Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
The engines of ferroptosis
The brakes of ferroptosis
The cascade responses of...
Ferroptosis in metabolic diseases
Methods and prospects for...
Conclusions
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2024; 14(15):5826-5852. doi:10.7150/thno.100080 This issue Cite
Review
Ferroptosis at the nexus of metabolism and metabolic diseases
Shuangwen Li1,#, Guixiang Zhang2,3,#, Jiankun Hu3,4, Yan Tian1, ![]() , Xianghui Fu1,
, Xianghui Fu1, ![]()
1. Department of Endocrinology and Metabolism, Department of Biotherapy, Center for Diabetes and Metabolism Research, State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University, Chengdu, 610041, Sichuan, China.
2. Division of Gastrointestinal Surgery, Department of General Surgery, West China Hospital, Sichuan University, Chengdu, 610041, Sichuan, China.
3. Gastric Cancer Center, West China Hospital, Sichuan University, Chengdu, 610041, Sichuan, China.
4. Department of General Surgery, West China Hospital, Sichuan University, Chengdu, 610041, Sichuan, China.
#Authors contributed equally to this work.
Received 2024-6-25; Accepted 2024-8-27; Published 2024-9-9
Abstract

Ferroptosis, an iron-dependent form of regulated cell death, is emerging as a crucial regulator of human physiology and pathology. Increasing evidence showcases a reciprocal relationship between ferroptosis and dysregulated metabolism, propagating a pathogenic vicious cycle that exacerbates pathology and human diseases, particularly metabolic disorders. Consequently, there is a rapidly growing interest in developing ferroptosis-based therapeutics. Therefore, a comprehensive understanding of the intricate interplay between ferroptosis and metabolism could provide an invaluable resource for mechanistic insight and therapeutic development. In this review, we summarize the important metabolic substances and associated pathways in ferroptosis initiation and progression, outline the cascade responses of ferroptosis in disease development, overview the roles and mechanisms of ferroptosis in metabolic diseases, introduce the methods for ferroptosis detection, and discuss the therapeutic perspectives of ferroptosis, which collectively aim to illustrate a comprehensive view of ferroptosis in basic, translational, and clinical science.
Keywords: Redox metabolism, Glucose catabolism, Lipid peroxidation, Ferroptosis cascade responses, Ferroptosis-related therapeutics
Introduction
As an emerging mode of programmed cell death, ferroptosis has attracted tremendous interest in the past decade due to its increasing importance in both physiology and pathology. In principle, ferroptosis is driven by iron-dependent lipid peroxidation, which results from a combinational outcome of aberrant cellular metabolism and imbalanced redox homeostasis. Abnormal metabolism of diverse nutrients, such as glucose, amino acids, lipids, and minerals, may potentiate lipid peroxidation and consequent ferroptosis by increasing the buildup of reactive oxygen species (ROS) or polyunsaturated fatty acids (PUFAs) [1]. Correspondingly, many key metabolic regulators, such as adenosine 5'-monophosphate-activated protein kinase (AMPK), mammalian target of rapamycin (mTOR), α-enolase (ENO), and 7-dehydrocholesterol (7-DHC) have been implicated in ferroptosis initiation and progression [2-6]. Conversely, several systems that are closely associated with the homeostasis of various amino acids and minerals comprise the resistance mechanisms against ferroptosis, including the system Xc-/glutathione (GSH)/glutathione peroxidase 4 (GPX4) system, GTP cyclohydrolase-1 (GCH1)/tetrahydrobiopterin (BH4) pathway, and coenzyme Q10 (CoQ10) [7-9]. Therefore, the inception and progression of ferroptosis are an integrated outcome of numerous dysregulated metabolic pathways under the control of both inducible and inhibitory molecules. Additionally, ferroptosis has been observed to exacerbate several pathological processes and human diseases, including obesity, type 2 diabetes (T2D) and its complications, and non-alcoholic fatty liver disease (NAFLD), by further disrupting metabolic homeostasis, amplifying the propagation of ferroptotic death signals, and initiating inflammatory responses [10-13]. Consequently, the implementation of ferroptosis-based therapies holds immense promise in catalyzing remarkable strides in the field of disease treatment.
Overall, ferroptosis appears to be a critical nexus for the integration of metabolism, redox biology, and pathophysiology. A better understanding of the interplay between ferroptosis and metabolism may provide new mechanistic insights and potential therapeutic strategies for the prevention and treatment of human diseases.
The engines of ferroptosis
It is widely accepted that labile iron accumulation tends to drive ferroptosis. Meanwhile, perturbations in metabolic pathways of fundamental energy substrates like glucose, lipids, and amino acids could augment the buildup of ROS and trigger the peroxidation of PUFAs, heightening the susceptibility to ferroptosis. In this section, we overview the functions and mechanisms of ferroptosis inducers and effectors, with a focus on the aberrant metabolism of iron, glucose, amino acids, and lipids (Figure 1).
Metabolic engines for ferroptosis. Lipid peroxidation plays a central role in the process of ferroptosis. Disruptions in the homeostasis of iron, glucose, amino acids, and lipids result in the accumulation of highly toxic ROS and/or PUFAs. Specifically, dysregulations in iron uptake, transport, storage, utilization, and export can lead to iron overload, which triggers excessive ROS production through the Fenton reaction. Furthermore, iron serves as a vital component of various oxidoreductases that are intricately involved in the redox system. The TCA cycle and OXPHOS interconnect metabolism of glucose, amino acids, and lipid to supply the necessary ROS and PUFAs for ferroptosis. However, the role of nutrient metabolism in ferroptosis extends beyond this. The production of NADPH, the synthesis of oxidoreductases, and the process of lipophagy are associated with ferroptosis. Arrows and vertical bars indicate positive and negative effects, respectively. Created with BioRender.com.
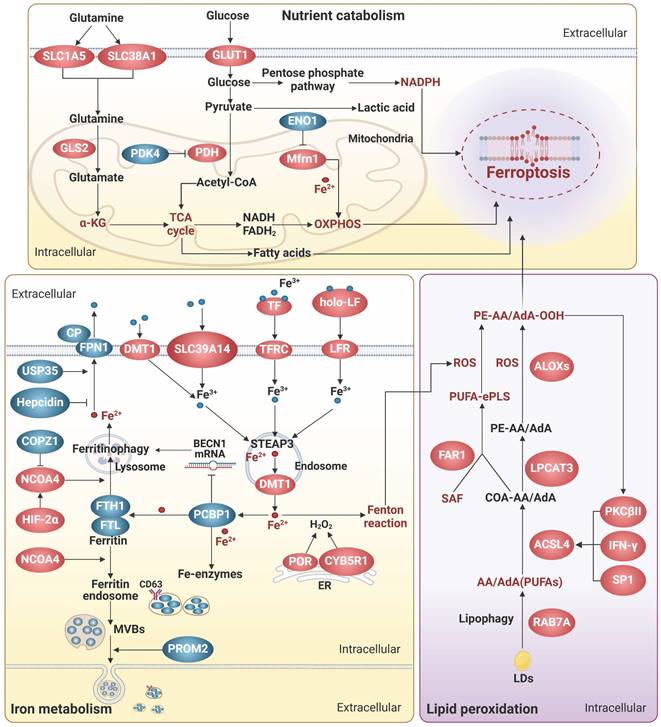
Iron metabolism
Iron homeostasis is a multi-step process involving iron transport, uptake, storage, and export, and is tightly regulated to avoid excess iron accumulation. Excess iron can contribute to ferroptosis by catalyzing the Fenton reaction or by functioning as the active core of oxidoreductases, including the nicotinamide adenine dinucleotide phosphate hydride (NADPH) oxidases (NOXs), xanthine oxidase, arachidonic acid lipoxygenases (ALOXs), and cytochrome P450 enzymes (Figure 1).
Iron transportation and absorption
Generally, iron absorption is accomplished by two ways: transferrin receptor (TFRC)-mediated endocytosis of iron-loaded transferrin (TF) and direct iron intake mediated by metal transporters. TF and TFRC have been extensively implicated in ferroptosis [14]. TF binds to free iron, forms a complex with TFRC on the cell surface, and transports iron via endocytosis. It is worth noting that lactoferrin (LF), another TF family member, can regulate ferroptosis beyond iron transportation. Iron free LF shows a superior anti-ferroptosis property by increasing the expression of GPX4, solute carrier family 7 member 11 (SLC7A11), and nuclear factor erythroid 2-related factor 2 (Nrf2) [15, 16]. Moreover, LF orchestrates the function of pivotal proteins within the iron metabolism network. For instance, Fe2+ needs to be catalyzed to Fe3+ by iron oxidizing enzymes, such as ceruloplasmin, before it can be transported out of the cell by ferritin 1 (FPN1). LF enhances the iron oxidase efficacy of ceruloplasmin, thereby augmenting the exportation of iron [17].
Non-transferrin-bound iron (NTBI) can directly enter cells through metal transporters, such as divalent metal transporter 1 (DMT1), SLC39A14, and SLC39A8. DMT1 functions as the major iron transporter for non-heme iron uptake in most cell types under NTBI conditions, while SLC39A14 and SLC39A8 act primarily in the liver, skeletal muscle, and pancreas [18-21]. As cytoplasmic NTBI is unstable and prone to generate free radicals, anachronistic increases in metal transporters may augment the propensity for ferroptosis [19-21]. Intriguingly, SLC39A14 and SLC39A8 are also responsible for the transportation of other metal ions, including Zn2+ and Mn2+, which potentially collaborate with iron to facilitate the process of ferroptosis [22].
Iron utilization and storage
Metals are utilized by transferring to their cognate proteins through the mediation of metallochaperones. Frataxin and poly(rC)-binding proteins (PCBPs) are crucial iron chaperones, which aid in delivering iron to iron-containing functional proteins for utilization or to ferritin for storage. Frataxin is responsible for facilitating iron-sulfur clusters (ISCs) assembly in mitochondria, while PCBPs primarily bind to ferrous iron and transport it to ferritin and other non-heme iron enzymes, including deoxyhypusine hydroxylase, prolyl hydroxylase 2, and prolyl and asparagyl hydroxylases, through direct protein-protein interactions [23, 24]. Additionally, PCBP1 can interact with BolA family member 2 (BolA2) and form a unique iron chaperone complex that transfers iron to the cell membrane for [2Fe-2S] cluster assembly, establishing a connection between the labile iron pool in the cell membrane and cellular ISCs distribution system [25]. As a result, deficiency in both frataxin and PCBPs severely disrupts cell-autonomous iron homeostasis, leading to labile iron-induced ferroptosis [23, 24]. Intriguingly, PCBP1 is also able to prevent ferroptosis triggered by ferritinophagy, a selective form of autophagy process that degrades ferritin, by functioning as an RNA-binding protein to destabilize beclin 1 (BECN1) [26]. Notably, the iron chaperone and RNA-binding activities of PCBP1 are separate and occur at different sites, which independently confer essential functions [27]. Therefore, a better understanding of the mechanism underlying the coordination of iron- and RNA-binding activities could provide valuable insights into the versatile roles of PCBP1 in diverse systems, including iron metabolism, immunity, cancer metastasis, and so on.
Excess free iron is stored in ferritin, a protein complex consisting of heavy-chain ferritin 1 (FTH1) and light-chain ferritin (FTL). The levels of ferritin subunits influence the rate of iron storage and the susceptibility to ferroptosis. Besides, overactive autophagy-lysosome-mediated degradation of ferritin and release of stored iron can lead to accumulation of labile iron, thereby triggering ferroptosis [28]. During this process, nuclear receptor cofactor 4 (NCOA4) acts as a core adaptor to bind and deliver FTH1 to autophagosomes, which are then fused to lysosomes with the aid of vesicle-associated membrane protein 8 [29]. Thus, interfering with NCOA4-FTH1 interactions, such as FTH1 O-GlcNacylation [30], and the E3 ligase HERC2-mediated NCOA4 degradation, can impede ferritinophagy and subsequently ferroptosis [29]. Recently, the vesicle trafficking gene SEC24B was identified as a novel driver of ferroptosis in microglia and macrophages [31]. Despite of its known function in autophagosome formation, SEC24B did not affect ferritin degradation or ferritinophagy, but may regulate iron homeostasis by modulating the labile iron pool through potential mechanisms including altering export of iron transporters.
Iron export
Intracellular iron can be expelled from cells either by the membrane transporter FPN1, which is the most classical way, or by being transported out of the cell as ferritin via multivesicular bodies (MVBs). FPN1 is the only known mammalian iron transport protein, and thus it is conceivable that its expression and activity might affect iron metabolism and ferroptosis. In line with this idea, hepcidin, the only known natural ligand of FPN1 thus far, can bind to the central cavity between the N and C domains of iron-loaded FPN1 and induce ligand-receptor complex ubiquitination, internalization, and degradation to reduce iron output, increasing ferroptosis sensitivity [32]. In contrast, ubiquitin-specific protease 35 maintains FPN1 protein stability by deubiquitination to prevent iron overload and ferroptosis [33].
Exosomes, a class of emerging extracellular vesicles (EVs) mediating inter-cellular and inter-organ communication, exist in various body fluids and are involved in numerous pathophysiological processes [34]. Given that iron limitation inhibits ferroptosis, it is imaginable that the generation and secretion of ferritin-containing exosomes could attenuate ferroptosis. Indeed, prominin 2 (PROM2), a pentaspan membrane glycoprotein, can act as a ferroptosis braker by facilitating the formation of ferritin-containing MVBs and exosomal release of iron [35]. Correspondingly, biocompatible hybrid nanoparticles composed of ferroptosis inducers and PROM2 siRNAs efficiently decreased iron efflux and overcome ferroptosis resistance in cancer cells, indicative of an attractive strategy for ferroptosis-based cancer therapy [36]. Similarly, the inhibitors against heat shock factor 1, an activator of PROM2 transcription, can sensitize cancer cells to ferroptosis-inducing drugs and alleviate the oncogenic and ferroptosis resistance programs [37]. Interestingly, CD63, a crucial regulator of exosome formation and secretion, is able to interact with NCOA4 and facilitate the exosomal export of iron, suggesting a role in ferroptosis [38]. Moreover, CD63 has been shown to be post-transcriptionally regulated by iron via the iron-responsive elements/iron regulatory proteins system [38], adding an additional layer of complexity to the interplay between exosomes, iron efflux, and ferroptosis.
Glucose metabolism
Glucose is the major carbon source for cellular biosynthesis and energy generation. Under aerobic conditions, acetyl-coenzyme A (acetyl-CoA), a key metabolite derived from glucose metabolism, enters the tricarboxylic acid (TCA) cycle and undergoes mitochondrial oxidative phosphorylation (OXPHOS) to generate highly efficient ATP. However, the process of OXPHOS is always accompanied by the production of toxic by-products such as superoxide anion radical, hydrogen peroxide, and hydroxyl radical, which are the main endogenous sources of ROS. In pancreatic ductal adenocarcinoma cells under high glucose conditions, SLC2A1-dependent glucose uptake and pyruvate oxidation-dependent fatty acid synthesis facilitate system Xc- inhibitor-induced ferroptosis, while pyruvate dehydrogenase kinase 4 (PDK4), a suppressor of the conversion of pyruvate into acetyl-CoA that is downregulated by SLC2A1, can repress pyruvate oxidation, and thus resist ferroptosis [39].
Under conditions of limited oxygen supply, lactate dehydrogenase (LDH) facilitates the conversion of pyruvate to lactate, which appears to prevent the supportive effects of aerobic glucose oxidation on ferroptosis. Further compelling evidence reveals that hypoxia-inducible factor-1α (HIF-1α) can enhance LDH expression and lactate production, leading to pH-dependent resistance to ferroptosis in tumor cells [40]. Moreover, heightened lactate uptake stimulates the generation of monounsaturated fatty acids (MUFAs) via activation of the sterol regulatory element-binding transcription factor 1 (SREBP1)/stearoyl CoA desaturase 1 (SCD1) axis [41]. The replacement of PUFAs in cell membrane by MUFAs is able to mitigate lipid peroxidation and ferroptosis [42]. Notably, lactate exists in two optical isomers, namely D-lactate and L-lactate. L-lactate is prevalent in mammals and serves as the primary carbohydrate fuel, signaling molecule for intercellular communication, and glycolytic precursor, but its implication in ferroptosis remains unknown. In contrast, the accumulation of D-lactate, whose concentration is very low in physiological serum, induced ferroptosis in tumor cells through increasing ROS and intracellular iron levels [43]. Correspondingly, phosphorylation of Yes-associated protein (YAP) that enhances the transcription of D-LDH can facilitate D-lactate catabolism in mitochondria and safeguard tumor cells against ferroptosis.
The glucose pentose phosphate pathway (PPP) serves as a vital source of NADPH that assumes a dualistic role in ferroptosis, based on its capacity to supply electrons to various ferroptosis-regulating molecules and engage in interactions with functional proteins. NADPH actively engages in ROS detoxification through delivering electrons to antioxidant proteins including GSH, BH4, thioredoxin, and CoQ10, NAD(P)H quinone oxidoreductase 1 (NQO1) [44, 45]. NADPH also supports the formation of ISCs and maintains iron homeostasis by providing electrons to ferredoxin2 [46]. However, it is worth noting that NQO1, in its interaction with the potential drugs like β-lapachone and 2-methoxy-6-acetyl-7-methylhomoquinone, can induce ROS production and drive ferroptosis, making it a target of interest in ferroptosis-focused cancer therapy [47]. Furthermore, catalyzed by oxidoreductases such as NADPH-cytochrome P450 reductase (POR) and NADH-cytochrome b5 reductase (CYB5R1), electrons from NAD(P)H are transferred to oxygen, resulting in the generation of hydrogen peroxide that disrupts membrane integrity during ferroptosis [48]. Additionally, NADPH is able to enhance the activity of the E3 ligase, membrane-associated ring-CH-type finger 6 (MARCHF6), which subsequently degrades of key ferroptosis inducers coenzyme A synthase long-chain family member 4 (ACSL4) and p53, and increases anti-ferroptosis proteins, including Nrf2, GPX4 and SLC7A11, thereby attenuating ferroptosis in cancer cells [49]. Interestingly, MARCHF6 can mediate the ubiquitination and degradation of prohormone pro-opiomelanocortin (POMC), resulting in reductions in NADPH consumption by NOX2/NOX4, ROS production, and ferroptosis in POMC neurons [50]. Altogether, these data suggest NADPH as a crucial intermediary mediator of ferroptosis, however its impact on ferroptosis is contingent upon specific substrates within a given microenvironment.
Additionally, a variety of metabolic enzymes related to glucose metabolism, such as ENO, creatine kinase B (CKB), myo-inositol oxygenase, and malic enzyme (ME), are involved in the regulation of ferroptosis. For example, all three of ENO isozymes, key glycolytic enzymes, are considered to be ferroptosis suppressors. α-enolase (ENO1), with a universal expression in adult tissues, can function as an RBP, beyond its metabolic activity, to degrade iron regulatory protein 1 (IRP1) mRNA, resulting in reduced expression of mitochondrial ferritin-1 (Mfrn1) and subsequent suppression of mitochondrial iron-triggered ferroptosis [4]. γ-enolase (ENO2) and β-enolase (ENO3), which display tissue-specific expression patterns, have been implicated in ferroptosis [51, 52], however the underlying mechanisms are elusive and await further investigation. CKB, a key enzyme in ATP production, can phosphorylate GPX4 at serine 104 and block its degradation by heat shock cognate 70, thereby preventing ferroptosis [53]. ME catalyzes the conversion of NAD(P)+ to NAD(P)H, and therefore deletion of ME1/2 reduces NADPH production, disrupting cellular redox homeostasis and increasing ferroptosis susceptibility [54, 55]. Conversely, myo-inositol oxygenase, a non-heme ferritin that catalyzes the conversion of myo-inositol to D-glucuronide, is able to promote ferritinophagy-induced ferroptosis by reducing NADPH and GSH levels [56].
In summary, the metabolic enzymes, free radicals, and intermediate metabolites that are involved in the metabolism of glucose, the fundamental energy source of life, play a more expansive and versatile role in the process of ferroptosis, suggesting ferroptosis as an ancestral mechanism of cell death that accompanies both the beginning and end of life.
Amino acid metabolism
Glutamine, glutamate, and arginine have been implicated in increasing sensitivity to ferroptosis. Inside cells, glutamine is deamidated by glutaminase (GLS) to produce glutamate, which is then converted to α-ketoglutarate (α-KG) by glutamate dehydrogenase 1. Glutamate and α-KG are the key intermediate metabolites in glutaminolysis-induced ferroptosis. Exogenous addition of glutamate can induce ferroptosis by inhibiting the uptake of cystine by system Xc- [57]. Elevated levels of endogenous glutamate result in increased cellular Ca2+ concentration, which subsequently phosphorylates and inhibits glutamine-fructose-6-phosphate transaminase (GFPT1) through activation of the adenylyl cyclase/protein kinase A axis. Decreased GFPT1 leads to destabilization of YAP, which reduces expression of its downstream target gene, FTH1, thereby reducing ferritin phagocytosis-triggered FTH1 transcriptional substitutions and enhancing sensitivity to ferroptosis [58]. α-KG can serve as an intermediate in TCA cycle or be converted to 2-hydroxyglutarate by malate dehydrogenase 1, and thus trigger the generation of mitochondrial ROS (mtROS) and ferroptosis [59]. Additionally, α-KG can activate p53-mediated ferroptosis by promoting oxidative DNA damage [59]. Hence, glutaminolysis is considered a crucial contributor to ferroptosis, and it is plausible that inhibiting glutamine transporters like SLC1A5 and SLC38A1 or key nodes of glutaminolysis such as GLS could alleviate ferroptosis [57, 60]. Argininosuccinate synthase 1 (ASS1) plays a role in reducing glutamine mitochondrial oxidative metabolism-induced ferroptosis by promoting reductive carboxylation of cytoplasmic glutamine [61]. Moreover, ASS1 also facilitates the synthesis of arginine succinate, which is further cleaved to produce arginine. Arginine can activate the mTOR complex 1 (mTORC1)/SREBP1/SCD5 axis, which enhances the de novo synthesis of MUFAs utilizing acetyl-CoA derived from the glutamine reductive pathway and alleviates ferroptosis susceptibility.
Notably, arginine may also contribute to ferroptosis. Arginine has been identified as an essential amino acid for the induction of ferroptosis in HT1080 cells exposed to erastin2 or cystine deprivation [62]. Furthermore, arginine has a role in sustaining fumarate biosynthesis through the urea cycle [63]. Fumarate, as a reactive α, β-unsaturated electrophilic metabolite, can covalently binds to GSH, leading to a decrease in intracellular GSH levels and ultimately ferroptosis. This may be due to the fact that energy metabolism exhibits diverse trajectories in accordance with the requirements of vital biological processes, consequently yielding distinct consequences on ferroptosis. This is also evident in glutamine metabolism, as it contributes to the production of molecules like GSH (Glu-Cys-Gly) and NADPH that could protect cells against ferroptosis [64].
Lipid peroxidation
Peroxidation of phospholipids containing PUFAs (PL-PUFAs) in cell membranes not only triggers liquid-liquid phase separation of the plasma membrane and impairs its physical and chemical functions, but also produces by-products such as 4-hydroxynonenal (4-HNE) and 4-hydroxyhexenal, which form adducts with biomolecules such as lipids, proteins, and DNA, ultimately resulting in ferroptosis [65]. Among them, phospholipids containing a single polyunsaturated fatty acyl tail at the sn1 position of the glycerol backbone (PL-PUFA1s) have been extensively studied, whereas phospholipids possessing diacyl-PUFA tails at both the sn1 and sn2 positions (PL-PUFA2s) have received less attention. Recent investigations have demonstrated that an increased intake of PUFAs in the diet leads to an elevation in PL-PUFA2s, which interact with the mitochondrial electron transport chain to trigger mtROS generation and subsequent lipid peroxidation, thereby inducing ferroptosis [66]. Furthermore, polyunsaturated ether phospholipids (PUFA-ePLs), particularly plasmalogens, owing to the presence of PUFAs at their Sn-2 position, can supply substrates for lipid peroxidation and facilitate ferroptosis induction [67]. Therefore, regulating the content of PL-PUFAs is crucial in the regulation of ferroptosis. Diacylglycerol acyltransferase (DGAT) can facilitate the incorporation of PUFAs into the sn-3 position of diacylglycerol, resulting in the formation of triacylglycerol (TAG), which is stored as lipid droplets (LDs) surrounded by monolayers of phospholipids within cells. This process effectively decreases the level of PL-PUFAs. Inducing cell cycle arrest can enhance the DGAT-mediated reshuffling of PUFAs from phospholipids to TAG, consequently reducing the cell susceptibility to ferroptosis [68]. In addition, the substitution of other types of lipids for PL-PUFAs undergoing peroxidation could attenuate ferroptosis. For example, 7-DHC, an intermediate in the cholesterol synthesis pathway, has recently been demonstrated as a natural inhibitor of ferroptosis [5, 6]. Due to the unsaturated B-ring structure, 7-DHC can sequester free radicals and reduce lipid peroxidation of phospholipids, thereby safeguarding cells from ferroptosis. Ergosterol, an analog of 7-DHC, also exhibits ferroptosis-inhibitory properties [5, 6], suggesting that the trapping of free radicals to impede ferroptosis may be a shared chemical characteristic among these unsaturated sterols.
The initiation of lipid peroxidation requires the catalysis of a series of enzymes, including ACSL4, lysophosphatidylcholine acyltransferase 3 (LPCAT3), and ALOXs. Here, we specifically delve into novel insights regarding their potential relevance to endocrine metabolism. The involvement of the protein kinase C (PKC) family in metabolic diseases has been well-documented. Recent studies have revealed that PKCs also participate in promoting ferroptosis by activating lipid peroxidation enzymes. PKCβII was shown to phosphorylate the Thr328 residue of ACSL4, initiating the biosynthesis of PUFA-containing lipids and promoting lipid peroxidation [69]. In turn, lipid peroxidation induces the activation of PKCβII, and thus amplifies ferroptosis through the lipid peroxidation-PKCβII-ACSL4 positive feedback loop. PKCα contributes to ferroptosis by phosphorylating the ALOX5 Ser663 [70]. LPCAT3 plays a crucial role in various processes including intestinal lipid absorption, hepatocyte lipoprotein assembly and secretion, adipocyte differentiation, and ferroptosis, due to its capability of incorporating unsaturated acyl groups into phospholipids [71]. Interestingly, LPCAT3, also known as membrane-bound O-acyltransferase-containing structural domain (MBOAT) 5, belongs to the MBOAT family. Recent genome-wide CRISPR screening identified another two MBOAT members, MBOAT1 and MBOAT2, as ferroptosis suppressors [72]. These two members can be transcriptionally induced by estrogenic and androgenic signaling, respectively, and mediate the effects of sex hormones on ferroptosis. Mechanistically, MBOAT1 and MBOAT2 hinder ferroptosis by selectively translocating MUFAs to the sn-2 position of phospholipids. Considering the significance of LPCAT3 and MBOAT1/2 in ferroptosis regulation, along with the similar enzymatic activities exhibited by other MBOAT family members, it is reasonable to speculate that other MBOAT members might also have regulatory effects on ferroptosis.
The brakes of ferroptosis
Cells have developed several efficient redox systems, including the system Xc-/GSH/GPX4, CoQ10, and GCH1/BH4 axis, which play crucial roles in cellular resistance to ferroptosis (Figure 2). Moreover, as imbalanced nutrient metabolism is related to induce ferroptosis, it is not surprising that cellular energy sensors such as AMPK and mTOR are involved in the regulation of ferroptosis. Additionally, it is important to recognize that cellular activities are intricately linked to mechanical signaling. Indeed, many mechanical checkpoints, such as integrin β1, focal adhesion kinase, YAP, and piezo-type mechanosensitive ion channel component 1 (Piezo1), have been shown to regulate ferroptosis.
Ferroptosis brakes. The antioxidant system represents the most extensively investigated pathway that inhibits ferroptosis. This system encompasses the axis of system Xc-/GSH/GPX4, the GCH1/BH4 axis, and the CoQ10 system. The system Xc-/GSH/GPX4 axis is a well-known and classical anti-ferroptosis pathway, in which system Xc- promotes the synthesis of GSH that in turn collaborates with GPX4 to convert lipid peroxidation products into alcohols, thereby effectively preventing ferroptosis. The transsulfuration pathway plays a role in providing cysteine for GSH synthesis, thereby conferring resistance against ferroptosis. The GCH1/BH4 axis, CoQ10 system, and VK also exhibit resistance against membrane phospholipid peroxidation through redox reactions. Energy metabolism regulatory systems, particularly AMPK and mTOR, also participate in the regulation of ferroptosis. Notably, AMPK displays a dual role in ferroptosis, which may be attributed to its diverse downstream targets. Interestingly, mechanotransduction appears to exert regulatory control over ferroptosis. Intercellular adhesion contacts can inhibit ferroptosis by activating the E-cadherin/Hippo/YAP signaling pathway. However, it remains unclear whether other forms of mechanical forces, such as solid stress, fluid pressure, stiffness, and physical microstructure, modulate ferroptosis. Arrows and vertical bars indicate positive and negative effects, respectively. Created with BioRender.com.
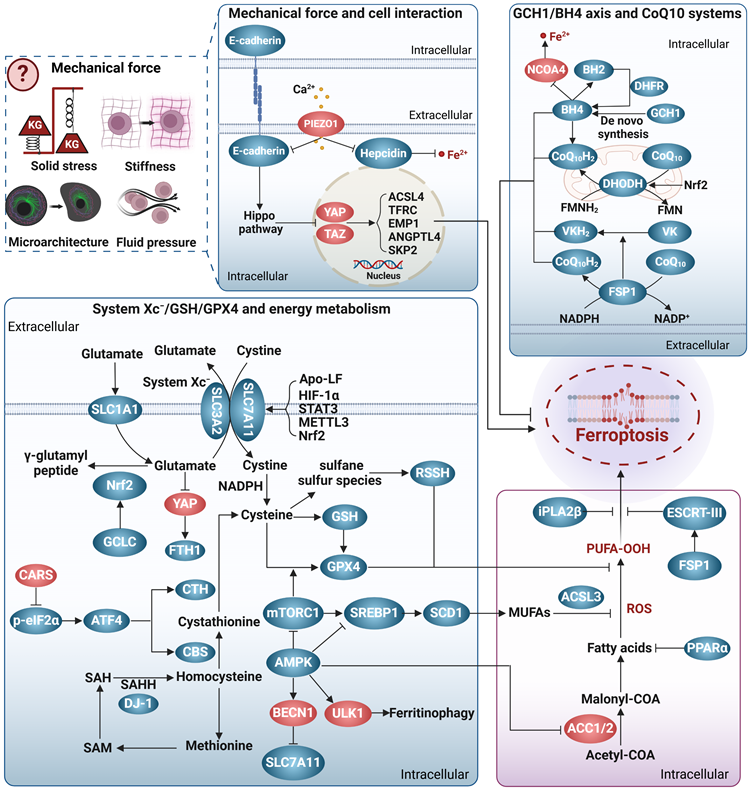
System Xc-/GSH/GPX4
System Xc- is a cystine/glutamate transporter consisting of SLC7A11 and SLC3A2, which prevents ferroptosis by several ways. Firstly, it transports glutamate out of the cell, thereby preventing glutamate accumulation and subsequent ferroptosis [58]. Secondly, it can supply cystine to the cell to support the synthesis of GSH, which cooperates with GPX4 in the reduction of the membrane phospholipid hydroperoxides to non-toxic lipids, thus protecting the cell from the ferroptosis caused by lipid peroxidation [73]. Additionally, cysteine, the product of cystine reduction, serves as a source of sulfane sulfur in persulfides and polysulfides [74]. Hydropersulfides (RSSH), in particular, exhibit even greater antioxidant capacity than alpha-tocopherol (the active ingredient in vitamin E), providing enhanced protection against ferroptosis [75]. Thirdly, system Xc- also participates in the transport of other amino acids, presenting an additional mechanism against ferroptosis. SLC7A11 is responsible for the transport of kynurenine into cell [76]. Kynurenine is further metabolized to generate 3-hydroxykynurenine and 3-hydroxy-o-cyclo-anisoleic acid, both of which possess ROS scavenging and ferroptosis resisting activities [76]. Interestingly, the competing use of SLC7A11 by kynurenine and cystine can be regarded as a temporary state of cystine deprivation or “pseudo-starvation”, necessitating the amplification of SLC7A11 transcription to fulfill the cellular requisites for amino acid absorption. This implies that the targeting of SLC7A11 has the capacity to concomitantly provoke nutrient deprivation and ferroptosis in energy-intensive tumors. Interestingly, p53, a frequently mutated tumor suppressor, is a repressor of SLC7A11 transcription. It has indicated that p53 mutation-triggered SLC7A11 overexpression can suppress ROS-induced ferroptosis and facilitate tumor metabolic reprogramming [77]. Similarly, certain mutations in several genes, such as HJV, KDM5C, IDH1/2, and KRAS, have been linked to disease progression through modulating ferroptosis sensitivity and metabolism [78, 79]. These observations suggest an emerging interplay among gene mutations, ferroptosis, and disease progression, which warrants further investigation.
The selenoprotein GPX4 is a pivotal enzyme in defense against ferroptosis. Nevertheless, metabolic disorders like selenium deficiency, copper accumulation, hyperglycemia, and hyperlipidemia have the potential to impact the expression and/or activity of GPX4, consequently leading to the induction of ferroptosis [80-83]. More importantly, not all GPX4 isoforms possess the ability to inhibit ferroptosis. Depending on their cellular localization, three distinct isoforms of GPX4 exist: cytoplasmic (cGPX4), mitochondrial (mGPX4), and nuclear (nGPX4). Among them, cGPX4 is widely recognized as a key factor in protecting against iron-induced mutations [81]. In cases where cells primarily experience damage from mitochondrial lipid peroxidation, the protective effect of mGPX4 surpasses that of cGPX4 [84]. nGPX4 is important for the integrity of sperm chromatin structure by modulating oxidation of cysteines in protamine [85]. Interestingly, Xie et al. recently identified an inducible GPX4 transcript (iGPX4), whose expression was significantly increased in the livers of NAFLD. The induction of iGPX4 promotes the oligomerization of cGPX4, which counteracts the anti-ferroptotic properties of cGPX4 and induces ferroptosis, thereby aggravating NAFLD [81]. Altogether, different GPX4 isoforms, together with their inter-translocation and interaction, greatly increase the complexity to dissect the role and mechanism underlying GPX4, and a better understanding would advance the GPX4-targeting strategies for ferroptosis and associated diseases.
CoQ10 systems
CoQ10 plays a crucial role as a versatile electron carrier in supporting the mitochondrial respiratory chain. Additionally, reduced CoQ10 (CoQ10H2) functions as a potent lipophilic antioxidant, protecting against lipid peroxidation and ferroptosis [8]. CoQ10 is synthesized within the inner mitochondrial membrane, which is typically impermeable. The dual localization of StAR-associated lipid transfer domain 7 (STARD7) in the cytoplasm and mitochondria enables it to regulate CoQ10 synthesis and intracellular distribution [86]. Specifically, mitochondrial STARD7 ensures CoQ10 synthesis, while cytoplasmic STARD7 binds to CoQ10 and facilitates its passage through the mitochondrial membrane, thereby maintaining a balance between normal mitochondrial respiration and resistance against ferroptosis.
In the context of ferroptosis regulation, CoQ10 is primarily reduced to CoQ10H2 through ferroptosis suppressor protein 1 (FSP1)/CoQ10 and dihydronicotinic acid dehydrogenase (DHODH)/CoQ10 [8, 84, 87]. Recent studies have indicated that selenium can also generate selenides that enhance CoQ10H2 production via sulfide quinone oxidoreductase [88]. The anti-ferroptosis effect of FSP1/CoQ10 or DHODH/CoQ10 has been demonstrated in various pathological models, and numerous proteins, non-coding RNAs, metabolites, and hormones have been identified as regulators of the FSP1/CoQ10 or DHODH/CoQ10 system [89-91]. Specifically, both FSP1 and DHODH mediate NAD(P)H-dependent CoQ10H2 production in a similar manner, but their action sites differ, with FSP1 located at the cytosolic membrane and DHODH at the mitochondrial membrane [8, 84, 87]. Subcellular localization is crucial for the anti-ferroptotic functions of FSP1 and DHODH. icFSP1, a new FSP1 inhibitor belonging to the 3-phenylquinazolinones class, has no role in FSP1 enzymatic activity, but induces FSP1 phase separation and leads to its separation from lipid membranes, thus hampering its anti-ferroptotic function [92]. Interestingly, the interaction between FSP1 and 4-hydroxy-2-nonenal can relocate FSP1 from the mitochondria to the nucleus and promote apoptosis [93]. Thus, proper subcellular localization appears crucial for FSP1 to exert specific biological functions. Whether the function of DHODH also exhibits site-selectivity remains unknown. Significantly, FSP1 also enables the reduction of vitamin K to hydroquinone, which effectively scavenges free radicals, underscoring the important role of FSP1 in supporting the antioxidant system against ferroptosis [94].
GCH1/BH4 axis
BH4 is an effective endogenous antioxidant, and GCH1 is the main checkpoint for de novo synthesis of BH4. The GCH1/BH4 axis is suggested to be a predictor of ferroptosis sensitivity [9], and may protect cells from ferroptosis by at least two pathways. BH4 acts as a coenzyme to mediate the conversion of phenylalanine to tyrosine and increases the generation of 4-OH-benzoate, a precursor for CoQ10 biosynthesis [95]. In this way, high levels of BH4 may promote de novo synthesis of CoQ10, and thus increase cellular tolerance to ferroptosis [9]. Alternatively, the GCH1/BH4 axis may prevent ferroptosis via modulating iron metabolism. In a follow-up study, patients with phenylketonuria who responded to BH4 treatment had significantly lower serum iron levels, implying a role in iron homeostasis [96]. Although the precise mechanisms underlying the regulation of GCH1/BH4 axis on iron metabolism are unclear, previous clues suggest that its inhibition could promote ferroptosis by activating ferritinophagy [97].
Energy metabolism
AMPK and mTOR serve as crucial regulators of cellular energy status and play important roles in modulating ferroptosis. AMPK has been shown to prevent ferroptosis in conditions like diabetic cardiomyopathy (DCM), diabetic nephropathy (DN), NAFLD, and pulmonary fibrosis by promoting Nrf2 transcription or translation [83, 98, 99]. AMPK also inhibits mitochondrial lipid peroxidation by activating forkhead box O 3a (FOX3A) signaling and blocking ACSL4 mitochondrial translocation, thereby reducing ferroptosis in cerebral ischemia-reperfusion injury and DCM [100, 101]. Additionally, AMPK phosphorylates acetyl-CoA carboxylase 1/2 (ACC1/2) to inhibit fatty acid synthesis and reduce ferroptosis susceptibility [102]. Nevertheless, recent evidence suggests a promotive effect of AMPK in ferroptosis. The synthesis of MUFAs is concurrently diminished by AMPK activation in certain conditions, which is not conductive to against ferroptosis [41]. Consistently, AMPK induces ferritinophagy and subsequent ferroptosis by relieving inhibitory phosphorylation on unc-51 like autophagy activating kinase 1 (ULK1) [103]. AMPK also phosphorylates BECN1 and facilitates the formation of the BECN1-SLC7A11 complex, thereby weakening the protective effect of system Xc-/GPX4/GSH [2].
mTOR exists in two multiprotein complexes, mTORC1 and mTORC2. In general, mTORC1 promotes anabolic processes and inhibits catabolism. To date, at least three pathways have been implicated in mTORC1-mediated ferroptosis repression. Firstly, mTORC1 can enhance the transcription, translation, and stability of regulators that resist ferroptosis, such as GPX4 [104]. Secondly, mTORC1 may protect against ferroptosis by promoting the synthesis of MUFAs via activating SCD [105]. Thirdly, mTORC1 inhibits specific types of autophagy that contribute to ferroptosis [106]. However, the degradation of cysteine-rich proteins such as albumin favors cellular cysteine replenishment, making the inhibitory effect of mTORC1 on autophagy unfavorable for ferroptosis resistance [107]. Compared to mTORC1, the function of mTORC2 in ferroptosis is poorly understood. mTORC2 is predominantly activated by growth signaling and nutrient limitation [108]. Recent studies suggest that the mechanisms by which cystine counteracts ferroptosis are partially attributed to mTOR2 activation [109]. More specifically, Wang et al. demonstrated that mTORC2 can antagonize ferroptosis by enabling the nuclear translocation of Nrf2 through the activation of protein kinase B (AKT)/glycogen synthase kinase 3 β (GSK3β) signaling cascade [110].
In conclusion, while the roles of AMPK and mTOR in ferroptosis have been extensively explored, the complex disease background and the diverse repertoire of their downstream targets have complicated the establishment of a clear regulatory network linking energy metabolism and ferroptosis. Further research is required for a comprehensive understanding of the relationship between ferroptosis and energy metabolism.
Mechanotransduction
Mechanosensory receptors within cells can detect mechanical stimuli and subsequently trigger signaling events that initiate biological responses. This process is known as mechanotransduction. Given the growing connection between metabolism and mechanical signaling, coupled with the recognition of mechanical signaling as a potential modulator of ferroptosis [111], a comprehensive investigation of ferroptosis and mechanotransduction might yield valuable insights into elucidating the interrelationship among these three factors.
In general, cells cultured at higher densities or in three-dimensional (3D) configurations exhibit enhanced resistance to ferroptosis, probably due to increased cell-cell interactions and activated cadherins [9, 112-114]. Empirical evidence indicates that cadherin 2-mediated increased membrane tension can impede the process of ferroptosis [115]. This inhibition potentially stems from the impact of heightened membrane tension on cell membrane fluidity, as well as the regulation of the uptake and release of iron and other extracellular metabolites. Furthermore, the mechanical activation of E/N-cadherin can initiate the Hippo signaling pathway, resulting in the inhibition of the YAP/tafazzin axis. This axis is known to promote the transcription of various genes associated with ferroptosis, including ACSL4, TFRC, epithelial cell membrane protein 1, angiopoietin-like 4, and E3 ligase S-phase kinase-related protein 2 [112-114, 116, 117]. Conversely, an elevation in hepatic extracellular matrix stiffness stimulates activation of the integrin β1/focal adhesion kinase/YAP pathway, consequently promoting ferroptosis [118].
Additionally, cell crowding, stretching and lipid peroxidation can modify membrane tension and activate mechanosensitive ion channels, such as Piezo1. Piezo1 acts as a homeostatic regulator for cell number and distribution by initiating cell death in crowded areas, but enhances cell division in sparse areas [119]. Mechanistically, activation of Piezo1 leads to Ca2+ influx, which results in the degradation of VE-cadherin, reduced levels of GSH and GPX4, and increased cellular rupture, collectively intensifying ferroptosis [120-122]. Interestingly, Piezo1 also exerts an inhibitory effect on heme expression in macrophages and hepatocytes, thereby exacerbating cellular iron accumulation [123], indicating that mechanotransduction has the potential to impact ferroptosis through the modulation of metabolism. Investigating the interconnectedness of mechanotransduction, metabolism, and ferroptosis may unveil a novel pathway for the regulation of ferroptosis.
The cascade responses of ferroptotic cells
Ferroptotic signals in living organisms may continue with: (I) propagation of the death signal, (II) triggering immune responses, and (III) clearance by phagocytes (Figure 3), paving the way for the story of death.
The cascade reaction of ferroptotic cells. Ferroptosis signals can propagate both intracellularly and intercellularly, thereby expanding the scope of ferroptotic processes. The lipid peroxidation signal associated with ferroptosis can propagate continuously across the cellular membrane. An undefined intercellular ferroptosis signal propagates in waveforms that may be linked to the ferroptotic cells released toxic substances. Furthermore, DAMPs are released through nanopores in membrane of ferroptotic cells and are recognized by PRRs, leading to the induction of immune responses, and ultimately sterile inflammation in metabolic diseases. Ferroptotic cells also have the capability to emit PS, SAPE-OOH, or other “eat me” signals that stimulate phagocytosis. Created with BioRender.com.
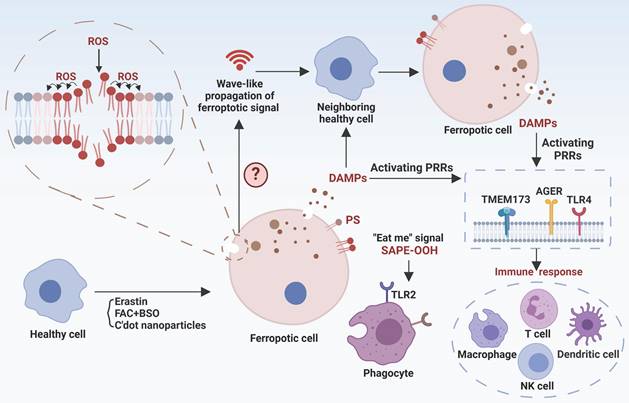
Ferroptotic death signal propagation
The dissemination of ferroptotic death signals has been observed in a wave-like manner throughout cell populations, influencing the fate of neighboring cells [124]. The unique spatiotemporal ferroptotic signals occur prior to cell rupture, and osmoprotectant-mediated inhibition of cell rupture slows this propagation, suggesting a potential involvement of the "substances" in triggering ferroptosis. However, the exact definition of the ferroptosis signal remains unclear, including the identification of specific “toxic substances” and the selection of target cells to establish distinct spatial patterns. Hence, it is essential to identify ferroptosis signaling types and their mode of action for comprehending the progression of ferroptosis. Interestingly, platelet-activating factor (PAF) and PAF-like phospholipids, which are released by ferroptotic cells and actively internalized by adjacent cells via endocytosis, induce membrane rupture and subsequent cellular demise, exemplifying the propagation of ferroptotic death signals [12]. EVs have recently been found to be an attractive transmitter for ferroptosis signals [125]. For example, EVs derived from erastin-induced ferrototic cells can migrate to neighboring cells and subsequently expedite the death of these recipient cells. However, not all forms of ferroptosis exhibit this specific spatiotemporal propagation pattern. Particularly, cells undergoing ferroptosis induced by GPX4 inhibition exhibit a pattern of single-cell ferroptosis, wherein the death signal is not propagated to neighboring cells [124]. In summary, the propagation of death signals is critical for biological, physiological, and pathogenic function of ferroptosis. Our current knowledge is almost certainly limited to the tip of the iceberg. Consequently, many exciting breakthroughs may lie ahead by using up-to-date technologies including single-cell omics, such as the discovery of potential systematic ferroptosis across distal tissues, the identification of spatiotemporal landscape and molecular mechanisms/checkpoints for ferroptosis propagation, and the dissection of different characteristics between single-cell and propagated multi-cell ferroptosis.
Ferroptosis-related immune responses
Ferroptosis-related immune responses encompass both immune cell ferroptosis and immune disturbances provoked by ferroptotic cells. On one hand, immune cells exhibit varying susceptibility to ferroptosis, and ferroptosis in these cells directly influences the immune microenvironment and is associated with disease processes. For instance, aberrations in neutrophils are closely associated with various autoimmune diseases, and neutrophil ferroptosis significantly contributes to neutropenia in systemic lupus erythematosus, exacerbating disease manifestations [126]. Antigen-specific memory CD4+ T cells play a crucial role in preventing microbial reinfection, and disturbances in their mTORC2 pathway result in aberrant accumulation of mtROS and subsequent ferroptosis [110]. Therefore, triggering ferroptosis in antigen-specific memory CD4+ T cells have the potential to alleviate certain autoimmune conditions. Conversely, inhibiting ferroptosis to prolong the persistence of antigen-specific memory CD4+ T cells may contribute to the long-term efficacy of vaccination. Furthermore, dysfunction and depletion of CD8+ T cells play significant roles in tumor immune evasion. CD36 can facilitate fatty acid uptake, triggering CD8+ T cells ferroptosis and reducing their immunosuppressive effects on tumors [127]. Interestingly, interferon-γ (IFN-γ) released by CD8+ T cells are able to downregulate the expression of two subunits of system Xc-, SLC7A11 and SLC3A2, while enhance ACSL4 expression in tumor cells, collectively promoting lipid peroxidation and ferroptosis [128, 129]. Thus, combining induction of tumor ferroptosis with immunotherapy holds promise as a strategy for tumor management. Indeed, arsenic trioxide (ATO) is a potent broad-spectrum cytotoxic agent with highly immunogenic properties. ATO has the ability to convert immunologically “cold” tumors into “hot” tumors by enhancing the infiltration of immune cells and promoting the secretion of IFN-γ and tumor necrosis factor-α by CD8+ T cells. Simultaneously, ATO activates multiple cell death pathways, including ferroptosis [130]. The development of an ATO-based cancer vaccine is anticipated to effectively prevent and intervene early in tumor progression. On the other hand, as ferroptotic cells release damage-associated molecular patterns (DAMPs), these molecules can bind to pattern recognition receptors (PRRs) on immune cells and alter their immune phenotypes. For example, DAMPs released by ferroptotic tumor cells can drive macrophage polarization towards the M2 phenotype in aggressive tumors, and ferroptotic neurons can activate CD4+ T cells, potentially promoting autoimmune encephalitis development [131, 132]. Moreover, myeloid-derived suppressor cells (PMN-MDSCs), crucial negative regulators of antitumor immunity, are particularly vulnerable to ferroptosis in the tumor microenvironment. Although ferroptosis reduces the presence of PMN-MDSCs, the release of oxygenated lipids induced by ferroptosis limits the anticancer activity of T cells [133]. Hence, ongoing efforts to translate pharmacological induction of ferroptosis in tumor cells into clinical practice should consider the impact of alterations in the immune milieu to ensure the specificity and minimal toxicity of the therapeutic approach.
Although recent studies have shed light on ferroptosis-associated immune disorders, it remains unclear whether immune cells have distinct mechanisms to sense, evade, transduce and release ferroptosis signals. Furthermore, current research focuses on ferroptosis-associated immune responses in tumors. Intriguingly, ferroptosis in immune or self-tissue cells has been shown to modulate the progression of autoimmune diseases [134], suggesting that autoimmune diseases may provide a valuable model for dissecting ferroptosis-associated immune responses.
Ferroptotic cells clearance
Prompt identification and removal of ferroptotic cells are essential to reduce the propagation of ferroptotic signals and DAMPs. The “find me” and “eat me” signals exposed by dying cells interact with a series of receptors on the surface of phagocytes to mediate the phagocytosis of cell corpses. Phosphatidylserine (PS) and oxygenated PS exposed on the membrane are common “eat-me” signals involved in the phagocytosis of apoptotic cells. PS is also presented on the surface of ferroptotic Jurkat cells as a phagocytic signal, but its clearance efficiency is not as high as that on apoptotic cells [135]. However, PS does not appear on the plasma membrane surface of ferroptotic HL60 cells and mouse embryonic fibroblasts. Therefore, different modalities of cell death may have their specific cell clearance signals. The oxidized phospholipid, 1-stearoyl-2-15-HpETE-sn-glycero-3-phosphatidylethanolamine (SAPE-OOH), has now been demonstrated to be a key “eat-me” signal of ferroptotic cells [136]. Mechanistically, SAPE-OOH can directly interact with Toll-like receptor 2 (TLR2) of macrophages, resulting in effective elimination of ferroptotic cells [136]. However, TLR2 deficiency was unable to abolish this elimination [136], suggesting alternative undocumented pathways. Notably, necroptic cells were found to evade phagocytic clearance by releasing “avoid me” signals, such as thromboxane that can disrupt macrophage OXPHOS and thus impair death cell clearance [137]. It remains unknown whether ferroptotic cells have similar “avoid me” signals.
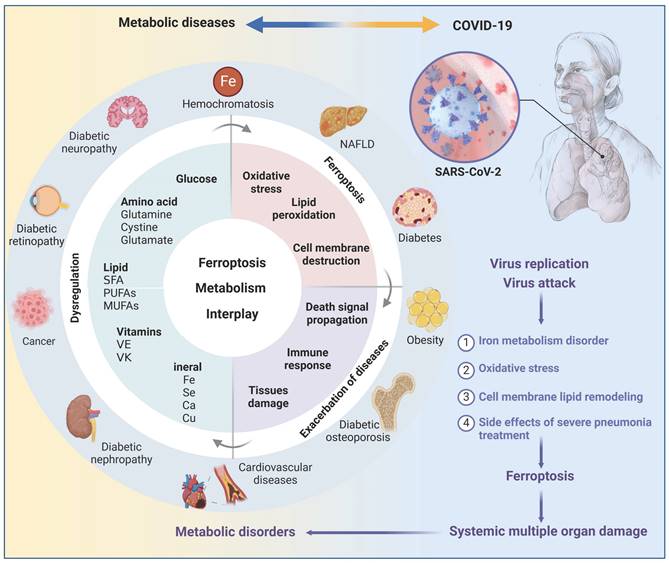
Ferroptosis in metabolic diseases
Metabolic disorders always provide conducive conditions for ferroptosis, and ferroptotic cells may induce chronic inflammation and pathological cell loss that could exacerbate metabolic disorders, including obesity, diabetes and its complications, and NAFLD (Figure 4). Coronavirus disease 2019 (COVID-19), a disastrous pandemic, may also have a close correlation with ferroptosis. Untangling the intricate relationship between dysregulated metabolism, ferroptosis, and disease progression would facilitate the development of effective defense and therapeutic strategies.
The interplay of ferroptosis and metabolism in human diseases. Disturbances in the metabolism of glucose, lipids, amino acids, and trace elements not only play a significant role in the pathogenesis of metabolic diseases such as obesity, diabetes, fatty liver, and hemochromatosis, but also serve as a crucial precondition for the occurrence of ferroptosis, suggesting a potential complementary relationship between metabolic diseases and ferroptosis. Metabolic dysregulation creates a favorable environment for ferroptosis, which further compromises tissue function and triggers inflammation, thereby exacerbating disease progression. It is crucial to note that obesity and diabetes are vital risk factors for COVID-19, and SARS-CoV-2 infection can induce ferroptosis. Thus, it is plausible that SARS-CoV-2 infection may cause metabolic imbalances in the microenvironment, subsequently trigger ferroptosis and result in secondary dysfunction across multiple organs. Created with BioRender.com.
Obesity
Obesity, characterized by a body mass index greater than 30 kg/m2, is a prominent risk factor for various noncommunicable diseases, such as T2D, cardiovascular disease, and cancer. Recent research indicates that obesity can nurture an environment conducive to ferroptosis in the liver, kidney, and cardiovascular system, by promoting lipid peroxidation and iron overload [138, 139]. Additionally, exosomes have an emerging role in obesity-associated ferroptosis, as exemplified by exosomal miR-140-5p derived from adipose tissue macrophages that induces ferroptosis in recipient cardiomyocytes by targeting SLC7A11 [140]. Paradoxically, obesity may confer a survival advantage to tumor cells by enabling resistance to ferroptosis. For example, adipose tissue-derived exosomes are enriched with microsomal triglyceride transfer protein (MTTP), which can increase GPX4 expression and decrease PUFAs levels, resulting in ferroptosis inhibition and drug resistance in colorectal cancer cells [141]. In addition to exosomes, chemerin, a multifunctional adipokine, enables tumor cells to evade ferroptosis by inhibiting fatty acid oxidation [142]. Of note, current research focuses on the impact of obese pathological environment on ferroptosis, but the role of ferroptosis in the development of obesity, such as adipocyte proliferation, differentiation, and function, remains to be fully understood.
NAFLD
NAFLD has recently been redefined as metabolic dysfunction-associated fatty liver disease (MASLD), with a global prevalence of 30% and an increasing trend. Cell death, including necroptosis, pyroptosis, cuproptosis, and ferroptosis, have been reported in NAFLD, with ferroptosis being considered the primary cell death mechanism in early steatohepatitis [143]. Two potent inhibitors of ferroptosis, namely Trolox (an antioxidant) and Deferiprone (an iron chelator), have demonstrated considerable potential in inhibiting cell death, inflammatory cytokine expression, and inflammatory cell infiltration in early stage non-alcoholic steatohepatitis (NASH) [143], highlighting the importance of ferroptosis in NAFLD pathogenesis and treatment.
Generally, NAFLD ferroptosis is closely associated with disrupted iron metabolism, and elevated serum ferritin levels is an independent predictor of advanced NAFLD [144], strongly suggesting a vicious cycle between ferroptosis and NAFLD. On the one hand, the microenvironment of NAFLD nourishes hepatic ferroptosis. As the main iron storage site, hepatocytes frequently experience disturbances in iron metabolism during liver injury. Meanwhile, hepatocytes often release excess iron to hepatic stellate cells via EVs, contributing to iron accumulation in these cells and the development of liver fibrosis [145]. Moreover, metabolic dysregulation in NAFLD hepatocytes leads to the formation of iron pools that triggers Fenton reaction-derived ferroptosis [146]. In addition, metabolites produced by the aberrant intestinal flora in individuals with NAFLD can also significantly induce the expression of TFRC and ACSL4 in the liver, enhancing the sensitivity of hepatocytes to ferroptosis [147]. Notably, NAFLD progression is often associated with deregulated expression/function of key ferroptosis regulators, such as downregulation of Nrf2, GPX4, and SLC7A11, increased m6A methylation of SLC7A11, and hyperoxidization of peroxiredoxin 3 (PRDX3) [11, 98, 148], which collectively promote hepatocyte ferroptosis.
On the other hand, growing evidence emphasizes the significance of ferroptosis in NAFLD development. NAFLD encompasses a spectrum of conditions, ranging from simple steatosis and steatohepatitis to fibrosis. Diverse mechanisms linked to ferroptosis influence multiple pathological changes of NAFLD, including glucolipid toxicity, insulin resistance (IR), inflammatory cell infiltration, and hepatocyte loss. Therefore, inhibition of hepatocyte ferroptosis holds promise for attenuating the progression of NAFLD. Interestingly, ferroptosis in hepatic stellate cells seems to attenuate liver fibrosis [149]. In this regard, a deeper understanding of the dynamics of ferroptosis and underlying mechanisms at different NAFLD stages and in distinct cell types is critical to advance the therapeutic use of ferroptosis in treating this disease.
T2D
T2D accounts for nearly 90% of the estimated 537 million cases of diabetes worldwide and its prevalence is rapidly increasing. The pathological environment of T2D can promote ferroptosis through enhancing ferroptosis engines and diminishing ferroptosis brakes. For instance, hyperglycemia may increase glucose auto-oxidation and mitochondrial dysfunction in T2D, resulting in excessive ROS generation. Increased circulating fatty acids, a hallmark of obesity-associated T2D, heightens the risk of lipid peroxidation and subsequent ferroptosis [150]. Moreover, T2D is associated with decreases in the expression and/or activity of various antioxidant enzymes, such as superoxide dismutase (SOD), GPX1, GPX4, thioredoxin reductase, thioredoxin reductase 2, glutathione, and peroxiredoxin [151]. Dysregulation of AMPK expression and function may also contribute to T2D-associated ferroptosis [83].
Ferroptosis exerts a notable impact on numerous T2D pathological alterations, including β cell dysfunction, IR, inflammation and lipotoxicity. Ferroptosis is prevalent in β cells enriched with arachidonic acid (AA), which is crucial for insulin secretion, however, it increases susceptibility to lipid peroxidation [152]. Experimental evidence underscores the significance of β cell ferroptosis in T2D, such as the impairment of glucose-stimulated insulin secretion [13]. Conversely, certain compounds that attenuate β cell ferroptosis, including quercetin and cryptochlorogenic acid, exhibit potential in ameliorating T2D [153, 154]. IR, a pivotal pathological hallmark of T2D, is partially due to ferroptosis. Inhibition of ferroptosis by liproxstatin-1 (a ferroptosis inhibitor) and ACSL4 knockout significantly alleviated hepatic and adipose IR, respectively [155, 156]. Notably, ferroptotic cells typically release a range of signaling molecules and lipotoxic products, such as prostaglandin E2, 4-HNE, and hydroxyeicosatetraenoic acids, which might contribute to inflammatory cascades, impair the function of metabolic tissues, and stimulate angiogenesis, thereby triggering T2D complications [157]. In conclusion, targeting ferroptosis may emerge as a promising strategy for impeding the progression of T2D through multifaceted mechanisms.
Diabetic nephropathies (DN)
DN is the most serious microvascular complication of diabetes and the leading cause of chronic kidney disease. Glucose uptake by renal units is primarily regulated by ambient glucose levels, making the kidneys vulnerable to damage from high circulating glucose concentrations. At the same time, the kidneys are rich in mitochondria due to their high energy demands, which further increases their susceptibility to damage from ROS [158]. Thus, there is an obvious elevation of ferroptosis in various types of renal cells under diabetic conditions.
A number of ferroptosis drivers and brakers that are deregulated in DN, together with various DN pathogenic factors, collectively modulate the initiation, progression, and elimination of ferroptosis. In cellular and animal models of DN, there is a decrease in ferroptosis suppressors (Nrf2, GPX4, SLC7A11, FTH1, etc.) but an increase in ferroptosis activators (ACSL4, TFRC, NOX1, PTGS2, etc.) [159]. Besides, critical drivers of DN, such as transforming growth factor-β1 (TGF-β1) and HIF 1α, may induce ferroptosis in renal cells, and thus accelerate DN progression [160, 161]. Specifically, TGF-β1 can repress GPX4 and SLC7A11 expression in renal tubular epithelial cells [160], whereas HIF1α is capable of activating heme oxygenase 1 (HO-1) that degrades heme and causes iron overload to promote lipid peroxidation and ferroptosis initiation [161]. Notably, HO-1 may also function as a downstream target of Nrf2, thus exerting antioxidant effects in DN ferroptosis [162]. In this regard, critical investigations using HO-1 deficient cell and animal models are required to clarify the functional outcome of HO-1 in DN development.
Consistently, ferroptosis presents a novel therapeutic avenue for DN. Numerous natural compounds, such as quercetin, ginkgolide B, and umbelliferone, have been extensively studied for their ability to enhance renal function and decelerate DN progression by attenuating ferroptosis [162-164]. Further refinement of the chemical structures of these natural products to improve their targeting and specificity for ferroptosis may increase their translational efficacy of clinical interventions.
Diabetic retinopathy (DR)
DR is a common microvascular complication of diabetes and is the leading cause of preventable blindness in the working-age population. Ferroptosis has recently been shown to be associated with the pathological process of DR. For instance, retinal pigment epithelial (RPE) cells play a crucial role in regulating the metabolic balance of retinal cell nutrients and maintaining the normal function of photoreceptor cells. Upon exposure to high glucose levels, extracellular glial maturation factor-β is increased and can induce ferroptosis in RPE cells by impeding autophagy-lysosomal degradation of ACSL4, contributing to the progression of DR [165]. Correspondingly, 1,8-Cineole is able to alleviate DR by suppressing RPE ferroptosis [166]. Moreover, high glucose levels may induce TRIM46 and promote GPX4 ubiquitination and subsequent ferroptosis in retinal capillary endothelial cells, exacerbating vascular permeability and capillary degeneration in DR [167].
Diabetic cerebral vascular complications
Patients with obesity-related T2D exhibit excess iron deposition in the brain, which is associated with an elevated risk of stroke, cognitive impairment, and development of dementia [168]. Hence, diabetes-induced neuronal ferroptosis may play a crucial role in the development of cerebrovascular complications associated with diabetes. Diabetic rats have reduced levels of FPN1 in the brain, leading to iron deposition and subsequent hippocampal ferroptosis that contributes to cognitive dysfunction [169]. Diabetes also impairs the proliferator-activated receptor γ (PPARγ) signaling pathway in peripheral neutrophils, resulting in decreased LF transcription and secretion [170]. This impairment contributes to iron deposition in neurons and, ultimately, neuronal ferroptosis. However, diabetic cerebrovascular lesions tend to have an insidious onset compared with other diabetic complications, emphasizing the significance of early detection and intervention. Advances in high-field magnetic resonance imaging techniques and reconstruction algorithms have shown promise in accurately mapping iron distribution, which contributes to the early diagnosis of diseases associated with ferroptosis and facilitates timely intervention [171]. Significantly, caveolin-1 and hydrogen sulfide have been demonstrated to alleviate ferroptosis by reducing iron accumulation and oxidative stress in neurons and cerebral microvascular endothelial cells, and their effectiveness has been observed in reducing the susceptibility to diabetes-related cognitive dysfunction and psychiatric disorders, respectively [172, 173].
DCM
Diabetes can cause alterations in the structure and function of the heart that raise the likelihood of developing heart failure. Glycolipotoxicity, mitochondrial dysfunction, and endoplasmic reticulum stress (ERS), have been identified as important contributors to the progression of DCM. Intriguingly, these mechanisms have also been implicated as significant pathways of cardiomyocyte ferroptosis in DCM, suggesting that controlling cardiomyocyte ferroptosis may be effective in delaying the onset and progression of DCM.
Specifically, in diabetic patients, increased insulin resistance causes a reduction in glucose uptake by the heart, leading to a shift towards utilizing fatty acids (FAs) as an energy source and advanced glycation end products (AGEs) accumulation [174]. These factors are directly implicated in cardiomyocyte ferroptosis. Elevated uptake of FAs by cardiomyocytes not only induces lipid overaccumulation and the production of lipotoxic metabolites, but also exacerbates the accumulation of ROS within the cytoplasm and mitochondria, thereby fostering the development of ferroptosis in cardiomyocytes [150]. Excess AGEs in cardiac tissues also elevate intracellular cytoplasmic ROS levels and subsequently trigger cardiomyocyte ferroptosis by downregulating SLC7A11 expression and reducing GSH levels [99]. Additionally, endoplasmic reticulum (ER) has been identified as a significant site of lipid peroxidation in ferroptosis, and the quantity and functionality of ER may influence the vulnerability to ferroptosis [175]. ERS-induced abnormalities in Ca2+ handling have been proposed as key factors contributing to DCM. Consistently, ERS appears to be a triggering factor for ferroptosis in cardiomyocytes during diabetic myocardial ischemia/reperfusion injury [176]. Communications between the ER and mitochondria through mitochondria-associated ER membranes (MAMs) play crucial roles in Ca2+ signaling, energy metabolism, and cell survival. Dysfunction of MAMs has been implicated in the development of DCM [177]. Interestingly, some molecular components of MAMs, such as voltage-dependent anion-selective channel 1 and mitochondrial protein 1/2, have been identified as important signaling molecules in ferroptosis [178, 179], suggesting that MAMs may represent an unexplored regulatory site for ferroptosis in DCM.
COVID-19
There have been over 760 million recorded COVID-19 cases worldwide, and there is still limited knowledge about the pathological process and prognosis of SARS-CoV-2 on different tissues. The rising incidence of new-onset diseases, including diabetes [180], among individuals with COVID-19 underscores the significance of investigating the pathological alterations following acute COVID-19. Growing evidence suggests that SARS-CoV-2 has the ability to induce ferroptosis in host cells via various mechanisms [181, 182]. Firstly, iron plays an important role in the replication of RNA viruses, including SARS-CoV-2. COVID-19 patients often have dysregulated iron metabolism, as manifested by elevated serum ferritin and hepcidin [183]. Correspondingly, reducing iron levels not only curbs RNA virus replication, but also has beneficial immunomodulatory effects on the host and enhances antiviral capabilities [184]. Secondly, hyperactive mitochondrial metabolic pathways are required to produce enough energy to support SARS-CoV-2 replication and reproduction, accompanied by excessive mtROS [185]. Meanwhile, SARS-CoV-2 can inhibit the synthesis of some antioxidant enzymes, such as SOD3 and GPX4, in host cells, exacerbating the oxidative stress state [186]. Thirdly, SARS-CoV-2 can reshape the cellular lipid membrane and promote the formation of replication organelle, providing the structural scaffold for the viral replication/transcription complex. Phospholipase A2 may participate in coronavirus replication organelle formation by cleaving the intact phospholipids on cellular and mitochondrial membranes to form lysophospholipids and unsaturated FAs [187]. Besides, the levels of AA, palmitic acid and stearic acid are upregulated in COVID-19 individuals, which significantly increase the risk of lipid peroxidation and are associated with poor outcomes [188]. These findings suggest that the disrupted metabolic environment triggered by SARS-CoV-2 may enhance vulnerability to ferroptosis in various organs. At the same time, ferroptosis could potentially serve as a trigger for the development of new-onset metabolic diseases in individuals affected by COVID-19 through various mechanisms, as mentioned in the T2D section. Therefore, it is reasonable to hypothesize that SARS-CoV-2 infection and ferroptosis form a vicious cycle to promote the progression of COVID-19 and its related metabolic diseases.
Methods and prospects for ferroptosis detection
Reliable methods for accurate identification of ferroptosis amidst various modes of cell death are crucial for effective disease management. A combination of molecular biology, advanced imaging techniques, and biomedical engineering has been employed to develop a series of assays for detecting ferroptosis in vitro and in vivo (Table 1). As an easy means, transmission electron microscopy (TEM) is widely used to directly visualize the morphological changes of ferroptotic cells, such as mitochondrial condensation or swelling, increased membrane density, reduced cristae, and outer membrane rupture, thereby providing valuable insights into the ultrastructure of ferroptotic cells [175]. However, these ultrastructural changes may occur in other types of cell death or cellular stress conditions. Therefore, the expression of ferroptotic signature genes, as well as the concentrations of certain metabolites, are always utilized to improve the detection of ferroptosis accuracy and dynamics. Due to the fact that ferroptosis is characterized by the peroxidation of PUFAs, liquid chromatography-mass spectrometry (LC-MS) is the preferred method for identifying peroxidized ferroptosis biomarkers. By combining MS-based lipidomics with proteomics, it is possible to characterize the ferroptotic covalent adducts of oxidative phosphatidylethanolamine (PE) with target proteins [189]. However, LC-MS cannot offer information on the intracellular distribution of lipid molecules. Recently, gas cluster ion beam secondary ion mass spectrometry (GCIB-SIMS) that is capable of mapping intact lipids in cellular biofilms, coupled with a 70 keV (H2O)n+ cluster ion beam to amplify the signal, has enabled not only sensitive detection of intact biomolecules but also in-situ chemical imaging in ferroptosis cells [190].
Methods of ferroptosis detection
| Targeting aspect | Specific materials | Targeting analyte | Technology/equipment | |
|---|---|---|---|---|
| Morphology | / | Mitochondrial | TEM | |
| / | Lysosomal | |||
| / | Nuclear | |||
| / | Plasma membrane | |||
| RNA expression of hallmark genes | Primers for corresponding genes | Hallmark genes | qRT-PCR | |
| / | RNA-seq | |||
| scRNA-seq | ||||
| scRandom-seq | ||||
| Protein expression of hallmark genes | Antibodies for corresponding proteins | Hallmark genes | Western blotting | |
| IHC | ||||
| Oxidative lipidomics | / | PE | LC-MS/MS | |
| / | PEox | (H2O)n-GCIB-SIMS | ||
| Probe | NBD-Pen | Lipid-derived radicals | LC/FL and HRMS/MS | |
| ROS | Probe | Mito-QL and MBI-OMe | HOCl | Fluorescence microscope |
| HP, BT-HP, QS-4 and BTFMB | H2O2 | |||
| Coum-HCy | ˙OH | |||
| Biothiol | Probe | CP2 and Coum-HCy | Cysteine | |
| CM-Mit | Mitochondrial thiol | |||
| RT | GSH | |||
| Fe2+ | Probe | FIP-1, Ac-MtFluNox, Lyso-RhoNox, ER-SiRhoNox, FeP and COU-LIP-1 | Fe2+ | |
| Heme | Probe | H-FluNox | Labile heme | |
| Fe3+ | Probe | DRhFe | Fe3+ | |
| Lipid peroxidation products | Probe | NBD-Pen, C11-BODIPY 581/591 and Liperfluo | Lipid peroxidation products | |
| pH | Probe | DSPI-3 | pH | |
| Viscosity | Probe | DSPI-3, L-Vis-1, LD-1, Ir-ER, MN-V and BDHT | Viscosity | |
| Polarity | Probe | CQPP and TPA-SO2 | Polarity | |
| Fe2+ | Probe | POSS-Art-DOTA | Fe2+ | PET/MRI |
| Art-Gd | MRI | |||
HOCl, hypochlorous acid; HRMS/MS, high-resolution tandem mass spectrometry; H2O2, hydrogen peroxide; IHC, Immunohistochemistry; LC/FL, online high-performance liquid chromatography-fluorometry; qRT-PCR, quantitative reverse transcription polymerase chain reaction; PEox, oxidized PE; ˙OH, hydroxyl radical; RNA-seq, RNA sequencing; ScRNA-seq, Single-cell RNA sequencing.
Overall, MS-based assays provide high specificity and sensitivity for the detection and quantification of various molecules involved in ferroptosis, but they require specialized equipment and sample preparation. Moreover, when the natural cellular environment is disrupted, certain indicators of ferroptosis, such as Fe2+, can be rapidly oxidized and therefore cannot be reliably quantified from biopsy samples. As a result, a variety of small molecule fluorescent probes have been widely developed to enable real-time monitoring of ferroptosis-related indicators, including ROS, cysteine, GSH, lipid peroxidation, Fe2+, and Fe3+ (Table 1). For example, the redox balance of Fe2+ and Fe3+ is dynamic during ferroptosis, representing an attractive hallmark. Lu et al. recently developed DNAzyme-based fluorescent turn-on sensors that are selective for either Fe2+ or Fe3+ and can monitor both simultaneously, revealing a decreased Fe3+/Fe2+ ratio during ferroptosis [191]. Additionally, specific fluorescent probes have been devised to assess the spatial, chemical, and physical microenvironments of ferroptotic cells, such as viscosity, polarity, and pH within subcellular organelles like the endoplasmic reticulum, mitochondria, and LDs (Table 1), enriching the landscape of evolving ferroptosis. Of note, due to the lack of a ferroptosis-specific biomarker, multifunctional fluorescent probes that can capture dynamic changes in various analytes are required to confirm and delineate the ferroptosis process. For instance, VPCPP, the near-infrared fluorescent probe that can simultaneously monitor local microviscosity, micropolarity, and carboxylesterase in living cells, revealed that sorafenib-induced ferroptosis led to an increase in the microviscosity and upregulation of carboxylesterase at the same time [192].
Nevertheless, most fluorescent probes for ferroptosis are limited by shallow tissue penetration depth, strong autofluorescence, and low signal-to-background ratios, which significantly impede their applications in vivo. Therefore, the development of sensors and imaging techniques that are more suitable for in vivo imaging is expected to facilitate the clinical translation of ferroptosis detection. Positron emission tomography (PET) and magnetic resonance imaging (MRI), two commonly non-invasive and quantitative imaging modalities for in vivo clinical testing, have been increasingly used for ferroptosis detection (Table 1). PET can provide quantitative 3D images for in vivo assessment of labile Fe2+ in ferroptotic cells [193], whereas MRI may offer advantages such as high resolution, absence of off-radiation, and deep tissue penetration, and is now more widely used in the diagnosis and treatment of ferroptosis-related diseases. For example, artemisinin-based probe (Art-Gd) is an MRI imaging probe specifically designed to target Fe2+ and exhibits promising capabilities for the early diagnosis of ferroptosis-related diseases. Building upon this, the POSS-Art-DOTA probe was developed, integrating PET and MRI to establish a dual-modality imaging technique that offers exceptional sensitivity and diagnostic precision [193, 194].
Despite of these advances, ideal approaches that can simultaneously detect multiple types of ferroptosis targets are still in their infancy. The combination of several advanced techniques, such as (H2O)n-GCIB-SIMS, C60-SIMS, desorption electrospray ionization, matrix-assisted laser desorption ionization, and artificial intelligence-assisted computational processing, has the potential to understand whether there are specific tissue compartments or cell types for ferroptosis, the metabolic landscape between these cells and the interconnected molecular networks between different cell populations. Improving the accuracy and safety of in vivo ferroptosis detection and accelerating its clinical translation would enable early detection of ferroptosis-related diseases and image-guided ferroptosis treatment.
Ferroptosis-related therapy
Targeting ferroptosis has emerged as a promising therapeutic approach. Multiple small molecule drugs have been shown to possess the capacity to modulate the previously mentioned “engines” or “brakes” of ferroptosis. These drugs can alleviate the ferroptosis-related physiological conditions like metabolic disorders (Figure 5), or combat tumor growth by inducing ferroptosis (Table 2). These studies with conventional drugs provide valuable information for the subsequent clinical translation of ferroptosis treatments. However, there are currently no clinical drugs specifically targeting ferroptosis, which is associated with the short history of ferroptosis research and the limited discovery of unique ferroptosis targets. Therefore, there is a pressing need to integrate new technologies to advance ferroptosis therapies, with nanomedicines showing significant promise.
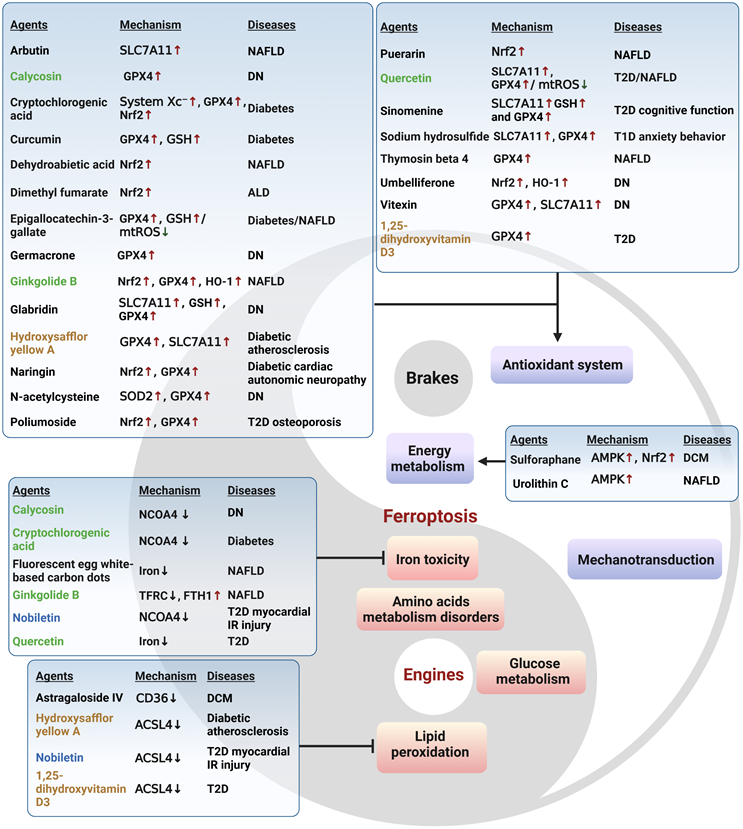
Molecular compounds that ameliorate metabolic diseases by inhibiting ferroptosis. Numerous natural products and experimental compounds have demonstrated efficacy in alleviating metabolic diseases by inhibiting ferroptosis, albeit its contribution and action mechanism are incompletely unknown. There is a summary of these molecular compounds targeting ferroptosis engines or brakes, potential mechanisms, and associated metabolic diseases. Compounds highlighted in green exhibit a dual function of acting as antioxidants and regulating iron levels. Those in blue have a dual function of inhibiting lipid peroxidation and regulating iron levels, while those in brown act as both antioxidants and inhibitors of lipid peroxidation. Arrows and vertical bars indicate positive and negative effects, respectively. Created with BioRender.com.
FDA-approved drugs relevant to ferroptosis induction
| Drugs | Target | Effects on ferroptosis | Clinical uses |
|---|---|---|---|
| Acetaminophen | Nrf2 inhibitor | Oxidative stress | Pain and fever |
| Cerivastatin | HMGR inhibitor | Inhibition of mevalonate synthesis | Hyperlipidemia |
| Dexamethasone | GSH inhibitor | Oxidative stress | Immunosuppressive therapy |
| Dimethyl fumarate | GPX4/GSH inhibitor | Oxidative stress | Multiple sclerosis |
| Fluvastatin | GPX4 inhibitor | Oxidative stress | Hyperlipidemia |
| Lapatinib | TF agonist; FPN1 inhibitor | Iron toxicity; Oxidative stress | Cancer |
| Leflunomide | DHODH inhibitor | Oxidative stress | Active rheumatoid arthritis |
| Lidocaine | SLC7A11 inhibitor | Oxidative stress | Heart disease |
| Metformin | SLC7A11 and Nrf2 inhibitor | Oxidative stress | Diabetes |
| Methotrexate | DHFR and GPX4 inhibitor | Oxidative stress | Cancer |
| Olaparib | SLC7A11 and AMPK/SCD1 inhibitor | Lipid peroxidation | Cancer |
| Orlistat | GPX4 inhibitor | Oxidative stress | Obesity |
| Regorafenib | System Xc- and GPX4 inhibitor | Oxidative stress | Cancer |
| Simvastatin | GPX4 inhibitor | Oxidative stress | Hyperlipidemia |
| Sorafenib | System Xc- inhibitor | Oxidative stress | Cancer |
| Sulfasalazine | System Xc- inhibitor | Oxidative stress | CIRD; IBD |
ARA, active rheumatoid arthritis; CIRD, chronic inflammatory rheumatic diseases; DHFR, dihydrofolate reductase; FDA, Food and drug administration; HMGR, 3-hydroxy-3-methylglutaryl-CoA-reductase; IBD, inflammatory bowel diseases.
There are various types of nanomedicines used to treat tumors by inducing ferroptosis, including drug delivery nanoparticles, active metal nanoparticles such as iron, manganese, copper and gold, single atom catalysts including selenium, nickel and iridium-based, multifunctional nanoparticles, and multifunctional magnetic nanomaterials [195-198]. Among these, multifunctional magnetic nanomaterials have the ability to release magnetic iron oxide nanoparticles (IO) that can act as T1-weighted MRI agents upon reaching the tumor site, and thus induce tumor ferroptosis, enabling MRI-guided ferroptosis therapy.
For instance, IO-BQR@PMEMA exhibits a dark T2 signal in normal tissues, but when it reaches the tumor tissue, IO results in a bright T1 signal by the combined effect of ROS and GSH. Consequently, the released Fe2+/3+ and the drug Brequinar act synergistically to promote ferroptosis in tumor through Fenton reaction and DHODH inhibition, respectively [197]. In conclusion, magnetic nanomaterials associated with ferroptosis offer new prospects for tumor therapeutics, and also provide a multi-scale for studying ferroptosis in the pathogenesis of diseases, such as metabolic disorders.
The application of nanomedicines in treating metabolic diseases by restraining ferroptosis is relatively lagging behind. Only a few studies have reported on nanoparticles carrying natural products (such as curcumin and puerarin) or ferroptosis inhibitors (such as liproxsatin-1 and SOD) for the treatment of metabolic diseases like diabetic osteoporosis, fatty liver, and diabetic wounds [199-202]. Significantly, while nanomedicines hold potential in the realms of disease diagnosis, treatment, and prevention, they are unlikely to swiftly supplant traditional medicines due to considerations surrounding in vivo absorption, metabolism, and long-term safety. Consequently, a prevailing approach during this transitional phase may involve the collaborative design and integration of traditional medicines with nanomedicines. A number of drugs approved for the treatment of metabolic diseases, including iron chelators, hypoglycemic agents, and hypolipidemic agents, have demonstrated significant efficacy in modulating ferroptosis in diverse disease conditions (Figure 6). Nanomedicines based on these drugs for treating ferroptosis in metabolic diseases have the potential to enhance safety, efficacy, and accelerate clinical translation.
The potential of approved drugs for metabolic disorders in modulating ferroptosis. A range of drugs currently used in clinical practice for treating metabolic disorders, such as hypolipidemic and hypoglycemic drugs, as well as iron chelators, have demonstrated the ability to modulate ferroptosis. Drugs shown in blue inhibit ferroptosis, and those in red promote ferroptosis. Drugs in orange have a dual effect on ferroptosis depending on the disease context. Arrows and vertical bars indicate positive and negative effects, respectively. Created with BioRender.com.
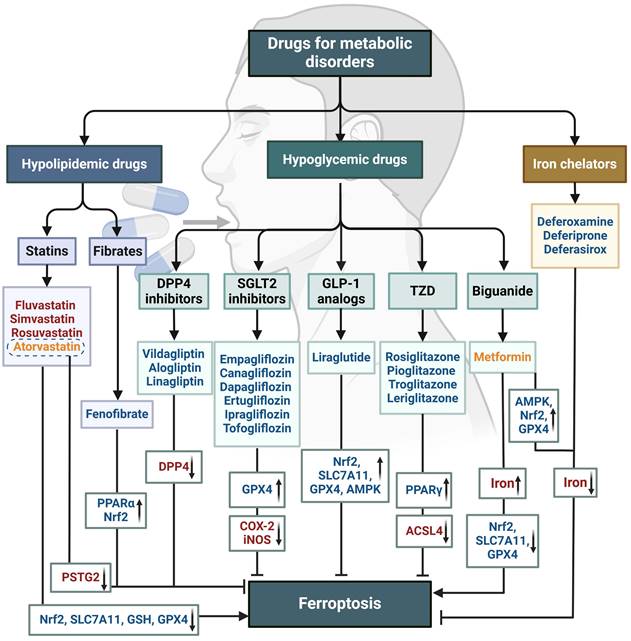
Conclusions
Metabolic dysregulation stands as a significant underpinning for ferroptosis development, which in turn exacerbates the progression of metabolic diseases through a cascade of reactions and cellular loss, perpetuating a detrimental cycle. Consequently, targeting ferroptosis has emerged as a novel strategy to impede the advancement of metabolic disorders. Nonetheless, numerous challenges persist in both mechanistic understanding and targeted therapeutics.
Numerous essential nutrients, such as glucose, FAs, glutamine, cystine, iron, and selenium, have been identified as crucial regulators of ferroptosis. However, the implications of various remaining nutrients, such as fructose, essential amino acids, and minerals (e.g. calcium, copper, and zinc), in ferroptosis remain unclear. Furthermore, the roles of key metabolic molecules (e.g. glutamine and NADPH) and regulatory nodes (e.g. AMPK) in ferroptosis regulation appear to be context-dependent, and their outcomes as either "engine" or "brake" of ferroptosis are frequently variable and await further dissection. Additionally, subcellular organelles including mitochondria, ER, and peroxisomes may serve as key sites for ferroptosis, but the mechanisms and outcomes of inter-organelle communication in orchestrating the progression of systematic ferroptosis are largely unknown.
The links between ferroptosis and the progression of metabolic diseases are widely acknowledged, the underlying mechanisms are incompletely known. To this end, further investigations into ferroptotic signaling pathways in diverse microenvironmental metabolisms are anticipated to provide clear and solid evidence. Intriguingly, the dysregulation of many hormones, such as insulin, glucagon, and sex hormones, is often observed in metabolic diseases, and emerging evidence indicates them as critical regulators of ferroptosis. For example, sex hormones and melatonin inhibit ferroptosis in tumor and neurons, respectively, while glucocorticoids can increase ferroptosis sensitivity. Therefore, it is of interest to investigate the potential of hormonal disorders in the synergistic development of metabolic diseases and ferroptosis.
Additionally, although the detrimental effects of metabolic dysregulation and ferroptosis on several systemic diseases are well documented, the involvement of these diseases in ferroptosis and metabolic disorders is emerging. For example, in aplastic anemia, ferroptosis of hematopoietic stem cells may exacerbate anemia and hypoxia, thereby disrupting systemic metabolic balance [203]. Furthermore, prolonged transfusion therapy in such patients might cause hemochromatosis that could induce systemic iron overload and ferroptosis in hepatocytes and β cells, resulting in liver injury and diabetes. It is interesting for future research to expand the potential involvement of hematopoietic diseases in the pathogenesis of ferroptosis and metabolic diseases.
Of note, different types of cell death, such as apoptosis, necroptosis, pyroptosis, autophagy, cuproptosis, and ferroptosis, may coexist and be coordinated in many physiological and pathological processes, adding another layer of complexity to ferroptosis research (Figure 7). Improvements on cell and animal models that mimic the sole and synthetical roles of distinct cell death modes, in combination with up-to-state technologies such as single-cell and spatiotemporal omics, could help to elucidate their entangled mechanisms and interplay, eventually enhancing our understanding of the trajectory of cell death during dynamic physiology and pathology.
Distinctions and interplay among cell death modes. Different types of cell death, including apoptosis, necroptosis, pyroptosis, autophagy, cuproptosis, and ferroptosis, may simultaneously occur and interact in various physiological and pathological processes. Morphological changes of different cell death modes are also briefly summarized. Arrows and vertical bars indicate positive and negative effects, respectively. Created with BioRender.com.
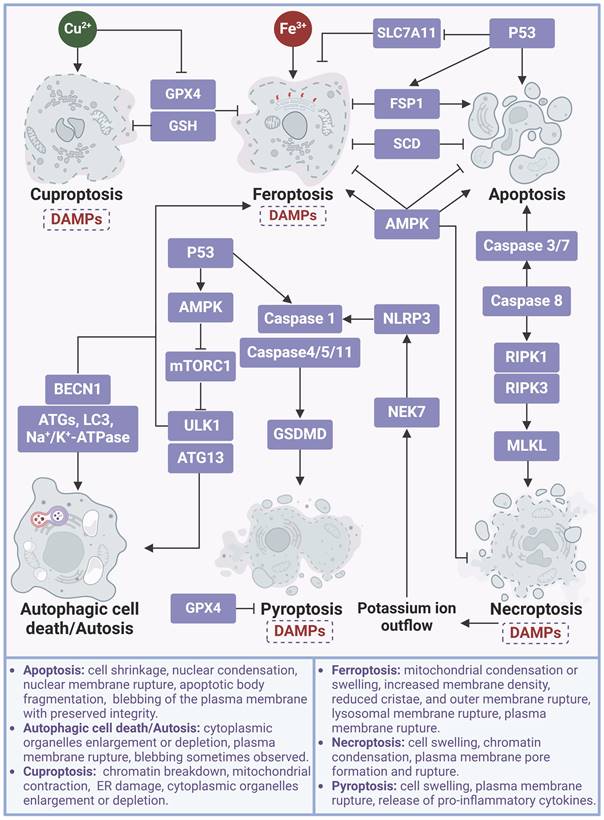
Abbreviations
AA: Arachidonic acid
Acetyl-CoA: Acetyl-coenzyme A
ACC1/2: Acetyl-CoA carboxylase 1/2
ACSL4: Coenzyme A synthase long-chain family member 4
AGEs: Advanced glycation end products
AKT: Protein kinase B
ALOXs: Arachidonic acid lipoxygenases
AMPK: Adenosine 5'-monophosphate-activated protein kinase
ASS1: Argininosuccinate synthase 1
ATO: Arsenic trioxide
BECN1: Beclin 1
BH4: Tetrahydrobiopterin
BolA2: BolA family member 2
cGPX4: Cytoplasmic glutathione peroxidase 4
CKB: Creatine kinase B
CoQ10: Coenzyme Q10
CoQ10H2: Reduced CoQ10
COVID-19: Coronavirus disease 2019
CYB5R1: NADH-cytochrome b5 reductase
DAMPs: Damage-associated molecular models
DCM: Diabetic cardiomyopathy
DGAT: Diacylglycerol acyltransferase
DHODH: Dihydroorotate dehydrogenase
DMT1: Divalent metal transporter 1
DN: Diabetic nephropathy
DR: Diabetic retinopathy
ENO: α-Enolase
ER: Endoplasmic reticulum
ERS: ER stress
EVs: Extracellular vesicles
FAs : Fatty acids
FOX3A: Forkhead box O 3a
FSP1: Ferroptosis suppressor protein 1
FTH1: Ferritin heavy chain
FTL: Ferritin light chain
FPN1: Ferroportin1
GCH1: GTP cyclohydrolase-1
GCIB-SIMS: Gas cluster ion beam secondary ion mass spectrometry
GFPT1: Glutamine-fructose-6-phosphate transaminase
GLS: Glutamate under the catalysis of glutaminase
GPX4: Glutathione peroxidase 4
GSH: Glutathione
GSK3β: Glycogen synthase kinase 3 β
HIF: Hypoxia-inducible factor
HO-1: Heme oxygenase 1
IFN-γ: Interferon-γ
iGPX4: Inducible GPX4 transcript
IONs: Iron oxide nanoparticles
IR: Insulin resistance
IRP1: Iron regulatory protein 1
ISCs: Iron-sulfur clusters
LC-MS: Liquid chromatography-mass spectrometry
LDH: Lactate dehydrogenase
LDs: Lipid droplets
LF: Lactoferrin
LPCAT3: Lysophosphatidylcholine acyltransferase 3
MAMs: Mitochondria-associated ER membranes
MARCHF6: Membrane associated ring-CH-type finger 6
MASLD: Metabolic dysfunction-associated fatty liver disease
MBOAT: Membrane bound O-acyltransferase domain containing
ME: Malic enzyme
Mfrn1: Mitochondrial ferritin-1
mGPX4: Mitochondrial GPX4
MRI: Magnetic resonance imaging
mTOR: Mammalian target of rapamycin
mTORC1: mTOR complex 1
mtROS: mitochondrial ROS
MUFAs: Monounsaturated fatty acids
MVBs: Multivesicular bodies
NADPH: Nicotinamide adenine dinucleotide phosphate hydride
NAFLD: Nonalcoholic fatty liver disease
NASH: Non-alcoholic steatohepatitis
NCOA4: Nuclear receptor coactivator 4
nGPX4: Nuclear GPX4
NOXs: NADPH oxidases
NQO1: NAD(P)H quinone oxidoreductase 1
Nrf2: Nuclear factor erythroid 2-related factor 2
NTBI: Non-TF-bound iron
OXPHOS: Oxidative phosphorylation
PAF: Platelet-activating factor
PCBPs: Poly rC Binding Proteins
PDK4: Pyruvate dehydrogenase kinase 4
PE: Phosphatidylethanolamine
PET: Positron emission tomography
Piezo1: Piezo-type mechanosensitive ion channel component 1
PKC: Protein kinase C
PL: Phospholipids
PMN-MDSCs: Myeloid-derived suppressor cells
POMC: Prohormone pro-opiomelanocortin
POR: NADPH-cytochrome P450 reductase
PPAR: Proliferator-activated receptor
PPP: Pentose phosphate pathway
PRDX3: Hyperoxidization of peroxiredoxin 3
PROM2: Prominin 2
PRRs: Pattern recognition receptors
PS: Phosphatidylserine
PTGS2: Prostaglandin endoperoxide synthase 2
PUFAs: Polyunsaturated fatty acids
PUFA-ePLs: Polyunsaturated ether phospholipids
ROS: Reactive oxygen species
RPE: Retinal pigment epithelial
RSSH: Hydropersulfides
TAG: Triacylglycerol
TCA: Tricarboxylic acid
TF: Transferrin
TFRC: Transferrin receptor
TGF-β1: Transforming growth factor-β1
TLR: Toll-like receptor
T2D: Type 2 diabetes
SAPE-OOH: 1-stearoyl-2-15-HpETE-sn-glycero-3 phosphatidylethanolamine
SCD: Steroyl CoA desaturase
SLC7A11: Solute carrier family 7 member 11
SOD: Superoxide dismutase
SREBP1: Sterol regulatory element binding transcription factor 1
STARD7: StAR-related lipid transfer domain containing 7
ULK1: Unc-51 like autophagy activating kinase 1
YAP: Yes-associated protein
3D: Three-dimensional
4-HNE: 4-hydroxynonenal
7-DHC: 7-dehydrocholesterol
α-KG: α-ketoglutarate
Acknowledgements
This work was supported by the National Science and Technology Major Project (2023ZD0507500), the National Natural Science Foundation of China (92157205, 81970561, and 82172986), and the 1.3.5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (ZYJD23005).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2021;12:599-620
2. Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J. et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc(-) Activity. Curr Biol. 2018;28:2388-99 e5
3. Zhang Y, Swanda RV, Nie L, Liu X, Wang C, Lee H. et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat Commun. 2021;12:1589
4. Zhang T, Sun L, Hao Y, Suo C, Shen S, Wei H. et al. ENO1 suppresses cancer cell ferroptosis by degrading the mRNA of iron regulatory protein 1. Nat Cancer. 2022;3:75-89
5. Freitas FP, Alborzinia H, Dos Santos AF, Nepachalovich P, Pedrera L, Zilka O. et al. 7-Dehydrocholesterol is an endogenous suppressor of ferroptosis. Nature. 2024;626:401-10
6. Li Y, Ran Q, Duan Q, Jin J, Wang Y, Yu L. et al. 7-Dehydrocholesterol dictates ferroptosis sensitivity. Nature. 2024;626:411-8
7. Liu Y, Wan Y, Jiang Y, Zhang L, Cheng W. GPX4: The hub of lipid oxidation, ferroptosis, disease and treatment. Biochim Biophys Acta Rev Cancer. 2023;1878:188890
8. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I. et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693-8
9. Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Muller C, Zandkarimi F. et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent Sci. 2020;6:41-53
10. Sha W, Hu F, Xi Y, Chu Y, Bu S. Mechanism of Ferroptosis and Its Role in Type 2 Diabetes Mellitus. J Diabetes Res. 2021;2021:9999612
11. Jiang T, Xiao Y, Zhou J, Luo Z, Yu L, Liao Q. et al. Arbutin alleviates fatty liver by inhibiting ferroptosis via FTO/SLC7A11 pathway. Redox Biol. 2023;68:102963
12. Zhang Q, Sun T, Yu F, Liu W, Gao J, Chen J. et al. PAFAH2 suppresses synchronized ferroptosis to ameliorate acute kidney injury. Nat Chem Biol. 2024;20:835-846
13. Bruni A, Pepper AR, Pawlick RL, Gala-Lopez B, Gamble AF, Kin T. et al. Ferroptosis-inducing agents compromise in vitro human islet viability and function. Cell Death Dis. 2018;9:595
14. Feng H, Schorpp K, Jin J, Yozwiak CE, Hoffstrom BG, Decker AM. et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020;30:3411-23 e7
15. Zakharova ET, Sokolov AV, Pavlichenko NN, Kostevich VA, Abdurasulova IN, Chechushkov AV. et al. Erythropoietin and Nrf2: key factors in the neuroprotection provided by apo-lactoferrin. Biometals. 2018;31:425-43
16. Zhang Z, Lu M, Chen C, Tong X, Li Y, Yang K. et al. Holo-lactoferrin: the link between ferroptosis and radiotherapy in triple-negative breast cancer. Theranostics. 2021;11:3167-82
17. Sokolov AV, Ageeva KV, Pulina MO, Zakharova ET, Vasilyev VB. Effect of lactoferrin on oxidative features of ceruloplasmin. Biometals. 2009;22:521-9
18. Coffey R, Knutson MD. The plasma membrane metal-ion transporter ZIP14 contributes to nontransferrin-bound iron uptake by human beta-cells. Am J Physiol Cell Physiol. 2017;312:C169-C75
19. Yu Y, Jiang L, Wang H, Shen Z, Cheng Q, Zhang P. et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood. 2020;136:726-39
20. Zhang T, Wang S, Hua D, Shi X, Deng H, Jin S. et al. Identification of ZIP8-induced ferroptosis as a major type of cell death in monocytes under sepsis conditions. Redox Biol. 2024;69:102985
21. Tan Q, Zhang X, Li S, Liu W, Yan J, Wang S. et al. DMT1 differentially regulates mitochondrial complex activities to reduce glutathione loss and mitigate ferroptosis. Free Radic Biol Med. 2023;207:32-44
22. Aschner M, Skalny AV, Martins AC, Sinitskii AI, Farina M, Lu R. et al. Ferroptosis as a mechanism of non-ferrous metal toxicity. Arch Toxicol. 2022;96:2391-417
23. Du J, Zhou Y, Li Y, Xia J, Chen Y, Chen S. et al. Identification of Frataxin as a regulator of ferroptosis. Redox Biol. 2020;32:101483
24. Protchenko O, Baratz E, Jadhav S, Li F, Shakoury-Elizeh M, Gavrilova O. et al. Iron Chaperone Poly rC Binding Protein 1 Protects Mouse Liver From Lipid Peroxidation and Steatosis. Hepatology. 2021;73:1176-93
25. Patel SJ, Frey AG, Palenchar DJ, Achar S, Bullough KZ, Vashisht A. et al. A PCBP1-BolA2 chaperone complex delivers iron for cytosolic [2Fe-2S] cluster assembly. Nat Chem Biol. 2019;15:872-81
26. Lee J, You JH, Roh JL. Poly(rC)-binding protein 1 represses ferritinophagy-mediated ferroptosis in head and neck cancer. Redox Biol. 2022;51:102276
27. Patel SJ, Protchenko O, Shakoury-Elizeh M, Baratz E, Jadhav S, Philpott CC. The iron chaperone and nucleic acid-binding activities of poly(rC)-binding protein 1 are separable and independently essential. Proc Natl Acad Sci U S A. 2021;118:e2104666118
28. Tian P, Xu Z, Guo J, Zhao J, Chen W, Huang W. et al. Hypoxia causes trophoblast cell ferroptosis to induce miscarriage through lnc-HZ06/HIF1alpha-SUMO/NCOA4 axis. Redox Biol. 2024;70:103073
29. Anandhan A, Dodson M, Shakya A, Chen J, Liu P, Wei Y. et al. NRF2 controls iron homeostasis and ferroptosis through HERC2 and VAMP8. Sci Adv. 2023;9:eade9585
30. Yu F, Zhang Q, Liu H, Liu J, Yang S, Luo X. et al. Dynamic O-GlcNAcylation coordinates ferritinophagy and mitophagy to activate ferroptosis. Cell Discov. 2022;8:40
31. Ryan SK, Zelic M, Han Y, Teeple E, Chen L, Sadeghi M. et al. Microglia ferroptosis is regulated by SEC24B and contributes to neurodegeneration. Nat Neurosci. 2023;26:12-26
32. Billesbolle CB, Azumaya CM, Kretsch RC, Powers AS, Gonen S, Schneider S. et al. Structure of hepcidin-bound ferroportin reveals iron homeostatic mechanisms. Nature. 2020;586:807-11
33. Tang Z, Jiang W, Mao M, Zhao J, Chen J, Cheng N. Deubiquitinase USP35 modulates ferroptosis in lung cancer via targeting ferroportin. Clin Transl Med. 2021;11:e390
34. Liu J, Zhang Y, Tian Y, Huang W, Tong N, Fu X. Integrative biology of extracellular vesicles in diabetes mellitus and diabetic complications. Theranostics. 2022;12:1342-72
35. Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, Zhu LJ. et al. Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev Cell. 2019;51:575-86 e4
36. Wang Y, Chen Q, Song H, Zhang Y, Chen H, Liu P. et al. A Triple Therapeutic Strategy with Antiexosomal Iron Efflux for Enhanced Ferroptosis Therapy and Immunotherapy. Small. 2022;18:e2201704
37. Brown CW, Chhoy P, Mukhopadhyay D, Karner ER, Mercurio AM. Targeting prominin2 transcription to overcome ferroptosis resistance in cancer. EMBO Mol Med. 2021;13:e13792
38. Yanatori I, Richardson DR, Dhekne HS, Toyokuni S, Kishi F. CD63 is regulated by iron via the IRE-IRP system and is important for ferritin secretion by extracellular vesicles. Blood. 2021;138:1490-503
39. Song X, Liu J, Kuang F, Chen X, Zeh HJ 3rd, Kang R. et al. PDK4 dictates metabolic resistance to ferroptosis by suppressing pyruvate oxidation and fatty acid synthesis. Cell Rep. 2021;34:108767
40. Yang Z, Su W, Wei X, Qu S, Zhao D, Zhou J. et al. HIF-1alpha drives resistance to ferroptosis in solid tumors by promoting lactate production and activating SLC1A1. Cell Rep. 2023;42:112945
41. Zhao Y, Li M, Yao X, Fei Y, Lin Z, Li Z. et al. HCAR1/MCT1 Regulates Tumor Ferroptosis through the Lactate-Mediated AMPK-SCD1 Activity and Its Therapeutic Implications. Cell Rep. 2020;33:108487
42. Ubellacker JM, Tasdogan A, Ramesh V, Shen B, Mitchell EC, Martin-Sandoval MS. et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature. 2020;585:113-8
43. Lv M, Gong Y, Liu X, Wang Y, Wu Q, Chen J. et al. CDK7-YAP-LDHD axis promotes D-lactate elimination and ferroptosis defense to support cancer stem cell-like properties. Signal Transduct Target Ther. 2023;8:302
44. Culbertson B, Garcia K, Markett D, Asgharian H, Chen L, Fish L. et al. A sense-antisense RNA interaction promotes breast cancer metastasis via regulation of NQO1 expression. Nat Cancer. 2023;4:682-98
45. Ding CC, Rose J, Sun T, Wu J, Chen PH, Lin CC. et al. MESH1 is a cytosolic NADPH phosphatase that regulates ferroptosis. Nat Metab. 2020;2:270-7
46. Han Y, Zhang YY, Pan YQ, Zheng XJ, Liao K, Mo HY. et al. IL-1beta-associated NNT acetylation orchestrates iron-sulfur cluster maintenance and cancer immunotherapy resistance. Mol Cell. 2023;83:1887-902 e8
47. Yu J, Zhong B, Zhao L, Hou Y, Ai N, Lu JJ. et al. Fighting drug-resistant lung cancer by induction of NAD(P)H:quinone oxidoreductase 1 (NQO1)-mediated ferroptosis. Drug Resist Updat. 2023;70:100977
48. Yan B, Ai Y, Sun Q, Ma Y, Cao Y, Wang J. et al. Membrane Damage during Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Mol Cell. 2021;81:355-69 e10
49. Nguyen KT, Mun SH, Yang J, Lee J, Seok OH, Kim E. et al. The MARCHF6 E3 ubiquitin ligase acts as an NADPH sensor for the regulation of ferroptosis. Nat Cell Biol. 2022;24:1239-51
50. Mun SH, Lee CS, Kim HJ, Kim J, Lee H, Yang J. et al. Marchf6 E3 ubiquitin ligase critically regulates endoplasmic reticulum stress, ferroptosis, and metabolic homeostasis in POMC neurons. Cell Rep. 2023;42:112746
51. Lu D, Xia Q, Yang Z, Gao S, Sun S, Luo X. et al. ENO3 promoted the progression of NASH by negatively regulating ferroptosis via elevation of GPX4 expression and lipid accumulation. Ann Transl Med. 2021;9:661
52. Ye H, Wang R, Wei J, Wang Y, Zhang X, Wang L. Bioinformatics Analysis Identifies Potential Ferroptosis Key Gene in Type 2 Diabetic Islet Dysfunction. Front Endocrinol (Lausanne). 2022;13:904312
53. Wu K, Yan M, Liu T, Wang Z, Duan Y, Xia Y. et al. Creatine kinase B suppresses ferroptosis by phosphorylating GPX4 through a moonlighting function. Nat Cell Biol. 2023;25:714-25
54. Fang X, Zhang J, Li Y, Song Y, Yu Y, Cai Z. et al. Malic Enzyme 1 as a Novel Anti-Ferroptotic Regulator in Hepatic Ischemia/Reperfusion Injury. Adv Sci (Weinh). 2023;10:e2205436
55. Liu F, Jiang LJ, Zhang YX, Xu ST, Liu SL, Ye JT. et al. Inhibition of miR-214-3p attenuates ferroptosis in myocardial infarction via regulating ME2. Biochem Biophys Res Commun. 2023;661:64-74
56. Deng F, Sharma I, Dai Y, Yang M, Kanwar YS. Myo-inositol oxygenase expression profile modulates pathogenic ferroptosis in the renal proximal tubule. J Clin Invest. 2019;129:5033-49
57. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell. 2015;59:298-308
58. Zhang X, Yu K, Ma L, Qian Z, Tian X, Miao Y. et al. Endogenous glutamate determines ferroptosis sensitivity via ADCY10-dependent YAP suppression in lung adenocarcinoma. Theranostics. 2021;11:5650-74
59. Cai Y, Lv L, Lu T, Ding M, Yu Z, Chen X. et al. alpha-KG inhibits tumor growth of diffuse large B-cell lymphoma by inducing ROS and TP53-mediated ferroptosis. Cell Death Discov. 2023;9:182
60. Luo M, Wu L, Zhang K, Wang H, Zhang T, Gutierrez L. et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. 2018;25:1457-72
61. Hu Q, Dai J, Zhang Z, Yu H, Zhang J, Zhu X. et al. ASS1-Mediated Reductive Carboxylation of Cytosolic Glutamine Confers Ferroptosis Resistance in Cancer Cells. Cancer Res. 2023;83:1646-65
62. Conlon M, Poltorack CD, Forcina GC, Armenta DA, Mallais M, Perez MA. et al. A compendium of kinetic modulatory profiles identifies ferroptosis regulators. Nat Chem Biol. 2021;17:665-74
63. Guo X, Guo Y, Li J, Liu Q, Wu H. Arginine Expedites Erastin-Induced Ferroptosis through Fumarate. Int J Mol Sci. 2023;24:14595
64. Ma Z, Ye W, Wang J, Huang X, Huang J, Li X. et al. Glutamate dehydrogenase 1: A novel metabolic target in inhibiting acute myeloid leukaemia progression. Br J Haematol. 2023;202:566-77
65. Balakrishnan M, Kenworthy AK. Lipid Peroxidation Drives Liquid-Liquid Phase Separation and Disrupts Raft Protein Partitioning in Biological Membranes. J Am Chem Soc. 2024;146:1374-87
66. Qiu B, Zandkarimi F, Bezjian CT, Reznik E, Soni RK, Gu W. et al. Phospholipids with two polyunsaturated fatty acyl tails promote ferroptosis. Cell. 2024;187:1177-1190 e18
67. Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P. et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature. 2020;585:603-8
68. Lee H, Horbath A, Kondiparthi L, Meena JK, Lei G, Dasgupta S. et al. Cell cycle arrest induces lipid droplet formation and confers ferroptosis resistance. Nat Commun. 2024;15:79
69. Zhang HL, Hu BX, Li ZL, Du T, Shan JL, Ye ZP. et al. PKCbetaII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat Cell Biol. 2022;24:88-98
70. Li K, Wang M, Huang ZH, Wang M, Sun WY, Kurihara H. et al. ALOX5 inhibition protects against dopaminergic neurons undergoing ferroptosis. Pharmacol Res. 2023;193:106779
71. Wang B, Tontonoz P. Phospholipid Remodeling in Physiology and Disease. Annu Rev Physiol. 2019;81:165-88
72. Liang D, Feng Y, Zandkarimi F, Wang H, Zhang Z, Kim J. et al. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell. 2023;186:2748-64 e22
73. Xia C, Xing X, Zhang W, Wang Y, Jin X, Wang Y. et al. Cysteine and homocysteine can be exploited by GPX4 in ferroptosis inhibition independent of GSH synthesis. Redox Biol. 2024;69:102999
74. Filipovic MR, Zivanovic J, Alvarez B, Banerjee R. Chemical Biology of H2S Signaling through Persulfidation. Chem Rev. 2018;118:1253-337
75. Barayeu U, Schilling D, Eid M, Xavier da Silva TN, Schlicker L, Mitreska N. et al. Hydropersulfides inhibit lipid peroxidation and ferroptosis by scavenging radicals. Nat Chem Biol. 2023;19:28-37
76. Fiore A, Zeitler L, Russier M, Gross A, Hiller MK, Parker JL. et al. Kynurenine importation by SLC7A11 propagates anti-ferroptotic signaling. Mol Cell. 2022;82:920-32 e7
77. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H. et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57-62
78. Shen X, Dong P, Kong J, Sun N, Wang F, Sang L. et al. Targeted single-cell RNA sequencing analysis reveals metabolic reprogramming and the ferroptosis-resistant state in hematologic malignancies. Cell Biochem Funct. 2023;41:1343-56
79. Zheng Q, Li P, Zhou X, Qiang Y, Fan J, Lin Y. et al. Deficiency of the X-inactivation escaping gene KDM5C in clear cell renal cell carcinoma promotes tumorigenicity by reprogramming glycogen metabolism and inhibiting ferroptosis. Theranostics. 2021;11:8674-91
80. Xue Q, Yan D, Chen X, Li X, Kang R, Klionsky DJ. et al. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy. 2023;19:1982-96
81. Tong J, Li D, Meng H, Sun D, Lan X, Ni M. et al. Targeting a novel inducible GPX4 alternative isoform to alleviate ferroptosis and treat metabolic-associated fatty liver disease. Acta Pharm Sin B. 2022;12:3650-66
82. Xue Q, Yan D, Chen X, Li X, Kang R, Klionsky DJ. et al. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy. 2023;19:1982-1996
83. Lu Q, Yang L, Xiao JJ, Liu Q, Ni L, Hu JW. et al. Empagliflozin attenuates the renal tubular ferroptosis in diabetic kidney disease through AMPK/NRF2 pathway. Free Radic Biol Med. 2023;195:89-102
84. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H. et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586-90
85. Pfeifer H, Conrad M, Roethlein D, Kyriakopoulos A, Brielmeier M, Bornkamm GW. et al. Identification of a specific sperm nuclei selenoenzyme necessary for protamine thiol cross-linking during sperm maturation. FASEB J. 2001;15:1236-8
86. Deshwal S, Onishi M, Tatsuta T, Bartsch T, Cors E, Ried K. et al. Mitochondria regulate intracellular coenzyme Q transport and ferroptotic resistance via STARD7. Nat Cell Biol. 2023;25:246-57
87. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688-92
88. Lee N, Park SJ, Lange M, Tseyang T, Doshi MB, Kim TY. et al. Selenium reduction of ubiquinone via SQOR suppresses ferroptosis. Nat Metab. 2024;6:343-358
89. Tian Y, Xie Y, Guo Z, Feng P, You Y, Yu Q. 17beta-oestradiol inhibits ferroptosis in the hippocampus by upregulating DHODH and further improves memory decline after ovariectomy. Redox Biol. 2023;62:102708
90. Cui W, Guo M, Liu D, Xiao P, Yang C, Huang H. et al. Gut microbial metabolite facilitates colorectal cancer development via ferroptosis inhibition. Nat Cell Biol. 2024;26:124-37
91. Song Z, Jia G, Ma P, Cang S. Exosomal miR-4443 promotes cisplatin resistance in non-small cell lung carcinoma by regulating FSP1 m6A modification-mediated ferroptosis. Life Sci. 2021;276:119399
92. Nakamura T, Hipp C, Santos Dias Mourao A, Borggrafe J, Aldrovandi M, Henkelmann B. et al. Phase separation of FSP1 promotes ferroptosis. Nature. 2023;619:371-7
93. Miriyala S, Thippakorn C, Chaiswing L, Xu Y, Noel T, Tovmasyan A. et al. Novel role of 4-hydroxy-2-nonenal in AIFm2-mediated mitochondrial stress signaling. Free Radic Biol Med. 2016;91:68-80
94. Mishima E, Ito J, Wu Z, Nakamura T, Wahida A, Doll S. et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature. 2022;608:778-83
95. Pierrel F, Burgardt A, Lee JH, Pelosi L, Wendisch VF. Recent advances in the metabolic pathways and microbial production of coenzyme Q. World J Microbiol Biotechnol. 2022;38:58
96. Brantley KD, Douglas TD, Singh RH. One-year follow-up of B vitamin and Iron status in patients with phenylketonuria provided tetrahydrobiopterin (BH4). Orphanet J Rare Dis. 2018;13:192
97. Hu Q, Wei W, Wu D, Huang F, Li M, Li W. et al. Blockade of GCH1/BH4 Axis Activates Ferritinophagy to Mitigate the Resistance of Colorectal Cancer to Erastin-Induced Ferroptosis. Front Cell Dev Biol. 2022;10:810327
98. Liu P, Anandhan A, Chen J, Shakya A, Dodson M, Ooi A. et al. Decreased autophagosome biogenesis, reduced NRF2, and enhanced ferroptotic cell death are underlying molecular mechanisms of non-alcoholic fatty liver disease. Redox Biol. 2023;59:102570
99. Wang X, Chen X, Zhou W, Men H, Bao T, Sun Y. et al. Ferroptosis is essential for diabetic cardiomyopathy and is prevented by sulforaphane via AMPK/NRF2 pathways. Acta Pharm Sin B. 2022;12:708-22
100. Zhong S, Chen W, Wang B, Gao C, Liu X, Song Y. et al. Energy stress modulation of AMPK/FoxO3 signaling inhibits mitochondria-associated ferroptosis. Redox Biol. 2023;63:102760
101. Chen Z, Li S, Liu M, Yin M, Chen J, Li Y. et al. Nicorandil alleviates cardiac microvascular ferroptosis in diabetic cardiomyopathy: Role of the mitochondria-localized AMPK-Parkin-ACSL4 signaling pathway. Pharmacol Res. 2024;200:107057
102. Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L. et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. 2020;22:225-34
103. Qin X, Zhang J, Wang B, Xu G, Yang X, Zou Z. et al. Ferritinophagy is involved in the zinc oxide nanoparticles-induced ferroptosis of vascular endothelial cells. Autophagy. 2021;17:4266-85
104. Cai J, Ye Z, Hu Y, Ye L, Gao L, Wang Y. et al. Fatostatin induces ferroptosis through inhibition of the AKT/mTORC1/GPX4 signaling pathway in glioblastoma. Cell Death Dis. 2023;14:211
105. Zeng F, Ye L, Zhou Q, He Y, Zhang Y, Deng G. et al. Inhibiting SCD expression by IGF1R during lorlatinib therapy sensitizes melanoma to ferroptosis. Redox Biol. 2023;61:102653
106. Han D, Jiang L, Gu X, Huang S, Pang J, Wu Y. et al. SIRT3 deficiency is resistant to autophagy-dependent ferroptosis by inhibiting the AMPK/mTOR pathway and promoting GPX4 levels. J Cell Physiol. 2020;235:8839-51
107. Armenta DA, Laqtom NN, Alchemy G, Dong W, Morrow D, Poltorack CD. et al. Ferroptosis inhibition by lysosome-dependent catabolism of extracellular protein. Cell Chem Biol. 2022;29:1588-600 e7
108. Kazyken D, Magnuson B, Bodur C, Acosta-Jaquez HA, Zhang D, Tong X. et al. AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci Signal. 2019;12:eaav3249
109. Wang G, Chen L, Qin S, Zheng Y, Xia C, Yao J. et al. Cystine Induced-mTORC2 Activation through Promoting Sin1 Phosphorylation to Suppress Cancer Cell Ferroptosis. Mol Nutr Food Res. 2022;66:e2200186
110. Wang Y, Tian Q, Hao Y, Yao W, Lu J, Chen C. et al. The kinase complex mTORC2 promotes the longevity of virus-specific memory CD4+ T cells by preventing ferroptosis. Nat Immunol. 2022;23:303-17
111. Ke W, Liao Z, Liang H, Tong B, Song Y, Li G. et al. Stiff Substrate Induces Nucleus Pulposus Cell Ferroptosis via YAP and N-Cadherin Mediated Mechanotransduction. Adv Healthc Mater. 2023;12:e2300458
112. Yang WH, Ding CC, Sun T, Rupprecht G, Lin CC, Hsu D. et al. The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep. 2019;28:2501-8 e4
113. Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR. et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature. 2019;572:402-6
114. Bao Z, Hua L, Ye Y, Wang D, Li C, Xie Q. et al. MEF2C silencing downregulates NF2 and E-cadherin and enhances Erastin-induced ferroptosis in meningioma. Neuro Oncol. 2021;23:2014-27
115. Chen X, Song X, Li J, Zhang R, Yu C, Zhou Z. et al. Identification of HPCAL1 as a specific autophagy receptor involved in ferroptosis. Autophagy. 2023;19:54-74
116. Yang WH, Lin CC, Wu J, Chao PY, Chen K, Chen PH. et al. The Hippo Pathway Effector YAP Promotes Ferroptosis via the E3 Ligase SKP2. Mol Cancer Res. 2021;19:1005-14
117. Gu Y, Wu S, Fan J, Meng Z, Gao G, Liu T. et al. CYLD regulates cell ferroptosis through Hippo/YAP signaling in prostate cancer progression. Cell Death Dis. 2024;15:79
118. Zhao Y, Ye Z, Liu Y, Zhang J, Kuermanbayi S, Zhou Y. et al. Investigating the Role of Extracellular Matrix Stiffness in Modulating the Ferroptosis Process in Hepatocellular Carcinoma Cells via Scanning Electrochemical Microscopy. Anal Chem. 2024;96:1102-11
119. Gudipaty SA, Lindblom J, Loftus PD, Redd MJ, Edes K, Davey CF. et al. Mechanical stretch triggers rapid epithelial cell division through Piezo1. Nature. 2017;543:118-21
120. Guo XW, Zhang H, Huang JQ, Wang SN, Lu Y, Cheng B. et al. PIEZO1 Ion Channel Mediates Ionizing Radiation-Induced Pulmonary Endothelial Cell Ferroptosis via Ca(2+)/Calpain/VE-Cadherin Signaling. Front Mol Biosci. 2021;8:725274
121. Wang S, Li W, Zhang P, Wang Z, Ma X, Liu C. et al. Mechanical overloading induces GPX4-regulated chondrocyte ferroptosis in osteoarthritis via Piezo1 channel facilitated calcium influx. J Adv Res. 2022;41:63-75
122. Hirata Y, Cai R, Volchuk A, Steinberg BE, Saito Y, Matsuzawa A. et al. Lipid peroxidation increases membrane tension, Piezo1 gating, and cation permeability to execute ferroptosis. Curr Biol. 2023;33:1282-94 e5
123. Ma S, Dubin AE, Zhang Y, Mousavi SAR, Wang Y, Coombs AM. et al. A role of PIEZO1 in iron metabolism in mice and humans. Cell. 2021;184:969-82 e13
124. Riegman M, Sagie L, Galed C, Levin T, Steinberg N, Dixon SJ. et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat Cell Biol. 2020;22:1042-8
125. Wang LL, Mai YZ, Zheng MH, Yan GH, Jin JY. A single fluorescent probe to examine the dynamics of mitochondria-lysosome interplay and extracellular vesicle role in ferroptosis. Dev Cell. 2024;59:517-528.e3
126. Li P, Jiang M, Li K, Li H, Zhou Y, Xiao X. et al. Glutathione peroxidase 4-regulated neutrophil ferroptosis induces systemic autoimmunity. Nat Immunol. 2021;22:1107-17
127. Ma X, Xiao L, Liu L, Ye L, Su P, Bi E. et al. CD36-mediated ferroptosis dampens intratumoral CD8+ T cell effector function and impairs their antitumor ability. Cell Metab. 2021;33:1001-12 e5
128. Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK. et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270-4
129. Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y. et al. CD8+T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell. 2022;40:365-78 e6
130. Chen J, Jin Z, Zhang S, Zhang X, Li P, Yang H. et al. Arsenic trioxide elicits prophylactic and therapeutic immune responses against solid tumors by inducing necroptosis and ferroptosis. Cell Mol Immunol. 2023;20:51-64
131. Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ. et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. 2020;16:2069-83
132. Luoqian J, Yang W, Ding X, Tuo QZ, Xiang Z, Zheng Z. et al. Ferroptosis promotes T-cell activation-induced neurodegeneration in multiple sclerosis. Cell Mol Immunol. 2022;19:913-24
133. Kim R, Hashimoto A, Markosyan N, Tyurin VA, Tyurina YY, Kar G. et al. Ferroptosis of tumour neutrophils causes immune suppression in cancer. Nature. 2022;612:338-46
134. Zhang D, Li Y, Du C, Sang L, Liu L, Li Y. et al. Evidence of pyroptosis and ferroptosis extensively involved in autoimmune diseases at the single-cell transcriptome level. J Transl Med. 2022;20:363
135. Kloditz K, Fadeel B. Three cell deaths and a funeral: macrophage clearance of cells undergoing distinct modes of cell death. Cell Death Discov. 2019;5:65
136. Luo X, Gong HB, Gao HY, Wu YP, Sun WY, Li ZQ. et al. Oxygenated phosphatidylethanolamine navigates phagocytosis of ferroptotic cells by interacting with TLR2. Cell Death Differ. 2021;28:1971-89
137. Hosseini Z, Marinello M, Decker C, Sansbury BE, Sadhu S, Gerlach BD. et al. Resolvin D1 Enhances Necroptotic Cell Clearance Through Promoting Macrophage Fatty Acid Oxidation and Oxidative Phosphorylation. Arterioscler Thromb Vasc Biol. 2021;41:1062-75
138. Zhu M, Peng L, Huo S, Peng D, Gou J, Shi W. et al. STAT3 signaling promotes cardiac injury by upregulating NCOA4-mediated ferritinophagy and ferroptosis in high-fat-diet fed mice. Free Radic Biol Med. 2023;201:111-25
139. Yang Y, Chen J, Gao Q, Shan X, Wang J, Lv Z. Study on the attenuated effect of Ginkgolide B on ferroptosis in high fat diet induced nonalcoholic fatty liver disease. Toxicology. 2020;445:152599
140. Zhao X, Si L, Bian J, Pan C, Guo W, Qin P. et al. Adipose tissue macrophage-derived exosomes induce ferroptosis via glutathione synthesis inhibition by targeting SLC7A11 in obesity-induced cardiac injury. Free Radic Biol Med. 2022;182:232-45
141. Zhang Q, Deng T, Zhang H, Zuo D, Zhu Q, Bai M. et al. Adipocyte-Derived Exosomal MTTP Suppresses Ferroptosis and Promotes Chemoresistance in Colorectal Cancer. Adv Sci (Weinh). 2022;9:e2203357
142. Tan SK, Mahmud I, Fontanesi F, Puchowicz M, Neumann CKA, Griswold AJ. et al. Obesity-Dependent Adipokine Chemerin Suppresses Fatty Acid Oxidation to Confer Ferroptosis Resistance. Cancer Discov. 2021;11:2072-93
143. Tsurusaki S, Tsuchiya Y, Koumura T, Nakasone M, Sakamoto T, Matsuoka M. et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019;10:449
144. Kowdley KV, Belt P, Wilson LA, Yeh MM, Neuschwander-Tetri BA, Chalasani N. et al. Serum ferritin is an independent predictor of histologic severity and advanced fibrosis in patients with nonalcoholic fatty liver disease. Hepatology. 2012;55:77-85
145. Gao H, Jin Z, Bandyopadhyay G, Wang G, Zhang D, Rocha KCE. et al. Aberrant iron distribution via hepatocyte-stellate cell axis drives liver lipogenesis and fibrosis. Cell Metab. 2022;34:1201-13 e5
146. Zhao M, Jin Z, Xia C, Chen S, Zeng L, Qin S. et al. Inhibition of free heme-catalyzed Fenton-like reaction prevents non-alcoholic fatty liver disease by hepatocyte-targeted hydrogen delivery. Biomaterials. 2023;301:122230
147. Liu S, Gao Z, He W, Wu Y, Liu J, Zhang S. et al. The gut microbiota metabolite glycochenodeoxycholate activates TFR-ACSL4-mediated ferroptosis to promote the development of environmental toxin-linked MAFLD. Free Radic Biol Med. 2022;193:213-26
148. Cui S, Ghai A, Deng Y, Li S, Zhang R, Egbulefu C. et al. Identification of hyperoxidized PRDX3 as a ferroptosis marker reveals ferroptotic damage in chronic liver diseases. Mol Cell. 2023;83:3931-9 e5
149. Luo P, Liu D, Zhang Q, Yang F, Wong YK, Xia F. et al. Celastrol induces ferroptosis in activated HSCs to ameliorate hepatic fibrosis via targeting peroxiredoxins and HO-1. Acta Pharm Sin B. 2022;12:2300-14
150. Ni X, Duan L, Bao Y, Li J, Zhang X, Jia D. et al. Circ_005077 accelerates myocardial lipotoxicity induced by high-fat diet via CyPA/p47PHOX mediated ferroptosis. Cardiovasc Diabetol. 2024;23:129
151. Styskal J, Van Remmen H, Richardson A, Salmon AB. Oxidative stress and diabetes: what can we learn about insulin resistance from antioxidant mutant mouse models? Free Radic Biol Med. 2012;52:46-58
152. Jacobson DA, Weber CR, Bao S, Turk J, Philipson LH. Modulation of the pancreatic islet beta-cell-delayed rectifier potassium channel Kv2.1 by the polyunsaturated fatty acid arachidonate. J Biol Chem. 2007;282:7442-9
153. Li D, Jiang C, Mei G, Zhao Y, Chen L, Liu J. et al. Quercetin Alleviates Ferroptosis of Pancreatic beta Cells in Type 2 Diabetes. Nutrients. 2020;12:2954
154. Zhou Y. The Protective Effects of Cryptochlorogenic Acid on beta-Cells Function in Diabetes in vivo and vitro via Inhibition of Ferroptosis. Diabetes Metab Syndr Obes. 2020;13:1921-31
155. Killion EA, Reeves AR, El Azzouny MA, Yan QW, Surujon D, Griffin JD. et al. A role for long-chain acyl-CoA synthetase-4 (ACSL4) in diet-induced phospholipid remodeling and obesity-associated adipocyte dysfunction. Mol Metab. 2018;9:43-56
156. Tong J, Lan XT, Zhang Z, Liu Y, Sun DY, Wang XJ. et al. Ferroptosis inhibitor liproxstatin-1 alleviates metabolic dysfunction-associated fatty liver disease in mice: potential involvement of PANoptosis. Acta Pharmacol Sin. 2023;44:1014-28
157. Dong L, Wang H, Chen K, Li Y. Roles of hydroxyeicosatetraenoic acids in diabetes (HETEs and diabetes). Biomed Pharmacother. 2022;156:113981
158. Sagoo MK, Gnudi L. Diabetic nephropathy: Is there a role for oxidative stress? Free Radic Biol Med. 2018;116:50-63
159. Wu Y, Zhao Y, Yang HZ, Wang YJ, Chen Y. HMGB1 regulates ferroptosis through Nrf2 pathway in mesangial cells in response to high glucose. Biosci Rep. 2021;4:BSR20202924
160. Kim S, Kang SW, Joo J, Han SH, Shin H, Nam BY. et al. Characterization of ferroptosis in kidney tubular cell death under diabetic conditions. Cell Death Dis. 2021;12:160
161. Feng X, Wang S, Sun Z, Dong H, Yu H, Huang M. et al. Ferroptosis Enhanced Diabetic Renal Tubular Injury via HIF-1alpha/HO-1 Pathway in db/db Mice. Front Endocrinol (Lausanne). 2021;12:626390
162. Jin T, Chen C. Umbelliferone delays the progression of diabetic nephropathy by inhibiting ferroptosis through activation of the Nrf-2/HO-1 pathway. Food Chem Toxicol. 2022;163:112892
163. Feng Q, Yang Y, Qiao Y, Zheng Y, Yu X, Liu F. et al. Quercetin Ameliorates Diabetic Kidney Injury by Inhibiting Ferroptosis via Activating Nrf2/HO-1 Signaling Pathway. Am J Chin Med. 2023;51:997-1018
164. Chen J, Ou Z, Gao T, Yang Y, Shu A, Xu H. et al. Ginkgolide B alleviates oxidative stress and ferroptosis by inhibiting GPX4 ubiquitination to improve diabetic nephropathy. Biomed Pharmacother. 2022;156:113953
165. Liu C, Sun W, Zhu T, Shi S, Zhang J, Wang J. et al. Glia maturation factor-beta induces ferroptosis by impairing chaperone-mediated autophagic degradation of ACSL4 in early diabetic retinopathy. Redox Biol. 2022;52:102292
166. Liu Z, Gan S, Fu L, Xu Y, Wang S, Zhang G. et al. 1,8-Cineole ameliorates diabetic retinopathy by inhibiting retinal pigment epithelium ferroptosis via PPAR-gamma/TXNIP pathways. Biomed Pharmacother. 2023;164:114978
167. Zhang J, Qiu Q, Wang H, Chen C, Luo D. TRIM46 contributes to high glucose-induced ferroptosis and cell growth inhibition in human retinal capillary endothelial cells by facilitating GPX4 ubiquitination. Exp Cell Res. 2021;407:112800
168. Fernandez-Real JM, Manco M. Effects of iron overload on chronic metabolic diseases. Lancet Diabetes Endocrinol. 2014;2:513-26
169. Hao L, Mi J, Song L, Guo Y, Li Y, Yin Y. et al. SLC40A1 Mediates Ferroptosis and Cognitive Dysfunction in Type 1 Diabetes. Neuroscience. 2021;463:216-26
170. Xiao Z, Shen D, Lan T, Wei C, Wu W, Sun Q. et al. Reduction of lactoferrin aggravates neuronal ferroptosis after intracerebral hemorrhagic stroke in hyperglycemic mice. Redox Biol. 2022;50:102256
171. Nabavizadeh A, Bagley SJ. Exploiting iron metabolism as a therapeutic vulnerability in glioblastoma. Clin Cancer Res. 2024;30:255-256
172. Tang W, Li Y, He S, Jiang T, Wang N, Du M. et al. Caveolin-1 alleviates diabetes-associated cognitive dysfunction through modulating neuronal ferroptosis-mediated mitochondrial homeostasis. Antioxid Redox Signal. 2022;37:867-886
173. Wang Y, Wang S, Xin Y, Zhang J, Wang S, Yang Z. et al. Hydrogen sulfide alleviates the anxiety-like and depressive-like behaviors of type 1 diabetic mice via inhibiting inflammation and ferroptosis. Life Sci. 2021;278:119551
174. Murtaza G, Virk HUH, Khalid M, Lavie CJ, Ventura H, Mukherjee D. et al. Diabetic cardiomyopathy - A comprehensive updated review. Prog Cardiovasc Dis. 2019;62:315-26
175. Stockwell BR. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185:2401-21
176. Li W, Li W, Leng Y, Xiong Y, Xia Z. Ferroptosis Is Involved in Diabetes Myocardial Ischemia/Reperfusion Injury Through Endoplasmic Reticulum Stress. DNA Cell Biol. 2020;39:210-25
177. Wu S, Lu Q, Ding Y, Wu Y, Qiu Y, Wang P. et al. Hyperglycemia-Driven Inhibition of AMP-Activated Protein Kinase alpha2 Induces Diabetic Cardiomyopathy by Promoting Mitochondria-Associated Endoplasmic Reticulum Membranes In Vivo. Circulation. 2019;139:1913-36
178. Niu B, Lei X, Xu Q, Ju Y, Xu D, Mao L. et al. Protecting mitochondria via inhibiting VDAC1 oligomerization alleviates ferroptosis in acetaminophen-induced acute liver injury. Cell Biol Toxicol. 2022;38:505-30
179. Li C, Liu J, Hou W, Kang R, Tang D. STING1 Promotes Ferroptosis Through MFN1/2-Dependent Mitochondrial Fusion. Front Cell Dev Biol. 2021;9:698679
180. Khunti K, Del Prato S, Mathieu C, Kahn SE, Gabbay RA, Buse JB. COVID-19, Hyperglycemia, and New-Onset Diabetes. Diabetes Care. 2021;44:2645-55
181. Liu L, Du J, Yang S, Zheng B, Shen J, Huang J. et al. SARS-CoV-2 ORF3a sensitizes cells to ferroptosis via Keap1-NRF2 axis. Redox Biol. 2023;63:102752
182. Han Y, Zhu J, Yang L, Nilsson-Payant BE, Hurtado R, Lacko LA. et al. SARS-CoV-2 Infection Induces Ferroptosis of Sinoatrial Node Pacemaker Cells. Circ Res. 2022;130:963-77
183. Zhou C, Chen Y, Ji Y, He X, Xue D. Increased Serum Levels of Hepcidin and Ferritin Are Associated with Severity of COVID-19. Med Sci Monit. 2020;26:e926178
184. Dalamaga M, Karampela I, Mantzoros CS. Commentary: Could iron chelators prove to be useful as an adjunct to COVID-19 Treatment Regimens? Metabolism. 2020;108:154260
185. de Las Heras N, Martin Gimenez VM, Ferder L, Manucha W, Lahera V. Implications of Oxidative Stress and Potential Role of Mitochondrial Dysfunction in COVID-19: Therapeutic Effects of Vitamin D. Antioxidants (Basel). 2020;9:897
186. Wang Y, Huang J, Sun Y, Stubbs D, He J, Li W. et al. SARS-CoV-2 suppresses mRNA expression of selenoproteins associated with ferroptosis, endoplasmic reticulum stress and DNA synthesis. Food Chem Toxicol. 2021;153:112286
187. Snider JM, You JK, Wang X, Snider AJ, Hallmark B, Zec MM. et al. Group IIA secreted phospholipase A2 is associated with the pathobiology leading to COVID-19 mortality. J Clin Invest. 2021;131:e149236
188. Barberis E, Timo S, Amede E, Vanella VV, Puricelli C, Cappellano G. et al. Large-Scale Plasma Analysis Revealed New Mechanisms and Molecules Associated with the Host Response to SARS-CoV-2. Int J Mol Sci. 2020;21:8623
189. Amoscato AA, Anthonymuthu T, Kapralov O, Sparvero LJ, Shrivastava IH, Mikulska-Ruminska K. et al. Formation of protein adducts with Hydroperoxy-PE electrophilic cleavage products during ferroptosis. Redox Biol. 2023;63:102758
190. Sparvero LJ, Tian H, Amoscato AA, Sun WY, Anthonymuthu TS, Tyurina YY. et al. Direct Mapping of Phospholipid Ferroptotic Death Signals in Cells and Tissues by Gas Cluster Ion Beam Secondary Ion Mass Spectrometry (GCIB-SIMS). Angew Chem Int Edit. 2021;60:11784-8
191. Wu Y, Torabi SF, Lake RJ, Hong S, Yu Z, Wu P. et al. Simultaneous Fe(2+)/Fe(3+) imaging shows Fe(3+) over Fe(2+) enrichment in Alzheimer's disease mouse brain. Sci Adv. 2023;9:eade7622
192. Qi YL, Wang HR, Chen LL, Yang B, Yang YS, He ZX. et al. Multifunctional Fluorescent Probe for Simultaneously Detecting Microviscosity, Micropolarity, and Carboxylesterases and Its Application in Bioimaging. Anal Chem. 2022;94:4594-601
193. Nijiati S, Zeng FT, Zuo CC, Zhang QY, Du C, Shi CR. et al. Fe(II)-Targeted PET/19F MRI Dual-Modal Molecular Imaging Probe for Early Evaluation of Anticancer Drug-Induced Acute Kidney Injury. Mol Pharmaceut. 2023;20:5185-94
194. Zeng FT, Nijiati S, Liu YTY, Yang QQ, Liu XM, Zhang QY. et al. Ferroptosis MRI for early detection of anticancer drug- induced acute cardiac/kidney injuries. Sci Adv. 2023;9:eadd8539
195. Cheng J, Li L, Jin D, Dai Y, Zhu Y, Zou J. et al. Boosting Ferroptosis Therapy with Iridium Single-Atom Nanocatalyst in Ultralow Metal Content. Adv Mater. 2023;35:e2210037
196. Cheng J, Li L, Jin D, Zhang Y, Yu W, Yu J. et al. A non-metal single atom nanozyme for cutting off the energy and reducing power of tumors. Angew Chem Int Ed Engl. 2024;63:e202319982
197. Fan Q, Xiong W, Zhou H, Yang J, Feng J, Li Z. et al. An AND Logic Gate for Magnetic-Resonance-Imaging-Guided Ferroptosis Therapy of Tumors. Adv Mater. 2023;35:e2305932
198. Zeng F, Tang L, Zhang Q, Shi C, Huang Z, Nijiati S. et al. Coordinating the Mechanisms of Action of Ferroptosis and the Photothermal Effect for Cancer Theranostics. Angew Chem Int Ed Engl. 2022;61:e202112925
199. Fu J, Zhang P, Sun Z, Lu G, Cao Q, Chen Y. et al. A combined nanotherapeutic approach targeting farnesoid X receptor, ferroptosis, and fibrosis for nonalcoholic steatohepatitis treatment. Acta Pharm Sin B. 2024;14:2228-46
200. Zhang W, Song Q, Bi X, Cui W, Fang C, Gao J. et al. Preparation of Pueraria lobata Root-Derived Exosome-Like Nanovesicles and Evaluation of Their Effects on Mitigating Alcoholic Intoxication and Promoting Alcohol Metabolism in Mice. Int J Nanomedicine. 2024;19:4907-21
201. Li F, Mao Z, Du Y, Cui Y, Yang S, Huang K. et al. Mesoporous MOFs with ROS scavenging capacity for the alleviation of inflammation through inhibiting stimulator of interferon genes to promote diabetic wound healing. J Nanobiotechnology. 2024;22:246
202. Li Y, Cai Z, Ma W, Bai L, Luo E, Lin Y. A DNA tetrahedron-based ferroptosis-suppressing nanoparticle: superior delivery of curcumin and alleviation of diabetic osteoporosis. Bone Res. 2024;12:14
203. Guo R, Kong J, Tang P, Wang S, Sang L, Liu L. et al. Unbiased Single-Cell Sequencing of Hematopoietic and Immune Cells from Aplastic Anemia Reveals the Contributors of Hematopoiesis Failure and Dysfunctional Immune Regulation. Adv Sci (Weinh). 2024;11:e2304539
Author contact
![]() Corresponding authors: Xianghui Fu, Ph.D. and Yan Tian. Ph.D. E-mail: xfuedu.cn (X. F); ytianedu.cn (Y. T).
Corresponding authors: Xianghui Fu, Ph.D. and Yan Tian. Ph.D. E-mail: xfuedu.cn (X. F); ytianedu.cn (Y. T).