Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results and discussion
Conclusions
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(18):5088-5105. doi:10.7150/thno.26225 This issue Cite
Research Paper
Hypoxia-responsive lipid-poly-(hypoxic radiosensitized polyprodrug) nanoparticles for glioma chemo- and radiotherapy
Lei Hua1,2,3*, Zhen Wang1*, Liang Zhao1*, Honglin Mao1, Guanghui Wang4, Kairuo Zhang2, Xuejiao Liu1, Dongmei Wu5, Yuanlin Zheng5, Jun Lu5 ![]() , Rutong Yu1,2,3
, Rutong Yu1,2,3 ![]() , Hongmei Liu1,2,3
, Hongmei Liu1,2,3 ![]()
1. Institute of Nervous System Diseases, Xuzhou Medical University, Xuzhou, 221002, PR China;
2. Department of Neurosurgery, Brain Hospital, Affiliated Hospital of Xuzhou Medical University, Xuzhou, 221002, PR China;
3. Jiangsu Center for the Collaboration and Innovation of Cancer Biotherapy, Cancer Institute, Xuzhou Medical University, Xuzhou, 221002, PR China;
4. Department of Gynaecology and Obstetrics, Affiliated Hospital of Xuzhou Medical University, Xuzhou, 221002, PR China;
5. Key Laboratory for Biotechnology on Medicinal Plants of Jiangsu Province, School of Life Science, Jiangsu Normal University, Xuzhou, 221116, PR China.
*These authors contributed equally to this work.
Received 2018-3-21; Accepted 2018-9-19; Published 2018-10-6
Abstract

Treatment of malignant glioma is a challenge facing cancer therapy. In addition to surgery, and chemotherapy, radiotherapy (RT) is one of the most effective modalities of glioma treatment. However, there are two crucial challenges for RT facing malignant glioma therapy: first, gliomas are known to be resistant to radiation due to their intratumoral hypoxia; second, radiosensitizers may exhibit a lack of target specificity, which may cause a lower concentration of radiosensitizers in tumors and toxic side effects in normal tissues. Thus, novel angiopep-2-lipid-poly-(metronidazoles)n (ALP-(MIs)n) hypoxic radiosensitizer-polyprodrug nanoparticles (NPs) were designed to enhance the radiosensitizing effect on gliomas.
Methods: In this study, different degrees and biodegradabilites of hypoxic radiosensitizer MIs-based polyprodrug (P-(MIs)n) were synthesized as a hydrophobic core. P-(MIs)n were mixed with DSPE-PEG2000, angiopep-2-DSPE-PEG2000 and lecithin to self-assemble ALP-(MIs)n through a single-step nanoprecipitation method. The ALP-(MIs)n encapsulate doxorubicin (DOX) (ALP-(MIs)n/DOX) and provoke the release of DOX under hypoxic conditions for glioma chemo- and radiotherapy. In vivo glioma targeting was tested in an orthotopic glioma using live animal fluorescence/bioluminescence imaging. The effect on sensitization to RT of ALP-(MIs)n and the combination of chemotherapy and RT of ALP-(MIs)n/DOX for glioma treatment were also investigated both in vitro and in vivo.
Results: ALP-(MIs)n/DOX effectively accumulated in gliomas and could reach the hypoxic glioma site after systemic in vivo administration. These ALP-(MIs)n showed a significant radiosensitizing effect on gliomas and realized combination chemotherapy and RT for glioma treatment both in vitro and in vivo.
Conclusions: In summary, we constructed a lipid-poly-(hypoxic radiosensitized polyprodrug) nanoparticles for enhancing the RT sensitivity of gliomas and achieving the combination of radiation and chemotherapy for gliomas.
Keywords: glioma, radiosensitized polyprodrug, chemo- and radiotherapy, hypoxia-responsive, blood-brain barrier
Introduction
Glioblastoma multiforme (GBM) represents the most aggressive subgroup of malignant gliomas with a median survival of 14.6 months with surgery, radiation, and chemotherapy [1-4]. RT has long been the standard adjuvant approach for glioblastoma, and it remains the primary treatment modality for unresectable glioblastoma. However, there are two crucial challenges of RT for malignant glioma therapy: first, gliomas are known to be more resistant to radiation due to their intratumoral hypoxia [5-7]; second, the lack of target specificity of radiosensitizers may cause lower radiosensitizer concentration in tumor and toxic side effects to normal tissues. Therefore, the development of new approaches to improve RT is necessary for glioma treatment.
Hypoxic cells are common in tumors and are resistant to RT [8, 9]. Hypoxia occurs in the core region of the glioma and is recognized as a major cause of failure of RT. Accumulating evidence shows that hypoxia causes glioma tumoral cells to become radioresistant, and the extent of hypoxia in malignant gliomas before RT is closely correlated with overall survival of patients [10, 11]. Adams and Cooke introduced the concept of hypoxic cell radiosensitizers, which are chemicals that mimic oxygen and thereby enhance radiation damage. These researchers demonstrated that the efficiency of radiosensitization is directly related to electron affinity. Because these drugs mimic the effect of oxygen, they should not increase the toxic effects of RT to the well-oxygenated normal tissues surrounding the tumor [12, 13]. Nitroimidazoles such as 5-nitroimidazole, metronidazole or flagyl have been proposed as hypoxic cell radiosensitizers since they were found to mimic the effect of oxygen during the radiochemical process [14, 15]. MIs as specific radiosensitizers of hypoxic cells have been used in controlled trials to evaluate the possible enhancement of the radiation effect in patients with a malignant glioma [16-19]. However, the results of these clinical trials have generally been disappointing. The most important factor underlying the failure of MIs to achieve a useful advantage is undoubtedly the low plasma concentrations that are achievable with the permitted dose of this neurotoxic drug [20-22]. Hence, the major challenge facing the use of nitroimidazoles for hypoxic cell radiosensitization is development of a method to elevate the drug concentration in the tumor and to minimize their toxic side effects on normal tissues.
Nanocarriers, which have been considered to be emerging vehicles in the field of cancer therapy, can overcome these obstacles [23, 24]. Nanocarriers were designed to overcome hurdles such as the side effects of chemotherapy drugs, to increase the intratumor concentration of the drug and to achieve glioma targeting [25]. Various nanocarriers have been designed to deliver drugs for cancer-targeted therapy, such as liposomes [26-28], micelles [29] and polymersomes [30, 31]. A hypoxic radiosensitizer-prodrug liposome (MLP) was developed in our previous study. MLP was shown to enhance the glioma targeting ability, minimize toxic side effects to normal tissues and increase the RT treatment effects on gliomas. However, assistance lipid molecules were used to comprise the MLP and resulted in a low MIs loading content. Moreover, Shen Y et al. reported that the use of nanoparticles to deliver therapeutic drugs introduces excessive inert material and they may increase toxicity in patients [32].
Recently, nitroimidazoles have been widely utilized in the development of drug carriers due to their hypoxia responsiveness and hydrophobic properties [27, 33, 34]. Gu et al. developed hypoxia-responsive vesicles formed from 2-nitroimidazole-conjugated hyaluronic acid for insulin delivery [33]. Further, 2-nitroimidazole-modified CP [34] and carboxymethyl dextran [35] as hypoxia-responsive nanoparticles were created to deliver doxorubicin (DOX) in order to enhance its drug release rate and increase anticancer therapy effectiveness. Thus, biodegradable nitroimidazole polymers were synthesized as the hydrophobic cores of nanocarriers to increase nitroimidazoles loading in this study.
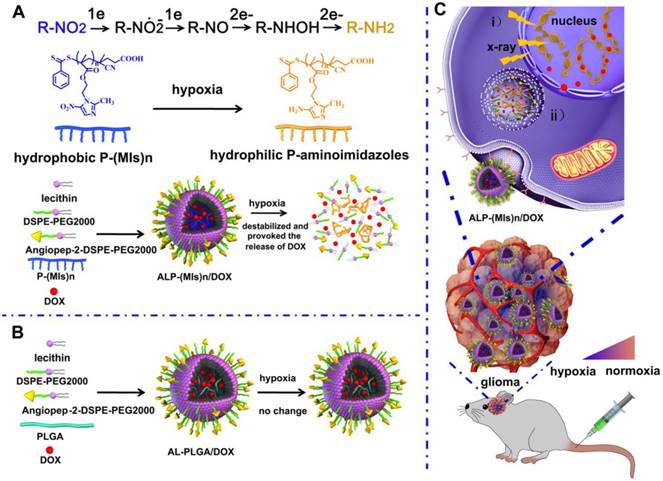
In this study, we designed a novel angiopep-2-lipid-poly(MIs)n (ALP-(MIs)n), which demonstrated radiosensitization effects on hypoxic tumor cells. The ALP-(MIs)n comprises three distinct functional components: 1) a hydrophobic P-(MIs)n core where poorly water-soluble drugs can be encapsulated; 2) nitro groups of the hydrophobic P-(MIs)n core that are converted into hydrophilic amino groups under low oxygen conditions to mimic the oxygen-increased sensitization to RT and to provoke the release of poorly water-soluble drugs; 3) a lipid monolayer at the interface of the core and the shell to modify angiopep-2 (a specific ligand for low-density lipoprotein receptor-related protein-1 (LRP-1) expressed in brain microvascular endothelial cells (BMEC), which primarily form the BBB, and human glioma cells [36-38]), thereby enhancing drug deposition in gliomas. In this paper, DOX, a widely used anticancer drug, was used as a model drug combination with chemotherapy and radiation for glioma treatment. These features make ALP-(MIs)n/DOX an excellent carrier for the specific delivery of DOX concurrent with chemotherapy and RT. Most importantly, the effect of ALP-(MIs)n on sensitization to RT and the combination of chemotherapy and RT using ALP-(MIs)n/DOX for glioma treatment were also investigated both in vitro and in vivo.
After tail vein injection, ALP-(MIs)n/DOX entered the glioma and were later internalized into cells by endocytosis. And then, ALP-(MIs)n/DOX destabilized and rapidly released DOX in the cytoplasm, due to the conversion of hydrophobic P-(MIs)n core to hydrophilic poly-aminoimidazoles under the hypoxic environment via a single-electron reduction catalyzed by intracellular nitroreductases [39, 40]. Meanwhile, MIs increased the radiosensitivity of radio-resistant hypoxic cells, enhancing the DNA damage induced by ionizing radiation, because of their electron affinity. The released DOX transported to the nucleus to kill the tumor cells and, when combined with the P-(MIs)n radiosensitization effects, enhanced the effect of chemoradiotherapy against gliomas (Scheme 1).
Schematic of the hypoxia-responsive and hypoxia RT sensitization ALP-(MIs)n drug-delivery system. (A) Mechanism of ALP-(MIs)n RT sensitization and DOX release under hypoxic condition and formation of ALP-(MIs)n/DOX. Six electrons are transferred in the complete reduction of nitro (R-NO2) to amine (R-NH2) under hypoxic conditions via a single-electron reduction catalyzed by a series of intracellular nitroreductases. (B) Formation of AL-PLGA/DOX as the control group. (C) Schematic illustrating ALP-(MIs)n applications: i) hypoxic cell radiosensitizer; ii) hypoxia-responsive release of DOX into the cytoplasm, and then transports it to the nucleus to kill tumor cells.
Materials and methods
Materials
2-(dimethylamino)ethyl methacrylate (DMAEMA, Aldrich, 98%) was brought from Sigma Aldrich (Saint Louis, MO, USA) and was purified by passing through an alumina column. 4, 4'-Azobis(4-cyanovaleric acid) (V-501, Aldrich, 99%) was obtained from J&K Scientific Ltd (Beijing, China) and was purified by precipitation in methyl alcohol followed by drying under vacuum at 25 oC overnight. The chain-transfer agent (4-cyanopentanoic acid dithiobenzoate, CPADB) for RAFT polymerization was also got from J&K Scientific Ltd (Beijing, China). 1,2-dioleoylsn-glycero-3-phosphoethanolamine-n-[poly(ethyleneglycol)]2000 (DSPE-PEG2000) were purchased from Shanghai Advance Vehicle Technology Pharmaceutical L.T.D (Shanghai, China). DSPE-PEG2000-PDP was purchased from Xian Ruixi Biological Technology Co., Ltd (Xian, China). Angiopep-2 (TFFYGGSRGKRNNFKTEEY) was purchased from GL Biochem Ltd (Shanghai, China). DOX and D-Luciferin potassium salt were got from Dalian Meilun Biotech Co., Ltd (Dalian, China). Poly-lactic-co-glycolic acid (PLGA), 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT), 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) were purchased from Sigma Aldrich (Saint Louis, MO, USA). γ-H2AX, and Ki67 antibodies were obtained from CellSignaling Technology, Inc. (Danvers, MA, USA). Hypoxyprobe-1 Plus Kit was purchased from hypoxyprobe, Inc. (Burlington, MA, USA). In Situ Cell Death Detection Kit was purchased from Roche (Mannheim, Germany).
Nanoparticle preparation and characterization
Preparation of MI-MA
Metronidazole (MI, 8.55 g, 50 mmol), methacrylic acid (MA, 6.45 g, 75 mmol) and 4-dimethylaminopyridine (DMAP, 3.05 g, 25 mmol) were dissolved in 100 mL of dry dichloromethane (DCM) in an oven-dried 250 mL three-necked round-bottom flask attached to a 100 mL slow-addition apparatus. Next, the mixture was stirred under argon flow for 1 h, then dicyclohexyl carbodiimide (DCC, 20.6 g, 100 mmol, dissolved in 50 mL DCM) was added dropwise. The mixture was stirred at 30 oC overnight. The mixture was cooled down to room temperature and filtered. The filter cake was washed with DCM (50.0 × 3 mL). The filtrate was combined and washed with water (50.0 × 3 mL). And then, the organic layer was dried with anhydrous Na2SO4, and concentrated under reduced pressure. The product was subjected to silica gel column chromatography PE/EA (v/v = 1/1) to afford MI-MA (10.9 g, yield 91.5%) as a white solid. 1H NMR (300 MHz, DMSO-d6, ppm): δ 8.04 (s, 1H), 5.95 (s, 1H), 5.68 (s, 1H), 4.70 (m, 2H), 4.49 (m, 2H), 2.50 (s, 3H), 1.82 (s, 3H). 13C NMR (DMSO-d6) : δ = 14.35, 18.28, 45.03, 63.09, 126.70 133.61, 135.78, 139.03, 151.91, 166.50 ppm. MS (m/z): 240.09 [M + H]+ calcd for C10H13N3O4 239.09, found 239.09.
Preparation of Poly-(MIs)n
The polymerization was carried out in a baked Schlenk tube under argon protection. MI-MA (718 mg, 300 mmol), V-501 (4.8 mg, 30 mg), CPADB (16.8 mg, 60 mmol) and DMSO (1 mL) were added to tube. The solution was deoxygenated by three standard freeze pump thaw cycles. Then, the reaction tube was placed into an oil bath preset at 70 oC. After maintained at 70 oC for predetermined time, the reaction tube was placed into a cooling water bath. The resultant polymer after participation in methanol appeared as orangered oil, and dried under vacuum at 25 oC for 24 h.
Preparation of angiopep-2-DSPE-PEG2000
DSPE-PEG2000-PDP and agiopep-2 with the same mol were added to the DMSO. The reaction mixture was stirred gently at room temperature for 36 h. The released 2-pyridinethione was measured to characterize the obtained product. The resolution was lyophilized to obtain the final product angiopep-2-DSPE-PEG2000. The ALP-(MIs)n NPs size were adjusted based on the total lipid (lecithin: DSPE-PEG2000: angiopep-2-DSPE-PEG2000)/P-(MIs)n weight ratio of 1; 3.8.
Preparation of ALP-(MIs)n and ALP-(MIs)n/DOX
DSPE-PEG2000 (0.44 mg), lecithin (0.13 mg), angiopep-2-DSPE-PEG2000 (0.06 mg), P-(MIs)n (2.40 mg) were completely solubilized in DMSO. The mixtures were added together and vortexed vigorously for 2 min, and dripped into water solution under vigorous stirring conditions for 20 min followed by gently stirring for 2 h at room temperature to form ALP-(MIs)n.
2.0 mg of DOX.HCl was dissolved in 1 mL DMSO, and 0.05 mL of triethylamine was then added into the solution to remove hydrochloride [41]. DSPE-PEG2000 (0.44 mg), lecithin (0.13 mg), angiopep-2-DSPE-PEG2000 (0.06 mg), P-(MIs)n (2.40 mg) and DOX (0.53 mg) were completely solubilized in DMSO. DOX with 15% weight of ALP-(MIs)n/DOX were added to prepare ALP-(MIs)n/DOX. The mixtures were added together and vortexed vigorously for 2 min, and dripped into water solution under vigorous stirring conditions for 20 min followed by gently stirring for 2 h at room temperature to form ALP-(MIs)n/DOX.
And then, ALP-(MIs)n and ALP-(MIs)n/DOX were purified by being dialyzed against PBS (pH 7.4) for 12 h in order to remove the free DOX and DMSO. The AL-PLGA and AL-PLGA/DOX were prepared with the similar described above.
DOX loading and characterization of ALP-(MIs)n and ALP-(MIs)n/DOX
The experimental method was performed as previously described [27]. The DOX were measured using a UV-vis spectrophotometer (Bio Tek Synergy2) at 480 nm. The loading efficiency and content of DOX were calculated as:
Loading efficiency (%) = (weight of loaded drug)/(weight of drug in feed) × 100%;
Loading content (%) = (weight of loaded drug)/(total weight of NPs) × 100%.
The loading efficiency and content of DOX into the ALP-(MIs)n/DOX were found to be 96.0% and 14.4 wt%, respectively.
The condition of hypoxia was generated by the direct depletion of oxygen in sealed environment using the glucose (10 Mm), glucose oxidase (2 units mL-1) and catalase (120 units mL-1) dual enzyme system as previously described [42]. NADPH (100 mM) was added as electron donor.
Hydrodynamic diameter, poly-dispersity (PDI), and zeta potential of the ALP-(MIs)n, AL-PLGA, ALP-(MIs)n/DOX, and AL-PLGA/DOX were determined by dynamic light scattering (DLS) using a ZEN3600 Zetasizer NanoZS (Malvern Instruments Ltd., Massachusetts, U.S.A.). Hydrodynamic diameter and PDI of the ALP-(MIs)n, and AL-PLGA, which were incubated in hypoxic conditions for 15 min, were determined by DLS.
For transmission electron microscopy (TEM) characterization, ALP-(MIs)n, AL-PLGA under normoxic and hypoxic conditions (for 15 min) were dropped onto a TEM copper grid and then stained with 2% (w: v) phosphotungstic acid for 1 min. After air-drying, the samples were observed by TEM.
Cell lines
C6-GFP-Luci cells were constructed by lentivirus transduction and could stably express firely luciferase protein. Mice glioma cell line C6 and C6-GFP-Luci cells were grown in Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% fetal bovine serum (Hyclone). Cell cultures under normoxic conditions (pO2: 21%) were maintained in a humidified incubator at 37 ℃ in 5% CO2 and 95% air. Hypoxic conditions (pO2: 2%) were produced by placing cells in a hypoxia incubator (Thermo Scientific HERAcell 150i) in a mixture of 2% O2, 5%CO2 and 93% N2, as previously described [27].
Cellular DOX uptake and intracellular release of DOX
C6 cells were seeded in 12-well plates at the density of 1 × 105 cells/well at 37 °C and in 5% CO2. After cultured for 24 h, C6 cells were pretreated with 200 µM of free angiopep-2 before treated with ALP-(MIs)n/DOX. The cells were harvested, and cell uptake determined from DOX fluorescence per cell using a BD FACSCalibur flow cytometer (Bedford, MA) and FlowJo software for analysis. C6 cells were incubated with the LP-(MIs)n/DOX and ALP-(MIs)n/DOX for 4 h. The DOX fluorescence intensity was analyzed by OLYMPUS TH4-200 fluorescence microscopy and flow cytometry. C6 cells were seeded in 12-well plates at the density of 1 × 105 cells/well and incubated with either free DOX or the ALP-(MIs)n/DOX and AL-PLGA/DOX under 2% oxygen concentrations (normoxia, 2%) conditions for 2 h. The cellular release of DOX from ALP-(MIs)n/DOX and AL-PLGA/DOX was tested by OLYMPUS TH4-200 fluorescence microscopy and flow cytometry.
Colony formation assay
The experimental method was performed as previously described [43]. In order to examine the effects of ALP-(MIs)n and AL-PLGA on the radiosensitivity of C6 cells, C6 cells were seeded in 6-well plates in triplicate at densities of 600, 800, 1000, 2000, and 4000 per well and were correspondingly exposed to 0, 2, 4, 6 and 8 Gy IR. 24 h later, cells were treated with NPs at the dose of 0.18 mg mL-1 MIs or PLGA for 5 h. After 5 h, cells were irradiated with 0, 2, 4, 6 and 8 Gy, respectively. For 1 h later, fresh complete DMEM were added after removing the existed culture medium and then incubated for another 14 days. Cell clones were fixed with 4% paraformaldehyde and stained with crystal violet after 14 days. All colonies with exceeding 50 cells were calculated. The plating efficiency was calculated as dividing the number of colonies. The radiosensitivity was evaluated by the sensitization enhancement ratio (SER). As our previously reported, the radiosensitivity enhancement ratio was calculated by several parameters; For instance, the surviving fraction at 2 Gy (SF2), radiation dose (D), mean lethal dose (D0), quasi-threshold dose (Dq). Above all, the sensitization enhancement ratio (SER) was calculated as dividing the D0 of control group by the D0 of experimental group [43].
Cell immunofluorescence for H2AX
The experimental method was performed as previously described [44]. C6 cells were seeded at 10 000 cells per well in a 12-well plate for 24 h before treatment. Cells were treated by incubation with AL-PLGA, ALP-(MIs)25, and ALP-(MIs)48 4 h under hypoxic condition (pO2: 2%) and ALP-(MIs)25 under normoxic condition, followed by irradiation with 2 Gy. Cells were stained with an anti-γ-H2AX antibody (red) and DAPI (blue) 24 h after RT. Cells were analyzed using fluorescence microscopy and photographed (Olympus, Japan).
Animals
Male ICR mice with 18-20 g weight were purchased from Beijing HFK Bioscience Co., Ltd. (Beijing, China). All animals received care according to the guidelines in the Guide for the Care and Use of Laboratory Animals. All procedures were approved by Xuzhou Medical University of China Animal Care and Use Committee. Glioma-bearing ICR mice were prepared by intracranial injection (striatum, 1.8 mm right lateral to the bregma and 3 mm of depth) of 1 × 105 C6-GFP-Luci cells suspended in 5 µL of serum-free media into ICR mice, as previously described [45].
In vivo distribution of ALP-(MIs)n/DOX and AL-PLGA/DOX
The experimental method was performed as previously described [46]. ALP-(MIs)n/DOX and AL-PLGA/DOX in glioma model nude mice: glioma model nude mice were first injected with freshly prepared luciferin substrate and imaged with the Xenogen IVIS Spectrum optical imaging device to prove to have similar volume tumors in the brain, at the 7 th day after implantation. After that, glioma model ICR mice were injected intravenously with ALP-(MIs)n/DOX and AL-PLGA/DOX through the tail vein at the dose of 3 mg kg-1 DOX and 14.1 mg kg-1 P-(MIs)n (n = 25, 48) per animal. Then at 24 h after administration, the mice were sacrificed by cervical dislocation under anesthesia with isoflurane. And then, the glioma model brains as well as other principal organs (heart, liver, spleen, lung and kidneys) were excised carefully and visualized under the in vivo real-time fluorescence imaging system. The concentrations of DOX from brains were measured by a microplate reader (Bio Tek Synergy H4 Hybrid Reader) with excitation at 480 nm and emission at 570 nm.
Examination of tissue hypoxia pimonidazole
The experimental method was performed as previously described [27]. Pimonidazole hydrochloride (HypoxyprobeTM-1) as a hypoxic staining probe was used to stain the hypoxic tissue. The slices were treated according to the manufacturer's instruction and sections immunofluorescence was evaluated by fluorescence microscopy (Olympus, Japan).
Blood pharmacokinetics
Either DOX.HCl (at 3 mg kg-1) or ALP-(MIs)n/DOX (at DOX concentration of 3 mg kg-1) was intravenously injected into ICR mice through the tail vein to determine pharmacokinetics [27]. A blood sample (40 μL) was collected from the tail vein at different time points post-injection and mixed with K3-EDTA (0.5 μL, an anticoagulation agent). Fluorescence was measured using a microplate reader (Bio Tek Synergy H4 Hybrid Reader) with excitation at 480 nm and emission at 570 nm. The dataset was analysed using PKSlover software.
In vivo antiglioma efficiency
The experimental method was performed as previously described [46]. Glioma C6 cells were transformed with luciferase gene (C6-GFP-Luci). The real-time fluorescence imaging analysis was used to examine the therapeutic efficiency of different types of formulation from 10 to 27 days after tumor implantation. After 10 days of xenograft glioma, mice were randomly divided into 8 groups (n = 10) to test the antiglioma effect, which mice treated with PBS, PBS + RT, LP-(MIs)25/DOX + RT, LP-(MIs)48/DOX + RT, ALP-(MIs)25/DOX, ALP-(MIs)48/DOX, ALP-(MIs)25/DOX + RT, and ALP-(MIs)48/DOX + RT. To further evaluate the radiosensitivity effect, mice were randomly divided into 9 groups (n = 10), which received PBS, PBS+RT, AL-PLGA + RT, free DOX + RT, ALP-(MIs)25 + RT, ALP-(MIs)48 + RT, AL-PLGA/DOX + RT, ALP-(MIs)25/DOX + RT and ALP-(MIs)48/DOX + RT, after 10 days of xenograft glioma. At days 12, 14 and 16 after implantation, DOX (3 mg kg-1) and P-(MIs)25, P-(MIs)48, PLGA (14.1 mg kg-1) per dose were injected via the tail vein and radiation therapy with 2 Gy RT per dose . Radiation treatment was performed by using X-ray (0.3 Gy minute-1 and 6MV) with whole brain RT. The mice were injected with 15 mg kg-1 of freshly prepared luciferin substrate and imaged using the IVIS kinetic imaging system (Caliper Life Sciences, Hopkinton, MA, USA). Tumor growth was monitored by live bioluminescence imaging with the Xenogen IVIS Spectrum optical imaging device (Caliper Life Sciences) at different time points. The relative tumor inhibition rate was calculated using the following formulas: The relative tumor inhibition rate = the tumor bioluminescence intensity of 27 days/the tumor bioluminescence intensity of 10 days.
Mice were euthanized when they became moribund and the date recorded. Kaplan-Meier survival curves were plotted for each group. In the whole study, mice were weighted regularly.
Toxicity evaluation
C6-bearing ICR mice received injections of PBS, PBS + RT, free DOX + RT, AL-PLGA + RT, ALP-(MIs)25 + RT, ALP-(MIs)48 + RT, AL-PLGA/DOX + RT, ALP-(MIs)25/DOX + RT, ALP-(MIs)48/DOX + RT via the tail vein containing DOX (3 mg kg-1) and P-(MIs)n, PLGA (14.1 mg kg-1) per dose with 2 Gy RT per dose on days 12, 14 and 16. One day after the last treatment, two mice of per group were sacrificed, and sections of the main organs (brain, heart, liver, spleen, lung, kidneys) were stained with hematoxylin and eosin.
In vivo biocompatibility
To evaluate the biocompatibility in vivo toxicity of the ALP-(MIs)n/DOX. PBS, free DOX, ALP-(MIs)25/DOX and ALP-(MIs)48/DOX were administered intravenously to healthy tumor-free ICR mice at a single dose of PBS, 3 mg kg-1 of free DOX, 14.1 mg kg-1 of P-(MIs)25 and P-(MIs)48 (n = 3). The hemanalysis and biochemical analyses were performed on blood withdrawn from the mice on 24 h post drug treatment. The blood samples were centrifuged at 5000 rpm for 10 min to separate the plasma. The plasma was then submitted to the Nanjing Biomedical Research Institute Affiliated to Nanjing University for analysis of ALT, AST, TBIL, BUN and CREA levels, which studied the samples by 7020 Japan Hitachi fully automatic biochemical analyzer.
Immunohistology
The brain sections containing the tumor were incubated with 0.3% triton X-100 followed by 10% goat serum and were then incubated overnight with TdT-dependent dUTP-biotin nick end labeling (TUNEL) and Ki67 primary antibody at 4 ℃. Cell nuclei were stained with DAPI.
Statistical analyses
Statistical analyses were performed using SPSS version 13.0 and analyzed using one-way ANOVA. The experimental results were given in the format of mean ± SD in the figures (*p< 0.05, **p< 0.01, ***p< 0.001).
Results and discussion
Synthesis and characterization of P-(MIs)n
In this study, P-(MIs)n were prepared according to the steps shown in Figure S1A. First, MI-MA was synthesized. The formation of MI-MA was confirmed by 1H NMR and 13C NMR spectroscopy, which revealed all of the characteristic peaks and integration values of MI-MA, as seen in Figure S2A-B. The resultant MI-MA was further examined with high-resolution mass spectrometry for the determination of its mass and molecular formula. The results were consistent with the expected formula of MI-MA (Figure S2C). These data verify that MI-MA had been synthesized. Second, P-(MIs)n was prepared by RAFT polymerization. P-(MIs)25 and P-(MIs)48 were obtained. The 1H NMR spectra fully confirmed the chemical structures, and the polymerization degree (DP) of P-(MIs)n was calculated using 1H NMR end group analysis (Figure S3A-B). The GPC results showed that P-(MIs)25 and P-(MIs)48 had a narrow distribution of molecular weights with PDIs of 1.12 and 1.15 (Table S1). DSPE-PEG2000-PDP was chemically modified with angiopep-2 via thiol-disulfide exchange (Figure S1B) [47]. The absorbance peak at 343 nm, assigned to the released 2-pyridinethione, indicated that angiopep-2 had conjugated to DSPE-PEG2000-PDP to form angiopep-2-DSPE-PEG2000. There was no change in the absorbance peak at 343 nm after 30 h, indicating that the thiol exchange reaction was approximately completed (Figure S4) [48].
Fabrication and characterization of ALP-(MIs)n/DOX
After their successful synthesis, P-(MIs)25 and P-(MIs)48, DSPE-PEG2000, angiopep-2-DSPE-PEG2000, DOX and lecithin were allowed to self-assemble into ALP-(MIs)n/DOX via a single-step nanoprecipitation [49, 50]. For comparison, we used a control PLGA as a hydrophobic core to form AL-PLGA/DOX without hypoxia-responsive or radiosensitization effects on the hypoxic tumor cells. To achieve the optimal ALP-(MIs)n pharmacokinetic properties in vivo, the nanoparticle size (diameter, nm) and PDI were adjusted. The nanosize and PDI could be determined by adjusting the molar ratio of lecithin, DSPE-PEG2000, angiopep-DSPE-PEG2000, and the weight ratio of total lipids (lecithin + DSPE-PEG2000 + angiopep-DSPE-PEG2000) and the polymer mass. As shown in Table S2-S10, experiments have shown that the molar ratio of lecithin, DSPE-PEG2000 and angiopep-2-DSPE-PEG2000 (7: 2.8: 0.2) and the weight ratio of total lipids (lecithin + DSPE-PEG2000 + angiopep-2-DSPE-PEG2000) and polymer mass (1: 3.8), which we choose ultimately achieved the smallest size and a narrow PDI value. The average diameters of ALP-(MIs)25, ALP-(MIs)48 and AL-PLGA were approximately 88.81 ± 0.98 nm, 76.76 ± 2.31 nm, and 80.38 ± 1.07 nm, and the average PDI of ALP-(MIs)25, ALP-(MIs)48 and AL-PLGA were 0.23 ± 0.02, 0.19 ± 0.02, and 0.21 ± 0.02 nm, respectively (Figure 1A). The average diameters of ALP-(MIs)25/DOX, ALP-(MIs)48/DOX and AL-PLGA/DOX were similar to those of ALP-(MIs)25, ALP-(MIs)48 and AL-PLGA at 79.52 ± 0.70 nm, 61.66 ± 1.28 nm, and 76.71 ± 2.83 nm, and the average PDI of ALP-(MIs)25, ALP-(MIs)48 and AL-PLGA were 0.23 ± 0.02, 0.21 ± 0.02, and 0.26 ± 0.04 nm, respectively (Figure 1B). The zeta potential of these NPs was spread in the range of -17 mV and -30 mV. The DOX loading content of ALP-(MIs)25/DOX, ALP-(MIs)48/DOX and AL-PLGA/DOX was approximately 14.4%, and the MI loading content of ALP-(MIs)25/DOX and ALP-(MIs)48/DOX was approximately 67.2%. Cytotoxicity was evaluated in C6 cells using an MTT assay. As shown in Figure S5, the viability of the cells was greater than 85% with ALP-(MIs)25, ALP-(MIs)48 and AL-PLGA concentrations up to 800 μg mL-1, demonstrating the fairly low cytotoxicity of these NPs. The stability of ALP-(MIs)25, ALP-(MIs)48, AL-PLGA, ALP-(MIs)25/DOX, ALP-(MIs)48/DOX and AL-PLGA/DOX NPs were investigated in DMEM containing 10% FBS. As shown in Figure 1C, there were no significant increases in the size of ALP-(MIs)25, ALP-(MIs)48, AL-PLGA, ALP-(MIs)25/DOX, ALP-(MIs)48/DOX and AL-PLGA/DOX NPs over six days. This result suggests that ALP-(MIs)25, ALP-(MIs)48, AL-PLGA, ALP-(MIs)25/DOX, ALP-(MIs)48/DOX and AL-PLGA/DOX had a good colloidal stability in 10% FBS. These results are important because the negative surface charge should enhance colloidal stability and prolong the NPs residence time in the body by reducing protein adsorption and aggregation.
(A) Size distribution of AL-PLGA, ALP-(MIs)25, and ALP-(MIs)48 under normoxic conditions. (B) Size distribution of AL-PLGA/DOX, ALP-(MIs)25/DOX, and ALP-(MIs)48/DOX under normoxic conditions. (C) The stability of ALP-(MIs)25, ALP-(MIs)48, AL-PLGA, ALP-(MIs)25/DOX, ALP-(MIs)48/DOX and AL-PLGA/DOX NPs were investigated in DMEM containing 10% FBS over six days. (D-F) Size distribution of AL-PLGA, ALP-(MIs)25, and ALP-(MIs)48 under normoxic and hypoxic conditions. (G) The appearance change of AL-PLGA/DOX, ALP-(MIs)25/DOX, and ALP-(MIs)48/DOX under hypoxic conditions. (H) TEM images of ALP-(MIs)25 and AL-PLGA under normoxic and under hypoxic conditions. (I) Absorption spectra of ALP-(MIs)25, ALP-(MIs)48 and AL-PLGA when incubated under hypoxic and normoxic conditions for 2 h. (J) Hypoxic-responsive drug release from AL-PLGA/DOX, ALP-(MIs)25/DOX, and ALP-(MIs)48/DOX.
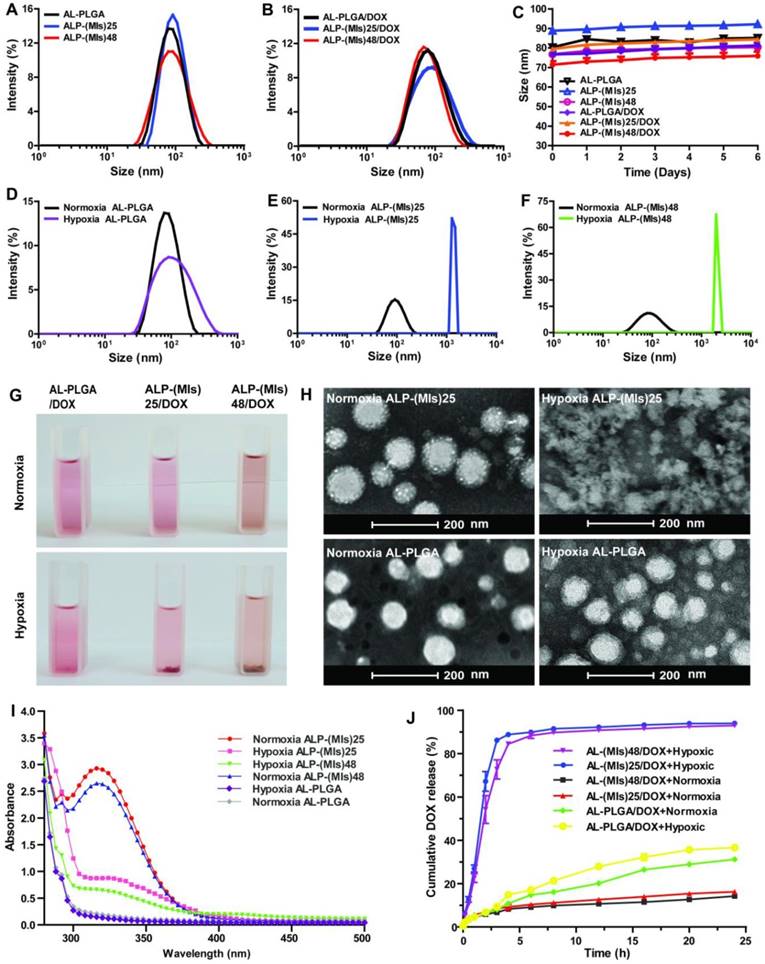
To test the hypoxic sensitivity of P-(MIs)n, the changes in size of ALP-(MIs)25, ALP-(MIs)48 and AL-PLGA were measured under normoxia (21% oxygen concentration) and hypoxia (2% oxygen concentration) using DLS. As shown in Figure 1D-F, ALP-(MIs)25 and ALP-(MIs)48 exhibited considerable swelling. The size of AL-PLGA did not change significantly under hypoxic conditions in comparison to that of AL-PLGA under normoxic conditions. To further confirm the size change of ALP-(MIs)25 and ALP-(MIs)48, ALP-(MIs)25/DOX and ALP-(MIs)48/DOX were incubated under hypoxic and normoxic conditions and were photographed. As shown in Figure 1G, the size of ALP-(MIs)25/DOX and ALP-(MIs)48/DOX increased and precipitated under hypoxic conditions. There was no red precipitation in the AL-PLGA/DOX control group under hypoxic conditions, and the size of AL-PLGA/DOX did not change significantly under hypoxic conditions in comparison to that of AL-PLGA/DOX under normoxic conditions. To further confirm the change in ALP-(MIs)n morphology, we selected ALP-(MIs)25 as treatment group and AL-PLGA as a control for characterization under normoxic and hypoxic conditions using TEM. The TEM images suggested that the AL-PLGA and ALP-(MIs)25 were spherical NPs with a unimodal size distribution under normoxic conditions (Figure 1H). In sharp contrast, the conformation of ALP-(MIs)25 was disassembled, and the conformation of AL-PLGA remained unchanged under hypoxic conditions. These results support our prediction that ALP-(MIs)n would experience destabilization and conformational change due to the conversion of the MI nitro group to an amino group under hypoxic conditions.
The nitro group of MI is converted to an amino group under low-oxygen conditions, as previously reported [27]. To confirm the conversion of NO2 to NH2, there was a characteristic peak of MI at 325 nm under normoxic conditions. Meanwhile, under hypoxic conditions, the characteristic peak of MI at 325 nm completely disappeared, indicating that the nitro group of MI is converted to an amino group under low-oxygen conditions. To further confirm the conversion of the MI nitro group to an amino group in ALP-(MIs)n, the OD value was measured. There is a characteristic peak of P-(MIs)n at 325 nm under normoxic condition. As shown in Figure 1I, the characteristic peak of ALP-(MIs)n at 325 nm was seen to completely disappear under hypoxic conditions. The result suggests that the nitro groups of ALP-(MIs)n were converted to amino groups under hypoxic conditions.
We predicated that ALP-(MIs)n have great potential for hypoxia-responsive controlled drug release. Thus, the release mechanism of DOX from ALP-(MIs)25/DOX was investigated under both hypoxic and normoxic conditions at pH 7.4 PBS and 37 ℃. Under hypoxic conditions, ALP-(MIs)25/DOX and ALP-(MIs)48/DOX released DOX within 4 h (Figure 1J, blue and purple squares), suggesting that ALP-(MIs)n/DOX selectively dissociates and releases DOX under hypoxia. However, no significant DOX release was found from ALP-PLGA/DOX under hypoxic or normoxic conditions (Figure 1J, yellow and green squares). These results support our prediction that ALP-(MIs)n would experience destabilization and conformational change due to the conversion of the MI nitro group to an amino group under hypoxic conditions.
For glioma therapy, selective uptake by glioma cells is an important factor that affects the ultimate therapeutic efficacy. Angiopep-2, a specific ligand for LRP-1 that is expressed in the BBB and human glioma cells, was modified on the surface of LP-(MIs)25 to enhance the glioma distribution of NPs. Beliveau et al. reported that hypoxia was associated with a statistically significant increase in LRP1 protein expression in glioma cells [51].
Angiopep-2-conjugated DSPE-PEG2000 was used to decorate LP-(MIs)25 to enhance their glioma-targeting ability (denoted as ALP-(MIs)25). To evaluate the glioma-targeting ability of ALP-(MIs)25, C6 cells were incubated with LP-(MIs)25 and ALP-(MIs)25/DOX under normoxic conditions. As shown in Figure 2A-B, DOX fluorescence intensity clearly increased in the ALP-(MIs)25/DOX-treated group under normoxic conditions. To verify the above results, C6 cells were cultured with ALP-(MIs)25/DOX and ALP-(MIs)25/DOX + free angiopep-2 under hypoxic and normoxic conditions for 4 h before being collected and analyzed using flow cytometry. As shown in Figure S6, DOX fluorescence intensity clearly increased in the ALP-(MIs)25/DOX-treated group under hypoxic conditions and this elevation could be rescued by pretreating with free angiopep-2. The aforementioned results reveal that the angiopep-2-modified LP-(MIs)25/DOX enhanced cellular uptake by the receptor-mediated transcytosis (RMT) strategy. Additionally, compared with the ALP-(MIs)25/DOX-treated group under normoxic conditions, DOX fluorescence intensity was elevated in the ALP-(MIs)25/DOX-treated group under hypoxic conditions. After 4 h incubation, more DOX can enter into nuclei under hypoxic conditions (Figure S7). This result is in accordance with the literature reported that the hypoxic environment could enhance the uptake of ALP-(MIs)25/DOX by upregulation of LRP1 [51].
(A) Intracellular release of DOX from LP-(MIs)25/DOX and ALP-(MIs)25/DOX. Samples were incubated with C6 cells under normoxic and hypoxic conditions for 4 h. Scale bar: 50 μm. (B) Cellular uptake of LP-(MIs)25/DOX and ALP-(MIs)25/DOX was analyzed using flow cytometry after 4 h incubation under normoxic and hypoxic conditions. (C) Intracellular release of DOX from AL-PLGA/DOX and ALP-(MIs)25/DOX. Samples were incubated with C6 cells under normoxic and hypoxic conditions for 4 h. Scale bar, 50 μm. (D) Cellular uptake of AL-PLGA/DOX and ALP-(MIs)25/DOX was analyzed using flow cytometry after 4 h incubation under normoxic and hypoxic conditions.
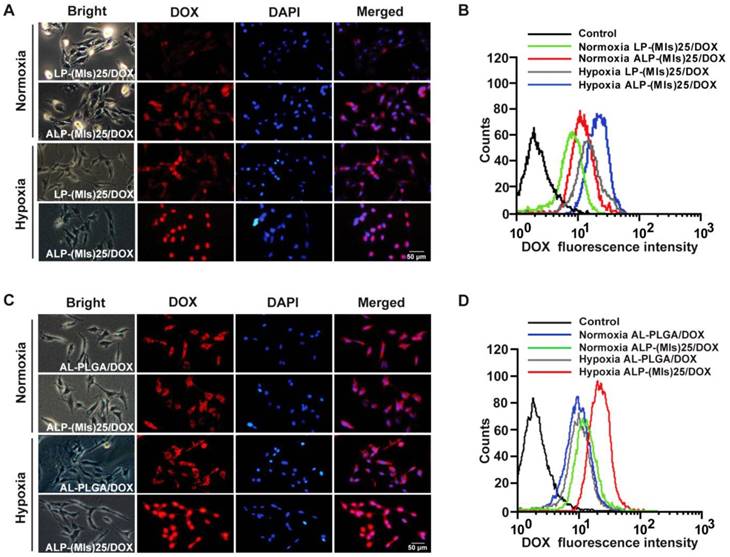
We have already demonstrated that ALP-(MIs)25 is completely destabilized under hypoxia. The P-(MIs)n core was then expected to provoke the rapid release of DOX when present in a hypoxic environment. DOX fluorescence was examined in C6 cells incubated under normoxia (21% oxygen concentration) and hypoxia (2% oxygen concentration) for 4 h. As shown in Figure 2C-D, ALP-(MIs)25 displayed higher DOX fluorescence and most of DOX accumulated into nuclear under hypoxic conditions than under normoxic conditions. As a control, the DOX fluorescence of AL-PLGA/DOX was similar under normoxic and hypoxic conditions. These results demonstrate that the conformation of ALP-(MIs)25 was destabilized under hypoxia due to conversion of the hydrophobic P-(MIs)25 core to hydrophilic poly-aminoimidazoles via a series of selective bioreductions, triggering the release of DOX.
Radiosensitization by ALP-(MIs)n in vitro
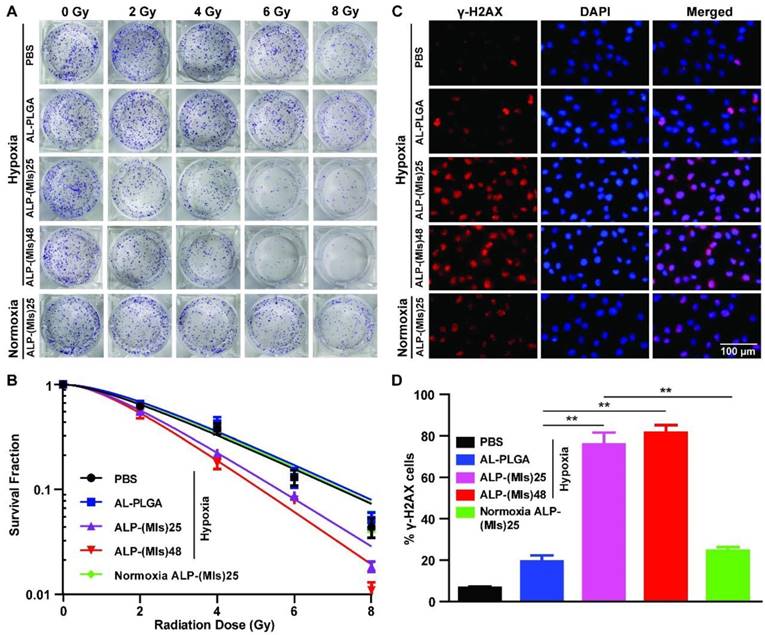
Hypoxia can induce conversion of the hydrophobic P-(MIs)n core to hydrophilic poly-aminoimidazoles through the transfer of six electrons. Due to their electron affinity, MIs increase the radiosensitivity of radio-resistant hypoxic cells, enhancing the DNA damage induced by ionizing radiation. DNA damage arising from ionizing radiation triggers cell apoptosis if DNA repair proteins fail to repair the damage [52]. To test whether ALP-(MIs)n could increase the RT sensitivity of hypoxic tumor cells, a colony-forming assay and immunofluorescence of phospho-Histone H2AX were conducted as the standard method to evaluate RT-induced apoptosis. As observed from cell survival in response to ionizing radiation depicted in Figure 3A-B, ALP-(MIs)n, including ALP-(MIs)25, and ALP-(MIs)48, were all observed to have an RT sensitization function in C6 cells under hypoxic conditions (2% oxygen concentration), as indicated by their colony formation capacity and the downward shift of the survival curves following the administration of both ALP-(MIs)n and X-rays. Meanwhile, AL-PLGA under hypoxia and ALP-(MIs)25 under normoxia (21% oxygen concentration) as the control groups showed no obvious radiosensitizing effect on C6 cells; the SER of ALP-(MIs)25 and ALP-(MIs)48 in C6 cells were 1.34 and 1.46. The degree of DNA damage caused by RT can be assessed using γ-H2AX immunohisto-chemistry staining, a biomaker for DSBs [53-55]. Compared with AL-PLGA under hypoxic conditions and ALP-(MIs)25 under normoxic conditions, the administration of ALP-(MIs)25 and ALP-(MIs)48 in conjunction with RT significantly increased γ-H2AX under hypoxic conditions because ALP-(MIs)n acts as a hypoxic radiosensitizer to enhance the DNA damage caused by radiation (Figure 3C-D). These results demonstrate that ALP-(MIs)n can effectively enhance the therapeutic X-ray effects and are as potential radiosensitizers. Given that ALP-(MIs)25 and ALP-(MIs)48 all had a good effect on RT sensitization and a nanosize of less than 80 nm, we chose the two drugs with different degrees of polymerization to continue the subsequent in vivo experiments due to their beneficial effects for targeting gliomas.
Radiosensitization by ALP-(MIs)n in vitro. (A) Representative images of clonogenic survival assays of C6 cells cultured with AL-PLGA, ALP-(MIs)25, and ALP-(MIs)48 under hypoxic condition (pO2: 2%) and ALP-(MIs)25 under normoxic condition following treatment with 0, 2, 4, 6 and 8 Gy. (B) Clonogenic survival curves of C6 cells cultured with AL-PLGA, ALP-(MIs)25, and ALP-(MIs)48 under hypoxic condition (pO2: 2%) and ALP-(MIs)25 under normoxic condition following treatment with 0, 2, 4, 6 and 8 Gy. (C) Immunocytochemical analysis of γ-H2AX expressed by C6 cells. Cells were treated by incubation with AL-PLGA, ALP-(MIs)25, and ALP-(MIs)48 for 4 h under hypoxic condition (pO2: 2%) and ALP-(MIs)25 under normoxic condition followed by irradiation with 2 Gy using a dose rate of 0.3 Gy min-1. Cells were stained with an anti-γ-H2AX antibody (red) and DAPI (blue) 24 h after RT. Scale bar, 100 μm. (D) Quantitation of percentage of γ-H2AX positive cells. (Mean ± SD, n = 5, **p < 0.01).
Tumor therapy monitoring in mice
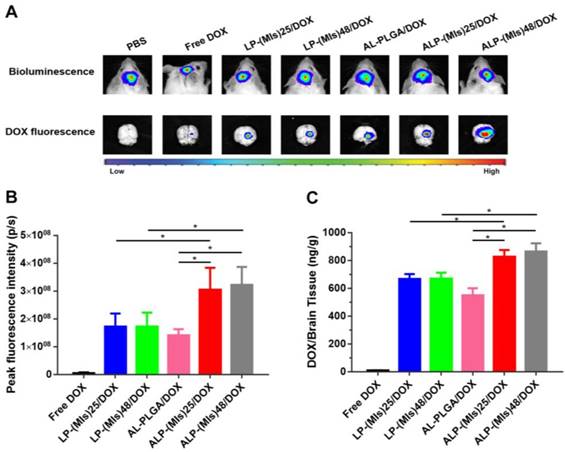
For glioma-therapy monitoring, orthotopic mouse models of C6-GFP-Luci were developed in ICR mice. Despite the structural integrity of the BBB partly broken in glioma, the BBB surrounding the proliferating cells at the tumor's edge remains intact [56, 57]. ALP-(MIs)n/DOX was anticipated to be able to enter the glioma through active targeting due to the angiopep-2 modification of ALP-(MIs)n/DOX. To evaluate the glioma targeting properties of ALP-(MIs)n/DOX, we applied an in vivo fluorescence imaging technique to examine the distribution of ALP-(MIs)n/DOX after intravenous injection into an orthotopic implantation model of C6-GFP-Luci glioma cells in ICR mice. As shown in Figure 4A, at 7 days after orthotopic implantation of C6-GFP-Luci glioma cells in ICR mice, the luciferase signal was detected, confirming the presence of a brain glioma with approximately the same volume. Free DOX, LP-(MIs)25/DOX, LP-(MIs)48/DOX, AL-PLGA/DOX, ALP-(MIs)25/DOX and ALP-(MIs)48/DOX were then injected via tail vein at 24 h post injection, gliomas were carefully excised and visualized using an in vivo real-time fluorescence imaging system. As shown in Figure 4A-B, compared with free DOX and LP-(MIs)n/DOX, the strongest DOX fluorescence was observed in the gliomas of mice that had received ALP-(MIs)n/DOX, indicating angiopep-2 modified ALP-(MIs)n/DOX improvement the BBB crossing and targeting glioma abilities due to the angiopep-2 effect. We also found more DOX fluorescence in the ALP-(MIs)25/DOX and ALP-(MIs)48/DOX groups than in the AL-PLGA/DOX group 24 h after injection due to the conformation of ALP-(MIs)25 and ALP-(MIs)48 destabilizing in the hypoxic tumor microenvironment, triggering intracellular DOX release. Approximately no DOX fluorescence was observed in animals that received an injection of free DOX into their bloodstream because it was rapidly filtered and excreted via the kidneys into the urine. Therefore, free DOX showed less tumor accumulation than DOX encapsulated in the NPs. To confirm the tumor distribution of free DOX, LP-(MIs)25/DOX, LP-(MIs)48/DOX, ALP-(MIs)25/DOX, ALP-(MIs)48/DOX, and AL-PLGA/DOX, the DOX content in excised mouse brains was measured (Figure 4C). The results from the concentration of DOX analysis were consistent with the results of DOX fluorescence intensity (Figure 4B). This similar result indicates that the DOX fluorescence observed in the imaging experiments is of released DOX, as DOX fluorescence is quenched in the nanoparticle due to intermolecular π-π stacking and hydrophobic interactions [58, 59]. Once released from the NPs, the fluorescence of DOX is restored and concentration dependent.
DOX glioma distribution 24 h after intravenous injection of mice with either free DOX, LP-(MIs)25/DOX, LP-(MIs)48/DOX, ALP-(MIs)25/DOX, ALP-(MIs)48/DOX, and AL-PLGA/DOX. (A) Bioluminescence of luciferase-expressing tumor cells 10 min after injection with luciferin solution and fluorescence images of excised mouse brains. (B) Quantitative analysis of DOX fluorescence intensity in excised mouse brains. (Mean ± SD, n = 4, *p < 0.05) (C) Mean DOX concentration in excised mouse brains. (Mean ± SD, n = 4, *p < 0.05)
In above in vitro experiments, ALP-(MIs)25/DOX was demonstrated to sensitize glioma cells to RT under hypoxic conditions. Thus, it is essential for ALP-(MIs)25/DOX reaching hypoxic regions in glioma to enhance the therapeutic effect of RT. The ALP-(MIs)25/DOX distribution in the C6 orthotopic transplantation model was assessed using an immunofluorescence assay. Hypoxic tissues were stained with a FITC-labeled monoclonal antibody and were found to contain a large amount of imbedded ALP-(MIs)25/DOX (Figure S8). ALP-(MIs)25/DOX was thus able to effectively reach the hypoxic tumor site after systemic in vivo administration, validating the feasibility of the in vivo anti-glioma efficacy test by combining RT with ALP-(MIs)25/DOX.
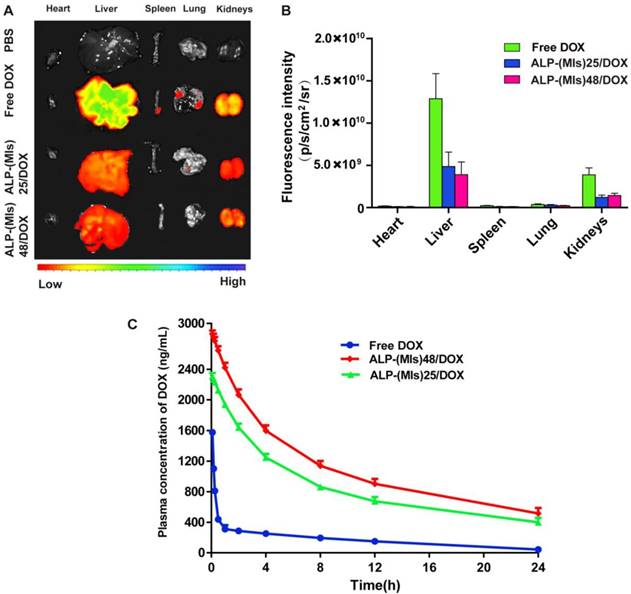
We also evaluated the pharmacokinetics and biodistribution of free DOX, ALP-(MIs)25/DOX, and ALP-(MIs)48/DOX after intravenous injection into C6-GFP-Luci-bearing ICR mice by quantitatively detecting the amounts of DOX in the plasma and different tissues, including tissues of the heart, liver, spleen, lung, kidneys and brain. The major organs were harvested at 4 h after injection, and the biodistribution is shown in Figure 5A-B. Free DOX, ALP-(MIs)25/DOX and ALP-(MIs)48/DOX showed high accumulation in the liver and kidneys. However, liver and kidney accumulations of free DOX were more than 2.64-fold and 3.18-fold higher than those of ALP-(MIs)25/DOX. The DOX distribution of ALP-(MIs)25/DOX was approximately the same as that of ALP-(MIs)48/DOX. As shown in Figure 5C, free DOX was rapidly cleared from the blood, with a half-life (t1/2) of approximately 0.21 h. In contrast, the half-lives (t1/2) of ALP-(MIs)25/DOX and ALP-(MIs)48/DOX were approximately 5.88 h and 5.91 h due to their nano size and PEG shell extending the blood circulation time of DOX. Together, these findings demonstrate that ALP-(MIs)25/DOX and ALP-(MIs)48/DOX can successfully deliver DOX and target the tumor in vivo, extensively prolonging the serum retention time of DOX.
(A) Fluorescence images of free DOX, ALP-(MIs)25/DOX and ALP-(MIs)48/DOX tissue distribution (at a DOX concentration of 3 mg kg-1) in intravenously injected C6-GFP-Luci-bearing ICR mice. (B) Quantitative analysis of DOX in tissues. (C) Blood retention kinetics of free DOX, ALP-(MIs)25/DOX and ALP-(MIs)48/DOX in ICR mice (DOX concentration of 3 mg kg-1). All the data of blood retention kinetics were analyzed by Drug and Statistics (DAS) 3.23 software (mean ± SD, n = 4).
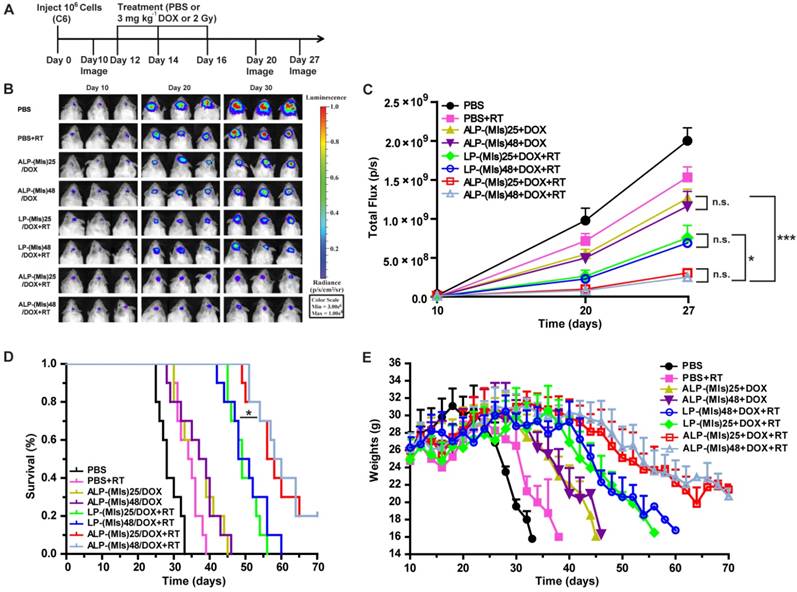
Finally, the antiglioma efficacy of ALP-(MIs)n/DOX was estimated in orthotopic C6-GFP-Luci-bearing ICR mice (Figure 6). C6 cells were genetically modified to express the luciferase enzyme, and bioluminescence imaging was used to measure in vivo growth. Mice were treated with different groups after confirming the presence of brain glioma at 10 days using in vivo imaging. P-(MIs)n had been proven to increase the radiosensitivity of hypoxic cells in vitro. As shown in Figure 6B-C, ALP-(MIs)n/DOX + RT had significant effects on inhibiting glioma growth compared with LP-(MIs)n/DOX + RT, and ALP-(MIs)n/DOX groups (n = 25, 48). Survival of C6-GFP-Luci glioma-bearing mice receiving different treatments is presented in a Kaplan-Meier plot (Figure 6D). For mice treated with ALP-(MIs)25/DOX + RT (57 days) and ALP-(MIs)48/DOX + RT (59 days), the median survival times were significantly different than those of mice treated with LP-(MIs)25/DOX + RT (49 days) and LP-(MIs)48/DOX + RT (49.5 days) groups (P < 0.05). The body weights of mice treated with ALP-(MIs)25/DOX + RT and ALP-(MIs)48/DOX + RT decreased more slowly than those of mice treated with LP-(MIs)25/DOX + RT, LP-(MIs)48/DOX + RT, ALP-(MIs)25/DOX, and ALP-(MIs)48/DOX (Figure 6E). Compared with LP-(MIs)n/DOX + RT, ALP-(MIs)n/DOX + RT had a strong inhibitory action on gliomas, since angiopep-2-modified LP-(MIs)n/DOX effectively accumulated in the glioma and improved the curative effect of glioma. ALP-(MIs)n/DOX + RT significantly inhibited the growth of gliomas compared with ALP-(MIs)n/DOX without RT. However, these results cannot directly demonstrate that the anti-glioma effect was caused by the combination of P-(MIs)n and DOX with RT.
In vivo efficacy in the C6-GFP-Luci glioma mouse model. (A) C6-GFP-Luci-bearing mice received three injections of PBS, PBS + RT, ALP-(MIs)25/DOX, ALP-(MIs)48/DOX, LP-(MIs)25/DOX + RT, LP-(MIs)48/DOX + RT, ALP-(MIs)25/DOX + RT, and ALP-(MIs)48/DOX + RT at a dose of 3 mg kg-1 DOX and 14.1 mg kg-1 P-(MIs) on days 12, 14, and 16, with 2 Gy RT. (B) Bioluminescence signal change correlating to tumor growth over time following inoculation. (C) Quantification of the tumor bioluminescence signal. (n = 5 mice per group); *p < 0.05, ***p < 0.001, one-way ANOVA; n.s. indicates no statistical significance. (D) Kaplan-Meier survival curves for the mice (n = 10). *p < 0.05, one-way ANOVA. (E) Body weight change. Data are presented as the mean ± SD.
To further investigate the radiosensitization effect of P-(MIs)n, the polymer PLGA without a radiosensitizing effect was used as the control group. As shown in Figure 7B, D, the PBS, AL-PLGA, free DOX, ALP-(MIs)25, ALP-(MIs)48, AL-PLGA/DOX, ALP-(MIs)25/DOX, and ALP-(MIs)48/DOX formulations with RT significantly inhibited glioma growth compared with the PBS group (glioma inhibition rate of 236.4) as a negative control. In contrast, as the polymer PLGA does not have a radiosensitizing effect, the glioma inhibition rate of the AL-PLGA + RT group (172.3) was approximately the same as that of the PBS + RT group (184.5). Both the glioma inhibition rates of ALP-(MIs)25 (83.1) and ALP-(MIs)48 (79.2) presented a remarkably higher inhibition efficacy towards glioma growth than the PBS + RT group and the AL-PLGA + RT group (p < 0.01), which suggests that P-(MIs)25 and P-(MIs)48 possess a radiosensitizing effect.
In vivo efficacy in the C6-GFP-Luci glioma mouse model. (A) C6-GFP-Luci-bearing mice received three injections of PBS, PBS + RT, free DOX + RT, AL-PLGA + RT, ALP-(MIs)25 + RT, ALP-(MIs)48 + RT, AL-PLGA/DOX + RT group, ALP-(MIs)25/DOX + RT and ALP-(MIs)48/DOX + RT at a dose of 3 mg kg-1 DOX and 14.1 mg kg-1 P-(MIs) on days 12, 14, and 16, with 2 Gy RT. (B) Bioluminescence signal change correlating to tumor growth over time following inoculation. (C) Quantification of the tumor bioluminescence signal (n = 5 mice per group); ** p < 0.01, one-way ANOVA; n.s. indicates no statistical significance. (D) Relative tumor inhibitory rate for each treatment; ** p < 0.01, one-way ANOVA; n.s. indicates no statistical significance. (E) Representative H&E-stained images of coronal sections from mouse brains with orthotopic tumors. (F) Ki67 staining of coronal sections from mouse brains with orthotopic tumors. Scale bar, 50 µm. (G) TUNEL staining of coronal sections from mouse brains with orthotopic tumors. Scale bar, 50 µm. (H) Kaplan-Meier survival curves for the mice (n = 10), *** p < 0.001, one-way ANOVA. (I) Body weight change. Data are presented as the mean ± SD.
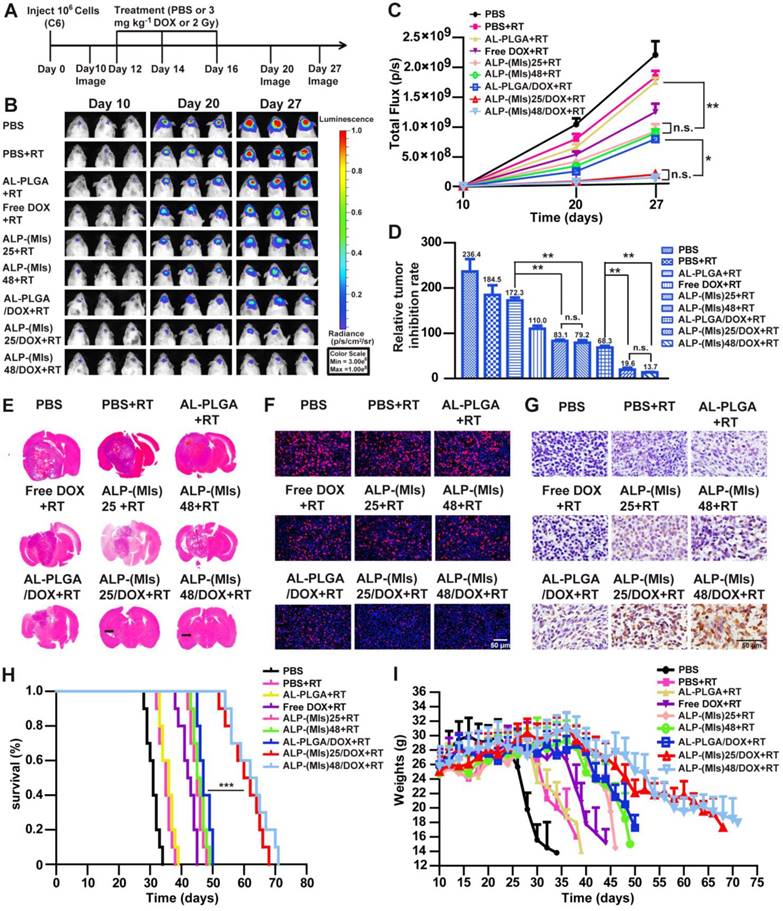
Using the same P-(MIs)25 and P-(MIs)48 doses for mice, there were no significant differences (p > 0.05) in glioma inhibition rates. These results show that the polymerization degree of P-(MIs)n did not affect the radiosensitizing effect of MTZ due to the formation of P-(MIs)n by biodegradable ester linkages between MTZ monomers. The glioma inhibition rates in the AL-PLGA/DOX + RT, ALP-(MIs)25/DOX + RT and ALP-(MIs)48/DOX + RT groups were 68.3, 19.6 and 13.7, respectively. Moreover, compared with the glioma inhibition rates determined for ALP-(MIs)25 (83.1), the ALP-(MIs)48 (79.2), AL-PLGA/DOX + RT (68.3), ALP-(MIs)25/DOX + RT and ALP-(MIs)48/DOX + RT groups clearly demonstrated the strongest inhibition of glioma growth (p < 0.01), suggesting that the combination of ALP-(MIs)25/DOX and ALP-(MIs)48/DOX with RT enhanced the inhibition of glioma growth. As shown in Figure 7E, at 18 days after glioma transplantation, the transplanted gliomas in ALP-(MIs)25 + RT and ALP-(MIs)48 + RT-treated mice were visibly smaller than in the AL-PLGA + RT-treated mice. Based on H&E staining of coronal sections of mouse brains, the smallest glioma volumes were observed in ALP-(MIs)25/DOX + RT and ALP-(MIs)48/DOX + RT-treated mice. To further evaluate the antiglioma effects of these NPs on tumor growth, Ki67 and TUNEL were used to show cell proliferation and apoptosis in xenografts, respectively.
We also observed a decrease of proliferation and an enhancement of apoptosis in ALP-(MIs)25 + RT, ALP-(MIs)48 + RT, ALP-(MIs)25/DOX + RT and ALP-(MIs)48/DOX + RT-treated mice (Figure 7F-G). Collectively, these results suggest the following: 1) ALP-(MIs)25 and ALP-(MIs)48 effectively enhanced the therapeutic X-ray effects and were as potential radiosensitizers; and 2) the combination of DOX, hypoxic RT sensitization (P-(MIs)n) and RT in ALP-(MIs)25/DOX + RT and ALP-(MIs)48/DOX + RT-treated mice effectively inhibited glioma growth.
To further estimate the antitumor efficacy, the C6-GFP-Luci-bearing ICR mice were monitored for body weight changes and median survival times. As shown in Figure 7H, the median survival times for mice treated with PBS, PBS + RT, AL-PLGA + RT, free DOX + RT, ALP-(MIs)25 + RT, ALP-(MIs)48 + RT, AL-PLGA/DOX + RT, ALP-(MIs)25/DOX + RT and ALP-(MIs)48/DOX + RT were 31, 35, 36, 42.5, 45, 45.5, 47, 61 and 63 days, respectively. The median survival times for the mice treated with ALP-(MIs)25 + RT and ALP-(MIs)48 + RT were longer than those of PBS, PBS + RT and AL-PLGA + RT, suggesting that ALP-(MIs)25 and ALP-(MIs)48 improved the efficacy of the radiotherapy. The groups that received ALP-(MIs)25/DOX + RT and ALP-(MIs)48/DOX + RT displayed the longest survival times among all the groups, with high statistical significance compared with the ALP-(MIs)25 + RT, ALP-(MIs)48 + RT and AL-PLGA/DOX + RT groups, indicating that the combination of chemotherapy and RT enhanced the inhibition of glioma growth. The dominance of ALP-(MIs)25 + RT, ALP-(MIs)48 + RT, ALP-(MIs)25/DOX + RT and ALP-(MIs)48/DOX + RT was also reflected by comparing the changes in body weight. The body weights of mice treated with ALP-(MIs)25 + RT, ALP-(MIs)48 + RT, ALP-(MIs)25/DOX + RT and ALP-(MIs)48/DOX + RT slowly decreased, while all other groups lost weight rapidly (Figure 7I). Collectively, our results verified that ALP-(MIs)25 and ALP-(MIs)48 were potent radiosensitizers and drug carriers for the delivery of hydrophobic chemotherapeutics, achieving concurrent RT and chemotherapy for glioma treatment.
Tissue-specific toxicity was examined by histological analysis of various tissues (heart, liver, spleen, lung or kidneys). As shown in Figure S9, H&E-stained images of major organs revealed no noticeable tissue damage or obvious changes in morphology in any of the organs in the ALP-(MIs)25, ALP-(MIs)48, ALP-(MIs)25/DOX and ALP-(MIs)48/DOX groups compared with the PBS group. The in vivo results suggest that the ALP-(MIs)25, ALP-(MIs)48, ALP-(MIs)25/DOX and ALP-(MIs)48/DOX groups were biocompatible and had potentially positive biological applications with few side effects. To further assess the potential in vivo toxicity of NPs on the physiology of ICR mice, a serum biochemistry assay was performed on four groups, i.e., the PBS control group, the free DOX group, the ALP-(MIs)25/DOX group, and the ALP-(MIs)48/DOX group (Figure S10). Liver function (AST, ALT, TBIL) and renal function (CREA, BUN) showed no obvious changes for the ALP-(MIs)25/DOX and ALP-(MIs)48/DOX groups compared with the PBS control group 24 h after intravenous injection, indicating that the physiological function and health conditions were not affected by ALP-(MIs)n. In contrast, the free DOX group elicited a remarkable increase in AST, ALT and TBIL in the liver due to the high concentration of DOX that was accumulated in the liver leading to potential liver toxicity. The liver status is mainly reflected by the level of ALT. These results suggest that the DOX encapsulated in the NPs (ALP-(MIs)25/DOX group, and ALP-(MIs)48/DOX group) does not alter liver function or renal function while exerting more control over drug loading and release compared to free DOX.
Conclusions
We successfully developed a new hypoxia-responsive polyprodrug NP with RT hypoxic sensitization effects for targeted glioma therapy. The RT hypoxic sensitization effects of ALP-(MIs)n polyprodrug NPs can deliver hydrophobic chemotherapy, achieving the effect of concurrent chemotherapy and radiation and inducing the release of hydrophobic chemotherapeutics under hypoxic conditions. The in vitro and in vivo results demonstrate that these ALP-(MIs)n polyprodrug NPs can efficiently target gliomas and lead to significant inhibition of glioma growth. Therefore, our ALP-(MIs)n polyprodrug NPs may represent a powerful new strategy for enhancing the RT sensitivity of gliomas and achieving combination of radiation and chemotherapy for gliomas.
Abbreviations
CPADB: 4-cyanopentanoic acid dithiobenzoate; DAPI: 4′,6-diamidino-2-phenylindole dihydrochloride; DCC: dicyclohexyl carbodiimide; DCM: dichloromethane; DLS: dynamic light scattering; DMAEMA: 2-(dimethylamino)ethyl methacrylate; DMAP: 4-dimethylaminopyridine; DOX: Doxorubicin; DSPE-PEG2000: 1, 2-dioleoylsn-glycero-3-phosphoethanolamine-n-[poly(ethyleneglycol)] 2000; GBM: Glioblastoma multiforme; LRP-1: low-density lipoprotein receptor-related protein-1; MA: methacrylic acid; MI: Metronidazole; MTT: 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; PLGA: Poly-lactic-co-glycolic acid; RT: Radiotherapy; SER: sensitization enhancement ratio; TUNEL: TdT-dependent dUTP-biotin nick end labeling.
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (81772665, 81502153, 81472345, 81302175), Natural Science Foundation of Jiangsu Province (BK20150221), China Postdoctoral Science Foundation funded project (2016M591926; 2017T100409), Jiangsu Province, Key Research & Development Plan of Jiangsu Province (BE2016646), Jiangsu provincial Commission of Health and Family Planning (Q201608), Jiangsu Provincial Medical Youth Talent (2016786) and Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX17_1733).
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492-507
2. Sukhdeo K, Hambardzumyan D, Rich JN. Glioma development: where did it all go wrong? Cell. 2011;146:187-88
3. Nicholas S, Mathios D, Ruzevick J, Jackson C, Yang I, Lim M. Current trends in glioblastoma multiforme treatment: radiation therapy and immune checkpoint inhibitors. Brain Tumor Res Treat. 2013;1:2-8
4. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987-96
5. Bernsen HJ, Rijken PF, Peters H, Raleigh JA, Jeuken JW, Wesseling P. et al. Hypoxia in a human intracerebral glioma model. J Neurosurg. 2000;93:449-54
6. Brown JM. Tumor hypoxia in cancer therapy. Methods Enzymol. 2007;435:297-321
7. Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro-oncology. 2005;7:134-53
8. Hockel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst. 2001;93:266-76
9. Zhou J, Schmid T, Schnitzer S, Brune B. Tumor hypoxia and cancer progression. Cancer Lett. 2006;237:10-21
10. Evans SM, Judy KD, Dunphy I, Jenkins WT, Hwang WT, Nelson PT. et al. Hypoxia is important in the biology and aggression of human glial brain tumors. Clin Cancer Res. 2004;10:8177-84
11. Siegal T, Charbit H, Paldor I, Zelikovitch B, Canello T, Benis A. et al. Dynamics of circulating hypoxia-mediated miRNAs and tumor response in patients with high-grade glioma treated with bevacizumab. J Neurosurg. 2016;125:1008-1015
12. Adams GE, Flockhart IR, Smithen CE, Stratford IJ, Wardman P, Watts ME. Electron-affinic sensitization. VII. A correlation between structures, one-electron reduction potentials, and efficiencies of nitroimidazoles as hypoxic cell radiosensitizers. Radiat Res. 1976;67:9-20
13. Adams GE, Cooke MS. Electron-affinic sensitization. I. A structural basis for chemical radiosensitizers in bacteria. Int J Radiat Biol Relat Stud Phys Chem Med. 1969;15:457-71
14. Overgaard J. Hypoxic radiosensitization: Adored and ignored. J Clin Oncol. 2007;25:4066-74
15. Feketeova L, Postler J, Zavras A, Scheier P, Denifl S, O'Hair RAJ. Decomposition of nitroimidazole ions: experiment and theory. Phys Chem Chem Phys. 2015;17:12598-607
16. Cyb AF, Chmelevskaja ZI, Kudrjavceva GT. Clinical experiences with the use of metronidazole in the radiotherapy of cancer patients. Radiobiol Radiother. 1985;26:339-42
17. Eyre HJ, Ohlsen JD, Frank J, LoBuglio AF, McCracken JD, Weatherall TJ. et al. Randomized trial of radiotherapy versus radiotherapy plus metronidazole for the treatment metastatic cancer to brain. A Southwest Oncology Group study. J Neurooncol. 1984;2:325-30
18. Urtasun RC, Miller JD, Frunchak V, Koziol D, Band PR, Chapman JD. et al. Radiotherapy pilot trials with sensitizers of hypoxic cells: metronidazole in supratentorial glioblastomas. Br J Radiol. 1977;50:602-3
19. Voronina SS, Pelevina, II. Increasing the effectiveness of tumor radiotherapy with metronidazole. Med Radiol. 1977;22:37-43
20. Frytak S, Moertel CH, Childs DS. Neurologic toxicity associated with high-dose metronidazole therapy. Ann Intern Med. 1978;88:361-2
21. Urtasun RC, Sturmwind J, Rabin H, Band PR, Chapman JD. Letter: "High-dose" metronidazole: a preliminary pharmacological study prior to its investigational use in clinical radiotherapy trials. Br J Radiol. 1974;47:297-9
22. Urtasun R, Band P, Chapman JD, Feldstein ML, Mielke B, Fryer C. Radiation and high-dose metronidazole in supratentorial glioblastomas. N Engl J Med. 1976;294:1364-7
23. Li Y, Xiao K, Zhu W, Deng W, Lam KS. Stimuli-responsive cross-linked micelles for on-demand drug delivery against cancers. Adv Drug Deliv Rev. 2014;66:58-73
24. Mura S, Nicolas J, Couvreur P. Stimuli-responsive nanocarriers for drug delivery. Nature Materials. 2013;12:991-1003
25. Chen Y, Zhang M, Jin H, Li D, Xu F, Wu A. et al. Glioma dual-targeting nanohybrid protein toxin constructed by intein-mediated site-specific ligation for multistage booster delivery. Theranostics. 2017;7:3489-503
26. Deng C, Zhang Q, Fu Y, Sun X, Gong T, Zhang Z. Coadministration of oligomeric hyaluronic acid-modified liposomes with tumor-penetrating peptide-iRGD enhances the antitumor efficacy of doxorubicin against melanoma. ACS Appl Mater Interfaces. 2017;9:1280-92
27. Liu H, Xie Y, Zhang Y, Cai Y, Li B, Mao H. et al. Development of a hypoxia-triggered and hypoxic radiosensitized liposome as a doxorubicin carrier to promote synergetic chemo-/radio-therapy for glioma. Biomaterials. 2017;121:130-43
28. Zhao Y, Ren W, Zhong T, Zhang S, Huang D, Guo Y. et al. Tumor-specific pH-responsive peptide-modified pH-sensitive liposomes containing doxorubicin for enhancing glioma targeting and anti-tumor activity. J Control Release. 2016;222:56-66
29. Huang Y, Liu W, Gao F, Fang X, Chen Y. c(RGDyK)-decorated Pluronic micelles for enhanced doxorubicin and paclitaxel delivery to brain glioma. Int J Nanomedicine. 2016;11:1629-41
30. Wang X, Liu G, Hu J, Zhang G, Liu S. Concurrent block copolymer polymersome stabilization and bilayer permeabilization by stimuli-regulated "traceless" crosslinking. Angew Chem Int Ed Engl. 2014;53:3138-42
31. Qian X, Long L, Shi Z, Liu C, Qiu M, Sheng J. et al. Star-branched amphiphilic PLA-b-PDMAEMA copolymers for co-delivery of miR-21 inhibitor and doxorubicin to treat glioma. Biomaterials. 2014;35:2322-35
32. Shen Y, Jin E, Zhang B, Murphy CJ, Sui M, Zhao J. et al. Prodrugs forming high drug loading multifunctional nanocapsules for intracellular cancer drug delivery. J Am Chem Soc. 2010;132:4259-65
33. Yu J, Zhang Y, Ye Y, DiSanto R, Sun W, Ranson D. et al. Microneedle-array patches loaded with hypoxia-sensitive vesicles provide fast glucose-responsive insulin delivery. Proc Natl Acad Sci USA. 2015;112:8260-5
34. Qian C, Yu J, Chen Y, Hu Q, Xiao X, Sun W. et al. Light-activated hypoxia-responsive nanocarriers for enhanced anticancer therapy. Adv Mater. 2016;28:3313-20
35. Thambi T, Deepagan VG, Yoon HY, Han HS, Kim SH, Son S. et al. Hypoxia-responsive polymeric nanoparticles for tumor-targeted drug delivery. Biomaterials. 2014;35:1735-43
36. Gonias SL, Campana WM. LDL receptor-related protein-1: a regulator of inflammation in atherosclerosis, cancer, and injury to the nervous system. Am J Pathol. 2014;184:18-27
37. Demeule M, Currie JC, Bertrand Y, Che C, Nguyen T, Regina A. et al. Involvement of the low-density lipoprotein receptor-related protein in the transcytosis of the brain delivery vector angiopep-2. J Neurochem. 2008;106:1534-44
38. Gao S, Tian H, Xing Z, Zhang D, Guo Y, Guo Z. et al. A non-viral suicide gene delivery system traversing the blood brain barrier for non-invasive glioma targeting treatment. J Control Release. 2016;243:357-69
39. Edwards DI. Nitroimidazole drugs-action and resistance mechanisms. I. Mechanisms of action. J Antimicrob Chemother. 1993;31:9-20
40. Kang L, Fan B, Sun P, Huang W, Jin M, Wang Q. et al. An effective tumor-targeting strategy utilizing hypoxia-sensitive siRNA delivery system for improved anti-tumor outcome. Acta Biomater. 2016;44:341-54
41. Zhu Z, Gao N, Wang H, Sukhishvili SA. Temperature-triggered on-demand drug release enabled by hydrogen-bonded multilayers of block copolymer micelles. J Control Release. 2013;171:73-80
42. Baumann RP, Penketh PG, Seow HA, Shyam K, Sartorelli AC. Generation of oxygen deficiency in cell culture using a two-enzyme system to evaluate agents targeting hypoxic tumor cell. Radiat Res. 2008;170:651-60
43. Yao H, Qiu H, Shao Z, Wang G, Wang J, Yao Y. et al. Nanoparticle formulation of small DNA molecules, Dbait, improves the sensitivity of hormone-independent prostate cancer to radiotherapy. Nanomedicine. 2016;12:2261-71
44. Liu H, Cai Y, Zhang Y, Xie Y, Qiu H, Hua L. et al. Development of a hypoxic radiosensitizer-prodrug liposome delivery DNA repair inhibitor Dbait combination with radiotherapy for glioma therapy. Adv Healthc Mater. 2017:6 (12)
45. Li Y, Baiyang L, Leran B, Zhen W, Yandong X, Baixiang D. et al. Reduction-responsive PEtOz-SS-PCL micelle with tailored size to overcome blood-brain barrier and enhance doxorubicin antiglioma effect. Drug Deliv. 2017;24:1782-90
46. Liu HM, Zhang YF, Xie YD, Cai YF, Li BY, Li W. et al. Hypoxia-responsive ionizable liposome delivery siRNA for glioma therapy. Int J Nanomedicine. 2017;12:1065-83
47. Lee SH, Mok H, Lee Y, Park TG. Self-assembled siRNA-PLGA conjugate micelles for gene silencing. J Control Release. 2011;152:152-8
48. Liu H, Li Y, Mozhi A, Zhang L, Liu Y, Xu X. et al. SiRNA-phospholipid conjugates for gene and drug delivery in cancer treatment. Biomaterials. 2014;35:6519-33
49. Chan JM, Zhang L, Yuet KP, Liao G, Rhee JW, Langer R. et al. PLGA-lecithin-PEG core-shell nanoparticles for controlled drug delivery. Biomaterials. 2009;30:1627-34
50. Zhang L, Chan JM, Gu FX, Rhee JW, Wang AZ, Radovic-Moreno AF. et al. Self-assembled lipid-polymer hybrid nanoparticles: a robust drug delivery platform. ACS Nano. 2008;2:1696-702
51. Bertrand Y, Currie JC, Poirier J, Demeule M, Abulrob A, Fatehi D. et al. Influence of glioma tumour microenvironment on the transport of ANG1005 via low-density lipoprotein receptor-related protein 1. Br J Cancer. 2011;105:1697-707
52. Thoms J, Bristow RG. DNA repair targeting and radiotherapy: a focus on the therapeutic ratio. Semi Radiat Oncol. 2010;20:217-22
53. Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008;36:5678-94
54. Mariotti LG, Pirovano G, Savage KI, Ghita M, Ottolenghi A, Prise KM. et al. Use of the gamma-H2AX assay to investigate DNA repair dynamics following multiple radiation exposures. PloS One. 2013;8:e79541
55. Zhang J, He Y, Shen X, Jiang D, Wang Q, Liu Q. et al. γ-H2AX responds to DNA damage induced by long-term exposure to combined low-dose-rate neutron and γ-ray radiation. Mutat Res Genet Toxicol Environ Mutagen. 2016;795:36-40
56. Gao X, Yue Q, Liu Y, Fan D, Fan K, Li S. et al. Image-guided chemotherapy with specifically tuned blood brain barrier permeability in glioma margins. Theranostics. 2018;8:3126-37
57. Shergalis A, Bankhead RA, Luesakul U, Muangsin N, Neamati N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacological Reviews. 2018;70:412-45
58. Lee JH, Chen KJ, Noh SH, Garcia MA, Wang H, Lin WY. et al. On-demand drug release system for in vivo cancer treatment through self-assembled magnetic nanoparticles. Angew Chem Int Ed Engl. 2013;52:4384-8
59. Fulop Z, Gref R, Loftsson T. A permeation method for detection of self-aggregation of doxorubicin in aqueous environment. Int J Pharm. 2013;454:559-61
Author contact
![]() Corresponding authors: Hongmei Liu, PhD. Institute of Nervous System Diseases, Xuzhou Medical University, Xuzhou, 221002, PR China. Phone: 86-0516-85587335; Fax: 86-0516-85587335; liuhongmei816com. Rutong Yu, PhD, MD. Institute of Nervous System Diseases, Xuzhou Medical University, Xuzhou, 221002, PR China. Phone: 86-0516-85587335; Fax: 86-0516-85587335; yu.rutongcom. Jun Lu, PhD. Key Laboratory for Biotechnology on Medicinal Plants of Jiangsu Province, School of Life Science, Jiangsu Normal University, Xuzhou, 221116, PR China. Phone: 86-0516-83500348; Fax: 86-0516-83500348; lu-jun75com
Corresponding authors: Hongmei Liu, PhD. Institute of Nervous System Diseases, Xuzhou Medical University, Xuzhou, 221002, PR China. Phone: 86-0516-85587335; Fax: 86-0516-85587335; liuhongmei816com. Rutong Yu, PhD, MD. Institute of Nervous System Diseases, Xuzhou Medical University, Xuzhou, 221002, PR China. Phone: 86-0516-85587335; Fax: 86-0516-85587335; yu.rutongcom. Jun Lu, PhD. Key Laboratory for Biotechnology on Medicinal Plants of Jiangsu Province, School of Life Science, Jiangsu Normal University, Xuzhou, 221116, PR China. Phone: 86-0516-83500348; Fax: 86-0516-83500348; lu-jun75com