Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(15):3624-3637. doi:10.7150/thno.21408 This issue Cite
Research Paper
Presenilin Mutation Suppresses Lung Tumorigenesis via Inhibition of Peroxiredoxin 6 Activity and Expression
Mi Hee Park1*, Hyung-Mun Yun2*, Chul Ju Hwang1, Sang Ick Park3, Sang Bae Han1, Dae Youn Hwang4, Do-Young Yoon5, Sanghyeon Kim6, Jin Tae Hong1 ![]()
1. College of Pharmacy and Medical Research Center, Chungbuk National University, 12, Gaeshin-dong, Heungduk-gu, Cheongju, Chungbuk 361-763, Republic of Korea;
2. Department of Oral and Maxillofacial Pathology, School of Dentistry, Kyung Hee University, 1 Heogi-Dong, Dongdaemun-Gu, Seoul, 130-701, Republic of Korea;
3. Center for Biomedical Sciences, Korea National Institute of Health, Cheongju 28159, Republic of Korea;
4. Department of Biomaterials Science, College of Natural Resources and Life Science/Life and Industry Convergence Research Institute, Pusan National University, Miryang-si, Gyeongsangnam‑do 627‑706, Republic of Korea;
5. Department of Bioscience and Biotechnology, Bio/Molecular Informatics Center, Konkuk University, Gwangjin-gu, Seoul 143-701, Republic of Korea;
6. Stanley Brain Research Laboratory, Stanley Medical Research Institute, 9800 Medical Center Drive, Rockville, MD 20850.
* These authors equally contributed to this work.
Received 2017-6-10; Accepted 2017-6-24; Published 2017-9-1
Abstract

Some epidemiological studies suggest an inverse correlation between cancer incidence and Alzheimer's disease (AD). In this study, we demonstrated experimental evidences for this inverse relationship. In the co-expression network analysis using the microarray data and GEO profile of gene expression omnibus data analysis, we showed that the expression of peroxiredoxin 6 (PRDX6), a tumor promoting protein was significantly increased in human squamous lung cancer, but decreased in mutant presenilin 2 (PS2) containing AD patient. We also found in animal model that mutant PS2 transgenic mice displayed a reduced incidence of spontaneous and carcinogen-induced lung tumor development compared to wildtype transgenic mice. Agreed with network and GEO profile study, we also revealed that significantly reduced expression of PRDX6 and activity of iPLA2 in these animal models. PS2 mutations increased their interaction with PRDX6, thereby increasing iPLA2 cleavage via increased γ-secretase leading to loss of PRDX6 activity. However, knockdown or inhibition of γ-secretase abolished the inhibitory effect of mutant PSs. Moreover, PS2 mutant skin fibroblasts derived from patients with AD showed diminished iPLA2 activity by the elevated γ-secretase activity. Thus, the present data suggest that PS2 mutations suppress lung tumor development by inhibiting the iPLA2 activity of PRDX6 via a γ-secretase cleavage mechanism and may explain the inverse relationship between cancer and AD incidence.
Keywords: Presenilin, Alzheimer's disease, PRDX6, iPLA2, lung cancer.
Introduction
The PS homologs PS1 and PS2 function as the catalytic subunits of β- and γ-secretase [1, 2]. PS mutations are critical for the generation of β-amyloids and increased β- and γ-secretase activity that account for most early-onset familial AD [3, 4]. In addition, PS participate in several signaling pathways that regulate cell survival and tumorigenesis [5, 6], however, the definitive functions of PSs in tumor development have not yet been elucidated. It was previously reported that PS/γ-secretase-generated-amyloid precursor protein intracellular domain (AICD) regulates epidermal growth factor receptor (EGFR) transcription by directly binding the EGFR promoter, resulting in the tumor suppressive effect [7]. Similarly, germline mutations of PS2 (R62H and R71W) induced breast cancer growth [8], however, other studies have shown that PS1 mutant overexpression promotes cellular apoptosis [9]. Notably, skin and carcinogen-induced brain tumorigenesis were higher in PS1 knockout and in mutant PS1 mice [10, 11], respectively. Although the connection between PSs and tumor development is controversial, a recent epidemiological study demonstrated that older adults with prevalent clinical AD developed cancer at a slower rate when compared to older adults without AD [12]. Conversely, PS2 knockout mice exhibited higher lung tumor development [13], suggesting that mutant PS may have an anti-oncogenic function.
Lung cancer is the leading cause of cancer related mortality worldwide [14]. Proteomic analysis suggests that the expression of PRDXs increased in human lung cancer cell lines and tissues [15]. Thus, the roles of these new genes should be elucidated. PRDXs are thiol-specific antioxidant proteins consisting of six isoforms (PRDX1-PRDX6) and are expressed in mammals, yeast, and bacteria [16]. PRDXs are classified based on having either one (1-Cys) or two (2-Cys) conserved cysteine residues [17]. PRDXs regulate the cellular redox state by breaking down hydrogen peroxide, suggesting that they may also participate in signal transduction, lung tumor progression, and stress resistance [18]. Specifically, PRDX6 is elevated in several lung pathologies, including lung cancer, mesothelioma, and sarcoidosis [19]. Among the six members, PRDX6 is the only peroxiredoxin that exhibits inducible phospholipase A2 (iPLA2) and glutathione peroxidase activity [20]. It was found that glioblastoma and Lewis lung carcinoma progression were attenuated more in iPLA2α knockout mice than in wildtype controls, and iPLA2 knockout murine pulmonary microvascular endothelial (MPMEC) cells treated with lysophosphatidylcholine and lysophosphatidic acid displayed increased cell proliferation and invasive migration when compared to untreated cells [21, 22]. Moreover, another study reported that iPLA2 knockout mice developed 43% fewer lung tumors than wildtype mice did [22]. Further, human NSCLC lines with oncogenic KRAS mutations synthesize higher levels of prostaglandin E2 (PGE2) compared to NSCLC lines with wildtype KRAS or non-transformed lung epithelial cells resulting from iPLA2 and COX2 induction [23]. Our recent studies also showed that PRDX6 overexpression promotes lung tumor development via increased iPLA2 activity [23, 24]. Thus, these data indicate that the iPLA2 activity of PRDX6 could be a critical factor in lung tumor development.
The γ-secretase protease complex consisting of PS1, nicastin, anterior pharynx-defective phenotype-1 (APH-1), and PS enhancer-2 (PEN-2) cleaves several transmembrane proteins, such as CD44, Notch-1, EphB2, amyloid β-precursor protein (APP), and E-cadherin [25]. Interestingly, of the 55 known γ-secretase substrates, more than 25% contain a GXXXG pentapeptide in their transmembrane domain [26]. In AD, this GXXXG motif in APP is critical for the generation of Aβ42 [27]. Specifically, the iPLA2 active site (GXSXG; 30-34aa) is classified as a phospholipase motif [28] and could be subject to cleavage by γ-secretase, thereby losing its phospholipase activity. Moreover, other groups have evaluated that surfactant protein A (SP-A) inhibits PRDX6 iLPA2 activity through a direct protein-protein interaction, as increased iPLA2 activity is found in SP-A knockout cells or those treated with SP-A inhibitors [29]. Further, γ-secretase activity is elevated in mutant PS2 cells, suggesting that the PRDX6 iPLA2 activity can be inhibited by γ-secretase in mutant PSs cells, but not in wildtype cells. Based on these findings, we hypothesized that mutant PSs could play a critical role in lung cancer inhibition via increased γ-secretase activity and subsequent cleavage-dependent iPLA2 inhibition, which could explain the reverse relationship between AD and cancer.
Results
Effect of mutant PS2 on PRDX6 expression
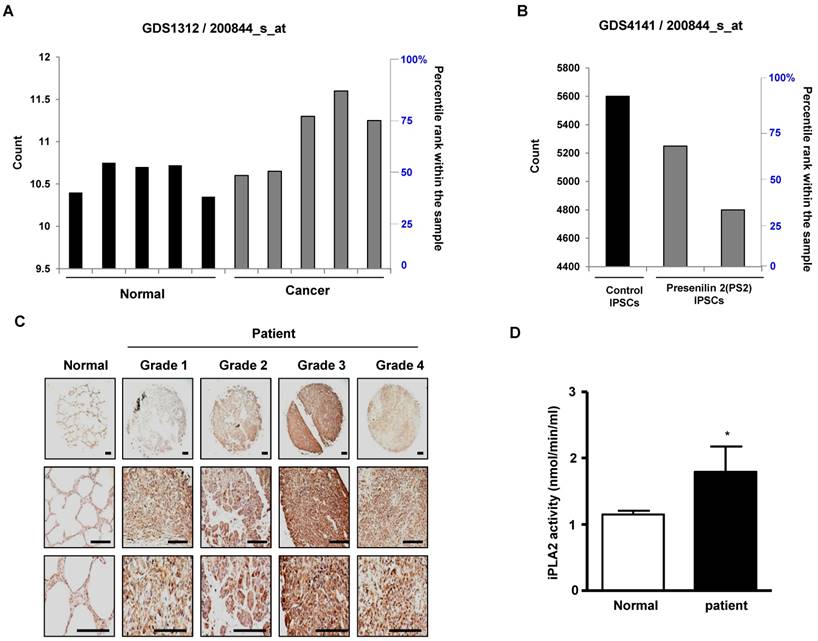
First, we observed a reduced incidence of spontaneous lung cancer in mutant PS2 (N141I) transgenic mice when compared to that in wildtype PS2 transgenic mice (Fig. 1A). Proteomic analysis of harvested lung tumor tissue revealed significantly lower PRDX6 expression (571-fold) in PS2 transgenic mice than that in wildtype transgenic mice (Fig. 1B and Table 1). Next, we analyzed the PRDX6 in squamous lung cancer patient samples and AD patient sample with PS2 mutation. In the data using GDS4141/200844 probe, the average percentile of PRDX6 expression in normal sample was 53%, but in cancer patient sample was 68% (Fig. 2A). Moreover, in the data using same probe, the average percentile of PRDX6 expression pattern in control induced pluripotent stem (iPS) cells was 90%, but in AD patient derived iPS cells containing mutant PS2 was 52.5% (Fig.2B). These data suggest that PRDX6 is critical factor for development of lung cancer in AD patient.
Effect of PS2 mutation on spontaneous development of lung tumor. (A) Spontaneous lung tumor incidence was determined in over 18 month-aged non transgenic mice, PS2 wildtype transgenic mice and mutant PS2 transgenic mice (n=20). (B) Proteomics analysis of spontaneously induced lung tumor tissues of PS2 wildtype transgenic mice and mutant PS2 transgenic mice as described in materials and methods.
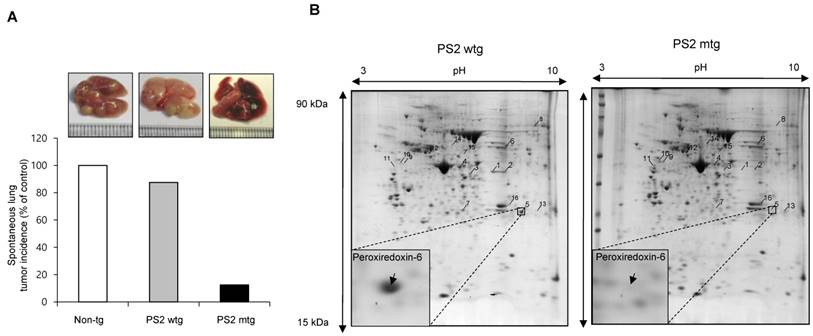
The result of proteomics analysis. 16 proteins with more than 3 fold changes were identified and listed.
| No | protein identified | Fold | MOWSE score | Sequence coverage | NCBI accession number | Molecular mass (kDa) | pI |
|---|---|---|---|---|---|---|---|
| 1 | Chitinase 3-like 4 precursor | -596.8 | 89 | 30% | gi|251823804 | 44.95 | 5.8 |
| 2 | Chitinase 3-like 4 precursor | -10.9 | 69 | 22% | gi|251823804 | 44.95 | 5.8 |
| 3 | Chitinase 3-like 4 precursor | -7.1 | 82 | 24% | gi|251823804 | 44.95 | 5.8 |
| 4 | Chitinase 3-like 4 precursor | -3 | 66 | 22% | gi|251823804 | 44.95 | 5.8 |
| 5 | Peroxiredoxin-6 | -571.7 | 76 | 44% | gi|3219774 | 24.86 | 5.7 |
| 6 | Fibrinogen beta chain precursor | -3.9 | 81 | 29% | gi|33859809 | 54.72 | 6.7 |
| 7 | Ig kappa chain V region | -264.2 | 73 | 16% | gi|110433 | 11.76 | 8.0 |
| 8 | Msn protein | -3.3 | 109 | 27% | gi|116283288 | 51.88 | 8.8 |
| 9 | Vimentin | 96.6 | 92 | 25% | gi|2078001 | 51.53 | 5.0 |
| 10 | Vimentin | 62.8 | 112 | 23% | gi|2078001 | 51.53 | 5.0 |
| 11 | Vimentin | 3.7 | 88 | 26% | gi|2078001 | 51.53 | 5.0 |
| 12 | Vimentin | 3.7 | 208 | 38% | gi|2078001 | 51.53 | 5.0 |
| 13 | Similar to Stromal antigen 1 | 6 | 79 | 10% | gi|149260258 | 128.83 | 5.5 |
| 14 | Lymphocyte cytosolic protein 1 | 5.2 | 211 | 37% | gi|31543113 | 70.11 | 5.2 |
| 15 | T-cell specific GTPase | 5 | 82 | 31% | gi|158749628 | 47.09 | 5.5 |
| 16 | Indolethylamine N-methyltransferase | 3.7 | 84 | 47% | gi|6678281 | 29.44 | 6.0 |
PRDX6 expression in lung cancer patient and AD patient samples. A, After search the GEO profile, we analyzed the PRDX6 expression pattern in gene expression omnibus data set GDS1312 containing five human lung cancer samples and normal controls. B, We analyzed the PRDX6 expression pattern in gene expression omnibus data set GDS4141 containing mutant PS2 containing samples and normal controls. This data analyzed in the data from human iPS (induced pluripotent stem) cells including normal and AD patient. The GEO Profiles database stores gene expression profiles derived from curated GEO DataSets. Each Profile displays the expression level of one gene across all samples within a DataSet. Experimental context is provided in the bars along the bottom of the charts making it possible to see at a glance whether a gene is differentially expressed across different experimental conditions. 200844_s_at means probe number and each GSM number means GEO data id. C, Human normal lung or cancer sections (grade 1-3) were processed and analyzed by immunohistochemistry for detection of positive cells for PRDX6. D, Human normal lung or cancer samples were analyzed by iPLA2 activity of PRDX6. The results (Fig.1D) are expressed as mean ± SD from three independent experiments. *P< 0.05 indicates significant difference from normal group.
Co-expression network analysis of PRDX6 expression and lung tumorigenesis
To verify the role of PRDX6 in the tumorigenesis of human lung cancer, we performed co-expression network analysis using the microarray data from both squamous lung cancer biopsy specimens and paired normal specimens from 5 patients. Of the 61 co-expression modules, C_M19 module which included PRDX6 gene was significantly associated with human squamous lung cancer (Supplementary Table 1 and 2). The eigengene values in this module were significantly up-regulated in the cancer specimens as compared to normal samples (Supplementary Fig. 1A), and cytoskeleton organization, RNA processing, cellular protein complex assembly and methylation were significantly enriched in this module (Supplementary Fig. 1B Supplementary Table 3). This result suggests that up-regulation of PRDX6 may significantly contribute to lung cancer development through regulation of epigenetic gene expression such as cytoskeleton organization, RNA processing, cellular protein complex assembly and methylation.
PRDX6 expression in lung cancer patient samples
To verify the high expression of PRDX6 in lung cancer, we determined the protein level of PRDX6 in a human lung cancer tissue array. Results showed that, compared with normal tissue, PRDX6 is more overexpressed in lung cancer tissues depend on tumor stage (Fig. 2C). These results indicated that PRDX6 might be a critical molecule in lung cancer development. Because previous data indicated that iPLA2 activity is critical indicator for PRDX6 activity, we also determined iPLA2 activity in lung cancer patient samples. Our data showed that iPLA2 activity was about a 50% increase in lung cancer patient samples (Fig. 2D).
Effect of mutant PS2 on proliferation, PRDX6 expression, iPLA2 and γ-secretase activity, and PRDX6 colocalization in the fibroblasts derived from AD patients
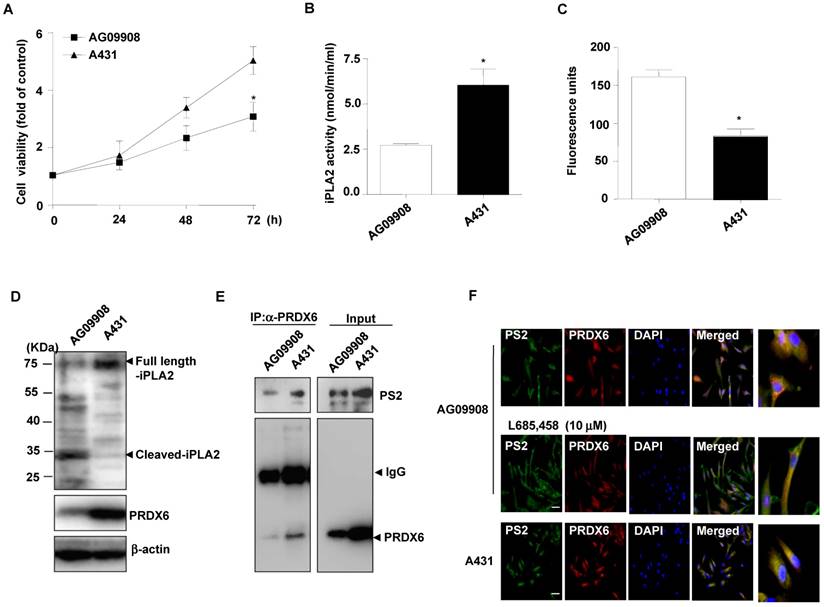
Additional analysis on cell proliferation, PRDX6 expression and iPLA2 activity in skin fibroblasts (AG09908) derived from AD patients with mutations in PS2 was studied. Notably, we found that cell proliferation (Fig. 3A), and iPLA2 activity (Fig. 3B) were attenuated in PS2 mutant fibroblasts. Moreover, the GXSXG phospholipase (PLA2) motif in PRDX6 is the catalytic site for its iPLA2 activity. This site also exists in iPLA2 protein. The GXSXG site is a helix-dimerization motif significantly affected by γ-secretase [27] known to be increased by PSs mutation. Thereafter, we questioned whether the increased γ-secretase activity observed in PS2 mutant cells is attributed to cleavage-dependent iPLA2 inhibition. As expected, γ-secretase activity was markedly elevated (Fig. 3C) and cleaved iPLA2 was increased (Fig. 3D). Moreover, immunoprecipitation analysis indicated that PRDX6 and PS2 co-localization was increased in AG09908 cells (Fig. 3E). Accompanied with immunoprecipitation analysis, we confirmed that co-localization was stronger in AG09908 cells compared to wildtype counterparts, but reversed by treatment of γ-secretase inhibitor (L685,458, 10 μm) (Fig. 3F). Our data suggest that co-localization of PRDX6 and PS2 was dramatically increased in fibroblasts derived from AD patients, but reversed by γ-secretase inhibitor.
Effect of mutant PS2 on cell proliferation, PRDX6 expression, iPLA2 activity, iPLA2 cleavage, γ-secretase activity, and PS2/PRDX6 colocalization in AD patient-derived skin fibroblast. A, After skin fibroblast cells from an AD (N141) patient (AG09908) and non-mutated epithelial cells (A431) cells were cultured in 96-well plates (1 × 104 cells per well), cell viability was analyzed by MTT assay after 0, 24, 48, 72 hr. The results are expressed as mean±s.d. of three experiments with each experiment performed in triplicate. *P<0.05 compared with the non-mutated epithelial cells. The levels of iPLA2 activities (B) and γ-secretase activity (C) were measured in A431 and AG09908 after 24 hr. D, A431 and AG09908 cell lysates were analyzed by Western blotting. Samples (20 μg) were resolved on SDS-PAGE, and detected with antibodies against iPLA2, PRDX6 and β-actin. E, AG09908 and A431 cells were cultured in 100mm dish (5 × 106 cells) for 24 hr, and cell lysates were immunoprecipitated with PRDX6 and analyzed with western blotting by PS2 and PRDX6 antibody. F, AG09908 and A431 cells were plated in 8 chamber slide (5 × 104 cells per well) and treated with γ-secretase inhibitor (L685, 458, 10 μM) for 24 hr, and the colocalization of PS2 and PRDX6 was analyzed with IF staining.
Mutant PS2 transgenic mice are resistant to urethane-induced lung tumorigenesis
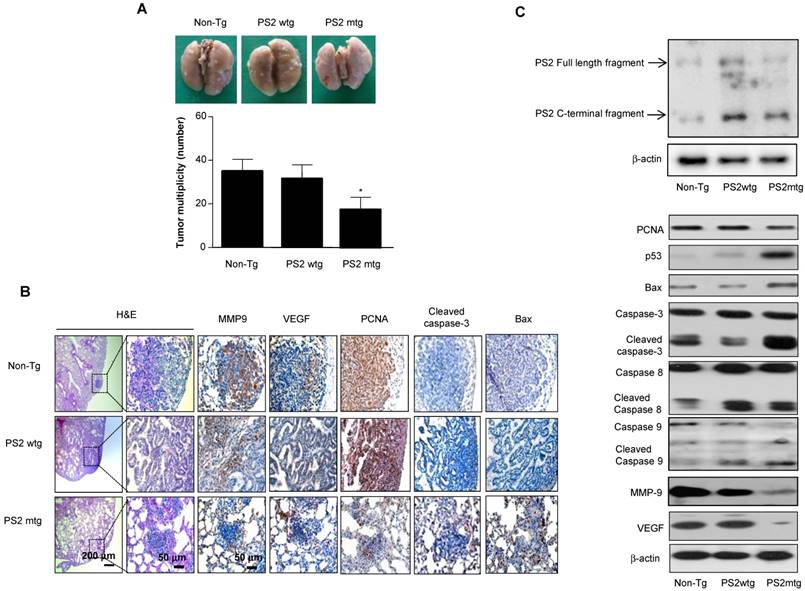
To determine whether mutant PSs contributes to lung tumorigenesis, wildtype or PS transgenic mice were subjected to urethane-induced carcinogenesis. Several lung tumors were observed in wildtype PS2 transgenic mice and non-transgenic controls; however, this number was significantly reduced in mutant PS2 transgenic mice (Fig. 4A). Tumor multiplicity was 17.3 ± 6.5 in mutant PS2 transgenic mice, but 33.6 ± 6.5 and 36.5 ± 5.7 in wildtype PS2 transgenic and non-transgenic controls, respectively. H&E staining showed well-differentiated lung adenocarcinomas in wildtype PS2 transgenic and non-transgenic controls, whereas tumors in mutant PS2 transgenic were far less progressed (hyperplasia or early adenomas) (Fig. 4B). Immunohistochemical and Western blot analysis showed significantly lower proliferation (PCNA), metastasis (matrix metalloproteinase-9, MMP-9), and angiogenic (vascular endothelial growth factor, VEGF) marker expression in mutant PS2 lung tissue, whereas expression of the apoptotic markers caspase-3, caspase-8, caspase-9, p53, and Bax were markedly higher when compared to controls (Fig 4B and C, Table 2).
Apoptotic and proliferative protein expression in tumor tissues
| Carcinogen induction model (PS2) | Metastasis model (PS2) | |||||
|---|---|---|---|---|---|---|
| Non-Tg | Tg- wt | Tg-mut | Vector | WT | MT | |
| PRDX6 | 72.5 ± 3.5 | 90.4 ± 2.2 | 20.6 ± 1.5* | 43.2 ± 3.2 | 77.8 ± 3.5 | 25.4 ± 1.8* |
| PCNA | 80.2 ± 4.2 | 76.5 ± 3.8 | 40.5 ± 4* | 86.5 ± 3.4 | 87.4 ± 2.2 | 48.2 ± 3.6* |
| Caspase-3 | 2.5 ± 1.2 | 3.6 ± 0.8 | 48 ± 3.2* | 3.2 ± 1.4 | 1.2 ± 0.5 | 30.5 ± 2.8* |
| Bax | 5.4 ± 1.5 | 4.2 ± 1.2 | 55.8 ± 2.4* | 20.8 ± 1.6 | 30.5 ± 2.5 | 80.5 ± 3.4* |
| MMP9 | 85.4 ± 3.7 | 88.6 ± 2.6 | 50.2 ± 3* | 74.2 ± 2.5 | 68.6 ± 1.2 | 5.2 ± 2.2* |
| VEGF | 65.4 ± 3.2 | 76.4 ± 2.5 | 40.5 ± 3.5* | 53 ± 2.4 | 57.8 ± 1.8 | 15.2 ± 1.5* |
Tg, transgenic; wt, wildtype; mut, mutant.
The results are expressed as mean ± SD.
*P < 0.05 compared with wildtype PS counterparts or vector controls.
Effect of mutant PS2 on lung tumor development. A, Tumors were induced by a single intraperitoneal injection of 1 mg/g urethane once a week for 10 weeks. Mice were euthanized at time points up to 7 months after injection of carcinogen. At the time of killing, lungs were lavaged, perfused and fixed in ice-cold Bouin's fixative solution for 24 h. After fixation, lungs were used for surface tumor number and diameter measurements. The results are expressed as mean±s.d. The results are expressed as mean ± SD. *P<0.05 compared with wildtype PS2 transgenic mice (n=20). B, Lung tissues were processed and stained with haematoxylin and eosin or analyzed by immunohistochemistry for detection of positive cells for MMP9, VEGF, PCNA, Cleaved caspase-3 and Bax. C, Carcinogen induced lung tissue extracts were analyzed by western blotting. Samples (20 μg) were resolved on SDS-PAGE, and detected with antibodies against PS2, PCNA, p53, Bax, Cleaved caspase-3, -8 and -9, MMP-9, VEGF and β-actin. The experiments shown in Figure 1 were repeated in triplicate with similar results. *P<0.05 compared with the wild-type mice.
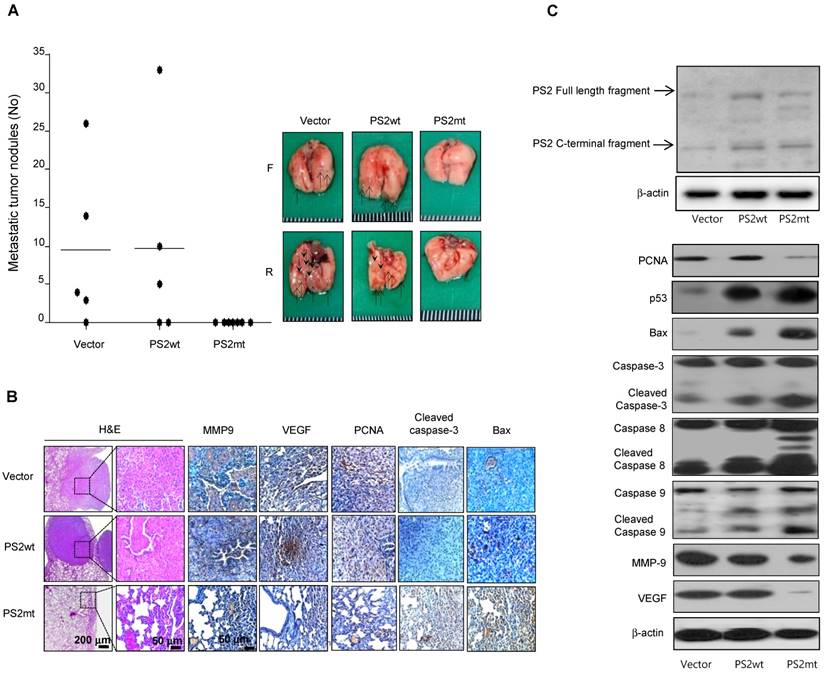
Reduced lung cancer metastasis by mutant PS2
The number of surface lung metastases was significantly lower in mice injected with mutant PS2 expressing lung cancer cells than that observed in vector or wildtype PS2 (Fig. 5A). Actually, in the mice injected with mutant PS2 expressing lung cancer cells, the number of observed tumor nodules was zero, but in the immunohistochemical staining, lung metastases were hyperplasias. H&E staining indicated that lung tumors in mice injected with vector or wildtype PS2 expressing lung cancer cells were well-differentiated lung adenocarcinomas, whereas hyperplasia was found in the mice injected with mutant PS2 expressing lung cancer cells, similar to that found in the carcinogen-induced tumor model (Fig. 5B). Moreover, immunohistochemical and Western blot analysis showed that proliferation, metastasis, and angiogenic marker expression was significantly lower, but apoptotic markers were markedly higher in the lung of mice injected with mutant PS2 expressing lung cancer cells than those in vector or wildtype PS2 counterparts (Fig. 5B and 5C, Table 2).
Effect of mutant PS2 on development of lung metastasis. A, A549 cells expressing vector, wildtype PS2 or mutant PS2 were injected into the lateral tail veins of nude mice (2 × 106 cells in /100 μL phosphate-buffered saline per animal). After 8 weeks, animals were sacrificed and the tumor lung metastases were counted on the lung surface. B, Lung tissues were processed and stained with haematoxylin and eosin or analyzed by immunohistochemistry for detection of positive cells for MMP9, VEGF, PCNA, Cleaved caspase-3 and Bax. C, Metastasized lung tissue extracts were analyzed by western blotting. Samples (20 μg) were resolved on SDS-PAGE, and detected with antibodies against PS2, PCNA, p53, Bax, Cleaved caspase-3, -8 and -9, MMP-9, VEGF and β-actin. The experiments shown in Figure 1 were repeated in triplicate with similar results. *P<0.05 compared with the wild-type mice.
Effect of mutant PS2 on PRDX6 expression and activity
Analysis PRDX6 expression in carcinogen-induced by immunohistochemistry revealed markedly lower levels in mutant PS2 transgenic mice than in the wildtype PS2 transgenic or non-transgenic controls (Fig. 6A, left panel). Similar findings were also observed by Western blotting (Fig. 6A, right panel). Since PRDX6 possesses pro-tumorigenic glutathione peroxidase and iPLA2 activity, we examined whether these functions were altered by PS2 mutation. Notably, both glutathione peroxidase and iPLA2 activity were lowered in mutant PS2 transgenic mice as compared to that in wildtype PS2 transgenic or non-transgenic counterparts (Fig. 6B).
Effect of mutant PS2 on PRDX6 expression, iPLA2 and γ-secretase activity, and iPLA2 cleavage. A, Effect of mutant PS2 on the expression of PRDX6 expression in urethane-induced tumors from non-transgenic, wildtype PS2, and mutant PS2 mice. Lung sections were analyzed by immunohistochemistry and their protein extracts were analyzed by Western blotting. Each image and band is representative of three mice. Effect of PS2 on PRDX6 glutathione peroxidase and iPLA2 activity (B), γ-secretase activity (C) iPLA2 cleavage (D) respectively, in urethane-induced tumors from non-transgenic, wildtype PS2, and mutant PS2 transgenic mice. *P< 0.05 indicates significant difference from wild PS2 transgenic mice group. E, Effect of mutant PS2 on the colocalization and interaction of PRDX6 and PS2. Localization of PRDX6 (red) and PS2 (green) was observed by confocal microscopy after immunofluorescence staining in lung tumor tissues of urethane induced wildtype PS2 and mutant PS2 transgenic mice (E, upper panel), and PS2 lung cancer cells transfected with wildtype PS2 or mutant PS2 (H, upper panel). Immunoprecipitation of lung tumor tissues of urethane induced wildtype PS2 and mutant PS2 transgenic mice (E, lower panel), and PS2 lung cancer cells transfected with wildtype PS2 or mutant PS2 (H, lower panel). Each image and band is representative of three independent mice experiments. F, Effect of γ-secretase inhibition on mutant PS2-induced cell growth inhibition, and activity of iPLA2 and the cleavage of iPLA2. Lung cancer cells were cultured into 24-well plates (5×104 cells per well) and transfected with wildtype or mutant PS2 plasmid with/without treatment of PEN2 siRNA (100 nM) or DAPT (2 μM) for 24 h. Cells were harvested and assayed cell viability by MTT method (F), assayed iPLA2 activity (G) and determined cleavage of iPLA2 by Western blotting (I). The results are expressed as mean ± SD from three independent experiments. *P< 0.05 indicates significant difference from wild PS2 transfected group. #P< 0.05 indicates significant difference from mutant PS2 transfected group.
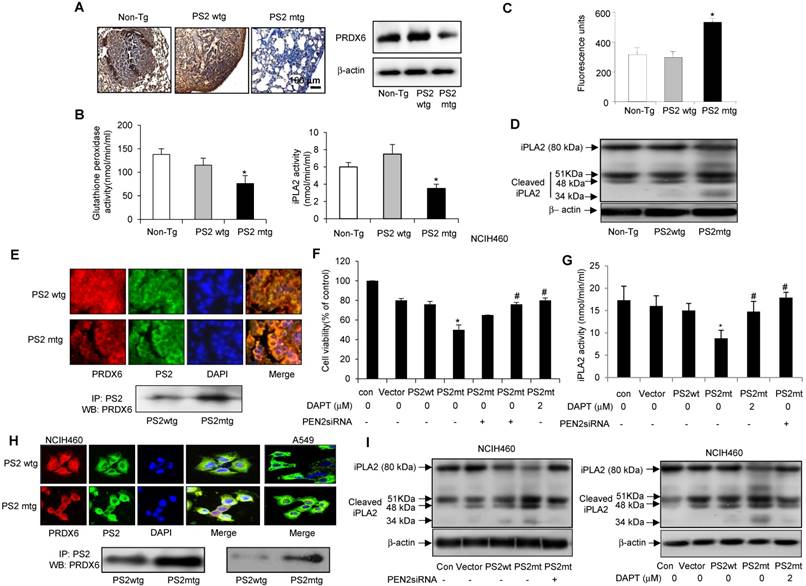
Effect of mutant PS2 on the γ-secretase activity and iPLA2 cleavage
In human patient samples, we showed that γ-secretase activity was markedly and cleaved iPLA2 was increased in AD patient. Agreed with human patient data, increased iPLA2 cleavage was observed in carcinogen-induced and metastatic lung tumors isolated from mutant PS2 transgenic mice (Fig. 6D), consistent with the increased γ-secretase activity (Fig. 6C). Thus, these data suggest that the iPLA2 motif in PRDX6 is cleaved by γ-secretase, which is elevated in the presence mutant PS2.
Colocalization and interaction of PRDX6 and PS2
We then investigated the mechanism by which PS2 mutation alters iPLA2 activity. Since γ-secretase cleaves membrane phospholipids, we hypothesized that PRDX6 could localized to membrane in complex with PS2, where γ-secretase could then cleave PRDX6 in its GXSXG phospholipase motif. Interestingly, PRDX6 localized to the cytosol in wildtype PS lung tissue, but translocated to the plasma membrane in tumor tissue (Fig. 6E, upper panel). Immunoprecipitation analysis indicated that PRDX6 and PS2 colocalization was increased in carcinogen-induced lung tumors isolated from mutant PS2 transgenic mice (Fig. 6E, lower panel). This increased co-localization was also observed in NCIH460 and A549 lung cancer cells transfected with mutant PS2 (Fig. 6H, upper panel). Moreover, Immunoprecipitation analysis showed that the mutant PS2 proteins had a stronger affinity for PRDX6 as compared to wildtype counterparts (Fig. 6H, lower panel). Inhibition of γ-secretase activity with PEN2 (γ-secretase) siRNA or the inhibitor DAPT (2 μM) showed rescued cell proliferation (Fig. 6F), iPLA2 activity (Fig. 6G) and iPLA2 cleavage (Fig. 6I). Thus, these data suggest that the co-localization of PRDX6 and PS2 is elevated in the presence mutant PS2, resulting alters iPLA2 activity by increased γ-secretase activity.
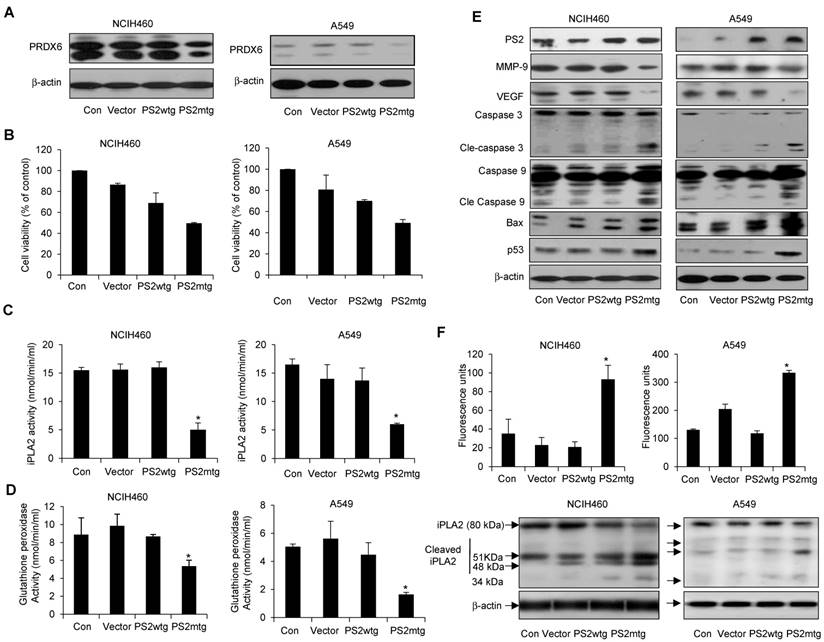
Effect of mutant PS2 on PRDX6 expression, proliferation, iPLA2 and γ-secretase activity, and apoptotic protein expression in lung cancer cells
To further investigate whether the physiological effect of mutant PS2 in lung cancer, NCIH460 and A549 lung cancer cells were transfected with vector alone, wildtype or mutant PS2 plasmid. As expected, mutant PS2 cells inhibited PRDX6 expression (Fig. 7A), cell viability (Fig. 7B) as well as iPLA2 (Fig. 7C) and glutathione peroxidase activity (Fig. 7D), when compared wildtype PS2 transfected cells or vector alone transfected cell. Similar to our previous findings, mutant PS2 lung cancer cells showed elevated levels cleaved caspase-3 and caspase-9, Bax, and p53, whereas VEGF and MMP-9 were decreased (Fig. 7E). Moreover, iPLA2 cleavage was significantly higher in mutant PS2 lung cancer cells (Fig. 7F, lower panel) compared to vector alone or wildtype PS2 transfected cells consistent with the higher γ-secretase activity (Fig. 7F upper panel).
Effect of mutant PS2 on cell proliferation, PRDX6 expression, glutathione and iPLA2 activity, iPLA2 cleavage, and γ-secretase activity, and protein expression in lung cancer cells. Lung cancer cells with exogenous wildtype or mutant PS expression were examined for PRDX6 expression (A), cell proliferation (B), iPLA2 activity (C) and glutathione peroxidase activity (D). (E) Lung cancer cells were transfected with the vector, wildtype PS2, or mutant PS2 plasmid for 24h. Cell extracts were analyzed by Western blotting. (F) iPLA2 cleavage was monitored as described in Figure 5. Each band is representative of three independent experiments. The results are expressed as mean ± SD from three independent experiments. *P< 0.05 indicates significant difference from wild PS2 transfected group.
Discussion
In population based cohort study, patients with cancer had a 43% lower risk of developing AD, whereas those with AD had a 69% lower risk of being admitted to the hospital for cancer [12]. Although the relationship between AD and cancer incidence has been theorized, it has not been conclusively shown with experimental data. Here, we showed that spontaneous and carcinogen-induced lung tumor development and metastasis were significantly inhibited, and the expression of PRDX6 was downregulated in mutant PSs transgenic mice. Accompanied with this data, the expression of PRDX6 was upregulated in squamous lung cancer patient samples, but lowered in AD patient sample with PS2 mutation. Moreover, this finding was confirmed by the decreased proliferation of lung cancer cells transfected with mutant PS2 (N141I), as well as mutant PS2 skin fibroblasts derived from patients with AD. These data support the epidemiological observation of lower cancer prevalence in AD patients, albeit through an unclear mechanism.
Recently, we found that PRDX6 overexpression increased the growth and metastasis of xenograft and urethane-induced lung tumors, indicating that PRDX6 may regulate lung tumor progression. Moreover, PRDX6 expression in these samples associated with that of PCNA, VEGF, and MMP-9, were inversely correlated with cleaved caspase-3, caspase-8, caspase-9, Bax, and p53 levels. Therefore, the decreased PRDX6 resulting from PSs mutation may regulate the expression of genes involved in cell proliferation, metastasis and apoptosis, thereby attenuating lung tumor development in mouse models of AD. Similarly, in vitro studies showed that mutant PS2 lung cancer cells and mutant PS2 patient-derived fibroblasts exhibit lower PRDX6 expression, proliferation, and proliferative gene expression. We also found that PRDX6 gene was significantly associated with human squamous lung cancer in co-expression network analysis using the microarray data from both squamous lung cancer biopsy specimens and paired normal specimens. The major biological processes (Gene ontology) including cytoskeletal assembly, RNA processing, cellular protein complex assembly and methylation significantly enriched in the genes of the co-expression module. These co-expressed genes are reported to be involved in lung cancer development. It has been suggested that cytoskeleton assembly is upregulated in metastatic and invasive cancer cells [30, 31]. Goehe et al. demonstrated that RNA processing factor is specifically phosphorylated in non-small cell lung cancer (NSCLC), thereby promotes expression of the anti-apoptotic proteins and contributing to tumorigenesis [32]. Recent study also demonstrates that lung cancer susceptibility genes might be regulated by methylation changes in response to smoking by genome-wide association studies (GWASs) [33]. PRDX6 exhibits glutathione peroxidase and phospholipase A2 (PLA2) activity (24). Increased glutathione peroxidase activity enhances cholangiocarcinoma growth [34], lung metastases [35], and H460 human lung cancer cell growth [36], whereas iPLA2 activation contributes to lung metastasis [37], as well as A549 and H460 human lung cancer cell invasion. In the present study, we identified that mutant PS2 attenuated PRDX6 expression, and iPLA2 and glutathione peroxidase activity. Moreover, iPLA2 activity was significantly lower in mutant PS2 skin fibroblasts. Thus, iPLA2 and glutathione peroxidase inhibition could limit the inhibitory effect of mutant PSs in lung tumor development. Based on these studies, our results suggest that PRDX6 is important for the development of lung cancer, and mutant PSs in AD patient could inhibit lung tumorigenesis. However, the mechanism of PS mutation-mediated PRDX6 downregulation remains unclear.
PRDX6 is mostly localized to the cytosol [17], its activity may be regulated in response to In the present study, we found that PRDX6 localized the plasma membrane of cells harboring mutant PS2 in vitro and in vivo, but was retained in the cytosol of wildtype PS2 cells. Thus, PS mutations may alter PRDX6 localization, thereby attenuating iPLA2 activity leading to inhibition of cell proliferation. PS is a member of the γ-secretase enzymatic complex. Several reports demonstrate that γ-secretase activity is regulated by its interaction with enzymatic components in specific subcellular compartments. For example, the p24 protein family member TMP21 is expressed in intracellular compartments where it binds PS, leading to changes in γ-secretase activity and Aβ generation [38]. Moreover, iPLA2 activity can be inhibited by surfactant protein A (SP-A) through direct protein-protein interaction [29]. Thus, it is possible that the increased interaction between PRDX6 and mutant PSs in the plasma membrane may have a substantial effect on PRDX6 activity. In fact, we found that mutant PS bound PRDX6 with a higher affinity compared to the wildtype protein. Mechanistically, the change in PRDX6 activity could result from the γ-secretase-mediated cleavage of the GXSXG phospholipase motif in the PRDX6 iPLA2 catalytic site. Correspondingly, our data showed that γ-secretase activity and iPLA2 cleavage were increased in mutant PS transgenic mice lung tumor tissues and transfected cancer cells, which could be returned to normal levels in the presence of PEN-2 siRNA or the γ-secretase inhibitor DAPT. Further, γ-secretase activity and the cleavage of iPLA2 cleavage were also significantly increased in PS mutant fibroblasts. Thus, the increased interaction between mutant PSs and PRDX6 likely favors the γ-secretase-mediated cleavage of the iPLA2 in PRDX6, which results in diminished lung tumor growth. However, lung tissue from lung tumor patients exhibited higher PRDX6 expression and iPLA2 activity, as well as and full-length iPLA2, and showed no differences in γ-secretase activity or PSs/PRDX6 affinity when compared to normal lung tissues, indicating that other mechanisms may be at play. The Notch receptor is a single-pass transmembrane protein critical for the initiation of several cancers [39]. Previous studies identified Notch as a γ-secretase substrate expressed in ~40% of NSCLC tumors. Moreover, expression of dominant-negative Notch or γ-secretase inhibitor treatment inhibits lung cancer growth in vitro and in vivo [40, 41]. Thus, γ-secretase-dependent Notch signaling could play a role in oncogenesis; however, no significant differences in expression were observed between wildtype and mutant PS tumor tissues and cells (data not shown). Although Notch does not contain a GXSXG motif, indicating that the tumor suppressor effect of PSs is γ-secretase dependent mechanism by the cleavage of iPLA2 that is the catalytic center of PRDX6.
Many epidemiological studies suggest an inverse correlation between cancer development and neurodegenerative disorders, such as Parkinson's disease [42, 43] and Huntington's disease [44], which could also be present in patients of AD. Our results showed that PS mutations may could play a critical role in the suppressing lung cancer development in AD patients, and support the inverse association AD development and cancer incidence.
Materials and Methods
Co-expression network analysis
We constructed co-expression networks using publicly available microarray data from both squamous lung cancer biopsy specimens and paired normal specimens from 5 patients. The microarray data (GSE3268) from a previous lung cancer study were downloaded from the GEO database (http://www.ncbi.nlm.nih.gov/geo/). The normalized microarray data were used to generate gene co-expression networks using WGCNA.
Functional annotation
DAVID (http://david.abcc.ncifcrf.gov/home.jsp) was used to identify the biological processes that were significantly enriched in the genes included in the co-expression modules. P-values less than 0.05 were considered significant.
GEO profiling
After search the GEO profile, we analyzed the PRDX6 expression pattern in gene expression omnibus data set GDS1312 containing human lung cancer samples and normal controls. We analyzed the PRDX6 expression pattern in gene expression omnibus data set GDS4141 containing mutant PS2 containing samples and normal controls.
Carcinogenesis protocols
PS2 wildtype transgenic and PS2 mutant (N141I) knock-in transgenic and non-transgenic mice were obtained from Dr. Hwang [45] and were maintained in accordance with the guidelines prescribed by the Chungbuk National University Animal Care Committee (Chungbuk National University, Korea, CBNUA-436-12-02). Tumors were induced in 18-20-week-old mice by a single i.p. injection of 1 mg/g urethane (ethyl carbamate; Sigma-Aldrich) once a week for 10 weeks. Mice were progressively euthanized for up to 7 months after the injection procedure. At the time of sacrifice, lungs were lavaged, perfused, fixed in ice-cold Bouin's fixative solution (Sigma-Aldrich) for 24 h, and then paraffin-embedded to assess surface tumor number and diameter. Tumors on the lung surface were enumerated by at least two experienced readers in a blinded manner under a dissecting microscope. Diameters were measured using digital calipers (Tracer, Fisher Scientific, Waltham, MA, USA). At the end of the experiment, the animals were euthanized and tumors were separated from the surrounding muscle.
Lung metastasis model
Eight-week-old male BALB/C nude mice were purchased from Orient-Bio (Gyunggi-do, Korea). The mice were maintained in accordance with the guidelines prescribed by the Chungbuk National University Animal Care Committee (Chungbuk National University, Korea, CBNUA-436-12-02). The mice were held for 4 days after arriving before they were injected with cells. A549 cells were injected into the lateral tail veins of nude mice (2 × 106 cells in /100 μL phosphate-buffered saline per animal). After 8 weeks, animals were sacrificed and the tumor lung metastases were counted on the lung surface. Metastases were counted in all lobes of the lung, except for the middle lobe where the primary tumors localized. Data are presented as the number of tumor nodules per lung.
Cell culture
NCIH460, A549 human lung cancer cells and normal skin fibroblast were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). Skin fibroblast cells prepared from a patient with AD patient (N141I) were obtained from Coriell Institute (Cat #AG09908, NJ, USA). Cells were grown in RPMI1640 or MEM with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C in a 5% CO2 humidified atmosphere.
Co-Immunoprecipitation
AG09908, normal skin fibroblast, A549 and NCIH460 cells were gently lysed for 1 h on ice and then centrifuged at 14,000 rpm for 15 min at 4 °C, and the supernatant was collected. The soluble lysates were incubated with anti-PRDX6 antibody (Novus Biologicals, Littleton, CO, USA) at 4 °C, followed by Protein A/G beads (Santa Cruz Biotechnology), and then washed 3 times. Immune complexes were eluted by boiling for 10 min at 95 °C in SDS sample buffer and immunoblotted with anti-PRDX6 (1:2000) or anti-PS2 (1:1000) antibodies.
Immunohistochemistry
All specimens were formalin-fixed and paraffin-embedded. Hematoxylin and eosin (H&E) staining and immunohistochemistry were performed as described previously (Hwang et al, 2002). Tissue sections were blocked for 30 min with 3% normal horse serum diluted in PBS. The sections were then blotted and incubated with primary mouse MMP9, PCNA, Ki-67, and Bax monoclonal antibodies diluted 1:200 in blocking serum for 4 h at room temperature, or primary rabbit anti-VEGF and cleaved caspase-3 polyclonal antibody diluted 1:100 dilution in blocking serum overnight at 4 oC. The next day, the slides were washed three times for 5 minutes each in PBS and incubated in biotinylated anti-mouse or anti-rabbit antibodies for 2 h, and then washed again in PBS. The avidinbiotin-peroxidase complexes were formed (ABC, Vector Laboratories, Inc., Burlingame, CA) and the peroxidase reaction developed with diaminobenzidine and peroxide. The tissue sections were then counterstained with hematoxylin, mounted with aqua-mount, and evaluated using a light microscope (200 × magnification, Olympus, Tokyo, Japan).
Immunoprecipitation
Protein A/G agarose beads (Santa Cruz Biochemicals) were saturated in wash buffer (50 mM Tris-HCl pH 7.4, 100 mM KCl, 0.1% NP-40) for 2 h at 4 °C and conjugated with anti-PS1 or anti-PS2 for 4 h at 4 °C. Cytosol extracts were incubated with antibody-coupled beads for 4 h at 4 °C in immunoprecipitation buffer (50 mM Tris-HCl pH 7.4, 100 mM KCl, 5 mM MgCl2, 0.1% NP-40, 10% glycerol). The protein-bound beads were washed in wash buffer and protein was eluted by boiling for 2 min in SDS sample buffer.
Immunofluorescence staining
Fixed cells and tissues were permeabilized with 0.1% Triton X-100 in PBS for 2 min, blocked in 5% bovine serum albumin in PBS at room temperature for 2 h, and then incubated with anti-rabbit PRDX6 polyclonal antibody for PRDX6 (1:200 dilution) or PS2 goat polyclonal antibody (1:200 dilution) overnight at 4 °C. After washing in ice-cold PBS, samples were incubated with anti-rabbit Alexa Fluor 568 and anti-goat Alexa Fluor 488 (1:100 dilution, Molecular Probes Inc., Eugene, OR, USA) for 2 h at room temperature. Immunofluorescence images were acquired using a confocal laser scanning microscope (TCS SP2, Leica Microsystems AG, Wetzlar, Germany) equipped with a 630× oil immersion objective.
Western blot analysis
Western blot analysis was done as described previously (Hwang et al, 2002). The membranes were immunoblotted with the following antibodies: mouse monoclonal anti-Bax (1:500 dilution, Santa Cruz Biotechnology Inc., Dallas, TX, USA), mouse monoclonal anti-PCNA (1:1000 dilution, Cell Signaling Technology, Inc. Beverly, MA, USA), rabbit polyclonal anti-PRDX6, anti-iPLA2, anti-caspase-3, and anti-cleaved caspase-3 (1:1000 dilution, Cell Signaling Technology, Inc. Beverly, MA, USA). The blots were then incubated with the corresponding horseradish peroxidase-conjugated anti-rabbit and anti-mouse IgG (1:2000 dilution, Santa Cruz Biotechnology Inc.). Immunoreactive proteins were detected by enhanced chemiluminescence and subjected to densitometric analysis using MyImage (SLB, Seoul, Korea), and quantified in Labworks 4.0 software (UVP Inc., Upland, CA, USA).
Two-dimensional (2D) gel electrophoresis
Lung tissues were suspended in 0.5 mL sample buffer (40 mM Tris, 7 M urea [Merck, Darmstadt, Germany], 2 M thiourea [Sigma, St. Louis, MO, USA], 4% CHAPS [Sigma], 60 mM 1,4-dithioerythritol [Merck], 1 mM EDTA [Merck], 0.5% v/v IPG buffer pH 4-7, 1 mM phenylmethylsulfonyl fluoride [Sigma], and a protease inhibitor cocktail tablet [Roche, Basel, Switzerland]). The suspension was sonicated for approximately 30 s and centrifuged at 12,000 ×g for 1 h. The protein concentration of the supernatant was determined with by Bradford assay. Samples (500 μg) were applied to Immobiline Drystrips (pH 4-7, 18 cm, Amersham, Little Chalfont, UK). After rehydration for 12 h, the proteins were focused for a total of 57,000 Vh. The second separation was performed on 7.5-17.5% linear gradient polyacrylamide gels. The gels were fixed with 40% methanol containing 5% phosphoric acid for 12 h and then stained with colloidal Coomassie blue (Novex, San Diego, CA, USA) for 48 h. Molecular mass was determined with a 10-200-kDa standard protein marker (Gibco, Basel, Switzerland) on the right side of select gels. Isoelectric point (pI) values were determined using a guide supplied by the manufacturer of the IPG strips. The gels were then destained with water, scanned using a GS-800 imaging densitometer (Bio-Rad), and the images analyzed with PDQuest software (Bio-Rad). Matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) analysis was used to identify protein bands (ProteomTech Inc., Emeryville, CA, USA).
GPX and PLA2 enzymatic activity
PLA2, iPLA2, and GPX activity were measured according to the manufacturer's recommendations (Cayman Chemicals, Ann Arbor, MI).
γ-secretase assay
Enzyme activity levels were quantified as previously described [46].
Statistics
The data were analyzed using the GraphPad Prism 4 v.4.03 software (GraphPad Software, La Jolla, CA, USA). Data are presented as mean ± SD. Statistical differences were assessed by one-way analysis of variance (ANOVA) with Tukey's post-hoc test for multiple comparisons. P < 0.05 was considered statistically significant.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (MRC, 2017R1A5A2015541).
Study Approval
All animal experiments were performed in accordance with the guidelines prescribed by the Chungbuk National University Animal Care Committee (Chungbuk National University, Korea, CBNUA-436-12-02).
Supplementary Material
Supplementary figure and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bertram L, Tanzi RE. Thirty years of Alzheimer's disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci. 2008;9:768-778
2. Bertram L, Lill CM, Tanzi RE. The Genetics of Alzheimer Disease: Back to the Future. Neuron. 2010;68:270-281
3. Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N. et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2:864-870
4. Gandy S. The role of cerebral amyloid β accumulation in common forms of Alzheimer disease. J Clin Invest. 2005;115:1121-1129
5. Rocher-Ros V, Marco S, Mao JH, Gines S, Metzger D, Chambon P. et al. Presenilin modulates EGFR signaling and cell transformation by regulating the ubiquitin ligase Fbw7. Oncogene. 2010;29:2950-2961
6. Kang DE, Soriano S, Xia X, Eberhart CG, De Strooper B, Zheng H. et al. Presenilin Couples the Paired Phosphorylation of β-Catenin Independent of Axin: Implications for β-Catenin Activation in Tumorigenesis. Cell. 2002;110:751-762
7. Zhang YW, Wang R, Liu Q, Zhang H, Liao FF, Xu H. Presenilin/γ-secretase-dependent processing of β-amyloid precursor protein regulates EGF receptor expression. Proc Natl Acad Sci U S A. 2007;104:10613-10618
8. To MD, Gokgoz N, Doyle TG, Donoviel DB, Knight JA, Hyslop PS. et al. Functional characterization of novel presenilin-2 variants identified in human breast cancers. Oncogene. 2006;25:3557-3564
9. Kovacs DM, Mancini R, Henderson J, Na SJ, Schmidt SD, Kim TW. et al. Staurosporine-Induced Activation of Caspase-3 Is Potentiated by Presenilin 1 Familial Alzheimer's Disease Mutations in Human Neuroglioma Cells. J Neurochem. 1999;73:2278-2285
10. Xia X, Qian S, Soriano S, Wu Y, Fletcher AM, Wang XJ. et al. Loss of presenilin 1 is associated with enhanced β-catenin signaling and skin tumorigenesis. Proc Natl Acad Sci U S A. 2001;98:10863-10868
11. Serrano J, Fernandez AP, Martinez-Murillo R, Martinez A. High sensitivity to carcinogens in the brain of a mouse model of Alzheimer's disease. Oncogene. 2010;29:2165-2171
12. Driver JA, Beiser A, Au R, Kreger BE, Splansky GL, Kurth T. et al. Inverse association between cancer and Alzheimer's disease: results from the Framingham Heart Study. BMJ. 2012;344:e1442
13. Yun HM, Park MH, Kim DH, Ahn YJ, Park KR, Kim TM. et al. Loss of presenilin 2 is associated with increased iPLA2 activity and lung tumor development. Oncogene. 2014;33:5193-5200
14. Jagadeeswaran R, Surawska H, Krishnaswamy S, Janamanchi V, Mackinnon AC, Seiwert TY. et al. Paxillin Is a Target for Somatic Mutations in Lung Cancer: Implications for Cell Growth and Invasion. Cancer Res. 2008;68:132-142
15. Granville CA, Dennis PA. An Overview of Lung Cancer Genomics and Proteomics. Am J Respir Cell Mol Biol. 2005;32:169-176
16. Rhee SG, Chae HZ, Kim K. Peroxiredoxins: A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543-1552
17. Wood ZA, Schröder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28:32-40
18. Lehtonen ST, Svensk AM, Soini Y, Pääkkö P, Hirvikoski P, Kang SW. et al. Peroxiredoxins, a novel protein family in lung cancer. Int J Cancer. 2004;111:514-521
19. Vázquez-Medina JP, Dodia C, Weng L, Mesaros C, Blair IA, Feinstein SI. et al. The phospholipase A2 activity of peroxiredoxin 6 modulates NADPH oxidase 2 activation via lysophosphatidic acid receptor signaling in the pulmonary endothelium and alveolar macrophages. FASEB J. 2016;30:2885-2898
20. Zhou S, Sorokina EM, Harper S, Li H, Ralat L, Dodia C. et al. Peroxiredoxin 6 homodimerization and heterodimerization with glutathione S-transferase pi are required for its peroxidase but not phospholipase A2 activity. Free Radic Biol Med. 2016;94:145-156
21. Linkous AG, Yazlovitskaya EM, Hallahan DE. Cytosolic Phospholipase A2 and Lysophospholipids in Tumor Angiogenesis. J Natl Cancer Inst. 2010;102:1398-1412
22. Meyer AM, Dwyer-Nield LD, Hurteau GJ, Keith RL, O'Leary E, You M. et al. Decreased lung tumorigenesis in mice genetically deficient in cytosolic phospholipase A2. Carcinogenesis. 2004;25:1517-1524
23. Heasley LE, Thaler S, Nicks M, Price B, Skorecki K, Nemenoff RA. Induction of Cytosolic Phospholipase A2 by Oncogenic Ras in Human Non-small Cell Lung Cancer. J Biol Chem. 1997;272:14501-14504
24. Jo M, Yun HM, Park KR, Park MH, Kim TM, Pak JH. et al. Lung tumor growth-promoting function of peroxiredoxin 6. Free Radic Biol Med. 2013;61:453-463
25. Fortini ME. Gamma-Secretase-mediated proteolysis in cell-surface-receptor signalling. Nat Rev Mol Cell Biol. 2002;3:673-684
26. Beel AJ, Sanders CR. Substrate specificity of gamma-secretase and other intramembrane proteases. Cell Mol Life Sci. 2008;65:1311-1334
27. Munter LM, Voigt P, Harmeier A, Kaden D, Gottschalk KE, Weise C. et al. GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Aβ42. EMBO J. 2007;26:1702-1712
28. Sagi SA, Lessard CB, Winden KD, Maruyama H, Koo JC, Weggen S. et al. Substrate Sequence Influences γ-Secretase Modulator Activity, Role of the Transmembrane Domain of the Amyloid Precursor Protein. J Biol Chem. 2011;286:39794-39803
29. Wu YZ, Manevich Y, Baldwin JL, Dodia C, Yu K, Feinstein SI. et al. Interaction of Surfactant Protein A with Peroxiredoxin 6 Regulates Phospholipase A2 Activity. J Biol Chem. 2006;281:7515-7525
30. Yamaguchi H, Condeelis J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta. 2007;1773:642-652
31. Sahai E. Mechanisms of cancer cell invasion. Curr Opin Genet Dev. 2005;15:87-96
32. Goehe RW, Shultz JC, Murudkar C, Usanovic S, Lamour NF, Massey DH. et al. hnRNP L regulates the tumorigenic capacity of lung cancer xenografts in mice via caspase-9 pre-mRNA processing. J Clin Invest. 2010;120:3923-3939
33. Helin K, Dhanak D. Chromatin proteins and modifications as drug targets. Nature. 2013;502:480-488
34. Chen Q, Li W, Wan Y, Xia X, Wu Q, Chen Y. et al. Amplified in breast cancer 1 enhances human cholangiocarcinoma growth and chemoresistance by simultaneous activation of Akt and Nrf2 pathways. Hepatology. 2012;55:1820-1829
35. Conti A, Rodriguez GC, Chiechi A, Blazquez RMD, Barbado V, Krènacs T. et al. Identification of Potential Biomarkers for Giant Cell Tumor of Bone Using Comparative Proteomics Analysis. Am J Pathol. 2011;178:88-97
36. Wang L, Chanvorachote P, Toledo D, Stehlik C, Mercer RR, Castranova V. et al. Peroxide Is a Key Mediator of Bcl-2 Down-Regulation and Apoptosis Induction by Cisplatin in Human Lung Cancer Cells. Mol Pharmacol. 2008;73:119-127
37. McHowat J, Gullickson G, Hoover RG, Sharma J, Turk J, Kornbluth J. Platelet-activating factor and metastasis: calcium-independent phospholipase A(2)β deficiency protects against breast cancer metastasis to the lung. Am J Physiol Cell Physiol. 2011;300:C825-C832
38. Chen F, Hasegawa H, Schmitt-Ulms G, Kawarai T, Bohm C, Katayama T. et al. TMP21 is a presenilin complex component that modulates gamma-secretase but not epsilon-secretase activity. Nature. 2006;440:1208-1212
39. Das I, Craig C, Funahashi Y, Jung KM, Kim TW, Byers R. et al. Notch Oncoproteins Depend on γ-Secretase/Presenilin Activity for Processing and Function. J Biol Chem. 2004;279:30771-30780
40. Haruki N, Kawaguchi KS, Eichenberger S, Massion PP, Olson S, Gonzalez A. et al. Dominant-Negative Notch3 Receptor Inhibits Mitogen-Activated Protein Kinase Pathway and the Growth of Human Lung Cancers. Cancer Res. 2005;65:3555-3561
41. Konishi J, Kawaguchi KS, Vo H, Haruki N, Gonzalez A, Carbone DP. et al. γ-Secretase Inhibitor Prevents Notch3 Activation and Reduces Proliferation in Human Lung Cancers. Cancer Res. 2007;67:8051-8057
42. Rugbjerg K, Friis S, Lassen CF, Ritz B, Olsen JH. Malignant melanoma, breast cancer and other cancers in patients with Parkinson's disease. Int J Cancer. 2012;131:1904-1911
43. Plun-Favreau H, Lewis PA, Hardy J, Martins LM, Wood NW. Cancer and Neurodegeneration: Between the Devil and the Deep Blue Sea. PLoS Genet. 2010;6:e1001257
44. Liu X, Hemminki K, Försti A, Sundquist J, Sundquist K, Ji J. Cancer risk and mortality in asthma patients: A Swedish national cohort study. Acta Oncol. 2015;54:1120-1127
45. Hwang DY, Chae KR, Kang TS, Hwang JH, Lim CH, Kang HK. et al. Alterations in behavior, amyloid β-42, caspase-3, and Cox-2 in mutant PS2 transgenic mouse model of Alzheimer's disease. FASEB J. 2002;16:805-813
46. Farmery MR, Tjernberg LO, Pursglove SE, Bergman A, Winblad B, Näslund J. Partial Purification and Characterization of γ-Secretase from Post-mortem Human Brain. J Biol Chem. 2003;278:24277-24284
Author contact
![]() Corresponding author: Dr. Jin Tae Hong, College of Pharmacy and Medical Research Center, Chungbuk National University, 48 Gaeshin-dong, Heungduk-gu, Cheongju, Chungbuk 361-763, Korea; Tel: 82-043-261-2813, Fax: 82-043-268-2732, Email: jinthongac.kr
Corresponding author: Dr. Jin Tae Hong, College of Pharmacy and Medical Research Center, Chungbuk National University, 48 Gaeshin-dong, Heungduk-gu, Cheongju, Chungbuk 361-763, Korea; Tel: 82-043-261-2813, Fax: 82-043-268-2732, Email: jinthongac.kr