Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2024; 14(10):3827-3842. doi:10.7150/thno.95633 This issue Cite
Research Paper
Adeno-associated-virus-mediated delivery of CRISPR-CasRx induces efficient RNA knockdown in the mouse testis
Kai Li†, Mengzhen Li†, Yanyun Luo†, Dingfeng Zou, Yahui Li, Xinyu Mang, Zexuan Zhang, Pengyu Li, Yan Lu, Shiying Miao, Wei Song ![]()
Department of Biochemistry and Molecular Biology, State Key Laboratory of Common Mechanism Research for Major Diseases, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100005, China.
†These authors contributed equally to this work.
Received 2024-2-24; Accepted 2024-6-3; Published 2024-6-17
Abstract

Rationale: In male mammals, many developmental-stage-specific RNA transcripts (both coding and noncoding) are preferentially or exclusively expressed in the testis, where they play important roles in spermatogenesis and male fertility. However, a reliable platform for efficiently depleting various types of RNA transcripts to study their biological functions during spermatogenesis in vivo has not been developed.
Methods: We used an adeno-associated virus serotype nine (AAV9)-mediated CRISPR-CasRx system to knock down the expression of exogenous and endogenous RNA transcripts in the testis. Virus particles were injected into the seminiferous tubules via the efferent duct. Using an autophagy inhibitor, 3-methyladenine (3-MA), we optimized the AAV9 transduction efficiency in germ cells in vivo.
Results: AAV9-mediated delivery of CRISPR-CasRx effectively and specifically induces RNA transcripts (both coding and noncoding) knockdown in the testis in vivo. In addition, we showed that the co-microinjection of AAV9 and 3-MA into the seminiferous tubules enabled long-term transgene expression in the testis. Finally, we found that a promoter of Sycp1 gene induced CRISPR-CasRx-mediated RNA transcript knockdown in a germ-cell-type-specific manner.
Conclusion: Our results demonstrate the efficacy and versatility of the AAV9-mediated CRISPR-CasRx system as a flexible knockdown platform for studying gene function during spermatogenesis in vivo. This approach may advance the development of RNA-targeting therapies for conditions affecting reproductive health.
Keywords: AAV9-CRISPR-CasRx, RNA knockdown, 3-MA, germ-cell-specific transgene, spermatogenesis
Introduction
The testis has a highly complex transcriptome, in terms of the number of coding or non-coding genes expressed [1]. Single-cell RNA sequencing (scRNA-seq) of both human and murine germ cells has revealed that these cells exhibit distinct transcriptional signatures during spermatogenesis [2-6]; however, whether these widespread transcriptional products play a functional role in spermatogenesis and fertilization requires further experimental investigation in vivo. Although the CRISPR/Cas9 system, a widely used technology for genome engineering, is a valuable tool for studying gene function in vivo during spermatogenesis, the permanent deletion of targeted genes may result in embryonic lethality or overt phenotypic changes; these factors may obscure genuine biological processes and features of gene expression regulation [7, 8]. Additionally, the application of the CRISPR/Cas9 system for gene knockout studies of spermatogenesis is limited by certain genomic loci of non-coding genes, such as overlapping DNA regulatory elements [9, 10]. As such, there is an unmet need for additional loss-of-function tools to interrogate gene function during spermatogenesis in vivo.
Local ''RNA knockdown'' using tools such as RNA interference (RNAi) or antisense oligonucleotides (ASOs) provides a highly complementary approach for depleting RNA, including long non-coding RNA (lncRNA), messenger RNA (mRNA), and circular RNA [11-13]. In our previous reports, we established a strategy to identify functional lncRNAs during spermatogenesis in vivo using the adeno-associated virus (AAV)-mediated delivery of short-hairpin RNAs (shRNAs) [14]. In addition, an ASO-based approach has been used to successfully knockdown lncRNA in the mouse testes [15]. More recently, CRISPR-Cas13d, has been employed as a novel RNA-guided RNA-targeting system, to efficiently deplete RNA transcripts in mammalian cell lines and an animal model. Cas13d, a class 2 type VI single-effector CRISPR ribonuclease, can be directed to cleave single-stranded RNA via an engineered spacer within the guide RNA (gRNA), which is complementary to the sequence of its RNA targets. Among the known members of the Cas13d family, the Cas13d derived from Ruminococcus flavefaciens XPD3002 (CasRx), demonstrates superior RNA knockdown efficiency and specificity to those of RNAi [8, 16-21]. Additionally, its small size (~966 amino acids) allows for convenient packaging into the AAV for transgene delivery in vivo [16]. The AAV9-mediated CRISPR-CasRx system has recently been used to achieve the functional knockdown of RNA transcripts in a mouse model and, therefore, has therapeutic potential in the treatment of certain hereditary diseases [22-24]. Given the preliminary success of this strategy, we sought to test whether AAV9-mediated CRISPR-CasRx could be delivered to the testis to induce endogenous and exogenous knockdown of RNA targets, and consequently, accelerate and simplify the process of gene function interrogation in this organ in vivo.
Results
Optimization of AAV9-mediated gene transduction of germ cells in vivo
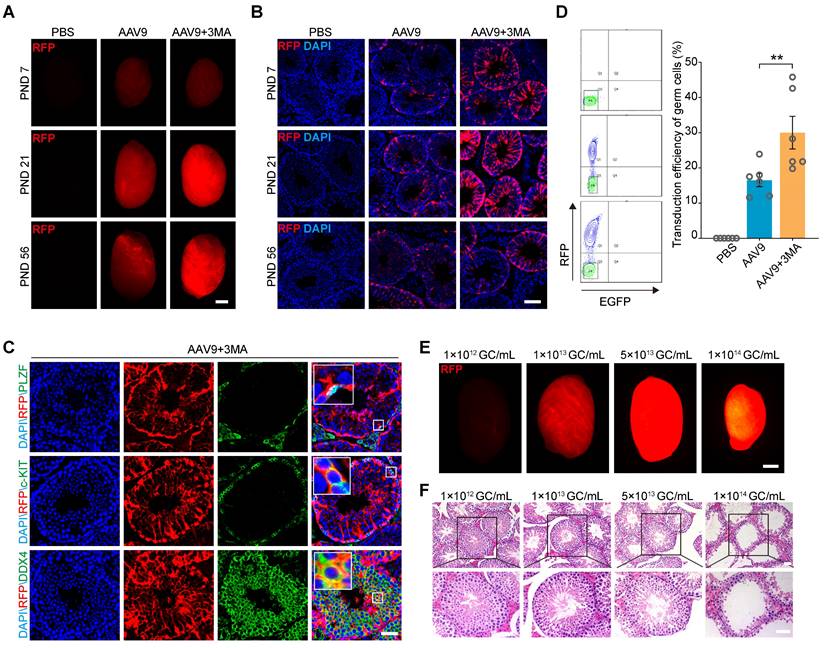
The tubular injection of AAV9 transduces both germ and Sertoli cells; however, the transduction efficiency of germ cells remains suboptimal [25]. Inhibiting autophagy can increase the efficiency of exogenous DNA electroporation or mumps virus (MuV) infection into male germ cells [26, 27]. Thus, to optimize AAV9-mediated delivery of CRISPR-CasRx to the testis in vivo and subsequently improve the AAV9 transduction efficiency of this organ, we co-injected AAV9-CMV-RFP with 3-methyladenine (3-MA), a widely used autophagy inhibitor, into the seminiferous tubules of testis on postnatal days (PND) 7, 21, and 56. We then checked the fluorescence intensity of the red florescence protein (RFP) reporter by both fluorescence stereomicroscopy and immunostaining 4 weeks after each microinjection. The results showed that the co-injection of AAV9-CMV-RFP and 3-MA substantially increased RFP expression in the testis from PND 21 onward (Figure 1A-B). In order to identify the expression characteristics of AAV9-CMV-RFP during mouse spermatogenesis, we tested its expression in spermatogenic cells by immunofluorescence staining. We observed the co-localization of the RFP signal with that of PLZF (a marker for undifferentiated spermatogonia), c-KIT (a marker for differentiating spermatogonia) and DDX4 (a marker for spermatogenic cell), suggesting that AAV9-mediated gene transfer had occurred in the germ cells (Figure 1C). To quantify the transduction efficiency of co-injecting AAV9 and 3-MA into spermatogenic cells, we depleted the somatic cells and quantified the percentage of RFP+ germ cells using flow cytometry. The results showed that the transduction efficiency of AAV9+3-MA in spermatogenic cells was significantly higher than that of AAV9 alone (AAV9+3-MA, mean ~30%; AAV9 alone, mean ~16.5%; P < 0.01) (Figure 1D). Because the AAV9 vector can be administered at relatively high doses, we next evaluated how testis transduction efficiency varied at different AAV9 titers. Stereomicroscopic fluorescence imaging showed that a titer of 1-5 × 1013 genome copies (GC)/mL resulted in high RFP expression; moreover, the hematoxylin and eosin (H&E) staining of testicular sections showed that spermatogenesis was not significantly affected at this AAV9 dose (Figure 1E-F). Although a titer of 1 × 1014 GC/mL elicited the strongest RFP fluorescence, it induced marked developmental defects in spermatogenesis, such as testicular shrinkage and germ cell loss. In view of this, we selected a titer of 1 × 1013 GC/mL AAV9 for subsequent transgene delivery experiments. Our findings suggest that 3-MA improves AAV9 transduction efficiency in germ cells in vivo and that AAV9 is safe to use within the specified titer range.
Co-injection of AAV9 and 3-MA increase the transduction efficiency of spermatogenic cells in vivo. (A) Stereomicroscopic fluorescence imaging of representative testes (postnatal days [PND] 7, 21, and 56) 4 weeks after the microinjection of PBS, AAV9-CMV-RFP, or AAV9-CMV-RFP+3-MA. RFP: red fluorescence protein reporter gene. Scale bar: 1 mm. (B) Immunostaining of testicular sections for RFP (red) derived from testes (PND 7, 21, and 56) microinjected with PBS, AAV9-CMV-RFP, or AAV9-CMV-RFP+3-MA. Nuclei were stained with DAPI (blue). Scale bar: 100 μm. (C) Immunostaining of testicular sections from testes co-injected with AAV9-CMV-RFP and 3-MA for RFP (red), c-KIT (green, top), and DDX4 (green, bottom). Nuclei were stained with DAPI (blue). Scale bar: 50 μm. (D) Flow cytometric analysis of the percentage of RFP+ germ cells in testes 4 weeks after microinjection of PBS, AAV9-CMV-RFP, or AAV9-CMV-RFP+3-MA. Left: representative flow cytometry contour plot; Right: quantification of the percentage of RFP+ germ cells; Data are presented as the mean ± SEM (n = 6). P-values were determined using the two-tailed Student's t-test; **P < 0.01. (E) Stereomicroscopic fluorescence imaging of representative testes co-injected with AAV9-CMV-RFP and 3-MA at the indicated AAV titer. Scale bar: 1 mm. (F) H&E staining of testicular sections from testes co-injected with AAV9-CMV-RFP and 3-MA at the indicated AAV titer. Scale bar: 50 μm.
In vivo kinetics and safety assessment of AAV9 and 3-MA co-injection into the testis
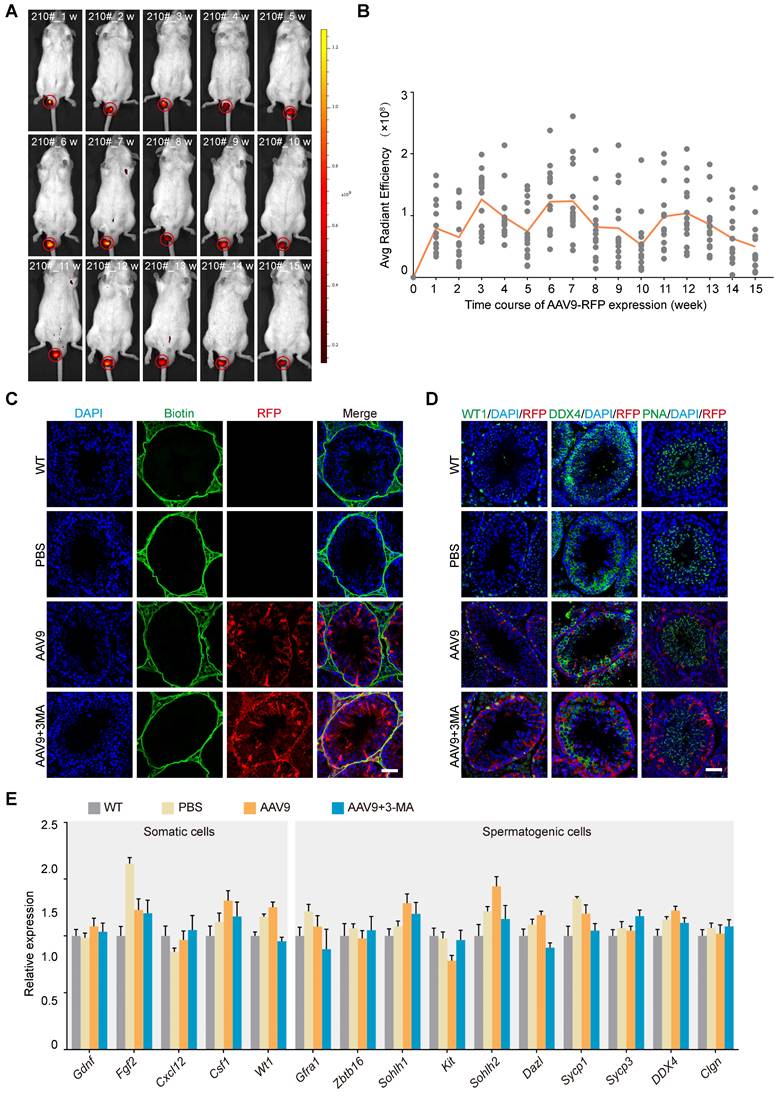
Our next objective was to characterize the AAV9 transduction kinetics in the mouse testis. To this end, we investigated the tissue expression profile of AAV9-CMV-RFP using stereomicroscopy and western blotting after microinjecting AAV9 and 3-MA into the seminiferous tubules of the testis. The results showed that RFP expression was localized to the mouse testis and negligible in other organs (Figure S1A-B). We then tested whether AAV9-mediated transgene expression persisted long-term by tracking the dynamics of the RFP signal in the mouse testis over time. Continuous in vivo bioluminescence imaging and a semi-quantitative analysis of radiant efficiency revealed that RFP expression was detected 1 week after microinjection, peaked after 3-4 weeks, and then gradually decreased until week 5. Notably, we observed that RFP expression assumed a rhythmic profile in the mouse testis, whereby its expression rose again on week 6, before gradually declining in week 7. Interestingly, this 5-week cycle aligned with the spermatogenic wave pattern of mice and was maintained for at least 15 weeks, suggesting that AAV9 induced long-term transgene expression in the testis (Figure 2A-B). AAV9 penetrates the tight junctions of the blood-testis barrier (BTB) without perturbing their structure; in addition, it exerts no apparent toxicity on somatic and spermatogenic cells. We therefore next assessed whether AAV9 co-injection with 3-MA impaired the integrity of the BTB and induced potential toxic effects on germline developmental progression in vivo. To this end, we performed a biotin tracer experiment in the testis to test the integrity of the BTB and immunostaining for DDX4, PNA (a post-meiotic germ cell marker) and WT1 (a Sertoli cell marker) 3 weeks after AAV9 and 3-MA co-injection. The biotin tracer experiment revealed that the biotin was localized to the interstitial area and the basement membrane, and did not enter the lumen of the seminiferous tubules. This finding suggested that the co-injection of AAV9 and 3-MA had no effect on the integrity of the BTB (Figure 2C). In addition, the expression of DDX4, PNA, and WT1 was not significantly different between the AAV9+3-MA and AAV9 alone groups, indicating that the combination treatment did not hamper germline developmental progression in vivo (Figure 2D). Next, we examined the number and morphological development of mature spermatozoa. The results showed that there were no significant differences in the number of mature spermatozoa among WT group, AAV9 group and AAV9+3MA group (Figure S1C). Immunostaining for PNA showed the normal morphology of mature spermatozoa from testis treated with AAV9+3MA (Figure S1D). We then examined the expression levels of genes encoding several representative cytokines from the testicular microenvironment (e.g., Gdnf, Fgf2, Cxcl12, Csf1), as well as that of functional spermatogenic genes (e.g., Zbtb16, Sohlh1, Kit, Dazl, Sycp1, Sycp3, Ddx4, Clgn), using quantitative real-time PCR (qRT-PCR). We found that the expression levels of these genes were largely unaltered following AAV9 and 3-MA co-injection (Figure 2E), suggesting that this combination had little effect on the testicular microenvironment and endogenous gene expression in spermatogenic cells. Taken together, these findings demonstrate that AAV9 and 3-MA co-injection provided long-term transgene expression without adversely affecting germline development in vivo.
In vivo kinetics and safety assessment of the co-injection of AAV9 and 3-MA into the testis. (A) Representative sequential fluorescence imaging of the RFP signal (in mouse 210#) at indicated time-points (weeks 1-15) after co-injection of AAV9-CMV-RFP+3-MA, determined using an in vivo imaging system (IVIS). Dark red and yellow indicate low and high RFP expression, respectively, following AAV9-CMV-RFP administration. (B) Kinetics of AAV9-CMV-RFP expression in the mouse testis in vivo at the indicated time-points. Average (Avg) radiant efficiency was determined from the fluorescence imaging results. Sequential fluorescence imaging data were generated from each mouse at each time-point (n = 15). (C) Results of the biotin tracer experiment to determine BTB integrity. The mouse testes (at PND 21) were first microinjected with PBS, AAV9-CMV-RFP, or AAV9-CMV-RFP+3-MA and then interstitially injected with biotin (green). Samples were recovered 30 min after biotin microinjection (n = 3). Scale bar: 50 μm. (D) Immunostaining of testicular sections for WT1 (green, left), DDX4 (green, middle), and PNA (green, right) from testes microinjected with PBS, AAV9-CMV-RFP, or AAV9-CMV-RFP+3-MA. Nuclei were stained with DAPI (blue). Scale bar: 50 μm. (E) qRT-PCR analysis of representative functional gene expression in somatic and spermatogenic cells isolated from testes microinjected with PBS, AAV9-CMV-RFP, or AAV9-CMV-RFP+3-MA. Data were normalized to Actb expression and are presented as the mean ± SEM (n = 3).
AAV9-mediated CRISPR-CasRx induces reporter mRNA knockdown in the testis in vivo
CRISPR-CasRx offers an effective approach for knocking down specific RNA transcripts in individual mammalian cells (e.g., the neuron) or organs (e.g., the liver), as well as on a systemic level (i.e., in the embryo) [8, 16, 19]. Meanwhile, AAV is a promising vehicle for in vivo transgene delivery and gene therapy [16]. Thus, we next determined the RNA-targeting capabilities of the AAV9-mediated delivery of CRISPR-CasRx into the mouse testis. To achieve this, we first constructed an AAV expression vector encoding the tdTomato fluorescence reporter (AAV9-EFS-tdTomato). Next, we designed a set of three 30-nucleotide gRNAs targeting the coding sequence of tdTomato mRNA; an expression vector carrying tdTomato-non-targeting gRNAs (AAV9-EFS-CasRx-NTG) was used as a negative control. We then constructed the all-in-one AAV expression vector which contained a gRNA array as well as an mRNA encoding the CasRx protein (AAV9-EFS-CasRx-tdTomato) (Figure 3A). To examine the efficacy of the gRNAs at knocking down the expression of tdTomato, we co-transfected human embryonic kidney (HEK) 293T cells with AAV9-EFS-tdTomato and AAV9-EFS-CasRx-tdTomato or AAV9-EFS-CasRx-NTG. Fluorescence imaging and western blotting showed that the expression of tdTomato decreased dramatically following the transfection of tdTomato-targeting gRNA but not after the transfection of the NTG (Figure S2A-B). The above vectors were then packaged into AAV9 and used in bilateral testicular microinjection experiments, whereby one testis of a 3-week-old mouse was co-injected (into the seminiferous tubule) with AAV9-EFS-tdTomato and AAV9-EFS-CasRx-NTG, while the contralateral testis received AAV9-EFS-tdTomato and AAV9-EFS-CasRx-tdTomato. The CasRx-mediated knockdown efficiency of tdTomato was then examined 1 week after the co-injection. Fluorescence stereomicroscopy showed that the mean fluorescence intensity (MFI) of the testis co-injected with AAV9-EFS-tdTomato and AAV9-EFS-CasRx-tdTomato was ~50% lower than that of the control testis (Figure 3B-C); Western blotting further confirmed that CRISPR-CasRx caused a 2-fold reduction in tdTomato protein expression in the testis (Figure 3D). These findings demonstrate that AAV9-mediated CRISPR-CasRx effectively lowers tdTomato protein level in the testis in vivo.
AAV9-mediated CRISPR-CasRx specifically targets reporter mRNA expression in the mouse testis in vivo. (A) Schematic illustration of reporter mRNA knockdown by AAV9-mediated CRISPR-CasRx in vivo. Vector#1 (AAV9-EFS-tdTomato) encodes the tdTomato reporter, driven by the EFS promoter; Vector#2 (AAV9-EFS-CasRx-tdTomato) encodes CasRx and gRNAs. (B) Stereomicroscopic fluorescence imaging of representative testes microinjected with AAV9-EFS-CasRx-tdTomato or AAV9-EFS-CasRx-NTG together with AAV9-EFS-tdTomato. Scale bar: 1 mm. (C) Mean fluorescence intensity (MFI) of tdTomato in testes microinjected with either AAV9-EFS-CasRx-tdTomato or AAV9-EFS-CasRx-NTG together with AAV9-EFS-tdTomato. Data were normalized to the tdTomato-non-targeting condition (n = 6). (D) Western blotting analysis of tdTomato protein expression following AAV9-EFS-CasRx-tdTomato treatment. GAPDH was served as the loading control. Data from two biologically independent samples are shown. (E) Representative flow cytometry dot plot showing the percentage of tdTomato-positive cells in the testes microinjected with AAV9-EFS-CasRx-tdTomato or AAV9-EFS-CasRx-NTG together with AAV9-EFS-tdTomato. (F and G) Flow cytometric analysis of the tdTomato MFI and the percentage of tdTomato+ cells from the testes microinjected with AAV9-EFS-CasRx-tdTomato or AAV9-EFS-CasRx-NTG together with AAV9-EFS-tdTomato. Data were normalized to the tdTomato-non-targeting condition (n = 3). (H) Immunostaining of testicular sections for tdTomato (red) and DDX4 (green). Nuclei were stained with DAPI (blue). Scale bar: 100 μm. (I) qRT-PCR analysis of tdTomato mRNA knockdown efficiency by individual position-matched gRNAs and siRNAs. Data were normalized to Gapdh expression (n >3). (J) Volcano plots of differential transcript levels between tdTomato-targeting and NTG CasRx (left) or tdTomato-targeting and non-targeting siRNA (right) as determined by RNA sequencing. All data are presented as the mean ± SEM. P-values were determined using the two-tailed Student's t-test; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
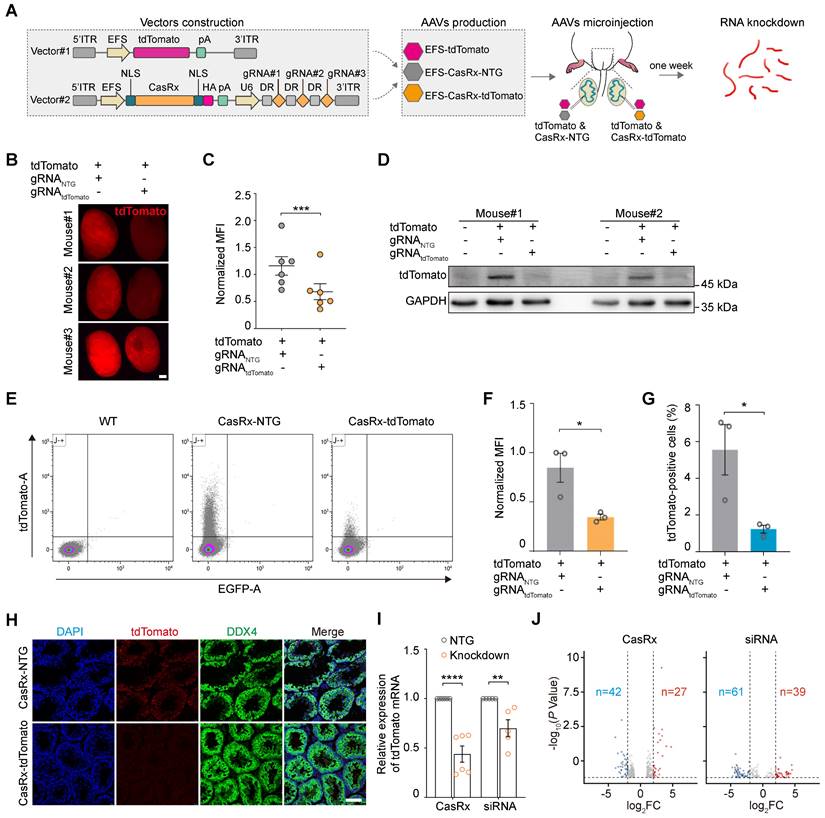
We next used flow cytometry to quantify tdTomato fluorescence intensity and further evaluate the knockdown efficiency and specificity of CRISPR-CasRx in the testis in vivo (Figure 3E). We found that CRISPR-CasRx significantly altered the level of tdTomato expression in testicular cells. Notably, the administration of AAV9-EFS-CasRx-tdTomato reduced tdTomato fluorescence intensity by ~50% (Figure 3F), while the percentage of tdTomato+ testicular cells was decreased by ~75% (Figure 3G). Additionally, immunostaining showed that there was no substantial difference in DDX4 expression between the testis of the AAV9-EFS-CasRx-NTG- and AAV9-EFS-CasRx-tdTomato-treated groups of mice, other than the marked reduction in tdTomato expression (Figure 3H). These findings confirm that AAV9-mediated CRISPR-CasRx achieved effective knockdown of tdTomato expression in the testis in vivo.
To further compare the efficiency and specificity of RNA knockdown achieved by CRISPR-CasRx and siRNA, we designed position-matched gRNA and siRNA to target reporter tdTomato mRNA (Figure S2C-E). We observed that compared to siRNA, CRISPR-CasRx showed higher efficiency of RNA knockdown (CRISPR-CasRx, mean ~60%; siRNA, mean ~30%) in the testis in vivo (Figure 3I). We next performed RNA-seq to test the possible off-target effects of CRISPR-CasRx and siRNA. Differential expression analysis indicated that CRISPR-CasRx exerts fewer off-target effects than that of siRNA in the testis in vivo (Figure 3J). Importantly, tdTomato was the most significantly downregulated transcript (foldchange > 5), whereas the transcriptional levels of typical functional spermatogenic genes remained largely unchanged in CRISPR-CasRx condition (Figure S2F-G). Collectively, these results suggest that AAV9-mediated CRISPR-CasRx is a highly effective and safe RNA knockdown tool for use in the testis in vivo.
AAV9-mediated CRISPR-CasRx effectively silences endogenous functional spermatogenic genes in the testis in vivo
To better understand the roles of specific genes during spermatogenesis, we next evaluated the ability of the AAV9-mediated CRISPR-CasRx system to silence endogenous spermatogenic genes. Our previous work showed that the shRNA-mediated knockdown of Sycp3 mRNA led to spermatogenic defects [14]; thus, we decided to target Sycp3 mRNA to investigate its potential application within the CRISPR-CasRx system in testis in vivo. To identify gRNAs targeting Sycp3 mRNA, we transfected HEK293T cells with an expression vector encoding Sycp3 (EFS-Sycp3), as well as an all-in-one vector expressing CasRx and gRNAs (AAV9-EFS-CasRx-Sycp3 or AAV9-EFS-CasRx-NTG). qRT-PCR and western blotting results showed a significant decrease (~90%) in Sycp3 expression 2 days after transfection (Figure 4A; Figure S3A). We then packaged the above vectors in AAV9 and microinjected them into each testis of a 3-week-old mouse. We found that, 3 weeks after the microinjection, the testis treated with AAV9-EFS-CasRx-Sycp3 was smaller and weighed significantly less (~25%) than that treated with the CasRx-NTG control (Figure 4B-C). Immunofluorescence staining for SYCP3 revealed a significant decrease in the number of SYCP3+ cells and the corresponding fluorescence intensity of the CasRx-Sycp3-treated testes, suggesting that CasRx successfully knocked down Sycp3 in the testis in vivo (Figure 4D). H&E staining of testicular sections revealed that the CasRx-Sycp3-treated testes exhibited histological defects, characterized by a thinner seminiferous epithelium and germ cell loss, compared to the control testes (Figure 4E). To further characterize these spermatogenic defects, we quantified the number of the spermatocyte populations (pre-leptotene, leptotene and zygotene spermatocytes [PL/L/Z]; pachytene and diplotene spermatocytes [P/Di]) based on morphological analysis of testicular sections [28]. The result showed a significant decrease in the number of pachytene and diplotene spermatocytes (P/Di) (Figure 4E). Additionally, we performed immunofluorescence staining for PNA and WT1 of testicular sections from CasRx-Sycp3- and CasRx-NTG-treated mice. Although we found no significant difference in the number of Sertoli cells between these two groups of mice, the number of post-meiotic germ cells was substantially reduced in the testes of CasRx-Sycp3-treated mice, indicating spermatogenesis dysfunction in these animals (Figure 4F; Figure S3B).
Transcriptome editing of Sycp3 and lncRNA-Eif2c5 by AAV9-mediated CRISPR-CasRx in the testis in vivo. (A) Western blotting analysis of SYCP3 protein expression in HEK293T cells after CRISPR-CasRx gene editing. GFP was used as transfection control. β-Tubulin was used as a loading control. (B and C) Comparison of the size (B) and weight (C) of testes from control (CasRx-NTG) and SYCP3-depleted (CasRx-Sycp3) mice (n = 5). Scale bar: 1 mm. (D) Immunostaining of testicular sections from the control (CasRx-NTG) and SYCP3-depleted (CasRx-Sycp3) mice for SYCP3 (green). Nuclei were stained with DAPI (blue). Scale bar: 50 μm. Left: representative images; Right: number of stained cells per tubule (n = 10). (E) H&E staining of testicular sections from the control (CasRx-NTG) and SYCP3-depleted (CasRx-Sycp3) mice. Scale bar: 50 μm. Left: representative images; Right: number of Sub-stages of the prophase I spermatocytes (pre-leptotene, leptotene and zygotene spermatocytes [PL/L/Z]; pachytene and diplotene spermatocytes [P/DI]) with staining per tubule (n = 10). (F) Immunostaining of testicular sections from the control (CasRx-NTG) and SYCP3-depleted (CasRx-Sycp3) mice for PNA (red). Nuclei were stained with DAPI (blue). Scale bar: 50 μm. (G) qRT-PCR analysis of lncRNA-Eif2c5 knockdown efficiency in HEK293T cells transfected with vectors expressing Eif2c5, CasRx, and gRNAs. Data were normalized to Actb expression (n = 3). (H and I) The size (H) and weight (I) of testes from the control (CasRx-NTG) and Eif2c5-depleted (CasRx-Eif2c5) mice (n =12). (J) RNAscope analysis of lncRNA-Eif2c5 (red) targeted by AAV9-mediated CRISPR-CasRx in vivo. Nuclei were stained with DAPI (blue). Scale bar: 50 μm. Left: representative images; Right: quantification of Eif2c5 punctate signal per tubule. (K) H&E staining of testicular sections from the control (CasRx-NTG) and Eif2c5-depleted (CasRx-Eif2c5) mice. Scale bar: 50 μm. (L) Immunostaining of testicular sections from the control (CasRx-NTG) and Eif2c5-depleted (CasRx-Eif2c5) mice for WT1 (green) and PNA (red). Nuclei were stained with DAPI (blue). Scale bar: 50 μm. All data are presented as the mean ± SEM. P-values were determined using the two-tailed Student's t-test; *P < 0.05; **P < 0.01; ***P < 0.001.
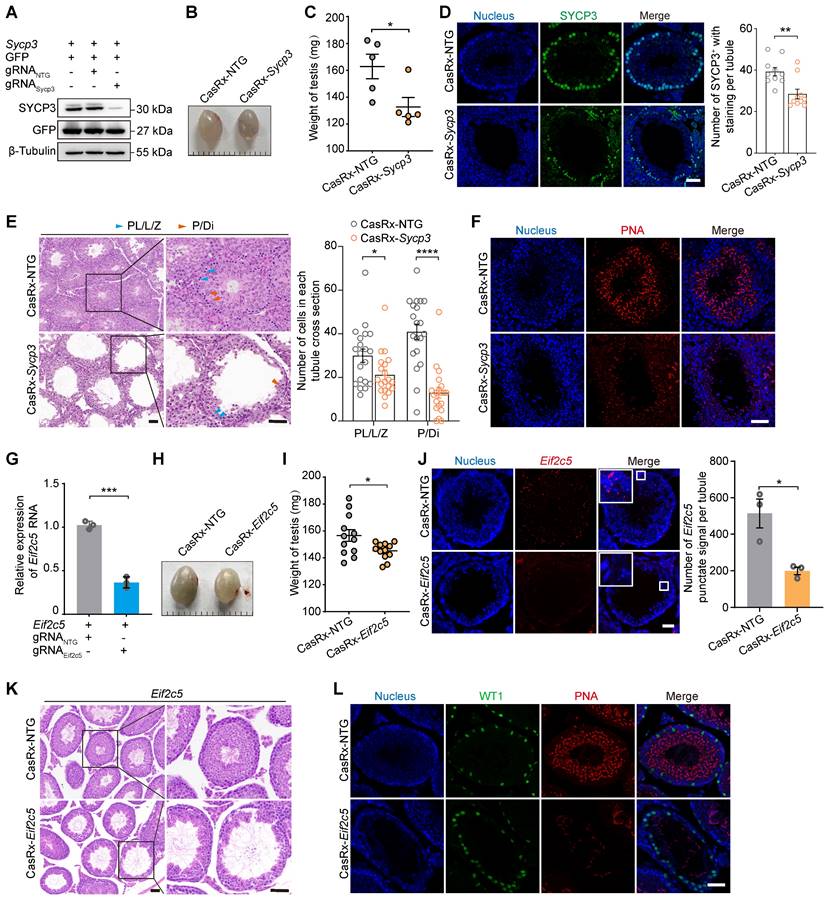
RNAi-based methods (siRNA or shRNA) are often more effective against RNA localized to the cytoplasm than that localized to the nucleus [29]. Therefore, we next sought to determine whether CRISPR-CasRx could achieve specific and efficient knockdown of nuclear RNA in the testis in vivo. To this end, we targeted a nuclear lncRNA of metastasis-associated lung adenocarcinoma transcript 1 (Malat1), a gene previously reported to have no impact on reproductive development [30]. We used qRT-PCR to show that CRISPR-CasRx decreased lncRNA-Malat1 levels by ~70% in GC1-spg cells and by ~50% in the testis in vivo, indicating that this approach achieved successful knockdown of an RNA localized to the nucleus (Figure S3C-D). Our previous work had revealed the important regulatory role of lncRNA-Eif2c5 in mouse spermatogenesis [12]. We therefore designed gRNAs targeting lncRNA-Eif2c5 and examined how well CRISPR-CasRx knocked down its expression using qRT-PCR; we observed a ~60% decrease in HEK293T cells and a ~40% decrease in the testis in vivo (Figure 4G; Figure S3E). Additionally, we found that CasRx-mediated Eif2c5 knockdown (CasRx-Eif2c5) reduced testis weight by ~25% relative to that of the CasRx-NTG control (Figure 4H-I). We next performed RNAscope in situ hybridization to evaluate the knockdown efficiency of Eif2c5 and found that CRISPR-CasRx visibly decreased the Eif2c5 punctate signal (~60%) (Figure 4J). H&E staining of testicular sections revealed that the CasRx-Eif2c5-treated testes exhibited severe morphological defects and had markedly fewer germ cells in their seminiferous tubules than the control testes (Figure 4K). Immunofluorescence staining of testicular sections from mice injected with CasRx-Eif2c5 for PNA showed a significant reduction in the number of post-meiotic germ cells, indicative of disrupted spermatogenesis (Figure 4L). Collectively, these results demonstrate that the CRISPR-CasRx system can be delivered to the testis to effectively knock down endogenous spermatogenic genes, mRNA, and lncRNA, and therefore be used as a tool for studying gene function during spermatogenesis in vivo.
AAV-mediated expression of the Sycp1 promoter drives transgene expression in meiotic germ cells in vivo
The targeted delivery of genes to specific cell types is a critical aspect of AAV9-mediated gene therapy and regulation [31]. Our next objective was therefore to screen germ-cell-specific gene promoters, which drive transgene expression, in distinct germ cell types in vivo. To this end, we generated four independent AAV expression vectors carrying germ-cell-specific gene promoter fragment fused to an RFP-reporter-encoding gene to generate AAV9-Stra8-RFP, AAV9-Hspa2-RFP, AAV9-Sycp1-RFP, and AAV9-Pgk2-RFP [32-35]. These vectors were then packaged into AAV9 and used in testicular microinjection experiments, which measured the pattern of RFP expression in distinct germ cell types (Figure 5A). A strong RFP fluorescence signal was detected by fluorescence stereomicroscopy in the testis treated with AAV9-Stra8-RFP, AAV9-Hspa2-RFP, and AAV9-Sycp1-RFP at 3 weeks after microinjection; meanwhile, only weak RFP fluorescence intensity was observed in the testes treated with AAV9-Pgk2-RFP (Figure 5B). To further determine whether these promoters could drive transgene expression specifically in germ cells, we examined the distribution of RFP fluorescence in testicular sections subjected to immunofluorescence staining for RFP and WT1. Notably, we observed that RFP expression driven by AAV9-Sycp1-RFP was germ cell-specific, mainly in elongated spermatids and sperm at 3 weeks after microinjection. In contrast, AAV9-Stra8-RFP, AAV9-Hspa2-RFP and AAV9-Pgk2-RFP unexpectedly induced RFP expression in both germ and Sertoli cells (Figure 5C). Next, we characterized the temporal and spatial expression pattern of RFP fluorescence, driven by AAV9-Sycp1-RFP, during germ cell development in vivo. The fluorescence stereomicroscopy results showed a gradual increase in RFP fluorescence intensity in the testis on day 7 after microinjection of AAV9-Sycp1-RFP, which peaked on day 14 and gradually decrease until day 21 (Figure 5D). Immunofluorescence staining for DDX4 and WT1 showed that AAV9-Sycp1-RFP initiated RFP expression from day 5 to day 21 after microinjection, which persisted in spermatocytes, round spermatids, and elongating spermatids (Figure 5E).
AAV9 induces the Sycp1 promoter to drive transgene expression in germ cells in vivo. (A) Schematic illustration of the process used to screen for germ-cell-specific gene promoters that drive RFP reporter gene expression in distinct germ cell types in vivo; four independent germ-cell-specific gene promoter (i.e., Stra8, Hspa2, Sycp1, and Pgk2) were individually fused to the RFP reporter gene, packaged into AAV9, and then microinjected into the seminiferous tubule of testis of 3-week-old mice. (B) Stereomicroscopic fluorescence imaging of representative testes microinjected with AAV9-Stra8-RFP, AAV9-Hspa2-RFP, AAV9-Sycp1-RFP, or AAV9-Pgk2-RFP. Scale bar: 1 mm. (C) Immunostaining of testicular sections from testes received microinjection with AAV9-Stra8-RFP、AAV9-Hspa2-RFP、AAV9-Sycp1-RFP and AAV9-Pgk2-RFP for WT1 (green) and RFP (red). Nuclei were stained with DAPI (blue). Scale bar: 50 μm. (D) Representative stereomicroscopic fluorescent images of testes on days 5, 7, 14, 21, and 28 after microinjection of AAV9-Sycp1-RFP. Scale bar: 1 mm. (E) Immunostaining of testicular sections on days 5, 7, 14, 21, and 28 after microinjection of AAV9-Sycp1-RFP for WT1 (yellow), DDX4 (green), and RFP (red). Nuclei were stained with DAPI (blue). Scale bar: 50 μm.
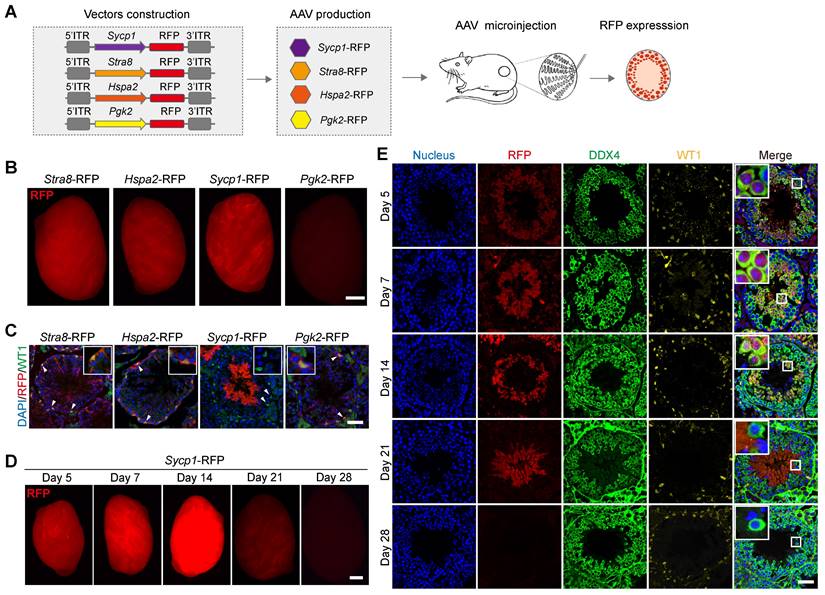
We next wondered whether the AAV9-mediated germ-cell-specific transgene expression was driven by the Sycp1 promoter; Thus, we generated an AAV expression vector carrying the Sycp1 promoter fragment fused to a Cre recombinase gene (AAV9-Sycp1-Cre), packaged it into AAV9, and microinjecting it into the testis of 3-week-old Ai9 mice (Rosa26-CAG-LSL-tdTomato); we then characterized the temporal and spatial expression pattern of tdTomato fluorescence in the testis (Figure S4A). Using fluorescence stereomicroscopy, we observed that tdTomato fluorescence intensity gradually increased at day 7 post-microinjection, peaked on day 21, and significantly decreased by day 28 (Figure S4B). Next, we performed immunofluorescence staining of testicular sections for DDX4 and WT1 to determine the distribution of tdTomato fluorescence in distinct germ cell types. The tdTomato fluorescence was continuously detected from day 7 to day 21 after microinjection and persisted in spermatocytes, round spermatids, and elongating spermatids (Figure S4C). Taken together, these results demonstrate that the AAV9-mediated delivery of the germ-cell-specific Sycp1 promoter could successfully drive transgene expression specifically in germ cells.
The germ-cell-specific Sycp1 promoter controls CRISPR-CasRx efficiently and induces reporter mRNA knockdown in germ cells in vivo
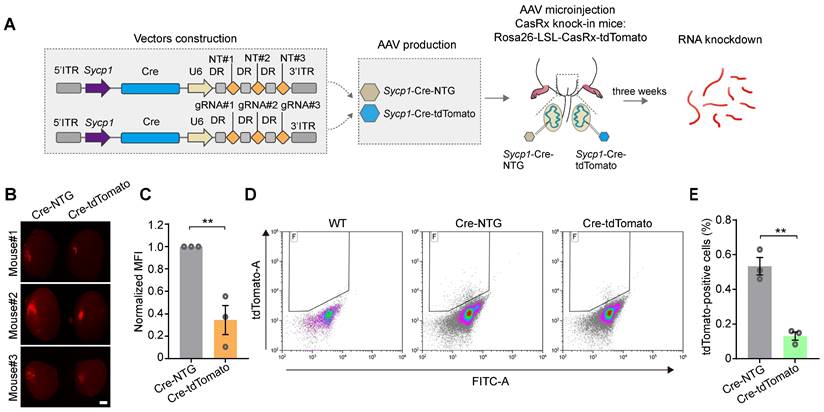
We next aimed to test the ability of the Sycp1 promoter to initiate the CRISPR-CasRx-mediated knockdown of RNA specifically in germ cells in vivo. We therefore constructed a tdTomato-targeting vector (AAV9-Sycp1-Cre-tdTomato) and the AAV9-Sycp1-Cre-NTG control, before microinjecting them into the testes of 3-week-old Cre-dependent CasRx knock-in mice (Rosa-CAG-LSL-CasRx-tdTomato-WPRE); in these assays, the CasRx was linked to tdTomato via a self-cleaving P2A peptide (Figure 6A). The knockdown efficiency of CasRx was determined by measuring germ-cell-specific tdTomato expression 3 weeks after microinjection. Fluorescence stereomicroscopy showed that the MFI of the testes treated with AAV9-Sycp1-Cre-tdTomato was visibly reduced (by ~60%) relative to that of the AAV9-Sycp1-Cre-NTG control (Figure 6B-C). Moreover, flow cytometry showed that the AAV9-Sycp1-Cre-tdTomato vector induced a ~5-fold decrease in the percentage of tdTomato+ germ cells relative to that in the control group (Figure 6D-E). Together, our results suggest that the Sycp1-promoter-initiating CRISPR-CasRx system effectively knocks down target genes in a germ-cell-type-specific manner.
The AAV9- and Sycp1-mediated induction of the CRISPR-CasRx system achieves RNA knockdown specifically in germ cells. (A) Schematic illustration of reporter RNA knockdown by Sycp1-induced CRISPR-CasRx. Vector#3 carrying the Sycp1 promoter encodes the Cre recombinase and non-targeting gRNAs (AAV9-Sycp1-Cre-NTG); Vector#4 carrying the Sycp1 promoter encodes the Cre recombinase and tdTomato-targeting gRNAs (AAV9-Sycp1-Cre-tdTomato). The testes from a Cre-dependent CasRx knock-in mice (Rosa-CAG-LSL-CasRx-tdTomato-WPRE) were microinjected with AAV9-Sycp1-Cre-NTG or AAV9-Sycp1-Cre-tdTomato. (B) Representative stereomicroscopic fluorescent images of testes from CasRx-knock-in mice microinjected with AAV9-Sycp1-Cre-NTG or AAV9-Sycp1-Cre-tdTomato. Scale bar: 1 mm. (C) Mean fluorescence intensity (MFI) of tdTomato in testes of CasRx knock-in mice microinjected with AAV9-Sycp1-Cre-NTG or AAV9-Sycp1-Cre-tdTomato. Data were normalized to the tdTomato-non-targeting condition (n = 3). (D and E) Representative flow cytometry dot plots (D) and the corresponding quantification of the percentage of tdTomato+ cells (E) in the testes of CasRx knock-in mice microinjected with AAV9-Sycp1-Cre-NTG or AAV9-Sycp1-Cre-tdTomato. Data were normalized to the tdTomato-non-targeting condition (n = 3). All data are presented as the mean ± SEM. P-values were determined using the two-tailed Student's t-test; *P < 0.05; **P < 0.01.
Discussion
In the present study, we optimized the in vivo AAV9-mediated delivery of CRISPR-CasRx into the testis and showed that the system induced effective exogenous and endogenous RNA depletion in this organ without adverse effects.
AAV has been identified as a versatile vehicle for transgene delivery because of its ability to transduce a variety of cell types, its lack of apparent pathogenicity, its ability to achieve long-term transgene expression without integrating into the host genome, and its overall safety [36]. Previous reports have established that AAV9 preferentially infects Sertoli cells; however, its ability to transduce spermatogenic cell in the testis, particularly spermatids, remains limited [25]. A possible explanation is that autophagy plays a protective role in spermatogenic cells. Autophagy is an intracellular catabolic process, which degrades and recycles proteins and organelles, thereby facilitating cellular homeostasis and survival [37]. Autophagy also plays a significant role in host defense against microbial infection, including antiviral defense [38]. As such, autophagy combats infections with RNA and DNA viruses by degrading viral components. Additionally, spermatogenesis itself is accompanied by increased levels of autophagy, which may potentially further reduce the efficiency of transducing spermatogenic cells with AAV9 [39]. Given these factors, we speculated that inhibiting autophagy during spermatogenesis may improve the AAV9 transduction efficiency of spermatogenic cells. To test this hypothesis, we used the common autophagy inhibitor 3-MA to show that inhibiting autophagy indeed improved the in vivo AAV9 transduction efficiency of spermatogenic cells. Additional experiments will be required to understand the mechanisms underlying this 3-MA-mediated improvement in AAV9 transduction efficiency, which could also be applicable to organs other than the testis.
Long-term gene modulation and safety are important considerations for future clinical investigation of AAV-mediated transgene delivery. Hence, it was essential to study the transduction kinetics, organ distribution, and safety of AAV9 and 3-MA co-administration in vivo. Our findings demonstrated that AAV9-mediated transgene expression could be detected at 1 week and peaked at 3-4 weeks after microinjection. Additionally, AAV9 and 3-MA co-injection induced efficient and long-term gene expression (> 15 weeks) in the testis, which followed a rhythmic expression pattern. We surmised that this 5-weekly cycling of gene expression synchronized with the spermatogenic wave of mice. Importantly, the co-injection of AAV9 and 3-MA did not substantially influence the integrity of the BTB or the testicular microenvironment. These results suggested that AAV9 and 3-MA could be used efficiently and safely to deliver transgenes into the testis in vivo.
Spermatogenesis is a tightly controlled process, which relies on the precise regulation of specific transcriptional programs at each stage of development [40]. Previous studies have proposed the role of lncRNAs in spermatogenesis and emphasized the significance of experimentally disrupting RNA activity in vivo as a means of understanding their biological functions in this process [41, 42]. Despite these promising findings, the functional roles of many lncRNAs in spermatogenesis remain unclear, partly due to the lack of effective tools to perturb lncRNA activity (and particularly that of nuclear lncRNAs) in the testis in vivo. The CRISPR-CasRx system provides a highly specific and efficient method to deplete specific RNA transcripts in plants, Drosophila, mammalian cell lines, as well as in murine neurons, liver, and embryos [8, 16, 24, 43, 44]. Despite the broad applicability of the CRISPR-CasRx system, the ability to perturb RNA function in the testis in vivo requires further evaluation. In this study, we generated an all-in-one AAV delivery vehicle comprising a gRNAs array and an mRNA encoding the CasRx protein, and demonstrated that AAV9-mediated CRISPR-CasRx induced the degradation of specific RNA transcripts, which exhibits superior RNA knockdown efficiency and specificity to those of RNAi in the testis in vivo. To rule out the possibility that RNA transcript depletion arose as a result of decreased cell numbers, caused by the disruption of functional gene expression, we tested the knockdown efficiency and specificity of CasRx for a tdTomato fluorescence reporter transiently overexpressed in testis in vivo. The rationale for selecting tdTomato was that, theoretically, its expression would not have a significant effect on cell maintenance. We showed that the AAV9-mediated CRISPR-CasRx system successfully knocked down tdTomato reporter mRNA expression without significantly altering the expression levels of typical functional genes in the testis. Furthermore, by targeting the nuclear lncRNA-Eif2c5, we established that the CRISPR-CasRx system could effectively knock down both cytoplasmic and nuclear RNAs and was therefore a useful tool for studying gene function in vivo during germline development. In this regard, the AAV9-mediated CRISPR-CasRx system is a promising molecular tool for transcriptome engineering and offers several benefits when used in the testis in vivo. For example, the versatility and simplicity of this tool enable the large-scale screening of functional RNAs involved in spermatogenesis before investing considerable effort in investigating the molecular functions of a select few candidates in a knockout animal model. Moreover, local RNA knockdown but not permanent DNA knockout is ideal for studying target genes, the deletion of which may lead to early embryonic lethality, as well as minimizing multiple off-target effects which could result from the deletion of an entire gene locus (e.g., in the case of lncRNAs). Moreover, the robust RNA cleavage property of CRISPR-CasRx makes it a highly promising method for the treatment of testicular infections caused by single-stranded RNA viruses, such as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) or Zika virus. Infections with these RNA viruses may induce inflammation, damage testicular tissue, and perturb spermatogenesis [45-47]. Recently, CRISPR-Cas13 has been shown to effectively interfere with multiple SARS-CoV-2 variants in vitro [48]. Our study validated the fact that AAV9-mediated CRISPR-CasRx effectively targeted RNA transcripts in the testis in vivo, further showing the potential of CRISPR-CasRx in RNA-targeting therapies.
The cell-type-specific activation of the CRISPR system has been an ongoing challenge, due to the need to ensure both the safety and efficacy of its in vivo application [49]. One strategy for achieving germ-cell-type-specific, CRISPR-mediated gene editing involves employing a germ-cell-specific gene promoter. We selected four genes, Stra8, Hspa2, Sycp1 and Pgk2, for the screening of germ-cell-specific promoters. Among them, the promoter of Stra8, Hspa2 and Pgk2 genes induced RFP expression in both germ and Sertoli cells. We tend to believe that such result was due to a partial promoter fragment instead of the complete promoter sequence as well as absence of the negative cis element, which led to non-specific expression of initiating RFP gene. Our work revealed that the promoter of a germ-cell-specific gene, Sycp1, successfully drove transgene expression in meiotic germ cells in vivo. Moreover, we showed that an AAV9 vector carrying the Sycp1 promoter was able to maintain long-term transgene expression (up to 3 weeks) in the testis in vivo. The ability to efficiently induce target gene knockdown in a germ-cell-type-specific manner underscores the potential of this approach as a therapeutic application in vivo.
In summary, we have optimized the AAV9-mediated delivery of the CRISPR-CasRx system to the testis and demonstrated the feasibility of this approach in efficiently and safely knocking down endogenous and exogenous RNA expression in the testis in vivo. Moreover, we achieved germ-cell-specific, CRISPR-CasRx-mediated RNA knockdown by including the Sycp1 promoter in our vector construct. We envisage that these developments will facilitate transcriptome-related investigations and the development of RNA-targeting therapies for reproduction health conditions.
Methods
Plasmid construction
All AAV expression vectors used for packaging in AAV9 were constructed using an AAV-based cloning vector backbone (pAAV.H1.CMV.RFP). To construct the all-in-one AAV expression vector for the pre-gRNAs array and the mRNA-encoding CasRx protein (AAV9-EFS-CasRx-U6-gRNA), the NLS-CasRx-NLS and DNA sequences were PCR-amplified from the plasmids expressing CasRx (Addgene, #109049) or pre-gRNA scaffold (Addgene, #109054), respectively. These cassettes were then cloned into an AAV-based cloning vector using single-step Gibson assembly with the NEBuilder® HiFi DNA Assembly Master Mix (NEB, Cat# E2621L), according to the manufacturer's instructions. The sequences of oligonucleotides used to construct the gRNAs array were synthesized by Beijing Tianyi Huiyuan Biotechnology and then cloned into the linearized AAV9-EFS-CasRx-U6-gRNA plasmid using the BplI restriction site. The gRNAs were designed using the cas13design tool [18]. The sequences of gRNAs used in this study are provided in Table S1.
To construct the AAV reporter plasmids carrying the germ-cell-specific gene promoter fused to an RFP reporter (AAV9-Stra8-RFP, AAV9-Hspa2-RFP, AAV9-Sycp1-RFP, and AAV9-Pgk2-RFP), the following promoter fragments were PCR-amplified from mouse genomic DNA: -772/+102 of Sycp1, -371/+29 of Stra8, -932/-225 of Hspa2, and -515/0 of Pgk2. Next, these fragments were individually cloned into the AAV-based cloning vector using single-step Gibson assembly. The primer sequences for the germ-cell-specific gene promoters are listed in Table S2.
To construct the AAV expression vector AAV9-Sycp1-Cre-U6-gRNAs, the U6-DRs-gRNAs DNA sequence was PCR-amplified from the plasmid expressing the pre-gRNA scaffold (Addgene, #109054); meanwhile, the cyclization recombination enzyme (Cre) DNA sequence was PCR-amplified from the genomic DNA of the Cre-transgenic mouse. These cassettes were then cloned into the linearized AAV9-Sycp1-RFP plasmid via BamHI and XhoI using single-step Gibson assembly. The primer sequences used for Cre amplification are listed in Table S2.
Cell culture and transfections
HEK293T cells and GC1-spg cells were obtained from the Cell Resource Center, Peking Union Medical College. HEK293T cells and GC1-spg cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Corning) containing 10% fetal bovine serum (FBS), supplemented with 1% penicillin-streptomycin, in a humidified 5% CO2 incubator at 37 °C.
To determine the knockdown efficiency of the tdTomato reporter, Sycp3, and lncRNA-Eif2C5, mediated by CRISPR-CasRx, an average of 5 × 105 HEK293T cells were seeded into 6-well plates and transfected with 1 μg of AAV expression vector expressing the target gene and 1.5 μg of the all-in-one AAV CRISPR-CasRx expression vector (AAV9-EFS-CasRx-U6-gRNA). Transfection was achieved using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer's protocol. The transfected cells were harvested 48 h post-transfection and subjected to qRT-PCR, western blotting, and fluorescence imaging.
To determine the knockdown efficiency of lncRNA-Malat1 by CRISPR-CasRx, an average of 5 × 105 GC1-spg cells were seeded into 6-well plates and infected with a lentivirus expressing CasRx and gRNAs targeting lncRNA-Malat1. GC1-spg cells were harvested 48 h post-infection and sorted by flow cytometry for qRT-PCR analysis.
AAV9 packaging
AAV-HEK293 cells (HEK293 cells stably expressing the E1 gene region) were obtained from the Cell Resource Center. AAV-HEK293 cells were cultured in DMEM containing 10% FBS supplemented with 1% penicillin-streptomycin in a humidified 5% CO2 incubator at 37 °C. AAV-HEK293 cells were plated in 150-mm culture dishes and grown to reach 70% confluency. The AAV vector plasmids, pAAV2/9, and the adenoviral helper plasmids, were transfected at a ratio of 1:1:1 in serum-free Opti-MEM (Gibco, Cat# 31985070) using PEI Max at a DNA:PEI ratio of 1:2. The culture cells and supernatant were collected 120 h after transfection. Subsequently, the crude AAV9 fraction was treated with 50 units/mL UltraNuclease (YEASEN, Cat# 20156) for 30 min at 37 °C, and then with 5 M NaCl at 50 °C for 30 min. The pellet debris was then removed by centrifugation at 12,000 ×g for 30 min. Next, AAVs were purified by gradient centrifugation using 9, 6, 5, and 5 mL of 15%, 25%, 40%, and 54% iodixanol, respectively. After ultracentrifugation at 350,000 ×g for 90 min, 4 mL of the transparent layer were extracted into PBS and the viral particles were obtained. AAV titers (GC/mL) were determined by qRT-PCR. After viral titration (>1013 GC/mL), the AAV was aliquoted and stored at -80 °C until used.
Animals and microinjection
For the in vivo experiments, male CD-1/ICR mice were purchased from Beijing Sibeifu Biotechnology Co. Ltd. Ai9 mice (Rosa26-CAG-LSL-tdTomato) were obtained from the Shanghai Model Organisms Center. CasRx knock-in mice (Rosa-CAG-LSL-CasRx-tdTomato-WPRE) were a gift from Professor Yu Nie, Peking Union Medical College [50]. Male mice (3-week-old) were used in the AAV9 or siRNA microinjection experiments performed in this study, unless otherwise specified. Microinjection was conducted as described in our previous study [14]. Briefly, ~10 µL of viral particles containing 0.04% Trypan blue were injected via the efferent duct into the seminiferous tubules. To improve transduction efficiency of AAV9, 3-MA (at a final concentration of 10 mM) was co-injected with the virus. A dual-virus model was employed to confirm the knockdown efficiency and specificity of the CRISPR-CasRx system for the exogenous reporter tdTomato. The dual-viral particle ratio of AAV9-EFS-tdTomato to AAV9-EFS-CasRx-NTG or AAV9-EFS-CasRx-tdTomato was 1:2. For siRNA microinjection, siRNA pool (Ribobio, Guangzhou, China) chemically modifed with 2'-OM (2-methoxyethyl) and cholesterol was used for the exogenous reporter tdTomato knockdown:
tdTomato siRNAs (target sequences): 5'-AGCGCGTGATGAACTTCGA-3', 5'-AAGAGTTCATGCGCTTCAA-3', 5'-CACGAGTTCGAGATCGAGG-3'.
Microinjection of siRNA (100 µM) was operated as the CRISPR-CasRx condition. The ratio between AAV9-EFS-tdTomato and siRNA pool was 1:2. All experimental procedures were approved by the Animal Experimental Ethical Inspection of Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences and Peking Union Medical College.
Tracer experiment
The tracer experiment was performed as previously described [25]. Approximately 15 μL Sulfo-NHS-LC-biotin (7.5 mg/mL; Thermo Fisher Scientific, Cat# A39257) was interstitially injected into the testis 3 weeks after the AAV injection. The mice were euthanized 30 min post-injection and their testes were immediately collected, and fixed in 4% paraformaldehyde (PFA) overnight at 4 °C. The testis samples were then embedded in the Tissue-Tek OCT compound (Sakura Finetek, Cat# 4583) for 7-μm-thick cryosections. The sections were incubated with Alexa Fluor 488-conjugated streptavidin (Thermo Fisher Scientific, Cat# S11223) and counterstained using Fluoroshield mounting medium (Sigma-Aldrich, Cat# F6057). Fluorescent images were captured using a Zeiss LSM780 confocal microscope.
In vivo animal imaging
In vivo image acquisition and analysis were performed weekly after the AAV injections using the Xenogen IVIS spectrum imaging system. Each mouse was anesthetized with 2% isoflurane and placed in the prone position in the bioluminescent imager. The fluorescent intensity was measured and quantified at different time-points. Spectral unmixing was used for image reconstruction. The region of interest (ROI), encompassing the whole testis, was manually outlined on the fused images for use in further analyses of radiant efficiency and to compare different time-points. Each mouse was imaged on a weekly basis for up to 15 weeks after AAV microinjection.
RNA extraction and quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from the testes 3-4 weeks after AAV microinjection using the TRIzol reagent (Thermo Fisher Scientific, Cat# 15596018) according to the manufacturer's instructions. cDNA was synthesized using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Cat# K1622) and processed for qRT-PCR using the PerfectStart® Green qPCR SuperMix (TransGen, Cat# AQ601) on the CFX Connect System. Relative expression levels of target genes were quantified using the delta-delta Ct (ΔΔCt) method. The data were normalized to Gapdh or Actb expression. Primer sequences used in this study are listed in Table S3.
Western blotting
The samples lysates were prepared in 1 × sodium dodecyl sulfate buffer supplemented with protease inhibitor cocktail. Protein concentration was measured using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Cat# 23227). Protein samples were separated by SDS-PAGE electrophoresis using 10% polyacrylamide gels (EpiZyme Biotechnology, Cat# PG112) and then transblotted onto polyvinylidene difluoride (PVDF) membranes. After blocking with 5% skimmed milk for 1 h at room temperature (RT), the membranes were incubated with the primary antibodies overnight at 4 °C. After a washing step with 1× TBST, the membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h at RT. Protein blots were visualized using the Fusion FX EDGE chemiluminescence imaging system.
RNA sequencing
Sequencing libraries were generated using the VAHTS mRNA-seq v2 Library Prep Kit for Illumina by following manufacturer's recommendations. Next, library quantification was performed using the Qubit high sensitivity assay, while quality control was conducted using the Agilent 2100 bioanalyzer or Fragment Analyzer 5300; the final library has an average size of ~350 bp. The libraries were sequenced on an Illumina NovaSeq platform to generate 150-bp paired-end reads. Sequence reads were mapped to the mouse genome (version mm10) using the HISAT2 (v2.0.0). Differentially expressed genes were defined as those with an adjusted P-value (Padj) < 0.05 and a log2 fold change (log2FC) > 2 using the DEGSeq R package (v1.20.0).
Flow cytometry
Testicular cells were isolated from the testes received microinjection of AAV9 as previously described [14, 51]. In brief, the testes were decapsulated and incubated with collagenase type IA (0.5 mg/mL) at 37 °C for 15 min to separate the interstitial cells and seminiferous tubules. After a mincing step, the seminiferous tubules were digested with collagenase type IV (1 mg/mL) and DNase I (500 μg/mL) at 37 °C for 15 min with gentle oscillation. The collagenase digestion process was stopped using DMEM containing 10% FBS. For germ cell isolation, the testicular cells were cultured in F12/DMEM containing 10% FBS for 6 h at 37 °C and then recovered by collecting nonadherent cells. The cell suspensions were then filtered through a 40-mm nylon mesh and re-suspended in PBS containing 2% FBS for flow cytometry analysis and RFP or tdTomato-positive cell sorting on a MA900 Multi-Application Cell Sorter (SONY). A total of 105 events were recorded per sample and the data were analyzed using Kaluza Analysis Software (BECKMAN).
Immunofluorescence staining and the RNAscope in situ hybridization assay
Testes from AAV9-microinjected mice were dissected, and fixed in 4% PFA overnight at 4 °C. After cryoprotection in sucrose-PBS solution (10% sucrose for 1 h, 20% sucrose for 1 h, and 30% sucrose overnight) at 4 °C the testes were embedded in the Tissue-Tek OCT compound and sectioned into 7-μm-thick cryosections for subsequent staining. After a blocking step in 5% bovine serum albumin with 0.1% Triton X-100 for 1 h at RT, the cryosections were incubated with primary antibodies overnight at 4 °C. After washing with PBS, the sections were incubated with an AlexaFluor™-conjugated secondary antibody for 1 h at RT. Subsequently, the sections were mounted using Fluoroshield mounting medium (Sigma-Aldrich, Cat# F6057) and the fluorescent images were visualized using a confocal microscope (LSM780). Number of cells with staining were calculated using image analysis software Image J. The primary antibodies and secondary antibodies used in this study are listed in Table S4.
For RNAscope in situ hybridization, the Multiplex Fluorescent Manual Assay Kit and RNAscope® Probe-Ms-Eif2c5 were purchased from Advanced Cell Diagnostics (ACD; Hayward, CA, USA). The pretreatment of testicular sections, target retrieval, protease plus treatment, target probe hybridization, signal amplification, counterstaining, and mounting were performed according to the manufacturer's instructions. Fluorescent signals were detected on a Zeiss LSM780 confocal microscope. Quantification of Eif2c5 punctate signal per tubule were calculated using image analysis software Image J.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 7. All experiments were independently repeated at least three times, and experimental data were represented as mean ± SEM. Statistically significant differences (P < 0.05) were assessed using two-tailed Student's t-tests as indicated in figures and figure legends.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This study was supported by grants from the National Key Research and Development Program of China (grant number: 2022YFA0806302), the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (grant numbers: 2021-I2M-1-019 and 2021-I2M-1-066), the National Natural Science Foundation of China (grant numbers: 92268111, 32370910, 31970794, and 32000586), and the State Key Laboratory Special Fund (grant number: 2060204).
Author contributions
W.S. and K.L. conceptualized and supervised the project. K.L., M.L. and Y.L. conducted the experiments and data analysis; M.L. and Y.L. performed testicular microinjection; D.Z., Y.L., X.M., Z.Z., P.L. and Y.L. provided technical assistance; S.M. provided critical suggestions on this work. W.S., K.L., M.L. and Y.L. wrote the draft.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Soumillon M, Necsulea A, Weier M, Brawand D, Zhang X, Gu H. et al. Cellular source and mechanisms of high transcriptome complexity in the mammalian testis. Cell Rep. 2013;3:2179-2190
2. Green CD, Ma Q, Manske GL, Shami AN, Zheng X, Marini S. et al. A Comprehensive Roadmap of Murine Spermatogenesis Defined by Single-Cell RNA-Seq. Dev Cell. 2018;46:651-677.e10
3. Hermann BP, Cheng K, Singh A, Roa-De La Cruz L, Mutoji KN, Chen IC. et al. The Mammalian Spermatogenesis Single-Cell Transcriptome, from Spermatogonial Stem Cells to Spermatids. Cell Rep. 2018;25:1650-1667.e8
4. Nie X, Munyoki SK, Sukhwani M, Schmid N, Missel A, Emery BR. et al. Single-cell analysis of human testis aging and correlation with elevated body mass index. Dev Cell. 2022;57:1160-1176.e5
5. Wang M, Liu X, Chang G, Chen Y, An G, Yan L. et al. Single-Cell RNA Sequencing Analysis Reveals Sequential Cell Fate Transition during Human Spermatogenesis. Cell Stem Cell. 2018;23:599-614.e4
6. Xia B, Yan Y, Baron M, Wagner F, Barkley D, Chiodin M. et al. Widespread Transcriptional Scanning in the Testis Modulates Gene Evolution Rates. Cell. 2020;180:248-262.e21
7. Miyata H, Castaneda JM, Fujihara Y, Yu Z, Archambeault DR, Isotani A. et al. Genome engineering uncovers 54 evolutionarily conserved and testis-enriched genes that are not required for male fertility in mice. Proc Natl Acad Sci U S A. 2016;113:7704-7710
8. Kushawah G, Hernandez-Huertas L, Abugattas-Nunez Del Prado J, Martinez-Morales JR, DeVore ML, Hassan H. et al. CRISPR-Cas13d Induces Efficient mRNA Knockdown in Animal Embryos. Dev Cell. 2020;54:805-817.e7
9. Bassett AR, Akhtar A, Barlow DP, Bird AP, Brockdorff N, Duboule D. et al. Considerations when investigating lncRNA function in vivo. Elife. 2014;3:e03058
10. Ransohoff JD, Wei Y, Khavari PA. The functions and unique features of long intergenic non-coding RNA. Nat Rev Mol Cell Biol. 2018;19:143-157
11. Tang XE, Tan SX, Hoon S, Yeo GW. Pre-existing adaptive immunity to the RNA-editing enzyme Cas13d in humans. Nat Med. 2022;28:1372-1376
12. Li K, Zhong S, Luo Y, Zou D, Li M, Li Y. et al. A long noncoding RNA binding to QKI-5 regulates germ cell apoptosis via p38 MAPK signaling pathway. Cell Death Dis. 2019;10:699
13. Legnini I, Di Timoteo G, Rossi F, Morlando M, Briganti F, Sthandier O. et al. Circ-ZNF609 Is a Circular RNA that Can Be Translated and Functions in Myogenesis. Mol Cell. 2017;66:22-37.e9
14. Li K, Xu J, Luo Y, Zou D, Han R, Zhong S. et al. Panoramic transcriptome analysis and functional screening of long noncoding RNAs in mouse spermatogenesis. Genome Res. 2021;31:13-26
15. Chen ZH, Ling L, Shi XL, Li W, Zhai HC, Kang ZL. et al. Microinjection of antisense oligonucleotides into living mouse testis enables lncRNA function study. Cell Biosci. 2021;11:213
16. Konermann S, Lotfy P, Brideau NJ, Oki J, Shokhirev MN, Hsu PD. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell. 2018;173:665-676.e14
17. Li SQ, Li X, Xue W, Zhang L, Yang LZ, Cao SM. et al. Screening for functional circular RNAs using the CRISPR-Cas13 system. Nat Methods. 2021;18:51-59
18. Wessels HH, Mendez-Mancilla A, Guo XY, Legut M, Daniloski Z, Sanjana NE. Massively parallel Cas13 screens reveal principles for guide RNA design. Nat Biotechnol. 2020;38:722-727
19. He BB, Peng WB, Huang J, Zhang H, Zhou YS, Yang XL. et al. Modulation of metabolic functions through Cas13d-mediated gene knockdown in liver. Protein Cell. 2020;11:518-524
20. Granados-Riveron JT, Aquino-Jarquin G. CRISPR-Cas13 Precision Transcriptome Engineering in Cancer. Cancer Res. 2018;78:4107-4113
21. Cheng R, Zheng XM, Wang YM, Ma X, Liu X, Xu WJ. et al. Modification of alternative splicing in bovine somatic cell nuclear transfer embryos using engineered CRISPR-Cas13d. Sci China Life Sci. 2022;65:2257-2268
22. Zheng ZW, Li G, Cui C, Wang F, Wang XH, Xu ZJ. et al. Preventing autosomal-dominant hearing loss in Bth mice with CRISPR/CasRx-based RNA editing. Signal Transduct Target Ther. 2022;7:79
23. Guo Y, Han L, Han S, Tang HH, Wang SY, Cui C. et al. Specific knockdown of Htra2 by CRISPR-CasRx prevents acquired sensorineural hearing loss in mice. Mol Ther Nucleic Acids. 2022;28:643-655
24. Powell JE, Lim CKW, Krishnan R, McCallister TX, Saporito-Magrina C, Zeballos MA. et al. Targeted gene silencing in the nervous system with CRISPR-Cas13. Sci Adv. 2022;8:eabk2485
25. Watanabe S, Kanatsu-Shinohara M, Ogonuki N, Matoba S, Ogura A, Shinohara T. In Vivo Genetic Manipulation of Spermatogonial Stem Cells and Their Microenvironment by Adeno-Associated Viruses. Stem Cell Reports. 2018;10:1551-1564
26. Wang LY, Liu C, Wei HF, Ouyang YC, Dong MZ, Zhang RD. et al. Testis electroporation coupled with autophagy inhibitor to treat non-obstructive azoospermia. Mol Ther Nucleic Acids. 2022;30:451-464
27. Wu H, Zhao X, Wang F, Jiang Q, Shi LL, Gong ML. et al. Mouse Testicular cell Type-specific antiviral response against Mumps Virus replication. Front Immunol. 2017;8:117
28. Dai JQ, Voloshin O, Potapova S, Camerini-Otero RD. Meiotic Knockdown and Complementation Reveals Essential Role of RAD51 in Mouse Spermatogenesis. Cell Rep. 2017;18:1383-1394
29. Lennox KA, Behlke MA. Cellular localization of long non-coding RNAs affects silencing by RNAi more than by antisense oligonucleotides. Nucleic Acids Res. 2016;44:863-877
30. Zhang B, Arun G, Mao YS, Lazar Z, Hung GN, Bhattacharjee G. et al. The lncRNA Malat1 Is Dispensable for Mouse Development but Its Transcription Plays a cis-Regulatory Role in the Adult. Cell Rep. 2012;2:111-123
31. Powell SK, Samulski RJ, McCown TJ. AAV Capsid-Promoter Interactions Determine CNS Cell Selective Gene Expression In Vivo. Mol Ther. 2020;28:1373-1380
32. Nayernia K, Li MY, Jaroszynski L, Khusainov R, Wulf G, Schwandt I. et al. Stem cell based therapeutical approach of male infertility by teratocarcinoma derived germ cells. Hum Mol Genet. 2004;13:1451-1460
33. Inselman AL, Nakamura N, Brown PR, Willis WD, Goulding EH, Eddy EM. Heat Shock Protein 2 Promoter Drives Cre Expression in Spermatocytes of Transgenic Mice. Genesis. 2010;48:114-120
34. Vidal F, Sage J, Cuzin F, Rassoulzadegan M. Cre expression in primary spermatocytes: A tool for genetic engineering of the germ line. Mol Reprod Dev. 1998;51:274-280
35. Yang ZS, Yoshioka H, McCarrey JR. Sequence-specific promoter elements regulate temporal-specific changes in chromatin required for testis-specific activation of the Pgk2 gene. Reproduction. 2013;146:501-516
36. Li CW, Samulski RJ. Engineering adeno-associated virus vectors for gene therapy. Nat Rev Genet. 2020;21:255-272
37. Wang M, Zeng L, Su P, Ma L, Zhang M, Zhang YZ. Autophagy: a multifaceted player in the fate of sperm. Hum Reprod Update. 2022;28:200-231
38. Richetta C, Faure M. Autophagy in antiviral innate immunity. Cell Microbiol. 2013;15:368-376
39. Huang Q, Liu YH, Zhang SY, Yap YT, Li W, Zhang D. et al. Autophagy core protein ATG5 is required for elongating spermatid development, sperm individualization and normal fertility in male mice. Autophagy. 2021;17:1753-1767
40. Luk ACS, Chan WY, Rennert OM, Lee TL. Long noncoding RNAs in spermatogenesis: insights from recent high-throughput transcriptome studies. Reproduction. 2014;147:R131-R141
41. Sarropoulos I, Marin R, Cardoso-Moreira M, Kaessmann H. Developmental dynamics of lncRNAs across mammalian organs and species. Nature. 2019;571:510-514
42. Trovero MF, Rodriguez-Casuriaga R, Romeo C, Santinaque FF, Francois M, Folle GA. et al. Revealing stage-specific expression patterns of long noncoding RNAs along mouse spermatogenesis. RNA Biol. 2020;17:350-365
43. Mahas A, Aman R, Mahfouz M. CRISPR-Cas13d mediates robust RNA virus interference in plants. Genome Biol. 2019;20:263
44. Buchman A, Brogan DJ, Sun RC, Yang T, Hsu P, Akbari OS. Programmable RNA Targeting Using CasRx in Flies. CRISPR J. 2020;3:164-176
45. Ma XX, Guan CH, Chen R, Wang YY, Feng SL, Wang RS. et al. Pathological and molecular examinations of postmortem testis biopsies reveal SARS-CoV-2 infection in the testis and spermatogenesis damage in COVID-19 patients. Cell Mol Immunol. 2021;18:487-489
46. Ma WQ, Li SH, Ma SQ, Jia LN, Zhang FC, Zhang Y. et al. Zika Virus Causes Testis Damage and Leads to Male Infertility in Mice. Cell. 2016;167:1511-1524.e10
47. Chen X, Ding JL, Liu M, Xing K, Ye P, Min JX. et al. A Systemic Review and Meta-analysis of the Effect of SARS-CoV-2 Infection on Sperm Parameters. Research. 2022;2022:9835731
48. Zeng LP, Liu YX, Nguyenla XH, Abbott TR, Han MT, Zhu YY. et al. Broad-spectrum CRISPR-mediated inhibition of SARS-CoV-2 variants and endemic coronaviruses in vitro. Nat Commun. 2022;13:2766
49. Alyami MZ, Alsaiari SK, Li YY, Qutub SS, Aleisa FA, Sougrat R. et al. Cell-Type-Specific CRISPR/Cas9 Delivery by Biomimetic Metal Organic Frameworks. J Am Chem Soc. 2020;142:1715-1720
50. Li JC, Zhu DK, Hu SS, Nie Y. CRISPR-CasRx knock-in mice for RNA degradation. Sci China Life Sci. 2022;65:2248-2256
51. Zhu WW, Liu P, Yu LL, Chen QY, Liu ZH, Yan KQ. et al. p204-Initiated Innate Antiviral Response in Mouse Leydig Cells. Biol Reprod. 2014;91:8
Author contact
![]() Corresponding author: Wei Song, email: songweipumc.edu.cn.
Corresponding author: Wei Song, email: songweipumc.edu.cn.