Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
The progress of mRNA therapy in...
Challenges in mRNA therapy
Delivery strategies for mRNA...
mRNA NPs targeting immune cells
Clinical status of mRNA-based...
Conclusion, prospects, and...
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2024; 14(14):5528-5550. doi:10.7150/thno.93745 This issue Cite
Review
Advances in targeted delivery of mRNA into immune cells for enhanced cancer therapy
Linzhuo Huang1,2,3, Zhiquan Huang1,2,3, Yuxuan Zhang1,2,3, Chunhao Lin1,2,3, Zixuan Zhao4, Rong Li4, Phei Er Saw1,2,3 ![]() , Xiaoding Xu1,2,3
, Xiaoding Xu1,2,3 ![]()
1. Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Medical Research Center, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou 510120, P. R. China.
2. Guangzhou Key Laboratory of Medical Nanomaterials, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou 510120, P. R. China.
3. Nanhai Translational Innovation Center of Precision Immunology, Sun Yat-Sen Memorial Hospital, Foshan 528200, P. R. China.
4. The Second Affiliated Hospital, Hengyang Medical School, University of South China, Hengyang 421001, P. R. China.
Received 2023-12-30; Accepted 2024-6-6; Published 2024-9-3
Abstract

Messenger RNA (mRNA) therapy has been applied to the treatment of various human diseases including malignant tumors. Increasing evidences have shown that mRNA can enhance the efficacy of cancer immunotherapy by modulating the functions of immune cells and stimulating their activity. However, mRNA is a type of negatively charged biomacromolecules that are susceptible to serum nucleases and cannot readily cross the cell membrane. In the past few decades, various nanoparticles (NPs)-based delivery systems have been rationally designed and developed to facilitate the intracellular uptake and cytosolic delivery of mRNA. More importantly, by means of the specific recognition between the targeting ligands decorated on NP surface and receptors specifically expressed on immune cells, these mRNA delivery systems could be functionalized to target immune cells to further enhance the mRNA-based cancer immunotherapy. In this review, we briefly introduced the advancements of mRNA in cancer therapy, discussed the challenges faced by mRNA delivery, and systematically summarized the recent development in NPs-based mRNA delivery systems targeting various types of immune cells for cancer immunotherapy. The future development of NPs-mediated targeted mRNA delivery and their challenges in clinical translation are also discussed.
Keywords: Messenger RNA (mRNA), immune cells, nanoparticles (NPs), targeted delivery, cancer immunotherapy
Introduction
Messenger RNA (mRNA) is a single-stranded ribonucleic acid transcribed from DNA that has the capability to encode nearly all proteins. Unlike siRNA or CRISPR interference, which operate on the principle of reducing intracellular gene expression, mRNA is primarily used to compensate for genetic mutations or deficiencies, thereby elevating the levels of specific proteins [1]. Increasing research demonstrates significant advantages of mRNA therapy. In comparison to DNA-based therapies, mRNA can exert its effects without needing to enter the cell nucleus, and it carries a relatively high transfection efficiency and safety due to the absence of the risk of random insertional mutations [2]. Additionally, in contrast to transient protein or peptide drug therapies, mRNA allows for sustained translation, resulting in a prolonged therapeutic effect [3]. Therefore, over the past few decades, mRNA has been used in various biological applications, including protein replacement therapy, tissue engineering, gene editing, cancer vaccines, etc [4]. We briefly summarize the applications of mRNA beyond immunotherapy (Table 1), and the application in cancer immunotherapy will be described in detail in the next section. In the field of cancer immunotherapy, mRNA can complement immune-related deficient proteins, modulate the immune system, and improve the efficacy of cancer immunotherapy. What is even more crucial is that, in immunotherapy, each type of immune cell requires specific mRNA to carry out its unique and vital immune functions [5]. Therefore, it is essential to deliver mRNA to specific subsets of immune cells.
The application of mRNA
| Application | Agent | Disease/Condition | Reference |
|---|---|---|---|
| Protein replacement therapy | PTEN mRNA | subcutaneous PTEN-mutated melanoma and orthotopic PTEN -null prostate tumor models | [6] |
| KDM6A mRNA | bladder cancer | [7] | |
| TP53 mRNA | hepatocellular carcinoma and non-small-cell lung cancer | [8] | |
| Gene editing | Cas9 mRNA | Lung-, spleen- and liver-targeted selective organ targeting (SORT) lipid nanoparticles | [9] |
| mRNA/single-guide RNA | tissue-selective mRNA delivery and CRISPR-Cas9 gene editing in spleen, liver and lungs. | [10] |
To function effectively, mRNA needs to be delivered to cellular compartments. However, mRNA is sensitive to enzymatic degradation and faces challenges entering the cell cytoplasm [11]. To overcome these obstacles, mRNA molecules can be encapsulated in nanoparticle carriers. In the field of immunotherapy, to achieve optimal therapeutic effects and minimize side effects, it is crucial to deliver mRNA to specific subsets of immune cells, ensuring the uptake of genetic material and the expression of proteins [5]. For instance, delivering mRNA to dendritic cells (DCs) enhances antigen presentation capabilities, thereby improving anti-tumor immune responses. Delivering mRNA to T and natural killer (NK) cells enhances their cytotoxic effects; Delivering mRNA to tumor-associated macrophages (TAMs) reverses immune suppression, and delivering mRNA to B cells enhances adaptive immune functions, etc [12]. Therefore, research on immune cell-targeted delivery based on mRNA therapy is crucial, as it will contribute to advancing precision medicine. Surface modification of nanoparticles or the selection of nanoparticles with specific targeting functions can enhance the immune cells targeting, enabling the delivery of higher concentrations of mRNA to specific immune cell populations. Optimizing these delivery systems will drive progress in mRNA-based immunotherapy. In this review, we first introduced the progress of mRNA in cancer immunology, providing a brief overview of the challenges faced by mRNA delivery and existing delivery systems. Subsequently, we described the advancements in immune cell targeting methods based on these cancer immunotherapy targets and mRNA delivery systems. Finally, we summarized the potential strategies for mRNA targeted delivery and discussed the prospects and challenges of designing nanoparticles to enhance targeted immunotherapy.
The progress of mRNA therapy in cancer immunology
Cancer immunotherapy is a treatment strategy based on the ability of the immune system to recognize and eliminate cancer cells. The success of a series of clinical drugs targeting cancer immunotherapy demonstrates the potential of the human immune system to fight cancer. Tisagenlecleucel is an autologous CD19-targeted CAR T cell recently approved by the U.S. Food and Drug Administration (FDA), the European Union (EU) and Japan for pediatric or young adult patients with B-ALL [13]; Aldesleukin, a recombinant form of human IL-2, was first approved by the FDA for the treatment of metastatic renal cell carcinoma (RCC) in 1992, and subsequently for the treatment of melanoma in 1998 [14]; Imiquimod (formulated as a 5% topical cream) is widely used in dermatology for the treatment of condyloma acuminatum and may be used to relieve patients with superficial basal cell carcinoma (BCC) [15]. This approach was approved by the FDA in 2004. Immune checkpoint blockade (ICB) antibodies have revolutionized cancer treatment, significantly reducing the tumor burden in difficult-to-treat cancer patients [16]. However, the success of ICB in solid tumors is limited, with only 13% of patients responding to various cancer types [17]. Additionally, most responsive patients eventually experience relapse due to immunotherapy resistance [18]. Therefore, there is an urgent need to improve the response rate of immunotherapy for all cancer types. mRNA can be employed in cancer immunotherapy through various treatment modalities, including cancer vaccines, adoptive T-cell therapy, therapeutic antibodies, and immunomodulatory proteins, to mobilize tumor-specific anti-tumor immune responses (Table 2) [19]. In recent years, with the development of biotechnology and molecular medicine, mRNA has shown tremendous potential in cancer immunotherapy, as evidenced by recent preclinical and clinical results (Figure 1).
The application of mRNA in cancer immunotherapy
| Agent | Disease/Condition | Reference | |
|---|---|---|---|
| Cancer vaccines | luciferase and carcinoembryonic antigen (CEA) mRNA | colon carcinoma | [20] |
| gp100 mRNA | melanoma | [21] | |
| prostate-specific antigen (PSA) mRNA to DC | prostate cancer | [22] | |
| mRNA coding for the melanoma associated antigens | melanoma | [24] | |
| tumor-associated antigens (TAAs) mRNA | triple negative breast cancer | NCT02316457 | |
| personalized mRNA vaccines | melanoma | [25] | |
| Adoptive T-cell therapy | chimeric antigen receptor (CAR) mRNA to T cells | acute lymphoblastic leukemia | [28] |
| c-Met-CAR mRNA | metastatic breast cancer | [29] | |
| mRNA-encoded antibodies | BiTE encoding mRNA with 1-methylpseudouridine (RiboMAB) simultaneously targets CD3 and one of three tumor-associated antigens (TAAs) | ovarian cancer, gastric adenocarcinoma | [26] |
| mRNA encoding bispecifically binds and neutralizes CCL2 and CCL5 (BisCCL2/5i) | primary liver cancer, liver metastasis of colorectal and pancreatic cancers | [30] | |
| mRNA-encoded immunomodulatory proteins | mRNA encoding four tumor-regressing cytokines (IL-12 single chain, IFN-α, GM-CSF, and IL-15) | colorectal cancer, melanoma | [27] |
| mRNAs encoding OX40L, IL-36γ, and IL-23 | hepatoma, colon carcinoma | [31] |
The history of important discoveries in mRNA biology and developments in mRNA therapy and immunotherapy
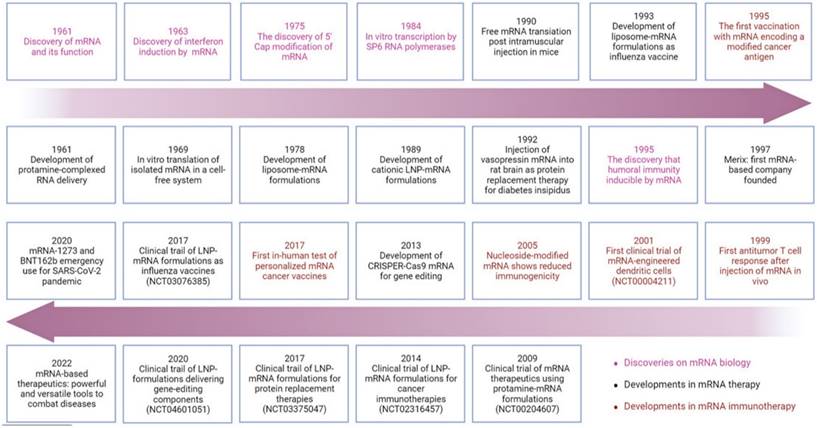
Cancer vaccines have both preventive and therapeutic potential, stimulating and enhancing existing immune responses against tumor antigens. The first mRNA cancer vaccine designed in 1995 demonstrated successful induction of humoral immune responses against encoded tumor-associated antigens (TAAs) in mice [20]. In 1999, Zhou and colleagues reported the vaccination using mRNA targeting the tumor-specific antigen gp100 to induce T-cell responses [21]. The first clinical trial of prostate-specific antigen (PSA) RNA-loaded DCs vaccine (NCT00004211) began in 2001 [22]. However, these initial attempts did not significantly propel the widespread application of mRNA therapy in clinical experiments. It wasn't until 2005 that Karikó and Weissman, first reported that mRNA synthesized with pseudouridine instead of uridine could greatly reduce immunogenicity, thereby avoiding recognition and clearance by the immune system [23]. This groundbreaking work led them to receive the Nobel Prize in 2023. Subsequently, an increasing number of clinical trials of mRNA vaccines are underway, such as NCT00204607 and NCT02316457[24]. It is noteworthy that scientists reported the first application of personalized mRNA vaccines in humans in 2017 for the treatment of melanoma [25]. This study indicated that leveraging individual mutations could pave the way for personalized immunotherapy for cancer patients.
Adoptive T-cell therapy, based on chimeric antigen receptor (CAR) in cancer treatment, has been effectively applied in the treatment of liquid tumors but faces challenges in solid tumors. By surface conjugation, specific antibodies target immune cells to deliver mRNA, thereby reprogramming T cells, enabling the possibility of CAR expression in vivo.
mRNA-encoded antibodies, such as bispecific T-cell engagers (BiTE), bridge the gap between tumor cells and T cells, inducing target-dependent T-cell activation. For instance, BiTE encoding mRNA with 1-methylpseudouridine (RiboMAB) simultaneously targets CD3 and one of three TAAs: claudin-6 (CLDN6), claudin 18.2 (CLDN18.2), or epithelial cell adhesion molecule (EpCAM), which has been shown to induce effective T-cell activation and targeted cancer cell lysis at low concentrations [26].
mRNA-encoded immunomodulatory proteins, including cytokines, toll-like receptors (TLR), chemokines, and costimulatory ligands, can reprogram the tumor immune microenvironment. Hotz et al. investigated the anti-tumor effects of intratumoral injection of mRNA encoding four tumor-regressing cytokines (IL-12 single chain, IFN-α, GM-CSF, and IL-15) [27].
Challenges in mRNA therapy
Despite the significant role of mRNA therapy in cancer immunotherapy, its application is constrained. The major challenges of naked mRNA-based therapy are its biological properties and physical properties, including instability, immunogenicity, large size and negative charge.
Biological properties
mRNA instability. As there is a large amount of RNA enzyme existence in the cell, the mRNA is easily degraded [32]. In addition, the mRNA's secondary structure dsRNA antigen can activate Oligoadenylate synthetases (OASs, the members of IFN-stimulating genes) to produce oligopolyamine to induce Ribonuclease L (RNase-L) to degrade mRNA [33]. A variety of chemical modifications can be performed on the mRNA skeleton structure to improve the stability of mRNA, such as modifying the 5' cap structure, optimizing the 5' UTR sequence and adding an appropriate length of poly (A) tail [34, 35].
mRNA Immunogenicity. When mRNA enters the endosome or cytoplasm, the antigen is detected by pattern recognition receptors (PRRs), including TLR3, TLR7, TLR8, and retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), and these receptors recognize mRNA and stimulate downstream pathways to produce type I interferon and pro-inflammatory cell factors [36]. Reducing the immunogenicity of mRNA is essential for enhancing its therapeutic and vaccine applications. Several strategies can be employed for this purpose. One approach is to carefully select nucleotide sequences with lower CpG content, as CpG-rich regions can trigger immune responses [37]. Additionally, incorporating chemically modified nucleotides, such as pseudo-uridine and 5-methylcytidine, can minimize the immunogenic potential of mRNA [38]. Avoiding the formation of stable secondary RNA structures is also crucial, as these structures may resemble viral RNA and trigger immune responses [39]. Optimizing the selection of promoter and terminator sequences can further reduce the immunogenicity of mRNA [40]. By employing these approaches and carefully designing mRNA sequences, the risk of immune recognition can be lowered, enhancing the safety and efficacy of mRNA-based therapies and vaccines [41].
Physical properties
Efficient in vitro and in vivo delivery of mRNA requires overcoming various barriers. The size of mRNA (300-5,000 kDa, 1-15 kb) is significantly larger than that of siRNA and miRNA analogs (13-15 kDa), antisense oligonucleotides (4-10 kDa) [42]. Moreover, mRNA is a negatively charged single-stranded polynucleotide, and it is difficult for naked mRNA to pass through the negatively charged cell membrane.
Delivery strategies for mRNA therapy
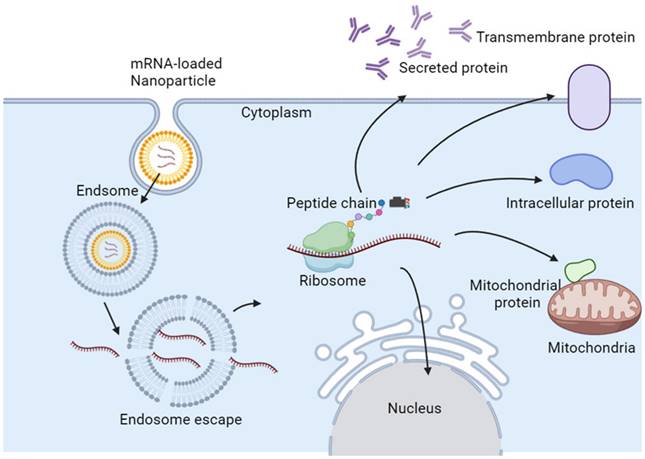
In the case of direct injection of mRNA, only 0.01% of the mRNA can enter the target cell, and most of the mRNA is trapped in the endosome of the target cell and subsequently degraded [43]. Eventually, only a few mRNAs escape from the endosome and reach the ribosome for protein translation. Therapeutic mRNAs require more efficient and safer delivery methods, which are critical for enabling promising transformational therapies [44]. Therefore, suitable mRNA nanoparticles are necessary for efficient mRNA delivery into most types of cells. Typically, mRNA nanoparticles are ingested by endocytosis, and then mRNA is released from endosomes, where lysosomes will initiate translation and produce any type of protein, including secreted, transmembrane, intracellular, and intramitochondrial proteins (Figure 2) [45].
Proposed mechanism of endosomal escape and action of mRNA NPs.
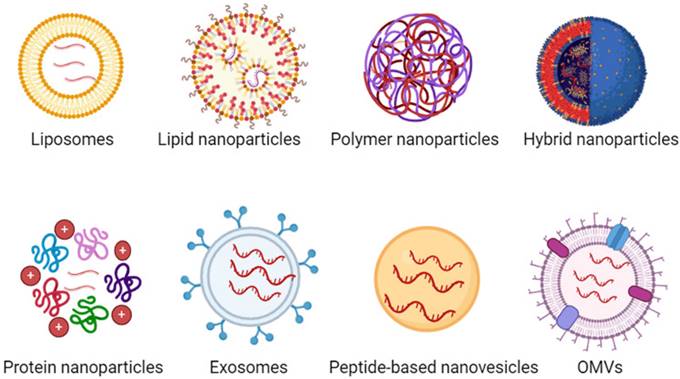
In recent years, various nanoparticles (NPs) have been developed for in vivo mRNA delivery, including both viral and non-viral carriers. While viral carriers can achieve high levels of transfection in the host, they come with potential immunogenicity and toxicity issues. Therefore, there is an urgent need for non-viral carriers with low immunogenicity and high safety for mRNA delivery. Currently, a variety of non-viral carriers are used for mRNA delivery, such as liposomes, lipid nanoparticles (LNPs), polymer nanoparticles, lipid-polymer hybrid nanoparticles, protein nanoparticles, exosomes, peptide-based nanovesicles, outer membrane vesicles (OMVs) (Figure 3). In addition, we have included table 3 representing the pros and cons of each mRNA NPs.
Various types of NPs used for mRNA delivery, including liposomes, LNPs, polymer NPs, lipid-polymer hybrid NPs, protein NPs, exosomes, peptide-based nanovesicles, outer membrane vesicles (OMVs).
The pros and cons of each mRNA NPs
| Nanoparticles carrier | Pros | Cons |
|---|---|---|
| Liposomes | Good biocompatibility; Multifunctionality; Can encapsulate hydrophilic and hydrophobic drugs at the same time. | Complex preparation process; Less amenable to scalability |
| Lipid nanoparticles | Encapsulation of mRNA using lipid bilayers prevents enzymatic degradation in the somatic circulation; Simple chemical synthesis of lipid-related components; Robust encapsulation capabilities. | The reticuloendothelial system (RES) or multiple organs can remove LNP from somatic circulation limiting its effectiveness. |
| Polymer nanoparticles | Forms stable complexes with RNA through electrostatic interactions, thus resisting degradation and promoting cellular uptake; Highly modifiable (easily functionalized, optimized drug release kinetics); Robust nucleic acid loading capacity. | Have the cytotoxicity; High molecular weight polymers are prone to aggregation in vivo. |
| Hybrid nanoparticles | Diverse structures; Better stability and biocompatibility. | Complexity of design and synthesis; Poor biodegradability; High production cost. |
| Protein nanoparticles | Good biocompatibility, adjustability and biodegradability | Low encapsulation efficiency; Endotoxin-induced toxicity; Abrupt drug release. |
| Exosomes | Good biocompatibility | Complexity of extraction |
| Peptide-based nanovesicles | High drug loading capacity; Good biocompatibility; Strong customizability | Poor stability; High production costs; Prone to immune reactions |
| OMVs | Strong immunogenicity; Multifunctional | High production costs; Poor stability; Unclear mechanisms |
To gain comprehensive views of various drug delivery platforms for mRNA therapy, readers should read several of the more comprehensive reviews previously published on the topic of mRNA nanodrugs [46]. Researchers summarize the types of delivery vectors for mRNA vaccines [47], mRNA delivery vectors for cancer therapy [48-50], types of lipid-associated mRNA delivery vectors [51]. LNPs are currently the most intensively studied and clinically advanced mRNA delivery vehicles [52]. Among them, cationic and ionizable LNPs are widely used. LNPs typically consist of cationic or ionizable lipids, cholesterol, auxiliary lipids, and polyethylene glycol (PEG)-modified lipids [53]. The negatively charged phosphate backbone of mRNA molecules can be efficiently attracted to the positively charged headgroups of cationic lipids through electrostatic interactions, thereby enhancing the encapsulation efficiency of mRNA [54]. In clinical research, LNPs have become the state-of-the-art approach for synthetic RNA therapy targeting a range of diseases, such as Patisiran, BNT162b2, and mRNA-1273 [55, 56].
Most importantly, owing to the significance of immunotherapy, numerous NPs have been designed and developed for mRNA-based cancer immunotherapy. This mRNA-based cancer immunotherapy includes indirect therapy via delivering mRNA into tumor cells and direct therapy via delivering mRNA into immune cells [57]. In this review, we focus on the researches of targeted delivery of mRNA into immune cells for cancer therapy.
mRNA NPs targeting immune cells
Targeted administration of agents to specific cell subpopulations allows therapeutic agents to concentrate their effects on the target cells, thereby enhancing the therapeutic effect. In addition, immune cells themselves exert their anti-tumor functions through specific mechanisms of action. Therefore, targeted delivery of mRNA to specific immune cells has important application prospects. Specifically, DCs has the key function of initiating T-cell immunity, and DCs can be effectively activated and enhance immunotherapy by tumor-specific antigen (TSA), tumor-associated antigen (TAA), and immune adjuvants (TLR agonists, STING agonists, and C-type lectin receptor (CLR) agonists) [58-61]; Co-stimulatory factors OX40 and 4-1BB mRNA can stimulate the proliferation and expansion of CD8+ T cells and enhances T cell-mediated anti-tumor immune responses [62]; NK cells also have unique stimulatory receptors (NKG2D and NKp46) to promote NK cells activation and killing of tumor cells [63]. In this section, we will elaborate on the design strategies for delivering mRNA NPs to specific immune cells for the purpose of precision immune cell targeting. Therefore, we comprehensively summarize the specific information on delivering mRNA to different cell types, including animal models, administration routes, injection routes, dosing amount, dosing times, final results (Table 4).
mRNA NPs targeting different immune cells.
| Immune cells | Animal models | Administration routes | Injection routes | Dosing amount | Dosing times | Results | Reference |
|---|---|---|---|---|---|---|---|
| DCs | TC-1 subcutaneous tumor model | Day 5, 10, 15 | Intravenous | 10 μg mRNA | Three | Systemic LPR treatment improved the median survival time of TC-1-inoculated mice and was even superior in controlling tumor growth. | [64] |
| TC-1, B16F0, EG7-OVA subcutaneous tumor model | Day 7, 9 | Intradermal | 56 μl of LPR | Two | MART1 and OVA triMN-LPR triggered a significant delay in the B16F0 and EG7 tumor growth. | [65] | |
| B16-OVA, B16 subcutaneous tumor model | Day 4, 8, 12 | Subcutaneous | 10 μg mRNA | Three | C1 mRNA vaccine with a self-antigen and model antigen inhibited the growth of tumors. | [66] | |
| / | Twice of two week | Intranasal | 10 μg mRNA | Two | CP 2k/mRNA induced significantly higher titers of IgG1 and IgG2a than naked mRNA. | [67] | |
| LL2 orthotopic tumor model | Day 4, 11, 18 | Subcutaneous | 10 μg mRNA | Three | DOTAP/DP7-C/neoantigen mRNA complexes exert a better antitumor effect | [68] | |
| T cells | E0771 tumor model | Intravenous | mCherry mRNA (0.6 mg) . | One | aCD3-LNPs transfected 2-7% of circulating T cells and 2-4% of splenic T cells respectively. | [69] | |
| Ai6 mice carrying a Cre reporter allele | Every 24 h | Intravenous | 10 μg Cre mRNA | Three or five | The sequential administrations of the targeted mRNA-LNPs resulted in increasing Cre-induced genetic recombination with increased number of injections in both the spleen and lymph nodes. | [70] | |
| A mouse model of heart failure | / | Intravenous | 10 μg LNP | One | Marked functional improvements were observed in injured mice. | [71] | |
| LNCaP C42 orthotopic tumor model | Day 0, 7, 14, 21, 28, 35 | Intravenous | 50 μg mRNA | Six | CAR-encoding or TCR-encoding mRNA particles can genetically reprogram circulating T cells to induce antitumor responses. | [72] | |
| A20, CT26, or B16F10 subcutaneous tumor model | Day 5, 7, 9, 11, 13, 15 | Intratumoral | 10 μg mRNA | Six | PL1 nanoparticles delivering the costimulatory OX40 mRNA could enhance the immunotherapeutic effects of anti-OX40 Ab therapy in different mouse models. | [73] | |
| NK cells | Orthotopic HCC tumor model | Day 22, 29 | Intravenous | 2×10 6 cells | Two | CAR-DLNP mNK cell therapy decreased tumor proliferation and increased tumor cell apoptosis. | [74] |
| Macrophages | Gliomas tumor model | 3 doses/week for 3 weeks | Retro-orbital | 30 µg mRNA | Nine | Nanoparticles can deliver genes encoding master regulators of macrophage polarization to re-program immunosuppressive macrophages into tumor-clearing phenotypes. | [75] |
| B16F10 tumor model | / | Intravenous | 0.6 mg/kg mRNA | One | CD11bhi macrophage-tropism of Lipid 16 would increase the mRNA delivery to a solid tumor. | [76] | |
| B cells | C57BL/6 mice | / | Intravenous | 0.75 ~ 2.25 mg/kg mRNA | One | Approximately 60 pg of luciferase protein was produced per million B cells at the highest OF-Deg-Lin LNP dose. | [77] |
| B16F10 tumor model | Every other day | Intratumoral | 2 μg IL-12 mRNA or 6 μg IL-27 mRNA | Six | Intratumoral administration of IL-12 and IL-27 mRNAs by DAL-LNP promoted sustained inhibition of B16F10 melanoma growth without causing significant toxicity. | [78] | |
| Neutrophils | MC38 tumor model | / | Intratumoral | 5 μg of OX40L mRNA | One | The three most abundant myeloid cell types within tumors are macrophages, monocytes, and granulocytes; all expressed OX40L above control mRNA-dosed tumors. | [31] |
| B16F10 tumor model | Days 3, 6,10 | Subcutaneous | 10 μg mRNA | Three | Treatment of B16F10 melanoma tumors with lipid nanoparticles containing mRNA coding for the tumor-associated antigens gp100 and TRP2 resulted in tumor shrinkage and extended the overall survival of the treated mice. | [79] |
mRNA NPs targeting DCs
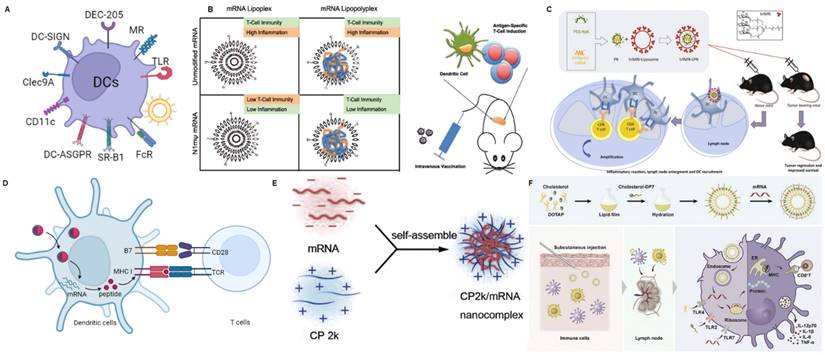
DCs are antigen-presenting cells (APCs) present in all tissues. DCs are primarily responsible for the uptake, internalization, and processing of antigens, and subsequently, they deliver processed antigen peptides to naïve T cells, serving as a bridge between innate and adaptive immunity [80]. The presence of various specific receptors on the surface of DCs provides natural targets for the delivery vehicles aimed at targeting DCs [81]. Here, we will discuss some different methods for DC-targeted delivery systems and related mRNA delivery systems (Figure 4A).
mRNA nanoparticles targeting DCs. (A) Schematic illustration of various receptors expressed on DCs that could be employed to design DCs-targeted NP-mediated delivery systems; (B) Schematic diagram of DCs targeting mRNA lipopolyplexes to exert anti-tumor immunity. Adapted with permission from [64], copyright 2018 ACS Publications; (C) Schematic diagram of DCs targeting antigen-encoding mRNA-loaded triMN-LPR as an anti-tumor vaccine formulation. Adapted with permission from [65], copyright 2018 Elsevier; (D) Schematic illustration of antigen presentation using antigen-encoding mRNA formulated with lipid nanoparticles. Reproduced with permission [66]; (E) Schematic diagram of loading mRNA vaccines using self-assembled nanoparticles formulated with CP 2k. Adapted with permission from [67], copyright 2016 Elsevier; (F) Schematic representation of DP7-C modified liposomes enhancing immune response to neoantigen encoding mRNA complexes. Adapted with permission from [68], copyright 2020 Elsevier.
C-type lectin receptor (CLR) family
Many receptors used in targeted research belong to the CLR family. CLRs are a family of lectins that recognize carbohydrates in a calcium-dependent manner, and their carbohydrate recognition domains (CRDs) share primary structural homology [82]. The N-terminus of the I-type CLR group is located extracellularly and mainly includes DEC205 (LY75/ CD205) and mannose receptor (MR/CD206/Clec13D). Most CLRs are of the II-type, with their amino terminus located intracellularly, which primarily includes Clec9A (CD370/DNGR-1) and DC-SIGN (Clec4L/CD209) [83].
MR is expressed on macrophages, endothelial cells, smooth muscle cells of the trachea, mature and immature moDC, and human peripheral blood CD1c+ DC [84]. Kramer and colleagues prepared amphiphilic block copolymer micelles containing antigenic peptides and adjuvants through the hydrolysis of polymaleic anhydride (HPMA) and lauryl methacrylate to induce desired antigen-specific T-cell responses [85]. Among them, mannose and trimannose are introduced into the hydrophilic corona as units targeting MR and DC-SIGN. Similarly, Zhu and colleagues developed mannose-functionalized lipid-hybrid polymersomes (MAN-IMO-PS) for co-delivery of ovalbumin antigen within the core, the hydrophobic membrane-embedded TLR7/8 agonist imiquimod, and the TLR4 agonist monophosphoryl lipid A on the lipid layer to induce synergistic anti-tumor immune responses [86].
Based on mannose, there have been a series of studies on targeted delivery of mRNA. Hybrid lipid-shell-polymer core mRNA nanoparticles (LPRs) may be valuable alternatives to lipid-based mRNA nanocomplexes (LRs), as they combine improved stability and reduced cytotoxicity [87]. Therefore, researchers have developed the tri-mannosylated LPR nanoplatform to efficiently target the delivery of Fluc mRNA to DC cells (Figure 4B) [64]. Subsequently, the authors validated the ability of this nano system to stimulate CD8 T-cell immune responses by delivering ovalbumin (OVA) and human papillomavirus 16 (HPV16) mRNA, achieving high anti-tumor efficacy. Moreover, the delivery of mRNA modified with N1-methylpseudouridine could reduce adverse inflammatory reactions. This study establishes that the LPR platform possesses excellent immunogenicity and improved inflammatory response modulation.
Human DC-specific intercellular adhesion molecule 3 grabbing non-integrin (DC-SIGN) is primarily expressed on the surface of immature dendritic cells (iDCs), with lower levels of expression on mature dendritic cells (mDCs) and macrophages [88]. DC-SIGN has a CRD to bind ligands with high mannose and fucose structures, including Lewis(Le)-type antigens and host glycoproteins [89]. DC-SIGN recognizes carbohydrate structures to facilitate antigen uptake, processing, and presentation through MHC II molecules, thereby enhancing T-cell responses [90]. For example, research achieved specific DC-SIGN targeting by using a multivalent liposomal formulation containing the glycan LeX to deliver adjuvants and tumor antigens to induce immune responses [91].
Based on DC-SIGN, Moignic group has demonstrated the capability of targeting with a lipid-polymer-RNA lipopolyplex functionalized with a tri-antenna of α-d-mannopyranoside (triMN-LPR), possessing binding affinity to DC-SIGN and CD207 (Langerin) on DCs (Figure 4C) [65]. After intradermal injection of mRNA NPs encoding the papillomavirus E7 antigen to C57BL/6 mice, draining lymph nodes exhibited activated DCs, significant gene expression of CCR7 and CXCR4 at the injection site, and E7-specific T cell responses. In the E7-expressing TC1 tumor model, triMN-LPR NP resulted in significant long-term survival compared to control PBS injections.
One of the most well-known methods for targeting DCs is to use specific antibodies against DEC-205, which is expressed in both mouse and human DCs. DEC-205 has ten carbohydrate recognition domains with a molecular weight of 205 kDa [92]. It is a phagocytic receptor that mediates antigen uptake and is highly expressed in DCs and thymic epithelial cells [92]. To improve the selective distribution of nanoparticles, antibodies or other targeting moieties can be incorporated into the nanoparticles. For example, Katakowski et al. conjugated the reduced anti-DEC205 single-chain variable antibody fragments (scFv) to the maleimide groups of DSPE-PEG-MAL in LNPs by simple mixing [93]. They demonstrated that the anti-DEC205 scFv-modified LNPs preferentially target DEC205+ DCs.
Clec9A is highly expressed in type 1 conventional dendritic cells (cDC1) in both humans and mice [94]. Clec9A-targeted antigen delivery also promotes MHC-II antigen presentation to CD4+ T cells [94]. Zeng et al. prepared functionalized nanoemulsions that encapsulated tumor antigens to target Clec9A (Clec9A-TNE), which can stimulate therapeutically effective tumor-specific immunity [95].
CD11c
In addition, some targeting approaches for DCs have been used for delivering drug/biological/antigen. Although they are not yet used for mRNA delivery, this could serve as inspiration for mRNA delivery. CD11c (CD18) is a type of leukocyte integrin receptor, which is primarily expressed on the surface of DCs [96]. Antigens can be internalized through these receptors, facilitating antigen capture and processing by DCs [96]. One study suggests that Fab fragments targeting CD11c conjugated with model antigen OVA can induce a significant T-cell response compared to other targets binding DCs, including CD205, TLR2 or FccRII/III [97]. Another study indicates that encapsulating OVA antigen in PLGA-NPs modified with targeted antibodies, such as αCD40, αCD11c, and αDEC-205, can effectively enhance the internalization of NPs by DCs and IL-12 release [98]. CD11c and DEC-205 receptors are considered to play a crucial role in the process of antigen capture and presentation and are almost exclusively expressed in DCs [99]. Researchers anchored ScFv targeting DC markers, such as CD11c and DEC-205, to tumor-derived plasma membrane vesicles (PMVs) or lipid vesicles containing antigens, which can efficiently target the receptors on the surface of DCs [100].
Scavenger receptor (SRs)
SRs can recognize modified low-density lipoproteins (LDL), either through oxidized LDL or acetylated LDL. DC-asialoglycoprotein receptor (DC-ASGPR) is a lectin-like SR. Li and colleagues used DC-ASGPR to deliver self-antigens (PSA) and exogenous antigens (hemagglutinin 1, HA1) to DCs, leading to the generation of antigen-specific CD4+ T cell responses that produce IL-10 [101]. Scavenger Receptor Class B Type 1 (SR-B1) is one of the SRs that facilitates the uptake of cholesterol esters from circulating lipoproteins. Yuan and colleagues leveraged the high expression of scavenger receptor SR-B1 on mDCs and used nanoparticles (α-Ap-FNP) with SR-B1-targeting capabilities to directly transport tumor antigen peptides to DCs in lymph nodes, which resulted in significant inhibition of tumor growth [102].
Fc receptors (FcRs)
FcRs bind to the constant domains of antibodies, acting as connectors between humoral immune responses and cellular immune responses. FcRs are present in various immune cells, such as monocytes, macrophages, DCs, and neutrophils [103]. Kawamura and colleagues used FcγR-targeted liposomes to deliver OVA, which was 2-5 times better than non-targeted liposomes [104]. Similarly, another study showed that gold nanoparticles and liposomes targeted at DCs via FcγR (the receptor for the IgG Fc segment) were effective antigen delivery carriers, which can induce a stronger immune response compared to non-targeted nanoparticles or naked antigens [105].
Toll-like receptors (TLRs)
The immune system needs to distinguish between self-structures and foreign substances to detect pathogens and function. PRRs can distinguish self from non-self and recognize specific microbial-associated molecular patterns (MAMPs), which activate immune signals, acting as mediators between the innate and adaptive immune systems [106]. TLRs are a type of PRR and are evolutionarily conserved proteins and are expressed on various APCs, such as DCs, B cells, and macrophages [107]. Therefore, antigen-delivery vehicles modified with TLR ligands can generate excellent immune responses by targeting APCs and TLR-mediated APC stimulation. Li and colleagues designed cancer nanovaccines (BTs) prepared by fusing bacterial OMVs and tumor cell membranes (TCM) [108]. Pathogenic adjuvants from bacteria can promote DC targeting, DC maturation, and antigen presentation.
Other DCs-targeting strategies based the physiochemical characteristics of NPs
With the advancement of nanotechnology, there have been extensive researches on optimizing the formulations of NPs to achieve the goal of DCs-targeted mRNA delivery [9]. For example, Zhang et al. discovered LNPs with a 12-carbon tail, referred to as C1, which effectively delivers antigen-coding mRNA to DCs (Figure 4D) [66]. Additionally, C1 can stimulate the expression of inflammatory cytokines in DCs by activating the TLR4 signaling pathway. This nano-vaccine exhibited promising anti-tumor efficacy in vivo therapies. Therefore, this minimalistic mRNA nano-vaccine offers a multifunctional platform for the development of personalized vaccines. Li et al. have developed a cationic cyclodextrin-polyethylenimine 2k conjugate (CP 2k) polymer-based intranasal mRNA vaccine delivery system for the treatment of HIV-1(Figure 4E) [67]. The first cationic polymer capable of serving as a safe and effective intranasal mRNA vaccine carrier to overcome the nasal epithelial barrier has been provided. The authors observed significantly higher luciferase expression when transfecting CP 2k/mRNA (encoding luciferase mRNA) complexes into DC2.4 murine DCs, with an N/P ratio of 16, compared to using polyethylenimine (PEI) nanoparticles with a molecular weight of 25 kDa. Zhang et al. used a cholesterol-modified cationic peptide DP7 (VQWRIRVAVIRK)(DP7-C), with transmembrane structure and immunostimulatory properties to modify DOTAP liposomes, creating a universal mRNA delivery system (Figure 4F) [68]. DOTAP liposomes modified with DP7-C (DOTAP/DP7-C) acted as carriers for mRNA and efficiently delivered mRNA to different types of DCs in vitro. As an immunostimulant, DOTAP/DP7-C liposomes were more effective than DOTAP liposomes in stimulating the maturation of DCs, the production of CD103+ DCs (which aids in antigen presentation), and the secretion of proinflammatory cytokines both in vitro and in vivo.
mRNA NPs targeting T cells
T cells are an integral component of the adaptive immune system, playing critical roles in defending against pathogen invasion, mediating anti-tumor immunity, establishing immunological memory, assisting B cells with antibody production, and regulating the activities of other immune cells. The activation and differentiation of T cells are precisely controlled by T cell receptor (TCR) signaling, costimulatory signals, and cytokines [109]. By modulating the stimulatory signals, T cells can be “triggered” to produce perforin and granzymes to lyse tumor cells. The secretion of cytokines may induce tumor cell apoptosis through Fas-FasL interaction. Recent studies also reveal a new anti-tumor mechanism of T cells by promoting tumor ferroptosis [110]. Activating endogenous T cells of cancer patients to suppress malignancy has been a central topic in tumor immunology research, but intracellular targeting of T cells remains a major challenge. Next, we will briefly discuss the targeting strategies for T-cell delivery (Figure 5A).
mRNA nanoparticles targeting T cells. (A) Schematic illustration of various receptors expressed on T cells that could be employed to design T cells-targeted NP-mediated delivery systems; (B) Schematic diagram of the preparation of mCherry mRNA for targeted delivery to T cells using αCD3 F(ab')2. Adapted with permission from [69], copyright 2022 Elsevier; (C) Schematic diagram of targeted delivery of mRNA to CD4+ T cells. Adapted with permission from [70]; (D) Schematic representation of delivery of FAP CAR mRNA using CD5-targeted LNPs. Adapted with permission from [71], copyright 2022 American Association for the Advancement of Science; (E) Schematic representation of delivery disease-specific CAR or TCR transiently expressed nucleic acid (IVT mRNA) using CD8-targeted nanoparticles. Adapted with permission from [72], copyright 2020 Springer Nature; (F) Schematic illustration of enhanced tumor immunotherapy via phospholipid nanoparticles (PL1) delivery of OX40 mRNA combined with anti-OX40 antibodies. Adapted with permission from [73], copyright 2021 Springer Nature.
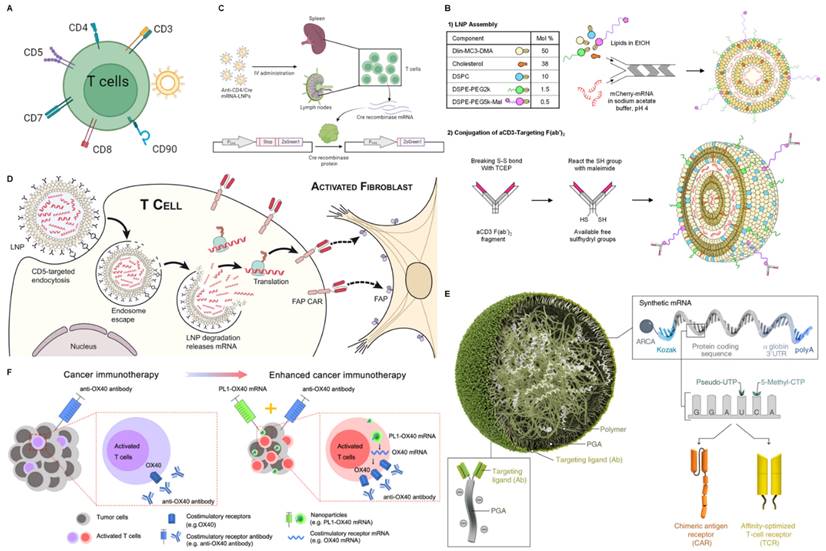
CD3
The use of CD3-specific antibodies to target T cells provides an exciting approach to achieving T cell-specific delivery. Although anti-CD3 mAbs have been shown to deplete T cells while promoting in vivo anergy and extensive cytokine release, F(ab')2 fragments of anti-CD3ε can partially avoid this effect due to the lack of the Fc antibody portion [111]. In addition, antibody engineering can optimize potency and affinity [112].
Based on the advantages of antibody engineering, CD3 bispecific antibodies have been extensively explored clinically and are a major research focus. Cheng et al. modified the surface of endogenous exosomes with two different scFVs, generating SMART-Exos that simultaneously target T cell surface CD3 and cancer cell-associated epidermal growth factor receptor (EGFR), which redirected and activated cytotoxic T cells to kill the cancer cells [113]. Duwa and co-workers designed bis-R848-PLGA-NP containing dual-specific nanoparticles conjugated with two antibodies to sequentially target CD3 and the tumor-specific protein PD-L1 to enhance T cell cytotoxicity [114]. Apart from that, there are also studies involving artificial APCs. Researchers utilize DC membranes with azide-functionalized loaded imiquimod and modify anti-CD3ε antibodies through click chemistry for stimulating T cells, which can potentiate cancer immunotherapy [115]. Based on the CD3 target, there have also been many studies on targeted delivery of mRNA. For example, researchers evaluated the use of anti-CD3-targeted LNP for direct in situ transfection of T cells, rather than ex vivo administration of CD3 antibody or engineered therapeutic cells (Figure 5B) [69]. Here, LNP packaged mCherry mRNA or Fluc mRNA in lipid-based nanoparticles and targeted T cells using αCD3 F(ab')2. In vitro, aCD3-LNP transfection infected and activated ~97% of Jurkat cells. In vivo, aCD3-LNP transfected 2-7% of circulating T cells and 2-4% of spleen T cells, causing transient activation, exhaustion, migration, cytokine release, and phenotypic changes. Huang et al. designed a novel liver-targeted ionizable lipid nanoparticle delivery system for mRNA encoding the B7H3×CD3 BiTE to exert potent anti-tumor activity against Acute Myelogenous Leukemia (AML) and melanoma [116].
CD4
CD4 is a transmembrane glycoprotein expressed on the surface of T helper cells. It serves as a co-receptor that assists the TCR in activating the T cell when bound to the β2 domain of major histocompatibility complex II (MHC II) on antigen-presenting cells. The presence of CD4 also contributes to T-cell signaling and trafficking [109]. Therefore, CD4 is an appealing target for modulating T-cell-mediated immunity. Ramishetti et al. achieved specific delivery of siRNA to mouse CD4 T cells by modifying lipid nanoparticles with a monoclonal antibody against the CD4 receptor on the T cell surface, silencing CD45 by targeting T cells to deliver siCD45 [117]. McHugh et al. used anti-CD4 antibody-conjugated biodegradable nanoparticles loaded with TGF-β and IL-2 to induce the expansion of CD4+ Treg cells in vitro to directly improve clinical therapies for inflammatory and cell-mediated diseases [118].
Based on the CD4 target, Tombácz et al. achieved efficient, specific in vitro and in vivo mRNA delivery by using CD4 antibody-bound LNP to specifically target CD4+ cells and interfere with mRNA (Figure 5C) [70]. After systemic administration in mice, radiolabeled mRNA-LNP accumulated in the spleen, providing a promising tool for in vivo T cell manipulation.
CD5
CD5 is a 67 kDa type I transmembrane glycoprotein belonging to the conserved scavenger receptor cysteine-rich (SRCR) family of receptors. It is also a pan-T cell marker, regularly expressed on normal T cells and about 85% of T cell malignancies, as well as some B cell malignancies [119]. Researchers connected effector T cells expressing CD5 with B lymphoma target cells expressing CD19 using the bispecific antibody HD37xT5.16 (CD19xCD5), enabling activated effector T cells to interact with target cells and induce cell apoptosis [120].
Similarly, studies are achieving targeted delivery of mRNA to T cells through the CD5 target. Rurik and co-workers designed a CAR mRNA encoding for fibroblast activation protein (FAP, a marker of activated fibroblasts), and packaged it into CD5-targeted LNPs, termed “targeted antibody/LNP-mRNA cargo” or CD5/LNP-FAPCAR) (Figure 5D) [71]. CD5 is naturally expressed by T cells and a small subset of B cells and is not required for T cell effector function [121]. Treatment with the targeted modified mRNA LNPs reduced fibrosis and restored cardiac function after injury [71].
CD8
CD8 is a transmembrane glycoprotein that serves as a co-receptor for the TCR. It is expressed on the surface of cytotoxic T cells. The CD8 co-receptor binds to MHC class I molecules on antigen-presenting cells to recognize and kill cells displaying antigenic peptides bound to MHC class I. After TCR activation, CD8+ cytotoxic T cells release perforin, granzymes, and cytokines like IFN-γ to induce apoptosis of the target cell. Therefore, CD8 plays a crucial role in the cytotoxic function and anti-tumor immunity mediated by CD8+ T cells. Schmid and co-workers targeted compound delivery to specific leukocyte subpopulations to enhance the therapeutic index using anti-CD8a F(ab')2 fragments against CD8 T cells [122]. These modified nanoparticles could bind to CD8+ T cells and deliver drugs to play a better anti-tumor role. In addition, Parayath et al. reported injectable nanoparticles for delivery of in vitro transcribed (IVT) CAR or TCR mRNA to reprogram circulating T cells to recognize disease-associated antigens (Figure 5E). Surface-anchored targeting ligands selectively bind nanoparticles to T cells and initiate rapid receptor-induced endocytosis for their internalization, which is achieved by conjugating anti-CD8 antibodies with polyglutamic acid (PGA) [72]. The resulting mRNA nanocarriers could be lyophilized for long-term storage.
Other targeting strategies
Besides the markers expressed on the surface of T cells described above, some other molecules specifically expressed on T cells including CD7[123], CD90 (Thy1.1) [124]. For example, Lee et al. used chitosan nanoparticles modified with scFvCD7 to deliver siCD4 to T cells to enhance the ability to bind to T cells and higher silencing efficiency of CD4 compared to unmodified chitosan nanoparticles [125]. Zheng et al. used PEGylated liposomes targeted respectively to the unique cell surface antigen CD90 on transferred T cells, which provided highly specific targeting with liposome binding to over 90% of cells after a single injection [126]. Similarly, researchers prepared PEGylated immunoliposomes delivering the TGF-β inhibitor SB525334 (TGF-βI) and compared targeting using the internalizing receptor CD90 versus the non-internalizing receptor CD45 to achieve T cell-targeted delivery [127]. These potential targets used for targeted delivery of other therapeutic agents can serve as references for future targeted delivery of mRNA to T cells.
Other T cell-targeting strategies based the physiochemical characteristics of NPs
Besides the strategies based on the ligand-receptor interaction described above, there are also some T cell-targeting delivery strategies based on inherent physiochemical characteristics of NPs. Researchers have shown that maleimide-functionalized NPs covalently coupled to free thiol groups on T cell membrane proteins can effectively deliver compounds to T cells [128]. For example, Dong et al. designed a biomimetic phospholipid nanoparticle, PL1, which could deliver the costimulatory receptor OX40 to T cells and synergize with an agonistic anti-OX40 antibody to mediate effective antitumor treatment (Figure 5F) [73]. Based on the chemical structure of natural cell membrane components, phospholipids, and glycolipids, the authors designed and synthesized a library of biomimetic materials comprising biomimetic heads (phosphate or sugar heads), ionizable amino cores, and multiple hydrophobic tails. The screened PL1 nanoparticles not only efficiently delivered co-stimulatory receptor mRNA to T cells in vitro but also effectively delivered it to T cells within tumors in vivo, providing valuable delivery materials for modulating T cell function.
mRNA NPs targeting NK cells
NK cells, as an important component of the innate immune system, play a vital role in eliminating senescent cells and pathogenic microbes. With tumor cells downregulating MHC expression to evade adaptive immunity, they become more susceptible to NK cell cytotoxicity [129]. Mechanistically, NK cells play a critical role in the first line of defense against cancer, mediating antitumor effects via two pathways: direct cytotoxicity by releasing perforin and granzymes or antibody-dependent cell-mediated cytotoxicity (ADCC) through death receptor ligands [130]. NK cells also participate in tumor cell clearance by secreting cytokines or mobilizing DCs, macrophages, T cells, and other immune cells, which are emerging as promising candidates to be attractive targets for cancer immunotherapy [131]. Here, we will discuss some different methods for NK-targeted delivery systems and related mRNA delivery systems (Figure 6A).
mRNA nanoparticles targeting NK cells. (A) Schematic illustration of various receptors expressed on NK cells that could be employed to design NK cells-targeted NP-mediated delivery systems; (B) Structural schematic of CL1H6-LNP for mRNA delivery. Adapted with permission from [137], copyright 2023 Elsevier; (C) Mechanism schematic and advantages of CART for targeted delivery of mRNA to NK cells. Adapted with permission from [138], copyright 2020 Elsevier; (D) Optimized LNP-based mRNA targeting NK cells showed higher transfection efficiency and higher overall eGFP expression than electroporation or polymeric nanoparticles. Adapted with permission from [139], copyright 2023 Elsevier; (E) Schematic representation of the process of generating anti-GPC3-CAR NK cells in mouse primary NK cells (mNK cells) through DLNPs-mediated transfection of mRNAs [74], copyright 2024 John Wiley and Sons.
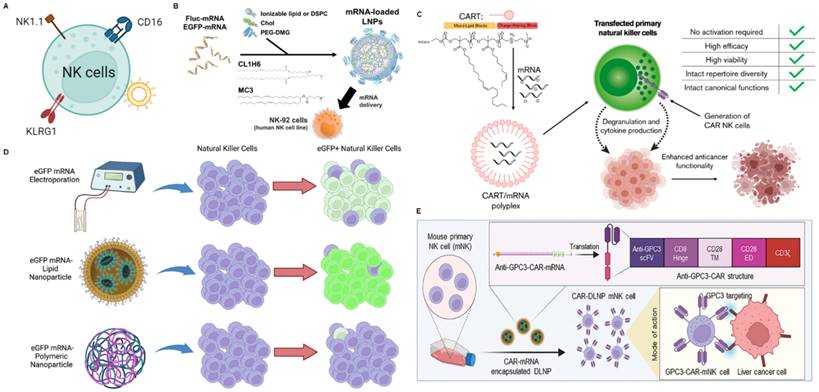
Cluster of differentiation 16 (CD16/FcγRIIIA), NK1.1, killer cell lectin-like receptor G1 (KLRG1) are specifically expressed in NK cells and have been used as targets for targeted delivery, providing reference significance for targeted delivery of mRNA. Based on CD16, researchers developed a bispecific Au nanoparticle that was dual-conjugated with IgG anti-HIVgp120 and IgG anti-human CD16 antibodies to enhance intercellular contact between HIV-expressing cells and NK cells [132]. To further enhance NK cell-mediated antitumor activity, trifunctional NK cell engagers (NKCEs) have recently been designed and produced. A study reported a trispecific NKCE platform, including α-CD16, α-4-1BB, α-EGFR and epirubicin (EPI), which facilitated the recruitment and activation of NK cells, which can ultimately promote NK cell recruitment and activation to eradicate these cancer cells [133]. Based on NK1.1, Chandrasekaran et al. designed functionalized liposomes for NK cells, which were decorated with anti-mouse NK1.1 antibody and tumor necrosis factor-α related apoptosis-inducing ligand (TRAIL) that initiated cell apoptosis through interacting with death receptors on cancer cells [134]. Based on KLRG1, Jiang et al. developed an NK cell-targeting immunomodulating nano-adaptor (imNA) to promote NK cells engagement of tumor cells for better exertion of tumor cytotoxicity, including αFc-NP with anti-KLRG1 antibody and anti-PDL1 antibody [135]. Compared to free antibodies, this strategy significantly reduced lung metastatic melanoma formation.
Current researches on targeting NK cells to deliver mRNA are mainly based on the physical and chemical characteristics of nanoparticles. For example, Nakamura and co-workers developed an LNP composed of CL1H6 (CL1H6-LNP) for effective siRNA delivery to NK-92 cells, achieving low cytotoxicity and efficient gene silencing [136]. Subsequently, they used CL1H6-LNP to deliver mRNA to NK cells (Figure 6B) [137]. The study indicated that LNP exhibited significantly higher mRNA expression intensity, primarily attributed to its high affinity with NK-92 cells and rapid, robust fusion with endosomal membranes. Thus, CL1H6-LNP serves as a non-viral vector that can regulate the function of NK-92 cells by delivering mRNA and thereby promote tumor immunotherapy. Wilk and co-workers reported that a cost-effective, easily synthesizable non-viral charge-altering releasable transporter (CART) can effectively transfect mRNA into primary human NK cells, independent of NK cell activation (Figure 6C) [138]. Compared to electroporation, CART is more efficient in transfecting NK cells, better preserves cell viability, and minimizes reshaping of NK cell phenotype and function. More importantly, the authors produced anti-CD19 human CAR NK cells in vitro by CART-mediated transfection of mRNA encoding anti-human CD19-41BB-CD3ζ CAR (hCAR). These CAR NK cells exhibited potent cytotoxicity and enhanced activation of CD19+ target cells compared to their untransfected counterparts. This predicts that CAR NK prepared by CART-mediated mRNA transfection greatly facilitates tumor immunotherapy. Douka prepared polymer and LNPs for the delivery of enhanced green fluorescent protein (eGFP)-mRNA into NK cells, including triblock co-polymer pHDePA, the homopolymer pHPMA-DEAE, and PEGylated forms of pDMAEMA (Figure 6D) [139]. By optimizing the lipid components and mRNA encapsulation methods, a promising lipid-complex-based mRNA formulation for NK cell transfection was identified, successfully delivering eGFP-mRNA to KHYG-1 cells. This NP-based mRNA delivery is a promising strategy for further development of novel NK cell therapies. For example, NK cells were engineered to overexpress CAR mRNAs encoding activating receptors (CD16 or CXCR4) to enhance their tumor targeting and cytotoxicity, thereby facilitating tumor immunotherapy [140]. Recently, it was reported that a DOTAP-functionalized lipid nanoparticle could deliver CAR mRNA to NK cells via lectin-mediated endocytosis and enhanced the ability of killing tumor cells via the extracellular signal-regulated kinase/Mitogen-Activated Protein Kinase pathway modulation and mitochondrial dynamics changes [74]. More importantly, the study achieved therapeutic effects of orthotopic HCC tumor model by injecting mouse-derived engineered anti-glypican 3 (GPC3)-CAR-NK cells [74]. The strategy for modifying CAR-NK enhance tumor immunotherapy.
mRNA NPs targeting macrophages
Macrophages are a type of white blood cell that belong to the innate immune system and play various roles in maintaining tissue homeostasis [141]. Macrophages have two main phenotypes and the balance between M1 and M2 macrophages in the tumor microenvironment is crucial. Tumors often exploit the immunomodulatory functions of macrophages to create an immune-suppressive environment that supports their growth and survival. Strategies aiming to repolarize tumor-associated macrophages into the M1 phenotype have been studied as a potential approach to enhance anti-tumor immune responses [142]. As the non-specific delivery method could inevitably lead to adverse systemic effects, the strategy commonly used by researchers is to use cell surface markers to modify specific targeting ligands for the targeted delivery of therapeutic agents. Examples of TAM markers widely used for ligand-targeted delivery include mannose receptor (MR), folate receptor (FR-β), and scavenger receptor (MARCO, SR-B1) (Figure 7A) [143, 144].
mRNA NPs targeting macrophages. (A) Schematic illustration of various receptors expressed on macrophages that could be employed to design macrophages-targeted NP-mediated delivery systems; (B) Schematic illustration of nanoparticles with mannose receptor targeting loaded with mRNA encoding reprogramming transcription factors. Adapted with permission from [75], copyright 2019 Springer Nature; (C) Schematic diagram of the structure and mechanism of action of a series of macrophage-targeting nanoparticles with different carbohydrate modifications; Adapted with permission from [150], copyright 2020 Elsevier; (D) Schematic of screening and structural of LNPs with macrophage targeting and liver targeting. Adapted with permission from [76], copyright 2023 John Wiley and Sons; (E) Schematic illustration of lipid nanoparticles encapsulating CAR mRNA for targeted delivery to macrophages. Adapted with permission from [169], copyright 2022 American Chemical Society.
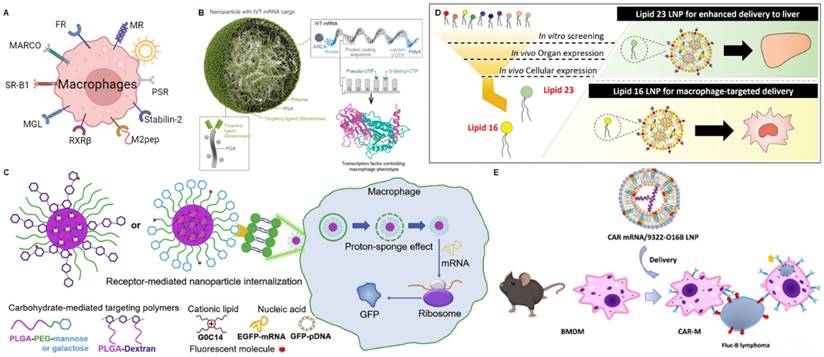
Mannose receptor (MR)
MR is a type I transmembrane protein belonging to the C-type lectin family and is primarily located on the surface of macrophages and iDCs [145]. The expression of MR is further upregulated when these monocytes extravasate from the circulation and are exposed to factors in the tumor microenvironment around blood vessels [146]. Due to the high expression of CD206 in TAMs, strategies that optimize cargo absorption through these receptors can be highly effective. Li et al. synthesized mannose-modified porous hollow iron oxide nanoparticles (PHNP) for loading the PI3Kγ small molecule inhibitor (3-methyladenine, 3-MA) to target TAMs and activate the immune response [147]. Chen et al. developed a series of PEG conjugate nanocarriers with varying numbers of mannose units and identified the optimal structural configuration for targeting macrophage-like J774.E cells [148]. Ye et al. established a siRNA conjugate platform to reduce the expression of Marburg virus (MARV) for prolonged survival benefit [149]. This nanoplatform contains a hexavalent mannose conjugate that can target macrophages and DCs, and GalNAc-siRNA conjugates that can achieve liver cell targeting through the asialoglycoprotein receptor (ASGPR).
Based on the discussed targeted techniques for macrophages, various mRNA delivery systems targeting macrophages have been developed. Zhang described a TAM-targeted nanoparticle that can deliver ex vivo transcribed mRNA encoding M1 program transcription factors to induce anti-tumor immunity (Figure 7B) [75]. These particles are composed of polyglutamic acid (PGA) functionalized with Di-mannose coated mRNA-PbAE complexes, which target macrophages expressing the MR with an M2-like phenotype. Meanwhile, nanoparticles encoding mRNA for interferon regulatory factor 5 (IRF5) significantly reduced tumor progression in ovarian cancer, melanoma lung metastasis, or glioblastoma models.
Chen et al. prepared a series of nanoparticles targeting macrophages using cationic lipid compounds G0-C14 and different carbohydrate modifications on poly(lactide-co-glycolide) (PLGA) or poly(lactide-co-glycolide)-b-poly(ethylene glycol) (PLGA-PEG) through self-assembly (Figure 7C) [150]. The carbohydrate modifications include mannose, lactose, maltose, and a mixture of mannose and lactose. EGFP messenger RNA (mRNA) was used as reporter genes to assess NP-mediated gene transfection in macrophages. Macrophage engulfment experiments showed that more carbohydrate-modified nanoparticles were internalized by Raw 264.7 cells compared to nanoparticles without carbohydrate modification. Compared to nanoparticles modified with other carbohydrate modifications, mannose-modified nanoparticles exhibit superior targeting capability to macrophages. This provides a potential technical platform for delivering biologics and therapeutic genes to macrophages in inflamed areas. Recently, Tang et al. also discovered a dual-targeting nano-delivery system that simultaneously targets pulmonary macrophages and tumor cells by DSPE-PEG-Mannose and HA for mRNA delivery [151]. This finding provides guidance for the development of vaccines or drugs for pulmonary-related diseases.
Folate receptor (FR)
In addition to the examples mentioned above, researchers have also identified other potential targeting ligands for macrophage-targeted mRNA delivery. FR is a 38 kDa glycosylphosphatidylinositol (GPI)-anchored protein [152]. The FR family comprises four members, including FRα (FOLR1), FRβ (FOLR2), FRγ (FOLR3), and FRδ (FOLR4). FRα and FRβ are anchored to the cell membrane via GPI, and they are overexpressed in tumor cells and TAMs [153]. A study indicated that folate can be used to target TAMs in a murine ovarian cancer xenograft model via liposomes [154]. Another study found that the overexpression of FRβ in TAMs is associated with poor prognosis in lung cancer. Therefore, the author utilized folate-modified liposomes (F-PLP) to deliver a plasmid containing BIM-S (a cell death mediator that interacts with BCL-2) to target lung cancer cells and FRβ-positive macrophages in the tumor microenvironment for the significant suppression of in vivo tumor growth [155].
Scavenger receptors (SR)
Macrophage surface scavenger receptors primarily include the following categories: Class A SR, mainly SR-A1 and macrophage receptor with collagenous structure (MARCO), which can recognize the lipopolysaccharides of Gram-positive bacteria; Class B SR, mainly CD36 and SR-B1, which can recognize oxidized LDL [156]. These receptors are widely distributed on the surface of macrophages and play a crucial role in the biological functions of macrophages by the recognition of various ligands.
MARCO has been identified as a gene that is overexpressed in the tumor microenvironment (TME) and is associated with poor prognosis in human breast cancer [157]. A study indicated that the anti-MARCO antibody has the potential to repolarize MARCO-positive TAMs [158]. Another study found that negatively charged immune-modifying microparticles (IMPs) can be taken up by inflammatory monocytes via the MARCO on macrophages [159].
SR-B1 participates in the phagocytosis of apoptotic cells by recognizing oxidized phospholipids displayed on the cell membrane when cells undergo apoptosis [160]. Wang and colleagues synthesized high-density lipoprotein-mimetic peptides-phospholipid scaffolds (HPPS) modified with apoA-1 mimetic peptide (R4F) that could target peripheral monocytes via the SR-B1 receptor [161]. Kuninty and co-workers designed engineered nanoliposomes containing peroxidized phospholipids that could be recognized and internalized by the SR-B1 for the delivery of STAT6 inhibitor (AS1517499), zoledronic acid, or cell wall lipopeptides to inhibit the premetastatic microenvironment and tumor growth [162].
Other targeting ligands
Besides the targeted ligands described above, some other molecules specifically expressed on macrophages including macrophage galactose-type lectin (MGL) [163], retinoid X receptor β (RXRβ) [164], stabilin-2 [165], and phosphatidylserine receptors (PSR) [156], have been employed as targeting ligands to design and develop macrophage-targeted NPs-based delivery systems. For example, based on the specific recognition between peptide sequence CRTLTVRKC (denoted S2P peptide) and stabilin-2 [166], Tao et al. recently developed a S2P peptide-modified polymeric NPs for macrophage-targeted delivery of Camk2γ siRNA and the treatment of atherosclerosis [167]. Although these macrophage-targeted delivery systems have not been designed for mRNA delivery, they definitely provide the potential strategies and guidelines for the future development of macrophage-targeted mRNA delivery techniques.
Other macrophage-targeting strategies based the physiochemical characteristics of NPs
In addition to the ligand-receptor interaction to achieve macrophage-targeted mRNA delivery, it has been found that the macrophage-targeted mRNA delivery could be also achieved by optimizing the physiochemical characteristics of NPs. For example, Naidu et al. synthesized a new library of ionizable lipids by modifying the hydrophobic tails and linker regions, incorporating other helper lipids, and using microfluidic mixing techniques to form stable LNPs (Figure 7D) [76]. Subsequently, they conducted rigorous in vitro and in vivo screening experiments to identify lipids suitable for cell-type-specific targeting of macrophages (Lipid 16) and high-quality lipids for liver targeting (Lipid 23). This study demonstrated that their structural modules could drive cell-specificity, opening new pathways for the development of efficient mRNA therapies using amino-lipid-based LNPs. The FDA approval of two anti-CD19 chimeric antigen receptor (CAR) T cell therapies has spurred increased research interest in CAR therapies [168]. Therefore, Ye et al. screened for optimal mRNA and lipid formulations to deliver mRNA coding for anti-CD19 CAR into primary mouse macrophages for the treatment of B-cell lymphoma (Figure 7E) [169]. The authors utilized the RAW264.7 cell line as a macrophage model and identified an optimized LNP formulation (9322-O16B/Chol/DOPE, 16:10:1, w/w) for delivering anti-CD19-eGFP mRNA. This improved CAR mRNA delivery system holds potential clinical applications for cell therapy.
mRNA NPs targeting B cells
B cells can regulate immune function by recognizing antigens, differentiating into plasma cells, producing antigen-specific antibodies, acting as APCs, and secretion of cytokines [170]. B cell dysregulation is associated with autoimmune diseases such as systemic lupus erythematosus, rheumatoid arthritis, and cancer [171]. Hence, the use of nanoparticles to modulate B cells is a promising strategy.
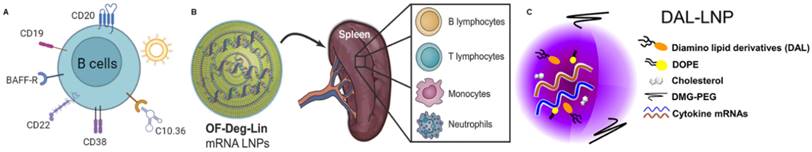
Currently, the targeting strategies for B cells primarily rely on receptors that are specific to B cell expression, including CD20 [172], CD19 [173], B-cell activating factor receptor (BAFF-R) [174], CD22 [175], CD38 [176], etc. (Figure 8A). For instance, poly(lactic-co-glycolic acid) (PLGA) nanoparticles with a core coated with a poly-L-arginine layer and dual-targeting outer layers of CD20 and CD44 antibodies have been developed for delivering siBCL-2 to lymphoblastic leukemia and B lymphoma cells [177]. Satake and co-workers discovered that the conjugation of superparamagnetic iron oxide nanoparticles (SPIO NPs) with anti-CD22 mAb enhances the delivery efficacy of siRNA therapy in acute lymphoblastic leukemia (ALL) [178]. Puente and co-workers reported the first study using CD38 as a target for a drug delivery system to treat multiple myeloma (MM) [179]. They modified polymer chitosan nanoparticles loaded with bortezomib (BTZ) with αCD38 mAb to enhance targeting efficacy and therapeutic outcomes.
mRNA NPs targeting B cells. (A) Schematic illustration of various receptors expressed on B cells that could be employed to design B cell-targeted NP-mediated delivery systems; (B) Schematic diagram of OF-Deg-Lin mRNA LNP targeting various immune cells including B lymphocytes in the spleen; Adapted with permission from [77], copyright 2017 John Wiley and Sons; (C) Schematic diagram of DAL-LNP loaded with mRNA. Adapted with permission from [78], copyright 2022 Elsevier.
Aptamers are a class of single-stranded oligonucleotides showing high specificity [180]. In addition, aptamers are generally easier to obtain than antibodies with low expense and high stability. Researchers reported a modular nanostructure that delivers fluorescent RNA aptamers (50-80 kDa, 175-250 nt) to target cells by recognizing various human B cell cancer cell lines and transferrin receptor-expressing cells [181]. The C10.36 aptamer is a compact G-quadruplex DNA that internalizes into B-cell cancer cell lines upon binding to an unknown cell surface molecule [182]. Although these B cell-targeted delivery systems were not designed for mRNA delivery, they undoubtedly provide potential strategies and guidelines for the future development of B-cell-targeted mRNA delivery technology.
Based on the physical and chemical characteristics of nanoparticles, there are also some applications in the research of B-cell-targeted mRNA delivery. Fenton and co-workers designed a LNP delivery system that can encapsulate mRNA and transfect B lymphocytes in the spleen, inducing protein expression in B cells (Figure 8B) [77]. While LNPs can be transiently observed in the liver and other organs, this LNP can induce the expression of more than 85% of the protein in the spleen. These results suggest that OF-Deg-Lin mRNA LNPs, as delivery vehicles, can significantly enhance protein expression in B lymphocytes in the spleen. It also demonstrates the significant advantages of nanomaterials in achieving organ and cell targeting. Liu et al. developed a novel mRNA delivery vehicle, DAL4-LNP, for delivering cytokines mRNA (Figure 8C) [78]. By intratumorally injection of DAL4-LNP loaded with GFP-coding mRNA (DAL4-LNP-GFP) into B16F10 tumors, the authors observed that LNP can selectively deliver mRNA targeting to CD19 B cells. By flow cytometry analysis, approximately 98% of GFP-positive immune cells are B cells.
mRNA NPs targeting neutrophils
Granulocytes make up the largest proportion of white blood cells. They enter the bloodstream and survive for several hours before leaving and dying [183]. Granulocytes include eosinophils, neutrophils, and basophils and are distinguished by granule staining. Neutrophils are a type of phagocyte found in the blood and are one of the first responders in the early stages of inflammation caused by bacterial infection and environmental changes [184]. They can be rapidly recruited to sites of tissue damage and exert antibacterial and inflammatory functions through phagocytosis, degranulation, neutrophil extracellular traps (NETs), and antigen presentation [185]. Therefore, targeting neutrophils could be a new therapeutic approach to treat inflammatory diseases and cancer (Figure 9A).
mRNA nanoparticles targeting neutrophils. (A) Schematic illustration of various receptors expressed on neutrophils that could be employed to design neutrophils-targeted NP-mediated delivery systems; (B) After intratumoral injection of 5 μg of mRNA encoding OX40L, OX40L mRNA can be delivered to a variety of bone marrow cells, including granulocytes. Adapted with permission from [31], copyright 2019 The American Association for the Advancement of Science; (C) Schematic diagram of the synthesis of lipid nanoparticles containing mRNA and the expression of Cre mRNA in various immune cells including neutrophils. Adapted with permission from [79], copyright 2017 American Chemical Society.
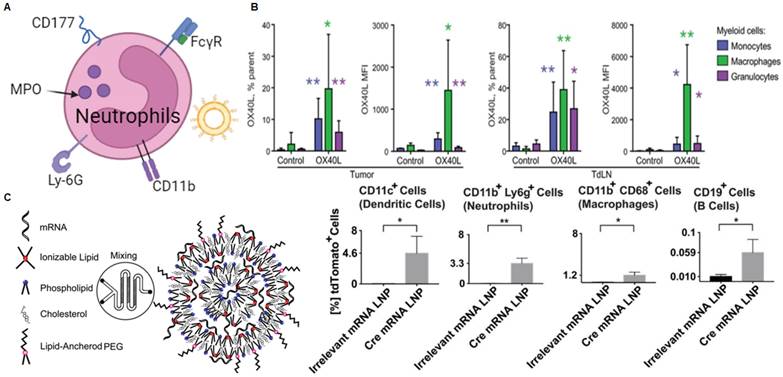
Some other molecules specifically expressed on neutrophils, including Fcγ receptor III (FcγRIII) [186], CD177 [187], Myeloperoxidase (MPO) [188], Ly-6G [189], CD11b (integrin αM, called Mac-1) [190]. For example, Wang et al. internalized resveratrol-loaded albumin particles into neutrophils adhering to inflamed endothelial surfaces through the FcγRIII receptor, which can be used to prevent lung injury [191]. Researchers have utilized phage display technology to identify peptide sequences that specifically bind to CD177. They found that modifying liposomes with neutrophil-specific peptides enhances neutrophil-specific delivery and the ability to alter neutrophil function, thus potentially treating various diseases [192]. Tang et al. self-assembled a ligand known as bis-5HT, which has two serotonin (5-hydroxytryptamine or 5-HT) terminal ends, with poly(propylene glycol)-poly(ethylene glycol)-carboxyl (PLGA-PEG-COOH) to create MPO and neutrophil targeting nanoparticles to improve tumor therapy[193]. Although these neutrophil-targeted delivery systems are used to deliver other therapeutic agents, they provide potential strategies and guidelines for the future development of neutrophil-targeted mRNA delivery technology.
While there are few reports on neutrophil-targeted mRNA delivery, various combinations of cytokine therapy and LNP-related mRNA vaccines have been confirmed to deliver mRNA to neutrophils. A study described a delivery system encapsulated in LNP for intratumoral delivery of IL-23/IL-36γ/OX40L trimeric mRNA, which can achieve complete remission (CR) of tumors (Figure 9B) [31]. Specifically, after intertumoral injection of 5 μg of mRNA encoding OX40L, the authors observed the expression of OX40L in three of the most abundant bone marrow cells, including macrophages, monocytes, and granulocytes. This suggests that the strategy can effectively deliver mRNA to granulocytes to exert its effects. Similarly, researchers developed LNPs for mRNA vaccine delivery to induce CD8 T-cell cytotoxicity (Figure 9C) [79]. Specifically, the authors found that the screened B-11 LNPs can be taken up by various immune cells, including DCs, macrophages, neutrophils, and B cells. Flow cytometry analysis revealed that 3.3% of neutrophils in the inguinal lymph nodes expressed the mRNA.
Clinical status of mRNA-based therapeutics
To date, there have been more than 1,000 clinical trials of mRNA-based cancer therapy. LNPs account for a large portion of these, most of which are in clinical phases I and II. We summarize the results of the current clinical trials of mRNA therapy, which suggest that mRNA therapy will be a promising strategy, and that further research and development will advance antitumor therapy (Table 5).
Current clinical trials of mRNA-nanoparticle therapy against cancer.
| Cancer type | mRNA | Nanoparticle carrier | Phase | NCT number | Reference |
|---|---|---|---|---|---|
| Metastatic non-small cell lung cancer | BI 1361849 | LNPs | I/II | NCT03164772 | https://clinicaltrials.gov/ct2/show/NCT03164772 |
| Malignant solid tumors | BNT113 | Liposomes | II | NCT04534205 | https://clinicaltrials.gov/ct2/show/NCT04534205 |
| Squamous cell carcinoma, head and neck neoplasm, cervical neoplasm, penile neoplasms malignant | HARE-40 | LNPs | I/II | NCT03418480 | https://clinicaltrials.gov/ct2/show/NCT03418480 |
| Melanoma | Lipo-MERIT | LNPs | I | NCT02410733 | https://clinicaltrials.gov/ct2/show/NCT02410733 |
| Melanoma, colon cancer, gastrointestinal cancer, genitourinary cancer, hepatocellular cancer | mRNA-4650 | LNPs | I/II | NCT03480152 | https://clinicaltrials.gov/ct2/show/NCT03480152 |
| Non-small cell lung cancer, pancreatic neoplasms, colorectal neoplasms | mRNA-5671/V941 | LNPs | I | NCT03948763 | https://clinicaltrials.gov/ct2/show/NCT03948763 |
| Adult glioblastoma | RNA-LPs | Liposomes | I | NCT04573140 | https://clinicaltrials.gov/ct2/show/NCT04573140 |
| Melanoma/colorectal cancer | RO7198457 | LNPs | II | NCT03815058 | https://clinicaltrials.gov/ct2/show/NCT03815058 |
| BNT122 | NCT04486378 | ||||
| Metastatic neoplasm | SAR441000 | LNPs | I | NCT03871348 | https://clinicaltrials.gov/ct2/show/NCT03871348 |
| Triple negative breast cancer | TNBC-MERIT | Liposomes | I | NCT02316457 | https://clinicaltrials.gov/ct2/show/NCT02316457 |
| Ovarian cancer | W_ova1 | Liposomes | I | NCT04163094 | https://clinicaltrials.gov/ct2/show/NCT04163094 |
| Non-small cell lung cancer | CV9202 | Protamine | I/II | NCT03164772 | https://clinicaltrials.gov/ct2/show/NCT03164772 |
| Melanoma/non-small cell lung cancer | mRNA-4157 | LNPs | I/II/III | NCT03897881, NCT03313778 | https://clinicaltrials.gov/ct2/show/NCT03313778 |
Conclusion, prospects, and challenges
Immunotherapy has revolutionized cancer treatment, including therapies like immune checkpoint inhibitors (ICI) and adoptive cell therapies. However, due to tumor heterogeneity, patient's benefit rates need to be improved. Furthermore, short half-lives of immunotherapeutic agents and adverse autoimmune reactions pose significant challenges in cancer immunotherapy. Recent studies have highlighted the therapeutic potential of mRNA therapy in various applications. However, challenges such as mRNA instability and immunogenicity must be addressed to enhance mRNA effectiveness. Therefore, the selection of suitable mRNA delivery vehicles is crucial. Furthermore, for the treatment to be effective, mRNA molecules must reach target cells and produce sufficient target proteins. Overcoming various biological and pharmacological obstacles in clinical applications is essential. Targeted delivery plays a crucial role, and guiding the delivery of mRNA macromolecules to immune cells using nanoparticles is significant, including T cells, DCs, NK cells, macrophages, B cells, and neutrophils. In this review, we systematically summarize existing mRNA targeting delivery nanoparticles for immune cells and potential methods for mRNA delivery targeting. This aims to enable the production of long-lasting therapeutic drugs while minimizing off-target toxicity to the greatest extent.
mRNA nanotechnology-mediated immunotherapy has been widely applied in preclinical and clinical research for cancer treatment. Recent achievements highlight the potential of mRNA as a breakthrough treatment for various disease. Despite the promising prospects and remarkable success of mRNA technology in cancer immunotherapy, there are challenges in advancing mRNA as a therapeutic immune drug in clinical practice. Firstly, it is essential to deepen our understanding of the relationship between modified mRNA constructs and their stability, translation, and immune regulation. High-throughput methods are attractive strategies that can establish correlations between mRNA performance and its numerous structural variants accurately and rapidly. Additionally, tumors have complex microenvironments and heterogeneity, and intercellular interactions affect tumor progression. Emerging techniques such as single cell sequencing and spatial transcriptomics help us understand the interaction mechanisms between mRNA and the patient's TME, providing a basis and support for developing the next generation of mRNA nanoparticles. Furthermore, optimizing mRNA delivery platforms is necessary. mRNA delivery platforms enhance the stability of exposed mRNA. Designing mRNA delivery platforms with high loading capacity and enhanced mRNA translation efficiency is crucial for improving safety and efficacy. In the context of targeting immune cells in tumor sites, nanoparticles encapsulating mRNA encoding immune-stimulating proteins can be passively delivered to tumors through the enhanced permeability and retention (EPR) effect. However, their effectiveness is often limited by multiple physical barriers. Therefore, designing nanoparticles that actively target mRNA molecules to specific cells with high efficiency is paramount. It is worth noting that the physicochemical properties of these biomaterials may affect the activation of immune pathways, or these interactions might alter the function of tissues or cells involved in immune regulation, which remains an unresolved issue. It's worth noting that introducing targeting modules into NPs typically involves multiple steps of synthesis, purification, and characterization, significantly increasing the complexity, cost, and regulatory challenges of production. Furthermore, the targeting capability of functionalized nanoparticles in a biological environment may be diminished or lost due to the "protein corona" effect [194]. Therefore, the use of targeting molecules to functionalize NP-mRNA should be carefully considered.
Finally, most LNP-mRNA therapies for cancer patients are still in the early stages of clinical trials. Positive therapeutic results from these clinical studies will support promising candidate drugs' progression to the next evaluation stage. More importantly, studying clinical data will provide valuable insights into optimizing suitable mRNA delivery methods, thereby promoting the translation of mRNA nanotechnology into clinical research. In conclusion, driving the development of mRNA delivery technology in tumor immunotherapy requires more effort. We look forward to continuous innovation and optimization of mRNA technology, bringing novel and effective results to life sciences and medical research.
Acknowledgements
L.H. and Z.H. contributed equally to this work. The work was supported by the National Natural Science Foundation of China (82173392, 81874226, 82001822), the grants from Guangdong Science and Technology Department (2024B1515040006) and Guangzhou Science and Technology Bureau (20210303004), Guangdong Basic and Applied Basic Research Foundation (2022A1515110065), the Key Research and Development Program of Hunan Province (2021SK2019), the Natural Science Foundation of Hunan Province (2023JJ50149), and the “Three million for Three Years” Project of the High-level Talent Special Funding Scheme of Sun Yat-Sen Memorial Hospital.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Crunkhorn S. Regulatory watch: enhanced chance of success for protein replacement therapies. Nat Rev Drug Discov. 2013;12:414
2. Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines - a new era in vaccinology. Nat Rev Drug Discov. 2018;17:261-279
3. Deal CE, Carfi A, Plante OJ. Advancements in mRNA encoded antibodies for passive immunotherapy. Vaccines. 2021;9:108
4. Han G, Noh D, Lee H, Lee S, Kim S, Yoon HY. et al. Advances in mRNA therapeutics for cancer immunotherapy: From modification to delivery. Adv Drug Deliv Rev. 2023;199:114973
5. Shi J, Huang MW, Lu ZD, Du XJ, Shen S, Xu CF. et al. Delivery of mRNA for regulating functions of immune cells. J Control Release. 2022;345:494-511
6. Islam MA, Xu Y, Tao W, Ubellacker JM, Lim M, Aum D. et al. Restoration of tumour-growth suppression in vivo via systemic nanoparticle-mediated delivery of PTEN mRNA. Nat Biomed Eng. 2018;2:850-864
7. Kong N, Zhang R, Wu G, Sui X, Wang J, Kim NY. et al. Intravesical delivery of KDM6A-mRNA via mucoadhesive nanoparticles inhibits the metastasis of bladder cancer. Proc Natl Acad Sci U S A. 2022;119:e2112696119
8. Kong N, Tao W, Ling X, Wang J, Xiao Y, Shi S. et al. Synthetic mRNA nanoparticle-mediated restoration of p53 tumor suppressor sensitizes p53-deficient cancers to mTOR inhibition. Sci Transl Med. 2019;11:eaaw1565
9. Cheng Q, Wei T, Farbiak L, Johnson LT, Dilliard SA, Siegwart DJ. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat Nanotechnol. 2020;15:313-320
10. Liu S, Cheng Q, Wei T, Yu X, Johnson LT, Farbiak L. et al. Membrane-destabilizing ionizable phospholipids for organ-selective mRNA delivery and CRISPR-Cas gene editing. Nat Mater. 2021;20:701-710
11. Wadhwa A, Aljabbari A, Lokras A, Foged C, Thakur A. Opportunities and challenges in the delivery of mRNA-based vaccines. Pharmaceutics. 2020;12:102
12. Zhang Y, Liu C, Wu C, Song L. Natural peptides for immunological regulation in cancer therapy: mechanism, facts and perspectives. Biomed Pharmacother. 2023;159:114257
13. Prasad V. Immunotherapy: Tisagenlecleucel - the first approved CAR-T-cell therapy: implications for payers and policy makers. Nat Rev Clin Oncol. 2018;15:11-12
14. Melero I, Castanon E, Alvarez M, Champiat S, Marabelle A. Intratumoural administration and tumour tissue targeting of cancer immunotherapies. Nat Rev Clin Oncol. 2021;18:558-576
15. Stary G, Bangert C, Tauber M, Strohal R, Kopp T, Stingl G. Tumoricidal activity of TLR7/8-activated inflammatory dendritic cells. J Exp Med. 2007;204:1441-1451
16. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069-1086
17. Morotti M, Albukhari A, Alsaadi A, Artibani M, Brenton JD, Curbishley SM. et al. Promises and challenges of adoptive T-cell therapies for solid tumours. Br J Cancer. 2021;124:1759-1776
18. Fang J, Lin L, Cao Y, Tan J, Liang Y, Xiao X. et al. Targeting the CD24-Siglec10 Axis: a potential strategy for cancer immunotherapy. BIO Integration. 2024;5:997
19. Beck JD, Reidenbach D, Salomon N, Sahin U, Tureci O, Vormehr M. et al. mRNA therapeutics in cancer immunotherapy. Mol Cancer. 2021;20:69
20. Conry RM, LoBuglio AF, Wright M, Sumerel L, Pike MJ, Johanning F. et al. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res. 1995;55:1397-1400
21. Zhou WZ, Hoon DS, Huang SK, Fujii S, Hashimoto K, Morishita R. et al. RNA melanoma vaccine: induction of antitumor immunity by human glycoprotein 100 mRNA immunization. Hum Gene Ther. 1999;10:2719-2724
22. Heiser A, Coleman D, Dannull J, Yancey D, Maurice MA, Lallas CD. et al. Autologous dendritic cells transfected with prostate-specific antigen RNA stimulate CTL responses against metastatic prostate tumors. J Clin Invest. 2002;109:409-417
23. Karikó K, Buckstein M, Ni H, Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165-175
24. Weide B, Pascolo S, Scheel B, Derhovanessian E, Pflugfelder A, Eigentler TK. et al. Direct injection of protamine-protected mRNA: results of a phase 1/2 vaccination trial in metastatic melanoma patients. J Immunother. 2009;32:498-507
25. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M. et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222-226
26. Stadler CR, Bahr-Mahmud H, Celik L, Hebich B, Roth AS, Roth RP. et al. Elimination of large tumors in mice by mRNA-encoded bispecific antibodies. Nat Med. 2017;23:815-817
27. Hotz C, Wagenaar TR, Gieseke F, Bangari DS, Callahan M, Cao H. et al. Local delivery of mRNA-encoded cytokines promotes antitumor immunity and tumor eradication across multiple preclinical tumor models. Sci Transl Med. 2021;13:eabc7804
28. Billingsley MM, Singh N, Ravikumar P, Zhang R, June CH, Mitchell MJ. Ionizable lipid nanoparticle-mediated mRNA delivery for human CAR T cell engineering. Nano Lett. 2020;20:1578-1589
29. Tchou J, Zhao Y, Levine BL, Zhang PJ, Davis MM, Melenhorst JJ. et al. Safety and efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol Res. 2017;5:1152-1161
30. Wang Y, Tiruthani K, Li S, Hu M, Zhong G, Tang Y. et al. mRNA delivery of a bispecific single-domain antibody to polarize tumor-associated macrophages and synergize immunotherapy against liver malignancies. Adv Mater. 2021;33:e2007603
31. Hewitt SL, Bai A, Bailey D, Ichikawa K, Zielinski J, Karp R. et al. Durable anticancer immunity from intratumoral administration of IL-23, IL-36gamma, and OX40L mRNAs. Sci Transl Med. 2019;11:eaat9143
32. Tsui NB, Ng EK, Lo YM. Stability of endogenous and added RNA in blood specimens, serum, and plasma. Clin Chem. 2002;48:1647-1653
33. Li Y, Banerjee S, Wang Y, Goldstein SA, Dong B, Gaughan C. et al. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc Natl Acad Sci U S A. 2016;113:2241-2246
34. Zhang H, Zhang L, Lin A, Xu C, Li Z, Liu K. et al. Algorithm for optimized mRNA design improves stability and immunogenicity. Nature. 2023;621:396-403
35. Leppek K, Byeon GW, Kladwang W, Wayment-Steele HK, Kerr CH, Xu AF. et al. Combinatorial optimization of mRNA structure, stability, and translation for RNA-based therapeutics. Nat Commun. 2022;13:1536
36. Li D, Wu M. Pattern recognition receptors in health and diseases. Signal Transduct Target Ther. 2021;6:291
37. Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R. et al. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374:546-549
38. Nance KD, Meier JL. Modifications in an Emergency: The role of N1-methylpseudouridine in COVID-19 vaccines. ACS Cent Sci. 2021;7:748-756
39. Dias Junior AG, Sampaio NG, Rehwinkel J. A balancing act: MDA5 in antiviral immunity and autoinflammation. Trends Microbiol. 2019;27:75-85
40. Wang S, Farfan-Arribas DJ, Shen S, Chou TH, Hirsch A, He F. et al. Relative contributions of codon usage, promoter efficiency and leader sequence to the antigen expression and immunogenicity of HIV-1 Env DNA vaccine. Vaccine. 2006;24:4531-4540
41. Gao M, Zhang Q, Feng XH, Liu J. Synthetic modified messenger RNA for therapeutic applications. Acta Biomater. 2021;131:1-15
42. Fenton OS, Kauffman KJ, McClellan RL, Appel EA, Dorkin JR, Tibbitt MW. et al. Bioinspired alkenyl amino alcohol ionizable lipid materials for highly potent in vivo mRNA delivery. Adv Mater. 2016;28:2939-2943
43. Sahin U, Kariko K, Tureci O. mRNA-based therapeutics-developing a new class of drugs. Nat Rev Drug Discov. 2014;13:759-780
44. Kowalski PS, Rudra A, Miao L, Anderson DG. Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol Ther. 2019;27:710-728
45. Yang W, Cao J, Cheng H, Chen L, Yu M, Chen Y. et al. Nanoformulations targeting immune cells for cancer therapy: mRNA therapeutics. Bioact Mater. 2023;23:438-470
46. Saiding Q, Zhang Z, Chen S, Xiao F, Chen Y, Li Y. et al. Nano-bio interactions in mRNA nanomedicine: Challenges and opportunities for targeted mRNA delivery. Adv Drug Deliv Rev. 2023;203:115116
47. Chen S, Huang X, Xue Y, Álvarez-Benedicto E, Shi Y, Chen W. et al. Nanotechnology-based mRNA vaccines. Nat Rev Methods Primers. 2023;3:63
48. Liu C, Shi Q, Huang X, Koo S, Kong N, Tao W. mRNA-based cancer therapeutics. Nat Rev Cancer. 2023;23:526-543
49. Xiao Y, Tang Z, Huang X, Chen W, Zhou J, Liu H. et al. Emerging mRNA technologies: delivery strategies and biomedical applications. Chem Soc Rev. 2022;51:3828-3845
50. Zhong Y, Du S, Dong Y. mRNA delivery in cancer immunotherapy. Acta Pharm Sin B. 2023;13:1348-1357
51. Zhang W, Jiang Y, He Y, Boucetta H, Wu J, Chen Z. et al. Lipid carriers for mRNA delivery. Acta Pharm Sin B. 2023;13:4105-4126
52. Huang X, Kong N, Zhang X, Cao Y, Langer R, Tao W. The landscape of mRNA nanomedicine. Nat Med. 2022;28:2273-2287
53. Zhang Y, Sun C, Wang C, Jankovic KE, Dong Y. Lipids and lipid derivatives for RNA delivery. Chem Rev. 2021;121:12181-12277
54. Hald Albertsen C, Kulkarni JA, Witzigmann D, Lind M, Petersson K, Simonsen JB. The role of lipid components in lipid nanoparticles for vaccines and gene therapy. Adv Drug Deliv Rev. 2022;188:114416
55. Hoy SM. Patisiran: first global approval. Drugs. 2018;78:1625-1631
56. Kaplonek P, Cizmeci D, Fischinger S, Collier AR, Suscovich T, Linde C. et al. mRNA-1273 and BNT162b2 COVID-19 vaccines elicit antibodies with differences in Fc-mediated effector functions. Sci Transl Med. 2022;14:eabm2311
57. Murciano-Goroff YR, Warner AB, Wolchok JD. The future of cancer immunotherapy: microenvironment-targeting combinations. Cell Res. 2020;30:507-519
58. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature. 2001;413:732-738
59. Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529-1531
60. Crunkhorn S. Strengthening the sting of immunotherapy. Nat Rev Drug Discov. 2020;20:589
61. Borriello F, Poli V, Shrock E, Spreafico R, Liu X, Pishesha N. et al. An adjuvant strategy enabled by modulation of the physical properties of microbial ligands expands antigen immunogenicity. Cell. 2022;185:614-629
62. Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellström KE. et al. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med. 1997;3:682-685
63. Page A, Chuvin N, Valladeau-Guilemond J, Depil S. Development of NK cell-based cancer immunotherapies through receptor engineering. Cell Mol Immunol. 2024;21:315-331
64. Van der Jeught K, De Koker S, Bialkowski L, Heirman C, Tjok Joe P, Perche F. et al. Dendritic cell targeting mRNA lipopolyplexes combine strong antitumor T-cell immunity with improved inflammatory safety. ACS nano. 2018;12:9815-9829
65. Le Moignic A, Malard V, Benvegnu T, Lemiegre L, Berchel M, Jaffres PA. et al. Preclinical evaluation of mRNA trimannosylated lipopolyplexes as therapeutic cancer vaccines targeting dendritic cells. J Control Release. 2018;278:110-121
66. Zhang H, You X, Wang X, Cui L, Wang Z, Xu F. et al. Delivery of mRNA vaccine with a lipid-like material potentiates antitumor efficacy through Toll-like receptor 4 signaling. Proc Natl Acad Sci U S A. 2021;118:e2005191118
67. Li M, Zhao M, Fu Y, Li Y, Gong T, Zhang Z. et al. Enhanced intranasal delivery of mRNA vaccine by overcoming the nasal epithelial barrier via intra- and paracellular pathways. J Control Release. 2016;228:9-19
68. Zhang R, Tang L, Tian Y, Ji X, Hu Q, Zhou B. et al. DP7-C-modified liposomes enhance immune responses and the antitumor effect of a neoantigen-based mRNA vaccine. J Control Release. 2020;328:210-221
69. Kheirolomoom A, Kare AJ, Ingham ES, Paulmurugan R, Robinson ER, Baikoghli M. et al. In situ T-cell transfection by anti-CD3-conjugated lipid nanoparticles leads to T-cell activation, migration, and phenotypic shift. Biomaterials. 2022;281:121339
70. Tombacz I, Laczko D, Shahnawaz H, Muramatsu H, Natesan A, Yadegari A. et al. Highly efficient CD4+ T cell targeting and genetic recombination using engineered CD4+ cell-homing mRNA-LNPs. Mol Ther. 2021;29:3293-3304
71. Rurik JG, Tombacz I, Yadegari A, Mendez Fernandez PO, Shewale SV, Li L. et al. CAR T cells produced in vivo to treat cardiac injury. Science. 2022;375:91-96
72. Parayath NN, Stephan SB, Koehne AL, Nelson PS, Stephan MT. In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nat Commun. 2020;11:6080
73. Li W, Zhang X, Zhang C, Yan J, Hou X, Du S. et al. Biomimetic nanoparticles deliver mRNAs encoding costimulatory receptors and enhance T cell mediated cancer immunotherapy. Nat Commun. 2021;12:7264
74. Shin HE, Han J-H, Park JD, Park M, Han J, Kang M-H. et al. Enhancing CAR-NK cells against solid tumors through chemical and genetic fortification with DOTAP-functionalized lipid nanoparticles. Adv Funct Mater. 2024 2315721
75. Zhang F, Parayath NN, Ene CI, Stephan SB, Koehne AL, Coon ME. et al. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat Commun. 2019;10:3974
76. Naidu GS, Yong SB, Ramishetti S, Rampado R, Sharma P, Ezra A. et al. A combinatorial library of lipid nanoparticles for cell type-specific mRNA delivery. Adv Sci (Weinh). 2023;10:e2301929
77. Fenton OS, Kauffman KJ, Kaczmarek JC, McClellan RL, Jhunjhunwala S, Tibbitt MW. et al. Synthesis and biological evaluation of ionizable lipid materials for the in vivo delivery of messenger RNA to B lymphocytes. Adv Mater. 2017;29:1606944
78. Liu JQ, Zhang C, Zhang X, Yan J, Zeng C, Talebian F. et al. Intratumoral delivery of IL-12 and IL-27 mRNA using lipid nanoparticles for cancer immunotherapy. J Control Release. 2022;345:306-313
79. Oberli MA, Reichmuth AM, Dorkin JR, Mitchell MJ, Fenton OS, Jaklenec A. et al. Lipid nanoparticle assisted mRNA delivery for potent cancer immunotherapy. Nano Lett. 2017;17:1326-1335
80. Sabado RL, Balan S, Bhardwaj N. Dendritic cell-based immunotherapy. Cell Res. 2017;27:74-95
81. Tacken PJ, de Vries IJ, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat Rev Immunol. 2007;7:790-802
82. van Kooyk Y, Rabinovich GA. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat Immunol. 2008;9:593-601
83. Jung SY, Kim SS, Kim YI, Chung HY, Kim SH, Yeo SG. Expression, distribution, and role of C-type lectin receptors in the human and animal middle ear and eustachian tube: a review. Molecules. 2018;23:734
84. Chatterjee B, Smed-Sörensen A, Cohn L, Chalouni C, Vandlen R, Lee B-C. et al. Internalization and endosomal degradation of receptor-bound antigens regulate the efficiency of cross presentation by human dendritic cells. Blood. 2012;120:2011-2020
85. Kramer S, Langhanki J, Krumb M, Opatz T, Bros M, Zentel R. HPMA-based nanocarriers for effective immune system stimulation. Macromol Biosci. 2019;19:e1800481
86. Zhu D, Hu C, Fan F, Qin Y, Huang C, Zhang Z. et al. Co-delivery of antigen and dual agonists by programmed mannose-targeted cationic lipid-hybrid polymersomes for enhanced vaccination. Biomaterials. 2019;206:25-40
87. Rezaee M, Oskuee RK, Nassirli H, Malaekeh-Nikouei B. Progress in the development of lipopolyplexes as efficient non-viral gene delivery systems. J Control Release. 2016;236:1-14
88. Geijtenbeek TB, Torensma R, van Vliet SJ, van Duijnhoven GC, Adema GJ, van Kooyk Y. et al. Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that supports primary immune responses. Cell. 2000;100:575-85
89. Geijtenbeek TB, van Vliet SJ, Engering A, t Hart BA, van Kooyk Y. Self- and nonself-recognition by C-type lectins on dendritic cells. Annu Rev Immunol. 2004;22:33-54
90. Svajger U, Anderluh M, Jeras M, Obermajer N. C-type lectin DC-SIGN: an adhesion, signalling and antigen-uptake molecule that guides dendritic cells in immunity. Cell Signal. 2010;22:1397-1405
91. Boks MA, Ambrosini M, Bruijns SC, Kalay H, van Bloois L, Storm G. et al. MPLA incorporation into DC-targeting glycoliposomes favours anti-tumour T cell responses. J Control Release. 2015;216:37-46
92. Jiang W, Swiggard WJ, Heufler C, Peng M, Mirza A, Steinman RM. et al. The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature. 1995;375:151-155
93. Katakowski JA, Mukherjee G, Wilner SE, Maier KE, Harrison MT, DiLorenzo TP. et al. Delivery of siRNAs to dendritic cells using DEC205-targeted lipid nanoparticles to inhibit immune responses. Mol Ther. 2016;24:146-155
94. Poulin LF, Reyal Y, Uronen-Hansson H, Schraml BU, Sancho D, Murphy KM. et al. DNGR-1 is a specific and universal marker of mouse and human Batf3-dependent dendritic cells in lymphoid and nonlymphoid tissues. Blood. 2012;119:6052-6062
95. Zeng B, Middelberg AP, Gemiarto A, MacDonald K, Baxter AG, Talekar M. et al. Self-adjuvanting nanoemulsion targeting dendritic cell receptor Clec9A enables antigen-specific immunotherapy. J Clin Invest. 2018;128:1971-1984
96. Wang F, Ullah A, Fan X, Xu Z, Zong R, Wang X. et al. Delivery of nanoparticle antigens to antigen-presenting cells: from extracellular specific targeting to intracellular responsive presentation. J Control Release. 2021;333:107-128
97. Castro FV, Tutt AL, White AL, Teeling JL, James S, French RR. et al. CD11c provides an effective immunotarget for the generation of both CD4 and CD8 T cell responses. Eur J Immunol. 2008;38:2263-2273
98. Cruz LJ, Rosalia RA, Kleinovink JW, Rueda F, Lowik CW, Ossendorp F. Targeting nanoparticles to CD40, DEC-205 or CD11c molecules on dendritic cells for efficient CD8(+) T cell response: a comparative study. J Control Release. 2014;192:209-218
99. Joshi MD, Unger WJ, Storm G, van Kooyk Y, Mastrobattista E. Targeting tumor antigens to dendritic cells using particulate carriers. J Control Release. 2012;161:25-37
100. van Broekhoven CL, Parish CR, Demangel C, Britton WJ, Altin JG. Targeting dendritic cells with antigen-containing liposomes: a highly effective procedure for induction of antitumor immunity and for tumor immunotherapy. Cancer Res. 2004;64:4357-4365
101. Li D, Romain G, Flamar AL, Duluc D, Dullaers M, Li XH. et al. Targeting self- and foreign antigens to dendritic cells via DC-ASGPR generates IL-10-producing suppressive CD4+ T cells. J Exp Med. 2012;209:109-121
102. Qian Y, Jin H, Qiao S, Dai Y, Huang C, Lu L. et al. Targeting dendritic cells in lymph node with an antigen peptide-based nanovaccine for cancer immunotherapy. Biomaterials. 2016;98:171-183
103. Bournazos S, Gupta A, Ravetch JV. The role of IgG Fc receptors in antibody-dependent enhancement. Nat Rev Immunol. 2020;20:633-643
104. Kawamura K, Kadowaki N, Suzuki R, Udagawa S, Kasaoka S, Utoguchi N. et al. Dendritic cells that endocytosed antigen-containing IgG-liposomes elicit effective antitumor immunity. J Immunother. 2006;29:165-174
105. Cruz LJ, Rueda F, Cordobilla B, Simon L, Hosta L, Albericio F. et al. Targeting nanosystems to human DCs via Fc receptor as an effective strategy to deliver antigen for immunotherapy. Mol Pharm. 2011;8:104-116
106. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22:240-273
107. Kokkinopoulos I, Jordan WJ, Ritter MA. Toll-like receptor mRNA expression patterns in human dendritic cells and monocytes. Mol Immunol. 2005;42:957-968
108. Li M, Zhou H, Jiang W, Yang C, Miao H, Wang Y. Nanovaccines integrating endogenous antigens and pathogenic adjuvants elicit potent antitumor immunity. Nano Today. 2020;35:101007
109. Shah K, Al-Haidari A, Sun J, Kazi JU. T cell receptor (TCR) signaling in health and disease. Signal Transduct Target Ther. 2021;6:412
110. Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK. et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270-274
111. Perruche S, Zhang P, Liu Y, Saas P, Bluestone JA, Chen W. CD3-specific antibody-induced immune tolerance involves transforming growth factor-beta from phagocytes digesting apoptotic T cells. Nat Med. 2008;14:528-535
112. Labrijn AF, Janmaat ML, Reichert JM, Parren P. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov. 2019;18:585-608
113. Zhu AX, Finn RS, Edeline J, Cattan S, Ogasawara S, Palmer D. et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. 2018;19:940-952
114. Duwa R, Pokhrel RH, Banstola A, Pandit M, Shrestha P, Jeong JH. et al. T-cell engaging poly(lactic-co-glycolic acid) nanoparticles as a modular platform to induce a potent cytotoxic immunogenic response against PD-L1 overexpressing cancer. Biomaterials. 2022;291:121911
115. Xiao P, Wang J, Zhao Z, Liu X, Sun X, Wang D. et al. Engineering nanoscale artificial antigen-presenting cells by metabolic dendritic cell labeling to potentiate cancer immunotherapy. Nano Lett. 2021;21:2094-2103
116. Huang C, Duan X, Wang J, Tian Q, Ren Y, Chen K. et al. Lipid nanoparticle delivery system for mRNA encoding B7H3-redirected bispecific antibody displays potent antitumor effects on malignant tumors. Adv Sci (Weinh). 2023;10:e2205532
117. Ramishetti S, Kedmi R, Goldsmith M, Leonard F, Sprague AG, Godin B. et al. Systemic gene silencing in primary T lymphocytes using targeted lipid nanoparticles. ACS nano. 2015;9:6706-6716
118. McHugh MD, Park J, Uhrich R, Gao W, Horwitz DA, Fahmy TM. Paracrine co-delivery of TGF-beta and IL-2 using CD4-targeted nanoparticles for induction and maintenance of regulatory T cells. Biomaterials. 2015;59:172-181
119. Dalloul A. CD5: a safeguard against autoimmunity and a shield for cancer cells. Autoimmun Rev. 2009;8:349-353
120. Tita-Nwa F, Moldenhauer G, Herbst M, Kleist C, Ho AD, Kornacker M. Cytokine-induced killer cells targeted by the novel bispecific antibody CD19xCD5 (HD37xT5.16) efficiently lyse B-lymphoma cells. Cancer Immunol Immunother. 2007;56:1911-1920
121. Soldevila G, Raman C, Lozano F. The immunomodulatory properties of the CD5 lymphocyte receptor in health and disease. Curr Opin Immunol. 2011;23:310-318
122. Schmid D, Park CG, Hartl CA, Subedi N, Cartwright AN, Puerto RB. et al. T cell-targeting nanoparticles focus delivery of immunotherapy to improve antitumor immunity. Nat Commun. 2017;8:1747
123. Gomes-Silva D, Srinivasan M, Sharma S, Lee CM, Wagner DL, Davis TH. et al. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood. 2017;130:285-296
124. Bradley JE, Ramirez G, Hagood JS. Roles and regulation of Thy-1, a context-dependent modulator of cell phenotype. BioFactors. 2009;35:258-265
125. Lee J, Yun KS, Choi CS, Shin SH, Ban HS, Rhim T. et al. T cell-specific siRNA delivery using antibody-conjugated chitosan nanoparticles. Bioconjug Chem. 2012;23:1174-1180
126. Zheng Y, Stephan MT, Gai SA, Abraham W, Shearer A, Irvine DJ. In vivo targeting of adoptively transferred T-cells with antibody- and cytokine-conjugated liposomes. J Control Release. 2013;172:426-435
127. Zheng Y, Tang L, Mabardi L, Kumari S, Irvine DJ. Enhancing adoptive cell therapy of cancer through targeted delivery of small-molecule immunomodulators to internalizing or noninternalizing receptors. ACS nano. 2017;11:3089-3100
128. Stephan MT, Stephan SB, Bak P, Chen J, Irvine DJ. Synapse-directed delivery of immunomodulators using T-cell-conjugated nanoparticles. Biomaterials. 2012;33:5776-5787
129. Bald T, Krummel MF, Smyth MJ, Barry KC. The NK cell-cancer cycle: advances and new challenges in NK cell-based immunotherapies. Nat Immunol. 2020;21:835-847
130. Shimasaki N, Jain A, Campana D. NK cells for cancer immunotherapy. Nat Rev Drug Discov. 2020;19:200-218
131. Myers JA, Miller JS. Exploring the NK cell platform for cancer immunotherapy. Nat Rev Clin Oncol. 2021;18:85-100
132. Astorga-Gamaza A, Vitali M, Borrajo ML, Suarez-Lopez R, Jaime C, Bastus N. et al. Antibody cooperative adsorption onto AuNPs and its exploitation to force natural killer cells to kill HIV-infected T cells. Nano Today. 2021;36:101056
133. Au KM, Park SI, Wang AZ. Trispecific natural killer cell nanoengagers for targeted chemoimmunotherapy. Sci Adv. 2020;6:eaba8564
134. Chandrasekaran S, Chan MF, Li J, King MR. Super natural killer cells that target metastases in the tumor draining lymph nodes. Biomaterials. 2016;77:66-76
135. Jiang CT, Chen KG, Liu A, Huang H, Fan YN, Zhao DK. et al. Immunomodulating nano-adaptors potentiate antibody-based cancer immunotherapy. Nat Commun. 2021;12:1359
136. Nakamura T, Nakade T, Yamada K, Sato Y, Harashima H. The hydrophobic tail of a pH-sensitive cationic lipid influences siRNA transfection activity and toxicity in human NK cell lines. Int J Pharm. 2021;609:121140
137. Nakamura T, Nakade T, Sato Y, Harashima H. Delivering mRNA to a human NK cell line, NK-92 cells, by lipid nanoparticles. Int J Pharm. 2023;636:122810
138. Wilk AJ, Weidenbacher NL, Vergara R, Haabeth OAW, Levy R, Waymouth RM. et al. Charge-altering releasable transporters enable phenotypic manipulation of natural killer cells for cancer immunotherapy. Blood Adv. 2020;4:4244-4255
139. Douka S, Brandenburg LE, Casadidio C, Walther J, Garcia BBM, Spanholtz J. et al. Lipid nanoparticle-mediated messenger RNA delivery for ex vivo engineering of natural killer cells. J Control Release. 2023;361:455-469
140. Carlsten M, Levy E, Karambelkar A, Li L, Reger R, Berg M. et al. Efficient mRNA-based genetic engineering of human NK cells with high-affinity CD16 and CCR7 augments rituximab-induced ADCC against lymphoma and targets NK cell migration toward the lymph node-associated chemokine CCL19. Front Immunol. 2016;7:105
141. Mosser DM, Hamidzadeh K, Goncalves R. Macrophages and the maintenance of homeostasis. Cell Mol Immunol. 2021;18:579-587
142. Huang L, Xu R, Li W, Lv L, Lin C, Yang X. et al. Repolarization of macrophages to improve sorafenib sensitivity for combination cancer therapy. Acta Biomater. 2023;162:98-109
143. Paulos CM, Turk MJ, Breur GJ, Low PS. Folate receptor-mediated targeting of therapeutic and imaging agents to activated macrophages in rheumatoid arthritis. Adv Drug Deliv Rev. 2004;56:1205-1217
144. Sun X, Gao D, Gao L, Zhang C, Yu X, Jia B. et al. Molecular imaging of tumor-infiltrating macrophages in a preclinical mouse model of breast cancer. Theranostics. 2015;5:597-608
145. Taylor PR, Gordon S, Martinez-Pomares L. The mannose receptor: linking homeostasis and immunity through sugar recognition. Trends Immunol. 2005;26:104-110
146. Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A. et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer cell. 2011;19:512-526
147. Li K, Lu L, Xue C, Liu J, He Y, Zhou J. et al. Polarization of tumor-associated macrophage phenotype via porous hollow iron nanoparticles for tumor immunotherapy in vivo. Nanoscale. 2020;12:130-144
148. Chen P, Zhang X, Jia L, Prud'homme RK, Szekely Z, Sinko PJ. Optimal structural design of mannosylated nanocarriers for macrophage targeting. J Control Release. 2014;194:341-349
149. Ye X, Holland R, Wood M, Pasetka C, Palmer L, Samaridou E. et al. Combination treatment of mannose and GalNAc conjugated small interfering RNA protects against lethal Marburg virus infection. Mol Ther. 2023;31:269-281
150. Chen Q, Gao M, Li Z, Xiao Y, Bai X, Boakye-Yiadom KO. et al. Biodegradable nanoparticles decorated with different carbohydrates for efficient macrophage-targeted gene therapy. J Control Release. 2020;323:179-190
151. Tang Z, You X, Xiao Y, Chen W, Li Y, Huang X. et al. Inhaled mRNA nanoparticles dual-targeting cancer cells and macrophages in the lung for effective transfection. Proc Natl Acad Sci U S A. 2023;120:e2304966120
152. Samadian H, Hosseini-Nami S, Kamrava SK, Ghaznavi H, Shakeri-Zadeh A. Folate-conjugated gold nanoparticle as a new nanoplatform for targeted cancer therapy. J Cancer Res Clin Oncol. 2016;142:2217-2229
153. Varghese B, Vlashi E, Xia W, Ayala Lopez W, Paulos CM, Reddy J. et al. Folate receptor-beta in activated macrophages: ligand binding and receptor recycling kinetics. Mol Pharm. 2014;11:3609-3616
154. Turk MJ, Waters DJ, Low PS. Folate-conjugated liposomes preferentially target macrophages associated with ovarian carcinoma. Cancer Lett. 2004;213:165-172
155. Tie Y, Zheng H, He Z, Yang J, Shao B, Liu L. et al. Targeting folate receptor beta positive tumor-associated macrophages in lung cancer with a folate-modified liposomal complex. Signal Transduct Target Ther. 2020;5:6
156. Platt N, Haworth R, Darley L, Gordon S. The many roles of the class A macrophage scavenger receptor. Int Rev Cytol. 2002;212:1-40
157. Elomaa O, Kangas M, Sahlberg C, Tuukkanen J, Sormunen R, Liakka A. et al. Cloning of a novel bacteria-binding receptor structurally related to scavenger receptors and expressed in a subset of macrophages. Cell. 1995;80:603-609
158. Eisinger S, Sarhan D, Boura VF, Ibarlucea-Benitez I, Tyystjarvi S, Oliynyk G. et al. Targeting a scavenger receptor on tumor-associated macrophages activates tumor cell killing by natural killer cells. Proc Natl Acad Sci U S A. 2020;117:32005-32016
159. Getts DR, Terry RL, Getts MT, Deffrasnes C, Muller M, van Vreden C. et al. Therapeutic inflammatory monocyte modulation using immune-modifying microparticles. Sci Transl Med. 2014;6:219ra7
160. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787-795
161. Wang J, Peng X, Wei J, Dai Y, Huang S, Deng D. et al. In situ phagocyte-mediated deep tumor penetration assisted by ApoA-1 mimetic peptide-modified silicasome. Nano Today. 2023;50:101864
162. Kuninty PR, Binnemars-Postma K, Jarray A, Pednekar KP, Heinrich MA, Pijffers HJ. et al. Cancer immune therapy using engineered ‛tail-flipping' nanoliposomes targeting alternatively activated macrophages. Nat Commun. 2022;13:4548
163. van Vliet SJ, Saeland E, van Kooyk Y. Sweet preferences of MGL: carbohydrate specificity and function. Trends Immunol. 2008;29:83-90
164. Tang T, Wei Y, Kang J, She ZG, Kim D, Sailor MJ. et al. Tumor-specific macrophage targeting through recognition of retinoid X receptor beta. J Control Release. 2019;301:42-53
165. Zhou B, Weigel JA, Fauss L, Weigel PH. Identification of the hyaluronan receptor for endocytosis (HARE). J Biol Chem. 2000;275:37733-37741
166. Lee GY, Kim JH, Oh GT, Lee BH, Kwon IC, Kim IS. Molecular targeting of atherosclerotic plaques by a stabilin-2-specific peptide ligand. J Control Release. 2011;155:211-217
167. Tao W, Yurdagul A Jr, Kong N, Li W, Wang X, Doran AC. et al. siRNA nanoparticles targeting CaMKIIgamma in lesional macrophages improve atherosclerotic plaque stability in mice. Sci Transl Med. 2020;12:eaay1063
168. Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ. et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378:449-459
169. Ye Z, Chen J, Zhao X, Li Y, Harmon J, Huang C. et al. In vitro engineering chimeric antigen receptor macrophages and T cells by lipid nanoparticle-mediated mRNA delivery. ACS Biomater Sci Eng. 2022;8:722-733
170. Rawlings DJ, Metzler G, Wray-Dutra M, Jackson SW. Altered B cell signalling in autoimmunity. Nat Rev Immunol. 2017;17:421-436
171. Sabatino JJ Jr, Probstel AK, Zamvil SS. B cells in autoimmune and neurodegenerative central nervous system diseases. Nat Rev Neurosci. 2019;20:728-745
172. Katchi T, Liu D. Diagnosis and treatment of CD20 negative B cell lymphomas. Biomark Res. 2017;5:1-5
173. Nadler LM, Anderson KC, Marti G, Bates M, Park E, Daley JF. et al. B4, a human B lymphocyte-associated antigen expressed on normal, mitogen-activated, and malignant B lymphocytes. J Immunol. 1983;131:244-250
174. Rahman ZS, Rao SP, Kalled SL, Manser T. Normal induction but attenuated progression of germinal center responses in BAFF and BAFF-R signaling-deficient mice. J Exp Med. 2003;198:1157-1169
175. Moyron-Quiroz JE, Partida-Sanchez S, Donis-Hernandez R, Sandoval-Montes C, Santos-Argumedo L. Expression and function of CD22, a B-cell restricted molecule. Scand J Immunol. 2002;55:343-351
176. Liu Q, Kriksunov IA, Graeff R, Munshi C, Lee HC, Hao Q. Crystal structure of human CD38 extracellular domain. Structure. 2005;13:1331-1339
177. Choi KY, Correa S, Min J, Li J, Roy S, Laccetti KH. et al. Binary targeting of siRNA to hematologic cancer cells in vivo using layer-by-layer nanoparticles. Adv Funct Mater. 2019;29:1900018
178. Satake N, Duong C, Chen C, Barisone GA, Diaz E, Tuscano J. et al. Targeted therapy with MXD3 siRNA, anti-CD22 antibody and nanoparticles for precursor B-cell acute lymphoblastic leukaemia. Br J Haematol. 2014;167:487-499
179. de la Puente P, Luderer MJ, Federico C, Jin A, Gilson RC, Egbulefu C. et al. Enhancing proteasome-inhibitory activity and specificity of bortezomib by CD38 targeted nanoparticles in multiple myeloma. J Control Release. 2018;270:158-176
180. Xiang D, Shigdar S, Qiao G, Wang T, Kouzani AZ, Zhou SF. et al. Nucleic acid aptamer-guided cancer therapeutics and diagnostics: the next generation of cancer medicine. Theranostics. 2015;5:23-42
181. Porciani D, Cardwell LN, Tawiah KD, Alam KK, Lange MJ, Daniels MA. et al. Modular cell-internalizing aptamer nanostructure enables targeted delivery of large functional RNAs in cancer cell lines. Nat Commun. 2018;9:2283
182. Opazo F, Eiden L, Hansen L, Rohrbach F, Wengel J, Kjems J. et al. Modular assembly of cell-targeting devices based on an uncommon G-quadruplex aptamer. Mol Ther Nucleic Acids. 2015;4:e251
183. Nemeth T, Sperandio M, Mocsai A. Neutrophils as emerging therapeutic targets. Nat Rev Drug Discov. 2020;19:253-275
184. Herrero-Cervera A, Soehnlein O, Kenne E. Neutrophils in chronic inflammatory diseases. Cell Mol Immunol. 2022;19:177-191
185. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS. et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532-1535
186. Coxon A, Cullere X, Knight S, Sethi S, Wakelin MW, Stavrakis G. et al. Fc gamma RIII mediates neutrophil recruitment to immune complexes. a mechanism for neutrophil accumulation in immune-mediated inflammation. Immunity. 2001;14:693-704
187. Bouvain P, Ding Z, Kadir S, Kleimann P, Kluge N, Tiren Z-B. et al. Non-invasive mapping of systemic neutrophil dynamics upon cardiovascular injury. Nat Cardiovasc Res. 2023;2:126-143
188. Liu YW, Li S, Dai SS. Neutrophils in traumatic brain injury (TBI): friend or foe? J Neuroinflammation. 2018;15:1-18
189. Wang JX, Bair AM, King SL, Shnayder R, Huang YF, Shieh CC. et al. Ly6G ligation blocks recruitment of neutrophils via a beta2-integrin-dependent mechanism. Blood. 2012;120:1489-1498
190. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159-175
191. Wang Z, Li J, Cho J, Malik AB. Prevention of vascular inflammation by nanoparticle targeting of adherent neutrophils. Nat Nanotechnol. 2014;9:204-210
192. Vols S, Kaisar-Iluz N, Shaul ME, Ryvkin A, Ashkenazy H, Yehuda A. et al. Targeted nanoparticles modify neutrophil function in vivo. Front Immunol. 2022;13:1003871
193. Tang L, Wang Z, Mu Q, Yu Z, Jacobson O, Li L. et al. Targeting neutrophils for enhanced cancer theranostics. Adv Mater. 2020;32:e2002739
194. Salvati A, Pitek AS, Monopoli MP, Prapainop K, Bombelli FB, Hristov DR. et al. Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule corona adsorbs on the surface. Nat Nanotechnol. 2013;8:137-143
Author contact
![]() Corresponding authors: caipeiesysu.edu.cn; xuxiaod5sysu.edu.cn.
Corresponding authors: caipeiesysu.edu.cn; xuxiaod5sysu.edu.cn.