Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. TFs and epigenetic regulators...
3. Therapeutic targeting of...
4. Therapeutic targeting of...
5. Perspective
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2025; 15(8):3345-3367. doi:10.7150/thno.107908 This issue Cite
Review
Supercharging CAR-T cells through transcriptional and epigenetic armoring
Diyuan Qin1,2†, Yanna Lei3†, Pei Shu4†, Yugu Zhang3, Yuin-Han Loh2, Yongsheng Wang3,5 ![]() , Qijing Li2,6,7
, Qijing Li2,6,7 ![]()
1. Cancer Center, Clinical Trial Center, West China Hospital, Sichuan University, Chengdu, Sichuan, China.
2. Institute of Molecular and Cell Biology (IMCB), Agency for Science, Technology and Research (A ∗ STAR), Singapore 138673, Singapore.
3. Division of Thoracic Tumor Multimodality Treatment, Cancer Center, West China Hospital, Sichuan University, Chengdu, Sichuan, China.
4. Division of Abdominal Tumor Multimodality Treatment, Cancer Center, West China Hospital, Sichuan University, Chengdu, Sichuan, China.
5. Cancer Center, National Medical Products Administration Key Laboratory for Clinical Research and Evaluation of Innovative Drugs, West China Hospital, Sichuan University, Chengdu, Sichuan, China.
6. Singapore Immunology Network (SIgN), Agency for Science, Technology and Research (A*STAR), Singapore 138648, Singapore.
7. Department of Microbiology and Immunology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 117545, Singapore.
†These authors contributed equally to this work and share first authorship
Received 2024-11-30; Accepted 2025-2-10; Published 2025-2-18
Abstract

Inspired by the remarkable success of CAR-T therapy in hematologic malignancies, research is increasingly focused on adapting this treatment for solid tumors. However, CAR-T efficacy remains limited due to its exhaustion and shortened persistence. Transcription factors and epigenetic modifications play pivotal roles in modulating T cell differentiation and functionality, which have been leveraged in numerous strategies to promote the formation of long-lasting memory cells with stem-like properties and supercharging CAR-T performance. This review highlights pivotal transcriptional factors, such as c-Jun and FOXO1, which enhance and sustain T cell effector function, diminishes exhaustion, and epigenetic regulators like TET2 and DNMT3A, whose knockout promotes memory T subsets formation. We explore their interconnections, downstream targets, biological impacts, and the potential application risks of certain candidates, providing a comprehensive theoretical framework for supercharging CAR-T therapies through transcriptional and epigenetic interventions.
Keywords: CAR-T cells, transcriptional regulation in immunotherapy, epigenetic modification, exhaustion and memory differentiation, tumor microenvironment
1. Introduction
Chimeric antigen receptor (CAR) T cells are genetically engineered to express a receptor on cell membrane that consists of an antibody fragment capable of specifically recognizing tumor antigens, coupled with intracellular costimulatory and activation domains that trigger T cell activation and function. This innovation facilitates precise tumor antigen targeting, robust costimulatory signaling, and the production of effector molecules, such as interferon-γ, granzyme B, and perforin, that effectively eradicate tumor cells. CAR-T therapy has achieved significant clinical success in treating hematologic malignancies [1-4]. However, their therapeutic efficacy in solid tumors remains suboptimal due to various inhibitory factors [5, 6].
The immunosuppressive cells and cytokines in the tumor microenvironment [7], along with abnormal tumor vasculature and metabolic competition, directly suppress CAR-T cell activity [8-11]. Additionally, chronic exposure to tumor antigens frequently induces T cell exhaustion, characterized by diminished proliferation and attenuated effector cytokine production, which compromises durable memory T cells formation, undermines sustained antitumor responses and ultimately results in tumor relapse and treatment failure [12].
T cell differentiation status directly influences the antitumor efficacy of immunotherapy [13-15]. Memory T cells are key players in maintaining long-term protective immunity, enabling the immune system to launch faster and more robust reactions to previously encountered antigens [16, 17]. The persistence of durable memory T cells is closely associated with improved CAR-T efficacy and extended disease remission [18-21]. Numerous studies are seeking strategies to prolong T cell persistence while alleviating exhaustion, both of which are crucial for enhancing antitumor responses [12, 22].
Recent advances in research have revealed that T cell memory differentiation is governed by the interaction of transcription factors (TFs) and epigenetic modifications. TFs orchestrate gene regulatory networks and serve as convergence points for signaling pathways driving T-cell differentiation [23, 24]. Epigenetic modifications such as DNA methylation, histone acetylation, and chromatin remodeling regulate gene expression and are also essential for T cell differentiation.
Importantly, TFs and epigenetic regulation are tightly interdependent: TFs recruit epigenetic complexes to modulate chromatin state, while epigenetic changes alter chromatin accessibility and impact TFs activity [25-27]. Together, they coregulate gene expression, forming a core mechanism that governs T cell commitment in response to environmental signals. Targeting TFs and epigenetic regulators offers a promising strategy to boost CAR-T cell expansion, stemness and resistance to exhaustion, ultimately strengthening CAR-T efficacy [12].
This review aims to summarize and highlight the roles of crucial TFs and epigenetic regulators in CAR-T cells, as well as their connection and impact on therapeutic outcomes, underscoring the potential of targeting TFs and epigenetic mechanisms to advance adoptive cell immunotherapy.
2. TFs and epigenetic regulators cooperate in T cell differentiation
Elucidating the mechanisms governing T cell fate decisions is significant for optimizing immunotherapies by precisely modulating T cell states. Advances in next-generation sequencing (NGS) technologies, such as ATAC-seq and scRNA-seq, enable comprehensive and detailed analysis of transcriptional changes across various T cell subsets. Numerous critical TFs and epigenetic regulators contributing to T cell memory differentiation have been identified.
TFs cooperate with epigenetic regulators to coordinate T cell differentiation. They directly control downstream gene transcription while shaping the epigenetic landscape through their intrinsic epigenetic regulatory functions or by recruiting chromatin-modifying enzymes, such as histone acetyltransferases or methyltransferases. Interferon regulatory factor 4 (IRF4) is crucial for driving naïve CD8+ T cells activation, expansion and effector differentiation. Together with basic leucine zipper ATF-like transcription factor (BATF) and Jun family members, IRF4 drives key TFs expression such as Runt-related transcription factor 3 (Runx3), T-box expressed in T cells (T-bet) and B-lymphocyte-induced maturation protein 1 (Blimp-1) while upregulating the expression of effector cytokines and receptors, which are essential for T cell effector functions and differentiation [28, 29]. Prior to the first division of naïve T cells, Runx3 initiates chromatin accessibility changes that open memory T cell-specific cis-regulatory regions. These regions are highly enriched with binding motifs for bZIP, IRF and Prdm1, leading to IRF4 and Blimp-1 upregulation and facilitating early effector and memory precursor cell differentiation [30]. Blimp-1 further recruits histone-modifying enzymes, including histone deacetylase 2 (HDAC2) and G9a, to target loci such as Il2ra and Cd27, causing increased histone H3K9 trimethylation and decreased histone H3 acetylation, which leads to the downregulation of target gene transcription [31]. Other TFs, such as lymphoid enhancer-binding factor 1 (LEF1) and T cell factor 1 (TCF1), display HDAC activity during T cell development, inhibiting histone acetylation in T cells to establish an epigenetic signature conducive to CD8+ T cell identity [32].
Moreover, epigenetic modifications also affect TFs expression. The upregulation of de novo methyltransferase DNA (cytosine-5)-methyltransferase 3A (DNMT3A) in activated T cells leads to Ccr7, Tcf7, and Sell methylation, which results in decreased gene transcription [33, 34].
T cell activation triggers dynamic variations in chromatin accessibility across differentiation stages, resulting in unique chromatin accessibility profiles. These changes are closely tied to the binding of specific TFs, reflecting the identical function and development states of T cells. For instance, TCF1 binding sites are prevalent in memory and naïve T cells, basic leucine zipper domain protein (bZIP), T-box and IRF motifs are enriched in effector and memory subsets, while chromatin regions enriched with nuclear receptor subfamily 4A (NR4A) and nuclear factor of activated T cells (NFAT) motifs are hallmarks of exhausted T cells [35].
Yu et al. explored TFs crucial for effector or memory CD8+ T cell commitment in Listeria infected mice. Effector T cell subsets are enriched with motifs binding T-bet, BATF, sterol regulatory element binding protein 2 (SREBP2) and AP-1 [36], which are pivotal for CD8+ T cells activation and functional capacity [29, 37-39]. Conversely, motifs associated with TCF1, LEF1, and E2A are predominantly enriched in naïve, memory precursor, and memory T cell subsets, correlating with their roles in maintaining stem-like properties and long-term immune memory [40-42].
Therefore, decoding the regulatory networks between TFs and epigenetic regulators will aid in understanding T cell differentiation. This knowledge provides a foundation for fine-tuning T cell differentiation, thereby optimizing T cell-based immunotherapies.
3. Therapeutic targeting of transcription factors in CAR-T therapy
T cell differentiation and functionality are orchestrated by complex gene regulatory networks, with TFs playing a pivotal role in this process. In our review, we categorized key TFs into two groups based on their impact: those promoting memory differentiation and those inducing exhaustion. Memory-promoting TFs, such as c-Jun [43], FOXO1 [44, 45] and FOXP1 [46], play critical roles in maintaining T cell stemness and resilience against exhaustion. The overexpression of these TFs has been shown to enhance the proliferative capacity, improve antitumor activity, and limit terminal differentiation in CAR-T cells. In contrast, TFs such as BATF [47], thymocyte selection-associated high mobility group box (TOX) [48], Blimp-1 [49] and the NR4A family [50] are linked to T cell exhaustion and dysfunction. Knockout of these TFs has been demonstrated to downregulate exhaustion-associated genes, promote memory T cell formation, and strengthen antitumor efficacy (Table 1).
Summary of transcriptional and epigenetic regulators involved in T cell effector functions and memory formation
| Transcriptional and Epigenetic Modifications | Regulators | Biological roles | Interventions | Impacts on T cells | Refs. |
|---|---|---|---|---|---|
| Memory relative TFs | c-Jun | Correlates with various biological processes Drives T cell activation genes expression Enhances T cell cytotoxicity and effector functions | Overexpression | Enhances cytokine secretion Promotes memory phenotypes Diminishes exhaustion | [43, 51] |
| TCF7 | Involves in Wnt signaling regulation Correlates with T cell development and differentiation | Overexpression | Promotes memory differentiation Diminishes exhaustion | [40, 52] | |
| FOXO1 | Participants in regulating cellular processes Regulated by PI3K-AKT signaling pathway Regulates the homeostasis and survival of naïve T cells and functions of dendritic cell, macrophages and B cells | Overexpression | Promotes memory differentiation Diminishes exhaustion Boost CAR-T cell expansion and persistence Enhances cytokine secretion | [44, 45] | |
| STAT5 | Activity is predominantly regulated by IL-2 cytokine family Regulates T cell homeostasis and immune function Rejuvenates exhausted CD8+ T cells by counteracting TOX | Overexpression | Diminishes exhaustion Boost CAR-T cell expansion and persistence Enhances cytokine secretion | [53-55] | |
| FOXP1 | Correlates with immune responses, organ development and cancer pathogenesis Regulates the quiescence and homeostasis of naïve T cells | Knockout | Impairs tumor suppression Diminishes proliferative capacity Downregulates stemness-associated genes Regulates checkpoints of stem-like to effector transition in CAR T cells | [46] | |
| KLF2 | Participants in regulating cellular processes Correlates with functionality and homeostasis of immune cells and T cells migration | Knockout | Impairs effector differentiation, Enhanced the exhaustion program and upregulated Tox Regulates checkpoints of stem-like to effector transition in CAR T cells | [46] | |
| Exhaustion-inducing TFs | BATF | Negatively regulates AP-1-mediated T cell effector functions Drive the differentiation of progenitor T cells into CX3CR1⁺ effector cells The effects on T cell-mediated antitumor responses remain inconsistent and context dependent. | Overexpression or Knockout | Represents dual Effect on CAR-T cells Correlates with T cells exhaustion, memory differentiation, and cytotoxicity. | [47, 56, 57] |
| TOX | Correlates with ontogeny of innate lymphoid cells, NK cells, and T cells Regulates T cells dysfunction | Knockout | Increases cytokine expression Decreases expression of inhibitory receptors Enhances the cytolytic activity | [58, 59] | |
| NR4A | Participants in regulating cellular processes Regulates T cells dysfunction Associates with chronic inflammation, altered immune cell responses, and cancer development. | Knockout | Decreases inhibitory receptor expression Increases cytokine production Enhances effector functions | [50, 60] | |
| Blimp-1 | Induces terminal differentiation in both B and T cell lineage Associates with immune suppression and formation of short-lived effector cells | Knockout | Maintains the memory phenotype Enhances cytokine polyfunctionality | [49, 61-64] | |
| IKZF3 | Participants in lymphopoiesis and differentiation Promotes tumor progression Facilitates T follicular helper (Tfh) cell commitment | Knockout | Enhances cytokine signaling, chemotaxis, and adhesion pathways Boosts effector molecule secretion Reverse T cell exhaustion | [65-67] | |
| Epigenetic regulator | TET2 | Mediating DNA demethylation Related to myeloid malignancies Directs T cells differentiation | Knockout | Promotes memory differentiation Diminishes exhaustion Increases CAR-T persistence | [68-71] |
| DNMT3A | De novo methyltransferases Mediates T cell Th1 and Th2 polarization Regulates the proportion of memory precursor effector cells | Knockout | Promotes memory differentiation Enhances CAR-T cells expansion Increases cytokine production | [72-75] | |
| HDACs | Removing acetyl groups from lysine residues on histone and non-histone proteins Participants in a variety of physiological and pathological processes Involves in various cellular physiological processes | HDAC inhibitor | Promotes memory differentiation Diminishes exhaustion Enhances CAR-T cells expansion | [76-78] | |
| SUV39H1 | Histone lysine methyltransferase, induces heterochromatin formation and gene transcriptional silencing Modulates T cell activity and lineage commitment Promotes the differentiation and expansion of effector T cells | Knockout | Promotes memory differentiation Diminishes exhaustion Optimizes the long-term functional persistence | [79, 80] | |
| ASXL1 | Participants in physiological processes Related to myeloid malignancies Regulates T cell differentiation | Knockout | Promotes memory differentiation Diminishes exhaustion Synergized with anti-PD-L1 blockade | [81] |
3.1 Transcription factors positively correlated with functional CAR-T cells
Numerous TFs have been identified that support memory phenotype maintenance and boost CAR-T cell effector functionality. Overexpressing these TFs in CAR-T cells has demonstrated potential in counteracting exhaustion and upgrading immunotherapies.
3.1.1 c-Jun
c-Jun is a member of the nuclear transcription activator protein-1 (AP-1) family (Figure 1), which possesses a conserved basic DNA-binding domain and a bZIP region. The bZIP mediates subunits from the Jun, Fos, Maf or activating transcription factor (ATF) families form AP-1 homodimers or heterodimers, enabling specific binding to target DNA sequences by relying on the basic domain. The composition and complex abundance of the AP-1 dimer dictate its specificity and function in gene activation or repression and regulating critical processes, including activation, proliferation, transformation, migration, inflammation and apoptosis [82-85].
Family members of AP-1 and their roles in regulating gene transcription. (A) Members of the AP-1 TF family. Active AP-1 is a dimeric transcription factor complex (Jun, Fos, ATF, BATF, JDP2) that binds DNA with partner-specific preferences and interacts with other TFs to form ternary complexes targeting composite DNA motifs. (B) Overexpressed c-Jun, together with c-Fos, promotes high-affinity AP-1 dimer formation, reducing the formation of dimers involving other subunits. The c-Jun/c-Fos dimer complexes with NFAT drive the expression of genes critical for T cell activation, including cytokines associated with STAT signaling, such as IL-2, IL-4, and IL-12. In contrast, under exhaustion-inducing conditions, the BATF/JUN heterodimer forms a complex with IRF4. This complex binds to specific DNA motifs, promoting the transcription of genes associated with T cell exhaustion, thereby contributing to reduced T cell functionality. Figure was created in BioRender.com.
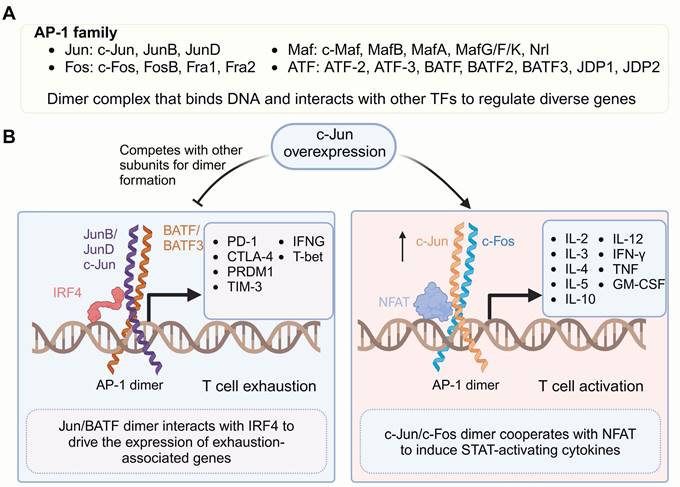
Jun and Fos proteins represent the core components of AP-1 and are its most extensively studied subunits. The heterodimer complex formed by Jun/Fos has a stronger DNA-binding affinity compare to the Jun/Jun homodimer, thereby resulting in greater transcriptional activation potential [86], which drives the transcription of IL-2, a crucial cytokine essential for T-cell activation and expansion [87]. Moreover, AP-1 dimers can associate with other TFs to form ternary complexes, enabling them to bind specific DNA motifs and modulate gene expression. Specifically, the Jun/Fos heterodimer has been shown to form a ternary complex with NFAT, which drives CD8+ T cell activation and serials of cytokines secretion, such as IL-3, IL-4, IL-5, IL-10, IL-12, interferon γ (IFN-γ), tumor necrosis factor (TNF) and granulocyte-macrophage colony-stimulating factor (GM-CSF) [88-90], which cooperate to sustain T cell effector functions. However, with prolonged antigen stimulation and the absence of costimulatory signals, AP-1 is progressively downregulated, leaving NFAT alone to drive T cell exhaustion and promote the upregulation of inhibitory receptors, including cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), LAG3, programmed cell death protein 1 (PD-1) and TIM-3 [91-94].
In 2019, Mackall and colleagues discovered that c-Jun overexpression strengthens CAR-T cells efficacy and counteracts dysfunction [43, 95]. HA-28z CAR-T cells served as a model to explore T cell exhaustion, revealing the pivotal role of AP-1-associated bZIP/IRF families in this process. c-Jun outperforms other AP-1 subunits, forming c-Jun/c-Fos dimers and reducing JunB/BATF dimerization as well as downregulating JunB, BATF, and BATF3 protein levels. c-Jun overexpression prevents T cells excessively expressing immunoregulatory receptors and terminal differentiation, resulting in an increased memory population and enhanced antitumor activity. In a leukemia mouse model, Feng and colleagues showed that c-Jun triggers the extracellular signal-regulated kinase1/2 (ERK1/2) cascade, boosts the co-stimulatory signaling [51], increases IL-2 and IFN-γ secretion, and positively regulates cell activation and T cell effector functions without upregulating exhaustion-related genes. A phase I clinical trial in which c-Jun overexpressing CD33 CAR-T cells were used to treat acute myeloid leukemia confirmed that c-Jun significantly boost CAR-T effector activity. Among all four patients who successfully underwent CAR-T therapy, substantial CAR-T cells expansion and clinical benefit were observed [51]. These results highlight the significant potential of c-Jun in augmenting CAR-T cell therapeutic efficacy.
However, c-Jun overexpression significantly activates CAR-T cells, which may trigger massive cytokines release and pose a risk of severe therapeutic side effects. In the aforementioned clinical trial, the study was prematurely halted due to severe adverse events, including grade 4 cytokine release syndrome (CRS) and infection, observed in the second patient [51]. Moreover, a clinical trial of receptor tyrosine kinase-like orphan receptor 1 (ROR1) CAR-T cells overexpressing c-Jun revealed a potential risk of treatment-related pneumonia in cancer patients with lung metastases. One participant experienced Grade 5 respiratory failure [96]. Therefore, while the overexpression of c-Jun can enhance CAR-T effector activity, it is crucial and necessary to be aware of the associated potential toxic side effects.
3.1.2 TCF1
TCF7 is located on chromosome 5 and encodes the human transcription factor TCF1. Among the reported isoforms, full-length TCF1 contains an N-terminal domain that interacts with β-catenin [52], which is involved in Wnt signaling regulation. This process is followed by a histone deacetylase (HDAC) activity segment, which is essential for defining CD8+ T cell lineage and preserving a less differentiation state. The C-terminal region includes a high-mobility-group (HMG) box DNA-binding domain, which governs gene transcription by binding to enhancers and promoters [32]. In contrast, the short isoform lacks the β-catenin-binding domain but retains the HMG and HDAC activity domains [97]. Interestingly, the TCF1 homolog LEF1 also has similar DNA-binding domains and HDAC activity sequences, positioning it as a critical TF closely associated with T cell development and differentiation [40, 52, 98].
Recent advances have highlighted TCF1 as a versatile regulator, exerting context-dependent functions that shape CAR-T cell phenotypes across varying conditions and stages of T cell differentiation. Upon chronic antigen exposure, CD8+ T cells differentiate into TCF1high progenitor-like subsets and TCF1low terminal subsets [52]. TCF1 suppresses several pro-exhaustion factors, such as Blimp-1, TIM-3, Tbx21, ID2 and Runx3, while maintaining T cell stemness by upregulating memory-related genes, like EOMES, cMyb and interferon-stimulated gene (ISG), while reinforcing the TCF1-Bcl6 axis [99]. TCF1 is essential for the memory-subset formation and serves as a predictor of the responses to immune checkpoint blockade (ICB) and tumor-infiltrating lymphocyte (TIL) therapy [52, 100]. Considering its significant role in T cell immunity, leveraging TCF1 and its downstream transcriptional pathways holds potential to strengthen the antitumor effectiveness of CAR-T cells (Figure 2).
Structure of TCF1 and strategies for regulating T cell memory differentiation. (A) TCF1 has two isoforms: a full-length isoform (containing the β-catenin interacting domain, HDAC activity segment, and HMG DNA-binding domain) and a short isoform lacking the β-catenin binding domain. (B) Strategies such as regnase-1 deficiency, DFO treatment to mitigate lipid peroxidation and ferroptosis, and BCL2L1 overexpression for optimizing CAR-T therapy by modulating TCF1 expression to promote T cell memory differentiation. HDAC: histone deacetylase; HMG: high-mobility-group box; DFO: deferoxamine. Figure was created in BioRender.com.Transcriptomic analysis showed that CAR-T cells from complete responders with chronic lymphocytic leukemia (CLL) were enriched with memory-promoting genes, such as TCF7 and LEF1 [18]. Moreover, Zheng and colleagues suggest that upregulating TCF1 expression could help to form a desirable immunological output [101]. Regnase-1 was identified as a critical regulator of CAR-T cell differentiation, directly suppressing Tcf7 and hindering the development of immune-supporting precursor exhausted T (Tpex) cells. Regnase-1 deficiency could promote TCF1 expression, thereby enhancing CAR-T cells persistence and achieving sustained tumor control. Teshima and colleagues used the iron chelator deferoxamine (DFO) during the production of mouse CAR-T cells to reduce lipid peroxidation and inhibit ferroptosis [102]. This not only increased the proportion of TCF1+TIM-3- progenitor subsets, which are known to be linked to the longevity of CAR-T but also enhanced CAR-T cell cytotoxicity and in vivo antitumor effects. Another study involved overexpressing the BCL2 Like 1 (BCL2L1) gene in B-cell maturation antigen (BCMA) targeting CAR-T cells [103]. These BCMA_BCL2L1 CAR-T cells exhibited higher TCF1 and lower TOX/TOX2 expression, showed an increased presence of naïve T cells and less-differentiated T cells, such as early activated T cells and precursor T cells, which improved the functional versatility of CAR-T cells and boosted their proliferation and cytotoxicity under chronic antigen stimulation in vitro, achieving excellent tumor clearance in a multiple myeloma (MM) xenograft model.
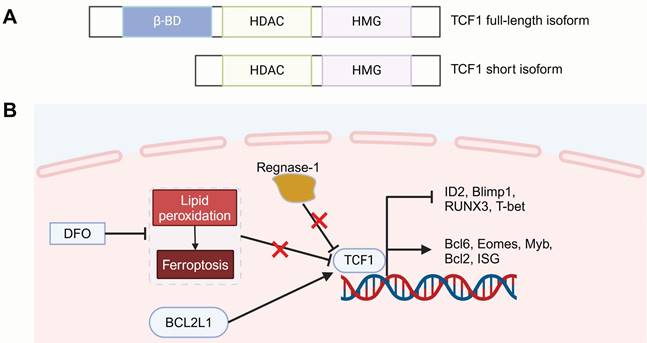
However, the strategy of enhancing the CAR-T memory phenotype by directly overexpressing TCF1 remains debatable. Weber and colleagues directly overexpressed the TCF7 gene, which indeed increased oxidative phosphorylation (OXPHOS) and metabolic adaptability in CAR-T cells but failed to promote the expansion of memory subsets [45]. Transcriptome sequencing revealed that TCF1 overexpressing cells exhibited high levels of TOX, CD39 and NR4A family exhaustion-related TFs. Further mechanistic studies are needed to uncover the regulatory networks involving TCF1 and its role in T cell memory formation and subsequently adapt it to optimize CAR-T antitumor efficacy.
3.1.3 FOXO1
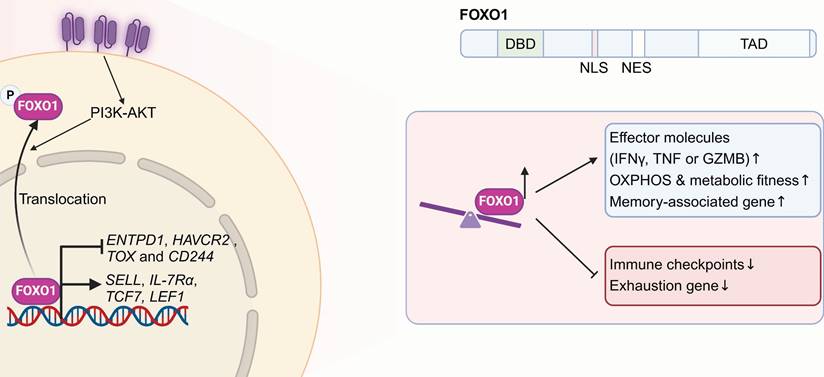
Forkhead box protein O1 (FOXO1), belonging to the forkhead TF family, participants in regulating cell metabolism, the regulation of cell division, programmed cell death, genomic repair processes, and the response to oxidative stress [104]. Structurally, FOXO1 contains a conserved DNA-binding domain (DBD) that facilitates its interaction with specific target genes, a nuclear localization signal (NLS) responsible for its nuclear import, a nuclear export signal (NES) that regulates its cytoplasmic translocation, and a transactivation domain (TAD) that mediates its transcriptional activity. Two independent papers published in 2024 highlighted FOXO1 as a vital regulator of CAR-T cell stemness, efficacy as well as metabolic fitness [44, 45]. FOXO1 activity is primarily controlled through protein modifications, including phosphorylation, acetylation, ubiquitylation and methylation [105]. The PI3K-AKT signaling pathway serves as a primary regulator of FOXO1 activity by promoting its phosphorylation through AKT. This phosphorylation exposes the NES on FOXO1, facilitating its translocation from the nucleus to the cytoplasm, thereby hindering its transcriptional activity [106]. The functions of FOXO1 within immune cells are also progressively being elucidated. FOXO1 regulates the homeostasis and survival of naïve T cells by controlling interleukin 7 receptor (IL-7R), L-selectin (CD62L) and C-C chemokine receptor type 7 (CCR7) [107, 108]. Moreover, FOXO1 also regulates the activity and functions of dendritic cells [109], macrophages [110] and B cells [111].
Here, both research teams aimed to boost CAR-T cell expansion and persistence in vivo, pinpointing FOXO1 as a critical target for resisting T cell exhaustion and enhancing therapeutic efficacy (Figure 3) [44, 45]. Compared with TCF7 or ID3, FOXO1 overexpression proved to be the most effective in maintaining the stemness of human Lewis Y CAR-T cells. It significantly reduced chromatin accessibility at regions rich in AP-1 and NF-κB specific motifs, thereby promoting a quiescent T cell state by inhibiting AP-1 activity. FOXO1 overexpression in CAR-T cells promoted a CD45RA+CD62L+ stem-like phenotype, metabolic fitness, and enhanced cytokine secretion by promoting the expression of memory-differential genes (SELL, IL-7Rα, TCF7, and LEF1) and suppression of exhaustion-related genes (CD39, HAVCR2, TOX, and CD244). In both leukemia and solid tumors, FOXO1 overexpression prolonged CAR-T cell persistence and strengthened tumor control capabilities. Furthermore, transcriptomic analyses of CAR-T clinical data revealed marked enrichment of FOXO1 regulon downstream genes in pre-infusion CAR-T products from CLL patients with complete or partial responses, correlating with in vivo CAR-T expansion and overall survival. Epigenetic profiling further demonstrated that T cells from patients with durable CAR-T responses were enriched with FOXO1-associated epigenetic signatures. Thus, FOXO1 overexpression presents a promising approach for improving the effectiveness of T cell-based cancer immunotherapies.
Overexpressing FOXO1 promotes CAR T cell memory formation, improves metabolic adaptability, and boosts antitumor effectiveness. The FOXO1 structure includes a conserved DNA-binding domain (DBD), nuclear localization (NLS) and export signals (NES) for nuclear-cytoplasmic transport, and a transactivation domain (TAD) for transcriptional activity. The PI3K-AKT pathway regulates FOXO1 by promoting its AKT-mediated phosphorylation, exposing its NES and driving nuclear export, which inhibits transcriptional activity. FOXO1 overexpression in CAR-T cells promotes a stem-like phenotype, metabolic fitness, and enhanced cytokine secretion by upregulating memory-associated genes (SELL, IL-7Rα, TCF7 and LEF1) and suppressing exhaustion-related genes (CD39, HAVCR2, TOX, CD244). Figure was created in BioRender.com.
3.1.4 STAT5
Signal transducers and activators of transcription (STAT) typically refer to two closely related isoforms, STAT5a and STAT5b, which exhibit 94% structural similarity [112]. STAT5 takes part in numerous vital cellular processes[113, 114], especially in regulating T cell homeostasis as well as immune function (Figure 4) [115, 116]. The activity of STAT5 proteins is predominantly regulated by cytokines from the IL-2 family, such as IL-2, IL-4, IL-7, IL-9, and IL-15. These cytokines utilize a shared gamma chain (γ(c)) within their receptor complexes [117-119] to induce downstream signals.
Schematic of STAT5-mediated T cell regulatory mechanisms. The IL-2 cytokine family promotes STAT5 expression, while STAT5 counteracts TOX to regulate T cell proliferation, cytokine secretion, and memory formation, ultimately enhancing CAR-T antitumor efficacy. Figure was created in BioRender.com.
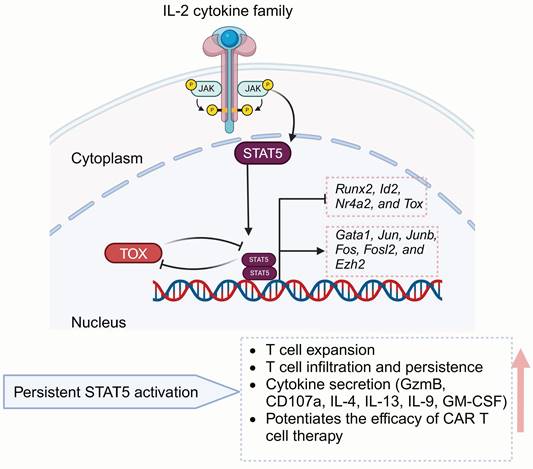
STAT5 functionally rejuvenates exhausted CD8+ T cells by counteracting TOX and reshaping the terminal differentiation epigenic landscape, thereby enhancing the response to ICB treatment [53, 54]. CD4+ T cells also closely influence T cell effectiveness via inflammatory cytokines releasing and orchestrating multiple immunologic effector mechanisms [120, 121]. IL-7, a gamma-chain cytokine, is widely utilized in ex vivo T cell cultures in CAR-T manufacturing due to the ability to enhance the stemness and polyfunctionality of CD4+ T cells through STAT5 activation [122, 123]. Directly modulating STAT5 could also enhance CAR-T cells polyfunctionality. Ding et al. engineered CD4+ T cells to express a variant of STAT5 with constitutive activity (CASTAT5) [55], demonstrating that these modified cells possess enhanced expansion potential following adoptive transfer and secrete multiple helper T cell (Th) cytokines, including Granzyme B (GzmB), CD107a, IL-4, IL-9, IL-13 and GM-CSF. Moreover, CASTAT5 significantly promotes CD4+ T cell tumor infiltration, which supports CAR-T cell proliferation and persistence, while enhancing overall effector function in a CD8+ T cell-dependent manner. Next-generation sequencing revealed that CASTAT5-producing CD4+ T cells undergo extensive remodeling of their transcriptional and epigenetic landscapes. STAT5A activates genes such as Junb, Jun, Fos, Fosl2, Ezh2 and Gata1, while inhibiting Id2, Nr4a2, Runx2, and Tox. CASTAT5 expression also benefits CD8+ T cells, and its co-expression in both CD4+ and CD8+ T cells results in the most pronounced antitumor effects. Thus, sustained STAT5 activation in CAR-T cells presents a promising strategy to improve therapeutic effectiveness.
3.1.5 FOXP1 and KLF2
Epigenomic and transcriptomic analyses have identified forkhead box P1 (FOXP1) and Kruppel-like factor 2 (KLF2) as key regulators of CD8+ CAR-T cell stemness and effector functionality, respectively [46]. Zhu and colleagues uncovered the heterogeneous chromatin states of CAR T cells among solid tumors and leukemia. They clarified gene networks governing CAR-T differentiation, exhaustion and dysfunction, identifying master TFs that govern CAR-T fate and therapeutic outcomes. These two TFs exhibit reciprocal regulation, where FOXP1 deficiency reduces KLF2 expression, and vice versa (Figure 5) [124].
FOXP1 and KLF2 are master transcription factors that govern CAR-T cell fate and therapeutic efficacy. FOXP1 preserves the quiescence and homeostasis of naïve T cells, limiting their transition into effector cells while supporting CAR-T cell stemness and enhancing antitumor activity. KLF2 acts as a pivotal TF within effector CD8+ CAR-T cell networks, promoting effector differentiation and enhancing resistance to exhaustion through suppressing exhaustion-related TFs and genes. Figure was created in BioRender.com.
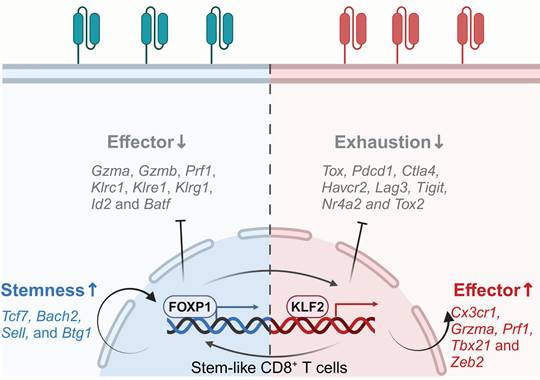
FOXP1, part of the Foxp subgroup within the forkhead TF family, is pivotal in immune responses, organ development and cancer pathogenesis [125-127], while also maintaining the quiescence and homeostasis of naïve T cells, partly by antagonizing FOXO1 interaction with shared forkhead-binding sites within the IL-7Rα enhancer, thus repressing IL-7Rα expression and MEK-Erk signaling. Consequently, FOXP1 deficiency leads to increased IL-7R expression in T cells, enabling enhanced proliferation upon IL-7 stimulation [126]. Zhu and colleagues showed that FOXP1 deletion in CD8+ CAR-T cells downregulates stemness-associated genes such as Tcf7, Bach2, Sell, and Btg1, while upregulating effector-related genes including Gzma, Gzmb, Prf1, Klrc1, Klre1, Klrg1, Id2 and Batf. CD8+ CAR-T lacking FOXP1 impair tumor suppression and diminish the proliferative capacity and lower the expression of the memory-related markers [46]. These findings validate FOXP1's role in supporting CAR-T cell stemness and enhancing anti-tumor activity.
KLF2, a member of the Krüppel-like TF family, regulates the differentiation, migration, functionality and homeostasis of immune cells, such as T and B lymphocytes, NK cells and monocytes [128-131], and participates in various biological processes in cancers [132, 133]. KLF2 governs T cell trafficking by upregulating the lipid-binding receptor sphingosine-1-phosphate (S1P) and the selectin CD62L. In the absence of KLF2, impaired peripheral migration causes CD4/CD8 single-positive T cells to be retained within the thymus, resulting in a significant reduction of T cells in the peripheral blood, lymph nodes and spleen [134, 135]. Besides regulating T cell migration, KLF2 facilitates the establishment of effector phenotypes in T cells. Knockout of KLF2 reduces the abundance of T effector-like cells, downregulates genes associated with effector characteristics, such as Gzma, Klrc1, Klrd1, Zeb2, Prf1, Tbx21, and Cx3cr1. Furthermore, it also upregulates inhibitory receptors expression, including Pdcd1, Ctla4, Tigit, Havcr2 and Lag3, while promoting exhaustion-related TFs like TOX2 and NR4A2. Additionally, it amplifies TOX-induced transcriptional signatures and downregulates TOX-repressed genes. Chromatin accessibility analysis revealed that T cells lacking KLF2 exhibited enhanced accessibility at the Pdcd1 and Tox gene loci, as well as at the NFAT and AP-1 TF binding sites. These data suggest that KLF2 promotes T cell differentiation into effector subsets and inhibits exhaustion by regulating T cell transcription and epigenetics [46].
3.2 Transcription factors associated with CAR-T cells exhaustion
T cells exhaustion poses a significant hurdle for CAR-T therapy in solid tumors. Extensive research has focused on uncovering critical regulators responsible for T cell dysfunction [136, 137]. Several exhaustion-inducing TFs have emerged as key targets, such as BATF, TOX, NR4A and Blimp-1. Recent studies suggest that knocking out these regulators holds significant potential for prolonging CAR-T cell functionality.
3.2.1 BATF
BATF belongs to the AP-1 TF family and negatively regulates AP-1-mediated T cell effector functions by competing with Fos subunits for Jun binding, which leads to transcriptionally repressive Jun/BATF heterodimer formation [138, 139] (Figure 1). The Jun/BATF dimer further binds to IRF4 or IRF8 and specifically recognizes AP-1-IRF composite elements (AICEs) within target genes, facilitating CD8+ T cell exhaustion [138, 140]. BATF also plays a positive transcriptional regulatory role in influencing T cell commitment and supporting the functional maturation of dendritic cells and B cells [141-145]. However, the role of BATF in T cell function remains controversial because of conflicting findings from different research groups. Studies have shown that BATF can either promote effector function or drive T cell exhaustion, depending on the specific immune context [29, 47, 56, 146-148].
In viral infection models, chronic HIV infection has been shown to elevate BATF expression in T cells, leading to exhaustion and impairing the body's antiviral response [147]. Interestingly, some other research also indicates that elevated BATF expression can enhance T cell effector functions in both chronic and acute viral infections. Xin et al. demonstrated that in a Clone 13 strain of lymphocytic choriomeningitis virus (LCMV) chronic infection model, IL-21 secreted by CD4+ helper T cells drives sustained BATF expression in CD8+ T cell subsets through the STAT3 signaling pathway. BATF, together with IRF4, supports Blimp-1 expression to maintain CD8+ T cell effector functions [148]. Haining et al. revealed that BATF is crucial for naïve T cell activation and differentiation into effector cells. A mouse model of acute infection with the Armstrong strain of LCMV showed that BATF deficiency impairs early T cell activation, reducing T-bet, Runx3, and Blimp-1 upregulation and limiting inflammatory cytokine receptor expression, which hinders the transition of naïve T cells into effector cells [29].
Likewise, in tumor models, the effects of BATF on T cell-mediated antitumor responses remain inconsistent and context-dependent. Seo et al. proved that BATF cooperate with IRF4 could steer CAR-T cells away from exhaustion and toward an effector-like phenotype [56]. They showed that CAR-T cells overexpressing BATF exhibited increased effector cytokines secretion, enhanced proliferation, decreased levels of exhaustion-associated TOX, increased expansion potential and tumor rejection. Additionally, BATF serves as a crucial target of Regnase-1, which dose-dependently suppresses the 3' UTR of Batf, thereby influencing mitochondrial fitness and effector functions [146]. Regnase-1 deletion reprogrammed T cells to enhance BATF function and mitochondrial metabolism, resulting in greater T cell persistence and survival, thereby improving antitumor effectiveness in both blood and solid tumor models. However, Zhang et al., using a CAR-T exhaustion model induced by chronic antigen stimulation combined with clustered regularly interspaced short palindromic repeats (CRISPR) screening, demonstrated that BATF promotes T cell exhaustion via direct binding to exhaustion-associated genes and increasing their expression [47]. Knocking out BATF, rather than overexpressing it, upregulated T cell activation-associated genes while suppressing exhaustion-related genes, promoting the differentiation of central memory T cells, and enhancing tumor rejection.
A deeper investigation into the conflicting effects of BATF on T cell differentiation and function across studies is crucial for achieving a more precise and effective optimization of CAR-T therapy.
Zhang found that the differences in tumor-killing models employed in these studies play a significant role. In Zhang's study, BATF was shown to drive T cell exhaustion under conditions of low T cell-to-tumor cell ratios and repeated tumor antigen stimulation, which is an exhaustion-inducing microenvironment. In this context, BATF deficiency reduced CAR-T exhaustion and enhanced their cytotoxicity. They also found that in a non-exhaustion context characterized by a high CAR-T-to-tumor ratio and transient tumor stimulation, BATF knockout had no impact on exhaustion or memory formation, while BATF overexpression enhanced CAR-T cytotoxicity, which aligns with Seo et al.'s observations [47]. Notably, Seo's study utilized a short-term co-culture cytotoxicity assay without repeated tumor antigen stimulation, minimizing exhaustion-inducing factors [56]. However, the conclusion by Zhang et al. that the exhaustion context determines BATF's regulation of T cell function appears to be inaccurate. As previously noted, even in chronic viral infection models under exhaustion conditions, BATF's impact on T cell exhaustion varies significantly and is sometimes contradictory, as observed in studies of chronic HIV infection and Xin et al.'s chronic infection model [147, 148].
Further exploration of the regulatory mechanisms of BATF in T cell function is needed. Studies have reported that in infection models, CD8+ T cells differentiate into three distinct subsets: progenitors (Ly108⁺TCF-1⁺), terminally exhausted (Ly108⁻CX3CR1⁻) and cytotoxic effector cells (CX3CR1⁺) [149, 150]. Chen et al. investigated the gene regulatory networks of these subsets and found that BATF expression induced by chronic infection binds to the regulatory regions of Tbx21 and Klf2, enhancing their expression and facilitating the transition of TCF-1⁺ progenitor cells into CX3CR1⁺ effector cells [57]. This study suggests that increasing BATF expression may drive the differentiation of progenitor T cells into CX3CR1⁺ effector cells, boosting CD8⁺ T cell effector functions. However, as chronic infection progresses, Chen et al. observed that terminally exhausted CD8⁺ T cells gradually increase 30 days after LCMV infection [57]. At this stage, BATF overexpression no longer enhances effector functions but may instead collaborate with IRF4 and NFAT to drive exhaustion [140].
Therefore, the discrepancies in BATF's effects on T cell differentiation and function across studies may stem from differences in the stages of T cell differentiation (progenitor-effector-exhaustion) induced by chronic stimulation in different models. For example, in the HIV infection model, chronic T cell exhaustion persists for several months or years, while in the LCMV chronic infection model, exhaustion occurs within 30 days. The latter contains a higher proportion of progenitor T cells, allowing BATF overexpression to enhance effector functions.
Additionally, the cytokine composition during CAR-T culture plays a pivotal role in shaping T cell metabolism and T cell exhaustion phenotypes. For instance, the addition of IL-21 into culture promotes oxidative phosphorylation, reduces BATF expression, and inhibits exhaustion, whereas IL-2 and IL-12 synergistically induce a glycolytic metabolic shift, favoring exhaustion phenotypes [151]. What's more, high IL-2 doses are known to upregulate STAT5, activate tryptophan hydroxylase 1, and drive aryl hydrocarbon receptor nuclear translocation, resulting in T cell exhaustion [152]. The cytokines used in CAR-T culture procedures vary among these studies. Zhang's study used a high IL-2 dose (400 IU/mL) for CAR-T culture [47], in contrast to Seo's 100 IU/mL [56], which potentially increased the likelihood of T cell exhaustion, which may have further contributed to the observed differences in the role of BATF.
In conclusion, BATF's role in CAR-T cells is highly context and T cell differentiation stage dependent. Understanding these factors is crucial for harnessing BATF's potential in CAR-T therapy. Further mechanistic research is necessity to better understand how to regulate BATF for optimized applications in CAR-T therapy.
3.2.2 TOX and NR4A
The TOX protein is part of the HMG-box family and interacts with DNA in a sequence-nonspecific fashion [153, 154]. TOX plays a vital role in the ontogeny of innate lymphoid cells, NK cells, and T cells [155-157]. In 2019, Alfei et al. found that TOX decreases T cell effector function and drives, as well as sustains exhaustion status during persistent infections [156]. Further research has demonstrated that TOX orchestrates transcriptional networks associated with T cell exhaustion by altering chromatin accessibility and modulating the activity of gene enhancers and promoters, ultimately driving irreversible exhaustion [158].
TOX is upregulated during prolonged stimulation and is highly expressed in CD8+ TILs due to sustained tumor antigen interactions [48, 159, 160]. This makes TOX a valuable biomarker for assessing T cell exhaustion and predicting patient responses to anti-PD-1 immunotherapy [58, 59]. In hepatocellular carcinoma (HCC), TOX facilitates PD-1 endocytic recycling, thereby resulting in sustained high levels of PD1 expression. TOX downregulation led to increased T cell infiltration, reduced dysfunction and an improved response to anti-PD1 therapy [59]. Targeting TOX has shown promise in enhancing T cell infiltration and reversing dysfunction in exhausted T cells.
Similarly, the NR4A family of orphan nuclear receptors, including NR4A1, NR4A2, and NR4A3, are crucial mediators of T cell exhaustion. Knockout of NR4a1/2 increases the proportion of TCF1+ exhaustion precursor CD8+ T cells, boosts both glycolysis as well as oxidative phosphorylation, and enhances T cell persistence within the tumor microenvironment [60]. This makes NR4A an appealing target for reinvigorating CAR-T cells by promoting early memory differentiation while limiting exhaustion.
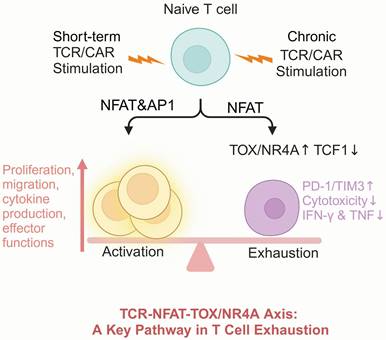
TOX and NR4A are both integral components of the TCR (T cell receptor) -NFAT (nuclear factor of activated T cells)-TOX/NR4A axis, a critical signaling pathway that governs T cell fate upon TCR activation [161, 162]. Under conditions of chronic TCR/CAR stimulation, limited AP-1/NFAT interactions would induce a hyporesponsive state in T cells [163]. Thus, NFAT initiates a cascade that drives the expression of TOX and NR4A. This pathway forms a positive feedback loop in which TOX and NR4A enhance each other's expression, reinforcing inhibitory receptors expression and chromatin accessibility remodeling, ultimately hampering T cell function [48]. While NFAT plays a central role in orchestrating this axis, its broad involvement in T cell activation and differentiation complicates its potential as a therapeutic target [163-165]. For instance, theoretically reducing NFAT activity could mitigate exhaustion by attenuating TOX/NR4A expression; however, NFAT knockout has been proved to impair T cell memory differentiation and diminish effector function [166, 167].
Thus, directly targeting NFAT could inadvertently disrupt essential immune functions, making it an unsuitable candidate for precise modulation. Recent attention has shifted to TOX and NR4A as actionable targets within the TCR-NEAT-TOX/NR4A axis (Figure 6). Targeting TOX and NR4A in CAR-T therapy offers a viable method to improve T cell durability and functionality.
T cell exhaustion is regulated through the TCR-NFAT-TOX/NR4A pathway. Upon TCR activation through antigen recognition, downstream AP-1 expression is upregulated, and together with NFAT, which promotes the transcription of T cell effector genes. Under chronic TCR stimulation, the intracellular levels of NFAT and AP-1 decrease, leading to increased expression of TOX and NR4A, suppression of TCF1, and promotion of the exhausted T cell phenotype. Figure was created in BioRender.com.
Seo et al. showed that TOX protein is highly induced in dysfunctional CAR-T cells which expression closely correlated with higher PD-1 expression and impaired cytolytic activity and cytokine production [48]. CAR-T cells with dual genetic ablation of TOX and TOX2 exhibited improved antitumor activity and downregulation of inhibitory receptors. In contrast to their wild-type counterparts, TOX/TOX2-KO CAR-T cells revealed attenuated expression of Nr4a1 and Nr4a2, indicating the underlying interactions among NFAT, NR4A, and TOX in driving T cell exhaustion.
Chen and colleagues also explored the function of NR4A TFs in CAR-T cells [50]. Consistent with other studies, they found that NR4A family members show significant upregulation in exhausted CAR-T cells, with their expression positively correlates with PD-1 and TIM3 levels. NR4A triple KO CAR-T cells exhibited enhanced effector potential and greater tumor control capabilities.
Taken together, these findings highlight the prominent role of the TCR-NFAT-TOX/NR4A axis in mediating CAR-T dysfunction and underscore the therapeutic potential of targeting TOX and NR4A to improve CAR-T efficacy in solid tumors.
3.2.3 Blimp-1
B lymphocyte-induced maturation protein-1 (Blimp-1), which is encoded by the PR domain zinc finger protein-1 (PRDM1), acts as a key epigenetic factor in T-cell exhaustion [168, 169]. Blimp-1 has been reported to induce terminal differentiation in both B and T cell lineages and is recognized as a key suppressor of memory T cells during viral infections [49, 61]. It is also associated with the formation of short-lived effector cells [31] and has been implicated in immune suppression within acute myeloid leukemia (AML) [62]. In T cells from AML patients, Blimp-1 is upregulated and associated with terminally differentiated effector T cells. These Blimp-1+ T cells exhibit functional defects, such as reduced cytokine production and impaired cytotoxicity. Moreover, Blimp-1 directly upregulates PD-1 and TIGIT expression through specific binding with their promoters.
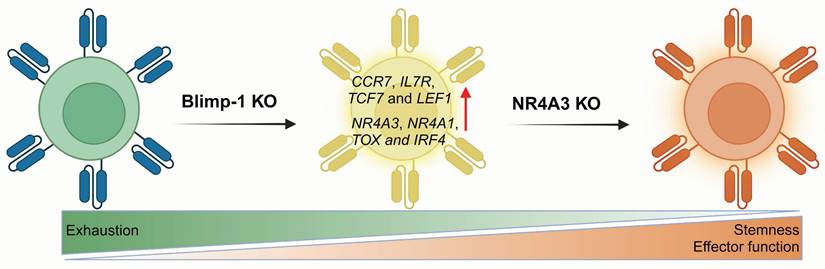
In 2022, Oshikawa et al. examined how PRDM1 knockout affects the characteristics of CD19 CAR-T cells [63]. They found that PRDM1 deficiency could help maintain the memory phenotype and enhance cytokine polyfunctionality as well as the therapeutic response. Genome-wide gene expression profiling revealed that PRDM1-knockout CAR-T cells markedly increased the expression of memory-associated genes, including CCR7, IL7R, TCF7 and LEF1. However, they also noted an increase in TOX expression, which is relative to T cell exhaustion. Subsequently, Jung and colleagues investigated the potential of PRDM1 knockout to boost the efficacy of CAR-T therapy in solid tumors [64]. Although PRDM1 single knockout enhanced early memory differentiation and CAR-T cell polyfunctionality, which was driven primarily by TCF7 upregulation. In vivo experiments showed that PRDM1 knockout only slightly improved leukemia control in mice. Further research revealed that PRDM1 deficiency triggered negative feedback driven by enhanced calcineurin-NFAT signaling, leading to T cell dysfunction. This process triggers compensatory upregulation of several T cell exhaustion genes such as NR4A3, NR4A1, TOX and IRF4, which diminishs the antitumor effectiveness of T cells. They further knocked out NR4A3, the most significantly upregulated gene in the feedback loop, confirming that dual PRDM1/NR4A3 knockout boosts long-term antitumor activity by preserving both stemness and effector function, achieving superior antitumor efficacy compared with that of PRDM1 deletion alone (Figure 7).
Blimp-1 is involved in the regulation of T cell memory differentiation. Compared to Blimp-1 deletion alone, dual knockout of Blimp-1 and NR4A3 synergistically reduces T cell exhaustion and enhances antitumor responses across multiple murine models, yielding superior outcomes. Figure was created in BioRender.com.
Therefore, targeting a single exhaustion-related molecule alone may be insufficient to fully reverse T cell exhaustion. Multiplex gene-editing strategies offer a promising approach to counteract the compensatory upregulation of exhaustion genes observed with single-gene knockouts, thereby facilitating the formation of stem-like T cell subsets and improving therapeutic outcomes.
3.2.4 IKZF3
Ikaros family zinc finger 3 (IKZF3) is a member of the IKAROS family and is crucial for lymphopoiesis and differentiation [170]. Additionally, IKZF3 promotes tumor progression, and its high expression in intestinal-type gastric cancer is linked to a poor prognosis [171]. IKZF3 possesses four zinc finger domains, which are critical for recognizing the common DNA motif a/gGGAA. Mutations in the second zinc finger domain of IKZF3 have been identified as potential drivers of CLL. These mutations alter IKZF3's DNA-binding properties, leading to CLL transformation via excessive B-cell receptor (BCR) signaling and increased expression of NK-κB target genes [172]. In mouse models, IKZF3 knockout has been demonstrated to result in B cell lymphoma formation [173].
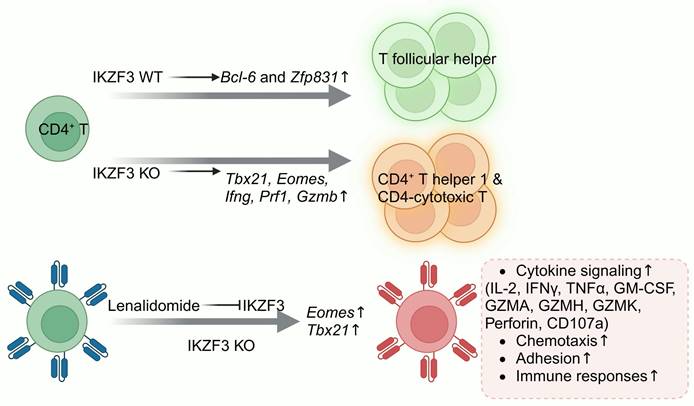
Research on IKZF3 function in CD4+ T cells has shown that IKZF3 facilitates T follicular helper (Tfh) cell commitment by inducing the TFs Bcl-6 and Zfp831 (Figure 8). Knockout of IKZF3 in mice reduces the proportion of antigen-specific Tfh cells, significantly increases Tbx21, Eomes, Ifng, Prf1 and Gzmb expression; promotes Th1 and CD4 cytotoxic T lymphocyte (CTL) differentiation; and boosts the granzyme B, IFN-γ, and perforin secretion in response to antigenic stimulation [174]. Lenalidomide, an oral administered immunomodulator, has been approved by the FDA for treating multiple myeloma and myelodysplastic syndromes. Lenalidomide modulates T cell function through the induction of the ubiquitination and subsequent degradation of IKZF1 and IKZF3. Zhu et al. proved that lenalidomide treatment of HER2 CAR-T and CD133 CAR-T cells increased effector cytokines secretion, such as IL-2, IFNγ, tumor necrosis factor α (TNFα) and GM-CSF, and increased specific cytotoxicity [65]. Hay and colleagues also discovered that lenalidomide treatment reversed T cell exhaustion in CAR-T cells and enhanced their responsiveness to immune checkpoint inhibitors [66]. Zhu and Chi collaborated to knock out IKZF3 in HER2 and CD133 CAR-T cells, which indeed enhanced antitumor activity. Transcriptomic analysis showed that IKZF3 knockout enhanced cytokine signaling, chemotaxis, and adhesion pathways, boosting effector molecule secretion. However, it also upregulated several pro-apoptotic genes, such as BCL2L11 and cyclin dependent kinase inhibitor 2a (CDKN2A), thus slightly increasing CAR-T cell apoptosis [67].
Schematic of IKZF3 regulation in T cell differentiation. IKZF3 promotes CD4+ T follicular helper cell differentiation. Upon IKZF3 knockout, CD4+ T cells shift toward Th1 and CD4-CTL differentiation. In CAR-T cells, treatment with lenalidomide or IKZF3 knockout enhances the transcription of Eomes and Tbx21, boosting cytokine secretion, chemotactic function, adhesion capacity, and immune responsiveness. Figure was created in BioRender.com.
Overall, modulating IKZF3 expression through different approaches could improve effector function, reverse T cell exhaustion, and propose a new approach to boost CAR-T therapy efficacy against solid tumors. However, it is crucial to consider that IKZF3 also participates in the orchestration of lymphocyte development, and its knockout may pose a potential risk of tumorigenesis.
4. Therapeutic targeting of epigenetic regulators in CAR-T therapy
As discussed above, transcription factors play pivotal roles in regulating T cell function as well as memory and exhaustion differentiation. Similarly, epigenetic modifications also exert a widespread influence over T cell biology (Table 1). Epigenetic modifications in T cells, such as CpG dinucleotide methylation in DNA promoter regions [175] and histone acetylation and methylation of nucleosomes, contribute to shaping T cell differentiation states and functional capacities [176, 177]. Researchers have modulated T cell differentiation by focusing on epigenetic regulators, including DNA demethylases, DNA methyltransferases and histone deacetylases, etc, to support memory T cell generation while reducing or avoiding T cell exhaustion (Figure 9).
Schematic of epigenetic regulators influencing DNA and chromatin landscapes to modulate T cell differentiation and function. (A) TET2 mediates DNA demethylation, promoting target gene transcription. TET2 knockout enhances T cell memory differentiation and persistence, resisting exhaustion from chronic stimulation. (B) DNMT3A expression increases post-T cell activation, leading to Tcf7 methylation. DNMT3A knockout elevates TCF-7 and LEF1 expression, boosting T cell effector activity, memory formation, and sensitivity to anti-PD-L1 treatment. (C) HDACs facilitate the removal of acetyl moieties from lysine residues on histone and non-histone proteins, inhibiting gene transcription. HDAC inhibition or knockout enhances effector gene expression, suppresses exhaustion markers, increases the expression of the TFs Eomes, TCF4, LEF1, and activates Wnt/β-catenin signaling. (D) SUV39H1 primarily mediates H3K9me3, inducing heterochromatin formation and gene silencing. SUV39H1 knockout increases the transcription of memory-associated CAR-T genes (TCF7, LEF1, IL7R, and SELL), enhancing metabolic adaptability and effector function. (E) ASXL1 facilitates the removal of ubiquitin from H2AK119Ub. ASXL1 deficiency or knockout preserves more ubiquitinated gene sites, opening chromatin regions that support T cell stemness and functionality and enhancing T cell maintenance and responsiveness to ICB. ASXL1: additional sex combs-like 1; HDAC: histone deacetylase; PR-DUB: polycomb repressive deubiquitinase; BAP1: BRCA1 associated protein. Figure was created in BioRender.com.
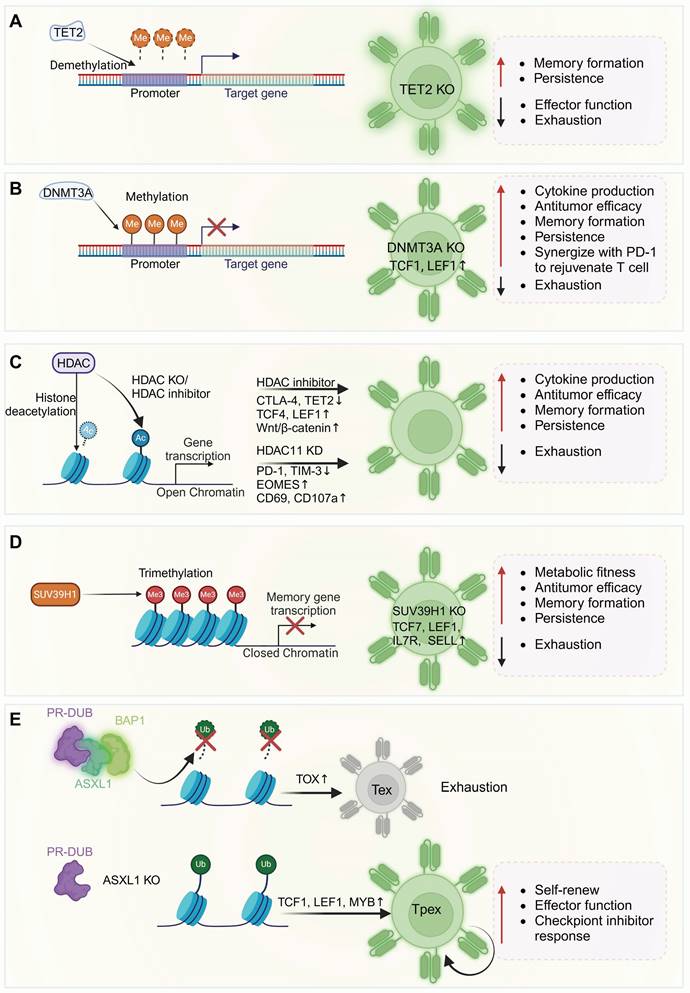
4.1 TET2
Tet methylcytosine dioxygenase 2 (TET2), a member of the TET family of epigenetic regulators, oxidizes 5-methylcytosine (5mC) in DNA, thus mediating DNA demethylation. TET2 mutations frequently occur in the hematopoietic and immune systems, leading to myelodysplasia and related myeloid malignancies [178], and approximately 15% of myeloid cancers harbor somatic TET2 mutations [179]. Research on the role of TET2 in T cells revealed that after LCMV infection, TET2-deficient mice exhibited rapid expansion of memory gp33+ CD8+ T cells, indicating that TET2 critically directs CD8+ T cells differentiation into either effector or memory subsets [68].
Considering TET2's pivotal role in epigenetic regulation, an escalating trend in research is the exploration of strategies to modulate TET2 activity to enhance T cell-based immunotherapy. An intriguing case reported by Carl June's team involved a chronic lymphocytic leukemia patient in one CD19 CAR-T clinical trial [69]. After two infusions of CTL019, the patient achieved rapid CLL cell clearance and complete remission. TCR repertoire analysis of the patient's peripheral blood CD8+ T cells indicated that although T cells were polyclonal early during treatment, 94% of the CD8+ T cells originated from a single clone by two months after the second CAR-T cell infusion.
NGS analysis uncovered a missense mutation at position 1879 (glutamic acid to glutamine) in the catalytic domain of the patient's TET2 gene, along with a loss-of-function heterozygous mutation due to CAR segment insertion in one allele. These combined mutations caused TET2 dysfunction, reshaped the epigenetic landscape of CAR-T cells, reduced or eliminated chromatin accessibility for genes related to T cell effector function and exhaustion (IFNG, NOTCH2, CD28, ICOS, and PRDM1), which promoted memory T cell development and prolonged CAR-T persistence in vivo. Fraietta and colleagues applied a targeted integration strategy to insert CD19 CAR into TET2, generating TET2 knockout CAR-T cells with more central memory T cells subsets. Upon chronic antigen stimulation, TET2-deficient CD8+ T cells showed a greater tendency to adopt a CCR7+ CD45RO+ memory phenotype with elevated TCF1 expression, preventing CAR-T cells from progressing to terminal exhaustion [70].
Notably, the Sadelain team found that TET2 knockout could improve CAR-T therapeutic efficacy, although this effect enhancement was dependent on the characteristics of the CAR. CAR-T cells containing the 4-1BB intracellular domain improved tumor treatment outcomes in mice after TET2 knockout, whereas CD28 signaling CAR-T cells exhibited no notable differences in cell differentiation or antitumor efficacy before versus after TET2 knockout. Further studies found that the enhancement of T cell memory phenotype and therapeutic efficacy due to TET2 deficiency depended on the epigenetic state of increased BATF3 expression, which ultimately led to antigen-independent sustained proliferation of TET2 biallelic knockout CAR-T cells [71]. Nonetheless, it is crucial to recognize that TET2 deficiency-induced epigenetic alterations could potentially trigger somatic malignant transformation [180]; therefore, careful consideration is required when TET2 knockout strategies are employed to enhance CAR-T therapy.
4.2 DNMT3A
DNMT3A and DNMT3B are the major de novo methyltransferases in mammals [181]. After TCR-mediated activation triggered by antigen stimulation, DNMT3A transcription is upregulated over 30-fold, mediating T cell polarization toward Th1 or Th2 through the methylation regulation of IFN-γ and IL-4 [72]. Furthermore, DNMT3A-mediated Tcf7 methylation in early effector T cells plays a critical role in regulating the proportion of memory precursor effector cells and restricting the development of TCF1hi CD127+KLRG1- memory precursor T cells [73].
Therefore, de novo DNA methylation programming reinforces T cell exhaustion and creates a stable internal barrier to T cell reactivation in response to PD-1 blockade. DNMT3A deficiency results in reduced methylation at the IFNγ, Myc, Tcf7, and Tbx21 loci, resisting terminal differentiation and enhancing responsiveness to anti-PD-L1 therapy. Pretreatment with low doses of the DNA demethylating agent decitabine for two weeks enhanced the T cell response to PD-L1 blockade in Tramp-C2 tumor-bearing mice, inducing the expansion of tumor-infiltrating CD8+ T cells [74].
The research team further validated the impact of DNMT3A among several human CAR-T models targeting solid tumors [75]. They demonstrated that DNMT3A deficiency enhanced CAR-T cell expansion during constant stimulation and sustained their cytokine secretion and tumor cell lysis. After chronic stimulation, DNMT3A KO CAR-T cells exhibited higher TCF1 and LEF1 expressions, with epigenetic profiles resembling those of stem-cell-like memory or naïve T cells, ultimately increasing in vivo antitumor activity across multiple solid tumor models.
4.3 HDAC
Histone deacetylases (HDACs) are a group of enzymes that catalyze removing acetyl groups from lysine residues on histone and non-histone proteins. HDACs are classified into four major classes based on sequence homology, comprising a total of 18 enzymes. HDACs are integral to a variety of physiological and pathological processes, exerting significant influence on their regulation. Recently, several studies have reported that HDAC expression or activity modification via inhibitors or genetic editing enhances CAR-T antitumor efficacy and persistence [76-78].
Treatment of B7-H3 CAR-T cells with the pan-HDAC inhibitor, vorinostat (SAHA), downregulated CTLA-4 and TET2 expression [78]. Moreover, combining SAHA with B7-H3 CAR-T cells exhibited excellent tumor suppression effects in orthotopic patient-derived xenograft (PDX) and metastatic xenograft models. A high-throughput drug screening platform was employed by Zhu and colleagues to evaluate the effects of 370 epigenetic regulatory compounds on CAR-T cell dysfunction and phenotype modulation in vitro [76]. Ultimately, they identified two inhibitors for class I HDACs, chidamide and M344. Mechanistic analysis revealed that these inhibitors decreased HDAC1 expression as well as enhanced histone H3K27 acetylation, which upregulated TCF4 and LEF1 while elevating intracellular β-catenin expression, reinforcing the Wnt/β-catenin signaling cascade. Consequently, they promoted T cell memory subset differentiation both in vitro and in vivo, counteracted T cells exhaustion, and improved tumor suppression efficacy. Zhang et al. knocked down HDAC11 in NKG2D CAR-T cells via shRNA interference, which significantly enhanced intracellular Eomes expression and cell expansion. Additionally, it reduced exhaustion markers (TIM-3, PD-1) expression, facilitated the formation of Tcm subsets, and prolonged the survival in prostate cancer-bearing mice [77].
It is important to note that HDACs are widely involved in various cellular physiological processes [182]; therefore, strategies that enhance CAR-T therapy by targeting HDACs carry certain risks. Targeting HDAC inhibition may produce direct toxicity; for example, as reported by Lei et al., the pan-HDAC inhibitor SAHA has shown dose-limiting toxicity in clinical trials [183], although they used low-dose SAHA to treat CAR-T cells, achieving a balance between therapeutic efficacy and the drug's safe dosage is challenging because lower doses may reduce efficacy, whereas higher doses increase toxicity [184, 185]. Employing selective HDAC inhibitors or targeted genetic modifications to regulate the expression or function of specific HDACs strengthens targeting specificity to some extent, helping to reduce the side effects caused by pan-HDAC modification. However, long-term monitoring of cellular developmental safety remains necessary.
4.4 SUV39H1
The histone methyltransferase suppressor of variegation 3-9 homolog 1 (SUV39H1) is primarily responsible for the trimethylation of histone H3 at lysine 9 (H3K9me3), which induces heterochromatin formation and gene transcriptional silencing [186-189]. SUV39H1 is pivotal in modulating T cell activity and lineage commitment, which suppresses memory- and stemness-related genes in terminal differentiation effector CD8+ T cells. Depletion of SUV39H1 in mice leads to reduced H3K9me3 levels in CD8+ T cells, which manifests as diminished production of effector cytokines (granzyme B and IFN-γ), impaired proliferation, and compromised elimination of Listeria infection. Additionally, the reduced gene methylation including Il7r (CD127), Sell (CD62L), Ccr7, and Cxcr4, leads to increased transcription, contributing to an increased fraction of stem-like memory T cells [190]. While SUV39H1 knockout impairs T cell effector response during infection, Niborski further found that in tumor models of B16F10-OVA and MCA-101 fibrosarcoma, SUV39H1-deficient mice demonstrated stronger antitumor activity in tumor-infiltrating lymphocytes (TILs) when combined with PD-1 ICB, compared to wild-type controls. SUV39H1 deficiency appeared to delay or prevent TIL exhaustion [79].
Jain et al. reported that SUV39H1 knockout in human CAR-T cells led to a slight reduction in the production of effector cytokines such as TNFα, IFNγ, IL2 and granzyme B. However, after multiple rounds of tumor cell co-culture, SUV KO 1928z CAR-T cells exhibited enhanced mitochondrial activity and improved metabolic fitness, while maintaining chromatin accessibility at loci (KLF2, LEF1, TCF7) associated with T cell memory and effector functionality and decreasing accessibility at effector function and immune checkpoint gene loci. These modifications promoted CAR-T cell proliferation and sustained functionality under chronic antigen stimulation, reduced the proportion of exhausted subsets, and ultimately improved the antitumor efficacy and prolonged protection of CAR-T cells [80].
The effects of SUV39H1 knockout on T cell effector functions have demonstrated variability across studies, likely due to differences in disease models (tumor vs. virus), T cell activation methods (TCR vs. CAR), and therapeutic approaches (PD-1 blockade vs. T cell therapy). Overall, SUV39H1 deficiency leads to increased chromatin accessibility in T cells, particularly at loci related to memory differentiation genes such as TCF7, LEF1, IL7R, and SELL, thereby promoting the differentiation of memory T cell subsets and supporting sustained T cell effector function. This finding offers a novel therapeutic strategy for enhancing CAR-T antitumor effectiveness.
Notably, while Jain et al. observed no signs of malignant expansion or transformation in SUV39H1-knockout CAR-T cells during a 200-day monitoring period [80], Niborski found that when Suv39h1-KO T cells were transplanted into B6xDBA/2 F1 recipient mice, they experienced more severe graft-versus-host disease (GvHD) compared to those receiving WT B6 bone marrow. In fact, 78% of the mice infused with Suv39h1-deficient T cells died from GvHD [79]. Therefore, further studies with longer durations are required to confirm the safety of SUV39H1 knockout with respect to the potential for excessive T cell proliferation.
4.5 ASXL1
The epigenetic regulator additional sex combs-like 1 (ASXL1) plays a key role in physiological processes. It stabilizes the polycomb repressive deubiquitinase (PR-DUB) complex and facilitates the recruitment of its partner, BRCA1 associated protein (BAP1), to mediate the deubiquitination of histone H2A at lysine 119 (H2AK119Ub). ASXL1 mutations are frequently detected in various myeloid malignancies [191, 192].
In a clinical trial conducted by O'Connell et al., which combined PD-L1 immune checkpoint inhibitors with the hypomethylating agent guadecitabine in relapsed or refractory myelodysplastic syndrome (MDS) and chronic myelomonocytic leukemia (CMML) patients, a correlation was noted between long-term survival in MDS patients and the presence of ASXL1 mutations in their T cells [193]. Building on this observation, Kang et al. recently investigated the impact of ASXL1 in regulating the gradual transfer of multipotent progenitor exhausted T cells to terminally differentiated T cells [81]. The study revealed that ASXL1-knockout T cells generated a larger population of TCF1+ stem-like cells in a chronic LCMV infection model, resulting in more than 1 year of long-term persistence, enhanced effector function, and lower levels of the exhaustion TF Tox. However, ASXL1 does not rely on DNA methylation to regulate CD8+ T cell differentiation, as ASXL1-deficient T cells displayed no significant site-specific or genome-wide methylation differences compared to wild-type T cells. However, ATAC-seq revealed >5200 differentially accessible regions between ASXL1 KO and WT CD8+ T cells, particularly with increased accessibility to stem and memory-related genes like Tcf7, Lef1 and Myb in ASXL1-deficient T cells. Additionally, CD8+ T cells with Asxl1 knockout presented elevated levels of H3K9ac, H3K4me3 and H3K27me3. The histone profile of ASXL1 KO CD8+ T cells closely matched that of Tpex and Teff subsets. Furthermore, ASXL1 loss destabilized the PR-DUB complex, reducing its deubiquitination function and leading to higher H2AK119Ub levels in ASXL1 KO P14 T cells compared to WT P14 T cells. In mouse tumor models, Asxl1-deficient T cells exhibited strong effector activity and responsiveness to anti-PD-L1 treatment, contributing to improved tumor control. Among cancer patients receiving ICB therapy, those with ASXL1 insertion mutations showed longer survival, as ASXL1 disruption provided CD8+ T cells with a selective survival benefit after PD-L1 treatment.
Thus, targeting ASXL1 offers considerable potential for enhancing the prolonged survival and enduring efficacy of T cell therapies.
5. Perspective
Optimizing CAR-T therapeutic effects through transcriptional and epigenetic modulation has shown substantial translational potential. Gene editing and small molecule inhibitors allow precise modulation of key regulators, reshaping gene networks involved in T cell memory differentiation and effector functions in preclinical models. The optimization strategies primarily focus on two directions. The first involves enhancing and sustaining T cell effector functions to improve tumor cell killing, as seen in c-Jun overexpression, which enhances T cell activation, promotes IL-2 and IFN-γ secretion, reduces exhaustion markers, and collectively improves CAR-T therapy [43, 51]. A similar strategy involves KLF2, which drives CAR-T cells toward effector subset differentiation instead of exhaustion [67]. The second direction focuses on promoting T cell memory differentiation and extending persistence, both of which are critical for achieving durable CAR-T responses [18-21]. A notable example is TET2 deficiency, identified through clinical trials, which enhances long-term T cell persistence. CAR-T cells lacking TET2 tend to differentiate into CCR7⁺ CD45RO⁺ memory subsets [69]. Additionally, FOXO1 overexpression can also enhance CAR-T stemness, prolonging T cell persistence [45].
It is crucial to recognize that these optimization strategies are double-edged swords. Besides the improved CAR-T efficacy, those strategies may also introduce side effects and risks that must be addressed to enable successful clinical trials. First, the enhanced functionality of modified CAR-T cells may elevate the risk of "on-target, off-tumor" toxicity. For example, in clinical trials of c-Jun-overexpressing CAR-T cells [51, 96], while c-Jun boosts CAR-T cytotoxicity, it also increases cytokine secretion and activation signals, potentially leading to CRS and unintended damage to normal tissues expressing low levels of tumor-associated antigens. Moreover, owing to inherent differences between preclinical mouse models and humans, many preclinical CAR-T data derived from mice have limited applicability in humans. Differences in antigen expression and the higher tolerance of mice to toxicities compared to humans make it difficult to predict CRS and "on-target, off-tumor" side effects. Mitigating these risks requires a comprehensive evaluation of human antigen expression patterns and distribution across tissues using tools like single-cell RNA sequencing, combined with the cautious selection of CAR-T targets to minimize or avoid potential toxicities [194].
Another widely discussed challenge is the tumorigenic risk of CAR-T cells, as gene editing could potentially introduce somatic mutations leading to malignant transformation. Theoretically, the gene editing steps in CAR-T therapy preparation may introduce random mutations. As a result, the FDA has issued a boxed warning for approved BCMA- and CD19-targeting CAR-T therapies due to the risk of T cell malignancy post CAR-T therapy [195]. Interestingly, follow-up studies revealed that the incidence of second primary malignancies (SPMs) among CAR-T-treated cancer survivors was statistically comparable to that observed with standard therapies [196]. Additionally, a recent analysis of a French CAR-T patient database reported that among 3,066 patients treated with CAR-T and followed for up to 17.7 months, only one case of T cell malignancy occurred, representing an incidence rate of 0.03%. This indicates that the risk of malignant transformation from random mutations is very low [197]. However, transcription factors like IKZF3 and TET2, due to their roles in T cell lineage commitment and tumorigenesis, may theoretically pose a higher risk of tumor development when manipulated [173, 178, 179]. While no definitive evidence of tumorigenesis has been observed in current studies on TET2- and IKZF3-modified CAR-T cells, rigorous post-treatment monitoring of CAR-T pharmacokinetics and gene editing outcomes remains critical to identify and manage potential malignant transformations. In summary, comprehensive risk assessments and proactive safety measures tailored to the mechanisms and characteristics of CAR-T modifications are essential to ensure the clinical safety of these enhanced therapies.
Furthermore, as part of translational research, the feasibility of producing GMP-grade CAR-T therapies must be evaluated after proof-of-concept validation and before transitioning to human studies. Fortunately, existing GMP-grade CAR-T production technologies can accommodate both gene overexpression and knockout modifications [198]. Thus, production-related challenges are currently within a solvable scope.
However, it is important to recognize that the transcriptional and epigenetic targets discussed in this review exhibit diverse mechanisms of action and play broad roles in regulating T cell development, function, and differentiation. The intricate interactions between these targets and their downstream signaling pathways are not yet fully elucidated. Additionally, the limited availability of real-world clinical trial data makes it challenging to identify the most effective optimization strategy for enhancing CAR-T efficacy. Further mechanistic studies are still needed to establish a comprehensive, precise, and safe foundation for future clinical trials.
Abbreviations
AICE: AP-1-IRF composite elements; AML: acute myeloid leukemia; AP-1: activator protein 1; ASXL1: additional sex combs like 1; ATAC-seq: assay for transposase-accessible chromatin using sequencing; ATF: activating transcription factor; BAP1: BRCA1 associated protein 1; BATF: basic leucine zipper atf-like transcription factor; BCL2L1: BCL2-like 1; Bcl6: B-cell lymphoma 6; BCMA: B-cell maturation antigen; BCR: B-cell receptor; Blimp-1: B-lymphocyte-induced maturation protein 1; bZIP: basic leucine zipper domain protein; CAR-T: chimeric antigen receptor t-cell therapy; CCR7: C-C chemokine receptor type7; CDKN2A: cyclin dependent kinase inhibitor 2a; CLL: chronic lymphocytic leukemia; CMML: chronic myelomonocytic leukemia; CRISPR: clustered regularly interspaced short palindromic repeats; CRS: cytokine release syndrome; CTL: cytotoxic t lymphocyte; CTLA-4: cytotoxic t-lymphocyte-associated protein 4; CX3CR1: CX3C motif chemokine receptor 1; DFO: Deferoxamine; DNMT3: DNA (cytosine-5)-methyltransferase 3; E2A: T-cell E2A transcription factor; Eomes: Eomesodermin; ERK: extracellular signal-regulated kinase; Ezh2: enhancer of zeste homolog 2; FDA: Food and drug administration; FOS: fos proto-oncogene; FOXO1: forkhead box o1; FOXP1: forkhead box p1; G9A: histone-lysine n-methyltransferase g9a; Gata1: Gata binding protein 1; GM-CSF: granulocyte-macrophage colony-stimulating factor; GZMA: granzyme a; GZMB: granzyme b; H2AK119Ub: monoubiquitinated histone h2a at lysine 119; H3K9me3: histone h3 lysine 9 trimethylation; HAVCR2: hepatitis a virus cellular receptor 2; HCC: hepatocellular carcinoma; HDAC: histone deacetylase; HER2: human epidermal growth factor receptor 2; HMG: high mobility group; ICB: immune checkpoint blockade; ICOS: inducible t-cell costimulator; ID2: inhibitor of DNA binding 2; IFNγ: interferon γ; IKZF3: Ikaros family zinc finger 3; IL2RA: interleukin 2 receptor alpha; IL-7R: interleukin 7 receptor; IRF4: interferon regulatory factor 4; ISG: interferon-stimulated gene; JUN: Jun proto-oncogene; KLF2: Kruppel-like factor 2; KLRC1: killer cell lectin like receptor c1; KLRD1: killer cell lectin like receptor d1; KLRE1: killer cell lectin like receptor e1; KLRG1: killer cell lectin like receptor g1; LAG3: lymphocyte activation gene 3; LCMV: lymphocytic choriomeningitis virus; LEF1: lymphoid enhancer-binding factor 1; MAF: musculoaponeurotic fibrosarcoma oncogene; MDS: myelodysplastic syndromes; MEK: mitogen-activated protein kinase kinase; MM: multiple myeloma; NFAT: nuclear factor of activated t cells; NF-KB: nuclear factor kappa-light-chain-enhancer of activated b cells; NGS: next-generation sequencing; NR4A: nuclear receptor subfamily 4 group a; OXPHOS: oxidative phosphorylation; PD-1: programmed cell death protein 1; PI3K: phosphoinositide 3-kinase; PRDM1: PR domain zinc finger protein 1; PR-DUB: polycomb repressive-deubiquitinase complex; PRF1: perforin 1; ROR1: receptor tyrosine kinase-like orphan receptor 1; RUNX3: Runt-related transcription factor 3; S1P: sphingosine-1-phosphate; scRNA-seq: single-cell RNA sequencing; SELL: L-selectin; SPMs: second primary malignancies; SREBP2: sterol regulatory element binding protein 2; STAT: signal transducer and activator of transcription; SUV39H1: suppressor of variegation 3-9 homolog 1; T-BET: T-box expressed in T cells; TCF1: T cell factor 1; TCR: T cell receptor; Teff: Effector T cell; TET2: Tet methylcytosine dioxygenase 2; TF: transcription factor; Tfh: Follicular helper T cell; Th: T helper cell; TIL: tumor-infiltrating lymphocyte; TIM-3: T cell immunoglobulin and mucin-domain-containing-3; TNF: tumor necrosis factor; TOX: thymocyte selection-associated high mobility group box; Tpex: progenitor-exhausted t cell; ZEB2: zinc finger e-box binding homeobox 2; Zfp831: zinc finger protein 831.
Acknowledgements
DY.Q., YN.L., and P.S. contributed equally to this work. DY.Q. and YN.L. were responsible for outlining the review, drafting the manuscript, and creating the figures. P.S contributed to revising the manuscript and figure design. YG.Z. organized and curated the literature. YH.L., YS.W. and QJ.L. contributed to the conceptual framework and revision of the manuscript.
This work was supported by the China Postdoctoral Science Foundation (No. 2021M702346), the Post-Doctor Research Project, West China Hospital, Sichuan University (No. 2021HXBH092), the Fundamental Research Funds for the Central Universities (No. 2023SCU12051), Sichuan Science and Technology Program (2023YFS0003), National Key Research and Development Program of China (2024YFA1306400), the Agency for Science, Technology and Research (A∗STAR) under its Industry Alignment Fund-Pre-Positioning Program (H23J2a0095) and Competitive Research Programme (CRP29-2022-0032).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP. et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med. 2019;380:45-56
2. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J. et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396:839-52
3. Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak O. et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N Engl J Med. 2017;377:2545-54
4. Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S. et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24:20-8
5. Kankeu Fonkoua LA, Sirpilla O, Sakemura R, Siegler EL, Kenderian SS. CAR T cell therapy and the tumor microenvironment: Current challenges and opportunities. Mol Ther Oncolytics. 2022;25:69-77
6. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11:69
7. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423-37
8. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A. et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016;24:657-71
9. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD. et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell. 2015;162:1229-41
10. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R. et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell. 2015;162:1217-28
11. Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4:762-74
12. Bulliard Y, Andersson BS, Baysal MA, Damiano J, Tsimberidou AM. Reprogramming T cell differentiation and exhaustion in CAR-T cell therapy. J Hematol Oncol. 2023;16:108
13. Giles JR, Globig AM, Kaech SM, Wherry EJ. CD8(+) T cells in the cancer-immunity cycle. Immunity. 2023;56:2231-53
14. Wang Q, Qin Y, Li B. CD8(+) T cell exhaustion and cancer immunotherapy. Cancer Lett. 2023;559:216043
15. Liu Y, Sperling AS, Smith EL, Mooney DJ. Optimizing the manufacturing and antitumour response of CAR T therapy. Nat Rev Bioeng. 2023;1:271-85
16. Omilusik KD, Goldrath AW. The origins of memory T cells. Nature. 2017;552:337-9
17. Restifo NP, Gattinoni L. Lineage relationship of effector and memory T cells. Curr Opin Immunol. 2013;25:556-63
18. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S. et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. 2018;24:563-71
19. Biasco L, Izotova N, Rivat C, Ghorashian S, Richardson R, Guvenel A. et al. Clonal expansion of T memory stem cells determines early anti-leukemic responses and long-term CAR T cell persistence in patients. Nat Cancer. 2021;2:629-42
20. Wang X, Popplewell LL, Wagner JR, Naranjo A, Blanchard MS, Mott MR. et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood. 2016;127:2980-90
21. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF. et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17:1290-7
22. Ye B, Stary CM, Li X, Gao Q, Kang C, Xiong X. Engineering chimeric antigen receptor-T cells for cancer treatment. Mol Cancer. 2018;17:32
23. Papavassiliou KA, Papavassiliou AG. Transcription Factor Drug Targets. J Cell Biochem. 2016;117:2693-6
24. Bushweller JH. Targeting transcription factors in cancer - from undruggable to reality. Nat Rev Cancer. 2019;19:611-24
25. Jiang H, Li G. Transcription factors direct epigenetic reprogramming at specific loci in human cancers. Front Genet. 2023;14:1234515
26. Huang Q, Ma C, Chen L, Luo D, Chen R, Liang F. Mechanistic Insights Into the Interaction Between Transcription Factors and Epigenetic Modifications and the Contribution to the Development of Obesity. Front Endocrinol (Lausanne). 2018;9:370
27. Gray SM, Kaech SM, Staron MM. The interface between transcriptional and epigenetic control of effector and memory CD8(+) T-cell differentiation. Immunol Rev. 2014;261:157-68
28. Yao S, Buzo BF, Pham D, Jiang L, Taparowsky EJ, Kaplan MH. et al. Interferon regulatory factor 4 sustains CD8(+) T cell expansion and effector differentiation. Immunity. 2013;39:833-45
29. Kurachi M, Barnitz RA, Yosef N, Odorizzi PM, DiIorio MA, Lemieux ME. et al. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat Immunol. 2014;15:373-83
30. Wang D, Diao H, Getzler AJ, Rogal W, Frederick MA, Milner J. et al. The Transcription Factor Runx3 Establishes Chromatin Accessibility of cis-Regulatory Landscapes that Drive Memory Cytotoxic T Lymphocyte Formation. Immunity. 2018;48:659-74 e6
31. Shin HM, Kapoor V, Guan T, Kaech SM, Welsh RM, Berg LJ. Epigenetic modifications induced by Blimp-1 Regulate CD8(+) T cell memory progression during acute virus infection. Immunity. 2013;39:661-75
32. Xing S, Li F, Zeng Z, Zhao Y, Yu S, Shan Q. et al. Tcf1 and Lef1 transcription factors establish CD8(+) T cell identity through intrinsic HDAC activity. Nat Immunol. 2016;17:695-703
33. Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE. et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8(+) T cells. Immunity. 2011;35:400-12
34. Youngblood B, Hale JS, Kissick HT, Ahn E, Xu X, Wieland A. et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature. 2017;552:404-9
35. Scott-Browne JP, Lopez-Moyado IF, Trifari S, Wong V, Chavez L, Rao A. et al. Dynamic Changes in Chromatin Accessibility Occur in CD8(+) T Cells Responding to Viral Infection. Immunity. 2016;45:1327-40
36. Yu B, Zhang K, Milner JJ, Toma C, Chen R, Scott-Browne JP. et al. Epigenetic landscapes reveal transcription factors that regulate CD8(+) T cell differentiation. Nat Immunol. 2017;18:573-82
37. Rincon M, Flavell RA. AP-1 transcriptional activity requires both T-cell receptor-mediated and co-stimulatory signals in primary T lymphocytes. EMBO J. 1994;13:4370-81
38. Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP. et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol. 2013;14:489-99
39. Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR. et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol. 2005;6:1236-44
40. Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, Xue HH. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity. 2010;33:229-40
41. D'Cruz LM, Lind KC, Wu BB, Fujimoto JK, Goldrath AW. Loss of E protein transcription factors E2A and HEB delays memory-precursor formation during the CD8+ T-cell immune response. Eur J Immunol. 2012;42:2031-41
42. Zhou X, Xue HH. Cutting edge: generation of memory precursors and functional memory CD8+ T cells depends on T cell factor-1 and lymphoid enhancer-binding factor-1. J Immunol. 2012;189:2722-6
43. Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z. et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature. 2019;576:293-300
44. Chan JD, Scheffler CM, Munoz I, Sek K, Lee JN, Huang YK. et al. FOXO1 enhances CAR T cell stemness, metabolic fitness and efficacy. Nature. 2024;629:201-10
45. Doan AE, Mueller KP, Chen AY, Rouin GT, Chen Y, Daniel B. et al. FOXO1 is a master regulator of memory programming in CAR T cells. Nature. 2024;629:211-8
46. Zhu Z, Lou G, Teng XL, Wang H, Luo Y, Shi W. et al. FOXP1 and KLF2 reciprocally regulate checkpoints of stem-like to effector transition in CAR T cells. Nat Immunol. 2024;25:117-28
47. Zhang X, Zhang C, Qiao M, Cheng C, Tang N, Lu S. et al. Depletion of BATF in CAR-T cells enhances antitumor activity by inducing resistance against exhaustion and formation of central memory cells. Cancer Cell. 2022;40:1407-22 e7
48. Seo H, Chen J, Gonzalez-Avalos E, Samaniego-Castruita D, Das A, Wang YH. et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc Natl Acad Sci U S A. 2019;116:12410-5
49. Nutt SL, Fairfax KA, Kallies A. BLIMP1 guides the fate of effector B and T cells. Nat Rev Immunol. 2007;7:923-7
50. Chen J, Lopez-Moyado IF, Seo H, Lio CJ, Hempleman LJ, Sekiya T. et al. NR4A transcription factors limit CAR T cell function in solid tumours. Nature. 2019;567:530-4
51. Zuo S, Li C, Sun X, Deng B, Zhang Y, Han Y. et al. C-JUN overexpressing CAR-T cells in acute myeloid leukemia: preclinical characterization and phase I trial. Nat Commun. 2024;15:6155
52. Raghu D, Xue HH, Mielke LA. Control of Lymphocyte Fate, Infection, and Tumor Immunity by TCF-1. Trends Immunol. 2019;40:1149-62
53. Beltra JC, Abdel-Hakeem MS, Manne S, Zhang Z, Huang H, Kurachi M. et al. Stat5 opposes the transcription factor Tox and rewires exhausted CD8(+) T cells toward durable effector-like states during chronic antigen exposure. Immunity. 2023;56:2699-718 e11
54. IL-2-STAT5 Activity Antagonizes TOX and Reverses CD8+ T Cell Exhaustion. Cancer Discov. 2023: OF1.
55. Ding ZC, Shi H, Aboelella NS, Fesenkova K, Park EJ, Liu Z. et al. Persistent STAT5 activation reprograms the epigenetic landscape in CD4(+) T cells to drive polyfunctionality and antitumor immunity. Sci Immunol. 2020;5:eaba5962
56. Seo H, Gonzalez-Avalos E, Zhang W, Ramchandani P, Yang C, Lio CJ. et al. BATF and IRF4 cooperate to counter exhaustion in tumor-infiltrating CAR T cells. Nat Immunol. 2021;22:983-95
57. Chen Y, Zander RA, Wu X, Schauder DM, Kasmani MY, Shen J. et al. BATF regulates progenitor to cytolytic effector CD8(+) T cell transition during chronic viral infection. Nat Immunol. 2021;22:996-1007
58. Kim K, Park S, Park SY, Kim G, Park SM, Cho JW. et al. Single-cell transcriptome analysis reveals TOX as a promoting factor for T cell exhaustion and a predictor for anti-PD-1 responses in human cancer. Genome Med. 2020;12:22
59. Wang X, He Q, Shen H, Xia A, Tian W, Yu W. et al. TOX promotes the exhaustion of antitumor CD8(+) T cells by preventing PD1 degradation in hepatocellular carcinoma. J Hepatol. 2019;71:731-41
60. Srirat T, Hayakawa T, Mise-Omata S, Nakagawara K, Ando M, Shichino S. et al. NR4a1/2 deletion promotes accumulation of TCF1(+) stem-like precursors of exhausted CD8(+) T cells in the tumor microenvironment. Cell Rep. 2024;43:113898
61. Rutishauser RL, Martins GA, Kalachikov S, Chandele A, Parish IA, Meffre E. et al. Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity. 2009;31:296-308
62. Zhu L, Kong Y, Zhang J, Claxton DF, Ehmann WC, Rybka WB. et al. Blimp-1 impairs T cell function via upregulation of TIGIT and PD-1 in patients with acute myeloid leukemia. J Hematol Oncol. 2017;10:124
63. Yoshikawa T, Wu Z, Inoue S, Kasuya H, Matsushita H, Takahashi Y. et al. Genetic ablation of PRDM1 in antitumor T cells enhances therapeutic efficacy of adoptive immunotherapy. Blood. 2022;139:2156-72
64. Jung IY, Narayan V, McDonald S, Rech AJ, Bartoszek R, Hong G. et al. BLIMP1 and NR4A3 transcription factors reciprocally regulate antitumor CAR T cell stemness and exhaustion. Sci Transl Med. 2022;14:eabn7336
65. Wang Z, Zhou G, Risu N, Fu J, Zou Y, Tang J. et al. Lenalidomide Enhances CAR-T Cell Activity Against Solid Tumor Cells. Cell Transplant. 2020;29:963689720920825
66. Hay J, Acklam F, Turner D, Barbour M, Singh P, Grant C. et al. Abstract 5586: Demonstrating restoration of T cell function in exhausted T cells with IKZF3 small molecule inhibitor, Lenalidomide. Cancer Res. 2022;82:5586
67. Zou Y, Liu B, Li L, Yin Q, Tang J, Jing Z. et al. IKZF3 deficiency potentiates chimeric antigen receptor T cells targeting solid tumors. Cancer Lett. 2022;524:121-30
68. Carty SA, Gohil M, Banks LB, Cotton RM, Johnson ME, Stelekati E. et al. The Loss of TET2 Promotes CD8(+) T Cell Memory Differentiation. J Immunol. 2018;200:82-91
69. Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ. et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. 2018;558:307-12
70. Dimitri AJ, Baxter AE, Chen GM, Hopkins CR, Rouin GT, Huang H. et al. TET2 regulates early and late transitions in exhausted CD8(+) T cell differentiation and limits CAR T cell function. Sci Adv. 2024;10:eadp9371
71. Jain N, Zhao Z, Feucht J, Koche R, Iyer A, Dobrin A. et al. TET2 guards against unchecked BATF3-induced CAR T cell expansion. Nature. 2023;615:315-22
72. Gamper CJ, Agoston AT, Nelson WG, Powell JD. Identification of DNA methyltransferase 3a as a T cell receptor-induced regulator of Th1 and Th2 differentiation. J Immunol. 2009;183:2267-76
73. Ladle BH, Li KP, Phillips MJ, Pucsek AB, Haile A, Powell JD. et al. De novo DNA methylation by DNA methyltransferase 3a controls early effector CD8+ T-cell fate decisions following activation. Proc Natl Acad Sci U S A. 2016;113:10631-6
74. Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P. et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell. 2017;170:142-57 e19
75. Prinzing B, Zebley CC, Petersen CT, Fan Y, Anido AA, Yi Z. et al. Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Sci Transl Med. 2021;13:eabh0272
76. Zhu M, Han Y, Gu T, Wang R, Si X, Kong D. et al. Class I HDAC inhibitors enhance antitumor efficacy and persistence of CAR-T cells by activation of the Wnt pathway. Cell Rep. 2024;43:114065
77. Zhang H, Yao J, Ajmal I, Farooq MA, Jiang W. shRNA-mediated gene silencing of HDAC11 empowers CAR-T cells against prostate cancer. Front Immunol. 2024;15:1369406
78. Lei X, Ou Z, Yang Z, Zhong J, Zhu Y, Tian J. et al. A Pan-Histone Deacetylase Inhibitor Enhances the Antitumor Activity of B7-H3-Specific CAR T Cells in Solid Tumors. Clin Cancer Res. 2021;27:3757-71
79. Niborski LL, Gueguen P, Ye M, Thiolat A, Ramos RN, Caudana P. et al. CD8+T cell responsiveness to anti-PD-1 is epigenetically regulated by Suv39h1 in melanomas. Nat Commun. 2022;13:3739
80. Jain N, Zhao Z, Koche RP, Antelope C, Gozlan Y, Montalbano A. et al. Disruption of SUV39H1-Mediated H3K9 Methylation Sustains CAR T-cell Function. Cancer Discov. 2024;14:142-57
81. Kang TG, Lan X, Mi T, Chen H, Alli S, Lim SE. et al. Epigenetic regulators of clonal hematopoiesis control CD8 T cell stemness during immunotherapy. Science. 2024;386:eadl4492
82. Papavassiliou AG, Musti AM. The Multifaceted Output of c-Jun Biological Activity: Focus at the Junction of CD8 T Cell Activation and Exhaustion. Cells. 2020;9:2470
83. Schnoegl D, Hiesinger A, Huntington ND, Gotthardt D. AP-1 transcription factors in cytotoxic lymphocyte development and antitumor immunity. Curr Opin Immunol. 2023;85:102397
84. Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240-6
85. Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:E131-6
86. Atsaves V, Leventaki V, Rassidakis GZ, Claret FX. AP-1 Transcription Factors as Regulators of Immune Responses in Cancer. Cancers (Basel). 2019;11:1037
87. McGuire KL, Iacobelli M. Involvement of Rel, Fos, and Jun proteins in binding activity to the IL-2 promoter CD28 response element/AP-1 sequence in human T cells. J Immunol. 1997;159:1319-27
88. Cockerill PN, Bert AG, Jenkins F, Ryan GR, Shannon MF, Vadas MA. Human granulocyte-macrophage colony-stimulating factor enhancer function is associated with cooperative interactions between AP-1 and NFATp/c. Mol Cell Biol. 1995;15:2071-9
89. Feske S, Draeger R, Peter HH, Eichmann K, Rao A. The duration of nuclear residence of NFAT determines the pattern of cytokine expression in human SCID T cells. J Immunol. 2000;165:297-305
90. Macian F, Lopez-Rodriguez C, Rao A. Partners in transcription: NFAT and AP-1. Oncogene. 2001;20:2476-89
91. Mognol GP, Gonzalez-Avalos E, Ghosh S, Spreafico R, Gudlur A, Rao A. et al. Targeting the NFAT:AP-1 transcriptional complex on DNA with a small-molecule inhibitor. Proc Natl Acad Sci U S A. 2019;116:9959-68
92. Pereira RM, Hogan PG, Rao A, Martinez GJ. Transcriptional and epigenetic regulation of T cell hyporesponsiveness. J Leukoc Biol. 2017;102:601-15
93. Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V. et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670-84
94. Macian F, Garcia-Cozar F, Im SH, Horton HF, Byrne MC, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002;109:719-31
95. LaFleur MW, Miller BC, Sharpe AH. Prevention of CAR-T-cell dysfunction. Nat Biomed Eng. 2020;4:16-7
96. Lyell Immunopharma Reports Dose-dependent Clinical Activity from Phase 1 Trial of LYL797, a ROR1-targeted CAR-T Cell Product Candidate Enhanced with its Proprietary Anti-exhaustion Technology. 26 June 2024. https://ir.lyell.com/
97. Gullicksrud JA, Li F, Xing S, Zeng Z, Peng W, Badovinac VP. et al. Differential Requirements for Tcf1 Long Isoforms in CD8(+) and CD4(+) T Cell Responses to Acute Viral Infection. J Immunol. 2017;199:911-9
98. Escobar G, Mangani D, Anderson AC. T cell factor 1: A master regulator of the T cell response in disease. Sci Immunol. 2020;5:eabb9726
99. Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M. et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol. 2016;1:eaai8593
100. Krishna S, Lowery FJ, Copeland AR, Bahadiroglu E, Mukherjee R, Jia L. et al. Stem-like CD8 T cells mediate response of adoptive cell immunotherapy against human cancer. Science. 2020;370:1328-34
101. Zheng W, Wei J, Zebley CC, Jones LL, Dhungana Y, Wang YD. et al. Regnase-1 suppresses TCF-1+ precursor exhausted T-cell formation to limit CAR-T-cell responses against ALL. Blood. 2021;138:122-35
102. Harada S, Hashimoto D, Saito Y, Miyajima T, Li W, Senjo H. et al. Ferroptosis Inhibition Generates TCF-1 + CAR-T Cells with Enhanced Persistence and Cytotoxicity. Blood. 2023;142:97 -
103. Poorebrahim M, Lee H, Benaoudia S, Maity R, Ahn S, Leblay N. et al. Bclxl Prevents Progressive Exhaustion in BCMA CAR T Cells with Maintenance of High TCF1 Expressing T Cells. Blood. 2023;142(Suppl 4):454
104. Ebrahimnezhad M, Natami M, Bakhtiari GH, Tabnak P, Ebrahimnezhad N, Yousefi B. et al. FOXO1, a tiny protein with intricate interactions: Promising therapeutic candidate in lung cancer. Biomed Pharmacother. 2023;169:115900
105. Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol. 2013;14:83-97
106. Calissi G, Lam EW, Link W. Therapeutic strategies targeting FOXO transcription factors. Nat Rev Drug Discov. 2021;20:21-38
107. Kerdiles YM, Beisner DR, Tinoco R, Dejean AS, Castrillon DH, DePinho RA. et al. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat Immunol. 2009;10:176-84
108. Fabre S, Carrette F, Chen J, Lang V, Semichon M, Denoyelle C. et al. FOXO1 regulates L-Selectin and a network of human T cell homing molecules downstream of phosphatidylinositol 3-kinase. J Immunol. 2008;181:2980-9
109. Dong G, Wang Y, Xiao W, Pacios Pujado S, Xu F, Tian C. et al. FOXO1 regulates dendritic cell activity through ICAM-1 and CCR7. J Immunol. 2015;194:3745-55
110. Cabrera-Ortega AA, Feinberg D, Liang Y, Rossa C Jr, Graves DT. The Role of Forkhead Box 1 (FOXO1) in the Immune System: Dendritic Cells, T Cells, B Cells, and Hematopoietic Stem Cells. Crit Rev Immunol. 2017;37:1-13
111. Szydlowski M, Jablonska E, Juszczynski P. FOXO1 transcription factor: a critical effector of the PI3K-AKT axis in B-cell development. Int Rev Immunol. 2014;33:146-57
112. Kanai T, Seki S, Jenks JA, Kohli A, Kawli T, Martin DP. et al. Identification of STAT5A and STAT5B target genes in human T cells. PLoS One. 2014;9:e86790
113. Li YJ, Zhang C, Martincuks A, Herrmann A, Yu H. STAT proteins in cancer: orchestration of metabolism. Nat Rev Cancer. 2023;23:115-34
114. Wang W, Lopez McDonald MC, Hariprasad R, Hamilton T, Frank DA. Oncogenic STAT Transcription Factors as Targets for Cancer Therapy: Innovative Strategies and Clinical Translation. Cancers (Basel). 2024;16:1387
115. Burchill MA, Goetz CA, Prlic M, O'Neil JJ, Harmon IR, Bensinger SJ. et al. Distinct effects of STAT5 activation on CD4+ and CD8+ T cell homeostasis: development of CD4+CD25+ regulatory T cells versus CD8+ memory T cells. J Immunol. 2003;171:5853-64
116. Hand TW, Cui W, Jung YW, Sefik E, Joshi NS, Chandele A. et al. Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proc Natl Acad Sci U S A. 2010;107:16601-6
117. Russell SM, Keegan AD, Harada N, Nakamura Y, Noguchi M, Leland P. et al. Interleukin-2 receptor gamma chain: a functional component of the interleukin-4 receptor. Science. 1993;262:1880-3
118. Wong GL, Manore SG, Doheny DL, Lo HW. STAT family of transcription factors in breast cancer: Pathogenesis and therapeutic opportunities and challenges. Semin Cancer Biol. 2022;86:84-106
119. Lin JX, Leonard WJ. The role of Stat5a and Stat5b in signaling by IL-2 family cytokines. Oncogene. 2000;19:2566-76
120. Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The central role of CD4(+) T cells in the antitumor immune response. J Exp Med. 1998;188:2357-68
121. Corthay A, Skovseth DK, Lundin KU, Rosjo E, Omholt H, Hofgaard PO. et al. Primary antitumor immune response mediated by CD4+ T cells. Immunity. 2005;22:371-83
122. Zhou J, Jin L, Wang F, Zhang Y, Liu B, Zhao T. Chimeric antigen receptor T (CAR-T) cells expanded with IL-7/IL-15 mediate superior antitumor effects. Protein Cell. 2019;10:764-9
123. Ding ZC, Liu C, Cao Y, Habtetsion T, Kuczma M, Pi W. et al. IL-7 signaling imparts polyfunctionality and stemness potential to CD4(+) T cells. Oncoimmunology. 2016;5:e1171445
124. Casucci M, Bonini C, Ruggiero E. Epigenetic checkpoints regulate the fate and function of CAR-T cells. Nat Immunol. 2024;25:4-6
125. Kaminskiy Y, Kuznetsova V, Kudriaeva A, Zmievskaya E, Bulatov E. Neglected, yet significant role of FOXP1 in T-cell quiescence, differentiation and exhaustion. Front Immunol. 2022;13:971045
126. Feng X, Wang H, Takata H, Day TJ, Willen J, Hu H. Transcription factor Foxp1 exerts essential cell-intrinsic regulation of the quiescence of naive T cells. Nat Immunol. 2011;12:544-50
127. Kim JH, Hwang J, Jung JH, Lee HJ, Lee DY, Kim SH. Molecular networks of FOXP family: dual biologic functions, interplay with other molecules and clinical implications in cancer progression. Mol Cancer. 2019;18:180
128. Winkelmann R, Sandrock L, Porstner M, Roth E, Mathews M, Hobeika E. et al. B cell homeostasis and plasma cell homing controlled by Kruppel-like factor 2. Proc Natl Acad Sci U S A. 2011;108:710-5
129. Weinreich MA, Takada K, Skon C, Reiner SL, Jameson SC, Hogquist KA. KLF2 transcription-factor deficiency in T cells results in unrestrained cytokine production and upregulation of bystander chemokine receptors. Immunity. 2009;31:122-30
130. Jha P, Das H. KLF2 in Regulation of NF-kappaB-Mediated Immune Cell Function and Inflammation. Int J Mol Sci. 2017;18:2383
131. He J, Chen D, Xiong W, Hou X, Quan Y, Yang M. et al. Eomesodermin spatiotemporally orchestrates the early and late stages of NK cell development by targeting KLF2 and T-bet, respectively. Cell Mol Immunol. 2024;21:662-73
132. Lu Y, Qin H, Jiang B, Lu W, Hao J, Cao W. et al. KLF2 inhibits cancer cell migration and invasion by regulating ferroptosis through GPX4 in clear cell renal cell carcinoma. Cancer Lett. 2021;522:1-13
133. Ma Z, Wei K, Yang F, Guo Z, Pan C, He Y. et al. Tumor-derived exosomal miR-3157-3p promotes angiogenesis, vascular permeability and metastasis by targeting TIMP/KLF2 in non-small cell lung cancer. Cell Death Dis. 2021;12:840
134. Kuo CT, Veselits ML, Leiden JM. LKLF: A transcriptional regulator of single-positive T cell quiescence and survival. Science. 1997;277:1986-90
135. Carlson CM, Endrizzi BT, Wu J, Ding X, Weinreich MA, Walsh ER. et al. Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature. 2006;442:299-302
136. Ahn T, Bae EA, Seo H. Decoding and overcoming T cell exhaustion: Epigenetic and transcriptional dynamics in CAR-T cells against solid tumors. Mol Ther. 2024;32:1617-27
137. Zeng Z, Wei F, Ren X. Exhausted T cells and epigenetic status. Cancer Biol Med. 2020;17:923-36
138. Murphy TL, Tussiwand R, Murphy KM. Specificity through cooperation: BATF-IRF interactions control immune-regulatory networks. Nat Rev Immunol. 2013;13:499-509
139. Echlin DR, Tae HJ, Mitin N, Taparowsky EJ. B-ATF functions as a negative regulator of AP-1 mediated transcription and blocks cellular transformation by Ras and Fos. Oncogene. 2000;19:1752-63
140. Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC. et al. Transcription Factor IRF4 Promotes CD8(+) T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity. 2017;47:1129-41 e5
141. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M. et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097-100
142. Schraml BU, Hildner K, Ise W, Lee WL, Smith WA, Solomon B. et al. The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature. 2009;460:405-9
143. Betz BC, Jordan-Williams KL, Wang C, Kang SG, Liao J, Logan MR. et al. Batf coordinates multiple aspects of B and T cell function required for normal antibody responses. J Exp Med. 2010;207:933-42
144. Glasmacher E, Agrawal S, Chang AB, Murphy TL, Zeng W, Vander Lugt B. et al. A genomic regulatory element that directs assembly and function of immune-specific AP-1-IRF complexes. Science. 2012;338:975-80
145. Li P, Spolski R, Liao W, Wang L, Murphy TL, Murphy KM. et al. BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature. 2012;490:543-6
146. Wei J, Long L, Zheng W, Dhungana Y, Lim SA, Guy C. et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature. 2019;576:471-6
147. Quigley M, Pereyra F, Nilsson B, Porichis F, Fonseca C, Eichbaum Q. et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat Med. 2010;16:1147-51
148. Xin G, Schauder DM, Lainez B, Weinstein JS, Dai Z, Chen Y. et al. A Critical Role of IL-21-Induced BATF in Sustaining CD8-T-Cell-Mediated Chronic Viral Control. Cell Rep. 2015;13:1118-24
149. Zander R, Schauder D, Xin G, Nguyen C, Wu X, Zajac A. et al. CD4(+) T Cell Help Is Required for the Formation of a Cytolytic CD8(+) T Cell Subset that Protects against Chronic Infection and Cancer. Immunity. 2019;51:1028-42 e4
150. Hudson WH, Gensheimer J, Hashimoto M, Wieland A, Valanparambil RM, Li P. et al. Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1(+) Stem-like CD8(+) T Cells during Chronic Infection. Immunity. 2019;51:1043-58 e4
151. Zhang M, Kong J, Yin F, Shi J, Li J, Qiu Z. et al. Optimizing CAR-T cell Culture: Differential effects of IL-2, IL-12, and IL-21 on CAR-T cells. Cytokine. 2024;184:156758
152. Liu Y, Zhou N, Zhou L, Wang J, Zhou Y, Zhang T. et al. IL-2 regulates tumor-reactive CD8(+) T cell exhaustion by activating the aryl hydrocarbon receptor. Nat Immunol. 2021;22:358-69
153. Aliahmad P, Seksenyan A, Kaye J. The many roles of TOX in the immune system. Curr Opin Immunol. 2012;24:173-7
154. O'Flaherty E, Kaye J. TOX defines a conserved subfamily of HMG-box proteins. BMC Genomics. 2003;4:13
155. Aliahmad P, Kaye J. Development of all CD4 T lineages requires nuclear factor TOX. J Exp Med. 2008;205:245-56
156. Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P. et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature. 2019;571:265-9
157. Aliahmad P, de la Torre B, Kaye J. Shared dependence on the DNA-binding factor TOX for the development of lymphoid tissue-inducer cell and NK cell lineages. Nat Immunol. 2010;11:945-52
158. Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP. et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature. 2019;571:211-8
159. Scott AC, Dundar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M. et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature. 2019;571:270-4
160. Liang C, Huang S, Zhao Y, Chen S, Li Y. TOX as a potential target for immunotherapy in lymphocytic malignancies. Biomark Res. 2021;9:20
161. Gaud G, Lesourne R, Love PE. Regulatory mechanisms in T cell receptor signalling. Nat Rev Immunol. 2018;18:485-97
162. Seo W, Jerin C, Nishikawa H. Transcriptional regulatory network for the establishment of CD8(+) T cell exhaustion. Exp Mol Med. 2021;53:202-9
163. Martinez GJ, Pereira RM, Aijo T, Kim EY, Marangoni F, Pipkin ME. et al. The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity. 2015;42:265-78
164. Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205-32
165. Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5:472-84
166. Xu T, Keller A, Martinez GJ. NFAT1 and NFAT2 Differentially Regulate CTL Differentiation Upon Acute Viral Infection. Front Immunol. 2019;10:184
167. Gabriel CH, Gross F, Karl M, Stephanowitz H, Hennig AF, Weber M. et al. Identification of Novel Nuclear Factor of Activated T Cell (NFAT)-associated Proteins in T Cells. J Biol Chem. 2016;291:24172-87
168. Hwang S, Cobb DA, Bhadra R, Youngblood B, Khan IA. Blimp-1-mediated CD4 T cell exhaustion causes CD8 T cell dysfunction during chronic toxoplasmosis. J Exp Med. 2016;213:1799-818
169. Sawant DV, Yano H, Chikina M, Zhang Q, Liao M, Liu C. et al. Adaptive plasticity of IL-10(+) and IL-35(+) T(reg) cells cooperatively promotes tumor T cell exhaustion. Nat Immunol. 2019;20:724-35
170. Kuehn HS, Chang J, Yamashita M, Niemela JE, Zou C, Okuyama K. et al. T and B cell abnormalities, pneumocystis pneumonia, and chronic lymphocytic leukemia associated with an AIOLOS defect in patients. J Exp Med. 2021;218:e20211118
171. Cui Z, Liang H, Luo R, Huang W, Yuan W, Zhang L. et al. IKZF3 amplification predicts worse prognosis especially in intestinal-type gastric cancer. J Cancer Res Clin Oncol. 2024;150:363
172. Wang JH, Avitahl N, Cariappa A, Friedrich C, Ikeda T, Renold A. et al. Aiolos regulates B cell activation and maturation to effector state. Immunity. 1998;9:543-53
173. Lazarian G, Yin S, Ten Hacken E, Sewastianik T, Uduman M, Font-Tello A. et al. A hotspot mutation in transcription factor IKZF3 drives B cell neoplasia via transcriptional dysregulation. Cancer Cell. 2021;39:380-93 e8
174. Read KA, Jones DM, Pokhrel S, Hales EDS, Varkey A, Tuazon JA. et al. Aiolos represses CD4(+) T cell cytotoxic programming via reciprocal regulation of T(FH) transcription factors and IL-2 sensitivity. Nat Commun. 2023;14:1652
175. Scharer CD, Barwick BG, Youngblood BA, Ahmed R, Boss JM. Global DNA methylation remodeling accompanies CD8 T cell effector function. J Immunol. 2013;191:3419-29
176. Northrop JK, Wells AD, Shen H. Cutting edge: chromatin remodeling as a molecular basis for the enhanced functionality of memory CD8 T cells. J Immunol. 2008;181:865-8
177. Araki Y, Wang Z, Zang C, Wood WH 3rd, Schones D, Cui K. et al. Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity. 2009;30:912-25
178. Mullighan CG. TET2 mutations in myelodysplasia and myeloid malignancies. Nat Genet. 2009;41:766-7
179. Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A. et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360:2289-301
180. Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O. et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20:25-38
181. Jeltsch A. Molecular enzymology of mammalian DNA methyltransferases. Curr Top Microbiol Immunol. 2006;301:203-25
182. Shvedunova M, Akhtar A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat Rev Mol Cell Biol. 2022;23:329-49
183. Subramanian S, Bates SE, Wright JJ, Espinoza-Delgado I, Piekarz RL. Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals (Basel). 2010;3:2751-67
184. Li W, Fu Y, Wang W. A real-world pharmacovigilance study investigating the toxicities of histone deacetylase inhibitors. Ann Hematol. 2024;103:3207-17
185. Shah RR. Safety and Tolerability of Histone Deacetylase (HDAC) Inhibitors in Oncology. Drug Saf. 2019;42:235-45
186. Melcher M, Schmid M, Aagaard L, Selenko P, Laible G, Jenuwein T. Structure-function analysis of SUV39H1 reveals a dominant role in heterochromatin organization, chromosome segregation, and mitotic progression. Mol Cell Biol. 2000;20:3728-41
187. Maison C, Quivy JP, Probst AV, Almouzni G. Heterochromatin at mouse pericentromeres: a model for de novo heterochromatin formation and duplication during replication. Cold Spring Harb Symp Quant Biol. 2010;75:155-65
188. Fodor BD, Shukeir N, Reuter G, Jenuwein T. Mammalian Su(var) genes in chromatin control. Annu Rev Cell Dev Biol. 2010;26:471-501
189. Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17:487-500
190. Pace L, Goudot C, Zueva E, Gueguen P, Burgdorf N, Waterfall JJ. et al. The epigenetic control of stemness in CD8(+) T cell fate commitment. Science. 2018;359:177-86
191. Asada S, Fujino T, Goyama S, Kitamura T. The role of ASXL1 in hematopoiesis and myeloid malignancies. Cell Mol Life Sci. 2019;76:2511-23
192. Gao X, You X, Droin N, Banaszak LG, Churpek J, Padron E. et al. Role of ASXL1 in hematopoiesis and myeloid diseases. Exp Hematol. 2022;115:14-9
193. O'Connell CL, Baer MR, Orskov AD, Saini SK, Duong VH, Kropf P. et al. Safety, Outcomes, and T-Cell Characteristics in Patients with Relapsed or Refractory MDS or CMML Treated with Atezolizumab in Combination with Guadecitabine. Clin Cancer Res. 2022;28:5306-16
194. Zhang Y, Li Y, Cao W, Wang F, Xie X, Li Y. et al. Single-Cell Analysis of Target Antigens of CAR-T Reveals a Potential Landscape of "On-Target, Off-Tumor Toxicity". Front Immunol. 2021;12:799206
195. FDA Requires Boxed Warning for T cell Malignancies Following Treatment with BCMA-Directed or CD19-Directed Autologous Chimeric Antigen Receptor (CAR) T cell Immunotherapies. 18 April 2024. https://www.fda.gov/
196. Tix T, Alhomoud M, Shouval R, Cliff ERS, Perales MA, Cordas Dos Santos DM. et al. Second Primary Malignancies after CAR T-Cell Therapy: A Systematic Review and Meta-analysis of 5,517 Lymphoma and Myeloma Patients. Clin Cancer Res. 2024;30:4690-700
197. Dulery R, Guiraud V, Choquet S, Thieblemont C, Bachy E, Barete S. et al. T cell malignancies after CAR T cell therapy in the DESCAR-T registry. Nat Med. 2025: [Epub ahead of print].
198. Dias J, Garcia J, Agliardi G, Roddie C. CAR-T cell manufacturing landscape-Lessons from the past decade and considerations for early clinical development. Mol Ther Methods Clin Dev. 2024;32:101250
Author contact
![]() Corresponding authors: Yongsheng Wang: wangysedu.cn; Qijing Li: li_qijinga-star.edu.sg.
Corresponding authors: Yongsheng Wang: wangysedu.cn; Qijing Li: li_qijinga-star.edu.sg.