Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Pathologic features and...
3. The role of macrophages in...
4. Macrophage-targeted diagnosis...
5. Macrophage-targeted...
6. Conclusions and future...
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2025; 15(4):1570-1588. doi:10.7150/thno.105256 This issue Cite
Review
Macrophage-based pathogenesis and theranostics of vulnerable plaques
Fei Fang1, Erxiang Wang1, Mengjia Fang2, Hongyan Yue1, Hanqiao Yang1, Xiaoheng Liu1, ![]()
1. Institute of Biomedical Engineering, West China School of Basic Medical Sciences & Forensic Medicine, Sichuan University, Chengdu 610041, China.
2. College of Veterinary Medicine, Northwest A&F University, Yangling 712100, Shaanxi, China.
Received 2024-10-15; Accepted 2024-12-9; Published 2025-1-2
Abstract

Vulnerable plaques, which are high-risk features of atherosclerosis, constitute critical elements in the disease's progression due to their formation and rupture. Macrophages and macrophage-derived foam cells are pivotal in inducing vulnerability within atherosclerotic plaques. Thus, understanding macrophage contributions to vulnerable plaques is essential for advancing the comprehension of atherosclerosis and devising novel therapeutic and diagnostic strategies. This review provides an overview of the pathological characteristics of vulnerable plaques, emphasizes macrophages' critical role, and discusses advanced strategies for their diagnosis and treatment. It aims to present a comprehensive macrophage-centered perspective for addressing vulnerable plaques in atherosclerosis.
Keywords: vulnerable plaque, macrophage, theranostics, nanoparticles
1. Introduction
Atherosclerosis is a chronic inflammatory disease of blood vessels, representing a major cause of cardiovascular disease (CVD) on a global scale. This condition is marked by the development of atheroma and fibrous plaques within the intimal layer of blood vessels [1]. Although age-related mortality from atherosclerosis has declined in recent years, it remains a leading global cause of death [2]. Atherosclerosis progresses via complex mechanisms involving lipid accumulation, fibrous tissue proliferation, and plaque formation. These plaques result in thickening of the arterial lining, lumen narrowing, and arterial wall hardening, culminating in restricted blood flow to vital organs. During disease progression, plaques may detach from the arterial wall, forming thrombi that can migrate to other organs and cause severe complications, including strokes and myocardial infarction [3]. Atherosclerotic plaques prone to thrombosis or rupture, referred to as unstable or vulnerable plaques, are characterized by a lipid-rich necrotic core and a thin fibrous cap infiltrated by macrophages [4]. Vulnerable plaques substantially elevate the risk of thrombus formation, leading to strokes and heart attacks [5, 6]. Vulnerable plaques substantially elevate the risk of thrombus formation, leading to strokes and heart attacks [7].
The progression of atherosclerosis involves a complex interplay of cellular mechanisms, including inflammation of endothelial cells (ECs), phenotypic changes in vascular smooth muscle cells (VSMCs), macrophage inflammatory responses, lipid transport dysfunction, and immune cell activation [8]. Macrophages are pivotal in the development and progression of atherosclerosis, particularly in the growth and rupture of plaques [9]. Within atherosclerotic plaques, macrophages exhibit diverse phenotypes, including classical M1 and M2 types as well as other subtypes such as Mox and M4 [10, 11]. From a traditional perspective, inflammatory M1 macrophages release pro-inflammatory factors that promote extracellular matrix (ECM) remodeling and VSMC proliferation, leading to lipid-laden plaque formation beneath the endothelium [12]. Foam cells, which are lipid- and cholesterol-rich macrophages with impaired lipid metabolism, contribute to plaque rupture [13]. Under normal conditions, macrophages phagocytose oxidized low-density lipoprotein (ox-LDL) through scavenger receptors (SR), such as LOX-1, CD36, and SR-A, which degrade cholesteryl esters into free cholesterol and free fatty acids within lysosomes. Free cholesterol is subsequently exported via ATP-binding cassette (ABC) transporters ABCA1, ABCG1, and SR-BI [14]. However, this critical balance of cholesterol transport is disrupted in atherosclerotic conditions. Upregulated SR increases ox-LDL uptake, while suppressed ABCA1 and ABCG1 expression reduces cholesterol efflux, resulting in foam cell formation [14]. Foam cells secrete matrix-degrading enzymes, facilitating fibrous cap detachment from the endothelium and thrombus formation [15]. The presence of foam cells in plaques is strongly linked to increased plaque rupture risk [16]. Given macrophages' pivotal role in vulnerable plaques, comprehensively understanding their pathological mechanisms is essential for effective atherosclerosis management.
This review aims to summarize the main pathological processes, in vivo mouse models, and macrophage-based insights into atherosclerotic vulnerable plaques. Additionally, it examines current strategies targeting macrophages or foam cells for the diagnosis and treatment of vulnerable plaques. This work provides a macrophage-centered perspective on diagnosing and treating vulnerable plaques, contributing to a deeper understanding of their pathophysiology and management.
2. Pathologic features and animal model
2.1. Pathologic features
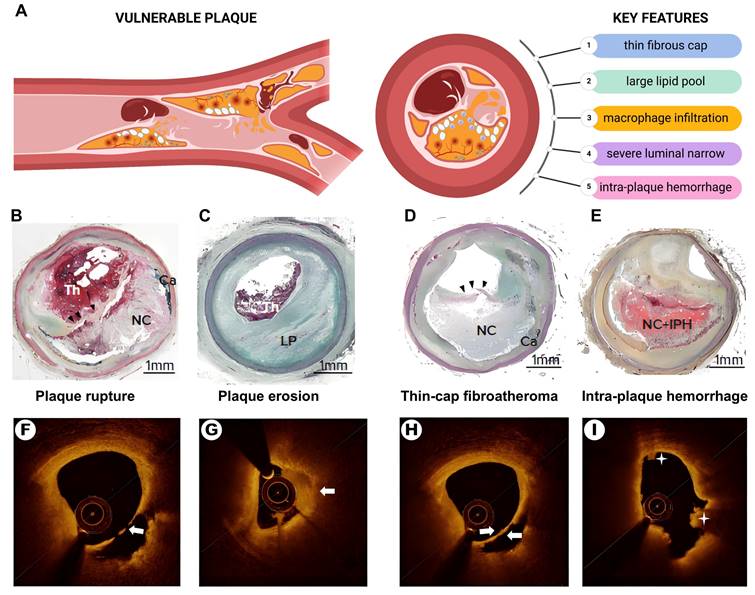
The concept of vulnerable plaques, characterized by their propensity for rupture and thrombosis, was introduced by Muller et al. in the 1980s [17]. Vulnerable plaques exhibit distinct anatomical, biological, and pathophysiological features. Autopsy studies have revealed key histological features linked to an increased risk of plaque rupture and acute coronary events (Figure 1). Key features include: 1) a thin fibrous cap (<65 μm); 2) a large lipid core (>40% of the plaque area); 3) pronounced macrophage infiltration; 4) endothelial cell ablation with platelet aggregation; and 5) severe luminal narrowing (>90%) [18-20]. Secondary pathological features include neovascularization, vascular remodeling, superficial nodal calcification, endothelial dysfunction, enlarged necrotic cores, inflammation, and intraplaque hemorrhage.
The pathologic features of vulnerable plaques. (A) The Schematic diagram of vulnerable plaque and its main pathological features. (B) plaque rupture. (C) plaque erosion with a large lipid pool. (C) Thin-cap fibroatheroma (black arrowhead). (E) intra-plaque hemorrhage. (F) Optical coherence tomography (OCT)-plaque rupture. (G) OCT-plaque erosion (white arrow shows thrombus). (H) Thin-cap fibroatheroma (white arrows show thin-cap fibroatheroma). (I) Intra-plaque hemorrhage (asterisk). Th = thrombus; NC = necrotic core; Ca = calcification; LP = lipid pool; IPH = intra-plaque hemorrhage. Source: Panels B-E: Sakamoto et al., 2022 [21]. Adapted with permission from US Cardiology. Panels F-I: Xiaoxiao et al., 2021 [22]. Adapted with permission from Springer Nature. Panel A created with BioRender.com.
2.2. Animal models
Transgenic ApoE-/- and LDLr-/- mice are widely used as models for experimental atherosclerosis research due to their accessibility and reproducibility. They are widely used as models for experimental atherosclerosis research due to their accessibility and reproducibility. Three primary methods are used in mouse models to induce plaque rupture. The first approach involves surgical disruption of the fibrous cap. For instance, Robert et al. used round-tipped tweezers to gently compress the abdominal aorta plaque of ApoE-/- mice [23]. Similarly, Jacob et al. punctured the carotid plaque with a 9-0 needle, causing rupture [24] (Figure 2A). The second method exploits hemodynamics alterations to destabilize plaque. For example, researchers performed incomplete ligation near the right carotid artery bifurcation in 12- to 14-week-old male mice to establish stable lesions. They then positioned conical polyethylene cuffs (300 and 150 μm diameters) over the ligature for 4 days, promoting collagen degradation and plaque rupture [25, 26] (Figure 2B). Jan et al. accelerated atherosclerosis in ApoE-/- and LDLr-/- mice by inserting a 0.3 mm inner diameter annular silicone ring into the common carotid artery for 3 weeks. After ring removal, adenovirus containing tumor suppressor protein p53 was injected into the distal artery and left to soak for 10 minutes. This approach targeted the delivery of p53 to VSMCs in the fibrous cap, inducing apoptosis within one day and triggering plaque rupture [27] (Figure 2C). Peter et al. created a vulnerable plaque model by double ligating the carotid artery at 1 mm and 4 mm from the upper bifurcation, following the insertion of a 150-µm needle beneath the vessel (Figure 2D) [28]. This method produces two types of plaque in the right carotid artery: a stable plaque between the ligature points and a vulnerable plaque near the second ligature point. Finally, Jin et al. developed a spontaneous plaque rupture model with luminal thrombosis by partially ligating the left carotid artery and renal arteries in ApoE-/- mice. Simultaneously, they activated the renin-angiotensin system to induce local stress changes (Figure 2E) [29].
Schematic diagram of the scheme for constructing a vulnerable plaque model in mice. (a) Exogenous mechanical force destroys the plaque structure and causes plaque rupture, such as puncturing the plaque with a needle or squeezing the plaque with tweezers. (b) Plaques were formed after ligation, and plaque rupture was induced by changing hemodynamics by implanting a conical cuff. (c) After the implantation of a silicone collar induced plaque formation, plaque rupture was induced by the instillation of a virus that promotes cell apoptosis. (d) Tandem stenosis induces stable plaque and vulnerable plaque models by changing hemodynamic parameters. (e) Unilateral carotid and renal artery ligation induce hypertension and plaque rupture. (f) Transgenic animal hybrids.CA = carotid artery; AA = abdominal aorta; LCA = left carotid artery; RCA = right carotid artery, DA = descending aorta, LICA = left internal carotid artery, LOA = left occipital artery, LECA = left external carotid artery, STA = superior thyrotropin artery, LRA = left renal artery. Figure created with BioRender.com.
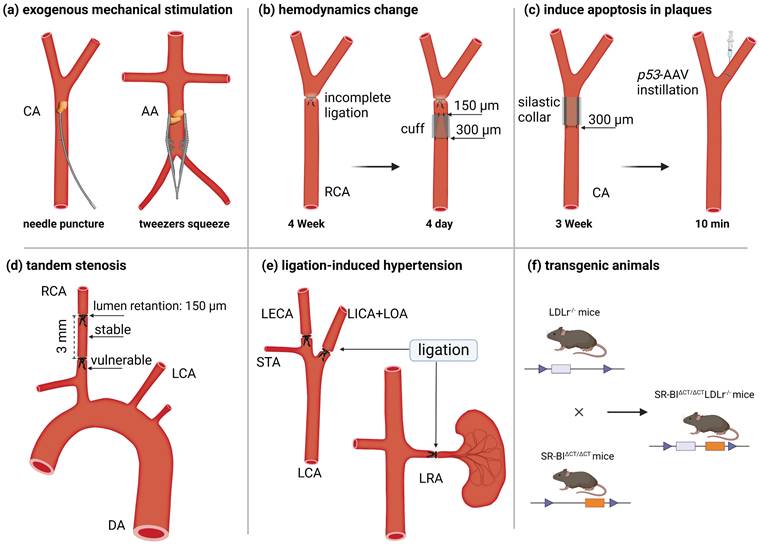
The third method involves engineering transgenic mouse models specifically designed to develop vulnerable plaques. For example, Tian et al. generated vulnerable plaque model mice by crossbreeding Fbn1C1039G+/- mice with LDLr-/- mice [30] (Figure 2F). Wu et al. induced vulnerable plaques in NR1D1-/- mice on a C57BL/6 background by partially ligating the left renal and carotid arteries [31]. Recently, Shamsuzzaman et al. created an advanced atherosclerotic vulnerable plaque model by knocking out SR-BI in LDLr-/- mice and administering a high-fat diet for 26 weeks [32].
3. The role of macrophages in vulnerable plaques
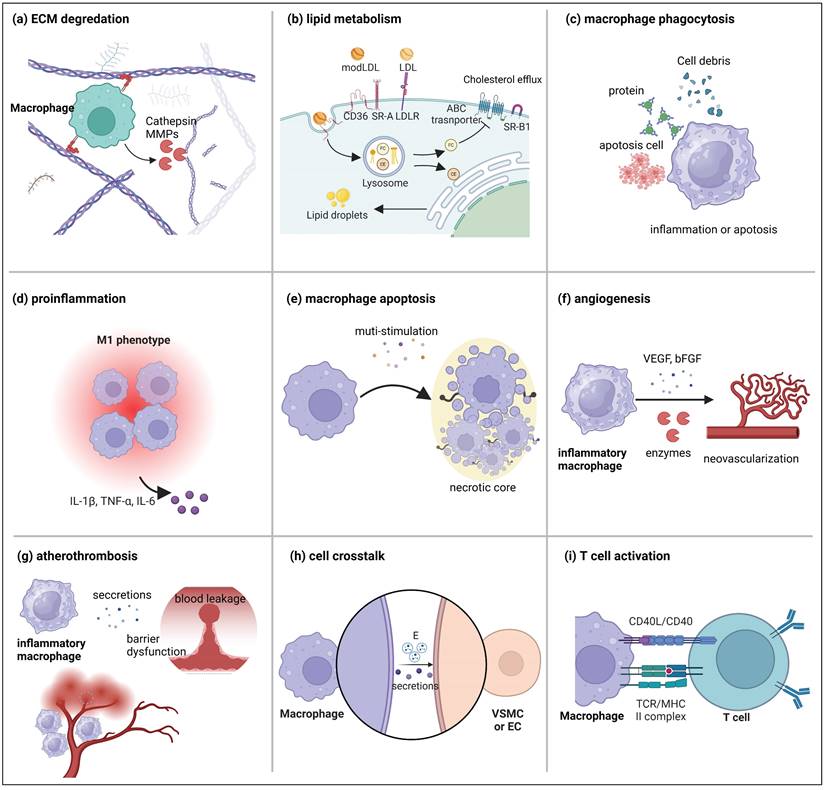
Although the role of macrophages in atherosclerotic plaque formation has been extensively reviewed elsewhere [33], this section emphasizes their contribution to plaque destabilization. This discussion highlights key macrophage functions in vulnerable plaques, including ECM remodeling, foam cell formation, inflammation, necrotic core formation, intra-plaque angiogenesis, intra-plaque hemorrhage, and interactions with other cell types (Figure 3).
The leading role of the macrophage in vulnerable plaque. (a) Macrophages release enzymes such as MMPs and cathepsin to degrade the ECM. (b) Macrophage trafficking and metabolism of ox-LDL in plaques. (C) Macrophages phagocytize cell debris, proteins, and apoptotic cell components. (d) Macrophages secrete inflammatory factors to promote plaque inflammation. (e) Macrophage apoptosis in plaques. (f) Macrophages secrete VEGC and bFGF to promote angiogenesis. (g) Macrophages promote intraplaque hemorrhage. (h) Macrophages promote the phenotypic transformation of VSMCs and endothelial dysfunction. (i) Macrophages activate T cells through antigen presentation. Figure created with BioRender.com.
3.1. ECM remodeling
The ECM is a complex, vital structure surrounding cells, crucial for maintaining tissue integrity and offering cellular support and protection [34]. It comprises collage, non-collagen proteins, elastin, proteoglycans, and glycosaminoglycans. Collagen, a key component, provides tissue strength and flexibility, which is critical in safeguarding tissue integrity from damage [35]. Studies indicate that ECM composition in vulnerable plaques undergoes significant changes, notably a marked reduction in collagen fiber content, a hallmark characteristic [36]. The reduction in collagen is primarily attributed to the macrophages, the predominant cell types in plaques responsible for ECM degradation. Inflammatory macrophages and macrophage-derived foam cells within plaques secrete enzymes that degrade the ECM, contributing to plaque rupture [37] (Figure 3A).
Matrix metalloproteinases (MMPs) are key enzymes that regulate atherosclerotic plaque stability. Studies dating back to 1994 have shown increased expression of MMPs and heightened matrix degradation activity in vulnerable regions of human atherosclerotic plaques [38]. Macrophage-associated MMPs that regulate plaque stability include collagenases (MMP-1, MMP-8, MMP-13), gelatinase (MMP-9), stromelysin (MMP-3, MMP-10), and other MMPs (MMP-7, MMP-12). Studies confirm that T cell-mediated adaptive immunity activates macrophages via CD40 signals, which upregulate the expression of collagenases MMP-1, MMP-8, and MMP-13 [39]. mRNA analysis of plaques from carotid endarterectomy patients showed that MMP-1 expression in plaques with thin fibrous caps was 7.8 times higher than in those with thick fibrous caps [40]. Recent studies also show that MMP-1a co-expresses with PAR1 (protease-activated receptor-1) on monocytes, promoting monocyte/macrophage infiltration [41].
The primary gelatinase secreted by macrophages and foam cells in plaques is MMP-9, also called gelatinase B. Studies have shown that MMP-9 co-localizes with macrophages in unstable carotid plaques [42]. Moreover, overexpression of the rabbit MMP-9 gene under the control of the scavenger receptor A (SR-A) enhancer/promoter resulted in more significant plaque burden and increased monocyte/macrophage infiltration [43]; in contrast, ApoE/MMP-9 double knockout mice exhibited reduced aortic plaque burden, macrophage infiltration, and collagen content [44]. MMP-3 (stromelysin 1) and MMP-10 (stromelysin 2) are among the earliest stromelysins identified in atherosclerotic plaques, with their expression localized to macrophages. Studies have shown that induced hypoxia upregulates MMP-3 and MMP-10 expression in macrophages via HIF-1α signaling, affecting the stability of mouse plaques [45].
In addition to MMPs, cysteine protease cathepsins (CTSs) play a crucial role in the ECM remodeling of atherosclerotic plaques. The cysteine protease family comprises 11 human members, including cathepsins B, C, H, F, K, L, O, S, V, W, and X/Z. Notably, cathepsins B [46], C [47], K [48], L [49], S [50], and V [51] have all been identified in atherosclerotic plaques. Cathepsin K (CTSK) is a potent type I collagen-degrading enzyme, and its expression is significantly upregulated in advanced plaques [48, 52, 53]. A recent study elucidated that macrophages, particularly foam cells, are the primary producers of CTSK [54]. Mechanistic studies have revealed that cholesterol loading in macrophages activates Toll-like receptor (TLR) signaling, leading to sustained phosphorylation of p38 mitogen-activated protein kinase and induction of CTSK expression [55].
3.2. Foam cell and lipid core
The key features of vulnerable plaques include a prominent lipid core and a high density of foam cells. The lipids within the plaque predominantly originate from the circulating blood. Under pathological conditions, the function of vascular ECs is impaired, leading to increased endothelial permeability. Circulating LDL infiltrates the vascular intima, which is oxidized and modified into ox-LDL.
Macrophages recruited and differentiated from the circulation can uptake ox-LDL via SR, subsequently transporting it to lysosomes where cholesterol esters undergo hydrolysis into free cholesterol and fatty acids [14, 56] (Figure 3B). For instance, M1 macrophages upregulate the expression of SR, including LOX-1, CD36, and SR-A, which exacerbate cholesterol absorption and ox-LDL [14, 56]. Moreover, the impairment of the macrophage reverse cholesterol transport (RCT) cholesterol efflux pathway mediated by ABCA1 and ABCG1, leads to the accumulation of free cholesterol (FRC) in macrophages, ultimately contributing to foam cell formation [57]. These FRCs spontaneously self-assemble and form cholesterol crystals (CCs). CCs can be released into the cytosol, inducing the complement cascade and activating the nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) inflammasome, triggering inflammation via IL-1β production and causing macrophage apoptosis [58]. Studies have shown that CCs are present at higher levels in the plaques of patients with acute coronary syndrome, indicating an association with plaque vulnerability and instability [59, 60]. Therefore, many strategies currently focus on reducing the risk of plaque rupture by inhibiting foam cell formation [61, 62].
3.3. Pro-inflammation
Atherosclerosis is primarily characterized by a sustained imbalance between pro-inflammatory and anti-inflammatory factors. This imbalance contributes to the lesions deterioration, leading to macrophage death and impaired resolution of inflammation [63, 64]. Macrophages play a crucial role in the progression of atherosclerosis. Inflammatory and immune responses are initiated when macrophages recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) binding to their pattern recognition receptors. This interaction produces various pro-inflammatory molecules, including cytokines like interleukin-α, interleukin-β, and TNF (Figure 3D) [65].
Macrophage phenotypes at inflammatory sites are diverse, with differentiation into specific subtypes occurring in response to microenvironmental changes. In atheromatous plaques, macrophages are classified into M1 (pro-inflammatory phenotype) and M2 (anti-inflammatory phenotype) macrophages [66]. Different subtypes of macrophages exhibit similar or opposing physiological functions and effects on the atherosclerotic environment. At rupture sites in vulnerable plaques, M1 phenotype macrophages contribute to plaque destabilization [67]. Macrophages in plaques can be activated by IFN-γ, TNF-α, lipopolysaccharide, and TLR4 [68]. Notably, M1 macrophages express various pro-inflammatory factors, including TNF-α, IL-1β, IL-12, IL-23, and chemokines such as CXCL9, CXCL10, and CXCL11. These factors can activate Th1 and Th17 cells, producing elevated levels of nitric oxide (NO) and reactive oxygen species (ROS), contributing to severe vascular inflammation and exacerbating atherosclerosis. Additionally, macrophage derivatives, such as 25-hydroxycholesterol (25-HC), have been demonstrated to promote the development of vascular inflammation and atherosclerosis [69]. Inhibiting the macrophage inflammatory response represents a crucial strategy for treating atherosclerosis. For example, a recent study showed that suppressing macrophage inflammasome activation with desmosterol prevented vascular inflammation and disease progression [70]. In addition, some miRNAs, including miR-375 [71], miR-30c-5p [72] and microRNA-99a/146b/378a [73] have been shown to inhibit macrophage inflammation and thus suppress the progression of atherosclerosis.
The inflammatory response mediated by macrophages is integral to the entire development stages of atherosclerosis. A deeper understanding of the pathological role of macrophages in this inflammatory response may offer valuable insights for potential atherosclerosis treatments.
3.4. Form necrotic core
An accumulation of lipids and cellular debris characterizes the necrotic core. The accumulation of necrotic cells and their contents contributes to forming and enlarging this core within plaques [74]. In advanced human atherosclerotic plaques, 80% of necrotic cores exceed 1 mm², comprising over 10% of the total plaque area. Notably, in 65% of plaque ruptures, the necrotic core accounted for more than 25% of the area, indicating a significant correlation between core size and plaque vulnerability [75]. Furthermore, the necrotic core in advanced plaques exhibits increased cholesterol clefts, abundant cellular debris, loss of ECM, and elevated tissue factor levels [76].
The prevalence of macrophage debris within the necrotic core has led to the hypothesis that plaque necrosis results directly from post-apoptotic macrophage death [77] (Figure 3E). Macrophages programmed cell death in atherosclerotic plaques encompasses apoptosis, necroptosis, pyroptosis, and ferroptosis. These forms of programmed cell death are interconnected components, with individual pathways closely linked and capable of compensating for one another [78]. Macrophage death in advanced plaques triggers a series of physiological consequences, including the formation of apoptotic bodies and the release of cytokines and DAMPs, which contribute to plaque instability [79, 80]. One prominent DAMP released in substantial quantities by late-stage macrophages is the HMGB-1 (high mobility group box 1) protein [81]. Empirical studies indicate that HMGB-1 expression increases as atherosclerosis progresses. Neutralizing HMGB-1 inhibits atherosclerosis by reducing the accumulation of immune cells and migrating macrophages in the aortic plaques of ApoE-/- mice [81, 82]. Mechanistic studies demonstrate that HMGB-1 interacts with various receptors, including RAGE (receptor for advanced glycation end products), and activates NF-κB (nuclear factor-κB) signaling to induce inflammatory response. This process promotes the further development of plaques [83]. Generally, macrophage death significantly influences the enlargement of the necrotic core. A comprehensive exploration of the mechanisms underlying programmed macrophage death could lead to the development of new therapeutic strategies for atherosclerosis.
3.5. Regulate neovascularization
A key distinction between vulnerable and stable plaques is the increased prevalence of neovascularization in the former, resulting in frequent plaque hemorrhages. The primary mechanism underlying this process is the induction of endothelial cell sprouting [84].
In vulnerable plaques, pathological neovascularization exhibits distinct morphological and functional characteristics, including abnormal vascular permeability, lack of a basement membrane and peripheral cells, and disorganized morphology [85, 86]. Additionally, neovascularization in plaques often causes leakage and the release of intra-plaque erythrocytes, resulting in hemorrhage. Several studies suggest that neovascularization promotes macrophage infiltration in vulnerable plaques under pathological conditions [87] (Figure 3F). In other tissues, however, macrophages of various phenotypes have been shown to regulate angiogenesis [88, 89]. However, clear evidence is lacking to demonstrate that macrophages directly regulate angiogenesis within plaques. Studies suggest a link between hypoxia-inducible transcription factors and macrophages in human atherosclerotic plaques and intra-plaque angiogenesis [90]. Furthermore, Li et al. conducted non-invasive imaging of 32 subjects with carotid artery stenosis using FDG positron emission tomography and dynamic contrast-enhanced magnetic resonance imaging (MRI) before carotid endarterectomy. They discovered a strong association between the extent of neovascularization, plaque macrophage infiltration, and the expression of plaque major histocompatibility complex (MHC) II, a marker of plaque inflammation [91].
In conclusion, macrophages have a role in intraplaque neovascularization, and targeting macrophages may be necessary for preventing intraplaque hemorrhage and stabilizing plaques.
3.6. Regulate intraplaque hemorrhage
Most intraplaque hemorrhage (IPH) is associated with centripetal neovascularization originating from the epicardium towards the plaque and increased vascular wall permeability [92, 93]. Recent studies have identified IPH as a contributing factor in forming vulnerable plaques [94], which are hallmark features of such plaques [95].
The correlation between IPH and macrophages plays a crucial role in the development of vulnerable plaques. For example, IPH can attract and release MMPs, destabilizing the plaque and accelerating rupture [96, 97]. Beyond its effects on macrophage activity and plaque development, IPH is also subject to regulation. CD163, a marker of M2-type macrophages, has been demonstrated to promote angiogenesis and increase vascular permeability [98], while also being linked to atherosclerotic inflammation. IPH is strongly associated with thrombus formation, which exacerbates plaque instability [99], ultimately leading to conditions such as cerebral hemorrhage and stroke [100]. Therefore, studying IPH could represent a breakthrough in treating vulnerable plaques. (Figure 3G).
3.7. Interact with other cell types within plaque
3.7.1. VSMC phenotypic transformation
Most VSMCs exhibit a contractile phenotype in healthy vessels but can be converted to a synthetic phenotype in response to pathological injury. Historically, VSMCs within vulnerable plaques were considered entirely beneficial to the disease due to their ability to produce ECM, thus stabilizing the fibrous cap. However, recent discoveries have revealed that VSMCs can undergo a phenotypic transformation, influenced by various factors, leading to the emergence of osteoblast-like VSMCs, macrophage-like VSMCs, mesenchymal cell-like VSMCs, and other phenotypes, ultimately contributing to plaque instability [101].
In the atherosclerotic microenvironment, macrophages and VSMCs are in close proximity and interact with each other through direct contact or the release of soluble factors and active carriers, significantly influencing the progression of the disease [102] (Figure 3H). Dysfunctional macrophages within the plaque release pro-inflammatory mediators such as TNF-α, IL-1β, and IL-6, which lead to VSMC dedifferentiation, ECM degradation, and fibrous cap thinning [103]. In vitro studies have demonstrated that co-culturing phorbol myristate acetate (PMA)-activated macrophages with human aortic VSMCs significantly reduces the synthesis of collagen type I and type III in VSMCs while simultaneously increasing the expression of matrix metalloproteinases (MMP1/9) [104]. This diminishes plaque stability from two key perspectives: first, by reducing collagen synthesis, and second, by degrading the ECM components of the fibrous cap. Another study showed that inflammatory signaling (NLRP3) was activated in VSMCs after co-culturing VSMCs with ox-LDL-induced monocytes [105]. NLRP3 has previously been shown to play a critical role in atherosclerosis and to facilitate the transformation of VSMCs into a macrophage-like phenotype. Additionally, extracellular vesicles (EVs) may represent a novel paradigm for interacting with macrophages and VSMCs. Although clear evidence of a regulatory effect of macrophage-derived EVs on VSMCs is lacking, sequencing and analysis of these vesicles indicated that their enriched contents may be associated with the migration and phenotypic transformation of VSMCs.
3.7.2. Endothelial dysfunction
In addition to VSMCs, macrophages can also regulate the function of vascular endothelial cells in various ways (Figure 3H). For example, a study found that CD163 macrophages promoted vascular cell adhesion molecule (VCAM) expression and inflammation in plaques by inhibiting prolyl hydroxylase and activating hypoxia-inducible factor 1-alpha (HIF-1α) [98]. Recently, a new study identified macrophage-promoted endothelial inflammation as a crucial factor in cholesterolemia-induced atherosclerosis [106]. Their single-cell RNA-seq study of human atherosclerotic lesions revealed that sterol 27-hydroxylase (CYP27A1) was highly expressed in foam cells. CYP27A1 catalyzes the conversion of cholesterol into 27-hydroxycholesterol (27HC) and cholesteryl esters. Macrophage-derived CYP27A1-generated 27HC drives vascular inflammation and promotes atherosclerosis through estrogen receptor (ER) α ligands [106]. In addition to the paracrine system, macrophages can regulate endothelial cell function by secreting EVs. For example, studies have shown that miRNAs enriched in macrophage-derived EVs can inhibit ECs proliferation[107] and damage[108].
3.7.3. Activate T cell
Crosstalk between macrophages and T cells is essential for the progression of inflammatory atherosclerosis [109] (Figure 3I). Inflammation in atherosclerosis is initiated by releasing IL-1β, TNF, CCL2 and CCL5 from macrophages in response to innate immune activation. This inflammatory process is further promoted by the recruitment of monocytes, dendritic cells, and effector T cells (Th1 cells, Th17 cells, and Th2 cells). Effector T cells release inflammatory factors, amplifying the inflammatory response [110]. Although macrophages can activate T cells as antigen-presenting cells, which is a fundamental aspect of acquired immune response activation [111], there is limited research on the mechanisms by which macrophages activate T cells in atherosclerosis. One study demonstrated that macrophage CD40 deficiency resulted in a bias towards the M2 phenotype, and CD40-/-ApoE-/- mice had reduced CD4+ and CD8+ T cells, suggesting that macrophage CD40 deficiency can regulate T cell activation [112]. Additionally, when miR-33-inhibited macrophages (which upregulated M2 phenotype markers) and naive T cells were co-cultured for 6 days, FoxP3+CD4+ Treg cells increased significantly, indicating a link between macrophages and innate adaptive immunity in atherosclerosis [113, 114].
4. Macrophage-targeted diagnosis for vulnerable plaques
4.1. Diagnostic imaging tools
Atherosclerotic plaque rupture and subsequent thrombosis are important causes of acute coronary syndrome (ACS), highlighting the essential need for the detection of vulnerable plaques in the management of atherosclerosis and CVD. A variety of both invasive and non-invasive imaging techniques have been developed for vulnerable plaque detection. Non-invasive methods include coronary angiography (CTA), MRI, and PET (positron emission tomography), while invasive methods include coronary angiography, intravascular ultrasound, optical coherence tomography, and near-infrared spectroscopy. The literature has extensively reviewed diagnostic imaging techniques for vulnerable plaques elsewhere [115]. This section will specifically focus on imaging methods that target the unique characteristics of macrophages within vulnerable plaques.
Although less commonly utilized, MRI is an effective modality for characterizing crucial features of vulnerable carotid atherosclerotic plaques. The clinical application of commercial dextran-coated ultra-small superparamagnetic iron oxide nanoparticles (USPIOs), with a particle size of 30 to 50 nm, as T2 contrast agents has been reported for identifying atherosclerotic plaques in humans [116]. Studies indicate that USPIOs are effectively phagocytosed by macrophages in vivo, resulting in detectable focal signal loss on MRI, which correlates with the extent of inflammation in atherosclerotic plaques. Consequently, USPIO-enhanced molecular MRI has been used to quantify the macrophage burden in plaques [117] and to evaluate plaques at risk of rupture in patients [118].
PET is a non-invasive imaging technique that uses contrast agents to evaluate plaque stability. PET radiotracers can identify activated macrophages and necrotic core microcalcifications, markers of vulnerable plaques. These contrast agents comprise 18F-sodium fluoride (18F-NaF, a marker of necrotic core microcalcifications), 18F-fluorodeoxyglucose (18F-FDG; a marker of activated macrophages), technetium-99m-annexin V (a marker of apoptosis cells), 18F-galacto RGD (a marker of angiogenesis), and 68Ga-DOTATATE (activated macrophages marker) [119]. Among these, 68Ga-DOTATATE is notable for its specific recognition of inflammatory macrophages. Due to the specific expression of the SST2 gene by pro-inflammatory macrophages in vulnerable carotid plaques, 68Ga-DOTATATE serves as a tracer for vulnerable plaques, targeting inflammatory macrophages specifically [120]. OCT is an invasive coronary artery imaging technique with an axial resolution of approximately 10-20 μm, excels in accurately measuring the true thickness of the fibrous cap. OCT can accurately differentiate between lipid and fibrous tissue and enables the visualization of macrophages, neovascularization, plaque rupture, and thrombosis [121]. Preclinical studies have demonstrated that combined contrast agents can image macrophage burden in vulnerable plaques. These contrast agents include iodinated and gold nanoparticles [122, 123].
Non-invasive optical imaging techniques, such as optical fluorescence and bioluminescence imaging, can be used to identify vulnerable atherosclerotic plaques. Since most of this research is still preclinical biomedical imaging research, we will discuss this method in the following paragraphs.
4.2. Novel targets
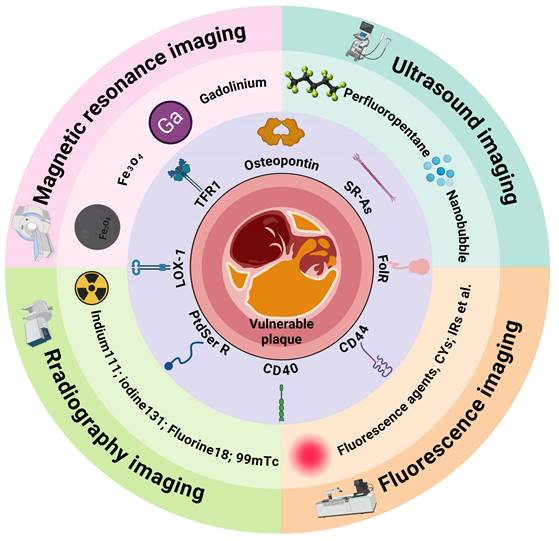
Given the central role of macrophages in plaque deterioration, their associated pathological processes are crucial targets for diagnostic imaging of vulnerable plaques [124]. In recent years, nanotechnology-based imaging techniques targeting macrophages have enabled an accurate plaque vulnerability assessment (Figure 4). This section will primarily summarize the imaging strategies of nanoplatforms targeting macrophages in vulnerable plaques (Table 1).
A schematic overview of the novel targets and the imaging agents used to diagnose vulnerable atherosclerotic plaques. TFR1 = transferrin receptor 1; SR-As = scavenger receptors A; FolR = folate receptor; PSR = phosphatidylserine receptor. Figure created with BioRender.com.
Macrophage-based vulnerable plaque imaging system
| Targets | Ligand | Carrier | Imaging agents | Imaging type | Year |
|---|---|---|---|---|---|
| Osteopontin | Osteopontin antibody | Fe3O4 NP | Fe3O4/Cy-5.5 | Fluorescence/MRI | 2017 [125] |
| Osteopontin antibody | NaGdF4:Yb,Er@NaGdF4 | NaGdF4 | MRI | 2017 [145] | |
| Osteopontin antibody | Ti3C2 nanosheets | Indocyanine green | PA | 2020 [126] | |
| Osteopontin antibody | Mesoporous silicon NP | Au NP | US/Fluorescence/ Photothermal | 2023 [146] | |
| Osteopontin antibody | mSiO2 NP | NIR persistent luminescent material | Fluorescence | 2024 [127] | |
| antiOPN peptide | Human serum albumin NP | IR780 | Fluorescence/MRI | 2022 [147] | |
| SR-As | Dextran sulfate | PLGA NP | Fe3O4/perfluoropentane | US/MRI | 2019 [148] |
| Dextran sulfate | PLGA-PEG-PLGA NP | Fe3O4/DiR/perfluoropentane | Fluorescence/MRI | 2022 [128] | |
| Dextran sulfate | Prussian blue-PEI | Rhodamine/Gd | Fluorescence/MRI | 2022 [149] | |
| PP1 peptide | Mesoporous silica NP | IR820/iron oxide NP | Fluorescence/MRI | 2021 [129] | |
| PP1 peptide | PLGA NP | Fe3O4/perfluoropentane | US/MRI | 2021 [150] | |
| Transferrin receptor 1 | High- Ferritin | Ferritin nanocages | 99mTc | Radiography | 2018 [132] |
| PtdSer receptor | Phosphatidylserine | Phosphatidylserine liposome | Indocyanine green | Fluorescence | 2019 [138] |
| Annexin V | Nanobubble | - | US | 2022 [151] | |
| Target | Ligand | Carrier | Imaging agents | Imaging type | Year |
| Peptide (CLIKKPF) | Cyclodextrin | Lipid target probes | Fluorescence | 2024 [152] | |
| Folate receptor | Folate | - | FITC | Fluorescence | 2012 [153] |
| Folate | - | 111indium-EC0800 | Radiography | 2014 [154] | |
| Folate | - | 18F | Radiography | 2018 [155] | |
| Folate | Human serum albumin | 131I | Radiography | 2023 [143] | |
| Folate | Pd@Au | Pd/Au | CT/PA | 2020 [156] | |
| LOX-1 | LOX-1 antibody | - | 99mTc | Radiography | 2008 [157] |
| LOX-1 antibody | - | 111indium/Gd/DiL | SPECT/CT/MRI/ Fluorescence | 2010 [158] | |
| LOX-1 antibody | Fe3O4 NP | Fe3O4 | MRI | 2014 [159] | |
| CD44 | Hyaluronic acid | Hybrid liposomal cerasomes | Gadodiamide | MRI | 2022 [160] |
| oxidized dextran | - | Lipid probe/iodinated contrast | Fluorescence/CT | 2024 [161] | |
| CD40 | Fe3O4 NP | Cy5.5/iron oxide | Fluorescence/MRI | 2023 [162] | |
| Phagocytosis | - | Bovine serum albumin | Cy-3-NO2/Mito-NIRHP | PA | 2018 [163] |
| - | DSPE-PEG NP | Semiconducting polymer | PA | 2021 [144] | |
| - | Amino acid NP | NIR dye | Fluorescence | 2024 [164] |
Abbreviations (NP: nanoparticles; MRI: magnetic resonance imaging; PA: photoacoustic; US: ultrasonic; NIR: near-infrared fluorophores; FITC: Fluorescein Isothiocyanate)
4.2.1. Osteopontin
Osteopontin (OPN) is a secreted protein that functions as a chemotactic cytokine, facilitating the adhesion, migration, and activation of foam macrophages. Studies have demonstrated foamy macrophages in human atherosclerotic lesions overexpression OPN, a finding confirmed through histological and molecular analyses [125]. Consequently, several strategies have been developed to evaluate plaque vulnerability by targeting OPN in foam macrophages. For instance, Zhen et al. conjugated OPN antibodies to Ti3C2 nanoplates and loaded them with indocyanine green (ICG) to enable photoacoustic imaging of vulnerable plaques [126]. The OPN Ab/Ti3C2/ICG nanoprobes demonstrated exceptional targeting capabilities for foam cells and vulnerable atherosclerotic plaques in both in vitro and in vivo studies [126]. More recently, Zhang et al. developed an atherosclerosis-targeted nanoprobe utilizing near-infrared (NIR) persistent luminescent nanoparticles (PLNPs) to detect early vulnerable plaques [127]. The surface conjugation of anti-OPN antibodies significantly enhanced the nanoprobe's targeting ability for foam cells. Upon intravenous administration in atherosclerosis model mice, this sensitive nanoprobe successfully detected atherosclerotic plaques prior to ultrasound (US) and MRI, achieving a signal-to-noise ratio (SNR) as high as 5.72 [127].
4.2.2. Scavenger receptors A (SR-As)
SR-A is a transmembrane glycoprotein receptor abundantly expressed on activated macrophages within vulnerable plaques. Consequently, several nano-delivery strategies have been developed to visualize vulnerable plaques by targeting SR-A. For example, Chen et al. conjugated SR-A-targeted dextran sulfate to PLGA-PEG-PLGA nanoparticles and loaded them with fluorescein DiR (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide) and Fe3O4. This nanocarrier targets macrophages in vulnerable aortic arch plaques, enabling near-infrared fluorescence (NIRF) and MRI of these plaques [128]. Another commonly utilized SR-A-targeted ligand is the PP1 peptide (LSLERFLRCWSDAPAK), known for its high affinity and specificity for SR-A. Zhang et al. synthesized mesoporous silica nanoparticles loaded with NIR dye IR820 and Fe3O4, subsequently conjugating them with the PP1 peptide. Fe3O4 was a magnetic core for T2 and T2*-weighted MR imaging, while IR820 was used for optical imaging. This carrier, featuring two composite imaging elements, enables dual-modality imaging of vulnerable atherosclerotic plaques [129].
4.2.3. Transferrin receptor 1
Transferrin receptor 1 (TFR1, CD71) is a type II transmembrane glycoprotein that binds transferrin and plays a crucial role in cellular iron uptake by interacting with iron-bound transferrin. Studies have demonstrated that the expression of the transferrin receptor is markedly elevated in vulnerable plaques, coinciding with the accumulation of heavy-chain ferritin (HFn) in infiltrating macrophages within human atherosclerotic plaques [130, 131]. To address this, Yan et al. developed radiolabeled bioengineered 99mTc-H-Ferritin nanocages to target TFR1 and visualize vulnerable plaques. Their study revealed that the iron-storing nanocage HFn is selectively absorbed by infiltrating macrophages in high-risk and early atherosclerotic plaques, enabling highly sensitive imaging of vulnerable and early active plaques [132]. The OX26 antibody has also been utilized as a ligand for targeting TFR1 in tumor delivery and imaging [133, 134]. However, this strategy has not yet been implemented in vulnerable atherosclerotic plaques.
4.2.4. Phosphatidylserine receptor
Under normal conditions, most phosphatidylserine (PtdSer) is localized to the inner leaflet of the cell membrane. However, as atherosclerosis advances, extensive apoptosis leads to the translocation of PtdSer to the outer layer of the cell membrane [135]. Macrophages express PtdSer receptors on their surface, enabling them to recognize and eliminate apoptotic cells [136]. This makes PtdSer receptors an ideal target for imaging vulnerable plaques. Mikako et al. loaded PEG-modified PtdSer liposomes with radioactive 111In, enabling PET imaging of macrophages in atherosclerotic plaques [137]. In addition, they designed a cathepsin B (CTSB)-responsive peptide-IGG fluorescent probe, which was encapsulated into PtdSer liposomes [138]. This carrier can target macrophages infiltrating plaques and emits fluorescence upon CTSB activation, facilitating NIRF imaging [138].
4.2.5. Folate receptor
The folate receptor beta (FolR-β) expression in activated macrophages [139] indicates its potential as a specific marker for vulnerable plaques. Researchers have employed folate-based radiopharmaceuticals (such as 111In-EC0800) combined with high-resolution animal single-photon emission computed tomography (SPECT/CT) for distinguishing between stable and vulnerable plaques [140]. Additionally, nano-molecules have been developed by conjugating the fluorescent contrast agent fluorescein isothiocyanate (FITC) with folate ligands to evaluate plaque vulnerability [141]. Furthermore, different research employed aluminum fluoride-18-labeled folic acid to visualize in vivo inflammation of atherosclerotic plaques through positron emission tomography [142]. In a recent study, Guo et al. developed a range of radio-iodinated albumin compounds targeted at folate receptors to image vulnerable plaques [143]. Among these compounds, [131I]-IBNHF stands out as a promising radiotracer for SPECT imaging of atherosclerosis [143].
Additionally, several macrophage-specific receptors, such as lectin-like oxidized low-density lipoprotein receptor 1 (LOX-1), CD44, and CD40, have been utilized as targets for imaging vulnerable plaques.
4.2.6. Phagocytosis
In addition to the previously mentioned macrophage-specific targets, macrophage phagocytosis can also be utilized to image vulnerable plaques. The semiconductor polymer-based nanoparticles (RSPN) developed by Zhang et al. enable highly specific and reliable imaging of O2•- in vulnerable plaques [144]. RSPN nanoparticles demonstrate excellent photoacoustic imaging capabilities and can be phagocytosed by macrophages in vivo and in vitro, allowing for accurate assessment of pneumonia-induced atherosclerotic vulnerable plaques [144].
5. Macrophage-targeted therapeutic strategy for stabilizing vulnerable plaques
Given the pivotal role of macrophages in the progression of vulnerable plaques, research efforts increasingly focus on modulating macrophage function as a therapeutic strategy for treating atherosclerosis and stabilizing vulnerable plaques. Present strategies aiming to regulate macrophage function to improve the stability of vulnerable plaques mainly involve inhibiting macrophage inflammation, modulation of lipid metabolism in macrophages and foam cells, and promotion of macrophage exocytosis (Figure 5).
Macrophage targeted therapeutic strategy for the treatment of vulnerable atherosclerotic plaques. Figure created with BioRender.com.
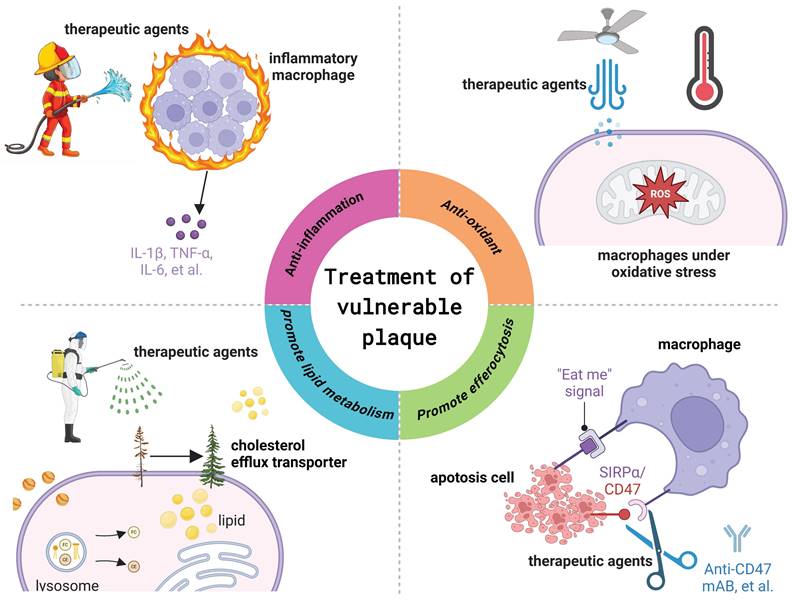
5.1. Anti-inflammation
There is a widespread consensus that atherosclerosis represents a chronic inflammatory malady that impacts blood vessels [52]. Consequently, anti-inflammatory therapy is an indispensable approach for managing atherosclerosis. For instance, the CANTOS study led by Dr. Paul Ridker demonstrated that subcutaneous injection of 300 mg of canakinumab, an IL-1β monoclonal antibody, every three months could reduce high-sensitivity C-reactive protein levels by 41% from baseline and lower the risk of cardiovascular events, the primary endpoint, by 14% [165]. This study provides direct evidence supporting the inflammatory hypothesis of atherosclerosis and serves as a foundation for the development of targeted anti-inflammatory therapies. Another notable case is colchicine, which has been approved by the U.S. Food and Drug Administration for the anti-inflammatory treatment of cardiovascular disease. A multinational, randomized, double-blind, placebo-controlled phase 3 clinical trial involving 5,522 patients with chronic coronary heart disease revealed that colchicine, when added to high-dose statins and other standard preventive therapies (0.5 mg daily), reduced the overall risk of cardiovascular death, spontaneous myocardial infarction, ischemic stroke, or ischemia-driven coronary artery revascularization by 31% compared to the placebo group [166]. Additionally, a recent prospective, single-center, randomized, double-blind clinical trial involving 128 patients with acute coronary syndrome found that 12 months of colchicine therapy (0.5 mg daily) significantly improved optical coherence tomography (OCT) parameters, including fibrous cap thickness (FCT), lipid arcs, and macrophage infiltration [167]. Macrophages are the primary effector cells in the inflammatory process within plaques and play a crucial role in innate and adaptive immunity [168]. Therefore, various medications are delivered to plaque macrophages to exert anti-inflammatory effects and stabilize plaques, including rapamycin [169], ibrutinib [170], and quercetin [171], among others.
Additionally, the repolarization of macrophage phenotypes has emerged as a novel approach for the anti-inflammatory treatment of atherosclerosis. Given the phenotypic flexibility of macrophages within plaques, reprogramming inflammatory M1 macrophages into anti-inflammatory M2 phenotypes can assist in inhibiting atherosclerosis and enhancing plaque stability [172, 173]. For example, Zhang et al. devised ROS-responsive dextran-g-PBMEO NPs with biomimetic modification of macrophage membranes to reprogram macrophage phenotypes by delivering the anti-inflammatory drug kaempferol specifically to plaques [174]. Their study confirmed that KPF@MM-NPs treatment significantly reduced inflammatory-related proteins, decreasing p-IκB levels by 3.2-fold and p-p65 levels by 1.2-fold in RAW264.7 macrophages compared to the model group. In vivo experiments further demonstrated that KPF@MM-NPs reduced the aortic plaque area to approximately 2.81% of the total aortic tissue area. Additionally, the treatment increased the collagen content and the number of VMCs surrounding the plaque, indicating improved plaque stability [174]. Recently, EVs have shown great potential in disease treatment due to their rich content and natural immunogenicity [175]. Our group stimulated endothelial cells to produce EVs with high anti-inflammatory activity (LSS-EVs) through hydrodynamics, which can significantly inhibit the progression of atherosclerosis by reprogramming macrophages to the anti-inflammatory M2 phenotype in vivo [173].
5.2. Anti-oxidant
Numerous studies have demonstrated that oxidative stress promotes atherosclerosis [176]. The accumulation of ROS represents a primary pathological consequence of oxidative stress. ROS can induce macrophage activation, cell apoptosis, and oxidative damage to biological macromolecules (e.g., lipids, proteins, DNA) [149]. ROS and its oxidative products can activate various signaling pathways involving functional signaling molecules, leading to changes in vascular cells and impacting vascular function and structure. Therefore, clearing ROS within atherosclerotic plaques is crucial for treating atherosclerosis. Gu et al. identified that Prussian blue nanozyme (PBNZ) demonstrates outstanding enzymatic activity and effectively eliminates ROS [177]. Consequently, they meticulously designed a PBNZ-based nanoparticle (PBNZ@PP-Man) to stabilize vulnerable plaques [177]. Their study revealed that PBNZ@PP-Man was internalized by macrophages via mannose receptor-mediated endocytosis, effectively alleviating inflammation by clearing ROS and enhancing endocytosis [177]. In in vivo animal experiments, the plaque area at the arterial root of mice treated with PBNZ@PP-Man decreased by 56.6% and collagen production was 2.6 times higher than that of the untreated control group [177].
He et al. recently developed two-dimensional black phosphorus nanosheets functionalized with Resolvin D1 (BPNSs@PEG-S2P/R) to effectively eliminate ROS within plaques to treat atherosclerosis [178]. Their research revealed that BPNSs@PEG-S2P/R efficiently accumulated within the diseased macrophages of atherosclerotic plaques aided by the S2P peptide. Furthermore, Resolvin D1 was released under ROS stimulation to eliminate ROS and suppress the ROS-induced inflammatory response in macrophages. Moreover, BPNSs@PEG-S2P/R can eliminate ROS within plaques in vivo, while increasing collagen content in the aortic peduncle reduces the incidence of ruptured or buried fibrous caps in the brachiocephalic artery region of mice. Additionally, it increases fibrous cap thickness.
5.3. Regulate lipid metabolism
Under normal conditions, macrophages can process substantial amounts of lipids and cholesterol in the blood vessel wall without significantly overburdening the metabolic decomposition process [179]. In the context of atherosclerosis, the capacity of macrophage lipid metabolism diminishes, leading to disrupted cholesterol and lipid metabolism, as well as cell apoptosis and necrosis. These necrotic macrophages are a significant factor influencing plaque stability. Therefore, restoring macrophage lipid transport capacity can help prevent the progression of atherosclerosis and maintain plaque stability. ABCA1, ABCG1, and SR-B1 play critical roles in cholesterol reverse transport. Liver X receptor (LXR) agonists (such as GW3965 and T0901317) are among the commonly used drugs for regulating macrophage lipid metabolism in atherosclerosis. For example, the LXR agonist GW3965 can upregulate the reverse cholesterol transporter ABCA1 expression in the small intestine and peripheral macrophages, elevate circulating HDL levels, and prevent atherosclerotic cardiovascular disease [180]. Yu et al. utilized DSPE-PEG nanoparticles to encapsulate the LXR agonist GW3965, aiming to mitigate plaque formation by upregulating ABCA1, predominantly expressed in macrophages, to enhance intracellular phospholipid and cholesterol efflux while not influencing hepatic lipid metabolism [181]. Recently, Chen et al. identified the significant role of Epsins in regulating lipid metabolism and transport in macrophages [182]. They observed that Epsins bind to CD36 and enhance lipid uptake by promoting CD36 endocytosis and recycling [182]. Conversely, Epsins facilitate ABCG1 degradation via lysosomes, impeding ABCG1-mediated cholesterol efflux and reverse cholesterol transport. Building on this model, they developed siRNA-loaded nano-therapies to inhibit Epsins, thereby halting inflammation and expediting the regression of atherosclerosis.
5.4. Promote efferocytosis
The accumulation of apoptotic cells in the necrotic core is a prominent feature of atherosclerotic plaques. Typically, apoptotic cells are eliminated through a process known as endocytosis, which is triggered by "eat me" ligands, signifying "entering the grave" [183]. Conversely, cells may overexpress "do not eat me" ligands to evade elimination. Research has demonstrated that the CD47 protein, a member of the "do not eat me" family of molecules, impedes the phagocytosis of apoptotic cells through its interaction with the signal regulatory protein α (SIRPα) receptor on phagocytes [184]. Thus, blocking the CD47 signal is a crucial strategy for controlling the elimination of apoptotic cells in atherosclerotic plaques. For example, Kojima et al. administered anti-CD47 antibodies and demonstrated an improvement in exocytosis within plaques and a reduction in the formation of necrotic cores [185]. To improve the bioavailability of CD47 antibody therapy and mitigate side effects, Chen et al. encapsulated CD47 antibodies in mesoporous silica nanoparticles and modified them with platelet membranes to prolong blood circulation time [186]. They discovered that this biomimetic carrier displayed immune evasion properties in both in vitro and in vivo settings, significantly reducing the plaque area of the aortic foot from 46.7 ± 2% to 16.6 ± 1.7% and increasing the VSMC content of the aortic foot from 22.1 ± 2.3% to 55.2 ± 4.1% [186]. Furthermore, Flores et al. employed nanotubes loaded with a small molecule inhibitor targeting the anti-phagocyte CD47-SIRPα signaling axis (SWNT-SHP1i) to induce phagocytosis at the plaque site [187]. Their findings validated the accumulation of SWNT-SHP1i in atherosclerotic plaques, reactivation of lesion phagocytosis, reduction of necrotic core and accumulation of apoptotic bodies in atherosclerotic mice, and enhancement of plaque stability [187]. Recently, Jarr et al. discovered that statins can enhance programmed cell elimination by restraining the NF-κB1 and inhibiting the expression of the pivotal "do not eat me" molecule, CD47 [188]. Statins enhance the anti-atherogenic effects of macrophage phagocytosis in an additive fashion, irrespective of any lipid-lowering effects [188].
6. Conclusions and future perspectives
Atherosclerosis is a widespread global disease with high prevalence across all regions. It also serves as an underlying factor for other cardiovascular diseases, including stroke and heart attack. Vulnerable plaques represent high-risk manifestations of atherosclerosis and studying them can lead to breakthroughs in diagnosing and treating atherosclerosis. We reviewed the primary pathological characteristics of vulnerable plaques and summarized the methodologies for constructing in vivo mouse models of vulnerable plaques. Modifying the plaque's local hemodynamic environment is a widely employed strategy for constructing vulnerable plaques. Among these methods, the right carotid artery tandem ligation model is a promising standard model for inducing vulnerable plaques in mice, as it can form stable and vulnerable plaques in the adjacent tissues of the same individual. This is significant for preclinical studies on vulnerable plaque pathology and therapeutic diagnostics. In addition, there are reports that transgenic ApoE-/- mice can develop vulnerable plaques. However, there may be potential drawbacks regarding model cost and stability. Despite exploring several relatively mature vulnerable plaque construction models in mouse models, there is still a lack of atherosclerosis and vulnerable plaque models in large animals.
Macrophages and macrophage-derived foam cells play an essential role in forming and rupturing vulnerable plaques. For example, macrophages can regulate ECM remodeling, lipid accumulation, inflammatory response, necrotic core formation, neovascularization, intraplaque hemorrhage, and the phenotypic transformation of smooth muscle cells. However, the formation of stable plaques is also accompanied by pathological processes such as lipid accumulation, inflammation, and the formation of necrotic cores. Therefore, it is still difficult to clearly define the difference between the functions of macrophages in stable plaques and those in vulnerable plaques. Since plaque rupture is a multifactorial pathological process, exploring the interaction and synergy between macrophages and other factors influencing vulnerable plaques is necessary.
Considering the important role of macrophages in vulnerable plaques, macrophage-based therapeutic diagnostics represent a logical and highly promising strategy for identifying and preventing the development of vulnerable plaques. Most macrophage-based diagnostic strategies for vulnerable plaques are in the preclinical research stage. These studies primarily focus on screening targets of macrophages in vulnerable plaques and innovating contrast agents. Currently, the reported macrophage targets that can identify vulnerable plaques include OPN, SR-As, transferrin receptor 1, phosphatidylserine, folate receptor, and others. Nanocarriers that bind to these targets and load-effective imaging agents can be used to assess the risk of vulnerable plaques. However, it should be noted that most of these studies compare imaging differences between vulnerable plaques and healthy tissues while ignoring the comparison with stable plaques. Therefore, developing strategies for achieving high-resolution imaging of vulnerable plaques that differ from stable plaques is essential.
Similarly to the imaging strategy for vulnerable plaques, the nano-drug delivery strategy is also in the preclinical research stage. Presently, strategies aimed at targeting macrophages to prevent and stabilize vulnerable plaques focus on inhibiting macrophage inflammation antioxidation, regulating macrophage cholesterol metabolism, and promoting macrophage endocytosis. Nevertheless, these pharmacological concepts for treating vulnerable plaques are nearly identical to the conventional pharmacological concepts for inhibiting the progression of atherosclerosis. Consequently, an in-depth exploration of the pathological mechanisms underlying vulnerable plaque formation, development, and rupture will significantly advance drug development targeting vulnerable plaques.
To conclude, this review outlines the pathologic features and animal models of vulnerable atherosclerosis plaques and summarizes the pivotal role of macrophages in their development. Furthermore, we also discuss the targeted diagnosis and treatment strategies for vulnerable plaques, focusing on the role of macrophages. Gaining a deeper understanding of the pathological mechanisms underlying vulnerable plaques and developing appropriate experimental animal models will significantly advance the field of effective treatment and diagnosis of atherosclerosis and simultaneously address related clinical issues.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant numbers 32301089 and 12372315) and the Sichuan Science and Technology Program (Grant numbers 2024NSFSC1703).
Author contributions
F.F. and E.W. conceived and drafted the manuscript. M. F., H.Y., and X.L. critically reviewed the manuscript and provided important intellectual content.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Björkegren JLM, Lusis AJ. Atherosclerosis: Recent developments. Cell. 2022;185:1630-45
2. Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW. et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation. 2021;143:e254-e743
3. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317-25
4. Ambrose JA, Sharma AV. Identifying and Treating Vulnerable Atherosclerotic Plaques. Am J Cardiol. 2023;205:214-22
5. Finn AV, Nakano M, Narula J, Kolodgie FD, Virmani R. Concept of vulnerable/unstable plaque. Arterioscler Thromb Vasc Biol. 2010;30:1282-92
6. Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006;47:C13-8
7. Hafiane A. Vulnerable Plaque, Characteristics, Detection, and Potential Therapies. J Cardiovasc Dev Dis. 2019;6:26
8. Libby P. The changing landscape of atherosclerosis. Nature. 2021;592:524-33
9. Song B, Bie Y, Feng H, Xie B, Liu M, Zhao F. Inflammatory Factors Driving Atherosclerotic Plaque Progression New Insights. J Transl Int Med. 2022;10:36-47
10. Fang F, Xiao C, Li C, Liu X, Li S. Tuning macrophages for atherosclerosis treatment. Regen Biomater. 2022;10:rbac103
11. Blagov AV, Markin AM, Bogatyreva AI, Tolstik TV, Sukhorukov VN, Orekhov AN. The Role of Macrophages in the Pathogenesis of Atherosclerosis. Cells. 2023;12:522
12. Badimon L, Vilahur G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J Intern Med. 2014;276:618-32
13. Yuan Y, Li P, Ye J. Lipid homeostasis and the formation of macrophage-derived foam cells in atherosclerosis. Protein Cell. 2012;3:173-81
14. Chistiakov DA, Melnichenko AA, Myasoedova VA, Grechko AV, Orekhov AN. Mechanisms of foam cell formation in atherosclerosis. J Mol Med (Berl). 2017;95:1153-65
15. De Meyer GRY, Zurek M, Puylaert P, Martinet W. Programmed death of macrophages in atherosclerosis: mechanisms and therapeutic targets. Nat Rev Cardiol. 2024;21:312-25
16. Gianopoulos I, Daskalopoulou SS. Macrophage profiling in atherosclerosis: understanding the unstable plaque. Basic Res Cardiol. 2024;119:35-56
17. Muller JE, Tofler GH, Stone PH. Circadian variation and triggers of onset of acute cardiovascular disease. Circulation. 1989;79:733-43
18. Naghavi M, Libby P, Falk E, Casscells SW, Litovsky S, Rumberger J. et al. From Vulnerable Plaque to Vulnerable Patient. Circulation. 2003;108:1664-72
19. Naghavi M, Libby P, Falk E, Casscells SW, Litovsky S, Rumberger J. et al. From Vulnerable Plaque to Vulnerable Patient. Circulation. 2003;108:1772-8
20. Weng S-T, Lai Q-L, Cai M-T, Wang J-J, Zhuang L-Y, Cheng L. et al. Detecting vulnerable carotid plaque and its component characteristics: Progress in related imaging techniques. Front Neurol. 2022;13:982147
21. Sakamoto A, Cornelissen A, Sato Y, Mori M, Kawakami R, Kawai K. et al. Vulnerable Plaque in Patients with Acute Coronary Syndrome: Identification, Importance, and Management. US Cardiology Review. 2022;16:e01 2022
22. Zhao X, Wang Y, Chen R, Li J, Zhou J, Liu C. et al. Triglyceride glucose index combined with plaque characteristics as a novel biomarker for cardiovascular outcomes after percutaneous coronary intervention in ST-elevated myocardial infarction patients: an intravascular optical coherence tomography study. Cardiovasc Diabetol. 2021;20:131
23. Reddick RL, Zhang SH, Maeda N. Aortic atherosclerotic plaque injury in apolipoprotein E deficient mice. Atherosclerosis. 1998;140:297-305
24. Bentzon JF, Sondergaard CS, Kassem M, Falk E. Smooth Muscle Cells Healing Atherosclerotic Plaque Disruptions Are of Local, Not Blood, Origin in Apolipoprotein E Knockout Mice. Circulation. 2007;116:2053-61
25. Fasolo F, Jin H, Winski G, Chernogubova E, Pauli J, Winter H. et al. Long Noncoding RNA MIAT Controls Advanced Atherosclerotic Lesion Formation and Plaque Destabilization. Circulation. 2021;144:1567-83
26. Jin H, Li DY, Chernogubova E, Sun C, Busch A, Eken SM. et al. Local Delivery of miR-21 Stabilizes Fibrous Caps in Vulnerable Atherosclerotic Lesions. Mol Ther. 2018;26:1040-55
27. von der Thüsen JH, van Vlijmen BJM, Hoeben RC, Kockx MM, Havekes LM, van Berkel TJC. et al. Induction of Atherosclerotic Plaque Rupture in Apolipoprotein E-/- Mice After Adenovirus-Mediated Transfer of p53. Circulation. 2002;105:2064-70
28. Noonan J, Bobik A, Peter K. The tandem stenosis mouse model: Towards understanding, imaging, and preventing atherosclerotic plaque instability and rupture. Br J Pharmacol. 2022;179:979-97
29. Jin S-x, Shen L-h, Nie P, Yuan W, Hu L-h, Li D-d. et al. Endogenous Renovascular Hypertension Combined With Low Shear Stress Induces Plaque Rupture in Apolipoprotein E-Deficient Mice. Arterioscler Thromb Vasc Biol. 2012;32:2372-9
30. Wang X, Fu Y, Xie Z, Cao M, Qu W, Xi X. et al. Establishment of a Novel Mouse Model for Atherosclerotic Vulnerable Plaque. Front Cardiovasc Med. 2021;8:642751
31. Wu Z, Liao F, Luo G, Qian Y, He X, Xu W. et al. NR1D1 Deletion Induces Rupture-Prone Vulnerable Plaques by Regulating Macrophage Pyroptosis via the NF-κB/NLRP3 Inflammasome Pathway. Oxid Med Cell Longev. 2021;2021:5217572
32. Shamsuzzaman S, Deaton RA, Salamon A, Doviak H, Serbulea V, Milosek VM. et al. Novel Mouse Model of Myocardial Infarction, Plaque Rupture, and Stroke Shows Improved Survival With Myeloperoxidase Inhibition. Circulation. 2024;150:687-705
33. Bobryshev YV, Nikiforov NG, Elizova NV, Orekhov AN. Macrophages and Their Contribution to the Development of Atherosclerosis. Results Probl Cell Differ. 2017;62:273-98
34. Karamanos NK, Theocharis AD, Piperigkou Z, Manou D, Passi A, Skandalis SS. et al. A guide to the composition and functions of the extracellular matrix. Febs j. 2021;288:6850-912
35. Holm Nielsen S, Jonasson L, Kalogeropoulos K, Karsdal MA, Reese-Petersen AL, Auf dem Keller U. et al. Exploring the role of extracellular matrix proteins to develop biomarkers of plaque vulnerability and outcome. J Intern Med. 2020;287:493-513
36. Olejarz W, Łacheta D, Kubiak-Tomaszewska G. Matrix Metalloproteinases as Biomarkers of Atherosclerotic Plaque Instability. Int J Mol Sci. 2020;21:3946
37. Wågsäter D, Björk H, Zhu C, Björkegren J, Valen G, Hamsten A. et al. ADAMTS-4 and -8 are inflammatory regulated enzymes expressed in macrophage-rich areas of human atherosclerotic plaques. Atherosclerosis. 2008;196:514-22
38. Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94:2493-503
39. Khandkar C, Madhavan MV, Weaver JC, Celermajer DS, Karimi Galougahi K. Atherothrombosis in Acute Coronary Syndromes—From Mechanistic Insights to Targeted Therapies. Cells. 2021;10:865
40. Morgan AR, Rerkasem K, Gallagher PJ, Zhang B, Morris GE, Calder PC. et al. Differences in Matrix Metalloproteinase-1 and Matrix Metalloproteinase-12 Transcript Levels Among Carotid Atherosclerotic Plaques With Different Histopathological Characteristics. Stroke. 2004;35:1310-5
41. Fletcher EK, Wang Y, Flynn LK, Turner SE, Rade JJ, Kimmelstiel CD. et al. Deficiency of MMP1a (Matrix Metalloprotease 1a) Collagenase Suppresses Development of Atherosclerosis in Mice. Arterioscler Thromb Vasc Biol. 2021;41:e265-e79
42. Inducer DEMM. Matrix Metalloproteinase 2 Is Associated With Stable and Matrix Metalloproteinases 8 and 9 With Vulnerable Carotid Atherosclerotic Lesions. Stroke. 2006;37:253-9
43. Chen Y, Waqar AB, Nishijima K, Ning B, Kitajima S, Matsuhisa F. et al. Macrophage-derived MMP-9 enhances the progression of atherosclerotic lesions and vascular calcification in transgenic rabbits. J Cell Mol Med. 2020;24:4261-74
44. Luttun A, Lutgens E, Manderveld A, Maris K, Collen D, Carmeliet P. et al. Loss of Matrix Metalloproteinase-9 or Matrix Metalloproteinase-12 Protects Apolipoprotein E-Deficient Mice Against Atherosclerotic Media Destruction but Differentially Affects Plaque Growth. Circulation. 2004;109:1408-14
45. Wei S, Isagawa T, Eguchi M, Sato D, Tsukano H, Miyata K. et al. Febuxostat, a Xanthine Oxidase Inhibitor, Decreased Macrophage Matrix Metalloproteinase Expression in Hypoxia. Biomedicines. 2020;8:470
46. Zhao CF, Herrington DM. The function of cathepsins B, D, and X in atherosclerosis. Am J Cardiovasc Dis. 2016;6:163-70
47. Herías V, Biessen EAL, Beckers C, Delsing D, Liao M, Daemen MJ. et al. Leukocyte Cathepsin C Deficiency Attenuates Atherosclerotic Lesion Progression by Selective Tuning of Innate and Adaptive Immune Responses. Arterioscler Thromb Vasc Biol. 2015;35:79-86
48. Fang F, Feng T, Li J, Zhang H, Wang Q, Chen Y. et al. Cathepsin K contributed to disturbed flow-induced atherosclerosis is dependent on integrin-actin cytoskeleton-NF-κB pathway. Genes Dis. 2023;10:583-95
49. Li W, Kornmark L, Jonasson L, Forssell C, Yuan X-M. Cathepsin L is significantly associated with apoptosis and plaque destabilization in human atherosclerosis. Atherosclerosis. 2009;202:92-102
50. Rodgers KJ, Watkins DJ, Miller AL, Chan PY, Karanam S, Brissette WH. et al. Destabilizing Role of Cathepsin S in Murine Atherosclerotic Plaques. Arterioscler Thromb Vasc Biol. 2006;26:851-6
51. Yasuda Y, Li Z, Greenbaum D, Bogyo M, Weber E, Brömme D. Cathepsin V, a Novel and Potent Elastolytic Activity Expressed in Activated Macrophages. J Biol Chem. 2004;279:36761-70
52. Fang F, Ni Y, Yu H, Yin H, Yang F, Li C. et al. Inflammatory endothelium-targeted and cathepsin responsive nanoparticles are effective against atherosclerosis. Theranostics. 2022;12:4200-20
53. Faber BCG, Cleutjens KBJM, Niessen RLJ, Aarts PLJW, Boon W, Greenberg AS. et al. Identification of Genes Potentially Involved in Rupture of Human Atherosclerotic Plaques. Circ Res. 2001;89:547-54
54. Nakagawa K, Nakashima Y. Pathogenesis of human atheroma necrotic core: degradation of connective tissue fibers and possible involvement of cathepsin K. Transl Med Commun. 2024;9:26
55. Sun Y, Ishibashi M, Seimon T, Lee M, Sharma SM, Fitzgerald KA. et al. Free Cholesterol Accumulation in Macrophage Membranes Activates Toll-Like Receptors and p38 Mitogen-Activated Protein Kinase and Induces Cathepsin K. Circ Res. 2009;104:455-65
56. Kataoka H, Kume N, Miyamoto S, Minami M, Moriwaki H, Murase T. et al. Expression of lectinlike oxidized low-density lipoprotein receptor-1 in human atherosclerotic lesions. Circulation. 1999;99:3110-7
57. Lien CC, Chen CH, Lee YM, Guo BC, Cheng LC, Pan CC. et al. The phosphatase activity of soluble epoxide hydrolase regulates ATP-binding cassette transporter-A1-dependent cholesterol efflux. J Cell Mol Med. 2019;23:6611-21
58. Russo M, Jang I-K. Cholesterol crystals in atherosclerotic plaques: A future target to reduce the risk of plaque rupture? Int J Cardiol Heart Vasc. 2022;365:30-1
59. Araki M, Park S-J, Dauerman HL, Uemura S, Kim J-S, Di Mario C. et al. Optical coherence tomography in coronary atherosclerosis assessment and intervention. Nat Rev Cardiol. 2022;19:684-703
60. Nishimura S, Ehara S, Hasegawa T, Matsumoto K, Yoshikawa J, Shimada K. Cholesterol crystal as a new feature of coronary vulnerable plaques: An optical coherence tomography study. J Cardiol. 2017;69:253-9
61. Li Y, Zhang L, Ren P, Yang Y, Li S, Qin X. et al. Qing-Xue-Xiao-Zhi formula attenuates atherosclerosis by inhibiting macrophage lipid accumulation and inflammatory response via TLR4/MyD88/NF-κB pathway regulation. Phytomedicine. 2021;93:153812
62. Zou J, Xu C, Zhao Z-W, Yin S-H, Wang G. Asprosin inhibits macrophage lipid accumulation and reduces atherosclerotic burden by up-regulating ABCA1 and ABCG1 expression via the p38/Elk-1 pathway. J Transl Med. 2022;20:337
63. Tabas I, Glass CK. Anti-Inflammatory Therapy in Chronic Disease: Challenges and Opportunities. Science. 2013;339:166-72
64. Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10:36-46
65. Libby P. Inflammation in Atherosclerosis-No Longer a Theory. Clin Chem. 2021;67:131-42
66. Cochain C, Zernecke A. Macrophages in vascular inflammation and atherosclerosis. Pflugers Arch. 2017;469:485-99
67. Wang J, Kang Z, Liu Y, Li Z, Liu Y, Liu J. Identification of immune cell infiltration and diagnostic biomarkers in unstable atherosclerotic plaques by integrated bioinformatics analysis and machine learning. Front Immunol. 2022;13:956078
68. Saradna A, Do DC, Kumar S, Fu QL, Gao P. Macrophage polarization and allergic asthma. Transl Res. 2018;191:1-14
69. Canfrán-Duque A, Rotllan N, Zhang X, Andrés-Blasco I, Thompson BM, Sun J. et al. Macrophage-Derived 25-Hydroxycholesterol Promotes Vascular Inflammation, Atherogenesis, and Lesion Remodeling. Circulation. 2023;147:388-408
70. Zhang X, McDonald JG, Aryal B, Canfrán-Duque A, Goldberg EL, Araldi E. et al. Desmosterol suppresses macrophage inflammasome activation and protects against vascular inflammation and atherosclerosis. Proceedings of the National Academy of Sciences. 2021;118:e2107682118
71. Qiu Y, Xu J, Yang L, Zhao G, Ding J, Chen Q. et al. MiR-375 silencing attenuates pro-inflammatory macrophage response and foam cell formation by targeting KLF4. Exp Cell Res. 2021;400:112507
72. Ceolotto G, Giannella A, Albiero M, Kuppusamy M, Radu C, Simioni P. et al. miR-30c-5p regulates macrophage-mediated inflammation and pro-atherosclerosis pathways. Cardiovasc Res. 2017;113:1627-38
73. Bouchareychas L, Duong P, Covarrubias S, Alsop E, Phu TA, Chung A. et al. Macrophage Exosomes Resolve Atherosclerosis by Regulating Hematopoiesis and Inflammation via MicroRNA Cargo. Cell Rep. 2020;32:107881
74. Puylaert P, Zurek M, Rayner KJ, De Meyer GRY, Martinet W. Regulated Necrosis in Atherosclerosis. Arterioscler Thromb Vasc Biol. 2022;42:1283-306
75. Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the Vulnerable Plaque. J Am Coll Cardiol. 2006;47:C13-C8
76. Martinet W, Schrijvers DM, De Meyer GRY. Necrotic cell death in atherosclerosis. Basic Res Cardiol. 2011;106:749-60
77. Seimon T, Tabas I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. J Lipid Res. 2009;50:S382-S7
78. Bedoui S, Herold MJ, Strasser A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nat Rev Mol Cell Biol. 2020;21:678-95
79. Boada-Romero E, Martinez J, Heckmann BL, Green DR. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol. 2020;21:398-414
80. Xiao Z, Liu M, Yang F, Liu G, Liu J, Zhao W. et al. Programmed cell death and lipid metabolism of macrophages in NAFLD. Front Immunol. 2023;14:1118449
81. Kalinina N, Agrotis A, Antropova Y, DiVitto G, Kanellakis P, Kostolias G. et al. Increased Expression of the DNA-Binding Cytokine HMGB1 in Human Atherosclerotic Lesions. Arterioscler Thromb Vasc Biol. 2004;24:2320-5
82. Kanellakis P, Agrotis A, Kyaw TS, Koulis C, Ahrens I, Mori S. et al. High-Mobility Group Box Protein 1 Neutralization Reduces Development of Diet-Induced Atherosclerosis in Apolipoprotein E-Deficient Mice. Arterioscler Thromb Vasc Biol. 2011;31:313-9
83. de Souza AWS, Westra J, Limburg PC, Bijl M, Kallenberg CGM. HMGB1 in vascular diseases: Its role in vascular inflammation and atherosclerosis. Autoimmun Rev. 2012;11:909-17
84. Eelen G, Treps L, Li X, Carmeliet P. Basic and Therapeutic Aspects of Angiogenesis Updated. Cir Res. 2020;127:310-29
85. Kavurma MM, Rayner KJ, Karunakaran D. The walking dead: macrophage inflammation and death in atherosclerosis. Curr Opin Lipidol. 2017;28:91-8
86. Hansson GK, Libby P, Tabas I. Inflammation and plaque vulnerability. J Intern Med. 2015;278:483-93
87. Moreno PR, Purushothaman M, Purushothaman KR. Plaque neovascularization: defense mechanisms, betrayal, or a war in progress. Ann N Y Acad Sci. 2012;1254:7-17
88. Takeda Y, Costa S, Delamarre E, Roncal C, Leite de Oliveira R, Squadrito ML. et al. Macrophage skewing by Phd2 haplodeficiency prevents ischaemia by inducing arteriogenesis. Nature. 2011;479:122-6
89. Spiller KL, Anfang RR, Spiller KJ, Ng J, Nakazawa KR, Daulton JW. et al. The role of macrophage phenotype in vascularization of tissue engineering scaffolds. Biomaterials. 2014;35:4477-88
90. Sluimer JC, Gasc J-M, van Wanroij JL, Kisters N, Groeneweg M, Sollewijn Gelpke MD. et al. Hypoxia, Hypoxia-Inducible Transcription Factor, and Macrophages in Human Atherosclerotic Plaques Are Correlated With Intraplaque Angiogenesis. J Am Coll Cardiol. 2008;51:1258-65
91. Taqueti VR, Di Carli MF, Jerosch-Herold M, Sukhova GK, Murthy VL, Folco EJ. et al. Increased Microvascularization and Vessel Permeability Associate With Active Inflammation in Human Atheromata. Circ Cardiovasc Imaging. 2014;7:920-9
92. Michel JB, Thaunat O, Houard X, Meilhac O, Caligiuri G, Nicoletti A. Topological determinants and consequences of adventitial responses to arterial wall injury. Arterioscler Thromb Vasc Biol. 2007;27:1259-68
93. Finn AV, Nakano M, Polavarapu R, Karmali V, Saeed O, Zhao X. et al. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J Am Coll Cardiol. 2012;59:166-77
94. Cai Y, Pan J, Li Z. Mathematical modeling of intraplaque neovascularization and hemorrhage in a carotid atherosclerotic plaque. Biomed Eng Online. 2021;20:42
95. Michel JB, Virmani R, Arbustini E, Pasterkamp G. Intraplaque haemorrhages as the trigger of plaque vulnerability. Eur Heart J. 2011;32:1977-85 85a, 85b, 85c
96. Leclercq A, Houard X, Philippe M, Ollivier V, Sebbag U, Meilhac O. et al. Involvement of intraplaque hemorrhage in atherothrombosis evolution via neutrophil protease enrichment. J Leukoc Biol. 2007;82:1420-9
97. Li B, Lu M, Wang H, Sheng S, Guo S, Li J. et al. Macrophage Ferroptosis Promotes MMP2/9 Overexpression Induced by Hemin in Hemorrhagic Plaque. Thromb Haemost. 2023;124:568-80
98. Guo L, Akahori H, Harari E, Smith SL, Polavarapu R, Karmali V. et al. CD163+ macrophages promote angiogenesis and vascular permeability accompanied by inflammation in atherosclerosis. J Clin Invest. 2018;128:1106-24
99. Liu X, Ma J, Ma L, Liu F, Zhang C, Zhang Y. et al. Overexpression of tissue factor induced atherothrombosis in apolipoprotein E-/- mice via both enhanced plaque thrombogenicity and plaque instability. J Mol Cell Cardiol. 2019;127:1-10
100. Li J, Zhang P, Chen H, Qing T, Wang Y, Zhou J. et al. Vulnerable Carotid Plaque is Associated with an Increased Risk of Intracerebral Hemorrhage in Individuals at High Risk of Stroke: A Chinese Population-based Cohort Study. Curr Neurovasc Res. 2023;20:244-53
101. Cao G, Xuan X, Hu J, Zhang R, Jin H, Dong H. How vascular smooth muscle cell phenotype switching contributes to vascular disease. Cell Commun Signal. 2022;20:180
102. Beck-Joseph J, Tabrizian M, Lehoux S. Molecular Interactions Between Vascular Smooth Muscle Cells and Macrophages in Atherosclerosis. Front Cardiovasc Med. 2021;8:737934
103. Yurdagul A. Crosstalk Between Macrophages and Vascular Smooth Muscle Cells in Atherosclerotic Plaque Stability. Arterioscler Thromb Vasc Biol. 2022;42:372-80
104. Butoi E, Gan AM, Tucureanu MM, Stan D, Macarie RD, Constantinescu C. et al. Cross-talk between macrophages and smooth muscle cells impairs collagen and metalloprotease synthesis and promotes angiogenesis. Biochim Biophys Acta Mol Cell Res. 2016;1863:1568-78
105. Miteva K, Burger F, Baptista D, Roth A, Da Silva RF, Mach F. et al. Effect of monocytes on NLRP3 inflammasome activation in vascular smooth muscle cells phenotypic switch and foam cells formation in atherosclerosis. Atherosclerosis. 2020;315:e20
106. Yu L, Xu L, Chu H, Peng J, Sacharidou A, Hsieh H-h. et al. Macrophage-to-endothelial cell crosstalk by the cholesterol metabolite 27HC promotes atherosclerosis in male mice. Nature communications. 2023;14:4101
107. Xu Z, Wang X, Zheng J, Peng L, Zhou Y, Song Y. et al. Extracellular-vesicle containing miRNA-503-5p released by macrophages contributes to atherosclerosis. Aging. 2021;13:12239-57
108. Liu P, Wang S, Wang G, Zhao M, Du F, Li K. et al. Macrophage-derived exosomal miR-4532 promotes endothelial cells injury by targeting SP1 and NF-κB P65 signalling activation. J Cell Mol Med. 2022;26:5165-80
109. Susser LI, Rayner KJ. Through the layers: how macrophages drive atherosclerosis across the vessel wall. J Clin Invest. 2022;132:e157011
110. Witztum JL, Lichtman AH. The Influence of Innate and Adaptive Immune Responses on Atherosclerosis. Annu Rev Pathol. 2014;9:73-102
111. Lahoute C, Herbin O, Mallat Z, Tedgui A. Adaptive immunity in atherosclerosis: mechanisms and future therapeutic targets. Nat Rev Cardiol. 2011;8:348-58
112. Lutgens E, Lievens D, Beckers L, Wijnands E, Soehnlein O, Zernecke A. et al. Deficient CD40-TRAF6 signaling in leukocytes prevents atherosclerosis by skewing the immune response toward an antiinflammatory profile. J Exp Med. 2010;207:391-404
113. Ouimet M, Ediriweera HN, Gundra UM, Sheedy FJ, Ramkhelawon B, Hutchison SB. et al. MicroRNA-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J Clin Invest. 2015;125:4334-48
114. Song M, Xu S, Zhong A, Zhang J. Crosstalk between macrophage and T cell in atherosclerosis: Potential therapeutic targets for cardiovascular diseases. Clin Immunol. 2019;202:11-7
115. Gaba P, Gersh BJ, Muller J, Narula J, Stone GW. Evolving concepts of the vulnerable atherosclerotic plaque and the vulnerable patient: implications for patient care and future research. Nat Rev Cardiol. 2023;20:181-96
116. Li X, Wu M, Li J, Guo Q, Zhao Y, Zhang X. Advanced targeted nanomedicines for vulnerable atherosclerosis plaque imaging and their potential clinical implications. Front Pharmacol. 2022;13:906512
117. Tang Tjun Y, Howarth Simon PS, Miller Sam R, Graves Martin J, Patterson Andrew J, Jean-Marie UK-I. et al. The ATHEROMA (Atorvastatin Therapy: Effects on Reduction of Macrophage Activity) Study. J Am Coll Cardiol. 2009;53:2039-50
118. Kwiecinski J, Lassen ML, Slomka PJ, Azhdarinia A. Advances in Quantitative Analysis of 18F-Sodium Fluoride Coronary Imaging. Mol Imaging. 2021;2021:8849429
119. Strauss HW, Narula J. Imaging Vulnerable Plaque. J Am Coll Cardiol. 2017;69:1792-4
120. Tarkin Jason M, Joshi Francis R, Evans Nicholas R, Chowdhury Mohammed M, Figg Nichola L, Shah Aarti V. et al. Detection of Atherosclerotic Inflammation by 68Ga-DOTATATE PET Compared to [18F]FDG PET Imaging. J Am Coll Cardiol. 2017;69:1774-91
121. Tearney Guillermo J, Regar E, Akasaka T, Adriaenssens T, Barlis P, Bezerra Hiram G. et al. Consensus Standards for Acquisition, Measurement, and Reporting of Intravascular Optical Coherence Tomography Studies. J Am Coll Cardiol. 2012;59:1058-72
122. Ding J, Wang Y, Ma M, Zhang Y, Lu S, Jiang Y. et al. CT/fluorescence dual-modal nanoemulsion platform for investigating atherosclerotic plaques. Biomaterials. 2013;34:209-16
123. Si-Mohamed SA, Sigovan M, Hsu JC, Tatard-Leitman V, Chalabreysse L, Naha PC. et al. In Vivo Molecular K-Edge Imaging of Atherosclerotic Plaque Using Photon-counting CT. Radiology. 2021;300:98-107
124. Chen W, Schilperoort M, Cao Y, Shi J, Tabas I, Tao W. Macrophage-targeted nanomedicine for the diagnosis and treatment of atherosclerosis. Nat Rev Cardiol. 2022;19:228-49
125. Qiao H, Wang Y, Zhang R, Gao Q, Liang X, Gao L. et al. MRI/optical dual-modality imaging of vulnerable atherosclerotic plaque with an osteopontin-targeted probe based on Fe3O4 nanoparticles. Biomaterials. 2017;112:336-45
126. Ge X, Cui H, Kong J, Lu S-Y, Zhan R, Gao J. et al. A Non-Invasive Nanoprobe for In Vivo Photoacoustic Imaging of Vulnerable Atherosclerotic Plaque. Adv Mater. 2020;32:2000037
127. Luo X, Shi J, Wang R, Cao L, Gao Y, Wang J. et al. Near-Infrared Persistent Luminescence Nanoprobe for Early Detection of Atherosclerotic Plaque. ACS Nano. 2024;18:6500-12
128. Hou J, Zhou J, Chang M, Bao G, Xu J, Ye M. et al. LIFU-responsive nanomedicine enables acoustic droplet vaporization-induced apoptosis of macrophages for stabilizing vulnerable atherosclerotic plaques. Bioact Mater. 2022;16:120-33
129. Wu M, Li X, Guo Q, Li J, Xu G, Li G. et al. Magnetic mesoporous silica nanoparticles-aided dual MR/NIRF imaging to identify macrophage enrichment in atherosclerotic plaques. Nanomed-Nanotechnol. 2021;32:102330
130. Li W, Xu L-H, Forssell C, Sullivan JL, Yuan X-M. Overexpression of Transferrin Receptor and Ferritin Related to Clinical Symptoms and Destabilization of Human Carotid Plaques. Exp Biol Med. 2008;233:818-26
131. Kraml P, Potocková J, Koprivová H, Stípek S, Crkovská J, Zima T. et al. Ferritin, oxidative stress and coronary atherosclerosis. Vnitr Lek. 2004;50:197-202
132. Liang M, Tan H, Zhou J, Wang T, Duan D, Fan K. et al. Bioengineered H-Ferritin Nanocages for Quantitative Imaging of Vulnerable Plaques in Atherosclerosis. ACS Nano. 2018;12:9300-8
133. Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin Immunol. 2006;121:144-58
134. Monsalve Y, Tosi G, Ruozi B, Belletti D, Vilella A, Zoli M. et al. PEG-g-chitosan nanoparticles functionalized with the monoclonal antibody OX26 for brain drug targeting. Nanomedicine (Lond). 2015;10:1735-50
135. Chen Y, Huang Y, Li Q, Luo Z, Zhang Z, Huang H. et al. Targeting Xkr8 via nanoparticle-mediated in situ co-delivery of siRNA and chemotherapy drugs for cancer immunochemotherapy. Nat Nanotechnol. 2023;18:193-204
136. Segawa K, Nagata S. An Apoptotic 'Eat Me'; Signal: Phosphatidylserine Exposure. Trends Cell Biol. 2015;25:639-50
137. Ogawa M, Uchino R, Kawai A, Kosugi M, Magata Y. PEG modification on 111In-labeled phosphatidyl serine liposomes for imaging of atherosclerotic plaques. Int J Nucl Med Biol. 2015;42:299-304
138. Narita Y, Shimizu K, Ikemoto K, Uchino R, Kosugi M, Maess MB. et al. Macrophage-targeted, enzyme-triggered fluorescence switch-on system for detection of embolism-vulnerable atherosclerotic plaques. J Control Release. 2019;302:105-15
139. Paulos CM, Turk MJ, Breur GJ, Low PS. Folate receptor-mediated targeting of therapeutic and imaging agents to activated macrophages in rheumatoid arthritis. Adv Drug Deliv Rev. 2004;56:1205-17
140. Winkel LC, Groen HC, van Thiel BS, Müller C, van der Steen AF, Wentzel JJ. et al. Folate receptor-targeted single-photon emission computed tomography/computed tomography to detect activated macrophages in atherosclerosis: can it distinguish vulnerable from stable atherosclerotic plaques? Mol Imaging. 2014 13
141. Jager NA, Westra J, van Dam GM, Teteloshvili N, Tio RA, Breek JC. et al. Targeted folate receptor β fluorescence imaging as a measure of inflammation to estimate vulnerability within human atherosclerotic carotid plaque. J Nucl Med. 2012;53:1222-9
142. Silvola JMU, Li XG, Virta J, Marjamäki P, Liljenbäck H, Hytönen JP. et al. Aluminum fluoride-18 labeled folate enables in vivo detection of atherosclerotic plaque inflammation by positron emission tomography. Sci Rep. 2018;8:9720
143. Wen X, Zeng X, Liu J, Zeng X, Zhang Y, Cheng X. et al. In Vivo Comparative Study of Radioiodinated Folate Receptor Targeting Albumin Probes for Atherosclerosis Plaque Imaging. Bioconjug Chem. 2023;34:2387-97
144. Ma Y, Xu L, Yin B, Shang J, Chen F, Xu J. et al. Ratiometric Semiconducting Polymer Nanoparticle for Reliable Photoacoustic Imaging of Pneumonia-Induced Vulnerable Atherosclerotic Plaque in Vivo. Nano Letters. 2021;21:4484-93
145. Qiao R, Qiao H, Zhang Y, Wang Y, Chi C, Tian J. et al. Molecular Imaging of Vulnerable Atherosclerotic Plaques in Vivo with Osteopontin-Specific Upconversion Nanoprobes. ACS Nano. 2017;11:1816-25
146. Wang S, Zhang X, Liu Y, Wang Y, Sun N, Yin J. et al. Osteopontin targeted non-invasive nanoprobes with amplified surface plasmon resonance for photothermally enhanced multimodal precision imaging of vulnerable atherosclerotic plaques. Chem Eng J. 2023;471:144766
147. Xu M, Mao C, Chen H, Liu L, Wang Y, Hussain A. et al. Osteopontin targeted theranostic nanoprobes for laser-induced synergistic regression of vulnerable atherosclerotic plaques. Acta Pharm Sin B. 2022;12:2014-28
148. Ye M, Zhou J, Zhong Y, Xu J, Hou J, Wang X. et al. SR-A-Targeted Phase-Transition Nanoparticles for the Detection and Treatment of Atherosclerotic Vulnerable Plaques. ACS Appl Mater Interfaces. 2019;11:9702-15
149. Dai Y, Sha X, Song X, Zhang X, Xing M, Liu S. et al. Targeted Therapy of Atherosclerosis Vulnerable Plaque by ROS-Scavenging Nanoparticles and MR/Fluorescence Dual-Modality Imaging Tracing. Int J Nanomedicine. 2022;17:5413-29
150. Gao B, Xu J, Zhou J, Zhang H, Yang R, Wang H. et al. Multifunctional pathology-mapping theranostic nanoplatforms for US/MR imaging and ultrasound therapy of atherosclerosis. Nanoscale. 2021;13:8623-38
151. Ma X, Wang J, Li Z, Zhou X, Liang X, Wang J. et al. Early Assessment of Atherosclerotic Lesions and Vulnerable Plaques in vivo by Targeting Apoptotic Macrophages with AV Nanobubbles. Int J Nanomedicine. 2022;17:4933-46
152. He Z, Chen Q, Duan X, Zhong Y, Zhu L, Mou N. et al. Reactive oxygen species-responsive nano-platform with dual-targeting and fluorescent lipid-specific imaging capabilities for the management of atherosclerotic plaques. Acta Biomater. 2024;181:375-90
153. Jager NA, Westra J, van Dam GM, Teteloshvili N, Tio RA, Breek J-C. et al. Targeted Folate Receptor β Fluorescence Imaging as a Measure of Inflammation to Estimate Vulnerability Within Human Atherosclerotic Carotid Plaque. J Nucl Med. 2012;53:1222-9
154. Winkel LCJ, Groen HC, van Thiel BS, Müller C, van der Steen AFW, Wentzel JJ. et al. Folate Receptor-Targeted Single-Photon Emission Computed Tomography/Computed Tomography to Detect Activated Macrophages in Atherosclerosis: Can It Distinguish Vulnerable from Stable Atherosclerotic Plaques? Mol Imaging. 2014;13:7290.2013.00061
155. Silvola JMU, Li X-G, Virta J, Marjamäki P, Liljenbäck H, Hytönen JP. et al. Aluminum fluoride-18 labeled folate enables in vivo detection of atherosclerotic plaque inflammation by positron emission tomography. Sci Rep. 2018;8:9720
156. Guo Z, Yang L, Chen M, Wen X, Liu H, Li J. et al. Molecular imaging of advanced atherosclerotic plaques with folate receptor-targeted 2D nanoprobes. Nano Res. 2020;13:173-82
157. Ishino S, Mukai T, Kuge Y, Kume N, Ogawa M, Takai N. et al. Targeting of Lectinlike Oxidized Low-Density Lipoprotein Receptor 1 (LOX-1) with <sup>99m</sup>Tc-Labeled Anti-LOX-1 Antibody: Potential Agent for Imaging of Vulnerable Plaque. J Nucl Med. 2008;49:1677-85
158. Li D, Patel AR, Klibanov AL, Kramer CM, Ruiz M, Kang B-Y. et al. Molecular Imaging of Atherosclerotic Plaques Targeted to Oxidized LDL Receptor LOX-1 by SPECT/CT and Magnetic Resonance. Circ Cardiovasc Imaging. 2010;3:464-72
159. Wen S, Liu D-F, Cui Y, Harris SS, Chen Y-c, Li KC. et al. In vivo MRI detection of carotid atherosclerotic lesions and kidney inflammation in ApoE-deficient mice by using LOX-1 targeted iron nanoparticles. Nanomed-Nanotechnol. 2014;10:639-49
160. Ma Q, Wu S, Yang L, Wei Y, He C, Wang W. et al. Hyaluronic Acid-Guided Cerasome Nano-Agents for Simultaneous Imaging and Treatment of Advanced Atherosclerosis. Adv Sci. 2023;10:2202416
161. Wang Y, Chen Z, Zhu Q, Chen Z, Fu G, Ma B. et al. Aiming at early-stage vulnerable plaques: A nanoplatform with dual-mode imaging and lipid-inflammation integrated regulation for atherosclerotic theranostics. Bioact Mater. 2024;37:94-105
162. Wu Q, Pan W, Wu G, Wu F, Guo Y, Zhang X. CD40-targeting magnetic nanoparticles for MRI/optical dual-modality molecular imaging of vulnerable atherosclerotic plaques. Atherosclerosis. 2023;369:17-26
163. Gao W, Li X, Liu Z, Fu W, Sun Y, Cao W. et al. A Redox-Responsive Self-Assembled Nanoprobe for Photoacoustic Inflammation Imaging to Assess Atherosclerotic Plaque Vulnerability. Anal Chem. 2019;91:1150-6
164. Han G-M, Liu B, Wang C-Y, Wang D-X, Li Q-N, Cai Q-L. et al. Diagnosis and Vulnerability Risk Assessment of Atherosclerotic Plaques Using an Amino Acid-Assembled Near-Infrared Ratiometric Nanoprobe. Anal Chem. 2024;96:10380-90
165. Ridker Paul M, Everett Brendan M, Thuren T, MacFadyen Jean G, Chang William H, Ballantyne C. et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377:1119-31
166. Ridker PM, Bhatt DL, Pradhan AD, Glynn RJ, MacFadyen JG, Nissen SE. Inflammation and cholesterol as predictors of cardiovascular events among patients receiving statin therapy: a collaborative analysis of three randomised trials. Lancet. 2023;401:1293-301
167. Yu M, Yang Y, Dong S-L, Zhao C, Yang F, Yuan Y-F. et al. Effect of Colchicine on Coronary Plaque Stability in Acute Coronary Syndrome as Assessed by Optical Coherence Tomography: The COLOCT Randomized Clinical Trial. Circulation. 2024;150:981-93
168. Moroni F, Ammirati E, Norata GD, Magnoni M, Camici PG. The Role of Monocytes and Macrophages in Human Atherosclerosis, Plaque Neoangiogenesis, and Atherothrombosis. Mediators Inflamm. 2019;2019:7434376
169. Dou Y, Guo J, Chen Y, Han S, Xu X, Shi Q. et al. Sustained delivery by a cyclodextrin material-based nanocarrier potentiates antiatherosclerotic activity of rapamycin via selectively inhibiting mTORC1 in mice. J Control Release. 2016;235:48-62
170. Wang H, Zhao R, Peng L, Yu A, Wang Y. A Dual-Function CD47-Targeting Nano-Drug Delivery System Used to Regulate Immune and Anti-Inflammatory Activities in the Treatment of Atherosclerosis. Adv Healthc Mater. 2024;13:2400752
171. Gao C, Liu C, Chen Q, Wang Y, Kwong CHT, Wang Q. et al. Cyclodextrin-mediated conjugation of macrophage and liposomes for treatment of atherosclerosis. J Control Release. 2022;349:2-15
172. Xu M, Ren C, Zhou Y, Heger Z, Liang X, Adam V. et al. Enhanced macrophage polarization induced by COX-2 inhibitor-loaded Pd octahedral nanozymes for treatment of atherosclerosis. Chin Chem Lett. 2023;34:107585
173. Li C, Fang F, Wang E, Yang H, Yang X, Wang Q. et al. Engineering extracellular vesicles derived from endothelial cells sheared by laminar flow for anti-atherosclerotic therapy through reprogramming macrophage. Biomaterials. 2025;314:122832
174. Zhao J, Ling L, Zhu W, Ying T, Yu T, Sun M. et al. M1/M2 re-polarization of kaempferol biomimetic NPs in anti-inflammatory therapy of atherosclerosis. J Control Release. 2023;353:1068-83
175. Fang F, Yang J, Wang J, Li T, Wang E, Zhang D. et al. The role and applications of extracellular vesicles in osteoporosis. Bone Res. 2024;12:4
176. Wu C, Mao J, Wang X, Yang R, Wang C, Li C. et al. Advances in treatment strategies based on scavenging reactive oxygen species of nanoparticles for atherosclerosis. J Nanobiotechnology. 2023;21:271
177. He H, Han Q, Wang S, Long M, Zhang M, Li Y. et al. Design of a Multifunctional Nanozyme for Resolving the Proinflammatory Plaque Microenvironment and Attenuating Atherosclerosis. ACS Nano. 2023;17:14555-71
178. He Z, Chen W, Hu K, Luo Y, Zeng W, He X. et al. Resolvin D1 delivery to lesional macrophages using antioxidative black phosphorus nanosheets for atherosclerosis treatment. Nat Nanotechnol. 2024;19:1386-98
179. Sukhorukov VN, Khotina VA, Chegodaev YS, Ivanova E, Sobenin IA, Orekhov AN. Lipid Metabolism in Macrophages: Focus on Atherosclerosis. Biomedicines. 2020;8:262
180. Collins JL, Fivush AM, Watson MA, Galardi CM, Lewis MC, Moore LB. et al. Identification of a Nonsteroidal Liver X Receptor Agonist through Parallel Array Synthesis of Tertiary Amines. J Med Chem. 2002;45:1963-6
181. Yu M, Amengual J, Menon A, Kamaly N, Zhou F, Xu X. et al. Targeted Nanotherapeutics Encapsulating Liver X Receptor Agonist GW3965 Enhance Antiatherogenic Effects without Adverse Effects on Hepatic Lipid Metabolism in Ldlr Mice. Adv Healthc Mater. 2017;6:1700313
182. Cui K, Gao X, Wang B, Wu H, Arulsamy K, Dong Y. et al. Epsin Nanotherapy Regulates Cholesterol Transport to Fortify Atheroma Regression. Cir Res. 2023;132:e22-e42
183. Arandjelovic S, Ravichandran KS. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol. 2015;16:907-17
184. Jaiswal S, Jamieson CHM, Pang WW, Park CY, Chao MP, Majeti R. et al. CD47 Is Upregulated on Circulating Hematopoietic Stem Cells and Leukemia Cells to Avoid Phagocytosis. Cell. 2009;138:271-85
185. Kojima Y, Volkmer J-P, McKenna K, Civelek M, Lusis AJ, Miller CL. et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature. 2016;536:86-90
186. Chen L, Zhou Z, Hu C, Maitz MF, Yang L, Luo R. et al. Platelet Membrane-Coated Nanocarriers Targeting Plaques to Deliver Anti-CD47 Antibody for Atherosclerotic Therapy. Research. 2022;2022:9845459
187. Flores AM, Hosseini-Nassab N, Jarr K-U, Ye J, Zhu X, Wirka R. et al. Pro-efferocytic nanoparticles are specifically taken up by lesional macrophages and prevent atherosclerosis. Nat Nanotechnol. 2020;15:154-61
188. Jarr K-U, Ye J, Kojima Y, Ye Z, Gao H, Schmid S. et al. The pleiotropic benefits of statins include the ability to reduce CD47 and amplify the effect of pro-efferocytic therapies in atherosclerosis. Nat Cardiovasc Res. 2022;1:253-62
Author contact
![]() Corresponding author: Xiaoheng Liu: liuxiaohgedu.cn.
Corresponding author: Xiaoheng Liu: liuxiaohgedu.cn.