Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
History and Radiolabeling...
Actinium-225 based TAT Agents...
Clinical Studies Testing...
TAT in the hormone-sensitive PCa...
Current and Future Combination...
Challenges and Future...
Other Alpha Particle Emitting...
Conclusion
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2024; 14(7):2969-2992. doi:10.7150/thno.96403 This issue Cite
Review
Actinium-225 targeted alpha particle therapy for prostate cancer
Anil P. Bidkar1, Luann Zerefa1, Surekha Yadav1, Henry F. VanBrocklin1,2, Robert R. Flavell1,2,3, ![]()
1. Department of Radiology and Biomedical Imaging, University of California San Francisco, CA-94107, USA.
2. UCSF Helen Diller Family Comprehensive Cancer Center, San Francisco, CA-94107, USA.
3. Department of Pharmaceutical Chemistry, University of California, San Francisco, CA-94107, USA.
Received 2024-3-19; Accepted 2024-5-1; Published 2024-5-11
Abstract

Targeted alpha particle therapy (TAT) has emerged as a promising strategy for the treatment of prostate cancer (PCa). Actinium-225 (225Ac), a potent alpha-emitting radionuclide, may be incorporated into targeting vectors, causing robust and in some cases sustained antitumor responses. The development of radiolabeling techniques involving EDTA, DOTA, DOTPA, and Macropa chelators has laid the groundwork for advancements in this field. At the forefront of clinical trials with 225Ac in PCa are PSMA-targeted TAT agents, notably [225Ac]Ac-PSMA-617, [225Ac]Ac-PSMA-I&T and [225Ac]Ac-J591. Ongoing investigations spotlight [225Ac]Ac-hu11B6, [225Ac]Ac-YS5, and [225Ac]Ac-SibuDAB, targeting hK2, CD46, and PSMA, respectively. Despite these efforts, hurdles in 225Ac production, daughter redistribution, and a lack of suitable imaging techniques hinder the development of TAT. To address these challenges and additional advantages, researchers are exploring alpha-emitting isotopes including 227Th, 223Ra, 211At, 213Bi, 212Pb or 149Tb, providing viable alternatives for TAT.
Keywords: Actinium-225, targeted alpha therapy, prostate cancer, alpha particle therapy
Introduction
Prostate cancer (PCa) is the most common non-cutaneous malignancy among men, and a significant threat to public health. PCa is projected to be diagnosed in approximately 299,010 men within the United States in 2024, accounting for approximately 29% of all malignancies [1]. The lethality of prostate cancer stems from its capacity to metastasize, leading to significant health challenges and loss of life. Despite the advances in diagnostic techniques, including prostate-specific antigen (PSA) screening, a substantial proportion of patients still present with aggressive forms of the disease, necessitating better and alternative therapeutic strategies. Effective management of prostate cancer involves a diverse array of treatment modalities tailored to specific characteristics and disease stage [2]. For localized or early-stage prostate cancer, treatment options may include active surveillance, cryosurgery, brachytherapy or external beam radiotherapy, radical prostatectomy (surgical removal of the prostate gland), hormone therapy, or other focal therapies aimed at targeting specific areas of the prostate [2,3]. However, in metastatic disease, curative options become limited, and the challenges of treatment increase. Conventional therapies, such as surgery, radiation, and hormone therapy, may effectively control localized disease or delay progression, but they often are not cancer-specific and come with significant side effects, which may not be suitable for all patients.
The urgency for more precise and effective treatment options has driven the emergence of targeted therapies in oncology. These therapies aim to exploit specific molecular features of cancer cells to deliver highly potent and selective treatments. Prostate-Specific Membrane Antigen (PSMA), a type II transmembrane glycoprotein has revolutionized diagnostic, prognostic, and therapeutic approaches [4]. With its elevated and selective overexpression in prostate cancer cells, PSMA stands as an important target for precision medicine strategies. The ectodomain of PSMA exhibits glutamate carboxypeptidase enzymatic activity, mediating the cleavage of N-acetylated-L-aspartyl-L-glutamate (NAAG), thereby potentially influencing tumor microenvironment modulation [5]. Its overexpression is attributed to androgen receptor signaling and other molecular mechanisms and substantiates its potential as a therapeutic target. PSMA-targeted imaging using radiopharmaceuticals has revolutionized prostate cancer detection [6]. PSMA targeted PET imaging is now standard of care in prostate cancer.
More recently, PSMA targeted radiopharmaceutical therapy has emerged as a powerful therapeutic modality in advanced metastatic castration resistant prostate cancer. The phase II TheraP study compared [177Lu]Lu-PSMA-617 with cabazitaxel in metastatic castration-resistant prostate cancer [7]. [177Lu]Lu-PSMA-617 showed higher PSA responses (66% vs 37%) and fewer severe adverse events (33% vs 53%) compared to cabazitaxel [8]. The findings suggest [177Lu]Lu-PSMA-617 as a promising and well-tolerated alternative to cabazitaxel for this patient population. Subsequently, the VISION trial marked a substantial advancement in the ongoing battle against mCRPC. This pivotal international effort was a randomized, open-label phase III trial that successfully recruited 831 men diagnosed with PSMA-positive mCRPC [9]. The participants were divided into two treatment groups: one receiving [177Lu]Lu-PSMA-617 in conjunction with the standard of care (n=551), and the other undergoing the standard of care alone (n=280). The groundbreaking results of the trial revealed a significant improvement in progression-free and overall survival for those treated with [177Lu]Lu-PSMA-617 alongside standard of care compared to those receiving standard of care alone.
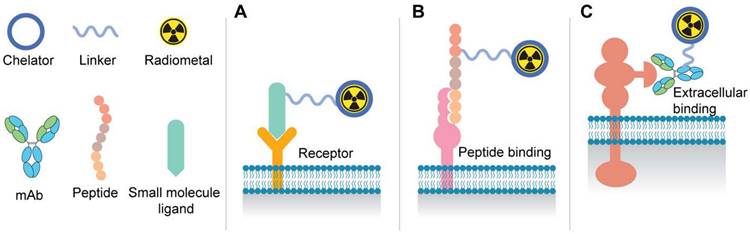
Figure 1 depicts the most common components of the various radioligand modalities including small molecules, peptides, and antibody-based agents for radiotheranostics. Positron Emission Tomography (PET) radiotracers, such as [68Ga]Ga-PSMA-11 and [18F]-DCFPyL, exhibit high specificity and sensitivity in identifying metastatic lesions and guiding treatment decisions, thereby circumventing the limitations of conventional imaging techniques [10]. Recently, the FDA has approved [177Lu]Lu-PSMA-617 (Pluvicto) for patients with advanced metastatic castration-resistant prostate cancer (mCRPC) previously treated with chemotherapy and anti-androgen therapy [11]. However, the TheraP study, as well as the phase III VISION study, further indicated that 17% and 30% of patients had inherent resistance to the [177Lu]Lu-PSMA-617, respectively [12].
Schematic illustration of radioligand therapy molecules: A. Binding of small molecule PSMA ligands to receptor active site on the cell surface. B. Representation of the use of peptide-based targeting agents. C. Tumor-specific binding of antibodies to the extracellular domain of surface proteins.
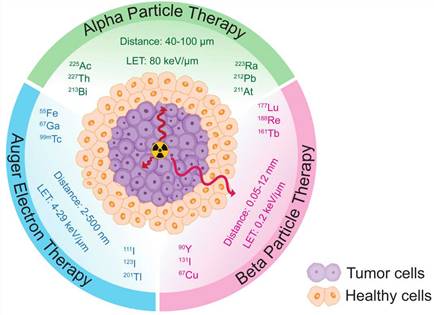
Targeted alpha therapy (TAT) harnesses the unique properties of alpha particle radiation for cancer treatment. Alpha particles are high-energy (LET= 80 keV/µm), highly charged particles that possess a short range (40-100 µm), making them ideal for localized and precise targeting of cancer cells (Figure 2) [13]. Unlike beta particles (0.2 keV/µm, 0.05-12 mm), alpha particles deposit a significant amount of energy over a very short distance, resulting in potent and localized damage to tumor cells. This characteristic allows for targeted delivery and reduced cross-fire effect to surrounding healthy tissues.
Schematic comparison of the distance traveled and Linear Energy Transfers (LETs) of α, β particles, and Auger electrons in tumor and healthy tissues.
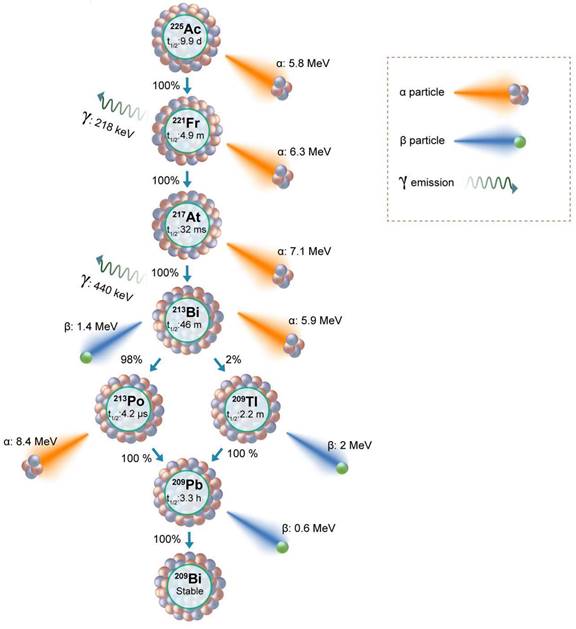
Actinium-225 (225Ac) is a promising alpha-emitting radionuclide for prostate cancer targeted radiotherapy. 225Ac has a half-life of 9.92 days, during which it decays through a series of short-lived alpha and beta-emitting daughters (Figure 3). This decay chain results in the release of four net alpha particles per decay event, maximizing the therapeutic potential of 225Ac. In this review paper, we aim to provide a comprehensive understanding of Actinium-225-based targeted alpha therapy for prostate cancer treatment. By exploring the principles of alpha particle radiation and its advantages in cancer therapy, we seek to emphasize the significance of targeted therapies in prostate cancer treatment. Furthermore, we will delve into the properties of 225Ac, the radiolabeling techniques utilized for its efficient delivery, and its role in preclinical and clinical studies. Additionally, we will evaluate the efficacy, safety considerations, and challenges associated with 225Ac-based TAT for prostate cancer.
Decay scheme of 225Ac showing daughter isotopes, alpha, and beta particle emissions along with the energies.
History and Radiolabeling Methods of 225Ac
Actinium-225 production
Actinium-225 was first identified in 1947 by a team at Argonne National Laboratory and a Canadian research group led by A. C. English (Figure 4) [14,15]. It wasn't until 1993 that the possible use of 225Ac and 213Bi as radioimmunotherapy (RIT) agents was suggested by Geerlings et al. (Figure 4) [16]. 225Ac, characterized by a relatively long half-life, undergoes a decay process involving six short-lived radionuclide daughters, ultimately reaching the near-stable state of 209Bi (half-life: approximately 19 quintillion years) [17]. The four net alpha particles, two beta emissions, and two gamma emissions generated from 225Ac decay render it as an important radioisotope for therapeutic applications (see Figure 3) [18]. The 225Ac decays (alpha particle emission with Eα of 5.8 MeV, t1/2=9.9 days) to 221Fr (Eα 6.3 MeV, t1/2=4.8 min), and 217At (Eα 7.1 MeV t1/2=33 ms) followed by 213Bi (Eα 5.8 MeV, 45.6 min). 213Bi decays with branched beta decay (branching ratio 98%, Eβ 1.4 MeV) to alpha emitter 213Po, and alpha decay (2%, Eα 5.9 MeV) to 209Tl. Finally, both 213Po (8.4 MeV, t1/2= 3.7 µs) and 209Tl decay to 209Pb (Eβ 1.4 MeV), followed by stable 209Bi [18]. Two gamma emissions from 225Ac daughters, 221Fr (218 keV) and 213Bi (440 keV) are utilized for imaging applications. Additionally, the alpha particles have limited penetration (shorter path length, 40-100 µm), making them highly effective in delivering targeted radiation to localized cancer cells while minimizing damage to surrounding healthy tissues to reduce off-target toxicity. Among the daughters, 213Bi has gained more attention, and TATs based on 213Bi are under preclinical evaluation [19].
Chronological sequence of significant milestones spanning from the initial identification of 225Ac to its current role in prostate cancer.
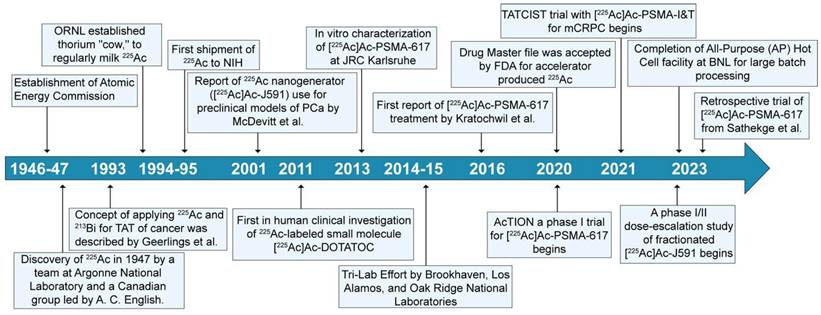
The approval of 223RaCl2 (Xofigo) by the FDA in 2013 for managing prostate cancer bone metastases in castration-resistant prostate cancer patients sparked a resurgence of interest in TATs. Despite the successful use of 223RaCl2, incorporation of 223Ra into targeted agents was not established due to its challenging chelation chemistry [20]. However, other α-emitting radionuclides, including 227Th, 225Ac, 213Bi, 212Bi, 212Pb, and 211At, have more suitable chemical properties for attachment to targeting vectors. The use of these radioisotopes requires the formation of a thermodynamically stable and kinetically inert complex with a chelator. Limited knowledge of the Ac3+ ion coordination chemistry made it difficult to find optimum chelators for a stable complex. Additionally, with a large ionic radius (112 pm, 1.12 Å) and low charge-to-ionic ratio, Ac3+ tends to display weak electrostatic interactions with the chelator [20]. The daughter isotopes from 225Ac also experience recoil energy due to the conservation of momentum, which leads to a break in the coordination and subsequent release of the daughter isotope from the chelator [21]. Therefore, finding a chelator providing sufficient stability has been an active challenge in the field of TAT with 225Ac. Additionally, chelator development research was slowed due to limited access to a regular supply of 225Ac.
Chelation chemistry
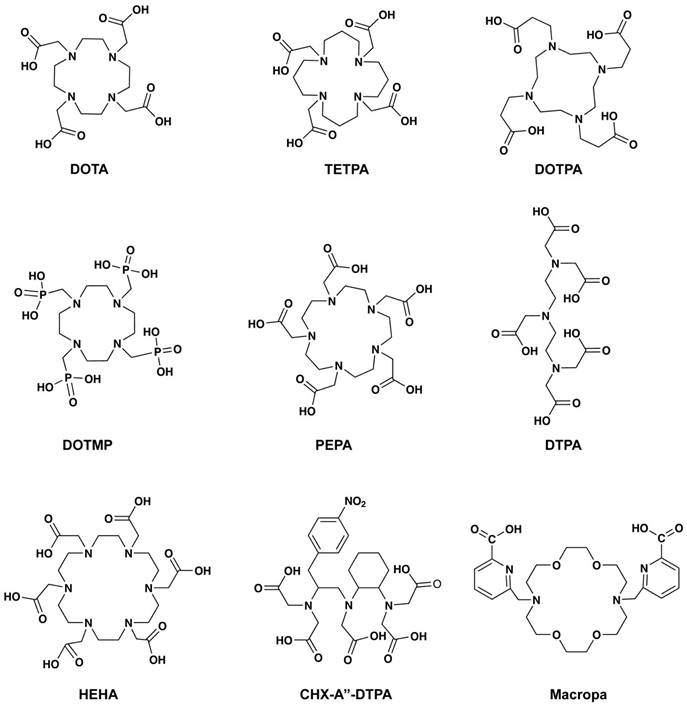
Early studies in 1999 reported by Davis et al. compared the biodistribution of the 225Ac-acetate with radiolabeled chelators such as ethylene diamine tetraacetic acid (EDTA), 1, 4, 7, 10, 13- pentaazacyclopentadecane-N, N′, N″, N‴, N⁗-pentaacetic acid (PEPA), and cyclohexyl diethylenetriamine pentaacetic acid (CHX-DTPA) (Table 1, Figure 5) [22]. The radiolabeling of EDTA, PEPA, and CHX-PEPA carried out in 1M NH4OH (pH=5) resulted in 80-90% radiochemical yield. CHX-DTPA and PEPA conjugates showed reduced uptake in the liver compared to the 225Ac-acetate and EDTA, showing in-vivo stability. In 1999, another study by Deal et al. reported the development of 1,4,7,10,13,16-hexaazacyclohexadecane-N,N′, N′′,N′′′,N′′′′,N′′′′′- hexa acetic acid (HEHA) for 225Ac complexation [23]. The use of the HEHA-mAb antibody conjugate resulted in labeling yield of 60-85% with a specific activity of 200-400 μCi/mg protein, in 0.15 M NH4OAc (pH= 4 to 7, 30 min, 37°C) [24]. Following this, McDevitt et al. studied diethylenetriaminepentaacetic acid (DTPA), 1,4,7,10- tetraazacyclododecane- 1,4,7,10-tetraacetic acid (DOTA), 1,4,8,11- tetraazacyclotetradecane- 1,4,8,11-tetraacetic acid (TETA), 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetrapropionic acid (DOTPA), 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetramethylene-phosphonic acid (DOTMP), and 1,4,8,11-tetraazacyclotetradecane-1,4,8,11-tetrapropionic acid (TETPA) for their radiolabeling and stability characteristics (Figure 5). Among the studied chelators, DOTA showed promising radiolabeling (3 M NH4OAc, 37°C, 1.0 mL reaction volume) with improved stability in the 25% serum samples (90% intact after 10 days) [25]. Significant improvement of radiolabeling yields and stability was seen with 18-membred macrocycle macropa. The monoclonal antibody trastuzumab and a PSMA ligand (RPS-070) were radiolabeled at room temperature in 5 minutes, with 99% retention of 225Ac after 7 days [26]. Recent studies demonstrate ongoing efforts to use additional macrocyclic chelators including crown, macropid, macrodipa, and py-macrodipa [27].
Radiolabeling conditions and radiochemical yields for chelators used for 225Ac complexes.
| Chelators | Buffers | pH | Temperature | Radiochemical Yield | Reference |
|---|---|---|---|---|---|
| EDTA, PEPA, CHX-PEPA | 1M NH4OH | 5 | 40°C for 30 min | 80-90% | [22] |
| HEHA | 0.15M NH4OH | 4 to 7 | 37°C for 30 min | 60-85% | [24] |
| DTPA, TETA, DOTPA | 3 M NH4Ac | 6 | 37°C for 0, 30, 60, and 120 min | Very low | [25] |
| DOTA and DOTMP | 3M NH4Ac | 6 | 0, 30, 60, and 120 min | 100% and 78% | [25] |
| Macropa | 0.15M NH4Ac | 5.5 to 6 | 37°C for 5 min | 96% | [26] |
Chemical structures of the chelators used for 225Ac radiolabeling.
Since the early 1990s, extraction of 225Ac from 229Th has assumed a pivotal role in the production of both 225Ac and 213Bi. Subsequently, clinical and preclinical studies have depended on the 225Ac derived through the decay of 229Th [19]. Globally, there exist three sources of 229Th facilitating the generation of 225Ac of clinical relevance. These sources are strategically located at the Directorate for Nuclear Safety and Security of the JRC of the European Commission in Karlsruhe, Germany, the Oak Ridge National Laboratory (ORNL) in the United States, and the Institute of Physics and Power Engineering (IPPE) in Obninsk, Russia. The origins of the 229Th sources trace back to aged 233U, initially synthesized for weaponry applications via neutron irradiation of natural 232Th.
The escalating demand for 225Ac has drawn the attention of researchers, medical practitioners, and the global patient population, all awaiting the provision of this crucial radionuclide. Their focus revolves around its subsequent application in tailoring patient-specific therapeutic regimens through radiolabeling methodologies. There has been a global response to produce 225Ac to meet the potential future demands fueled by TAT. A comprehensive update on 225Ac production is provided later in this review.
Actinium-225 based TAT Agents for Prostate Cancer Tumor Models
Preclinical studies evaluating small molecule TAT agents
PSMA is a prime target for radioligand therapy (RLT). Radionuclides like 177Lu and 225Ac, conjugated with PSMA-targeting ligands, enable selective irradiation of tumor cells while minimizing damage to healthy tissues. Since the alpha particle possesses high LET and shorter path length, TAT with 225Ac may demonstrate enhanced therapeutic efficacy. A range of α-emitting radionuclides, namely 149Tb, 211At, 212Pb/212Bi, 225Ac, and 227Th, are currently undergoing rigorous assessment for PSMA-targeted alpha particle therapy. PSMA is also a unique target in that it accommodates bulky groups conjugated to the molecule that engages the protein binding site. This property has been accessed to develop a variety imaging and therapeutic PSMA targeted agents.
The utilization of mouse models in PCa research has yielded valuable insights into cancer mechanisms, therapeutic strategies, and treatment advancements. However, the choice of model often limits its clinical applicability. Subcutaneous xenograft models facilitate convenient tracking of tumors and responses but cannot mimic metastatic progression. On the other hand, orthotopic and systemic models offer a clinically relevant context despite being more challenging to establish and monitor. In the context of PSMA-targeted alpha therapy within a murine metastatic prostate cancer model, the timing of treatment emerged as a pivotal factor influencing therapeutic outcomes [28]. Stuparu et al. showed that administering [225Ac]Ac-PSMA-617 in the early stage of C4-2 metastatic tumors holds promise for treating disease with significant survival benefit [28]. The use of systemic models is preferable due to their ability to replicate clinical conditions better and provide a precise assessment of treatment responses and interactions.
Due to its relatively low molecular weight, PSMA is readily eliminated through the renal excretion pathway. While a high clearance rate is advantageous for imaging radiotracers, it may inadvertently lead to diminished radiation dose deposition within tumors. To enhance the radiation dose targeted at tumors, modifications are being explored in PSMA ligands to prolong their circulation and tumor retention times. A study by Meyer et al. demonstrated that incorporating an extended linker with supplementary naphthyl groups enhances ligand binding to albumin, thereby extending circulating half-life [29]. Using a similar strategy, Busslinger et al. developed ibuprofen containing ligand [225Ac]Ac-SibuDAB, which showed similar radiolabeling and hematological effect as [225Ac]Ac-PSMA-617, whereas the tumor uptake of the [225Ac]Ac-SibuDAB doubled at 48 h as compared to [225Ac]Ac-PSMA-617 (64 ± 11% IA/g vs. 31 ± 3% IA/g), resulting in improved therapeutic response and overall survival in animal models [30]. Another study with macropa chelator conjugated to albumin binding unit (4-(p-iodophenyl)butyrate) and one or two PSMA ligands also showed that the uptake of an albumin-bound [225Ac]Ac-PSMA molecule was four times higher than the non-albumin binding counterpart [31]. A similar albumin-binding PSMA-targeting ligand, RPS-074, labeled with 225Ac, achieved complete remission in 6 out of 7 LNCaP xenografts with a single administration of 148 kBq [32].
Antibody-based Radioimmunotherapy agents for Preclinical Tumors of PCa
Radioimmunotherapy (RIT) combines the precision of monoclonal antibodies with the robust cell-destructive potential of radiation therapy. Large molecules like monoclonal antibodies with higher molecular weights typically exhibit extended circulation within the bloodstream and prolonged bodily retention. By incorporating 225Ac, characterized by its long half-life, onto RIT agent, the therapy gains the added advantage of sustained radiation emission over an extended timeframe. The selection of the chelator for radioimmunotherapy becomes essential, as it plays a crucial role in securely binding and holding the radioactive isotope to the monoclonal antibody. Seminal work by McDevitt et al. demonstrated that the internalizing anti-PSMA antibody [225Ac]Ac-J591 acts as a nanogenerator, producing a series of α particles for maximum radiation dose [33]. In vitro experiments conducted in this study revealed that internally localized [225Ac]Ac-J591 in LNCaP cells retained the daughter isotopes along with their corresponding α emissions. However, it was noted that daughters produced by [225Ac]Ac-J591 bound to the cell surface were likely to be transferred to other sites. Additionally, [225Ac]Ac-J591 exhibited a significant anti-tumor response in LNCaP tumor models, leading to improved survival rates and a substantial decrease in PSA levels. This agent is now in clinical trials as outlined below.
The significance of tumor PSMA expression is underscored by its far-reaching implications for both diagnostic and therapeutic strategies. The absence or downregulation of PSMA directly impacts the efficacy of PSMA-targeted approaches [34]. Consequently, a pressing need has arisen to explore alternative targets that can facilitate the development of efficacious theranostic strategies. In response to this, notable progress has been achieved through the development of antibodies that target prostate cancer regardless of their PSMA status. One instance of this approach is the development of a full-length IgG antibody targeting CD46, as demonstrated by Su et al [35]. CD46 protein was found to be overexpressed on PCa tissues, and the anti-CD46 antibody effectively binds to PCa irrespective of PSMA expression [35]. For TAT of prostate cancer, the CD46 targeting [225Ac]Ac-DOTA-YS5 antibody, labeled with 225Ac, was harnessed [36]. The therapeutic agent proved efficacious in reducing tumor volume, exhibiting efficacy against both cell-derived (22Rv1 and DU145) and patient-derived adenocarcinoma (LTL-484) and neuroendocrine prostate cancer (LTL-545) models. Bidkar et al. also showed that fractionation of [225Ac]Ac-DOTA-YS5 into multiple smaller doses of 0.125 µCi results in a better therapeutic response with lesser toxicity. In addition to this, a surrogate imaging probe [89Zr]Zr-DFO-YS5 has been developed to image CD46-positive tumors [37]. Recently, Bobba et al. utilized a PEGylated Macropa chelator (18-membered macrocyclic) conjugated to an anti-CD46 antibody, demonstrating highly efficient radiolabeling (96% yield) at room temperature within a short timeframe of 5 minutes [38]. This approach surpassed the performance of DOTA conjugates and exhibited superior antitumor efficacy. The CD46 target has proven effective for the tumor-targeted delivery of radiation. In a related study, Li et al. demonstrated the therapeutic efficacy of a similar alpha emitting isotope, 212Pb, conjugated to an anti-CD46 antibody ([212Pb]Pb-TCMC-YS5) [39]. This novel construct showed promising therapeutic effects with a single dose in both subcutaneous mCRPC cell line-derived and patient-derived xenograft models.
The hK2 is a serine protease expressed in prostate tissue that has an 80% amino acid sequence similar to PSA. The antibody hu11B6 targets hK2, an epitope expressed exclusively on prostate tissue and cancer in vivo. Additionally, it is regulated by the androgen receptor (AR) activity. McDevitt et al. prepared the [225Ac]Ac-hu11B6 (with DOTA chelator), which delivers radiation specifically to cancer cells, thereby initiating a DNA damage response (DDR) [40]. Subsequently, the DDR causes upregulation of the hK2, resulting in feed-forward alpha particle radiotherapy. The [225Ac]Ac-hu11B6 therapy took advantage of the unique phenomenon of increasing expression of AR and hK2 from alpha particle-induced DNA damage in the LNCaP-AR tumor model. The hK2 targeting antibody has also been utilized to make an imaging agent [89Zr]Zr-hu11B6 to use as a surrogate reporter for [225Ac]Ac-hu11B6 and beta particle therapy agent [177Lu]Lu-m11B6 [40,41]. The authors also studied the expression of AR-governed biomarkers in treatment-naïve tumors vs. relapsing tumors. Genes under the control of androgen receptor (AR), namely FOLH1, KLK3, PMEPA1, and SPOCK1, displayed a down-regulated pattern, whereas a subset of androgen-repressed genes, including PMP22, CAMK2N1, and UGT2B17, demonstrated an up-regulated expression [42]. Unlike the loss of PSMA in treatment-emergent neuroendocrine/small cell prostate cancer, hK2 (KLK2) expression was intact in the tumors of mice from [225Ac]Ac-hu11B6 treatment. The hK2 targeting alpha particle RIT agent ([225Ac]Ac-DOTA-h11B6) is currently under phase I clinical trial [43].
Since the expression of hK2 and PSA is regulated due to AR activity, it has led to the utilization of PSA as a target for RIT using an antibody. Veach et al. compared the therapeutic efficiency of two types of antibodies targeting PSA: high linear energy transfer (LET) [225Ac]Ac-hu5A10 with an LET of approximately 100 keV/µm, and low LET [90Y]Y-hu5A10 with a LET of about 0.2 keV/µm [44]. This comparison conducted in LNCaP-AR xenografts showed that both the high LET and low LET antibodies resulted in improved median survival when compared to saline control (32 days). Notably, the high LET antibody achieved a significantly higher median survival (188 days) compared to the low LET antibody, showcasing the superiority of [225Ac]Ac-hu5A10. Moreover, the study found that a substantial percentage of animals exhibited a complete response to the high LET [225Ac]Ac-hu5A10 (38.9%), whereas the low LET [90Y]Y-hu5A10 achieved a complete response in only 11.1% of animals.
In the context of addressing solid tumors, an additional emerging therapeutic strategy involves inhibiting tumor angiogenesis. An intriguing approach revolves around vascular endothelial (VE) cadherin, a molecule exclusive to vascular endothelial cells that are consistently expressed throughout the vascular network. VE plays a pivotal role in the establishment of adherens junctions between neighboring endothelial cells. A monoclonal antibody, E4G10, has been developed to specifically bind to an epitope exposed solely to the monomeric, unengaged form of VE-cadherin [45]. This epitope becomes concealed upon transdimerization, leading to the formation of inter-cellular junctions. This distinctive feature permits the precise targeting of endothelial cells within nascent tumor vasculature, presenting a selective strategy to hinder tumor growth. Using this approach, Jaggi et al. designed [225Ac]Ac-E4G10 antibody targeting neovasculature, which showed a significant reduction of LNCaP subcutaneous tumor xenografts.
While RIT agents effectively target tumors, a notable concern is the prolonged circulation time, particularly in the context of 225Ac delivery. Optimizing the circulation duration is crucial, and in certain cases, a faster clearance would be beneficial for improved therapeutic outcomes.
Clinical Studies Testing 225Ac TAT Agents in Prostate Cancer Patients
[225Ac]Ac-PSMA-617
In a first description of the use of [225Ac]Ac-PSMA-617 for patients, Kratochwil et al. presented two cases of mCRPC patients who had previously received either conventional chemotherapy or [177Lu]Lu-PSMA-617 treatment [46]. Notably, the first patient exhibited the disappearance of PSMA-positive lesions on PSMA PET/CT and a remarkable decline in PSA levels from over 3,000 ng/mL to 0.26 ng/mL within two months of treatment of [225Ac]Ac-PSMA-617. Subsequent consolidation therapy with [225Ac]Ac-PSMA-617 led to further PSA reduction and a complete PSMA PET/CT imaging response. The second patient, having exhausted conventional treatments and [177Lu]Lu-PSMA-617 regimens, entered the study with elevated PSA levels at 419 ng/mL. Treatment with [225Ac]Ac-PSMA-617 across three cycles led to a full response in this patient, with PSA levels reaching below 0.1 ng/mL. These findings underscored the potential of [225Ac]Ac-PSMA-617 in mCRPC or [177Lu]Lu-PSMA-617-resistant patients. More recently, Lawal et al. published a study involving 106 patients with skeletal metastasis, revealing that 80% of them exhibited a favorable antitumor response to [225Ac]Ac-PSMA-617 [47].
Though response can be favorable in patients treated with [225Ac]Ac-PSMA-617, toxicities including xerostomia present a significant challenge. Despite favorable PSA responses, severe xerostomia resulted in discontinuation of [225Ac]Ac-PSMA-617 therapy for 10% (4/40) [48] and 23% (6/26) [49] patients, reported independently. Another study reported the grade I-II xerostomia for patients treated with [225Ac]Ac-PSMA-617 [50]. Thus, a decrease of PSA in the first few cycles could be followed by a de-escalation of [225Ac]Ac-PSMA-617 dose to manage the xerostomia, simultaneously inhibiting tumor growth. The small molecule PSMA ligands show uptake in salivary glands leading to radiation-induced xerostomia. Various strategies are being employed to reduce the uptake of salivary gland hypofunction after RLT or other radiation therapies. Among these options are the external cooling of the glands [51] and the potential use of botulinum toxin A injections to reduce PSMA-targeting compound uptake by lowering salivary gland metabolism [52]. Another option involves investigating polyglutamate as a PSMA-binding competitor, with promising initial clinical outcomes [53]. Restoring salivary gland function is a third approach, with ongoing investigations into gene transfer of the aquaporin-1 gene [54]. Sour stimulation and sialendoscopy with dilatation, saline irrigation, and steroid injections have also been explored, demonstrating beneficial effects on radiation-induced inflammation and quality of life [55]. However, even with sialendoscopic support following multiple cycles of TAT, salivary gland function remained diminished, and xerostomia persisted. In addition to this, a combination of low-activity [225Ac]Ac-PSMA-617 with full-activity doses [177Lu]Lu-PSMA-617 shows improved efficacy and good tolerability in heavily pre-treated mCRPC patients. The approach appeared to enhance treatment response and reduce the xerostomia severity, suggesting that 'tandem therapy' could be an effective strategy [56]. Furthermore, the replacement of Glu from the PSMA ligand (Lys-urea-Glu) with Asp or Aad (L-2-aminoadipic acid) has led to the development of a new class of radiotracer probe or therapy agent aimed at reducing salivary gland or kidney uptake. Using this strategy, Kuo et al. designed the albumin-binding PSMA targeting agent [177Lu]Lu-HTK03149 (Glu replaced with Aad), resulting in significantly higher tumor uptake (145% higher absorbed dose) than [177Lu]Lu-PSMA-617, along with lower uptake in salivary glands (0.23 vs 1.78 %IA/g) and kidneys (79.7 vs 7.67%IA/g) at 1h post-injection [57].
Presently, [225Ac]Ac-PSMA-617 treatment is often being used as a final resort for mCRPC, after extensive pretreatment involving ADT, chemotherapy, and radioligand therapy with 177Lu-PSMA-617. This heavy pretreatment contributes to tumor resistance while also rendering patients more susceptible to adverse effects and toxicities. Chemotherapy-naïve patients with advanced metastatic prostate carcinoma respond to [225Ac]Ac-PSMA-617 effectively. The treatment-naïve patients displayed a decline of over 90% in their serum PSA levels after undergoing three or four treatment cycles [58]. Conversely, patients whose disease had progressed after receiving two or more previous treatments experienced a less favorable decline in PSA levels following the [225Ac]Ac-PSMA-617 treatment. The de-escalation from a fixed activity dose of 8 MBq for the first cycle to 7 MBq to 4 MBq was achieved for the next cycles with positive responses and reduced xerostomia. Using this regimen, a >50% PSA decrease was seen in 91% of patients [50]. Molecular changes in the tumor cells including the mutations in the DNA damage repair genes, are considered the factors associated with the resistance to TAT [59]. A recent multicenter study conducted by Sathekge et al. investigated the treatment outcomes and adverse effects of [225Ac]Ac-PSMA-617 in mCRPC patients who had undergone with one or more previous lines of treatment [60]. The study revealed that 70% of patients showed any decline in PSA, with 57% of patients showing a decline of 50% or more. The most common side effect observed was xerostomia, followed by bone marrow toxicity [60].
Owing to its longer path, beta particles cross the tumor-affected area and affect the surrounding healthy cells (sometimes termed the crossfire effect), potentially resulting in toxicity. [177Lu]Lu-PSMA-617 delivery to bone metastasis may result in significant toxicity to bone marrow leading to hematological complexities. Conversely, alpha particles from [225Ac]Ac-PSMA-617 with a shorter range and high LET are considered more useful for micro-metastatic tumors [61]. Therefore, [225Ac]Ac-PSMA-617 has been an effective treatment for brain metastasis of mCRPC [62].
J951 for PSMA Targeted Theranostics
In addition to the developments of the small molecule PSMA targeting ligands, various antibodies targeting PSMA have been studied, which mitigate salivary gland toxicity [63-65]. Among these, 7E11, a PSMA intracellular domain binding, and J951, an extracellular domain binding antibody, have been extensively tested for clinical applications (Table 2) [63]. Since then, [89Zr]Zr-huJ591 has been shown to localize and detect metastatic lesions in prostate cancer patients, while the β or α particle emitter-loaded antibodies are also being tested clinically [66]. A single dose or fractionated dose of [177Lu]Lu-J591 showed efficacy in mCRPC patients, with a decline in PSA levels [67,68]. Dose escalation studies (NCT04576871, NCT03276572, and NCT04506567) are being performed for single and fractionated doses of [225Ac]Ac-J591 in mCRPC patients [69,70]. The phase I dose escalation of the [225Ac]Ac-J591 involved 32 patients, dosed at 13.3 to 93.3 KBq/kg [71]. The results from this study demonstrated that one patient showed dose-limiting toxicity (grade ≥3 nonhematologic or grade 4 hematological) with 80 KBq/kg dose and hematologic adverse events (grade 3) were in correlation with administered radioactivity. This trial demonstrated safety in administering a single dose of [225Ac]Ac-J591 to patients with pretreated progressive mCRPC. The safety profile, dosimetry, and therapeutic potential of the combination of the small molecule [177Lu]Lu-PSMA I&T and the [225Ac]Ac-J591 are also being studied to utilize the complimentary benefits of two therapies in mCRPC patients [72].
Current or active clinical trials with 225Ac based monotherapy or combination therapy.
| Monotherapy | |||||
|---|---|---|---|---|---|
| Therapy | Trial number | Patients | Previous therapy | Parameters | References |
| [225Ac]Ac-J591 | NCT03276572 | mCRPC | anti-Androgen therapy | Dose escalation | [69] |
| [225Ac]Ac-J591 | NCT04506567 | mCRPC | anti-Androgen therapy | Fractionation, MTD | [70] |
| [225Ac]Ac-PSMA-I&T | NCT05219500 | mCRPC | anti-Androgen therapy | Safety and Efficacy | [73] |
| [225Ac]Ac-PSMA-617 | NCT04597411 | PCa | Naïve and Pretreatment with 177Lu-PSMA-617 | Dose escalation | [74] |
| [225Ac]Ac-PSMA-617 | NCT05567770 | Hormone-sensitive metastatic PCA | Naïve and prior curative-intent treatment to the prostate | Dose-limiting toxicities, MTD | [75] |
| Combinations | |||||
| [225Ac]Ac-J591 + 177Lu-PSMA I&T | NCT04886986 | mCRPC | Anti-androgen therapy, Chemotherapy | Dose-limiting toxicity, dose escalation | [72] |
| [225Ac]Ac-J591 + Pembrolizumab | NCT04946370 | mCRPC | anti-Androgen therapy | Safety, dose-limiting toxicity | [76] |
TAT in the hormone-sensitive PCa setting
Various factors such as significant tumor load, low hemoglobin levels, reduced performance condition, and a history of several past therapies all influence RLT outcome and tolerance [77]. Consequently, it is hypothesized that PSMA-RLT may be advantageous for patients in an earlier disease stage. A recent prospective phase I study demonstrated promising results, where ten patients in the early stages of hormone-sensitive prostate cancer underwent two cycles of [177Lu]Lu-PSMA-617 RLT [78,79]. Ongoing Phase 3 trials, including PSMAfore (NCT04689828), SPLASH (NCT04647526), and ECLIPSE (NCT05204927), are actively evaluating the benefits of [177Lu]Lu-PSMA-based RLT compared to transitioning to androgen receptor targeted therapy (ARTT). A recent press release from the SPLASH study demonstrated improved radiographic progression-free survival in mCRPC patients who progressed on ARTT and were then treated with RLT rather than switching to ARTT. While these investigations mostly focus on beta emitters, a corresponding concept for alpha emitters is emerging, while research in this field is still in its early stages. In a study evaluating the tandem utilization of [177Lu]Lu-PSMA-617 and [225Ac]Ac-PSMA-617 in patients with early-stage hormone-sensitive metastatic prostate cancer, 85% of patients exhibited a PSA response of ≥50% following PSMA-RLT [80]. This raises hope that future studies evaluating 225Ac based RLT alone or in combination with beta emitters might be applied in early prostate cancer.
Current and Future Combination Therapy Approaches in PCa Treatment
Research in the field of targeted radioligand therapy led to clinical approval of agents such as [177Lu]Lu-DOTATATE, 131I-metaiodobenzylguanidine, Bexxar, and Zevalin, for treating conditions like neuroendocrine tumors (NETs), neuroblastoma, and non-Hodgkin lymphoma. Monotherapy, though the cornerstone, has been challenged by the adaptive plasticity exhibited by prostate cancer cells [81]. These cells, adept at reconfiguring their signaling networks, often render single-agent interventions ineffective over time. Given the limitations of monotherapy, various combination therapy strategies are being explored, including enhancing tumor perfusion to enable optimal radiopharmaceutical distribution, upregulating target receptors to augment cellular uptake, combining nuclide therapy with DNA-damaging drugs, inhibiting essential processes like DNA damage repair for radiosensitization, and incorporating immune checkpoint inhibitors (Figure 6). One of the approaches is to combine mAb and the small molecule with two different therapeutic radioisotopes targeting the same protein on the cell surface. In this context, the use of mAb ([225Ac]Ac-J591) and [177Lu]Lu-PSMA-I&T showed improved binding and higher radiation dose delivery compared to a single agent, resulting in a synergistic therapeutic efficiency in xenograft models. Clinical studies are in progress on mCRPC patients to find the maximum tolerated dose for the combination treatment of PSMA targeting [225Ac]Ac-J591 and [177Lu]Lu-PSMA-I&T (NCT04886986) [70,72]. As TAT is known to produce DNA damage as well as activation of immunogenic response, the use of [225Ac]Ac-J591 in combination with pembrolizumab (PD-1 inhibitor) is being tested in PCa patients (NCT04946370) [76].
Summary of possible combination therapy approaches to enhance the therapeutic effect of TAT.
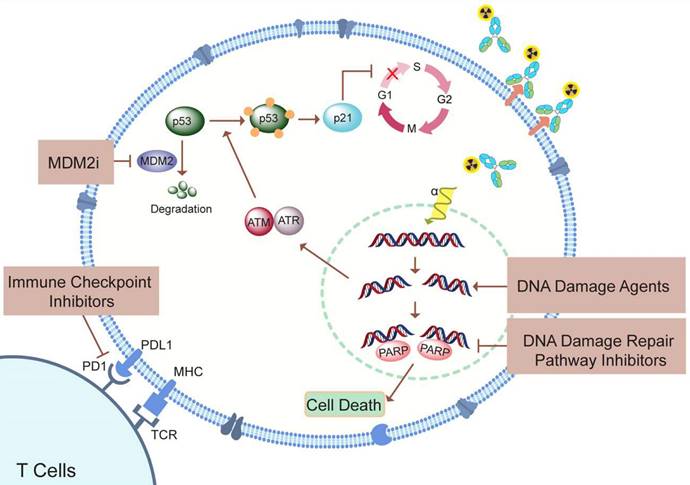
Since the poly (ADP-ribose) polymerase (PARP) enzymes are the first responder to DNA damage, alpha particle therapy may be effectively combined with the PARP inhibitors (PARPi) (Figure 6). 225Ac, an alpha particle-emitting radionuclide, contributes to this process by inducing highly localized, dense ionization along its path, resulting in complex DNA double-strand breaks. PARPi disrupt DNA repair pathways, particularly in cancers with BRCA mutations, leading to the accumulation of lethal DNA damage. By concurrently inhibiting PARP and exposing cancer cells to the high LET radiation from 225Ac, a dual assault on DNA repair mechanisms is orchestrated. The combination of beta particle therapies, with PARPi, like Olaparib, has been a subject of research interest [82,83]. A recent phase I study specifically investigated the safety profile of combining Olaparib with 223Ra in patients with mCRPC, indicating the growing interest and exploration of this combination in clinical settings [84].
Challenges and Future Perspectives
Actinium-225 production
The main source of 225Ac has been the stocks of 229Th extracted from 233U. The milking of 225Ac derived from 229Th remains insufficient to facilitate the widespread and routine demand. Due to this, alternative approaches to produce 225Ac at a large scale are being explored. The accelerator produced 225Ac has potential to meet current demand. Firstly, the high-energy proton irradiation (>100 MeV) of 232Th produces a large quantity of 225Ac, but it concurrently yields other radionuclides through spallation and fission reactions that need to be separated [18,85]. Notably, a longer-lived 227Ac isotope, with a half-life of 21.8 years, co-produced at a rate of 0.1-0.2% relative to 225Ac activity, is the main limitation. In addition to this, efforts are being made to produce 225Ac through proton irradiation of 226Ra targets. The most promising approach is the use of medium energy proton (20 MeV) accelerators. The irradiation of 226Ra targets via the reaction 226Ra(p,2n)225Ac presents distinct merits compared to the 232Th spallation process [86]. It can be performed in medium-sized cyclotrons, making it cost-effective without compromising the yield. Proton irradiation of 226Ra targets yields pure 225Ac without co-production of 227Ac. Although short-lived impurities like 226Ac (with a half-life of 29 hours) and 224Ac (with a half-life of 2.9 hours) are formed, their presence is manageable. Another approach to produce the 225Ac is via photonuclear 226Ra(ɣ,n)225Ra reaction, where generated 225Ra decays to 225Ac [87]. This approach will produce the 225Ac without impurities, however in lower yield [87].
In response to the growing worldwide demand and the critical scarcity of this isotope, the United States Department of Energy's Isotope Program (DOE IP) has initiated a comprehensive array of initiatives. These initiatives include strategic facility utilization and judicious allocation of funding, spanning various avenues for the synthesis of 225Ac [88]. Of particular significance among these ventures is the "Ac-225 Tri-Lab Effort," a synergistic collaboration undertaken by three national laboratories: Oak Ridge National Laboratory (ORNL), Brookhaven National Laboratory (BNL), and Los Alamos National Laboratory (LANL). This collaborative endeavor delves into the feasibility of generating 225Ac via accelerator-based methodologies, thereby contributing substantively to addressing the augmented global need. TRIUMF, a Canadian physics laboratory, has also been working towards production of 225Ac and other isotopes. Due to the high costs associated with each batch of 225Ac production, the current approach for synthesizing 225Ac at TRIUMF involves in-house production using the Isotope Separator and Accelerator (ISAC) Facility [19]. At TRIUMF, the 225Ac is generated through the irradiation of Uranium and Thorium targets with protons, and after additional purification steps, it is employed in TAT studies.
Quality control of 225Ac radiopharmaceuticals
The radiolabeling reactions and percentage purity of 225Ac compounds are primarily monitored using radio-thin layer chromatography (radio-TLC). The continuous generation of daughter isotopes (213Bi and 221Fr) makes it challenging to accurately estimate the radiochemical yield immediately after the run. 221Fr, with half-life of 4.9 minutes, reaches secular equilibrium in approximately 1 hr, while 213Bi (t1/2=45.6 minutes) take hours. Therefore, it is advisable to read the radio-TLCs after attaining secular equilibrium, approximately 4 to 5 hours after the completion of radio-TLC [89]. International Atomic Energy Agency (IAEA) also recommends radio-TLC as a quality control test for 225Ac labeled peptides [90,91]. A study by Yang et al. reported that additional quality control tests are needed to use of any radiopharmaceutical with 225Ac [91]. The [225Ac]Ac-crown-αMSH molecule synthesized by Yang et al showed radiolysis due to the high-energy alpha particles, which was not observed on radio-TLC but was evident on radio-high performance liquid chromatography (radio-HPLC). Therefore, the combination of delayed iTLC for radiolabeling, HPLC for the integrity of radiopharmaceuticals, and gamma spectroscopy for the determination of free 225Ac are recommended [91]. Additionally, retention times of the 213Bi and 221Fr, determined from fraction collection and gamma counting, could be used to calculate the radiochemical purity using HPLC [91]. Furthermore, the use of ROS scavengers like ascorbic acid or reducing the time between purification and dose injection could help reduce radiolysis [91,92]. Since the daughters 213Bi and 221Fr take different times to reach secular equilibrium with 225Ac, quantification of 225Ac using 213Bi and 221Fr before equilibrium or in case of 213Bi redistribution is challenging. In this regard, to determine the 225Ac amount, Seone et al. developed a multi-time point gamma counting of 213Bi (before secular equilibrium), and single time-point gamma counting of 213Bi and 221Fr considering no secular equilibrium [93]. Along with this, one major quality control concern related to accelerator produced 225Ac via spallation of 232Th is an impurity of long lived 227Ac. Various purification methods are being tested to separate the 227Ac from 225Ac [18,85,94].
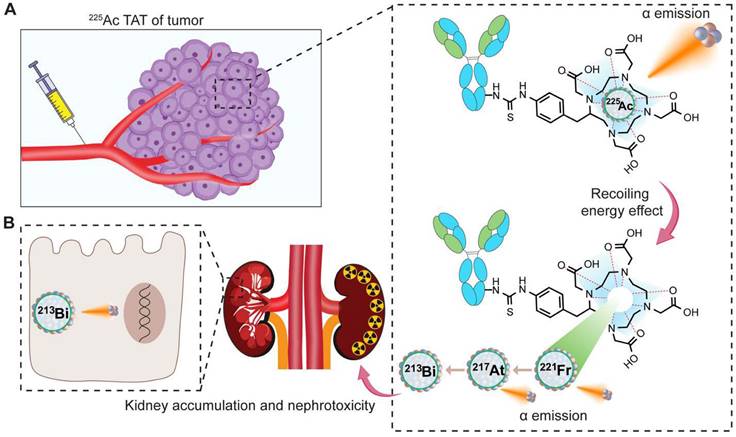
Recoiling energy, toxicity, and side effects
The toxicity associated with 225Ac-based TAT may arise in part from the recoil effect. Although significant progress has been made in developing chelators that create stable and inert complexes with 225Ac, the challenge of the recoil effect remains [20]. This recoil effect results in the release of the 221Fr isotope from the chelate as a result of the alpha emission from 225Ac, abiding by the principles of momentum conservation. During the decay process to produce four net alpha particles, the energy released due to the recoil of daughter nuclides exceeds that of more than 10,000 chemical bonds, thereby leading to the separation of daughter nuclides from the chelator (Figure 7A) [21]. This leads to the distribution of daughter isotopes to non-targeted tissues causing significant toxicity to healthy organs and compromising therapeutic efficacy. Various preclinical and clinical studies report that redistribution of daughter 213Bi into the kidney from 225Ac radioligand molecules can be observed, in some cases causing nephrotoxicity (Figure 7B) [36,50,95,96].
The decay process of 225Ac, leading to the emission of alpha particles, induces recoiling energy, subsequently causing the redistribution of daughter isotopes. A. Upon injection of the 225Ac-based TAT radiopharmaceutical, accumulation occurs in the tumor tissue. The 225Ac in circulation or tumor tissues decays, producing alpha particles along with recoiled 221Fr and 213Bi. B. The redistribution of the 213Bi contributes to nephrotoxicity.
To address the toxicity associated with recoiled daughters, researchers are investigating novel strategies (Figure 8). One approach involves facilitating the rapid uptake and internalization of alpha emitters within the target tissue (Figure 8A) [97]. The use of high affinity and rapidly internalizing molecules may decrease the delivery of daughter isotopes to healthy organs [21,97,98]. Secondly, the encapsulation of the nuclide within nanoparticles could minimize damage to normal cells (Figure 8B). Preclinical studies have explored the feasibility of loading radioisotopes in nanoformulations. Nanozeolites were successfully used as a carrier for 224Ra and 225Ra, along with their daughter radionuclides [99]. Similarly, surface-modified, PSMA-targeted liposomes also demonstrated the usability of nanoformulations for the delivery of 225Ac to prostate cancer cells [100]. In another approach, the layered nanoparticles consisting of 225Ac-doped {La0.5Gd0.5}PO4@GdPO4@Au were prepared to trap the daughters inside the core after decay [101]. These gold-coated, layered nanoparticles, labeled with 225Ac, help retain 213Bi in the target tissue minimizing the kidneys uptake over time compared to the non-coated or non-targeted nanoparticles [101]. Nano formulations allow site-specific and controlled delivery of the payload to target site. Thus, liposomes were utilized to deliver 225Ac and its radioactive daughters to ovarian cancer cells. The daughter isotopes were successfully retained in the liposome core, however the encapsulation efficiency was low [102]. Similarly, folate-F(ab′)2 decorated liposomes allow targeted 223Ra delivery [103]. In addition to targeting efficiency, the subcellular distribution of therapeutic molecules is crucial. A comparison was conducted to evaluate the cytotoxicity of PSMA and PSMA antibody-decorated liposomes loaded with 225Ac [104]. The PSMA-decorated liposomes were three times more effective in inducing cell death, attributed to the delivery of the isotope to the perinuclear space. Ensuring stringent quality control measures is imperative in the development of nanocarriers for 225Ac delivery. Stability studies under relevant physiological conditions are essential to ascertain the long-term viability and performance of the nanocarriers. Addressing quality control and stability issues is paramount to guarantee the safety and efficacy of 225Ac-loaded nanocarriers, which could pave the way for their future translation into clinical applications [104-106].
Strategies to mitigate the toxicity of recoiled daughters. A. Use of rapid uptake and internalizing targets, B. Encapsulation of the TAT radionuclide within nanoparticles, C. pretargeting, D. Intra-tumoral administration of radioactivity via injection.
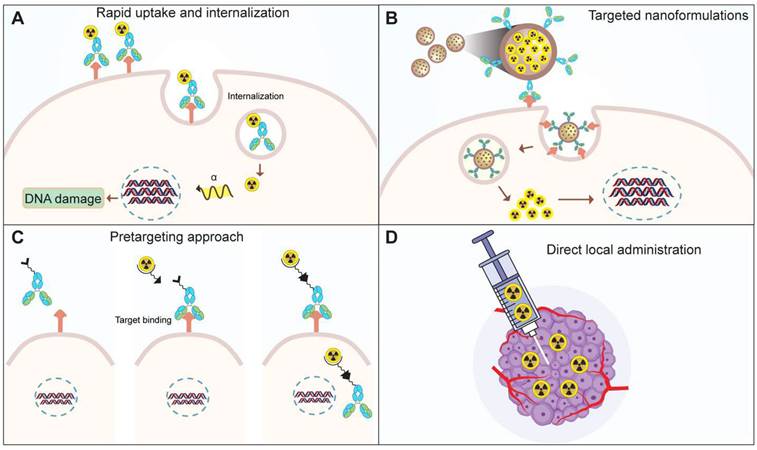
Extensive research on radionuclide carriers, primarily antibodies, has predominantly focused on IgG-type antibodies. However, the commercial viability of radiolabeled antibodies faces challenges due to hematological and other toxicities associated with the prolonged circulation of antibodies [107]. To overcome these challenges, alternative strategies including antibody fragments, recombinant proteins, and pre-targeting approaches have been explored. A noteworthy strategy is a pretargeting, where tumor cells are initially targeted with an unlabeled antibody [108]. Subsequent administration of a radiolabeled compound binds to the antibody, facilitating targeted delivery (Figure 8C). Unbound radiolabeled molecules are swiftly excreted, contributing to a more precise and efficient therapeutic approach [109]. Hapuarachchige et. al studied a pretargeting approach on PC3 cells where an anti-PSMA 5D3 mAb and its F(ab')2 fragments with trans-cyclooctene (TCO) were used as a pretargeting component [110]. Subsequently, a drug delivery component, human serum albumin loaded with the DM1 anti-tubulin agent functionalized with PEGylated tetrazine (PEG4-Tz), was employed. The results demonstrated enhanced specific killing of PSMA(+) cells. Similarly, van Rij et al. employed the trivalent bispecific antibody TF12, composed of two anti-TROP-2 Fab fragments and one anti-histamine-succinyl-glycine (HSG) Fab fragment, for pretargeted theranostics. The delivery of the therapeutic agent, a radiolabeled hapten peptide ([177Lu]Lu-IMP288), demonstrated efficacy in treating TROP-2-expressing PC3 tumors [111]. PET imaging using the same pair, [68Ga]Ga-IMP288 and TF12, enabled the detection of PCa tumors with improved contrast, showing lower bladder or kidney uptake compared to the commonly observed pattern with [68Ga]Ga-PSMA or FDG [111]. Similar to this, a theranostic pair, using proteus-DOTA as a carrier for 225Ac and 111In was developed [112]. This carrier was captured in vivo by a single-chain variable fragment (scFv) attached to the cell surface. This pretargeting study revealed high tumor accumulation and low uptake in normal tissues. The results were reproduced in solid human cancer xenograft models, including colorectal cancer, breast cancer, and neuroblastoma.
By injecting a radiolabeled molecule locally into the tumor tissue, redistribution of daughter isotopes can be minimized to some extent (Figure 8D) [21]. The dose escalation study by Krolicki et. al for [225Ac]Ac-DOTA-SP have shown that the local injections of a radiopharmaceutical were safe for glioblastoma patient when administered at 10, 20 and 30 MBq per cycle for 6 cycles (2 month interval) [113]. No hematological or renal toxicities were reported from this study [113]. Similar to this, in a pilot study by Cordier et al. in five patients of gliomas were locally injected with [213Bi]Bi-DOTA-substance P. The [213Bi]Bi-DOTA-substance P treatment was found to be well-tolerated without toxicity [114]. While direct administration is suitable for short half-life isotopes, caution is crucial for long half-life isotopes. The direct local administration route has been employed in the treatment of lung and pancreatic cancers in vivo [115,116]. The prolonged presence of long-lived isotopes in the body increases the risk of cumulative radiation exposure to healthy tissues. Therefore, meticulous consideration and potentially modified administration strategies are essential to ensure the balance between therapeutic efficacy and minimizing adverse effects when dealing with long half-life isotopes.
Radiobiological considerations, dose optimization, and fractionation
As the evidence supporting the utilization of 225Ac and other high-LET radioisotopes in cancer therapy accumulates, the consideration of the relative biological effectiveness (RBE) value becomes imperative [117]. This significance is particularly highlighted when patients undergo one or more forms of radiation therapy, such as a combination of EBRT followed by TAT, or when patients resistant to [177Lu]Lu-PSMA-617 treatment are enrolled for [225Ac]Ac-PSMA-617 therapy. The high RBE values characteristic of high-LET isotopes stem from their induction of DNA double-strand breaks (DSBs), ultimately leading to cell death. Despite technological advancements allowing for escalated tumor doses, the current approach often involves prescribing the same dose uniformly to all patients, this could lead to underdosage or overdosage for specific patients. Consideration of the distribution and volume of metastases can help tailor TAT treatments to individual patients, optimizing efficacy and minimizing toxicity. Thus, studying the tumor cells before and after the radiation dose would help prescribe an individual radiation dosage. Moreover, the bystander effects of alpha therapy, which can trigger immune responses and non-targeted tumor-cell killing, may offer promising therapeutic benefits.
In the context of therapeutic dose vs toxicity, fractionation of a large therapeutic dose of 225Ac into smaller manageable doses could enhance safety. This fractionation strategy serves several purposes. Firstly, it helps reduce the potential for toxicity by allowing healthy tissues to recover between doses. Secondly, it provides a more precise and controlled approach to treatment, optimizing the therapeutic ratio, and maximizing the impact on cancer cells while minimizing harm to surrounding normal tissues. Thirdly, it aligns with radiobiological principles, taking advantage of the fact that cancer cells may be more susceptible to cumulative radiation damage over multiple exposures. The fractionated dose strategy for 225Ac has been utilized in prostate cancer and multiple myeloma with promising results [36,70,118,119]. While it is widely acknowledged that fractionation of therapy offers advantages, it is important to consider potential disadvantages as well. Fractionation, although beneficial, can present challenges such as complex implementation strategies, treatment interruption, increased cost, and the potential for delayed tumor regression.
225Ac Imaging techniques
The four alpha particles from the decay chain of 225Ac imparts a high therapeutic efficacy; however, current studies indicate the hurdles of targeting affected tissue with the entire decay chain. Thus, it becomes imperative to confirm the presence of the daughter isotopes at the target tissue after the injections of 225Ac-based radiopharmaceuticals. In preclinical studies, the ex-vivo biodistribution analysis of 213Bi and 221Fr is difficult to complete due to their short half-lives. The gamma emissions of daughters, including the 440 keV (213Bi) and 218 keV (221Fr) peaks for SPECT imaging, remain valuable for dosimetry assessment and therapy response evaluation; however, the low therapeutic 225Ac activity administered, typically 100 kBq/kg, result in limited gamma emissions for imaging [120]. Alternatively, much progress has been made in the small-scale organ dosimetry for tumor models used in preclinical studies, which can be extrapolated to human subjects [121]. This involves ex-vivo autoradiography of alpha particle decay events, generating a real-time dose-rate map that provides information about the spatial distribution of the radioactivity in the tissue [121]. Traditional collimator-based SPECT imaging has challenges to image gamma emission above 300 keV, therefore utilization of 440 keV from 213Bi is more difficult. To overcome this, a new approach to combine the coded aperture imager (for lower keV) and Compton imager (for higher keV) was utilized to visualize and quantify the daughters from 225Ac [122].
Quantitative single photon emission computed tomography (SPECT) of gamma emissions at 440 keV and 218 keV peaks allows image-based dosimetry to determine the absorbed dose. Although posttherapy imaging is challenging for alpha emitters, one study presented a case of mCRPC treated with [225Ac]Ac-PSMA-617. The posttherapy scans, utilizing 3 different energies 78, 218, and 440 keV showed successful targeted therapy, and distribution was visualized [123]. Imaging with 3 major photopeaks (78, 218, and 440 keV) enhanced image quality compared to the previously reported 440 and 218 keV photopeaks [124]. Recently, a quantitative SPECT imaging for [225Ac]Ac-PSMA-I&T therapy for mCRPC patients was demonstrated with image-based estimations of the absorbed dose [125]. These prior and ongoing studies indicate promise for direct imaging of the daughters of 225Ac.
Other Alpha Particle Emitting Isotopes for TAT in PCa
Thorium-227
Targeted Thorium-227 conjugates (TTCs) represent another class of therapeutic radiopharmaceuticals meticulously designed for precision in TAT. The TTC process involves binding the alpha-emitting Thorium-227 (227Th) to a chelator, strategically linked to a monoclonal antibody (MAb) or small molecules engineered for tumor targeting [126]. 227Th, belonging to the actinide series (half-life of 18.7 days), follows a decay scheme featuring a cascade of alpha (five alpha particles) and beta emissions, culminating in the formation of the stable isotope 207Pb (Figure 9) [127]. The alpha particle emitted during the decay of 227Th possesses a mean energy of 5.9 MeV, contributing to a total energy of 34 MeV from all the five alpha particles in the decay series. Additionally, 227Th serves as the precursor to Radium-223 (Xofigo), an alpha-emitting radionuclide globally approved for clinical use in CRPC. In contrast to 223Ra, the precursor radionuclide 227Th demonstrates the ability to form stable chelator complexes, making it well-suited for targeted radioimmunotherapy. The extended half-life of 227Th not only streamlines practical handling and administration but also proves advantageous for achieving elevated radiation doses at the tumor site. This prolonged half-life contributes to enhanced efficiency in the production process and allows for optimized scheduling in therapeutic applications.
Schematic presentation of the decay series of Thorium-227 and Radium-223.
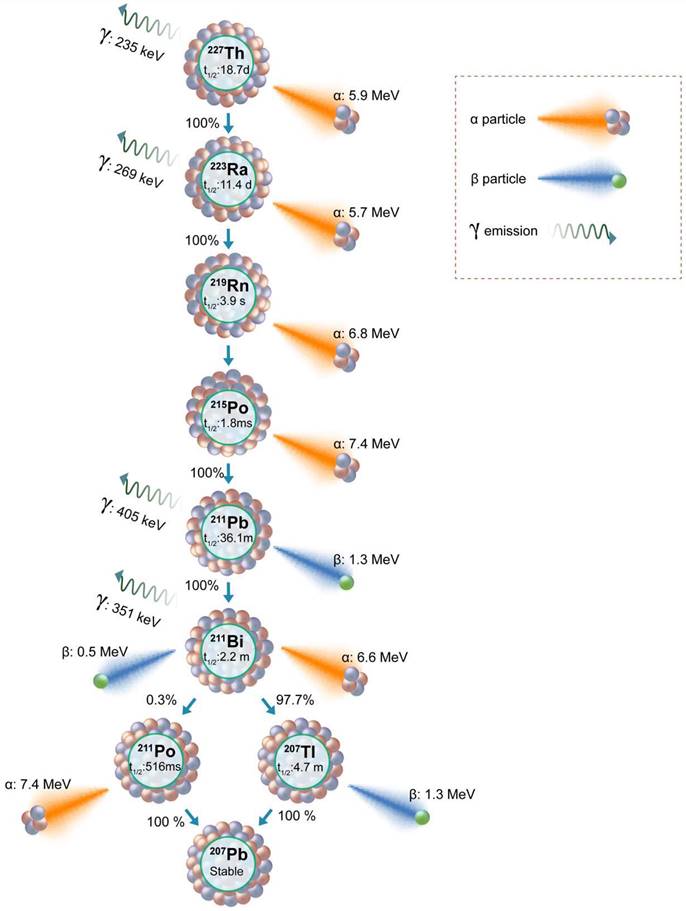
The radiolabeling of 227Th has posed a substantial hurdle in the progress of TTC. Initial investigations utilizing DOTA necessitated elevated temperatures (60oC) for efficient radiolabeling, a condition poorly suited for macromolecules like antibodies [128,129]. This underscores the demand for innovative methodologies to improve radiolabeling efficiency, particularly when dealing with larger biological entities. To overcome this challenge, Ramdahl et al. introduced a solution by employing HOPO (2,3-hydroxypyridinone) for the chelation of 227Th at room temperature, offering a promising avenue for addressing the issues in the context of TTC development [130]. Recently, Hammer et al. reported the use of PSMA-TTC for the treatment of preclinical models of PCa [131]. PSMA-TTC was synthesized by conjugating the HOPO chelator to PSMA targeting full-length antibody (BAY 2315158) for radiolabeling. This PSMA-TTC exhibited selective binding and internalization in the cells expressing PSMA, resulting in DNA damage and apoptosis from alpha particle-mediated toxicity. PSMA TTC was also effective for the treatment of AR-sensitive (LNCaP and ST1273) and AR-resistant (KUCaP-1) in-vivo tumor models. The conjugate was specifically effective in inhibiting cancer progression and abnormal bone growth in a bone metastasis model [131].
Given that PCa predominantly depends on androgens for growth, concurrently addressing androgen receptor signaling alongside PSMA-TTC treatment emerges as a promising approach for prostate cancer therapy. Hammer et al. expanded their investigations to assess the synergies between PSMA-TTC and darolutamide, an androgen receptor inhibitor [132]. The combined therapy involving PSMA-TTC and darolutamide exhibited synergistic effectiveness in the in-vitro as well as in-vivo tumor models. AR inhibition from darolutamide increased the PSMA expression thereby increasing the tumor-targeted delivery of PSMA-TTC and the therapeutic response [132]. Currently, the clinical trial to test the safety, tolerability, pharmacokinetics, and antitumor action of a PSMA-TTC is ongoing (NCT03724747). In addition to this, a combination of the PARP inhibitor (Olaparib) and PSMA-TTC is also being tested with synergistic anti-tumor results in various tumor models [133]. In addition to this, development of novel peptidomimetic small molecules is also being designed to target PSMA for the delivery of 227Th. A recent study by Böhnke et al. involved the synthesis of mono- and multimeric (di-, tri-, and tetramers) PSMA-targeting small molecules with modified carboxylated HOPO for the treatment of PCa [134]. The synthesized carboxy-HOPO exhibits heightened hydrophobicity and demonstrates robust labeling efficacy at room temperature. Among the studied forms of PSMA TTC, both monomeric and dimeric configurations exhibited exceptional tumor-to-background ratios with minimal accumulation in the kidneys. In contrast, the larger size of the trimer and tetramer led to increased accumulation in the liver [134]. Despite challenges in radiolabeling, innovative approaches like HOPO chelation at room temperature and synergistic treatments with androgen receptor inhibitors offer exciting avenues for further development and clinical exploration.
Radium-223
The ALSYMPCA trial played a pivotal role in establishing the therapeutic merit of Radium-223 (Xofigo), leading to its regulatory approval and emergence as a groundbreaking intervention for individuals with bone metastases. Radium-223's remarkable ability to selectively target bone metastases has led to improved overall survival rates and a significant delay in the development of symptomatic skeletal lesions [135]. As illustrated in Figure 9 decay scheme, 227Th undergoes alpha particle emission to transform into 223Ra, initiating a shared decay pathway between 227Th and 223Ra with the remaining daughter isotopes. 223Ra has a half-life of 11.4 days, resulting in a net of four alpha particles and two beta particles. Injection of the 223Ra and 89Sr resulted in increased bone uptake in the osseous site, without affecting the normal bone marrow suggesting its bone-seeking property [136]. Subsequent preclinical investigations consistently validated the bone-targeting efficacy of 223Ra chloride in metastatic tumors [137]. The clinical trials focused on toxicity and dose estimations and therapeutic effects of administration of Radium-223 to prostate cancer patients proved the efficacy for use in humans [138-140]. The unique bone-targeting properties of 223Ra, demonstrated through preclinical investigations and clinical trials, indicate its efficacy and safety in treating prostate cancer patients.
Astiatine-211
Astatine-211 (211At) stands out as a compelling TAT radioisotope, presenting a judicious balance of favorable characteristics [141]. With a half-life of 7.2 hours, 211At is endowed with an optimal temporal window for therapeutic applications. Importantly, unlike the other TAT isotopes, its decay series exhibit an absence of toxic daughters, a crucial consideration in ensuring the safety and efficacy of cancer treatments. Furthermore, 211At offers the practical advantage of feasible production in clinically relevant amounts, enhancing its accessibility for potential clinical applications. These attributes collectively position 211At as a promising contender in the expanding range of alpha-emitting radionuclides for targeted therapies. The decay process of 211At involves two branches (Figure 10). The first decay branch (42% decay) leads to the generation of 207Bi (t½ = 32.9 years) and the emission of an α-particle (5.9 MeV). The 207Bi subsequently decays by electron capture to 207Pb. The second decay branch (58% decay) facilitated by electron capture produces 211Po (t½ = 516 milliseconds), which subsequently undergoes decay to 207Pb, emitting an α-particle (7.4 MeV).
Decay scheme of Astatine-211.
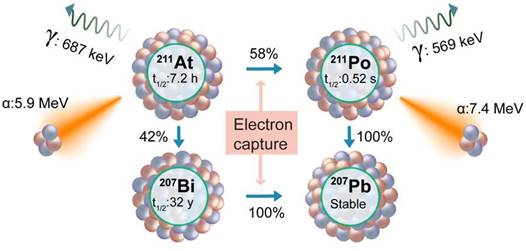
There are a few ongoing and planned clinical trials with 211At for other malignancies; however, the development of a TAT agent with 211At for PCa treatment is still in the preclinical stage. Early reports of development of PSMA-targeted (2S)-2-(3-(1-carboxy-5-(4-211At-astatobenzamido) pentyl)ureido)-pentanedioic acid ([211At]At-6) show that [211At]At-6 exhibited significant tumor growth delay and improved survival in a PSMA-positive xenograft as well as a micrometastatic model [142]. Similarly, Vaidyanathan et al. also studied Glu-urea based PSMA ligands containing a trialkyl stannyl group labeled with 211At by electrophilic astatodestannylation [143]. These urea-based molecules showed tumor accumulation with various degrees of dehalogenation resulting in thyroid, kidney, and stomach accumulation [142,143]. The radiolabeling method utilized in these studies results in lower molar activity. To address this challenge, a new methodology has been presented by Shirakami et al. for the synthesis of PSMA-targeted ligands [144]. This innovative approach entails a substitution reaction wherein dihydroxyboryl groups are replaced with 211At on PSMA ligands. Although the synthesized molecules ([211At]At-PSMA1, [211At]At-PSMA5, and [211At]At-PSMA6) show high tumor retention; due to dehalogenation, as well as due to PSMA expression at proximal tubules, significant toxicity was seen in healthy organs including kidneys [145,146]. In addition to this, peptide formulation labeled with the 211At has been tested to achieve TAT by targeting bombesin receptor [147]. The bombesin targeting [211At]At-AB-3 molecules failed to accumulate in the tumor to achieve sufficient concentration, possibly due to stability and dehalogenation issues.
To address potential PSMA negativity and heterogeneity, Back et al. tested the anti-PSCA A11 minibody to achieve targeted delivery of the 211At to prostate stem cell antigen (PSCA) [148]. PSCA is overexpressed in localized as well as metastatic prostate cancer. [211At]At-A11 showed significant therapeutic action for both subcutaneous as well as tibia tumor models. Interestingly, the blocking of sodium iodine importer with sodium perchlorate significantly reduced the uptake of free 211At in healthy organs, minimizing the off-target toxicity. In summary, 211At shows promise for Targeted TAT in PCa with favorable features, optimal timing, and no toxic byproducts.
Bismuth-213
The integration of Bismuth-213 (213Bi, half-life= 45.6 minutes) into TAT gained significant traction through the pioneering efforts of the Memorial Sloan Kettering Cancer Center in developing a 225Ac/213Bi generator. This generator, a pivotal advancement, enabled on-site production of 213Bi, thereby improving accessibility for its application in the treatment of patients with leukemia [149,150]. Early investigations using 213Bi-labeled J591 ([213Bi]Bi-J591) demonstrated encouraging outcomes in terms of targeted binding and effective tumor cell eradication in in-vitro cells, spheroids, and in-vivo tumor models [151-153]. These findings affirmed the viability of 213Bi in TAT, particularly for metastatic microtumors, where high doses of radiation per cell are crucial for achieving complete eradication of tumor cells, additionally, highlighting that larger solid tumors show response but the complete regression may not be achieved [154,155]. In addition to PSMA targeting with J591, another potential target investigated for the delivery of 213Bi is the plasminogen activator inhibitor type 2 (PAI2) protein, known to be overexpressed on prostate cancer cells. The [213Bi]Bi-PAI2 conjugate demonstrated induced cell death through mechanisms involving DNA damage and apoptosis, while concurrently exhibiting low or negligible toxicity [154]. Yong Li and colleagues proposed the concept of multiple targeted radioimmunotherapy conjugates, wherein multiple proteins were targeted using 213Bi radiolabeled vectors for respective proteins [155-157]. The cocktails of 213Bi-labeled monoclonal antibodies against C595, BLCA-38, and J591, along with [213Bi]Bi-PAI2, were tested on PCa cells, suggesting that the cocktails exhibited superior efficacy compared to monotherapies. However, for the personalized application of these combinations, information on target expression is imperative to determine the combination that would work effectively [155,156,158].
In addition to the high molecular weight macromolecules, utilizing small molecular weight vectors like PSMA or nanobodies for 213Bi delivery offers distinct advantages [159]. These vectors, due to their compact size, enhance tissue penetration, exhibit rapid clearance from non-target sites, and efficiently distribute within tumors. Additionally, the biological half-life of the vector aligns with that of 213Bi, resulting in rapid blood clearance reducing radiation exposure, and lowering the risk of hematologic toxicity [160]. In this context, the first-in-human study with [213Bi]Bi-PSMA-617 showed a remarkable response (PSA decreased to 43 µg/mL from 237 µg/L) when treated with two cycles of a combined 592 MBq dose [161,162]. Additionally, patients resistant to [177Lu]Lu-PSMA-617 exhibited a substantial anti-tumor response with minimal hematological toxicity when treated with [213Bi]Bi-DOTATOC [163]. An additional advantage of 213Bi, in comparison to 225Ac, is its absence of daughter isotopes, eliminating concerns related to recoiling. This characteristic simplifies the dosimetry calculations and ensures a more controlled and predictable radiation delivery in targeted alpha particle therapy. A key challenge in advancing 213Bi-based TAT lies in the availability of a reliable 225Ac/213Bi generator.
The successful establishment and operation of a reliable 225Ac/213Bi generator are indispensable for securing a sustainable and accessible supply of 213Bi, thereby facilitating the ongoing advancement and widespread adoption of this promising therapeutic modality [18,164]. The growing number of sites dedicated to accelerator-produced 225Ac holds promise in addressing and resolving this challenge, offering a potential solution to ensure the availability and accessibility of 213Bi for medical applications.
Lead-212
An additional noteworthy in situ α-emitting radiometal is Lead-212 (212Pb, 10.6 days half-life). Its decay involves α and β- particle emission to produce stable 208Pb. Utilizing 212Pb offers the advantage of employing the 203Pb as an imaging surrogate, facilitating imaging and dosimetry estimations. In clinical settings, 212Pb labeled trastuzumab has been tested in ovarian cancer patients with peritoneal carcinomatosis, and it demonstrated notable antitumor activity with good tolerability [165,166]. Similarly, in a preclinical study, a single intravenous dose of [212Pb]Pb-trastuzumab resulted in a substantial reduction in tumor growth with prolonged survival in mice with PC3 tumors, importantly, without loss of body weight [167]. The first human study with PSMA targeted 212Pb involved the use of urea-based ligands with p-SCN-Bn-TCMC or DO3AM chelators [168]. Two patients with mCRPC were injected with 203Pb labeled CA012, and dosimetry estimates were acquired, revealing an estimated effective dose of 6-7 mSv from 250-300 MBq of [203Pb]Pb-CA012. In-vivo studies have also explored the use of 212Pb for labeling PSMA-directed small molecules or biomarker-specific antibodies for the treatment of preclinical prostate cancer tumors [159].
Terbium-149
Terbium-149 (149Tb) is an isotope with favorable properties for theranostics. 149Tb has relatively short half-life (t½ = 4.1 hours) and decays by electron capture (76%), alpha (17%), and positron (7%) emissions. The alpha particles from 149Tb possess 25-28 μm path length, with 140-142 keV/μm LET [169]. The shorter half-life of 149Tb is considered optimal for delivery with PSMA ligands with faster excretion [169]. Utilizing the positrons emissions from 149Tb, Müller and coworkers developed “alpha-PET” with 149Tb labeled PSMA-617 ([149Tb]Tb-PSMA-617) for treatment and imaging of PSMA expressing PC3-PIP xenografts [170]. [149Tb]Tb-PSMA-617 was selectively internalized in PC3-PIP tumor xenografts and was successfully imaged with PET/CT scans. A single dose (6 MBq) or fractionated (2 x 3MBq) treatment of [149Tb]Tb-PSMA-617 resulted in delayed tumor growth, and improved survival compared to untreated control [170]. Considering the lower half life and faster excretion, multiple cycles of 149Tb labeled agents would be beneficial, however; this would require in house production. In addition to the supply, another concern for 149Tb is about the long-lived daughter isotopes. The daughter isotopes 149Gd (t½ =9.3 days), 145Sm (t½ = 340 days) 145Eu (t½ =93 days) from 149Tb decay require more long term safety studies [171]. Various preclinical studies have shown the use of 149Tb alpha therapy for treatment of pancreatic [169], breast [172], and hematological malignancies [173]; suggesting great potential for 149Tb-based radiopharmaceutical research.
Conclusion
Recent and notable clinical outcomes in the treatment of advanced-stage cancers with beta- and alpha-emitting radiopharmaceuticals have sparked increased interest in radiopharmaceutical therapy. The approval of [177Lu]Lu-PSMA-617, coupled with emerging evidence in receptor-targeted therapy that capitalizes on the unique properties of cancer cells, underscores the potential for developing novel therapeutic strategies. Actinium-225, known for high LET, longer half-life, and shorter path length has emerged as an important alpha emitter for various malignancies, including prostate cancer. Therefore, [225Ac]Ac-PSMA-617 therapy results have been considered as one of the promising options for treatment- or [177Lu]Lu-PSMA-617- resistant patients. To enhance the therapeutic efficacy of PSMA-based radionuclide therapy, diverse strategies are being explored in clinical settings. These strategies encompass developing refined patient selection methods, escalated radiation damage through dosimetry-guided dose selection, or utilization of α-emitters in place of β-emitters. Additionally, combined approaches aimed at overcoming radioresistance mechanisms, whether intrinsic or acquired, are being investigated. These multidimensional strategies involve novel hormonal agents, PARP inhibitors, and immunotherapy, all geared toward enhancing the effectiveness of PSMA-based radionuclide therapy. However, challenges persist as a vast number of mCRPC patients show no response to [177Lu]Lu-PSMA-617 RLT. Heterogeneity in PSMA expression, mechanisms of resistance, and the potential impact of PSMA-targeted therapies on non-prostate tissues necessitate further investigation. While [177Lu]Lu-PSMA-617 and [225Ac]Ac-PSMA-617 treatments have enhanced the overall prognosis for patients, the emergence of resistance within tumors has remained an issue, leading to ongoing disease progression. The length of responses to RLT is frequently brief, even among patients who are initially responsive, and the underlying reasons for resistance remain unknown. Inadequate radiation dose delivery has been established as a factor contributing to resistance, particularly for RLT with β-emitters. Diffuse marrow infiltration is a typical post-treatment progression pattern with [177Lu]Lu-PSMA-617, which could be caused by insufficient radiation dose distribution to small-volume illness. Given that [177Lu]Lu-PSMA-617 treatment failure is frequently related to the advancement of micrometastatic illness, the use of other radionuclides with shorter route lengths may be a viable approach. Single traversals of alpha particles or Auger electrons can trigger cytotoxic double-stranded DNA breaks in the nucleus, thereby bypassing the constraints associated with micrometastatic disease resistance shown with [177Lu]Lu-PSMA-617.
The successful development of radiopharmaceuticals relies on efficient chelators and tumor-specific targeting vectors. Chelators tested for radiolabeling of 225Ac are EDTA, PEPA, TETPA, DTPA, DOTA, and Macropa, among others. Among these, the DOTA and Macropa are the most favored to produce higher radiolabeling yield with suitable stability. The review brought attention to the significance of biomarkers in prostate cancer, with a particular focus on the commonly used biomarker PSMA. Despite its conventional use, ongoing studies to develop alternative PSMA ligands, peptides, or albumin modifications underscore the continuous exploration and refinement of diagnostic approaches related to PSMA. As research advances, the identification and utilization of biomarkers will likely play a pivotal role in enhancing diagnostic accuracy and tailoring more effective therapeutic interventions. In this regard, various new biomarkers are being tested for PSMA-negative prostate cancer, most notable novel biomarkers being tested are CD46, human KLK3, and VE-cadherin. DLL3, CDCP1 or GRPR are a few other promising targets that can be used for alpha therapy in PSMA-negative tumors.
The review highlights challenges in 225Ac-based therapies, including isotope production, recoiling energy, daughter redistribution, and the need for improved imaging techniques. These hurdles represent crucial areas for further research and development to unlock the full potential of 225Ac-based therapies. To improve the efficacy of current TAT agents, various combination approaches were discussed including combinations with PARP inhibitors, anti-androgens, and immune modulators. We also shed light on alternative isotopes, such as 227Th, 223Ra, 211At, 213Bi, 212Pb, and 149Tb suggesting a diversified landscape for potential alpha-emitting radionuclides in TAT for prostate cancer. These alternatives provide avenues for further research, offering potential solutions to some of the obstacles associated with 225Ac. While the application of actinium-225 in prostate cancer therapy holds tremendous potential, further research and development are vital to address the current limitations and optimize its use. The evolving landscape of alpha-particle therapy, with 225Ac at its forefront, signifies a paradigm shift in the way we approach and treat prostate cancer. As research continues to advance, the prospect of realizing the full therapeutic potential of actinium-225 in clinical settings becomes an exciting and achievable goal.
Acknowledgements
A. P. Bidkar acknowledges funding from the Early Investigator award from the Prostate Cancer Research Program of the Congressionally Directed Medical Research Program of the U.S. Department of Defense (HT9425-23-1-0139). R.R. Flavell acknowledges funding from Translational Science awards from the Prostate Cancer Research Program of the Congressionally Directed Medical Research Program of the U.S. Department of Defense (W81XWH-21-1-0792, W81XWH-20-1-0292), and R01 CA279203.
Data availability
Additional data are available from the corresponding author upon reasonable request.
Competing Interests
R.R.F. has filed patent application number 63/344,537 entitled “Radioimmunoconjugates and Therapeutic Uses Thereof.”
References
1. Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. 2024;74:12-49
2. Sandhu S, Moore CM, Chiong E, Beltran H, Bristow RG, Williams SG. Prostate cancer. The Lancet. 2021;398:1075-90
3. Hopstaken JS, Bomers JGR, Sedelaar MJP, Valerio M, Fütterer JJ, Rovers MM. An Updated Systematic Review on Focal Therapy in Localized Prostate Cancer: What Has Changed over the Past 5 Years? Eur Urol. 2022;81:5-33
4. Murthy V, Sonni I, Jariwala N. et al. The Role of PSMA PET/CT and PET/MRI in the Initial Staging of Prostate Cancer. Eur Urol Focus. 2021;7:258-66
5. Conway RE, Petrovic N, Li Z, Heston W, Wu D, Shapiro LH. Prostate-Specific Membrane Antigen Regulates Angiogenesis by Modulating Integrin Signal Transduction. Mol Cell Biol. 2006;26:5310-24
6. Farolfi A, Calderoni L, Mattana F. et al. Current and Emerging Clinical Applications of PSMA PET Diagnostic Imaging for Prostate Cancer. J Nucl Med. 2021;62:596-604
7. Sartor O, de Bono J, Chi KN. et al. Lutetium-177-PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N Engl J Med. 2021;385:1091-103
8. Hofman MS, Emmett L, Sandhu S. et al. Overall survival with [177Lu]Lu-PSMA-617 versus cabazitaxel in metastatic castration-resistant prostate cancer (TheraP): secondary outcomes of a randomised, open-label, phase 2 trial. Lancet Oncol. 2024;25:99-107
9. Morris MJ, De Bono JS, Chi KN. et al. Phase III study of lutetium-177-PSMA-617 in patients with metastatic castration-resistant prostate cancer (VISION). J Clin Oncol. 2021;39:LBA4-LBA4
10. Evangelista L, Maurer T, van der Poel H. et al. [68Ga]Ga-PSMA Versus [18F]PSMA Positron Emission Tomography/Computed Tomography in the Staging of Primary and Recurrent Prostate Cancer. A Systematic Review of the Literature. Eur Urol Oncol. 2022;5:273-82
11. Hennrich U, Eder M. [177Lu]Lu-PSMA-617 (PluvictoTM): The First FDA-Approved Radiotherapeutical for Treatment of Prostate Cancer. Pharmaceuticals. 2022;15:1292
12. Gafita A, Marcus C, Kostos L, Schuster DM, Calais J, Hofman MS. Predictors and Real-World Use of Prostate-Specific Radioligand Therapy: PSMA and Beyond. Am Soc Clin Oncol Educ Book. 2022:366-82
13. Poty S, Francesconi LC, McDevitt MR, Morris MJ, Lewis JS. α-Emitters for Radiotherapy: From Basic Radiochemistry to Clinical Studies—Part 1. J Nucl Med. 2018;59:878-84
14. English AC, Cranshaw TE, Demers P. et al. The (4n+1) Radioactive Series. Phys Rev. 1947;72:253-4
15. Hagemann F, Katzin LI, Studier MH, Ghiorso A, Seaborg GT. The (n+1$) Radioactive Series: The Decay Products of 223^U. Phys Rev. 1947;72:252-252
16. Geerlings MW, Kaspersen FM, Apostolidis C, Der Hout RV. The feasibility of 225 Ac as a source of α-particles in radioimmunotherapy. Nucl Med Commun. 1993;14:121
17. Pommé S, Marouli M, Suliman G. et al. Measurement of the 225Ac half-life. Appl Radiat Isot. 2012;70:2608-14
18. Morgenstern A, Apostolidis C, Kratochwil C, Sathekge M, Krolicki L, Bruchertseifer F. An Overview of Targeted Alpha Therapy with 225Actinium and 213Bismuth. Curr Radiopharm. 2018;11:200-8
19. Robertson AKH, Ramogida CF, Schaffer P, Radchenko V. Development of 225Ac Radiopharmaceuticals: TRIUMF Perspectives and Experiences. Curr Radiopharm. 11: 156-72.
20. Thiele NA, Wilson JJ. Actinium-225 for Targeted α Therapy: Coordination Chemistry and Current Chelation Approaches. Cancer Biother Radiopharm. 2018;33:336-48
21. de Kruijff RM, Wolterbeek HT, Denkova AG. A Critical Review of Alpha Radionuclide Therapy-How to Deal with Recoiling Daughters? Pharm Basel Switz. 2015;8:321-36
22. Davis IA, Glowienka KA, Boll RA. et al. Comparison of 225actinium chelates: tissue distribution and radiotoxicity. Nucl Med Biol. 1999;26:581-9
23. Deal KA, Davis IA, Mirzadeh S, Kennel SJ, Brechbiel MW. Improved in Vivo Stability of Actinium-225 Macrocyclic Complexes. J Med Chem. 1999;42:2988-92
24. Chappell LL, Deal KA, Dadachova E, Brechbiel MW. Synthesis, Conjugation, and Radiolabeling of a Novel Bifunctional Chelating Agent for 225Ac Radioimmunotherapy Applications. Bioconjug Chem. 2000;11:510-9
25. McDevitt MR, Ma D, Simon J, Frank RK, Scheinberg DA. Design and synthesis of 225Ac radioimmunopharmaceuticals. Appl Radiat Isot. 2002;57:841-7
26. Thiele NA, Brown V, Kelly JM. et al. An Eighteen-Membered Macrocyclic Ligand for Actinium-225 Targeted Alpha Therapy. Angew Chem Int Ed. 2017;56:14712-7
27. Hassan M, Bokhari TH, Lodhi NA, Khosa MK, Usman M. A review of recent advancements in Actinium-225 labeled compounds and biomolecules for therapeutic purposes. Chem Biol Drug Des. 2023;102:1276-92
28. Stuparu AD, Meyer CAL, Evans-Axelsson SL. et al. Targeted alpha therapy in a systemic mouse model of prostate cancer - a feasibility study. Theranostics. 2020;10:2612-20
29. Meyer C, Prasad V, Stuparu A. et al. Comparison of PSMA-TO-1 and PSMA-617 labeled with gallium-68, lutetium-177 and actinium-225. EJNMMI Res. 2022;12:65
30. Busslinger SD, Tschan VJ, Richard OK, Talip Z, Schibli R, Müller C. [225Ac]Ac-SibuDAB for Targeted Alpha Therapy of Prostate Cancer: Preclinical Evaluation and Comparison with [225Ac]Ac-PSMA-617. Cancers. 2022;14:5651
31. Reissig F, Zarschler K, Novy Z. et al. Modulating the pharmacokinetic profile of Actinium-225-labeled macropa-derived radioconjugates by dual targeting of PSMA and albumin. Theranostics. 2022;12:7203-15
32. Kelly JM, Amor-Coarasa A, Ponnala S. et al. A Single Dose of 225Ac-RPS-074 Induces a Complete Tumor Response in an LNCaP Xenograft Model. J Nucl Med. 2019;60:649-55
33. McDevitt MR, Ma D, Lai LT. et al. Tumor therapy with targeted atomic nanogenerators. Science. 2001;294:1537-40
34. Bakht MK, Derecichei I, Li Y. et al. Neuroendocrine differentiation of prostate cancer leads to PSMA suppression. Endocr Relat Cancer. 2019;26:131-46
35. Su Y, Liu Y, Behrens CR. et al. Targeting CD46 for both adenocarcinoma and neuroendocrine prostate cancer. JCI Insight. 2018;3:e121497
36. Bidkar AP, Wang S, Bobba KN. et al. Treatment of Prostate Cancer with CD46-targeted 225Ac Alpha Particle Radioimmunotherapy. Clin Cancer Res. 2023;29:1916-28
37. Wang S, Li J, Hua J. et al. Molecular Imaging of Prostate Cancer Targeting CD46 Using ImmunoPET. Clin Cancer Res. 2021;27:1305-15
38. Bobba KN, Bidkar AP, Wadhwa A. et al. Development of CD46 targeted alpha theranostics in prostate cancer using 134Ce/225Ac-Macropa-PEG4-YS5. Theranostics. 2024;14:1344-60
39. Li J, Huang T, Hua J. et al. CD46 targeted 212Pb alpha particle radioimmunotherapy for prostate cancer treatment. J Exp Clin Cancer Res. 2023;42:61
40. McDevitt MR, Thorek DLJ, Hashimoto T. et al. Feed-forward alpha particle radiotherapy ablates androgen receptor-addicted prostate cancer. Nat Commun. 2018;9:1629
41. Vilhelmsson Timmermand O, Larsson E, Ulmert D, Tran TA, Strand SE. Radioimmunotherapy of prostate cancer targeting human kallikrein-related peptidase 2. EJNMMI Res. 2016;6:27
42. Bicak M, Lückerath K, Kalidindi T. et al. Genetic signature of prostate cancer mouse models resistant to optimized hK2 targeted α-particle therapy. Proc Natl Acad Sci U S A. 2020;117:15172-81
43. Morris MJ, Sartor AO, Wong JYC. et al. Phase 1 study of JNJ-69086420, an actinium-225-labeled antibody targeting human kallikrein-2, for advanced prostate cancer. J Clin Oncol. 2022;40:TPS206-TPS206
44. Veach DR, Storey CM, Lückerath K. et al. PSA-Targeted Alpha-, Beta-, and Positron-Emitting Immunotheranostics in Murine Prostate Cancer Models and Nonhuman Primates. Clin Cancer Res Off J Am Assoc Cancer Res. 2021;27:2050-60
45. Jaggi JS, Henke E, Seshan SV. et al. Selective Alpha-Particle Mediated Depletion of Tumor Vasculature with Vascular Normalization. PLOS ONE. 2007;2:e267
46. Kratochwil C, Bruchertseifer F, Giesel FL. et al. 225Ac-PSMA-617 for PSMA-Targeted α-Radiation Therapy of Metastatic Castration-Resistant Prostate Cancer. J Nucl Med Off Publ Soc Nucl Med. 2016;57:1941-4
47. Lawal IO, Morgenstern A, Vorster M. et al. Hematologic toxicity profile and efficacy of [225Ac]Ac-PSMA-617 α-radioligand therapy of patients with extensive skeletal metastases of castration-resistant prostate cancer. Eur J Nucl Med Mol Imaging. 2022;49:3581-92
48. Kratochwil C, Bruchertseifer F, Rathke H. et al. Targeted α-Therapy of Metastatic Castration-Resistant Prostate Cancer with 225Ac-PSMA-617: Swimmer-Plot Analysis Suggests Efficacy Regarding Duration of Tumor Control. J Nucl Med Off Publ Soc Nucl Med. 2018;59:795-802
49. Feuerecker B, Tauber R, Knorr K. et al. Activity and Adverse Events of Actinium-225-PSMA-617 in Advanced Metastatic Castration-resistant Prostate Cancer After Failure of Lutetium-177-PSMA. Eur Urol. 2021;79:343-50
50. Sathekge M, Bruchertseifer F, Vorster M. et al. mCRPC Patients Receiving 225Ac-PSMA-617 Therapy in the Post-Androgen Deprivation Therapy Setting: Response to Treatment and Survival Analysis. J Nucl Med Off Publ Soc Nucl Med. 2022;63:1496-502
51. Langbein T, Chaussé G, Baum RP. Salivary Gland Toxicity of PSMA Radioligand Therapy: Relevance and Preventive Strategies. J Nucl Med. 2018;59:1172-3
52. Baum RP, Langbein T, Singh A. et al. Injection of Botulinum Toxin for Preventing Salivary Gland Toxicity after PSMA Radioligand Therapy: an Empirical Proof of a Promising Concept. Nucl Med Mol Imaging. 2018;52:80-1
53. Paganelli G, Sarnelli A, Severi S. et al. Dosimetry and safety of 177Lu PSMA-617 along with polyglutamate parotid gland protector: preliminary results in metastatic castration-resistant prostate cancer patients. Eur J Nucl Med Mol Imaging. 2020;47:3008-17
54. Baum BJ, Zheng C, Cotrim AP. et al. Transfer of the AQP1 cDNA for the correction of radiation-induced salivary hypofunction. Biochim Biophys Acta BBA - Biomembr. 2006;1758:1071-7
55. Rathke H, Kratochwil C, Hohenberger R. et al. Initial clinical experience performing sialendoscopy for salivary gland protection in patients undergoing 225Ac-PSMA-617 RLT. Eur J Nucl Med Mol Imaging. 2019;46:139-47
56. Khreish F, Ebert N, Ries M. et al. 225Ac-PSMA-617/177Lu-PSMA-617 tandem therapy of metastatic castration-resistant prostate cancer: pilot experience. Eur J Nucl Med Mol Imaging. 2020;47:721-8
57. Kuo H-T, Lin K-S, Zhang Z. et al. What a difference a methylene makes: replacing Glu with Asp or Aad in the Lys-urea-Glu pharmacophore of PSMA-targeting radioligands to reduce kidney and salivary gland uptake. Theranostics. 2022;12:6179-88
58. Sathekge M, Bruchertseifer F, Knoesen O. et al. 225Ac-PSMA-617 in chemotherapy-naive patients with advanced prostate cancer: a pilot study. Eur J Nucl Med Mol Imaging. 2019;46:129-38
59. Kratochwil C, Giesel FL, Heussel C-P. et al. Patients Resistant Against PSMA-Targeting α-Radiation Therapy Often Harbor Mutations in DNA Damage-Repair-Associated Genes. J Nucl Med Off Publ Soc Nucl Med. 2020;61:683-8
60. Sathekge MM, Lawal IO, Bal C. et al. Actinium-225-PSMA radioligand therapy of metastatic castration-resistant prostate cancer (WARMTH Act): a multicentre, retrospective study. Lancet Oncol. 2024;25:175-83
61. Haberkorn U, Giesel F, Morgenstern A, Kratochwil C. The Future of Radioligand Therapy: α, β, or Both? J Nucl Med. 2017;58:1017-8
62. Sathekge MM, Bruchertseifer F, Lawal IO. et al. Treatment of brain metastases of castration-resistant prostate cancer with 225Ac-PSMA-617. Eur J Nucl Med Mol Imaging. 2019;46:1756-7
63. Liu H, Moy P, Kim S. et al. Monoclonal antibodies to the extracellular domain of prostate-specific membrane antigen also react with tumor vascular endothelium. Cancer Res. 1997;57:3629-34
64. Chang SS, Reuter VE, Heston WD, Bander NH, Grauer LS, Gaudin PB. Five different anti-prostate-specific membrane antigen (PSMA) antibodies confirm PSMA expression in tumor-associated neovasculature. Cancer Res. 1999;59:3192-8
65. Pan M-H, Gao D-W, Feng J. et al. Biodistributions of 177Lu- and 111In- labeled 7E11 Antibodies to Prostate-Specific Membrane Antigen in Xenograft Model of Prostate Cancer and Potential Use of 111In-7E11 as a Pretherapeutic Agent for 177Lu-7E11 Radioimmunotherapy. Mol Imaging Biol MIB Off Publ Acad Mol Imaging. 2009;11:159-66
66. Pandit-Taskar N, O'Donoghue JA, Beylergil V. et al. 89Zr-huJ591 immuno-PET imaging in patients with advanced metastatic prostate cancer. Eur J Nucl Med Mol Imaging. 2014;41:2093-105
67. Tagawa ST, Milowsky MI, Morris M. et al. Phase II Study of Lutetium-177-Labeled Anti-Prostate-Specific Membrane Antigen Monoclonal Antibody J591 for Metastatic Castration-Resistant Prostate Cancer. Clin Cancer Res. 2013;19:5182-91
68. Tagawa ST, Batra J, Vallabhajosula S. et al. Final results of 2-dose fractionation of 177Lu-J591 for progressive metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2016;34:5022-5022
69. Tagawa ST, Osborne J, Niaz MJ. et al. Dose-escalation results of a phase I study of 225Ac-J591 for progressive metastatic castration resistant prostate cancer (mCRPC). J Clin Oncol. 2020;38:114-114
70. Nauseef JT, Osborne J, Gregos P. et al. Phase I/II study of 225Ac-J591 plus 177Lu-PSMA-I&T for progressive metastatic castration-resistant prostate cancer. J Clin Oncol. 2022;40:TPS5100-TPS5100
71. Tagawa ST, Thomas C, Sartor AO. et al. Prostate-Specific Membrane Antigen-Targeting Alpha Emitter via Antibody Delivery for Metastatic Castration-Resistant Prostate Cancer: A Phase I Dose-Escalation Study of 225Ac-J591. J Clin Oncol. 2023 JCO.23.00573
72. Tagawa ST, Fung E, Niaz MO. et al. Abstract CT143: Results of combined targeting of prostate-specific membrane antigen (PSMA) with alpha-radiolabeled antibody 225Ac-J591 and beta-radiolabeled ligand 177Lu-PSMA I&T: preclinical and initial phase 1 clinical data in patients with metastatic castration-resistant prostate cancer (mCRPC). Cancer Res. 2022;82:CT143
73. Excel Diagnostics and Nuclear Oncology Center. Targeted Alpha Therapy With 225Actinium-PSMA-I&T of Castration-resISTant Prostate Cancer (TATCIST) [Internet]. clinicaltrials.gov; 2023 Mar [cited 31 December 2022]. Report No.: NCT05219500. Available at:. https://clinicaltrials.gov/study/NCT05219500
74. AcTION: A Phase I Study of [225Ac]Ac-PSMA-617 in Men With PSMA-positive Prostate Cancer With or Without Prior [177Lu]Lu-PSMA-617 Radioligand Therapy [Internet]. clinicaltrials.gov; 2023 Dec [cited 31 December 2022]. Report No.: NCT04597411. Available at:. https://clinicaltrials.gov/study/NCT04597411
75. Weill Medical College of Cornell University. ACTInium-J591 Radionuclide Therapy in PSMA-Detected Metastatic HOrmone-Sensitive Recurrent Prostate CaNcer [Internet]. clinicaltrials.gov; 2023 Aug [cited 31 December 2022]. Report No.: NCT05567770. Available at:. https://clinicaltrials.gov/study/NCT05567770
76. Tagawa ST, Osborne J, Dallos M. et al. Phase I/II trial of pembrolizumab and AR signaling inhibitor +/- 225Ac-J591 for chemo-naive metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2022;40:TPS216-TPS216
77. Barber TW, Singh A, Kulkarni HR, Niepsch K, Billah B, Baum RP. Clinical Outcomes of 177 Lu-PSMA Radioligand Therapy in Earlier and Later Phases of Metastatic Castration-Resistant Prostate Cancer Grouped by Previous Taxane Chemotherapy. J Nucl Med. 2019;60:955-62
78. Privé BM, Peters SMB, Muselaers CHJ. et al. Lutetium-177-PSMA-617 in Low-Volume Hormone-Sensitive Metastatic Prostate Cancer: A Prospective Pilot Study. Clin Cancer Res. 2021;27:3595-601
79. Peters SMB, Privé BM, De Bakker M. et al. Intra-therapeutic dosimetry of [177Lu]Lu-PSMA-617 in low-volume hormone-sensitive metastatic prostate cancer patients and correlation with treatment outcome. Eur J Nucl Med Mol Imaging. 2022;49:460-9
80. Banda A, Privé BM, Allach Y. et al. PSMA-RLT in Patients with Metastatic Hormone-Sensitive Prostate Cancer: A Retrospective Study. Cancers. 2022;15:297
81. Yehya A, Ghamlouche F, Zahwe A. et al. Drug resistance in metastatic castration-resistant prostate cancer: an update on the status quo. Cancer Drug Resist. 2022;5:667-90
82. Cullinane C, Waldeck K, Kirby L. et al. Enhancing the anti-tumour activity of 177Lu-DOTA-octreotate radionuclide therapy in somatostatin receptor-2 expressing tumour models by targeting PARP. Sci Rep. 2020;10:10196
83. Feijtel D, Reuvers T, Van Tuyll-van Serooskerken C. et al. In Vivo Efficacy Testing of Peptide Receptor Radionuclide Therapy Radiosensitization Using Olaparib. Cancers. 2023;15:915
84. Pan E, Xie W, Ajmera A. et al. A Phase I Study of Combination Olaparib and Radium-223 in Men with Metastatic Castration-Resistant Prostate Cancer (mCRPC) with Bone Metastases (COMRADE). Mol Cancer Ther. 2023;22:511-8
85. Robertson AKH, McNeil BL, Yang H. et al. 232Th-Spallation-Produced 225Ac with Reduced 227Ac Content. Inorg Chem. 2020;59:12156-65
86. Apostolidis C, Molinet R, McGinley J, Abbas K, Möllenbeck J, Morgenstern A. Cyclotron production of Ac-225 for targeted alpha therapy11Dedicated to Prof. Dr. Franz Baumgärtner on the occasion of his 75th birthday. Appl Radiat Isot. 2005;62:383-7
87. Morgenstern A, Apostolidis C, Bruchertseifer F. Supply and Clinical Application of Actinium-225 and Bismuth-213. Semin Nucl Med. 2020;50:119-23
88. Zimmermann R. Is Actinium Really Happening? J Nucl Med. 2023;64:1516-8
89. Perron R, Gendron D, Causey PW. Construction of a thorium/actinium generator at the Canadian Nuclear Laboratories. Appl Radiat Isot. 2020;164:109262
90. Quality Control in the Production of Radiopharmaceuticals [Internet]. Vienna: INTERNATIONAL ATOMIC ENERGY AGENCY; 2018. (TECDOC Series). Available at: https://www.iaea.org/publications/13422/quality-control-in-the-production-of-radiopharmaceuticals.
91. Yang H, Zhang C, Yuan Z. et al. Synthesis and Evaluation of a Macrocyclic Actinium-225 Chelator, Quality Control and In Vivo Evaluation of 225 Ac-crown-αMSH Peptide. Chem - Eur J. 2020;26:11435-40
92. Reissig F, Bauer D, Zarschler K. et al. Towards Targeted Alpha Therapy with Actinium-225: Chelators for Mild Condition Radiolabeling and Targeting PSMA-A Proof of Concept Study. Cancers. 2021;13:1974
93. Castillo Seoane D, De Saint-Hubert M, Ahenkorah S. et al. Gamma counting protocols for the accurate quantification of 225Ac and 213Bi without the need for a secular equilibrium between parent and gamma-emitting daughter. EJNMMI Radiopharm Chem. 2022;7:28
94. Jalloul W, Ghizdovat V, Stolniceanu CR. et al. Targeted Alpha Therapy: All We Need to Know about 225Ac's Physical Characteristics and Production as a Potential Theranostic Radionuclide. Pharmaceuticals. 2023;16:1679
95. Satapathy S, Sood A, Das CK, Mittal BR. Evolving role of 225Ac-PSMA radioligand therapy in metastatic castration-resistant prostate cancer—a systematic review and meta-analysis. Prostate Cancer Prostatic Dis. 2021;24:880-90
96. Satapathy S, Mittal BR, Sood A. et al. Health-Related Quality-of-Life Outcomes with Actinium-225-Prostate-Specific Membrane Antigen-617 Therapy in Patients with Heavily Pretreated Metastatic Castration-Resistant Prostate Cancer. Indian J Nucl Med IJNM Off J Soc Nucl Med India. 2020;35:299-304
97. Feng Y, Meshaw R, Zhao X-G, Jannetti S, Vaidyanathan G, Zalutsky MR. Effective Treatment of Human Breast Carcinoma Xenografts with Single-Dose 211At-Labeled Anti-HER2 Single-Domain Antibody Fragment. J Nucl Med. 2023;64:124-30
98. Sathekge MM, Bruchertseifer F, Vorster M, Morgenstern A, Lawal IO. Global experience with PSMA-based alpha therapy in prostate cancer. Eur J Nucl Med Mol Imaging. 2021;49:30-46
99. Piotrowska A, Leszczuk E, Bruchertseifer F, Morgenstern A, Bilewicz A. Functionalized NaA nanozeolites labeled with 224,225Ra for targeted alpha therapy. J Nanoparticle Res Interdiscip Forum Nanoscale Sci Technol. 2013;15:2082
100. Bandekar A, Zhu C, Jindal R, Bruchertseifer F, Morgenstern A, Sofou S. Anti-prostate-specific membrane antigen liposomes loaded with 225Ac for potential targeted antivascular α-particle therapy of cancer. J Nucl Med Off Publ Soc Nucl Med. 2014;55:107-14
101. McLaughlin MF, Woodward J, Boll RA. et al. Gold Coated Lanthanide Phosphate Nanoparticles for Targeted Alpha Generator Radiotherapy. PLOS ONE. 2013;8:e54531
102. Sofou S, Kappel BJ, Jaggi JS, McDevitt MR, Scheinberg DA, Sgouros G. Enhanced Retention of the α-Particle-Emitting Daughters of Actinium-225 by Liposome Carriers. Bioconjug Chem. 2007;18:2061-7
103. Henriksen G, Schoultz BW, Michaelsen TE, Bruland ØS, Larsen RH. Sterically stabilized liposomes as a carrier for alpha-emitting radium and actinium radionuclides. Nucl Med Biol. 2004;31:441-9
104. Zhu C, Bandekar A, Sempkowski M. et al. Nanoconjugation of PSMA-Targeting Ligands Enhances Perinuclear Localization and Improves Efficacy of Delivered Alpha-Particle Emitters against Tumor Endothelial Analogues. Mol Cancer Ther. 2016;15:106-13
105. Mdanda S, Ngema LM, Mdlophane A, Sathekge MM, Zeevaart JR. Recent Innovations and Nano-Delivery of Actinium-225: A Narrative Review. Pharmaceutics. 2023;15:1719
106. Pratt EC, Shaffer TM, Grimm J. Nanoparticles and Radiotracers: Advances toward Radio-Nanomedicine. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2016;8:872-90
107. Bailly C, Bodet-Milin C, Rousseau C, Faivre-Chauvet A, Kraeber-Bodéré F, Barbet J. Pretargeting for imaging and therapy in oncological nuclear medicine. EJNMMI Radiopharm Chem. 2017;2:6
108. Altai M, Membreno R, Cook B, Tolmachev V, Zeglis BM. Pretargeted Imaging and Therapy. J Nucl Med. 2017;58:1553-9
109. Hapuarachchige S, Artemov D. Theranostic Pretargeting Drug Delivery and Imaging Platforms in Cancer Precision Medicine. Front Oncol. 2020;10:1131
110. Hapuarachchige S, Huang CT, Donnelly MC. et al. Cellular Delivery of Bioorthogonal Pretargeting Therapeutics in PSMA-Positive Prostate Cancer. Mol Pharm. 2020;17:98-108
111. van Rij CM, Lütje S, Frielink C. et al. Pretargeted immuno-PET and radioimmunotherapy of prostate cancer with an anti-TROP-2 x anti-HSG bispecific antibody. Eur J Nucl Med Mol Imaging. 2013;40:1377-83
112. Cheal SM, McDevitt MR, Santich BH. et al. Alpha radioimmunotherapy using 225Ac-proteus-DOTA for solid tumors - safety at curative doses. Theranostics. 2020;10:11359-75
113. Królicki L, Bruchertseifer F, Kunikowska J. et al. Dose escalation study of targeted alpha therapy with [225Ac]Ac-DOTA-substance P in recurrence glioblastoma - safety and efficacy. Eur J Nucl Med Mol Imaging. 2021;48:3595-605
114. Cordier D, Forrer F, Bruchertseifer F. et al. Targeted alpha-radionuclide therapy of functionally critically located gliomas with 213Bi-DOTA-[Thi8,Met(O2)11]-substance P: a pilot trial. Eur J Nucl Med Mol Imaging. 2010;37:1335-44
115. Cooks T, Schmidt M, Bittan H. et al. Local Control of Lung Derived Tumors by Diffusing Alpha-Emitting Atoms Released From Intratumoral Wires Loaded With Radium-224. Int J Radiat Oncol. 2009;74:966-73
116. Horev-Drori G, Cooks T, Bittan H. et al. Local control of experimental malignant pancreatic tumors by treatment with a combination of chemotherapy and intratumoral 224Radium-loaded wires releasing alpha-emitting atoms. Transl Res. 2012;159:32-41
117. Pouget J-P, Constanzo J. Revisiting the Radiobiology of Targeted Alpha Therapy. Front Med. 2021;8:692436
118. Jurcic JG, Ravandi F, Pagel JM. et al. Phase I trial of α-particle therapy with actinium-225 (225Ac)-lintuzumab (anti-CD33) and low-dose cytarabine (LDAC) in older patients with untreated acute myeloid leukemia (AML). J Clin Oncol. 2015;33:7050-7050
119. Wadhwa A, Wang S, Patiño-Escobar B. et al. CD46-Targeted Theranostics for PET and 225Ac-Radiopharmaceutical Therapy of Multiple Myeloma. Clin Cancer Res. 2024;30:1009-21
120. Robertson AKH, Ramogida CF, Rodríguez-Rodríguez C. et al. Multi-isotope SPECT imaging of the 225Ac decay chain: feasibility studies. Phys Med Biol. 2017;62:4406
121. Peter R, Sandmaier BM, Dion MP. et al. Small-scale (sub-organ and cellular level) alpha-particle dosimetry methods using an iQID digital autoradiography imaging system. Sci Rep. 2022;12:17934
122. Frame E, Bobba K, Gunter D. et al. Coded aperture and Compton imaging for the development of 225Ac-based radiopharmaceuticals. Med Phys. 2023;50:6454-68
123. Usmani S, Rasheed R, Al kandari F, Marafi F, Naqvi SAR. 225Ac Prostate-Specific Membrane Antigen Posttherapy α Imaging: Comparing 2 and 3 Photopeaks. Clin Nucl Med. 2019;44:401
124. Vatsa R, Sood A, Vadi SK. et al. 225Ac-PSMA-617 Radioligand Posttherapy Imaging in Metastatic Castrate-Resistant Prostate Cancer Patient Using 3 Photopeaks. Clin Nucl Med. 2020;45:437
125. Delker A, Schleske M, Liubchenko G. et al. Biodistribution and dosimetry for combined [177Lu]Lu-PSMA-I&T/[225Ac]Ac-PSMA-I&T therapy using multi-isotope quantitative SPECT imaging. Eur J Nucl Med Mol Imaging. 2023;50:1280-90
126. Frantellizzi V, Cosma L, Brunotti G. et al. Targeted Alpha Therapy with Thorium-227. Cancer Biother Radiopharm. 2020;35:437-45
127. Baranowska-Kortylewicz J, Sharp JG, McGuire TR, Joshi S, Coulter DW. Alpha-Particle Therapy for Multifocal Osteosarcoma: A Hypothesis. Cancer Biother Radiopharm. 2020;35:418-24
128. Dahle J, Borrebæk J, Melhus KB. et al. Initial evaluation of 227Th-p-benzyl-DOTA-rituximab for low-dose rate α-particle radioimmunotherapy. Nucl Med Biol. 2006;33:271-9
129. Heyerdahl H, Abbas N, Brevik EM, Mollatt C, Dahle J. Fractionated Therapy of HER2-Expressing Breast and Ovarian Cancer Xenografts in Mice with Targeted Alpha Emitting 227Th-DOTA-p-benzyl-trastuzumab. PLOS ONE. 2012;7:e42345
130. Ramdahl T, Bonge-Hansen HT, Ryan OB. et al. An efficient chelator for complexation of thorium-227. Bioorg Med Chem Lett. 2016;26:4318-21
131. Hammer S, Hagemann UB, Zitzmann-Kolbe S. et al. Preclinical Efficacy of a PSMA-Targeted Thorium-227 Conjugate (PSMA-TTC), a Targeted Alpha Therapy for Prostate Cancer. Clin Cancer Res. 2020;26:1985-96
132. Hammer S, Schlicker A, Zitzmann-Kolbe S. et al. Darolutamide Potentiates the Antitumor Efficacy of a PSMA-targeted Thorium-227 Conjugate by a Dual Mode of Action in Prostate Cancer Models. Clin Cancer Res. 2021;27:4367-78
133. Schatz CA, Hammer S, Wengner A, Hagemann UB, Mumberg D, Scholz A. Abstract 1393: PSMA-Targeted Thorium Conjugate (BAY 2315497) and olaparib combination show synergistic anti-tumor activity in prostate cancer models. Cancer Res. 2021;81:1393
134. Böhnke N, Indrevoll B, Hammer S. et al. Mono- and multimeric PSMA-targeting small molecule-thorium-227 conjugates for optimized efficacy and biodistribution in preclinical models. Eur J Nucl Med Mol Imaging. 2024;51:669-80
135. Parker C, Nilsson S, Heinrich D. et al. Alpha Emitter Radium-223 and Survival in Metastatic Prostate Cancer. N Engl J Med. 2013;369:213-23
136. Henriksen G, Fisher DR, Roeske JC, Bruland OS, Larsen RH. Targeting of osseous sites with (alpha)-emitting (232)Ra: Comparison with the (beta)-emitter (89)Sr in Mice. J Nucl Med. 2003;44:252-9
137. Henriksen G, Breistøl K, Bruland ØS, Fodstad Ø, Larsen RH. Significant Antitumor Effect from Bone-seeking, α-Particle-emitting 223Ra Demonstrated in an Experimental Skeletal Metastases Model1. Cancer Res. 2002;62:3120-5
138. Nilsson S, Larsen RH, Fosså SD. et al. First Clinical Experience with α-Emitting Radium-223 in the Treatment of Skeletal Metastases. Clin Cancer Res. 2005;11:4451-9
139. Nilsson S, Franzén L, Parker C. et al. Bone-targeted radium-223 in symptomatic, hormone-refractory prostate cancer: a randomised, multicentre, placebo-controlled phase II study. Lancet Oncol. 2007;8:587-94
140. Sartor O, Coleman R, Nilsson S. et al. Effect of radium-223 dichloride on symptomatic skeletal events in patients with castration-resistant prostate cancer and bone metastases: results from a phase 3, double-blind, randomised trial. Lancet Oncol. 2014;15:738-46
141. Chakravarty R, Lan X, Chakraborty S, Cai W. Astatine-211 for PSMA-targeted α-radiation therapy of micrometastatic prostate cancer: a sustainable approach towards precision oncology. Eur J Nucl Med Mol Imaging. 2023;50:1844-7
142. Kiess AP, Minn I, Vaidyanathan G. et al. (2S)-2-(3-(1-Carboxy-5-(4-211At-Astatobenzamido)Pentyl)Ureido)-Pentanedioic Acid for PSMA-Targeted α-Particle Radiopharmaceutical Therapy. J Nucl Med. 2016;57:1569-75
143. Vaidyanathan G, Mease RC, Minn I. et al. Synthesis and preliminary evaluation of 211At-labeled inhibitors of prostate-specific membrane antigen for targeted alpha particle therapy of prostate cancer. Nucl Med Biol. 2021;94-95:67-80
144. Shirakami Y, Watabe T, Obata H. et al. Synthesis of [211At]4-astato-L-phenylalanine by dihydroxyboryl-astatine substitution reaction in aqueous solution. Sci Rep. 2021;11:12982
145. Mease RC, Kang CM, Kumar V. et al. An Improved 211At-Labeled Agent for PSMA-Targeted α-Therapy. J Nucl Med. 2022;63:259-67
146. Watabe T, Kaneda-Nakashima K, Shirakami Y. et al. Targeted α-therapy using astatine (211At)-labeled PSMA1, 5, and 6: a preclinical evaluation as a novel compound. Eur J Nucl Med Mol Imaging. 2023;50:849-58
147. Aoki M, Zhao S, Takahashi K. et al. Preliminary Evaluation of Astatine-211-Labeled Bombesin Derivatives for Targeted Alpha Therapy. Chem Pharm Bull (Tokyo). 2020;68:538-45
148. Bäck TA, Jennbacken K, Hagberg Thulin M. et al. Targeted alpha therapy with astatine-211-labeled anti-PSCA A11 minibody shows antitumor efficacy in prostate cancer xenografts and bone microtumors. EJNMMI Res. 2020;10:10
149. Imam SK. Advancements in cancer therapy with alpha-emitters: a review. Int J Radiat Oncol. 2001;51:271-8
150. McDevitt MR, Finn RD, Ma D, Larson SM, Scheinberg DA. Preparation of α-Emitting 213Bi-Labeled Antibody Constructs for Clinical Use. J Nucl Med. 1999;40:1722-7
151. McDevitt MR, Barendswaard E, Ma D. et al. An α-Particle Emitting Antibody ([213Bi]J591) for Radioimmunotherapy of Prostate Cancer1. Cancer Res. 2000;60:6095-100
152. Ballangrud ÅM, Yang W-H, Charlton DE. et al. Response of LNCaP Spheroids after Treatment with an α-Particle Emitter (213Bi)-labeled Anti-Prostate-specific Membrane Antigen Antibody (J591)1. Cancer Res. 2001;61:2008-14
153. Li Y, Tian Z, Rizvi SMA, Bander NH, Allen BJ. In vitro and preclinical targeted alpha therapy of human prostate cancer with Bi-213 labeled J591 antibody against the prostate specific membrane antigen. Prostate Cancer Prostatic Dis. 2002;5:36-46
154. Li Y, Rizvi S, Ranson M, Allen B. 213Bi-PAI2 conjugate selectively induces apoptosis in PC3 metastatic prostate cancer cell line and shows anti-cancer activity in a xenograft animal model. Br J Cancer. 2002.
155. Li Y, Abbas Rizvi SM, Blair (Nee Brown) JM. et al. Antigenic expression of human metastatic prostate cancer cell lines for in vitro multiple-targeted α-therapy with 213Bi-conjugates. Int J Radiat Oncol. 2004;60:896-908
156. Wang J, Abbas Rizvi SM, Madigan MC. et al. Control of prostate cancer spheroid growth using 213 Bi-labeled multiple targeted α radioimmunoconjugates: Targeted Alpha Therapy for Human Prostate Cancer. The Prostate. 2006;66:1753-67
157. Li Y, Song E, Abbas Rizvi SM. et al. Inhibition of Micrometastatic Prostate Cancer Cell Spread in Animal Models By 213Bilabeled Multiple Targeted α Radioimmunoconjugates. Clin Cancer Res. 2009;15:865-75
158. Rizvi SMA, Li Y, Song EY. et al. Preclinical studies of bismuth-213 labeled plasminogen activator inhibitor type 2 (PAI2) in a prostate cancer nude mouse xenograft model. Cancer Biol Ther. 2006;5:386-93
159. Banerjee SR, Lisok A, Minn I. et al. Preclinical Evaluation of 213Bi- and 225Ac-Labeled Low-Molecular-Weight Compounds for Radiopharmaceutical Therapy of Prostate Cancer. J Nucl Med. 2021;62:980-8
160. Nonnekens J, Chatalic KLS, Molkenboer-Kuenen JDM. et al. 213 Bi-Labeled Prostate-Specific Membrane Antigen-Targeting Agents Induce DNA Double-Strand Breaks in Prostate Cancer Xenografts. Cancer Biother Radiopharm. 2017;32:67-73
161. Sathekge M, Knoesen O, Meckel M, Modiselle M, Vorster M, Marx S. 213Bi-PSMA-617 targeted alpha-radionuclide therapy in metastatic castration-resistant prostate cancer. Eur J Nucl Med Mol Imaging. 2017;44:1099-100
162. Kratochwil C, Schmidt K, Afshar-Oromieh A. et al. Targeted alpha therapy of mCRPC: Dosimetry estimate of 213Bismuth-PSMA-617. Eur J Nucl Med Mol Imaging. 2018;45:31-7
163. Kratochwil C, Giesel FL, Bruchertseifer F. et al. 213Bi-DOTATOC receptor-targeted alpha-radionuclide therapy induces remission in neuroendocrine tumours refractory to beta radiation: a first-in-human experience. Eur J Nucl Med Mol Imaging. 2014;41:2106-19
164. Makvandi M, Dupis E, Engle JW. et al. Alpha-Emitters and Targeted Alpha Therapy in Oncology: from Basic Science to Clinical Investigations. Target Oncol. 2018;13:189-203
165. Meredith RF, Torgue JJ, Rozgaja TA. et al. Safety and Outcome Measures of First-in-Human Intraperitoneal α Radioimmunotherapy With 212Pb-TCMC-Trastuzumab. Am J Clin Oncol. 2018;41:716
166. Meredith RF, Torgue J, Azure MT. et al. Pharmacokinetics and Imaging of 212Pb-TCMC-Trastuzumab After Intraperitoneal Administration in Ovarian Cancer Patients. Cancer Biother Radiopharm. 2014;29:12-7
167. Tan Z, Chen P, Schneider N. et al. Significant systemic therapeutic effects of high-LET immunoradiation by 212Pb-trastuzumab against prostatic tumors of androgen-independent human prostate cancer in mice. Int J Oncol. 2012;40:1881-8
168. dos Santos JC, Schäfer M, Bauder-Wüst U. et al. Development and dosimetry of 203Pb/212Pb-labelled PSMA ligands: bringing “the lead” into PSMA-targeted alpha therapy? Eur J Nucl Med Mol Imaging. 2019;46:1081-91
169. Müller C, Vermeulen C, Köster U. et al. Alpha-PET with terbium-149: evidence and perspectives for radiotheragnostics. EJNMMI Radiopharm Chem. 2017;1:5
170. Umbricht CA, Köster U, Bernhardt P. et al. Alpha-PET for Prostate Cancer: Preclinical investigation using 149Tb-PSMA-617. Sci Rep. 2019;9:17800
171. Naskar N, Lahiri S. Theranostic Terbium Radioisotopes: Challenges in Production for Clinical Application. Front Med. 2021;8:675014
172. Miederer M, Seidl C, Beyer G-J. et al. Comparison of the Radiotoxicity of Two Alpha-Particle-Emitting Immunoconjugates, Terbium-149 and Bismuth-213, Directed against a Tumor-Specific, Exon 9 Deleted (d9) E-Cadherin Adhesion Protein. Radiat Res. 2003;159:612-20
173. Beyer G-J, Miederer M, Vranješ-Đurić S. et al. Targeted alpha therapy in vivo: direct evidence for single cancer cell kill using 149Tb-rituximab. Eur J Nucl Med Mol Imaging. 2004;31:547-54
Author contact
![]() Corresponding author: Robert R. Flavell, Department of Radiology and Biomedical Imaging, University of California, San Francisco, CA-94107, tel: 415-353-3638; email: robert.flavelledu.
Corresponding author: Robert R. Flavell, Department of Radiology and Biomedical Imaging, University of California, San Francisco, CA-94107, tel: 415-353-3638; email: robert.flavelledu.