Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results and discussion
Conclusion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(11):3111-3125. doi:10.7150/thno.24613 This issue Cite
Research Paper
Dual-functional protein for one-step production of a soluble and targeted fluorescent dye
Yunjie Xiao1,*, Qian Zhang2,*, Yanyan Wang3,*, Bin Wang1, Fengnan Sun2, Ziyu Han3, Yaqing Feng4, Haitao Yang1,5, Shuxian Meng2 ![]() , Zefang Wang1
, Zefang Wang1 ![]()
1. School of Life Sciences, Tianjin University, Tianjin 300072, China.
2. School of Chemical Engineering and Technology, Tianjin University, Tianjin 300072, China.
3. College of Precision Instrument and Opto-electronics Engineering, Tianjin University, Tianjin 300072, China.
4. Collaborative Innovation Center of Chemical Science and Engineering, Tianjin University, Tianjin 300072, China.
5. Tianjin International Joint Academy of Biotechnology and Medicine, Tianjin 300457, China.
* These authors contributed equally to this work.
Received 2017-12-28; Accepted 2018-3-17; Published 2018-4-30
Abstract

Low water solubility and poor selectivity are two fundamental limitations that compromise applications of near-infrared (NIR) fluorescent probes.
Methods: Here, a simple strategy that can resolve these problems simultaneously was developed by using a novel hybrid protein named RGD-HFBI that is produced by fusion of hydrophobin HFBI and arginine-glycine-aspartic acid (RGD) peptide. This unique hybrid protein inherits self-assembly and targeting functions from HFBI and RGD peptide respectively.
Results: Boron-dipyrromethene (BODIPY) used as a model NIR dye can be efficiently dispersed in the RGD-HFBI solution by simple mixing and sonication for 30 min. The data shows that self-assembled RGD-HFBI forms a protein nanocage by using the BODIPY as the assembly template. Cell uptake assay proves that RGD-HFBI/BODIPY can efficiently stain αvβ3 integrin-positive cancer cells. Finally, in vivo affinity tests fully demonstrate that the soluble RGD-HFBI/BODIPY complex selectively targets and labels tumor sites of tumor-bearing mice due to the high selectivity of the RGD peptide.
Conclusion: Our one-step strategy using dual-functional RGD-HFBI opens a novel route to generate soluble and targeted NIR fluorescent dyes in a very simple and efficient way and may be developed as a general strategy to broaden their applications.
Keywords: NIR fluorescent probes, RGD peptide, self-assembly, protein nanocage
Introduction
Near-infrared (NIR) fluorescent probes have become powerful tools for non-invasive monitoring of various biologically important processes in vivo [1-7]. NIR probes have the advantages of deep tissue penetration, minimum photodamage and minimum interference from auto-fluorescence by biomolecules present in living systems [8, 9]. Accordingly, there is considerable interest in developing novel NIR fluorescent probes to satisfy the increasing demands of their applications including diagnostics and therapeutics [10-12]. However, NIR fluorescent probes do suffer from several major limitations including low water solubility and poor selectivity, which dramatically compromise their applications in diagnostics and therapeutics [6, 13, 14].
Up to now, tremendous efforts have been made to overcome the drawbacks mentioned above of NIR fluorescent probes, but in two separate steps [15-18]. In the first step, numerous polar groups like sulfonate, glycol, and carboxylate are linked to the core structures of NIR dyes to gain solubility in aqueous solutions [19, 20]. Alternatively, a hydrophilic shell such as a phospholipid monolayer could be assembled onto the surface of NIR probes to increase their water solubility [21, 22]. After solving the solubility problem, the next step is to improve the selectivity of NIR fluorescent probes. The current predominant strategy is through conjugation of specific ligands to the NIR probes, such as proteins, peptides and small molecules [23-27]. These ligands have enough binding affinity to recognize and bind the target molecules resulting in high selectivity of the conjugated NIR probes. After these two-step modifications, NIR fluorescent probes do obtain high water solubility and selectivity. However, we have to admit that this two-step strategy is not only somewhat complicated and time-consuming, but also requires careful selection of conjugation or labeling techniques to prevent any disruption of the intrinsic structure of the NIR fluorescent probes [28, 29], thereby compromising their exciting properties. Therefore, a one-step strategy that can render NIR fluorescent probes soluble and selective is highly desired to facilitate their applications.
To meet the requirement of a one-step strategy, one has to find a material that can render NIR fluorescent probes soluble and selective simultaneously. Hydrophobins are small-secreted proteins produced by filamentous fungi [30-32]. Strikingly, they expose most of their hydrophobic amino acids on the protein surface, forming a “hydrophobic patch” [33, 34], rather than burying their hydrophobic amino acids into their inner structures like normal proteins. This novel property makes hydrophobins natural amphiphilic molecules that can self-assemble at various hydrophobic/hydrophilic interfaces producing, a stable and well-ordered protein film 10 nm in thickness and reversing the features of the interface coated by them [35-40]. Moreover, hydrophobins with new properties and functionalities can be produced in a controlled manner by protein engineering [41, 42]. Therefore, we can easily produce an engineered hydrophobin with target selectivity by adding a specific ligand through recombinant DNA techniques. We intentionally chose the RGD tripeptide as the ligand in our study. We believe the hybrid hydrophobin-RGD fusion protein preserving dual abilities can render NIR fluorescent probes soluble and selective simultaneously.
To test our hypothesis, we firstly produced and purified hybrid RGD-HFBI fusion protein in Pichia pastoris (P. pastori). Then we systemically investigated the solubility and targeting abilities of RGD-HFBI fusion protein by studying its interaction with a synthesized NIR BODIPY derivative in different environments, including cell culture and animal models. Our results show that self-assembled RGD-HFBI fusion protein can readily modify NIR fluorescent probes with high solubility and selectivity in one step by simple mixing and sonication. To our knowledge, this is the first demonstration of modification of NIR fluorescent dyes by biomolecules with dual functions. Furthermore, our one-step strategy using a self-assembled hydrophobin hybrid opens a novel route to generate solubilized and targeted NIR fluorescent dyes in a very simple and efficient way and may be developed as a general strategy for broadening their applications.
Methods
Production of recombinant RGD-HFBI fusion protein
RGD-HFBI protein was constructed by fusion of RGD peptide and hydrophobin HFBI with a flexible linker (GGGGSGGGGS). The plasmid pPIC9-rgd-hfbI was transformed into P. pastoris GS115 His- cells (Invitrogen, Beijing, China) by electroporation. To obtain positive clones with high-level protein expression, selected P. pastoris positive clones were cultured in 25 mL of buffered minimal glycerol at 300 rpm at 30 °C until the OD600 reached 6.0. Then, the medium was exchanged with 100 mL buffered minimal methanol, and protein expression was induced by 0.5% (v/v) methanol at 28 °C for 96 h at 250 rpm. The supernatant was collected immediately after 96 h induction. Finally, 16% Tricine-SDS-PAGE was employed to analyze the protein content in the supernatant.
Large-scale cultivation (1 L) was performed after the clone with the highest level of RGD-HFBI expression was selected. When the cultivation was over, the culture was centrifuged at 7500 × g to get the supernatant. The resulting supernatant was concentrated by ultrafiltration with a 3 kDa molecular weight cut-off (Millipore, China). After ultrafiltration, the supernatant was lyophilized for reversed-phase high-performance liquid chromatography (RP-HPLC) purification. A Vydac C4 reversed-phase column (4.6 × 250 mm, GRACE, China) was used for purification of RGD-HFBI. Lyophilized protein powder was dissolved in 40% acetonitrile containing 0.1% TFA. Then, the supernatant was loaded and eluted with a 20-70% (v/v) acetonitrile gradient containing 0.1% TFA with a flow rate of 1 mL/min. The elution was monitored by UV absorption at 215 nm. All the fractions from RP-HPLC were collected. The purified RGD-HFBI was identified by 16% Tricine-SDS-PAGE.
Characterization of recombinant RGD-HFBI fusion protein
The self-assembly ability of RGD-HFBI was characterized by transmission electron microscopy (TEM) and water contact angle (WCA) measurements. For the TEM, 3 μL of RGD-HFBI (or native HFBI) solution in water (200 μg/mL) was placed onto the surface of a carbon-coated formvar film on a copper grid (Zhongjingkeyi Technology, Beijing, China) and left to dry overnight at room temperature. Then, the grid was examined by field emission TEM at 200 kV (JEM-2100F, JEOL, Japan).
WCA measurements of purified RGD-HFBI (or native HFBI) on hydrophobic and hydrophilic surfaces were performed using pieces of polystyrene and mica sheets, respectively. Both surfaces were coated with 20 μL (0.02 mg/mL) RGD-HFBI solution and incubated at room temperature for 30 min. After removing the solution gently, all the sheets were dried in a nitrogen stream and kept at room temperature overnight. WCA was measured with a 5 μL water droplet on the modified surfaces at room temperature. At least three water droplet readings were analyzed on different areas of the sample surfaces. The surface rinsed with water was also under the evaluation of WCA measurements.
Synthesis and characterizations of the BODIPY derivative
All solvents and starting materials were commercially available and were used without further purification (unless specially mentioned). Silica gel for column chromatography (CC) was 300-400 mesh. 13C and 1H NMR spectra were recorded on a Bruker AV400 MHz spectrometer in CDCl3 or with tetramethylsilane as a reference. Electrospray ionization-MS spectra were determined with a Shimadzu LCMS-2020 instrument. The UV-Vis spectra of dyes in DMSO solution were measured using Shimadzu UV-1800 in a 10 mm quartz cell spectrometer.
Methyl 4-formylbenzoate (0.864 g, 5.27 mmol) and 32 mL CH2Cl2 were added to a 100 mL round-bottom flask, and 1 mL (10.0 mmol) 2, 4-Dimethylpyrrole was added. The mixture was stirred in the dark for 3 h at room temperature. Nitrogen gas was introduced for 30 min to remove dissolved oxygen in the flask. Then, 0.08 mL CF3COOH was added with a syringe slowly under the nitrogen atmosphere. Samples were taken every 30 min during the reaction process for thin-layer chromatography (TLC) analysis. The nitrogen atmosphere was removed when the raw material 2, 4-dimethylpyrrole disappeared (about 4 h). DDQ (2, 3-dichloro-5, 6-dicyano-1, 4-benzoquinone) (1.2 g, 5.28 mmol) was dissolved in 5 mL CH2Cl2 and 5 mL THF. The resultant mixture was added to the reaction flask with constant-voltage funnels and was stirred in the dark for 1 h at room temperature. Then, 8.6 mL triethylamine was added by dropping, and the reaction was kept for 5 min under the condition of the ice bath. Next, 8.6 mL boron trifluoride etherate was added by dropping, and the reaction liquid was stirred in the dark for 3 h without the ice bath. After that, the mixture was washed with water and extracted with dichloromethane. The product was isolated by column chromatography using CH2Cl2/hexane = 1/2 as eluent. The second orange-yellow band was taken to recrystallize with CH2Cl2 and C6H12. Finally, BOD (210 mg) was obtained in the above reaction. 1H NMR (400 MHz, CDCl3): δ 8.18 (d, J = 8.0 Hz, 2H), 7.41 (d, J = 8.0 Hz, 2H), 5.99 (s, 2H), 3.97 (s, 3H), 2.56 (s, 6H), 1.36 (s, 6H). MS (ESI): [M]+ calcd for C21H21BF2N2O2, 382.1660, found [M + H] = 383.1, [M + Na] = 405.1, [M + K] = 421.1. 13C NMR (400 MHz, CDCl3) δ 166.44, 155.96, 142.86, 139.80, 130.76, 130.35, 128.34, 121.48, 52.39, 14.60, 14.50.
BOD (20 mg, 0.0524 mmol) and N-ethyl-3-formyl-dibenzyl (42.7 mg, 0.17 mmol) were dissolved in benzene. 0.1 mL catalyst piperidine and 0.1 mL acetic acid were injected with a syringe at the same time. The mixture was heated to 90 °C and stirred for 12 h. The product was isolated by column chromatography using CH2Cl2/hexane = 2/1 as the eluent. The blue band was taken to recrystallize with CH2Cl2 and C6H12. Finally, the dark green solid (18.7 mg) was in the above reaction. 1H NMR (400 MHz, CDCl3): δ 8.18 (d, J = 8.0 Hz, 2H), 7.57-7.60 (m, 2H), 7.36-7.44 (m, 6H), 7.14-7.19 (m, 6H), 7.07-7.10 (m, 4H), 6.96-6.97 (m, 2H), 6.60 (s, 2H), 3.97 (s, 3H), 3.82-3.87 (q, J = 6.8 Hz, 4H), 3.20 (s, 8H), 1.40 (s, 6H), 1.18 (t, J = 6.8 Hz, 6H). MS (ESI): [M]+ calcd for C55H51BF2N4O2 848.4230, found 848.0673. 13C NMR (400 MHz, CDCl3) δ 166.56, 153.04, 148.78, 136.43, 135.50, 133.03, 130.21, 129.89, 129.59, 129.25, 129.02, 126.45, 126.09, 122.98, 120.90, 119.88, 116.97, 52.39, 45.63, 33.02, 31.77, 14.77, 13.95.
Preparation of RGD-HFBI/BODIPY complex
Four concentrations of RGD-HFBI solution (50, 100, 150 and 200 μg/mL) were prepared by using phosphate buffers (pH 7.4). Then, 1 mg of BODIPY was dissolved in four concentrations of RGD-HFBI solutions respectively. The resultant mixtures were dispersed by ultrasonic agitation (120 W) for 30 min in iced water, and then nitrogen gas was introduced into the dispersion for 5 min to remove the free HFBI-RGD. Finally, the resultant dispersion was subjected to ultra-centrifugation at 9000 × g for 30 min at 4 °C, and 80% of the supernatant was collected and lyophilized for further analysis.
Characterization of RGD-HFBI/BODIPY complex
Dynamic light scattering and zeta potential measurements were carried out using a Zetasizer Nano ZS (Malvern Instrument, Malvern, Worcestershire, UK). TEM images were obtained by field emission TEM at 200 kV (JEM-2100F, JEOL, Japan). The chemical compositions of BODIPY and HFBI/BODIPY were analyzed using X-ray photoelectron spectroscopy (XPS) Apparatus (PHI-5300). The experiment conditions were as follows: the energy of excitation source monochromatic Mg-Kα radiation was 1253.6 eV, and the survey scan range was 0-1100 eV. Fourier-transform infrared (FTIR) spectra were obtained on a BRUKER IFS 55 FTIR system using the KBr disk method. The transmittance spectra were recorded at a resolution of 2 cm-1 between 4000 and 400 cm-1.
Cytotoxicity measurement
Cell viability measurements were performed by MTT analysis. MCF-7 (8 × 103 cells per well) and HeLa (7 × 103 cells per well) cells were cultured in DMEM medium with 10% fetal bovine serum. U-87MG (8 × 103 cells per well) cell was cultured in RPMI 1640 medium with 10% fetal bovine serum. Cells were seeded in 96-well flat-bottomed plates and incubated for 24 h at 37 °C under 5% CO2. After 24 h of cell attachment, the plates were washed with 100 µL of PBS per well. The cells were then cultured in a medium with ten different concentrations (0, 0.1, 1, 5, 10, 25, 50, 100, 250 and 500 µM) of RGD-HFBI/BODIPY, HFBI/BODIPY and BODIPY for 48 h. Cells in a culture medium without fluorescent dyes were used as the control. MTT (10 µL, 5 mg/mL) in PBS was subsequently added to each well. The plates were then incubated at 37 °C for 4 h in a 5% CO2 humidified incubator. The medium was carefully removed, and the products were lysed in 200 µL of DMSO. The plate was shaken for 10 min, and the absorbance was measured at 490 nm and 570 nm using a microplate reader (EnSpire Multilabel Reader, PerkinElmer).
Flow cytometry analysis
The cell binding and internalization were analyzed by flow cytometry. U-87MG, MCF-7 and Hela cells were grown separately in 6-well plates with a seeding concentration of 3 × 105 cells per well for 24 h. After that, cells were incubated with RGD-HFBI/BODIPY, HFBI/BODIPY and BODIPY for 4 h. The concentration of all the probes was 10 μM. Afterwards, the plates were washed with 1 mL of PBS (pH 7.4) per well three times to remove probes that were still in the medium, and then the cells underwent digestion with trypsin. Next, the cells were collected by centrifugation and resuspended in 1 mL of PBS, which was repeated 3 times. Finally, 2 mL of PBS was used to resuspend the cells. The cellular uptake of the fluorescence was measured on a BD FACSCanto II flow cytometer (BD Biosciences, USA). Approximately 10,000 events were counted for each sample.
Confocal microscopy analysis
The MCF-7 (αvβ3 integrin negative), U-87MG (high αvβ3 integrin expression) and HeLa (low αvβ3 integrin expression) cells were grown separately on glass coverslips in 48-well plates with a seeding concentration of 1 × 103 cells per well for 24 h. After that, cells were incubated with RGD-HFBI/BODIPY, HFBI/BODIPY and BODIPY at a concentration 10 μM, and then cultured for 4 h. Afterwards, the plates were washed with 100 μL of PBS per well three times to remove probes that were still in the medium. Before confocal microscopy (UltraVIEW VoX, PerkinElmer) imaging, all the samples were stained with DAPI for 5 min and washed with 100 μL of PBS per well three times. The excitation wavelengths for RGD-HFBI/BODIPY, HFBI/BODIPY and BODIPY were 405 nm and 633 nm.
Stability assays of RGD-HFBI/BODIPY complex in vivo and in vitro
For the in vivo assay, blood samples from rats were directly drawn by venipuncture at different time points (2, 4, 6, 8, 24, 48 and 72 h) after administration of RGD-HFBI/BODIPY complex. Then, the amounts of RGD-HFBI/BODIPY complex and free RGD-HFBI in serum were measured by RP-HPLC. The ratio between the amount of RGD-HFBI/BODIPY complex and free RGD-HFBI was considered an indicator of the serum stability of the complex. For the in vitro assay, the RGD-HFBI/BODIPY complex was added to rat serum directly. The remaining protocol was the same as the in vivo assay.
In vivo imaging in animal tumor models
Female nude mice (4-6 weeks) were obtained from the Institute of Radiation Medicine Chinese Academy of Medical Sciences. The U-87MG tumor and HeLa tumor models were generated via subcutaneous injection of 5.0 × 106 tumor cells in the left forelimb of the mice. Caliper measurements of the perpendicular axes of the tumor were performed to monitor the tumor growth. The mouse weights were also measured. The treatment was initiated when the tumor reached a diameter of approximately 8.0 mm (3-4 weeks after inoculation). The HFBI-RGD/BODIPY and HFBI/BODIPY fluorescent probes (17.7 nM, 200 μL) were injected via the tail vein. The mice were anesthetized with oxygen/air mixture containing 2% isoflurane and the in vivo images were captured at different points of time (2, 4, 6, 8, 24, 48 and 72 h). All images were collected with a Cri Maestro In vivo imaging system (Cri, Woburn, MA), with an excitation wavelength of 605 nm and emission wavelengths from 615 nm to 800 nm. To eliminate interference from the background, ex vivo imaging procedures were carried out. Thereafter, tumor-bearing mice were sacrificed, and then tumor and main organs (heart, liver, spleen, lung and kidneys) were harvested for isolated organ imaging that was performed with the same instrument under the same conditions as the in vivo imaging. All experimental procedures were reviewed and approved by the Animal Use and Care Committee for Research and Education of the Institute of Radiation Medicine Chinese Academy of Medical Sciences (Tianjin, China) due to the lack of animal facility in Tianjin University, and were in accordance with the guidelines provided by the National Institute of Health.
Statistical analysis
The assays were performed at least in triplicate on separate occasions. The data collected in this study are expressed as the mean value ± standard deviation. In all cases, p < 0.05 was considered significant.
Results and discussion
Production and characterization of recombinant RGD-HFBI fusion protein
Figure 1A shows the amphiphilic structure of native hydrophobin HFBI. The sun-yellow part represents the “hydrophobic patch” composed of about 80% of the hydrophobic residues of this protein [33, 43]. This hydrophobic patch is the basis for binding of HFBI to various hydrophobic surfaces by strong hydrophobic interactions [44]. In Figure 1B, a RGD motif is linked with the N terminus of HFBI protein via a flexible linker, and the resultant hybrid protein was named RGD-HFBI. Accordingly, we proposed that RGD-HFBI containing two distinct biological domains owns dual functions based on its structural properties. The first function derived from the HFBI part was the ability to self-assemble on different hydrophobic surfaces to make them hydrophilic. Another one inherited from the RGD motif was to facilitate recognition and binding to the target αvβ3 integrin protein on the surface of tumor angiogenic endothelial cells [45, 46]. Based on those two functions, we proposed that RGD-HFBI has the abilities to impart hydrophobic materials with solubility and targeting behaviors. To test our hypothesis, we firstly produced the RGD-HFBI protein in P. pastoris system. After several rounds of selection, a clone with the highest amount of protein production was picked to produce RGD-HFBI at large scale (Figure S1). Figure 1C shows the purification results of RGD-HFBI and native HFBI with RP-HPLC system. The retention volume of RGD-HFBI was quite close to that of native HFBI, indicating the similar polarities of those two proteins. Fractions corresponding to the predominant peak were lyophilized and analyzed by Tricine-SDS-PAGE. A single predominant band suggested the high purity of RGD-HFBI after RP-HPLC purification (Figure 1D).
(A) X-ray crystal structure of the amphiphilic hydrophobin HFBI [PDB ID 2FZ6]. The sun-yellow part in the surface of HFBI represents the hydrophobic patch that is the basis for binding to various hydrophobic surfaces by strong hydrophobic interactions. (B) A RGD motif is linked with the N terminus of HFBI protein via a flexible linker and the resultant hybrid protein was named RGD-HFBI, which owns dual functions based on its structural properties. The first function derived from the HFBI part is the ability to self-assemble and bind onto different hydrophobic surfaces. The second function inherited from the RGD motif is to facilitate recognition and binging to the target αvβ3 integrin protein on the surface of tumor angiogenic endothelial cells. (C) Purification results of RGD-HFBI and native HFBI with an RP-HPLC system. (D) Tricine-SDS-PAGE results of the purified RGD-HFBI and native HFBI. (E) Self-assembled RGD-HFBI film and native HFBI film on TEM copper grids. (F) WCA measurements of polystyrene and mica before and after modification with RGD-HFBI and native HFBI.
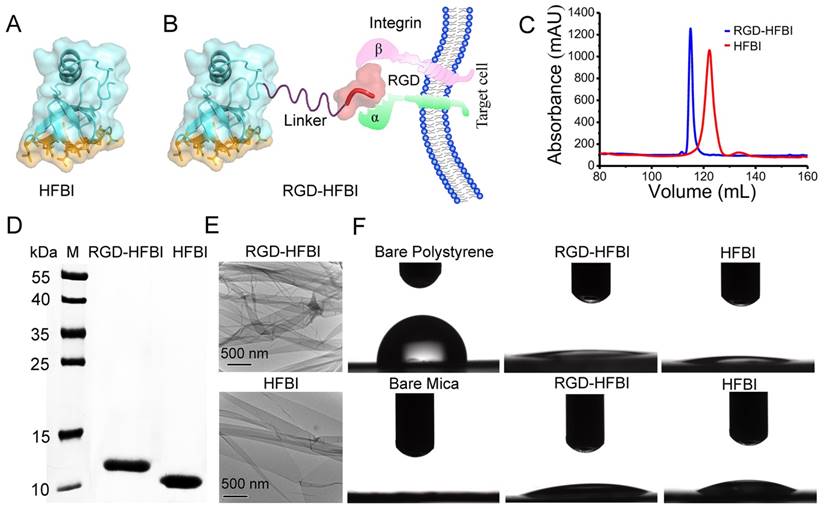
After the production and purification process, the self-assembly ability of RGD-HFBI was characterized by TEM and WCA measurements [47, 48]. Figure 1E shows a self-assembled RGD-HFBI film on a TEM copper grid. Like the native HFBI film, the RGD-HFBI film could form wrinkles or folds, implying the flexibility or elasticity of this film [49-51]. In our study, a surface is considered to be hydrophobic if its WCA is more than 60° while hydrophilic if it is lower. The WCA of bare and modified polystyrene and mica are shown in Figure 1F. The WCA of the bare polystyrene was about 87°, suggesting its natural hydrophobic property. However, the WCA of the polystyrene sharply decreased to 11° after modification with RGD-HFBI. The reversal of the surface property of polystyrene from hydrophobic to hydrophilic confirmed the self-assembly of RGD-HFBI onto the polystyrene surface. Several studies have proposed that when amphiphilic hydrophobin self-assembles on a hydrophobic surface, the “hydrophobic patch” exposed on its surface adheres onto the hydrophobic surface via strong hydrophobic interactions [52, 53]. Therefore, the hydrophilic part of hydrophobin is displayed to the outside, converting the hydrophobic surface into a hydrophilic one [54]. When RGD-HFBI covered the mica surface, the WCA increased from 4° to 17°. The hydrophobicity of mica surface was slightly improved. These results were very similar with those obtained from the native HFBI. Furthermore, we found that the WCAs on the polystyrene and mice surfaces little changed after extensive washing by pure water, indicating the stability of the assembled RGD-HFBI films (Figure S2). In summary, we concluded that RGD-HFBI fusion protein has a self-assembly ability like native HFBI and, therefore, can potentially modify hydrophobic materials.
The synthesis and characterization of BODIPY dye
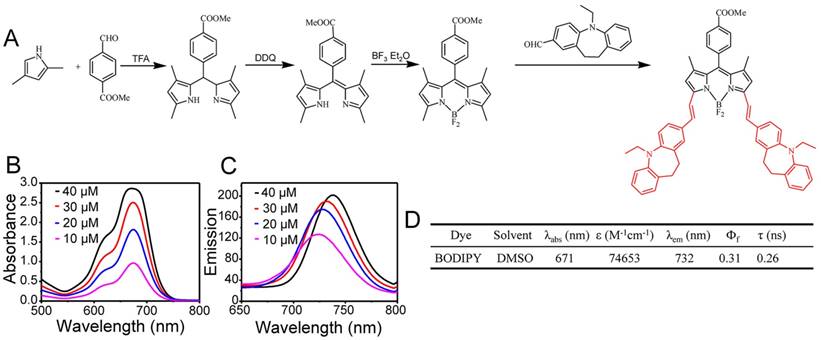
After the production of RGD-HFBI, we then designed and synthesized a new long-wavelength BODIPY dye that would be used as the model fluorescent dye in our following studies to test if RGD-HFBI could impart solubility and targeting abilities to hydrophobic NIR fluorescent probes. The design and synthesis of the BODIPY derivative are shown in Figure 2A. Briefly, the benzyl derivative was introduced to the 3- and 5-methyl sites through the Knoevenagel reaction. Then, an ethyl group was incorporated into BODIPY through the benzyl to enhance the solubility of the dye in non-polar solvents to facilitate preparation and characterization of this derivative. With a few steps, the BODIPY dye was obtained with a reasonable yield that was about 50% and was characterized by 13C NMR (Figure S3), 1H NMR (Figure S4) and MS (ESI) (Figure S5). We then dissolved this BODIPY derivative in DMSO at different concentrations to investigate its absorption and emission spectra. As shown in Figure 2B, the absorption spectrum of this dye contains a narrow spectral band with a major peak in the visible region at 671 nm. This BODIPY derivative in DMSO shows an emission spectrum (Figure 2C) with a maximum at 732 nm (excitation wavelength), with what could be Rayleigh and Tyndall scattering in the emission spectrum. The photophysical data of this BODIPY dye are summarized in Figure 2D.
(A) Synthetic procedure for the synthesis of the BODIPY derivative. (B-C) Absorption and fluorescence spectra of the BODIPY dye at different concentrations (10, 20, 30 and 40 μM) in DMSO. (D) Photophysical properties of the BODIPY dye.
RGD-HFBI disperses BODIPY in a concentration dependent manner
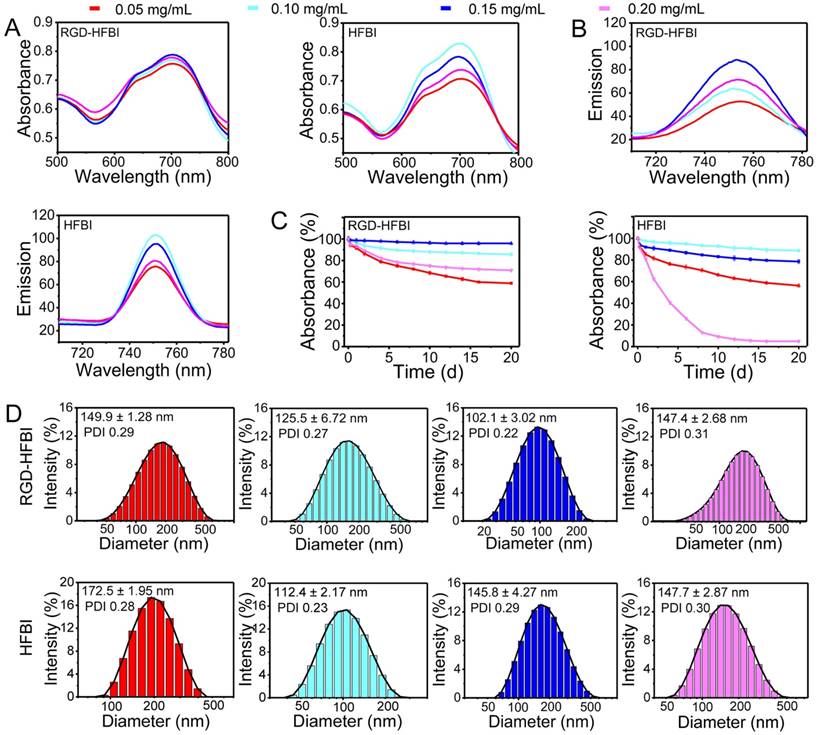
Considering that RGD-HFBI protein owns a self-assembly ability, we proposed that RGD-HFBI could modify hydrophobic BODIPY dye to gain solubility. To test this idea, we tried to disperse BODIPY using RGD-HFBI solutions at different concentrations. Figure 3A-B show results of absorption and emission spectra of the BODIPY in RGD-HFBI solutions at different concentrations. BODIPY gave the highest intensity both in the absorption and emission spectra when the protein concentration was at 0.15 mg/mL. The photophysical data of RGD-HFBI-treated BODIPY dye are summarized in Table 1. Both the fluorescence quantum yield and lifetime were increased to the maximum when BODIPY was dispersed in 0.15 mg/mL RGD-HFBI solution. Although we could not understand the detailed mechanism by which RGD-HFBI acted at this stage, these results give us clues that protein concentration might have a crucial impact on the dispersion of BODIPY. Therefore, the long-term stability of each sample was examined over 20 days. Figure S6 shows that the protein concentration had an obvious influence on the stability of the dispersed solutions. We quantified the stability of each sample by measuring its absorption at 699 nm. As shown in Figure 3C, there was almost no change in absorbance of the 0.15 mg/mL RGD-HFBI-treated sample within 20 days, indicating that a stable dispersion was formed immediately after sonication. For other samples, we found that the normalized absorptions kept on decreasing and finally stabilized after two weeks. The zeta potential of 0.15 mg/mL RGD-HFBI-treated BODIPY was around 40 mV indicating its relatively high stability [55] (Table 2). Regarding the other groups, the low zeta potential values suggested the instability of those solutions. Finally, the size of each sample was measured. As shown in Figure 3D, the smallest size of BODIPY was about 100 nm when BODIPY was dispersed in 0.15 mg/mL RGD-HFBI solution. The size of nanoparticles has a great effect on their behaviors like cell uptake and tumor-targeting ability. Normally, smaller sizes obtain better biodistribution and targeting results in vivo [56, 57]. Taking all the results together, we demonstrated that 0.15 mg/mL HFBI-RGD could disperse BODIPY effectively. When the protein concentration was less than 0.15 mg/mL, we believed that there was not enough hydrophobin to disperse the excessive BODIPY. However, when protein concentrations were above 0.15 mg/mL, the extra RGD-HFBI protein might compromise the stability of the resultant dispersions [58]. Therefore, we chose 0.15 mg/mL-treated BODIPY in the following studies. In addition, the best dispersion result of BODIPY was obtained by using 0.1 mg/mL native HFBI in the control group. This result strengthened the fact that hydrophobin concentration was a key factor influencing the outcome of the dispersion result as well.
Photophysical properties of RGD-HFBI- and native HFBI-treated BODIPY dye
| Dye | Dispersant | λabs (nm) | ε (M-1cm-1) | λem (nm) | Фf | τ (ns) |
|---|---|---|---|---|---|---|
| BODIPY | 0.05 mg/mL HFBI | 699 | 75385 | 753 | 0.32 | 1.04 |
| BODIPY | 0.10 mg/mL HFBI | 699 | 77796 | 753 | 0.39 | 1.36 |
| BODIPY | 0.15 mg/mL HFBI | 699 | 78793 | 753 | 0.37 | 1.34 |
| BODIPY | 0.20 mg/mL HFBI | 699 | 77302 | 753 | 0.33 | 1.28 |
| BODIPY | 0.05 mg/mL RGD-HFBI | 699 | 76842 | 753 | 0.34 | 1.07 |
| BODIPY | 0.10 mg/mL RGD-HFBI | 699 | 79861 | 753 | 0.38 | 1.33 |
| BODIPY | 0.15 mg/mL RGD-HFBI | 699 | 78181 | 753 | 0.41 | 1.38 |
| BODIPY | 0.20 mg/mL RGD-HFBI | 699 | 76762 | 753 | 0.32 | 1.31 |
Zeta potential of RGD-HFBI- and native HFBI-treated BODIPY dye
| Formulation | Concentration (mg/mL) | Zeta Potential (mV) |
|---|---|---|
| RGD-HFBI/BODIPY | 0.05 | 18.7 ± 1.51 |
| 0.10 | 28.4 ± 2.13 | |
| 0.15 | 39.9 ± 2.42 | |
| 0.20 | 22.5 ± 1.12 | |
| HFBI/BODIPY | 0.05 | 17.8 ± 1.25 |
| 0.10 | 32.3 ± 3.15 | |
| 0.15 | 26.9 ± 2.08 | |
| 0.20 | 8.93 ± 0.23 |
(A-B) Absorption and fluorescence spectra of the BODIPY dye solubilized in RGD-HFBI and native HFBI solution at different concentrations (0.05, 0.1, 0.15 and 0.2 mg/mL). (C) Stability of RGD-HFBI- and native HFBI-dispersed BODIPY solution over 20 days. (D) Particles size of RGD-HFBI- and native HFBI-treated BODIPY dye.
Characterization of RGD-HFBI/BODIPY complex
After solubilizing the BODIPY in RGD-HFBI solution, we used TEM to see if there were morphological changes of BODIPY. As shown in Figure 4A, we saw an irregular aggregate of BODIPY in pure water with a dimension of about 5 μm, which illustrated its intrinsic hydrophobic property. When BODIPY was dissolved in DMSO, the shape sharply changed to round, and the size was dramatically decreased to 600 nm (Figure S7). However, we found that BODIPY formed well-dispersed particles with a round shape and uniform size of about 108 nm (Figure S7) when solubilized in RGD-HFBI solution. This result was very similar to that obtained from the dynamic light scattering experiment. Furthermore, the size of RGD-HFBI-treated BODIPY was reduced by 50 times when compared with the huge aggregate in water. These results showed clearly that RGD-HFBI could disperse BODIPY efficiently. Moreover, we found that RGD-HFBI/BODIPY formed a core-shell heterostructure with BODIPY dye in the core area and the RGD-HFBI layer taking the shape of the BODIPY template. As shown in Figure 4A, a clear interface could be observed between the BODIPY core and the RGD-HFBI shell or film. The average thickness of the RGD-HFBI film was about 10 nm, which was similar to that of HFBI film formed on the BODIPY core, suggesting that RGD motif did not disturb the assembly of HFBI protein [59]. In addition, we found that there were plenty of small holes in the RGD-HFBI shell. Those holes obviously made the RGD-HFBI shell like an empty hollow “protein nanocage” with an embedded BODIPY core. Unlike the classic protein nanocages composed of multiple protein subunits that self-assemble with excellent precision forming a hollow caged structure of nanometer size, RGD-HFBI itself could self-assemble into a protein nanocage by using the BODIPY as the assembly template [60, 61]. So far, protein-based nanocages have been functionalized and applied into many applications including drug delivery, imaging and theranostics in cancer [62-64]. RGD-HFBI protein presents numerous reactive groups such as hydroxyls, amines, thiols, carboxylic acids and others. Therefore, it could provide active sites for easy further surface modification for different purposes. Figure 4B shows the RGD-HFBI film self-assembled on the mica surface that was observed with atomic force microscopy. We could see clearly small holes in the RGD-HFBI film, indicating that the self-assembly of RGD-HFBI protein was independent of the BODIPY dye. Therefore, RGD-HFBI has the potential to form protein nanocages by using other hydrophobic cores as the self-assembly template.
(A) TEM images of BODIPY dissolved in different solvents (H2O, DMSO, HFBI and RGD-HFBI). The red dashed line represents the RGD-HFBI shell or film. (B) Atomic force microscopy images of self-assembled RGD-HFBI (native HFBI) film on a mica surface. (C) Wide XPS spectra of RGD-HFBI/BODIPY, HFBI/BODIPY and BODIPY, and high-resolution spectra of F1s, B1s and O1s. (D) FTIR spectra of RGD-HFBI/BODIPY, HFBI/BODIPY and BODIPY.
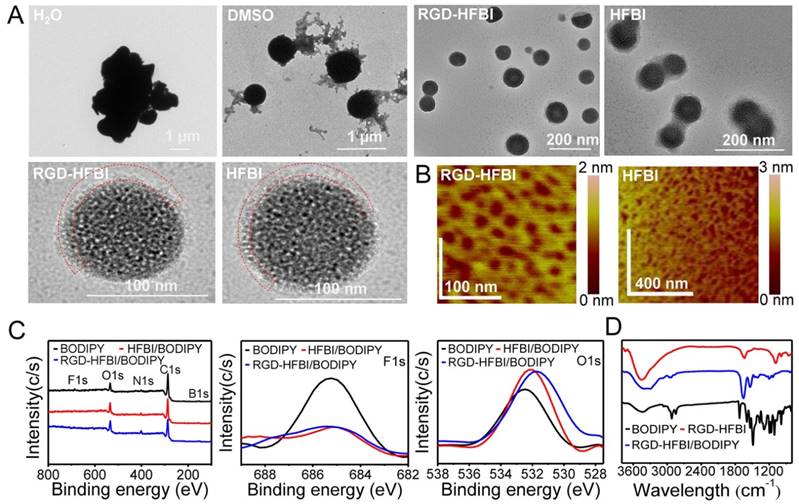
To further confirm the self-assembly of RGD-HFBI onto the BODIPY, chemical compositions of BODIPY before and after RGD-HFBI modification were analyzed by XPS. The full XPS spectra (Figure 4C) showed an overall change in the F1s, O1s, N1s, C1s and B1s of the BODIPY, HFBI-modified BODIPY and RGD-HFBI-modified BODIPY. The F1s (685 eV) peak was characteristic for BODIPY dye according to its chemical composition. The intensity of this elemental peak was remarkably decreased in the narrow spectrum of RGD-HFBI-modified BODIPY, suggesting that RGD-HFBI protein self-assembled and covered the surface of BODIPY. Furthermore, increased content of oxygen (Figure 4C) also confirmed the self-assembly of RGD-HFBI protein onto the surface of BODIPY. Next, FTIR (Figure 4D) was used to characterize the RGD-HFBI/BODIPY complex as the shape and frequency of the amide I band was modulated by the secondary structure of a protein [65]. The FTIR spectrum of RGD-HFBI protein showed a sharp peak at 1637 cm-1 involved in the vibration bands of amide I, which was caused by C=O stretching vibrations of peptide linkages. This result indicated RGD-HFBI contained mainly beta-sheet (1637 cm-1) structure. Hakanpaa et al. reported that native HFBI protein contains four beta-sheets based on its three-dimensional crystal structure [33]. The coherence between the secondary structures of RGD-HFBI and native HFBI illustrated that the RGD motif did not affect the correct protein folding of HFBI. For the BODIPY dye, strong peaks at 2848 and 2919 cm-1 were attributed to the C-H vibrations of the benzene bonds, and a peak at 1728 cm-1 resulted from C=O vibration. Moreover, sharp peaks at 1486, 1538 and 1594 cm-1 corresponded to v (C=C) stretching vibrations in benzene. Regarding the RGD-HFBI/BODIPY complex, the FTIR spectrum consisted of both characteristic peaks of HFBI/BODIPY and BODIPY without detectable chemical bond formation. This result suggested that RGD-HFBI adsorbed onto the surface of BODIPY dye non-covalently without protein structure changes. Furthermore, This finding was quite consistent with the previous reports that there was no secondary structure change during the binding process when class II hydrophobin assembled physically onto hydrophobic surfaces [66]. The FTIR results also strengthened the idea that physical hydrophobic force was the major driving force for the interaction between the hydrophobic part of HFBI and BODIPY dye, resulting in the formation of RGD-HFBI/BODIPY complex.
Analysis of biocompatibility, cell binding and cellular uptake in vitro
After we obtained the RGD-HFBI/BODIPY complex, we were eager to know whether this complex could be used in biolabeling or bioimaging from the safety point of view. Therefore, biological compatibility of this complex was an important issue we should consider in the next step. The biocompatibility of RGD-HFBI/BODIPY was evaluated by classical MTT assay. Three different cancer cell lines were used in our assays, including U-87MG (high αvβ3 integrin expression), HeLa (low αvβ3 integrin expression) and MCF-7 cells (without αvβ3 integrin expression) [67, 68]. As shown in Figure 5A, the viabilities of the three cell lines were not affected by the exposure of different concentrations of RGD-HFBI/BODIPY complex for 48 h, indicating its high biocompatibility and low cytotoxicity.
(A) MTT assay results of three different cancer cell lines including U-87MG (high αvβ3 integrin expression), HeLa (low αvβ3 integrin expression) and MCF-7 cells (without αvβ3 integrin expression). Those three cell lines were treated with different concentrations (0, 0.1, 1, 5, 10, 25, 50, 100, 250 and 500 µM) of RGD-HFBI/BODIPY, HFBI/BODIPY and BODIPY for 48 h. (B) Flow cytometry analysis of U-87MG, Hela and MCF-7 cells incubated with RGD-HFBI/BODIPY, HFBI/BODIPY and BODIPY for 4 h. The concentration of all probes was 10 μM and cells without treatments were used as the blank controls (red lines). (C) Confocal microscopy images of U-87MG, Hela and MCF-7 cells treated with RGD-HFBI/BODIPY, HFBI/BODIPY, BODIPY and synthetic RGD peptide.
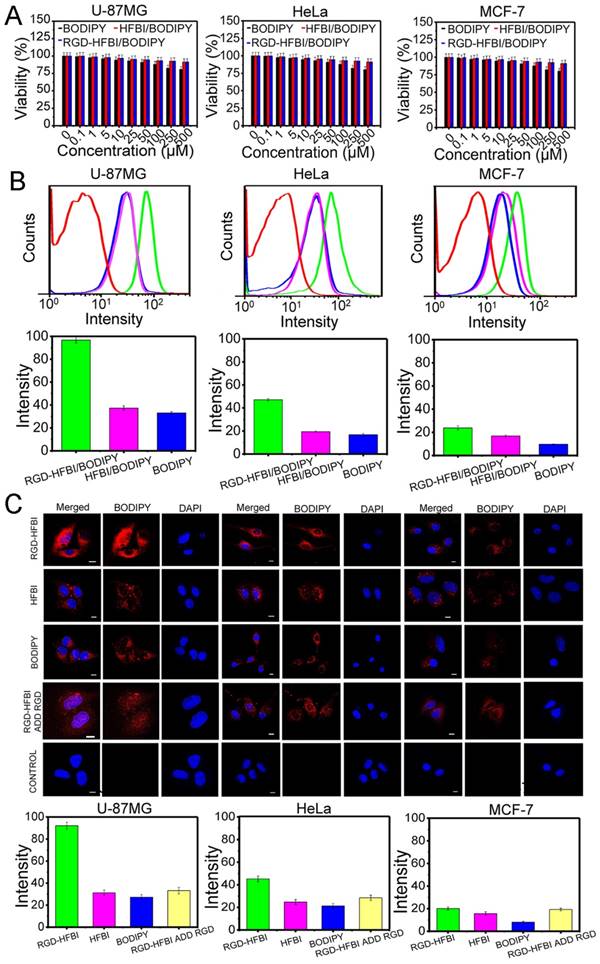
HFBI/BODIPY showed low cytotoxicity towards all the tested cell lines as well. However, DMSO-dissolved BODIPY showed slight cytotoxicity at high concentrations (250 and 500 μM), indicating BODIPY itself had detectable cytotoxicity at high concentrations. From the above results, we saw clearly that hydrophobin modification could increase the biocompatibility of BODIPY dye. Aimanianda et al. reported that surface hydrophobin prevented immune recognition of airborne fungal spores in the human body [69]. Their results implied that people could generate hydrophobin-based nanoparticles containing embedded diagnostic and therapeutic molecules that have to be transported to a specific body location without being recognized by the host immune system. Therefore, we considered that RGD-HFBI might mask most of the immunogenicity of BODIPY, resulting in its low cytotoxicity. This consideration will be addressed in future studies using immunocompetent animals. Nevertheless, we proved that the RGD-HFBI/BODIPY complex has the potential to be applied in the living cell system for bioimaging due its good biocompatibility.
Next, flow cytometry analysis (Figure 5B) was performed to quantify the cell binding and cellular uptake. The uptakes of RGD-HFBI/BODIPY complex were investigated by using the same three cell lines used in the MTT assays. As expected, the cell-associated fluorescence was highest in U-87MG cells. The fluorescence in HeLa cells was decreased about one-fold compared with the U-87MG cells. Regarding the negative control MCF-7 cells, we observed the lowest fluorescence that might come from nonspecific cellular uptake of RGD-HFBI/BODIPY complex. The relatively small size (about 100 nm) of RGD-HFBI/BODIPY complex might contribute to this phenomenon [70, 71]. These results demonstrated that integrin expression did have an obvious effect on the cellular uptake of RGD-HFBI/BODIPY complex and also suggested the biological activity of the RGD motif in the hybrid RGD-HFBI was preserved very well. In our flow cytometry analysis, HFBI-modified BODIPY and DMSO-dissolved BODIPY were used as controls. All three types of cells treated by those two BODIPYs with different solvents showed much lower cell-associated fluorescence than that obtained from RGD-HFBI/BODIPY. These results indicated the functionality of RGD motif of RGD-HFBI protein was a key factor in regulating the specific cell binding and cellular uptake of BODIPY as well. In order to further evaluate the cellular uptake of RGD-HFBI/BODIPY complex, we used confocal microscopy analysis to directly observe the interactions between RGD-HFBI/BODIPY complex and target cells. As shown in Figure 5C, we got very similar results as that obtained from flow cytometry analysis. U-87MG cells and HeLa cells with αvβ3 integrin expression showed higher fluorescence than that from the MCF-7 cells that did not have any αvβ3 integrin expression on their cell surfaces. All cells treated with HFBI-modified BODIPY and DMSO-dissolved BODIPY showed low fluorescence at the same level due to the lack of RGD. Moreover, the overall fluorescence was obviously decreased by adding the synthetic RGD peptide both in the U-87MG and HeLa cells. These results obviously demonstrated that the RGD motif remarkably improved the specific cell binding and cellular uptake of RGD-HFBI/BODIPY complex.
In vivo imaging in the tumor model
Before we did in vivo imaging using tumor models, we evaluated the stability of RGD-HFBI/BODIPY complex in vitro and in vivo. The stability of RGD-HFBI/BODIPY complex was determined by whether the RGD-HFBI on the shell of the complex could resist protease degradation in serum both in vivo and in vitro. Figure S8 shows that the RGD-HFBI/BODIPY complex was relatively stable both in vivo and in vitro. For example, only about 11% of RGD-HFBI/BODIPY was degraded after 72 h in vivo. This result was consistent with the previous reports that self-assembled hydrophobin could resist protease degradation both in vitro and in vivo [72-75]. After the stability of RGD-HFBI/BODIPY complex was confirmed, we did in vivo imaging using different tumor models. Figure 6A shows typical NIR fluorescence images of athymic nude mice bearing subcutaneous U-87MG glioblastoma tumor after intravenous injection of 17.7 nmol of RGD-HFBI/BODIPY. The subcutaneous U-87MG tumor could be clearly delineated from the surrounding background tissue at all the time points. We saw limited but distinguishable fluorescence at 2 h, indicating that RGD-HFBI/BODIPY reached the tumor site at this time point. Accumulation of fluorescence was observed with time, increasing until the maximum fluorescence occurred at 24 h. After that, the fluorescence in the tumor site went down, suggesting the washing-out of RGD-HFBI/BODIPY, but still remained distinguishable at 72 h. For the control HFBI/BODIPY group, we did not observe any fluorescence with high intensity in the tumor sites at all the time points (Figure 6C). The weak fluorescence in this group could be attributed to the enhanced permeability and retention (EPR) effect. However, we believed that effective accumulation of fluorescence in the RGD-HFBI/BODIPY group not only depended on the EPR effect, but also relied on the active targeting effect of the RGD motif of the RGD-HFBI protein.
(A) NIR fluorescence images of nude mice bearing U-87MG tumor after intravenous injection of RGD-HFBI/BODIPY and HFBI/BODIPY. (B) The ex vivo optical images of organs (heart, liver, spleen, lung and kidney) and tumors of the U-87MG tumor-bearing mice sacrificed at three time points (24, 48 and 72 h) after administration. (C) Quantitation of in vivo fluorescence intensity of U-87MG tumor after administration of RGD-HFBI/BODIPY and HFBI/BODIPY in the tumor-bearing mice at different time points (2, 4, 6, 8, 24, 48 and 72 h). (D) Quantitation of ex vivo fluorescence intensity of harvested tumors of the U-87MG tumor-bearing mice sacrificed at three time points (24, 48 and 72 h) after administration. (E) NIR fluorescence images of nude mice bearing HeLa tumor after intravenous injection of RGD-HFBI/BODIPY and HFBI/BODIPY. (F) The ex vivo optical images of organs (heart, liver, spleen, lung and kidney) and tumors of the HeLa tumor-bearing mice sacrificed at three time points (24, 48 and 72 h) after administration. (G) Quantitation of in vivo fluorescence intensity of HeLa tumor after administration of RGD-HFBI/BODIPY and HFBI/BODIPY in the tumor-bearing mice at different time points (2, 4, 6, 8, 24, 48 and 72 h). (H) Quantitation of in vitro fluorescence intensity of harvested tumors of the HeLa tumor-bearing mice sacrificed at three time points (24, 48 and 72 h) after administration.
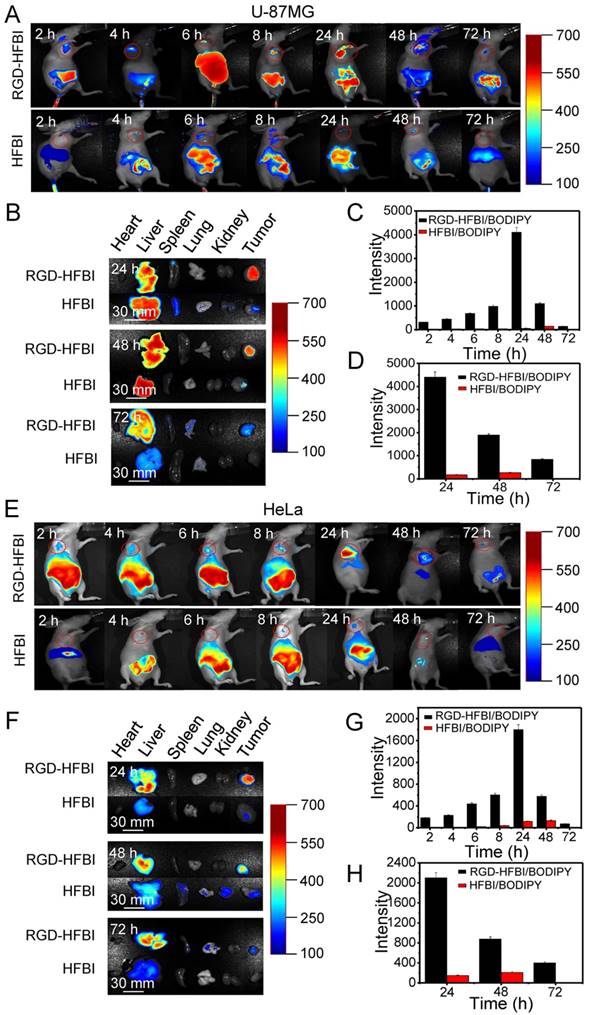
Therefore, the above results did reveal the specific affinity between the RGD-HFBI/BODIPY and the U-87MG glioblastoma tumor. To further verify the accumulation of RGD-HFBI/BODIPY, the mice were sacrificed at three time points (24, 48 and 72 h), and dominant organs were selected for in vitro imaging (Figure 6B). For the RGD-HFBI/BODIPY-treated mice, the tumor tissue showed distinct fluorescence at all three time points, suggesting BODIPY accumulation in the tumor due to specific targeting of the RGD-HFBI/BODIPY (Figure 6D). Regarding the control group, limited fluorescence was observed in the tumor tissue, and strong fluorescence signals were detected only in the liver due to nonspecific uptake and retention of HFBI/BODIPY.
We also did NIR fluorescence analysis of athymic nude mice bearing subcutaneous HeLa tumors. As shown in Figure 6E-H, the results were quite similar with those obtained from the U-87MG tumor cell. The only difference was the fluorescence obtained from HeLa cells was relatively weaker than that obtained from the U-87MG tumor cell at all the time points. This result is quite consistent with the results obtained from in vitro cell culture assay. Taken together, the in vivo affinity tests fully demonstrated the high specificity of RGD-HFBI/BODIPY due to the high selectivity and strong affinity of the RGD molecules.
Conclusion
In this paper, we showed a simple and reliable one-step strategy to produce water-soluble and targeted BODIPY dye by designing a novel hybrid protein named RGD-HFBI. This novel protein has dual biological functions according to our design. The first function of this protein is to self-assemble on hydrophobic surfaces including the hydrophobic BODIPY we used in this work. The outcome of the self-assembly of RGD-HFBI on different hydrophobic surfaces is a change in surface properties. In our study, RGD-HFBI could readily self-assemble on the BODIPY dye by simple mixing and sonication for 30 min, resulting in the efficient solubilization of this hydrophobic dye. It was notable that a protein nanocage was formed during the self-assembly process, which means the performance of this protein could be improved by further reasonable modifications. We also found that physical hydrophobic interaction could be the major driven force for the interaction of RGD-HFBI and BODIPY. The second function of this protein is to target specific cell lines by using the classic RGD motif in the protein. In our study, we proved that RGD-HFBI could efficiently guide the BODIPY dye to specifically label the U-87MG cancer cell in cell culture and in animal experiments. Furthermore, the good biocompatibility and prolonged circulation time of RGD-HFBI/BODIPY might facilitate future application of this dye. In our study, we only used a synthesized BODIPY dye to prove the dual functions of RGD-HFBI. We believed that RGD-HFBI could be applied to other hydrophobic fluorescent probes or even other hydrophobic materials that suffer from solubility and targeting problems. Therefore, our one-step strategy may be developed as a general strategy for biolabeling and bioimaging to broaden their applications.
Abbreviations
BODIPY: Boron-dipyrromethene; CC: column chromatography; DDQ: 2, 3-Dichloro-5, 6-dicyano-1, 4-benzoquinone; EPR: enhanced permeation and retention; FTIR: Fourier-transform infrared; NIR: near-infrared; RGD: arginine-glycine-aspartic acid; RP-HPLC: reversed-phase high-performance liquid chromatography; TEM: transmission electron microscopy; TLC: thin-layer chromatography; WCA: water contact angle; XPS: X-ray photoelectron spectroscopy.
Acknowledgements
Zefang Wang acknowledges the support of the National Natural Science Foundation of China (No. 81601593). Shuxian Meng acknowledges the support of National Natural Science Foundation of China (No. 21676187). Yanyan Wang acknowledges the support of the National Natural Science Foundation of China (No. 61501320). Yunjie Xiao, Qian Zhang and Yanyan Wang contributed equally to this work.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Guo Z, Park S, Yoon J, Shin I. Recent progress in the development of near-infrared fluorescent probes for bioimaging applications. Chem Soc Rev. 2014;43:16-29
2. Sato R, Kozuka J, Ueda M, Mishima R, Kumagai Y, Yoshimura A. et al. Intracellular protein-labeling probes for multicolor single-molecule imaging of immune receptor-adaptor molecular dynamics. J Am Chem Soc. 2017;139:17397-404
3. Zhong Y, Ma Z, Zhu S, Yue J, Zhang M, Antaris AL. et al. Boosting the down-shifting luminescence of rare-earth nanocrystals for biological imaging beyond 1500 nm. Nat Commun. 2017;8:737
4. Evans CL. New near-infrared dyes light up deep tissue imaging. Sci Transl Med. 2017;9:eaam6065
5. Frangioni JV. In vivo near-infrared fluorescence imaging. Curr Opin Chem Biol. 2003;7:626-34
6. Lou Z, Li P, Han K. Redox-responsive fluorescent probes with different design strategies. Acc Chem Res. 2015;48:1358-68
7. Yu F, Li P, Wang B, Han K. Reversible near-infrared fluorescent probe introducing tellurium to mimetic glutathione peroxidase for monitoring the redox cycles between peroxynitrite and glutathione in vivo. J Am Chem Soc. 2013;135:7674-80
8. Yuan L, Lin W, Zheng K, He L, Huang W. Far-red to near infrared analyte-responsive fluorescent probes based on organic fluorophore platforms for fluorescence imaging. Chem Soc Rev. 2013;42:622-61
9. Luo S, Zhang E, Su Y, Cheng T, Shi C. A review of NIR dyes in cancer targeting and imaging. Biomaterials. 2011;32:7127-38
10. Li Y, Xu D, Ho SL, Li H, Yang R, Wong M. A theranostic agent for in vivo near-infrared imaging of beta-amyloid species and inhibition of beta-amyloid aggregation. Biomaterials. 2016;94:84-92
11. Hyun H, Owens EA, Wada H, Levitz A, Park G, Park MH. et al. Cartilage-specific near-infrared fluorophores for biomedical imaging. Angew Chem Int Ed Engl. 2015;54:8648-52
12. Hyun H, Park MH, Owens EA, Wada H, Henary M, Handgraaf HJ. et al. Structure-inherent targeting of near-infrared fluorophores for parathyroid and thyroid gland imaging. Nat Med. 2015;21:192-7
13. Amiot CL, Xu S, Liang S, Pan L, Zhao J. Near-infrared fluorescent materials for sensing of biological targets. Sensors (Basel). 2008;8:3082-105
14. Yuan L, Lin W, Zhao S, Gao W, Chen B, He L. et al. A unique approach to development of near-infrared fluorescent sensors for in vivo imaging. J Am Chem Soc. 2012;134:13510-23
15. Verwilst P, Kim HR, Seo J, Sohn NW, Cha SY, Kim Y. et al. Rational design of in vivo tau tangle-selective near-infrared fluorophores: expanding the BODIPY universe. J Am Chem Soc. 2017;139:13393-403
16. Han C, Yang H, Chen M, Su Q, Feng W, Li F. Mitochondria-targeted near-infrared fluorescent off-on probe for selective detection of cysteine in living cells and in vivo. ACS Appl Mater Interfaces. 2015;7:27968-75
17. Zhang J, Yang M, Li C, Dorh N, Xie F, Luo F. et al. Near-infrared fluorescent probes based on piperazine-functionalized BODIPY dyes for sensitive detection of lysosomal pH. J Mater Chem B. 2015;3:2173-84
18. Wang D, Fan J, Gao X, Wang B, Sun S, Peng X. Carboxyl BODIPY dyes from bicarboxylic anhydrides: one-pot preparation, spectral properties, photostability, and biolabeling. J Org Chem. 2009;74:7675-83
19. Kim J, Kim Y. A water-soluble sulfonate-BODIPY based fluorescent probe for selective detection of HOCl/OCl- in aqueous media. Analyst. 2014;139:2986-9
20. Zhu S, Zhang J, Vegesna G, Luo F, Green SA, Liu H. Highly water-soluble neutral BODIPY dyes with controllable fluorescence quantum yields. Org Lett. 2011;13:438-41
21. Hu R, Law WC, Lin G, Ye L, Liu J, Liu J. et al. PEGylated phospholipid micelle-encapsulated near-infrared PbS quantum dots for in vitro and in vivo bioimaging. Theranostics. 2012;2:723-33
22. Liu F, Deng D, Chen X, Qian Z, Achilefu S, Gu Y. Folate-polyethylene glycol conjugated near-infrared fluorescence probe with high targeting affinity and sensitivity for in vivo early tumor diagnosis. Mol Imaging Biol. 2010;12:595-607
23. Ha Y, Choi HK. Recent conjugation strategies of small organic fluorophores and ligands for cancer-specific bioimaging. Chem Biol Interact. 2016;248:36-51
24. Patalag LJ, Jones PG, Werz DB. BOIMPYs: rapid access to a family of red-emissive fluorophores and NIR dyes. Angew Chem Int Ed Engl. 2016;55:13340-4
25. Becker A, Hessenius C, Licha K, Ebert B, Sukowski U, Semmler W. et al. Receptor-targeted optical imaging of tumors with near-infrared fluorescent ligands. Nat Biotechnol. 2001;19:327-31
26. Kanduluru AK, Srinivasarao M, Low PS. Design, synthesis, and evaluation of a neurokinin-1 receptor-targeted near-IR dye for fluorescence-guided surgery of reuroendocrine cancers. Bioconjug Chem. 2016;27:2157-65
27. Haque A, Faizi MS, Rather JA, Khan MS. Next generation NIR fluorophores for tumor imaging and fluorescence-guided surgery: a review. Bioorg Med Chem. 2017;25:2017-34
28. Achilefu S. The insatiable quest for near-infrared fluorescent probes for molecular imaging. Angew Chem Int Ed Engl. 2010;49:9816-8
29. Zhong Y, Ma Z, Zhu S, Yue J, Zhang M, Antaris AL. et al. Boosting the down-shifting luminescence of rare-earth nanocrystals for biological imaging beyond 1500 nm. Nat Commun. 2017;8:737
30. Bayry J, Aimanianda V, Guijarro JI, Sunde M, Latge JP. Hydrophobins-unique fungal proteins. PLoS Pathog. 2012;8:e1002700
31. Wosten HA. Hydrophobins: multipurpose proteins. Annu Rev Microbiol. 2001;55:625-46
32. Linder MB, Szilvay GR, Nakari-Setala T, Penttila ME. Hydrophobins: the protein-amphiphiles of filamentous fungi. FEMS Microbiol Rev. 2005;29:877-96
33. Hakanpaa J, Szilvay GR, Kaljunen H, Maksimainen M, Linder M, Rouvinen J. Two crystal structures of Trichoderma reesei hydrophobin HFBI-the structure of a protein amphiphile with and without detergent interaction. Protein Sci. 2006;15:2129-40
34. Sunde M, Kwan AH, Templeton MD, Beever RE, Mackay JP. Structural analysis of hydrophobins. Micron. 2008;39:773-84
35. Laaksonen P, Kainlauri M, Laaksonen T, Shchepetov A, Jiang H, Ahopelto J. et al. Interfacial engineering by proteins: exfoliation and functionalization of graphene by hydrophobins. Angew Chem Int Ed Engl. 2010;49:4946-9
36. Qin M, Wang L, Feng X, Yang Y, Wang R, Wang C. et al. Bioactive surface modification of mica and poly (dimethylsiloxane) with hydrophobins for protein immobilization. Langmuir. 2007;23:4465-71
37. Valo HK, Laaksonen PH, Peltonen LJ, Linder MB, Hirvonen JT, Laaksonen TJ. Multifunctional hydrophobin: toward functional coatings for drug nanoparticles. ACS Nano. 2010;4:1750-8
38. Pigliacelli C, Maiolo D, Nonappa N, Haataja JS, Amenitsch H, Michelet C. et al. Efficient encapsulation of fluorinated drugs in the confined space of water-dispersible fluorous supraparticles. Angew Chem Int Ed Engl. 2017;56:16186-90
39. Hahl H, Vargas JN, Griffo A, Laaksonen P, Szilvay G, Lienemann M. et al. Pure protein bilayers and vesicles from native fungal hydrophobins. Adv Mater. 2017;29:1602888
40. Gravagnuolo AM, Morales-Narváez E, Matos CRS, Longobardi S, Giardina P, Merkoçi A. On-the-spot immobilization of quantum dots, graphene oxide, and proteins via hydrophobins. Adv Funct Mater. 2015;25:6084-92
41. Malho JM, Arola S, Laaksonen P, Szilvay GR, Ikkala O, Linder MB. Modular architecture of protein binding units for designing properties of cellulose nanomaterials. Angew Chem Int Ed Engl. 2015;54:12025-8
42. Janssen MI, van Leeuwen MB, Scholtmeijer K, van Kooten TG, Dijkhuizen L, Wosten HA. Coating with genetic engineered hydrophobin promotes growth of fibroblasts on a hydrophobic solid. Biomaterials. 2002;23:4847-54
43. Kallio JM, Linder MB, Rouvinen J. Crystal structures of hydrophobin HFBII in the presence of detergent implicate the formation of fibrils and monolayer films. J Biol Chem. 2007;282:28733-9
44. Goldian I, Jahn S, Laaksonen P, Linder M, Kampf N, Klein J. Modification of interfacial forces by hydrophobin HFBI. Soft Matter. 2013;9:10627-39
45. Mas-Moruno C, Fraioli R, Rechenmacher F, Neubauer S, Kapp TG, Kessler H. αvβ3- or α5β1-integrin-selective peptidomimetics for surface coating. Angew Chem Int Ed Engl. 2016;55:7048-67
46. Avraamides CJ, Garmy-Susini B, Varner JA. Integrins in angiogenesis and lymphangiogenesis. Nat Rev Cancer. 2008;8:604-17
47. Grunér MS, Szilvay GR, Berglin M, Lienemann M, Laaksonen P, Linder MB. Self-assembly of class II hydrophobins on polar surfaces. Langmuir. 2012;28:4293-300
48. Macindoe I, Kwan AH, Ren Q, Morris VK, Yang W, Mackay JP. et al. Self-assembly of functional, amphipathic amyloid monolayers by the fungal hydrophobin EAS. Proc Natl Acad Sci USA. 2012;109:E804-11
49. Stanimirova RD, Gurkov TD, Kralchevsky PA, Balashev KT, Stoyanov SD, Pelan EG. Surface pressure and elasticity of hydrophobin HFBII layers on the air-water interface: rheology versus structure detected by AFM imaging. Langmuir. 2013;29:6053-67
50. Pham CLL, Rey A, Lo V, Soulès M, Ren Q, Meisl G. et al. Self-assembly of MPG1, a hydrophobin protein from the rice blast fungus that forms functional amyloid coatings, occurs by a surface-driven mechanism. Sci Rep. 2016;6:25288
51. Magarkar A, Mele N, Abdel-Rahman N, Butcher S, Torkkeli M, Serimaa R. et al. Hydrophobin film structure for HFBI and HFBII and mechanism for accelerated film formation. PLoS Comput Biol. 2014;10:e1003745
52. Moldovan C, Thompson D. Molecular dynamics of the "hydrophobic patch" that immobilizes hydrophobin protein HFBII on silicon. J Mol Model. 2011;17:2227-35
53. Peng C, Liu J, Zhao D, Zhou J. Adsorption of hydrophobin on different self-assembled monolayers: the role of the hydrophobic dipole and the electric dipole. Langmuir. 2014;30:11401-11
54. Lumsdon SO, Green J, Stieglitz B. Adsorption of hydrophobin proteins at hydrophobic and hydrophilic interfaces. Colloids Surf B Biointerfaces. 2005;44:172-8
55. Ostolska I, Wisniewska M. Application of the zeta potential measurements to explanation of colloidal Cr2O3 stability mechanism in the presence of the ionic polyamino acids. Colloid Polym Sci. 2014;292:2453-64
56. Hao Y, Huang Y, He Y, Peng J, Chen L, Hu X. et al. The evaluation of cellular uptake efficiency and tumor-targeting ability of MPEG-PDLLA micelles: effect of particle size. RSC Adv. 2016;6:13698-709
57. He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31:3657-66
58. Wang Z, Wang Y, Huang Y, Li S, Feng S, Xu H. et al. Characterization and application of hydrophobin-dispersed multi-walled carbon nanotubes. Carbon. 2010;48:2890-8
59. Hakanpaa J, Paananen A, Askolin S, Nakari-Setala T, Parkkinen T, Penttila M. et al. Atomic resolution structure of the HFBII hydrophobin, a self-assembling amphiphile. J Biol Chem. 2004;279:534-9
60. Bhaskar S, Lim S. Engineering protein nanocages as carriers for biomedical applications. NPG Asia Mater. 2017;9:e371
61. Votteler J, Ogohara C, Yi S, Hsia Y, Nattermann U, Belnap DM. et al. Designed proteins induce the formation of nanocage-containing extracellular vesicles. Nature. 2016;540:292-5
62. Schoonen L, van Hest JC. Functionalization of protein-based nanocages for drug delivery applications. Nanoscale. 2014;6:7124-41
63. Lin X, Xie J, Niu G, Zhang F, Gao H, Yang M. et al. Chimeric ferritin nanocages for multiple function loading and multimodal imaging. Nano Lett. 2011;11:814-9
64. Truffi M, Fiandra L, Sorrentino L, Monieri M, Corsi F, Mazzucchelli S. Ferritin nanocages: a biological platform for drug delivery, imaging and theranostics in cancer. Pharmacol Res. 2016;107:57-65
65. Kong J, Yu S. Fourier transform infrared spectroscopic analysis of protein secondary structures. Acta Biochim Biophys Sin (Shanghai). 2007;39:549-59
66. Meister K, Bäumer A, Szilvay GR, Paananen A, Bakker HJ. Self-assembly and conformational changes of hydrophobin classes at the air-water interface. J Phys Chem Lett. 2016;7:4067-71
67. Ye Y, Chen X. Integrin targeting for tumor optical imaging. Theranostics. 2011;1:102-26
68. Taherian A, Li X, Liu Y, Haas TA. Differences in integrin expression and signaling within human breast cancer cells. BMC Cancer. 2011;11:293
69. Aimanianda V, Bayry J, Bozza S, Kniemeyer O, Perruccio K, Elluru SR. et al. Surface hydrophobin prevents immune recognition of airborne fungal spores. Nature. 2009;460:1117-21
70. Win KY, Feng S. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials. 2005;26:2713-22
71. Shang L, Nienhaus K, Nienhaus GU. Engineered nanoparticles interacting with cells: size matters. J Nanobiotechnology. 2014;12:5
72. Zhao L, Xu H, Li Y, Song D, Wang X, Qiao M. et al. Novel application of hydrophobin in medical science: a drug carrier for improving serum stability. Sci Rep. 2016;6:26461
73. Askolin S, Nakari-Setälä T, Tenkanen M. Overproduction, purification, and characterization of the Trichoderma reesei hydrophobin HFBI. Appl Microbiol Biotechnol. 2001;57:124-30
74. Wang X, Permentier HP, Rink R, Kruijtzer JA, Liskamp RM, Wösten HA. et al. Probing the self-assembly and the accompanying structural changes of hydrophobin SC3 on a hydrophobic surface by mass spectrometry. Biophys J. 2004;87:1919-28
75. Wang X, Graveland-Bikker JF, de Kruif CG, Robillard GT. Oligomerization of hydrophobin SC3 in solution: from soluble state to self-assembly. Protein Sci. 2004;13:810-21
Author contact
![]() Corresponding authors: Prof. Zefang Wang; School of Life Sciences, Tianjin University, Tianjin 300072, China; Tel: +86-22-27403096; E-mail: zefangwangedu.cn; Prof. Shuxian Meng, School of Chemical Engineering and Technology, Tianjin University, Tianjin 300072, China; Tel: +86-1303433353; E-mail: msxmailedu.cn.
Corresponding authors: Prof. Zefang Wang; School of Life Sciences, Tianjin University, Tianjin 300072, China; Tel: +86-22-27403096; E-mail: zefangwangedu.cn; Prof. Shuxian Meng, School of Chemical Engineering and Technology, Tianjin University, Tianjin 300072, China; Tel: +86-1303433353; E-mail: msxmailedu.cn.