Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2024; 14(12):4555-4569. doi:10.7150/thno.96322 This issue Cite
Research Paper
Impact of TP53 loss-of-function alterations on the response to PSMA radioligand therapy in metastatic castration-resistant prostate cancer patients
Peter H.J. Slootbeek1, María Victoria Luna-Velez2, Bastiaan M. Privé3, Maarten J. van der Doelen2, Iris S.H. Kloots1, Samhita Pamidimarri Naga1, Hilde E. Onstenk1, James Nagarajah3,4, Harm Westdorp1, Inge M. van Oort2, Leonie I. Kroeze5, Jack. A. Schalken2, Haiko J. Bloemendal1, Niven Mehra1, ![]()
1. Department of Medical Oncology, Radboud university medical center, Nijmegen, The Netherlands.
2. Department of Urology, Radboud university medical center, Nijmegen, The Netherlands.
3. Department of Nuclear Medicine, Radboud university medical center, Nijmegen, The Netherlands.
4. Roentgeninstitut Duesseldorf, Duesseldorf, Germany.
5. Department of Pathology, Radboud university medical center, Nijmegen, The Netherlands.
Received 2024-2-1; Accepted 2024-7-11; Published 2024-8-1
Abstract

Rationale: PSMA-targeting radioligand therapy (PSMA-RLT) has shown promise in metastatic castration-resistant prostate cancer (mCRPC), particularly in PSMA-avid tumours. However, predicting response remains challenging. Preclinical data suggests aberrant p53-signalling as a predictor of poor response.
Methods: The patient population of this pre-planned retrospective cohort study consists of 96 patients with mCRPC who underwent treatment with PSMA-RLT and were molecularly profiled by whole-genome sequencing and or targeted next-generation sequencing. Response to PSMA-RLT was assessed per molecular subtype, including TP53-mutational status.
Results: Patients with TP53 loss-of-function alterations had a shorter median progression-free survival (3.7 versus 6.2 months, P<0.001), a lower median PSA change (-55% vs. -75%, P=0.012) and shorter overall survival from initiation of PMSA-RLT (7.6 vs. 13.9 months, P=0.003) compared to TP53-wildtype patients. Pathogenic alterations in AR, MYC, BRCA1, or BRCA2 as well as in genes linked to the PI3K or MAPK pathways or genes involved in homologous recombination repair, were not associated with response. Only lactate dehydrogenase was, alongside TP53-status, significantly associated with response. Transcriptome analysis of 21 patients, identified six p53 signalling genes whose low expression was associated to a shorter progression-free survival (P<0.05).
Conclusion: TP53 loss-of-function may serve as a prognostic factor for PSMA-RLT outcomes in patients with mCRPC.
Keywords: Castration-resistant prostate cancer, Precision oncology, TP53, Theranostics, Radioligand therapy
Introduction
Although the armamentarium for metastatic castration-resistant prostate cancer (mCRPC) has substantially expanded over the last decade, patients almost inevitably progress on all registered treatment lines, resulting in a median life expectancy of less than three years [1-3]. In the search to further broaden the treatment options of mCRPC patients, radioligand therapy (RLT) has gained momentum. The most common cell-surface protein used to guide radiopharmaceuticals towards prostate cancer cells is the prostate specific membrane antigen (PSMA) [4, 5]. As PSMA is overexpressed in prostate cancer cells compared to benign tissue, the therapeutical radiation dose is accumulated at the tumour site, limiting radiation damage to non-PSMA-expressing tissues and reducing damage to healthy tissues.
PSMA ligands can be labelled with radioisotopes such as the beta-emitter lutetium-177 (177Lu) or the alpha-emitter actinium-225 (225Ac) [6, 7]. The VISION trial led to the EMA and FDA approval of 177Lu-PSMA post-taxane, based on improved progression-free survival (PFS) and overall survival (OS) and while the final results of the PSMAfore study are pending, approval of 177Lu-PSMA for taxane-naive patients is anticipated, as the trial presented a significantly prolonged PFS [8, 9]. 225Ac-PSMA has not reached the phase 3 trial stage, but several phase 2 trials are currently ongoing (NCT03276572, NCT04506567, NCT05219500, NCT04597411). Tandem therapy with 177Lu-PSMA and 225Ac-PSMA has shown promising results, even after progression on single-agent 177Lu-PSMA. This is currently investigated in a phase 2 trial (NCT04886986) [10, 11].
For newly approved therapies in an all-comer population, such as PSMA-RLT, an unmet need is the identification of biomarkers that guide physicians to select responsive patients more optimally. As PSMA avidity strongly influences response, the landmark papers of LuPSMA, TheraP and VISION excluded patients with PSMA low or negative lesions based on relative uptake compared to the liver, a threshold maximum standardised uptake value (SUVmax) per lesion or mismatch with FDG-PET [8, 12-14]. Yet, post-hoc analyses of the VISION and TheraP showed that there are still many responders with intermediate PSMA uptake [15, 16]. Hence, exploring additional biomarkers is warranted.
Preclinical evidence supports p53 (encoded by TP53) signalling as an important biomarker candidate. In a study by Stuparu et al., global proteomics and phosphoproteomics were used to investigate the molecular changes induced by PSMA-RLT in mice [17]. Transcription factor enrichment analysis revealed that p53 was the most upregulated transcription factor post 177Lu-PSMA RLT and the third most upregulated post 225Ac-PSMA RLT. Additionally, kinase-substrate enrichment analysis showed increased activity of ATM and ATR in mice treated with RLT, and increased activity of CHK2 (encoded by Chek2) in 177Lu-PSMA treated mice. Interestingly, ATM, ATR and CHK2 are all involved in the stabilization and activation of p53 in response to ionizing radiation [18]. To further confirm these findings, the authors assessed the impact of Tp53 status on PSMA-RLT responsiveness in mice. They found that PSMA-RLT was effective in mice with wild-type Tp53 tumours but much less in mice with Tp53 knock-out tumours, with no significant reduction in tumour growth compared to untreated mice. From literature evaluating the genetic background of mCRPC patients treated with PSMA-RLT, TP53 status could not be evidently validated as a biomarker associated with response [19-21].
In this pre-planned retrospective cohort study, we hypothesised that mCRPC patients with loss-of-function alterations in TP53 would respond worse to PSMA-RLT when compared to patients with wild-type TP53. To test this hypothesis, we performed a comprehensive molecular characterization of 96 patients with mCRPC and evaluated the response to PSMA-RLT per molecular subtype, including TP53-mutational status. Lastly, transcriptome analysis was performed to identify signalling pathways and constituent genes associated to biochemical progression and the loss of p53 signalling.
Methods
Patient population and study design
The patient population of this pre-defined retrospective cohort study consisted of all patients known in the outpatient clinics of Medical Oncology or Nuclear Medicine at the Radboudumc, treated with 177Lu-PSMA or 225Ac-PSMA, from January 1, 2016, to May 1, 2023. Follow-up data were collected until November 1, 2023. Eligible patients previously underwent next-generation sequencing of tumour tissue (fresh or archived) or had residual tumour tissue from earlier biopsies. Different consents were allowed to be included in this study, all specified in study protocol, evaluated by the Medical Review Ethics Committee Oost-Nederland, The Netherlands (CMO-2022-16040). The study population in part overlaps with the study populations of previous publications from our centre with different research questions [22, 23].
The pre-planned primary research objective was to compare PFS on PSMA-RLT between patients with pathogenic TP53 alterations and patients without pathogenic TP53 alterations. The secondary endpoints were PSA response and overall survival per TP53 status. Patients were classified as TP53 mutated (TP53m) if they had a bi-allelic loss of TP53, a relevant splice-site mutation, a mutation in TP53 with a truncating effect or a missense mutation with a non-functional transcriptional activity according to The TP53 Database (R20, July 2019): https://tp53.isb-cgc.org [24]. Patients with non-deleterious alterations in TP53 or mutations with a partially functional transcriptional activity were included in the TP53 wild-type (TP53wt) subgroup.
PFS on PSMA-RLT was defined as the time from first administration of PSMA-RLT until radiologic or clinical progression including death or censoring at end of follow-up if treatment was still ongoing. PSA responses were assessed as maximal decline according to the Prostate Cancer Clinical Trials Working Group (PCWG3) criteria and dichotomised by ≥50% PSA decline (PSA50) [25]. Biochemical PFS was defined as the time from first administration of PSMA-RLT until ≥25% PSA increase from the nadir or baseline if PSA did not decline, censoring at next-systemic therapy, end of follow-up or death.
Molecular analysis
All patients underwent targeted or whole-genome sequencing (WGS) on primary or metastatic tissue by a non-profit institute (Hartwig Medical Foundation; WGS), by a fee for service provider (Foundation Medicine; Foundation One CDx) and/or in-house using a commercially available targeted sequencing panel containing 523 cancer-related genes (Illumina; True Sight Oncology 500) [26]. To compare the relative impact of TP53 loss-of-function alterations to presumed hyperactivation of canonical oncogenic pathways (AR, PI3K, MAPK, MYC) or impairment of homologous recombination repair (HRRm), all patients were sequenced for at least the following genes: TP53, AR, RB1, PTEN, AKT1, AKT2, AKT3, PIK3CA, PIK3CB, PIK3R1, BRAF, MAP2K1, MAP2K2, MAP2K4, MAP3K1, MYC, ATM, BARD1, BRCA1, BRCA2, BRIP1, CDK12, CHEK1, CHEK2, FANCA, FANCL, NBN, PALB2, PPP2R2A, RAD51B, RAD51C, RAD51D, RAD54L. To ensure consistency in pathogenicity reporting, all external sequencing reports were re-assessed based on guidelines from the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [27, 28]. Genes with six copies or more according to the reporting service or calculated inhouse were considered amplified [26]. Genes with no copies were reported as loss.
Transcriptome analysis
The transcriptional activity of p53 was explored with a gene-set enrichment analysis (GSEA). DESeq2 (v1.38.3) was used to normalise and log2-transform the raw count data from RNA sequencing of 21 mCRPC patients treated with PSMA-RLT [29]. For the enrichment analysis, patients were divided into groups to calculate fold change transcript expression between patients with and without a PSA50 and between TP53m and TP53wt. Log2 fold change values were calculated with the R package apeglm (v1.14.0) with the adaptive shrinkage estimator “ashr”, and used as input for the GSEA [30]. GSEA was performed using the molecular signatures database (MSigDB) hallmark gene set collection (v7.5.1) with the fgsea R package (v1.27.0) [31, 32]. Expression heatmaps of the normalised, log2-transformed data were created with ComplexHeatmap (v2.10.0) [33].
Statistical analyses
Time-to-event data were compared using Cox proportionate hazard models and visualised with Kaplan-Meier curves. Multivariable Cox proportionate hazard models were used to assess the impact of different molecular subgroups simultaneously and to test the impact of TP53m status on response relative to the line of therapy, time from androgen deprivation to mCRPC, type of isotope used, and the baseline laboratory values: prostate specific antigen (PSA), lactate dehydrogenase (LDH), haemoglobin (HB), alkaline phosphate (ALP). The proportional hazards assumption was tested with the Schoenfeld Test. To investigate the impact of single genes within the TP53 signature from the GSEA on PFS, the median value of the normalised and log2-transformed expression of each gene was used to separate patients into two groups, 50% highest and 50% lowest expression, which were compared using a log-rank test. To compare the baseline characteristics and biochemical outcomes of the subgroups, categorical variables were analysed using the Pearson Chi-Square or Fisher's Exact Test. Continuous variables were assessed using the nonparametric Mann-Whitney U test. All statistical tests were two-sided, with P values <0.05 considered statistically significant. All statistical tests and data visualization were performed in R (version 4.1.3) with RStudio (version 2022.02.1). A statistician was consulted during the analyses.
Results
Patient cohort
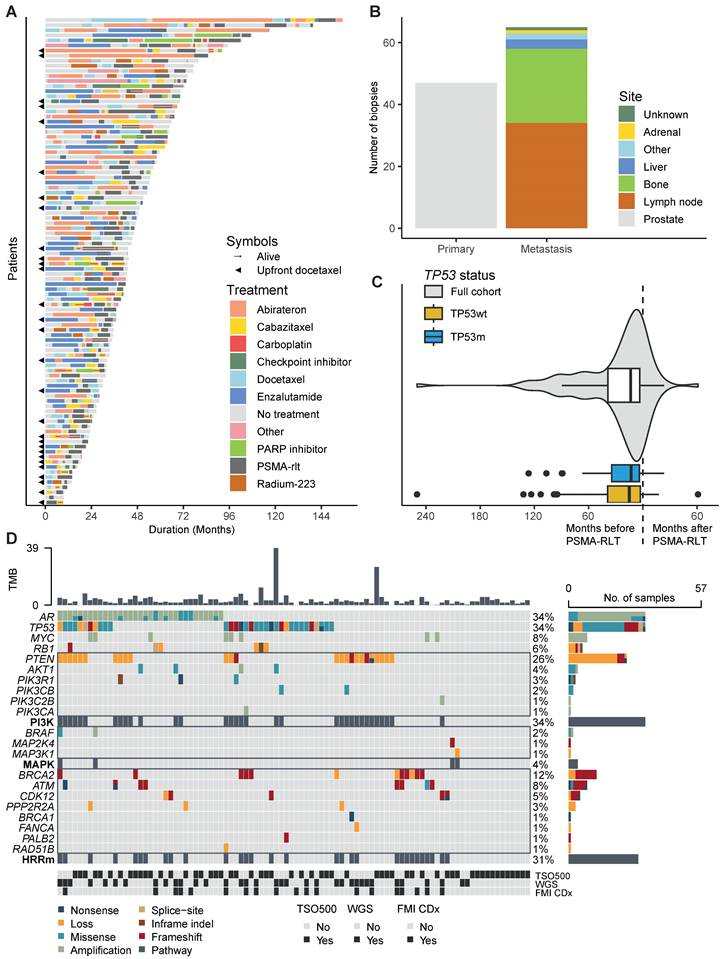
In total, 96 mCRPC patients were included in the study population. Patients were treated with a median of four systemic treatment lines for mCRPC before start of PSMA-RLT. The complete therapy sequence from mCRPC until last follow-up for each patient is presented in Figure 1A. Among the 96 patients, we analysed 112 tumour samples. Almost half of the samples were prostate tissue (42%), 30% were tissue from lymph nodes and 21% from bone (Figure 1B). The median time between obtaining the tissue and initiation of PSMA-RLT was 13.5 months (interquartile range 3.4 - 38.6, Figure 1C). Thirteen biopsies were taken after initiation of PSMA-RLT. The most frequently pathogenically altered genes were AR (34%) and TP53 (34%), followed by PTEN (26%) and BRCA2 (12%) (Figure 1D). Baseline characteristics did not significantly differ between the 33 patients (34%) in the TP53m subgroups and 63 patients (66%) in the TP53wt subgroup (Table 1). Sixty-seven patients received 177Lu-PSMA as single-agent, seven patients received 225Ac-PSMA as single-agent, and thirteen patients received tandem therapy with 177Lu-PSMA and 225Ac-PSMA.
A. Swimmerplot presenting the order and duration of systemic life-prolonging therapies for castration-resistant prostate cancer until death or last follow-up. The colour scheme represents therapies, and the symbols indicate if patients received upfront docetaxel or were alive at last follow-up. B. Barchart showing the sites from which biopsies were taken. C. Violin- and boxplots showing the timing of biopsies relative to the initiation of PSMA-RLT. D. Oncoplot presenting the genetic aberrations. The colour of the boxes represents the effect of the alteration, sorted by pathway. The tumour mutational burden (TMB) is presented at the top and at the bottom the different sequencing methods are presented. Abbreviations: FMI CDx, FoundationOne® companion diagnostic; HRRm, homologous recombination repair mutated (including loss); PSMA-RLT, prostate-specific membrane antigen-targeting radioligand therapy; TSO500, TruSight Oncology 500; WGS, whole genome sequencing.
Diagnostic, baseline, and treatment variables of the study population
| Variable | N | Missing | All | TP53wt | TP53m | P-value |
|---|---|---|---|---|---|---|
| Number of patients (valid %) or Median [interquartile range] | ||||||
| Diagnostic variables | ||||||
| ISUP-GGS | 95 | 1 | 0.733 | |||
| 1 | 9 (9.5) | 7 (11.3) | 2 (6.1) | |||
| 2 | 13 (13.7) | 8 (12.9) | 5 (15.2) | |||
| 3 | 10 (10.5) | 6 (9.7) | 4 (12.1) | |||
| 4 | 23 (24.2) | 17 (27.4) | 6 (18.2) | |||
| 5 | 40 (42.1) | 24 (38.7) | 16 (48.5) | |||
| Metastatic at diagnosis | 95 | 1 | 0.832 | |||
| No | 50 (52.6) | 32 (51.6) | 18 (54.6) | |||
| Yes | 45 (47.4) | 30 (48.4) | 15 (46.5) | |||
| Initial PSA level (µg/L) | 83 | 13 | 41.00 [10.8 - 131.0] | 48.00 [21.4 - 142.0] | 16.75 [8.5 - 86.5] | 0.053 |
| Age at initial diagnosis, years | 96 | 0 | 61.8 [56.2 - 67.0] | 61.6 [55.3 - 67.0] | 63.3 [58.5 - 66.2] | 0.287 |
| Age at CRPC, years | 96 | 0 | 66.3 [60.4 - 72.2] | 66.1 [60.3 - 72.1] | 66.3 [62.7 - 73.1] | 0.459 |
| Time to CRPC, months | 96 | 0 | 15.5 [10.0 - 30.0] | 16.0 [10.5 - 29.0] | 14.0 [9.7 - 31.7] | 0.948 |
| At start of PSMA-RLT | ||||||
| Line of therapy for CRPC | 96 | 0 | 4 [3 - 5] | 4 [3 - 5] | 4 [3 - 5] | 0.903 |
| PSMA-PET characteristics | ||||||
| SUVmax | 80 | 16 | 55.1 [29.8 - 73.6] | 57.6 [30.6 - 71.1] | 54.1 [29.6 - 84.8] | 0.904 |
| Bone metastases | 93 | 3 | 85 (91.4) | 54 (90.0) | 31 (93.9) | 0.707 |
| bone only | 20 (21.5) | 15 (25.0) | 5 (15.2) | |||
| Visceral metastases | 93 | 3 | 27 (29.0) | 18 (30.0) | 9 (27.3) | 0.816 |
| Laboratory variables | ||||||
| PSA (µg/L) | 94 | 2 | 233.0 [75.6 - 551.5] | 270.00 [79.6 - 794.7] | 163.76 [59.3 - 528.5] | 0.280 |
| ALP (U/L) | 89 | 7 | 138.0 [90.0 - 291.0] | 140.5 [91.8 - 310.3] | 136.0 [91.5 - 266.0] | 0.711 |
| LDH (U/L) | 87 | 9 | 258.0 [211.0 - 355.2] | 249.5 [207.0 - 349.3] | 272.0 [219.5 - 394.5] | 0.338 |
| HB (mmol/L) | 85 | 11 | 7.4 [6.5 - 8.2] | 7.3 [6.5 - 8.2] | 7.7 [6.5 - 8.2] | 0.887 |
| PSMA-RLT | ||||||
| Type of radioligand | 96 | 0 | 0.279 | |||
| 177Lutetium | 76 (79.2) | 52 (82.5) | 24 (72.7) | |||
| 225Actinium | 7 (7.3) | 5 (7.9) | 2 (6.1) | |||
| Tandem | 13 (13.5) | 6 (9.5) | 7 (21.2) | |||
| Cycles of PSMA-RLT | 96 | 0 | 3 [2 - 5] | 4 [2 - 6] | 3 [2 - 3] | 0.028 |
| ≥4 cycles | 42 (43.8) | 34 (54.0) | 8 (24.2) | |||
| Concurrent therapy | 96 | 0 | 0.006 | |||
| Enzalutamide | 10 (10.42) | 2 (3.17) | 8 (24.24) | |||
| Abiraterone | 7 (7.29) | 5 (7.94) | 2 (6.06) | |||
| None | 79 (82.29) | 56 (88.89) | 23 (69.70) | |||
P-values in bold are considered significant. Abbreviations: ALP, alkaline phosphatase; CRPC, castration-resistant prostate cancer; HB, haemoglobin; ISUP-GGS, International Society of Urological Pathology Gleason grading system; LDH, lactate dehydrogenase; PSA, prostate specific antigen; PSMA-RLT, prostate-specific membrane antigen-targeting radioligand therapy; SUVmax, maximal standardised uptake value; TP53m, TP53 mutated; TP53wt, TP53 wildtype.
TP53m patients received less cycles of PSMA-RLT compared to TP53wt patients (median 3 versus 4 cycles, P=0.028; Supplementary Table 1). Only one in four TP53m patients received four or more cycles (Table 1). In total, 89 patients received 177Lu-PSMA with a median total activity of 22.2GBq; 16.5GBq for the TP53m subgroups and 24.0GBq for the TP53wt subgroup (P=0.110). The median total activity for the 20 patients receiving 225Ac-PSMA was 14.3MBq; 8.0MBq for the TP53m patients and 20.0MBq the TP53wt patients (P=0.025).
Progression-free survival
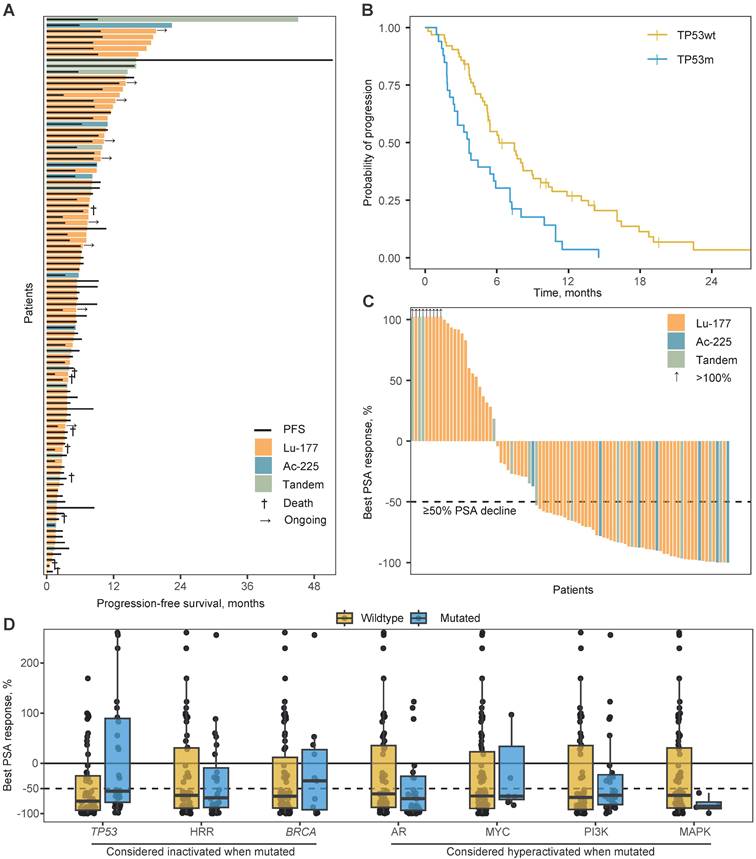
The median PFS on PSMA-RLT for the total population was 5.4 months (95% confidence interval [CI] 4.8 - 7.5) and was not impacted by type of RLT (P=0.432; Figure 2A). The TP53m subgroup had a significantly shorter PFS when compared to the TP53wt subgroup (median 3.7 versus 6.2 months; hazard ratio [HR] 2.2, 95%CI 1.4 - 3.5; P<0.001; Figure 2B). The hazard ratio for 177Lu-treated patients was 2.3 (95%CI 1.4 - 3.7; P<0.001) and for 255Ac-treated patients 2.0 (95%CI 0.7 - 5.4; P=0.177). HRRm, deleterious alterations in BRCA1 or BRCA2 specifically (BRCAm), as well as activating alterations in AR, MYC or key genes in the PI3K and MAPK pathway, were univariably not associated with PFS on PSMA-RLT (Table 2). In a multivariable analysis, only TP53-status was significantly associated with PFS (Table 2). Even when corrected for prognostic factors or possible confounders, TP53 status remained significantly associated with PFS (P=0.005). Only baseline LDH level was also significantly associated (P=0.001) with PFS (Supplementary Table 2). Notably, SUVmax did not show a significant association with PFS (P=0.703).
A. Swimmerplot presenting time on treatment per type of radioligand by coloured bars. The black lines indicate radiographic or clinical progression-free survival. B. Kaplan-Meijer curves for the progression-free survival per TP53-mutational status. C. Waterfallplot presenting the best prostate specific antigen (PSA) response from baseline per type of radioligand. D. Boxplot with individual points presenting the best PSA response per mutational status for canonical oncogenic or tumour suppressive pathways. Per boxplot: center line, median; box limits, upper and lower quartiles; from box to largest and smallest point within box + 1.5x interquartile range. Abbreviations: Ac-225, actinium-225; HHR, homologous recombination repair; Lu-177, lutetium-177; PSA, prostate specific antigen.
Univariable and multivariable analysis of potential prognostic molecular subgroups for progression-free survival on PSMA-RLT.
| Molecular subgroup | Effect of alteration | Univariable analysis | Multivariable analysis | |||
|---|---|---|---|---|---|---|
| HR [95%CI] | P-value | HR [95%CI] | P-value | |||
| TP53 | Inactivation | 2.21 [1.40-3.49] | <0.001 | 2.53 [1.52-4.22] | <0.001 | |
| AR | Hyperactivation | 0.86 [0.70-1.06] | 0.155 | 0.85 [0.69-1.05] | 0.130 | |
| MYC | Hyperactivation | 1.18 [0.57-2.45] | 0.661 | 0.61 [0.26-1.45] | 0.263 | |
| PI3K | Hyperactivation | 1.13 [0.72-1.78] | 0.597 | 1.13 [0.70-1.82] | 0.628 | |
| BRCA1/2 | Inactivation | 1.04 [0.57-1.88] | 0.905 | 0.90 [0.43-1.89] | 0.776 | |
| HRR | Inactivation | 0.96 [0.62-1.49] | 0.847 | 1.07 [0.61-1.88] | 0.811 | |
| MAPK | Hyperactivation | 1.05 [0.38-2.89] | 0.921 | 0.99 [0.33-3.02] | 0.987 | |
P-values in bold are considered significant. Abbreviations: CI, confidence interval; HR, hazard ratio; HRR, homologous recombination repair.
Exploratory analyses for progression-free survival
Patients with a molecular signature of aggressive variant prostate cancer (AVPC, n = 12), comprised of loss-of-function alterations in at least two of the three genes: TP53, RB1, PTEN, had a shorter PFS on PSMA-RLT (HR 1.8; 95%CI 1.0 - 3.4; Supplementary Figure 1). However, with a lower hazard ratio as TP53-status alone, suggesting TP53 loss-of-function drives the poor response on PSMA-RLT in AVPC patients, especially since all AVPC patients were also TP53m. By combining TP53-status with loss-of-function alterations in the genes encoding for the key activators and stabilisers of p53 (ATM, CHEK1, and CHEK2), an additional 11 patients were considered as having impaired p53 signalling: seven patients due to alterations in ATM and four due to alterations in CHEK2. The 44 patients with impaired p53 signalling generally had a shorter PFS (HR 1.7; 95%CI 1.1 - 2.5; Supplementary Figure 1). This effect was not as pronounced as when the subgroups were formed based on TP53-status alone, suggesting that TP53 is the main driver of a shorter PFS.
Although HRRm, or specifically BRCAm, was not associated with PFS in the full study population, HRRm might still be associated with PFS in patients treated with 225Ac-PSMA. 225Ac emits alpha-radiation, which is much more potent in inflicting double-stranded DNA breaks that are reliant on homologous recombination for error-free restoration when compared to beta-radiation. However, an exploratory analysis with only the 20 patients who received 225Ac-PSMA did not show an association between HRRm (n = 5) or BRCAm-status (n = 4) and PFS on 225Ac-PSMA (HRRm: HR 1.4; 95%CI 0.5 - 3.9; BRCAm: HR 1.2 95%CI 0.4 - 3.7).
Biochemical response
The median PSA change for the total cohort was -65% (interquartile range -0.89 - 0.26) with 61% of patients having a PSA50 (Figure 2C). The median PSA change was significantly more beneficial for TP53wt patients when compared to TP53m patients (-75% vs. -55%; P=0.012; Figure 2D). The proportion of patients with a PSA50 did not significantly differ (65% vs. 53%, respectively; P=0.400). At 12 weeks after initiation of PSMA-RLT, the median PSA change was -56% for TP53wt patients and -36% for TP53m patients (P=0.064). A PSA50 was witnessed by 56% versus 45% of patients, respectively (P=0.451). Notably, evaluation at 12 weeks was hampered due to missing PSA values for 24 of the 96 patients (25%). None of the other genetic subgroups was statistically significant associated with either median PSA response or PSA50 (Figure 2D; Supplementary Table 3). Notably, all four patients with presumed hyperactivation of the MAPK pathway, did reach a PSA50.
Biochemical progression-free survival (exploratory)
The median biochemical PFS (bPFS) of the total population was with 4.3 months (95%CI 4.0 - 5.9 months) approximately one month shorter than the radiologic/clinical PFS. The bPFS for the TP53m subgroup was shorter when compared to the TP53wt subgroup (3.1 vs. 5.5 months; HR 1.8; 95%CI 1.1 - 2.8; Supplementary Figure 2) and remained significant when corrected for PSA at initiation of PSMA-RLT (HR 1.7; 95%CI 1.1 - 2.8). In a multivariable analysis, TP53m was the sole molecular subgroup significantly associated with bPFS (HR 2.5; 95%CI 1.5 - 4.2; Supplementary Table 4).
Overall survival
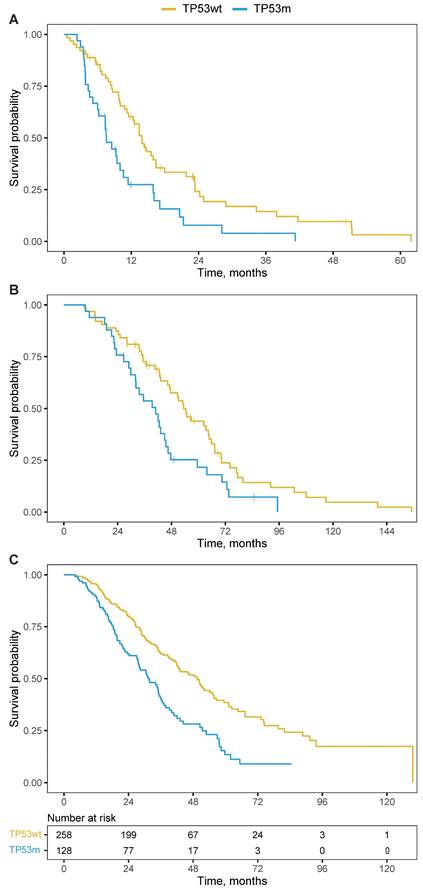
The TP53m subgroup had a significantly shorter OS when compared to the TP53wt subgroup. From initiation of PSMA-RLT, the median OS was 7.6 versus 13.9 months (HR 2.0; 95%CI 1.3 - 3.2; P=0.003; Figure 3A) and remained significant when corrected for the line of treatment in which PSMA-RLT was initiated (HR 2.1; 95%CI 1.3 - 3.3; P=0.003). From moment of castration-resistance, the OS was 40.9 months for the TP53m subgroup and 53.4 months for the TP53wt subgroup (HR 1.7; 95%CI 1.1 - 2.8; P=0.019; Figure 3B).
Kaplan-Meier curves for overall survival per TP53-mutational status. A. from initiation of PSMA-RLT. B. from castration-resistance. C. from castration-resistance for a non-PSMA-RLT-treated cohort, including a table presenting the number of patients at risk. Abbreviations: TP53wt, TP53 wildtype; TP53m, TP53 mutated.
Beyond PSMA-RLT
The prognostic power of TP53 loss-of-function alterations is well known and observed for several therapies for mCRPC. To validate the importance of TP53 mutational status beyond PSMA-RLT, we constructed a cohort of 386 mCRPC patients sequenced with the same inhouse targeted sequencing panel or whole-genome sequencing as the main study population but did not receive PSMA-RLT. The median OS from moment of castration-resistance was 41.2 months (95%CI 35.9 - 48.4). The TP53m patients (n=128) had a median OS of 31.7 months compared to 49.5 months for the 258 TP53wt patients (HR 1.9; 95%CI 1.5 - 2.5; P<0.001; Figure 3C). This is in line with the OS difference in the patients treated with PSMA-RLT.
Enrichment analysis in CRPC patients treated with PSMA-RLT
For 21 out of 96 patients from the main analysis, RNA sequencing was performed on tissues obtained before initiation PSMA-RLT. One patient (study ID 28) received 177Lu-PSMA followed by 225Ac-PSMA with tissue obtained in between. For this specific analysis, we ensured that all RNA sequencing was performed on pre-treatment tissue, and therefore for study ID 28 we only assessed response to the second PSMA-RLT (225Ac-PSMA).
Enrichment analysis
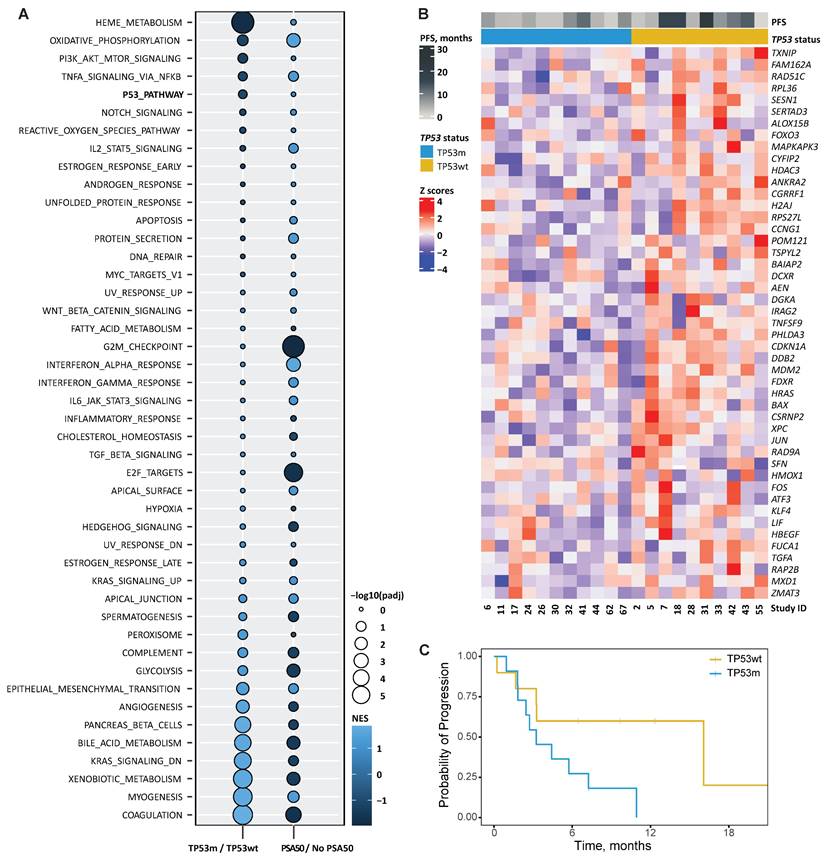
For the GSEA, we used the dichotomic endpoint PSA50 instead of PFS, as this generally correlates with PFS in mCRPC [34, 35]. Figure 4A visualises the GSEA based on TP53m over TP53wt and PSA50 over no PSA50. Several signatures were found commonly enriched in patients with PSA50 and TP53wt status. Among these, we found signatures involved in transcription factor activity, like NF-kB signalling in response to TNFα, the PI3K/AKT/MTOR pathway, NOTCH signalling, the p53 pathway, IL2/STAT5 signalling, an androgen responsive and an early oestrogen responsive gene signature, and MYC signalling. In contrast, besides E2F signalling and genes downregulated by KRAS activation, signatures commonly enriched in TP53m patients without PSA50 were constituted mainly by genes involved in biological processes like the development of skeletal muscle, genes encoding components of the blood coagulation system, genes associated with metabolism of xenobiotics, and bile acids and salts, genes encoding components of the complement immune system, and genes regulating glycolysis (Figure 4A). The expression of the 47 genes driving the enrichment in the TP53m/TP53wt comparison is visualised in Figure 4B. Their expression separated patients based on PFS, in line with the main analysis. TP53m patients in this subset had a significantly shorter PFS on PSMA-RLT (P=0.045; Figure 4C).
A. Bubble plot showing hallmarks of cancer signatures enriched (light blue) or decreased (dark blue) when comparing gene expression of patients with and without TP53 loss-of-function alterations (TP53m/TP53wt, respectively) and with and without ≥50% PSA decline (PSA50). The colour in the graph represents the normalised enrichment score (NES) and the size the false discovery rate-adjusted P-value (padj). B. Heatmap showing the relative change in mRNA expression of genes from the signature HALLMARK P53 PATHWAY that were enriched in the comparison TP53m/TP53wt (n=47) across patients. Rows show Z scores of normalised, log2-transformed values. Progression-free survival (PFS) and TP53 status for each patient is depicted. C. Kaplan-Meier curves per TP53-mutational status for the progression-free survival on PSMA-RLT for the 21 patients who underwent RNA sequencing.
p53 pathway genes association with PFS
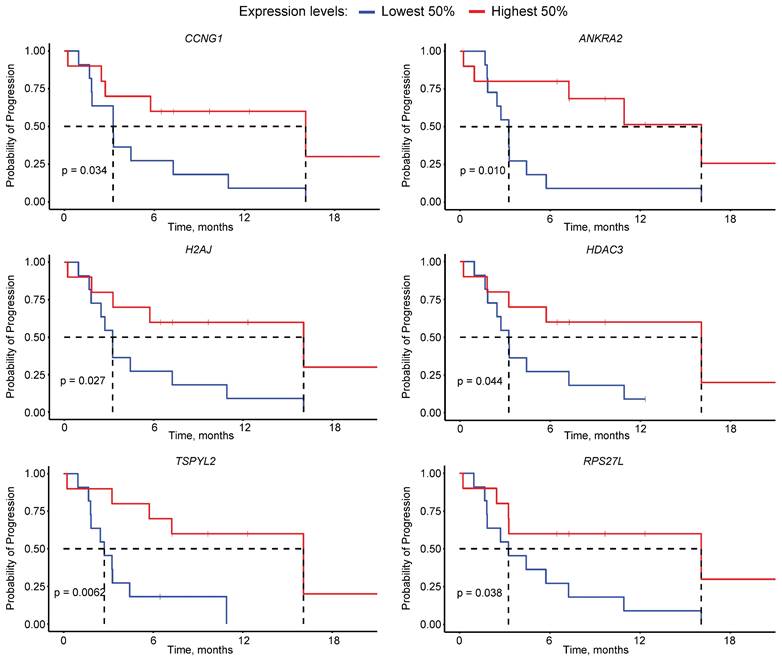
To identify possible drivers of poor outcome to PSMA-RLT among the target genes of p53, we first selected the 18 genes commonly down-regulated in TP53m patients without PSA50 (Supplementary Table 5). Survival analysis determined that the expression of six of these genes, namely CCNG1, ANKRA2, H2AJ, HDAC3, TSPYL2 and RPS27L, significantly affected the PFS (P<0.05), where the low expression of each gene was independently associated with a short PFS (Figure 5). From these, CCNG1, ANKRA2 and RPS27L are known p53 targets [36], whereas TSPYL2 is vital for effective p53 activation [37]. The p53 target genes FUCA1, RAP2B and SESN1 showed a similar trend but did not reach statistical significance (Supplementary Figure 3) [37].
Kaplan-Meier curves for the progression-free survival based on the expression of CCNG1, ANKRA2, H2AJ, HDAC3, TSPYL2 and RPS27L.
Discussion
In this pre-defined retrospective cohort study, we showed that mCRPC patients with TP53 loss-of-function alterations generally respond worse to PSMA-RLT in terms of PFS, biochemical response, and OS when compared to patients without TP53 loss-of-function alterations. In multivariable analyses with other canonical oncogenic pathways and HRRm, TP53 status was independently associated with response. In multivariable analyses with known prognostic factors, LDH was significantly associated with response alongside TP53 status. To our knowledge, this is the largest published molecularly profiled mCRPC population treated with PSMA-RLT.
Previous, mostly small, retrospective studies have failed to establish consensus regarding TP53 mutational status as predictor for response to PSMA-RLT. Vanwelkenhuyzen et al. included 46 mCRPC patients who received 177Lu-PSMA and analysed blood for qualitative circulating tumour DNA analysis. In the 39 patients with detectable circulating tumour DNA, TP53 mutational status was not associated with 177Lu-PSMA outcomes. Notably, the seven patients without detectable circulating tumour DNA were classified as lacking (TP53) genetic alterations. Another study, which included only 15 molecularly profiled mCRPC patients, identified two patients with a TP53 alteration, both did not respond to PSMA-RLT [20]. Kratochwil et al. described that six out of seven poor responders were associated with enhanced p53 signalling: 3/7 harboured a TP53 alteration, 2/7 a ATM alteration and 2/7 a CHEK2 alteration (one with a concurrent TP53 alteration) [21].
It was considered that the observed difference in PFS per TP53 status might not solely be attributed to TP53, but instead could be attributed to the presence of AVPC, characterised by compound genomic alterations in RB1, TP53, and/or PTEN [38]. AVPC, which exhibits features of small cell (neuroendocrine) prostate cancer, can lead to PSMA suppression, potentially reducing the effectiveness of PSMA-RLT [39-41]. However, our data suggests that TP53 status is a stronger predictor of PSMA-RLT outcomes than the molecular signature of AVPC.
The impact of TP53m on response to PSMA-RLT was compared to other genetic subgroups or prognostic variables. Apart from TP53m, none of the other genetic subgroups was associated with response to PSMA-RLT. In contrast to our findings, the aforementioned Vanwelkenhuyzen et al. identified pathogenic alterations in the PI3K pathway as most strongly associated with a shorter PFS [19]. De Giorgi et al., found AR amplifications to be linked with a shorter PFS [19, 42]. In our cohort, among 29 patients with AR amplifications, the median PFS was 5.4 months compared to 7.1 months for those without AR amplifications but did not reach statistical significance (P=0.51, data not presented). Raychaudhuri et al. reported a significantly higher PSA50 rate for patients with HRRm [43]. However, in our cohort, HRRm did not appear to have any discernible impact on the response to PSMA-RLT. Handke et al. conducted a transcriptome analysis on 23 patients, revealing an association between PD-L2 expression and response to PSMA-RLT. In our subgroup of 21 patients with available transcriptome data, however, PD-L2 expression did not correlate with PFS (P=0.64, data not presented). The only known prognostic variable, alongside TP53 status, significantly associated with response on PSMA-RLT was LDH. As described in two large meta-analyses, high LDH levels are associated with shorter OS and PFS across therapies for mCRPC [44, 45]. For PSMA-RLT specifically, LDH is more strongly associated with progression on 177Lu-PSMA than ALP or PSA [46, 47].
As p53 is a transcription factor, its functionality can be measured by the expression of its target genes. Within a subset of 21 patients, we identified gene expression signatures associated with both TP53 mutational status and biochemical response. While the KRAS pathway was enriched in TP53m patients without PSA50, a TNFα signature was enriched in TP53wt patients with PSA50. Although the KRAS gene is not commonly aberrant in metastatic prostate cancer (7%), deregulation of RAS proteins signalling has been reported and has tumour-promoting activity [48]. Depending on the biological context, TNFα can have two distinct roles in prostate cancer. In androgen-dependent tumours, TNFα signalling can drive the progression to castration-resistance [49]. On the other hand, and in line with our findings, in mCRPC TNFα has demonstrated to have an anti-tumour activity, by being effective in destroying tumour vasculature and stimulating anti-tumour immunity. Moreover, TNFα sensitises prostate cancer cells to ionizing radiation [50].
The enrichment analysis also identified 18 genes constituting the MSigDB p53 signature from the GSEA, whose transcript expression was markedly lower in TP53m patients without a PSA50. The low expression of six of these genes, namely CCNG1, ANKRA2, H2AJ, HDAC3, TSPYL2 and RPS27L, resulted in a significantly poorer PFS in our cohort of mCRPC patients. High expression of TSPYL2 and RPS27L correlate with better cancer prognosis across various cancer types [37, 51]. These genes are involved in inducing senescence, which in prostate cancer, upon ionizing radiation, is mainly mediated through p53 [37, 52, 53]. Additionally, TSPYL2 regulates p53 acetylation and p53-dependent cell death, potentially contributing to its tumour-suppressing activity [37, 54]. CCNG1 and ANKRA2 expression changes in response to ionizing radiation exposure, potentially serving as biomarkers [55]. In contrast to our results, lower expression of HDAC3 led to increased sensitivity to ionizing radiation in preclinical models [56].
Few patients, presumed to have a loss-of-function alteration in TP53, did show relatively high target gene expression. Downregulation of p53-mediated signalling requires inadequate p53 tetramerization, through homozygous loss or pathogenic mutations, even without loss of heterozygosity due to the dominant negative effect of most TP53 mutations [57, 58]. These discrepancies may be due to functional tetramerization by amplification of the wildtype allele or mutational exceptions.
Our analyses consistently identify TP53m as prognostic for poor response to PSMA-RLT. However, it may also have predictive value. The comparable OS deficit of TP53m patients in the populations treated with and without PSMA-RLT suggests that TP53 status is prognostic rather than predictive for response on PSMA-RLT. Yet, the rationale for TP53 alterations as a predictive factor cannot be overseen. Evidence from preclinical studies indicates p53 upregulation in response to PSMA-RLT and reduced sensitivity in TP53-/- tumours [17]. Additionally, TP53 loss-of-function alterations are often suggested as drivers of resistance to ionizing radiation, suggesting a predictive role [59-61]. The predictive value of TP53 mutations may extend to other therapies for mCRPC, with conflicting findings regarding response to taxanes or ARSIs based on TP53 status [62-65]. Preliminary data from the first prospective trial evaluating standard of care treatment based on TP53 status have not shown differences in responses to ARSIs or taxanes [66].
This study has several limitations that should be acknowledged. Firstly, due to its retrospective nature, there is missing data, leading to possible bias and reducing the power of the multivariable models. While this is the largest published population of its kind, the relatively low patient number means that this study may be underpowered to find associations with less prevalent molecular subgroups. Additionally, the cohort is heterogeneous as patients received different radionuclides and different PSMA ligands (PSMA-I&T or PSMA-617). The lack of standardised guidelines for PSMA-RLT administration throughout most of the inclusion period and delivery problems led to varying dosages per cycle and different number of administered cycles, which may have influenced treatment outcomes. In some cases, disease progression may have occurred due to postponed cycles, and patients experienced repeated responses after receiving subsequent cycles. Further limitations include imbalanced characteristics between TP53wt and TP53m patients, such as concurrent ARSI, and variations in biopsy timing relative to PSMA-RLT initiation. Although TP53 alterations are well-established as early and truncal events [67, 68], patients who underwent molecular profiling solely on archived primary tissue from localised prostate cancer are at small risk of underrepresentation of TP53 alterations due to intratumoural heterogeneity [69]. Also, the SUVmean of all lesions probably offers a more accurate assessment than the SUVmax of the hottest lesion for measuring PSMA expression [12, 70-74].
Conclusion
This study, describing the largest cohort of PSMA-RLT treated and molecularly profiled patients with mCRPC, confirms the preclinical indication that TP53 loss-of-function alterations are indicators for an unfavourable response on PSMA-RLT. No other canonical oncogenic or tumour suppressive pathway was associated with PSMA-RLT response. These results underscore the potential of molecular tumour profiling of mCRPC patients to personalise treatment plans with the goal of limiting unnecessary toxicities and improving OS and quality of life.
Abbreviations
177Lu: lutetium-177; 225Ac: actinium-225; ARSI: androgen receptor signalling inhibitor; AVPC: aggressive variant prostate cancer; bPFS: biochemical PFS; BRCAm: BRCA1 or BRCA2 mutated; CI: confidence interval; GSEA: gene-set enrichment analysis; HR: hazard ratio; HRRm: impairment of homologous recombination repair; LDH: lactate dehydrogenase; mCRPC: metastatic castration-resistant prostate cancer; OS: overall survival; PFS: progression-free survival; PSA50: ≥50% PSA decline; PSMA: prostate specific membrane antigen; RLT: radioligand therapy; SUVmax: maximum standardised uptake values; TP53m: TP53 mutated; TP53wt: TP53 wild-type; WGS: whole-genome sequencing.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
Funding
This research was in part supported by funding from the “KWF (Young investigator grant 14806 to MVLV)” as well as “VGZ - betaalbaar beter” and “Radboud Oncologie Fonds”.
Author contributions
PS, MVLV and NM conceived the study. PS, BP, MvdD, IK, HO gathered the data. PS, MVLV performed the bioinformatics and statistical analyses. PS, SPN, LK analysed the molecular data. PS and MVLV made the figures. PS, MVLV wrote the manuscript. JN, HW, IvO, LK, JS, HB, NM provided their expertise. NM supervised the project. All authors read and approved the manuscript.
Data availability
Data are available for bona fide researchers who request it from the authors (https://doi.org/10.17026/LS/QANPJ6).
Competing Interests
Maarten J. van der Doelen: Research support: 'Bayer, Janssen-Cilag'. Travel support: 'Astellas'. James Nagarajah: Research support: 'ABX, Novartis'. Advisory role: 'ITM, POINT biopharma, CURIUM, Novartis, Bayer'. Inge M. van Oort: Advisory role: 'Bayer, Astellas, Janssen, MSD/AstraZeneca'. Research support: 'Astellas, Janssen, Bayer'. Jack. A. Schalken: Speaker honorarium: 'Astellas, Bayer'. Niven Mehra: Advisory role: 'Roche, MSD, BMS, Bayer, Astellas, Janssen'. Research support: 'Astellas, Janssen, Pfizer, Roche and Sanofi' Genzyme'. Travel support: 'Astellas, MSD'. The remaining authors declare no conflict of interest.
References
1. Francini E, Gray KP, Shaw GK, Evan CP, Hamid AA, Perry CE. et al. Impact of new systemic therapies on overall survival of patients with metastatic castration-resistant prostate cancer in a hospital-based registry. Prostate Cancer Prostatic Dis. 2019;22:420-7
2. van den Bergh GPA, Kuppen MCP, Westgeest HM, Mehra N, Gerritsen WR, Aben KKH. et al. Incidence and survival of castration-resistant prostate cancer patients with visceral metastases: results from the Dutch CAPRI-registry. Prostate Cancer Prostatic Dis. 2022;26:162-9
3. Chowdhury S, Bjartell A, Lumen N, Maroto P, Paiss T, Gomez-Veiga F. et al. Real-World Outcomes in First-Line Treatment of Metastatic Castration-Resistant Prostate Cancer: The Prostate Cancer Registry. Target Oncol. 2020;15:301-15
4. Sweat SD, Pacelli A, Murphy GP, Bostwick DG. Prostate-specific membrane antigen expression is greatest in prostate adenocarcinoma and lymph node metastases. Urology. 1998;52:637-40
5. Wright GL Jr, Grob BM, Haley C, Grossman K, Newhall K, Petrylak D. et al. Upregulation of prostate-specific membrane antigen after androgen-deprivation therapy. Urology. 1996;48:326-34
6. Kratochwil C, Bruchertseifer F, Giesel FL, Weis M, Verburg FA, Mottaghy F. et al. 225Ac-PSMA-617 for PSMA-Targeted alpha-Radiation Therapy of Metastatic Castration-Resistant Prostate Cancer. J Nucl Med. 2016;57:1941-4
7. Kratochwil C, Giesel FL, Stefanova M, Benesova M, Bronzel M, Afshar-Oromieh A. et al. PSMA-Targeted Radionuclide Therapy of Metastatic Castration-Resistant Prostate Cancer with 177Lu-Labeled PSMA-617. J Nucl Med. 2016;57:1170-6
8. Sartor O, De Bono J, Chi KN, Fizazi K, Herrmann K, Rahbar K. et al. Lutetium-177-PSMA-617 for metastatic castration-resistant prostate cancer. N Engl J Med. 2021;385:1091-103
9. Sartor O, Castellano Gauna DE, Herrmann K, de Bono JS, Shore ND, Chi KNN. et al. LBA13 Phase III trial of [177Lu]Lu-PSMA-617 in taxane-naive patients with metastatic castration-resistant prostate cancer (PSMAfore). Ann Oncol. 2023;34:S1324-S5
10. Khreish F, Ebert N, Ries M, Maus S, Rosar F, Bohnenberger H. et al. 225Ac-PSMA-617/177Lu-PSMA-617 tandem therapy of metastatic castration-resistant prostate cancer: pilot experience. Eur J Nucl Med Mol Imaging. 2020;47:721-8
11. Rosar F, Hau F, Bartholomä M, Maus S, Stemler T, Linxweiler J. et al. Molecular imaging and biochemical response assessment after a single cycle of [(225)Ac]Ac-PSMA-617/[(177)Lu]Lu-PSMA-617 tandem therapy in mCRPC patients who have progressed on [(177)Lu]Lu-PSMA-617 monotherapy. Theranostics. 2021;11:4050-60
12. Seifert R, Seitzer K, Herrmann K, Kessel K, Schäfers M, Kleesiek J. et al. Analysis of PSMA expression and outcome in patients with advanced Prostate Cancer receiving (177)Lu-PSMA-617 Radioligand Therapy. Theranostics. 2020;10:7812-20
13. Hofman MS, Emmett L, Sandhu S, Iravani A, Joshua AM, Goh JC. et al. [177Lu] Lu-PSMA-617 versus cabazitaxel in patients with metastatic castration-resistant prostate cancer (TheraP): a randomised, open-label, phase 2 trial. Lancet. 2021;397:797-804
14. Hofman MS, Violet J, Hicks RJ, Ferdinandus J, Thang SP, Akhurst T. et al. [177Lu]-PSMA-617 radionuclide treatment in patients with metastatic castration-resistant prostate cancer (LuPSMA trial): a single-centre, single-arm, phase 2 study. Lancet Oncol. 2018;19:825-33
15. Kuo P, Hesterman J, Rahbar K, Kendi AT, Wei XX, Fang B. et al. [68Ga]Ga-PSMA-11 PET baseline imaging as a prognostic tool for clinical outcomes to [177Lu]Lu-PSMA-617 in patients with mCRPC: A VISION substudy. J Clin Oncol. 2022;40:5002 -
16. Buteau JP, Martin AJ, Emmett L, Iravani A, Sandhu S, Joshua AM. et al. PSMA and FDG-PET as predictive and prognostic biomarkers in patients given [<sup>177</sup>Lu]Lu-PSMA-617 versus cabazitaxel for metastatic castration-resistant prostate cancer (TheraP): a biomarker analysis from a randomised, open-label, phase 2 trial. Lancet Oncol. 2022;23:1389-97
17. Stuparu AD, Capri JR, Meyer CAL, Le TM, Evans-Axelsson SL, Current K. et al. Mechanisms of Resistance to Prostate-Specific Membrane Antigen-Targeted Radioligand Therapy in a Mouse Model of Prostate Cancer. J Nucl Med. 2021;62:989-95
18. Hirao A, Cheung A, Duncan G, Girard P-M, Elia AJ, Wakeham A. et al. Chk2 Is a Tumor Suppressor That Regulates Apoptosis in both an Ataxia Telangiectasia Mutated (ATM)-Dependent and an ATM-Independent Manner. Mol Cell Biol. 2002;22:6521-32
19. Vanwelkenhuyzen J, Van Bos E, Van Bruwaene S, Lesage K, Maes A, Üstmert S. et al. AR and PI3K Genomic Profiling of Cell-free DNA Can Identify Poor Responders to Lutetium-177-PSMA Among Patients with Metastatic Castration-resistant Prostate Cancer. European Urology Open Science. 2023;53:63-6
20. Satapathy S, Das CK, Aggarwal P, Sood A, Parihar AS, Singh SK, Mittal BR. Genomic characterization of metastatic castration-resistant prostate cancer patients undergoing PSMA radioligand therapy: A single-center experience. Prostate. 2023;83:169-78
21. Kratochwil C, Giesel FL, Heussel C-P, Kazdal D, Endris V, Nientiedt C. et al. Patients Resistant Against PSMA-Targeting α-Radiation Therapy Often Harbor Mutations in DNA Damage-Repair-Associated Genes. J Nucl Med. 2020;61:683-8
22. Privé BM, Slootbeek PH, Laarhuis BI, Naga SP, van Der Doelen MJ, van Kalmthout LW. et al. Impact of DNA damage repair defects on response to PSMA radioligand therapy in metastatic castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. 2022;25:71-8
23. van der Doelen MJ, Mehra N, van Oort IM, Looijen-Salamon MG, Janssen MJR, Custers JAE. et al. Clinical outcomes and molecular profiling of advanced metastatic castration-resistant prostate cancer patients treated with 225Ac-PSMA-617 targeted alpha-radiation therapy. Urologic Oncology: Seminars and Original Investigations. 2021;39:729.e7-e16
24. de Andrade KC, Lee EE, Tookmanian EM, Kesserwan CA, Manfredi JJ, Hatton JN. et al. The TP53 Database: transition from the International Agency for Research on Cancer to the US National Cancer Institute. Cell Death Differ. 2022;29:1071-3
25. Scher HI, Morris MJ, Stadler WM, Higano C, Basch E, Fizazi K. et al. Trial design and objectives for castration-resistant prostate cancer: updated recommendations from the Prostate Cancer Clinical Trials Working Group 3. J Clin Oncol. 2016;34:1402
26. Kroeze LI, de Voer RM, Kamping EJ, von Rhein D, Jansen EA, Hermsen MJ. et al. Evaluation of a hybrid capture-based pan-cancer panel for analysis of treatment stratifying oncogenic aberrations and processes. J Mol Diagn. 2020;22:757-69
27. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-23
28. Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S. et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19:4-23
29. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550
30. Zhu A, Ibrahim JG, Love MI. Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics. 2018;35:2084-92
31. Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417-25
32. Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, Sergushichev A. Fast gene set enrichment analysis. BioRxiv. 2016: 060012.
33. Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847-9
34. Matsubara N, Chi KN, Özgüroğlu M, Rodriguez-Antolin A, Feyerabend S, Fein L. et al. Correlation of Prostate-specific Antigen Kinetics with Overall Survival and Radiological Progression-free Survival in Metastatic Castration-sensitive Prostate Cancer Treated with Abiraterone Acetate plus Prednisone or Placebos Added to Androgen Deprivation Therapy: Post Hoc Analysis of Phase 3 LATITUDE Study. Eur Urol. 2020;77:494-500
35. Hammerer P, Al-Batran S-E, Windemuth-Kieselbach C, Keller M, Hofheinz R-D. PSA response to cabazitaxel is associated with improved progression-free survival in metastatic castration-resistant prostate cancer: the non-interventional QoLiTime study. World J Urol. 2018;36:375-81
36. Fischer M. Census and evaluation of p53 target genes. Oncogene. 2017;36:3943-56
37. Magni M, Buscemi G, Maita L, Peng L, Chan SY, Montecucco A. et al. TSPYL2 is a novel regulator of SIRT1 and p300 activity in response to DNA damage. Cell Death Differ. 2019;26:918-31
38. Aparicio AM, Shen L, Tapia ELN, Lu J-F, Chen H-C, Zhang J. et al. Combined tumor suppressor defects characterize clinically defined aggressive variant prostate cancers. Clin Cancer Res. 2016;22:1520-30
39. Montironi R, Cimadamore A, Lopez-Beltran A, Scarpelli M, Aurilio G, Santoni M. et al. Morphologic, Molecular and Clinical Features of Aggressive Variant Prostate Cancer. Cells. 2020;9:1073
40. Bakht MK, Derecichei I, Li Y, Ferraiuolo R-M, Dunning M, Oh SW. et al. Neuroendocrine differentiation of prostate cancer leads to PSMA suppression. Endocr Relat Cancer. 2019;26:131-46
41. Bronsert P, Reichel K, Ruf J. Loss of PSMA Expression in Non-neuroendocrine Dedifferentiated Acinar Prostate Cancer. Clin Nucl Med. 2018;43:526-8
42. De Giorgi U, Sansovini M, Severi S, Nicolini S, Monti M, Gurioli G. et al. Circulating androgen receptor gene amplification and resistance to 177Lu-PSMA-617 in metastatic castration-resistant prostate cancer: results of a Phase 2 trial. Br J Cancer. 2021;125:1226-32
43. Raychaudhuri R, Mo G, Haffner MC, Morrissey C, Ha G, Yu EY. et al. PSMA expression and response to 177Lu-PSMA-617 (LuPSMA) in men with vs. without DNA damage repair (DDR) mutations. J Clin Oncol. 2023;41:5055 -
44. Mori K, Kimura S, Parizi MK, Enikeev DV, Glybochko PV, Seebacher V. et al. Prognostic Value of Lactate Dehydrogenase in Metastatic Prostate Cancer: A Systematic Review and Meta-analysis. Clin Genitourin Cancer. 2019;17:409-18
45. Li F, Xiang H, Pang Z, Chen Z, Dai J, Chen S. et al. Association between lactate dehydrogenase levels and oncologic outcomes in metastatic prostate cancer: A meta-analysis. Cancer Medicine. 2020;9:7341-51
46. Ferdinandus J, Violet J, Sandhu S, Hicks RJ, Ravi Kumar AS, Iravani A. et al. Prognostic biomarkers in men with metastatic castration-resistant prostate cancer receiving [177Lu]-PSMA-617. Eur J Nucl Med Mol Imaging. 2020;47:2322-7
47. Rathke H, Holland-Letz T, Mier W, Flechsig P, Mavriopoulou E, Röhrich M. et al. Response Prediction of <sup>177</sup>Lu-PSMA-617 Radioligand Therapy Using Prostate-Specific Antigen, Chromogranin A, and Lactate Dehydrogenase. J Nucl Med. 2020;61:689-95
48. Min J, Zaslavsky A, Fedele G, McLaughlin SK, Reczek EE, De Raedt T. et al. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat Med. 2010;16:286-94
49. Tse BWC, Scott KF, Russell PJ. Paradoxical roles of tumour necrosis factor-alpha in prostate cancer biology. Prostate Cancer. 2012;2012:128965 -
50. Kimura K, Bowen C, Spiegel S, Gelmann EP. Tumor Necrosis Factor-α Sensitizes Prostate Cancer Cells to γ-Irradiation-induced Apoptosis1. Cancer Res. 1999;59:1606-14
51. Huang C-J, Yang S-H, Lee C-L, Cheng Y-C, Tai S-Y, Chien C-C. Ribosomal protein S27-like in colorectal cancer: a candidate for predicting prognoses. PLoS One. 2013;8:e67043-e
52. Faget DV, Ren Q, Stewart SA. Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer. 2019;19:439-53
53. Kallenbach J, Atri Roozbahani G, Heidari Horestani M, Baniahmad A. Distinct mechanisms mediating therapy-induced cellular senescence in prostate cancer. Cell Biosci. 2022;12:200
54. Zhang X, Wu X, Yao W, Wang Y-H. A tumor-suppressing role of TSPYL2 in thyroid cancer: Through interacting with SIRT1 and repressing SIRT1/AKT pathway. Exp Cell Res. 2023;432:113777 -
55. Hall J, Jeggo PA, West C, Gomolka M, Quintens R, Badie C. et al. Ionizing radiation biomarkers in epidemiological studies - An update. Mutation Research/Reviews in Mutation Research. 2017;771:59-84
56. Bhaskara S, Knutson SK, Jiang G, Chandrasekharan MB, Wilson AJ, Zheng S. et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell. 2010;18:436-47
57. Boettcher S, Miller PG, Sharma R, McConkey M, Leventhal M, Krivtsov AV. et al. A dominant-negative effect drives selection of <i>TP53</i> missense mutations in myeloid malignancies. Science. 2019;365:599-604
58. Gencel-Augusto J, Lozano G. p53 tetramerization: At the center of the dominant-negative effect of mutant p53. Genes Dev. 2020;34:1128-46
59. Robinson D, Van Allen Eliezer M, Wu Y-M, Schultz N, Lonigro Robert J, Mosquera J-M. et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell. 2015;161:1215-28
60. Mirzayans R, Andrais B, Scott A, Wang YW, Murray D. Ionizing Radiation-Induced Responses in Human Cells with Differing TP53 Status. Int J Mol Sci. 2013;14:22409-35
61. Kong X, Yu D, Wang Z, Li S. Relationship between p53 status and the bioeffect of ionizing radiation (Review). Oncol Lett. 2021;22:661
62. De Laere B, Oeyen S, Mayrhofer M, Whitington T, van Dam P-J, Van Oyen P. et al. TP53 Outperforms Other Androgen Receptor Biomarkers to Predict Abiraterone or Enzalutamide Outcome in Metastatic Castration-Resistant Prostate Cancer. Clin Cancer Res. 2019;25:1766-73
63. Maughan BL, Guedes LB, Boucher K, Rajoria G, Liu Z, Klimek S. et al. p53 status in the primary tumor predicts efficacy of subsequent abiraterone and enzalutamide in castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. 2018;21:260-8
64. Annala M, Vandekerkhove G, Khalaf D, Taavitsainen S, Beja K, Warner EW. et al. Circulating Tumor DNA Genomics Correlate with Resistance to Abiraterone and Enzalutamide in Prostate Cancer. Cancer Discov. 2018;8:444-57
65. Graf RP, Fisher V, Mateo J, Gjoerup OV, Madison RW, Raskina K. et al. Predictive Genomic Biomarkers of Hormonal Therapy Versus Chemotherapy Benefit in Metastatic Castration-resistant Prostate Cancer. Eur Urol. 2022;81:37-47
66. Grönberg H, Lindberg J, De Laere B, Crippa A, Discacciati A, Karlsson CT. et al. LBA86 Androgen receptor pathway inhibitors or taxanes for patients with metastatic castration-resistant prostate cancer: A direct comparison in ProBio, a randomized, outcome-adaptive, biomarker-driven platform trial. Ann Oncol. 2023;34:S1327
67. Abida W, Armenia J, Gopalan A, Brennan R, Walsh M, Barron D. et al. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precis Oncol. 2017:1-16
68. van de Haar J, Hoes LR, Roepman P, Lolkema MP, Verheul HMW, Gelderblom H. et al. Limited evolution of the actionable metastatic cancer genome under therapeutic pressure. Nat Med. 2021;27:1553-63
69. Warner EW, Van der Eecken K, Murtha AJ, Kwan EM, Herberts C, Sipola J. et al. Multiregion sampling of de novo metastatic prostate cancer reveals complex polyclonality and augments clinical genotyping. Nature Cancer. 2024;5:114-30
70. Karimzadeh A, Heck M, Tauber R, Solaris E, Nekolla S, Knorr K. et al. The Impact of PSMA PET-Based Eligibility Criteria Used in the Prospective Phase II TheraP Trial in Metastatic Castration-Resistant Prostate Cancer Patients Undergoing Prostate-Specific Membrane Antigen-Targeted Radioligand Therapy. J Nucl Med. 2023;64:jnumed.122.265346
71. Current K, Meyer C, Magyar CE, Mona CE, Almajano J, Slavik R. et al. Investigating PSMA-Targeted Radioligand Therapy Efficacy as a Function of Cellular PSMA Levels and Intratumoral PSMA Heterogeneity. Clin Cancer Res. 2020;26:2946-55
72. Gafita A, Marcus C, Kostos L, Schuster DM, Calais J, Hofman MS. Predictors and Real-World Use of Prostate-Specific Radioligand Therapy: PSMA and Beyond. American Society of Clinical Oncology Educational Book. 2022:366-82
73. Ferdinandus J, Eppard E, Gaertner FC, Kürpig S, Fimmers R, Yordanova A. et al. Predictors of Response to Radioligand Therapy of Metastatic Castrate-Resistant Prostate Cancer with 177Lu-PSMA-617. J Nucl Med. 2017;58:312-9
74. Gafita A, Calais J, Grogan TR, Hadaschik B, Wang H, Weber M. et al. Nomograms to predict outcomes after 177Lu-PSMA therapy in men with metastatic castration-resistant prostate cancer: an international, multicentre, retrospective study. Lancet Oncol. 2021;22:1115-25
Author contact
![]() Corresponding author: Dr Niven Mehra; PO Box 9101, 6500 HB Nijmegen, The Netherlands; Tel.: +31 24 361 0354; Fax: +31 24 361 5025; E-mail: Niven.Mehranl; OrcID: https://orcid.org/0000-0002-4794-1831.
Corresponding author: Dr Niven Mehra; PO Box 9101, 6500 HB Nijmegen, The Netherlands; Tel.: +31 24 361 0354; Fax: +31 24 361 5025; E-mail: Niven.Mehranl; OrcID: https://orcid.org/0000-0002-4794-1831.