Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2025; 15(4):1285-1303. doi:10.7150/thno.102228 This issue Cite
Research Paper
Discovery of indole analogue Tc3 as a potent pyroptosis inducer and identification of its combination strategy against hepatic carcinoma
Xiao Hu1*, Xiaomei Tang1*, Xiaoman Tian1, Xing Lv2, Yuanyuan Zhang1, Yingyue Pang1, Weilong Deng1, Yali Wang1, Changliang Shan1 ![]() , Luqing Shang1
, Luqing Shang1 ![]()
1. State Key Laboratory of Medicinal Chemical Biology, College of Pharmacy, Nankai University, Tianjin 300353, People's Republic of China.
2. Asymchem Pharmaceuticals (Tianjin) Co., Ltd., Tianjin 300457, People's Republic of China.
*These authors contributed equally to this work.
Received 2024-8-12; Accepted 2024-11-25; Published 2025-1-1
Abstract

Rationale: Hepatic carcinoma, one of the most malignant cancers in the world, has limited success with immunotherapy and a poor prognosis in patients. While pyroptosis is considered as a promising immunotherapy strategy for tumors, it still suffers from a lack of effective inducers.
Methods: We designed, synthesized and screened an indole analogue, Tc3, featuring a 2, 4-thiazolidinedione substituted indole scaffold. Western blotting, qPCR and immunofluorescence were employed to detect the levels of pyroptosis pathway induced by Tc3. RNA sequencing was used to identify the mechanisms of Tc3 in hepatic carcinoma. To validate anti-tumor effect of Tc3, we used CDXs and PDXs mouse models in vivo. Then, the syngeneic effects of Tc3 with cisplatin and anti-PD-1 antibody were verified via western blotting, immunofluorescence, flow cytometry and ELISA.
Results: Treatment with Tc3 notably inhibited the growth of hepatic carcinoma both in vitro and in vivo. Mechanistically, Tc3 inhibited the function of PRDX1 and up-regulated excessive ROS. Then, Tc3 induced gasderminE-mediated pyroptosis by activating the endoplasmic reticulum stress. Tumor cells with high expression of GSDME achieved better responses to Tc3-therapy. Tc3 also improved the efficacy of cisplatin against hepatic carcinoma. Additionally, superior synergistic treatment was observed when Tc3 was combined with anti-PD-1 antibody. Notably, Tc3 activated the tumor immune microenvironment (TIME) and enhanced CD8+ T cell infiltration in hepatic carcinoma.
Conclusions: Collectively, we identified Tc3 as a promising and effective compound for treating hepatic carcinoma and established its synergistic therapeutic strategy as a pyroptosis inducer.
Keywords: hepatic carcinoma, Tc3, pyroptosis, gasdermin E, synergy effect
Introduction
Hepatocellular carcinoma and intrahepatic ductal carcinoma account for over 90% of hepatic carcinoma cases, making it one of the most aggressive and malignant primary tumors globally [1, 2]. Conventional therapeutic strategies, including chemotherapy, surgery, and radiotherapy, have shown limited success in treating hepatic carcinoma. Currently, systemic therapies with molecular targeted drugs, such as sorafenib or lenvatinib, are the preferred treatment options, and immune checkpoint blockers have revolutionized the treatment of solid tumors by relieving the restricted T cells [3-6]. However, frequent side effects and resistance to monotherapy lead to poor clinical prognosis and patient survival in hepatic carcinoma [7, 8]. Therefore, it is imperative to seek innovative and effective combinatorial treatment strategies to supplement monotherapy, such as pyroptosis inducers.
Nitrogen-containing heterocyclic moieties, such as benzimidazoles, benzothiazoles, indoles, acridines, oxadiazoles, imidazoles, and thiazoles, are common in natural products and pharmaceuticals [9]. Among them, thiazole and Indole nuclei are considered fundamental scaffolds for anti-tumor drug research, as illustrated by clinically approved indole-based anticancer agents (e.g., panobinostat, alectinib, sunitinib, and nintedanib), and thiazole-containing drugs (e.g., tiazofurin, dasatinib, dabrafenib, patellamide A, and epothilone) [10, 11]. In recent years, the hybridization of pharmacophoric subunits has been investigated as a new method to find potential anti-tumor agents. Therefore, we screened a library of approximately 1000 compounds featuring a thiazole-substituted indole scaffold and identified a small molecule (T1) with anti-tumor activity against HepG2 cells.
Pyroptosis is a caspase-dependent programmed cell death activated by stimuli, such as viruses, bacteria, toxins, and chemotherapy [12-15]. Cleavage of gasdermin (GSDM) prompts the oligomerization of its N-terminal and the formation of pores in the cell membrane, which leads to excessive cell swelling and bubble-like protrusions on the cell surface, ultimately causing cell death [16-18]. Executors of pyroptosis, such as caspase-1/11, can process and release interleukin-1β (IL-1β) through the pores formed by gasderminD (GSDMD), initiating an immune response and providing resistance to infections or toxins [16, 19]. Previous studies have shown that certain chemotherapy drugs, like 5-FU, induce pyroptosis mediated by cleaved gasderminE (GSDME, also called deafness autosomal dominant 5, DFNA5), and the mechanism of cell death can shift from apoptosis to pyroptosis depending on the GSDME level [20-22]. As a result of promoter methylation of the DFNA5 gene, the expression of GSDME in most mouse tumor cells is far lower than normal cells. Therefore, a combination therapy is realized using decitabine (DNA methyltransferase inhibitor) in conjunction with chemotherapeutics. It was reported that the co-delivery of metal thiosemicarbazone complexes and decitabin with the assistance of a nanodrug carrier liposome can achieve accurate chemotherapeutic pyroptosis of tumor cells [23, 24]. Moreover, the construction of nanoparticles containing indocyanine green (a photosensitizer for photothermal therapy) and a metal thiosemicarbazone compound was developed to reverse tumor cisplatin resistance through GSDME-mediated pyroptosis [25]. Resistance to immunotherapy can be attributed to heterogeneity in the tumor immune microenvironment (TIME). Hence, beyond the canonical and non-canonical pathways, the activation of GSDME is considered a promising approach to remodel the TIME and trigger durable anti-tumor immunity for cancer therapy [26]. However, research on potent GSDME agonists and pyroptosis inducers for hepatic carcinoma is limited.
Herein, we designed, synthesized, and evaluated a series of indole analogues optimized from T1, identifying Tc3 as the hit compound. Tc3 directly induced GSDME-mediated pyroptosis and inhibited the growth of hepatic carcinoma in vitro and in vivo. Based on these findings, we sought to explore the therapeutic potential and combinatorial strategy of Tc3, evaluating its efficacy and safety in cancer therapy.
Materials and methods
Chemistry synthesis of compounds
The concrete chemistry synthesis procedures of all compounds are listed in the chemical supporting information.
Reagents and biological resources
The reagents and biological resources are listed in the key resources table of supporting information.
Cell culture
Hepatic carcinoma cell lines (HepG2, SK-Hep1 and PLC-PRF5) were gifts from Dr. Tao Sun, Nankai university college of pharmacy. HEK-293T cell line was purchased from Pricella cell company (Wuhan, China), and used for lenti-virus package. Alpha mouse liver 12 cells (AML-12 cells) were gifts from Dr. Junfang Qin, Nankai university college of medical. Cell lines (HepG2, SK-Hep1, PLC-PRF5 and HEK-293T) were cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco) supplemented with 10% FBS (ExCell Bio), and maintained according to ATCC. AML-12 cells were cultured in special medium for AML-12 cells (Hycyte), and maintained according to ATCC.
Cell proliferation assays
For cell proliferation curve analysis, the hepatic carcinoma cells (HepG2, SK-Hep1 and PLC-PRF5) were seeded in 24-well plates respectively, then treated with Tc3 at concentration gradient and counted for four days. For colony formation, hepatic carcinoma cells were seeded in 6-well plates and treated with Tc3 for almost 10 days, and the cell colonies were photographed for statistical analysis.
LDH release assay
The hepatic carcinoma cells were seeded in 96-well plates and treated with indole analogues for three days. Of note, the compounds were added into DMEM medium supplemented with 1% FBS. The cellular supernatant was collected for LDH release detection by using LDH cytotoxicity assay kit (Beyotime, C0016). Statistical analysis was combined with the cell viability data tested by WST-1 cell proliferation and cytotoxicity assay kit (Beyotime, C0035).
TUNEL Cy3 apoptosis detection
Briefly, the hepatic carcinoma cells were incubated in 12-well plate, adhered to a round glass slide and then treated with Tc3. The fluorescence staining was conducted by using one-step TUNEL Cy3 apoptosis detection kit (APExBIO, K1134) according to the manufacturer's instructions. The cells images were captured by a confocal microscope (Leica, TCS SP8).
Wheat germ Agglutinin (WGA) staining
To examine the membrane integrity, the hepatic carcinoma cells were seeded in 12-well plate, adhered to a round glass slide and then treated with indole analogues reagents and the control medium. The cells were fixed by paraformaldehyde, stained by iFluorTM 488-Wheat Germ Agglutinin (WGA) Conjugate (AAT Bioquest, 25530, 1:200) and captured by a confocal microscope. The cells of PC (positive control) group were permeabilized by 0.3% TritonX-100 before the 488-WGA staining.
RNA sequencing analysis
HepG2 cells were treated with dimethyl sulfoxide (DMSO) or Tc3 (4 μM) for 24h. And the total RNA was isolated using TRIzol (Solarbio, R1100). Next, the samples were processed and the RNA libraries were constructed by APTBIO company (Shanghai, China). Paired-end reads were aligned to a reference genome index which created using HISAT2, with read assignments to each gene quantified with feature counts. Gene expression was quantified using Transcripts Per Million (TPM). We identified Differentially Expressed Genes (DEGs) using the DESeq2 package in R, defining DEGs as those with an absolute log2 fold change greater than 1 and an adjusted p-value less than 0.05. We performed Gene Ontology (GO) and KEGG pathway enrichment analyses using the "cluster Profiler" package in R. GO terms and KEGG pathways with an adjusted P-value less than 0.05 were statistically significant. Our results were visualized using the ggplot2 and fact extra packages in R.
Pharmacokinetic study of vein administration of Tc3
Chromatographic conditions employed an ultimate AQ-C18 column (2.1 mm × 50 mm, 5.0 μm) and its temperature was set to 40℃. The mobile phase consisted of methanol and 0.1% formic acid, with a gradient elution program: 0-0.01 min, methanol 15%, 0.01-2 min, methanol 95%, 2-3 min, methanol 15%. The flow rate was set to 0.6 mL/min, the elution time was 3 min and ESI-MS in negative mode MRM was used for quantitative analysis. Masslynx 4.1 software was used for data acquisition and instrument control.
Stock solutions of Tc3 (1.0 mg/mL) and JBHDN (0.1 mg/mL) were prepared in methanol. A series of working solutions (5, 20, 100, 200, 800, 1000 and 2000 ng/mL) were added into blank mouse blood to prepare the standard curves. The JBHDN stock solution was diluted with 5% trichloroacetic acid-methanol to 100 ng/mL. In the same manner as the standard curve, quality control (QC) samples at 10 ng/mL, 450 ng/mL, and 1500 ng/mL were prepared. Next, Balb/c mice were administered intravenously (10 mg/kg). After 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h, and 24 h, blood samples (20 μL) were collected from the tail vein of mice and mixed with internal standard JBHDN (100 ng/mL). The mixtures were then vortexed for 10 min at 4℃ and centrifuged at 5500 g for 10 min. The supernatant was mixed with 200 μL ultrapure water, and then injected into liquid chromatography-tandem mass spectrometry (LC-MS/MS) system for analysis.
Liver microsome metabolic stability assay
The intrinsic clearance of Tc3 in liver microsomes was determined at 1 μM along with positive control (12 μM of midazolam). The reaction media contained liver microsomes (0.5 mg/mL), 1.0 mM β-nicotinamide adenine dinucleotide phosphate (β-NADPH), 3 mM MgCl2 phosphate buffer. Microsomal suspensions were pre-incubated with Tc3 for 3 min at 37℃. The reaction was initiated with the addition of β-NADPH and samples were collected at 0, 5, 15, 30 and 60 min. The negative control was excluded of β-NADPH. Midazolam was incubated similarly as a positive control substrate. At each time point, an aliquot was mixed acetonitrile. Samples were centrifuged and then supernatant was mixed with ultrapure water for LC-MS/MS analysis.
Animals and tumor models
Balb/c nude mice (female, 6-week-old) were subcutaneously injected with 2 × 106 HepG2 cells on the left and right flanks to construct cell-derived xenografts (CDXs). The patient-derived xenografts (PDXs) samples were gifts from Dr Changliang Shan of Nankai University. C57 BL/6N mice (female, 5-week-old) were subcutaneously injected with 2 × 106 Hepa1-6 cells mixed with matrigel (ABW, 0827265) on the left and right flanks. The mice were inoculated with hepatic carcinoma CDXs or PDXs for at least four days, and treated (i.p.) by vehicle and Tc3. The body weight and tumor growth were recorded before every injection. The tumors were harvested and weighed after mice sacrificed. Tumor volume = 4π/3 × (Length/2) × (Width/2)2. All animal experiments were carried out in accordance with the recommendations of Requirements of the Ethical Review System of Biomedical Research Involving Human by National Health and Family Planning Commission of China and Nankai University Ethics Committee with written informed consent complying with the Declaration of Helsinki (IAEC ID: 2023-STDWLL-000249).
Drugs synergistic effect in vitro and in vivo
In vitro assay, the IC50 values of Tc3 and clinical drugs (cisplatin, oxaliplatin, gemcitabine, sorafenib, regorafenib, axitinib, lenvatinib and cabozantinib) were obtained. Then, the hepatic carcinoma cells were treated with Tc3 and the drugs mentioned above for synergistic effect test. In vivo assay, Balb/c nude mice inoculated with hepatic carcinoma CDXs were divided into four groups randomly (n = 5) and treated with vehicle, Tc3 (20 mg/kg), cisplatin (2 mg/kg), sorafenib (20 mg/kg) and combination of the two for 14 days. The body weight and tumor growth were recorded before every injection. The tumors were harvested and weighed after mice sacrificed. CDI = AB/(A×B). Where AB is the ratio of the two-drugs combination group to the control group and A or B is the ratio of the single group to the control group. CDI < 0.7 indicates a significantly synergistic effect, CDI < 1 indicates synergism, CDI = 1 indicates additivity, CDI > 1 indicates antagonism.
Anti-PD-1 antibody synergistic effect in vivo
C57 BL/6N mice inoculated with Hepa1-6 CDXs were divided into four groups randomly (n = 6) and treated with vehicle (DMSO and IgG antibody), Tc3 (10 mg/kg), anti-PD-1 antibody (10 mg/kg) and combination of the two for ten days. The body weight and tumor growth were recorded every two days and the tumors were harvested and weighed after mice sacrificed. The CDI value of tumor weight was calculated using formula mentioned before.
Flow cytometry analysis
The lymphocytes were isolated from the tumors and blocked by purified CD16/32 antibody. Next, the cells were stained with fluorescent-conjugated antibodies (FITC-CD11b, APC-F4/80, PE-CD86, Alexa Flour® 700-CD206, APC-Ly6G, FITC-CD45, APC-CD8a, Pacific BlueTM-IFN-γ). The fluorescence intensity was measured by a FACS Calibur, and data were analyzed with FlowJo software.
ELISA detection
Briefly, the Hepa1-6 CDXs from C57 BL/6N mice were homogenized and centrifuged via RIPA buffer (Solarbio, R0010). The supernatant was collected and diluted to a suitable concentration for cytokines detection using specific ELISA kits (Proteintech, Wuhan) according to the manufacturer's instructions.
Statistical analysis
All experiments were repeated three times. Data are presented as means ± SD and were analyzed using appropriate statistical tests as indicated in the figure legends with Graphpad Prsim 9 software. Differences were considered statistically significant at p < 0.05.
Results
Synthesis and discovery of indole analogue Tc3
We found a series of indole analogues with promising anti-hepatoma activity. Initially, compound T1 was identified for its anti-tumor activity against HepG2 cells (IC50 = 15.94 μM) through screening of in-house library of approximately 1000 compounds. To optimize the structure of T1, a series of derivatives with different substitutions at the R1, R2 and R3 sites were rationally designed and evaluated for their anti-hepatoma activities.
The results were as follows: (1) A hydrophilic group at R1 is essential for cellular inhibitory potency. Compounds Ta1-Ta5, with fatty acid groups, showed IC50 values greater than 50 μM (except Ta1 with 27.66 μM). In contrast, compounds Tb1-Tb9, with amino acid groups, had IC50 values below 10 μM (except Tb5 with 11.34 μM), with Tb7 being particularly potent (IC50 = 1.79 μM). (2) For R2, the substitution H with Br or OCH3 at the 5-position of the indole ring did not significantly influence anti-tumor activity, as shown by the similar IC50 values of Tb3, Tb4 and Tb9 (6.20, 9.23 and 7.47 μM, respectively). (3) For R3, introducing a 4-morpholinoaniline group led to Tc1, exhibiting a ∼3-fold increase in potency (IC50 = 5.13 μM). Further modifications on the amino group of Tc1 resulted in Tc2, Tc3, Tc4 and Tc5, showing∼2-fold, ∼12-fold,∼6.5-fold and ~3-fold increase in cellular potency, respectively (IC50 = 8.77, 1.37, 2.46 and 4.90 μM, respectively). Compounds Tc6-Tc10, which contain other substituted phenyl groups instead of the 4-morpholinoaniline, showed similar or worse activities, with IC50 values greater than 100 μM for Tc6 and Tc10, and 12.40, 15.15, and 10.78 μM for Tc7, Tc8, and Tc9 respectively. Introducing aliphatic groups at R3 (Tc11 and Tc12) resulted in decreased potency, with IC50 values of 20.87 and 25.02 μM, respectively (Figure 1).
Structure optimization process toward T1. Step1: we introduced different carboxylic acid residues at R1. Step 2: we fixed R2 with bromine or methoxy and explored suitable amino acid moieties at R1. Step 3: we introduced 4-morpholinoaniline at R3, explored suitable amino moieties at R1, then fixed R1 and optimized R3.
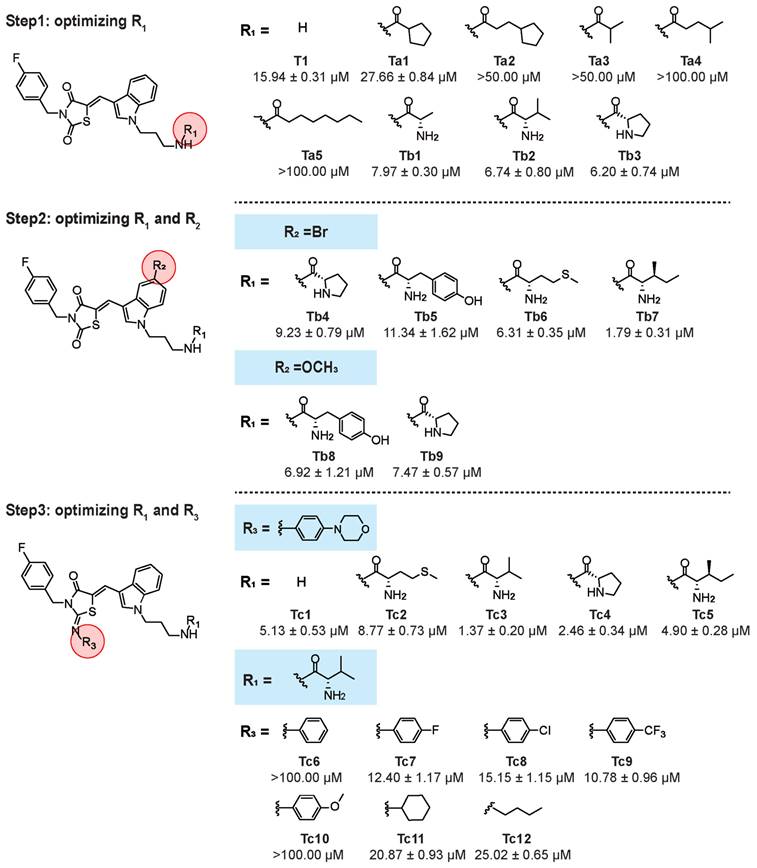
We further evaluated the anti-tumor activity of Tb7, Tc1, Tc3, and Tc4 against SK-Hep1 and PLC-PRF5 cells, confirming that Tc3 had the highest potency (IC50 = 1.64 and 1.79 μM, respectively) for hepatic carcinoma (Figure 2A). Tc3 also showed superior effects against hepatic carcinoma cells compared to other clinical chemotherapy drugs, including cisplatin, oxaliplatin, raltitrexed, etoposide, camptothecin, and gemcitabine at 5 μM (Figure 2B and S1A-B). Meanwhile, Tc3 outperformed clinical targeted drugs such as sorafenib, regorafenib, cabozantinib, lenvatinib, and axitinib at 3 μM (Figure 2C and S1C-D). We also confirmed the anti-tumor activity of Tc3 against mouse liver parenchymal cells (alpha mouse liver 12 cell, AML-12), as shown by the IC50 value at 6.14 μM (Figure S1E). Flow-cytometry analyses of annexin V and propidium iodide staining found that Tc3 could induce severe cell death of HepG2 cells at 4 μM (Figure 2D-F), which was not observed in AML-12 cells (Figure 2G-I). It indicated that compound Tc3 had selective toxicity in hepatic carcinoma cells and liver parenchymal cells. In addition, Tc3 were found to be remarkably resistant and stable to hydrolysis since it showed no changes in a phosphate buffer of PH 7.4 over 150 minutes (Figure S1F). Our findings revealed a moderate extent of Tc3 in mouse vitro metabolism with t1/2 of 116.0 min and a high extent of in human vitro metabolism of Tc3 with t1/2 of 49.5 min (2.72 min and 2.90 min for midazolam, respectively). CLint was calculated to be 0.0120 and 0.0280 mL/min/mg in mouse and human respectively (1.27 and 1.20 for midazolam). A middle level of pre-systemic metabolism in liver microsomes was observed (Figure S1G-H). Therefore, we explored the mechanism of Tc3's anti-tumor effect in greater depth, to highlight its potential as a useful agent for in vivo studies.
Cytotoxic effect of Tc3 on human hepatic carcinoma cells in vitro. (A) Chemical structures of Tb7, Tc1, Tc3, Tc4 and the IC50 values after treatment of HepG2, SK-Hep1 and PLC-PRF5 cells at indicated concentrations for 72 h. (B-C) Cell viability rates of HepG2 (B) and SK-Hep1 (C) cells treated with Tb7, Tc1, Tc3, Tc4 and other clinical chemotherapy drugs. (D-F) Flow cytometry of propidium iodide and annexin V-fluorescein isothiocyanate (FITC)-stained HepG2 cells (D). Statistical analysis of the early apoptotic cells (E) and late apoptotic cells (F). (G-I) Flow cytometry of propidium iodide and annexin V-fluorescein isothiocyanate (FITC)-stained AML-12 cells (G). Statistical analysis of the early apoptotic cells (H) and late apoptotic cells (I). Data are presented as mean ± SD of three independent biological experiments. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001, ns, no significant.
Tc3 induces pyroptosis in hepatic carcinoma cells
We further verified the anti-tumor effects of Tc3 in hepatic carcinoma cells. Tc3 significantly inhibited tumor proliferation, as demonstrated by cell growth curve analysis (Figure S2A-C) and colony formation assays (Figure S2D-E) in HepG2, SK-Hep1, and PLC-PRF5 cells. Additionally, Tc3 treatment reduced cell migration rates, as evidenced by wound scratch (Figure S2F-I) and transwell assays (Figure S2J-K). These results indicated that Tc3 effectively inhibits the growth and migration of hepatic carcinoma cells, making it a promising therapeutic agent.
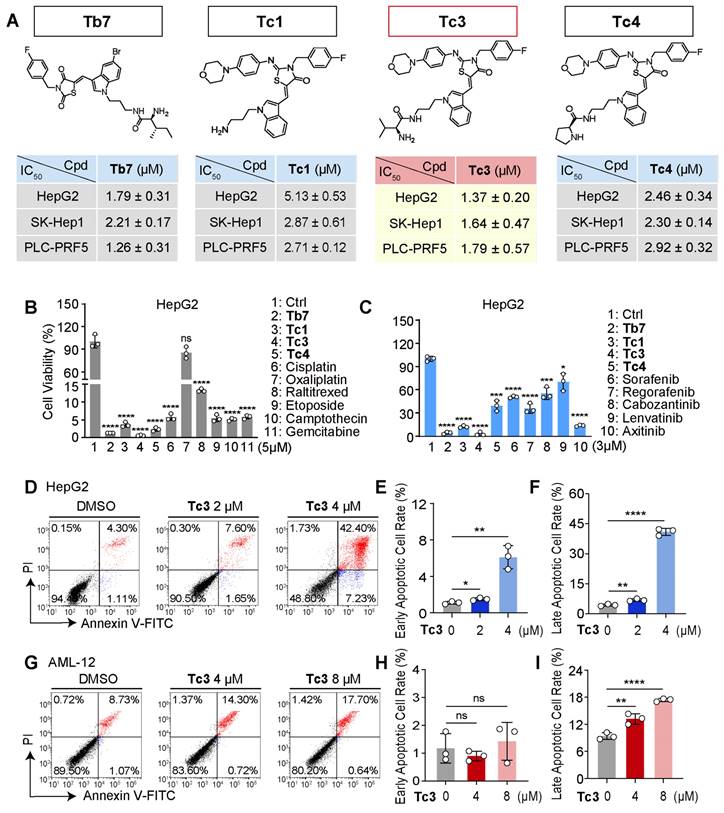
To investigate the mode of cell death induced by Tc3, we first observed the morphology of hepatic carcinoma cells treated with various indole analogues. As shown in Figure 3A, treatment with Tb7, Tc1, Tc3, and Tc4 induced the appearance of cytoplasmic vesicles to varying degrees. Consistent with the IC50 data, Tc3 caused pronounced membrane blebbing and rupture, leading to the damage of the integrity of the cell membrane (Figure 3C). Moreover, the LDH release levels were found all increased in the culture medium of cells treated with Tb7, Tc1, Tc3 and Tc4, indicating cell membrane disintegration and cell death. Tc3 treatment resulted in the highest LDH release, suggesting pore formation in the cell membrane characteristic of pyroptosis (Figure 3B). When cells were finally disintegrated via various forms of programmed cell death such as apoptosis and necrosis, the LDH release could also occurs. These observations led us to hypothesize that Tc3 might induce pyroptosis in hepatic carcinoma cells.
Tc3 induces pyroptosis and impairs cell membrane in vitro. (A) Representative microscopy images of HepG2 and SK-Hep1 cells after treatment with Tb7, Tc1, Tc3, Tc4 and control medium. Scale bars: 25 μm. (B) LDH release assay of HepG2 and SK-Hep1 cells treated with Tb7, Tc1, Tc3 and Tc4 at a concentration gradient (0-4 μM) (C) Representative fluorescence staining images of HepG2 and SK-Hep1 cells treated with Tb7, Tc1, Tc3, Tc4 and control medium. Cell membranes were labeled by 488-WGA and cell nuclei were labeled with DAPI. Scale bars: 25 μm. (D-E) Cell viability test and LDH release assay of HepG2 (D) and SK-Hep1 (E) cells treated with Tc3 (2 μM) alone or in combination with z-VAD-FMK (3 μM), 3-MA (1 mM) and DFOM (200 nM). (F-H) Representative fluorescence staining images of HepG2 and SK-Hep1 cells treated with Tc3. Apoptotic cells were labeled with dUTP-cy3, and cell nuclei were labeled with DAPI (F). Statistical analysis of dUTP-cy3 positive cells in HepG2 (G) and SK-Hep1 (H) cells. Scale bars: 200 μm. NC, negative control; PC, positive control. Data are presented as mean ± SD of three independent biological experiments. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001, ns, no significant.
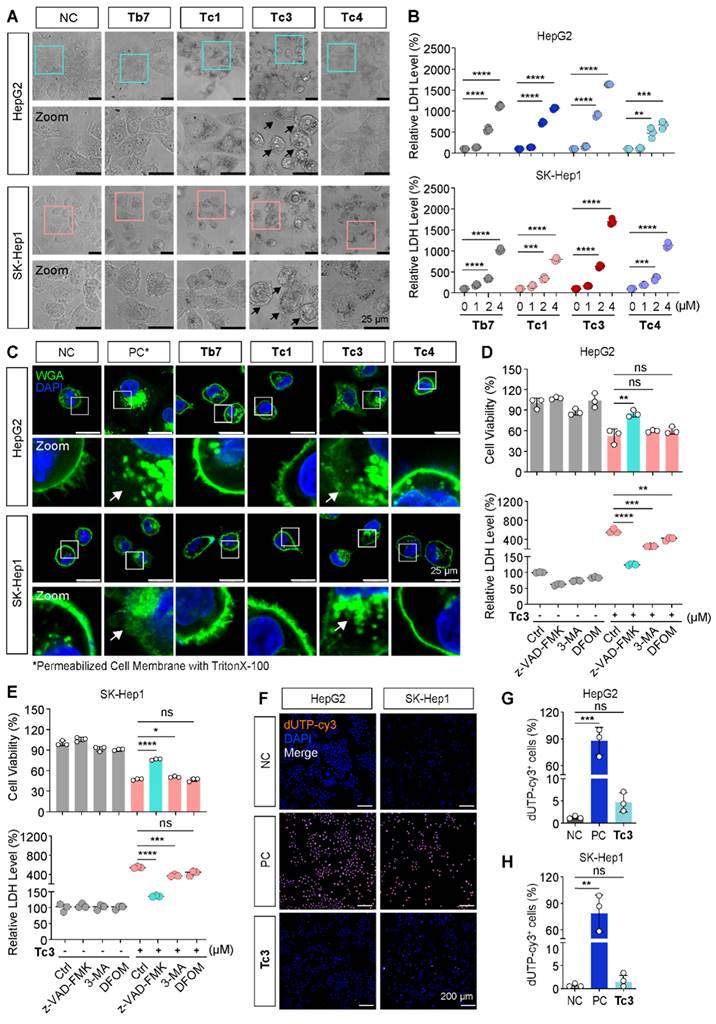
To further confirm the type of cell death induced by Tc3, we pre-treated hepatic carcinoma cells with z-VAD-FMK (a pan-caspase inhibitor), 3-Methyladenine (3-MA, an autophagy inhibitor), and Deferoxamine mesylate (DFOM, a ferroptosis inhibitor) before Tc3 treatment. Only z-VAD-FMK was able to rescue the cells from Tc3-induced death and reduce LDH release (Figure 3D-E). However, the RIPK1 inhibitor necrostatin-1 could also slightly rescue the cells death and the LDH release induced from Tc3 (Figure S2L-M). As inflammatory related cell death, pyroptosis and necroptosis both caused cell membrane rupture. In view of the activation of caspase8, it might prevent the initiate of necroptosis. TUNEL-Cy3 staining further confirmed that Tc3 induced caspase-mediated cell death, but not apoptosis (Figure 3F-H). As shown in Figure 2D, the most hepatic carcinoma cells treated with Tc3 were directly into late apoptotic stage. These data collectively demonstrate that Tc3 induces pyroptosis in hepatic carcinoma cells.
Tc3 activates GSDME-mediated pyroptosis of hepatic carcinoma cells
Members of the caspase and gasdermin families are crucial initiators and executors of pyroptosis. Currently, caspase1, caspase4/5, caspase3, and caspase8 have been identified as being related to pyroptosis, which can be activated by stimulants such as infections or toxins in humans [19]. We knocked down these pyroptosis-related caspases and gasdermins, and treated the stable knockdown cell lines with Tc3 (Figure S3). Our results demonstrated that the reduction of caspase3 and caspase8 partially rescued the hepatic carcinoma cell death rate and LDH release induced by Tc3. Correspondingly, the knockdown of GSDME (DFNA5) reduced the anti-tumor activity of Tc3 (Figure S4A-D and S5A-B). Western blotting results confirmed that the indole analogue Tc3 activated caspase3/8 and promoted the cleavage of GSDME, but not GSDMB or GSDMD. As a primary responder of Tc3, GSDME was relatively highly expressed in hepatic carcinoma cells, and the full-length GSDME could be up-regulated by Tc3, ensuring a better effect of Tc3 on hepatocarcinoma (Figure 4A and S4E). Other analogues, such as Tb7, Tc1, and Tc4, did not activate GSDME as expected (Figure S4F).
Tc3 activates the caspase8-caspase3-GSDME axis in hepatic carcinoma cells. (A) Immunoblot analysis showing activation of caspase3, caspase8 and GSDME in HepG2 and SK-Hep1 cells treated with Tc3. (B-D) Levels of GSDME, caspase8 and caspase3 in pLKO.1, shDFNA5 (B), shCaspase8 (C) and shCaspase3 (D) HepG2 cells treated with Tc3. (E) Dose-response curves for Tc3 inhibiting the growth of pLKO.1 and shDFNA5 HepG2 cells. (F) Representative microscopy images of pLKO.1, shDFNA5, pLVX-vector and pLVX-GSDME HepG2 cells treated with Tc3 and control medium. Scale bars: 25 μm. (G-H) Representative fluorescence staining images of pLKO.1, shDFNA5, pLVX-vector and pLVX-GSDME HepG2 cells after treatment with Tc3 (G). Apoptotic cells were labeled with dUTP-cy3, and cell nuclei were labeled with DAPI. Scale bars: 100 μm. Statistical analysis of dUTP-cy3 positive HepG2 cells (H). PC, positive control. (I-K) Colony formation assay of pLKO.1 and shDFNA5 of HepG2 and SK-Hep1 cells treated with Tc3 at various concentrations (I). Statistical analysis of colony counts for HepG2 (J) and SK-Hep1 (K) cells. (L-M) Wound scratch assay of pLKO.1 and shDFNA5 SK-Hep1 cells treated with Tc3 at various concentrations (L). Scale bars: 100 μm. Statistical analysis of the wound healing rate (M). Data are presented as mean ± SD of three independent biological experiments. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001, ns, no significant.
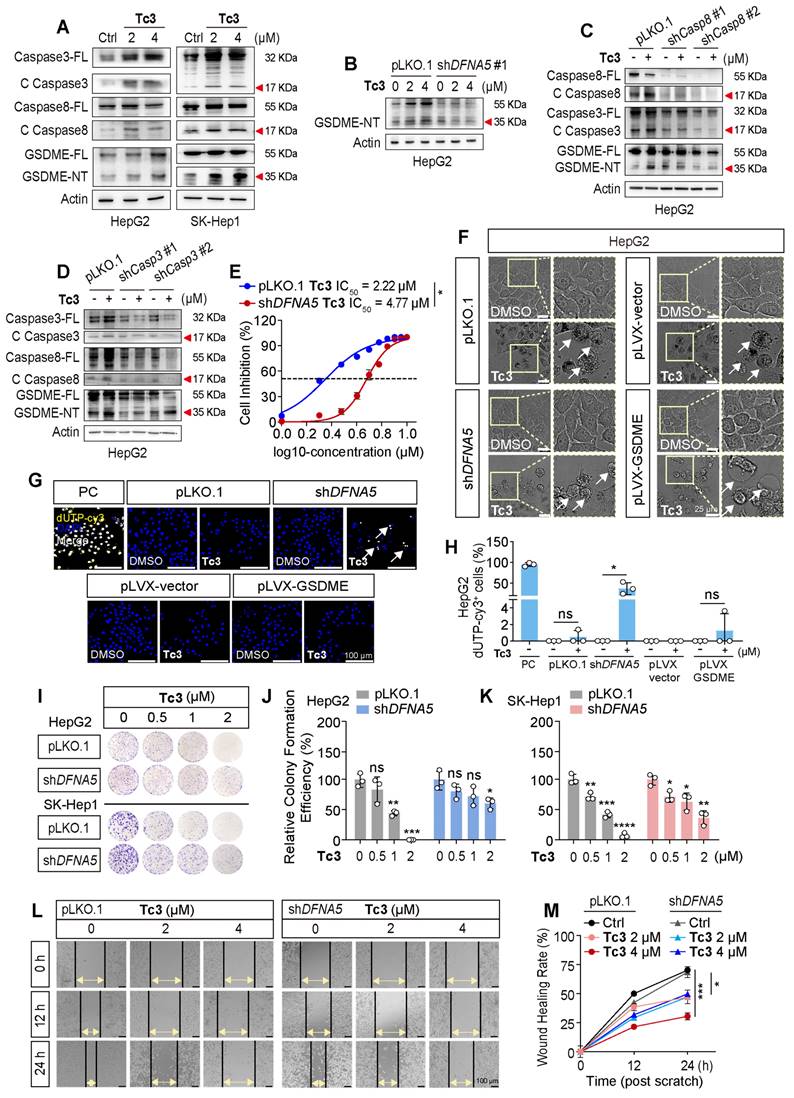
Of note, gasdermins were highest expressed in SK-Hep1 cells (liver sinusoidal endothelial immortal cells, LESCs). Therefore, SK-Hep1 cells were chosen as a representative GSDME over-expressed cell line for further study (Figure S5C). Additionally, GSDMA, GSDMC and PJVK were lowly expressed in hepatic carcinoma cells, especially GSDMA. Furthermore, the knockdown of caspase8 also restrained the activation of caspase3 and GSDME induced by Tc3, similarly to the reduction of caspase3 or GSDME, which further disturbed pyroptosis of cancer cells and LDH release (Figure 4B-D). As shown in Figure 4E, the IC50 value of Tc3 was increased from 2.22 μM to 4.77 μM after reducing the level of GSDME. We summarized that Tc3 activated the caspase8-caspase3-GSDME pathway and induced pyroptosis in cancer cells.
Previous studies have found that GSDME can convert caspase3-mediated apoptosis induced by TNF or chemotherapy drugs into pyroptosis, which is determined by the level of cellular GSDME. Here, we further knocked down or over-expressed GSDME in hepatic carcinoma cells to verify whether Tc3 could induce a change in the cell death mechanism (Figure S5D). Intriguingly, after reducing GSDME, the morphology of cancer cells treated with Tc3 showed shrinkage and became budding on cell membrane like apoptotic bodies.
Conversely, control or GSDME over-expressing cells exhibited swelling with bubble-like protrusions, and the inner cytoplasm became increased and gradually transparent when treated with Tc3 (Figure 4F and S5E). TUNEL-Cy3 staining also showed that Tc3 could promote apoptosis in a proportion of cancer cells when GSDME was knocked down (Figure 4G-H and S5F-G). The cell colony formation assay and cell scratch closure test demonstrated that Tc3 inhibited the proliferation (Figure 4I-K) and migration of hepatic carcinoma cells via activating GSDME (Figure 4L-M). The up-regulation of GSDME induced by Tc3 explains why the anti-tumor activity of Tc3 was rescued after the knockdown of GSDME, even though the cell death mechanism shifted from pyroptosis to apoptosis. These results indicate that the treatment of tumor types with high GSDME such as hepatic carcinoma was suitable for using Tc3, thereby inducing potent GSDME-mediated pyroptosis.
Tc3 triggers ER stress via PERK-EIF2α pathway for the induction of pyroptosis
To elaborate on the mechanism of pyroptosis induced by Tc3, we performed RNA sequencing analysis using total RNA samples from HepG2 cells. The sequence data identified 220 DEGs, with 182 genes up-regulated, and 38 genes down-regulated (Figure 5A). We primarily focused on analyzing the highly up-regulated genes in subsequent steps. Gene ontology (GO) analysis revealed “cellular response to topologically incorrect protein”, “response to endoplasmic reticulum stress” and “response to unfolded protein” as the top up-regulated biological processes upon Tc3 treatment. This indicated endoplasmic reticulum dysfunction and activation of the unfolded protein response (UPR) (Figure 5B). Moreover, Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis also demonstrated that the up-regulation of protein processing in the endoplasmic reticulum (Figure S6A). Consistently, hierarchical clustering of the top 20 most highly up-regulated genes following Tc3 treatment revealed that the prominence of endoplasmic reticulum stress (ER stress) related genes, such as HSPA5, DNAJC3, LOX, SQSTM1, HERPUD1, INSIG1, SCD, HYOU1, and HMGCS1 (Figure 5C). We next detected the mRNA levels of these representative genes in hepatoma cells treated with Tc3 via qRT-PCR, and confirmed the up-regulation of these ER stress related genes induced by Tc3 (Figure S6B).
Tc3 induces ER stress via the PERK-EIF2α pathway in hepatic carcinoma cells. (A) HepG2 cells were treated with Tc3 for RNA sequencing and a volcano plot of DEGs were generated (up-regulated genes shown in red; down-regulated genes shown in blue; non-regulated genes shown in gray) with|log2FC| ≥ 1 and q-value ≤ 0.05. (B) GO enrichment analysis identified “up-regulation of cellular response to topologically incorrect protein and regulation of response to endoplasmic reticulum stress” as a strongly associated biological processes in the Tc3 group. (C) Heatmap depicting the top 20 most significantly over-expressed genes in the Tc3 group compared to the control group. The color key corresponds to the row Z score (DMSO group shown in blue; Tc3-treated group shown in red). (D) Representative fluorescence staining images of HepG2 cells treated with Tc3 (4 μM) and 4-PBA (2 mM). Cell membranes were labeled with 488-WGA and cell nuclei were labeled with DAPI. PC, positive control, and cell membranes were permeabilized with 0.1% TritonX-100 in PC group. Scale bars: 50 μm. (E) Cell viability rate of HepG2 cells after treatment of Tc3 combined with 4-PBA. (F) Immunoblot analysis of the expression of caspase3, caspase8, GSDME, PERK, p-PERK, EIF2α, p-EIF2α, ATF6 and p-IRE1α in HepG2 cells treated with Tc3. (G) Schematic representation of the PERK-EIF2α pathway induced by Tc3. Data are presented as mean ± SD of three independent biological experiments. ***p < 0.001 and ****p < 0.0001, ns, no significant.
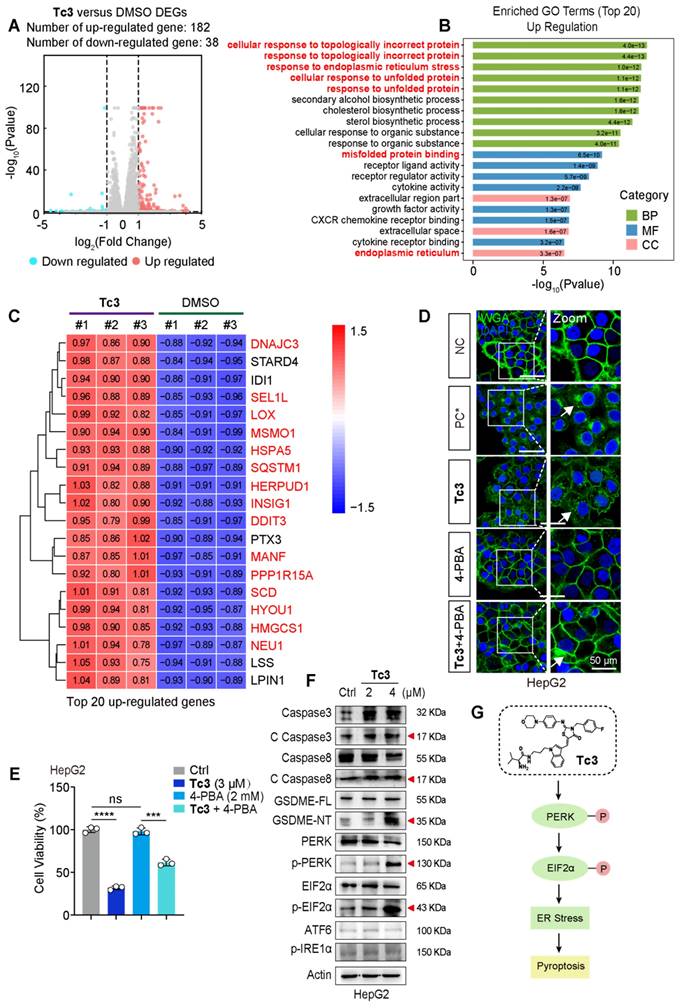
Previous studies have shown that excessive ER stress initiated by UPR can trigger cell death and inflammatory pathways [27]. Based on our RNA sequencing analysis, we hypothesized that Tc3-indcued GSDME-mediated pyroptosis might be modulated by the ER stress pathway. To verify this, hepatoma cells were treated with 4-PBA (an ER stress inhibitor) followed by Tc3 at an applicable concentration. The inhibitor 4-PBA could rescue cell viability and alleviate cell membrane rupture caused by Tc3 in hepatic carcinoma cells (Figure 5D-E and S6C). The responders of UPR and ER stress, including IRE1α, PERK, and ATF6, are typically activated to initiate downstream signaling pathways to reduce ER stress [28, 29]. Western blotting demonstrated that Tc3 activated protein kinase R-like endoplasmic reticulum kinase (PERK) but not other ER stress responders, and it also facilitated the phosphorylation of EIF2α (Figure 5F and S6D). These results indicate that Tc3 activates PERK-mediated ER stress, which subsequently leads to the activation of caspase3 and GSDME-mediated pyroptosis in hepatic carcinoma cells (Figure 5G).
Previous research also found that excessive accumulation of reactive oxygen species (ROS) could lead to oxidative stress inside cells, which further caused the organelle damage such as mitochondria or endoplasmic reticulum [28, 30]. The DCFH-DA probes of ROS indicated that the inside ROS were observably up-regulated by Tc3 (Figure S7A). It is notably that the accumulation of ROS is likely due to the suppression with its clearing system. Ruled out the ROS clearance system associated with ferroptosis like GPX4, we attempted to focus on an important member of the peroxidase reductase family, PRDX1, which had peroxidase activity and could remove peroxides such as hydrogen peroxide, organic hydrogen and peroxynitrite in cells [31, 32]. The expression of PRDX1 was obviously reduced in hepatic carcinoma cells treated with Tc3 (Figure S7B). We also proved that 4-PBA could disturb the activation of PERK-EIF2α-ATF4 pathway and the ER stress induced by Tc3, but not reverse the inhibition of Tc3 to PRDX1 (Figure S7C). The fluorescence staining was conducted to demonstrate Tc3 activated PERK by weakening PRDX1 function, resulting in impaired endoplasmic reticulum (Figure S7D-F). In addition, the knockdown of PRDX1 partially rescued the hepatic carcinoma cell death rate and LDH release induced by Tc3 (Figure S7G). As shown in Figure S7H, the reduction of PRDX1 directly restrained the activation of caspase3, PERK and EIF2α which induced by Tc3. Notably, the PRDX1 knockdown cells also impeded the up-regulation of ROS induced by Tc3 (Figure S7I-J). Collectively, the indole derivative Tc3 activated PERK-mediated ER stress via inhibiting the function of PRDX1 and up-regulating the inside ROS.
Tc3 inhibits the growth of hepatic carcinoma in vivo
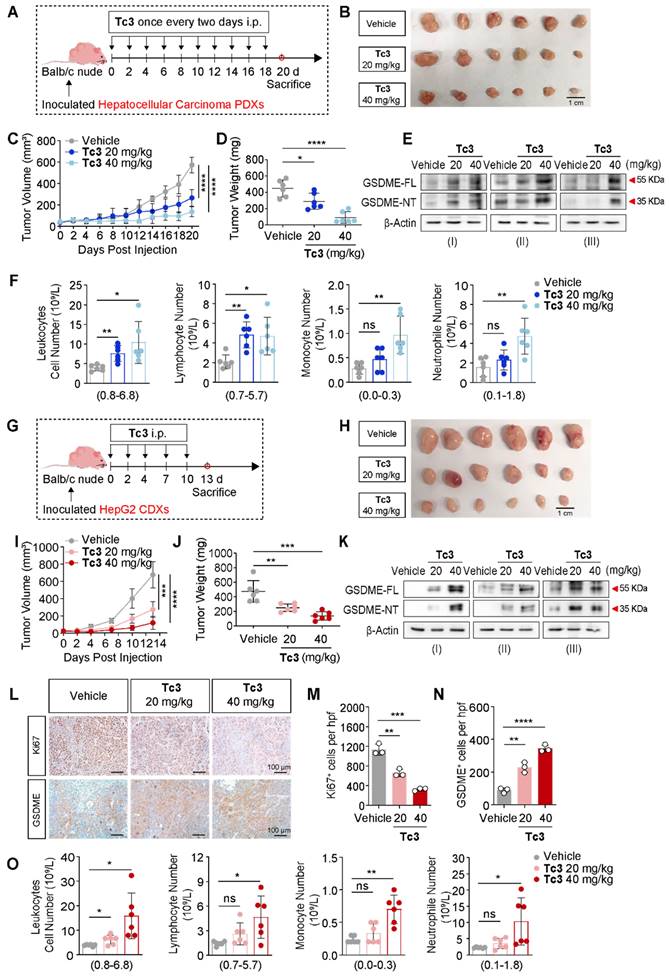
We next aimed to characterize the pharmacological properties of Tc3 in vivo. Pharmacokinetic profiling of Tc3 indicated effective venous absorption, characterized by a relatively long half-life elimination (t1/2 = 8.08 h). The plasma drug concentration of Tc3 ranged from 12.4-1417.0 ng/mL, and values of AUC0-t was 1524 h*ng/mL (Figure S8A). To carefully evaluate the safety of Tc3, we conducted a subchronic toxicity test in Balb/c mice (Figure S8B). Mice were treated with Tc3 at a concentration gradient (20, 40 and 60 mg/kg) by intraperitoneal injection (i.p.) every two days and were sacrificed on day 14. The nude mice all survived during the subchronic toxicity test. However, mice treated with the high dose (60 mg/kg) of Tc3 exhibited lower weight and worse health state (Figure S8C-D). As shown in Figure S8E, we investigated the toxicity of Tc3 via analyzing organ sections stained with hematoxylin-eosin (HE). Tc3 showed no significant toxicity to the heart, liver, spleen and stomach. However, Tc3 delivery in mice caused alveolar wall thickening and increased numbers of lymphocytes. In addition, the glomerular structure of the kidney was slightly damaged in mice treated with 60 mg/kg of Tc3. It is noteworthy that the immune cell infiltration of the jejunum increased and the crypts and villi were severely disrupted in mice injected with 60 mg/kg of Tc3. Thus, it is crucial to treat hepatocarcinoma xenografts with an appropriate dose of Tc3 to avoid bodily damage to the mice.
Further, we performed hepatocarcinoma xenograft treatment assay in Balb/c nude mice. In the hepatocarcinoma PDXs mouse model, Tc3 (20 mg/kg and 40 mg/kg) was injected (i.p.) ten times and the mice were sacrificed on day 20 (Figure 6A). Remarkably, Tc3 treatment significantly reduced the tumor size and weight (Figure 6B-D). And GSDME was up-regulated and activated by Tc3 in tumors, causing pyroptosis of cancer cells (Figure 6E), consistent with the results of blood routine examinations of the mice (Figure 6F). In the HepG2 cell line-derived xenografts (CDXs) mouse model, Tc3 (20 mg/kg and 40 mg/kg) was dosed (i.p.) five times and the mice were sacrificed (Figure 6G). The growth rate and weight of tumors treated with Tc3 were significantly smaller compared to the vehicle group (Figure 6H-J). Correspondingly, the levels of GSDME in tumors increased, and the caspase3-GSDME pathway was activated in the Tc3 treatment group compared to the vehicle group as shown by western blotting and immunohistochemical staining (IHC) assays (Figure 6K-N). Meanwhile, the levels of Ki67 decreased, which indicated the reduced growth rate of tumors (Figure 6L-M). Blood routine examinations of the mice showed that the immune system was activated and the inflammatory response was enhanced with increasing concentrations of Tc3, even in Blab/c nude mice with severe immune deficiency (Figure 6O). In conclusion, Tc3 exhibited significant anti-tumor activity via inducing pyroptosis and affecting the immune system of mice at a dose of 20 mg/kg.
Tc3 inhibits the growth of hepatic carcinoma PDXs and CDXs in vivo. (A) Schematic depicting the process of Tc3 treatment on tumors derived from hepatocarcinoma patients in Balb/c nude mice (n = 6). (B-D) Images of PDXs (B), tumor volumes (C) and tumor weight (D) from nude mice treated with Tc3 (20 mg/kg and 40 mg/kg) and control medium (n = 6). (E) Immunoblot analysis of GSDME activation in PDXs from three nude mice treated with Tc3 and control medium. (F) Blood routine examination of nude mice bearing hepatocarcinoma PDXs after Tc3 treatment, including statistical analysis of leukocytes, lymphocytes, monocytes and neutrophils. (G) Schematic depicting the process of Tc3 treatment on tumors derived from HepG2 cells in Balb/c nude mice (n = 6). (H-J) Images of HepG2 CDXs (H), tumor volumes (I) and tumor weight (J) from nude mice treated with Tc3 (20 mg/kg and 40 mg/kg) and control medium (n = 6). (K) Immunoblot analysis of GSDME activation in HepG2 CDXs from three nude mice treated with Tc3 and control medium. (L-N) Representative immunohistochemical (IHC) images of Ki67 and GSDME in HepG2 CDXs from nude mice treated with Tc3 (L). Scale bars: 100 μm. Statistical analysis of Ki67 (M) and GSDME (N) levels detected by IHC. (O) Blood routine examination of nude mice bearing HepG2 CDXs after Tc3 treatment, including statistical analysis of leukocytes, lymphocytes, monocytes and neutrophils. Data are presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001, ns, no significant.
Injection of Tc3 enhances the efficacy of cisplatin against hepatic carcinoma in vivo
To comprehensively assess the anti-tumor potency of Tc3, we compared it with several clinical chemotherapy drugs such as cisplatin, oxaliplatin, gemcitabine, and some first-line targeted anticancer drugs such as sorafenib, regorafenib, axitinib, lenvatinib, and cabozantinib, which were used for hepatocarcinoma treatment. The IC50 values of these drugs were determined to select appropriate concentrations. Noteworthy, Tc3 showed great anti-cancer potency among these clinical drugs in vitro (Figure 7A-B). Then, we attempted to evaluate their synergistic potential with Tc3 for hepatocarcinoma treatment to develop novel combination therapy.
Tc3 synergizes the anti-tumor effect of cisplatin in vivo. (A-B) Dose-response curves for Tc3 and other clinical drugs inhibiting the growth of HepG2 (A) and SK-Hep1 (B) cells. (C-D) Combination effects of Tc3 with cisplatin and sorafenib in HepG2 (C) and SK-Hep1 (D) cells. (E) Schematic depicting the process of Tc3 combined with sorafenib for treating HepG2 CDXs in Balb/c nude mice (n = 5). (F-H) Images of HepG2 CDXs (F), tumor volumes (G) and tumor weight (H) from nude mice treated with Tc3 (20 mg/kg) combined with sorafenib (20 mg/kg) (n = 5). (I) Schematic depicting the process of Tc3 combined with cisplatin for treating HepG2 CDXs in Balb/c nude mice (n = 5). (J-L) Images of HepG2 CDXs (J), tumor volumes (K) and tumor weight (L) from nude mice treated with Tc3 (20 mg/kg) combined with cisplatin (2 mg/kg) (n = 5). Data are presented as mean ± SD. ***p < 0.001 and ****p < 0.0001.
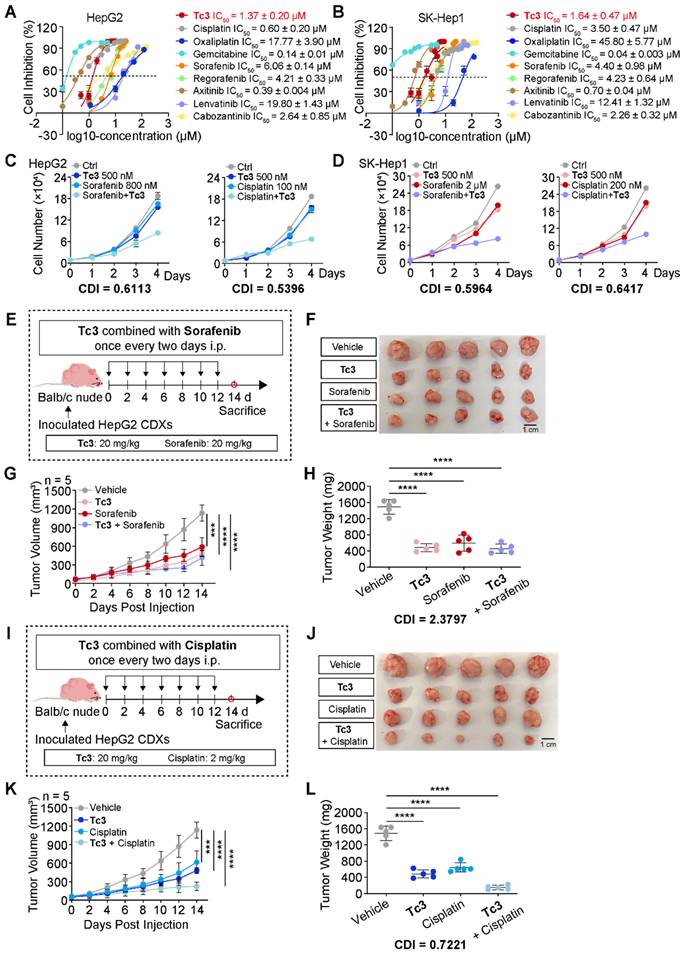
We next verified the combination therapy of Tc3 and some clinical drugs in hepatocarcinoma cells. Interestingly, only cisplatin and sorafenib exhibited a synergistically anti-tumor effects with Tc3 in HepG2 and SK-Hep1 cells (Figure 7C-D and S9A-L). Additionally, we detected the IC50 values of cisplatin and sorafenib in AML-12 cells, and verified the combination effect of cisplatin and sorafenib with Tc3, respectively (Figure S9M). As shown in Figure S9N-O, there exhibited no synergistic effects of cisplatin and sorafenib with Tc3 in AML-12 cells. To further confirm these findings in vivo, we employed the HepG2 CDXs mouse model. Contrary to the in vitro results, sorafenib did not show a synergistic anti-tumor effect with Tc3 in vivo (CDI=2.3797) (Figure 7E-H). This discrepancy may be due to the immune deficiency of Balb/c nude mice, which might impede Tc3's ability to activate the immune system. Intriguingly, cisplatin demonstrated a significant synergistic anti-tumor effect with Tc3 in vivo (CDI=0.7221) (Figure 7I-L). These results suggested that Tc3 is a highly promising candidate for enhancing the efficacy of clinical drugs, such as cisplatin, or as a standalone treatment for hepatocarcinoma.
Synergistic effect of Tc3 and anti-PD-1 on hepatic carcinoma via activating TIME
In addition to induce GSDME-mediated pyroptosis in tumor cells, Tc3 also has the capability to modulate and activate the immune response against hepatic carcinoma. We hypothesized that Tc3 inhibited tumor growth by activating anti-tumor immunity and it might have synergistic effect with anti-PD-1 antibody on hepatic carcinoma.
To better demonstrate the anti-tumor efficacy of Tc3, we employed a mouse model with Hepa1-6 xenografts. And mice were treated with Tc3 and anti-PD-1 antibody. Hepa1-6 cells mixed with extracellular matrix gel were injected into the subcutaneous tissues of C57 BL/6N mice, leading to the formation of suitably sized hepatic carcinoma CDXs within five days. Groups of C57 mice bearing tumors received injections of Tc3 (10 mg/kg), anti-PD-1 antibody (10 mg/kg) or the combination of two for five times, while control groups were dosed with DMSO or IgG antibodies (Figure 8A). A lower dosage of Tc3 at 10 mg/kg was used to prevent severe inflammatory effects resulting from GSDME-mediated pyroptosis. Notably, intraperitoneal injections of Tc3 alone or combined with anti-PD-1 antibodies significantly inhibited tumor growth compared to control groups. The combination of Tc3 and anti-PD-1 antibody exhibited a significant synergistic effect on the treatment of Hepa1-6 CDXs (CDI=0.6149) (Figure 8B-D). Pathological studies showed that Tc3 did not cause significant damage to the organs of the mice, including the liver, spleen, kidney and small intestine (Figure S10A).
Tc3 synergizes anti-PD-1 therapy to inhibit hepatoma in vivo. (A) Schematic depicting the process of Tc3 combined with anti-PD-1 antibody on tumors derived from Hepa1-6 cells in C57 BL/6N mice (n = 6). (B-D), Tumor volumes (B), tumor weight (C) and images of Hepa1-6 CDXs (D) from C57 mice treated with Tc3 (10 mg/kg) combined with anti-PD-1 antibody (10 mg/kg). (E) Immunoblot analysis of the activation of caspase3, caspase8 and GSDME in Hepa1-6 CDXs from C57 mice treated by Tc3 combined with anti-PD-1 antibody. (F) Representative fluorescence staining images showing the level and location of CD8, GSDME and caspase3 in tumor tissues. Scale bars: 100 μm. (G-I) Statistical analysis of flow cytometry data showing infiltration of IFNγ+CD8+ T cells (G), macrophages (H) and neutrophils (I) in tumor tissues derived from Hepa1-6 CDXs in C57 BL/6N mice (n=3). (J-L) Levels of cytokines IL-1α, IL-1β (J), IL-6, IL-10, IL-12, IL-18, IFNγ (K), TNFα, MCP-1, G-CSF and CXCL1 (L) in tumor tissues detected by ELISA (n=3). Data are presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
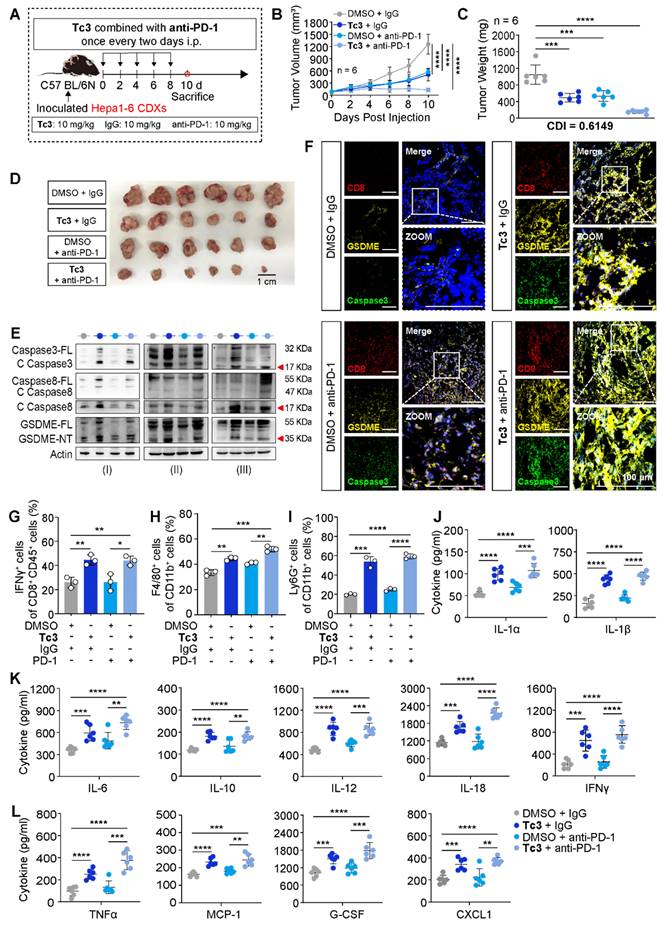
The caspase8-caspase3-GSDME pathway was activated by Tc3 in the tumors of C57 mice (Figure 8E). Given the synergistic effect of Tc3 and anti-PD-1 antibody, likely resulting from the activation of GSDME-mediated pyroptosis, we supposed that Tc3 stimulated the TIME of hepatocarcinoma, thereby enhancing the efficacy of anti-PD-1 antibody. Triple-labelled fluorescence staining and flow cytometry analysis were conducted to determine the cell types induced by Tc3. Injection of Tc3 significantly promoted the infiltration of activated CD8+ T cells (Figure 8F-G and S11A-B) and elevated the levels of cytokines (IL-1α, IL-1β, IL-6, IL-10, IL-12, IL-18, IFNγ, TNFα, MCP-1, G-CSF and CXCL1) (Figure 8J-L). Previous studies have reported that the inflammatory cytokines undergo secretion during pyroptosis of cells and activate the antitumor immunity in tumor microenvironment [33, 34]. These results indicated that Tc3 inhibits tumor growth primarily via activating CD8+ T cells and enhancing the activation of cytotoxic T lymphocytes induced by the anti-PD-1 antibody. Furthermore, the numbers of macrophages in tumors treated with Tc3 or Tc3 combined with anti-PD-1 antibodies were significantly higher than the control groups (Figure 8H and S10B). Compared to the M2 macrophages, the numbers of M1 macrophages were obviously up-regulated by Tc3, which could contribute to anti-tumor immunity (Figure S11C-D). Besides, the numbers of neutrophils were also higher in Tc3 treated groups than the control group (Figure 8I, S10C and S11E). Of note, the level of GSDME was markedly activated and up-regulated by Tc3 in tumors, co-localizing with activated caspase3 and infiltrative lymphocytes. Collectively, our findings suggest that Tc3 inhibits tumor growth by effectively enhancing anti-tumor immunity (Figure 9).
Graphical abstract of Tc3 in hepatic carcinoma cells. Tc3 significantly triggers endoplasmic reticulum stress, leading to GSDME-mediated pyroptosis. Additionally, Tc3 synergizes with cisplatin or anti-PD-1 antibody to inhibit hepatic carcinoma in vivo.
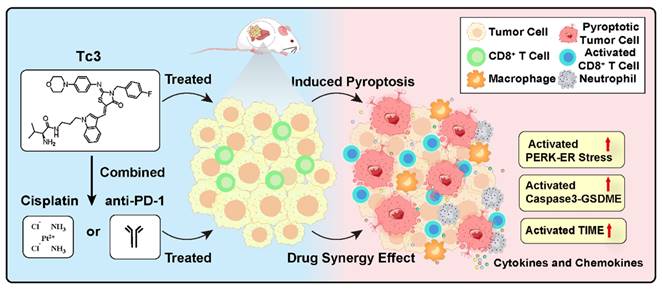
Discussion
In this study, we used T1 as the lead compound and introduced different functional groups at the amino side chain, confirming that amino acid residues can improve the anti-tumor activity. While substitution of H with Br or OCH3 at the 5-position of indole demonstrated comparable anti-tumor potency, Tb7, with a Br at the 5-position of indole and L-isoleucine at the amino side chain, exhibited a nearly 9-fold increase in potency compared to T1. Furthermore, the addition of a 4-morpholinoaniline group to T1 led to a significant enhancement in anti-tumor potency, with a nearly 3-fold improvement. Based on these observations, we synthesized four additional Tc1 derivatives (Tc2, Tc3, Tc4 and Tc5) with an amino acid group at the amino terminus. Compound Tc3 emerged as the most potent agent, with an IC50 of 1.37 μM (HepG2). Further modifications at the 2-position of 2,4-thiazolidinedione were planned to obtain more potent agents. However, none of these compounds showed any significant improvement in anti-tumor activity. While Tc3 showed slightly better potency than sorafenib, with IC50 values of 1.37 and 1.64 μM for HepG2 and SK-Hep1 cells respectively, compared to 6.06 and 4.40 μM for sorafenib (the first-line therapy for hepatocellular carcinoma), the rationale for these SAR trends is not yet clear. Further investigation on targets of Tc3 is essential to explore the chemical diversity in this series. We demonstrate here for the first time that the design of thiazole-substituted indole compound Tc3 as potent pyroptosis inducer, which has promising characteristics that could advance the treatment of hepatic carcinoma.
Pyroptosis as a novel inflammatory programmed cell death mediated by the gasdermins may act as a double-edged sword for tumors. The activation of pyroptosis leads to the release of inflammatory cytokines such as IL-1α and IL-1β, which induce chronic inflammation in tumor foci and are related to tumorigenesis and drug resistance [35, 36]. Some clinical chemotherapy drugs like cisplatin and 5-FU can activate pyroptosis and cause chemotherapy-induced tissue damage [21, 22]. On the contrary, inflammasomes induced by pyroptosis can facilitate the development of adaptive immune responses in the TIME, initiating immune cytotoxicity and inhibiting tumor cell proliferation, invasion and metastasis [37]. This indicates the potential of pyroptosis as a tumor treatment strategy, although the mechanism between pyroptosis and cancer is not clearly understood at present.
As a significant member of the gasdermin family, GSDME is conserved in even earlier metazoa which probably exerts a highly specific function for cell survival and propagation [21, 38]. In tumor cells, GSDME can inhibit cell proliferation and activate anti-tumor immunity, indicating that GSDME may function as a tumor suppressor [39-41]. However, GSDME assists tumor escape via activating GSDME-YBX1-mucin axis in pancreatic ductal adenocarcinoma, suggesting that the function of GSDME may regulate gene expression and serve as a tumor promoter independent of pyroptosis [21, 42]. Hence, the relationship between pyroptosis and cancer is complicated, and it is necessary to study the biochemical network of that to effectively exert the anti-tumor activity of pyroptosis. In this work, we attempt to activate GSDME and induce pyroptosis to inhibit hepatic carcinoma growth by Tc3. Additionally, it is also important to enliven the anti-tumor lymphocytes and cytokines in TIME via activated pyroptosis. Of note. Tc3 combined with anti-PD-1 antibody amplified the anti-tumor effect of tumor immunotherapy, attempting to balance the relationship of pyroptosis and tumor-therapy (Figure 8D-F). Following cleavage by caspase3, GSDME can induce tumor cell pyroptosis and enhance the anti-tumor immunity of T cells. It is reported that the level of GSDME is a key factor determining whether caspase3-activated cells undergo apoptosis or pyroptosis [43]. Although GSDME is often low-expressed in various tumor cells, the mRNA and protein level of GSDME remain normal in hepatic carcinoma cells according to our work, which is crucial for the anti-tumor effect of Tc3. GSDME was up-regulated in hepatic carcinoma cells by Tc3, indicating that Tc3 may overcome the restrictions of GSDME expression at the protein level.
Previous studies regarded that GSDME is silenced in many tumor cells, which can be achieved through epigenetic suppression or loss-of-function mutations in DFNA5 [39, 44]. It indicates that the combination of Tc3 with DNA-demethylating drugs such as distamine might potentially enhance the anti-tumor activity of Tc3 in other tumors. GSDME plays an important part in increasing the anti-tumor functions of tumor-infiltrating NK and CD8+ T cells, offering new inspiration for pyroptosis-mediated cancer treatments. Similarly, our data demonstrated that Tc3 could activate the TIME in mice and increase tumor-infiltrating lymphocytes, which is worthy of consideration as a potent pyroptosis inducer and GSDME activator for inhibiting hepatic carcinoma. The cellular morphology of hepatic carcinoma cells treated by Tc3 showed cytoplasmic vacuoles and larger endoplasmic reticulum, in addition to swelling cell membranes and bubble-like protrusions, indicating ER stress. RNA sequencing data also showed ER stress and UPR effects were emerged in hepatic carcinoma induced by Tc3. It is hypothesized that the reactive oxygen species (ROS) may be induced by Tc3, further stimulating the ER and triggering GSDME-mediated pyroptosis in hepatic carcinoma cells. However, the molecular mechanism and binding target of Tc3 remain unclear. Further investigation of Tc3 and its analogues is significant.
Taken together, we designed and developed a series of indole analogues based on compound T1 and evaluated the anticancer activity of 26 novel small molecules. The data in this study showed that Tc3 emerged as a promising lead compound, exhibiting potent induction of GSDME-mediated pyroptosis both in vitro and in vivo in hepatic carcinoma via activated PERK and the ER stress. Additionally, Tc3 significantly improved the anti-tumor potency of cisplatin in vivo. Moreover, Tc3 also had syngeneic effects with immune checkpoint inhibitor anti-PD-1 antibodies, by virtue of its induction of pyroptosis in hepatic carcinoma. Notably, Tc3 exhibited dual functions in cancer therapy. One is to motivate the pore-forming activity of GSDME to trigger tumor cell pyroptosis; another is to activate anti-tumor immune response. Specifically, this work first put forward thiazole-substituted indole Tc3 as pyroptosis inducer and identified its novel combination therapy for hepatic carcinoma. We provide a rationale to develop new therapeutic options to evoke the response of immunotherapy in hepatic carcinoma. Work along these lines is currently ongoing in our group.
Supplementary Material
Supplementary methods, figures and tables.
Abbreviations
CDXs: cell-derived xenografts; DEGs: differentially expressed genes; ER stress: endoplasmic reticulum stress; GSD: Gasdermin; HE: hematoxylin-eosin; LDH: lactate dehydrogenase; PDXs: patient-derived xenografts; PERK: protein kinase R-like ER kinase; TIME: tumor immune microenvironment; UPR: unfolded protein response.
Acknowledgements
We thank Dr. Qiao Wu from Xiamen university gifted us the plvx-GSDME plasmid.
Funding
This work was financially supported by the National Key Research and Development Program of China (grant no. 2022YFC2303300), the National Natural Science Foundation of China (grant no. 22177055, U23A20147), R&D Program of Guangzhou National Laboratory (grant no. GZNL2023A01002).
Author contributions
Luqing Shang and Changliang Shan designed and conceptualized this study. Xiao Hu, Xiaoman Tian and Yuanyuan Zhang performed the in vitro and in vivo experiments. Xiaomei Tang, Xing Lv, Yingyue Pang, Weilong Deng and Yali Wang designed and synthesized compounds mentioned above. Xiao Hu and Xiaomei Tang drafted the manuscript. All authors have read and approved the final manuscript.
Data and materials availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. For additional methods and supplemental figures please see the Supporting Information.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Arevalo JA, Vázquez-Medina JP. The role of peroxiredoxin 6 in cell signaling. Antioxidants. 2018;7:172
2. Kulik L, El-Serag HB. Epidemiology and management of hepatocellular carcinoma. Gastroenterology. 2019;156:477 -+
3. Yang XP, Yang C, Zhang S, Zhu AX, Geng HG, Bernards R. et al. Precision treatment in advanced hepatocellular carcinoma. Cancer Cell. 2024;42:1878-3686
4. Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol. 2018;15:599-616
5. Yang C, Zhang H, Zhang L, Zhu AX, Bernards R, Qin W. et al. Evolving therapeutic landscape of advanced hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2023;20:203-222
6. Ricke J, Schinner R, Seidensticker M, Gasbarrini A, van Delden OM, Amthauer H. et al. Liver function after combined selective internal radiation therapy or sorafenib monotherapy in advanced hepatocellular carcinoma. J Hepatol. 2021;75:1387-1396
7. Llovet JM, Pinyol R, Yarchoan M, Singal AG, Marron TU, Schwartz M. et al. Adjuvant and neoadjuvant immunotherapies in hepatocellular carcinoma. Nat Rev Clin Oncol. 2024;21:294-311
8. Llovet JM, Castet F, Heikenwalder M, Maini MK, Mazzaferro V, Pinato DJ. et al. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin Oncol. 2022;19:151-172
9. Akhtar J, Khan AA, Ali Z, Haider R, Yar MS. Structure-activity relationship (SAR) study and design strategies of nitrogen-containing heterocyclic moieties for their anticancer activities. Eur J Med Chem. 2017;125:143-189
10. Dadashpour S, Emami S. Indole in the target-based design of anticancer agents: a versatile scaffold with diverse mechanisms. Eur J Med Chem. 2018;150:9-29
11. Sharma PC, Bansal KK, Sharma A, Sharma D, Deep A. Thiazole-containing compounds as therapeutic targets for cancer therapy. Eur J Med Chem. 2020;188:112016
12. Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A. et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11:1136-1142
13. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97-111
14. Wan XP, Li JQ, Wang YP, Yu XF, He XJ, Shi JZ. et al. H7N9 virus infection triggers lethal cytokine storm by activating gasdermin E-mediated pyroptosis of lung alveolar epithelial cells. Natl Sci Rev. 2022;9:nwab137
15. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on cell death 2018. Cell Death Differ. 2018;25:486-541
16. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S. et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666-671
17. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J. et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111-116
18. Shi JJ, Gao WQ, Shao F. Pyroptosis: Gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42:245-254
19. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H. et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153-158
20. Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128
21. Wang Y, Gao W, Shi X, Ding J, Liu W, He H. et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99-103
22. Lage H, Helmbach H, Grottke C, Dietel M, Schadendorf D. DFNA5 (ICERE-1) contributes to acquired etoposide resistance in melanoma cells. FEBS Lett. 2001;494:54-59
23. Fan JX, Deng RH, Wang H, Liu XH, Wang XN, Qin R. et al. Epigenetics-based tumor cells pyroptosis for enhancing the immunological effect of chemotherapeutic nanocarriers. Nano Lett. 2019;19:8049-8058
24. Li WJ, Li SH, Xu G, Man XY, Yang TF, Zhang ZL. et al. Developing a ruthenium(III) complex to trigger gasdermin E-mediated pyroptosis and an immune response based on decitabine and liposomes: targeting inhibition of gastric tumor growth and metastasis. J Med Chem. 2023;66:13072-13085
25. Man XY, Sun ZW, Li SH, Xu G, Li WJ, Zhang ZL. et al. Development of a Pt(II) compound based on indocyanine green@human serum albumin nanoparticles: integrating phototherapy, chemotherapy and immunotherapy to overcome tumor cisplatin resistance. Rare Metals. 2024;43:6006-6022
26. Toyota M, Suzuki H, Maruyama R, Akino K, Watanabe Y, Sasaki Y. et al. Identification of DFNA5 as a target of epigenetic inactivation in gastric cancer. Proc Annu Meet Am Assoc Cancer Res. 2007;48:677
27. Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta Mol Cell Res. 2013;1833:3460-3470
28. Chen X, Cubillos-Ruiz JR. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer. 2021;21:71-88
29. Grootjans J, Kaser A, Kaufman RJ, Blumberg RS. The unfolded protein response in immunity and inflammation. Nat Rev Immunol. 2016;16:469-484
30. Rouschop KM, Dubois LJ, Keulers TG, van den Beucken T, Lambin P, Bussink J. et al. PERK/eIF2α signaling protects therapy resistant hypoxic cells through induction of glutathione synthesis and protection against ROS. Proc Natl Acad Sci U S A. 2013;110:4622-4627
31. Guan X, Ruan YY, Che XX, Feng WW. Dual role of PRDX1 in redox-regulation and tumorigenesis: past and future. Free Radic Biol Med. 2024;210:120-129
32. Neumann CA, Cao JX, Manevich Y. Peroxiredoxin 1 and its role in cell signaling. Cell Cycle. 2009;8:4072-4078
33. Dixon KO, Kuchroo VK. IL-18: throwing off the shackles to boost anti-tumor immunity. Cell Res. 2020;30:831-832
34. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99-109
35. Liu X, Lieberman J. A mechanistic understanding of pyroptosis: the fiery death triggered by invasive infection. Adv Immunol. 2017;135:81-117
36. Pezuk JA. Pyroptosis in combinatorial treatment to improve cancer patients' outcome, is that what we want? Ebiomedicine. 2019;41:17-18
37. Yu L, Xu Y, Pu ZJ, Kang H, Li M, Sessler JL. et al. Photocatalytic superoxide radical generator that induces pyroptosis in cancer cells. J Am Chem Soc. 2022;144:11326-11337
38. Jiang S, Zhou Z, Sun YY, Zhang TF, Sun L. Coral gasdermin triggers pyroptosis. Sci Immunol. 2020;5:eabd2591
39. Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X. et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature. 2020;579:415-420
40. Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. 2019;10:1689
41. Zhang L, Yang QC, Wang S, Xiao Y, Wan SC, Deng HX. et al. Engineering multienzyme-mimicking covalent organic frameworks as pyroptosis inducers for boosting antitumor immunity. Adv Mater. 2022;34:2108174
42. Lv J, Liu Y, Mo S, Zhou Y, Chen F, Cheng F. et al. Gasdermin E mediates resistance of pancreatic adenocarcinoma to enzymatic digestion through a YBX1-mucin pathway. Nat Cell Biol. 2022;24:364-372
43. Yokomizo K, Harada Y, Kijima K, Shinmura K, Sakata M, Sakuraba K. et al. Methylation of the DFNA5 Gene is frequently detected in colorectal cancer. Anticancer Res. 2012;32:1319-1322
44. Wang YY, Peng JY, Xie X, Zhang ZH, Li MX, Yang M. Gasdermin E-mediated programmed cell death: an unpaved path to tumor suppression. J Cancer. 2021;12:5241-5248
Author contact
![]() Corresponding authors: changliangshanedu.cn (Changliang Shan), shanglqedu.cn (Luqing Shang).
Corresponding authors: changliangshanedu.cn (Changliang Shan), shanglqedu.cn (Luqing Shang).