Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
The function of HSCs in chronic...
The mechanism of HSC activation...
Mechanism of metabolic...
Therapeutic implications
Future perspectives
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2025; 15(5):1715-1740. doi:10.7150/thno.106597 This issue Cite
Review
Metabolism of hepatic stellate cells in chronic liver diseases: emerging molecular and therapeutic interventions
Mengyao Yan1, Yimin Cui1,2 ![]() , Qian Xiang1,2
, Qian Xiang1,2 ![]()
1. Institute of Clinical Pharmacology, Peking University First Hospital, Beijing, China.
2. Department of Pharmacy Administration and Clinical Pharmacy, School of Pharmaceutical Sciences, Peking University Health Science Center, Beijing, China.
Received 2024-11-8; Accepted 2024-12-10; Published 2025-1-2
Abstract

Chronic liver diseases, primarily metabolic dysfunction-associated steatotic liver disease (MASLD), metabolic and metabolic dysfunction-associated alcoholic liver disease (MetALD), and viral hepatitis, can lead to liver fibrosis, cirrhosis, and cancer. Hepatic stellate cell (HSC) activation plays a central role in the development of myofibroblasts and fibrogenesis in chronic liver diseases. However, HSC activation is influenced by the complex microenvironments within the liver, which are largely shaped by the interactions between HSCs and various other cell types. Changes in HSC phenotypes and metabolic mechanisms involve glucose, lipid, and cholesterol metabolism, oxidative stress, activation of the unfolded protein response (UPR), autophagy, ferroptosis, senescence, and nuclear receptors. Clinical interventions targeting these pathways have shown promising results in addressing liver inflammation and fibrosis, as well as in modulating glucose and lipid metabolism and metabolic stress responses. Therefore, a comprehensive understanding of HSC phenotypes and metabolic mechanisms presents opportunities for novel therapeutic approaches aimed at halting or even reversing chronic liver diseases.
Keywords: hepatic stellate cells, cellular crosstalk, liver diseases, clinical treatment
Introduction
Hepatic stellate cells (HSCs) constitute approximately 15% of the total liver intrinsic cell population and about 30% of non-parenchymal cells. HSCs are not only the primary cell type involved in fibrosis during liver injury but also play a crucial role in liver regeneration and cancer progression [1]. Under normal conditions, HSCs exist in a quiescent state, do not express α-smooth muscle actin (α-SMA), and exhibit low levels of proliferation and collagen synthesis [2]. However, when the liver is subjected to inflammation or mechanical injury, HSCs become activated, and their phenotype transitions from quiescent to activated. Currently, HSCs are recognized for their high plasticity, which allows them to regulate energy and nutrient balance, inflammatory responses, immune functions, and liver growth in their activated state [3]. These processes rely on precise regulation of energy consumption and metabolic adaptability [4]. Recent studies have provided insights into the mechanisms governing HSC metabolism and their roles in liver homeostasis and the response to damage [5]. Given that the liver plays a pivotal role in regulating its metabolic processes, HSCs are also susceptible to disruptions in systemic metabolic regulation. Furthermore, HSCs serve as a model for metabolic homeostasis beyond the liver, primarily because they are liver-specific pericytes, akin to cells with fibrotic potential in other organs. Importantly, HSCs are an often-overlooked determinant of immune metabolism that supports liver function and inflammatory responses [6].
Chronic liver injury, primarily due to metabolic dysfunction-associated steatotic liver disease (MASLD), metabolic dysfunction-associated alcoholic liver disease (MetALD), or viral hepatitis, can lead to liver fibrosis, cirrhosis, and hepatocellular carcinoma [7]. Chronic liver damage promotes HSC activation, resulting in the accumulation of extracellular matrix proteins that disrupt the liver's architecture and impair its functionality [8]. Various signals that indicate cellular damage trigger HSC activation. These signals include pro-inflammatory cytokines produced by infiltrating immune cells, apoptotic bodies from hepatocytes, growth factor activation mediated by endothelial cells, and an increased burden of reactive oxygen species (ROS) [9]. Numerous paracrine and autocrine signaling loops, including fibrogenic signals such as transforming growth factor beta 1 (TGF-β1) and connective tissue growth factor (CTGF), amplify the response of HSCs to liver damage [10]. Besides, recent research has found that targeting Wnt/β-catenin signaling and lactate dehydrogenase A and targeting YAP has therapeutic promise for liver fibrosis through mediated HSC glycolysis, death susceptibility and senescence [11]. Single-cell RNA sequencing analyses reveals a high heterogeneity characterization and tightly interrelated network of HSCs and macrophage subpopulations in liver fibrosis [12].
This review has revealed that multiple cells are involved in the activation of HSC in chronic liver injury, mainly including hepatocytes, liver sinusoidal endothelial cells (LSECs) [13], platelets [14], and some immune cells such as macrophages, neutrophils, and various innate lymphocytes and unconventional T cell (UTC) populations [15]. This situation influences immunological processes by releasing chemokines and cytokines, or transdifferentiate into myofibroblasts that produce matrix [16, 17]. Changes in HSC phenotypes and functions include considerable modifications in transcriptional and protein synthesis, requiring metabolic adjustments in the metabolism of cellular substrates, such as glucose and lipid metabolism, and bearing similarities to the alterations linked to the Warburg effect in cancer cells [18, 19]. The activation of HSCs and their alterations in metabolism are also regulated by nuclear receptor signaling and metabolic stress responses. These changes in metabolism are essential for the inflammatory and fibrogenic activation of HSCs [20]. Therefore, altering these pathways may present chances for cutting-edge treatment strategies to stop or even reverse the advancement of chronic liver injury.
The function of HSCs in chronic livers diseases
Under healthy condition, HSC is located within the Disse space, closely adhering to the LSECs and hepatocytes. They constitute approximately 10% of all resident liver cells [21]. Under chronic liver diseases, activated HSCs express emerging molecular markers and release cellular signals that influence the critical cellular response to chronic liver damage[22]. The terms of 'initiation' and provide a useful framework for understanding the progression of HSC activation in chronic liver diseases. 'Initiation' means to early events that prompt cells to respond to a wide array of extracellular signals. The hallmarks of initiation include the rapid activation of growth factor receptors, the development of a contractile and fibrogenic phenotype, and the modulation of growth factor signaling. Events that further enhance the active phenotype are indicative of 'perpetuation' in chronic liver diseases, especially MASLD and MetALD [23, 24].
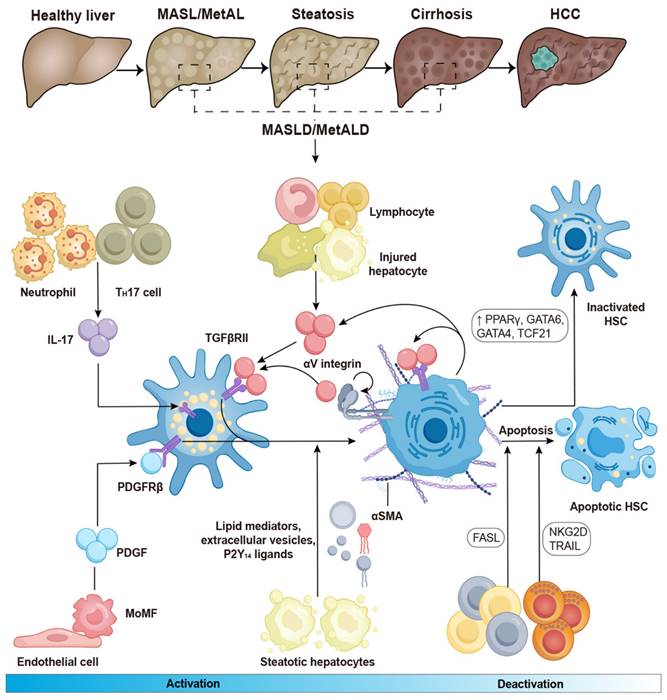
HSCs can be activated by initial stimuli during chronic liver diseases, such as a high-fat diet and excessive alcohol consumption, which may trigger to multiple receptors and factors. For instance, lymphocytes, monocytes, Kupffer cells, and damaged hepatocytes release TGF-β during the development of MASLD and MetALD [25, 26]. Under these conditions, neutrophils and T helper (TH)17 cells produce interleukin (IL)-17, which activates the TGF-β receptor II (TGF-βRII) and increases HSC susceptibility to TGF-β signaling [27, 28]. The αV subunits of integrins activate TGF-β in the extracellular matrix, leading to the contraction of activated HSCs and contributing to the development of liver fibrosis [29, 30]. Both endothelial cells and macrophages can release platelet-derived growth factor (PDGF), which binds to the platelet-derived growth factor receptor β (PDGFRβ) and promotes the activation of HSCs in in hepatic steatosis and fibrosis [31]. In the development of MASLD, steatotic hepatocytes produce lipid mediators, P2Y14 ligands, and extracellular vesicles, all of which contribute to HSC activation [32-34]. Collectively, these factors lead to increased proliferation, contractility, fibrogenesis, matrix degradation, and immune and inflammatory signaling, ultimately contributing to the formation of scars in chronic liver diseases (Figure 1).
The initiation and perpetuation of HSCs in chronic liver diseases. Conversion of quiescent HSCs to their activated state is triggered by in chronic liver diseases. This activation is characterized by distinct phenotypic changes, including fibrogenesis, increased contractility, proliferation, altered matrix degradation, chemotaxis, and enhanced immunological and inflammatory signaling. During the resolution of hepatic fibrosis, activated HSCs can be eliminated through three mechanisms: apoptosis, senescence, or reversion to an inactivated state. Adapted with permission from [35], Copyright 2021 Cell Press.
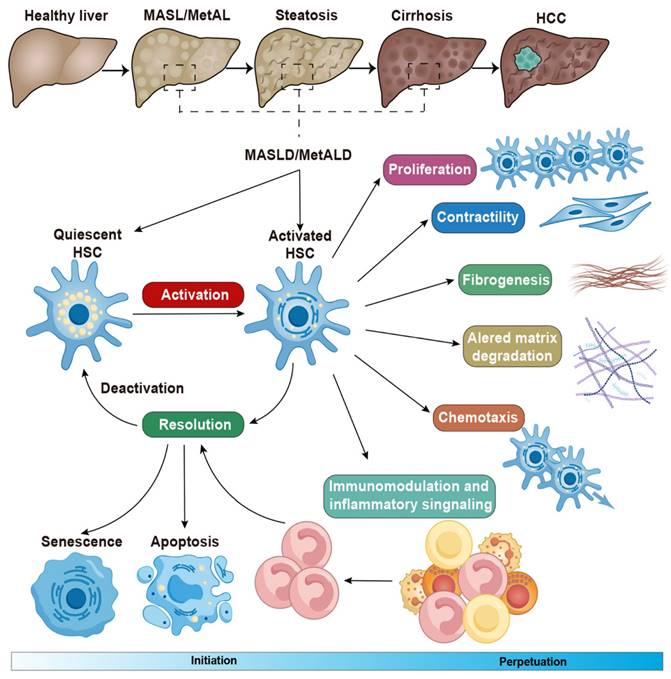
The above activation of quiescent HSCs contributes to fibrosis formation in chronic liver diseases, through their transdifferentiation into highly proliferative, extracellular matrix-producing activated hepatic stellate cells or myofibroblasts [35]. Once activated, HSCs lose intracellular fatty acids but the role of fatty acid oxidation, and result in steatosis in liver diseases [36]. Besides, these cells regulate hepatic immune homeostasis and inflammation, thereby contributing to worsening liver injury, mainly through the mechanisms of crucial signaling pathways, such as toll-like receptor (TLR) receptor, TGF-β and hedgehog mediated hepatic injury [16]. The activation of HSCs also affects oxidative stress in liver damage, result in tissue injury and trigger endoplasmic reticulum stress by the overproduction of proteins [37].
Different states of HSC perpetuation that distinguish between an early fibrogenic phenotype and a later inflammatory senescent phenotype are now integral to a more comprehensive understanding of the phase involved in the development of liver diseases [38]. Several molecules and signals can influence the progression of liver disease through HSC apoptosis, senescence, and reversion to a more quiescent, inactive state—three primary processes that contribute to HSC elimination [39, 40]. For example, liver γδ T cells and CD8+ T cells utilize the FAS-FAS ligand pathway to induce HSC apoptosis, while natural killer (NK) cells depend on NKG2D and tumor necrosis factor (TNF)-related apoptosis-inducing ligands to eliminate HSCs in chronic liver diseases [41]. Furthermore, the expression of peroxisome proliferator-activated receptor (PPAR)γ, GATA-binding factor 6 (GATA6), GATA4, and transcription factor 21 (TCF21) appears to play a role in regulating HSC reversion to an inactive state in liver fibrosis (Figure 2) [42-44].
The activation and deactivation of HSCs in chronic liver diseases. When profibrotic stimulation in chronic liver diseases activates HSCs, quiescent HSCs convert into myofibroblasts. This transformation results in a decrease in their vitamin A content, stimulation of α-SMA, and synthesis of collagen type I. The conversion of HSCs is induced by TGF-β, which is produced by infiltrating lymphocytes, monocytes/macrophages, and damaged hepatocytes. TGF-βRII is upregulated by IL-17, which is generated by TH17 cells and neutrophils, thereby increasing HSC sensitivity to TGF-β stimulation. When latent TGF-β binds to ECM proteins, it becomes inactive but can be released when activated HSCs contract through the action of αV integrin. In a feed-forward cycle, activated HSCs perpetuate their activation by producing TGF-β. HSC activation is also stimulated by PDGF, which is secreted by macrophages and endothelial cells. Additionally, HSC activation is sustained by lipid mediators, P2Y14 signaling, and extracellular vesicles released by injured hepatocytes. Following the resolution of fibrosis, HSCs undergo either apoptosis or revert to an inactive state, a process mediated by the overexpression of transcription factors such as TCF21, GATA4, GATA6, and PPARγ. Lymphocytes, including NK cells, γδ T cells, and CD8+ T cells can effectively eliminate activated HSCs and myofibroblasts by inducing apoptosis. FASL, Fas ligand; GATA 4/6, GATA-binding factor 4/6; IL-17, interleukin-17; MoMFs, monocyte-derived macrophages; NKG2D, NK receptor group 2 member D; PDGF, platelet-derived growth factor; PDGFRβ, platelet-derived growth factor receptor β; PPAR, peroxisome proliferator-activated receptor; α-SMA, α-smooth muscle actin; TCF21, transcription factor 21; TGF-βRII, TGF-β receptor II; TH 17, T helper 17; TRAIL, tumour necrosis factor-related apoptosis-inducing ligand. Adapted with permission from [8], Copyright 2023 Springer Nature.
The mechanism of HSC activation in chronic liver diseases
Under chronic liver diseases, multiple cell injury occurs, including hepatocyte cell death, LESCs injury and stimulation of immune cells. These cells secrete pro-inflammatory cytokines and chemokines, which ultimately activate HSCs. Then, the activation of quiescent HSCs leads to their transdifferentiation into highly proliferative, extracellular matrix-producing activated HSCs or myofibroblasts, which are key factors in progression of chronic liver diseases.
Multiple cells types influence HSC activation
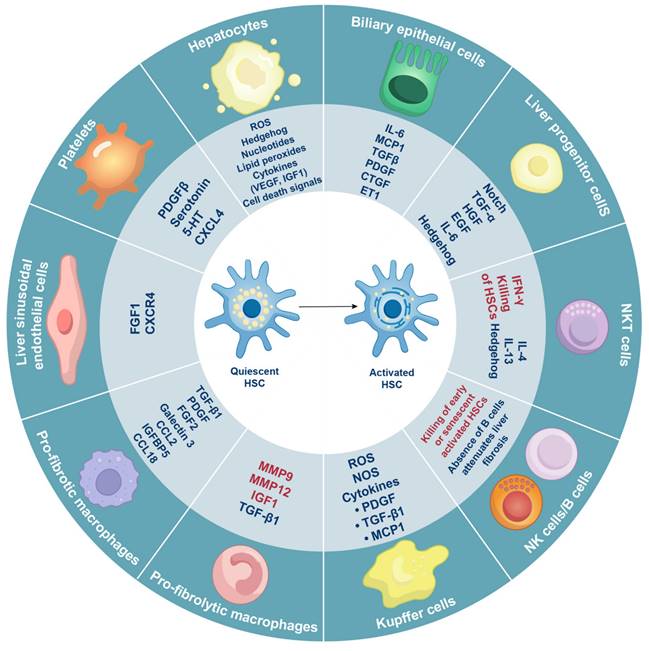
Even during homeostasis, liver tissue comprises a diverse array of cell types that are distributed throughout the parenchyma, with a higher concentration in the periportal regions. These cell types include hepatocytes, macrophages, biliary epithelial cells, liver progenitor cells, LESCs, NK cells, natural killer T (NKT) cells, platelets, B cells, mucosal-associated invariant T (MAIT) cells, γδ T cells, neutrophils, mast cells, and innate lymphoid cells. These various cell types converge on HSCs to promote HSC activation (Figure 3).
Multiple cell types influence the activation of HSCs in chronic liver diseases. Hepatocytes, macrophages, biliary epithelial cells, liver progenitor cells, LESCs, NK cells, NKT cells, platelets, and B cells can either stimulate (indicated in blue font) or inhibit (indicated in red font) HSC activation by releasing various hormones, cytokines, and other signaling molecules. CCL2/18, C-C motif ligand 2/18; CTGF, connective tissue growth factor; CXCL4, C-X-C motif ligand 4; CXCR4, C-X-C chemokine receptor 4; EGF, epidermal growth factor; ET1, endothelin-1; FGF1/2, fibroblast growth factor 1/2; HGF, hepatocyte growth factor; 5-HT, 5-hydroxytryptamine; IFN-γ, interferon-γ; IL-6/4/13, interleukin-6/4/13; IGF1, insulin-like growth factor 1; IGFBP5, insulin-like growth factor-binding protein-5; MCP1, monocyte chemoattractant protein 1; MMP9/12, Matrix metalloproteinase-9/12; PDGF, platelet-derived growth factor; ROS, reactive oxygen species; TGF-α/β, transforming growth factor-α/β; VEGF, vascular endothelial growth factor.
Hepatocytes affecting HSC activation
The destruction of hepatocytes occurs under certain conditions such as metabolic dysfunction-associated steatohepatitis (MASH) and viral infections, both of which are linked to parenchymal liver injury [45]. This process is facilitated by an expanding array of mediators, including nucleotides, hedgehog ligands, and reactive oxygen species (ROS) [46]. Damage-associated molecular patterns (DAMPs) secreted by dying hepatocytes can promote the activation of HSCs either directly or indirectly [47]. NACHT, LRR and PYD domains-containing protein 3 (NLRP3) is a crucial component of the inflammasome and acts as a downstream regulator of DAMPs [48]. Hepatocyte pyroptosis leads to the release of complex inflammatory particles, including the NLRP3 inflammasome, from within hepatocytes into the extracellular space. Mice with a constitutively active mutant NLRP3 exhibit severe liver inflammation, pyroptotic hepatocyte death, and HSC activation, which is accompanied by fibrosis [49]. In response to liver damage, hepatocytes produce IL33, which activates and expands liver-resident innate lymphoid cells (ILCs) [50]. The three known subsets of ILCs are ILC1, ILC2, and ILC3, with ILC2 being responsible for mediating liver damage that leads to HSC activation. Furthermore, the engulfment of apoptotic hepatocyte bodies by HSCs promotes their activation as the phagocytosis of these apoptotic bodies can enhance HSC activation [51].
Monocytes and macrophages affecting HSC activation
Hepatic monocytes can be broadly categorized into macrophages, which primarily include tissue-resident Kupffer cells and monocyte-derived macrophages (MoMFs) (Figure 4) [52]. Selective depletion of macrophages in mice, results in CD11b-diphtheria toxin transgenic mice, leads to reduced activation of HSC and fibrogenesis following liver injury induced by CCl4. This suggests that the removal of macrophages during sustained injury leads to a decrease in liver damage [53]. The recruited monocytes consist of two subsets: CD11c (-)/Ly6C (+) cells and CD11c (+)/Ly6C (-) cells. Ly6C (+) cells are inflammatory and recruited macrophages that contribute to advanced liver injury, while Ly6C (-) cells are known as 'alternative macrophages' that inhibit inflammatory responses and promote HSC activation. Ly6C (+) monocytes exhibit significantly higher levels of CC chemokine receptor (CCR)1 and CCR2, whereas Ly6C (-) cells show elevated expression of CCR5 and CX3C chemokine receptor 1 (CX3CR1). CCR1 primarily stimulates HSC activation by bone marrow-derived macrophages [54]. In models of bile duct ligation and CCl4 therapy, mice lacking CCR1 demonstrate decreased macrophage infiltration. Intrahepatic Ly6C (+) monocyte-derived macrophages are recruited in a CCR2-dependent manner during fibrogenesis triggered by CCl4 injection, indicating that CCR2 plays a crucial role in the accumulation of these macrophages. In CCR2-deficient mice, the recruitment of damaged Ly6C (+) monocytes is diminished, leading to decreased activation of HSC [55, 56]. Furthermore, in CCR2-deficient mice, a choline-deficient, L-amino acid-defined diet results in reduced infiltration of Ly6C (+) macrophages, consequently leading to less liver injury [57].
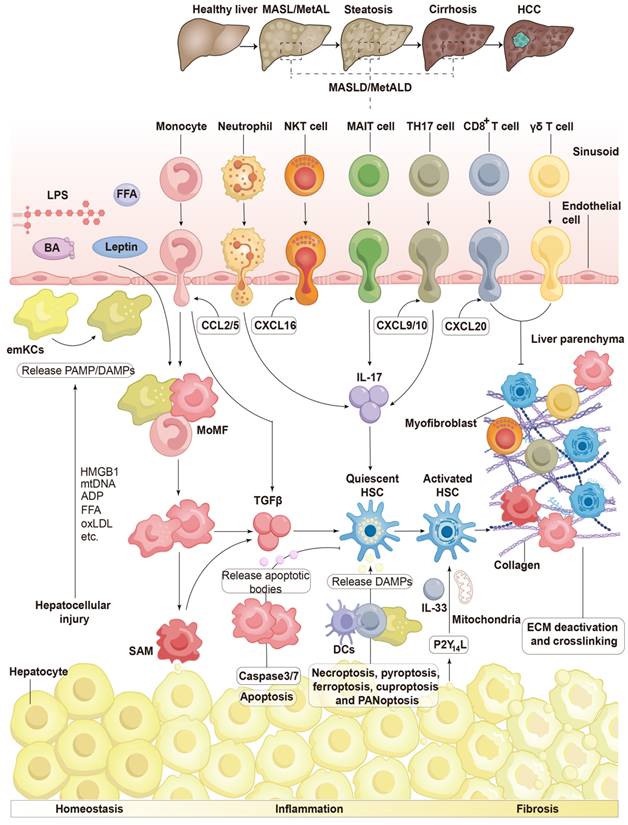
HSC activation is influenced by immune cells in chronic liver diseases. Chronic liver inflammation is initiated by hepatocellular damage, which releases DAMP and PAMP signals, including HMGB1, mtDNA, ADP, FFA, oxLDL, and others. These signaling molecules, along with cytokines and chemokines produced by activated emKCs, attract immune cells from the bloodstream, particularly resident macrophages to the liver, leading to significant phenotypic alterations. These immune cells respond promptly to disruptions in extrahepatic signals, primarily BAs and bacterial products such as LPS from the intestine, as well as lipid mediators like FFAs and leptin from adipose tissue and damaged hepatocytes, resulting in a pro-inflammatory response. Monocytes, driven by CCL2, are the first immune cells to arrive in the liver following injury. They release profibrogenic molecules, including TGF-β, and differentiate into MoMFs, which subsequently prolong the inflammatory response. MoMFs may further differentiate into SAM, which produce TGF-β and activate HSCs. Neutrophils may arrive early during the inflammatory response and promote fibrogenesis by producing IL-17, which stimulates HSCs. Auto-aggressive CD8+ T lymphocytes accelerate hepatocyte destruction. Injured hepatocytes emit danger signals, such as P2Y14L and alarmins, which include IL-33 and mitochondrial metabolites, potentially activating HSCs. Lymphocytes are attracted by various chemokines, including CXCL16 released by NKT cells, CXCL9 and CXCL10 released by conventional T cells, and CCL20 released by γδ T cells. TH17 cells and MAIT cells produce IL-17, which promotes fibrogenesis. However, certain CD8+ T cells, γδ T cells, and NK cells inhibit fibrosis formation by inducing apoptosis in myofibroblasts. The activation of caspase-3/7 in apoptotic hepatocytes leads to the production of apoptotic bodies, which can activate HSCs directly or indirectly through macrophage activation. DAMPs, particularly HMGB1, can be released through necroptosis, pyroptosis, ferroptosis, cuproptosis, and PANoptosis, causing macrophages, monocytes, and DCs to aggregate, secrete inflammatory factors, and further amplify the inflammatory response. ADP, adenosine diphosphate; BA, bile acid; CCL2/5, C-C motif ligand 2/5; CXCL, C-X-C motif ligand16; DCs, dendritic cells; DAMPs, damage-associated molecular patterns; ECM, extracellular matrix; emKCs, embryonic KCs; FFA, free fatty acids; HMGB1, high-mobility group box 1; HSCs, hepatic stellate cells; IL-33, interleukin-33; LPS, lipopolysaccharide; MAIT, Mucosal-associated invariant T; MoMF, monocyte-derived macrophage; NKT, natural killer T; oxLDL, oxidized low-density lipoprotein; PAMP, pathogen-associated molecular pattern; SAM, scar-associated macrophage; TGF, transforming growth factor; TH17, T helper 17. Adapted with permission from [8], Copyright 2023 Springer Nature.
Kupffer cells represent the largest resident macrophage population in the body and play a critical role in activating HSCs [58]. Bone marrow-derived monocytes (BMDMs) migrate to the damaged liver, where they promptly respond to inflammatory signals and differentiate into macrophages [59]. In the context of inflammation, a significant influx of BMDMs is responsible for producing MoMFs, which, in turn, expand the pool of liver macrophages. Although MoMFs and Kupffer cells differ in terms of phenotype and function, mouse models demonstrate that the two lineages exhibit remarkable plasticity [60]. Extensive research utilizing single-cell RNA sequencing has provided unprecedented insights into the heterogeneity of hepatic myeloid cells. One important finding is that MoMFs can replace Kupffer cells and adopt the phenotype of lipid-associated macrophages (LAMs) or scar-associated macrophages (SAMs), characterized by the expression of triggering receptor expressed on myeloid cells 2 (TREM2), CD9, and osteopontin, which may stimulate HSC activation [61, 62]. Spatial proteogenomics analyses have confirmed the persistence of these macrophage subtypes across various species, and single-cell RNA sequencing analyses conducted on human liver tissues have identified SAMs as a distinct group located in the fibrotic niche of cirrhotic livers, although many findings are still primarily based on mouse models. Notably, a hallmark of severe fibrosis in human liver biopsy samples across different etiologies is the accumulation of inflammatory MoMFs in portal regions, particularly surrounding ductular responses [63].
LSECs affecting HSC activation
In a healthy liver, the differentiation of LSECs inhibit HSC activation and promotes the transition of active HSCs to a quiescent state through the generation of nitric oxide (NO) derived from vascular endothelial growth factor (VEGF) [64]. Capillarization diminishes the ability of LSECs to inhibit HSC activation and is characterized by a lack of LSEC differentiation following liver injury [65]. In rats with thioacetamide-induced cirrhosis, the restoration of capillarization through the injection of BAY 60-2770, a soluble guanylate cyclase stimulator, leads to HSC quiescence. Endothelial nitric oxide synthase acts as the catalyst for the production of NO, which is the mechanism by which VEGF functions to activate soluble guanylate cyclase [66].
Given the circumstances surrounding liver damage, liver regeneration and fibrosis may be influenced by LSECs [67]. The differential expression of stromal cell-derived factor 1 receptors, C-X-C chemokine receptor (CXCR) 7, and CXCR4 following chronic liver damage indicates that angiocrine signals from LSECs promote HSC activation and regeneration immediately after injury. CXCR7 regulates regenerative pathways by inducing the expression of ID1, a known inhibitor of DNA-binding protein 1 [68]. Additionally, fibroblast growth factor receptor 1 (FGFR1) and CXCR4, which provide paracrine stimulation to HSCs, are key drivers of HSC activation and liver injury processes [69]. Regenerative pathways are restored in mice with LSEC-specific depletion of FGFR1 or CXCR4. The depletion of either FGFR1 or CXCR4 in LSECs uniquely facilitates the recovery of liver regenerative pathways in these mice [70].
Platelets affecting HSC activation
Platelets are a crucial cellular source of PDGFβ and TGF-β, which activate HSCs and influence various chronic liver diseases, including MASH, MetALD and hepatitis B and C [71, 72]. Platelets produce PDGFβ to activate HSCs and facilitate liver injury in multidrug resistance protein 2 (MDR2)/Abcb4-null mice, a model that promotes advanced biliary fibrosis. Depleting platelets or selectively inhibiting PDGFβ may reduce biliary fibrosis in patients with liver diseases [73]. Silencing Nogo-B suppresses the proliferation, fibrosis, and autophagy of HSCs while inducing cell cycle arrest and senescence of HSCs through the PDGFβ [74]. Gomisin D can inhibit HSC proliferation and activation, promote HSC apoptosis, and alleviate CCl4-induced liver injury by targeting PDGFRβ and regulating PDGFRβ signaling pathway [75].
Dendritic cells affecting HSC activation
DCs are a specialized type of antigen-presenting cell that primarily including plasmacytoid dendritic cells (pDCs) and conventional (classical) dendritic cells (cDC1s and cDC2s) [76]. Although mature DCs are enlarged in damaged liver tissue, the precise mechanisms by which they contribute to the activation of HSCs remain unclear. Several studies in murine models have demonstrated that inhibiting or depleting DC maturation and function reduces inflammation and, consequently, inhibits HSC activation and liver injury [77, 78]. However, other research has indicated that a reduction in DCs has no effect on liver damage [79]. Furthermore, in murine models of liver fibrosis, DCs have been shown to restrict inflammation and fibrogenesis. Specifically, cDC1s, particularly those expressing the CD103 (+) subtype, have been identified as a protective subset in mice with metabolic diseases [80]. More recent studies, however, suggest that cDC1s may also play a detrimental role in liver diseases [81]. These cells are widely distributed in humans with metabolic dysfunction-associated fatty liver disease (MAFLD) and in mice with steatohepatitis, and their presence correlates with fibrosis scores and disease severity [82]. In murine models, X-C motif chemokine receptor 1-expressing cDC1s exacerbate liver disease by inducing inflammatory reprogramming of CD8+ T cells [83].
T and B lymphocytes affecting HSC activation
In a healthy liver, T and B cells predominantly reside along the portal tracts. However, they often infiltrate the parenchyma during inflammatory responses [84]. In liver disorders triggered by antigens, such as autoimmune hepatitis and chronic viral hepatitis, adaptive immune cells play a crucial role [85].
For example, in chronic hepatitis B virus infection, T cell dysfunction is often attributed to insufficient priming of CD8+ T cells by hepatocytes. This process can be reversed through Kupffer cell-mediated cross-presentation in mice. While these inflammatory events contribute to the formation of fibrosis through their cytotoxic effects on hepatocytes, adaptive immunity may also influence hepatic fibrosis independently of specific antigens. This non-specific activation is particularly relevant to MAFLD, as it is driven by mediators released during metabolic processes linked with liver injury. These mediators primarily include acetate and extracellular adenosine 5' triphosphate (ATP), which may activate liver-resident CD8+ T cells characterized by high expression of CXCR6, PD-1, and perforin [86]. Consequently, these T cells perpetuate auto-aggression against hepatocytes in an MHC class I-independent manner, thereby accelerating the progression of steatohepatitis [87].
There is conflicting data regarding the role of CD8+ cytotoxic T cells in liver damage. This discrepancy may be attributed to a combination of auto-aggressive and protective T cell immunity [88]. The absence of CD8+ T cells has no effect on fibrosis formation in a mouse model of toxic liver fibrosis [89]. However, the activation of splenic CD8+ T cells in the same context exacerbates disease progression [90]. Conversely, in a mouse model of MASH induced by obesity, a reduction in CD8+ T cells leads to decreased activation of HSC and reduced liver inflammation. In this scenario, HSC may be directly activated by CD8+ T cells [91]. Furthermore, studies have shown that tissue-resident CD8+ T lymphocytes with a memory phenotype facilitate the resolution of hepatic fibrosis by inducing apoptosis in activated HSC. The adoptive transfer of CD8+ T lymphocytes inhibits the progression of fibrosis, while their absence hinders the resolution of fibrosis [92]. These contradictory findings suggest that the development and resolution of hepatic fibrosis may involve distinct populations of CD8+ T cells.
TH cells, including TH2 and TH17 cells, are closely associated with liver injury, particularly in the development of fibrosis [88, 93]. Numerous profibrogenic cytokines, such as IL-4 and IL-13, are released by TH2 cells, which activate fibroblasts and promote extracellular matrix remodeling [94]. In contrast, the cytokines IFN-γ and IL-12 produced by TH1 cells exhibit a somewhat antifibrotic effect [88]. TH17 cells contribute to liver fibrosis by producing IL-17 and IL-22, which stimulate the production of TGF-β in the liver, enhance TGF-β signaling in HSCs, and promote collagen and pro-inflammatory cytokine production by HSCs and Kupffer cells [95]. These processes ultimately lead to the progression of liver fibrosis. Furthermore, fibrosis formation is inhibited in mice lacking the IL-17 or IL-22 receptor, and the advancement of fibrosis can be halted in vivo by depleting IL-17 [96]. Notably, IL-22 may have protective effects against liver damage. Through the induction of HSC senescence, IL-22 agonism reduces liver fibrosis in mice treated with CCl4, protects hepatocytes from damage during acute hepatitis, and alleviates hepatic inflammation in mice with acute-on-chronic liver failure [97]. In a phase II clinical study, the IL-22 agonist F-652 has been shown to enhance hepatic regeneration and reduce hepatic inflammation in 18 individuals with severe alcoholic hepatitis [98].
Regulatory T (Treg) cells may prevent liver injury by releasing IL-10. As an anti-inflammatory cytokine, IL-10 reduces collagen I production, inhibits the activation of Kupffer cells, and decreases IL-17 production by TH17 cells, ultimately leading to reduced activation of HSCs [99]. Mice deficient in IL-10 are consistently more susceptible to liver injury [100]. Conversely, Treg cells may hinder the resolution of fibrosis in mice by suppressing the synthesis of matrix metalloproteinases by Kupffer cells, which contributes to the persistence of fibrosis [101].
B cells constitute only 5% of intrahepatic lymphocytes in a healthy human liver, representing a modest fraction of liver immune cells [102]. For example, mice lacking B cells do not develop fibrosis in either toxic or diet-induced models, but B cells that accumulate in the damaged liver exhibit an activated state and release pro-inflammatory cytokines, which promotes HSC activation. Data from individuals with MAFLD and mouse models indicate that gastrointestinal B cells may contribute to liver injury by secreting immunoglobulin A and activating Fcγ receptor signaling on myeloid cells [103]. Interestingly, certain types of B cells, such as regulatory B cells that produce IL-10, may also provide protection against HSC activation and inflammation associated with MAFLD [104].
Unconventional T cells affecting HSC activation
UTCs are a diverse collection of lymphocytes that include NKT cells, MAIT cells, and γδ T cells [105-108]. NKT cells appear to play dual roles in chronic liver damage. They can exacerbate liver injury through the production of profibrotic cytokines, such as IL-4 and IL-13, which may activate HSCs [109]. Under another conditions, NK cells may eliminate HSCs by expressing death receptor ligands, including Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand and Fas Ligand (also known as TNF ligand superfamily member 6), while simultaneously mitigating liver injury by producing IFN-γ. In addition to their direct influence on liver injury, NKT cells can polarize into profibrotic TH2 and TH1 cell responses by secreting cytokines like IL-4 and IFN-γ, which inhibit the activation of HSCs [110]. Furthermore, NK cells assist in the clearance of senescent activated HSCs, facilitating the resolution of fibrosis [111]. NKT cells exhibit a range of complex, context-dependent roles in liver fibrosis and HSC activation. Similar to NK cells, NKT cells produce IFN-γ and promptly eliminate activated HSCs, alleviating liver damage [112]. However, NKT cells also display profibrogenic characteristics. The migration of NKT cells is regulated by CXCR6, which is expressed by NKT cells, and its ligand, C-X-C motif ligand (CXCL) 16, produced by endothelial cells and macrophages. Mice deficient in CXCR6 demonstrate protection against liver injury, but the progression of liver injury resumes following the adoptive transfer of NKT cells into CXCR6-deficient mice [113].
The role of MAIT cells in the activation of HSCs remains incompletely understood. Recent research suggests that MAIT cells exhibit profibrogenic activities in chronically damaged livers. They can stimulate HSC activation through IL-17 or promote the pro-inflammatory polarization of macrophages. Studies have shown that the absence of MAIT cells inhibits the activation of HSCs and fibrosis formation in mice [114]. Functionally, MAIT cells have been proposed as the human equivalent of NKT cells [115].
γδ T cells have been shown to proliferate in these conditions. However, there is limited information regarding their specific role in the activation of HSCs and the development of chronic liver diseases [88]. γδ T cells exert profibrogenic effects by increasing the production of hepatic IL-17 in mice with liver fibrosis, which promotes the activation of HSCs. Additionally, γδ T cells induce apoptosis in activated HSCs and enhance NK cell-mediated cytotoxicity against these activated HSCs, thereby alleviating liver damage [116].
Neutrophils and mast cells affecting HSC activation
Neutrophils are present in various forms of chronic liver disease. By producing inflammatory cytokines, activating Kupffer cells, and attracting other immune cells, neutrophils can exacerbate hepatic inflammation. However, they do not appear to have a direct impact on the activation of HSCs [117]. Research conducted in mice suggests that neutrophils play a crucial role in the activation of HSCs and inflammation by facilitating the functional transition of macrophages from pro-inflammatory to restorative phenotypes. This effect is completely abolished upon neutrophil depletion [118].
Mast cells are innate myeloid cells that play a significant role in allergic reactions and the regulation of host-pathogen interactions in allergies and autoimmune liver diseases [119]. In a healthy liver, mast cells are relatively scarce. However, nearly all liver disorders exhibit mast cell infiltration, which appears to primarily contribute to profibrogenic activity [120]. The depletion of mast cells has been shown to reduce liver damage, while an increase in mast cell numbers is associated with severe fibrosis and inflammation in mouse models of MAFLD and MASH [121]. Similar outcomes have been observed in mouse models of cholestatic liver disease [122]. Furthermore, the severity of fibrosis in individuals with viral or alcoholic hepatitis correlates with the number of infiltrating mast cells. One of the key mediators released by activated mast cells is histamine. Given that both histamine inhibition and histamine receptor blockade inhibit the activation of HSCs, reduce liver damage and fibrosis formation in mouse models of cholestatic damage, it appears that histamine signaling plays a crucial role in mast cell-induced inflammation and the development of fibrosis [123].
Innate lymphoid cells affecting HSC activation
ILCs comprise several subsets, including lymphoid tissue inducer cells, NK cells, ILC1, ILC2, and ILC3. Hepatic ILCs constitute a significant portion of intrahepatic lymphocytes, primarily consisting of group 1 ILCs, which include NK cells and tissue-resident ILC1s [124]. The role of ILCs in the development of chronic liver disease remains an area of ongoing investigation and debate [125]. NK cells play a protective role in chronic liver disease in both mice and humans by eliminating activated HSCs, inducing apoptosis, and producing the antifibrotic cytokine IFN-γ. These effects contribute to the resolution of the extracellular matrix and promote a shift towards TH1 responses [126]. Other studies suggest that the protective effect of NK cells may be highly dependent on the specific NK cell subtype or the stage of disease progression. For instance, NK cells with high expression of CXCR3 appear to be functionally impaired in individuals with chronic hepatitis C virus infection, which subsequently exacerbates chronic liver injury [127]. In mice treated with CCl4, the antifibrotic effect of NK cells diminishes as the disease advances, potentially due to increased production of TGF-β by HSCs [112, 128].
All of these cell types, including hepatocytes, macrophages, biliary epithelial cells, liver progenitor cells, LESCs, NK cells, NKT cells, platelets, B cells, MAIT cells, γδ T cells, neutrophils, mast cells, and innate lymphoid cells, play a crucial role in chronic liver diseases. These various cell types converge upon HSCs to promote HSC activation, which contribute to release danger signals, extrahepatic signals and chemokines.
Inflammatory processes influence HSC activation
Danger signals released from dying cells
Damaged and dying cells produce soluble mediators that function as alarmins or DAMPs, signaling surrounding cells to respond to chronic liver injury (Figure 4) [129]. P2Y14 ligands have been shown to be released by dying hepatocytes, which interact with the P2Y14 receptor on HSCs, leading to their immediate activation in both mice and humans. Consequently, the absence of P2Y14 in several preclinical animal models has resulted in reduced liver injury. Injured hepatocytes can release the nuclear protein high-mobility group box 1 (HMGB1), which also promptly stimulates HSCs. Given that hepatocytes contain a high number of mitochondria due to their elevated metabolic activity, mitochondria-derived DAMPs are particularly abundant in the liver. These molecules primarily consist of mitochondrial DNA, which shares similarities with bacterial DNA [130]. In preclinical rodent models, mitochondria-derived DAMPs have been shown to stimulate HSC activation and the production of scar tissue. Furthermore, these DAMPs are elevated in patients with MASH and fibrosis [47].
Pattern recognition receptors enable both parenchymal and non-parenchymal cell types in the liver, including HSCs, LESCs, and Kupffer cells, to detect the release of danger signals in chronic liver diseases [131]. One significant response to these signals is the production of inflammasomes, which are protein complexes that have been conserved throughout evolution. These complexes are assembled through specific signaling pathways and cellular stress [49]. Inflammasomes process IL-1β, release IL-18, and ultimately promote pyroptosis in inflammatory cells, thereby initiating the inflammatory process [132].
IL-33, a cytokine associated with TH2 responses, is produced by stressed hepatocytes in both humans and animals suffering from chronic liver diseases [133]. The production of IL-33 leads to the recruitment of ILC2s, which subsequently produce IL-13, activating HSCs [134]. Furthermore, IL-33 may directly influence HSCs and promote the synthesis of extracellular matrix proteins. In cholestatic liver disorders, bile acids (BAs) accumulate in human and murine tissues, which can trigger inflammation by stimulating the production of pro-inflammatory mediators in hepatocytes and other cell types, including chemokines, cytokines, and adhesion molecules [135]. Harmful BAs can be detected by various non-parenchymal liver cells, such as HSCs and macrophages, through the receptor Takeda G protein-coupled receptor 5 (TGR5). This interaction may initiate fibrogenic responses, including cholangiocyte proliferation and ductular reactions [136, 137].
Extrahepatic signals
Extrahepatic mediators produced in locations such as the gut and adipose tissue significantly influence liver inflammation. This influence is particularly evident in MAFLD and MASH, where the progression of liver disease is affected by hormones from the gut and adipose tissue, microbiological metabolites, and dietary factors (Figure 4) [138, 139]. Given the physical and functional interconnection between the intestines and liver, the gut-liver axis can be disrupted in various clinical conditions, serving as an indicator of liver diseases [140]. When intestinal homeostasis is compromised, bacterial metabolic products, pathogen-associated molecular patterns (PAMPs), BAs, and nutrients can enter the liver via the portal vein [141]. Microbial compounds, such as lipopolysaccharides (LPS), exert a pro-inflammatory effect primarily mediated by liver-resident macrophages and are elevated in numerous liver disorders associated with increased intestinal permeability [142]. Additionally, liver inflammation can result from alterations in bacterial metabolites, including ethanol and fatty acid composition [143].
Gut dysbiosis alters the composition of BAs because gut bacteria modify secondary BAs before they are recycled to the liver. Pro-inflammatory microbes outcompete other species in the small intestine when there is a reduction in primary BAs, leading to an increase in toxic BAs, which exacerbates inflammation and damages liver cells [144]. Conversely, certain intrinsic or synthetic BAs that activate the farnesoid X receptor (FXR) in hepatocytes suppress the overexpression of inflammation-associated genes and promote cellular longevity [145]. Consequently, the effects on hepatic fibrosis and inflammation depend on the specific composition of the BA pool, which is closely linked to metabolic changes [146].
When insulin resistance is present, the flow of free fatty acids from adipose tissue to the liver is accelerated in obese individuals, exacerbating lipotoxicity and subsequent inflammation in chronic liver diseases. This process may be further aggravated by the liver, which can signal adipose tissue to enhance lipolysis [147]. Additionally, studies in mice have demonstrated that leptin released from adipose tissue activates liver-resident macrophages and increases their sensitivity to endotoxins, leading to heightened liver inflammation [148]. In in vitro experiments, leptin-activated Kupffer cells also induce the activation of HSCs, suggesting potential profibrogenic properties [149].
Chemokines and chemokine receptors
In chronic liver diseases, chemokines—also known as chemoattractant cytokines—play a crucial role in regulating the recruitment and localization of immune cells. C-C motif ligand (CCL)2, CCL3, CCL5, CXCL9, and CXCL10 are among the first chemokines produced following hepatic injury (Figure 4) [150]. These chemokines attract monocytes via CCR2 and CCR5, as well as T lymphocytes through CCR5 and CXCR3 [151]. The CCL2 and CCR2 pathway exerts a profibrogenic effect. CCR2 knockout mice exhibit significantly inactivate HSCs and less fibrosis compared to wild-type mice. Therefore, in animal models of MASH, therapeutic suppression of CCR2 results in a reduction of liver injury and inflammatory responses, while also inhibiting the infiltration of CCR2+ monocytes [152]. The common profibrogenic roles of CCL3 and CCL5 are supported by observations of decreased immune cell infiltration and alleviation of liver injury in mouse models due to their deficiency or antagonistic interactions [153]. In contrast, the fractalkine receptor CX3CR1 limits infiltration and regulates the survival and development of intrahepatic monocytes, thereby mitigating hepatic fibrogenesis in vivo [154].
CXCR3 and its ligands, including CXCL9, CXCL10, and CXCL11, play complex roles in the recruitment of various lymphocyte populations [155]. In individuals with chronic liver diseases, the levels of CXCL9 and CXCL10, both intrahepatic and peripheral, increase and correlate with the severity of the disease across different stages of fibrosis [156]. Preclinical studies have revealed conflicting roles for the CXCR3 axis. Mice lacking CXCR3 are less susceptible to activate HSCs and the induction of steatohepatitis compared to wild-type mice. However, they also exhibit more severe fibrosis following toxic liver injury. The finding that CXCR3 is involved in the recruitment of both pro-inflammatory and anti-inflammatory T cell subsets, such as TH1 cells and Treg cells, may help clarify these contradictory effects [157].
CXCR6 and its ligand CXCL16 play a significant role in the recruitment of NKT cells and other lymphocytes during chronic liver disease [158]. Mice deficient in CXCR6 demonstrate a reduced accumulation of NKT cells and inactivate HSCs and are protected against the progression of hepatic fibrosis in various experimental models [159]. A similar effect on fibrosis can be achieved through the therapeutic suppression of CXCL16, which also decreases intrahepatic levels of pro-inflammatory cytokines and macrophage infiltration [160]. However, in animal models, CXCR6+ NKT cells exhibit important anticancer properties, highlighting their dual role in chronic liver disorders. Importantly, CXCR6 is not exclusive to NKT cells. It is also present in various lymphocyte subsets, including CD8+ T cells that reside in the livers of patients with MASH. These cells contribute to auto-aggression against hepatocytes and display impaired tumor surveillance in MASH-affected mice [161].
CCL20 levels are elevated in mouse models and individuals with chronic liver diseases, correlating with the severity of the condition [162]. This chemokine is produced by both parenchymal and non-parenchymal cells and attracts specific γδ T cells via the CCR6 receptor. This interaction prompts activated HSCs to induce apoptosis, thereby limiting fibrosis [116]. Additionally, the pathways involving CCR7 and CCL21, as well as CXCR4 and CXCL12, are crucial for the recruitment of immune cells to the damaged liver. Given that these receptors are expressed by various cell types, including lymphocytes, myeloid cells, and stem cells, their precise roles in the disease process are highly context-dependent. However, overexpression of these ligands is frequently observed in numerous hepatic diseases [163].
All these cell types and inflammatory processes converge on HSCs to promote HSC activation, which contributes to the development of chronic liver diseases. After their activation, changes occur in the metabolic mechanisms of the cells themselves.
Mechanism of metabolic adaptations in activated HSCs
HSCs are the primary fibrogenic cell population in the liver. In their quiescent state, HSCs function as the liver's retinol-storing pericytes and regulate sinusoidal blood flow during homeostasis. However, in response to liver injury, various damage-associated, inflammatory, and metabolic signals trigger the loss of retinol droplets, leading to transdifferentiation into a phenotype resembling that of myofibroblasts and subsequent activation of HSCs. This activated phenotype is characterized by increased migration, contractility, proliferation, and inflammatory signaling, as well as heightened production of extracellular matrix components. Ultimately, these changes result in abnormal extracellular matrix deposition, contributing to the development of fibrosis and cirrhosis. Significant metabolic reprogramming is necessary to facilitate the energy-intensive phenotypic shift toward a myofibroblast-like state, characterized by enhanced glycolysis, mobilization of lipid droplets, cholesterol metabolism, and activation of stress response pathways (Figure 5).
Metabolic reprogramming in fibrogenic HSCs activation in chronic liver diseases. A, HSCs are activated by exosomes containing GLUT1 and PKM2. The overexpression of glucose transporters GLUT1, GLUT2, and GLUT4 facilitates excessive glucose uptake by HSCs, leading to increased glycolysis. Pyruvate, a byproduct of PKM2-catalyzed glycolysis, is largely diverted toward lactate synthesis, resulting in lactate accumulation within HSCs, despite the increased expression of the lactate transporter monocarboxylate transporter 4 (MCT4) and enhanced lactate efflux. In a separate metabolic pathway, pyruvate is converted to acetyl-CoA, which enhances the activity of the TCA cycle and releases lipids that are stored for β-oxidation. PKM2 regulates the expression of metabolic genes, which elevates ROS production and overall mitochondrial activity. Additionally, glutamine enters the cell via the ASCT1 and is metabolized to produce glutamate and α-ketoglutarate, which are subsequently integrated into the TCA cycle. Hedgehog signaling through leptin represents a significant pro-fibrotic signal that may reprogram glucose metabolism, primarily converging on the transcription factor HIF-1α. Since AGEs result from hyperglycemia, TGF-β signaling enhances HSC expression of RAGE, rendering activated HSCs more susceptible to global glucose metabolism. B, The lipid metabolism of activated HSCs is characterized by the loss of retinyl ester-containing cytoplasmic droplets. HSCs utilize enhanced fatty acid β-oxidation to degrade the contents of lipid droplets within the autolysosome. Lipid droplets are transported to the autolysosome, where they undergo degradation. The contents of lipid droplets are degraded in the autolysosome under LAL activity, and REH activity is also increased in the lysosome, releasing free retinol into the extracellular space. SREBP-1c and PPAR-γ are adipogenic markers of quiescent HSCs that are downregulated upon activation. The conversion of some intracellular retinol to retinoic acid facilitates elevated transcription of RARβ and RXRβ. C, Free cholesterol in activated HSCs is partially absorbed through PCSK9, which interacts with low-density LRP5 and LDLR, promoting the degradation of endosomes and lysosomes. Free cholesterol enhances the expression of TLR4 and is converted into cholesterol esters through ACAT1, sensitizing HSCs to TGF-β signaling. oxLDL increases the expression of profibrogenic genes such as TNF-α and IL-1β through the pJNK. D, Metabolic stress responses in HSCs lead to the conversion of latent TGF-β into activated TGF-β. Unfolded proteins and TGF-β signaling trigger ER stress responses via IRE1α, resulting in increased expression of fibrogenic genes through ASK1/JNK signaling and the canonical splicing of XBP1 into its active form. This process promotes the expression of C/EBPβ, COL1A1, TGF-β, and XBP1 in the cell nucleus, leading to increased collagen accumulation. TGF-β signaling activates NADPH oxidase in a self-replicating cycle, producing H2O2, which promotes fibrogenic gene expression and activates latent TGF-β in extracellular spaces. Reduced nuclear receptor signaling, such as that of LXR, FXR, or THRα, is a hallmark of HSC activation. Nevertheless, active HSCs also exhibit increased PPARβ/δ, which stimulates HSC proliferation. ACAT1, acyl-CoA:cholesterol acyltransferase 1; ADAM17, ADAM metallopeptidase domain 17; AGE, advanced glycation end product; ASCT2, alanine serine cysteine transporter 2; ASK1, apoptosis-signal-regulating kinase 1; C/EBPβ, CCAAT-enhancer-binding protein; COL1A1, collagen type 1 alpha 1; DNL, de novo lipogenesis; FXR, farnesoid X receptor; GLUT, glucose transporter protein 1; HIF-1α, hypoxia-inducible factor 1-α; H2O2, hydrogen peroxide; HSCs, hepatic stellate cells; IL, interleukin; IRE1α, inositol-requiring enzyme 1 alpha; LD, lipid droplet; LDL, low-density lipoprotein; LDLR, low-density lipoprotein receptor; LRP5, low-density lipoprotein-related protein 5; LXR, liver X receptor; MCT4, monocarboxylate transporter 4; NOX, NADPH oxidase; oxLDL, oxidized low-density lipoprotein; PCSK9, proprotein convertase subtilisin/kexin 9; PKM2, pyruvate kinase M2; PPAR, peroxisome proliferator-activated receptor; RAGE, receptor for advanced glycation end product; RARα/β, retinoic acid receptor α/β; REH, retinyl ester hydrolase; ROS, reactive oxygen species; SREBP-1c, sterol regulatory element-binding protein-1c; TGF, transforming growth factor; THR, thyroid hormone receptors; TLR4, toll-like receptor 4; TNF, tumor necrosis factor; TREM2, triggering receptor expressed on myeloid cells 2; XBP1s, IRE1α-X-box binding protein 1.
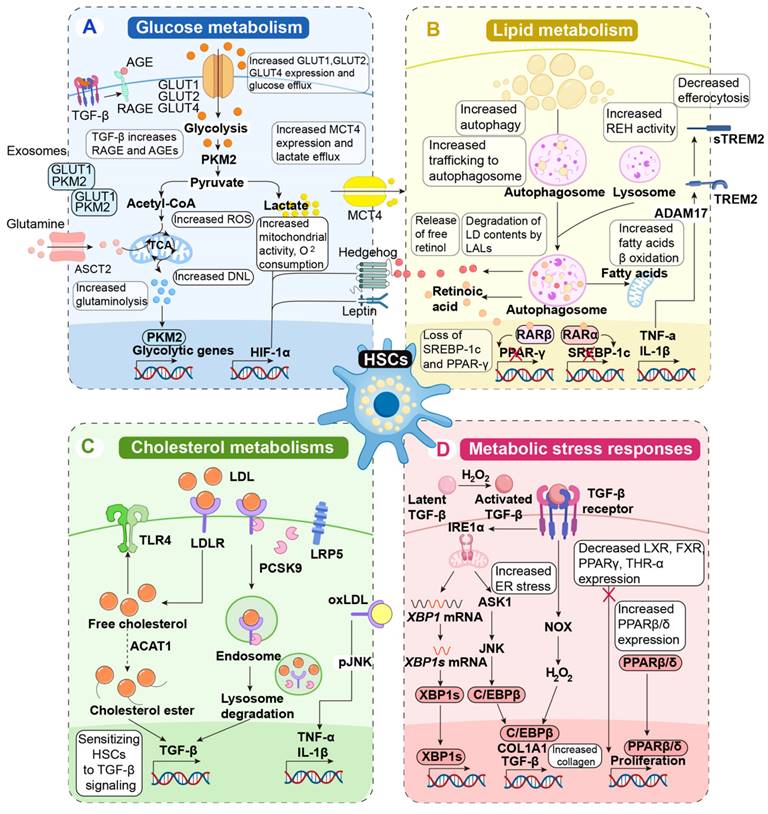
Glucose metabolism
The activation of HSCs primarily depends on the reprogramming of their glucose metabolism. Notably, alterations in glucose metabolism not only define the myofibroblast phenotype but also contribute to HSC activation [164].
In comparison to their quiescent counterparts, activated HSCs exhibit higher rates of glucose utilization, enhanced glucose transport capabilities, and increased glycolytic activity in response to elevated extracellular glucose levels or purinergic signaling [165]. This phenomenon is attributed to the upregulation of mRNA expression of glucose transporters, including glucose transporter protein 1 (GLUT1), GLUT2, and GLUT4 [166]. Notably, liver cancer cells, which primarily generate ATP through anaerobic metabolism, also demonstrate significant overexpression of GLUT1. GLUT1 represents a rate-limiting step in ATP synthesis [167]. Additionally, activated HSCs show increased mRNA expression of rate-limiting glycolytic enzymes, including hexokinase-2 (HK2), pyruvate kinase M2 (PKM2), and fructose-2,6-bisphosphatase-3 (PFKFB3) [168]. For example, the ablation of HK2 in HSCs completely inhibits CCl4-induced liver fibrosis in mice [169]. The activation of HSCs and the formation of fibrosis may be mitigated by blocking the nuclear translocation of PKM2, which is essential for metabolic transition in HSCs and resembles the metabolic reprogramming processes observed in liver macrophages. Furthermore, exosomes produced by activated HSCs contain GLUT1 and PKM2 proteins [170]. Hypoxia-inducible factor 1-α (HIF-1α) signaling enhances the production of exosomes by activated HSCs, particularly under hypoxic and inflammatory conditions [171].
The increased glycolysis observed in HSCs during cultivation is accompanied by the depletion of central carbon metabolites from the citric acid cycle. The activation of HSCs is characterized by high expression levels of pyruvate dehydrogenase kinase 3 (PDK3), which inhibits the conversion of pyruvate to acetyl coenzyme A (acetyl-CoA), thereby directing pyruvate toward lactate synthesis. Lactate plays a crucial role in HSC activation and the maintenance of myofibroblast phenotypes. Despite the upregulation of the lactate export pump, monocarboxylate transporter 4 (MCT4), the concentrations of lactate within activated HSCs remain elevated. Notably, inhibiting the intracellular accumulation of lactate reduces cell proliferation, decreases the expression of genes associated with the myofibroblast signature, and promotes lipid accumulation and lipogenic transcription [172].
Signaling pathways involving the interaction of hedgehog and leptin receptors play a crucial role in regulating glucose metabolism in HSCs [173]. Hedgehog signaling is increasingly recognized for its involvement in tissue repair following injury, with hepatocytes producing hedgehog ligands in response to hepatic damage. These ligands activate nearby HSCs through the expression of the HIF-1α gene, which modulates energy metabolism by activating specific genes, including those encoding glucose transporters and glycolytic enzymes [174]. Hypoxia triggers oxygen-independent ATP production processes, resulting in HIF-1α-mediated regulation of glucose metabolism, similar to the Warburg effect observed in activated HSCs [175]. Furthermore, activated HSCs enhance the activity of glutaminase, the rate-limiting enzyme responsible for the hydrolytic deamination of glutamine to glutamate, through hedgehog signaling. Recent findings suggest that targeting the amino acid transporter alanine-serine-cysteine transporter 2 (ASCT2) may significantly impact glutaminolysis in HSCs. In humans with the HSC line LX-2, pharmacological depletion of ASCT2 resulted in decreased levels of glutamate and α-ketoglutarate, which subsequently limited the rate of oxygen intake and HSC activation [176].
HSCs are also activated by advanced glycation end products (AGEs), which are formed through non-enzymatic interactions between proteins and sugars [177]. The stimulation of murine HSCs in culture by TGF-β leads to the development of the receptor for advanced glycation end products (RAGE), an effect that may be mitigated by inhibiting the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathway [178]. Cultured human HSCs exhibit increased expression of fibrogenic genes and produce higher levels of ROS when grown in a medium containing AGEs [179]. In vivo studies demonstrate that rats receiving intraperitoneal AGE injections develop more severe liver fibrosis following bile duct ligation compared to those not receiving AGE injections. Although AGE treatment is not directly associated with fibrosis, it does elevate the levels of the protein α-SMA. This research suggests that AGEs and hyperglycemia alone may not be sufficient to induce fibrosis. However, they could exacerbate fibrogenesis triggered by other factors in the presence of tissue damage [180].
Changes in the glucose metabolism of HSCs are also influenced by epigenetic regulation [181]. The histone methyltransferase G9a and DNA methyltransferase 1 (DNMT1) induce epigenetic modifications that shift HSC metabolism toward increased glycolysis. The profibrotic cytokine TGF-β stimulates the recruitment of these enzymes to chromatin in human HSCs [182]. Furthermore, in vitro models of hypoxia-driven and TGF-β1-driven activation demonstrate that the simultaneous inhibition of DNMT1 and G9a restores glycolytic rates to those observed in quiescent HSCs [183].
Lipid metabolism
Quiescent HSCs are characterized by the presence of vitamin A-rich lipid droplets, and the maintenance of this quiescent state requires both vitamin A metabolism and insulin signaling. Shortly after activation, HSCs release their lipid droplets, and over time, their lipid metabolism undergoes significant changes [184]. The elimination of lipid droplets through autophagy appears to be essential for HSC activation, and this metabolically demanding cellular response may depend on this process (Figure 5) [185]. Intracellular retinyl ester hydrolases (REHs) exhibit increased activity upon HSC activation, which subsequently triggers the release of retinol without a corresponding increase in the release of retinyl esters (REs). The absence of extracellular REH activity in activated HSCs supports the hypothesis that intracellular hydrolysis is responsible for retinol depletion [186]. Bile salt-independent REHs, including patatin-like phospholipase domain-containing protein 3, adipose triglyceride lipase, and lysosomal acid lipase (LAL), are recognized as enzymatic mediators of RE release from lipid droplets [187]. Notably, hormone-sensitive lipase, a key REH in liver adipocytes, is not significantly produced by HSCs and has minimal impact on lipid mobilization in these cells [188]. Activated HSCs demonstrate increased hydrolase activity alongside a diminished capacity for retinol esterification, as HSC activation leads to a rapid decrease in lecithin-retinol acyltransferase expression. Retinol is thought to play a crucial role in maintaining the quiescent phenotype of HSCs through its interactions with the nuclear receptors retinoic acid receptor (RARβ) and retinoid X receptor α (RXRα). This phenomenon is primarily mediated by sterol regulatory element-binding protein-1c (SREBP-1c) and PPARγ, two adipogenic transcription factors that reduce their expression [189].
De novo lipogenesis (DNL) suppression, achieved through the inhibition of 1-aminocyclopropane-1-carboxylate (ACC) or fatty acid synthetase (FASN), is currently being investigated as a potential treatment for steatotic liver diseases. Studies in murine models have demonstrated that this approach can reduce hepatic fibrosis [190]. Notably, multiple mechanisms contribute to these effects beyond the enhancement of hepatocyte metabolism. HSCs also exhibit increased expression of lipogenic pathway activity [191]. When DNL is inhibited via ACC1/2 or FASN suppression, fibrogenic gene expression is significantly diminished in vitro in primary HSCs derived from both rats and humans [192, 193]. This reduction is associated with decreased oxidative phosphorylation and glycolytic flow. However, the role of lipogenic pathways in non-metabolic hepatic fibrosis has not yet been established. Stearoyl-CoA desaturase-1 (SCD1) is the rate-limiting enzyme that converts saturated fatty acids into monounsaturated fatty acids and serves as a key regulator of fatty acid metabolic pathways [190]. In both human and animal models of hepatic fibrosis, activated HSCs express SCD1, which promotes both liver fibrosis and liver cancer. Furthermore, metabolism-induced HSC activation triggers the production of ADAM metallopeptidase domain 17 (ADAM17), which cleaves the cell surface TREM2 protein. This process activates receptors expressed on myeloid cells and encourages HSC activation by inhibiting efferocytosis in the presence of TNF-α and IL-1β [194].
Cholesterol metabolism
Cholesterol contributes to the pathophysiology of fibrosis formation in MASH because the accumulation of free cholesterol may activate HSCs [195]. Moreover, MASH can be more effectively induced in mice that are fed a cholesterol-rich diet [196]. When free cholesterol accumulates, TLR4 is upregulated in HSCs, making the cells more susceptible to activation by TGF-β [197]. The low-density lipoprotein receptor (LDLR) and miR-33a, a microRNA that regulates cholesterol metabolism, are both elevated upon HSC activation and influence the accumulation of free cholesterol in these cells. The endosomal-lysosomal degradation of TLR4 is impeded by signals mediated by LDLR and miR-33a. Consequently, this reduces the levels of bone morphogenetic protein and activin membrane-bound inhibitor, a pseudoreceptor for TGF-β, thereby increasing the susceptibility of HSCs to TGF-β [198].
The significance of acyl-CoA:cholesterol acyltransferase 1 (ACAT1), the enzyme responsible for catalyzing the conversion of free cholesterol into cholesterol esters, has been extensively studied [199]. The in vitro effects of free cholesterol on HSCs are significantly exacerbated, and liver fibrosis induced by CCl4 and bile duct ligation in mice is intensified by HSC-specific deletion of ACAT1. This pathway is dependent on the overexpression of TLR4, as the detrimental effects of ACAT1 deletion are mitigated in mice with conditional TLR4 deletion [200]. Additionally, due to its complex formation with proprotein convertase subtilisin/kexin 9 (PCSK9), LDL-related protein 5 (LRP5) partially mediates the uptake of low-density lipoprotein (LDL) cholesterol in HSCs [201]. Further research is needed to ascertain whether this cholesterol uptake pathway plays a role in HSC activation.
HSCs are rapidly activated by oxidized low-density lipoprotein (oxLDL). The uptake of oxLDL by HSCs is facilitated by scavenger receptors, specifically CD36 and lectin-like oxidized LDL receptor-1 (LOX-1). This process enhances the expression of profibrogenic genes, such as TNF-α and IL-1β, through the phosphorylation of c-Jun N-terminal kinases (JNK). Canonical Wnt signaling, recognized as an HSC activator, promotes the upregulation of LOX-1 expression [202].
Metabolic stress responses
Metabolic stress responses, which encompass elevated oxidative stress, activation of the unfolded protein response (UPR), autophagy, ferroptosis, senescence, and nuclear receptor signaling, are associated with the activation of HSCs.
Oxidative stress
Increased oxidative stress, a hallmark of activated HSCs, is crucial for collagen synthesis and fibrogenic activation [203]. The MAPK/ERK pathway is activated by several traditional HSC activators, such as TGF-β and PDGF, to the production of NADPH oxidase (NOX) enzymes [204]. Hydrogen peroxide (H2O2), produced by NOX enzymes, plays multiple roles in liver fibrogenesis. Additionally, H2O2 facilitates the binding of CCAAT-enhancer-binding protein (C/EBPβ) to its binding sites in the type I collagen and TGF-β1 promoter, promoting collagen generation and increasing levels of fibrogenic TGF-β1. These alterations in the redox state, characterized by elevated levels of ROS, are essential for the activation of latent TGF-β [205]. Consequently, in mouse models of liver disease, suppression or deletion of NOX enzymes reduces the progression of fibrosis and mitigates fibrotic responses in HSCs [206]. Furthermore, a recent study has demonstrated that ROS generation induced by excessive iron in HSCs in the MASH model promotes fibrogenic HSC activation. This effect can be reversed by N-acetylcysteine, an antioxidant therapy [207].
β-oxidation activity has been observed to be elevated during HSC activation. Liver HSCs significantly enhance the activity of carnitine palmitoyltransferase 1A (CPT1A), the rate-limiting enzyme in the β-oxidation of medium- and long-chain fatty acids, in both human and murine models of CCl4-induced and metabolism-driven liver fibrosis. The progression of fibrosis can be mitigated through genetic suppression, pharmacological inhibition of CPT1A, and HSC-specific knockdown of CPT1A in mice with choline-deficient models of liver fibrosis [36].
The UPR and ER stress
One of the initial steps in HSC activation and hepatic fibrosis is the generation of UPR and ER stress [208]. However, the ER stress pathway and the UPR alone are insufficient to fully activate HSCs. Evidence for this is provided by the reduced fibrosis formation observed in HSCs that overexpress the chaperone glucose-regulated protein, indicating that these processes are necessary for adequate HSC activation [209]. Furthermore, stimulation with TGF-β1 can directly induce the phosphorylation of inositol-requiring protein 1α (IRE1α), which promotes the activation of the TGF-β1 downstream effector transcription factor C/EBPβ in LX-2 cells through apoptosis-signal-regulating kinase 1 (ASK1) and the phosphorylation of JNK [210]. In parallel, TGF-β-induced overexpression of transport and Golgi organization 1, which facilitates HSC secretion of type I collagen, is mediated via the IRE1α-X-box binding protein 1 (XBP1s) pathway [211].
Autophagy, ferroptosis, and senescence
Autophagy, regulated by IRE1α-activated XBP1s, is closely associated with the activation of the ER stress pathway in HSCs [212]. Additionally, macrophages may promote HSC autophagy. For instance, LAMs produce prostaglandin E2 (PGE2), which triggers HSC autophagy through its receptor, the PGE2 receptor 4 (EP4) [34]. Autophagy in HSCs mobilizes lipid droplets, correlating with a reduction in retinol storage in stellate cells and an increase in the rate of β-oxidation [213]. These findings indicate that autophagy serves as an energy source for HSC activation. Consequently, inhibiting autophagy by suppressing the PGE2 receptor EP4 reduces liver fibrosis induced by methionine-choline deficiency. Moreover, HSC-specific genetic deletion of autophagy-related genes (ATG)5 or ATG7 contributes to the inhibition of fibrogenic activation and decreases fibrosis in murine models of liver fibrosis induced by CCl4 and thioacetamide. Furthermore, the loss of ATG7 negates the effects of IRE1α-XBP1 signaling, establishing autophagy as the primary facilitator of ER-stress-driven HSC activation [214].
Programmed cell death, which include apoptosis, pyroptosis, necroptosis, cuproptosis, ferroptosis, and PANoptosis, occurs in various cell types and has distinct effects on hepatic fibrosis [215]. Activated apoptotic proteases, such as caspase-3 and caspase-7, lead to the formation of apoptotic bodies, which can activate HSCs either directly or indirectly by stimulating macrophages. Hepatocyte death through mechanisms such as necroptosis, pyroptosis, ferroptosis, cuproptosis, and PANoptosis results in the aggregation of macrophages, monocytes, and DCs, leading to the release of DAMPs and inflammatory factors that further exacerbate the inflammatory response. Ferroptosis occurs in hepatocytes and promotes the formation of fibrosis. However, in HSCs, it exerts an anti-fibrotic effect. Hepatic fibrosis in mice, induced by bile duct ligation or CCl4, can be alleviated in vivo by the HSC-specific loss of ELAV-like RNA binding protein 1, a transcript essential for HSC ferroptosis, as well as by treatment with the ferroptosis inducers sorafenib or the anti-malarial medication dihydroartemisinin. Interestingly, sorafenib-induced ferroptosis does not occur in hepatocytes or macrophages, suggesting that it is exclusive to HSCs. Chronic iron overload, however, continues to stimulate hepatic fibrogenesis while inducing ferroptosis in both HSCs and hepatocytes.
The senescence of HSCs, a form of cell-cycle arrest in which the cells remain metabolically active, has been observed during the development of hepatic fibrosis and is initially associated with HSC deactivation [216]. However, through the senescence-associated secretory phenotype, which promotes inflammation and creates a pro-tumoral liver environment, HSC senescence appears to be a significant source of inflammatory and fibrogenic signals during the later stages of fibrosis. Senescent HSCs, identified by the expression of the urokinase plasminogen activator receptor (uPAR), are derived from active HSCs in both human and mouse models of MASLD-related fibrosis, according to advanced single-cell analyses. Although it may not be entirely specific to senescent HSCs, the regulation of senescence by uPAR-specific senolytic chimeric antigen receptor T cells has been demonstrated in mouse models of hepatic fibrosis [217]. Furthermore, hepatic fibrosis is reduced in both biliary and diet-induced mouse models of advanced hepatic fibrosis when cell-type-specific induction of senescence in HSCs is achieved through the conditional deletion of yes-associated protein (YAP) 1 [11]. This finding is consistent with previous research indicating that medication-induced suppression of YAP reduces CCl4-induced hepatic fibrosis in mice. However, other studies have found that YAP suppression has an opposing effect in ischemia-reperfusion injury, exacerbating hepatic fibrosis and delaying liver regeneration [218]. Therefore, before developing specific treatment strategies, it is essential to further elucidate the precise role of HSC senescence in the progression of hepatic fibrosis.
Nuclear receptors signaling
The expression levels of nuclear receptors in HSCs are relatively low, and their role in HSC activation remains unclear. While HSCs do not express PPARα and PPARγ, they do express PPARβ. The activation of HSCs leads to an increase in PPARβ expression, which, in turn, stimulates HSC proliferation in response to CCl4-induced hepatic fibrosis [219].
Liver X receptor (LXR) ligands inhibit HSC activation in in vitro studies, indicating that LXR-deficient mice are more susceptible to methionine and choline deficiency, as well as CCl4-induced hepatic dysfunction. The liver pathology observed in LXR mutant mice is not altered by bone marrow transplantation from wild-type donors, suggesting that HSCs are indeed the mediating factor for these effects. The absence of LXR modifies how HSCs process lipids and metabolize retinoid compounds, resulting in larger lipid droplets and an enhanced capacity to respond to retinoic acid. This ultimately drives HSCs toward a more activated state [220].
FXR is expressed at lower levels in HSC and hepatic myofibroblasts. Endogenous FXR ligands have been shown to reduce the fibrogenic response of HSCs, but this effect occurs only in the presence of a small heterodimer partner, known as small heterodimer partner (SHP) [221]. A different study finds no effect on HSC activation when rat or human HSC models are treated with obeticholic acid, a synthetic BA and FXR agonist. FXR signaling is predominantly observed in quiescent HSCs and diminishes upon fibrogenic activation [222]. The activation of HSCs is further exacerbated by whole-body FXR deletion [223]. The activation of HSCs is exacerbated by whole-body FXR deletion [220]. Despite this, obeticholic acid appears to have minimal impact on the activation process. Therefore, the role of this bile acid receptor in HSC activation must be elucidated through targeted manipulation of FXR activity in HSCs.
Thyroid hormone receptors (THR)-α and β are nuclear receptors primarily activated by thyroid hormones (THs) to mediate their cellular responses [224]. While THRβ is the mechanism through which THs exert their positive metabolic effects in hepatocytes, THRα is the predominant isoform in human and murine HSCs. THRα functions by inhibiting TGF-β signaling and fibrogenic activation in HSCs [225].
Therapeutic implications
Targeting liver inflammation and fibrosis
HSC metabolism is closely linked to the fibrotic response and hepatic inflammation. Currently, no medications targeting inflammatory or fibrogenic pathways have received regulatory approval for the treatment of liver damage. Several drugs, including the dual CCR2/5 inhibitor cenicriviroc and the ASK1 inhibitor selonsertib, have failed to demonstrate a reduction in fibrosis during Phase III clinical trials. This lack of efficacy is due to the complex nature of the underlying disease processes, which involve intricate interactions between inflammation, fibrogenic HSC activation, and hepatocellular damage [226]. Consequently, preventing liver injury is likely essential for therapeutic effectiveness, as targeting a single pathway downstream in the disease cascade may be insufficient. The significant effectiveness of substantial weight loss following bariatric surgery is evidenced by the fact that 56% of individuals achieve the primary objective of MASH resolution without worsening fibrosis at the one-year follow-up, compared to only 16% in the lifestyle intervention group [227]. Given that the metabolic responses of distinct cell types to liver damage overlap, there is an opportunity to target multiple cell types simultaneously and upstream in the disease cascade.
The development of innovative therapeutic options must take into account the etiology of the underlying liver disease. Given that macrophages play a significant role in the fibrosis associated with MAFLD and MASH, which have emerged as the most prevalent liver pathologies lacking effective treatment options worldwide, the majority of strategies discussed here focus on myeloid cells. However, different cell types may preferentially target other fibrotic liver diseases. For example, addressing T cell and other lymphocyte deficiencies in chronic hepatitis B virus (HBV) infection may represent the optimal treatment strategy to stimulate viral clearance and subsequently repair liver damage [228]. Nevertheless, targeting macrophages may also prove beneficial. In animal models of HBV infection, subsets of Kupffer cells may circumvent the tolerogenic potential of the liver environment by enhancing liver T cell immunity through the detection of IL-2 and the cross-presentation of hepatocellular antigens [229]. While managing auto-aggressive effector T cell responses requires further refinement, current techniques aim to increase the pool and functionality of Treg. T cells are also a significant focus in the treatment of autoimmune hepatitis [230]. Controlling the accumulation or activity of neutrophils presents a potential therapeutic avenue for MetALD, as neutrophil activity is a major characteristic that correlates with liver inflammation and drives the progression of liver injury [231].
The treatment of fibrosis regression is currently under investigation, with a focus on facilitating the conversion of activated HSCs to quiescent HSCs and promoting the degradation of the extracellular matrix. This process may be regulated by the interactions between neutrophils and macrophages [8]. Neutrophils can induce macrophages to switch to a restorative phenotype, which promotes liver tissue regeneration. In a study involving the accumulation of pro-inflammatory monocyte, the use of CCL2 inhibitors in mice demonstrates a transition of liver macrophages toward a recovery phenotype, thereby accelerating fibrosis regression [232]. In a modest study involving nine individuals with compensated hepatic cirrhosis, the adoptive transfer of ex vivo differentiated restorative macrophages improved hepatic fibrosis in animal models and marginally decreased the end-stage liver disease score [233]. Although this strategy appears safe in a small pilot study involving patients, it remains unclear whether it will be clinically effective and sustainable in the long term for the macrophage phenotype [234]. Mesenchymal stromal cells (MSCs) have been investigated for adoptive cell therapy due to their immunoregulatory properties. Clinical trials are currently being conducted on mice with liver disorders to determine whether MSCs derived from bone marrow and MSC-derived extracellular vesicles can reduce liver inflammation and fibrosis [235]. Preclinical models demonstrate a reduction in hepatic fibrosis following the adoptive transfer of chimeric antigen receptor T cells that target senescent hepatocytes via the senescence-associated urokinase-type plasminogen activator receptor [217].
The primary objective of therapies targeting the fibrogenesis process is to reduce or inhibit the activation of HSCs. TGF-β is considered a key target due to its significant role in HSC activation. However, pan-TGF-β blockade can lead to adverse effects, such as autoimmunity, because of its numerous systemic activities. These negative outcomes have resulted in the termination of several clinical trials involving monoclonal antibodies against TGF-β1 [236, 237]. A more prudent approach may involve regulating TGF-β locally at the site of action. Integrins containing αV subunits can activate latent TGF-β in the extracellular matrix, and the removal of this component has been shown to protect animals from fibrosis in various organs [238, 239]. Although there is currently limited information regarding the efficacy of αV integrin inhibitors in humans, several drugs, including abituzumab, a pan-αV-binding antibody, are under investigation [240].
Targeting glucose and lipid metabolism
One of the hallmarks of HSC activation is the increased glycolysis and the activation of lipogenic pathways, which reflect alterations in HSC metabolism associated with liver damage. Therefore, targeting these pathways may represent a promising strategy for treating hepatic injury, and several medications are currently undergoing preclinical or clinical research. Inhibiting rate-limiting glycolytic enzymes, such as hexokinase isoforms, is relatively straightforward, and this approach has demonstrated anti-fibrotic efficacy in animal models. PF-06835919 acts as an inhibitor of a rate-limiting enzyme in fructose metabolism and is currently in clinical trials for the treatment of multiple system lipodystrophies [241-243]. Emerging data suggest that PKM2, a key regulator that induces the Warburg effect and serves as a rate-limiting glycolytic enzyme, is a suitable therapeutic target. In vitro studies indicate that the allosteric PKM2 activator TEPP-46 positively impacts hepatic inflammation and fibrosis by stabilizing its tetrameric complexes and preventing nuclear translocation [244, 245]. Additionally, several medications targeting lipogenic pathways are currently under clinical investigation. In a human phase IIa trial, therapeutic suppression of ACC1 effectively reduces hepatic steatosis, fibrogenic gene expression, and liver inflammation in mouse models of MASH [246]. In this therapeutic context, blocking SCD1 is also being tested in patients with MASH, although it has resulted in a non-significant reduction in liver fat. However, no discernible effects on liver fibrosis or inflammation have been observed [247]. Another intriguing strategy involves the stimulation of AMP-activated protein kinase, a cellular energy sensor that inhibits anabolic pathways such as lipogenesis and may also confer beneficial cardiac effects [248, 249].
Targeting metabolic stress responses
Metabolic stress responses, including ER stress, the UPR, autophagy, ferroptosis, and nuclear receptor signaling, represent distinct therapeutic targets. Inhibiting IRE1α and its downstream mediator, caspase-2, has been shown to protect against liver steatosis and inflammation in animal models [250]. However, transient and low-quality activation of the ER stress system serves as an adaptive response in overweight individuals, and complete hepatic blockade can exacerbate liver damage. This consideration must be addressed when developing pharmaceutical therapies. The unique roles of autophagy and ferroptosis in HSCs may limit their applicability as treatment targets and necessitate further investigation. Deletion of ATG7 increases insulin resistance and ER stress, while overexpression of ATG5 extends longevity. Additionally, pharmacological strategies that activate autophagy have demonstrated protective effects against liver fibrosis, indicating that autophagy exerts a beneficial influence [251].
Special attention must be given to adverse reactions when addressing metabolic pathways, as drug activity is not limited to specific organs. For instance, PPAR agonists can effectively promote the morphology of anti-inflammatory macrophages and inhibit HSC activation. However, their clinical application is currently restricted due to cardiovascular issues, osteopenia, edema, fluid retention, and other adverse effects [252]. Emerging drug delivery strategies may enable cell-type-specific modulation of metabolic processes, potentially reducing undesirable off-target effects. For example, dendrimer-graphene nanostars have successfully delivered the non-thiazolidinedione PPARδ agonist GW1929 to macrophages in a mouse model of CCl4-induced liver damage, shifting macrophage morphologies toward an anti-inflammatory, alternative phenotype and significantly preventing fibrosis formation [253].
Nuclear receptor activation is widely regarded as the most advanced strategy for targeting HSC immunometabolism in liver fibrosis. Research indicates that the benefits of FXR agonists are primarily mediated through their effects on myeloid cells [254]. Obeticholic acid, an FXR agonist, is approved as a second-line therapy for primary biliary cholangitis [255], and both it and other FXR agonists, such as vonafexor are being investigated as treatments for MASLD fibrosis [256]. However, the research and development of obeticholic acid as an anti-fibrotic treatment for MASH has been halted, as the FDA did not grant fast-track approval based on the existing evidence of its effectiveness and safety. Some trials of vonafexor have reported increased mild to moderate pruritus and elevated LDL cholesterol levels, necessitating further exploration and larger trials to address the unmet medical need in MASH [257]. PPAR agonists, particularly those targeting PPARβ/δ, are well recognized for their efficacy in inducing anti-inflammatory macrophage polarization [258]. Lanifibranor, a pan-PPAR agonist, is in late-stage clinical development and has shown positive results in a Phase IIb trial [259]. Similarly, LXR activation promotes anti-inflammatory macrophage activation while suppressing HSC activation [260]. Additionally, the THRβ agonist resmetirom has recently been granted expedited approval by the FDA for the treatment of MASH fibrosis based on favorable findings from the registrational phase 3 MAESTRO-NASH study [261]. THRα is the predominant THR isoform in HSCs and innate immune cells, such as macrophages. Consequently, resmetirom exhibits both anti-inflammatory and anti-fibrotic properties, primarily mediated by hepatic metabolic changes rather than direct immune and metabolic actions [262]. Since resmetirom is now an approved drug for MASH, other THR beta agonists, such as sobetirome, eprotirome, and VK2809, will need to be evaluated to determine which drugs perform best in combination with resmetirom. Future real-world studies will also be necessary to assess the added benefits of ad-hoc combinations of resmetirom in patients with chronic liver diseases.
Future perspectives
HSC activation has opened up remarkable opportunities for novel therapies in patients with chronic liver diseases. The transition of HSCs from a quiescent state to a perpetuated phenotype is primarily characterized by fibrogenesis, contractility, proliferation, altered matrix degradation, chemotaxis, and immunological and inflammatory signaling. Equally important, various cell types—including hepatocytes, macrophages, biliary epithelial cells, liver progenitor cells, LESCs, NK cells, NKT cells, platelets, B cells, MAIT cells, γδ T cells, neutrophils, mast cells, and innate lymphoid cells—interact with HSCs to influence the progression of chronic liver diseases. Moreover, chronic liver diseases impact the metabolic and functional states of HSCs, which are largely regulated by glucose, lipid, and cholesterol metabolism, oxidative stress, UPR activation, autophagy, ferroptosis, senescence, and nuclear receptor activity. Therefore, a comprehensive understanding of the metabolic processes accompanying these differentiation pathways will be essential for developing targeted therapies.
Currently, numerous drugs targeting liver inflammation and fibrosis, as well as glucose and lipid metabolism and metabolic stress responses in HSC activation, are being investigated for the treatment of chronic liver diseases. For example, FXR, THR-β, and PPAR agonists regulate glucose, lipid, and bile acid metabolism, which positively influence chronic liver conditions such as MAFLD and MASH. By integrating emerging and state-of-the-art approaches, including single-cell [263] and molecule-resolution genome-wide molecular characterization [264], along with spatial (multi) metabolomics techniques [265], we may overcome the limitations of current strategies. This integration offers a comprehensive and multidimensional understanding of HSC metabolic processes, intercellular communication networks, and their relationships with metabolic cell states and disruptions in the steady-state cellular niche. Furthermore, in recent years, our understanding of cellular interactions in chronic liver disease, as well as the consequences of therapeutic targeting, has significantly expanded and extensively reviewed elsewhere. However, how intracellular metabolic regulation, especially in HSCs, largely enters these circuits remains to be determined.
Overall, it is crucial to attain a comprehensive understanding of the mechanisms that govern intracellular metabolic regulation of HSC in order to enhance diagnosis and treatment.
Abbreviations
ACAT1: acyl-CoA:cholesterol acyltransferase 1; ACC: 1-aminocyclopropane-1-carboxylate; acetyl-CoA: acetyl coenzyme A; ADAM17: ADAM metallopeptidase domain 17; ADP: adenosine diphosphate; AGE: advanced glycation end product; ASCT2: alanine serine cysteine transporter 2; ASK1: apoptosis-signal-regulating kinase 1; ATG5/7: autophagy-related 5/7; ATP: adenosine 5' triphosphate; BA: bile acid; BMDMs: bone marrow-derived monocytes; CCL: C-C motif ligand; CCl4: carbon tetrachloride; CCR: CC chemokine receptor; C/EBPβ: CCAAT-enhancer-binding protein; COL1A1: collagen type 1 alpha 1; CPT1A: carnitine palmitoyl transferase 1A; CTGF: connective tissue growth factor; CXCL: C-X-C motif ligand; CX3CR1: CX3C chemokine receptor 1; CXCR: C-X-C chemokine receptor; DAMPs: damage-associated molecular patterns; DCs: dendritic cells; DNL: de novo lipogenesis; DNMT: DNA methyltransferase; ECM: extracellular matrix; EGF: epidermal growth factor; emKCs: embryonic KCs; ERK: extracellular signal-regulated kinase; ET1: endothelin-1; FASL: Fas ligand; FASN: fatty acid synthetase; FFA: free fatty acids; FGF: fibroblast growth factor; FGFR1: fibroblast growth factor receptor 1; FXR: farnesoid X receptor; GATA: GATA-binding factor; GLUT: glucose transporter protein 1; HBV: hepatitis B virus; HGF: hepatocyte growth factor; HIF-1α: hypoxia-inducible factor 1-α; HK2: hexokinase-2; HMGB1: high-mobility group box 1; H2O2: hydrogen peroxide; HSCs: hepatic stellate cells; 5-HT: 5-hydroxytryptamine; IFN: interferon; IGF1: insulin-like growth factor 1; IGFBP5: insulin-like growth factor-binding protein-5; IL: interleukin; ILCs: innate lymphoid cells; IRE1α: inositol-requiring enzyme 1 alpha; JNK: c-Jun N-terminal kinases; LAL: lysosomal acid lipase; LAMs: lipid-associated macrophages; LD: lipid droplet; LDL: low-density lipoprotein; LDLR: low-density lipoprotein receptor; LOX-1: lectin-like oxidized LDL receptor-1; LPS: lipopolysaccharide; LRP5: low-density lipoprotein-related protein 5; LSECs: liver sinusoidal endothelial cells; LXR: liver X receptor; MAIT: Mucosal-associated invariant T; MASH: metabolic dysfunction-associated steatohepatitis; MASLD: metabolic dysfunction-associated steatotic liver disease; MAPK: mitogen-activated protein kinases; MetALD: metabolic dysfunction-associated alcoholic liver disease; MCT4: monocarboxylate transporter 4; MCP1: monocyte chemoattractant protein 1; MDR2: multidrug resistance protein 2; MMP: Matrix metalloproteinase-2; MoMF: monocyte-derived macrophage; MSCs: mesenchymal stromal cells; NK: natural killer; NKG2D: NK receptor group 2 member D; NKT: natural killer T; NLRP3: NACHT: LRR and PYD domainscontaining protein 3; NOS: nitric oxide ynthase; NOX: NADPH oxidase; oxLDL: oxidized low-density lipoprotein; PAMP: pathogen-associated molecular pattern; PCSK9: proprotein convertase subtilisin/kexin 9; pDCs: plasmacytoid dendritic cells; PDGF: platelet-derived growth factor; PDGFRβ: platelet-derived growth factor receptor β; PDK3: pyruvate dehydrogenase kinase 3; PFKFB3: fructose-2,6-bi-sphosphatase-3; PGE2: prostaglandin E2; PKM2: pyruvate kinase M2; PPAR: peroxisome proliferator-activated receptor; RAGE: receptor for advanced glycation end product; RARα/β: retinoic acid receptor α/β; REs: retinyl esters; REH: retinyl ester hydrolase; ROS: reactive oxygen species; RXRα: retinoid X receptor α; SAM: scar-associated macrophage; SCD1: stearoyl-CoA desaturase-1; SHP: small heterodimer partner; α-SMA: α-smooth muscle actin; SREBP-1c: sterol regulatory element-binding protein-1c; TCA: Trichloroacetic acid; TCF21: transcription factor 21; TGF: transforming growth factor; TGF-βRII: TGF-β receptor II; TGR5: Takeda G protein-coupled receptor 5; TH: T helper; THR: thyroid hormone receptors; THs: thyroid hormones; TLR: toll-like receptor; TNF: tumor necrosis factor; Treg: regulatory T; TREM2: triggering receptor expressed on myeloid cells 2; TRAIL: tumour necrosis factor-related apoptosis-inducing ligand; uPAR: urokinase plasminogen activated receptor; UPR: unfolded protein response; UTC: unconventional T cell; VEGF: vascular endothelial growth factor; XBP1s: IRE1α-X-box binding protein 1; YAP: yes-associated protein.
Acknowledgements
Funding
This work is supported by the National High Level Hospital Clinical Research Fund (Peking University First Hospital Science and Technology Achievement Transformation and Incubation Guidance Fund) grant 2023IR06, as well as the National Science Foundation of China grants No. 82073935 and 82274024.
Author contributions
Mengyao Yan performed reference searches, wrote the manuscript and created the tables and figures. Yimin Cui supervised, reviewed and edited the manuscript. Qian Xiang conceptualized, reviewed and edited the manuscript. The final version of the work has been read and approved by all authors.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Horn P, Tacke F. Metabolic reprogramming in liver fibrosis. Cell Metab. 2024;36:1439-55
2. Bogomolova A, Balakrishnan A, Ott M, Sharma AD. "The good, the bad, and the ugly" - about diverse phenotypes of hepatic stellate cells in the liver. Cell Mol Gastroenterol Hepatol. 2024;17:607-22
3. Breitkopf-Heinlein K, Martinez-Chantar ML. Targeting hepatic stellate cells to combat liver fibrosis: where do we stand? Gut. 2024;73:1411-3
4. Sun YD, Zhang H, Li YM, Han JJ. Abnormal metabolism in hepatic stellate cells: Pandora's box of MAFLD related hepatocellular carcinoma. Biochim Biophys Acta Rev Cancer. 2024;1879:189086
5. Bourebaba N, Marycz K. Hepatic stellate cells role in the course of metabolic disorders development - A molecular overview. Pharmacol Res. 2021;170:105739
6. Rani R, Gandhi CR. Stellate cell in hepatic inflammation and acute injury. J Cell Physiol. 2023;238:1226-36
7. Rinella ME, Lazarus JV, Ratziu V, Francque SM, Sanyal AJ, Kanwal F. et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. J Hepatol. 2023;79:1542-56
8. Hammerich L, Tacke F. Hepatic inflammatory responses in liver fibrosis. Nat Rev Gastroenterol Hepatol. 2023;20:633-46
9. Ezhilarasan D. Hepatic stellate cells in the injured liver: Perspectives beyond hepatic fibrosis. J Cell Physiol. 2022;237:436-49
10. Dewidar B, Meyer C, Dooley S, Meindl-Beinker AN. TGF-beta in hepatic stellate cell activation and liver fibrogenesis-updated 2019. Cells. 2019;8:1419
11. Du K, Maeso-Diaz R, Oh SH, Wang E, Chen T, Pan C. et al. Targeting YAP-mediated HSC death susceptibility and senescence for treatment of liver fibrosis. Hepatology. 2023;77:1998-2015
12. Cheng S, Zou Y, Zhang M, Bai S, Tao K, Wu J. et al. Single-cell RNA sequencing reveals the heterogeneity and intercellular communication of hepatic stellate cells and macrophages during liver fibrosis. MedComm (2020). 2023;4:e378
13. Poisson J, Lemoinne S, Boulanger C, Durand F, Moreau R, Valla D. et al. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J Hepatol. 2017;66:212-27
14. Meyer J, Balaphas A, Fontana P, Morel P, Robson SC, Sadoul K. et al. Platelet interactions with liver sinusoidal endothelial cells and hepatic stellate cells lead to hepatocyte proliferation. Cells. 2020;9:1243
15. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397-411
16. Carter JK, Friedman SL. Hepatic stellate cell-immune interactions in NASH. Front Endocrinol (Lausanne). 2022;13:867940
17. Liu Y, Zheng Y, Yang Y, Liu K, Wu J, Gao P. et al. Exosomes in liver fibrosis: The role of modulating hepatic stellate cells and immune cells, and prospects for clinical applications. Front Immunol. 2023;14:1133297
18. Zhang Z, Yao Z, Wang L, Ding H, Shao J, Chen A. et al. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy. 2018;14:2083-103
19. Xia S, Wang Z, Chen L, Zhou Y, Li Y, Wang S. et al. Dihydroartemisinin regulates lipid droplet metabolism in hepatic stellate cells by inhibiting lncRNA-H19-induced AMPK signal. Biochem Pharmacol. 2021;192:114730
20. Chang ML, Yang SS. Metabolic signature of hepatic fibrosis: from individual pathways to systems biology. Cells. 2019;8:1423
21. Cogliati B, Yashaswini CN, Wang S, Sia D, Friedman SL. Friend or foe? The elusive role of hepatic stellate cells in liver cancer. Nat Rev Gastroenterol Hepatol. 2023;20:647-61
22. Yin C, Evason KJ, Asahina K, Stainier DY. Hepatic stellate cells in liver development, regeneration, and cancer. J Clin Invest. 2013;123:1902-10
23. Kamm DR, McCommis KS. Hepatic stellate cells in physiology and pathology. J Physiol. 2022;600:1825-37
24. Lu JL, Yu CX, Song LJ. Programmed cell death in hepatic fibrosis: current and perspectives. Cell Death Discov. 2023;9:449
25. Batlle E, Massague J. Transforming growth factor-beta signaling in immunity and cancer. Immunity. 2019;50:924-40
26. Chen W. TGF-beta regulation of T cells. Annu Rev Immunol. 2023;41:483-512
27. Xia Y, Wang Y, Xiong Q, He J, Wang H, Islam M. et al. Neutrophil extracellular traps promote MASH fibrosis by metabolic reprogramming of HSC. Hepatology. 2024
28. You H, Wang X, Ma L, Zhang F, Zhang H, Wang Y. et al. Insights into the impact of hepatitis B virus on hepatic stellate cell activation. Cell Commun Signal. 2023;21:70
29. Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH. et al. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med. 2013;19:1617-24
30. Yokosaki Y, Nishimichi N. New Therapeutic Targets for Hepatic Fibrosis in the Integrin Family, alpha8beta1 and alpha11beta1, Induced Specifically on Activated Stellate Cells. Int J Mol Sci. 2021;22:12794
31. Gao J, Wei B, de Assuncao TM, Liu Z, Hu X, Ibrahim S. et al. Hepatic stellate cell autophagy inhibits extracellular vesicle release to attenuate liver fibrosis. J Hepatol. 2020;73:1144-54
32. Chen QT, Zhang ZY, Huang QL, Chen HZ, Hong WB, Lin T. et al. HK1 from hepatic stellate cell-derived extracellular vesicles promotes progression of hepatocellular carcinoma. Nat Metab. 2022;4:1306-21
33. Mederacke I, Filliol A, Affo S, Nair A, Hernandez C, Sun Q. et al. The purinergic P2Y14 receptor links hepatocyte death to hepatic stellate cell activation and fibrogenesis in the liver. Sci Transl Med. 2022;14:eabe5795
34. Cao Y, Mai W, Li R, Deng S, Li L, Zhou Y. et al. Macrophages evoke autophagy of hepatic stellate cells to promote liver fibrosis in NAFLD mice via the PGE2/EP4 pathway. Cell Mol Life Sci. 2022;79:303
35. Trivedi P, Wang S, Friedman SL. The power of plasticity-metabolic regulation of hepatic stellate cells. Cell Metab. 2021;33:242-57
36. Fondevila MF, Fernandez U, Heras V, Parracho T, Gonzalez-Rellan MJ, Novoa E. et al. Inhibition of carnitine palmitoyltransferase 1A in hepatic stellate cells protects against fibrosis. J Hepatol. 2022;77:15-28
37. Bai Y, Chen J, Zhang S, Xu G, Mao Z, Ding Y. et al. Inflammation-responsive cell membrane-camouflaged nanoparticles against liver fibrosis via regulating endoplasmic reticulum stress and oxidative stress. Adv Mater. 2024;36:e2310443
38. De Smet V, Eysackers N, Merens V, Kazemzadeh Dastjerd M, Halder G, Verhulst S. et al. Initiation of hepatic stellate cell activation extends into chronic liver disease. Cell Death Dis. 2021;12:1110
39. Gu L, Zhao C, Wang Y, Wang C, Yin X, Ye Q. et al. Senescence of hepatic stellate cells by specific delivery of manganese for limiting liver fibrosis. Nano Lett. 2024;24:1062-73
40. de Oliveira da Silva B, Ramos LF, Moraes KCM. Molecular interplays in hepatic stellate cells: apoptosis, senescence, and phenotype reversion as cellular connections that modulate liver fibrosis. Cell Biol Int. 2017;41:946-59
41. Weiskirchen R, Tacke F. Cellular and molecular functions of hepatic stellate cells in inflammatory responses and liver immunology. Hepatobiliary Surg Nutr. 2014;3:344-63
42. Boyer-Diaz Z, Aristu-Zabalza P, Andres-Rozas M, Robert C, Ortega-Ribera M, Fernandez-Iglesias A. et al. Pan-PPAR agonist lanifibranor improves portal hypertension and hepatic fibrosis in experimental advanced chronic liver disease. J Hepatol. 2021;74:1188-99
43. Cheng Y, Zhu X, Cheng F, Ji L, Zhou Y. Delta-like homolog1/GATA binding protein 2 axis mediates leptin inhibition of PPARgamma2 expression in hepatic stellate cells in vitro. Life Sci. 2018;192:183-9
44. Guan W, Cheng F, Wu H, Cao Q, Zhu X, Fan Y. et al. GATA binding protein 3 is correlated with leptin regulation of PPARgamma1 in hepatic stellate cells. J Cell Mol Med. 2017;21:568-78
45. Cheng Z, Chu H, Seki E, Lin R, Yang L. Hepatocyte programmed cell death: the trigger for inflammation and fibrosis in metabolic dysfunction-associated steatohepatitis. Front Cell Dev Biol. 2024;12:1431921
46. Li Y, Zhang T, Zhang J, Liu Q, Jia Q, Chen W. et al. Dually fibronectin/CD44-mediated nanoparticles targeted disrupt the Golgi apparatus and inhibit the hedgehog signaling in activated hepatic stellate cells to alleviate liver fibrosis. Biomaterials. 2023;301:122232
47. An P, Wei LL, Zhao S, Sverdlov DY, Vaid KA, Miyamoto M. et al. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat Commun. 2020;11:2362
48. Charan HV, Dwivedi DK, Khan S, Jena G. Mechanisms of NLRP3 inflammasome-mediated hepatic stellate cell activation: therapeutic potential for liver fibrosis. Genes Dis. 2023;10:480-94
49. Gaul S, Leszczynska A, Alegre F, Kaufmann B, Johnson CD, Adams LA. et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J Hepatol. 2021;74:156-67
50. Tan Z, Liu Q, Jiang R, Lv L, Shoto SS, Maillet I. et al. Interleukin-33 drives hepatic fibrosis through activation of hepatic stellate cells. Cell Mol Immunol. 2018;15:388-98
51. Tan F, Li X, Wang Z, Li J, Shahzad K, Zheng J. Clinical applications of stem cell-derived exosomes. Signal Transduct Target Ther. 2024;9:17
52. Wang Z, Du K, Jin N, Tang B, Zhang W. Macrophage in liver fibrosis: identities and mechanisms. Int Immunopharmacol. 2023;120:110357
53. Chu PS, Nakamoto N, Ebinuma H, Usui S, Saeki K, Matsumoto A. et al. C-C motif chemokine receptor 9 positive macrophages activate hepatic stellate cells and promote liver fibrosis in mice. Hepatology. 2013;58:337-50
54. Tada Y, Kasai K, Makiuchi N, Igarashi N, Kani K, Takano S. et al. Roles of macrophages in advanced liver fibrosis, identified using a newly established mouse model of diet-induced non-alcoholic steatohepatitis. Int J Mol Sci. 2022;23:13251
55. Zhang S, Wan D, Zhu M, Wang G, Zhang X, Huang N. et al. CD11b + CD43 hi Ly6C lo splenocyte-derived macrophages exacerbate liver fibrosis via spleen-liver axis. Hepatology. 2023;77:1612-29
56. Baeck C, Wei X, Bartneck M, Fech V, Heymann F, Gassler N. et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice. Hepatology. 2014;59:1060-72
57. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N. et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut. 2012;61:416-26
58. Guilliams M, Scott CL. Liver macrophages in health and disease. Immunity. 2022;55:1515-29
59. Beattie L, Sawtell A, Mann J, Frame TCM, Teal B, de Labastida Rivera F. et al. Bone marrow-derived and resident liver macrophages display unique transcriptomic signatures but similar biological functions. J Hepatol. 2016;65:758-68
60. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S. et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun. 2016;7:10321
61. Zhou L, Wang M, Guo H, Hou J, Zhang Y, Li M. et al. Integrated analysis highlights the immunosuppressive role of TREM2(+) macrophages in hepatocellular carcinoma. Front Immunol. 2022;13:848367
62. Timperi E, Gueguen P, Molgora M, Magagna I, Kieffer Y, Lopez-Lastra S. et al. Lipid-associated macrophages are induced by cancer-associated fibroblasts and mediate immune suppression in breast cancer. Cancer Res. 2022;82:3291-306
63. Guilliams M, Bonnardel J, Haest B, Vanderborght B, Wagner C, Remmerie A. et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell. 2022;185:379-96 e38
64. Qu J, Wang L, Li Y, Li X. Liver sinusoidal endothelial cell: An important yet often overlooked player in the liver fibrosis. Clin Mol Hepatol. 2024;30:303-25
65. Hammoutene A, Rautou PE. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J Hepatol. 2019;70:1278-91
66. Xie G, Wang X, Wang L, Wang L, Atkinson RD, Kanel GC. et al. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology. 2012;142:918-27 e6
67. Duan JL, Ruan B, Yan XC, Liang L, Song P, Yang ZY. et al. Endothelial Notch activation reshapes the angiocrine of sinusoidal endothelia to aggravate liver fibrosis and blunt regeneration in mice. Hepatology. 2018;68:677-90
68. Ding BS, Nolan DJ, Butler JM, James D, Babazadeh AO, Rosenwaks Z. et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature. 2010;468:310-5
69. Ding BS, Cao Z, Lis R, Nolan DJ, Guo P, Simons M. et al. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature. 2014;505:97-102
70. Huebert RC, Shah VH. Sinusoidal endothelial cells direct traffic at the intersection of regeneration and fibrosis. Hepatology. 2014;60:754-6
71. Borkham-Kamphorst E, Herrmann J, Stoll D, Treptau J, Gressner AM, Weiskirchen R. Dominant-negative soluble PDGF-beta receptor inhibits hepatic stellate cell activation and attenuates liver fibrosis. Lab Invest. 2004;84:766-77
72. Ghafoory S, Varshney R, Robison T, Kouzbari K, Woolington S, Murphy B. et al. Platelet TGF-beta1 deficiency decreases liver fibrosis in a mouse model of liver injury. Blood Adv. 2018;2:470-80
73. Yoshida S, Ikenaga N, Liu SB, Peng ZW, Chung J, Sverdlov DY. et al. Extrahepatic platelet-derived growth factor-beta, delivered by platelets, promotes activation of hepatic stellate cells and biliary fibrosis in mice. Gastroenterology. 2014;147:1378-92
74. Gao L, Zhuang Y, Liu Z. Nogo-B silencing expedites the senescence of platelet-derived growth factor-BB-induced human hepatic stellate cells via autophagy. Mol Biotechnol. 2024
75. Wang R, Liu F, Chen P, Li S, Gu Y, Wang L. et al. Gomisin D alleviates liver fibrosis through targeting PDGFRbeta in hepatic stellate cells. Int J Biol Macromol. 2023;235:123639
76. Wang B, Yang L, Yuan X, Zhang Y. Roles and therapeutic targeting of dendritic cells in liver fibrosis. J Drug Target. 2024;32:647-54
77. Xu Y, Liu F, He D, Han L, Zheng X, Hu M. et al. Monocyte-derived immature dendritic cells negatively regulate hepatic stellate cells in vitro by secreting IL-10. Immunobiology. 2023;228:152315
78. Mo C, Xie S, Zeng T, Lai Y, Huang S, Zhou C. et al. Ginsenoside-Rg1 acts as an IDO1 inhibitor, protects against liver fibrosis via alleviating IDO1-mediated the inhibition of DCs maturation. Phytomedicine. 2021;84:153524
79. Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH. et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58:1461-73
80. Heier EC, Meier A, Julich-Haertel H, Djudjaj S, Rau M, Tschernig T. et al. Murine CD103(+) dendritic cells protect against steatosis progression towards steatohepatitis. J Hepatol. 2017;66:1241-50
81. Xu F, Liu C, Dong Y, Wu W, Xu J, Yan Y. et al. Ablation of Cbl-b and c-Cbl in dendritic cells causes spontaneous liver cirrhosis via altering multiple properties of CD103(+) cDC1s. Cell Death Discov. 2022;8:142
82. Bernsmeier C, Albano E. Liver dendritic cells and NAFLD evolution: a remaining open issue. J Hepatol. 2017;66:1120-2
83. Wu Y, Li Q, Yan Y, Hao Y, Wang C, Liu B. et al. Gel-mediated recruitment of conventional type 1 dendritic cells potentiates the therapeutic effects of radiotherapy. Biomaterials. 2024;305:122470
84. Nellore A, Zumaquero E, Seifert M. T-bet + B cells in humans: protective and pathologic functions. Transplantation. 2024;108:1709-14
85. Longhi MS, Zhang L, Mieli-Vergani G, Vergani D. B and T cells: (Still) the dominant orchestrators in autoimmune hepatitis. Autoimmun Rev. 2024;23:103591
86. Northfield JW, Kasprowicz V, Lucas M, Kersting N, Bengsch B, Kim A. et al. CD161 expression on hepatitis C virus-specific CD8+ T cells suggests a distinct pathway of T cell differentiation. Hepatology. 2008;47:396-406
87. Dudek M, Pfister D, Donakonda S, Filpe P, Schneider A, Laschinger M. et al. Auto-aggressive CXCR6(+) CD8 T cells cause liver immune pathology in NASH. Nature. 2021;592:444-9
88. Zhang M, Zhang S. T Cells in Fibrosis and Fibrotic Diseases. Front Immunol. 2020;11:1142
89. Breuer DA, Pacheco MC, Washington MK, Montgomery SA, Hasty AH, Kennedy AJ. CD8(+) T cells regulate liver injury in obesity-related nonalcoholic fatty liver disease. Am J Physiol Gastrointest Liver Physiol. 2020;318:G211-G24
90. Hirose M, Leliavski A, de Assis LVM, Matveeva O, Skrum L, Solbach W. et al. Chronic inflammation disrupts circadian rhythms in splenic CD4+ and CD8+ T cells in mice. Cells. 2024;13:151
91. Burtis AEC, DeNicola DMC, Ferguson ME, Santos RG, Pinilla C, Kriss MS. et al. Ag-driven CD8+ T cell clonal expansion is a prominent feature of MASH in humans and mice. Hepatology. 2024
92. Koda Y, Teratani T, Chu PS, Hagihara Y, Mikami Y, Harada Y. et al. CD8(+) tissue-resident memory T cells promote liver fibrosis resolution by inducing apoptosis of hepatic stellate cells. Nat Commun. 2021;12:4474
93. Kokubo K, Onodera A, Kiuchi M, Tsuji K, Hirahara K, Nakayama T. Conventional and pathogenic Th2 cells in inflammation, tissue repair, and fibrosis. Front Immunol. 2022;13:945063
94. Kang SG, Lee SE, Choi MJ, Chang JY, Kim JT, Zhang BY. et al. Th2 cytokines increase the expression of fibroblast growth factor 21 in the liver. Cells. 2021;10:1298
95. Rolla S, Alchera E, Imarisio C, Bardina V, Valente G, Cappello P. et al. The balance between IL-17 and IL-22 produced by liver-infiltrating T-helper cells critically controls NASH development in mice. Clin Sci (Lond). 2016;130:193-203
96. Ma HY, Yamamoto G, Xu J, Liu X, Karin D, Kim JY. et al. IL-17 signaling in steatotic hepatocytes and macrophages promotes hepatocellular carcinoma in alcohol-related liver disease. J Hepatol. 2020;72:946-59
97. Xiang X, Feng D, Hwang S, Ren T, Wang X, Trojnar E. et al. Interleukin-22 ameliorates acute-on-chronic liver failure by reprogramming impaired regeneration pathways in mice. J Hepatol. 2020;72:736-45
98. Arab JP, Sehrawat TS, Simonetto DA, Verma VK, Feng D, Tang T. et al. An open-label, dose-escalation study to assess the safety and efficacy of IL-22 agonist F-652 in patients with alcohol-associated hepatitis. Hepatology. 2020;72:441-53
99. Liu L, Yan H, Wang Y, Xie Y, Jiang L, Zhao J. et al. Decreased absolute number of peripheral regulatory T cells in patients with idiopathic retroperitoneal fibrosis. Front Immunol. 2022;13:1012513
100. Nga HT, Moon JS, Tian J, Lee HY, Kim SH, Lee YS. et al. Interleukin-10 attenuates liver fibrosis exacerbated by thermoneutrality. Front Med (Lausanne). 2021;8:672658
101. Zhang X, Feng M, Liu X, Bai L, Kong M, Chen Y. et al. Persistence of cirrhosis is maintained by intrahepatic regulatory T cells that inhibit fibrosis resolution by regulating the balance of tissue inhibitors of metalloproteinases and matrix metalloproteinases. Transl Res. 2016;169:67-79 e1-2
102. Barrow F, Khan S, Wang H, Revelo XS. The emerging role of B cells in the pathogenesis of NAFLD. Hepatology. 2021;74:2277-86
103. Kotsiliti E, Leone V, Schuehle S, Govaere O, Li H, Wolf MJ. et al. Intestinal B cells license metabolic T-cell activation in NASH microbiota/antigen-independently and contribute to fibrosis by IgA-FcR signalling. J Hepatol. 2023;79:296-313
104. Chen R, Liu F, Xia L, Che N, Tian Y, Cao Y. et al. B10 cells decrease fibrosis progression following cardiac injury partially by IL-10 production and regulating hyaluronan secretion. J Leukoc Biol. 2022;111:415-25
105. Ottenhoff THM, Joosten SA. Mobilizing unconventional T cells. Science. 2019;366:302-3
106. Mehta H, Lett MJ, Klenerman P, Filipowicz Sinnreich M. MAIT cells in liver inflammation and fibrosis. Semin Immunopathol. 2022;44:429-44
107. Wang H, Park O, Gao B. NKT cells in liver fibrosis: controversies or complexities. J Hepatol. 2011;55:1166 author reply -7
108. Li H, Ding P, Peng B, Ming YZ. Cross-talk between hepatic stellate cells and T lymphocytes in liver fibrosis. Hepatobiliary Pancreat Dis Int. 2021;20:207-14
109. Nilsson J, Hornberg M, Schmidt-Christensen A, Linde K, Nilsson M, Carlus M. et al. NKT cells promote both type 1 and type 2 inflammatory responses in a mouse model of liver fibrosis. Sci Rep. 2020;10:21778
110. East JE, Kennedy AJ, Webb TJ. Raising the roof: The preferential pharmacological stimulation of Th1 and th2 responses mediated by NKT cells. Med Res Rev. 2014;34:45-76
111. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C. et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657-67
112. Shi J, Zhao J, Zhang X, Cheng Y, Hu J, Li Y. et al. Activated hepatic stellate cells impair NK cell anti-fibrosis capacity through a TGF-beta-dependent emperipolesis in HBV cirrhotic patients. Sci Rep. 2017;7:44544
113. Liepelt A, Wehr A, Kohlhepp M, Mossanen JC, Kreggenwinkel K, Denecke B. et al. CXCR6 protects from inflammation and fibrosis in NEMO(LPC-KO) mice. Biochim Biophys Acta Mol Basis Dis. 2019;1865:391-402
114. Bottcher K, Rombouts K, Saffioti F, Roccarina D, Rosselli M, Hall A. et al. MAIT cells are chronically activated in patients with autoimmune liver disease and promote profibrogenic hepatic stellate cell activation. Hepatology. 2018;68:172-86
115. Mabire M, Hegde P, Hammoutene A, Wan J, Caer C, Sayegh RA. et al. MAIT cell inhibition promotes liver fibrosis regression via macrophage phenotype reprogramming. Nat Commun. 2023;14:1830
116. Hammerich L, Bangen JM, Govaere O, Zimmermann HW, Gassler N, Huss S. et al. Chemokine receptor CCR6-dependent accumulation of gammadelta T cells in injured liver restricts hepatic inflammation and fibrosis. Hepatology. 2014;59:630-42
117. Cho Y, Szabo G. Two Faces of Neutrophils in Liver Disease Development and Progression. Hepatology. 2021;74:503-12
118. Calvente CJ, Tameda M, Johnson CD, Del Pilar H, Lin YC, Adronikou N. et al. Neutrophils contribute to spontaneous resolution of liver inflammation and fibrosis via microRNA-223. J Clin Invest. 2019;129:4091-109
119. Maurer M, Koberle M, Metz M, Biedermann T. Mast cells: Promoters of health and modulators of disease. J Allergy Clin Immunol. 2019;144:S1-S3
120. Xu X, Rivkind A, Pikarsky A, Pappo O, Bischoff SC, Levi-Schaffer F. Mast cells and eosinophils have a potential profibrogenic role in Crohn disease. Scand J Gastroenterol. 2004;39:440-7
121. Kennedy L, Meadows V, Sybenga A, Demieville J, Chen L, Hargrove L. et al. Mast cells promote nonalcoholic fatty liver disease phenotypes and microvesicular steatosis in mice fed a western diet. Hepatology. 2021;74:164-82
122. Kyritsi K, Kennedy L, Meadows V, Hargrove L, Demieville J, Pham L. et al. Mast cells induce ductular reaction mimicking liver injury in mice through mast cell-derived transforming growth factor beta 1 signaling. Hepatology. 2021;73:2397-410
123. Jones H, Hargrove L, Kennedy L, Meng F, Graf-Eaton A, Owens J. et al. Inhibition of mast cell-secreted histamine decreases biliary proliferation and fibrosis in primary sclerosing cholangitis Mdr2(-/-) mice. Hepatology. 2016;64:1202-16
124. Srivastava RK, Sapra L, Bhardwaj A, Mishra PK, Verma B, Baig Z. Unravelling the immunobiology of innate lymphoid cells (ILCs): Implications in health and disease. Cytokine Growth Factor Rev. 2023;74:56-75
125. Mikami Y, Takada Y, Hagihara Y, Kanai T. Innate lymphoid cells in organ fibrosis. Cytokine Growth Factor Rev. 2018;42:27-36
126. Weng HL, Feng DC, Radaeva S, Kong XN, Wang L, Liu Y. et al. IFN-gamma inhibits liver progenitor cell proliferation in HBV-infected patients and in 3,5-diethoxycarbonyl-1,4-dihydrocollidine diet-fed mice. J Hepatol. 2013;59:738-45
127. Kleczka A, Mazur B, Tomaszek K, Gabriel A, Dzik R, Kabala-Dzik A. Association of NK Cells with the severity of fibrosis in patients with chronic hepatitis C. Diagnostics (Basel). 2023;13:2187
128. Peng Y, Huang K, Shen L, Tao YY, Liu CH. Cultured mycelium cordyceps sinensis allevi notates CCl4-induced liver inflammation and fibrosis in mice by activating hepatic natural killer cells. Acta Pharmacol Sin. 2016;37:204-16
129. Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. 2020;20:95-112
130. Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol. 2013;59:583-94
131. Boller T, Felix G. A renaissance of elicitors: perception of microbe-associated molecular patterns and danger signals by pattern-recognition receptors. Annu Rev Plant Biol. 2009;60:379-406
132. Knorr J, Kaufmann B, Inzaugarat ME, Holtmann TM, Geisler L, Hundertmark J. et al. Interleukin-18 signaling promotes activation of hepatic stellate cells in mouse liver fibrosis. Hepatology. 2023;77:1968-82
133. He Y, Hwang S, Ahmed YA, Feng D, Li N, Ribeiro M. et al. Immunopathobiology and therapeutic targets related to cytokines in liver diseases. Cell Mol Immunol. 2021;18:18-37
134. Forkel M, Berglin L, Kekalainen E, Carlsson A, Svedin E, Michaelsson J. et al. Composition and functionality of the intrahepatic innate lymphoid cell-compartment in human nonfibrotic and fibrotic livers. Eur J Immunol. 2017;47:1280-94
135. Xun Z, Yao X, Ou Q. Emerging roles of bile acids in chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma. Cell Mol Immunol. 2023;20:1087-9
136. Keitel V, Haussinger D. Role of TGR5 (GPBAR1) in liver disease. Semin Liver Dis. 2018;38:333-9
137. Deutschmann K, Reich M, Klindt C, Droge C, Spomer L, Haussinger D. et al. Bile acid receptors in the biliary tree: TGR5 in physiology and disease. Biochim Biophys Acta Mol Basis Dis. 2018;1864:1319-25
138. Nie Q, Luo X, Wang K, Ding Y, Jia S, Zhao Q. et al. Gut symbionts alleviate MASH through a secondary bile acid biosynthetic pathway. Cell. 2024;187:2717-34 e33
139. Boesch M, Lindhorst A, Feio-Azevedo R, Brescia P, Silvestri A, Lannoo M. et al. Adipose tissue macrophage dysfunction is associated with a breach of vascular integrity in NASH. J Hepatol. 2024;80:397-408
140. Yan M, Man S, Sun B, Ma L, Guo L, Huang L. et al. Gut liver brain axis in diseases: the implications for therapeutic interventions. Signal Transduct Target Ther. 2023;8:443
141. Lopez-Yus M, Horndler C, Borlan S, Bernal-Monterde V, Arbones-Mainar JM. Unraveling adipose tissue dysfunction: Molecular mechanisms, novel biomarkers, and therapeutic targets for liver fat feposition. Cells. 2024;13:380
142. Jain H, Kumar A, Almousa S, Mishra S, Langsten KL, Kim S. et al. Characterisation of LPS+ bacterial extracellular vesicles along the gut-hepatic portal vein-liver axis. J Extracell Vesicles. 2024;13:e12474
143. Ralli T, Saifi Z, Tyagi N, Vidyadhari A, Aeri V, Kohli K. Deciphering the role of gut metabolites in non-alcoholic fatty liver disease. Crit Rev Microbiol. 2023;49:815-33
144. Sinha SR, Haileselassie Y, Nguyen LP, Tropini C, Wang M, Becker LS. et al. Dysbiosis-induced secondary bile acid deficiency promotes intestinal inflammation. Cell Host Microbe. 2020;27:659-70 e5
145. Xu H, Fang F, Wu K, Song J, Li Y, Lu X. et al. Gut microbiota-bile acid crosstalk regulates murine lipid metabolism via the intestinal FXR-FGF19 axis in diet-induced humanized dyslipidemia. Microbiome. 2023;11:262
146. Zhou J, Huang N, Guo Y, Cui S, Ge C, He Q. et al. Combined obeticholic acid and apoptosis inhibitor treatment alleviates liver fibrosis. Acta Pharm Sin B. 2019;9:526-36
147. Chang JS. Recent insights into the molecular mechanisms of simultaneous fatty acid oxidation and synthesis in brown adipocytes. Front Endocrinol (Lausanne). 2023;14:1106544
148. Monteiro LB, Prodonoff JS, Favero de Aguiar C, Correa-da-Silva F, Castoldi A, Bakker NVT. et al. Leptin signaling suppression in macrophages improves immunometabolic outcomes in obesity. Diabetes. 2022;71:1546-61
149. Geng Y, Wang J, Serna-Salas SA, Villanueva AH, Buist-Homan M, Arrese M. et al. Hepatic stellate cells induce an inflammatory phenotype in Kupffer cells via the release of extracellular vesicles. J Cell Physiol. 2023;238:2293-303
150. Wallace SJ, Tacke F, Schwabe RF, Henderson NC. Understanding the cellular interactome of non-alcoholic fatty liver disease. JHEP Rep. 2022;4:100524
151. Czaja AJ. Review article: chemokines as orchestrators of autoimmune hepatitis and potential therapeutic targets. Aliment Pharmacol Ther. 2014;40:261-79
152. Ambade A, Lowe P, Kodys K, Catalano D, Gyongyosi B, Cho Y. et al. Pharmacological inhibition of CCR2/5 signaling prevents and reverses alcohol-induced liver damage, steatosis, and inflammation in mice. Hepatology. 2019;69:1105-21
153. Nellen A, Heinrichs D, Berres ML, Sahin H, Schmitz P, Proudfoot AE. et al. Interference with oligomerization and glycosaminoglycan binding of the chemokine CCL5 improves experimental liver injury. PLoS One. 2012;7:e36614
154. Ni Y, Zhuge F, Ni L, Nagata N, Yamashita T, Mukaida N. et al. CX3CL1/CX3CR1 interaction protects against lipotoxicity-induced nonalcoholic steatohepatitis by regulating macrophage migration and M1/M2 status. Metabolism. 2022;136:155272
155. Van Raemdonck K, Van den Steen PE, Liekens S, Van Damme J, Struyf S. CXCR3 ligands in disease and therapy. Cytokine Growth Factor Rev. 2015;26:311-27
156. Zeremski M, Dimova R, Brown Q, Jacobson IM, Markatou M, Talal AH. Peripheral CXCR3-associated chemokines as biomarkers of fibrosis in chronic hepatitis C virus infection. J Infect Dis. 2009;200:1774-80
157. Romano A, Hou X, Sertorio M, Dessein H, Cabantous S, Oliveira P. et al. FOXP3+ regulatory T cells in hepatic fibrosis and splenomegaly caused by schistosoma japonicum: the spleen may be a major source of Tregs in subjects with splenomegaly. PLoS Negl Trop Dis. 2016;10:e0004306
158. Wang FT, Wu TQ, Lin Y, Jiao YR, Li JY, Ruan Y. et al. The role of the CXCR6/CXCL16 axis in the pathogenesis of fibrotic disease. Int Immunopharmacol. 2024;132:112015
159. Mossanen JC, Kohlhepp M, Wehr A, Krenkel O, Liepelt A, Roeth AA. et al. CXCR6 inhibits hepatocarcinogenesis by promoting natural killer T- and CD4(+) T-cell-dependent control of senescence. Gastroenterology. 2019;156:1877-89 e4
160. Wehr A, Baeck C, Heymann F, Niemietz PM, Hammerich L, Martin C. et al. Chemokine receptor CXCR6-dependent hepatic NK T Cell accumulation promotes inflammation and liver fibrosis. J Immunol. 2013;190:5226-36
161. Zhu H, Zhang Q, Chen G. CXCR6 deficiency ameliorates ischemia-reperfusion injury by reducing the recruitment and cytokine production of hepatic NKT cells in a mouse model of non-alcoholic fatty liver disease. Int Immunopharmacol. 2019;72:224-34
162. Affo S, Morales-Ibanez O, Rodrigo-Torres D, Altamirano J, Blaya D, Dapito DH. et al. CCL20 mediates lipopolysaccharide induced liver injury and is a potential driver of inflammation and fibrosis in alcoholic hepatitis. Gut. 2014;63:1782-92
163. Bonacchi A, Petrai I, Defranco RM, Lazzeri E, Annunziato F, Efsen E. et al. The chemokine CCL21 modulates lymphocyte recruitment and fibrosis in chronic hepatitis C. Gastroenterology. 2003;125:1060-76
164. Lee KC, Wu PS, Lin HC. Pathogenesis and treatment of non-alcoholic steatohepatitis and its fibrosis. Clin Mol Hepatol. 2023;29:77-98
165. Yin KL, Li M, Song PP, Duan YX, Ye WT, Tang W. et al. Unraveling the emerging niche role of hepatic stellate cell-derived exosomes in liver diseases. J Clin Transl Hepatol. 2023;11:441-51
166. Yan N. A glimpse of membrane transport through structures-advances in the structural biology of the GLUT glucose transporters. J Mol Biol. 2017;429:2710-25
167. Kang J, Heart E, Sung CK. Effects of cellular ATP depletion on glucose transport and insulin signaling in 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab. 2001;280:E428-35
168. Du D, Liu C, Qin M, Zhang X, Xi T, Yuan S. et al. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm Sin B. 2022;12:558-80
169. Rho H, Terry AR, Chronis C, Hay N. Hexokinase 2-mediated gene expression via histone lactylation is required for hepatic stellate cell activation and liver fibrosis. Cell Metab. 2023;35:1406-23 e8
170. Wan L, Xia T, Du Y, Liu J, Xie Y, Zhang Y. et al. Exosomes from activated hepatic stellate cells contain GLUT1 and PKM2: a role for exosomes in metabolic switch of liver nonparenchymal cells. FASEB J. 2019;33:8530-42
171. Sun J, Shi L, Xiao T, Xue J, Li J, Wang P. et al. microRNA-21, via the HIF-1alpha/VEGF signaling pathway, is involved in arsenite-induced hepatic fibrosis through aberrant cross-talk of hepatocytes and hepatic stellate cells. Chemosphere. 2021;266:129177
172. Karthikeyan S, Potter JJ, Geschwind JF, Sur S, Hamilton JP, Vogelstein B. et al. Deregulation of energy metabolism promotes antifibrotic effects in human hepatic stellate cells and prevents liver fibrosis in a mouse model. Biochem Biophys Res Commun. 2016;469:463-9
173. Du K, Hyun J, Premont RT, Choi SS, Michelotti GA, Swiderska-Syn M. et al. Hedgehog-YAP signaling pathway regulates glutaminolysis to control activation of hepatic stellate cells. Gastroenterology. 2018;154:1465-79 e13
174. Zhang F, Hao M, Jin H, Yao Z, Lian N, Wu L. et al. Canonical hedgehog signalling regulates hepatic stellate cell-mediated angiogenesis in liver fibrosis. Br J Pharmacol. 2017;174:409-23
175. Wang F, Chen L, Kong D, Zhang X, Xia S, Liang B. et al. Canonical Wnt signaling promotes HSC glycolysis and liver fibrosis through an LDH-A/HIF-1alpha transcriptional complex. Hepatology. 2024;79:606-23
176. Wang F, Li Z, Chen L, Yang T, Liang B, Zhang Z. et al. Inhibition of ASCT2 induces hepatic stellate cell senescence with modified proinflammatory secretome through an IL-1alpha/NF-kappaB feedback pathway to inhibit liver fibrosis. Acta Pharm Sin B. 2022;12:3618-38
177. Guimaraes EL, Empsen C, Geerts A, van Grunsven LA. Advanced glycation end products induce production of reactive oxygen species via the activation of NADPH oxidase in murine hepatic stellate cells. J Hepatol. 2010;52:389-97
178. Fehrenbach H, Weiskirchen R, Kasper M, Gressner AM. Up-regulated expression of the receptor for advanced glycation end products in cultured rat hepatic stellate cells during transdifferentiation to myofibroblasts. Hepatology. 2001;34:943-52
179. Leung C, Herath CB, Jia Z, Goodwin M, Mak KY, Watt MJ. et al. Dietary glycotoxins exacerbate progression of experimental fatty liver disease. J Hepatol. 2014;60:832-8
180. Owen T, Carpino G, Chen L, Kundu D, Wills P, Ekser B. et al. Endothelin receptor-a inhibition decreases ductular reaction, liver fibrosis, and angiogenesis in a model of cholangitis. Cell Mol Gastroenterol Hepatol. 2023;16:513-40
181. El Taghdouini A, van Grunsven LA. Epigenetic regulation of hepatic stellate cell activation and liver fibrosis. Expert Rev Gastroenterol Hepatol. 2016;10:1397-408
182. Barcena-Varela M, Paish H, Alvarez L, Uriarte I, Latasa MU, Santamaria E. et al. Epigenetic mechanisms and metabolic reprogramming in fibrogenesis: dual targeting of G9a and DNMT1 for the inhibition of liver fibrosis. Gut. 2021;70:388-400
183. Barcena-Varela M, Caruso S, Llerena S, Alvarez-Sola G, Uriarte I, Latasa MU. et al. Dual targeting of histone methyltransferase G9a and DNA-methyltransferase 1 for the treatment of experimental hepatocellular carcinoma. Hepatology. 2019;69:587-603
184. Yoneda A, Sakai-Sawada K, Niitsu Y, Tamura Y. Vitamin A and insulin are required for the maintenance of hepatic stellate cell quiescence. Exp Cell Res. 2016;341:8-17
185. Hernandez-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z. et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142:938-46
186. Pirazzi C, Valenti L, Motta BM, Pingitore P, Hedfalk K, Mancina RM. et al. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum Mol Genet. 2014;23:4077-85
187. Molenaar MR, Yadav KK, Toulmay A, Wassenaar TA, Mari MC, Caillon L. et al. Retinyl esters form lipid droplets independently of triacylglycerol and seipin. J Cell Biol. 2021;220:e202011071
188. Shajari S, Saeed A, Smith-Cortinez NF, Heegsma J, Sydor S, Faber KN. Hormone-sensitive lipase is a retinyl ester hydrolase in human and rat quiescent hepatic stellate cells. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864:1258-67
189. Hwang I, Lee EJ, Park H, Moon D, Kim HS. Retinol from hepatic stellate cells via STRA6 induces lipogenesis on hepatocytes during fibrosis. Cell Biosci. 2021;11:3
190. Matsukawa T, Yagi T, Uchida T, Sakai M, Mitsushima M, Naganuma T. et al. Hepatic FASN deficiency differentially affects nonalcoholic fatty liver disease and diabetes in mouse obesity models. JCI Insight. 2023;8:e161282
191. Scorletti E, Carr RM. A new perspective on NAFLD: Focusing on lipid droplets. J Hepatol. 2022;76:934-45
192. Ross TT, Crowley C, Kelly KL, Rinaldi A, Beebe DA, Lech MP. et al. Acetyl-CoA carboxylase inhibition improves multiple dimensions of NASH pathogenesis in model systems. Cell Mol Gastroenterol Hepatol. 2020;10:829-51
193. Harada N, Oda Z, Hara Y, Fujinami K, Okawa M, Ohbuchi K. et al. Hepatic de novo lipogenesis is present in liver-specific ACC1-deficient mice. Mol Cell Biol. 2007;27:1881-8
194. Wang X, He Q, Zhou C, Xu Y, Liu D, Fujiwara N. et al. Prolonged hypernutrition impairs TREM2-dependent efferocytosis to license chronic liver inflammation and NASH development. Immunity. 2023;56:58-77 e11
195. Rauchbach E, Zeigerman H, Abu-Halaka D, Tirosh O. Cholesterol induces oxidative stress, mitochondrial damage and death in hepatic stellate cells to mitigate liver fibrosis in mice model of NASH. Antioxidants (Basel). 2022;11:536
196. Horn CL, Morales AL, Savard C, Farrell GC, Ioannou GN. Role of cholesterol-associated steatohepatitis in the development of NASH. Hepatol Commun. 2022;6:12-35
197. Tomita K, Teratani T, Suzuki T, Shimizu M, Sato H, Narimatsu K. et al. Free cholesterol accumulation in hepatic stellate cells: mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology. 2014;59:154-69
198. Huang CF, Sun CC, Zhao F, Zhang YD, Li DJ. miR-33a levels in hepatic and serum after chronic HBV-induced fibrosis. J Gastroenterol. 2015;50:480-90
199. Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol. 2006;22:129-57
200. Tomita K, Teratani T, Suzuki T, Shimizu M, Sato H, Narimatsu K. et al. Acyl-CoA:cholesterol acyltransferase 1 mediates liver fibrosis by regulating free cholesterol accumulation in hepatic stellate cells. J Hepatol. 2014;61:98-106
201. Awan Z, Denis M, Bailey D, Giaid A, Prat A, Goltzman D. et al. The LDLR deficient mouse as a model for aortic calcification and quantification by micro-computed tomography. Atherosclerosis. 2011;219:455-62
202. Schneiderhan W, Schmid-Kotsas A, Zhao J, Grunert A, Nussler A, Weidenbach H. et al. Oxidized low-density lipoproteins bind to the scavenger receptor, CD36, of hepatic stellate cells and stimulate extracellular matrix synthesis. Hepatology. 2001;34:729-37
203. Banerjee P, Gaddam N, Chandler V, Chakraborty S. Oxidative stress-induced liver damage and remodeling of the liver vasculature. Am J Pathol. 2023;193:1400-14
204. Choi J, Corder NL, Koduru B, Wang Y. Oxidative stress and hepatic Nox proteins in chronic hepatitis C and hepatocellular carcinoma. Free Radic Biol Med. 2014;72:267-84
205. Garcia-Trevijano ER, Iraburu MJ, Fontana L, Dominguez-Rosales JA, Auster A, Covarrubias-Pinedo A. et al. Transforming growth factor beta1 induces the expression of alpha1(I) procollagen mRNA by a hydrogen peroxide-C/EBPbeta-dependent mechanism in rat hepatic stellate cells. Hepatology. 1999;29:960-70
206. Paik YH, Iwaisako K, Seki E, Inokuchi S, Schnabl B, Osterreicher CH. et al. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology. 2011;53:1730-41
207. Choi YY, Seok JI, Hwang JI, Kim DS. Co-administration of everolimus and N-acetylcysteine attenuates hepatic stellate cell activation and hepatic fibrosis. Am J Transl Res. 2020;12:2627-39
208. Maiers JL, Malhi H. Endoplasmic reticulum stress in metabolic liver diseases and hepatic fibrosis. Semin Liver Dis. 2019;39:235-48
209. Ji C, Kaplowitz N, Lau MY, Kao E, Petrovic LM, Lee AS. Liver-specific loss of glucose-regulated protein 78 perturbs the unfolded protein response and exacerbates a spectrum of liver diseases in mice. Hepatology. 2011;54:229-39
210. Bao X, Li J, Ren C, Wei J, Lu X, Wang X. et al. Aucubin ameliorates liver fibrosis and hepatic stellate cells activation in diabetic mice via inhibiting ER stress-mediated IRE1alpha/TXNIP/NLRP3 inflammasome through NOX4/ROS pathway. Chem Biol Interact. 2022;365:110074
211. Liang L, Zhang F, Feng N, Kuang B, Fan M, Chen C. et al. IRE1alpha protects against osteoarthritis by regulating progranulin-dependent XBP1 splicing and collagen homeostasis. Exp Mol Med. 2023;55:2376-89
212. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89-102
213. Kluwe J, Wongsiriroj N, Troeger JS, Gwak GY, Dapito DH, Pradere JP. et al. Absence of hepatic stellate cell retinoid lipid droplets does not enhance hepatic fibrosis but decreases hepatic carcinogenesis. Gut. 2011;60:1260-8
214. Ruart M, Chavarria L, Camprecios G, Suarez-Herrera N, Montironi C, Guixe-Muntet S. et al. Impaired endothelial autophagy promotes liver fibrosis by aggravating the oxidative stress response during acute liver injury. J Hepatol. 2019;70:458-69
215. Lenfant T, Costedoat-Chalumeau N. Hydroxychloroquine dose: balancing toxicity and SLE flare risk. Nat Rev Rheumatol. 2023;19:6-7
216. Zhou Y, Zhang L, Ma Y, Xie L, Yang YY, Jin C. et al. Secretome of senescent hepatic stellate cells favors malignant transformation from nonalcoholic steatohepatitis-fibrotic progression to hepatocellular carcinoma. Theranostics. 2023;13:4430-48
217. Yashaswini CN, Qin T, Bhattacharya D, Amor C, Lowe S, Lujambio A. et al. Phenotypes and ontogeny of senescent hepatic stellate cells in metabolic dysfunction-associated steatohepatitis. J Hepatol. 2024;81:207-17
218. Liu Y, Lu T, Zhang C, Xu J, Xue Z, Busuttil RW. et al. Activation of YAP attenuates hepatic damage and fibrosis in liver ischemia-reperfusion injury. J Hepatol. 2019;71:719-30
219. Hellemans K, Michalik L, Dittie A, Knorr A, Rombouts K, De Jong J. et al. Peroxisome proliferator-activated receptor-beta signaling contributes to enhanced proliferation of hepatic stellate cells. Gastroenterology. 2003;124:184-201
220. Beaven SW, Wroblewski K, Wang J, Hong C, Bensinger S, Tsukamoto H. et al. Liver X receptor signaling is a determinant of stellate cell activation and susceptibility to fibrotic liver disease. Gastroenterology. 2011;140:1052-62
221. Fickert P, Fuchsbichler A, Moustafa T, Wagner M, Zollner G, Halilbasic E. et al. Farnesoid X receptor critically determines the fibrotic response in mice but is expressed to a low extent in human hepatic stellate cells and periductal myofibroblasts. Am J Pathol. 2009;175:2392-405
222. Azizsoltani A, Hatami B, Zali MR, Mahdavi V, Baghaei K, Alizadeh E. Obeticholic acid-loaded exosomes attenuate liver fibrosis through dual targeting of the FXR signaling pathway and ECM remodeling. Biomed Pharmacother. 2023;168:115777
223. Garrido A, Kim E, Teijeiro A, Sanchez Sanchez P, Gallo R, Nair A. et al. Histone acetylation of bile acid transporter genes plays a critical role in cirrhosis. J Hepatol. 2022;76:850-61
224. Zwahlen J, Gairin E, Vianello S, Mercader M, Roux N, Laudet V. The ecological function of thyroid hormones. Philos Trans R Soc Lond B Biol Sci. 2024;379:20220511
225. Manka P, Coombes JD, Sydor S, Swiderska-Syn MK, Best J, Gauthier K. et al. Thyroid hormone receptor alpha modulates fibrogenesis in hepatic stellate cells. Liver Int. 2024;44:125-38
226. Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest. 2017;127:55-64
227. Lassailly G, Caiazzo R, Ntandja-Wandji LC, Gnemmi V, Baud G, Verkindt H. et al. Bariatric surgery provides long-term resolution of nonalcoholic steatohepatitis and regression of fibrosis. Gastroenterology. 2020;159:1290-301 e5
228. Bosch M, Kallin N, Donakonda S, Zhang JD, Wintersteller H, Hegenbarth S. et al. A liver immune rheostat regulates CD8 T cell immunity in chronic HBV infection. Nature. 2024;631:867-75
229. De Simone G, Andreata F, Bleriot C, Fumagalli V, Laura C, Garcia-Manteiga JM. et al. Identification of a Kupffer cell subset capable of reverting the T cell dysfunction induced by hepatocellular priming. Immunity. 2021;54:2089-100 e8
230. Longhi MS, Mieli-Vergani G, Vergani D. Regulatory T cells in autoimmune hepatitis: an updated overview. J Autoimmun. 2021;119:102619
231. Cho Y, Bukong TN, Tornai D, Babuta M, Vlachos IS, Kanata E. et al. Neutrophil extracellular traps contribute to liver damage and increase defective low-density neutrophils in alcohol-associated hepatitis. J Hepatol. 2023;78:28-44
232. Ehling J, Bartneck M, Wei X, Gremse F, Fech V, Mockel D. et al. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut. 2014;63:1960-71
233. Bernsmeier C, Pop OT, Singanayagam A, Triantafyllou E, Patel VC, Weston CJ. et al. Patients with acute-on-chronic liver failure have increased numbers of regulatory immune cells expressing the receptor tyrosine kinase MERTK. Gastroenterology. 2015;148:603-15 e14
234. Weiss E, de la Grange P, Defaye M, Lozano JJ, Aguilar F, Hegde P. et al. Characterization of blood immune cells in patients with decompensated cirrhosis including ACLF. Front Immunol. 2020;11:619039
235. Angioni R, Cali B, Vigneswara V, Crescenzi M, Merino A, Sanchez-Rodriguez R. et al. Administration of human MSC-derived extracellular vesicles for the treatment of primary sclerosing cholangitis: preclinical data in MDR2 knockout mice. Int J Mol Sci. 2020;21:8874
236. Ganapathy V, Ge R, Grazioli A, Xie W, Banach-Petrosky W, Kang Y. et al. Targeting the transforming growth factor-beta pathway inhibits human basal-like breast cancer metastasis. Mol Cancer. 2010;9:122
237. Sun T, Vander Heiden JA, Gao X, Yin J, Uttarwar S, Liang WC. et al. Isoform-selective TGF-beta3 inhibition for systemic sclerosis. Med. 2024;5:132-47 e7
238. Chen YW, Cheng PP, Yin YF, Cai H, Chen JZ, Feng MH. et al. Integrin alphaV mediated activation of myofibroblast via mechanoparacrine of transforming growth factor beta1 in promoting fibrous scar formation after myocardial infarction. Biochem Biophys Res Commun. 2024;692:149360
239. Yang WH, Deng YT, Hsieh YP, Wu KJ, Kuo MY. Thrombin activates latent TGFbeta1 via integrin alphavbeta1 in gingival fibroblasts. J Dent Res. 2016;95:939-45
240. Khanna D, Tashkin DP, Wells AU, Seibold JR, Wax S, Vazquez-Mateo C. et al. STRATUS: a phase II study of abituzumab in patients with systemic sclerosis-associated interstitial lung disease. J Rheumatol. 2021;48:1295-8
241. Saxena AR, Lyle SA, Khavandi K, Qiu R, Whitlock M, Esler WP. et al. A phase 2a, randomized, double-blind, placebo-controlled, three-arm, parallel-group study to assess the efficacy, safety, tolerability and pharmacodynamics of PF-06835919 in patients with non-alcoholic fatty liver disease and type 2 diabetes. Diabetes Obes Metab. 2023;25:992-1001
242. Zhu G, Li J, Lin X, Zhang Z, Hu T, Huo S. et al. Discovery of a novel ketohexokinase inhibitor with improved drug distribution in target tissue for the treatment of fructose metabolic disease. J Med Chem. 2023;66:13501-15
243. Gutierrez JA, Liu W, Perez S, Xing G, Sonnenberg G, Kou K. et al. Pharmacologic inhibition of ketohexokinase prevents fructose-induced metabolic dysfunction. Mol Metab. 2021;48:101196
244. Peng C, Yang P, Zhang D, Jin C, Peng W, Wang T. et al. KHK-A promotes fructose-dependent colorectal cancer liver metastasis by facilitating the phosphorylation and translocation of PKM2. Acta Pharm Sin B. 2024;14:2959-76
245. Zheng D, Jiang Y, Qu C, Yuan H, Hu K, He L. et al. Pyruvate kinase M2 tetramerization protects against hepatic stellate cell activation and liver fibrosis. Am J Pathol. 2020;190:2267-81
246. Zhang XJ, Ji YX, Cheng X, Cheng Y, Yang H, Wang J. et al. A small molecule targeting ALOX12-ACC1 ameliorates nonalcoholic steatohepatitis in mice and macaques. Sci Transl Med. 2021;13:eabg8116
247. Ratziu V, de Guevara L, Safadi R, Poordad F, Fuster F, Flores-Figueroa J. et al. Aramchol in patients with nonalcoholic steatohepatitis: a randomized, double-blind, placebo-controlled phase 2b trial. Nat Med. 2021;27:1825-35
248. Cusi K, Alkhouri N, Harrison SA, Fouqueray P, Moller DE, Hallakou-Bozec S. et al. Efficacy and safety of PXL770, a direct AMP kinase activator, for the treatment of non-alcoholic fatty liver disease (STAMP-NAFLD): a randomised, double-blind, placebo-controlled, phase 2a study. Lancet Gastroenterol Hepatol. 2021;6:889-902
249. Fouqueray P, Bolze S, Dubourg J, Hallakou-Bozec S, Theurey P, Grouin JM. et al. Pharmacodynamic effects of direct AMP kinase activation in humans with insulin resistance and non-alcoholic fatty liver disease: A phase 1b study. Cell Rep Med. 2021;2:100474
250. Dasgupta D, Nakao Y, Mauer AS, Thompson JM, Sehrawat TS, Liao CY. et al. IRE1A stimulates hepatocyte-derived extracellular vesicles that promote inflammation in mice with steatohepatitis. Gastroenterology. 2020;159:1487-503 e17
251. Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467-78
252. Kowdley KV, Bowlus CL, Levy C, Akarca US, Alvares-da-Silva MR, Andreone P. et al. Efficacy and safety of elafibranor in primary biliary cholangitis. N Engl J Med. 2024;390:795-805
253. Moreno-Lanceta A, Medrano-Bosch M, Simon-Codina B, Barber-Gonzalez M, Jimenez W, Melgar-Lesmes P. PPAR-gamma agonist GW1929 targeted to macrophages with dendrimer-graphene nanostars reduces liver fibrosis and inflammation. Pharmaceutics. 2023;15:1452
254. Shi T, Malik A, Yang Vom Hofe A, Matuschek L, Mullen M, Lages CS. et al. Farnesoid X receptor antagonizes macrophage-dependent licensing of effector T lymphocytes and progression of sclerosing cholangitis. Sci Transl Med. 2022;14:eabi4354
255. De Vincentis A, Ampuero J, Terracciani F, D'Amato D, Gerussi A, Cristoferi L. et al. Development and validation of a scoring system to predict response to obeticholic acid in primary biliary cholangitis. Clin Gastroenterol Hepatol. 2024;22:2062-74 e11
256. Adorini L, Trauner M. FXR agonists in NASH treatment. J Hepatol. 2023;79:1317-31
257. Ratziu V, Harrison SA, Loustaud-Ratti V, Bureau C, Lawitz E, Abdelmalek M. et al. Hepatic and renal improvements with FXR agonist vonafexor in individuals with suspected fibrotic NASH. J Hepatol. 2023;78:479-92
258. He L, Jhong JH, Chen Q, Huang KY, Strittmatter K, Kreuzer J. et al. Global characterization of macrophage polarization mechanisms and identification of M2-type polarization inhibitors. Cell Rep. 2021;37:109955
259. Francque SM, Bedossa P, Ratziu V, Anstee QM, Bugianesi E, Sanyal AJ. et al. A randomized, controlled trial of the pan-PPAR agonist lanifibranor in NASH. N Engl J Med. 2021;385:1547-58
260. Bonnardel J, T'Jonck W, Gaublomme D, Browaeys R, Scott CL, Martens L. et al. Stellate cells, hepatocytes, and endothelial cells imprint the Kupffer cell identity on monocytes colonizing the liver macrophage niche. Immunity. 2019;51:638-54 e9
261. Keam SJ. Resmetirom: first approval. Drugs. 2024;84:729-35
262. Harrison SA, Taub R, Neff GW, Lucas KJ, Labriola D, Moussa SE. et al. Resmetirom for nonalcoholic fatty liver disease: a randomized, double-blind, placebo-controlled phase 3 trial. Nat Med. 2023;29:2919-28
263. Yang W, He H, Wang T, Su N, Zhang F, Jiang K. et al. Single-cell transcriptomic analysis reveals a hepatic stellate cell-activation roadmap and myofibroblast origin during liver fibrosis in mice. Hepatology. 2021;74:2774-90
264. Jain I, Brougham-Cook A, Underhill GH. Effect of distinct ECM microenvironments on the genome-wide chromatin accessibility and gene expression responses of hepatic stellate cells. Acta Biomater. 2023;167:278-92
265. Khanal S, Liu Y, Bamidele AO, Wixom AQ, Washington AM, Jalan-Sakrikar N. et al. Glycolysis in hepatic stellate cells coordinates fibrogenic extracellular vesicle release spatially to amplify liver fibrosis. Sci Adv. 2024;10:eadn5228
Author contact
![]() Corresponding author: Yimin Cui, cui.pharmcom, Tel.: (010)66110802; Qian Xiang, xiang.pharmcom, Tel.: (010)66110802.
Corresponding author: Yimin Cui, cui.pharmcom, Tel.: (010)66110802; Qian Xiang, xiang.pharmcom, Tel.: (010)66110802.