Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Conclusion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2025; 15(4):1221-1237. doi:10.7150/thno.105163 This issue Cite
Research Paper
Transferrin-binding domain inserted-adenovirus hexon engineering enables systemic immune evasion and intratumoral T-cell activation
Dae Hoon Lee1,2, Youngtae Kwon3, Ki Hwan Um1,4, Jung Ki Yoo1,3, Wootae Ha3, Ki-Su Kim1,3,4, Jintak Cha1,4, Ha-Eun Cho5, Kyung Sun Park5, Min Jeong Kye3, Jin Woo Choi1,2,3,4 ![]()
1. Department of Pharmacology, College of Pharmacy, Kyung Hee University, Seoul 02447, Republic of Korea.
2. Department of Biomedical and Pharmaceutical Sciences, College of Pharmacy, Kyung Hee University, Seoul 02447, Republic of Korea.
3. R&D Center of Curigin Ltd. Seoul 04778, Republic of Korea.
4. Department of Regulatory Science, College of Pharmacy, Kyung Hee University, Seoul 02447, Republic of Korea.
5. Department of Laboratory Medicine, Kyung Hee University College of Medicine, Kyung Hee University Medical Center, Seoul 02447, Republic of Korea.
Received 2024-10-14; Accepted 2024-12-9; Published 2025-1-1
Abstract

Rationale: Adenovirus-based therapies have encountered significant challenges due to host immunity, particularly from pre-existing antibodies. Many trials have struggled to evade antibody response; however, the efficiency of these efforts was limited by the diversity of antibody Fv-region recognizing multiple amino acid sequences.
Methods: In this study, we developed an antibody-evading adenovirus vector by encoding a plasma-rich protein transferrin-binding domain. The coding sequence was employed from Neisseria Meningitides and inserted in the experimentally-optimized site within the adenovirus capsid protein.
Result: This engineered antibody-evading oncolytic adenovirus overcame the reduction in productivity and infectivity typically caused by the insertion of a foreign domain. We observed decreased immune recognition and compromised formation of anti-adenovirus antibodies. Furthermore, the anti-tumor efficacy was demonstrated both in vitro and in vivo, with increased recruitment of CD8+ T cells.
Conclusion: This novel antibody-evading strategy effectively evades neutralizing antibodies and innate immunity while boosting cytotoxic immunity by recruiting CD8+ T cells at the tumor site. Additionally, this strategy holds potential for application in other gene therapies and adenovirus vectors.
Keywords: systemic injectable viral vector, antibody evading viral vector, hexon engineered adenovirus, adenovirus, oncolytic virus
Introduction
Recently, diverse therapies utilizing viral vectors have emerged, primarily focusing on gene therapy and anti-cancer treatments. In the research field of cancer treatment using viral vectors, significant progress has been made since the US Food and Drug Administration (FDA) approved the first cancer therapeutic virus, talimogene laherparepvec (also known as T-VEC or Imlygic), for the local treatment of recurrent melanoma [1]. Notable subsequent approvals include adenoviral vectors nadofaragene firadenovec-vncg (also known as Adstiladrin) [2] and cretostimogene grenadenorepvec [3] for non-muscle invasive bladder cancer that no longer responds to standard therapy. Following these approvals, a diverse array of research efforts continues to advance anti-cancer viral therapy [4-6]. However, the application of these FDA-approved oncolytic viruses remains restricted to regions that can be directly injected, such as the skin via intratumoral injection or bladder via intravesical instillation. This administration strategy intends to avoid immune responses such as a cytokine release syndrome and an attack from neutralizing antibodies. In the field of gene therapy using viral vectors, the FDA has approved onasemnogene abeparvovec (Zolgensma) for the treatment of spinal muscular atrophy [7]. Similar to previous virotherapies, it may face challenges due to neutralizing antibodies in circulation from its second injection, which can diminish the efficacy of the treatment [8]. Consequently, most viral vector-based therapies still have a potential immune limitation and risk, depending on the specific condition of the patients.
Adenoviruses are known for their relatively low pathogenicity compared to other types of viruses in the field of oncolytic virotherapy [9]. They typically cause only mild infectious symptoms that are self-limiting [10]. In addition, in adverse effects, anti-adenoviral treatment with cidofovir can expect a rapid suppression of viral propagation [11]. Thus, adenovirus may be a safer option for therapeutic use. Despite the safety profile of oncolytic adenoviruses, pre-existing neutralizing antibodies to adenovirus are a significant challenge to compromise the therapeutic efficacy. These antibodies are commonly found across most people over the continents and are present in high concentrations in mouse serum [12-14]. While the high prevalence of antibodies suggests safety [15, 16], it still poses a hurdle for the effectiveness of oncolytic adenoviruses.
Numerous efforts to develop adenoviral vectors that can escape the immune system have focused on evading antibody recognition [17-19]. However, these approaches, which typically involve modifying only a few amino acids, may offer limited efficacy in evading an attack of antibody as anti-adenoviral antibodies target multiple position poly clonally, not just the modified sequence regions. Consequently, more comprehensive strategies for covering adenoviruses have gained prominence. One innovative strategy involves using plasma-rich proteins such as albumin, to surround and shield the virus [20]. This approach entails inserting an albumin-binding domain into the capsid protein of the adenovirus. Despite its novel concept, this strategy has limitations, primarily reduced infectivity caused by the insertion of the unnatural albumin-binding domain [20].
Thus, to preserve the natural character of adenovirus, the location of the sequence modification or domain insertion should be attentively designed with delicacy. The hypervariable regions (HVRs) of the adenovirus hexon protein may be an amenable region for modification, prompting extensive research into HVR modifications [21-23]. Additional key considerations include ensuring the structural change of capsid by the inserted domain, abundancy of the targeted plasma binding protein, and the binding affinity. The transferrin binding domain from transferrin binding protein A (TbpA) of Neisseria meningitidis emerged as a suitable candidate, as TbpA binds transferrin regardless of iron binding status, unlike transferrin binding protein B (TbpB) [24]. Our focus was thus on the transferrin binding domain of TbpA, particularly the loop 3 helix, which is critical for transferrin binding [25, 26]. We adopted this domain and successfully inserted it into the HVRs.
This antibody-evading adenoviral vector system can be utilized for the treatment of metastatic cancers via systemic administration and also serve as an efficient tool for gene delivery.
Results
Insertion site determination of for exogenous peptide
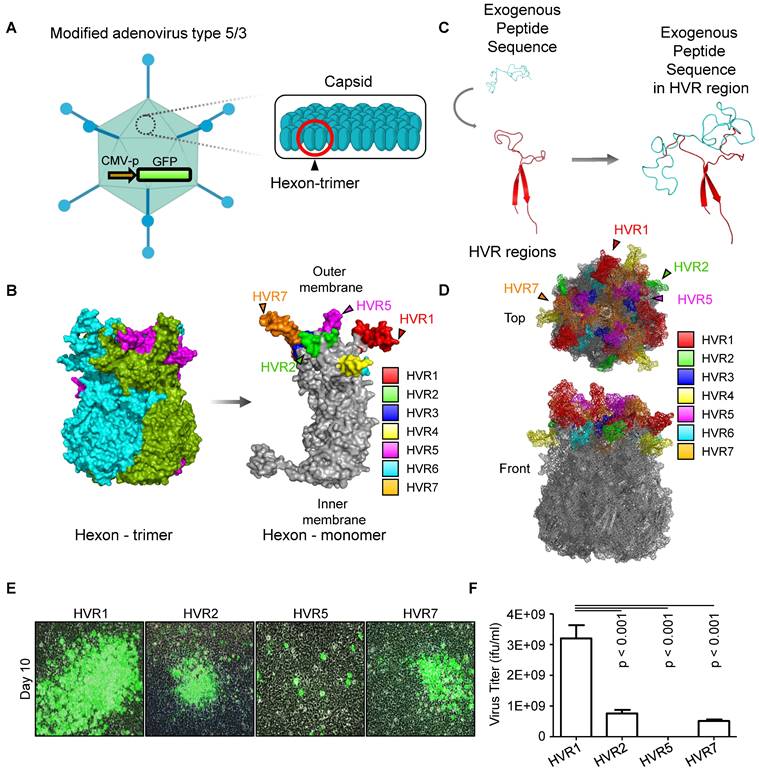
To establish a systemic injectable adenoviral vector, the optimal insertion site for exogenous peptides within the hexon protein needs to be experimentally determmined. Using a adenoviral vector, which is GFP-encoding oncolytic adenovirus serotype 5/3 (oAd5/3-GFP) having a basic backbone adenovirus, but GFP-expressed for visualization, we modified the hexon, unit protein of capsid (Figure 1A). Hexon protein exists as a trimer, with each monomer containing hypervariable regions 1-7 (HVR1-7), which are known to tolerate amino acid sequence modifications (Figure 1B). To evaluate spatial flexibility, an albumin-binding domain was used as the exogenous peptide first [27, 28], inserted into the bulged regions of each HVR domain (Figure 1C). Additionally, the HVRs (HVR1, 2, 5, and 7), which are the candidates for domain insertion, are indicated in the 3D hexon trimer model. (Figure 1D). We tested the remaining four HVRs for exogenous peptide insertion and measured the virus productivities (Figure 1, E and F). Viral replication-induced plaque was observed in HVR1, HVR2, and HVR7, but not in HVR5, with HVR1 showing a fourfold higher production yield compared to other sites.
Identification of hypervariable region (HVR) positioning for domain insertion within adenovirus type 5/3 hexon protein. (A) The adenovirus type 5/3 utilized in this study was engineered to express green fluorescent protein (GFP). The hexon protein, a component of the adenovirus capsid, was targeted and modified in this research. (B) The structure of the hexon protein (both trimer and monomer forms), a component of the adenovirus type 5/3 capsid, was illustrated, highlighting the hypervariable regions 1-7 (HVR1-7) in color. (C) Conceptual illustration of exogenous peptide insertion into HVR domains. (D) Visualization of HVR domains within the hexon trimer. (E and F) The albumin binding domain (ABD) was inserted into the tip regions of HVR1, 2, 5, and 7. The productivity of ABD-inserted viruses at these positions was analyzed to identify the optimal insertion site. (E) Representative images of plaque formation in virus production. At 10 days post-transfection of plasmid vectors into HEK-293 cells, plaque formation was confirmed via imaging. (F) Lysates from the cells in E were obtained through three freeze-thaw cycles, and the virus titer of each lysate was calculated.
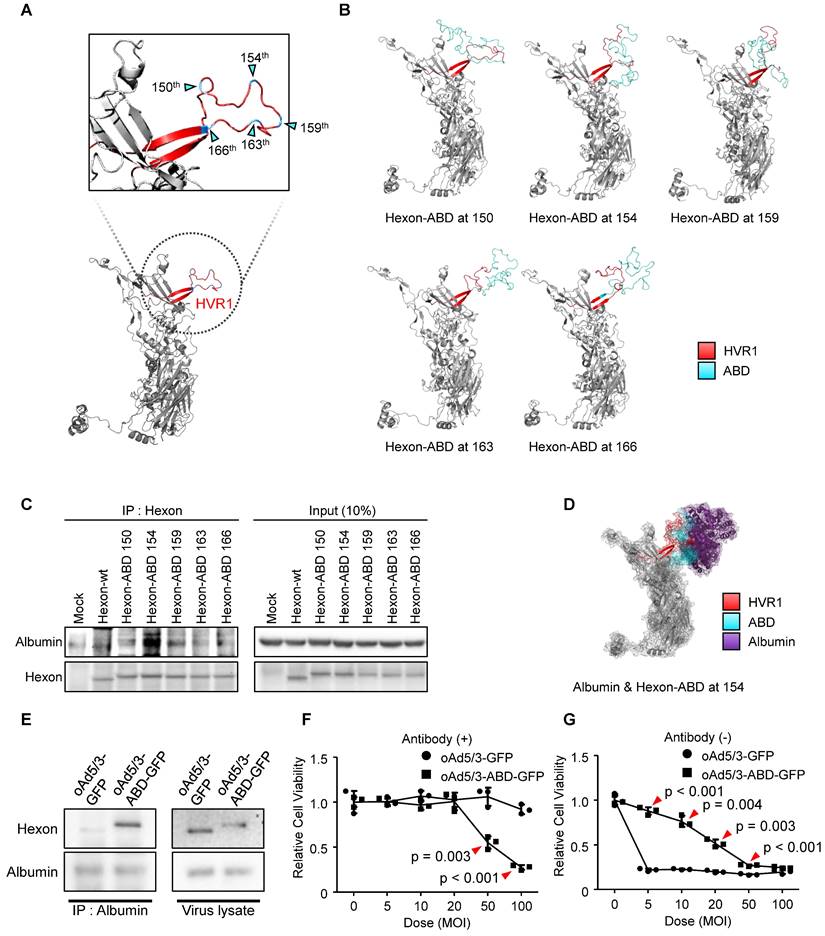
To determine the optimal insertion site within HVR1 at the single amino acid resolution, five sites within its bulged region were selected (Figure 2A). The hexon proteins were designed to bind albumin protein by insertion of albumin binding domain (ABD) at each selected site (Figure 2B). Binding affinities between albumin and hexon were evaluated to identify the optimal site using immunoprecipitation method (Figure 2C). While hexons with insertions at positions 150, 159, 163, and 166 exhibited weak binding, the hexon with insertion at position 154 showed significantly higher binding affinity (Figure 2C), and 3D modeling of this binding interaction was conducted and analyzed. (Figure 2D). Using this optimized position, an ABD-inserted oncolytic adenovirus serotype 5/3 (oAd5/3) was produced (named oAd5/3-ABD-GFP), and binding was verified (Figure 2E). Although oAd5/3-ABD-GFP maintained an oncolytic effect even in the presence of blocking antibodies, unlike oAd5/3-GFP (Figure 2F). The oncolytic activity appeared to be reduced by ABD insertion (Figure 2G).
Optimization for domain insertion position within hypervariable region 1 (HVR1). (A) Protein structure of the adenovirus type 5/3 hexon. The magnified region shows HVR1 (red) with potential insertion sites for the albumin binding domain (ABD) marked by cyan triangles. (B) 3D-models of hexon proteins with ABD inserted at specific amino acid positions (150th, 154th, 159th, 163th, and 166th). (C) Comparison of albumin binding affinities of ABD-inserted hexon proteins using an immunoprecipitation assay. Vectors expressing either wild-type hexon or ABD-inserted hexon proteins were transfected into HEK-293 cells. Hexon proteins were over-expressed and pulled down with anti-adenovirus type 5/3 hexon antibody in the presence of albumin. (D) 3D-structure of the interaction between hexon-ABD (at the 154th position) and albumin protein. (E) Immunoprecipitation assay to confirm the albumin binding ability of the oncolytic adenovirus type 5/3 (oAd5/3-ABD-GFP) expressing GFP. 1.0 x 1012 viral particles were mixed with 2μg albumin protein and pulled down with an anti-albumin antibody. (F) Cell viability assay comparing the cytotoxic effects of oAd5/3-ABD-GFP and oAd5/3-GFP on 1.0 x 104 A549 cells at various doses. (G) Cell viability assay assessing the antibody evasion capability of oAd5/3-ABD-GFP in the presence of 100ng/ml adenovirus neutralizing antibody on 1.0 x 104 A549 cells at various doses.
Establishment of antibody-evading viral vector via transferrin binding feature
Albumin interacts with various biomolecules and drugs, affecting their pharmacological actions [29-31]. This interaction may alter the concentration of active drugs, necessitating therapeutic drug monitoring in clinical settings [32, 33]. Moreover, ABD insertion impaired viral infectious ability (Figure 2G), prompting the search for alternative domains that bind plasma proteins.
Thus, we selected five plasma protein candidates to replace albumin, based on their concentrations in plasma [34] (Figure 3A). To evaluate potential cancer-specific delivery, receptor expressions for these plasma proteins were analyzed across 1019 human cancer cell lines using Human Protein Atlas [35-42] (Figure 3B). In addition, to exclude infection to normal cells we elicited cancer-specific receptors by the expression difference between cancerous- (n = 1019) and non-cancerous cells (n = 63) (Figure 3C). As a result, the transferrin receptor 1 (TFRC) emerged as the most highly expressed cancer-specific receptor.
Protein selection in human blood as a shielding protein for establishment of antibody-evading adenovirus. (A) The blood plasma is enriched with five primary proteins. Low-optimal and high-optimal concentration in blood plasma were represented with box. (B) Bar plot with mean and SEM for expression levels of receptors for albumin using normalized transcripts per million (nTPM), transferrin, transthyretin, haptoglobin, and hemopexin across 1019 cancer cell lines. Receptors: FCGRT (albumin), TFRC (transferrin), LRP2 (transthyretin), CD163 (haptoglobin), and CD91 (hemopexin). (C) Comparison of receptor expression in cancerous vs. non-cancerous cells. nTPM of Figure 3B (n = 1019) were devided by average nTPM of non-cancerous cells (n = 63). Data was illustrated using bar plot with mean and SEM. (D) 3D-models of hexon proteins with TBD inserted at specific positions (150th, 154th, 159th, 163th, and 166th). (E) Analysis of binding stability for TBD-transferrin interaction using root mean square deviation (RMSD). Mean and standard-deviation were represented. (F) Analysis of binding stability for TBD-transferrin interaction using electrostatic energy. Mean and standard-deviation were represented. (G) Comparison of 3D-binding models of ABD-albumin and TBD-transferrin. (H) Comparison of TBD-transferrin and ABD-albumin binding stability using RMSD. Mean and standard-deviation were represented. (I) Comparison of TBD-transferrin and ABD-albumin binding stability using electrostatic energy. Mean and standard-deviation were represented.
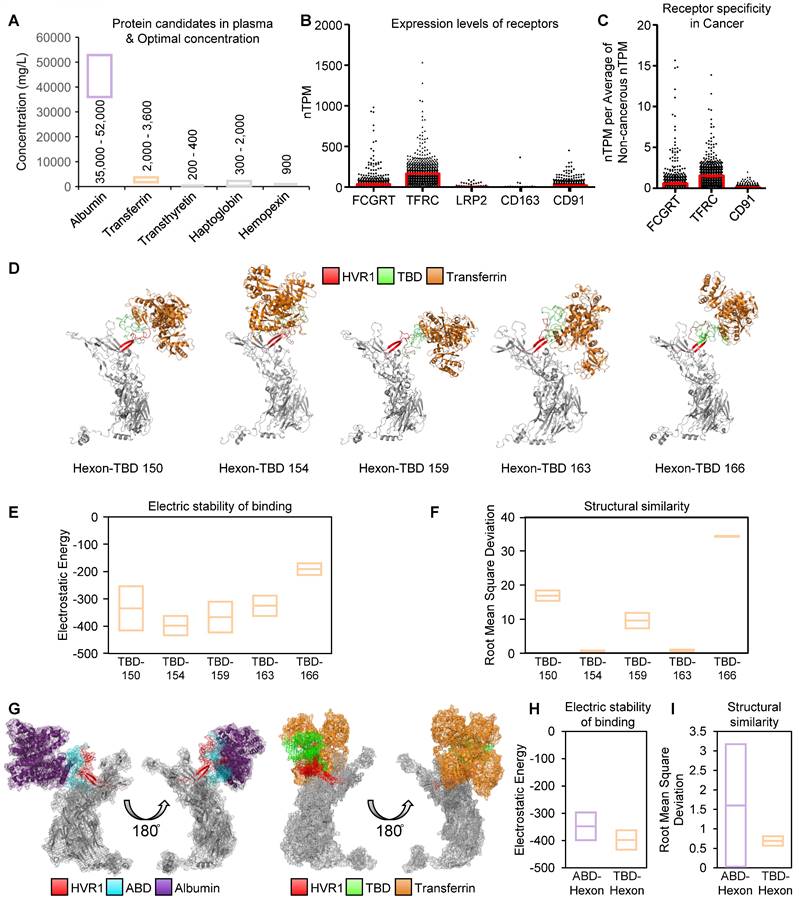
To establish a transferrin binding domain (TBD), we adopted the transferrin binding motif from the Neisseria species' TbpA protein, which binds human transferrin independently of ferrous binding, unlike TbpB [24]. The loop 3 helix of TbpA, critical for transferrin binding [25, 26], was investigated as a TBD. We optimized the insertion of TBD into oAd5/3 (named oAd5/3-TBD-GFP) by modeling interactions between modified hexon proteins and transferrin at multiple positions (150, 154, 159, 163, and 166) (Figure 3D). Molecular binding stability was assessed using electrostatic energy and root mean square deviation [43, 44] (Figure 3, E and F), with the 154th position showing the lowest scores and energy states, matching the optimal ABD insertion site. The binding stability of TBD-154 was the lowest one by electrostatic energy calculation (Figure 3E), and the TBD-154 model had the best structural similarity via the lowest value of root mean sqaure deviation (Figure 3F). 3D modeling revealed that TBD-transferrin binding was vertical, whereas ABD-albumin binding was horizontal, potentially causing interference between albumin molecules (Figure 3G). Binding affinities were predicted to be higher in TBD-inserted hexon with transferrin compared to ABD-inserted hexon with albumin (Figure 3, H and I).
Validation of oAd5/3-TBD-GFP construction
As illustrated in Figure 3, we developed oAd5/3-TBD-GFP, an adenovirus modified with a transferrin binding domain (TBD) inserted at the 154th position of the hexon protein. To check the physical properties of the engineered virus, a series of analyses were conducted. The size of oAd5/3-TBD-GFP was compared to that of oAd5/3-GFP using size exclusion-high-performance liquid chromatography (SEC-HPLC). The results indicated identical peak times for both, suggesting comparable sizes (Figure 4A). The surface charge properties, reflecting the outer membrane characteristics, were analyzed via ion exchange-high-performance liquid chromatography (IEC-HPLC). Unlike SEC-HPLC, the peak of oAd5/3-TBD-GFP appeared earlier than that of oAd5/3-GFP. This shift is attributable to the higher isoelectric point (relatively basic) of TBD compared to the viral hexon, which alters the elution profile (Figure 4B). According to the reference, the theoretical isoelectric points of the hexon and the TBD-inserted hexon were 5.17 and 5.25, respectively, while the isoelectric point of TBD alone was 8.06 [45, 46]. The peak shift observed in IEC-HPLC may be attributed to the exposure of the TBD on the outer membrane.
Property validation of oAd5/3-TBD-GFP for construction. (A) Size exclusion high-performance liquid chromatography (HPLC) analysis comparing the properties of oAd5/3-GFP and oAd5/3-TBD-GFP. (B) Ion exchange HPLC analysis comparing the properties of oAd5/3-GFP and oAd5/3-TBD-GFP. (C) Morphology of oAd5/3-GFP and oAd5/3-TBD-GFP visualized by cryo-EM. (D) Cryo-EM images of oAd5/3-GFP and oAd5/3-TBD-GFP after incubation with 1 μg/ml transferrin solution. Red arrows indicates white blob on the surface of virus. (E) The white layer size analysis of C and D conducted using ImageJ. (F) Immunoprecipitation assay to confirm transferrin binding ability of oAd5/3-TBD-GFP compared to oAd5/3-GFP. 1.0 x 1012 viral particles were mixed with 2μg transferrin protein and pulled down with an anti-transferrin antibody. (G) Cell viability assay comparing the cytotoxic effects of oAd5/3-TBD-GFP and oAd5/3-GFP on 1.0 x 104A549 cells at various doses. (H) Cell viability assay assessing the antibody evasion capability of oAd5/3-TBD-GFP in the presence of adenovirus neutralizing antibody on 1.0 x 104 A549 cells at various doses.
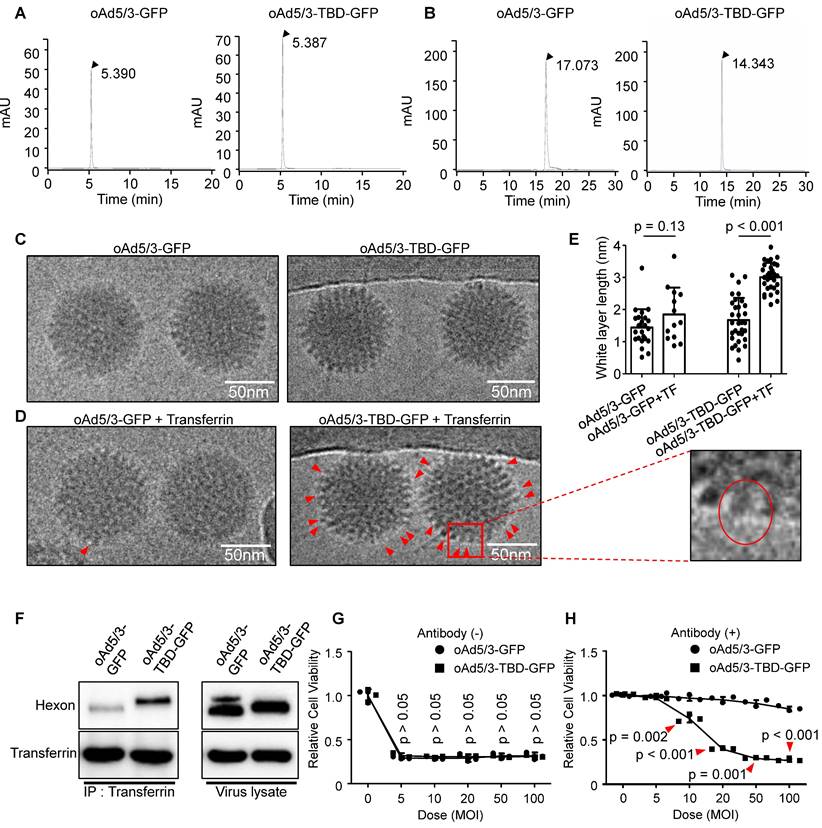
Further analysis involved visualizing virus establishment and transferrin binding using an electron microscope. The construction of oAd5/3-TBD-GFP was confirmed by its shape, which closely resembled that of oAd5/3-GFP (Figure 4C). The key acquired feature of oAd5/3-TBD-GFP was its ability to bind transferrin, as demonstrated by electron microscopy images showing transferrin binding (Figure 4D).
Since the non-tagged proteins appeared as white blobs [20, 47-49], the white layer was analyzed to assess the transferrin-virus interaction. Condensed white blobs were observed on the surface of oAd5/3-TBD-GFP, appearing to cover the virus, whereas no such layer was present on oAd5/3-GFP (Figure 4D). To quantify this interaction, the thickness of the white layer in Figure 4C and 4D was measured and analyzed (Figure 4E). In transferrin-incubated oAd5/3-GFP was similar in layer size to naïve oAd5/3-GFP, whereas transferrin-incubated oAd5/3-TBD-GFP showed an increase in layer size compared to naïve oAd5/3-TBD-GFP.
The interaction between oAd5/3-TBD-GFP and transferrin was re-validated through immunoprecipitation, confirming the virus's ability to evade antibodies via TBD-mediated shielding (Figure 4F). The oncolytic potential of oAd5/3-TBD-GFP remained unaffected by the TBD insertion (Figure 4G). Notably, in the presence of anti-adenovirus antibodies, oAd5/3-TBD-GFP sustained its oncolytic efficacy at multiplicities of infection (MOI) of 50 and 100 (Figure 4H).
Property comparison of oAd5/3-TBD-GFP with oAd5/3-ABD-GFP
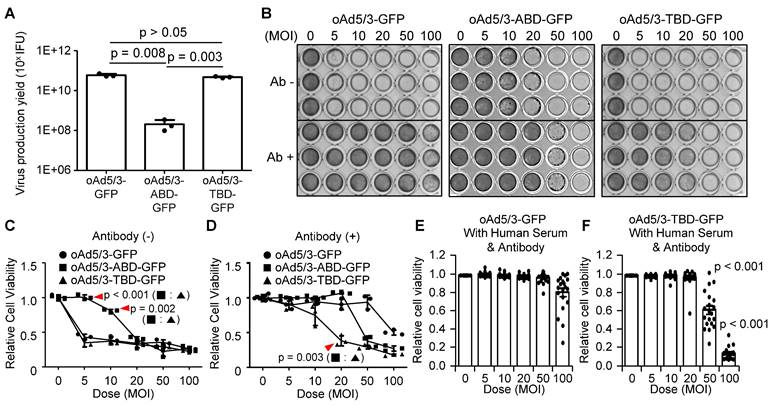
Finally to identify a clinically more useful antibody-evading virus, a comparative analysis was conducted between oAd5/3-ABD-GFP and oAd5/3-TBD-GFP. First, the yield of virus production was assessed. The productivity of oAd5/3-TBD-GFP was found to be 229 times higher than that of oAd5/3-ABD-GFP, indicating that ABD insertion partially impairs viral production (Figure 5A). Further, a comparison of viral cytotoxicity using crystal violet staining revealed that even though both viruses exhibited antibody-evading capabilities, the oncolytic capacity itself of oAd5/3-ABD-GFP was a little reduced compared with naïve oAd5/3 (Figure 5B). This finding was corroborated by cell viability assays (Figure 5, C and D). To evaluate the antibody-evading capability in human blood, 20 blood samples from healthy individuals were tested. The antibody evasion was assessed in 1% human serum media with added anti-adenovirus antibodies. In this setting, the infectivity of oAd5/3-GFP was entirely blocked across all doses (Figure 5E), whereas oAd5/3-TBD-GFP maintained its cytotoxicity (Figure 5F).
Property comparison of oAd5/3-TBD-GFP with oAd5/3-ABD-GFP. (A) Comparison of virus production yield between oAd5/3-GFP and oAd5/3-TBD-GFP. Viruses were produced in 1.0 x 109 HEK-293 cells, harvested after 48 h, and quantified. (B) Comparison of the oncolytic abilities of oAd5/3-GFP, oAd5/3-ABD-GFP, and oAd5/3-TBD-GFP using crystal violet staining on A549 cells infected at different multiplicities of infection (MOI) with or without adenovirus neutralizing antibody. (C and D) Comparison of the oncolytic abilities of oAd5/3-GFP, oAd5/3-ABD-GFP, and oAd5/3-TBD-GFP using cell viability assays in the absence (C) and presence (D) of 50ng/ml of neutralizing antibody on A549 cells infected at different MOIs. (E and F) Cell viability tests evaluating the antibody-evading ability of oAd5/3-GFP (E) and oAd5/3-TBD-GFP (F) in RPMI medium containing 1% human blood serum and 100ng/ml anti-adenovirus neutralizing antibodies. The assay used serum samples from 20 individuals The statistical analysis was calculated between 50 MOI of (E and F), and 100 MOI of (E and F) using two-tailed t-test.
Immune-refractory experiment of oAd5/3-TBD-GFP
Prior to initiating in vivo assessments of oAd5/3-TBD-GFP, the biodistribution profile of the viral construct was comprehensively analyzed. Spatiotemporal distribution studies demonstrated that intravenous administration of oAd5/3 in mice resulted in peak viral presence at the 24 h mark post-injection [50]. Consistent with previous findings, we subsequently evaluated the biodistribution of oAd5/3-TBD-GFP at this time point (Figure S1). Comparative analyses indicated no statistically significant differences in organ distribution between oAd5/3-TBD-GFP and the parental vector, oAd5/3-GFP. Although natural clearance of the adenovirus occurred within 24 h post-injection [50], further confirmation was obtained by assessing the vectors' sensitivity to the anti-adenoviral agent cidofovir (Figure S2) [51-54]. Both oAd5/3-TBD-GFP and oAd5/3-GFP exhibited comparable susceptibility to cidofovir, with no significant differences observed in their responsiveness.
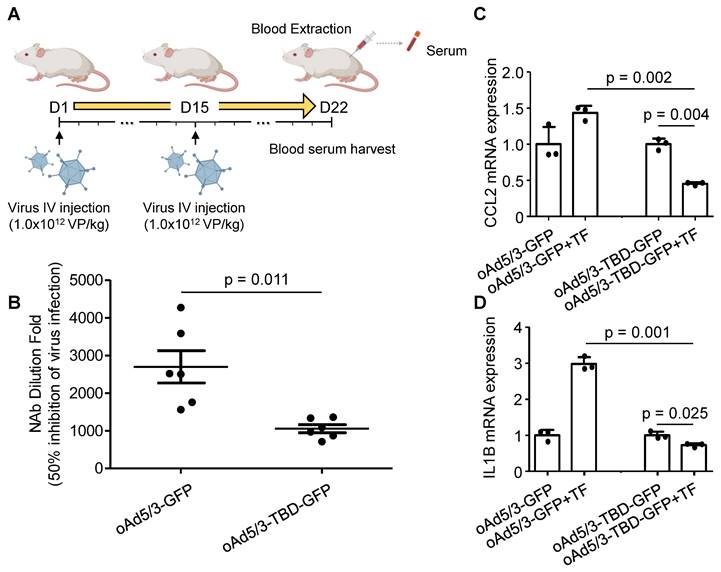
To assess the immune response in vivo, we measured anti-adenovirus neutralizing antibody production. BALB/c mice were intravenously administered oAd5/3-GFP and oAd5/3-TBD-GFP on days 1 and 15. Blood samples were collected on day 22 (Figure 6A). Analysis of serum samples revealed that the antibody titer induced by oAd5/3-GFP was significantly higher, at 2.56 times that of oAd5/3-TBD-GFP (Figure 6B). Given that antibody production reflects immune system activation, the primary immune response was further investigated. M1 macrophages, known to recognize pathogens via phagocytosis and secrete inflammatory cytokines such as CCL2 and IL-1B, were studied using differentiated U937 cells. In the presence of transferrin, CCL2 expression increased with oAd5/3-GFP but decreased with oAd5/3-TBD-GFP (Figure 6C). IL-1B expression mirrored the CCL2 response (Figure 6D). Transferrin addition caused an increase in cytokine expression for oAd5/3-GFP. Whereas no such effect was observed with oAd5/3-TBD-GFP.
oAd5/3-TBD-GFP evades recognition by the immune system in vivo. (A) A schedule for harvesting blood serum samples from BALB/c mice exposed to oAd5/3-GFP and oAd5/3-TBD-GFP. (B) The dilution fold of mouse blood serum required to inhibit 50% of virus infection. The serum dilution fold for 50% inhibition of GFP expression is indicated in the table. (C and D) Differentiated U937 cells into M1 macrophages were infected with oAd5/3-GFP and oAd5/3-TBD-GFP, with or without 2μg/ml transferrin (TF). After 4 h, the expression of CCL2 (C) and IL-1B (D) from M1 macrophages was analyzed.
Oncolytic efficacy of oAd5/3-TBD-GFP in a metastatic lung cancer model
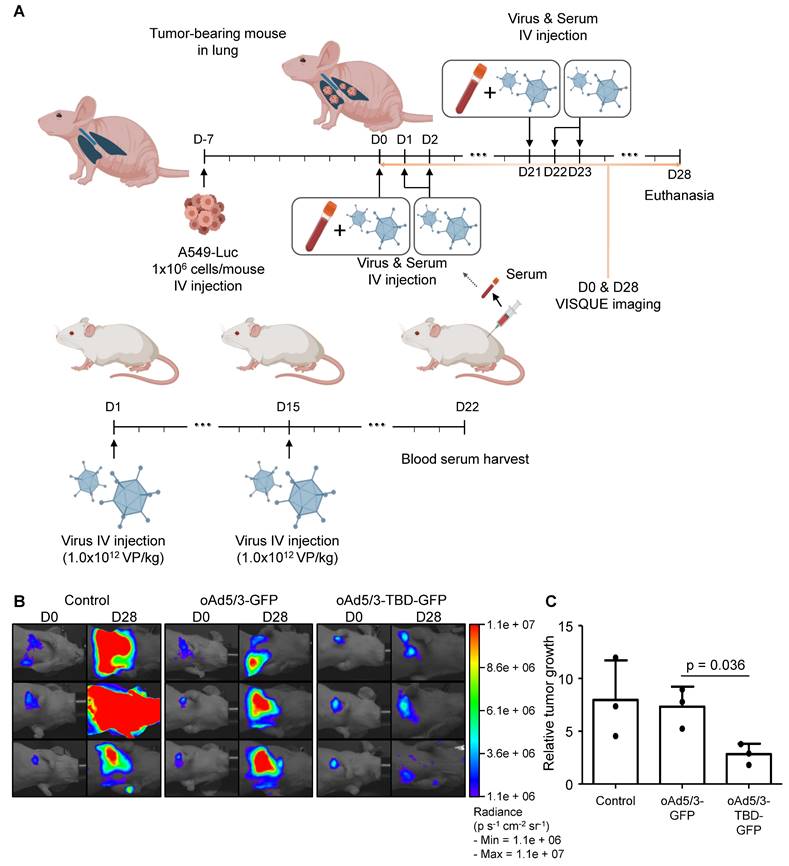
Prior to these in vivo studies, the antibody evasion potential of oAd5/3-TBD-GFP was assessed using mouse serum. The results confirmed that oAd5/3-TBD-GFP effectively circumvented antibody-mediated neutralization (Figure S3). To evaluate the oncolytic efficacy of oAd5/3-TBD-GFP, a metastatic lung cancer model was utilized to assess antibody evasion (Figure 7A). Tumor size was measured on day 28 using luciferase activity (Figure 7B). The oAd5/3-GFP treated group showed no significant regression in tumor growth. Conversely, the oAd5/3-TBD-GFP group exhibited significant tumor regression compared to oAd5/3-GFP treated groups. Tumor growth and statistical analysis are presented (Figure 7C). The therapeutic efficacy of oAd5/3-TBD-GFP was further investigated in a metastatic ovarian cancer model (Figure S4). The model was established via intraperitoneal injection of cancer cells, followed by intravenous administration of the virus. Tumor burden was monitored through bioluminescent imaging (Figure S4A). Notably, treatment with oAd5/3-TBD-GFP resulted in a substantial reduction in tumor growth, whereas the oAd5/3-GFP-treated group showed tumor progression akin to the control group (Figure S4B).
oAd5/3-TBD-GFP maintains oncolytic effect in the presence of antibodies in a metastatic lung cancer model in vivo. (A) A graphic illustrating the establishment of a metastatic lung cancer model with A549-luc cells and the production method for anti-adenovirus antibody from BALB/c mice (n = 3 per group). The administration route and schedule of A549-luc cells, virus, and serum are described. (B) Luciferase activities of A549-luc cells captured using a VISQUE instrument at 28 days after the first virus injection. (C) Graphs showing the calculation of A549-luc cancer cell growth based on VISQUE imaging and chemiluminescence. Tumor growth was normalized by calculating the ratio of intensity at day 28 (D28) relative to the baseline intensity at day 0 (D0) to minimize variability arising from differences in initial tumor size.
CD8+ T cell infiltration enhanced by oAd5/3-TBD-GFP through antibody evasion
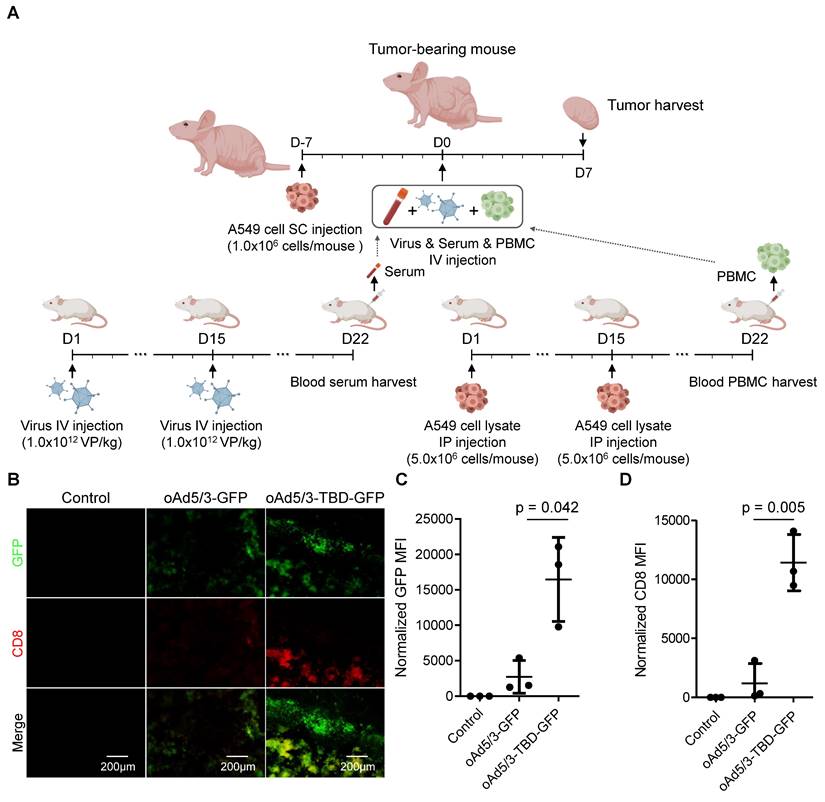
To further explore the immune response, we examined tumor-infiltrating CD8+ T cell populations. Serum containing specific antibodies against oAd5/3-GFP or oAd5/3-TBD-GFP, along with adapted peripheral blood mononuclear cells (PBMCs), were transplanted into a tumor-bearing nude mouse model (Figure 8A). On day 7, tumors were harvested and stained for CD8+ T cell markers. Both virus infection and CD8+ T cell presence were detected in the tumors of both treatment groups (Figure 8B). Expectedly, the levels of infection and CD8+ T cell recruitment were significantly higher in the oAd5/3-TBD-GFP group compared to the control (Figure 8, C and D). In contrast, there were no significant differences observed in CD4+ T cell recruitment between groups (Figure S5A-C).
CD8+ T cell infiltration is augmented in the oAd5/3-TBP-GDP treatment group in a xenograft mouse model by increased infection through antibody evasion. (A) A graphic illustrating the establishment of a lung cancer xenograft mouse model with A549 cells and the production method for anti-adenovirus antibody and A549-adapted PBMC from BALB/c mice. The administration route and schedule of A549 cells, virus, serum, and PBMC are described (n = 3 per group). (B) Representative images of GFP and CD8 expression in tumors from (A). Tumors were harvested and sectioned on days 7 to analyze virus infection and CD8+ T cell infiltration. Tumor sections were stained with CD8 antibody (red), and fluorescence images were captured, showing green for virus infection and red for CD8+ T cell infiltration. (C) Quantification of GFP expression from (B). (D) Quantification of CD8 expression from (B).
Discussion
Despite recent success in developing effective adenovirus-based oncolytic viruses [2, 4], upon systemic administration, the primary obstacle to the efficacy of the viral therapy has been the host immune response, particularly antibody recognition. The recent clinical trial, despite its noted advancements, continued to encounter challenges related to the immune response [55]. Due to the increase in neutralizing anti-adenovirus antibodies, the efficacy of the treatment might be compromised. Although adenovirus offers a safer profile compared to other viral vectors, the prevalence of anti-adenoviral antibodies is significantly higher [12-14]. This immune recognition hampers the delivery of intravenously administered adenoviruses to tumor cells, as they are often neutralized before reaching their target. For these reasons, several studies are focused on the bladder, which allows for more efficient non-systemic delivery to evade systemic immune reactions [2, 50]. To overcome this limitation, we engineered a novel adenovirus capable of infecting cells in the presence of antibodies through its interaction with human blood transferrin (Figure 5F).
In our research, adenovirus serotype 5/3 (oAd5/3), where the knob is replaced with that of adenovirus serotype 3 to enhance gene delivery and antitumor efficacy, was utilized as a basic backbone for modification [56-63]. The results related to virus neutralization indicated that the traditional oAd5/3 (oAd5/3-GFP) lost its infectivity in the presence of anti-adenovirus antibodies, resulting in reduced efficacy both in vitro and in vivo. However, oAd5/3-TBD-GFP successfully evaded attacks of antibody, including those from the innate immune system.
The concept of antibody-evading oAd5/3-TBD-GFP involves covering the virus with transferrin protein. When transferrin proteins coat oAd5/3-TBD-GFP, the innate immune system's recognition of the virus as a foreign antigen should be reduced. This theoretical process was validated by the decreased recognition by M1 macrophages (Figure 6, C and D). Sequentially, the innate immune system's antigen recognition leads to antibody production for the antigen, which was also observed to decrease (Figure 6, A and B). The basic concept of transferrin interaction aims to evade antibody attacks from both pre-existing antibodies (Figure 5, C-F, Figure 7B, and Figure 8B) and newly produced antibodies (Figure 6B). This immuno-silencing effect is thus demonstrated from a multi-dimensional perspective.
The previously developed albumin-binding domain (ABD)-based virus faced significant limitations for systemic delivery, including reduced infectivity and unpredictable interactions due to the high concentration and diverse nature of albumin (Figure 2G). In contrast, oAd5/3-TBD-GFP did not exhibit such reductions in infectivity (Figure 5, B and C) [20]. Additionally, oAd5/3-TBD-GFP demonstrated the ability to evade not only antibody-mediated neutralization but also broader immune recognition (Figure 6 and Figure 7), with a concomitant increase in CD8+ T cell recruitment (Figure 8).
As the oncolytic virus has the potential as a combinatorial regimen with immune checkpoint inhibitors, the challenge of oncolytic adenoviral therapy lies in balancing immune evasion and immune activation. The goal is to evade immunosurveillance during systemic delivery while enhancing immune response at the tumor site to maximize anti-cancer effects through immune cell recruitment. The engineered virus oAd5/3-TBD-GFP seemed to successfully achieve this balance. It evaded antigen recognition and antibody attacks during systemic circulation (Figure 5, Figure 6, and Figure 7) and promoted CD8+ T cell recruitment at the tumor site (Figure 8). Given that immune checkpoint inhibitors (ICIs) enhance the anti-cancer activity of CD8+ T cells, oAd5/3-TBD-GFP emerges as a promising combinatory partner for ICIs, providing a targeted approach to cancer therapy.
Recent advancements have concentrated on cytokine-armed oncolytic viruses, such as those encoding granulocyte-macrophage colony-stimulating factor (GM-CSF) and interferon-α [1, 2]. While these viruses demonstrate significant efficacy in specific indications, their ability to treat a broad range of cancer types remains limited, necessitating systemic administration. Thus, the development of a systemically injectable virus, like our oAd5/3-TBD-GFP, represents a significant leap forward. This virus not only functions as an oncolytic agent but also serves as a versatile gene therapy vector, capable of delivering a wide array of genetic materials.
Transferrin was chosen over albumin due to its lower serum concentration, which is sufficient to shield oAd5/3-TBD-GFP. A single viral particle of oAd5/3-TBD-GFP, comprising 720 hexon proteins, requires only 2μg of transferrin to coat 1 x 1012 viral particles, within the available transferrin concentration of 2,000-3,600μg/mL in blood. Moreover, leveraging transferrin receptor-mediated delivery pathways, as evidenced in brain delivery and cancer targeting studies [64, 65], suggests that oAd5/3-TBD-GFP may enhance tumor selectivity.
Taken together, the optimized insertion of the transferrin-binding domain into adenovirus may provide a new aspect to gene delivery expecting subsequent clinical implications.
Conclusion
The systemic injectable oncolytic adenovirus oAd5/3-TBD-GFP, engineered to evade antibody-mediated neutralization through the insertion of a transferrin-binding domain at the HVR1-154 position, was successfully developed. The antibody evasion capability of oAd5/3-TBD-GFP was validated both in vitro and in vivo. Additionally, oAd5/3-TBD-GFP exhibited reduced immunogenicity and enhanced tumor infiltration by CD8+ T cells, resulting in significant tumor size reduction in a metastatic lung cancer mouse model.
Methods
Production of oncolytic adenoviruses
The oncolytic adenovirus serotype 5/3 (oAd5/3)-GFP, oAd5/3-ABD-GFP, and oAd5/3-TBD-GFP were generated using adenovirus-producing plasmid vectors obtained from O.D.260 Inc. To ensure cancer cell-specific replication, the E1 promoter region was replaced with the human telomerase reverse transcriptase (hTERT) promoter for both oAd5/3-GFP and oAd5/3-TBD-GFP. For GFP expression, CMV promoter was used, and it is graphically described in Figure 1A. For the specific features of oAd5/3-TBD-GFP, the native hexon gene was substituted with a hexon gene bearing the transferrin binding domain. Albumin binding domain was following amino acid sequence; MGCSSHEHEHEDEAVDANSLAAAKETAL-YHLDRLGVADAYKDLIDKAKTVEGVKARYFEILHALPDDNEDEVDEQAEQQKTHVFGQA. Transferrin binding domain was following amino acid sequence; MDMTVPAFLTKAVFDANKKQAGSLPGNGKYAGNHKYGGLFTNGENGALVGAEYGT. These domains were incorporated at multiple insertion sites as illustrated in Figures 1-3, with the primary insertion point for the transferrin-binding domain (TBD) and albumin-binding domain (ABD) identified at the 154th amino acid position of the hexon protein.
Quantitative analysis of adenovirus titer
The concentration of adenovirus was determined using the infectious unit (IFU), calculated based on the ratio of adenovirus-infected cells to total cells in the field of view. HEK-293 (KCLB, 21573) cells were seeded into a 12-well plate at a density of 5×105 cells per each well. The adenovirus solution was serially diluted from 1:101 to 1:109. After 48 h of incubation with the virus diluent, cells were washed with PBS and fixed with -20℃ methanol. The concentration of adenovirus was then analyzed using the Adeno-X™ Rapid Titer Kit (Takara, 632250).
Receptor Expression analysis in cancerous and non-cancerous cell lines
Receptor expression levels across various cancerous and non-cancerous cell lines were obtained from The Human Protein Atlas and visualized using GraphPad Prism.
3D-structure modeling and binding affinity calculation
Following the incorporation of the domain into the hexon gene sequence, the corresponding amino acid sequence was used to generate a 3D structural model via the SWISS-MODEL server [66]. To evaluate protein-protein binding affinities, the 3D structures of the domain-modified hexon protein, albumin, and transferrin were obtained in PDB format. These structures were then uploaded into HADDOCK to calculate binding affinities and generate interaction models [44]. Visualization of protein structures was performed using PyMOL.
Cryogenic electron microscopy (Cryo-EM)
A 4 μL aliquot of virus sample, diluted in PBS, was applied to a hydrophilic grid (Quantifoil, R1.2/1.3, 300 mesh, EMS) prepared using a glow discharge system (PELCO easiGlow™, Ted Pella). The sample was blotted for 1.5 seconds at 4°C with 100% humidity, using a force setting of -3. Following vitrification in liquid ethane (Vitrobot Mark IV, FEI), the samples were analyzed at 120 kV using an electron microscope (Talos L120C, FEI).
Immunoprecipitation assay
2μg of transferrin (InVitria, 777TRF029) or 2μg albumin (Sigma, A1653) was added to 1.0 x 1012 viral particles of oAd5/3-GFP and oAd5/3-TBD-GFP (or oAd5/3-ABD-GFP), and the mixtures were incubated at room temperature for 2 h. For pull-down, 2μg of anti-transferrin antibody (Santa Cruz, sc-365871) or 2μg of anti-albimin antibody (Santa Cruz, sc-271605) was added to each tube, and protein was collected using protein A/G agarose beads (Santa Cruz, sc-2003).
Immunoblotting assay
5x SDS sample buffer (LPS solutions, CBS002) and 1x cell lysis buffer (Cell Signaling, 9803S) were utilized for SDS-PAGE. All samples were loaded onto SDS-polyacrylamide gels and electrophoresed at 80 V for 30 min and then at 120 V for 90 min. Proteins were transferred to polyvinylidene fluoride (PVDF) membranes (Merck, IPVH08100). The PVDF membranes were blocked with blocking buffer which is 5% skim milk in TBST buffer (LPS solutions, CBT007L). Primary antibody incubation was performed at a dilution fold of 1:1,000, followed by secondary antibody incubation at a dilution of 1:10,000, both in blocking buffer. All antibodies were diluted accordingly.
Antibody evasion ability test for oAd5/3-ABD-GFP or oAd5/3-TBD-GFP
To coat the virus with a coating protein, viruses were incubated in a 2μg/ml albumin solution or 2μg/ml transferrin solution state for 1 h at 4℃. The coated virus was then administered to cells, and antibodies were simultaneously diluted into the cell growth medium at a 1:1,000 dilution fold. In this experiment, anti-adenovirus antibodies (Abcam, ab6982) were used as the adenovirus neutralizing reagent.
For the method using human blood serum, blood samples were obtained from twenty voluntary blood donors (IRB no. KHUH2023-01-016-001). The blood samples were centrifuged at 1,500xg for 20 min, and serum samples were harvested. For the experiment on transferrin-mediated antibody evasion on the virus, blood serum containing transferrin was used instead of recombinant transferrin solution.
Cell viability assay
Cell viability was measured by trypan blue staining. Cells were harvested with 0.05% trypsin-EDTA solution after appropriate treatment. Harvested cells were stained with trypan blue solution (ThermoFisher, T10282) for 5 min, and the proportion of live and dead cells was measured automatically using the Countess 3 instrument (ThermoFisher, AMQAX2000). Each batch was measured three times.
Crystal violet staining
Cells were fixed with pre-chilled 100% methanol for 5 min at -20°C. Subsequently, a 1% crystal violet solution (Sigma, V5265) was added to the cells. After methanol fixation and crystal violet staining, cells were washed three times with PBS.
Purity confirmation of adenovirus through size exclusion-high-performance liquid chromatography (SEC-HPLC) and ion exchange-high-performance liquid chromatography (IEC-HPLC)
SEC-HPLC was performed using a 1290 Infinity II Prime HPLC (Agilent) and a TSKgel® G3000SWXL HPLC Column (MERCK). PBS was used for priming and washing steps. For IEC-HPLC, a Resource™ Q column (Cytiva) was utilized. Trizma-based buffer was used for priming and washing steps.
Animal experiments
Five-week-old BALB/c male mice and five-week-old BALB/c nude male mice were purchased from Orient Bio (Gyeonggi, Korea). All animal experiments were reviewed and approved by the Institutional Review Board (IRB, approval number: KHSASP-24-117) and the Institutional Animal Care and Use Committee (IACUC, approval number: CRG-RNDC02.01-02), and performed according to the criteria of the IRB and IACUC guidelines. Mice were maintained in pathogen-free facilities.
Assessment of neutralizing antibody titers in mouse serum
To determine the efficacy of neutralizing antibodies, 1.0 × 10⁴ A549 cells were seeded into 96-well plates. Serial 1:10 dilutions of mouse serum were prepared, and cells were infected with either oAd5/3-GFP or oAd5/3-TBD-GFP in the presence of the corresponding neutralizing serum. After 24 h, neutralizing antibody titers were quantified using the Fluoroskan™ FL Microplate Fluorometer (ThermoFisher). A standard curve was established using a commercial anti-adenovirus neutralizing antibody (Abcam, ab6982), which demonstrated that a 1:40,000 dilution inhibited 50% GFP expression for both viral constructs. Neutralizing antibody titers were then calculated based on this standard curve.
Antibody productivity test in vivo
For the antibody productivity test, 1.0 x 1012 viral particles (VP)/kg of oAd5/3-GFP and oAd5/3-TBD-GFP were injected intravenously into six-week-old BALB/c male mice on day 1 (n = 6 per group). To boost antibody production, 1.0 x 1012 VP/kg of oAd5/3-GFP and oAd5/3-TBD-GFP were reinjected intravenously on day 15. On day 22, all mice were euthanized, and blood was harvested.
Metastatic lung and ovarian cancer model and in vivo neutralization assay
To immunize BALB/c nude mice against human cells, materials derived from BALB/c mice were injected into BALB/c nude mice. 1.0 x 1012 VP/kg were injected into male BALB/c mice on day 1 through the tail vein. To boost antibody production, 1.0 x 1012 VP/kg were reinjected into male BALB/c mice on day 15 through the same injection point as before. On day 22, mice were euthanized, and serum was extracted from blood samples (n = 6). Each serum sample was mixed and titrated using the neutralizing antibody assay protocol.
For the metastatic lung cancer model, 1.0 x 106 A549-luc2 cells (ATCC) were injected intravenously into six-week-old BALB/c nude male mice (n = 3 per group) via tail vein. After 7 days (on day 0), a 20 μL mixture of serum, equivalent in potency to 10 μg of anti-adenovirus antibody (Abcam, ab6982), was administered via tail vein injections on days 0 and 21, immediately prior to viral administration. On days 0, 1, 2, 21, 22, and 23, 5 x 108 ifu of oAd5/3-GFP and oAd5/3-TBD-GFP were intravenously injected into the mice through tail vein.
For the metastatic ovarian cancer model, 1.0 × 10⁵ HeyA8-luc cells (kindly provided by Prof. Jung-Won Lee, Samsung Medical Center, Korea) were intraperitoneally injected into six-week-old BALB/c nude female mice (n = 3 per group) on day 0. On day 4, 20 µL of serum with neutralizing potency equivalent to 10 µg of anti-adenovirus antibody (Abcam, ab6982) was administered intravenouslly. Concurrently, mice received an intravenous injection of 5 × 10⁸ IFU of either oAd5/3-GFP or oAd5/3-TBD-GFP.
Bioluminescence imaging
For bioluminescence imaging, 3mg of luciferin (Merck, L6152) was intraperitoneally injected into mice. After 10 min, the mice were placed into the VISQUE InVivo Smart-LF instrument (VIEWWORKS, BI24001). Luciferase activity was then analyzed in units of radiance (p s-1 cm-2 sr-1).
Establishment of mouse model of CD4+ and CD8+ T cell infiltration
To induce immunity against A549 cells in BALB/c mice, 5.0 x 106 A549 cells, a human alveolar adenocarcinoma cell line, were injected into mice intraperitoneally on days 1 and 15. On day 22, mice were euthanized, and peripheral blood mononuclear cells (PBMCs) were isolated from blood samples. Immediately after PBMC isolation, 5.0 x 106 PBMCs were intravenously injected into BALB/c nude mice as a supply of T cell immunity on day 0 of the tumor-bearing nude mouse model. The protocol used in this article for PBMC transplatation is developted based on in vitro activated T cell transplantation protocol [67-74].
Subsequently, 1.0 x 106 A549 cells (KCLB, 10185) were subcutaneously xenografted into six-week-old male BALB/c nude mice. Seven days after A549 cell injection, 20μl of blood serum which is mentioned at establishment of metastatic lung cancer mouse model in method section, 5.0 x 106 PBMCs, and 5.0 x 108 ifu of viruses (oAd5/3-GFP and oAd5/3-TBD-GFP, with an equivalent volume of PBS used for the virus control group) were intravenously injected into the tumor-bearing BALB/c nude mouse model (n = 3 per group). At 7 days after the administrations, tumor-bearing nude mice were euthanized, and tumors were harvested and snap-frozen for sectioning.
Immunofluorescence staining
To analyze the expression of CD4 and CD8 in tumors, samples were sectioned and stained for the analysis of target proteins. Tumor samples were placed on slide-glass and fixed with -20℃ methanol for 10 min. Subsequently, a 5% bovine serum albumin solution in PBS was used for the blocking step. CD4 and CD8 antigen was stained using Alexa Fluor 546-conjugated anti-CD4 antibody (Santa Cruz, sc-19641 AF546) and Alexa Fluor 546-conjugated anti-CD8 antibody (Santa Cruz, sc-1177 AF546).
Statistical analysis
The statistical significance of the data was determined using a two-tailed t-test. Significance levels are indicated in each figure, with the compared groups marked by bars under the respective p-values.
Abbreviations
FDA: food and drug administration; T-VEC: talimogene laherparepvec; HVRs: hypervariable regions; TbpA: transferrin binding protein A; TbpB: transferrin binding protein B; oAd5/3: oncolytic adenovirus serotype 5/3; GFP: green fluorescent protein; ABD: albumin binding domain; FCGRT: Fc gamma receptor and transporter; TFRC: transferrin receptor 1; LRP2: low-density lipoprotein (LDL) receptor related protein 2; TBD: transferrin binding domain; SEC-HPLC: size exclusion-high-performance liquid chromatography; IEC-HPLC: ion exchange-high-performance liquid chromatography; MOI: multiplicities of infection; CCL2: C-C motif chemokine ligand 2; IL-1B: interleukin 1 beta; PBMCs: peripheral blood mononuclear cells; ICIs: immune checkpoint inhibitors; GM-CSF: granulocyte-macrophage colony-stimulating factor; hTERT: the human telomerase reverse transcriptase; IFU: infectious unit; PBS: phosphate buffered saline; Cryo-EM: cryogenic electron microscopy; SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis; PVDF: polyvinylidene fluoride; TBST: tris buffered saline with tween-20; EDTA: ethylene-diamine-tetraacetic acid; VP: viral particle.
Supplementary Material
Supplementary figures.
Acknowledgements
We thank the tool devised by BioRender for providing the techniques to create illustrations. Certain illustrations have been created using BioRender.com. And we thank the tool devised by HADDOCK for providing the technique to calculate protein binding. The FP7 WeNMR (project# 261572), H2020 West-Life (project# 675858), the EOSC-hub (project# 777536) and the EGI-ACE (project# 101017567) European e-Infrastructure projects are acknowledged for the use of their web portals, which make use of the EGI infrastructure with the dedicated support of CESNET-MCC, INFN-LNL-2, NCG-INGRID-PT, TW-NCHC, CESGA, IFCA-LCG2, UA-BITP, TR-FC1-ULAKBIM, CSTCLOUD-EGI, IN2P3-CPPM, CIRMMP, SURFsara and NIKHEF, and the additional support of the national GRID Initiatives of Belgium, France, Italy, Germany, the Netherlands, Poland, Portugal, Spain, UK, Taiwan and the US Open Science Grid.
This research was supported by a grant (No. 21153601-1) from the Ministry of Food and Drug Safety in 2021 and Korea Drug Development Fund funded by Ministry of Science and ICT, Ministry of Trade, Industry, and Energy, and Ministry of Health and Welfare, Republic of Korea (RS-2022-DD124198 and RS-2023-00219308). This research also was supported by the National Research Foundation of Korea (NRF) grant funded by the Ministry of Science and ICT, South Korea (MSIT) (NRF-2022R1A2C2009281 and RS-2023-00262386), and a grant (21153MFDS601) from Ministry of Food and Drug Safety in 2024. Also, this research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (RS-2024-00455520).
Author contributions
D. H. L., Y. K., K. H. U., J. K. Y., W. H., and K. K. manufactured virus. D. H. L., J. C., H. E. C., and K. S. P. performed the experiments. D. H. L. and J. W. C. analyzed the data. D. H. L., M. J. K., and J. W. C. prepared the manuscript. J.W.C. supervised the study.
Data availability
All data generated and analyzed or supporting the findings of this study are available from the corresponding authors upon reasonable request.
Competing Interests
D. H. L., Y. K., K. H. U., J. K. Y., W. H., K. K., and M. J. K. partially received research grants from Curigin. Ltd. J. C., H. C., K. S. P., and J. W. C. declares no competing interests associated with this manuscript.
References
1. Pol J, Kroemer G, Galluzzi L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology. 2016;5:e1115641
2. Lee A. Nadofaragene firadenovec: first approval. Drugs. 2023;83:353-7
3. Li R, Shah PH, Stewart TF, Nam JK, Bivalacqua TJ, Lamm DL. et al. Oncolytic adenoviral therapy plus pembrolizumab in BCG-unresponsive non-muscle-invasive bladder cancer: the phase 2 CORE-001 trial. Nat Med. 2024;30:2216-2223
4. Packiam VT, Lamm DL, Barocas DA, Trainer A, Fand B, Davis RL. et al. An open label, single-arm, phase II multicenter study of the safety and efficacy of CG0070 oncolytic vector regimen in patients with BCG-unresponsive non-muscle-invasive bladder cancer: Interim results. Urol Oncol. 2018;36:440-7
5. Shoushtari AN, Olszanski AJ, Nyakas M, Hornyak TJ, Wolchok JD, Levitsky V. et al. Pilot study of ONCOS-102 and pembrolizumab: remodeling of the tumor microenvironment and clinical outcomes in anti-PD-1-resistant advanced melanoma. Clin Cancer Res. 2023;29:100-9
6. Musher BL, Rowinsky EK, Smaglo BG, Abidi W, Othman M, Patel K. et al. LOAd703, an oncolytic virus-based immunostimulatory gene therapy, combined with chemotherapy for unresectable or metastatic pancreatic cancer (LOKON001): results from arm 1 of a non-randomised, single-centre, phase 1/2 study. Lancet Oncol. 2024;25:488-500
7. Day JW, Finkel RS, Chiriboga CA, Connolly AM, Crawford TO, Darras BT. et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. 2021;20:284-93
8. Day JW, Finkel RS, Mercuri E, Swoboda KJ, Menier M, van Olden R. et al. Adeno-associated virus serotype 9 antibodies in patients screened for treatment with onasemnogene abeparvovec. Mol Ther Methods Clin Dev. 2021;21:76-82
9. Rasa A, Alberts P. Oncolytic virus preclinical toxicology studies. J Appl Toxicol. 2023;43:620-48
10. Lynch JP, Kajon AE. Adenovirus: Epidemiology, Global spread of novel serotypes, and advances in treatment and prevention. Semin Respir Crit Care Med. 2016;37:586-602
11. Matthes-Martin S, Feuchtinger T, Shaw PJ, Engelhard D, Hirsch HH, Cordonnier C. et al. European guidelines for diagnosis and treatment of adenovirus infection in leukemia and stem cell transplantation: summary of ECIL-4 (2011). Transpl Infect Dis. 2012;14:555-63
12. Sun C, Zhang Y, Feng L, Pan W, Zhang M, Hong Z. et al. Epidemiology of adenovirus type 5 neutralizing antibodies in healthy people and AIDS patients in Guangzhou, southern China. Vaccine. 2011;29:3837-41
13. Nwanegbo E, Vardas E, Gao W, Whittle H, Sun H, Rowe D. et al. Prevalence of neutralizing antibodies to adenoviral serotypes 5 and 35 in the adult populations of The Gambia, South Africa, and the United States. Clin Diagn Lab Immunol. 2004;11:351-7
14. Ono R, Nishimae F, Wakida T, Sakurai F, Mizuguchi H. Effects of pre-existing anti-adenovirus antibodies on transgene expression levels and therapeutic efficacies of arming oncolytic adenovirus. Sci Rep. 2022;12:21560
15. Chen Y, Yu DC, Charlton D, Henderson DR. Pre-existent adenovirus antibody inhibits systemic toxicity and antitumor activity of CN706 in the nude mouse LNCaP xenograft model: implications and proposals for human therapy. Hum Gene Ther. 2000;11:1553-67
16. Dhar D, Spencer JF, Toth K, Wold WS. Pre-existing immunity and passive immunity to adenovirus 5 prevents toxicity caused by an oncolytic adenovirus vector in the syrian hamster model. Mol Ther. 2009;17:1724-32
17. Shin DH, Jiang H, Gillard AG, Kim D, Fan X, Singh SK. et al. Chimeric oncolytic adenovirus evades neutralizing antibodies from human patients and exhibits enhanced anti-glioma efficacy in immunized mice. Mol Ther. 2024;32:722-33
18. Flickinger JC, Singh J, Carlson R, Leong E, Baybutt TR, Barton J. et al. Chimeric Ad5.F35 vector evades anti-adenovirus serotype 5 neutralization opposing GUCY2C-targeted antitumor immunity. J Immunother Cancer. 2020;8:e001046
19. Atasheva S, Emerson CC, Yao J, Young C, Stewart PL, Shayakhmetov DM. Systemic cancer therapy with engineered adenovirus that evades innate immunity. Sci Transl Med. 2020;12(571):eabc6659
20. Rojas LA, Condezo GN, Moreno R, Fajardo CA, Arias-Badia M, San Martin C. et al. Albumin-binding adenoviruses circumvent pre-existing neutralizing antibodies upon systemic delivery. J Control Release. 2016;237:78-88
21. Vigant F, Descamps D, Jullienne B, Esselin S, Connault E, Opolon P. et al. Substitution of hexon hypervariable region 5 of adenovirus serotype 5 abrogates blood factor binding and limits gene transfer to liver. Mol Ther. 2008;16:1474-80
22. Yu B, Wang C, Dong J, Zhang M, Zhang H, Wu J. et al. Chimeric hexon HVRs protein reflects partial function of adenovirus. Biochem Biophys Res Commun. 2012;421:170-6
23. Ma J, Duffy MR, Deng L, Dakin RS, Uil T, Custers J. et al. Manipulating adenovirus hexon hypervariable loops dictates immune neutralisation and coagulation factor X-dependent cell interaction in vitro and in vivo. PLoS Pathog. 2015;11:e1004673
24. Boulton IC, Gorringe AR, Allison N, Robinson A, Gorinsky B, Joannou CL. et al. Transferrin-binding protein B isolated from Neisseria meningitidis discriminates between apo and diferric human transferrin. Biochem J. 1998;334(Pt 1):269-73
25. Noinaj N, Buchanan SK, Cornelissen CN. The transferrin-iron import system from pathogenic Neisseria species. Mol Microbiol. 2012;86:246-57
26. Cash DR, Noinaj N, Buchanan SK, Cornelissen CN. Beyond the crystal structure: insight into the function and vaccine potential of TbpA expressed by Neisseria gonorrhoeae. Infect Immun. 2015;83:4438-49
27. Garousi J, Lindbo S, Nilvebrant J, Astrand M, Buijs J, Sandstrom M. et al. ADAPT, a novel scaffold protein-based probe for radionuclide imaging of molecular targets that are expressed in disseminated cancers. Cancer Res. 2015;75:4364-71
28. Nilvebrant J, Astrand M, Georgieva-Kotseva M, Bjornmalm M, Lofblom J, Hober S. Engineering of bispecific affinity proteins with high affinity for ERBB2 and adaptable binding to albumin. PLoS One. 2014;9:e103094
29. Evans TW. Review article: albumin as a drug —biological effects of albumin unrelated to oncotic pressure. Aliment Pharmacol Ther. 2002;16(Suppl 5):6-11
30. Yamasaki K, Chuang VT, Maruyama T, Otagiri M. Albumin-drug interaction and its clinical implication. Biochim Biophys Acta. 2013;1830:5435-43
31. Sakaida I, Nakajima K, Okita K, Hori M, Izumi T, Sakurai M. et al. Can serum albumin level affect the pharmacological action of tolvaptan in patients with liver cirrhosis? A post hoc analysis of previous clinical trials in Japan. J Gastroenterol. 2015;50:1047-53
32. Zhang ZQ, Dong WC, Yang XL, Zhang JF, Jiang XH, Jing SJ. et al. The influence of plasma albumin concentration on the analysis methodology of free valproic acid by ultrafiltration and its application to therapeutic drug monitoring. Ther Drug Monit. 2015;37:776-82
33. Tayyab S, Feroz SR. Serum albumin: clinical significance of drug binding and development as drug delivery vehicle. Adv Protein Chem Struct Biol. 2021;123:193-218
34. Ahmed FE. Sample preparation and fractionation for proteome analysis and cancer biomarker discovery by mass spectrometry. J Sep Sci. 2009;32:771-98
35. Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G. et al. A pathology atlas of the human cancer transcriptome. Science. 2017;357(6352):eaan2507
36. Berglund L, Bjorling E, Oksvold P, Fagerberg L, Asplund A, Szigyarto CA. et al. A genecentric human protein atlas for expression profiles based on antibodies. Mol Cell Proteomics. 2008;7:2019-27
37. Uhlen M, Bjorling E, Agaton C, Szigyarto CA, Amini B, Andersen E. et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol Cell Proteomics. 2005;4:1920-32
38. Toh WH, Louber J, Mahmoud IS, Chia J, Bass GT, Dower SK. et al. FcRn mediates fast recycling of endocytosed albumin and IgG from early macropinosomes in primary macrophages. J Cell Sci. 2019;133(5):jcs235416
39. Kim H, Villareal LB, Liu Z, Haneef M, Falcon DM, Martin DR. et al. Transferrin receptor-mediated iron uptake promotes colon tumorigenesis. Adv Sci (Weinh). 2023;10:e2207693
40. Sousa MM, Norden AG, Jacobsen C, Willnow TE, Christensen EI, Thakker RV. et al. Evidence for the role of megalin in renal uptake of transthyretin. J Biol Chem. 2000;275:38176-81
41. Graversen JH, Madsen M, Moestrup SK. CD163: a signal receptor scavenging haptoglobin-hemoglobin complexes from plasma. Int J Biochem Cell Biol. 2002;34:309-14
42. Hvidberg V, Maniecki MB, Jacobsen C, Hojrup P, Moller HJ, Moestrup SK. Identification of the receptor scavenging hemopexin-heme complexes. Blood. 2005;106:2572-9
43. Honorato RV, Koukos PI, Jimenez-Garcia B, Tsaregorodtsev A, Verlato M, Giachetti A. et al. Structural biology in the clouds: the WeNMR-EOSC ecosystem. Front Mol Biosci. 2021;8:729513
44. van Zundert GCP, Rodrigues J, Trellet M, Schmitz C, Kastritis PL, Karaca E. et al. The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J Mol Biol. 2016;428:720-5
45. Bjellqvist B, Hughes GJ, Pasquali C, Paquet N, Ravier F, Sanchez JC. et al. The focusing positions of polypeptides in immobilized pH gradients can be predicted from their amino acid sequences. Electrophoresis. 1993;14:1023-31
46. Bjellqvist B, Basse B, Olsen E, Celis JE. Reference points for comparisons of two-dimensional maps of proteins from different human cell types defined in a pH scale where isoelectric points correlate with polypeptide compositions. Electrophoresis. 1994;15:529-39
47. Gasecka A, Nieuwland R, Budnik M, Dignat-George F, Eyileten C, Harrison P. et al. Randomized controlled trial protocol to investigate the antiplatelet therapy effect on extracellular vesicles (AFFECT EV) in acute myocardial infarction. Platelets. 2020;31:26-32
48. Shahnawaz Khan M, Tabrez S, Rehman MT, Alokail MS. Al (III) metal augment thermal aggregation and fibrillation in protein: Role of metal toxicity in neurological diseases. Saudi J Biol Sci. 2020;27:2221-6
49. Inforzato A, Baldock C, Jowitt TA, Holmes DF, Lindstedt R, Marcellini M. et al. The angiogenic inhibitor long pentraxin PTX3 forms an asymmetric octamer with two binding sites for FGF2. J Biol Chem. 2010;285:17681-92
50. Lee DH, Yoo JK, Um KH, Ha W, Lee SM, Park J. et al. Intravesical instillation-based mTOR-STAT3 dual targeting for bladder cancer treatment. J Exp Clin Cancer Res. 2024;43(1):170
51. Yoon HY, Cho HH, Ryu YJ. Adenovirus pneumonia treated with cidofovir in an immunocompetent high school senior. Respir Med Case Rep. 2019;26:215-8
52. Zhao J, Yap A, Wu E, Low CY, Yap J. Severe community acquired adenovirus pneumonia in an immunocompetent host successfully treated with IV cidofovir. Respir Med Case Rep. 2020;30:101037
53. Ko JH, Lim JU, Choi JY, Oh HS, Yoo H, Jhun BW. et al. Early cidofovir administration might be associated with a lower probability of respiratory failure in treating human adenovirus pneumonia: a retrospective cohort study. Clin Microbiol Infect. 2020;26(5):646.e9-646.e14
54. Doan ML, Mallory GB, Kaplan SL, Dishop MK, Schecter MG, McKenzie ED. et al. Treatment of adenovirus pneumonia with cidofovir in pediatric lung transplant recipients. J Heart Lung Transplant. 2007;26:883-9
55. Heo J, Liang JD, Kim CW, Woo HY, Shih IL, Su TH. et al. Safety and dose escalation of the targeted oncolytic adenovirus OBP-301 for refractory advanced liver cancer: Phase I clinical trial. Mol Ther. 2023;31:2077-88
56. Kanerva A, Hemminki A. Modified adenoviruses for cancer gene therapy. Int J Cancer. 2004;110:475-80
57. Kanerva A, Zinn KR, Chaudhuri TR, Lam JT, Suzuki K, Uil TG. et al. Enhanced therapeutic efficacy for ovarian cancer with a serotype 3 receptor-targeted oncolytic adenovirus. Mol Ther. 2003;8:449-58
58. Volk AL, Rivera AA, Kanerva A, Bauerschmitz G, Dmitriev I, Nettelbeck DM. et al. Enhanced adenovirus infection of melanoma cells by fiber-modification: incorporation of RGD peptide or Ad5/3 chimerism. Cancer Biol Ther. 2003;2:511-5
59. Zheng S, Ulasov IV, Han Y, Tyler MA, Zhu ZB, Lesniak MS. Fiber-knob modifications enhance adenoviral tropism and gene transfer in malignant glioma. J Gene Med. 2007;9:151-60
60. Ranki T, Sarkioja M, Hakkarainen T, von Smitten K, Kanerva A, Hemminki A. Systemic efficacy of oncolytic adenoviruses in imagable orthotopic models of hormone refractory metastatic breast cancer. Int J Cancer. 2007;121:165-74
61. Sarkioja M, Kanerva A, Salo J, Kangasniemi L, Eriksson M, Raki M. et al. Noninvasive imaging for evaluation of the systemic delivery of capsid-modified adenoviruses in an orthotopic model of advanced lung cancer. Cancer. 2006;107:1578-88
62. Kangasniemi L, Kiviluoto T, Kanerva A, Raki M, Ranki T, Sarkioja M. et al. Infectivity-enhanced adenoviruses deliver efficacy in clinical samples and orthotopic models of disseminated gastric cancer. Clin Cancer Res. 2006;12:3137-44
63. Guse K, Ranki T, Ala-Opas M, Bono P, Sarkioja M, Rajecki M. et al. Treatment of metastatic renal cancer with capsid-modified oncolytic adenoviruses. Mol Cancer Ther. 2007;6:2728-36
64. Johnsen KB, Burkhart A, Thomsen LB, Andresen TL, Moos T. Targeting the transferrin receptor for brain drug delivery. Prog Neurobiol. 2019;181:101665
65. Daniels TR, Bernabeu E, Rodriguez JA, Patel S, Kozman M, Chiappetta DA. et al. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim Biophys Acta. 2012;1820:291-317
66. Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296-W303
67. Yaguchi T, Kobayashi A, Inozume T, Morii K, Nagumo H, Nishio H. et al. Human PBMC-transferred murine MHC class I/II-deficient NOG mice enable long-term evaluation of human immune responses. Cell Mol Immunol. 2018;15:953-62
68. Holguin L, Echavarria L, Burnett JC. Novel humanized peripheral blood mononuclear cell mouse model with delayed onset of graft-versus-host disease for preclinical HIV research. J Virol. 2022;96:e0139421
69. Jensen SM, Meijer SL, Kurt RA, Urba WJ, Hu HM, Fox BA. Regression of a mammary adenocarcinoma in STAT6-/- mice is dependent on the presence of STAT6-reactive T cells. J Immunol. 2003;170:2014-21
70. Wesa AK, Herrem CJ, Mandic M, Taylor JL, Vasquez C, Kawabe M. et al. Enhancement in specific CD8+ T cell recognition of EphA2+ tumors in vitro and in vivo after treatment with ligand agonists. J Immunol. 2008;181:7721-7
71. Shu SY, Chou T, Rosenberg SA. Generation from tumor-bearing mice of lymphocytes with in vivo therapeutic efficacy. J Immunol. 1987;139:295-304
72. Salewski I, Gladbach YS, Kuntoff S, Irmscher N, Hahn O, Junghanss C. et al. In vivo vaccination with cell line-derived whole tumor lysates: neoantigen quality, not quantity matters. J Transl Med. 2020;18:402
73. Gonzalez FE, Gleisner A, Falcon-Beas F, Osorio F, Lopez MN, Salazar-Onfray F. Tumor cell lysates as immunogenic sources for cancer vaccine design. Hum Vaccin Immunother. 2014;10:3261-9
74. Ivanova DL, Thompson SB, Klarquist J, Harbell MG, Kilgore AM, Lasda EL. et al. Vaccine adjuvant-elicited CD8(+) T cell immunity is co-dependent on T-bet and FOXO1. Cell Rep. 2023;42:112911
Author contact
![]() Corresponding author: Jin Woo Choi; Tel: +82-2-961-9591, Email: jinwoo.chac.kr.
Corresponding author: Jin Woo Choi; Tel: +82-2-961-9591, Email: jinwoo.chac.kr.