Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
1. Mechano-Immunity in the...
2. Aging-Related...
3. Mechanical-Immune Imbalance...
4. Strategies for Modulating...
5. Conclusion
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2026; 16(13):7735-7764. doi:10.7150/thno.131071 This issue Cite
Review
Mechano-immune interactions in musculoskeletal aging: Mechanisms and translational perspectives
Fulin Zhou#, Xianglong Zhou#, Jiheng Xiao, Jianhui Xiang, Yang Liu, Weicheng Chen, Hanhong Fang, Haoran Zhou, Xiao Lv ![]() , Liming Xiong
, Liming Xiong ![]()
Department of Orthopedics, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei Province 430022, China.
# Contributed equally.
Received 2026-1-7; Accepted 2026-3-12; Published 2026-6-17
Abstract

Mechano-immunology is an interdisciplinary field that examines how mechanical forces—including endogenous tissue stresses and exogenous stimuli—modulate immune cell function in the maintenance of homeostasis and the progression of disease. As the primary load-bearing framework of the body, the musculoskeletal system depends on coordinated mechanical loading to sustain immune balance and tissue repair. With aging, reduced physical activity, progressive matrix stiffening, and impaired mechanosensing in immune-cells converge to disrupt this mechano-immune axis, undermining immune clearance, inflammatory regulation, and regenerative capacity within musculoskeletal tissues. These alterations collectively contribute to the onset and progression of osteoarthritis, osteoporosis, and other degenerative musculoskeletal disorders. This review summarizes current advances in understanding the mechanisms of mechano-immunology within the musculoskeletal system under physiological conditions and delineates its involvement in the initiation and progression of degenerative diseases. Emerging strategies, including exercise-based interventions, controlled mechanical stimulation, and biomaterial engineering approaches targeting mechano-immune pathways, are also discussed as potential means to restore musculoskeletal homeostasis during aging.
Keywords: aging, mechano-immunology, mechanobiology, musculoskeletal system, degenerative diseases, mechanotherapy
Introduction
With the acceleration of global population aging, the incidence of degenerative musculoskeletal diseases—including osteoporosis (OP), osteoarthritis (OA), intervertebral disc degeneration (IDD), and sarcopenia—has risen significantly. These conditions substantially impair quality of life among older adults and impose an increasing burden on healthcare systems and society [1-3]. Mechanical imbalance and chronic inflammation have traditionally been considered as primary contributors to disease progression. However, neither factor alone adequately explained the complex and multifaceted nature of age-related musculoskeletal degeneration [4-7]. The emerging discipline of mechano-immunology provides a unifying conceptual framework to address this gap. By elucidating the bidirectional interplay between mechanical inputs and immune cell behavior, mechano-immunology offers insight into how aging-associated alterations in tissue stiffness, mechanosensing capacity of immune cells, and mechanical load distribution reshape immune regulation and drive musculoskeletal degeneration [8].
In young individuals (Figure 1, left), mechanical homeostasis is maintained across multiple organ systems. Key tissues—including the heart, skeletal muscle, intervertebral discs (IVD), and bone—are characterized by relatively low stiffness, high elasticity, and efficient force transmission [9-12]. Under these conditions, physiological loading is translated into coordinated mechano-immune signaling that activates resident tissue cells and immune cells, promotes extracellular matrix (ECM) remodeling, and sustains regenerative pathways across tissues [13, 14]. In bone, appropriate microstrain stimulates the periosteum, tissue-resident macrophages, and osteoblasts, thereby supporting continuous bone remodeling.
Systemic mechanical remodeling across lifespan shapes tissue homeostasis and degeneration. This schematic summarizes age-associated alterations in mechanical properties across major organs and musculoskeletal tissues. In youth, balanced mechanical loading supports matrix remodeling, stem-cell activation, mechanosensitive signaling, and coordinated tissue repair. With aging, increased stiffness, impaired force-transfer, reduced mechanosensitivity, and altered osmotic and strain environments disrupt mechano-immune homeostasis, leading to inflammation, fibrosis, degeneration, and diminished regenerative capacity. Together, chronic mechanical loss drives the transition from mechanical homeostasis to mechanical imbalance across the lifespan.
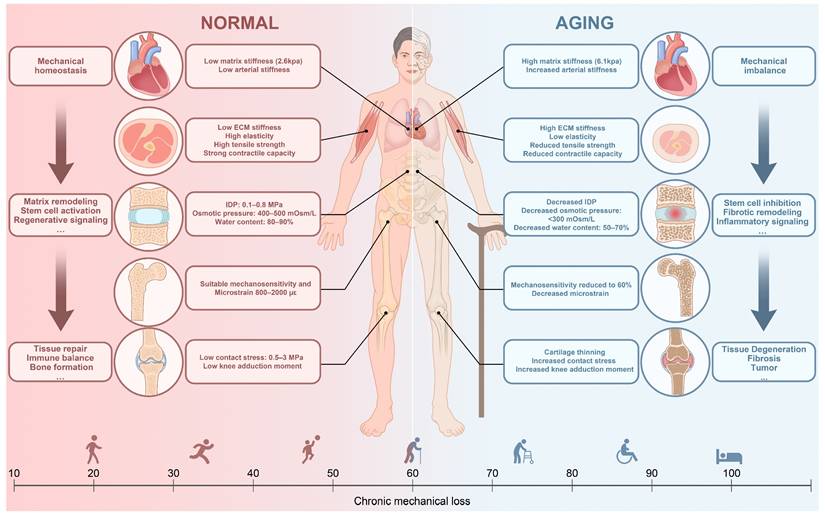
With aging (Figure 1, right), progressive mechanical remodeling occurs across organs and tissues. Increased in myocardial stiffness [15], heightened skeletal muscle rigidity accompanied by reduced elasticity and contractile capacity [16, 17], and a gradual decline in the efficiency of mechanical conduction in bone [18, 19] represent typical examples. These alterations disrupt the tightly regulated mechano-immune balance. At the cellular level, both innate and adaptive immune populations exhibit altered responsiveness to mechanical stimuli, with phenotypic shifts toward pro-inflammatory states that accelerate age-related pathology [20-22]. Concurrently, structural changes in the aging ECM reshape the mechanical microenvironment of immune cells, subjecting these cells to persistent aberrant mechanical stimuli [23, 24]. This age-related disruption of mechano-immune regulation contributes to bone loss, joint degeneration, annulus fibrosus rupture in IVD, and inflammation-associated sarcopenia, ultimately reinforcing a self-perpetuating cycle of degenerative musculoskeletal diseases.
Systematic elucidation of mechano-immune imbalance in the context of aging is crucial for clarifying the mechanisms underlying degenerative musculoskeletal diseases. A mechanistic framework centered on disrupted mechano-immune coupling provides deeper insight into disease initiation and progression. More importantly, this perspective informs potential clinical strategies. Integration of controlled mechanical loading—such as exercise, traction, or targeted biomechanical stimulation—with modulation of the immune microenvironment, including regulation of macrophage polarization and immunometabolic reprogramming, together with tissue engineering and regenerative medicine approaches, may facilitate restoration or reconstruction of mechano-immune coupling during the early stages of disease [25-27].
1. Mechano-Immunity in the Skeletal Muscle System
1.1 Mechanical Transduction in Immune Cells
Immune cells continuously perceive mechanical cues within the surrounding microenvironment, including ECM stiffness, compression, stretch, and shear stress (Figure 2, left; Table 1) [28-30]. Mechanotransduction is initiated at the plasma membrane through mechanosensitive structures such as integrins, T cell receptor (TCR), B cell receptor (BCR), piezo-type mechanosensitive ion channel component 1 (Piezo1), transient receptor potential vanilloid 4 (TRPV4), and two-pore-domain potassium (K2P) channels, including TREK-1/2 and TRAAK [31-37]. K2P channels are enriched in innate immune cells, particularly macrophages, where regulation of K+ flux contributes to modulation of inflammatory responses [38, 39]. Intrinsic mechanosensitivity positions these channels as potential mediators of macrophage responses to mechanical cues, although precise roles in macrophage mechanotransduction remain to be fully defined [38, 39].
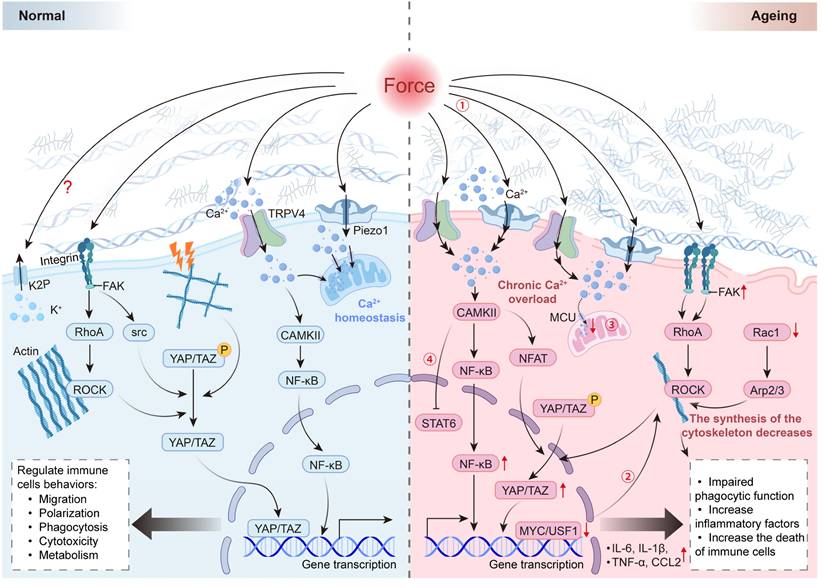
Schematic illustration of mechanotransduction cascades in normal and ageing immune cells. In normal immune cells, mechanical cues—including ECM stiffness, tension, compression, and shear stress—are sensed by mechanosensors such as integrins, Piezo1, and TRPV4. These inputs induce transient and well-controlled Ca²⁺ influx, cytoskeletal remodeling, and YAP/TAZ nuclear translocation, thereby maintaining NF-κB/STAT signaling homeostasis and supporting migration, phagocytosis, polarization, and metabolic activity. In contrast, aged immune cells exposed to stiffened ECM exhibit altered mechanosensing, leading to chronic Ca²⁺ overload, reduced mitochondrial buffering capacity, and aberrant activation of the ROCK-CAMKII-NF-κB/NFAT axis. These defects, together with diminished cytoskeletal synthesis and dysregulated YAP/TAZ activity, result in elevated inflammatory cytokines, impaired phagocytosis, and increased cell death, ultimately driving mechano-immune imbalance (①-④).
Summary of mechanosensing and mechanotransduction structures in immune cells.
| Category | Representative Molecules | Sensed Mechanical Stimuli | Signal Transduction Mechanism | Downstream Pathways | Reference |
|---|---|---|---|---|---|
| Adhesion receptors | Integrins (β1, β2, αvβ3, LFA-1) | ECM stiffness, tensile strain, shear stress | Force-induced clustering activates FAK, Src, and PLC, initiating phosphorylation cascades | MAPK, NF-κB, and YAP/TAZ activation; cytoskeletal remodeling | [35, 40, 41] |
| Immune receptors | TCR, BCR | Cell-cell traction, antigen stiffness | Receptor-ligand mechanical deformation triggers ITAM phosphorylation and Ca²⁺ influx | NFAT, NF-κB, ERK activation | [33, 34] |
| Mechanosensitive ion channels | Piezo1, TRPV4, K2P (TREK-1, TRAAK) | Membrane stretch, pressure, compression, osmotic and shear stress | Ion influx (Ca²⁺, Na⁺, K⁺) activates CaMKII, calcineurin, and PKC signaling | NFAT, NF-κB, YAP/TAZ; metabolic reprogramming | [31, 32, 39, 42] |
| Cytoskeletal network | Actin filaments, microtubules, intermediate filaments | Tensile/compressive stress, substrate rigidity | Transmission of mechanical tension via RhoA-ROCK and FAK pathways | Nuclear deformation; activation of YAP/TAZ, NF-κB | [46, 47] |
| Cytoskeleton-nucleus coupling | LINC complex (nesprin-SUN), lamins A/C | Cytoskeletal tension, substrate stiffness | Force transmission to nucleus; modulation of chromatin accessibility and nuclear stiffness | Transcriptional regulation; mechanosensitive gene expression | [43-45] |
ECM, extracellular matrix; FAK, focal adhesion kinase; PLC, phospholipase C; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor κB; YAP, Yes-associated protein; TAZ, transcriptional co-activator with PDZ-binding motif; TCR, T-cell receptor; BCR, B-cell receptor; ITAM, immunoreceptor tyrosine-based activation motif; CaMKII, calcium/calmodulin-dependent protein kinase II; PKC, protein kinase C; NFAT, nuclear factor of activated T cells; TRPV4, transient receptor potential vanilloid 4; TRPV2, transient receptor potential vanilloid 2; ASICs, acid-sensing ion channels; K2P, two-pore potassium channels; TREK-1, TWIK-related K⁺ channel 1; TRAAK, TWIK-related arachidonic acid-activated K⁺ channel; ROCK, Rho-associated kinase; LINC, linker of nucleoskeleton and cytoskeleton; SUN, Sad1/UNC-84 domain protein.
Activation of mechanosensors initiates two interelated signaling axes: biochemical cascades and cytoskeletal force transmission. Mechanical clustering of integrins promotes recruitment of focal adhesion kinase (FAK), Src, and phospholipase C (PLC), leading to activation of mitogen-activated protein kinase (MAPK) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathways [40, 41]. Concurrently, Ca²+ influx through Piezo1 and TRPV4 stimulates calmodulin-dependent kinase II (CaMKII) and calcineurin, facilitating nuclear translocation of nuclear factor of activated T cells (NFAT) and NF-κB [42].
In parallel, mechanical tension is propagated along the actin-microtubule cytoskeleton to the nucleus via the linker of nucleoskeleton and cytoskeleton (LINC) complex and lamins A/C, thereby modulating nuclear mechanics and chromatin organization [43-45]. Mechanosensitive transcriptional regulators, including yes-associated protein (YAP), transcriptional coactivator with PDZ-binding motif (TAZ), and NF-κB, integrate these inputs to reshape transcriptional programs[46, 47].
Collectively, these coordinated pathways regulate immune cell migration, differentiation, metabolic adaptation, and effector function. Under physiological mechanical conditions, such mechanisms support immune homeostasis, whereas persistent mechanical stress may amplify inflammatory responses and contribute to tissue dysfunction.
1.2 Mechano-Immunity in Musculoskeletal Homeostasis
1.2.1 Bone Homeostasis
Bone tissue serves not only as a core site of mechanical force transmission and metabolic activity but also as an essential niche for immune cell residence. Immune cell populations are regionally distributed within the periosteum and bone marrow, where distinct mechano-immune regulatory roles contribute to the maintenance of skeletal homeostasis (Table 2) [48, 49].
Summary of mechano-immune regulation mechanisms maintaining musculoskeletal homeostasis.
| Tissue | Immune Cell Type | Mechanical Stimulus | Mechanistic Pathway | Biological Effect | Ref. |
|---|---|---|---|---|---|
| Bone (Periosteum) | CD68⁺F4/80⁻ myeloid cells / macrophages (mouse) | Cyclic compression (0.1 Hz, 10 kPa, 1 h/day, 3 days) | Piezo1 activation induces differentiation of CD68⁺F4/80⁻ precursors into CD68⁺F4/80⁺ macrophages, leading to TGF-β1 secretion | Recruits bone progenitors and promotes cortical bone formation | [52] |
| BMDMs (mouse) | Shear stress (1 Pa, 2 h, orbital shaker) | Mechanical induction of periostin expression promotes macrophage M2 polarization and TGF-β release | Enhances angiogenesis and BMSC-mediated osteogenesis during bone repair | [53-55] | |
| Bone (Marrow) | BMDMs (rat) | Treadmill running (10 m/min, 30 min/day, 21 days) | Macrophage-derived reticulocalbin-2 (RCN2) activates Neuropilin-2 / Integrin β1 / cAMP-PKA signaling | Induces lipolysis in bone-marrow adipocytes and enhances osteogenesis and hematopoiesis | [61] |
| BMDMs (mouse) | Uniaxial strain (10%, 0.5 Hz, 0-10 h) | Mechanical stimulation triggers release of UCHL3-enriched exosomes, activating Smad1-RUNX signaling in BMSCs | Promotes mesenchymal stem cell osteogenesis | [63] | |
| BMDMs (mouse) | Tibial loading (2 Hz, 1-6 N, 120 cycles) | Piezo1 activation induces VEGFA and BMP-2 secretion | Enhances vessel-bone coupling and bone regeneration | [59] | |
| RAW264.7 cells | Cyclic stretch (10%, 0.5 Hz, 0-6 h) | Piezo1 activation suppresses p53 signaling, promoting M2 polarization and TGF-β1 secretion | Enhances osteogenic differentiation of BMSCs | [58] | |
| RAW264.7 cells | Cyclic stretch (10%, 0.5 Hz, 0-7 h) | Mechanical stimulation induces mitochondrial fission and CD200R-CD200 vesicular signaling | Upregulates Runx2, Osx, and ALP in MSCs, promoting osteogenesis | [64] | |
| γδ T cell-associated macrophages (mouse) | Orthodontic tooth movement | Mechanical stress induces macrophage IL-17A secretion, stimulating fibroblast RANKL expression and monocyte/neutrophil recruitment | Regulates bone remodeling | [66] | |
| dHL-60 cells (human) | Cyclic stretch (0.5 Hz, 8-12% strain, 6 h) | PI3K-AKT activation in neutrophil-lineage cells induces Oncostatin M secretion | Promotes bone regeneration | [67] | |
| Joint | Synovial macrophages (mouse) | Dynamic sinusoidal loading (1 N, 5 Hz, 6 min/day, 2 weeks) | Mechanical load suppresses PI3K/AKT and NF-κB pathways, promoting M2 polarization | Increases IL-10 and TGF-β, reduces MMP-13, and protects cartilage matrix | [73] |
| Synovial macrophages (human) | Cyclic strain (10%, 0.5 Hz, 24 h) with 1 nM LXA₄ | LXA4 promotes M2 polarization, increases IL-10 and TGF-β1, and suppresses TNF-α and IL-1β via NF-κB/NLRP3 inhibition | Attenuates inflammation and maintains synovial homeostasis | [74] | |
| Osteoclasts (mouse) | Dynamic load (1 N, 5 Hz, 5 min/day, 2 weeks) | Mechanical stimulation upregulates Wnt3a and downregulates NFATc1, RANKL, TNF-α, and cathepsin K | Suppresses osteoclastogenesis and preserves subchondral bone integrity | [75] | |
| Tendon | Macrophages (mouse) | Downhill treadmill running (-15°, 10 m/min, 20 min/session, 5 days/week, 5 weeks) | Eccentric loading promotes macrophage M2 polarization and TGF-β secretion | Enhances fibroblast proliferation and tendon-bone interface healing | [86] |
| Muscle | Regulatory T cells (mouse) | Voluntary wheel running | IL-6Rα signaling induces a highly functional muscle-resident Treg phenotype with elevated Areg, EGFR, and ST2 expression | Suppresses inflammation and promotes muscle regeneration | [81] |
| Muscle spindle macrophages (mouse) | Muscle contraction | Uptake of glutamine and conversion to glutamate modulate sensory nerve activity | Maintains muscle stretch reflex and neuromuscular balance | [82] | |
| Neutrophils (mouse) | Cyclic mechanical loading (1 Hz, 0.15-0.6 N, 10 min/day, 14 days) | Mechanical compression induces CXCL2, MMP-9, and CCL3 secretion, reducing neutrophil recruitment | Resolves inflammation and promotes muscle recovery | [78] | |
| Skeletal-muscle macrophages (human) | Endurance training | Exercise enhances macrophage M2 polarization, increases IL-4, and suppresses IL-6 expression | Promotes muscle hypertrophy and satellite-cell proliferation | [79, 80] | |
| Intervertebral Disc | - | - | - | - | - |
AKT, protein kinase B; ALP, alkaline phosphatase; BMDMs, bone marrow-derived macrophages; BMP-2, bone morphogenetic protein-2; CCL3, C-C motif chemokine ligand 3; CD68, cluster of differentiation 68; CXCL2, C-X-C motif chemokine ligand 2; cAMP, cyclic adenosine monophosphate; EGFR, epidermal growth factor receptor; F4/80, EGF-like module-containing mucin-like hormone receptor-like 1; IL, interleukin; IL-10, interleukin-10; IL-17A, interleukin-17A; LXA₄, lipoxin A₄; MMP-9, matrix metalloproteinase-9; NLRP3, NOD-like receptor family pyrin domain-containing 3; NFATc1, nuclear factor of activated T cells 1; NF-κB, nuclear factor κB; PI3K, phosphoinositide 3-kinase; PKA, protein kinase A; RANKL, receptor activator of NF-κB ligand; RCN2, reticulocalbin-2; RUNX, Runt-related transcription factor; Smad, mothers against decapentaplegic homolog; ST2, suppression of tumorigenicity 2; TGF-β, transforming growth factor beta; TNF-α, tumor necrosis factor alpha; UCHL3, ubiquitin carboxyl-terminal hydrolase L3; VEGFA, vascular endothelial growth factor A; Wnt3a, wingless-type integration site 3A.
The periosteum, forming the outer layer of bone, represents a highly metabolically active and mechanosensitive compartment enriched with macrophages and regulatory T cells (Tregs) [50, 51]. Mechanical loading has been shown to activate Piezo1 in CD68+F4/80- myeloid cells within the periosteum, driving differentiation into CD68+F4/80+ macrophages. These macrophages secrete transforming growth factor-β (TGF-β), facilitating recruitment of osteoprogenitor cells to the periosteal surface and promoting cortical bone deposition [52]. Periostin has emerged as a mechanoresponsive mediator linking mechanical stimuli to immune regulation during bone repair. Mechanical stimulation upregulates periostin expression in macrophages, promotes M2 polarization, and increases TGF-β secretion, thereby enhancing osteogenesis and angiogenesis within the periosteal microenvironment [53-55].
Within bone tissue, the marrow harbors diverse immune cell populations that actively participate in the regulation of skeletal homeostasis [56, 57]. Mechanical loading induces Piezo1-dependent Ca²⁺ influx and modulates p53 acetylation, promoting polarization of bone marrow macrophages (BMMs) toward an M2 phenotype. These macrophages secrete osteogenic and angiogenic mediators, including TGF-β1, vascular endothelial growth factor A (VEGFA), and bone morphogenetic protein 2 (BMP2), thereby stimulating differentiation of bone marrow stromal cells (BMSCs) [58, 59]. Mechanical cues further coordinate metabolic support for osteogenesis. Loading enhances BMM-derived reticulocalbin-2 (RCN2) expression, activating Neuropilin-2/Integrin β1-cyclic adenosine monophosphate-protein kinase A (cAMP-PKA) signaling to promote lipolysis in marrow adipocytes and provide metabolic substrates for BMSC-driven osteogenesis and hematopoietic activity [60, 61]. In parallel, mechanically stimulated BMMs release ubiquitin C-terminal hydrolase L3 (UCHL3)-containing exosomes that activate Smad1 signaling in BMSCs [62, 63], and transfer mitochondria via extracellular vesicles to enhance cellular metabolic capacity [64]. In cortical bone, physiological loading enhances osteocyte autophagy through mechanistic target of rapamycin complex 2 (mTORC2), increasing adenosine triphosphate (ATP) production and osteogenic signaling while suppressing osteoclast activity through regulation of colony-stimulating factor and receptor activator of NF-κB ligand/osteoprotegerin (RANKL/OPG) balance [65].
Beyond BMMs, additional immune cell subsets contribute to mechano-immune regulation in bone. γδT cells rapidly secrete interleukin-17A (IL-17A) during orthodontic tooth movement, stimulating fibroblasts to express RANKL and promote osteoclast activation. IL-17A also recruits and activates monocytes and neutrophils, thereby accelerating bone remodeling [66]. Conversely, mechanical loading induces c-Jun+ neutrophils to secrete oncostatin M, supporting bone regeneration [67].
Functional lymphatic vessels have recently been identified within bone tissue, where regulation of the immune microenvironment occurs through interstitial fluid drainage processes closely linked to mechanical stimulation [68, 69].
1.2.2 Joints
Within the joint, immune cells are predominantly localized in the synovial tissue. The intimal layer of the synovium consists of two major cell types: type A synoviocytes (macrophage-like) and type B synoviocytes (fibroblast-like). Type A synovial macrophages form a lining along the synovial cavity, establishing a semi-permeable immunological barrier responsible for recognition and clearance of cellular debris, apoptotic cells, and damage-associated molecular patterns (DAMPs) from synovial fluid. This barrier function restricts excessive infiltration of circulating immune cells and contributes to maintenance of immune homeostasis within the joint cavity [70-72].
Moderate mechanical stimulation represents a crucial regulatory factor in preserving this homeostatic state (Table 2). During early OA, mechanical-immune negative feedback mechanisms may attenuate disease progression. Appropriate mechanical loading has been shown to inhibit activation of the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT)/NF-κB signaling pathway in synovial macrophages, thereby limiting M1 polarization and reducing production of pro-inflammatory cytokines, such as interleukin-1 (IL-1) and tumor necrosis factor-α (TNF-α) [73]. Regular mechanical stimulation or exercise also elevates local and systemic levels of lipoxin A4. Under the synergistic action of mechanical cues and lipoxin A4 signaling, synovial macrophages shift toward M2 phenotype, characterized by secretion of anti-inflammatory cytokines such as interleukin-10 (IL-10) and TGF-β. These factors inhibit chondrocyte pyroptosis, promote tissue repair, and reinforce joint homeostasis [74]. Moreover, mechanical loading suppresses osteoclast activity in subchondral bone through upregulation of Wingless-type MMTV integration site family member 3A (Wnt3a), thereby reducing bone resorption and structural deterioration and preserving overall joint integrity [75].
1.2.3 Muscles
Under homeostatic conditions, skeletal muscle contains a relatively sparse yet functionally diverse population of resident immune cells, primarily tissue-resident macrophages with smaller subsets of T lymphocytes. These immune populations contribute to metabolic regulation, maintenance of satellite-cell quiescence and activation, as well as routine tissue turnover and repair [76, 77].
Mechanical forces serve as central upstream regulators of this immune milieu (Table 2). During acute muscle injury, physiological loading promotes interstitial fluid dynamics and facilitates clearance of pro-inflammatory cytokines, thereby limiting neutrophil influx and reducing early inflammatory mediators such as matrix metalloproteinase-9 (MMP-9) and C-C motif chemokine ligand 3 (CCL3) [78]. As the inflammatory phase resolves, sustained mechanical activity drives macrophage polarization toward an M2-like reparative phenotype, enhancing interleukin-4 (IL-4)-dependent regeneration, suppressing interleukin-6 (IL-6)-driven inflammation, and facilitating satellite-cell expansion and myofiber hypertrophy [79, 80]. Over longer durations, exercise-induced mechanical signaling expands an interleukin-6 receptor α (IL-6Rα)-dependent intramuscular regulatory T cell population that contributes to muscle regeneration and functional performance [81].
In addition to classical inflammation-regeneration dynamics, skeletal muscle contains a specialized neural-muscular-immune interface within muscle spindles. Spindle-associated macrophages metabolize myofiber-derived glutamine into glutamate during contraction or stretch, thereby modulating sensory afferent discharge. Depletion of these macrophages disrupts proprioceptive feedback and motor coordination, showing that immune cells act as metabolic intermediaries of mechanical signals and key regulators of neuromuscular homeostasis [82].
1.2.4 Tendons and Enthesis
The enthesis functions as the transitional interface between tendon and bone and is composed of four distinct zones—tendon, unmineralized fibrocartilage, mineralized fibrocartilage, and bone—enabling efficient transfer of tensile and compressive forces across tissues [83]. Within tendons, CD206⁺ M2-like resident macrophages are present in the inner connective tissue layer from the embryonic stage. Spatial maintenance of these macrophages is supported by colony-stimulating factor 1 derived from fibroblasts, underscoring an essential role in tendon development and homeostasis [84]. Following tendon injury, M1 macrophages predominate during the early inflammatory phase to facilitate clearance of necrotic tissue. Subsequent transition toward an M2 phenotype supports matrix remodeling through secretion of growth factors , thereby promoting tissue repair and limiting adhesion formation [85].
Mechanical loading represents a pivotal upstream regulator of immune homeostasis and dynamic ECM renewal in tendon tissue (Table 2). Under physiological tensile conditions, shifts in local cytokine profiles favor macrophage polarization toward an M2-like reparative phenotype, promoting secretion of anti-inflammatory and pro-regenerative mediators. Such responses facilitate coordinated ECM turnover and proper collagen fiber organization [86]. In contrast, excessive or aberrant mechanical loading sustains M1 macrophage activation, leading to disorganized ECM remodeling and chronic inflammatory states [87]. Similar patterns of mechanical-immune coupling are likely present at the enthesis; however, current evidence remains limited and does not yet permit definitive characterization of these mechanisms.
1.2.5 Intervertebral Discs
In contrast to synovial joints, IVD contain minimal immune cell infiltration under physiological conditions. The cartilaginous endplate and annulus fibrosus together establish a structural barrier that confers relative immune privilege, restricting penetration of circulating immune cells into the nucleus pulposus (NP) compartment [88, 89]. The chronically hypoxic and poorly vascularized microenvironment of the disc further constrains immune cell entry and activation [88, 89]. Available evidence indicates that only sparse macrophage-like or mononuclear-like cells are occasionally detected in the outer annulus fibrosus or adjacent endplate regions, where limited roles in local immune surveillance and ECM homeostasis have been proposed [90].
2. Aging-Related Mechanical-Immune Imbalance
2.1 ECM Aging Shapes the Mechano-Immune Niche
In healthy tissues, the ECM provides a compliant and structurally organized scaffold that supports normal cellular function. Aging is accompanied by lifelong exposure to subtle mechanical strain, metabolic perturbation, and oxidative microinjury. Repair processes are frequently incomplete, and cumulative microdamage progressively reshapes ECM architecture over decades. Collagen deposition increases, lysyl oxidase-mediated cross-linking intensifies, elastin undergoes fragmentation, and levels of proteoglycans and hyaluronic acid decline, while advanced glycation end products accumulate. These alternations collectively enhance matrix stiffness and tensile resistance, characteristic features of aged ECM. Elevated stiffness disrupts mechanotransduction in epithelial, endothelial, stromal, and immune cells, contributing to chronic low-grade inflammation and impaired regenerative capacity. Progressive accumulation of microdamage followed by maladaptive ECM remodeling represents a central biomechanical pathway in age-related tissue dysfunction [24, 91-93].
Within innate immune populations, particularly macrophages, ECM stiffening enhances integrin engagement and activation of mechanosensitive ion channels, converging on Rho-associated coiled-coil containing protein kinase (ROCK), es-associated protein/transcriptional coactivator with PDZ-binding motif (YAP/TAZ), signal transducer and activator of transcription 6 (STAT6), and NF-κB signaling pathways. [23, 94-97]. Sustained nuclear signaling alters chromatin accessibility and stabilized a pro-inflammatory transcriptional program characterized by increased production of IL-1, TNF-α, interferon-γ (IFN-γ), and TGF-β production. These mediators promote fibro-inflammatory activation and further matrix remodeling. [23, 98, 99]. Persistent inflammatory signaling also induces metabolic stress and cell death, resulting in release of DAMPs that amplify sterile inflammation. Through these interconnected mechanisms, ECM stiffening established a self-perpetuating mechanical-immune feedback loop that reinforces tissue degeneration [100-103].
Within adaptive immunity, increased matrix stiffness and architectural disorganization impair tissue surveillance and migratory capacity. Tissue-resident memory T (TRM) cells rely on CD103 and CD49a-mediated adhesion, CD69-dependent retention, and nuclear mechanosensing mechanisms that enable stepwise interstitial migration [104-108]. Age-associated ECM remodeling disrupts these processes, resulting in TRM mislocalization and functional exhaustion. Compromised positioning and reduced effector competence weaken surveillance against latent viral infection and transformed cells, thereby contributing to persistent low-grade inflammation [109-111]. Excessive ECM stiffness exceeding approximately 40 kPa has been shown to induce CD8+ T cell exhaustion through activation of the Piezo1-calmodulin-dependent kinase II (CaMKII) axis, leading to diminished IFN-γ and perforin secretion. Concurrently, GRHL3-RNF114-F-actin signaling reduces immune synapse tension, further limiting cytotoxic efficacy [112, 113].
Comparable mechanical-immune dysregulation is observed in degenerative musculoskeletal disorders. In OA, matrix stiffening potentiates macrophage-mediated synovial inflammation and fibrotic remodeling [94, 114-116]. In IDD, transient stiffening followed by matrix breakdown disrupts force transmission and perpetuates aseptic inflammation [117-119]. In OP, ECM degradation and trabecular deterioration heighten skeletal susceptibility to mechanical unloading, further destabilizing bone immune homeostasis (Figure 2, right) [120, 121].
2.2 Mechanobiological Dysfunction in Aging Immune Cells
Aging is accompanied by impaired mechanotransduction in tissue-resident matrix-producing and contractile cells, including cardiomyocytes, smooth muscle cells, and muscle stem cells. Attenuation of YAP/TAZ signaling has been linked to aberrant activation of the cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) pathway, accelerating ECM senescence and accumulation of apoptotic and senescent cells. These alterations foster a pro-inflammatory tissue microenvironment [122-124]. Immune cells, despite rapid turnover relative to many somatic cell types, also exhibit age-associated functional decline. Progressive accumulation of senescent immune populations contributes to dysregulated chronic inflammation and tissue fibrosis [125-127]. In addition to established metabolic and transcriptional reprogramming, emerging evidence highlights a less explored dimension of immunosenescence characterized by defects in mechanical sensing and force transduction. Disruption of mechanobiological responsiveness may represent a critical but underrecognized contributor to age-related immune dysfunction.
Accumulating evidence indicates that aging immune cells exhibit broad impairments in mechanosensing, cytoskeletal remodeling, and force execution. In aged macrophages, defective Rac1/Arp2/3-dependent F-actin polymerization, together with reduced expression of transcription factors such as MYC and upstream stimulatory factor 1 (USF1), compromises cytoskeletal integrity, adhesion capacity, and ECM remodeling. These alterations translate into reduced migration, phagocytosis, and efferocytosis [21, 22, 128]. Concurrent downregulation of mitochondrial calcium uniporter components, including MCU and MICU1, reduces mitochondrial Ca²+ buffering and disrupts cytosolic Ca²+ oscillatory patterns. Altered calcium homeostasis may heighten cellular sensitivity to mechanical stimuli, such that physiological levels of matrix stiffness or tissue tension trigger excessive Ca²⁺ influx through Piezo1 and TRPV4 channels, thereby aggravating inflammatory responses and tissue injury. Hyperglycemic conditions further amplify these effects [20, 129, 130]. Despite these observations, current research remains largely descriptive and lack systematic characterization of mechanical sensing properties and mechanotransductive responses in aged macrophages.
Comparable defects are evident in adaptive immune populations. Aged T cells exhibit disrupted F-actin dynamics and impaired nucleo-cytoskeletal coupling, leading to weakened immunological synapse force generation, reduced migratory efficiency, and elevated thresholds for TCR activation, ultimately diminishing cytotoxic and helper functions [131, 132]. Senescent dendritic cells demonstrate attenuated C-C motif chemokine receptor 7 (CCR7) signaling and defective cytoskeletal remodeling, limiting effective navigation through ECMs, homing to lymph nodes, and antigen presentation capacity [133, 134].
Collectively, these findings highlight that immunosenescence encompasses not only alterations in molecular signaling networks and metabolic pathways but also fundamental disruptions in mechanical sensing and force transduction. Such mechanobiological deficits may constitute a critical and underrecognized dimension of age-related immune dysfunction.
3. Mechanical-Immune Imbalance in Degenerative Musculoskeletal Diseases
3.1 Osteoporosis
Osteoporosis represents one of the most prevalent age-associated disorders affecting the elderly population [1]. Age-related skeletal deterioration reflects the convergence of two major mechanical deficits [135, 136]. Reduced mobility and physical activity diminish habitual mechanical loading, thereby suppressing osteoblastic bone formation and favoring osteoclastic resorption. In parallel, skeletal mechanosensation depends on osteocytes and the lacunocanalicular system (LCS), a specialized structural and fluidic network that transmits mechanical stimuli and coordinates mechano-immune signaling between the bone matrix and bone marrow. Advancing age is associated with quantitative reduction and structural degeneration of the LCS, leading to impaired mechanical signal propagation and a marked reduce in osteocyte-experienced fluid shear stress, which declines to approximately one-third of levels observed in young bone [18, 19]. Such mechanical insufficiency effectively produces a state of functional unloading for osteoblasts, osteoclasts, and marrow-resident immune cells. The combined effects of diminished external mechanical stimulation and intrinsic defects in mechanotransduction disrupt mechanical-immune homeostasis, driving chronic low-grade inflammation and progressive skeletal degeneration (Figure 3) [120, 121, 136].
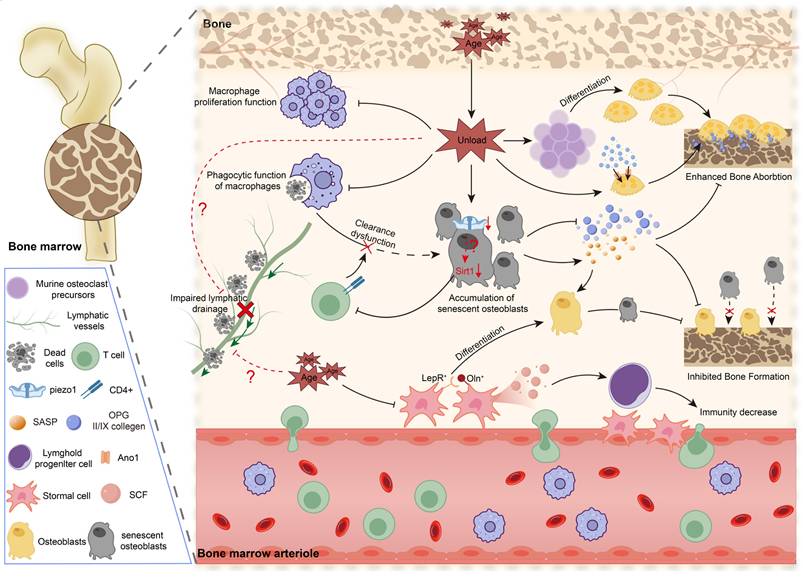
Mechanical unloading driven immune dysregulation in osteoporosis. Ageing-associated mechanical unloading disrupts bone-immune homeostasis by impairing macrophage proliferation and phagocytic clearance, promoting the accumulation of senescent osteoblasts, and enhancing osteoclast differentiation and bone resorption. Concurrently, lymphatic drainage may be further reduced under mechanical unloading and ageing, exacerbating inflammatory and metabolic stress within the bone marrow niche. These changes collectively diminish osteogenic potential, accelerate bone loss, and contribute to compromised immunity, underscoring the pivotal role of mechano-immune dysfunction in age-related osteoporosis.
3.1.1 Cellular Mechanosensory Dysfunction in the Aging Skeleton
In addition to degeneration of the LCS, aging compromises mechanosensing within stromal and osteolineage compartments, thereby destabilizing the immuno-bone axis. LepR+Oln+ osteogenic progenitors, short-lived and highly mechanoresponsive stromal cells, decline with advancing age and reduced physical activity. Loss of this population reduces osteogenic output and lymphopoietic support. Restoration through exercise in a Piezo1-dependent manner underscores a central role as a mechanical hub integrating skeletal and immune homeostasis [27]. Conversely, aging is associated with expansion of a LepR+Sca1+Cxcl9+ stromal subset enriched for interferon-response signatures. These cells form inflammatory perivascular niches that suppress lymphoid progenitor development and may further impair the mechanical responsiveness in adjacent osteogenic stromal populations [137].
Mechanosensory defects also extend to mature osteocytes. Reduced expression of Piezo1 in aged osteocytes, with declines of approximately 30-50%, is associated with a 40% reduction in OPG, thereby diminishing inhibitory control over osteoclast activity [138]. Similarly, decreased Piezo1 expression in osteoblasts results in type II and IX collagen deficiency, weakening integrin-mediated inhibitory signaling toward osteoclasts and increasing bone resorptive activity by approximately 2.1-fold [139]. Collectively, these alterations reflect progressive deterioration of cellular mechanosensing capacity that contributes to age-related skeletal fragility.
3.1.2 Mechanical Unloading Intensifies Immuno-Skeletal Imbalance
Senescent osteoblasts contribute substantially to the progression of osteoporosis. With advancing age, increased secretion of senescence-associated secretory phenotype (SASP) factors, including IL6, interleukin-1β (IL-1β), and MMPs, promotes osteoclastic bone resorption and induces adipogenic differentiation of BMSCs via paracrine mechanisms [140, 141]. Under physiological conditions, macrophages and T lymphocytes participate in clearance of senescent osteoblasts; however, immunosenescence compromises this surveillance system [142, 143]. Aging-associated downregulation of sirtuin 6 (SIRT6) in macrophages and apoptotic osteoblasts, results in CD47 overexpression and impaired efferocytosis [144, 145]. Concurrent reduction of sirtuin 1 (SIRT1) expression in osteoblasts diminishes chemokine production and CD4+ T cell recruitment, further weakening immune-mediated clearance [146]. Furthermore, senescent M1 macrophages and neutrophils secrete elevated levels of grancalcin and microRNA-enriched extracellular vesicles, which enhance adipogenic differentiation of BMSCs and propagate senescence-associated changes across multiple tissues and organs [147, 148].
In older individuals, reduced physical activity combined with degeneration of the LCS further exacerbates immunosenescence, accelerating chronic medullary inflammation and progressive bone loss. Evidence from microgravity models demonstrates that mechanical unloading impairs maturation of BMMs and disrupts cytoskeletal organization, resulting in reduced proliferation and significantly suppressed M1/M2 polarization capacity [149-151]. Mechanistic analyses indicate suppression of RAS/ERK/NF-κB signaling pathways in macrophages, thereby limiting proliferative and polarization responses, while concurrent activation of p53 signaling induces cell cycle arrest [150]. Observations from spaceflight studies further reveal markedly reduced phagocytic activity of monocyte-derived macrophages and neutrophils following return to Earth [152-154], accompanied by features consistent with immunosenescence such as functional exhaustion and increased production of pro-inflammatory cytokines [155]. Mechanical unloading also profoundly reduces SIRT1 expression in osteoblasts, likely secondary to diminished activation of the Piezo1-CaMKII-CREB axis [156, 157]. Reduced SIRT1 expression compromises chemokine-mediated recruitment of CD4+ T cells and impairs immune-mediated clearance of apoptotic or senescent osteoblasts [146, 158, 159]. Inefficient clearance promotes accumulation of dysfunctional cells within the bone marrow niche and sustains a chronic pro-inflammatory microenvironment. Collectively, these alterations establish a persistent inflammatory state closely resembling the medullary inflammation characteristic of age-related osteoporosis.
Conversely, absence of mechanical loading directly accelerates bone resorption [160]. Lack of mechanical stimulation increases the opening frequency of the anoctamin 1 (Ano1) channel in osteoclasts by approximately threefold [161], thereby enhancing resorptive activity through activation of the polycystin-1 (PC1)-TAZ signaling axis [162]. Mechanical deprivation also shifts hematopoietic lineage commitment toward osteoclast differentiation, further amplifying bone resorption [163]. Under conditions of extreme unloading, such as space microgravity, differentiation of osteoclast precursors is markedly increased, leading to heightened bone resorptive activity and accelerated skeletal calcium loss [164, 165].
3.1.3 Potential Skeletal Lymphatic Remodeling in Aging and Mechanical Unloading
Recent investigations have identified functional lymphatic vessels within bone tissue, challenging the longstanding view that the skeletal system lacks a lymphatic network. These vessels participate in interstitial fluid drainage, immune cell trafficking, and coordination of bone remodeling processes [166]. Despite these advances, adaptive responses of skeletal lymphatics to systemic aging and altered mechanical loading conditions remain insufficiently defined.
Aging is associated with reduced lymphangiogenesis and diminished sensitivity to vascular endothelial growth factor C (VEGF-C)/vascular endothelial growth factor receptor 3 (VEGFR3) signaling. Lymphatic vessels within bone similarly exhibit functional decline during aging, with potential consequences for osteoblast maintenance and immune cell dynamics [166-168]. In the aged bone microenvironment, accumulation of apoptotic osteocytes and impaired immune clearance have been documented. Decreased lymphatic density and function may exacerbate this condition by limiting cellular debris and inflammatory mediators, thereby sustaining a chronic inflammatory milieu and further disrupting skeletal homeostasis [144-146].
Mechanical cues function as critical regulators of lymphatic morphogenesis and maintenance. Fluid shear stress affects lymphatic endothelial cell morphology, arrangement, migration, and cell cycle progression, thereby regulating vessel dilation, structural remodeling, and functional capacity [68]. During progression of osteoporosis, mechanical unloading and ECM degradation may deprive biomechanical support for skeletal lymphatics, compromising structural stability and flow-dependent signaling. Experimental evidence indicates that chronic mechanical unloading, including limb immobilization or microgravity exposure, reduces lymphatic pump activity, drainage efficiency, and vessel density [169-171].
In age-related osteoporosis, skeletal lymphatics are therefore likely subjected to combined effects of systemic aging and reduced mechanical stimulation, predisposing to lymphatic stasis. Such maladaptive remodeling may favor accumulation of senescent cell, amplify chronic inflammatory bone loss, and further compromise skeletal homeostasis.
Taken together, these considerations position skeletal lymphatic remodeling as an underexplored but potentially critical axis linking aging, mechanical forces, and skeletal integrity.
3.2 Osteoarthritis
OA, the most prevalent form of degenerative joint disease and a leading contributor to global disability, affects more than 500 million individuals worldwide, with incidence rising sharply after 50 years of age [172, 173]. Aging is accompanied by progressive structural deterioration of joint components, particularly within the knee, including degeneration of articular cartilage and meniscal tissue. These changes are associated with increased joint contact stress, reduced skeletal muscle strength, and elevated knee varus moment, collectively contributing to mechanical imbalance [172-175]. Biomechanical alterations weaken the load-bearing and shock-absorbing properties of cartilage, leading to localized stress concentration and compromised force redistribution across the joint surface [176, 177]. Muscle atrophy, particularly involving the quadriceps and hamstrings, further destabilizes joint mechanics and amplifies medial compartment loading, thereby accelerating cartilage degeneration and subchondral bone sclerosis [178, 179]. At the cellular level, senescent chondrocytes exhibit impaired mechanosensory capacity, while dysregulated osteoclast-osteoblast coupling promotes ECM degradation and microarchitectural damage. These interrelated processes establish a self-reinforcing cycle characterized by stress concentration, microdamage accumulation, tissue dysfunction, and progressive mechanical overload, ultimately driving OA progression (Figure 4) [180].
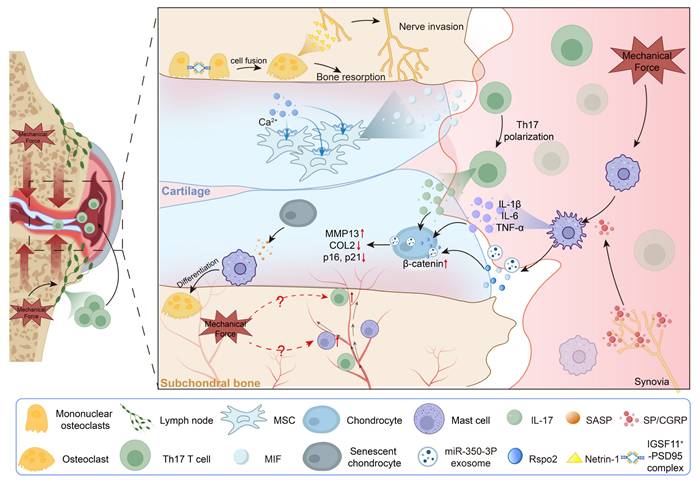
Mechanical loading drives immune-mediated synovial, cartilage and subchondral bone pathology. Mechanical loading promotes T-cell egress from lymph nodes and their subsequent recruitment into the synovium, where it drives macrophage M1 polarization and the production of IL-1β, IL-6, TNF-α, miRNAs, and Rspo2. This macrophage response is further amplified by SP/CGRP⁺ sensory afferents, reinforcing catabolic signaling to chondrocytes and inducing β-catenin activation, MMP13 upregulation, COL2 loss. Mechanical cues also stimulate cartilage MSCs to release MIF, promoting TH17 differentiation. Within the subchondral bone, loading triggers osteocyte fusion and netrin-1 production, facilitating sensory nerve ingrowth, while SASP factors from senescent chondrocytes enhance osteoclastogenesis and bone resorption.
3.2.1 Pathways of Immune Cell Infiltration in Osteoarthritis
Histologically, synovitis in OA is characterized by synovial lining hyperplasia, sublining fibrosis, and pronounced neovascularization. In response to locally elevated cytokines and adhesion molecules, leukocytes migrate from the vascular lumen into the synovial stroma. Macrophages and T lymphocytes constitute the predominant immune populations, whereas mast cells, B cells, and plasma cells are present at lower frequencies [70, 181, 182]. Persistent joint inflammation disrupts the balance of regulatory macrophage subsets, reduces phagocytic and anti-inflammatory capacity, and induces features of macrophage senescence in periarticular muscle tissue, thereby contributing to muscle atrophy and compromised joint stability [183, 184].
With disease progression, cartilage degradation and subchondral bone remodeling enable vascular invasion across the osteochondral junction, creating additional pathways for immune cell infiltration and dissemination of inflammatory mediators [182, 185]. Aging further intensifies this process. Senescent chondrocytes secrete SASP factors, such as IL-6, TNF-α, MMPs, and VEGF. These mediators facilitate neovascularization and promote upward vascular extension from subchondral bone into cartilage tissue [186-188], sustaining a chronic inflammatory milieu that disrupts synovial and adipose tissue homeostasis [189, 190]. Infiltration of pro-inflammatory macrophages, activated T lymphocytes, and angiogenic endothelial cells into the cartilage-bone interface establishes a self-perpetuating inflammatory circuit, accelerating cartilage erosion, vascular penetration, and nociceptive sensitization.
3.2.2 Mechanical Dysregulation Fuels Osteoarthritis Inflammation
During OA progression, infiltrating immune cells operate within a mechanically altered joint environment, generating a distinct mechano-immune pathogenic axis. Persistent mechanical imbalance activates pro-inflammatory signaling pathways that enhance immune cell recruitment and mediator release. Mechanical overload upregulates rap guanine nucleotide exchange factor 3 (Rapgef3) and activates the p65-NF-κB pathway in synovial macrophages, thereby inducing M1 polarization and enhancing secretion of pro-inflammatory cytokines [115]. M1-polarized macrophages subsequently release exosomes enriched in miR-350-3p, which are internalized by chondrocytes. These exosomes suppress nuclear receptor binding SET domain protein 1 (NSD1) expression, reduce histone H3 lysine 36 (H3K36) methylation, and accelerate chondrocyte senescence and ECM degradation [191]. In age-related OA, aberrant sensory nerve fiber ingrowth increases local release of neuropeptides, such as substance P and α- calcitonin gene-related peptide (αCGRP) [192]. Mechanical stress enhances macrophage responsiveness to these neuropeptides through downregulation of neurokinin-1 receptor and upregulation of αCGRP receptors, amplifying pro-inflammatory signaling, reinforcing M1 polarization, and intensifying joint inflammation and structural degeneration [193]. Mechanical strain also stimulates chondrocytes to secrete the CC chemokine ligand 5 (CCL5), which recruits macrophages and osteoclasts through CC chemokine receptor (CCR)-Akt2 signaling. This process induces subchondral bone loss and establishes a pathological remodeling environment characterized by osteoclast hyperactivation [194].
Abnormal mechanical loading also perturbs adaptive immune responses during OA progression. In murine models of mechanically induced knee OA, T cell populations were significantly increased in regional lymph nodes. Genetic deletion of TCRαβ substantially attenuated bilateral cartilage degeneration and osteophyte formation induced by mechanical stress. In contrast, pharmacological inhibition of T cell efflux from lymph nodes mitigated cartilage degradation without significantly affecting osteophyte development, indicating distinct contributions of T cell subsets and trafficking pathways to structural joint changes [116]. Additional studies have revealed that load sensing through Piezo1 in MSCs activates the STAT1-HK2 glycolytic pathway, inducing secretion of macrophage migration inhibitory factor (MIF). This cascade enhances recruitment and polarization of T helper 17 (Th17) cells, thereby amplifying OA-associated inflammation and bone destruction [195]. These findings support a model in which mechanical forces not only regulate T cell trafficking into the joint but also modulate intra-articular T-cell function to accelerate OA progression.
Subchondral bone similarly undergoes substantial immune infiltration during OA progression. Vascular invasion is accompanied by increase proportions of CD8⁺ T cells (18.84%), activated mast cells (17.37%), activated CD4⁺ memory T cells (9.12%), and diverse macrophage subsets [196, 197]. In other tissues, mast cells are highly responsive to mechanical stimulation, which can activate mechanosensitive channels such as Piezo1 and TRPV4, as well as integrin-mediated signaling pathways, leading to degranulation and release of inflammatory mediators. Alterations in matrix stiffness and cytoskeletal tension further modulate mast cell proliferation, migration, and immune responsiveness [197, 198]. However, the specific impact of mechanical forces on mast cell behavior within osteoarthritic joints remains poorly defined and may represent an underrecognized mechanism contributing to disease pathogenesis.
3.2.3 Mechanical Instability Models Expose Osteoarthritis Mechano-Immune Dynamics
Established animal models of mechanically induced OA include anterior cruciate ligament transection, meniscectomy, and destabilization of the medial meniscus (DMM). Each approach generates joint instability and abnormal mechanical loading, leading to progressive osteoarthritic changes [199, 200]. Among these models, DMM most closely resembles human primary OA, characterized by gradual progression and a relatively mild phenotype, thereby providing a suitable platform for investigating mechanical-immune coupling. Findings derived from these models collectively illustrate how mechanical imbalance drives synovial inflammation, subchondral bone remodeling, and sensory nerve ingrowth during disease development [200].
Mechanical instability rapidly induces synovial inflammatory responses. Early stages following DMM are marked by synovitis with accumulation of M1-polarized macrophages and elevated expression of IL-1β, IL-6, and TNF-α [201]. Mechanical loading further stimulates matrix-degrading enzymes, including ADAMTS4, MMP3, and MMP13, as well as nerve growth factor (NGF), thereby accelerating cartilage degradation [201]. Concentrated mechanical stress drives synovial macrophages to release R-spondin-2, which activates β-catenin signaling in chondrocytes and enhances expression of catabolic enzymes, promotes chondrocyte hypertrophy, and facilitates osteophyte formation [202]. In aged or chronically overloaded joints, increased cyclin-dependent kinase 8 (CDK8) activity in chondrocytes augments NF-κB/STAT1/3 signaling, induces SASP production, and promotes macrophage transition toward osteoclast-like phenotypes, thereby intensifying inflammation and matrix breakdown [203].
Subchondral bone demonstrates pronounced sensitivity to abnormal mechanical loading, with remodeling alterations frequently preceding overt cartilage degeneration [204]. DMM induces expansion of IgSF11⁺ osteoclasts, which enhance osteoclast differentiation and fusion through interactions involving postsynaptic density protein 95, thereby exacerbating bone resorption [205-207]. Within two weeks following DMM surgery, activation of the RANKL-UCHL1-sCD13 negative feedback pathway is detectable in osteoclast precursors, serving to restrain excessive resorptive activity during early OA. However, age-associated decline in UCHL1 expression may impair this regulatory loop, potentially accelerating disease progression [208-210]. In parallel, monocyte-derived pre-osteoclasts secrete elevated levels of platelet-derived growth factor-BB (PDGF-BB), inducing aberrant angiogenesis and sensory nerve ingrowth within the subchondral compartment [211]. Under mechanical stress, osteoclasts upregulate collagen type VI alpha 1 chain (COL6A1) to activate the EPAC/RAP1 pathway, further enhancing osteoclast differentiation and contributing to structural deterioration of subchondral bone [212].
As DMM-induced OA progresses into the chronic phase, coordinated mechanical-neurological-immune interactions drive persistent pain. Approximately eight weeks post-surgery, nociceptive neurons within the dorsal root ganglia (DRG) become primary responders to abnormal mechanical input, accompanied by marked infiltration of macrophages predominantly exhibiting an M1-polarized phenotype. These macrophages release pro-nociceptive mediators, such as IL-1β, TNF, reactive oxygen species (ROS), C-X-C motif chemokine ligand 10 (C-X-C) motif chemokine ligand 10 (CXCL10), and CXCL2, which enhance DRG neuronal excitability and facilitate pain transmission [213-216]. Sensory neurons further secrete NGF and fractalkine; fractalkine cleavage by cathepsin S activates spinal microglia, leading to central sensitization that synergizes with peripheral inflammation to maintain chronic hyperalgesia [217, 218]. Concurrently, abnormal mechanical loading enhances Netrin-1 secretion from osteoclasts, stimulating aberrant subchondral nerve ingrowth and establishing peripheral nociceptive foci [213].
In aged DMM models, articular cartilage degeneration, subchondral bone remodeling, and osteophyte formation occur earlier and with greater severity than in younger counterparts, suggesting that aging amplifies mechanically driven structural deterioration process induced by mechanical stimuli [219]. Although direct comparative data on temporal differences in inflammatory mediator profiles and macrophage polarization between aged and young mice remain limited, aging has been associated with increase propensity toward M1 polarization and reduced capacity for transition to reparative M2 phenotypes. This polarization bias is amplified under conditions of abnormal mechanical stress, accelerating inflammatory progression [220]. Consequently, mechanically induced synovitis and pro-inflammatory mediator release in aged individuals are likely to arise earlier and persist longer, thereby accelerating the immunopathological course of OA.
3.3. Intervertebral Disc Degeneration
IDD represents a principal contributor to chronic low back pain, affecting more than 80% of adults over the course of life and imposing substantial socioeconomic burden [221, 222]. Pathogenesis involves complex interplay among mechanical overload, inflammatory activation, oxidative stress, and ECM remodeling [223, 224]. Aging is accompanied by progressive degenerative alterations within the IVD, including dehydration of the NP, calcification of cartilaginous endplates, and disruption of collagen fiber organization. These changes reduce disc elasticity and alter physiological load distribution [12, 225-229]. Subsequent biomechanical consequences include decreased range of motion, loss of disc height, reduced proteoglycan content and osmotic pressure, increased matrix stiffness, and heightened susceptibility of the annulus fibrosus to fissure formation. Impaired mechanical buffering capacity disrupts homeostatic mechanotransductive signaling within disc cells, reinforcing a cycle of mechanical imbalance, inflammatory activation, and matrix degradation. This self-perpetuating cascade constitutes a central mechanism underlying age-related IDD progression (Figure 5) [12, 225-229].
Ageing-enhanced mechanical loading drives mechano-immune dysregulation in the intervertebral disc. Ageing increases mechanical loading within the intervertebral disc, amplifying mechano-immune responses across the nucleus pulposus, annulus fibrosus, and endplate. In the nucleus pulposus and annulus fibrosus, excessive force modulates macrophage activation, promoting pro-inflammatory cytokine release and accelerating disc degeneration. In the endplate, mechanical stimulation facilitates vascular ingrowth and immune-cell infiltration, further sustaining inflammation. Moreover, ageing-related osmotic fluctuations may intensify immune-cell activation and inflammatory signaling.
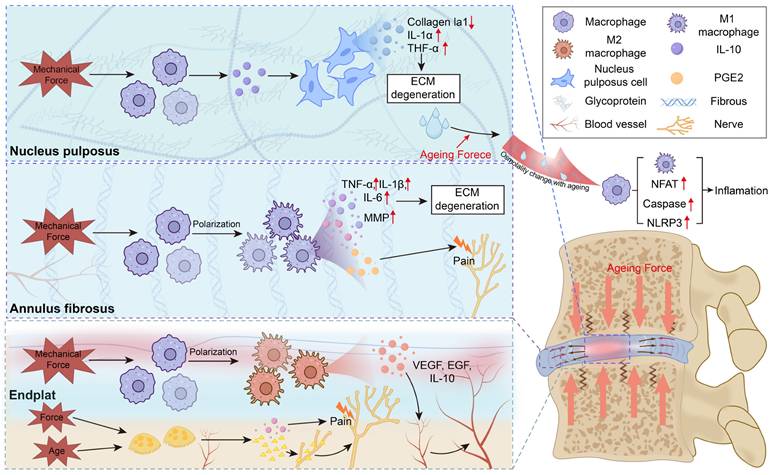
3.3.1 Aging and Abnormal Force Disrupt the Disc Immune Barrier
Loss of immune privilege in IDD is primarily driven by the combined effects of aging and aberrant mechanical stress. Aging impairs the synthetic activity of nucleus pulposus cells (NPCs) and annulus fibrosus cells (AFCs), resulting in reduced production of proteoglycan and type II collagen, increased deposition of type I and III collagen, and upregulation of catabolic enzymes, including MMPs and ADAMTS family members [225, 230-232]. These structural and metabolic alterations compromise maintenance of an immune-isolated microenvironment and lower intradiscal pressure, increasing susceptibility to stress concentration, endplate microfractures, and formation of Schmorl's nodes [12]. Concurrently, aging is associated with accumulation of M2-polarized macrophages within the cartilaginous endplate. Through secretion of IL-10, these macrophages promote pathological angiogenesis and contribute to endplate stiffening [233].
Within this mechanically compromised context, abnormal mechanical loading further disrupts barrier integrity. In NPCs and annulus AFCs, excessive compressive, tensile, or shear forces induce oxidative stress, cytoskeletal remodeling, and increased expression of matrix-degrading enzyme, culminating in ECM breakdown [234-236]. Chronic compression of the cartilaginous endplate promotes calcification, microdamage, and reduced permeability [237]. These biomechanical insults collectively compromise matrix integrity and trigger the release of DAMPs, including high mobility group box 1 (HMGB1), ATP, and collagen fragments. DAMPs then activate Toll-like receptor- and NOD-like receptor-dependent NF-κB signaling pathways, amplifying inflammatory responses and destabilizing local immune balance [238, 239].
Collectively, aging initiates mechanical disequilibrium within the intervertebral disc, creating a permissive environment for degeneration, while progressive structural degeneration further alters mechanical loading patterns. This bidirectional reinforcement establishes a self-amplifying cycle of structural failure, immune barrier disruption, and sustained inflammation in IDD [233, 240, 241].
3.3.2 Mechanical Stress Activates Disc Immune Cells
Following disruption of the cartilaginous endplate or annulus fibrosus, peripheral immune cells gain access to the intervertebral disc and are subsequently exposed to aberrant mechanical cues, including excessive compression, tensile strain, and shear stress. These altered biomechanical cues influence immune cell activation and polarization states. Mechanical overload enhances expression of matrix-degrading enzymes and, through immune-stromal crosstalk, impairs reparative capacity of NPCs and AFCs, thereby accelerating inflammatory amplification and ECM degradation.
Within the NP, mechanically induced degeneration has been associated with increased macrophage infiltration and upregulation of secreted phosphoprotein 1 (SPP1) [242]. Elevated SPP1 expression inhibits the protein kinase RNA-like endoplasmic reticulum kinase (PERK)/activating transcription factor 4 (ATF4)/IL-10 signaling axis, decreasing synthesis of NPC-derived matrix components and promoting ECM breakdown. Suppression of SPP1 restores PERK/ATF4/IL-10 signaling and mitigates disc injury [242].
Regional mechanical heterogeneity further shapes immune phenotypes in degenerative discs. The high-intensity zone (HIZ) associated with annular fissures is subjected to concentrated shear and circumferential stress and is frequently characterized by granulation tissue formation, inflammatory infiltration, and neovascularization [243, 244]. In contrast, Modic changes occurring at the endplate-marrow interface result from microdamage and abnormal axial loading, manifesting as marrow edema, fatty replacement, or sclerosis [245]. These region-specific mechanical environments contribute to distinct immune and stromal responses within the degenerating disc.
Within the annulus fibrosus, high-intensity zone lesions are characterized by pronounced M1 macrophage polarization, creating a mechanically reinforced pro-inflammatory niche. These macrophages produce IL-6, IL-8, MMPs, and nociceptive mediators such as NGF, thereby amplifying local inflammation, accelerating ECM degradation, and enhancing pain sensitization. Mechanical stress-induced M1 programming may engage activation of the Piezo1-YAP mechanotransduction axis, a pathway implicated in pro-inflammatory macrophage activation under conditions of elevated mechanical load or matrix stiffening [94, 246, 247].
In contrast, chronic mechanical overload and endplate microfractures contribute to Modic changes, where macrophage phenotypes vary according to lesion stage. M1 macrophages predominate in acute Type I lesions, whereas M2 subsets are more prominent in chronic Type II lesions [246, 248]. Early M2 responses aid debris clearance; however, prolonged M2 activity and associated SASP factors, including IL-10 and neurotrophic mediators, promote vascular and sensory nerve ingrowth, sustaining inflammation, pain, and endplate sclerosis [233, 249]. Mechanical instability, whether age-related or surgically induced, further accelerates endplate degeneration by enhancing osteoclast recruitment and resorptive activity. Osteoclast-mediated micro-porosity creates structural channels for sensory fiber extension, while secretion of Netrin-1 guides nerve ingrowth and prostaglandin E2 (PGE2) sensitizes nociceptors to mechanical stimulation [250].
Although mechanobiological-immune interactions in IDD remain incompletely defined, available evidence supports a model in which aging and abnormal mechanical loading progressively transform the relatively immune-privileged disc into a mechano-inflammatory niche. This altered environment reprograms infiltrating immune cells, compromises reparative capacity of NPCs and AFCs, and facilitates pathological neurovascular ingrowth.
3.3.3 Osmotic Imbalance Under Mechanical Stress May Accelerates IDD Inflammation
In the healthy intervertebral disc, osmotic pressure within the NP is maintained by proteoglycan content and associated fixed charge density, fluctuating dynamically within a physiological range of approximately 430-496 mOsm/L [251]. This osmotic environment undergoes predictable diurnal variation in response to posture and mechanical loading [251]. Aging is accompanied by progressive depletion of proteoglycans and reduced tissue hydration, lowering baseline osmolarity to approximately 300 mOsm/L and diminishing the intrinsic buffering and recovery capacity of the NP [119, 252, 253]. Concurrent sclerosis of the cartilaginous endplate compromises water regulation and nutrient transport, further accelerating osmotic decline [254, 255]. Under repetitive or sustained mechanical loading, the aging NP is therefore prone to more frequent and pronounced hypotonic fluctuations, leading to an unstable osmotic stress environment and limiting restoration of physiological balance during circadian cycles. Such osmotic dysregulation may represent an additional mechanobiological factor contributing to inflammatory activation and progression of intervertebral disc degeneration.
Osmotic fluctuations represent a critical factor in disruption IVD homeostasis. Hypoosmolar conditions activate pro-inflammatory signaling in NPCs via the TRPV4/Ca²⁺-NF-κB pathway, whereas hyperosmotic stress induces nuclear factor of activated T cells 5 (NFAT5) and NOD-like receptor family pyrin domain containing 3 (NLRP3) activation, both of which contribute to inflammatory amplification and ECM degradation [119, 252, 256]. Immune cells demonstrate similarly osmotic sensitivity. Hypoosmolarity favors M1 macrophage polarization and NLRP3 inflammasome activation, whereas hypertonic conditions enhance pro-inflammatory cytokine production through NFAT5 and caspase-1 signaling pathways [257-260].
Current investigations have largely focused on static hypo- or hyperosmotic conditions. In contrast, the impact of mechanically driven, periodic osmotic fluctuations on the immune-inflammatory microenvironment of the disc remains poorly understood. In aging intervertebral discs, diminished osmotic buffering capacity of the NP may expose infiltrating immune cells to recurrent osmotic stress, thereby amplifying inflammatory polarization and tissue-destructive activity. Whether distinct immune cell subsets, including macrophages, T cells, and neutrophils, exhibit differential sensitivity to dynamic osmotic fluctuations, and how intercellular interactions evolve under such stress, represent unresolved questions that warrant systematic investigation.
3.4 Enthesis Pathology
Enthesis-related injuries, such as rotator cuff tears and Achilles tendinopathy, represent some of the most prevalent musculoskeletal disorders, with incidence increasing markedly with advancing age. Epidemiological data indicate that structural or inflammatory abnormalities at the enthesis are present in more than 70% of middle-aged adults, and degeneration of enthesis fibrocartilage is strongly associated with pain, muscle weakness, and reduced mobility [261, 262].
The enthesis is characterized by a graded transitional architecture that enables efficient force transfer between tendon and bone. Repetitive mechanical loading or acute trauma disrupts this gradient structure, resulting in microtears, fibrocartilage disorganization, and compromised healing capacity [262, 263]. Aging further exacerbates structural vulnerability. The fibrocartilaginous transition zone becomes thinner and architecturally disordered, cellularity declines, and mineralization expands, collectively reducing elasticity and mechanical resilience [264, 265]. Increased collagen cross-linking, diminished viscoelastic properties, and dysfunction of resident stem or progenitor cells further lower the mechanical tolerance of the tendon-bone interface [264, 265]. Under these conditions, even physiological loading may induce microdamage and release DAMPs, such as high mobility group box 1 (HMGB1) and ATP. These signals activate macrophage-mediated inflammatory responses and promote M1 polarization, thereby contributing to chronic enthesis pathology [266-268].
The magnitude, pattern, and timing of mechanical stimulation critically determine the nature of the immune response at the enthesis. Controlled and appropriately timed loading favors resolution of inflammation by promoting macrophage transition toward an M2 phenotype and supporting coordinated ECM remodeling. In contrast, premature, excessive, or irregular mechanical stress prolongs the M1-dominant inflammatory phase and predisposes to fibrotic repair rather than functional regeneration [269, 270]. Aging attenuates this adaptive flexibility. Macrophages in aged tissues exhibit reduced capacity for M1-to-M2 phenotypic transition, resulting in sustained inflammation, progressive ECM degradation, and fibrotic remodeling of the tendon-bone interface [4, 271].
Such age-associated alterations define an “aging-mechanical-immune” axis, in which impaired mechanotransduction and unresolved inflammatory signaling reinforce perpetuate tendon-bone degeneration. Therapeutic strategies directed toward this axis—including optimized mechanical conditioning protocols, targeted modulation of macrophage polarization, and application of bioengineered scaffolds designed to restore physiological mechanical cues—may offer potential to re-establish enthesis homeostasis and improve tendon-to-bone healing in aging individuals.
3.5 Sarcopenia
Sarcopenia, characterized by progressive loss of skeletal muscle mass and functional capacity, represents a major contributor to frailty, falls, and disability in older adults. Prevalence ranges from 5-13% in individuals aged 60-70 years and approaches 50% in those older than 80 years [272, 273]. Although multifactorial in origin, immune dysregulation has emerged as a key component of pathogenesis. Aging reshapes the muscle immune microenvironment, defined by metabolic reprogramming of macrophages, impaired transition from pro- inflammatory to reparative phenotypes, and reduced Treg function [274].
Age-associated alterations in mechanosensitivity and polarization dynamics further increase susceptibility of skeletal muscle to degeneration during cycles of mechanical unloading and reloading, such as disuse, immobilization, or microgravity exposure. In young muscle, reloading after atrophy induces transient M1 macrophage activation to facilitate clearance of necrotic debris, followed by polarization toward an M2 phenotype that secretes insulin-like growth factor 1 (IGF-1) and IL-10, thereby promoting regeneration. In aged muscle, this M1-to-M2 transition is attenuated, delaying recovery and impairing regenerative efficiency [274-278]. Immobilization reduces the abundance of inducible nitric oxide synthase (iNOS)⁺ macrophages, suppresses iNOS expression, and disrupts satellite cell activation, collectively compromising myofiber regeneration. Prolonged mechanical unloading also sustains Ly-6C⁺ inflammatory macrophages and inhibits transition to anti-inflammatory phenotypes, leading to sustained TNF-α and IL-1β production that exacerbates inflammation and accelerate muscle atrophy [279]. Restoration of the pro-inflammatory-to-reparative shift, achieved through macrophage transplantation or targeted metabolic and immunomodulatory interventions, including transient hypoxia or metabolic reprogramming, has been shown to enhance muscle regeneration and functional recovery in aged, disused models [280, 281].
4. Strategies for Modulating Musculoskeletal Mechano-Immunity
4.1 Mechanical and Material-Based Interventions
Mechano-immunity plays a dual role in the musculoskeletal system, sustaining tissue homeostasis under physiological conditions while contributing to pathological progression in degenerative disorders. Therapeutic modulation of this axis has therefore emerged as a promising strategy. Application of controlled external mechanical stimuli may reshape the immune microenvironment during early disease stages or counteract aberrant mechanical signaling in advanced degeneration. Current approaches can be broadly categorized into three principal strategies: exercise-based interventions, exogenous physical stimulation modalities, and development of advanced biomaterials designed to restore or optimize mechanical-immune coupling. Among these, structured exercise regimens remain the most extensively studied and mechanistically characterized modality (Table 3).
Summary of mechanobiology-based immunomodulatory interventions for treating degenerative musculoskeletal diseases.
| Category | Mechanical Intervention Type | Animal / Cell Model | Cellular Target | Mechanistic Pathways | Biological Effects | Ref. |
|---|---|---|---|---|---|---|
| Exercise | Moderate treadmill running (10-20 m/min, 30 min/day, 3×/week, for 4 weeks) | MIA-induced OA rats | Synovial macrophages | Promotes M2 polarization via upregulation of IL-10 and TGF-β, and inhibition of NF-κB and NLRP3 activation | Reduced joint inflammation and chondrocyte pyroptosis, preserved cartilage matrix | [74, 285] |
| Low-intensity treadmill running (10 m/min, 15 min/day for 3 days) | SAMP8 spontaneous OA mice | Synovial macrophages | Decreases M1 macrophage infiltration and downregulates MCP-1 and TNF-α expression | Reduced synovitis and cartilage degradation | [286] | |
| Whole-body vibration+squat (35-40 Hz, 4 mm, 3×/week) | Elderly OA patients | CD4⁺ T cells | Modulates T-cell immune responsiveness | Potential slowing of osteoarthritis progression | [284] | |
| Endurance running (18 m/min, 90 min) | Exercise-induced muscle inflammation | Regulatory T cells (Tregs) | Expands muscle Tregs, suppresses IFN-γ production, and maintains mitochondrial electron transport chain function | Preserved mitochondrial metabolism and exercise capacity | [26] | |
| Endurance cycling (45 min, 4 days/week, 4 weeks) | Human participants | Macrophages | Increases IL-10 and decreases TNF-α expression; activates M2 polarization and interaction with muscle satellite cells | Enhanced M2 macrophage population, muscle hypertrophy, and repair | [79] | |
| Eccentric mechanical stimulation (downhill treadmill) | Mouse rotator cuff injury model | Macrophages | Increases M2 macrophage expression and decreases M1 macrophage expression | Enhanced fibrocartilage formation and proteoglycan content | [86] | |
| Moderate treadmill exercise (10 m/min, 20 min/day,2-8 weeks) | Mouse rotator cuff injury model | Macrophages | Activates the IL-4/JAK/STAT6 pathway, promoting M2 polarization (elevated CD206, Arg-1, IL-10) | Enhanced fibrocartilage formation and tendon-bone integration | [270] | |
| Treadmill running (12 m/min, 1 h/day, 8 weeks) | OVX osteoporotic mice | Osteoclast precursors | Muscle-derived L-BAIBA through SLC6A6 suppresses PI3K/AKT/NF-κB signaling and activates NRF2 | Decreased osteoclastogenesis and bone loss | [287] | |
| Voluntary running | Mouse model | LEPR⁺/Osteolectin⁺ stromal cells | Muscle-derived L-BAIBA suppresses PI3K/AKT/NF-κB signaling and activates NRF2 | Supports osteogenesis and lymphopoiesis | [27] | |
| Treadmill running (10 m/min, 30 min/day, 3 weeks) | Mouse model | Macrophages | RCN2 activates Neuropilin-2/Integrin β1-cAMP-PKA signaling | Promotes lipolysis, osteogenesis, and hematopoiesis | [61] | |
| External mechanical stimulation | Robot-actuated cyclic loading (0.15-0.3 N, 1 Hz, 10 min/day) with anti-inflammatory therapy | Aged mouse tibialis anterior muscle injury model | Macrophages, muscle stem cells | Combined loading and therapy suppress NF-κB activation and restore YAP/MRTF-A mechanotransduction | Enhanced muscle regeneration, vascularization, and contractile strength | [295] |
| Cyclic compressive loading (robotic actuator) | Mouse tibialis anterior muscle injury | Neutrophils | Mechanical loading accelerates neutrophil clearance and downregulates MMP-9 and CCL3 | Improved muscle healing and biomechanical strength | [296] | |
| LIPUS (1.5 MHz frequency, 1:4 duty cycle, and 30 mW/cm²) | Rat rotator cuff repair model | Macrophages | Promotes early macrophage accumulation followed by M2 transition | Inhibits osteoclastogenesis and enhances osteogenesis | [297] | |
| LIPUS (1.5 MHz frequency, 1:4 duty cycle, and 30 mW/cm²) | DMM-induced OA model | Macrophages | SQSTM1-dependent autophagic degradation of PKM2 reduces IL-1β maturation | Anti-inflammatory effect and improved joint function | [298] | |
| LIPUS (1.5 MHz frequency, 1:4 duty cycle, and 30 mW/cm²) | Rat ACLT post-traumatic OA model | Osteoclasts | Suppresses netrin-1 expression and osteoclast differentiation | Mitigates cartilage degradation and pain, preserves bone structure | [299] | |
| Low-magnitude high-frequency vibration (35 Hz, 0.3 g, 20 min/day) | OVX osteoporotic fracture model | Macrophages | Restores early pro-inflammatory response and accelerates M2 transition | Improved callus formation and fracture healing | [300] | |
| Nanovibrational stimulation (~40 nm, 1 kHz) | 3D human osteoblast/osteoclast co-culture | Osteoclast precursors | Akt-TGF-β1/BMP2 signaling mediates nanovibration response, reducing osteoclast activity | Enhanced osteogenesis and reduced osteoclastogenesis | [301] | |
| Distraction osteogenesis | Rat/rabbit/sheep models | Macrophages, T/B cells | Integrin-FAK-PI3K/AKT-MAPK-YAP/BMP2 pathways regulate M1-to-M2 transition | Controlled inflammation, neovascularization, and bone regeneration | [303] | |
| Scaffold-based mechanical modulation | 80 kPa polyacrylamide hydrogel | Mouse calvarial defect model | Macrophages | Inhibition of Piezo1 attenuates macrophage senescence and the pro-inflammatory phenotype, promotes M2 polarization through reduced IL-1β, IL-6, and TNF-α, and increased IL-10 and VEGF expression | Anti-inflammatory response, enhanced angiogenesis and osteogenesis, improved bone regeneration | [315] |
| Low-stiffness Ti2448 scaffold | Rabbit; RAW264.7, HUVECs, MC3T3-E1 | Macrophages | Active Piezo1/YAP axis modulates M2 polarization | Promotes angiogenesis & osteogenesis | [316] | |
| Nano-sheet array (Ti surface topography) | RAW264.7 cells | Macrophages | Activation of the integrin β2-FAK-PI3K-AKT-mTOR pathway promotes M2 polarization and upregulates TGF-β, BMP2, and VEGF | Suppressed inflammation, enhanced osteogenesis and angiogenesis | [317] | |
| Left-handed chiral nanohydroxyapatite (nHA) scaffolds | Tibial distraction osteogenesis model | Macrophages | L-chiral structures activate the integrin β1-FAK-TRPV4-Ca²⁺-STAT6 pathway to induce M2 polarization | Promoted vascularization and bone regeneration | [318] | |
| Aligned PCL nanofiber scaffold | Mouse Achilles tendon injury model | Macrophages | Scaffold alignment guides macrophage orientation, suppresses M1 polarization, and enhances M2 polarization with increased IL-10 and TGF-β secretion | Enhanced extracellular matrix synthesis and tenocyte activity, improved tendon healing | [319] | |
| Mechanosensitive materials | Injectable piezoelectric hydrogel (PVA/CNF/BTO@PDA) | Rabbit rotator cuff injury model | Macrophages | Piezoelectric microelectrical cues enhance M2 polarization (elevated CD206, reduced iNOS) and recruit bone marrow stem cells for zonal differentiation | Enhanced fibrocartilage and bone formation, improved tendon-bone healing strength | [320] |
| Janus asymmetric piezoelectric adhesive | Rat rotator cuff repair model | Macrophages | TRPV1-Ca²⁺-cAMP/PKA signaling promotes M2 polarization and stimulates VEGF and TGF-β secretion | Reduced local inflammation, enhanced angiogenesis and fibrocartilage regeneration | [321] | |
| Magnetized nanocomposite hydrogel | Rat calvarial defect model | Macrophages | Magnetic mechanical cues activate the podosome-Rho-ROCK signaling pathway, inducing time-dependent M1-to-M2 transition | Sequential immune modulation and improved bone formation | [322] | |
| Piezoelectric biomimetic periosteum (BaTiO₃/MWCNT/collagen) | Mouse cranial bone defect model | Macrophages | Ultrasound-activated Piezo1 triggers Ca²⁺ influx, suppresses NF-κB (TRAF6/TLR) signaling, and induces Arg1 and IL-10 expression | Enhanced osteogenic differentiation of bone marrow stem cells, improved bone healing | [290] | |
| BaTiO₃/Ti6Al4V piezoelectric scaffold | Sheep cervical bone defect model | Macrophages | Electrical stimulation inhibits MAPK/JNK signaling and enhances oxidative phosphorylation and ATP synthesis, leading to M2 polarization | Reduced inflammation and enhanced bone regeneration | [323] | |
| Perfusion bioreactor system with electrospun membranes | Rat calvarial defect model | Macrophages | Shear stress activates Piezo1-Ca²⁺-YAP signaling, promoting M2 polarization and secretion of miR-210-3p-enriched extracellular vesicles | Increased extracellular vesicle yield (~12.5-fold), enhanced bone regeneration and angiogenesis | [324] |
ACLT, anterior cruciate ligament transection; BAIBA, β-aminoisobutyric acid; BMP, bone morphogenetic protein; BTO, barium titanate; CCL3, chemokine (C-C motif) ligand 3; DMM, destabilization of the medial meniscus; ECM, extracellular matrix; FAK, focal adhesion kinase; IFN-γ, interferon-γ; IL, interleukin; JAK, Janus kinase; LIPUS, low-intensity pulsed ultrasound; LXA4, lipoxin A4; MCP-1, monocyte chemoattractant protein-1; MHC, major histocompatibility complex; MIA, monosodium iodoacetate; MWCNT, multi-walled carbon nanotube; NF-κB, nuclear factor-κB; NLRP3, NOD-like receptor protein 3; NRF2, nuclear factor erythroid 2-related factor 2; OA, osteoarthritis; OVX, ovariectomized; PCL, polycaprolactone; PKA, protein kinase A; PKM2, pyruvate kinase M2; PVA, polyvinyl alcohol; Rho, Ras homolog family; ROCK, Rho-associated protein kinase; SAMP8, senescence-accelerated mouse prone 8; SQSTM1, sequestosome-1/p62; STAT6, signal transducer and activator of transcription 6; TGF-β, transforming growth factor-β; Tregs, regulatory T cells; TRPV1/4, transient receptor potential vanilloid 1/4; VEGF, vascular endothelial growth factor; YAP, yes-associated protein
4.1.1 Exercise Therapy
Exercise confers protective effects on musculoskeletal tissues by optimizing biomechanical loading patterns and modulating the local immune microenvironment. Clinical evidence demonstrates that a 12-week Tai Chi program performed three times weekly improves pain and functional outcomes, accompanied by enhanced neuromuscular control, improved load distribution, and regulation of inflammatory and immune pathways [282]. Comparable benefits have been observed with structured yoga and resistance-based strengthening programs [283]. Whole-body vibration training (WBV; 35-40 Hz, 4 mm amplitude, 3 sessions per week) combined with squat exercises further augments T cell-mediated immune responses and delays progression of knee OA in older adults [284].
Preclinical studies provide mechanistic support for these findings. Moderate-intensity treadmill running (10-20 m/min, 30 minutes per day) increases intra-articular lipoxin A4 levels, promotes macrophage polarization toward an M2 phenotype, and reduces synovitis in mid-stage OA models [74, 285]. Low-intensity running similarly attenuates disease progression in aged mice by suppressing synovial inflammation and enhancing M2 polarization. [286]. Prolonged moderate running (18 m/min for 90 minutes) expands intramuscular Treg populations, restrains IFN-γ production, and preserves mitochondrial integrity [26]. Endurance training protocols (45 minutes per session, 4 days per week for 4 weeks) increase M2 and M2c macrophage subsets, stimulate myofiber hypertrophy, and enhance satellite cell accumulation via upregulation of hepatocyte growth factor (HGF)/insulin-like growth factor 1 (IGF1), alongside suppression of CCAAT/enhancer-binding protein β (C/EBPβ) and muscle RING finger protein through the TWEAK-FN14 signaling axis [79].
In tendon tissue, exercise-induced mechanical loading activates IL-4/JAK/STAT6 signaling, driving macrophage polarization toward an M2 phenotype and facilitating fibrocartilage regeneration. Eccentric loading protocols (10 m/min, 20 minutes per day, 2-8 weeks) reduce M1 macrophage prevalence and accelerate tendon-bone healing in rotator cuff injury models [86, 270].
Exercise also exerts systemic osteoimmune effects via the muscle-bone axis. A regimen of treadmill running (12 m/min, 1 hour per day, 8 weeks) enhances skeletal-immune regulation. Contracting muscle releases myogenic metabolites, such as L-β-aminoisobutyric acid (L-BAIBA), which suppress osteoclastogenesis and prevent bone loss in ovariectomized models [287]. Within the bone marrow niche, mechanical stimulation increases macrophage-derived RCN2, which interacts with neuregulin-2 and integrin β1 on adipocytes to activate the cAMP-PKA pathway, thereby promoting lipolysis and generating metabolic substrates that support osteogenesis [61]. Sustained exercise further preserves LEPR+ osteolectin+ matrix cells, maintaining pools of osteogenic and lymphoid progenitors and reinforcing skeletal-immune homeostasis [27].
Adults aged ≥75 years often experience declines in muscle strength, balance, and overall functional capacity, limiting tolerance for high-intensity exercise. Clinical evidence shows that low- to moderate-intensity activities—such as Tai Chi, walking, chair-based exercise, or structured multicomponent programs—performed for approximately 150 minutes per week (e.g., 30 minutes per session, 5 sessions per week) at moderate exertion level ( approximately 55-75% of maximal heart rate) safely improve muscular strength, balance, flexibility, and functional capacity in this age group [282, 288, 289]. Mechanistic studies further demonstrate that graded, low- to moderate-load mechanical stimuli effectively engage mechano-immune pathways, including promotion of anti-inflammatory macrophage polarization, expansion of T cell populations, and enhancement of tissue repair processes. These converging findings support the concept that appropriately prescribed, low- to moderate-intensity exercise in advanced age may exert therapeutic benefit through activation of mechano-immune mechanisms, thereby linking underlying physiological mechanisms with feasible clinical applications.
4.1.2 Exogenous Physical Stimulation
In addition to conventional exercise, externally applied mechanical stimuli are increasingly investigated as therapeutic “mechanical prescriptions” capable of reprogramming immune responses. Modalities such as low-intensity pulsed ultrasound (LIPUS), mechanical vibration, and controlled stretching activate cellular mechanosensors, including Piezo1 and TRPV4, with activation efficiency dependent on stimulus frequency, intensity, duty cycle, and duration. In vitro experiments indicate that LIPUS delivered at 38 kHz, 250 mW/cm², 20% duty cycle for 90 min optimally activates Piezo1 in macrophages and promotes anti-inflammatory polarization. In contrast, in vivo applications in preclinical and clinical settings commonly employ higher frequencies and lower intensities, such as 1 MHz, 30 mW/cm², 20% duty cycle for 10 minutes per day, which are sufficient to induce Piezo1-mediated Ca²⁺ influx in immune and osteolineage cells [290-292]. Low-amplitude, high-frequency vibration (20-40 Hz, 0.3-1.0 g, 10-20 minutes per session) has similarly been shown to gate Piezo1 and TRPV4 channels [293].
Distraction osteogenesis (DO) provides a clinically established example of mechanically guided tissue regeneration. Standard protocols typically involve a latency period of 5-7 days, a distraction rate of approximately 1 mm per day applied in multiple incremental steps, and a consolidation phase of at least 6-8 weeks, collectively promoting stable bone formation and neovascularization. However, direct characterization of mechanosensitive receptor activation, including Piezo1 and TRP channels, under these clinical parameters remains limited. Most mechanistic insights are derived from in vitro or animal models, whereas systematic quantification linking distraction rate, rhythm, and strain magnitudes to mechanoreceptor activation remains lacking [58, 294].
Therapeutic modulation of mechano-immune pathways through externally applied mechanical stimuli has been validated in multiple experimental models. Manual massage (10-30 minutes per day at 1 Hz) promotes macrophage polarization toward an M2 phenotype while suppressing M1 dominance in young murine models, accelerates muscle recovery, and enhances neutrophil clearance to preserve tissue homeostasis [78, 295, 296].
LIPUS similarly exploits mechanosensitive signaling pathways to regulate immune responses. Application of LIPUS facilitates tendon-bone interface repair by promoting anti-inflammatory M2 macrophages polarization [297]. In the DMM OA model, LIPUS enhances macrophage autophagic activity, facilitates degradation of pyruvate kinase M (PKM) and sequestosome 1 (SQSTM1), reduces mature IL-1β levels, and improves gait parameters while attenuating synovial inflammation [298]. Suppression of osteoclast activity and reduced Netrin-1 secretion further contribute to analgesic effects [299].
Mechanical vibration represents an additional modality with mechanotype-specific effects. Low-amplitude high-frequency vibration (LMHFV) promotes bone healing in ovariectomized osteoporotic fracture models by modulating macrophage polarization [300]. Nanovibrational stimulation has been demonstrated to inhibit osteoclastogenesis and enhance osteogenic differentiation, supporting bone regeneration under mechanically optimized conditions [301].
DO represents a classical endogenous bone engineering approach in which controlled mechanical distraction, applied at defined rates and frequencies, induces tissue regeneration [302]. Mechanical stretching during DO activates FAK/MAPK, PI3K/AKT, and Wingless-related integration site (Wnt) signaling pathways, shifts macrophage polarization toward anti-inflammatory phenotypes, and upregulates TGF-β, BMP, and IL-6 expression, thereby promoting angiogenesis and osteogenesis [303]. Mechanical stretching has also shown anti-inflammatory effects in intervertebral disc degeneration models, potentially through analogous mechanically regulated immune mechanisms [304, 305].
Clinical cohort studies indicate that early application of LIPUS is associated with favorable fracture healing outcomes in elderly patients, with advanced age not substantially diminishing therapeutic efficacy, supporting its relevance in geriatric bone repair [306]. WBV (20-40 Hz, 1-4 mm amplitude, 0.3-1.0 g, 10-20 minutes per session) improves muscle strength and balance in older adults. Notably, increases in bone mineral density are more pronounced in individuals with osteopenia or osteoporosis, whereas effects in healthy young adults are generally minimal, suggesting preferentially benefit in mechanically or metabolically compromised bone [307-309]. Clinical observations further indicate that bone regeneration can be achieved across age groups with DO, although prolonged consolidation periods are often required in elderly patients [310]. Collectively, WBV and DO remain clinically applicable in older populations under standard parameters; however, regenerative efficiency may be slightly lower compared with younger individuals, consistent with age-associated decline in mechanosensitivity. Integration of multiple physical modalities, including exercise combined with DO or WBV, may enhance regenerative responses and optimize outcomes in aging musculoskeletal tissues.
4.1.3 Mechanical Stimulation of Bionic Materials and Scaffolds
Micromotion fixation systems are designed to provide controlled sub-millimeter axial displacement at fracture sites, thereby maintaining local strain within a biologically permissive range. In contrast to rigid fixation, such systems preserve moderate interfragmentary micromotion, supporting periosteal mechanosensing and facilitating early immune regulation during bone repair [311-313]. Experimental evidence shows that intermediate compressive strain of approximately 5%, comparable to micromotion-induced strain, promotes a regenerative macrophage phenotype, suppresses M1-dominant inflammation, and enhances pro-angiogenic signaling. In contrast, excessive strain approaching 35% triggers maladaptive inflammatory responses and impairs mineralization processes [314]. Maintenance of strain within this optimal mechanobiological window likely enables coordinated macrophage-mediated angiogenic and osteogenic coupling, thereby improving fracture healing outcomes [314].
Bionic materials and scaffold-derived mechanical cues have emerged as important modulators of immune responses in musculoskeletal repair. Implants engineering with moderate stiffness (approximately 80 kPa) inhibit Piezo1 activity and downregulate the IL-17/TNF signaling axis, thereby reducing inflammation associated with senescent macrophage while enhancing osteogenesis and implant integration [315]. These findings suggest that rationally designed mechanical properties can alleviate local inflammatory activation and potentially mitigate features of immunosenescence. Low-modulus Ti2448 alloy similarly modulates macrophage Piezo1 via stiffness-dependent cues, activating the Piezo1-YAP pathway to promote M2 polarization, angiogenesis, and osteogenic differentiation [316]. Surface nano-sheet arrays further stimulate integrin β2/FAK-PI3K-AKT-mTOR signaling, driving macrophage polarization toward M2 phenotype and supporting bone regeneration [317]. Left-handed nanohydroxyapatite activates the integrin β1-FAK-TRPV4 axis to reprogram macrophage responses and enhance osteogenic activity [314]. In tendon repair models, ordered nanofiber topographies modulate macrophage-driven inflammatory responses, and when combined with tensile loading, further promote M2 polarization to improve structural and functional healing outcomes [318, 319].
Beyond intrinsic physicochemical characteristics, emerging bionic and piezoelectric materials are capable of converting mechanical stimuli into bioactive immunoregulatory signals. Piezoelectric hydrogels combined with structured mechanical training have been demonstrated to accelerate rotator cuff repair and improve mechanical strength of the repaired interface [320]. Piezoelectric bioadhesives subjected to mechanical stress activate TRPV1-Ca²+-cAMP signaling, thereby promoting macrophage polarization toward an M2 phenotype, enhancing angiogenesis, and facilitating fibrocartilage formation [321]. In bone regeneration models, magnetically responsive hydrogels exposed to static magnetic fields stimulate M2 macrophage differentiation and enhance osteogenesis through activation of the podosome/Rho/ROCK pathway [322]. Scaffolds integrating LIPUS with piezoelectric components further augment Piezo1-mediated mechanotransduction, promote M2 polarization, and support bone regeneration [290, 323]. Advanced bioreactor systems capable of delivering precisely controlled mechanical stimuli to macrophages have recently been developed to generate EVs enriched with defined bioactive cargo, including miR-210-3p. These mechanically conditioned vesicles markedly accelerate bone defect healing in preclinical models, offering a promising cell-free therapeutic approach for musculoskeletal regeneration [324].
Despite accumulating evidence that mechanical cues regulate immune programs during tissue repair, the mechanistic framework and translational potential of mechano-immunomodulation in chronic musculoskeletal disorders, such as osteoarthritis and sarcopenia, remain incompletely defined. Advancement in this field will likely require development of tissue-specific “mechanical prescriptions” that target distinct mechano-immune axes accordingly to pathological context. For synovial joints, multidirectional, low-impact movement strategies such as Tai Chi and yoga may optimize load redistribution and alleviate synovial inflammation [282, 283]. In skeletal muscle, combined endurance and resistance training protocols may restore myokine signaling and facilitate T cell- and macrophage-mediated regenerative processes [325, 326]. Comparable precision-based mechanical interventions tailored to bone, tendon, and intervertebral disc tissues may similarly re-establish local immune equilibrium and improve functional recovery. Refinement of such targeted strategies may provide a framework for integrating mechanobiology with immune modulation in the management of age-related musculoskeletal disease.
4.2 Precision Targeting of Mechano-Immune Signaling Networks
Although mechanical loading, structured exercise, and biomaterial-derived stiffness provide effective means for modulating mechano-immunity at the tissue level, a complementary and potentially more precise strategy involves direct targeting of mechanoreceptors and their downstream epigenetic regulatory elements.
4.2.1 Targeting Mechanosensitive Ion Channels in Immune Cells
Mechanosensitive ion channels, particularly Piezo1, represent promising molecular targets for modulation of immune cell mechanotransduction. Pharmacological inhibition of Piezo1 with the peptide GsMTx4 has been reported to attenuate disease progression in ankylosing spondylitis through regulation of macrophage autophagy [327]. Natural compounds and traditional Chinese medicine-derived formulations have also demonstrated modulatory effects on Piezo1 signaling. Guizhitongluo Tablet has been observed to reduce macrophage Piezo1 expression and Ca²⁺ influx, thereby suppressing NLRP3 inflammasome activation and pyroptosis, ultimately delaying atherosclerosis progression in murine models [328]. The alkaloid jatrorrhizine similarly inhibits Piezo1 channel activation and mitigates vascular inflammation [329]. These findings suggest that modulation of Piezo1-dependent mechanosensing pathways may provide a strategy for regulating mechano-immune responses in chronic inflammatory states. Bioactive compounds derived from herbal sources may offer comparatively favorable safety profiles while influencing mechanotransductive signaling. Nevertheless, targeted manipulation of immune cell Piezo1 in degenerative musculoskeletal disorders remains insufficiently investigated, and translation of such approaches into therapeutic application is currently at an early experimental stage.
4.2.2 Targeting Mechanosensitive Epigenetic Modifications
Aberrantly regulated enhancers have been identified in degenerative musculoskeletal disorders including osteoporosis, IDD, and osteoarthritis, implicating epigenetic dysregulation in disease pathogenesis [330-332]. Recent genomic analyses have delineated mechanosensitive enhancers responsive to ECM stiffness, including loci associated with SKP2, connective tissue growth factor (CTGF), and myosin heavy chain 9 (MYH9). These regulatory elements modulate transcriptional programs governing apoptosis, proliferation, migration, and cytoskeletal contractility, thereby establishing a direct molecular interface between mechanical cues and gene expression[333].
Despite these advances, therapeutic exploitation of mechanosensitive enhancers within immune cells in musculoskeletal tissues remain largely unexplored. Epigenetic modulation of enhancer activity has emerged as a promising strategy. Pharmacological agents targeting bromodomain and extraterminal (BET) proteins or DNA methyltransferases (DNMTs) have demonstrated the capacity to regulate disease-associated enhancer landscapes and have shown efficacy in oncologic and cardiovascular contexts. [334]. Translation of such approaches to mechano-immune regulation in musculoskeletal disorders warrants systematic investigation.
Future directions may include precise gene or epigenome editing to directly regulate mechanoresponsive enhancer elements, as well as biomaterial-based approaches, such as stiffness-tunable hydrogels. to alter the mechanical microenvironment and indirectly reshape enhancer activity. Integration of these approaches may provide a framework for precision-based intervention in degenerative musculoskeletal diseases.
4.2.3 Targeting the Bone Mechanical-Neural-Immune Axis
Intraosseous interoception has recently been recognized as a regulatory mechanism mediated by Aδ and C sensory fibers that detect mechanical forces and metabolic stress within bone tissue, thereby contributing to skeletal homeostasis [335]. Under pathological conditions, such as aging and diabetes, intraosseous sensory function declines, leading to impaired mechanosensory signaling and associated immune dysregulation [336]. Restoration of disrupted intraosseous interoception through advanced biomaterials strategies may therefore improve both bone metabolic activity and the local immune microenvironment.
Material-based strategies aimed at re-establishing intraosseous interoceptive function include natural or synthetic carriers delivering neurotrophic factors or neuropeptides, inorganic or metal-based biomaterials releasing bioactive ions, conductive composite materials, and cell-free biologic constructs. These platforms promote reconstruction of the intraosseous sensory network by activating nerve-bone signaling pathways, promoting nerve reinnervation, and facilitating transmission of mechanical signals. Detailed overviews of these strategies have been provided in recent reviews [337-339].
Collectively, precision targeting of mechano-immune networks across multiple regulatory layers—including mechanosensitive ion channels, epigenetic enhancer elements, and the bone mechanical-neural-immune axis—represents a multifaceted framework for restoring tissue homeostasis. Although most approaches remain at the preclinical stage, such strategies outline a conceptual basis for future translational therapies in degenerative musculoskeletal diseases.
5. Conclusion
Aging exerts profound effects on the musculoskeletal system through tightly interconnected mechanical and immune pathways. Progressive reductions in tissue elasticity, ECM integrity, and mechanosensory capacity of immune and stromal cells disrupt physiological mechano-immune signaling, fostering chronic inflammation and structural degeneration in bone, joints, skeletal muscle, tendon, and intervertebral disc tissues. These alterations establish self-reinforcing feedback loops in which mechanical imbalance intensifies immune dysfunction, while immunosenescence further impairs mechanotransductive responsiveness.
Recognition of these age-associated mechano-immune interactions provides a unifying framework for understanding the progression of osteoporosis, OA, IDD, and sarcopenia. This perspective also identifies potential therapeutic avenues. Restoration or recalibration of mechano-immune coupling through controlled mechanical stimulation, biomaterial-guided mechanoregulation, and targeted modulation of cellular mechanotransductive signaling networks may attenuate degenerative cascades, promote tissue repair, and improve functional outcomes in older populations. Continued integration of mechanobiology, immunology, and tissue engineering will be pivotal for translating principles of mechano-immunity into precision-based interventions for age-related musculoskeletal disorders.
Abbreviations
ADAMTS: a disintegrin and metalloproteinase with thrombospondin motifs; AFCs: annulus fibrosus cells; AKT: protein kinase B; BMMs: bone marrow macrophage; BMP2: bone morphogenetic protein 2; BMSCs: bone marrow stromal cells; CaMKII: calmodulin-dependent kinase II; CCL: CC chemokine ligand; DAMPs: damage-associated molecular patterns; DMM: destabilization of the medial meniscus; DRG: dorsal root ganglia; ECM: extracellular matrix; EVs: extracellular vesicles; FAK: focal adhesion kinase; IDD: intervertebral disc degeneration; IGF-1: insulin-like growth factor 1; IL-17A: interleukin-17A; K2P: two-pore potassium channels; LCS: lacunocanalicular system; LINC: linker of nucleoskeleton and cytoskeleton; LIPUS: low-intensity pulsed ultrasound; L-BAIBA: L-β-aminoisobutyric acid; MAPK: mitogen-activated protein kinase; MMP-9: matrix metalloproteinase-9; MSCs: mesenchymal stem cells; NGF: nerve growth factor; NFAT: nuclear factor of activated T cells; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; NLRP3: NOD-like receptor family pyrin domain containing 3; OA: osteoarthritis; OPG: osteoprotegerin; OP: osteoporosis; Piezo1: piezo-type mechanosensitive ion channel component 1; PDGF-BB: platelet-derived growth factor-BB; PI3K: phosphatidylinositol 3-kinase; RANKL: receptor activator of nuclear factor κB ligand; RCN2: reticulocalbin-2; Rho-ROCK: Ras homolog-Rho-associated coiled-coil kinase; SASP: senescence-associated secretory phenotype; SIRT6: sirtuin 6; STAT: signal transducer and activator of transcription; TCR: T cell receptor; TGF-β1: transforming growth factor-β1; Th17: T helper 17 cells; Tregs: regulatory T cells; TRPV4: transient receptor potential vanilloid 4; VEGF-C: vascular endothelial growth factor-C; VEGFR3: vascular endothelial growth factor receptor 3; WBV: whole body vibration; Wnt3a: Wingless-type MMTV integration site family member 3A; YAP/TAZ: Yes-associated protein/transcriptional coactivator with PDZ-binding motif; NPCs: nucleus pulposus cells.
Acknowledgements
This study was partly supported by the National Natural Science Foundation of China (82472437 to X.Li. and 82472513 to L.X.), Hubei Provincial Science and Technology Plan Project (2025BCB002) and Hubei Provincial Natural Science Foundation of China (2025AFB550).
Author Contributions
All authors discussed and wrote the article.
Artificial Intelligence (AI) Usage Statement
During the preparation of this manuscript, AI-assisted tools (ChatGPT) were used solely for language polishing to improve the fluency, grammar, and semantic accuracy of the English text, facilitating the conversion from the authors' native language into academic English. These tools were not used to generate scientific ideas, draft the manuscript, perform data analysis, interpret results, create figures, or formulate conclusions.
All scientific content, including the literature interpretation, data presentation, and final manuscript revision, was independently completed, reviewed, and approved by the authors. The authors take full responsibility for the accuracy, originality, and integrity of the work.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Salari N, Darvishi N, Bartina Y, Larti M, Kiaei A, Hemmati M. et al. Global prevalence of osteoporosis among the world older adults: a comprehensive systematic review and meta-analysis. Journal of orthopaedic surgery and research. 2021;16:669
2. Courties A, Kouki I, Soliman N, Mathieu S, Sellam J. Osteoarthritis year in review 2024: Epidemiology and therapy. Osteoarthritis Cartilage. 2024;32:1397-404
3. Diwan AD, Melrose J. Intervertebral disc degeneration and how it leads to low back pain. JOR Spine. 2023;6:e1231
4. Kushioka J, Chow SK, Toya M, Tsubosaka M, Shen H, Gao Q. et al. Bone regeneration in inflammation with aging and cell-based immunomodulatory therapy. Inflamm Regen. 2023;43:29
5. Motta F, Barone E, Sica A, Selmi C. Inflammaging and Osteoarthritis. Clin Rev Allergy Immunol. 2023;64:222-38
6. Shen L, Zong Y, Zhao J, Yang Y, Li L, Li N. et al. Characterizing the skeletal muscle immune microenvironment for sarcopenia: insights from transcriptome analysis and histological validation. Front Immunol. 2024;15:1414387
7. Wang Y, Hu B, Tian L, Li G, Xu B. Comprehensive profiling of immune cell infiltration and biomarker identification in intervertebral disc degeneration. Journal of orthopaedic surgery and research. 2025;20:829
8. Mittelheisser V, Gensbittel V, Bonati L, Li W, Tang L, Goetz JG. Evidence and therapeutic implications of biomechanically regulated immunosurveillance in cancer and other diseases. Nat Nanotechnol. 2024;19:281-97
9. Morgan EF, Unnikrisnan GU, Hussein AI. Bone Mechanical Properties in Healthy and Diseased States. Annu Rev Biomed Eng. 2018;20:119-43
10. Ateş F, Marquetand J, Zimmer M. Detecting age-related changes in skeletal muscle mechanics using ultrasound shear wave elastography. Scientific reports. 2023;13:20062
11. Lyon RC, Zanella F, Omens JH, Sheikh F. Mechanotransduction in cardiac hypertrophy and failure. Circ Res. 2015;116:1462-76
12. Vergroesen PP, Kingma I, Emanuel KS, Hoogendoorn RJ, Welting TJ, van Royen BJ. et al. Mechanics and biology in intervertebral disc degeneration: a vicious circle. Osteoarthritis Cartilage. 2015;23:1057-70
13. Du H, Bartleson JM, Butenko S, Alonso V, Liu WF, Winer DA. et al. Tuning immunity through tissue mechanotransduction. Nature reviews Immunology. 2023;23:174-88
14. Yan R, Li Y, Chen S, Zhu L, Zhou C, Chen J. et al. Mechanotransduction in Shaping Immunity: Pathways, Crosstalk, and Pathophysiological Relevance. Adv Sci (Weinh). 2025;12:e12164
15. Villemain O, Correia M, Mousseaux E, Baranger J, Zarka S, Podetti I. et al. Myocardial Stiffness Evaluation Using Noninvasive Shear Wave Imaging in Healthy and Hypertrophic Cardiomyopathic Adults. JACC Cardiovasc Imaging. 2019;12:1135-45
16. Stearns-Reider KM, D'Amore A, Beezhold K, Rothrauff B, Cavalli L, Wagner WR. et al. Aging of the skeletal muscle extracellular matrix drives a stem cell fibrogenic conversion. Aging Cell. 2017;16:518-28
17. Larsson L, Degens H, Li M, Salviati L, Lee YI, Thompson W. et al. Sarcopenia: Aging-Related Loss of Muscle Mass and Function. Physiol Rev. 2019;99:427-511
18. Tiede-Lewis LM, Dallas SL. Changes in the osteocyte lacunocanalicular network with aging. Bone. 2019;122:101-13
19. Niroobakhsh M, Laughrey LE, Dallas SL, Johnson ML, Ganesh T. Computational modeling based on confocal imaging predicts changes in osteocyte and dendrite shear stress due to canalicular loss with aging. Biomech Model Mechanobiol. 2024;23:129-43
20. Seegren PV, Harper LR, Downs TK, Zhao XY, Viswanathan SB, Stremska ME. et al. Reduced mitochondrial calcium uptake in macrophages is a major driver of inflammaging. Nature aging. 2023;3:796-812
21. Li Z, Jiao Y, Fan EK, Scott MJ, Li Y, Li S. et al. Aging-Impaired Filamentous Actin Polymerization Signaling Reduces Alveolar Macrophage Phagocytosis of Bacteria. J Immunol. 2017;199:3176-86
22. Moss CE, Johnston SA, Kimble JV, Clements M, Codd V, Hamby S. et al. Aging-related defects in macrophage function are driven by MYC and USF1 transcriptional programs. Cell Rep. 2024;43:114073
23. Vasse GF, Nizamoglu M, Heijink IH, Schlepütz M, van Rijn P, Thomas MJ. et al. Macrophage-stroma interactions in fibrosis: biochemical, biophysical, and cellular perspectives. J Pathol. 2021;254:344-57
24. Kroemer G, Maier AB, Cuervo AM, Gladyshev VN, Ferrucci L, Gorbunova V. et al. From geroscience to precision geromedicine: Understanding and managing aging. Cell. 2025;188:2043-62
25. Chen K, Griffin M, Henn D, Berryman KS, Sivaraj D, Kussie HC. et al. Targeting circulating mechanoresponsive monocytes and macrophages to reduce fibrosis. Nat Biomed Eng. 2025
26. Langston PK, Sun Y, Ryback BA, Mueller AL, Spiegelman BM, Benoist C. et al. Regulatory T cells shield muscle mitochondria from interferon-γ-mediated damage to promote the beneficial effects of exercise. Sci Immunol. 2023;8:eadi5377
27. Shen B, Tasdogan A, Ubellacker JM, Zhang J, Nosyreva ED, Du L. et al. A mechanosensitive peri-arteriolar niche for osteogenesis and lymphopoiesis. Nature. 2021;591:438-44
28. Cheng CK, Wang N, Wang L, Huang Y. Biophysical and Biochemical Roles of Shear Stress on Endothelium: A Revisit and New Insights. Circ Res. 2025;136:752-72
29. Bagnat M, Daga B, Di Talia S. Morphogenetic Roles of Hydrostatic Pressure in Animal Development. Annu Rev Cell Dev Biol. 2022;38:375-94
30. Linke JA, Munn LL, Jain RK. Compressive stresses in cancer: characterization and implications for tumour progression and treatment. Nature reviews Cancer. 2024;24:768-91
31. Lai A, Cox CD, Chandra Sekar N, Thurgood P, Jaworowski A, Peter K. et al. Mechanosensing by Piezo1 and its implications for physiology and various pathologies. Biol Rev Camb Philos Soc. 2022;97:604-14
32. Rosenbaum T, Islas LD. Molecular Physiology of TRPV Channels: Controversies and Future Challenges. Annu Rev Physiol. 2023;85:293-316
33. de Jesus M, Settle AH, Vorselen D, Gaetjens TK, Galiano M, Romin Y. et al. Single-cell topographical profiling of the immune synapse reveals a biomechanical signature of cytotoxicity. Sci Immunol. 2024;9:eadj2898
34. Wan Z, Xu C, Chen X, Xie H, Li Z, Wang J. et al. PI(4,5)P2 determines the threshold of mechanical force-induced B cell activation. J Cell Biol. 2018;217:2565-82
35. Pang X, He X, Qiu Z, Zhang H, Xie R, Liu Z. et al. Targeting integrin pathways: mechanisms and advances in therapy. Signal Transduct Target Ther. 2023Jan2;8(1):1
36. Türkaydin B, Schewe M, Riel EB, Schulz F, Biedermann J, Baukrowitz T. et al. Atomistic mechanism of coupling between cytosolic sensor domain and selectivity filter in TREK K2P channels. Nat Commun. 2024;15:4628
37. Berrier C, Pozza A, de Lacroix de Lavalette A, Chardonnet S, Mesneau A, Jaxel C. et al. The purified mechanosensitive channel TREK-1 is directly sensitive to membrane tension. J Biol Chem. 2013;288:27307-14
38. Sorum B, Docter T, Panico V, Rietmeijer RA, Brohawn SG. Tension activation of mechanosensitive two-pore domain K+ channels TRAAK, TREK-1, and TREK-2. Nat Commun. 2024;15:3142
39. Zhang Y, Fu J, Han Y, Feng D, Yue S, Zhou Y. et al. Two-Pore-Domain Potassium Channel TREK-1 Mediates Pulmonary Fibrosis through Macrophage M2 Polarization and by Direct Promotion of Fibroblast Differentiation. Biomedicines. 2023 11
40. Gao T, Maskalenko NA, Kabir S, Campbell KS, Wu J. Molecular basis of β2 integrin activation by talin unveils subunit-specific mechanisms of integrin signaling. Cell Rep. 2025;44:115607
41. Di X, Gao X, Peng L, Ai J, Jin X, Qi S. et al. Cellular mechanotransduction in health and diseases: from molecular mechanism to therapeutic targets. Signal Transduct Target Ther. 2023;8:282
42. Xiao B. Mechanisms of mechanotransduction and physiological roles of PIEZO channels. Nat Rev Mol Cell Biol. 2024;25:886-903
43. Saez A, Herrero-Fernandez B, Gomez-Bris R, Somovilla-Crespo B, Rius C, Gonzalez-Granado JM. Lamin A/C and the Immune System: One Intermediate Filament, Many Faces. Int J Mol Sci. 2020 21
44. Wang N, Tytell Jd Fau - Ingber DE, Ingber DE. Mechanotransduction at a distance: mechanically coupling the extracellular matrix with the nucleus. Nat Rev Mol Cell Biol. 2009Jan;10(1):75-82
45. Lee YL, Burke B. LINC complexes and nuclear positioning. Semin Cell Dev Biol. 2018 Oct:82:67-76
46. Wei Y, Hui VLZ, Chen Y, Han R, Han X, Guo Y. YAP/TAZ: Molecular pathway and disease therapy. MedComm (2020). 2023;4:e340
47. Scott KE, Fraley SI, Rangamani P. A spatial model of YAP/TAZ signaling reveals how stiffness, dimensionality, and shape contribute to emergent outcomes. Proc Natl Acad Sci U S A. 2021 118
48. Huang Z, Iqbal Z, Zhao Z, Liu J, Alabsi AM, Shabbir M. et al. Cellular crosstalk in the bone marrow niche. J Transl Med. 2024;22:1096
49. Li C, Fennessy P. The periosteum: a simple tissue with many faces, with special reference to the antler-lineage periostea. Biol Direct. 2021;16:17
50. Iglesias-Velazquez O, Gf Tresguerres F, I FT, Leco-Berrocal I, Lopez-Pintor R, Baca L. et al. OsteoMac: A new player on the bone biology scene. Ann Anat. 2024;254:152244
51. Batoon L, Millard SM, Raggatt LJ, Pettit AR. Osteomacs and Bone Regeneration. Current osteoporosis reports. 2017;15:385-95
52. Deng R, Li C, Wang X, Chang L, Ni S, Zhang W. et al. Periosteal CD68(+) F4/80(+) Macrophages Are Mechanosensitive for Cortical Bone Formation by Secretion and Activation of TGF-β1. Adv Sci (Weinh). 2022;9:e2103343
53. Takeshita S, Kikuno R, Tezuka K, Amann E. Osteoblast-specific factor 2: cloning of a putative bone adhesion protein with homology with the insect protein fasciclin I. Biochem J. 1993;294( Pt 1):271-8
54. Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19:179-92
55. Wang Z, Lin M, Pan Y, Liu Y, Yang C, Wu J. et al. Periostin(+) myeloid cells improved long bone regeneration in a mechanosensitive manner. Bone research. 2024;12:59
56. Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nature reviews Immunology. 2007;7:292-304
57. Okamoto K, Takayanagi H. Osteoimmunology. Cold Spring Harbor perspectives in medicine. 2019 9
58. Cai G, Lu Y, Zhong W, Wang T, Li Y, Ruan X. et al. Piezo1-mediated M2 macrophage mechanotransduction enhances bone formation through secretion and activation of transforming growth factor-β1. Cell Prolif. 2023;56:e13440
59. Liu H, Zhou H, Fan Y, Li J, Guo Z, Xu Q. et al. Macrophages regulate angiogenesis-osteogenesis coupling induced by mechanical loading through the Piezo1 pathway. J Bone Miner Res. 2025;40:725-37
60. Kim SP, Li Z, Zoch ML, Frey JL, Bowman CE, Kushwaha P. et al. Fatty acid oxidation by the osteoblast is required for normal bone acquisition in a sex- and diet-dependent manner. JCI Insight. 2017 2
61. Peng H, Hu B, Xie LQ, Su T, Li CJ, Liu Y. et al. A mechanosensitive lipolytic factor in the bone marrow promotes osteogenesis and lymphopoiesis. Cell Metab. 2022Aug2;34(8):1168-1182.e6
62. Ouyang L, Yan B, Liu Y, Mao C, Wang M, Liu N. et al. The deubiquitylase UCHL3 maintains cancer stem-like properties by stabilizing the aryl hydrocarbon receptor. Signal Transduct Target Ther. 2020;5:78
63. Pu P, Wu S, Zhang K, Xu H, Guan J, Jin Z. et al. Mechanical force induces macrophage-derived exosomal UCHL3 promoting bone marrow mesenchymal stem cell osteogenesis by targeting SMAD1. J Nanobiotechnology. 2023;21:88
64. Li Y, Yan Z, Dai Y, Cai H, Chen Y, Chen Y. et al. Mechanical Force Promotes Mitochondrial Transfer From Macrophages to BMSCs to Enhance Bone Formation. Cell Prolif. 2025: e70121.
65. Gao L, Liu G, Wu X, Liu C, Wang Y, Ma M. et al. Osteocytes autophagy mediated by mTORC2 activation controls osteoblasts differentiation and osteoclasts activities under mechanical loading. Arch Biochem Biophys. 2023;742:109634
66. Wald S, Leibowitz A, Aizenbud Y, Saba Y, Zubeidat K, Barel O. et al. γδT Cells Are Essential for Orthodontic Tooth Movement. J Dent Res. 2021;100:731-8
67. Sun Z, Chen Y, Ding P, Wang Z, Lin Z, Zhou B. et al. Mechanical force-induced oncostatin M secretion by Jun-positive neutrophils promotes craniofacial bone regeneration for midface hypoplasia treatment. Stem cell research & therapy. 2025;16:330
68. Angeli V, Lim HY. Biomechanical control of lymphatic vessel physiology and functions. Cell Mol Immunol. 2023;20:1051-62
69. Liu H, Liu L, Zhang Y, Tian Q, Ding Z, Chen J. et al. Lymphatic-immune interactions in the musculoskeletal system. Front Immunol. 2025;16:1578847
70. Knab K, Chambers D, Krönke G. Synovial Macrophage and Fibroblast Heterogeneity in Joint Homeostasis and Inflammation. Front Med (Lausanne). 2022;9:862161
71. Zhao K, Ruan J, Nie L, Ye X, Li J. Effects of synovial macrophages in osteoarthritis. Front Immunol. 2023;14:1164137
72. Zhang H, Cai D, Bai X. Macrophages regulate the progression of osteoarthritis. Osteoarthritis Cartilage. 2020;28:555-61
73. Zheng W, Li X, Li J, Wang X, Liu D, Zhai L. et al. Mechanical loading mitigates osteoarthritis symptoms by regulating the inflammatory microenvironment in a mouse model. Ann N Y Acad Sci. 2022;1512:141-53
74. Shen P, Jia S, Wang Y, Zhou X, Zhang D, Jin Z. et al. Mechanical stress protects against chondrocyte pyroptosis through lipoxin A(4) via synovial macrophage M2 subtype polarization in an osteoarthritis model. Biomed Pharmacother. 2022;153:113361
75. Li X, Yang J, Liu D, Li J, Niu K, Feng S. et al. Knee loading inhibits osteoclast lineage in a mouse model of osteoarthritis. Scientific reports. 2016;6:24668
76. Theret M, Rossi FMV, Contreras O. Evolving Roles of Muscle-Resident Fibro-Adipogenic Progenitors in Health, Regeneration, Neuromuscular Disorders, and Aging. Front Physiol. 2021;12:673404
77. Wang X, Zhou L. The Many Roles of Macrophages in Skeletal Muscle Injury and Repair. Front Cell Dev Biol. 2022;10:952249
78. Seo BR, Payne CJ, McNamara SL, Freedman BR, Kwee BJ, Nam S. et al. Skeletal muscle regeneration with robotic actuation-mediated clearance of neutrophils. Sci Transl Med. 2021;13:eabe8868
79. Walton RG, Kosmac K, Mula J, Fry CS, Peck BD, Groshong JS. et al. Human skeletal muscle macrophages increase following cycle training and are associated with adaptations that may facilitate growth. Scientific reports. 2019;9:969
80. Peck BD, Murach KA, Walton RG, Simmons AJ, Long DE, Kosmac K. et al. A muscle cell-macrophage axis involving matrix metalloproteinase 14 facilitates extracellular matrix remodeling with mechanical loading. FASEB J. 2022;36:e22155
81. Becker M, Joseph SS, Garcia-Carrizo F, Tom RZ, Opaleva D, Serr I. et al. Regulatory T cells require IL6 receptor alpha signaling to control skeletal muscle function and regeneration. Cell Metab. 2023;35:1736-51.e7
82. Yan Y, Antolin N, Zhou L, Xu L, Vargas IL, Gomez CD. et al. Macrophages excite muscle spindles with glutamate to bolster locomotion. Nature. 2025;637:698-707
83. Benjamin M, Toumi H, Ralphs JR, Bydder G, Best TM, Milz S. Where tendons and ligaments meet bone: attachment sites ('entheses') in relation to exercise and/or mechanical load. J Anat. 2006;208:471-90
84. Bautista CA, Srikumar A, Tichy ED, Qian G, Jiang X, Qin L. et al. CD206+ tendon resident macrophages and their potential crosstalk with fibroblasts and the ECM during tendon growth and maturation. Front Physiol. 2023;14:1122348
85. Wang Y, Lu X, Lu J, Hernigou P, Jin F. The role of macrophage polarization in tendon healing and therapeutic strategies: Insights from animal models. Front Bioeng Biotechnol. 2024;12:1366398
86. Peng Y, Diao L, Li F, Wang J, Yu Y, Jia S. et al. Eccentric Mechanical Stimulation Promotes Rotator Cuff Healing by Regulating Macrophage Polarization in a Murine Model. J Shoulder Elbow Surg. 2025
87. Blomgran P, Blomgran R, Ernerudh J, Aspenberg P. A possible link between loading, inflammation and healing: Immune cell populations during tendon healing in the rat. Scientific reports. 2016;6:29824
88. Feng P, Che Y, Gao C, Zhu L, Gao J, Vo NV. Immune exposure: how macrophages interact with the nucleus pulposus. Front Immunol. 2023;14:1155746
89. Sun Z, Liu B, Luo ZJ. The Immune Privilege of the Intervertebral Disc: Implications for Intervertebral Disc Degeneration Treatment. Int J Med Sci. 2020;17:685-92
90. Ren Q, Chen L, Ma Y, Huang Y, Wang S. Immune microenvironment in intervertebral disc degeneration: pathophysiology and therapeutic potential. Front Immunol. 2025;16:1563635
91. Lloyd SM, He Y. Exploring Extracellular Matrix Crosslinking as a Therapeutic Approach to Fibrosis. Cells. 2024 13
92. Sutherland TE, Dyer DP, Allen JE. The extracellular matrix and the immune system: A mutually dependent relationship. Science. 2023;379:eabp8964
93. Harper EI, Weeraratna AT. A Wrinkle in TIME: How Changes in the Aging ECM Drive the Remodeling of the Tumor Immune Microenvironment. Cancer Discov. 2023;13:1973-81
94. Atcha H, Jairaman A, Holt JR, Meli VS, Nagalla RR, Veerasubramanian PK. et al. Mechanically activated ion channel Piezo1 modulates macrophage polarization and stiffness sensing. Nat Commun. 2021;12:3256
95. Meli VS, Atcha H, Veerasubramanian PK, Nagalla RR, Luu TU, Chen EY. et al. YAP-mediated mechanotransduction tunes the macrophage inflammatory response. Sci Adv. 2020 6
96. Zhou X, Li W, Wang S, Zhang P, Wang Q, Xiao J. et al. YAP Aggravates Inflammatory Bowel Disease by Regulating M1/M2 Macrophage Polarization and Gut Microbial Homeostasis. Cell Rep. 2019;27:1176-89.e5
97. Veerasubramanian PK, Shao H, Meli VS, Phan TAQ, Luu TU, Liu WF. et al. A Src-H3 acetylation signaling axis integrates macrophage mechanosensation with inflammatory response. Biomaterials. 2021;279:121236
98. Xu Y, Ying L, Lang JK, Hinz B, Zhao R. Modeling mechanical activation of macrophages during pulmonary fibrogenesis for targeted anti-fibrosis therapy. Sci Adv. 2024;10:eadj9559
99. Hsieh JY, Keating MT, Smith TD, Meli VS, Botvinick EL, Liu WF. Matrix crosslinking enhances macrophage adhesion, migration, and inflammatory activation. APL bioengineering. 2019;3:016103
100. Tan H, Yang G, Zhu Y, He X, Yang L, Hu Y. et al. Mechanical Force Triggers Macrophage Pyroptosis and Sterile Inflammation by Disrupting Cellular Energy Metabolism. Int J Mol Sci. 2025 26
101. Zhao D, Wu Y, Zhuang J, Xu C, Zhang F. Activation of NLRP1 and NLRP3 inflammasomes contributed to cyclic stretch-induced pyroptosis and release of IL-1β in human periodontal ligament cells. Oncotarget. 2016;7:68292-302
102. Tan H, Wang S, He X, Yang G, Zhu Y, Yang S. et al. Microneedles Loaded with Nitric-Oxide Driven Nanomotors Improve Force-Induced Efferocytosis Impairment and Sterile Inflammation by Revitalizing Macrophage Energy Metabolism. ACS Nano. 2025;19:9390-411
103. Vasudevan SO, Behl B, Rathinam VA. Pyroptosis-induced inflammation and tissue damage. Semin Immunol. 2023;69:101781
104. Reilly EC, Lambert Emo K, Buckley PM, Reilly NS, Smith I, Chaves FA. et al. T(RM) integrins CD103 and CD49a differentially support adherence and motility after resolution of influenza virus infection. Proc Natl Acad Sci U S A. 2020;117:12306-14
105. Ferreira C, Barros L, Baptista M, Blankenhaus B, Barros A, Figueiredo-Campos P. et al. Type 1 T(reg) cells promote the generation of CD8(+) tissue-resident memory T cells. Nat Immunol. 2020;21:766-76
106. Holz LE, Chua YC, de Menezes MN, Anderson RJ, Draper SL, Compton BJ. et al. Glycolipid-peptide vaccination induces liver-resident memory CD8(+) T cells that protect against rodent malaria. Sci Immunol. 2020 5
107. Stolp B, Thelen F, Ficht X, Altenburger LM, Ruef N, Inavalli V. et al. Salivary gland macrophages and tissue-resident CD8(+) T cells cooperate for homeostatic organ surveillance. Sci Immunol. 2020 5
108. Ruef N, Martínez Magdaleno J, Ficht X, Purvanov V, Palayret M, Wissmann S. et al. Exocrine gland-resident memory CD8(+) T cells use mechanosensing for tissue surveillance. Sci Immunol. 2023;8:eadd5724
109. Moreau JF, Pradeu T, Grignolio A, Nardini C, Castiglione F, Tieri P. et al. The emerging role of ECM crosslinking in T cell mobility as a hallmark of immunosenescence in humans. Ageing Res Rev. 2017;35:322-35
110. Madissoon E, Oliver AJ, Kleshchevnikov V, Wilbrey-Clark A, Polanski K, Richoz N. et al. A spatially resolved atlas of the human lung characterizes a gland-associated immune niche. Nat Genet. 2023;55:66-77
111. Goplen NP, Wu Y, Son YM, Li C, Wang Z, Cheon IS. et al. Tissue-resident CD8(+) T cells drive age-associated chronic lung sequelae after viral pneumonia. Sci Immunol. 2020 5
112. Zhang J, Li J, Hou Y, Lin Y, Zhao H, Shi Y. et al. Osr2 functions as a biomechanical checkpoint to aggravate CD8(+) T cell exhaustion in tumor. Cell. 2024;187:3409-26.e24
113. Pang R, Sun W, Yang Y, Wen D, Lin F, Wang D. et al. PIEZO1 mechanically regulates the antitumour cytotoxicity of T lymphocytes. Nat Biomed Eng. 2024;8:1162-76
114. Huang K, Cai H. Matrix stiffness in osteoarthritis: from mechanism introduction to biomaterial-based therapies. Frontiers in endocrinology. 2025;16:1571502
115. Tang W, Yin JB, Lin RG, Wu CY, Huang JL, Zhu JJ. et al. Rapgef3 modulates macrophage reprogramming and exacerbates synovitis and osteoarthritis under excessive mechanical loading. iScience. 2025;28:112131
116. Wheeler TA, Antoinette AY, Bhatia E, Kim MJ, Ijomanta CN, Zhao A. et al. Mechanical loading of joint modulates T cells in lymph nodes to regulate osteoarthritis. Osteoarthritis Cartilage. 2024;32:287-98
117. Walter BA, Mageswaran P, Mo X, Boulter DJ, Mashaly H, Nguyen XV. et al. MR Elastography-derived Stiffness: A Biomarker for Intervertebral Disc Degeneration. Radiology. 2017;285:167-75
118. Tseng C, Chen B, Han Y, Wang K, Song Q, Shen H. et al. Advanced glycation end products promote intervertebral disc degeneration by transactivation of matrix metallopeptidase genes. Osteoarthritis Cartilage. 2024;32:187-99
119. Sadowska A, Kameda T, Krupkova O, Wuertz-Kozak K. Osmosensing, osmosignalling and inflammation: how intervertebral disc cells respond to altered osmolarity. Eur Cell Mater. 2018;36:231-50
120. Chen Y, Klein-Nulend J, Bravenboer N. Ageing-Related Changes in Ultrastructural Bone Matrix Composition and Osteocyte Mechanosensitivity. Current osteoporosis reports. 2025;23:35
121. Cunningham HC, Orr S, Murugesh DK, Hsia AW, Osipov B, Go L. et al. Differential bone adaptation to mechanical unloading and reloading in young, old, and osteocyte deficient mice. Bone. 2023;167:116646
122. Sladitschek-Martens HL, Guarnieri A, Brumana G, Zanconato F, Battilana G, Xiccato RL. et al. YAP/TAZ activity in stromal cells prevents ageing by controlling cGAS-STING. Nature. 2022;607:790-8
123. Poon IKH, Ravichandran KS. Targeting Efferocytosis in Inflammaging. Annu Rev Pharmacol Toxicol. 2024;64:339-57
124. Prata L, Ovsyannikova IG, Tchkonia T, Kirkland JL. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin Immunol. 2018;40:101275
125. Lv J, Zhang C, Liu X, Gu C, Liu Y, Gao Y. et al. An aging-related immune landscape in the hematopoietic immune system. Immun Ageing. 2024;21:3
126. Pappert M, Khosla S, Doolittle M. Influences of Aged Bone Marrow Macrophages on Skeletal Health and Senescence. Current osteoporosis reports. 2023;21:771-8
127. Sawaki D, Zhang Y, Mohamadi A, Pini M, Mezdari Z, Lipskaia L. et al. Osteopontin promotes age-related adipose tissue remodeling through senescence-associated macrophage dysfunction. JCI Insight. 2023 8
128. Linehan E, Dombrowski Y, Snoddy R, Fallon PG, Kissenpfennig A, Fitzgerald DC. Aging impairs peritoneal but not bone marrow-derived macrophage phagocytosis. Aging Cell. 2014;13:699-708
129. Adams S, Wuescher LM, Worth R, Yildirim-Ayan E. Mechano-Immunomodulation: Mechanoresponsive Changes in Macrophage Activity and Polarization. Ann Biomed Eng. 2019;47:2213-31
130. Johnson CD, Fischer D, Smith IM, Aranda-Espinoza H, Fisher JP. Hyperglycemic Conditions Enhance the Mechanosensitivity of Proinflammatory RAW264.7 Macrophages. Tissue Eng Part A. 2023;29:172-84
131. Cheung HT, Rehwaldt CA, Twu JS, Liao NS, Richardson A. Aging and lymphocyte cytoskeleton: age-related decline in the state of actin polymerization in T lymphocytes from Fischer F344 rats. J Immunol. 1987;138:32-6
132. González-Bermúdez B, Kobayashi H, Abarca-Ortega A, Córcoles-Lucas M, González-Sánchez M, De la Fuente M. et al. Aging is accompanied by T-cell stiffening and reduced interstitial migration through dysfunctional nuclear organization. Immunology. 2022;167:622-39
133. Zacca ER, Crespo MI, Acland RP, Roselli E, Núñez NG, Maccioni M. et al. Aging Impairs the Ability of Conventional Dendritic Cells to Cross-Prime CD8+ T Cells upon Stimulation with a TLR7 Ligand. PLoS One. 2015;10:e0140672
134. Grolleau-Julius A, Harning EK, Abernathy LM, Yung RL. Impaired dendritic cell function in aging leads to defective antitumor immunity. Cancer Res. 2008;68:6341-9
135. Chen JS, Cameron ID, Cumming RG, Lord SR, March LM, Sambrook PN. et al. Effect of age-related chronic immobility on markers of bone turnover. J Bone Miner Res. 2006;21:324-31
136. Birkhold AI, Razi H, Duda GN, Weinkamer R, Checa S, Willie BM. The influence of age on adaptive bone formation and bone resorption. Biomaterials. 2014;35:9290-301
137. Du L, Freitas-Cortez MA, Zhang J, Xue Y, Veettil RT, Zhao Z. et al. Periarteriolar niches become inflamed in aging bone marrow, remodeling the stromal microenvironment and depleting lymphoid progenitors. Proc Natl Acad Sci U S A. 2025;122:e2412317122
138. Li X, Zhang C, Bowman HH, Stambough JB, Stronach BM, Mears SC. et al. Piezo1 opposes age-associated cortical bone loss. Aging Cell. 2023;22:e13846
139. Wang L, You X, Lotinun S, Zhang L, Wu N, Zou W. Mechanical sensing protein PIEZO1 regulates bone homeostasis via osteoblast-osteoclast crosstalk. Nat Commun. 2020;11:282
140. Khosla S, Farr JN, Monroe DG. Cellular senescence and the skeleton: pathophysiology and therapeutic implications. J Clin Invest. 2022 132
141. Zhou Y, Xin X, Wang L, Wang B, Chen L, Liu O. et al. Senolytics improve bone forming potential of bone marrow mesenchymal stem cells from aged mice. NPJ Regen Med. 2021;6:34
142. Kale A, Sharma A, Stolzing A, Desprez PY, Campisi J. Role of immune cells in the removal of deleterious senescent cells. Immun Ageing. 2020;17:16
143. Gibon E, Loi F, Córdova LA, Pajarinen J, Lin T, Lu L. et al. Aging Affects Bone Marrow Macrophage Polarization: Relevance to Bone Healing. Regen Eng Transl Med. 2016;2:98-104
144. Sinder BP, Zweifler L, Koh AJ, Michalski MN, Hofbauer LC, Aguirre JI. et al. Bone Mass Is Compromised by the Chemotherapeutic Trabectedin in Association With Effects on Osteoblasts and Macrophage Efferocytosis. J Bone Miner Res. 2017;32:2116-27
145. Xu R, Xie H, Shen X, Huang J, Zhang H, Fu Y. et al. Impaired Efferocytosis Enables Apoptotic Osteoblasts to Escape Osteoimmune Surveillance During Aging. Adv Sci (Weinh). 2023;10:e2303946
146. Yang B, Zhang G, Zhu Y, Wang J, Feng X, Fei W. et al. Osteoblast-CD4(+) CTL Crosstalk Mediated by SIRT1/DAAM2 Axis Prevents Age-Related Bone Loss. Adv Sci (Weinh). 2025: e01170.
147. Hou J, Chen KX, He C, Li XX, Huang M, Jiang YZ. et al. Aged bone marrow macrophages drive systemic aging and age-related dysfunction via extracellular vesicle-mediated induction of paracrine senescence. Nature aging. 2024;4:1562-81
148. Li CJ, Xiao Y, Sun YC, He WZ, Liu L, Huang M. et al. Senescent immune cells release grancalcin to promote skeletal aging. Cell Metab. 2021;33:1957-73.e6
149. Ludtka C, Moore E, Allen JB. The Effects of Simulated Microgravity on Macrophage Phenotype. Biomedicines. 2021 9
150. Shi L, Tian H, Wang P, Li L, Zhang Z, Zhang J. et al. Spaceflight and simulated microgravity suppresses macrophage development via altered RAS/ERK/NFκB and metabolic pathways. Cell Mol Immunol. 2021;18:1489-502
151. Maier JA. Impact of simulated microgravity on cell cycle control and cytokine release by U937 cells. Int J Immunopathol Pharmacol. 2006;19:279-86
152. Kaur I, Simons ER, Castro VA, Ott CM, Pierson DL. Changes in monocyte functions of astronauts. Brain Behav Immun. 2005;19:547-54
153. Pecaut MJ, Mao XW, Bellinger DL, Jonscher KR, Stodieck LS, Ferguson VL. et al. Is spaceflight-induced immune dysfunction linked to systemic changes in metabolism? PLoS One. 2017;12:e0174174
154. Kaur I, Simons ER, Castro VA, Mark Ott C, Pierson DL. Changes in neutrophil functions in astronauts. Brain Behav Immun. 2004;18:443-50
155. Luxton JJ, McKenna MJ, Lewis A, Taylor LE, George KA, Dixit SM. et al. Telomere Length Dynamics and DNA Damage Responses Associated with Long-Duration Spaceflight. Cell Rep. 2020;33:108457
156. Qin L, He T, Chen S, Yang D, Yi W, Cao H. et al. Roles of mechanosensitive channel Piezo1/2 proteins in skeleton and other tissues. Bone research. 2021;9:44
157. Noriega LG, Feige JN, Canto C, Yamamoto H, Yu J, Herman MA. et al. CREB and ChREBP oppositely regulate SIRT1 expression in response to energy availability. EMBO Rep. 2011;12:1069-76
158. Liu L, Cheng Y, Wang J, Ding Z, Halim A, Luo Q. et al. Simulated Microgravity Suppresses Osteogenic Differentiation of Mesenchymal Stem Cells by Inhibiting Oxidative Phosphorylation. Int J Mol Sci. 2020 21
159. Bai Y, Wu Z, Leary SC, Fang C, Yu M, Genth H. et al. Focal Adhesion Kinase Alleviates Simulated Microgravity-Induced Inhibition of Osteoblast Differentiation by Activating Transcriptional Wnt/β-Catenin-BMP2-COL1 and Metabolic SIRT1-PGC-1α-CPT1A Pathways. Int J Mol Sci. 2025 26
160. Huang X, Zhu Y, Sun S, Gao X, Yang Y, Xu H. et al. Exercise maintains bone homeostasis by promoting osteogenesis through STAT3. Int J Biol Sci. 2023;19:2021-33
161. Sun W, Li Y, Li J, Tan Y, Yuan X, Meng H. et al. Mechanical stimulation controls osteoclast function through the regulation of Ca(2+)-activated Cl(-) channel Anoctamin 1. Communications biology. 2023;6:407
162. Huang M, Zhou J, Li X, Liu R, Jiang Y, Chen K. et al. Mechanical protein polycystin-1 directly regulates osteoclastogenesis and bone resorption. Sci Bull (Beijing). 2024;69:1964-79
163. Zhang P, Zhang C, Li J, Han J, Liu X, Yang H. The physical microenvironment of hematopoietic stem cells and its emerging roles in engineering applications. Stem cell research & therapy. 2019;10:327
164. Tamma R, Colaianni G, Camerino C, Di Benedetto A, Greco G, Strippoli M. et al. Microgravity during spaceflight directly affects in vitro osteoclastogenesis and bone resorption. FASEB J. 2009;23:2549-54
165. Stavnichuk M, Mikolajewicz N, Corlett T, Morris M, Komarova SV. A systematic review and meta-analysis of bone loss in space travelers. NPJ Microgravity. 2020;6:13
166. Biswas L, Chen J, De Angelis J, Singh A, Owen-Woods C, Ding Z. et al. Lymphatic vessels in bone support regeneration after injury. Cell. 2023;186:382-97.e24
167. Wagner J, Gulshan H, Sultan I, Antila S, Manickam N, Panthel J. et al. Age-related decline in cardiac lymphatic vessels causes cardiac edema and macrophage accumulation. European Heart Journal. 2024 45
168. Kataru RP, Park HJ, Shin J, Baik JE, Sarker A, Brown S. et al. Structural and Functional Changes in Aged Skin Lymphatic Vessels. Front Aging. 2022;3:864860
169. Kawashima T, Ji RC, Itoh Y, Agata N, Sasai N, Murakami T. et al. Morphological and biochemical changes of lymphatic vessels in the soleus muscle of mice after hindlimb unloading. Muscle Nerve. 2021;64:620-8
170. Hettrick H, Aviles F. Microgravity and Lymphatics: Why Space Programs Need Lymphedema Physiology Specialists. Lymphat Res Biol. 2023;21:262-9
171. Barnhart H, Aviles F Jr, Pannunzio J, Sirkis N, Hubbard C, Hardigan P. et al. Using noninvasive imaging to assess manual lymphatic drainage on lymphatic/venous responses in a spaceflight analog. NPJ Microgravity. 2024;10:93
172. Segal NA, Anderson DD, Iyer KS, Baker J, Torner JC, Lynch JA. et al. Baseline articular contact stress levels predict incident symptomatic knee osteoarthritis development in the MOST cohort. J Orthop Res. 2009;27:1562-8
173. Rudolph KS, Schmitt LC, Lewek MD. Age-related changes in strength, joint laxity, and walking patterns: are they related to knee osteoarthritis? Phys Ther. 2007;87:1422-32
174. Wilkinson DJ, Piasecki M, Atherton PJ. The age-related loss of skeletal muscle mass and function: Measurement and physiology of muscle fibre atrophy and muscle fibre loss in humans. Ageing Res Rev. 2018;47:123-32
175. Nagano H, Begg R. Ageing-Related Gait Adaptations to Knee Joint Kinetics: Implications for the Development of Knee Osteoarthritis. Applied Sciences. 2020
176. Bank RA, Bayliss MT, Lafeber FP, Maroudas A, Tekoppele JM. Ageing and zonal variation in post-translational modification of collagen in normal human articular cartilage. The age-related increase in non-enzymatic glycation affects biomechanical properties of cartilage. Biochem J. 1998;330( Pt 1):345-51
177. Stolz M, Gottardi R, Raiteri R, Miot S, Martin I, Imer R. et al. Early detection of aging cartilage and osteoarthritis in mice and patient samples using atomic force microscopy. Nat Nanotechnol. 2009;4:186-92
178. Sørensen TJ, Langberg H, Aaboe J, Bandholm T, Bliddal H, Henriksen M. The association between submaximal quadriceps force steadiness and the knee adduction moment during walking in patients with knee osteoarthritis. J Orthop Sports Phys Ther. 2011;41:592-9
179. Hutchison L, Grayson J, Hiller C, D'Souza N, Kobayashi S, Simic M. Relationship Between Knee Biomechanics and Pain in People with Knee Osteoarthritis: A Systematic Review and Meta-Analysis. Arthritis Care Res (Hoboken). 2023;75:1351-61
180. Jiang W, Chen H, Lin Y, Cheng K, Zhou D, Chen R. et al. Mechanical stress abnormalities promote chondrocyte senescence - The pathogenesis of knee osteoarthritis. Biomed Pharmacother. 2023;167:115552
181. Mathiessen A, Conaghan PG. Synovitis in osteoarthritis: current understanding with therapeutic implications. Arthritis Res Ther. 2017;19:18
182. Hopkins T, Garcia J, Hulme CH, Tins B, Perry J, Jermin P. et al. Increased synovitis and pro-inflammatory macrophage abundance are observed in the synovia of patients at risk of developing post-traumatic OA compared to those with established OA. Osteoarthr Cartil Open. 2025;7:100643
183. Philpott HT, Birmingham TB, Blackler G, Klapak JD, Knights AJ, Farrell EC. et al. Association of Synovial Innate Immune Exhaustion with Worse Pain in Knee Osteoarthritis. Arthritis Rheumatol. 2025;77:664-76
184. Xiang W, Zhang T, Li B, Li S, Zhang B, Fang S. et al. Senescent macrophages induce ferroptosis in skeletal muscle and accelerate osteoarthritis-related muscle atrophy. Nature aging. 2025
185. Hu Y, Chen X, Wang S, Jing Y, Su J. Subchondral bone microenvironment in osteoarthritis and pain. Bone research. 2021;9:20
186. Moses MA, Sudhalter J, Langer R. Identification of an inhibitor of neovascularization from cartilage. Science. 1990;248:1408-10
187. Loeser RF, Collins JA, Diekman BO. Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2016;12:412-20
188. Lu J, Zhang H, Cai D, Zeng C, Lai P, Shao Y. et al. Positive-Feedback Regulation of Subchondral H-Type Vessel Formation by Chondrocyte Promotes Osteoarthritis Development in Mice. J Bone Miner Res. 2018;33:909-20
189. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99-118
190. Arra M, Swarnkar G, Alippe Y, Mbalaviele G, Abu-Amer Y. IκB-ζ signaling promotes chondrocyte inflammatory phenotype, senescence, and erosive joint pathology. Bone research. 2022;10:12
191. Lin R, Yin J, Huang J, Zou L, Liu L, Tang W. et al. Macrophage-derived ectosomal miR-350-3p promotes osteoarthritis progression through downregulating chondrocyte H3K36 methyltransferase NSD1. Cell death discovery. 2024;10:223
192. Wan QQ, Qin WP, Ma YX, Shen MJ, Li J, Zhang ZB. et al. Crosstalk between Bone and Nerves within Bone. Adv Sci (Weinh). 2021;8:2003390
193. Muschter D, Beiderbeck AS, Späth T, Kirschneck C, Schröder A, Grässel S. Sensory Neuropeptides and their Receptors Participate in Mechano-Regulation of Murine Macrophages. Int J Mol Sci. 2019 20
194. Feng SY, Lei J, Li YX, Shi WG, Wang RR, Yap AU. et al. Increased joint loading induces subchondral bone loss of the temporomandibular joint via the RANTES-CCRs-Akt2 axis. JCI Insight. 2022 7
195. Zhou Y, Li M, Lin S, Zhu Z, Zhuang Z, Cui S. et al. Mechanical sensing protein PIEZO1 controls osteoarthritis via glycolysis mediated mesenchymal stem cells-Th17 cells crosstalk. Cell death & disease. 2025;16:231
196. Chen Z, Ma Y, Li X, Deng Z, Zheng M, Zheng Q. The Immune Cell Landscape in Different Anatomical Structures of Knee in Osteoarthritis: A Gene Expression-Based Study. Biomed Res Int. 2020;2020:9647072
197. Geurts J, Patel A, Hirschmann MT, Pagenstert GI, Müller-Gerbl M, Valderrabano V. et al. Elevated marrow inflammatory cells and osteoclasts in subchondral osteosclerosis in human knee osteoarthritis. J Orthop Res. 2016;34:262-9
198. Li H, Guo Z, Xiangdong Q. Role of mechanical stimulus in mast cell activation. Digital Medicine. 2024 10
199. Pond MJ, Nuki G. Experimentally-induced osteoarthritis in the dog. Ann Rheum Dis. 1973;32:387-8
200. Glasson SS, Blanchet TJ, Morris EA. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage. 2007;15:1061-9
201. Liao L, Zhang S, Zhao L, Chang X, Han L, Huang J. et al. Acute Synovitis after Trauma Precedes and is Associated with Osteoarthritis Onset and Progression. Int J Biol Sci. 2020;16:970-80
202. Zhang H, Lin C, Zeng C, Wang Z, Wang H, Lu J. et al. Synovial macrophage M1 polarisation exacerbates experimental osteoarthritis partially through R-spondin-2. Ann Rheum Dis. 2018;77:1524-34
203. Lin Z, Xu Y, Jiang H, Zeng W, Wang Y, Zhu L. et al. CDK8 mediated inflammatory microenvironment aggravates osteoarthritis progression. Journal of advanced research. 2025
204. Fang H, Huang L, Welch I, Norley C, Holdsworth DW, Beier F. et al. Early Changes of Articular Cartilage and Subchondral Bone in The DMM Mouse Model of Osteoarthritis. Scientific reports. 2018;8:2855
205. Kim GM, Kim J, Lee JY, Park MC, Lee SY. IgSF11 deficiency alleviates osteoarthritis in mice by suppressing early subchondral bone changes. Exp Mol Med. 2023;55:2576-85
206. Kim H, Takegahara N, Walsh MC, Middleton SA, Yu J, Shirakawa J. et al. IgSF11 regulates osteoclast differentiation through association with the scaffold protein PSD-95. Bone research. 2020;8:5
207. McDonald MM, Khoo WH, Ng PY, Xiao Y, Zamerli J, Thatcher P. et al. Osteoclasts recycle via osteomorphs during RANKL-stimulated bone resorption. Cell. 2021;184:1330-47.e13
208. Liang W, Feng R, Li X, Duan X, Feng S, Chen J. et al. A RANKL-UCHL1-sCD13 negative feedback loop limits osteoclastogenesis in subchondral bone to prevent osteoarthritis progression. Nat Commun. 2024;15:8792
209. Zhao D, Sha BX, Zeng LF, Liang GH, Huang HT, Pan JK. et al. Exploring and analyzing two aging related genes FPR1 and UCHL1 and their potential molecular mechanisms in aggravating lumbar disc herniation. Journal of orthopaedic surgery and research. 2024;19:841
210. Su Z, Xiong H, Liu Y, Pang J, Lin H, Zhang W. et al. Transcriptomic analysis highlights cochlear inflammation associated with age-related hearing loss in C57BL/6 mice using next generation sequencing. PeerJ. 2020;8:e9737
211. Su W, Liu G, Liu X, Zhou Y, Sun Q, Zhen G. et al. Angiogenesis stimulated by elevated PDGF-BB in subchondral bone contributes to osteoarthritis development. JCI Insight. 2020 5
212. Xu Q, Feng G, Tang Z, Wang R, Ma P, Yan J. et al. Mechanistic study of COL6A1-mediated subchondral bone remodeling in osteoarthritis via the EPAC/RAP1 axis. FASEB J. 2025;39:e70473
213. Zhu S, Zhu J, Zhen G, Hu Y, An S, Li Y. et al. Subchondral bone osteoclasts induce sensory innervation and osteoarthritis pain. J Clin Invest. 2019;129:1076-93
214. Miller RE, Tran PB, Das R, Ghoreishi-Haack N, Ren D, Miller RJ. et al. CCR2 chemokine receptor signaling mediates pain in experimental osteoarthritis. Proc Natl Acad Sci U S A. 2012;109:20602-7
215. Miotla Zarebska J, Chanalaris A, Driscoll C, Burleigh A, Miller RE, Malfait AM. et al. CCL2 and CCR2 regulate pain-related behaviour and early gene expression in post-traumatic murine osteoarthritis but contribute little to chondropathy. Osteoarthritis Cartilage. 2017;25:406-12
216. Raoof R, Martin Gil C, Lafeber F, de Visser H, Prado J, Versteeg S. et al. Dorsal Root Ganglia Macrophages Maintain Osteoarthritis Pain. J Neurosci. 2021;41:8249-61
217. Tran PB, Miller RE, Ishihara S, Miller RJ, Malfait AM. Spinal microglial activation in a murine surgical model of knee osteoarthritis. Osteoarthritis Cartilage. 2017;25:718-26
218. Clark AK, Malcangio M. Fractalkine/CX3CR1 signaling during neuropathic pain. Front Cell Neurosci. 2014;8:121
219. Huang H, Skelly JD, Ayers DC, Song J. Age-dependent Changes in the Articular Cartilage and Subchondral Bone of C57BL/6 Mice after Surgical Destabilization of Medial Meniscus. Scientific reports. 2017;7:42294
220. Becker L, Nguyen L, Gill J, Kulkarni S, Pasricha PJ, Habtezion A. Age-dependent shift in macrophage polarisation causes inflammation-mediated degeneration of enteric nervous system. Gut. 2018;67:827-36
221. Teraguchi M, Yoshimura N, Hashizume H, Muraki S, Yamada H, Minamide A. et al. Prevalence and distribution of intervertebral disc degeneration over the entire spine in a population-based cohort: the Wakayama Spine Study. Osteoarthritis Cartilage. 2014;22:104-10
222. Teraguchi M, Yoshimura N, Hashizume H, Yamada H, Oka H, Minamide A. et al. Progression, incidence, and risk factors for intervertebral disc degeneration in a longitudinal population-based cohort: the Wakayama Spine Study. Osteoarthritis Cartilage. 2017;25:1122-31
223. Karchevskaya AE, Poluektov YM, Korolishin VA. Understanding Intervertebral Disc Degeneration: Background Factors and the Role of Initial Injury. Biomedicines. 2023 11
224. Silwal P, Nguyen-Thai AM, Mohammad HA, Wang Y, Robbins PD, Lee JY. et al. Cellular Senescence in Intervertebral Disc Aging and Degeneration: Molecular Mechanisms and Potential Therapeutic Opportunities. Biomolecules. 2023 13
225. Vo NV, Hartman RA, Patil PR, Risbud MV, Kletsas D, Iatridis JC. et al. Molecular mechanisms of biological aging in intervertebral discs. J Orthop Res. 2016;34:1289-306
226. Fearing BV, Hernandez PA, Setton LA, Chahine NO. Mechanotransduction and cell biomechanics of the intervertebral disc. JOR Spine. 2018 1
227. Götschi T, Widmer J, Cornaz F, Kimenai J, Spirig JM, Snedeker JG. et al. Region- and degeneration dependent stiffness distribution in intervertebral discs derived by shear wave elastography. Journal of biomechanics. 2021;121:110395
228. Zehra U, Tryfonidou M, Iatridis JC, Illien-Jünger S, Mwale F, Samartzis D. Mechanisms and clinical implications of intervertebral disc calcification. Nat Rev Rheumatol. 2022;18:352-62
229. Menon RG, de Moura HL, Kijowski R, Regatte RR. Age and gender differences in lumbar intervertebral disk strain using mechanical loading magnetic resonance imaging. NMR Biomed. 2023;36:e4999
230. Yamagishi A, Nakajima H, Kokubo Y, Yamamoto Y, Matsumine A. Polarization of infiltrating macrophages in the outer annulus fibrosus layer associated with the process of intervertebral disc degeneration and neural ingrowth in the human cervical spine. Spine J. 2022;22:877-86
231. Freemont AJ, Peacock TE, Goupille P, Hoyland JA, O'Brien J, Jayson MI. Nerve ingrowth into diseased intervertebral disc in chronic back pain. Lancet. 1997;350:178-81
232. Lian C, Gao B, Wu Z, Qiu X, Peng Y, Liang A. et al. Collagen type II is downregulated in the degenerative nucleus pulposus and contributes to the degeneration and apoptosis of human nucleus pulposus cells. Mol Med Rep. 2017;16:4730-6
233. Fan Y, Zhang W, Huang X, Fan M, Shi C, Zhao L. et al. Senescent-like macrophages mediate angiogenesis for endplate sclerosis via IL-10 secretion in male mice. Nat Commun. 2024;15:2939
234. Chen CN, Chang HI, Yen CK, Liu WL, Huang KY. Mechanical Stretch Induced Osteogenesis on Human Annulus Fibrosus Cells through Upregulation of BMP-2/6 Heterodimer and Activation of P38 and SMAD1/5/8 Signaling Pathways. Cells. 2022 11
235. Chang HI, Chen CN, Huang KY. Mechanical Stretch-Induced NLRP3 Inflammasome Expression on Human Annulus Fibrosus Cells Modulated by Endoplasmic Reticulum Stress. Int J Mol Sci. 2022 23
236. Taiji R, Kang JD, Mizuno S. Cellular behavior and extracellular matrix turnover in bovine annulus fibrosus cells under hydrostatic pressure and deviatoric strain. J Orthop Res. 2024;42:1326-34
237. Crump KB, Alminnawi A, Bermudez-Lekerika P, Compte R, Gualdi F, McSweeney T. et al. Cartilaginous endplates: A comprehensive review on a neglected structure in intervertebral disc research. JOR Spine. 2023;6:e1294
238. Lu S, Li M, Cheng Z, Liang Y, Huang J, Huang J. et al. HMGB1-mediated macrophage regulation of NF-κB activation and MMP3 upregulation in nucleus pulposus cells: A critical mechanism in the vicious cycle of intervertebral disc degeneration. Cell Signal. 2025;127:111628
239. Luo J, Yang Y, Wang X, Chang X, Fu S. Role of Pyroptosis in Intervertebral Disc Degeneration and Its Therapeutic Implications. Biomolecules. 2022 12
240. Yamamoto Y, Kokubo Y, Nakajima H, Honjoh K, Watanabe S, Matsumine A. Distribution and Polarization of Hematogenous Macrophages Associated with the Progression of Intervertebral Disc Degeneration. Spine (Phila Pa 1976). 2022;47:E149-e58
241. Koike Y, Uzuki M, Kokubun S, Sawai T. Angiogenesis and inflammatory cell infiltration in lumbar disc herniation. Spine (Phila Pa 1976). 2003;28:1928-33
242. Yu XJ, Zou P, Li TQ, Bai XF, Wang SX, Guan JB. et al. Deciphering SPP1-related macrophage signaling in the pathogenesis of intervertebral disc degeneration. Cell Biol Toxicol. 2025;41:33
243. Aprill C, Bogduk N. High-intensity zone: a diagnostic sign of painful lumbar disc on magnetic resonance imaging. Br J Radiol. 1992;65:361-9
244. Yang L, Li W, Yang Y, Zhao H, Yu X. The correlation between the lumbar disc MRI high-intensity zone and discogenic low back pain: a systematic review and meta-analysis. Journal of orthopaedic surgery and research. 2023;18:758
245. Rahme R, Moussa R. The modic vertebral endplate and marrow changes: pathologic significance and relation to low back pain and segmental instability of the lumbar spine. AJNR Am J Neuroradiol. 2008;29:838-42
246. Li XC, Luo SJ, Wu F, Mu QC, Yang JH, Jiang C. et al. Investigation of macrophage polarization in herniated nucleus pulposus of patients with lumbar intervertebral disc herniation. J Orthop Res. 2023;41:1335-47
247. Mei F, Guo Y, Wang Y, Zhou Y, Heng BC, Xie M. et al. Matrix stiffness regulates macrophage polarisation via the Piezo1-YAP signalling axis. Cell Prolif. 2024;57:e13640
248. Li W, Djuric N, Mink C, Vleggeert-Lankamp CLA. Inflammation and macrophage polarization are associated with Modic change type in lumbar radiculopathy. Brain Spine. 2025;5:104249
249. Zhang SP, Tong M, Mo J, Dong ZY, Huang YF. M2 macrophages activate the IL-10/JAK2/STAT3 pathway to induce pathological microangiogenesis in the nucleus pulposus exacerbating intervertebral disc degeneration. Journal of orthopaedic surgery and research. 2025;20:532
250. Ni S, Ling Z, Wang X, Cao Y, Wu T, Deng R. et al. Sensory innervation in porous endplates by Netrin-1 from osteoclasts mediates PGE2-induced spinal hypersensitivity in mice. Nat Commun. 2019;10:5643
251. Sivan S, Neidlinger-Wilke C, Würtz K, Maroudas A, Urban JP. Diurnal fluid expression and activity of intervertebral disc cells. Biorheology. 2006;43:283-91
252. Johnson ZI, Shapiro IM, Risbud MV. Extracellular osmolarity regulates matrix homeostasis in the intervertebral disc and articular cartilage: evolving role of TonEBP. Matrix Biol. 2014;40:10-6
253. Wang C, Shi Z. [Research progress in creep characteristics of lumbar intervertebral disc]. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi. 2020;34:1624-9
254. Ashinsky BG, Bonnevie ED, Mandalapu SA, Pickup S, Wang C, Han L. et al. Intervertebral Disc Degeneration Is Associated with Aberrant Endplate Remodeling and Reduced Small Molecule Transport. J Bone Miner Res. 2020;35:1572-81
255. Rodriguez AG, Rodriguez-Soto AE, Burghardt AJ, Berven S, Majumdar S, Lotz JC. Morphology of the human vertebral endplate. J Orthop Res. 2012;30:280-7
256. Walter BA, Purmessur D, Moon A, Occhiogrosso J, Laudier DM, Hecht AC. et al. Reduced tissue osmolarity increases TRPV4 expression and pro-inflammatory cytokines in intervertebral disc cells. Eur Cell Mater. 2016;32:123-36
257. Yan Y, Ding Y, Ming B, Du W, Kong X, Tian L. et al. Increase in hypotonic stress-induced endocytic activity in macrophages via ClC-3. Mol Cells. 2014;37:418-25
258. Ip WK, Medzhitov R. Macrophages monitor tissue osmolarity and induce inflammatory response through NLRP3 and NLRC4 inflammasome activation. Nat Commun. 2015;6:6931
259. Chuang YC, Wu SY, Huang YC, Peng CK, Tang SE, Huang KL. Cell volume restriction by mercury chloride reduces M1-like inflammatory response of bone marrow-derived macrophages. Front Pharmacol. 2022;13:1074986
260. Rabolli V, Wallemme L, Lo Re S, Uwambayinema F, Palmai-Pallag M, Thomassen L. et al. Critical role of aquaporins in interleukin 1β (IL-1β)-induced inflammation. J Biol Chem. 2014;289:13937-47
261. Moshrif A, Abdel Noor R, Aly H, Mortada M, Hafez A. Aging and entheses: An ultrasonographic probing of degenerative enthesopathy in a cohort of 147 healthy subjects. Int J Rheum Dis. 2022;25:481-8
262. Tashjian RZ. Epidemiology, natural history, and indications for treatment of rotator cuff tears. Clin Sports Med. 2012;31:589-604
263. Deymier AC, An Y, Boyle JJ, Schwartz AG, Birman V, Genin GM. et al. Micro-mechanical properties of the tendon-to-bone attachment. Acta Biomater. 2017;56:25-35
264. Lacraz G, Rouleau AJ, Couture V, Söllrald T, Drouin G, Veillette N. et al. Increased Stiffness in Aged Skeletal Muscle Impairs Muscle Progenitor Cell Proliferative Activity. PLoS One. 2015;10:e0136217
265. Marcucci L, Reggiani C. Increase of resting muscle stiffness, a less considered component of age-related skeletal muscle impairment. Eur J Transl Myol. 2020;30:8982
266. Kwan KYC, Ng KWK, Rao Y, Zhu C, Qi S, Tuan RS. et al. Effect of Aging on Tendon Biology, Biomechanics and Implications for Treatment Approaches. Int J Mol Sci. 2023 24
267. Zhao G, Zhang J, Nie D, Zhou Y, Li F, Onishi K. et al. HMGB1 mediates the development of tendinopathy due to mechanical overloading. PLoS One. 2019;14:e0222369
268. Zhang C, Gu X, Zhao G, Wang W, Shao J, Zhu J. et al. Extracellular HMGB-1 activates inflammatory signaling in tendon cells and tissues. Ther Adv Chronic Dis. 2020;11:2040622320956429
269. Bedi A, Kovacevic D, Fox AJ, Imhauser CW, Stasiak M, Packer J. et al. Effect of early and delayed mechanical loading on tendon-to-bone healing after anterior cruciate ligament reconstruction. J Bone Joint Surg Am. 2010;92:2387-401
270. Liu Y, Wang L, Li S, Zhang T, Chen C, Hu J. et al. Mechanical stimulation improves rotator cuff tendon-bone healing via activating IL-4/JAK/STAT signaling pathway mediated macrophage M2 polarization. J Orthop Translat. 2022;37:78-88
271. Mahbub S, Deburghgraeve CR, Kovacs EJ. Advanced age impairs macrophage polarization. J Interferon Cytokine Res. 2012;32:18-26
272. Petermann-Rocha F, Balntzi V, Gray SR, Lara J, Ho FK, Pell JP. et al. Global prevalence of sarcopenia and severe sarcopenia: a systematic review and meta-analysis. J Cachexia Sarcopenia Muscle. 2022;13:86-99
273. Fernandes LV, Paiva AEG, Silva ACB, de Castro IC, Santiago AF, de Oliveira EP. et al. Prevalence of sarcopenia according to EWGSOP1 and EWGSOP2 in older adults and their associations with unfavorable health outcomes: a systematic review. Aging clinical and experimental research. 2022;34:505-14
274. Reidy PT, Dupont-Versteegden EE, Drummond MJ. Macrophage Regulation of Muscle Regrowth from Disuse in Aging. Exerc Sport Sci Rev. 2019;47:246-50
275. Reidy PT, McKenzie AI, Mahmassani ZS, Petrocelli JJ, Nelson DB, Lindsay CC. et al. Aging impairs mouse skeletal muscle macrophage polarization and muscle-specific abundance during recovery from disuse. Am J Physiol Endocrinol Metab. 2019;317:E85-e98
276. Ahmadi M, Karlsen A, Mehling J, Soendenbroe C, Mackey AL, Hyldahl RD. Aging is associated with an altered macrophage response during human skeletal muscle regeneration. Exp Gerontol. 2022;169:111974
277. Cui CY, Driscoll RK, Piao Y, Chia CW, Gorospe M, Ferrucci L. Skewed macrophage polarization in aging skeletal muscle. Aging Cell. 2019;18:e13032
278. Dumont N, Frenette J. Macrophages protect against muscle atrophy and promote muscle recovery in vivo and in vitro: a mechanism partly dependent on the insulin-like growth factor-1 signaling molecule. Am J Pathol. 2010;176:2228-35
279. Kohno S, Yamashita Y, Abe T, Hirasaka K, Oarada M, Ohno A. et al. Unloading stress disturbs muscle regeneration through perturbed recruitment and function of macrophages. J Appl Physiol (1985). 2012;112:1773-82
280. Ferrara PJ, Yee EM, Petrocelli JJ, Fix DK, Hauser CT, de Hart N. et al. Macrophage immunomodulation accelerates skeletal muscle functional recovery in aged mice following disuse atrophy. J Appl Physiol (1985). 2022;133:919-31
281. Fennel ZJ, Kosari N, Bourrant PE, Yee EM, Castro RJ, Kurian AS. et al. Macrophage metabolic rewiring rejuvenates muscle raman signatures and cellular remodeling during regrowth in aged mice. JCI Insight. 2025
282. Zhu SJ, Hinman RS, Nelligan RK, Li P, De Silva AP, Harrison J. et al. Online Unsupervised Tai Chi Intervention for Knee Pain and Function in People with Knee Osteoarthritis: The RETREAT Randomized Clinical Trial. JAMA Intern Med. 2025
283. Abafita BJ, Singh A, Aitken D, Ding C, Moonaz S, Palmer AJ. et al. Yoga or Strengthening Exercise for Knee Osteoarthritis: A Randomized Clinical Trial. JAMA Netw Open. 2025;8:e253698
284. Tossige-Gomes R, Avelar NC, Simão AP, Neves CD, Brito-Melo GE, Coimbra CC. et al. Whole-body vibration decreases the proliferativeb response of TCD4(+) cells in elderly individuals with knee osteoarthritis. Braz J Med Biol Res. 2012;45:1262-8
285. Oka Y, Murata K, Ozone K, Minegishi Y, Kano T, Shimada N. et al. Mild treadmill exercise inhibits cartilage degeneration via macrophages in an osteoarthritis mouse model. Osteoarthr Cartil Open. 2023;5:100359
286. Norimatsu K, Nakanishi K, Ijuin T, Otsuka S, Takada S, Tani A. et al. Effects of low-intensity exercise on spontaneously developed knee osteoarthritis in male senescence-accelerated mouse prone 8. Arthritis Res Ther. 2023;25:168
287. Huang ZW, Yu YP, He XR, Chen YB, Xiang X, Li HZ. et al. Exercise suppresses osteoclastogenesis by increasing the secretion of muscle-derived L-β-aminoisobutyric acid. J Sport Health Sci. 2025: 101077.
288. Ma N, Lu Z, Mei X, Ma L, Zhang Q. The effectiveness of exercise interventions on muscle strength and balance function in pre-frail older adults: a systematic review and Bayesian network meta-analysis. Frontiers in public health. 2025;13:1718120
289. Luo H, Zheng Z, Yuan Z, Hu H, Sun C. The effectiveness of multicomponent exercise in older adults with cognitive frailty: a systematic review and meta-analysis. Archives of public health = Archives belges de sante publique. 2024;82:229
290. Jiang T, Yu F, Zhou Y, Li R, Zheng M, Jiang Y. et al. Synergistic effect of ultrasound and reinforced electrical environment by bioinspired periosteum for enhanced osteogenesis via immunomodulation of macrophage polarization through Piezo1. Mater Today Bio. 2024;27:101147
291. Iacoponi F, Cafarelli A, Fontana F, Pratellesi T, Dumont E, Barravecchia I. et al. Optimal low-intensity pulsed ultrasound stimulation for promoting anti-inflammatory effects in macrophages. APL bioengineering. 2023;7:016114
292. Zhang G, Li X, Wu L, Qin YX. Piezo1 channel activation in response to mechanobiological acoustic radiation force in osteoblastic cells. Bone research. 2021;9:16
293. Zeng Y, Wu Z, Xiong M, Liang Z, Chen Z, Huang H. et al. Piezo1 promotes vibration-induced vascular smooth muscle injury by regulating the NF-κB/p65 axis. Communications biology. 2025;8:96
294. Adejuyigbe B, Gharpure M, Wahle CF, Kallini JR. Distraction Osteogenesis: A Comprehensive Review. 2024; 3: 503-16.
295. McNamara SL, Seo BR, Freedman BR, Roloson EB, Alvarez JT, O'Neill CT. et al. Anti-inflammatory therapy enables robot-actuated regeneration of aged muscle. Sci Robot. 2023;8:eadd9369
296. Saitou K, Tokunaga M, Yoshino D, Sakitani N, Maekawa T, Ryu Y. et al. Local cyclical compression modulates macrophage function in situ and alleviates immobilization-induced muscle atrophy. Clin Sci (Lond). 2018;132:2147-61
297. Xu Z, Li S, Wan L, Hu J, Lu H, Zhang T. Role of low-intensity pulsed ultrasound in regulating macrophage polarization to accelerate tendon-bone interface repair. J Orthop Res. 2023;41:919-29
298. Zhang B, Chen H, Ouyang J, Xie Y, Chen L, Tan Q. et al. SQSTM1-dependent autophagic degradation of PKM2 inhibits the production of mature IL1B/IL-1β and contributes to LIPUS-mediated anti-inflammatory effect. Autophagy. 2020;16:1262-78
299. Lee W, Georgas E, Komatsu DE, Qin YX. Daily low-intensity pulsed ultrasound stimulation mitigates joint degradation and pain in a post-traumatic osteoarthritis rat model. J Orthop Translat. 2024;44:9-18
300. Chow SK, Chim YN, Wang J, Zhang N, Wong RM, Tang N. et al. Vibration treatment modulates macrophage polarisation and enhances early inflammatory response in oestrogen-deficient osteoporotic-fracture healing. Eur Cell Mater. 2019;38:228-45
301. Kennedy IW, Tsimbouri PM, Campsie P, Sood S, Childs PG, Reid S. et al. Nanovibrational stimulation inhibits osteoclastogenesis and enhances osteogenesis in co-cultures. Scientific reports. 2021;11:22741
302. Hamdy RC, Rendon JS, Tabrizian MJBr. Distraction osteogenesis and its challenges in bone regeneration. 2012; 8: 177-204.
303. Yang S, Wang N, Ma Y, Guo S, Guo S, Sun H. Immunomodulatory effects and mechanisms of distraction osteogenesis. Int J Oral Sci. 2022;14:4
304. Han C, Ma XL, Wang T, Ma JX, Tian P, Zang JC. et al. Low magnitude of tensile stress represses the inflammatory response at intervertebral disc in rats. Journal of orthopaedic surgery and research. 2015;10:26
305. Shi S, Kang XJ, Zhou Z, He ZM, Zheng S, He SS. Excessive mechanical stress-induced intervertebral disc degeneration is related to Piezo1 overexpression triggering the imbalance of autophagy/apoptosis in human nucleus pulpous. Arthritis Res Ther. 2022;24:119
306. Zura R, Mehta S, Della Rocca GJ, Jones J, Steen RG. A cohort study of 4,190 patients treated with low-intensity pulsed ultrasound (LIPUS): findings in the elderly versus all patients. BMC musculoskeletal disorders. 2015;16:45
307. Tan X, Jiang G, Zhang L, Wang D, Wu X. Effects of Whole-Body Vibration Training on Lower Limb Muscle Strength and Physical Performance Among Older Adults: A Systematic Review and Meta-analysis. Archives of physical medicine and rehabilitation. 2023;104:1954-65
308. Zha DS, Zhu QA, Pei WW, Zheng JC, Wu SH, Xu ZX. et al. Does whole-body vibration with alternative tilting increase bone mineral density and change bone metabolism in senior people? Aging clinical and experimental research. 2012;24:28-36
309. de Oliveira RDJ, de Oliveira RG, de Oliveira LC, Santos-Filho SD, Sá-Caputo DC, Bernardo-Filho M. Effectiveness of whole-body vibration on bone mineral density in postmenopausal women: a systematic review and meta-analysis of randomized controlled trials. Osteoporosis international: a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA. 2023;34:29-52
310. Kao KC, Chang WC, Chang WL, Kan SA, Hsu KH, Huang HY. et al. Specific callus patterns in intermediate stage of distraction osteogenesis bring negative impact on consolidation. Journal of orthopaedic surgery and research. 2025;20:965
311. Bottlang M, Lesser M, Koerber J, Doornink J, von Rechenberg B, Augat P. et al. Far cortical locking can improve healing of fractures stabilized with locking plates. J Bone Joint Surg Am. 2010;92:1652-60
312. Han Z, Wu J, Deng G, Bi C, Wang J, Wang Q. Axial Micromotion Locking Plate Construct Can Promote Faster and Stronger Bone Healing in an Ovine Osteotomy Model. Front Bioeng Biotechnol. 2020;8:593448
313. Bottlang M, Shetty SS, Blankenau C, Wilk J, Tsai S, Fitzpatrick DC. et al. Advances in Dynamization of Plate Fixation to Promote Natural Bone Healing. J Clin Med. 2024 13
314. Petrousek SR, Kronemberger GS, O'Brien G, Hughes C, O'Rourke SA, Lally C. et al. Mechano-immunomodulation of macrophages influences the regenerative environment of fracture healing through the regulation of angiogenesis and osteogenesis. Acta Biomater. 2025;200:187-201
315. Bai L, Zhang Y, Wang J, Gai T, Ren X, Liu H. et al. Mechanotransduction-driven immunointegration at soft-hard tissue interfaces facilitates aged bone regeneration. Chem Eng J. 2025;514:163315
316. Tang Z, Wei X, Li T, Wu H, Xiao X, Hao Y. et al. Three-Dimensionally Printed Ti2448 with Low Stiffness Enhanced Angiogenesis and Osteogenesis by Regulating Macrophage Polarization via Piezo1/YAP Signaling Axis. Front Cell Dev Biol. 2021;9:750948
317. Zheng X, Chen L, Tan J, Miao J, Liu X, Yang T. et al. Effect of micro/nano-sheet array structures on the osteo-immunomodulation of macrophages. Regen Biomater. 2022;9:rbac075
318. Lou T, Wang X, Li J, Wang W, Han P, Yu S. et al. Chirality Regulates Bone Regeneration through Mechanoresponse and Immunoregulation. ACS Nano. 2025;19:7767-83
319. Schoenenberger AD, Tempfer H, Lehner C, Egloff J, Mauracher M, Bird A. et al. Macromechanics and polycaprolactone fiber organization drive macrophage polarization and regulate inflammatory activation of tendon in vitro and in vivo. Biomaterials. 2020;249:120034
320. Li X, Liu Y, Yang Q, Zhang W, Wang H, Zhang W. et al. Injectable Piezoelectric Hydrogel Promotes Tendon-Bone Healing via Reshaping the Electrophysiological Microenvironment and M2 Macrophage Polarization. ACS Appl Mater Interfaces. 2025;17:22210-31
321. Huang M, Li W, Sun Y, Dong J, Li C, Jia H. et al. Janus piezoelectric adhesives regulate macrophage TRPV1/Ca(2+)/cAMP axis to stimulate tendon-to-bone healing by multi-omics analysis. Bioactive materials. 2025;50:134-51
322. Huang D, Xu K, Huang X, Lin N, Ye Y, Lin S. et al. Remotely Temporal Scheduled Macrophage Phenotypic Transition Enables Optimized Immunomodulatory Bone Regeneration. Small. 2022;18:e2203680
323. Wu H, Dong H, Tang Z, Chen Y, Liu Y, Wang M. et al. Electrical stimulation of piezoelectric BaTiO3 coated Ti6Al4V scaffolds promotes anti-inflammatory polarization of macrophages and bone repair via MAPK/JNK inhibition and OXPHOS activation. Biomaterials. 2023;293:121990
324. Huang H, Xiao L, Fang L, Lei M, Liu Z, Gao S. et al. Static Topographical Cue Combined with Dynamic Fluid Stimulation Enhances the Macrophage Extracellular Vesicle Yield and Therapeutic Potential for Bone Defects. ACS Nano. 2025;19:8667-91
325. Liu M, Li J, Xu J, Chen Y, Chien C, Zhang H. et al. Graded Progressive Home-Based Resistance Combined with Aerobic Exercise in Community-Dwelling Older Adults with Sarcopenia: A Randomized Controlled Trial. Clin Interv Aging. 2024;19:1581-95
326. Shen Y, Shi Q, Nong K, Li S, Yue J, Huang J. et al. Exercise for sarcopenia in older people: A systematic review and network meta-analysis. J Cachexia Sarcopenia Muscle. 2023;14:1199-211
327. Zhao H, Yu X, Jiang M, Li Z, Zhao Y, Ren Y. et al. Piezo1-mediated autophagy promotes immune-inflammatory responses in ankylosing spondylitis. Cell death & disease. 2026;17:12
328. Pan X, Xu H, Ding Z, Luo S, Li Z, Wan R. et al. Guizhitongluo Tablet inhibits atherosclerosis and foam cell formation through regulating Piezo1/NLRP3 mediated macrophage pyroptosis. Phytomedicine: international journal of phytotherapy and phytopharmacology. 2024;132:155827
329. Li R, Xia J, Shi C, Zhang K, Qu Y, He G. et al. Direct pharmacological targeting of Piezo1 by Paeoniflorin: a novel therapeutic approach for renal fibrosis. Journal of advanced research. 2025
330. Ren P, Zheng C, Pang Y, Qiang Y, Sun S, Wang Q. et al. Single-cell analysis of the somatic mutational landscape in human chondrocytes during aging and in osteoarthritis. Nature aging. 2025;5:2417-31
331. Li G, Kang Y, Feng X, Wang G, Yuan Y, Li Z. et al. Dynamic changes of enhancer and super enhancer landscape in degenerated nucleus pulposus cells. Life science alliance. 2023 6
332. Chen XF, Duan YY, Jia YY, Dong QH, Shi W, Zhang Y. et al. Integrative high-throughput enhancer surveying and functional verification divulges a YY2-condensed regulatory axis conferring risk for osteoporosis. Cell genomics. 2024;4:100501
333. Cosgrove BD, Bounds LR, Taylor CK, Su AL, Rizzo AJ, Barrera A. et al. Mechanosensitive genomic enhancers potentiate the cellular response to matrix stiffness. Science. 2025;390:eadl1988
334. Claringbould A, Zaugg JB. Enhancers in disease: molecular basis and emerging treatment strategies. Trends in molecular medicine. 2021;27:1060-73
335. Xu M, Li Z, Thottappillil N, Cherief M, Zhu M, Xing X. et al. Mapping somatosensory afferent circuitry to bone identifies neurotrophic signals required for fracture healing. Science. 2026;391:eadr9608
336. Cherief M, Gomez-Salazar M, Kang M, Lee S, Ramesh S, Qin Q. et al. Reduced somatosensory innervation alters the skeletal transcriptome at a single cell level in a mouse model of type 2 diabetes. Bone research. 2025;13:67
337. Rao F, Wang Y, Zhang D, Lu C, Cao Z, Sui J. et al. Aligned chitosan nanofiber hydrogel grafted with peptides mimicking bioactive brain-derived neurotrophic factor and vascular endothelial growth factor repair long-distance sciatic nerve defects in rats. Theranostics. 2020;10:1590-603
338. Zhang W, Zhang Y, Ma H, Duan L, Zhang W, Ding L. et al. Biomaterial and Hydrogel Strategies for Regenerative Microenvironment Reconstruction in Peripheral Nerve Conduits. Gels (Basel, Switzerland). 2025 11
339. Bai L, Li J, Li G, Zhou D, Su J, Liu C. Skeletal interoception and prospective application in biomaterials for bone regeneration. Bone research. 2025;13:1
Author contact
![]() Corresponding authors: Xiao Lv, MD, PhD Department of Orthopedics, Union Hospital Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei Province 430022, China Email: lvxiaotjmucom. * Liming Xiong, MD, PhD Department of Orthopedics, Union Hospital Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei Province 430022, China, Email: xionglimingedu.cn.
Corresponding authors: Xiao Lv, MD, PhD Department of Orthopedics, Union Hospital Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei Province 430022, China Email: lvxiaotjmucom. * Liming Xiong, MD, PhD Department of Orthopedics, Union Hospital Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei Province 430022, China, Email: xionglimingedu.cn.