autophagy in antiviral defense in humans. Sci Immunol. 2020;5:eabc2691
42. Ye J, Zhang J, Zhu Y, Wang L, Jiang X, Liu B. et al. Targeting autophagy and beyond: deconvoluting the complexity of Beclin-1 from biological function to cancer therapy. Acta Pharm Sin B. 2023;13:4688-714
43. Dodson M, Liang Q, Johnson MS, Redmann M, Fineberg N, Darley-Usmar VM. et al. Inhibition of glycolysis attenuates 4-hydroxynonenal-dependent autophagy and exacerbates apoptosis in differentiated SH-SY5Y neuroblastoma cells. Autophagy. 2013;9:1996-2008
44. Moruno Manchon JF, Uzor NE, Finkbeiner S, Tsvetkov AS. SPHK1/sphingosine kinase 1-mediated autophagy differs between neurons and SH-SY5Y neuroblastoma cells. Autophagy. 2016;12:1418-24
45. Cheng G, Zhang Q, Pan J, Lee Y, Ouari O, Hardy M. et al. Targeting lonidamine to mitochondria mitigates lung tumorigenesis and brain metastasis. Nat Commun. 2019;10:2205
46. Li QX, Zhou X, Huang TT, Tang Y, Liu B, Peng P. et al. The Thr300Ala variant of ATG16L1 is associated with decreased risk of brain metastasis in patients with non-small cell lung cancer. Autophagy. 2017;13:1053-63
47. Shi LL, Chen Y, Xie MX, Chen QZ, Qiao XW, Cheng QH. et al. UBE2T/CDC42/CD276 signaling axis mediates brain metastasis of triple-negative breast cancer via lysosomal autophagy. J Immunother Cancer. 2025;13:e010782
48. Santana-Codina N, Muixí L, Foj R, Sanz-Pamplona R, Badia-Villanueva M, Abramowicz A. et al. GRP94 promotes brain metastasis by engaging pro-survival autophagy. Neuro Oncol. 2020;22:652-64
49. Kaverina N, Borovjagin AV, Kadagidze Z, Baryshnikov A, Baryshnikova M, Malin D. et al. Astrocytes promote progression of breast cancer metastases to the brain via a KISS1-mediated autophagy. Autophagy. 2017;13:1905-23
50. Ryu KJ, Lee KW, Park SH, Kim T, Hong K-S, Kim H. et al. Chaperone-mediated autophagy modulates snail protein stability: implications for breast cancer metastasis. Mol Cancer. 2024;23:227
51. Nawrocki ST, Espitia CM, Espinoza MJC, Gamble ME, Sureshkumar S, Chang M. et al. Targeting autophagy: a promising approach for the treatment of breast cancer brain metastases. Clin Transl Discov. 2024;4:e340
52. Talukdar S, Pradhan AK, Bhoopathi P, Shen XN, August LA, Windle JJ. et al. MDA-9/Syntenin regulates protective autophagy in anoikis-resistant glioma stem cells. Proc Natl Acad Sci U S A. 2018;115:5768-73
53. Xia Y, Xu F, Xiong M, Yang H, Lin W, Xie Y. et al. Repurposing of antipsychotic trifluoperazine for treating brain metastasis, lung metastasis and bone metastasis of melanoma by disrupting autophagy flux. Pharmacol Res. 2021;163:105295
54. Sugiarto S, Persson AI, Munoz EG, Waldhuber M, Lamagna C, Andor N. et al. Asymmetry-defective oligodendrocyte progenitors are glioma precursors. Cancer Cell. 2011;20:328-40
55. Liu C, Sage JC, Miller MR, Verhaak RGW, Hippenmeyer S, Vogel H. et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. 2011;146:209-21
56. Bergles DE, Roberts JD, Somogyi P, Jahr CE. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature. 2000;405:187-91
57. Osswald M, Jung E, Sahm F, Solecki G, Venkataramani V, Blaes J. et al. Brain tumour cells interconnect to a functional and resistant network. Nature. 2015;528:93-8
58. Weil S, Osswald M, Solecki G, Grosch J, Jung E, Lemke D. et al. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro Oncol. 2017;19:1316-26
59. Venkatesh HS, Morishita W, Geraghty AC, Silverbush D, Gillespie SM, Arzt M. et al. Electrical and synaptic integration of glioma into neural circuits. Nature. 2019;573:539-45
60. Venkataramani V, Tanev DI, Strahle C, Studier-Fischer A, Fankhauser L, Kessler T. et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature. 2019;573:532-8
61. Barron T, Yalçın B, Su M, Byun YG, Gavish A, Shamardani K. et al. GABAergic neuron-to-glioma synapses in diffuse midline gliomas. Nature. 2025;639:1060-8
62. Tetzlaff SK, Reyhan E, Layer N, Bengtson CP, Heuer A, Schroers J. et al. Characterizing and targeting glioblastoma neuron-tumor networks with retrograde tracing. Cell. 2025;188:390-411.e36
63. Krishna S, Choudhury A, Keough MB, Seo K, Ni L, Kakaizada S. et al. Glioblastoma remodelling of human neural circuits decreases survival. Nature. 2023;617:599-607
64. Zeng Q, Michael IP, Zhang P, Saghafinia S, Knott G, Jiao W. et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature. 2019;573:526-31
65. Neman J, Termini J, Wilczynski S, Vaidehi N, Choy C, Kowolik CM. et al. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc Natl Acad Sci U S A. 2014;111:984-9
66. Savchuk S, Gentry KM, Wang W, Carleton E, Biagi-Junior CAO, Luthria K. et al. Neuronal activity-dependent mechanisms of small cell lung cancer pathogenesis. Nature. 2025;646:1232-42
67. Onganer PU, Seckl MJ, Djamgoz MBA. Neuronal characteristics of small-cell lung cancer. Br J Cancer. 2005;93:1197-201
68. Yang D, Denny SK, Greenside PG, Chaikovsky AC, Brady JJ, Ouadah Y. et al. Intertumoral heterogeneity in SCLC is influenced by the cell type of origin. Cancer Discov. 2018;8:1316-31
69. Yang D, Qu F, Cai H, Chuang CH, Lim JS, Jahchan N. et al. Axon-like protrusions promote small cell lung cancer migration and metastasis. Elife. 2019;8:e50616
70. Chakraborty R, Nonaka T, Hasegawa M, Zurzolo C. Tunnelling nanotubes between neuronal and microglial cells allow bi-directional transfer of α-synuclein and mitochondria. Cell Death Dis. 2023;14:329
71. Scheiblich H, Eikens F, Wischhof L, Opitz S, Jüngling K, Cserép C. et al. Microglia rescue neurons from aggregate-induced neuronal dysfunction and death through tunneling nanotubes. Neuron. 2024;112:3106-3125.e8
72. Melwani PK, Pandey BN. Tunneling nanotubes: the intercellular conduits contributing to cancer pathogenesis and its therapy. Biochim Biophys Acta Rev Cancer. 2023;1878:189028
73. Lu WC, Xie H, Tie XX, Wang R, Wu AH, Shan FP. NFAT-1 hyper-activation by methionine enkephalin (MENK) significantly induces cell apoptosis of rats C6 glioma in vivo and in vitro. Int Immunopharmacol. 2018;56:1-8
74. Tuo Y, Tian C, Lu L, Xiang M. The paradoxical role of methionine enkephalin in tumor responses. Eur J Pharmacol. 2020;882:173253
75. Taylor KR, Barron T, Hui A, Spitzer A, Yalçin B, Ivec AE. et al. Glioma synapses recruit mechanisms of adaptive plasticity. Nature. 2023;623:366-74
76. Venkatesh HS, Johung TB, Caretti V, Noll A, Tang Y, Nagaraja S. et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell. 2015;161:803-16
77. Venkatesh HS, Tam LT, Woo PJ, Lennon J, Nagaraja S, Gillespie SM. et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature. 2017;549:533-7
78. Pan Y, Hysinger JD, Barron T, Schindler NF, Cobb O, Guo X. et al. NF1 mutation drives neuronal activity-dependent initiation of optic glioma. Nature. 2021;594:277-82
79. Yao M, Ventura PB, Jiang Y, Rodriguez FJ, Wang L, Perry JSA. et al. Astrocytic trans-differentiation completes a multicellular paracrine feedback loop required for medulloblastoma tumor growth. Cell. 2020;180:502-520.e19
80. Chen P, Wang W, Liu R, Lyu J, Zhang L, Li B. et al. Olfactory sensory experience regulates gliomagenesis via neuronal IGF1. Nature. 2022;606:550-6
81. Li H, Liu Y, Liu Y, Xu L, Sun Z, Zheng D. et al. Tumor-associated astrocytes promote tumor progression of sonic hedgehog medulloblastoma by secreting lipocalin-2. Brain Pathol. 2024;34:e13212
82. Qu F, Brough SC, Michno W, Madubata CJ, Hartmann GG, Puno A. et al. Crosstalk between small-cell lung cancer cells and astrocytes mimics brain development to promote brain metastasis. Nat Cell Biol. 2023;25:1506-19
83. Dong ZK, Wang YF, Li WP, Jin WL. Neurobiology of cancer: adrenergic signaling and drug repurposing. Pharmacol Ther. 2024;264:108750
84. Kobayashi H, Iida T, Ochiai Y, Malagola E, Zhi X, White RA. et al. Neuro-mesenchymal interaction mediated by a β2-adrenergic nerve growth factor feedforward loop promotes colorectal cancer progression. Cancer Discov. 2025;15:202-26
85. Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ. et al. Autonomic nerve development contributes to prostate cancer progression. Science. 2013;341:1236361
86. Sloan EK, Priceman SJ, Cox BF, Yu S, Pimentel MA, Tangkanangnukul V. et al. The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res. 2010;70:7042-52
87. Hayakawa Y, Sakitani K, Konishi M, Asfaha S, Niikura R, Tomita H. et al. Nerve growth factor promotes gastric tumorigenesis through aberrant cholinergic signaling. Cancer Cell. 2017;31:21-34
88. Renz BW, Tanaka T, Sunagawa M, Takahashi R, Jiang Z, Macchini M. et al. Cholinergic signaling via muscarinic receptors directly and indirectly suppresses pancreatic tumorigenesis and cancer stemness. Cancer Discov. 2018;8:1458-73
89. Gallo S, Vitacolonna A, Crepaldi T. NMDA receptor and its emerging role in cancer. Int J Mol Sci. 2023;24:2540
90. Li F, He C, Yao H, Zhao Y, Ye X, Zhou S. et al. Glutamate from nerve cells promotes perineural invasion in pancreatic cancer by regulating tumor glycolysis through HK2 mRNA-m6A modification. Pharmacol Res. 2023;187:106555
91. He S, Chen CH, Chernichenko N, He S, Bakst RL, Barajas F. et al. GFRα1 released by nerves enhances cancer cell perineural invasion through GDNF-RET signaling. Proc Natl Acad Sci U S A. 2014;111:E2008-2017
92. Ceyhan GO, Giese NA, Erkan M, Kerscher AG, Wente MN, Giese T. et al. The neurotrophic factor artemin promotes pancreatic cancer invasion. Ann Surg. 2006;244:274-81
93. Deborde S, Omelchenko T, Lyubchik A, Zhou Y, He S, McNamara WF. et al. Schwann cells induce cancer cell dispersion and invasion. J Clin Invest. 2016;126:1538-54
94. Aloe L, Rocco ML, Balzamino BO, Micera A. Nerve growth factor: role in growth, differentiation and controlling cancer cell development. J Exp Clin Cancer Res. 2016;35:116
95. Blondy S, Christou N, David V, Verdier M, Jauberteau MO, Mathonnet M. et al. Neurotrophins and their involvement in digestive cancers. Cell Death Dis. 2019;10:123
96. Zhang Y, Lin C, Liu Z, Sun Y, Chen M, Guo Y. et al. Cancer cells co-opt nociceptive nerves to thrive in nutrient-poor environments and upon nutrient-starvation therapies. Cell Metab. 2022;34:1999-2017.e10
97. Jiang T, Wang G, Liu Y, Feng L, Wang M, Liu J. et al. Development of small-molecule tropomyosin receptor kinase (TRK) inhibitors for NTRK fusion cancers. Acta Pharm Sin B. 2021;11:355-72
98. Jin M, Wang Y, Zhou T, Li W, Wen Q. Norepinephrine/β2-adrenergic receptor pathway promotes the cell proliferation and nerve growth factor production in triple-negative breast cancer. J Breast Cancer. 2023;26:268-85
99. Amaro F, Silva D, Reguengo H, Oliveira JC, Quintas C, Vale N. et al. β-adrenoceptor activation in breast MCF-10A cells induces a pattern of catecholamine production similar to that of tumorigenic MCF-7 cells. Int J Mol Sci. 2020;21:7968
100. Renz BW, Takahashi R, Tanaka T, Macchini M, Hayakawa Y, Dantes Z. et al. β2 adrenergic-neurotrophin feedforward loop promotes pancreatic cancer. Cancer Cell. 2018;33:75-90.e7
101. Banh RS, Biancur DE, Yamamoto K, Sohn ASW, Walters B, Kuljanin M. et al. Neurons release serine to support mRNA translation in pancreatic cancer. Cell. 2020;183:1202-1218.e25
102. Padmanaban V, Keller I, Seltzer ES, Ostendorf BN, Kerner Z, Tavazoie SF. Neuronal substance P drives metastasis through an extracellular RNA-TLR7 axis. Nature. 2024;633:207-15
103. Farooq MA, Ajmal I, Hui X, Chen Y, Ren Y, Jiang W. β2-adrenergic receptor mediated inhibition of T cell function and its implications for CAR-T cell therapy. Int J Mol Sci. 2023;24:12837
104. Capellino S, Claus M, Watzl C. Regulation of natural killer cell activity by glucocorticoids, serotonin, dopamine, and epinephrine. Cell Mol Immunol. 2020;17:705-11
105. Guyot M, Simon T, Panzolini C, Ceppo F, Daoudlarian D, Murris E. et al. Apical splenic nerve electrical stimulation discloses an anti-inflammatory pathway relying on adrenergic and nicotinic receptors in myeloid cells. Brain Behav Immun. 2019;80:238-46
106. Pan Y, Xiong M, Chen R, Ma Y, Corman C, Maricos M. et al. Athymic mice reveal a requirement for T-cell-microglia interactions in establishing a microenvironment supportive of Nf1 low-grade glioma growth. Genes Dev. 2018;32:491-6
107. Guo X, Pan Y, Xiong M, Sanapala S, Anastasaki C, Cobb O. et al. Midkine activation of CD8+ T cells establishes a neuron-immune-cancer axis responsible for low-grade glioma growth. Nat Commun. 2020;11:2177
108. Geiß C, Witzler C, Poschet G, Ruf W, Régnier-Vigouroux A. Metabolic and inflammatory reprogramming of macrophages by ONC201 translates in a pro-inflammatory environment even in presence of glioblastoma cells. Eur J Immunol. 2021;51:1246-61
109. Garofalo S, D’Alessandro G, Chece G, Brau F, Maggi L, Rosa A. et al. Enriched environment reduces glioma growth through immune and non-immune mechanisms in mice. Nat Commun. 2015;6:6623
110. Garofalo S, Porzia A, Mainiero F, Di Angelantonio S, Cortese B, Basilico B. et al. Environmental stimuli shape microglial plasticity in glioma. Elife. 2017;6:e33415
111. Laureys G, Gerlo S, Spooren A, Demol F, De Keyser J, Aerts JL. β₂-adrenergic agonists modulate TNF-α induced astrocytic inflammatory gene expression and brain inflammatory cell populations. J Neuroinflammation. 2014;11:21
112. Quan W, Xu CS, Ma C, Chen X, Yu DH, Li ZY. et al. Anti-tumor effects of telmisartan in glioma-astrocyte non-contact co-cultures: a critical role of astrocytic IL-6-mediated paracrine growth promotion. Int Immunopharmacol. 2024;139:112707
113. Li Y, Huang M, Wang M, Wang Y, Deng P, Li C. et al. Tumor cells impair immunological synapse formation via central nervous system-enriched metabolite. Cancer Cell. 2024;42:985-1002.e18
114. Layer N, Bunse L, Venkataramani V. Neural deception: breast cancer co-opts neuronal mechanisms to evade the immune system. Cancer Cell. 2024;42:936-8
115. Gabanyi I, Muller PA, Feighery L, Oliveira TY, Costa-Pinto FA, Mucida D. Neuro-immune interactions drive tissue programming in intestinal macrophages. Cell. 2016;164:378-91
116. Balood M, Ahmadi M, Eichwald T, Ahmadi A, Majdoubi A, Roversi K. et al. Nociceptor neurons affect cancer immunosurveillance. Nature. 2022;611:405-12
117. Lin Y, Liu Y, Gao Z, Jing D, Bi R, Cui X. et al. Beta-adrenergic receptor blocker propranolol triggers anti-tumor immunity and enhances irinotecan therapy in mice colorectal cancer. Eur J Pharmacol. 2023;949:175718
118. Chen M, Qiao G, Hylander BL, Mohammadpour H, Wang XY, Subjeck JR. et al. Adrenergic stress constrains the development of anti-tumor immunity and abscopal responses following local radiation. Nat Commun. 2020;11:1821
119. Zhao X, Li F, Cheng C, Bi M, Li J, Cong J. et al. Social isolation promotes tumor immune evasion via β2-adrenergic receptor. Brain Behav Immun. 2025;123:607-18
120. Qiao G, Chen M, Mohammadpour H, MacDonald CR, Bucsek MJ, Hylander BL. et al. Chronic adrenergic stress contributes to metabolic dysfunction and an exhausted phenotype in T cells in the tumor microenvironment. Cancer Immunol Res. 2021;9:651-64
121. Mohammadpour H, MacDonald CR, Qiao G, Chen M, Dong B, Hylander BL. et al. β2 adrenergic receptor-mediated signaling regulates the immunosuppressive potential of myeloid-derived suppressor cells. J Clin Invest. 2019;129:5537-52
122. Mohammadpour H, MacDonald CR, McCarthy PL, Abrams SI, Repasky EA. β2-adrenergic receptor signaling regulates metabolic pathways critical to myeloid-derived suppressor cell function within the TME. Cell Rep. 2021;37:109883
123. Mohammadpour H, Bucsek MJ, Hylander BL, Repasky EA. Depression stresses the immune response and promotes prostate cancer growth. Clin Cancer Res. 2019;25:2363-5
124. Cheng Y, Tang XY, Li YX, Zhao DD, Cao QH, Wu HX. et al. Depression-induced neuropeptide Y secretion promotes prostate cancer growth by recruiting myeloid cells. Clin Cancer Res. 2019;25:2621-32
125. Yang MW, Tao LY, Jiang YS, Yang JY, Huo YM, Liu DJ. et al. Perineural invasion reprograms the immune microenvironment through cholinergic signaling in pancreatic ductal adenocarcinoma. Cancer Res. 2020;80:1991-2003
126. Cavel O, Shomron O, Shabtay A, Vital J, Trejo-Leider L, Weizman N. et al. Endoneurial macrophages induce perineural invasion of pancreatic cancer cells by secretion of GDNF and activation of RET tyrosine kinase receptor. Cancer Res. 2012;72:5733-43
127. Yin T, Wang G, Wang L, Mudgal P, Wang E, Pan CC. et al. Breaking NGF-TrkA immunosuppression in melanoma sensitizes immunotherapy for durable memory T cell protection. Nat Immunol. 2024;25:268-81
128. Yu Q, Wang Y, Yi G, Yang W, Chen K, Tan X. et al. BDNF is a prognostic biomarker involved in immune infiltration of lung adenocarcinoma and is associated with brain metastasis. Immunology. 2023;168:320-30
129. Liu YN, Chen WY, Liu MK, Yeh HL, Chen WH, Jiang KC. et al. Immunosuppressive role of BDNF in therapy-induced neuroendocrine prostate cancer. Mol Oncol. 2024;18:1665-86
130. Zhang S, Huang H, Handley M, Griffin N, Bai X, Shan F. A novel mechanism of lung cancer inhibition by methionine enkephalin through remodeling the immune status of the tumor microenvironment. Int Immunopharmacol. 2021;99:107999
131. Wang X, Li S, Yan S, Shan Y, Wang X, Jingbo Z. et al. Methionine enkephalin inhibits colorectal cancer by remodeling the immune status of the tumor microenvironment. Int Immunopharmacol. 2022;111:109125
132. Zhang Y, Guo Y, Liu Z, Sun Y, Yang X, Chen M. et al. Cancer cells co-opt an inter-organ neuroimmune circuit to escape immune surveillance. Cell. 2025;188:6754-6773.e29
133. Mishra AK, ur Rasheed MS, Shukla S, Tripathi MK, Dixit A, Singh MP. Aberrant autophagy and parkinsonism: does correction rescue from disease progression? Mol Neurobiol. 2015;51:893-908
134. Tsopela V, Korakidis E, Lagou D, Kalliampakou KI, Milona RS, Kyriakopoulou E. et al. L-dopa decarboxylase modulates autophagy in hepatocytes and is implicated in dengue virus-caused inhibition of autophagy completion. Biochim Biophys Acta Mol Cell Res. 2024;1871:119602
135. Kuijpers M, Haucke V. Neuronal autophagy controls the axonal endoplasmic reticulum to regulate neurotransmission in healthy neurons. Autophagy. 2021;17:1049-51
136. Yang S, Park D, Manning L, Kargbo-Hill SE, Cao M, Xuan Z. et al. Presynaptic autophagy is coupled to the synaptic vesicle cycle via ATG-9. Neuron. 2022;110:824-840.e10
137. Nakamura Y, Sawai T, Kakiuchi K, Arawaka S. Neuronal activity promotes secretory autophagy for the extracellular release of α-synuclein. J Biol Chem. 2024;300:107419
138. Duplan E, Bernardin A, Goiran T, Leroudier N, Casimiro M, Pestell R. et al. α-synuclein expression in glioblastoma restores tumor suppressor function and rescues temozolomide drug resistance. Cell Death Dis. 2025;16:188
139. Zhi X, Li B, Li Z, Zhang J, Yu J, Zhang L. et al. Adrenergic modulation of AMPK-dependent autophagy by chronic stress enhances cell proliferation and survival in gastric cancer. Int J Oncol. 2019;54:1625-38
140. Dolma S, Selvadurai HJ, Lan X, Lee L, Kushida M, Voisin V. et al. Inhibition of dopamine receptor D4 impedes autophagic flux, proliferation, and survival of glioblastoma stem cells. Cancer Cell. 2016;29:859-73
141. Soll C, Jang JH, Riener MO, Moritz W, Wild PJ, Graf R. et al. Serotonin promotes tumor growth in human hepatocellular cancer. Hepatology. 2010;51:1244-54
142. Niture S, Gyamfi MA, Kedir H, Arthur E, Ressom H, Deep G. et al. Serotonin induced hepatic steatosis is associated with modulation of autophagy and notch signaling pathway. Cell Commun Signal. 2018;16:78
143. Hui KK, Tanaka M. Autophagy links MTOR and GABA signaling in the brain. Autophagy. 2019;15:1848-9
144. Liu Y, Zhang H, Wang Z, Wu P, Gong W. 5-Hydroxytryptamine1a receptors on tumour cells induce immune evasion in lung adenocarcinoma patients with depression via autophagy/pSTAT3. Eur J Cancer. 2019;114:8-24
145. Tang Z, Xue Z, Liu X, Zhang Y, Zhao J, Liu J. et al. Inhibition of hypoxic exosomal miR-423-3p decreases glioma progression by restricting autophagy in astrocytes. Cell Death Dis. 2025;16:265
146. Bai X, Cao X, Qu N, Huang H, Handley M, Zhang S. et al. Methionine enkephalin activates autophagy and stimulates tumour cell immunogenicity in human cutaneous squamous cell carcinoma. Int Immunopharmacol. 2021;96:107733
147. Cheng J, Liao Y, Dong Y, Hu H, Yang N, Kong X. et al. Microglial autophagy defect causes Parkinson disease-like symptoms by accelerating inflammasome activation in mice. Autophagy. 2020;16:2193-205
148. Li B, Liu Y, Chen D, Sun S. Comprehensive analysis of predictive value and the potential therapeutic target of NLRP3 inflammasome in glioma based on tumor microenvironment. Clin Immunol. 2024;261:109918
149. Mészáros Á, Molnár K, Fazakas C, Nógrádi B, Lüvi A, Dudás T. et al. Inflammasome activation in peritumoral astrocytes is a key player in breast cancer brain metastasis development. Acta Neuropathol Commun. 2023;11:155
150. Varveri A, Papadopoulou M, Papadovasilakis Z, Compeer EB, Legaki AI, Delis A. et al. Immunological synapse formation between T regulatory cells and cancer-associated fibroblasts promotes tumour development. Nat Commun. 2024;15:4988
151. Akalay I, Janji B, Hasmim M, Noman MZ, André F, De Cremoux P. et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer Res. 2013;73:2418-27
152. Li J, Kang R, Tang D. Cellular and molecular mechanisms of perineural invasion of pancreatic ductal adenocarcinoma. Cancer Commun (Lond). 2021;41:642-60
153. Becker KN, Pettee KM, Sugrue A, Reinard KA, Schroeder JL, Eisenmann KM. The cytoskeleton effectors rho-kinase (ROCK) and mammalian diaphanous-related (mDia) formin have dynamic roles in tumor microtube formation in invasive glioblastoma cells. Cells. 2022;11:1559
154. Kast DJ, Dominguez R. The cytoskeleton-autophagy connection. Curr Biol. 2017;27:R318-26
155. Joseph JV, Magaut CR, Storevik S, Geraldo LH, Mathivet T, Latif MA. et al. TGF-β promotes microtube formation in glioblastoma through thrombospondin 1. Neuro Oncol. 2022;24:541-53
156. Shen Y, Lu C, Song Z, Qiao C, Wang J, Chen J. et al. Ursodeoxycholic acid reduces antitumor immunosuppression by inducing CHIP-mediated TGF-β degradation. Nat Commun. 2022;13:3419
157. Li H, Li J, Chen L, Qi S, Yu S, Weng Z. et al. HERC3-mediated SMAD7 ubiquitination degradation promotes autophagy-induced EMT and chemoresistance in glioblastoma. Clin Cancer Res. 2019;25:3602-16
158. Portela M, Venkataramani V, Fahey-Lozano N, Seco E, Losada-Perez M, Winkler F. et al. Glioblastoma cells vampirize WNT from neurons and trigger a JNK/MMP signaling loop that enhances glioblastoma progression and neurodegeneration. PLoS Biol. 2019;17:e3000545
159. Colella B, Faienza F, Carinci M, D’Alessandro G, Catalano M, Santoro A. et al. Autophagy induction impairs wnt/β-catenin signalling through β-catenin relocalisation in glioblastoma cells. Cell Signal. 2019;53:357-64
160. Jung E, Osswald M, Ratliff M, Dogan H, Xie R, Weil S. et al. Tumor cell plasticity, heterogeneity, and resistance in crucial microenvironmental niches in glioma. Nat Commun. 2021;12:1014
161. Jung E, Osswald M, Blaes J, Wiestler B, Sahm F, Schmenger T. et al. Tweety-homolog 1 drives brain colonization of gliomas. J Neurosci. 2017;37:6837-50
162. Lai W, He Y, Zhou B, Wu Q, Wu H, Chen J. et al. Salidroside facilitates neuroprotective effects in ischemic stroke by promoting axonal sprouting through promoting autophagy. Phytomedicine. 2024;135:156208
163. Leung HH, Mansour C, Rousseau M, Nakhla A, Kiselyov K, Venkatachalam K. et al. Drosophila tweety facilitates autophagy to regulate mitochondrial homeostasis and bioenergetics in glia. Glia. 2024;72:433-51
164. Zhang K, Wang F, Zhai M, He M, Hu Y, Feng L. et al. Hyperactive neuronal autophagy depletes BDNF and impairs adult hippocampal neurogenesis in a corticosterone-induced mouse model of depression. Theranostics. 2023;13:1059-75
165. Martinelli S, Anderzhanova EA, Bajaj T, Wiechmann S, Dethloff F, Weckmann K. et al. Stress-primed secretory autophagy promotes extracellular BDNF maturation by enhancing MMP9 secretion. Nat Commun. 2021;12:4643
166. Jawhari S, Bessette B, Hombourger S, Durand K, Lacroix A, Labrousse F. et al. Autophagy and TrkC/NT-3 signaling joined forces boost the hypoxic glioblastoma cell survival. Carcinogenesis. 2017;38:592-603
167. Mazouffre C, Geyl S, Perraud A, Blondy S, Jauberteau MO, Mathonnet M. et al. Dual inhibition of BDNF/TrkB and autophagy: a promising therapeutic approach for colorectal cancer. J Cell Mol Med. 2017;21:2610-22
168. Zajicek AS, Ruan H, Dai H, Skolfield MC, Phillips HL, Burnette WJ. et al. Cylindromatosis drives synapse pruning and weakening by promoting macroautophagy through akt-mTOR signaling. Mol Psychiatry. 2022;27:2414-24
169. Crawley O, Opperman KJ, Desbois M, Adrados I, Borgen MA, Giles AC. et al. Autophagy is inhibited by ubiquitin ligase activity in the nervous system. Nat Commun. 2019;10:5017
170. Fu S, Sun H, Wang J, Gao S, Zhu L, Cui K. et al. Impaired neuronal macroautophagy in the prelimbic cortex contributes to comorbid anxiety-like behaviors in rats with chronic neuropathic pain. Autophagy. 2024;20:1559-76
171. Shi L, Yan H, An S, Shen M, Jia W, Zhang R. et al. SIRT5-mediated deacetylation of LDHB promotes autophagy and tumorigenesis in colorectal cancer. Mol Oncol. 2019;13:358-75
172. Wu S, Wei Y, Li J, Bai Y, Yin P, Wang S. SIRT5 represses neurotrophic pathways and Aβ production in Alzheimer’s disease by targeting autophagy. ACS Chem Neurosci. 2021;12:4428-37
173. Hai L, Hoffmann DC, Wagener RJ, Azorin DD, Hausmann D, Xie R. et al. A clinically applicable connectivity signature for glioblastoma includes the tumor network driver CHI3L1. Nat Commun. 2024;15:968
174. Hong DE, Yu JE, Yoo SS, Yeo IJ, Son DJ, Yun J. et al. CHI3L1 induces autophagy through the JNK pathway in lung cancer cells. Sci Rep. 2023;13:9964
175. Ye ZC, Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res. 1999;59:4383-91
176. Berendsen S, Varkila M, Kroonen J, Seute T, Snijders TJ, Kauw F. et al. Prognostic relevance of epilepsy at presentation in glioblastoma patients. Neuro Oncol. 2016;18:700-6
177. Piao Y, Lu L, de Groot J. AMPA receptors promote perivascular glioma invasion via beta1 integrin-dependent adhesion to the extracellular matrix. Neuro Oncol. 2009;11:260-73
178. Campbell SL, Robel S, Cuddapah VA, Robert S, Buckingham SC, Kahle KT. et al. GABAergic disinhibition and impaired KCC2 cotransporter activity underlie tumor-associated epilepsy. Glia. 2015;63:23-36
179. Conti L, Palma E, Roseti C, Lauro C, Cipriani R, de Groot M. et al. Anomalous levels of Cl- transporters cause a decrease of GABAergic inhibition in human peritumoral epileptic cortex. Epilepsia. 2011;52:1635-44
180. Blanchart A, Fernando R, Häring M, Assaife-Lopes N, Romanov RA, Andäng M. et al. Endogenous GABAA receptor activity suppresses glioma growth. Oncogene. 2017;36:777-86
181. Yu K, Lin C-CJ, Hatcher A, Lozzi B, Kong K, Huang-Hobbs E. et al. PIK3CA variants selectively initiate brain hyperactivity during gliomagenesis. Nature. 2020;578:166-71
182. Mauffrey P, Tchitchek N, Barroca V, Bemelmans AP, Firlej V, Allory Y. et al. Progenitors from the central nervous system drive neurogenesis in cancer. Nature. 2019;569:672-8
183. Amit M, Takahashi H, Dragomir MP, Lindemann A, Gleber-Netto FO, Pickering CR. et al. Loss of p53 drives neuron reprogramming in head and neck cancer. Nature. 2020;578:449-54
184. Gastaldi M, Scaranzin S, Pietro B, Lechiara A, Pesce G, Franciotta D. et al. Paraneoplastic neurological syndromes: transitioning between the old and the new. Curr Oncol Rep. 2022;24:1237-49
185. Dong L, He J, Luo L, Wang K. Targeting the interplay of autophagy and ROS for cancer therapy: an updated overview on phytochemicals. Pharmaceuticals (Basel). 2023;16:92
186. Balogh M, Zhang J, Gaffney CM, Kalakuntla N, Nguyen NT, Trinh RT. et al. Sensory neuron dysfunction in orthotopic mouse models of colon cancer. J Neuroinflammation. 2022;19:204
187. Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A. et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. 2016;533:493-8
188. Nguyen LD, Ehrlich BE. Cellular mechanisms and treatments for chemobrain: insight from aging and neurodegenerative diseases. EMBO Mol Med. 2020;12:e12075
189. Tao H, Wang C, Zou C, Zhu H, Zhang W. Unraveling the potential of neuroinflammation and autophagy in schizophrenia. Eur J Pharmacol. 2025;997:177469
190. Park HS, Kim CJ, Kwak HB, No MH, Heo JW, Kim TW. Physical exercise prevents cognitive impairment by enhancing hippocampal neuroplasticity and mitochondrial function in doxorubicin-induced chemobrain. Neuropharmacology. 2018;133:451-61
191. Geraghty AC, Gibson EM, Ghanem RA, Greene JJ, Ocampo A, Goldstein AK. et al. Loss of adaptive myelination contributes to methotrexate chemotherapy-related cognitive impairment. Neuron. 2019;103:250-265.e8
192. Shi Z, Yu P, Lin WJ, Chen S, Hu X, Chen S. et al. Microglia drive transient insult-induced brain injury by chemotactic recruitment of CD8+ T lymphocytes. Neuron. 2023;111:696-710.e9
193. Fan Y, Wang Y, Zhang J, Dong X, Gao P, Liu K. et al. Breaking bad: autophagy tweaks the interplay between glioma and the tumor immune microenvironment. Front Immunol. 2021;12:746621
194. Wang H, Sun P, Yuan X, Xu Z, Jiang X, Xiao M. et al. Autophagy in tumor immune escape and immunotherapy. Mol Cancer. 2025;24:85
195. New J, Thomas SM. Autophagy-dependent secretion: mechanism, factors secreted, and disease implications. Autophagy. 2019;15:1682-93
196. Monkkonen T, Debnath J. Inflammatory signaling cascades and autophagy in cancer. Autophagy. 2018;14:190-8
197. Shifman SG, O’Connor JL, Radin DP, Sharma A, Infante L, Ferraresso F. et al. Targeting autophagy and plasminogen activator inhibitor-1 increases survival and remodels the tumor microenvironment in glioblastoma. J Exp Clin Cancer Res. 2025;44:214
198. Dias-Carvalho A, Ferreira M, Reis-Mendes A, Ferreira R, Bastos ML, Fernandes E. et al. Chemobrain: mitoxantrone-induced oxidative stress, apoptotic and autophagic neuronal death in adult CD-1 mice. Arch Toxicol. 2022;96:1767-82
199. Abdel-Aziz AK, Mantawy EM, Said RS, Helwa R. The tyrosine kinase inhibitor, sunitinib malate, induces cognitive impairment in vivo via dysregulating VEGFR signaling, apoptotic and autophagic machineries. Exp Neurol. 2016;283:129-41
200. Luo N, Zhu W, Li X, Fu M, Zhang Y, Yang F. et al. Defective autophagy of pericytes enhances radiation-induced senescence promoting radiation brain injury. Neuro Oncol. 2024;26:2288-304
201. Yun EJ, Kim S, Hsieh JT, Baek ST. Wnt/β-catenin signaling pathway induces autophagy-mediated temozolomide-resistance in human glioblastoma. Cell Death Dis. 2020;11:771
202. Mitrakas AG, Kalamida D, Giatromanolaki A, Pouliliou S, Tsolou A, Kyranas R. et al. Autophagic flux response and glioblastoma sensitivity to radiation. Cancer Biol Med. 2018;15:260-74
203. Mravec B. Neurobiology of cancer: introduction of new drugs in the treatment and prevention of cancer. Int J Mol Sci. 2021;22:6115
204. Fumagalli C, Maurizi N, Marchionni N, Fornasari D. β-blockers: their new life from hypertension to cancer and migraine. Pharmacol Res. 2020;151:104587
205. Yuan D, Hu J, Ju X, Putz EM, Zheng S, Koda S. et al. NMDAR antagonists suppress tumor progression by regulating tumor-associated macrophages. Proc Natl Acad Sci U S A. 2023;120:e2302126120
206. Grossman SA, Ye X, Chamberlain M, Mikkelsen T, Batchelor T, Desideri S. et al. Talampanel with standard radiation and temozolomide in patients with newly diagnosed glioblastoma: a multicenter phase II trial. J Clin Oncol. 2009;27:4155-61
207. Ahmed EA, Alkuwayti MA, Ibrahim H-IM. Atropine is a suppressor of epithelial-mesenchymal transition (EMT) that reduces stemness in drug-resistant breast cancer cells. Int J Mol Sci. 2022;23:9849
208. Kuol N, Davidson M, Karakkat J, Filippone RT, Veale M, Luwor R. et al. Blocking muscarinic receptor 3 attenuates tumor growth and decreases immunosuppressive and cholinergic markers in an orthotopic mouse model of colorectal cancer. Int J Mol Sci. 2022;24:596
209. Prabhu VV, Morrow S, Rahman Kawakibi A, Zhou L, Ralff M, Ray J. et al. ONC201 and imipridones: anti-cancer compounds with clinical efficacy. Neoplasia. 2020;22:725-44
210. Peng T, Guo Y, Gan Z, Ling Y, Xiong J, Liang X. et al. Nerve growth factor (NGF) encourages the neuroinvasive potential of pancreatic cancer cells by activating the Warburg effect and promoting tumor derived exosomal miRNA-21 expression. Oxid Med Cell Longev. 2022;2022:8445093
211. Doz F, van Tilburg CM, Geoerger B, Højgaard M, Øra I, Boni V. et al. Efficacy and safety of larotrectinib in TRK fusion-positive primary central nervous system tumors. Neuro Oncol. 2022;24:997-1007
212. Schneider M, Vollmer L, Potthoff AL, Ravi VM, Evert BO, Rahman MA. et al. Meclofenamate causes loss of cellular tethering and decoupling of functional networks in glioblastoma. Neuro Oncol. 2021;23:1885-97
213. You F, Zhang C, Liu X, Ji D, Zhang T, Yu R. et al. Drug repositioning: using psychotropic drugs for the treatment of glioma. Cancer Lett. 2022;527:140-9
214. Yang YH, Liu JB, Gui Y, Lei LL, Zhang SJ. Relationship between autophagy and perineural invasion, clinicopathological features, and prognosis in pancreatic cancer. World J Gastroenterol. 2017;23:7232-41
215. Shi J, Xu J, Li Y, Li B, Ming H, Nice EC. et al. Drug repurposing in cancer neuroscience: from the viewpoint of the autophagy-mediated innervated niche. Front Pharmacol. 2022;13:990665
216. Kataura T, Sedlackova L, Sun C, Kocak G, Wilson N, Banks P. et al. Targeting the autophagy-NAD axis protects against cell death in Niemann-Pick type C1 disease models. Cell Death Dis. 2024;15:382
217. Wu H, Wang X, Liang H, Zheng J, Huang S, Zhang D. Enhanced efficacy of propranolol therapy for infantile hemangiomas based on a mesoporous silica nanoplatform through mediating autophagy dysfunction. Acta Biomater. 2020;107:272-85
218. Cao J, Cao F, Wang C, Jiao Z, You Y, Wang X. et al. ONC206 targeting ClpP induces mitochondrial dysfunction and protective autophagy in hepatocellular carcinoma cells. Neoplasia. 2024;55:101015
219. Bao S, Zhang Y, Zeng J, Zhang B, Wang H, Li X. et al. Innovative role of the antidepressant imipramine in esophageal squamous cell carcinoma treatment: promoting apoptosis and protective autophagy. Int Immunopharmacol. 2025;147:113969
220. Ordoñez R, Fernández A, Prieto-Domínguez N, Martínez L, García-Ruiz C, Fernández-Checa JC. et al. Ceramide metabolism regulates autophagy and apoptotic cell death induced by melatonin in liver cancer cells. J Pineal Res. 2015;59:178-89
221. Kong W, Zhu H, Zheng S, Yin G, Yu P, Shan Y. et al. Larotrectinib induces autophagic cell death through AMPK/mTOR signalling in colon cancer. J Cell Mol Med. 2022;26:5539-50
222. Compter I, Eekers DBP, Hoeben A, Rouschop KMA, Reymen B, Ackermans L. et al. Chloroquine combined with concurrent radiotherapy and temozolomide for newly diagnosed glioblastoma: a phase IB trial. Autophagy. 2021;17:2604-12
223. Petrosyan E, Fares J, Cordero A, Rashidi A, Arrieta VA, Kanojia D. et al. Repurposing autophagy regulators in brain tumors. Int J Cancer. 2022;151:167-80
224. Jeon HM, Oh YT, Shin YJ, Chang N, Kim D, Woo D. et al. Dopamine receptor D2 regulates glioblastoma survival and death through MET and death receptor 4/5. Neoplasia. 2023;39:100894
225. Lei M, Liu Q, Nie J, Huang R, Mei Y, Pan D. et al. Impact and mechanisms of action of BDNF on neurological disorders, cancer, and cardiovascular diseases. CNS Neurosci Ther. 2024;30:e70138
226. Li Z, Gao W, Fei Y, Gao P, Xie Q, Xie J. et al. NLGN3 promotes neuroblastoma cell proliferation and growth through activating PI3K/AKT pathway. Eur J Pharmacol. 2019;857:172423
Author contact
![]() Corresponding authors: Tel./Fax: 86-28-85164063. E-mail addresses: quyi-henrycom (Yi Qu); liubo2400com (Bo Liu); aprillee202com (Aizhuo Li).
Corresponding authors: Tel./Fax: 86-28-85164063. E-mail addresses: quyi-henrycom (Yi Qu); liubo2400com (Bo Liu); aprillee202com (Aizhuo Li).