Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results and Discussion
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2024; 14(13):5219-5234. doi:10.7150/thno.93654 This issue Cite
Research Paper
Amide naphthotube as a novel supramolecular sequestration agent for tetracaine and decamethonium
Cheng-Da Zhao1, Wei Cai2 ![]() , Wen-Jie Chen1, Huan Yao1, Song-Meng Wang3, Kailin Li2, Yan-Long Ma4
, Wen-Jie Chen1, Huan Yao1, Song-Meng Wang3, Kailin Li2, Yan-Long Ma4 ![]() , Li-Li Wang1
, Li-Li Wang1 ![]() , Liu-Pan Yang1
, Liu-Pan Yang1 ![]()
1. The Affiliated Nanhua Hospital, School of Pharmaceutical Science and School of Basic Medical Sciences, Hengyang Medical School, University of South China, Hengyang, 421001, China.
2. School of Pharmaceutical Sciences, Hunan University of Medicine, Huaihua, 418000, China.
3. Department of Chemistry, Southern University of Science and Technology, Xueyuan Blvd 1088, Shenzhen, 518055, China.
4. School of Chemistry and Chemical Engineering and Guangdong Cosmetics Engineering & Technology Research Center, Guangdong Pharmaceutical University, Zhongshan 528458, China.
Received 2023-12-27; Accepted 2024-7-30; Published 2024-8-19
Abstract

Rationale: Anesthetics are widely used for optimizing surgical conditions, postoperative pain management, and treating various chronic pain conditions. Tetracaine and decamethonium are representative drugs of local anesthetics and neuromuscular blocking agents, respectively. However, overdose and toxicity of the drugs always lead to serious adverse events. Thus, there is a strong demand for effective antidotes.
Methods: The binding interactions of amide naphthotubes with tetracaine and decamethonium were systematically studied using 1H NMR, ITC, and DFT calculations. The antidotal effects of amide naphthotube to tetracaine toxicity were assessed in vitro and in vivo, and the mechanism of detoxification was explored at a cellular level. Additionally, mouse models were established to evaluate the reversal activities of amide naphthotube on decamethonium-induced mortality and muscle relaxation, and the reversal mechanism was investigated through pharmacokinetic experiments.
Results: We have demonstrated that the anti-isomer of amide naphthotube exhibits significant binding affinities towards tetracaine (Ka = 1.89×107 M-1) and decamethonium (Ka = 1.01×107 M-1) in water. The host displayed good biocompatibility both in vitro and in vivo. The administration of amide naphthotube following tetracaine overdose in mouse models notably increased the overall survival rate, indicating its effective antidotal properties. The host could reverse the tetracaine-induced Na+ channels blockage at the cellular level. Moreover, the injection of amide naphthotube also reversed the mortality and paralysis induced by decamethonium in mouse models following a pharmacokinetic mechanism.
Conclusion: An emerging artificial receptor, amide naphthotube, has strong binding affinities towards tetracaine and decamethonium. It functions as a supramolecular antidote for tetracaine poisoning and a reversal agent for decamethonium by selectively sequestering these compounds in vivo.
Keywords: supramolecular sequestration, detoxification, tetracaine, decamethonium, amide naphthotube
Introduction
Medications are crucial tools in managing and conquering diseases, contributing to the increased lifespan and improved quality of life seen over the past century. However, medications also carry potential risks, as their toxic side effects can persist even after their beneficial effects have ceased. Moreover, the accumulation of exogenous environmental toxins and endogenous toxic metabolites can result in significant organ damage and threaten human health [1]. To address medication overdose and toxicity, pharmacodynamic and pharmacokinetic methods are utilized. Pharmacodynamic methods involve the competitive binding of small molecule antagonists to block and disrupt medication actions [2]. In contrast, pharmacokinetic approach methods seek to lower medication concentrations, deactivate their biological activity, and facilitate their elimination from the body [3, 4]. Comparatively, the latter approach does not necessitate an exact comprehension of the underlying mechanisms of drug action, and notable advancements have been achieved utilizing this methodology.
Since 2000, there has been rapid development in the field of aqueous molecular recognition based on synthetic hosts [5], laying a solid foundation for applications in the pharmaceutical industry [6, 7, 8, 9, 10, 11, 12, 13, 14, 15]. In this area, synthetic supramolecular sequestration agents are highly representative due to their easy modification, high thermal and chemical stability, and abiotic nature [16, 17, 18, 19, 20]. For instance, Sugammadex, a derivative of γ-cyclodextrin, can effectively antagonize muscle relaxants rocuronium and vecuronium by forming tight complexes and has achieved impressive commercial success with annual sales revenue exceeding $1 billion [21]. Besides, the application of other artificial hosts such as calixarenes [22], (acyclic)cucurbiturils [23, 24, 25], pillararenes [26, 27, 28] and biphen[n]arene [29] in this field have also been explored. The guests include neuromuscular blockers, anesthetics, toxic substances, and drugs of abuse [16-20]. Existing supramolecular sequestration agents are typically derived from modifying classic macrocyclic hosts. Developing new host systems to sequester complex or polar guests that are resistant to physiological salt and serum proteins is highly desirable.
Local anesthetics effectively and reversibly block sensory nerve impulses in specific areas, which is crucial in postoperative pain management and treatment of chronic pain conditions [30, 31, 32]. Tetracaine (TC, Figure 1A) is a potent ester-type drug commonly used for various types of anesthesia [33]. In recent years, with the emergence of the medical cosmetic industry, tetracaine has increasingly been applied in procedures such as cosmetic surgery, eyebrow tattooing, and tattooing [34]. However, incidents of TC poisoning, shock, and even death have occurred due to improper administration leading to bloodstream entry, excessive dosage, or allergic reactions, posing significant threats to public health [35, 36, 37, 38]. Still, there is no specific antidote for TC poisoning in clinical practice [39]. In addition, neuromuscular blocking agents are commonly used in anesthesia to relax skeletal muscle movements and improve surgical conditions. Rapid recovery after surgery is essential for muscle function and to prevent lingering effects. Decamethonium (C10) is a conventional neuromuscular blocking agent with a lengthy record of clinical and experimental applications. However, the restricted metabolic clearance in the body and the tendency to cause muscle pain and other adverse reactions, combined with the lack of a suitable antidote for clinical management, significantly limit the clinical use of C10 [40, 41, 42, 43]. Therefore, it is clinically significant to develop an effective antidote that can alleviate or reverse the toxicity of TC and C10.
Drug reversal by amide naphthotube. (A) Chemical structures of amide naphthotube and drug molecules. (B) Cartoon depicting supramolecular sequestration of TC and C10 in vivo.
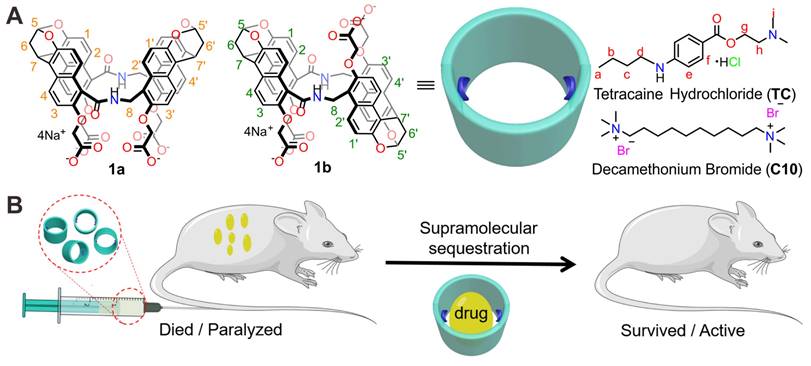
In recent years, amide naphthotubes (Figure 1A, 1a and 1b) have garnered significant attention in supramolecular chemistry due to their unique recognition capabilities in water [44]. Initially reported by Glass in 2004 for the selective sensing of lipids [45], our group has comprehensively studied the guest binding scope, recognition mechanism, physico-organic characteristics, and functional applications of amide naphthotubes [46, 47, 48]. Amide naphthotubes have the ideal properties for in vivo sequestration applications: Firstly, they have low cytotoxicity and have realized some biomedical applications [49, 50]; Secondly, carboxylate-substituted hosts are highly water-soluble and do not aggregate at high concentrations (350 mM) [50]; Thirdly, the hosts have deep hydrophobic cavities with buried hydrogen-bonding sites, allowing for selective recognition of hydrophilic and polar organic molecules, which is quite complementary to traditional macrocyclic receptors; Finally, their binding affinities to neutral organic molecules in aqueous environments generally unaffected by the presence of small biomolecules and inorganic salts [51]. Herein, we investigated and demonstrated that amide naphthotube could bind TC and C10 with binding affinities more than 107 M-1 in water, and exhibited good biocompatibility in mouse models. Moreover, the synthetic receptor could serve as a sequestration agent for TC and C10 via supramolecular pharmacokinetic approaches to reverse the toxicity of TC and the neuromuscular blocking effect of C10 (Figure 1B). More importantly, it may serve to meet the longstanding medical requirements of the healthcare community.
Methods
General methods
The reagents and guest molecules used in this study were commercially available and used without further purification unless stated otherwise. Ultrapure water was obtained from the Chuangchun pure water machine CCH-H200. 1H NMR spectra were recorded using a Bruker Avance 500 NMR spectrometer, and chemical shifts are reported in ppm with residual solvents or sodium methanesulfonate as internal standards. Amide naphthotubes 1a and 1b (Chemical Formula: C58H38N2Na4O18, Molecular Weight: 1142.8971 g/mol) were synthesized as described previously [52].
ITC experiments
ITC experiments were conducted in water or fetal bovine serum (FBS) at a certain temperature using a Malvern MicroCal PEAQ-ITC Automated instrument. Typically, 280 µL of the host solution in water or 100% FBS was placed in the sample cell, while 38 µL of the guest solution was loaded into the injection syringe. The titration consisted of an initial 0.4 µL injection followed by 18 consecutive 2 µL injections with a 120 s interval between injections. All solutions were degassed before titration. The data was analyzed using the instrument's internal software and fit to a "one set of binding sites" model.
Computational Methods
The quantum chemistry calculations were conducted using the Gaussian 16 package to optimize the structures of host-guest complexes. This was achieved through density functional theory (DFT) with calculations (ωB97xd/(ma)-def2-SVP for energy-minimizing, ωB97xd/ma-def2-TZVPP for single point energy) in water (PCM/SMD model) at 298 K [53, 54]. Additionally, an independent gradient model based on Hirshfeld partition (IGMH) analysis was carried out using the Multiwfn 3.8 (dev) program, and the molecular plots were visualized with the VMD 1.9.3 program [55, 56].
Hemolysis test
Fresh blood was collected from Sprague-Dawley rats from the orbital sinuses. The red blood cells (RBCs) were extracted by centrifugation and diluted to 2% (v/v) with saline. Various concentrations of 1b ranging from 12.5 to 800 μM were added to the 2% RBCs. Ultrapure water and saline served as the positive and negative controls, respectively. Following a 3-hour incubation at 37°C, the supernatants were obtained through centrifugation. The hemolysis rate was determined by measuring the A450 nm of the supernatants from different samples using the following equation:
Hemolysis ratio (%) = (ODsample - ODnegative control)/(ODpositive control - ODnegative control) × 100%
Toxicity evaluation of 1b with i.v. administration
Lethal 1b poisoning mouse model: Female Balb/c mice aged 8-10 weeks were randomly assigned to four groups (n = 8 for each group). The mice were i.v. injected with 1b saline solutions at 500, 750, and 1000 mg/kg. In the control group, mice received the same amount of saline. Their weight was measured every two days, and their behavior was observed daily for signs of illness. After 21 days, the animals were sacrificed, and major organs such as the heart, liver, kidneys, lungs, and spleen were collected and weighed to calculate the organ index.
Potential toxicity evaluation of 1b: Female Balb/c mice (8-10 weeks old) weighing 20-25 g were randomly divided into groups (n = 15 for each group). 1b/saline solutions (20 mg/mL) were prepared and filtered through a 0.22 µm membrane and then i.v. administered to mice at a dose of 100 mg/kg body weight. The control group received 50 µL saline/10 g body weight. Mice were monitored daily for weight changes and signs of illness. Major organs of three mice from each group were collected for histopathological analysis after 1, 3, 7, and 14 days. After 1 and 14 days, blood samples were collected for hematologic analysis (n = 6 for each group). The serum was separated for liver and renal function markers detection. The tissues were initially fixed in 4% paraformaldehyde, then submitted to Hunan AiFang Biological Co., Ltd., for paraffin embedding and sectioning. The sections were subsequently stained with H&E and examined using an optical microscope by the company. Whole blood cell (WBC), red blood cell (RBC), platelet (PLT), hemoglobin (HGB), alanine aminotransferase (ALT), aspartate aminotransferase (AST), creatinine (CRE) and urea were analyzed by Wuhan Servicebio Technology Co., Ltd.
The evaluation of the capacity of 1b for reversing TC toxicity
Cells experiments: AC16 cells were cultured in DMEM/F12 with 10% FBS, 1% penicillin, and 1% streptomycin at 37 °C with 5% CO2, and passaged every 2 days. They were seeded into 96-well plates at a density of 5-8×103 cells per well and cultured overnight. Then, different concentrations of TC, 1a, 1b, TC@1a and TC@1b mixture solution at various concentrations (25, 50, 100, 100, 200, 400, 800, and 1600 μM) were added to replace the culture medium for 24 h at 37 °C. After incubation, Cell Counting Kit-8 solution (10 μL per well) was added, and cells were incubated for 4 h at 37 °C. The optical density values at 450 nm were measured using a microplate reader to assess cell viability as a percentage of control cells. Finally, IC50 values were calculated using GraphPad Prism 8.0.2 software.
According to the IC50 value of TC, the concentration of TC was fixed at 400 μM, the cells were treated with a series of fresh medium containing different fixed molar ratios of TC at a minimum of 8 concentrations (1:0, 1:0.00625, 1:0.125, 1:0.25, 1:0.5, 1:1, 1:2 and 1:4) of TC and 1b for 24 h at 37 °C. The assay and cell viability were calculated as in the cell experiment above.
Lethal TC poisoning mouse model: the lethal dose of i.v. administered TC was calculated in mice (8-10 week old female Balb/c mice, weighing 20-25 g) using Dixon's up-and-down method. The initial dose of TC given to the first mouse was 9.0 mg/kg, and the standard dose ratio of TC given to the adjacent mouse was 0.9. If the animal survives, the subsequent animal receives a higher dose. If the animal dies, the following animal receives a lower dose. This process was repeated until at least seven crossover points were achieved. Probit analysis was employed to determine the LD50 (50% lethal dose) and LD95 (95% lethal dose) of TC, along with their corresponding CI (confidence intervals). Sequence diagrams and dose-response curves were generated using GraphPad Prism 9 software.
Assessment of the antidotal effects of 1b in a mouse model of lethal TC poisoning: Female Balb/c mice aged 8-10 weeks and weighing 20-25 g were randomly allocated to various groups (n = 8 for each group). Solutions of 1b/saline at concentrations of 4, 8, 12, and 16 mg/mL were prepared. The mice were initially i.v. injected with TC at 9.0 mg/kg, followed by i.v. administration of 1b at doses of 20, 40, 60, and 80 mg/kg. The control group received an i.v. injection of 100 µL of saline per 20 g. The mice were monitored for survival for 24 hours. Furthermore, the safety of mice with 100% survival was evaluated by recording their body weights and observing their behavior daily for any signs of illness.
Determination of the plasma membrane potential: AC16 cells were seeded into 12-well plates and cultured until reaching 80-90% confluence. Following removal of the culture medium, cells were exposed to HBSS (Hank's Balanced Salt Solution) containing 0, 100, 200, 400, 600, and 800 mM TC for 10 minutes at 37 °C. Subsequently, cells were collected and incubated with 200 nM Bis-(1,3-Dibutylbarbituric Acid)-Trimethine Oxanol (DiBAC4(3), Solarbio) for 20 minutes at 37 °C. The fluorescence intensity of cells was then assessed using flow cytometry. To determine the impact of 1b (ammonium salt) on plasma membrane potential, 25×104 AC16 cells were co-incubated with 400 µM TC and varying concentrations of 1b (0, 400, 800, and 2000 µM) or exclusively with 1b (2000 µM) in HBSS for 10 minutes at 37 °C. Plasma membrane potential was subsequently evaluated using the aforementioned methodology.
Determination of intracellular sodium levels: 3×105 AC16 cells were seeded in 6-well plates with 2 mL of medium. After 12 hours, the cell medium was replaced with 400 µM TC and varying concentrations of compound 1b (ammonium salt, 0, 400, 800, and 2000 µM) or 2000 µM 1b in HBSS for 10 minutes at 37 °C. Control cells were treated with 2 mL HBSS for 10 minutes at 37 °C. After washing with PBS, cells were lysed in 300 µL of 1% Triton X-100 in saline with a 1% proteinase inhibitor cocktail from Meilunbio. The lysates were centrifuged at 12,000 ×g for 15 minutes, and the supernatant was collected for sodium level determination (7600 Modular-Hitachi-Chemistry Analyzer) and total protein content BCA assays (Epizyme Biotech). The sodium-to-protein ratio was calculated, presenting results as relative intracellular sodium content compared to the control group.
The evaluation of the capacity of 1b for reversing C10 toxicity and muscle relaxation
Lethal C10 poisoning mouse model: the lethal dose of i.v. administered C10 was calculated using Dixon's up-and-down method similar to TC. The initial dose of C10 was 0.9 mg/kg. Subsequent doses for each mouse were adjusted by ±0.1 mg/kg based on the outcome of the preceding animal.
Assessment of the antidotal effects of 1b in a mouse model of lethal C10 poisoning: Female Balb/c mice (8-10 weeks old) weighing 20-25 g were randomly divided into groups (n = 8 for each group). Solutions of 1b/saline at concentrations of 0.4, 0.8, 1.2, and 1.6 mg/mL were prepared. The mice were first injected intravenously with C10 at a dose of 0.9 mg/kg, followed by i.v. administration of 1b at doses of 2, 4, 6, and 8 mg/kg. The control group was given a 100 µL saline i.v. injection per 20 g. The survival of the mice was monitored within 24 hours. To further assess the well-being of the mice with a 100% survival rate, their body weights were recorded and their behavior was observed daily for any signs of illness.
Determination of C10-induced paralysis dose in a mouse model: The movement behavior was conducted with the Rotarod rotor (ZB-200). Female Balb/c mice (8-9 weeks old) weighing 18-22 g were placed on the Rotarod rotor and trained three times for 30 minutes, three times a day for three consecutive days. On day 4, Dixon's up-and-down method was employed to explore the effective dose: The initial dose of C10 to the first mouse was 0.6 mg/kg, and the standard dose ratio of C10 given to the adjacent mouse was 0.8. If the former mouse had a response to injection (''response'' defined as the mice could not move on the rotor for 30 seconds after 30 seconds of C10 injection), the subsequent mouse received a decreased dose. Conversely, in the case of a negative response, the following mouse received an increased dose. Tests were conducted sequentially, with changes in mobility response transitioning between positive and negative until at least 7 crossover points were reached. Probit analysis was employed to determine the ED50 (50% effective dose) and ED95 (95% effective dose) of C10, along with their corresponding CI (confidence intervals). Sequence diagrams and dose-response curves were generated using GraphPad Prism 9 software.
Mobility recovery assessment in a mouse model: the trained mice for 4 days were i.v. administered with either 4 mg/kg or 6 mg/kg of 1b, or saline, 30 seconds after administering 0.53 mg/kg C10. The mice were then placed on the rotor, and the time taken to move on the rotor for 30 seconds successfully was recorded.
Pharmacokinetics studies
Male Kunming mice (30 ± 2 g) were used in the pharmacokinetic study. Drugs were injected through the tail vein of mice. Blood, liver, and kidney tissues were collected at certain intervals after administration with 6 mice at each time point. Blood samples were placed in heparin sodium tubes and centrifuged at 6000 rpm for 20 minutes at 4 oC to obtain plasma samples. Plasma, liver and kidney tissues were stored in a freezer at -80 oC and frozen until analysis was performed. The drug concentrations were determined by Liquid chromatography-mass spectrometry (Waters ACQUITY UPLC/Xevo TQ-S). The Masslynx 4.1 software was used to obtain the peak area of the tested compounds and internal standards in each plasma sample and calculate the standard curve formula. The following pharmacokinetic parameters were calculated in each group using the DAS 3.2.8 software: Cmax (maximal plasma level), tmax (time to peak concentration), AUC (0-48 h) (area under the curve between 0 and 48 h), AUC (0-∞) (area under the curve extrapolated until infinite), MRT (mean residence time), t1/2z (terminal elimination half-life), Vd (volume of distribution) and CL (total clearance). The detailed pharmacokinetics experiment procedure including materials and reagents, sample preparation, establishment and validation of analytical strategy, and LC-MS conditions are described in the Supporting Information.
Statistical analysis
Statistical analysis was performed using PASW Statistics 18.0 and GraphPad Prism 9.5.1 software. One-way ANOVA was utilized for experiments involving more than two groups, while two-tailed, unpaired Student's t-tests were conducted for comparisons between two sample sets. Statistical significance was defined as P < 0.05. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns indicates no statistically significant difference.
Study approval
Female Balb/c mice aged 8-10 weeks were procured from Changsha Tianqin Biotechnology Co. Ltd and approved by Hengyang Medical School, University of South China. All mice had ad libitum access to food and water during the experiments. The animal experiments adhered to ethical standards, approved by the Institutional Animal Care and Use Committee of the Center (NO: USC2023XS107, USC2023XS130, USC2024XS037), following the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care International.
Results and Discussion
Binding behaviors between the amide naphthotubes with TC and C10
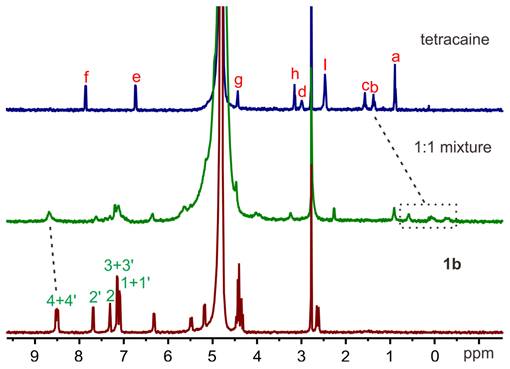
The complexation of amide naphthotubes with TC and C10 in aqueous environments was initially investigated through 1H-NMR spectroscopy. All the complexes exhibit rapid exchange dynamics between the unbound and bound states at the NMR timescale, resulting in observed chemical shifts that represent an ensemble average of these states. Consequently, many signals become broad or disappear into the baseline. For the complex of the hosts with TC (Figures 2 and S1), the aromatic signals of the host experience broadening and obvious chemical shifts. Moreover, some guest signals are no longer detectable, while methylene signals of the guest molecules shift towards approximately 0 ppm. For the complex of the hosts with C10 (Figures S2-S3), strongly shielded methyl signals between -1.5 ppm and -3.5 ppm confirmed the formation of the corresponding inclusion complexes. These NMR spectral alterations collectively provide evidence for the encapsulation of guest molecules within the host cavity.
The 1H NMR spectra (500 MHz,0.5 mM, 298 K) of TC (top), 1b (bottom) and their equimolar mixture (middle) in PB buffer (pD =7.4).
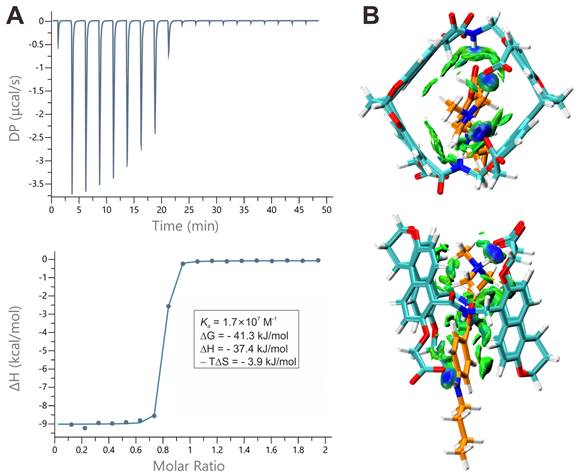
After confirming the inclusion binding of guests within hosts 1a and 1b by 1H NMR spectroscopy, we thus conducted ITC measurements to determine the binding thermodynamics for all complexes (Figures 3A, S4-S7). By titrating the solutions of the hosts with the guests and using the "one set of binding sites" model, we extracted the values of Ka, ΔH, -TΔS and the binding stoichiometry, presented in Table 1. The binding constants are all higher than 106 M-1 and the binding events are predominantly driven by favorable changes in enthalpy, which are attributed to the non-classical hydrophobic effects [57]. This effect involves the release of cavity waters with incomplete hydrogen bonding into the surrounding solvent upon complexation [58]. These association constants are much higher when compared to other classic molecular hosts, whose affinities are lower than 105 M-1 and 107 M-1 toward TC and C10 [59, 60, 61], respectively.
Binding behaviors of amide naphthotube and guests. (A) top: Raw ITC data obtained from titration of 1b with TC in water at 25 oC, bottom: apparent reaction heat obtained from the integration of calorimetric traces and fitted using the “one set of binding sites” model. (B) Energy-minimized structures and independent gradient models of TC@1b obtained by DFT (ωB97xd/(ma)-def2-SVP) calculations.
Association Constants and Thermodynamic Parameters as Determined by ITC Titrations.a
| Guest | Host | Solvent | Temperature | Ka (M-1) | ΔG (kJ/mol) | ΔH (kJ/mol) | -TΔS (kJ/mol) |
|---|---|---|---|---|---|---|---|
| TC | 1a | water | 25 oC | (1.32 ± 0.10) × 107 | -40.7 | -32.9 | -7.8 |
| 1b | water | 25 oC | (1.89 ± 0.24) × 107 | -41.5 | -37.1 | -4.4 | |
| 1b | water | 37 oC | (1.40 ± 0.04) × 107 | -42.4 | -40.2 | -2.2 | |
| 1b | FBS | 25 oC | (1.74 ± 0.17) × 105 | -29.9 | -23.1 | -6.8 | |
| C10 | 1a | water | 25 oC | (5.73 ± 0.16) × 106 | -38.6 | -21.0 | -17.6 |
| 1b | water | 25 oC | (1.01± 0.02) × 107 | -40.0 | -23.9 | -16.1 | |
| 1b | water | 37 oC | (9.56 ± 1.33) × 106 | -41.4 | -28.1 | -13.3 | |
| 1b | FBS | 25 oC | (1.44 ± 0.11) × 105 | -29.5 | -15.3 | -14.2 |
a All the titration experiments were repeated twice, and the averaged values with standard deviations are reported here.
To investigate the binding strength of amide naphthotube with TC and C10 under conditions mimicking biological environments, additional ITC experiments of 1b were conducted at 37 oC and in FBS, as 1b is a stronger binder than 1a (Figures S8-S11). At 37°C, the binding constants of 1b with TC and C10 show a slight decrease compared to 25°C, yet remains approximately 107 M-1. Moreover, the changes of ΔG for complex formation exhibit minimal variation (<1.5 kJ mol-1) across temperatures, contrasting with more pronounced alterations in ΔH and -TΔS (ca. 2.2-4.4 kJ mol-1). Notably, the opposing changes in ΔH and -TΔS compensate each other, thereby enabling the maintenance of strong binding affinities even at elevated temperatures, demonstrating an enthalpy-entropy compensation effect. Serum contains high concentrations of salts and diverse small biomolecules [62]. For example, some quaternary ammonium salts such as acetylcholine and choline present in the blood may bind the hosts competitively. The binding affinities between 1b and acetylcholine and choline were determined by NMR titrations (Figures S12-S15), and the values are four orders of magnitude lower than those for TC and C10. The binding constants of 1b towards TC and C10 in 100% FBS (Ka = 1.74 × 105 M-1, 1.44 × 105 M-1) are two orders of magnitude lower than that in water. This reduction is considered moderate, given the presence of a myriad of intricate components within the serum. Therefore, the strong binding of 1b under mimicking physiological conditions provides a foundational assurance for its detoxification efficacy in vivo.
DFT calculations for all the complexes at the level of theory of ωB97XD/def2-SVP (ma-def2-SVP basis set was used for anionic parts) in water (PCM) were then performed to elucidate the detailed interactions responsible for the strong binding (Figures 3B, S16-S18). Generally, both TC and C10 are well encapsulated inside the cavity of 1a and 1b. Independent gradient model based on Hirshfeld partition (IGMH) analysis reveals that C-H⋅⋅⋅π, N-H⋅⋅⋅π, π⋅⋅⋅π and electrostatic interactions between the guests and the hosts are involved in the complexes. Moreover, the optimized structures provide insights into the differences in binding constants of 1a and 1b. For TC, the deeper cavity of 1b results in a more enclosed inclusion than 1a, thus a slightly higher binding constant for 1b than 1a is observed. Moreover, there are two representative conformers for the host-guest complex for 1a and TC, the DFT calculations show the conformer with more favored electrostatic interactions of carboxylate groups with the positive Protonated tertiary amine is more energetically stable (Figure S16). Generally, 1a and 1b are very potent for binding TC and C10 but 1a is a slightly weaker host than 1b, so the latter biocompatibility and antidotal tests are generally based on 1b.
Biocompatibility evaluation of 1b in a mouse model
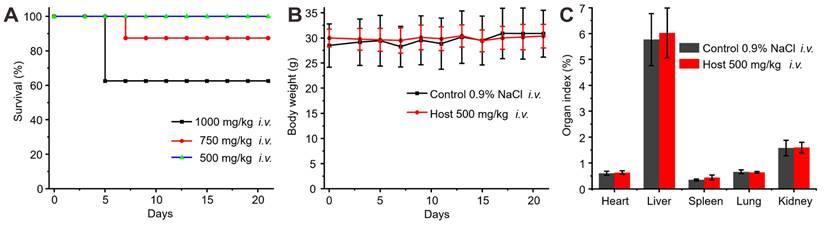
The biocompatibility of 1b was previously assessed in vitro and showed minimal cytotoxicity at concentrations up to 0.3 mM [49]. However, a systematic examination of its in vivo toxicity has been lacking. In this study, the in vivo biocompatibility of 1b was evaluated in mice following i.v. administration by monitoring changes in body weight, examining organ indices, hematology parameters, hepatic function markers, and conducting histopathological analysis of major organ tissues. The assessment of a drug's hemolytic effects is critical for ensuring its safety for i.v. administration. To evaluate the hemolytic activity of 1b, a Red Blood Cell Hemolysis test was conducted using blood samples obtained from Sprague-Dawley. The results indicated that the RBC hemolysis ratio for 1b remained below 0.5% across a concentration range of 10 to 800 µM (Figure S19), thus suggesting minimal hemolytic activity on erythrocytes. After confirming the minimal hemolytic activity of 1b, we began our next step, which involved assessing the in vivo biocompatibility of 1b. We initially determined the maximum tolerable dose of 1b in mice through i.v. administration (Figure 4A). Administering 1b at doses of 1000 mg/kg or 750 mg/kg resulted in approximately 40% or 10% mortality 4-8 days post-administration. After reducing the dose to 500 mg/kg, all the mice remained alive and showed no unusual behaviors or signs of illness throughout the 21-day experiment. Subsequent examination revealed a marginal reduction in body weights during the initial 7 days post-administration, followed by a consistent rise thereafter, as elucidated in Figure 4B. Notably, evaluation of organ indices in mice sacrificed on Day 21 following i.v. injection of 1b demonstrated no discernible disparities as compared to the control group, as illustrated in Figure 4C.
Toxicity assessment of i.v. administration of 1b. (A) Survival rates of mice after i.v. administration of varying doses of 1b. (B) Weight changes in mice after i.v. administration of 1b (500 mg/kg). (C) Organ indices of the mice 21 days after i.v. administration of 1b.
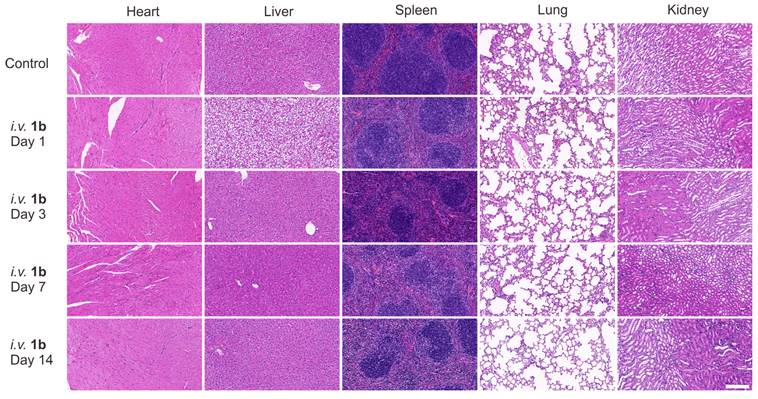
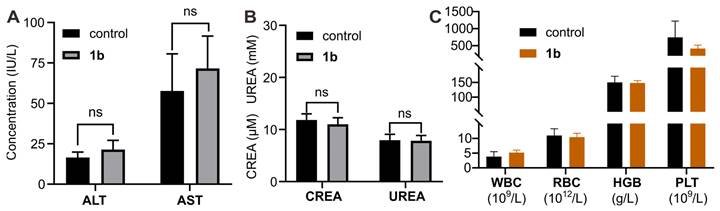
Furthermore, histological analysis was conducted following i.v. administration of 1b at 100 mg/kg doses (maximum possible dosage estimated according to the literature). Hematoxylin and eosin H&E staining was employed to assess tissue samples obtained through the mice sacrificed at 1, 3, 7, and 14 days post-administration (Figure 5). Generally, the histopathological sections of the majority of the major organs in the mice did not display discernible signs of injury or inflammatory cell infiltration, consistent with the findings in the control group. Specifically, mild hepatocyte edema was observed on the first day following 1b administration, indicative of a certain degree of associated hepatotoxicity. Nonetheless, the subsequent reduction in hepatic edema by the third day suggests the liver damage caused by 1b was reversible. Furthermore, the hepatic and renal function tests conducted on day 1 (Figure S20) and day 14 (Figures 6A and 6B) after 1b injection revealed no statistically significant differences in aspartate aminotransferase (AST), alanine aminotransferase (ALT), creatinine (CREA), and urea nitrogen (UREA) levels compared to the control group, indicating minimal impact on the hepatic and renal functions. Subsequent analysis of typical hematological parameters in the blood revealed values within the normal range, comparable to those of the control group, providing further evidence of the low cytotoxicity of 1b (Figure 6B). Taken together, these findings indicate the high i.v. biocompatibility of 1b in mice at a dose as high as 100 mg/kg, as demonstrated by the absence of toxicity in our systemic evaluations.
H&E stained sections of major organs with intravenous administration of 1b (100 mg/kg). Mice were sacrificed on day 1, 3, 7, 14 after the i.v. administration of 1b. Scale bar = 200 μm.
Toxicity evaluation of 1b with i.v. administration. (A) Hepatic and renal (B) function markers test on the blood samples collected from the mice on Day 14 after i.v. administration of 1b. (C) Hematological parameters of blood from mice that were sacrificed on Day 14 after i.v. administration of 1b.
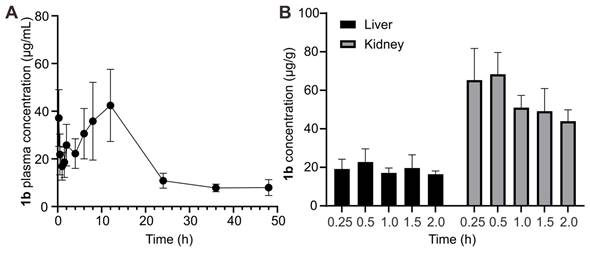
In addition to toxicity assessments evaluating the safety of 1b, understanding its plasma pharmacokinetic profile in vivo is crucial to assessing its clinical potential as a scavenger of TC and C10. Following i.v. administration (80 mg kg-1), plasma concentration of 1b at various intervals was measured based on UPLC-Xevo TQ-S MS/MS. The concentration-time profile was depicted in Figure 7A, and pharmacokinetic parameters were determined using non-compartmental modeling analysis with DAS 3.2.8 software and summarized in Table 2. After i.v. administration, the maximum plasma concentration (Cmax) was 43.02 μg/L. The area under the plasma concentration-time curve (AUC0-∞) value was 999.15μg/L/h. The half-life (t1/2) of 1b was 14.52±6.32 h. The relatively long t1/2 of 1b ensures that drugs captured by 1b are less prone to re-occurrence of toxicity or effects. The established LC-MS protocol also analyzed the tissue distribution of 1b in the liver and kidney. The result (Figure 7B) shows that the concentration of 1b in the kidney is higher than in the liver at all times. Meanwhile, the highest content was reached at 0.25 h in those two organs, hinting that 1b could be easily transferred into tissues.
Pharmacokinetics of 1b. (A) Plasma concentration-time plots of 1b after i.v. administration. (B) 1b concentrations in the liver and kidney at different time points.
Main pharmacokinetic parameters of 1b after i.v. administration
| Parameters | 1b |
|---|---|
| Tmax (h) | 10.04±4.80 |
| Cmax (μg/mL) | 43.02±15.04 |
| AUC0-t (μg/mL*h) | 900.08±297.87 |
| AUC0-∞ (μg/mL*h) | 999.15±272.92 |
| MRT0-t (h) | 16.03±0.41 |
| MRT0-∞ (h) | 22.27±4.96 |
| T1/2z (h) | 14.52±6.32 |
| CL (mL/h/g) | 0.09±0.02 |
In vitro relieving effects of 1b on TC-poisoning
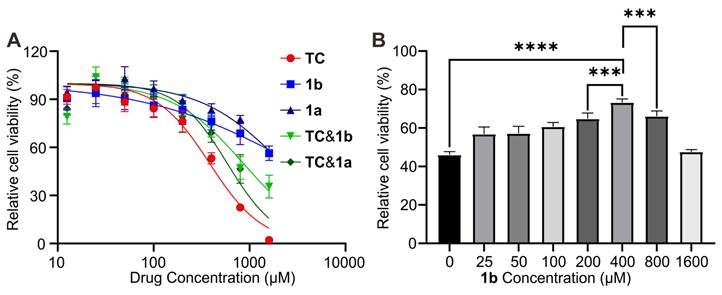
The capacity of 1b as a specific antidote for reversing TC toxicity was first investigated in vitro in Human myocytes AC16 cell, as TC is known to induce cardiac toxicity. The cytotoxicity of the drug was evaluated using the CCK-8 assay method. Figure 8A shows the cell viability after incubation of AC16 for 24 hours with these compounds at concentrations up to 1600 μM. The IC50 values were determined to be 383.7 μM, 2189 μM, and 2961 μM for TC, 1a, and 1b, respectively. Furthermore, when TC was mixed with 1a or 1b in a 1:1 ratio, the toxicity was effectively reduced, resulting in higher IC50 values of 586 μM and 862 μM for TC@1a and TC@1b, respectively. These findings provide evidence for the potential detoxification effect of the hosts on TC. We further investigated the impact of different dosages of 1b on reducing cellular toxicity with TC concentration fixed at 400 μM. The results demonstrated that the cell survival rate was the highest at a 1:1 ratio, indicating the optimal detoxification effect (Figure 8B).
In vitro relieving effects of 1b. (A) Cytotoxicity of TC, 1a, 1b, TC@1a, TC@1b against AC-16 cells after incubation for 24 h. (B). In vitro cytotoxic effect on AC16 cells after incubation for 24 h with fixed TC (400 μM) and different proportions of 1b at different concentrations. Data were represented as mean ± SD from six independent experiments.
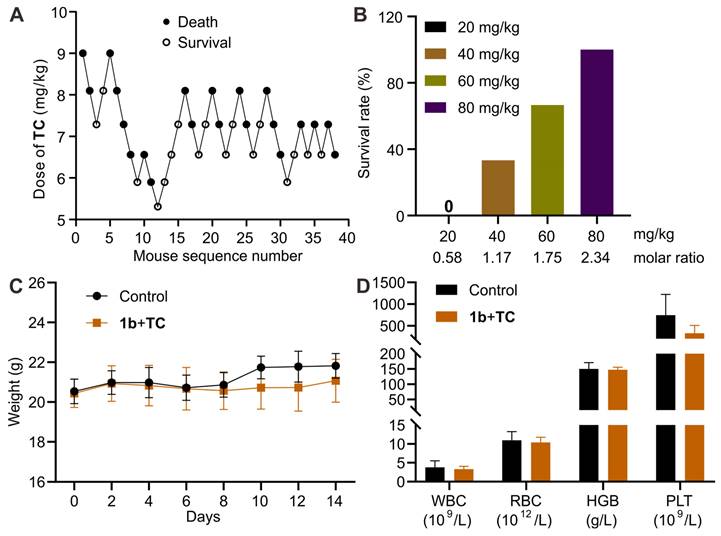
Improved survival in a lethal TC-poisoning mouse model
After confirming the detoxification effect of 1b in vitro, we performed experiments to assess its ability to reverse the TC toxicity in vivo. To select an appropriate dose of TC for this study, we first estimated its LD50 and LD95 using Dixon's up-and-down method (Figure 9A). The initial animal received a dose of 9.0 mg/kg. Subsequent dosages for the following animals were adjusted based on whether the preceding animal survived. Higher doses were administered if survival occurred and lower doses if the animal perished. Mortality rates were recorded for each dosage group. Probit analysis (Figure S21) was used to calculate the LD50 and LD95 values of TC as 6.9 mg/kg (95% CI, 6.1-7.5 mg/kg) and 8.7 mg/kg (95% CI, 7.9-12.9 mg/kg), respectively. Based on these findings, we chose the lethal dose of 9.0 mg/kg to evaluate the antidotal effects of 1b on TC-induced mortality. Mice received varying doses of 1b immediately after the i.v. administration of TC. As depicted in Figure 9B, the injection of 1b notably decreased TC-induced mortality, with all mice surviving at a 1b dose of 80 mg/kg (1b/TC = 2.34). This suggests that 1b can completely reverse TC-induced death when administered at a dose higher than 2.34 equiv. of TC. We then assessed the mice that survived after treatment with 1b following a lethal dose of TC. Their body weights remained similar to the control group during the 14-day follow-up period (Figure 9C), and their blood parameters indicated that values were comparable between the treatment and control groups (Figure 9D).
1b reversed TC-induced death in a mouse model. (A) Consecutive TC concentration following the Dixon up-and-down method for determining lethal dose. (B) Mice survival rates when treated with varying doses of 1b immediately after receiving a lethal dose of TC. (C) Changes in body weight following treatment with 1b after i.v administration of 9 mg/kg TC. (D) Hematological parameters of mice 14 days after treatment with 1b following the i.v. administration of a lethal dose of TC.
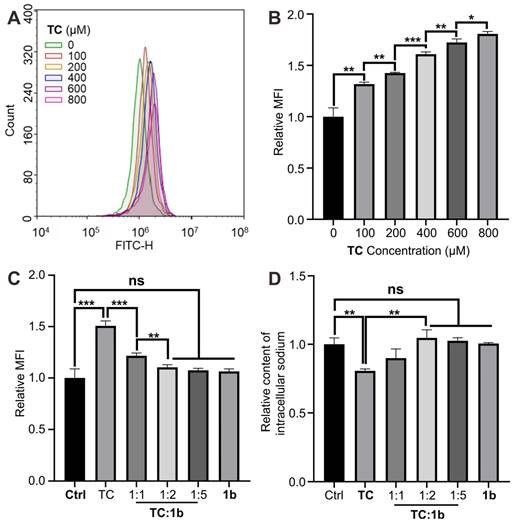
Reversal of TC-induced alterations in plasma membrane potential and restoration of blocked sodium channels in vitro
Voltage-gated Na+ channels are dynamic transmembrane proteins essential for initiating action potential upstrokes in excitable membranes. Local anesthetics selectively bind to specific sites within these channels, blocking Na+ currents and preventing normal depolarization [63]. This alters the plasma membrane potential, leading to conduction block and reducing excitability in neurons, cardiac tissue, and the central nervous system. To explore reversal mechanisms at the cellular level, we employed the Human myocytes AC16 cell line, on which Na+ channels are very easily blocked by local anesthesia. Using a potential-sensitive fluorescent probe, we measured relative plasma membrane potential changes, observing increased cellular fluorescence after the sodium channel was blocked (Figures 10A-10B). The results demonstrate a significant rise in mean fluorescence intensity during TC exposure. Co-incubation with TC and 1b at molar ratios of 1:1 and 1:2 respectively markedly reduced mean fluorescence intensity (MFI) compared to cells treated with TC alone, indicating that 1b reverses TC-induced plasma membrane potential changes at the cellular level and restored the normal depolarization of cells (Figure 10C). Further investigations revealed that 1b effectively restores TC's blocking of sodium ion channels (Figure 10D). Exposure to TC (400 µM) for 10 minutes resulted in approximately 20% intracellular sodium loss, which was fully reversed by co-incubation with TC and 1b (1:2). These results highlight that 1b restores Na+ channels blocked by TC. The cells return to normal action potential and excitation conduction.
1b reversed changes in plasma membrane potential and reduced intracellular sodium loss in vitro. (A and B) Typical flow cytometry curves (A) and quantitative analysis (B) for determining the plasma membrane potential of AC16 cells treated with TC for 10 minutes. (C) Quantitative analysis of the plasma membrane potential of AC16 cells treated with TC, 1b or co-incubated with TC and 1b at various molar ratios for 10 min. (D) Relative intracellular sodium content of AC16 cells treated with TC, 1b, or co-treated with TC and 1b at various molar ratios for 10 minutes. Data were represented as mean ± SD from three independent experiments.
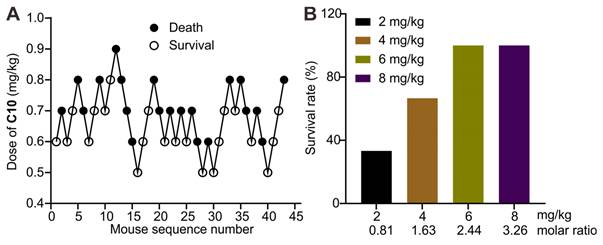
Improved survival in a lethal C10-poisoning mouse model
The application of 1b as a supramolecular sequestration agent for then conducted neuromuscular blocking agent C10. The evaluation of LD50 and LD95 of C10 in mice was performed to determine an appropriate dose for this preclinical study. Dose-mortality data for each mouse, obtained by the up-and-down method, are shown in Figure 11A. Probit analysis (Figure S22) yielded an LD50 of 0.69 mg/kg (95% CI, 0.65-0.75 mg/kg) and LD95 of 0.86 mg/kg (95% CI, 0.78-1.11). The lethal dose of 0.9 mg/kg was then used to evaluate the antidotal effects of synthetic receptors. Administering 1b at different doses immediately after the C10 injection revealed its potential to fully reverse C10-induced mortality at doses higher than 2.44 equiv. of C10 (Figure 11B). This effect is likely due to 1b's specific binding and sequestration of C10 in the blood and neuromuscular junctions, leading to the dissociation of C10 from the nicotinoyl acetylcholine receptor.
1b reversed C10-induced mortality in mice. (A) Consecutive C10 concentration following the Dixon up-and-down method for determining lethal dose. (B) Survival rates of mice treated with various doses of 1b immediately after the i.v. administration of a lethal dose of C10 (0.9 mg/kg).
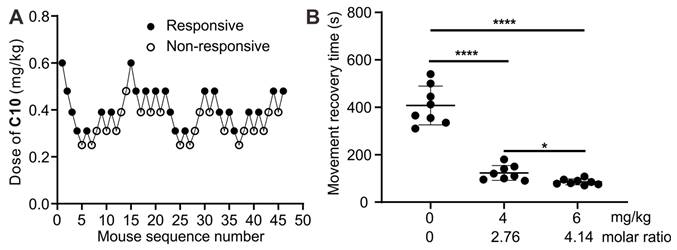
Reversal of paralysis in a mouse model
Subsequently, an assessment was conducted to investigate the potential of 1b to reverse the paralysis caused by C10 and expedite mobility restoration in a murine model. The half-effective dose (ED50) and 95% effective dose (ED95) of C10 for inducing muscle paralysis were determined using the up-and-down method (Figure 12A). According to probit analysis (Figure S23), the ED50 of C10 was 0.37 mg/kg (95% CI, 0.32-0.42 mg/kg), and the ED95 was 0.53 mg/kg (95% CI, 0.46-0.76 mg/kg). Therefore, we selected 0.53 mg/kg to assess the reversal effects of 1b on C10-induced paralysis. Immediately after i.v. administration of C10 to the mice, paralysis ensued, preventing movement on an accelerating rotor, necessitating prompt removal. Due to the in vivo metabolism of C10, the mice gradually regained mobility within minutes, allowing continuous movement on the rotor for over 30 seconds, similar to control mice. The average recovery time was approximately 402.8 seconds for mice given C10 (Figure 12b). In contrast, the average recovery time was reduced to 123.1 seconds and 85.1 seconds when mice were given 1b at doses of 4 mg/kg and 6 mg/kg, respectively, 30 seconds after the administration of C10. This observation was attributed to the complexation of C10 by 1b in the blood, thereby expediting neuromuscular recovery. These findings demonstrate the potential of 1b in reversing protracted paralysis induced by C10, especially in instances of misuse or overdose in clinical settings.
Reversal of paralysis in a mouse model. (A) Consecutive C10 concentration following the Dixon up-and-down method for determining effective dose. (B) movement recovery time of the mice when i.v. administered with 1b (4 mg/kg or 6 mg/kg) at 30 s after the administration of C10 (0.53 mg/kg).
Pharmacokinetic experiments
Furthermore, we conducted a quantitative analysis of C10 concentration in blood to elucidate the potential mechanism of action of reversal agent 1b. Sixty seconds after intravenous injection of C10, KM mice were randomly assigned to receive either saline or 1b. Blood samples were collected at predetermined intervals to determine the concentration of C10 using LC-MS. The plasma concentration-time profiles of C10 are depicted in Figure 13, and the key pharmacokinetic parameters are summarized in Table 3. The non-compartmental model was employed to characterize the pharmacokinetics of both formulations. C10 levels exhibited rapid decline during the initial 0.2 hours post-dose for both groups. After treatment with 1b, the maximum plasma concentration (Cmax) and the area under the plasma concentration-time curve (AUC0-∞) increased from 1305.40 ± 179.41 to 1633.02 ± 344.88 ng/mL, and from 293.66 ± 27.97 to 414.10 ± 80.89 ng/mL/h, respectively. Whereas, the total body clearance for the saline-treated Group and 1b-treated Group significantly decreased from 1.54 ± 0.16 to 1.12 ± 0.22 mL/h/kg. The pharmacokinetic findings generally indicate an increase in total C10 concentrations following administration of 1b. These observations are consistent with the pharmacokinetic model proposed for reversing neuromuscular blockade by Sugammadex [64, 65, 66]. Thus, the reversal of C10 by 1b follows a pharmacokinetic mechanism: 1b binds specifically and with high affinity to C10 through encapsulation in the plasma. Free C10 molecules in the plasma are sequestered by 1b, resulting in a rapid decrease in the concentration of free C10 in the plasma. This establishes a concentration gradient between free C10 in the neuromuscular junction and the plasma, causing free C10 molecules to migrate back to the plasma where they are encapsulated by 1b. Consequently, this process leads to a swift reversal of the C10 blockade.
Plasma pharmacokinetics of C10. C10 plasma concentration against time for both the saline- and 1b-treated groups.
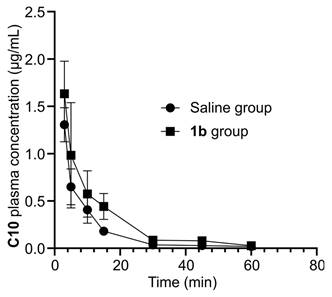
Main pharmacokinetic parameters of C10 for the saline- and 1b-treated groups
| Parameters | Saline-treated Group | 1b-treated Group |
|---|---|---|
| Tmax (h) | 0.05±0.00 | 0.05±0.00 |
| Cmax (ng/mL) | 1305.40±179.41 | 1633.02±344.88 |
| AUC0-t (ng/mL*h) | 284.16±26.43 | 406.02±77.15 |
| AUC0-∞ (ng/mL*h) | 293.66±27.97 | 414.10±80.89 |
| MRT0-t (h) | 0.11±0.02 | 0.15±0.02 |
| MRT0-∞ (h) | 0.16±0.04 | 0.17±0.03 |
| T1/2z (h) | 0.36±0.16 | 0.19±0.02 |
| Vz (mL/g) | 0.80±0.33 | 0.30±0.06 |
| CL (mL/h/g) | 1.54±0.16 | 1.12±0.22 |
Conclusions
In summary, we have investigated the molecular recognition properties of amide naphthotubes toward TC and C10 using a combination of ITC and DFT calculations. 1b possessed strong binding ability towards TC and C10 with Ka values of 1.89×107 M-1 and 1.01×107 M-1. The evaluation of the safety and biocompatibility of 1b in mice via i.v. administration suggests a good biocompatibility profile of 1b in vivo. Intravenous injection of 1b could contribute to reverse overdose TC or C10-induced mortality and reverse the NMB effect of C10 in mice models via direct host-guest encapsulation through a pharmacokinetics approach. Our systemic studies suggested that 1b may be used as an in vivo sequestrant for toxicant poisoning, offering a prospective clinical approach for mitigating the adverse effects of local anesthetics.
More generally, macrocycles with endo-functionalized cavities have several important implications: (a) This study marks the first comprehensive evaluation of the safety and biocompatibility of amide naphthotube when administered intravenously in mice. The favorable safety profile observed suggests that amide naphthotube holds promise for further research and potential clinical applications in the field of biomedicine. (b) Local anesthetics are commonly utilized for managing postoperative pain and treating various chronic pain conditions. However, the risk of local anesthetic systemic toxicity poses a potentially life-threatening concern as their use in healthcare settings grows. Our systemic studies indicate that supramolecular hosts have the potential to serve as supramolecular antidotes for mitigating the adverse effects of local anesthetic drugs, thus offering a promising clinical approach for the treatment of local anesthetic systemic toxicity. (c) Although decamethonium is no longer utilized in clinical practice, it was previously extensively employed and might be reintroduced with the availability of appropriate antagonists. (d) The development of host systems as sequestrants for more complex guests beyond hydrophobic cations and to withstand the effects of physiological salt and serum proteins remains a challenge in the field of supramolecular sequestrants [18]. The distinctive feature of amide naphthotube, which combines hydrogen-bonding sites within a hydrophobic cavity, suggests exceptionally strong binding not only with highly hydrophobic guest molecules but also potentially with hydrophilic molecules, thereby opening up new possibilities for supramolecular sequestration agents. In conclusion, we believe that this unique macrocyclic receptor enhances the repertoire of supramolecular sequestrants and holds promise for diverse future applications.
Supplementary Material
Supplementary figures, materials and methods, and tables.
Acknowledgements
This research was financially supported by National Natural Science Foundation of China (No. 22174059, 22201128 and 22301046), Natural Science Foundation of Hunan Province, China (No. 2022JJ40363 and 2022JJ40365), the Young Science and Technology Innovation Program of Hunan Province (No. 2022RC1230), the Excellent youth funding of Hunan Provincial Education Department (22B0460), and China Postdoctoral Science Foundation (No. 2022M721542).
Author contributions
L.P.Y., L.L.W. and Y.L.M. conceived and designed the experiments. C.D.Z. carried out most of the experimental work. W.J.C. contributed to host synthesis. S.M.W. performed the DFT calculations. H.Y. helped performing ITC experiments. K.L. performed the pharmacokinetic experiments supervised by W.C. L.P.Y., L.L.W., W.C. and Y.L.M. analyzed the data and wrote the manuscript, and all authors commented on it.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Heindel JJ, Blumberg B, Cave M, Machtinger R, Mantovani A. et al. Metabolism disrupting chemicals and metabolic disorders. Reprod Toxicol. 2017;68:3-33
2. Forray A, Sofuoglu M. Future pharmacological treatments for substance use disorders. Br J Clin Pharmacol. 2014;77:382-400
3. Andrae B, Bauer D, Gass P, Koller M, Worek F, Kubik S. Influence of cyclic and acyclic cucurbiturils on the degradation pathways of the chemical warfare agent VX. Org Biomol Chem. 2020;18:5218-27
4. Gorelick DA. Pharmacokinetic strategies for treatment of drug overdose and addiction. Future Med Chem. 2012;4:227-43
5. Escobar L, Ballester P. Molecular Recognition in Water Using Macrocyclic Synthetic Receptors. Chem Rev. 2021;121:2445-514
6. Zhou J, Rao L, Yu G, Cook TR, Chen X, Huang F. Supramolecular cancer nanotheranostics. Chem Soc Rev. 2021;50:2839-91
7. Zhou J, Yu G, Huang F. Supramolecular chemotherapy based on host-guest molecular recognition: a novel strategy in the battle against cancer with a bright future. Chem Soc Rev. 2017;46:7021-53
8. Song N, Lou XY, Ma L, Gao H, Yang YW. Supramolecular nanotheranostics based on pillarenes. Theranostics. 2019;9:3075-93
9. Yu G, Chen X. Host-guest chemistry in supramolecular theranostics. Theranostics. 2019;9:3041-74
10. Pan YC, Hu XY, Guo DS. Biomedical applications of calixarenes: state of the art and perspectives. Angew Chem Int Ed. 2021;60:2768-94
11. Zhou L, Cao S, Liu C, Zhang H, Zhao Y. Pillar[n]arene-based polymeric systems for biomedical applications. Coordin Chem Rev. 2023;491:215260
12. Zhu H, Li Q, Khalil-Cruz LE, Khashab NM, Yu G, Huang F. Pillararene-based supramolecular systems for theranostics and bioapplications. Sci China Chem. 2021;64:688-700
13. Ma X, Zhao Y. Biomedical applications of supramolecular systems based on host-guest interactions. Chem Rev. 2015;115:7794-839
14. Guo DS, Liu Y. Supramolecular Chemistry of p-sulfonatocalix[n]arenes and its biological applications. Acc Chem Res. 2014;47:1925-34
15. Yin H, Cheng Q, Bardelang D, Wang R. Challenges and opportunities of functionalized cucurbiturils for biomedical applications. JACS Au. 2023;3:2356-77
16. Yin H, Bardelang D, Wang R. Macrocycles and related hosts as supramolecular antidotes. Trends Chem. 2021;3:1-4
17. Yin H, Zhang X, Wei J, Lu S, Bardelang D, Wang R. Recent advances in supramolecular antidotes. Theranostics. 2021;11:1513-26
18. Deng CL, Murkli SL, Isaacs LD. Supramolecular hosts as in vivo sequestration agents for pharmaceuticals and toxins. Chem Soc Rev. 2020;49:7516-32
19. Macartney DH. Cucurbit[n]uril type hosts for the reversal of steroidal neuromuscular blocking agents. Future Med Chem. 2013;5:2075-89
20. Yu SB, Zhou W, Tian J, Ma D, Zhang DW, Li ZT. Host-guest chemistry for the design of drug antagonists. Sci China Chem. 2023;53:2345-56
21. Bom A, Bradley M, Cameron K, Clark J K, Egmond Jv, Feilden H. et al. A novel concept of reversing neuromuscular block: chemical encapsulation of rocuronium bromide by a cyclodextrin-based synthetic host. Angew Chem Int Ed Engl. 2002;41:265-70
22. Wang K, Guo DS, Zhang HQ, Li D, Zheng XL, Liu Y. Highly effective binding of viologens by p-sulfonatocalixarenes for the treatment of viologen poisoning. J Med Chem. 2009;52:6402-12
23. Ma D, Zhang B, Hoffmann U, Sundrup MG, Eikermann M, Isaacs L. Acyclic cucurbit[n]uril-type molecular containers bind neuromuscular blocking agents in vitro and reverse neuromuscular block in vivo. Angew Chem Int Ed. 2012;51:11358-62
24. Liu HK, Lin F, Yu SB, Wu Y, Lu S, Liu YY. et al. Highly Water-Soluble Cucurbit[8]uril Derivative as a BroadSpectrum Neuromuscular Block Reversal Agent. J Med Chem. 2022;65:16893-901
25. Zhang X, Xu X, Li S, Li L, Zhang J, Wang R. A Synthetic Receptor as a Specific Antidote for Paraquat Poisoning. Theranostics. 2019;9:633-45
26. Zhang X, Cheng Q, Li L, Shangguan L, Li C, Li S. et al. Supramolecular therapeutics to treat the side effects induced by a depolarizing neuromuscular blocking agent. Theranostics. 2019;9:3107-21
27. Chai Y, Chen L, Zhang Y, Zhao L, Meng Z, Chen J. et al. Reversing neuromuscular blocking agent decamethonium by carboxylatopillar[6]arene based on host-guest encapsulation. Chin Chem Lett. 2022;33:3003-6
28. Brockett AT, Xue W, King D, Deng CL, Zhai C, Shuster M. et al. Pillar[6]MaxQ: A potent supramolecular host for in vivo sequestration of methamphetamine and fentanyl. Chem. 2023;9:881-900
29. Zhao Y, Chen L, Chen J, Li J, Meng Q, Sue ACH. et al. Water-soluble terphen[3]arene macrocycle: a versatile reversal agent of neuromuscular blockers. Chem Commun. 2023;59:5858-61
30. Becker DE, Reed KL, Local anesthetics. review of pharmacological considerations. Anesth Prog. 2012;59:90-102
31. Gan TJ. Poorly controlled postoperative pain: prevalence, consequences, and prevention. J Pain Res. 2017;10:2287-98
32. O'Connor AB, Dworkin RH, Treatment of neuropathic pain. an overview of recent guidelines. Am J Med. 2009;122:S22-32
33. Pardo M, Miller RD. Basics of Anesthesia E-Book. Elsevier Health Sciences. 2017
34. Manna L, Gaudiano MC, Bartolomei M, Valvo L, Bertocchi P, Antoniella E. et al. A special case of medicine in disguise: Tattoo inks containing anaesthetics. Talanta. 2019;198:337-43
35. Kudoa K, Hinoa Y, Ikedaa N, Inoueb H, Takahashi S. Blood concentrations of tetracaine and its metabolite following spinal anesthesia. Forensic Sci Int. 2001;116:9-14
36. Hino Y, Kudo K, Kiyoshima A, Ikeda N. A sudden death following tetracaine-induced spinal anesthesia. Leg Med (Tokyo). 2002;4:55-9
37. Liu WQ, Bai R, Ma CL, Feng Y, Xie B, Dong MJ. et al. Metabolomics changes of serum and tissues in mice died of acute tetracaine poisoning. J Foren Med. 2021;37:166-74
38. Sun H, Wang A, Liu Z, Zhang X, Chang J. Advances of Tetracaine in Forensic to Toxicological Analysis. Forensic Science and Technology. in press, doi: 10.16467/j.1008-3650. 2023 0087
39. Wolfe RC, Spillars A. Local anesthetic systemic toxicity: reviewing updates from the American society of regional anesthesia and pain medicine practice advisory. J Perianesth Nurs. 2018;33:1000-5
40. Chen R, Sun Y, Li Y, Dou X, Dai M, Sun S. et al. Neuromuscular blocking agents and cancer: a narrative review. J Clin Pharm Ther. 2023;2023:5607134
41. Ball C, Westhorpe R. Muscle Relaxants-Decamethonium. Anaesth Intensive Care. 2005;33:709
42. Goswami LN, Olds TJ, Monk TG, Johnson QL, Dilger JP, Shanawaz MA. et al. Isomeric Carborane Neuromuscular Blocking Agents. ChemMedChem. 2019;14:1108-14
43. Brown TCK. Aspects of depolarizing neuromuscular blockers: decamethonium and suxamethonium. Paediatr Anaesth. 2013;23:868-70
44. Yang LP, Wang X, Yao H, Jiang W. Naphthotubes: macrocyclic hosts with a biomimetic cavity feature. Acc Chem Res. 2020;53:198-208
45. Shorthill BJ, Avetta CT, Glass TE. Shape-Selective Sensing of Lipids in Aqueous Solution by a Designed Fluorescent Molecular Tube. J Am Chem Soc. 2004;126:12732-33
46. Huang GB, Wang SH, Ke H, Yang LP, Jiang W. Selective recognition of highly hydrophilic molecules in water by endo-functionalized molecular tubes. J Am Chem Soc. 2016;138:14550-53
47. Yao H, Ke H, Zhang X, Pan SJ, Li MS, Yang LP. et al. Molecular Recognition of Hydrophilic Molecules in Water by Combining the Hydrophobic Effect with Hydrogen Bonding. J Am Chem Soc. 2018;140:13466-77
48. Zhao CD, Yao H, Li SY, Wang LL, Yang LP. Amide naphthotubes: Biomimetic macrocycles for selective molecular recognition. Chin. Chem. Lett. 2024;35:108879
49. Ma YL, Sun C, Li Z, Wang Z, Wei J, Cheng Q. et al. Biomimetic Recognition-Based Bioorthogonal Host-Guest Pairs for Cell Targeting and Tissue Imaging in Living Animals. CCS Chem. 2022;4:1977-89
50. Ma YL, Quan M, Lin XL, Cheng Q, Yao H, Yang XR. et al. Biomimetic Recognition of Organic Drug Molecules in Water by Amide Naphthotubes. CCS Chem. 2020;2:1078-92
51. Li MS, Dong YW, Pang XY, Chai H, Wang X, Jiang W. The Influence of Small Biomolecules, Salts and Buffers on the Molecular Recognition of Amide Naphthotube in Aqueous Solutions. Chem Eur J. 2023;29:e202202972
52. Ke H, Yang LP, Xie M, Chen Z, Yao H, Jiang W. Shear-induced Assembly of a Transient yet Highly Stretchable Hydrogel Based on Pseudopolyrotaxanes. Nat Chem. 2019;11:470-77
53. Chai JD, Head-Gordon M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys Chem Chem Phys. 2008;10:6615-20
54. Weigend F, Ahlrichs R, Balanced basis sets of split valence, triple zeta valence, quadruple zeta valence quality for H to Rn. Design and assessment of accuracy. Phys Chem Chem Phys. 2005;7:3297-305
55. Lu T, Chen Q. Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems. J Comput Chem. 2022;43:539-55
56. Lu T, Chen F. Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem. 2012;33:580-92
57. Cremer PS, Flood AH, Gibb BC, Mobley DL. Collaborative routes to clarifying the murky waters of aqueous supramolecular chemistry. Nat Chem. 2018;10:8-16
58. Biedermann F, Nau WM, Schneider HJ. The Hydrophobic Effect Revisited-Studies with Supramolecular Complexes Imply High-Energy Water as a Noncovalent Driving Force. Angew Chem Int Ed. 2014;53:11158-71
59. de Lima RAF, de Jesus MB, Cereda CMS, Tofoli GR, Cabeça LF, Mazzaro I. et al. Improvement of tetracaine antinociceptive effect by inclusion in cyclodextrins. J Drug Target. 2012;20:85-96
60. Li Q, Guo DS, Qian H, Liu Y. Complexation of p-Sulfonatocalixarenes with Local Anaesthetics Guests: Binding Structures, Stabilities, and Thermodynamic Origins. Eur J Org Chem. 2012:3962-71
61. Wyman IW, Macartney DH. Host-guest complexations of local anaesthetics by cucurbit[7]uril in aqueous solution. Org Biomol Chem. 2010;8:247-52
62. Krebs HA. Chemical Composition of Blood Plasma and Serum. Annu Rev Biochem. 1950;19:409-30
63. Körner J, Albani S, Eswaran VSB, Roehl AB, Rossetti G, Lampert A. Sodium Channels and Local Anesthetics-Old Friends With New Perspectives. Front Pharmacol. 2022;13:837088
64. Epemolu O, Bom A, Hope F, Mason R, Cert HN. Reversal of Neuromuscular Blockade and Simultaneous Increase in Plasma Rocuronium Concentration after the Intravenous Infusion of the Novel Reversal Agent Org25969. Anesthesiology. 2003;99:632-7
65. Sparr HJ, Vermeyen KM, Beaufort AM, Rietbergen H, Proost JH, Saldien V, Velik-Salchner C, Wierda JM. Early reversal of profound rocuronium-induced neuromuscular blockade by sugammadex in a randomized multicenter study: Efficacy, safety, and pharmacokinetics. Anesthesiology. 2007;106:935-43
66. Ploeger BA, Smeets J, Strougo A, Drenth HJ, Ruigt G, Houwing N, Danhof M. Pharmacokinetic-Pharmacodynamic Model for the Reversal of Neuromuscular Blockade by Sugammadex. Anesthesiology. 2009;110:95-105
Author contact
![]() Corresponding authors: Wei Cai, Yan-Long Ma, Li-Li Wang or Liu-Pan Yang. Email: 20120941161edu.cn (Wei Cai), mayanlong202106edu.cn (Yan-Long Ma), wanglledu.cn (Li-Li Wang), yanglpedu.cn (Liu-Pan Yang).
Corresponding authors: Wei Cai, Yan-Long Ma, Li-Li Wang or Liu-Pan Yang. Email: 20120941161edu.cn (Wei Cai), mayanlong202106edu.cn (Yan-Long Ma), wanglledu.cn (Li-Li Wang), yanglpedu.cn (Liu-Pan Yang).