Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Strategies for spatial...
3. ST for disease understanding...
4. ST for drug research
5. Challenges and prospects
6. Conclusion
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2024; 14(7):2946-2968. doi:10.7150/thno.95908 This issue Cite
Review
Spatial Transcriptomics: A Powerful Tool in Disease Understanding and Drug Discovery
Junxian Cao1,4, Caifeng Li1, Zhao Cui2, Shiwen Deng1,4, Tong Lei3, Wei Liu1 ![]() , Hongjun Yang1,2,4
, Hongjun Yang1,2,4 ![]() , Peng Chen1,4
, Peng Chen1,4 ![]()
1. Beijing Key Laboratory of Traditional Chinese Medicine Basic Research on Prevention and Treatment for Major Diseases, Experimental Research Center, China Academy of Chinese Medical Sciences, Beijing 100700, China.
2. Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing 100700, China
3. Institute of Basic Theory for Chinese Medicine, China Academy of Chinese Medical Sciences, Beijing 100700, China.
4. Analysis of Complex Effects of Proprietary Chinese Medicine, Hunan Provincial Key Laboratory, Yongzhou City, Hunan Province, China.
Received 2024-3-4; Accepted 2024-4-25; Published 2024-5-11
Abstract

Recent advancements in modern science have provided robust tools for drug discovery. The rapid development of transcriptome sequencing technologies has given rise to single-cell transcriptomics and single-nucleus transcriptomics, increasing the accuracy of sequencing and accelerating the drug discovery process. With the evolution of single-cell transcriptomics, spatial transcriptomics (ST) technology has emerged as a derivative approach. Spatial transcriptomics has emerged as a hot topic in the field of omics research in recent years; it not only provides information on gene expression levels but also offers spatial information on gene expression. This technology has shown tremendous potential in research on disease understanding and drug discovery. In this article, we introduce the analytical strategies of spatial transcriptomics and review its applications in novel target discovery and drug mechanism unravelling. Moreover, we discuss the current challenges and issues in this research field that need to be addressed. In conclusion, spatial transcriptomics offers a new perspective for drug discovery.
1. Introduction
Spatial transcriptomics (ST) is used to explore the spatial gene expression patterns of cells or tissues. It combines traditional histological techniques with high-throughput RNA sequencing to visualize and quantitatively analyse the transcriptome with spatial distribution in tissue sections [1]. In single-cell sequencing, cells are prone to dissociate in suspension, leading to the loss of positional information [2]. However, ST preserve the spatial information of RNA in tissue sections by mapping it to specific spatial locations and subsequently subjecting it to high-throughput sequencing, thus providing spatial information on the transcriptome [3]. ST can help uncover cellular and tissue heterogeneity, enabling a deeper understanding of the diversity within tissues, identification of signature genes for specific cell types, and exploration of intercellular interactions [4]. This technology offers a novel perspective for understanding the spatial variations and distributions of gene expression.
With the emergence of spatial transcriptomics, our ability to understand drug actions and discover new medications has been revolutionized. By integrating ST, gene expression profiles and spatial distribution data can be obtained while preserving the integrity of tissue structures [5]. This study allows for a deeper understanding of the processes and molecular mechanisms of drug action within the body, further advancing research into the dynamic panorama of drug effects. The application of ST in the field of drug research has significantly broadened our perspectives, paving the way for groundbreaking drug discoveries with far-reaching impacts.
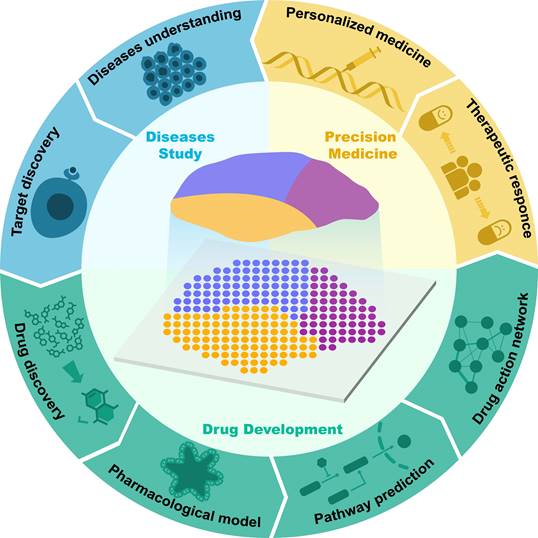
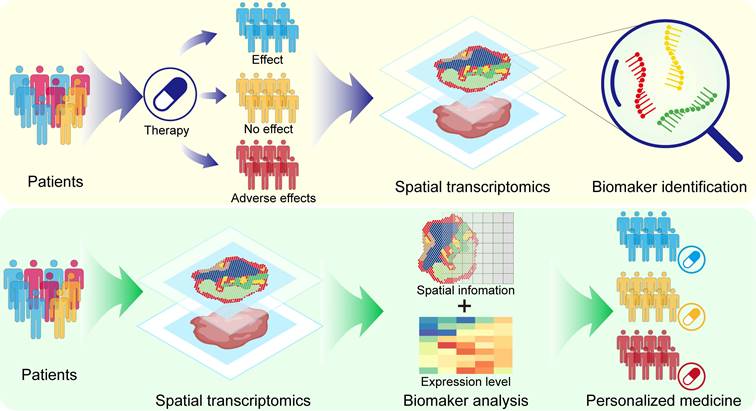
Currently, the applications of single-cell transcriptomics, proteomics, metabolomics, and genomics in drug discovery have been extensively reviewed [6-9]. However, most of the current reviews are still focused on discussing the application of ST in disease studies, and there is still a lack of comprehensive summaries concerning the application of ST in drug discovery and development. Therefore, in this article, we review the latest advancements in the use of ST in drug discovery and development. First, we outline the key technological and analytical research progress of current ST, encompassing methods based on microanatomy, in situ capture, and other approaches, while elucidating the strengths and limitations of each method. Subsequently, we introduce the applications of ST in disease research and target discovery and discuss the advancements and limitations of current ST analysis technologies. Additionally, we review classic cases of ST applications in drug development research, discussing their crucial contributions to new drug discovery, pharmacological model construction, drug action pathway research, and spatially dynamic drug action studies. We also discuss the application of ST in personalized therapy (Figure 1). Finally, we provide prospects for the future opportunities and challenges faced by ST in drug development.
Application of spatial transcriptomics in drug research and development.
2. Strategies for spatial transcriptomics
As omics technologies flourish, single-cell transcriptomics has greatly advanced drug discovery and development. However, single-cell transcriptomics relies on the isolation of individual cells, inevitably leading to the loss of the original spatial information within the studied samples. The emergence of ST overcomes this limitation, allowing scientists to obtain spatial distribution information on the transcriptome within research samples. The essence of ST lies in unravelling the spatial localization of detected RNA molecules. Since the concept of ST was first proposed by Joakim Lundeberg's team in 2016 [1], ST has integrated various technologies and developed diverse technical approaches, with both throughput and resolution rapidly increasing. Currently, ST has extensive applications in various fields, such as cancer research [10], developmental biology [11], pathology [12], and toxicology [13]. Notably, in 2021, ST was recognized as the "Method of the Year" by Nature Methods [14]. In this section, we provide a comprehensive review of mainstream ST research strategies and summarize the strengths and limitations of these methods (Figure 2 and Table 1).
Current mainstream spatial transcriptomics methods. ISC involves the specific binding of mRNA from corresponding cellular locations with spots or beads on slides carrying capture probes, followed by further NGS of the captured RNA molecules. ISC offers high sensitivity and specificity, enabling the detection of rare transcripts and precise spatial localization, while achieving single-cell resolution with this technology is currently challenging. Image-based methods, including ISH and ISS. ISH involves the specific hybridization of RNA in tissues with multiple fluorescently labelled short oligonucleotide probes, reflecting the abundance and spatial localization of specific gene transcripts. ISH detects low-abundance transcripts with high sensitivity and relatively high resolution, while the limited signal-to-noise ratio during imaging necessitates high-magnification imaging systems, limiting the area of detection. ISS involves the hybridization of padlock probes with cDNA, followed by RCA to form RCP. ISS allows for the sequencing of RNA molecules directly on tissue sections, providing higher-resolution data, while it has lower sensitivity, and padlock probes may introduce significant probe-specific biases, increasing the complexity of image processing.
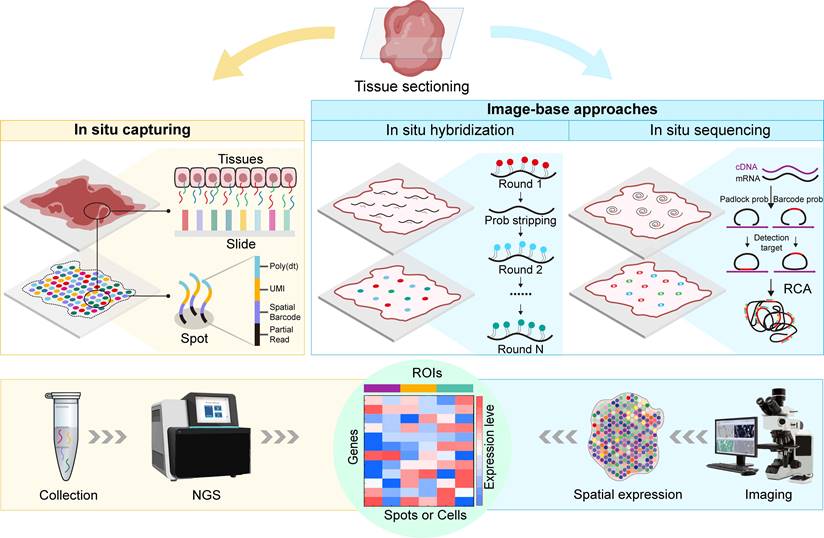
Comparison of different spatial transcriptomics methods.
| Method | Year | Resolution | Probes | Sample | Advantage | Limitation |
|---|---|---|---|---|---|---|
| In situ capture | ||||||
| ST | 2016 | 100 µm | Oligo probes | Fresh-frozen tissue | High throughput | Lower resolution |
| APEX-seq | 2019 | Subcellular (<10 µm) | Enzymatic tagging | Mammalian cells | Captures in situ activity; High temporal resolution | Requires specific enzyme for taggingLimited to cells with enzymatic activity |
| DSP | 2019 | 10 µm | DNA Oligo probes | FFPEFrozen tissue | Profile both RNA and proteinsFlexibility in ROI selection | Limited to predefined panelsSpecialized equipment needed |
| HDST | 2019 | 10-100 µm | Barcoded beads | Fresh-frozen tissue | High resolution Analyze large tissue areas | |
| Slide-seq | 2019 | 100-200 µm | Unique molecular identifiers (UMIs) | Fresh-frozen tissue | High resolutionHigh sensitivity | |
| DBIT-SEQ | 2020 | Subcellular (<10 µm) | Barcoded probes | Fresh-frozen tissue | High resolutionCaptures both mRNA and protein | |
| Seq-Scope | 2021 | Subcellular (<10 µm) | RNA probes | Fresh-frozen tissue | High resolutionHigh compatibility | |
| Slide-seqV2 | 2021 | 10-20 µm | Barcoded probes | Fresh-frozen tissue | Improved resolution over Slide-seq;Can detect low-abundance transcripts | |
| Stereo-seq | 2022 | Subcellular (<10 µm) | Expansion microscopy | Fresh-frozen tissue | 3D imaging capability | |
| Visium HD | 2024 | 55 µm | Bead-based | FFPE, frozen tissue | High throughputAnalyze large tissue areas | |
| Imaging-Based approaches | ||||||
| ISH | ||||||
| FISH | 1969 | 10-20 nm | DNA or RNA probes | Fixed cells or tissues | High specificity for DNA or RNA targets | Small number of targetsCross-hybridization and autofluorescence |
| smFISH | 2011 | Single-molecule | Oligo-dT probes | Fixed or live cells | Visualization of individual mRNA moleculesQuantitative analysis | Limited by the number of fluorophoresComplex imaging required |
| seqFISH | 2014 | Single-cell | Multiplexed probes | Fixed cells | Enables the detection of thousands of RNA molecules in a highly multiplexed manner | Complex image analysisHigher background signal |
| MERFISH | 2015 | Single-cell | Error-robust barcodes | Fixed cells | High multiplexing capabilityError correction | Requires high-quality imaging equipment and expertise |
| osmFISH | 2018 | Single-molecule | Multiplexed probes | Fixed cells | High multiplexingHigh sensitivity | Low signal-to-noise ratioRequire optimization for different samples |
| EEL FISH | 2022 | Single-cell | High-density probes | Fixed cells | High spatial resolutionLarge-scale gene expression analysis | |
| ISS | ||||||
| FISSEQ | 2014 | Subcellular (<10 µm) | Fluorescently labeled probes | Fixed cells | Captures all types of RNA in situSubcellular levels | Lower throughputSmall field of view |
| BaristaSeq | 2018 | Single-cell | Padlock probes | Fresh-frozen tissue | High amplification efficiencySequencing accuracy of at least 97% | Requires specific equipment and expertise |
| STARmap | 2018 | Subcellular (200-300 nm) | Optimized probes | Fresh-frozen tissue | High-resolution 3D intact-tissue sequencing | Complex sample preparationNot suitable for all types of samples |
| BARseq | 2019 | Single neuron level | Viral encoding RNA barcodes | Frozen tissue, fixed tissue | High throughputNeuronal markers | Only for neuron |
| MERSCOPE | 2022 | 100nm | Fluorescently labeled probes | FFPEFrozen tissue | High throughputNo sequencing required | |
| Xenium | 2022 | Subcellular (<10 µm) | Padlock probes | Fresh-frozen tissue | High sensitivity and specificitySupports customized gene panels | |
| Other approaches | ||||||
| LCM | 1996 | Cell-level | None | Frozen or FFPE tissue sections | High precision in isolating specific cell types | Lower throughputLimited by size and structure of the tissue |
| TSCS | 2016 | Single-cell | None | FFPE | Assesses genomic copy number variations | Lower sequencing depth |
| TIVA | 2014 | Single-cell | None | Live tissue | ST analysis in vivoCaptures dynamic gene expression | Limited by the number of photoactivatable tags |
| RAINBOW-seq | 2015 | ~1 micrometer | DNA probes | Fresh-frozen tissue | Enables multi-color imaging and quantitative analysis of mRNA | Complex probe design |
2.1 In situ capture (ISC)
Fundamentally, in situ capture (ISC) involves the in situ labelling of RNA molecules within designated regions of interest (ROIs) using spatial barcodes before library preparation, followed by the extraction and spatially resolved sequencing of the captured RNA molecules via next-generation sequencing (NGS). The ST strategy was initially proposed by Joakim Lundeberg's team, who applied this method to reveal spatial information on gene expression in mouse brain and human breast cancer tissues [1]. In this approach, the authors immobilize reverse-transcription oligo primers with spatial barcodes onto a chip and subsequently perform tissue sectioning and reverse transcription to obtain cDNA. The spatial positions were recorded by comparing the fluorescence signal of fluorescently labelled DNA with hematoxylin and eosin staining of the tissues. Subsequently, next-generation sequencing was performed ex situ, and the spatial barcode-containing RNA-seq data were aligned to the corresponding positions in the image to visualize the transcriptome information. In 2018, 10X Genomics acquired this technology and launched a commercial ST platform called "Visium Spatial Gene Expression", which improved the resolution from the original 100 µm spot diameter to 55 µm, reduced the spot-to-spot distance from 200 µm to 100 µm, and shortened the processing time [15].
In the Slide-seq method [16], microbeads covered with DNA barcodes are densely arrayed onto a glass slide, with each microbead bearing a distinct spatial identifier [17]. Subsequently, RNA from tissue sections can be captured on these microbeads, followed by sequencing of the barcode RNA library using single-cell RNA sequencing (scRNA-seq). The scRNA-seq results are then mapped onto the Slide-seq data, allowing for comprehensive integration of spatial and single-cell transcriptomics information with non-negative matrix factorization regression (NMFreg) [18]. The size of these microbeads is only 10 μm, allowing minimal lateral diffusion of mRNA from the tissue to the microbeads. This innovation enables the visualization of high-resolution, high-throughput ST. In 2020, the research team introduced a new generation of Slide-seq, named Slide-seqV2. Compared with its predecessor, Slide-seqV2 has undergone enhancements in library preparation, microbead synthesis, and array indexing. These improvements have resulted in a tenfold increase in RNA capture efficiency for Slide-seqV2, offering a substantial advancement over the first generation [19].
Shortly after the publication of Slide-seq, another technique utilizing even smaller barcode-bearing microbeads emerged, named high-definition spatial transcriptomics (HDST) [20]. HDST employs a similar principle as Slide-seq but features high-density microbeads with a diameter as small as 2 μm on the chip array. Traditional ST platforms mainly face the problem of insufficient spatial resolution, as multiple cells are captured in one spot. The latest ST platforms provide smaller and denser spots, which theoretically have the potential to capture more mRNA in an area, yielding an even higher spatial resolution [21]. However, higher spatial resolution does not necessarily improve the capacity to resolve individual cells. Two different cells can still be captured even if subcellular resolution (less than 10 μm) is achieved; therefore, the main challenge of current high spatial resolution platforms lies in cell separation and spot allocation issues [22].
In contrast to strategies involving the embedding of spatial information with microbeads, APEX-seq is an ST method that relies on the direct labelling of RNA with the peroxidase enzyme APEX2 [23]. APEX-seq enables the precise localization of endogenous RNA within individual live cells, achieving the remarkable resolution of single-nucleotide RNA sequence information [24]. Nevertheless, a drawback of APEX-seq lies in the requirement for APEX to be recombinantly expressed within target cells, thereby limiting its applicability within normal human tissues.
The Seq-Scope is a submicron resolution ST based on the Illumina sequencing platform that involves a two-round sequencing process [25]. In the first round, cDNA is solid-phase amplified to generate spatial barcodes on the capture array [26]. In the second round, mRNA molecules captured on the physical array are released and sequenced. This approach achieves a spatial resolution of 0.5 to 0.8 μm.
The progression of other in situ capture-based ST methods remains ongoing. Noteworthy examples include DBIT-SEQ [27], which leverages microfluidic barcode labelling for spatial transcriptomic sequencing. Pixel-seq achieves a high-density DNA chip with a resolution of 1 μm at lower cost by repetitive printing on the surface of a polyacrylamide gel [28]. Stereo-seq uses a randomly synthesized 25-nucleotide sequence as a spatial coordinate (coordinate identity, CID), which is amplified through rolling-circle replication to form DNA [29]. These DNBs are then attached to the Stereo-seq Chip, followed by sequencing of the DNBs on the chip using the DNBSEQ sequencer [30]. The Stereo-seq chip used for capturing mRNA has regularly arranged spots, with each spot having a diameter of approximately 220 nm and a center-to-center distance between spots of 500 nm. The GeoMx Digital Spatial Profiler (DSP) introduced by NanoString Technologies is a probe-based technology that attaches DNA oligos to antibodies or RNA, with each DNA oligo corresponding to a target [31]. It can detect up to 96 protein targets or more than 1000 RNA targets. DSP enables users to select ROIs based on tissue outlines on a single slide while maintaining spatial context with cellular-level resolution. The ongoing development of these methodologies holds promise for advancing our understanding of spatial gene expression patterns.
2.2 Imaging-based approaches
Imaging-based ST can be taxonomically classified into two primary categories based on the methodology employed for RNA detection: in situ sequencing (ISS) and in situ hybridization (ISH). The fundamental principle of ISH involves the utilization of complementary fluorescently labelled probes to perform in situ hybridization with target RNA, enabling both quantitative detection and spatial localization of the target RNA molecules [32]. The origins of ISH can be traced back to 1969 when Pardue and Gall pioneered the use of isotopically labelled nucleic acid probes to ascertain the abundance and spatial distribution of DNA and RNA within tissues and cells, a pioneering technique also known as fluorescence in situ hybridization (FISH) [33]. Early FISH approaches could only detect individual transcripts. Femino et al. combined FISH with digital microscopy imaging, targeting RNA molecules with oligonucleotide probes synthesized with five fluorescent dyes per molecule, thus achieving the detection of single RNA molecules [34]. While this method exhibits high sensitivity and achieves subcellular resolution, it is limited by its resolution, allowing the simultaneous measurement of only a few mRNA molecules. Subsequently, Raj et al. improved this method by using multiple short, single-labelled oligonucleotide probes to detect each mRNA type, achieving more efficient in situ hybridization, and they named this method single-molecule fluorescence in situ hybridization (smFISH) [35]. While smFISH offers high sensitivity and subcellular spatial resolution, it is limited in its capacity to target only a few genes simultaneously due to spectral overlap constraints in standard microscopy. Therefore, researchers have turned their attention to multiplexed smFISH techniques, such as sequential hybridization FISH (seqFISH) introduced in 2018, which involves multiple rounds of hybridization, imaging, and probe stripping, allowing for the sequential detection of individual transcripts [36]. However, repeated hybridization of multiple transcripts is expensive and time-consuming. Cyclic-Oroboros smFISH (osmFISH) is a cyclic hybridization method involving multiple rounds of hybridization with probe removal after each imaging round before the next hybridization [37]. The multiplexed error-robust fluorescence in situ hybridization (MERFISH) technique, introduced in 2015, uses a combination of FISH labelling and error-robust encoding, enabling the analysis of hundreds or even thousands of RNA molecules within cells [38]. Combining MERFISH with super-resolution microscopy significantly increases the total density of measurable RNA [39]. DNA microscopy is a novel imaging method introduced in 2019 that relies on molecular thermodynamic entropy imaging instead of optical devices for spatial capture through the encoding of physical images into DNA and the generation of physical images of the original transcripts with precise sequence information, enabling the observation of cells at the genomic level [40]. Recently developed electrically enhanced fluorescence in situ hybridization (EEL FISH) methods can be used to capture RNA on a glass surface via electrophoresis and enable imaging after tissue removal, thereby facilitating accelerated imaging while preserving single-cell resolution [41].
Current in situ sequencing (ISS) technology usually relies on RNA as a probe ligation template. ISS employs padlock probes that hybridize to target RNA in tissues, circularize after hybridization, and produce numerous copies of the original probe through rolling circle amplification (RCA), which are then sequenced and visualized using fluorescence or chromogenic detection methods [42,43]. With the aid of microfluidic platforms, ISS technology has achieved automation, significantly enhancing efficiency and enabling the simultaneous detection of thousands of RNA species [44]. However, ISS technology based on gap-fill padlock probes is limited to targeted sequencing. Fluorescent in situ RNA sequencing (FISSEQ) achieves non-targeted detection of more than 8,000 RNA species in fibroblasts by reading 27 bases of each transcript [45]. Spatially resolved transcript amplicon readout mapping (STARmap) combines ISS technology with hydrogel chemistry, utilizing dynamic annealing and cross-linking to reduce sequencing errors, enabling the simultaneous analysis of more than one thousand genes and providing three-dimensional images of complete tissues [46]. While sequencing technologies based on padlock probes have low efficiency in reading barcodes, BaristaSeq combines the Illumina sequencing platform with a simultaneous synthesis and sequencing strategy to enhance sequencing efficiency and accuracy [47]. Subsequently, researchers further developed BARseq based on this approach [48]. Moreover, many companies have also introduced commercial platforms based on image-based ST and applied them to research, including MERSCOPE [49], Xenxium [50], GeoMX, and CosMX [51].
2.3 Other approaches
In addition to the mainstream methods mentioned before, as an emerging research field, new research strategies in the field of ST continue to emerge. For example, microscopic isolation approaches involve cutting tissue regions under a microscope, isolating individual cells or entire target areas, and then combining them with NGS for ST [52]. Laser capture microdissection (LCM) uses a laser beam to cut tissue regions under a microscope for spatial transcriptomic analysis [52]. Geographical position sequencing (GEO-seq) technology integrates LCM with scRNA-seq to analyse the entire transcriptome of a small number of cells while preserving their original spatial positions through zip-code genes [53]. Tomo-seq involves freezing tissue for sectioning and performing ex vivo transcriptional amplification on each section, thereby obtaining three-dimensional spatial transcriptomic information [54]. Topographic single-cell sequencing (TSCS) technology involves placing tissue slices on glass slides, using an LCM system to capture individual cells from the tissue slices while concurrently recording their coordinates, and subsequently performing next-generation sequencing (NGS) and single-nucleus sequencing [55]. A spatial transcriptomic approach based on transcriptome in vivo analysis (TIVA) uses light-activated biotin tags to enter living cells through cell-penetrating peptides, photoactivates the target regions to capture mRNA, and then performs RNA-seq [56,57]. Only a few cells can be analysed at a time via this method, which limits its application in high-throughput ST. Additionally, researchers have developed an ST method named RAINBOW-seq, which is specifically designed for colony analysis [58]. While these methods may currently have various limitations, the field of ST is experiencing rapid growth, and over time, these technologies may prove to be unexpectedly valuable in specific research domains.
3. ST for disease understanding and novel target identification
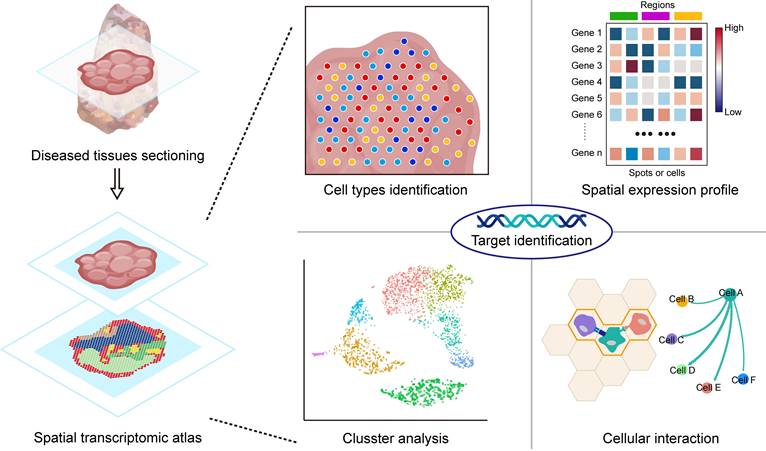
As multi-omics technologies continue to progress, the landscape of drug target discovery has evolved significantly [59]. Multi-omics approaches, such as genomics [60], proteomics [61], and transcriptomics [62], offer an in-depth perspective for unravelling the molecular mechanisms underlying diseases. In contrast to conventional transcriptomics, the emerging field of ST offers distinct advantages by preserving spatial information within transcripts [63]. Mapping the gene expression patterns in specific tissues and cell regions enables the identification of interactions between different cell types in different areas, including signalling pathways, cell adhesion, and cell-cell communication [64,65] (Figure 3). This innovative approach revealed genes exhibiting heightened expression levels in specific cells or subcellular structures within regions of interest, revealing potential targets for therapeutic intervention [66,67]. In the ensuing section, our focus centers on an extensive review of the applications of ST in the discovery of novel drug targets.
Application of spatial transcriptomics in target identification. By identifying cell types through spatial ST, it is possible to reveal the distribution of specific cell types within tissues and understand their function and state. The spatial expression profile depicts the expression of mRNAs in spatial dimensions, aiding in the identification of characteristic DEGs involved in disease processes. Clustering analysis helps in discovering specific cell populations associated with diseases that may be closely linked to the occurrence, progression, or prognosis of the disease. By analysing interactions between cells, key interactions involved in cell communication and signal transduction can be identified. Integrating this information can provide insights into the process of disease occurrence and reveal potential therapeutic targets.
3.1 Cancers
Tumor heterogeneity results in the high complexity and diversity of malignant tumors during their evolution, while drug resistance caused by tumor heterogeneity is a major challenge in current cancer research and antitumour drug discovery [68-70]. ST demonstrated excellent performance in identifying cell types within different tumor regions. Glioblastoma (GBM) is a common primary malignant tumor. However, current therapeutic modalities have shown limited efficacy in improving patient survival rates. ST analysis revealed the overexpression of EphA3 and ephrinA5 in certain regions of GBM tumors, with mutually exclusive expression patterns in different tumor regions, which are associated with the formation and induced differentiation of GBM cells [71]. EphrinA5 is a high-affinity ligand of EphA3, and EphA3 was identified as a target in GBM in a previous study [72]. Therefore, targeting the regulation of EphA3 and ephrinA5 could offer a more sustained and effective therapeutic approach. The integration of ST with single-cell transcriptomics has enabled the discovery and localization of novel cellular subtypes within tumors. Through scRNA-seq, a specific tumor-specific keratinocyte (TSK) has been identified as a distinct cell type unique to cutaneous squamous cell carcinoma, and the frontier heterogeneity of this cell cluster has been elucidated with ST, revealing its pivotal role as a hub for intercellular communication [73].
ST offers a significant advantage by facilitating comprehension of the spatial arrangement of diverse cell types within the tumor microenvironment (TME) and furnishing crucial spatial details concerning the transcriptomic landscape of tumor cells. This capability substantially enhances our understanding of cellular interactions and spatial impacts within the TME. Metastasis is a crucial factor contributing to high cancer mortality rates and is closely associated with drug resistance [74]. Therefore, identifying key genes and pathways involved in metastasis may reveal potential therapeutic targets for cancer treatment. Brain metastasis (BrMs) is the main component of malignant cancers of the central nervous system. To understand the molecular and cellular mechanisms of lung cancer brain metastasis, researchers have mapped a comprehensive transcriptional atlas of brain metastasis in non-small cell lung cancer with ST analysis on tumor samples from 44 patients [75]. Analysis of the ROIs revealed that the TME plays a pivotal role in metastasis and that extensive remodelling occurs during the metastatic process. Within the TME, there is a notable increase in fibrotic factors, fostering an immune-suppressed and fibrotic milieu that supports the development of BrMs. Elevated levels of fibrotic factors such as PDGFRβ, CXCR4, and TGFB1 are associated with promoting tumor growth and angiogenesis [76]. Consequently, the adjustment of treatment strategies based on the fibrotic state has become crucial, but targeting fibrosis-regulating factors in fibrotic BrMs has emerged as a potential therapeutic approach. Pancreatic ductal adenocarcinoma is a common digestive tract cancer known for its rapid metastasis and poor prognosis. Through whole-transcriptome digital spatial profiling, a neural-like progenitor molecular program present in cancer cells that persists after treatment has been identified. These unique receptor-ligand interactions confer resistance to therapies on these tumor cells [77]. This study revealed that this gene is a promising potential target for novel therapies.
3.2 Neurological diseases
Neurological disorders encompass a variety of cell types, and previous research utilizing scRNA-seq has offered novel insights into disease comprehension at the molecular level. As most complex neurological diseases involve interactions of different brain regions, ST can significantly advance target discovery by discerning transcriptional information and identifying cell types in distinct functional regions. For example, ST has been applied to elucidate the underlying mechanisms and identify potential therapeutic targets for conditions such as amyotrophic lateral sclerosis (ALS) [78], Alzheimer's disease (AD) [79], multiple sclerosis (MS) [80], and Parkinson's disease (PD) [81]. ST has been applied to construct a spatiotemporal map of ALS, wherein a discernible reduction in the expression module associated with various biological pathways, encompassing myelination, retrograde endocannabinoid signalling, and WNT signalling, was observed in spinal cord slices located proximal to the onset of symptoms [78]. This observation suggests the potential utility of myelination signalling modulators as a therapeutic avenue for ALS. Earlier research leveraged the ST to uncover novel targets within the hippocampal and olfactory bulb layers of AD mouse models [82]. Furthermore, subsequent studies employing ST identified differentially expressed genes in layer 2/3 of the middle temporal gyrus among postmortem individuals with and without AD [83]. Additionally, the use of the ST has provided insights into the treatment of progressive MS in conjunction with proteomic analysis. Through the tracking of changes in gene and protein pairs across spatially distributed stages of neurodegeneration, novel target candidates for progressive MS, including GPR37L1, TYRO3, SIRPA, and FGFR3, were identified [80]. Alterations in gene expression within distinct regions of the PD-affected hippocampus have also been revealed in the ST. Differential expression gene analyses based on ST imply that upregulated genes primarily contribute to processes such as neuron-to-neuron synapse function, vesicle-mediated transport in synapses, the calcium signalling pathway, and pathways associated with neurodegenerative diseases. Conversely, downregulated genes are predominantly linked to ATP metabolism and the GnRH signalling pathway [81]. The key genes identified in this study hold promise as potential novel therapeutic targets for PD treatment. In addition to its applications in the study of neurodegenerative diseases, ST analysis of cortical layers in a depression-like macaque model revealed discrete gene expression regions involved in the regulation of diverse emotional states, suggesting potential targets for the modulation of depressive emotions [84]. Furthermore, the ST has shed light on the complexity of schizophrenia-associated genes, revealing intricate intracellular signalling networks rather than isolated individual genes [85]. Thus, the exploration of multitarget therapeutic approaches for schizophrenia merits serious consideration. Moreover, the CCL5/CCR5 signalling pathway is a specific pathway in the hippocampal region of epileptic mice, and blocking CCL5/CCR5 signal transmission can alleviate hippocampal lesions, indicating that the CCL5/CCR5 signalling pathway is a potential therapeutic target for epilepsy [86].
3.3 Cardiovascular diseases
Cardiovascular disorders primarily comprise pathologies that impact the cardiac and vascular systems. In the field of cardiac diseases, ST has been harnessed for the purpose of identifying potential therapeutic targets for viral myocarditis [87], myocardial infarction (MI) [88], heart failure [89], and arrhythmogenic cardiomyopathy (ACM) [90]. ST enables the creation of detailed cardiac maps across various anatomical regions, advancing our understanding of cardiac development and disease at the molecular level [91]. For example, in the early stages after MI, mechanosensing genes such as Crsp3 are upregulated in the border zones of ST [88]. The downregulation of the Crsp3 gene exacerbates functional impairment after MI, while its upregulation improves this situation. Thus, CSRP3 may serve as a therapeutic target for myocardial infarction. Furthermore, ST also identified potential regulatory factors that influence myocardial cells and fibroblasts by comparing MI and normal cardiac tissue [92]. Cardiac fibrosis plays a crucial role in the etiology of heart failure. In early studies, through Tomo-seq of ischemic hearts, SOX9 was identified as a potential therapeutic target for mitigating cardiac fibrosis [93]. Recent ST analysis of mouse models revealed that the downregulation of Htra3 promotes cardiac fibrosis, and further integration with perturbation techniques and scRNA-seq analysis indicated that Htra3-TGF-β-IGFBP7 could be a therapeutic target for heart failure [94]. Additionally, the molecular mechanisms governing the progressive loss of cardiomyocytes and their replacement by adipose cells in ACM have been elucidated through Tomo-seq analysis [90]. This study identified ZBTB11 as a crucial factor responsible for inducing CM apoptosis. Considering this discovery, the inhibition of ZBTB11 has emerged as a promising therapeutic strategy for managing ACM.
In research on vascular diseases, ST has demonstrated utility in the identification of prospective targets associated with atherosclerosis and vasculitis [95,96]. Arterial atherosclerotic plaque rupture precipitates acute cardiovascular complications. ST analysis of the rupture-prone regions within vascular plaques has elucidated the molecular mechanisms underlying arterial atherosclerotic plaque rupture [96]. Moreover, novel therapeutic targets, including noteworthy candidates such as MMP9, have been identified based on spatial localization. In the field of Kawasaki disease vasculitis research, researchers have applied ST to analyse murine coronary arteritis tissue. Researchers have shown that focal infiltration of immune cells is associated with the activation of NLRP3 and the production of IL-1β and IL-18, which also regulate vascular smooth muscle phenotypic transition to the pathogenic type [95]. This compelling evidence supports the potential of NLRP3 as a promising therapeutic target for vasculitis in Kawasaki disease patients.
3.4 Infectious diseases
The application of ST in infectious diseases aims to gain further insights into cellular interactions and retain spatial information vital for deciphering the interplay between different cell types post infection. Recent research on coronavirus disease 2019 (COVID-19) to understand the cellular causes associated with lung damage and identify therapeutic targets is a notable application of ST approaches in infectious diseases. By comparing gene expression data from 46 areas of lung samples affected by COVID-19 across 3 patients, ST has been applied to describe the atlas of damage in lung tissue affected by COVID-19, encompassing cells, pathways, and genes [97]. Building upon this preceding study, the research team employed a combination of multi-omics analysis and machine learning techniques to delve deeper into their study. Through selective ST, they revealed signals associated with epithelial-mesenchymal transition and pinpointed LZTFL1 as a prospective therapeutic target [98]. Another significant application of ST in infectious diseases is its use in studying influenza infection. ST has been employed in combination with scRNA-seq and bulk RNA-seq to reveal substantial changes in gene expression patterns among various immune, endothelial, and epithelial cell types, delineating differences in the host response to influenza infection between young and aged mice [99]. The heightened levels of inflammation and fibrosis within the lungs of aged mice are clearly discernible through the perspective of ST. Furthermore, ST have highlighted the enrichment of Wfdc17 in fibrotic regions, which has been identified as a novel potential regulator following postinfluenza A virus infection. In the study of bacterial infections, ST has been used to map the spatial location of cell types within leprosy cell groups and the architecture of granulomeres [100]. Moreover, a specific set of genes encoding antimicrobial proteins was identified; these genes are differentially expressed in disseminated leprosy and are controlled by the actions of IFN-γ and IL-1β. In another example of bacterial infection, the integration of spatial and temporal transcriptomics was employed to analyse a mouse model of endotoxemia [101]. Through ST, the study revealed the spatial localization of the S3 proximal tubule subtype in the kidneys affected by endotoxemia. Additionally, temporal transcriptomic analysis revealed potential therapeutic targets for the treatment of human sepsis, including SOX9. ST also has applications in parasitic infectious diseases. Lymphatic filariasis is an ailment caused by nematodes, yet current drugs solely target the larvae, leaving the adults unaffected. Researchers have conducted ST analysis of the head region of adult parasites, establishing a gene expression map aimed at identifying potential novel drug and vaccine targets [102].
3.5 Other diseases
The kidney is a complex organ comprising a diverse array of structures with more than 20 distinct cell types in its mature state. The exploration of various intercellular communication networks and regional gene expression patterns within the kidney facilitates an enhanced understanding of renal diseases and the identification of potential therapeutic targets. Several studies employing ST in the field of kidney disease have undertaken comprehensive assessments of human and mouse model samples to identify potential therapeutic targets for kidney injury [103], chronic kidney disease [104], and kidney-related disorders [104-107]. For example, in renal transplantation, T-cell-mediated rejection initiates an immune attack on the transplanted kidney, resulting in renal injury. The utilization of ST to interrogate the transplant region represents a promising strategy for identifying prospective therapeutic targets associated with acute cellular rejection or other localized renal diseases [108]. In addition, renal fibrosis serves as a hallmark of the progressive progression of chronic kidney disease, yet the current therapeutic options for treating fibrosis are inadequate. The utilization of a strategy combining ST with scRNA-seq has elucidated the cellular origins and different roles of human renal myofibroblasts and their precursors [105]. Additionally, this study identified NKD2 as a specific therapeutic target for renal fibrosis involving myofibroblasts. Recently, ST has also been applied in the discovery of intrinsic TGF-β signalling that reduces proximal tubular mitochondrial injury and inflammation in chronic kidney disease. [104] Acute kidney injury exhibits regional specificity. The application of ST visually elucidates the re-expression of region-specific markers lost during the process of repair [103]. This method introduces a novel tool for identifying therapeutic targets in the context of acute kidney injury. In the field of diabetic kidney disease (DKD) research, researchers have employed ST and scRNA-seq to construct a renal atlas of DKD to unveil potential therapeutic targets for the treatment of DKD [107]. In renal papillary samples from patients with kidney stones, ST analysis revealed diffuse expression of MMP7 in the stone area and upregulation of MMP9 in the mineral deposition region, suggesting that these two molecules may serve as novel biomarkers for kidney stones [106].
Hepatic function in drug metabolism is pivotal, and investigating hepatic processes has considerable implications for drug discovery and development. ST reveals the spatial distribution of cellular phenotypes and transcriptional profiles in hepatic disorders, thereby aiding in the understanding of pathogenic mechanisms [109]. For example, analysing ST atlases of primary hepatocellular carcinoma can discern intratumoral diversities, encompassing predominant gene expression, functionalities, prognoses, and clonal origins of diverse cellular subpopulations, thereby facilitating the identification of therapeutic targets [110]. Additionally, the ST shows the spatial arrangement of distinct cellular populations within the liver, thereby contributing to the exploration of hepatic tissue architecture and function. For instance, the transcriptional differences between hepatocytes and nonparenchymal cells have been discerned by ST, revealing their respective contributions to hepatic homeostasis and delineating gene expression profiles and structural constituents across the murine hepatic zone [111]. Notably, in addition to scRNA-seq, ST has significant applications in the study of drug impacts on hepatic physiology, particularly drug-induced hepatotoxicity. For example, molecular distinctions between periportal and pericentral hepatocytes following exposure to acetaminophen have been elucidated by scRNA-seq, revealing the molecular underpinnings of acetaminophen-induced hepatic injury [112]. ST analysis of hepatic tissue sections after drug administration enables researchers to discern alterations in gene expression within specific hepatic regions induced by distinct pharmaceutical agents. Correlation of regional tissue damage assessments with spatial expression aids in elucidating the toxic mechanisms of drugs and the pathophysiology of hepatic injury. A study integrating single-nucleus RNA sequencing and ST revealed a spatially dependent dose-response relationship between liver lipid accumulation and inflammatory responses induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), elucidating the specificity of cellular phenotypes and transcriptional alterations in various hepatic regions induced by TCDD and the molecular mechanisms underlying TCDD-induced nonalcoholic fatty liver disease progression [113].
4. ST for drug research
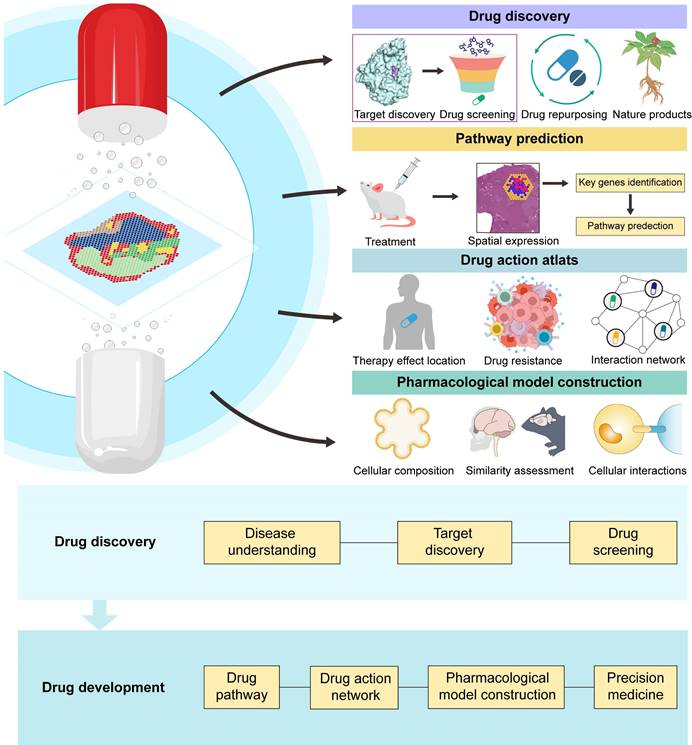
ST is driving incremental improvements in our pharmacological knowledge. This technology aids in identifying different cell types and regions associated with diseases, guiding the discovery of potential therapeutic drugs. Moreover, ST allows us to map changes in cells within specific areas and link them to their gene expression profiles. This helps identify genes and pathways involved in drug responses, offering a more precise understanding of how drugs work at the molecular level in distinct tissues or cell types. Although ST has not yet been widely applied in drug development, based on existing research, we can still foresee its enormous potential in future drug studies, particularly in pharmacology research. Hereafter, our discussion will focus on the application of this approach in drug discovery, pharmacological research, and precision medicine with several study cases, showing the utility of ST in drug development (Figure 4).
Application of spatial transcriptomics in drug discovery and drug development. ST aids in identifying new targets, facilitating drug screening. ST holds immense potential in drug repurposing and natural product drug discovery. ST utilizes spatial expression maps after drug treatment to identify key genes and predict treatment pathways. ST constructs a drug action atlas, enabling the localization of drug effects and the study of drug resistance and interactions. By analysing the spatial cell composition of pharmacological models, assessing model similarity to cases, and studying intercellular interactions, ST assists in building pharmacological research models. ST is being applied across various stages of drug discovery and development, and these applications are enhancing the probability of drug discovery success and improving therapeutic outcomes.
4.1 Drug discovery
Employing ST to pinpoint targets and then proceeding with drug identification and validation is a feasible approach for drug screening. Analysing unique gene expression patterns within affected regions and assessing cell interactions, along with leveraging established biological databases, enhances the precision of drug screening processes. For example, to study the prognosis and therapeutic strategies for cutaneous melanoma, researchers conducted an analysis of publicly available human cutaneous melanoma ST data from the 10x Genomics database, aiming to identify specific cellular types and gene expression patterns within distinct regions of melanoma [114]. Subsequently, the team employed single-cell transcriptomic analysis to validate characteristic genes associated with cancer-associated fibroblasts. Following this validation, they screened small molecular compounds from the CTD database capable of downregulating the gene expression of FBLN1 and COL5A1. Using the AutoDock Vina molecular docking method, they screened and performed molecular docking to identify potential drugs targeting FBLN1 and COL5A1, such as mifepristone and dexamethasone. Applying ST to discover new pathways influencing diseases can help expand the therapeutic scope of known drugs into novel areas of disease treatment, thereby granting these existing medications new application value. In a study with a mouse gastric tumor organoid model, researchers identified a set of key genes expressed in stromal cells located at the invasive front of tumors through ST analysis and discovered that MAPK signal transduction promotes tumor metastasis [115]. Based on these findings, researchers have attempted to use the MAPK kinase inhibitor trametinib to inhibit the development and invasion of gastric tumors. ST has also been applied in the discovery of natural active compounds. There are currently no effective pharmacological treatments for hepatic ischaemia-reperfusion injury (I/R) [116]. Researchers have conducted an analysis integrating ST with histopathological methods to scrutinize molecular and cellular changes within the injured hepatic area. Subsequently, leveraging region-specific gene expression variations, they queried the Connectivity MAP database for genes demonstrating I/R modulation and regional dependency characteristics, which led to the identification of the natural active compound celastrol as a potential therapeutic agent for I/R [117]. Further validation of celastrol's regulatory effects within relevant ROIs was also conducted using ST in this study. ST has been employed to identify potential drugs associated with breast cancer tumor tissue. Elevated levels of inner mitochondrial membrane protein (IMMT) were found to be coexpressed within the transformed sections of breast cancer tumor tissue by ST [118]. By leveraging the expression of IMMT, researchers have screened the Genomics of Drug Sensitivity in Cancer (GDSC) and Cancer Cell Line Encyclopedia (CCLE) databases, and pyridinol was identified as a potential drug candidate.
Advances in computer science and artificial intelligence have propelled the application of ST in drug screening. In the research of oral squamous cell carcinoma (OSCC), a study used the ST method to explore the gene expression variances within the tumor core (TC) and leading edge (LE) regions of OSCC and further identified potentially effective drugs that could impede the transfer from TC to LE [119]. Based on RNA velocity, Scvelo, a dynamic simulation model, was used to analyse cellular developmental trajectories and gene expression dynamics [120]. Dynamo is a machine learning technique used to predict cellular fate transitions following genetic perturbations [121]. By integrating both approaches, researchers characterized the developmental trajectories of cancer cells in the tumor core (TC) and edge (LE) regions using Dynamo to associate them with drug responses via the integration of gene-specific transcriptional dynamics. A total of 417 drugs were analysed, revealing 140 drugs demonstrating drug-gene interactions. This study has presented a novel approach to the application of ST in drug screening by integrating ST with the in silico technique. In another study, ST was used to examine the correlation between kidney injury and kidney renal clear cell carcinoma (KIRC), with a focus on alterations in the expression of the PGAM1 gene in both kidney injury and KIRC [122]. Q-omics, an integrative omics data mining software comprising data mining and visualization algorithms, was applied [123]. In this study, Q-omics was applied to screen the Genomics of Drug Sensitivity in Cancer (GDSC) database, and Pearson correlation coefficient analyses between PGAM1 expression levels and drug dosages were conducted to assess the sensitivity of various drugs in the context of high PGAM1 expression, identifying four drugs with potential therapeutic efficacy.
4.2 Pharmacological research
4.2.1 Drug pathway prediction and validation
The ST not only accelerated the discovery of new drugs by providing precision and spatial information but also aided in understanding drug mechanisms by predicting drug action pathways. Biological pathways and interactions within organisms are remarkably diverse and intricate, especially regarding drug actions involving various molecular interactions. Moreover, some drug mechanisms might involve biological processes that are not fully understood yet, further complicating pathway prediction. ST provides specific location information regarding drug actions within tissues, encompassing varied responses among different cell types and regions. This spatial resolution of cellular and tissue interaction information contributes to overcoming barriers in deciphering complex drug action mechanisms. The precise mechanism underlying the prolonged antidepressant effects of ketamine through facilitating myelin sheath formation remains elusive based on earlier research. With the aid of ST, researchers have investigated the enduring impact of ketamine treatment on the medial prefrontal cortex and hippocampus in mice, identified crucial DEGs involved in myelin sheath formation and revealed the pivotal role of the AMPAR pathway in mediating myelination [124]. This unveiling through ST shed light on the sustained antidepressant mechanism of ketamine. Similarly, in the field of vaccine therapeutics, ST revealed the CD4-dependent expression of interferon-stimulated genes in lung myeloid cells and epithelial cells and revealed that the Bacillus Calmette-Guérin vaccine induces CD4+ T cells to provide feedback to tissue myeloid cells and epithelial cells to achieve sustained broad-spectrum immune mechanisms [125].
Numerous traditional natural remedies have demonstrated significant therapeutic efficacy, yet their precise molecular mechanisms remain unclear. The application of ST has substantial potential for predicting the pathways of action of active constituents in natural remedies. For example, an ST analysis of a mouse brain revealed the molecular pathways influenced by Rhynchophylline, an alkaloid component of Uncaria known for its significant sleep-inducing effects, indicating that Rhynchophylline impacts genes associated with sleep regulation in specific brain regions [126]. Compared to single-component drugs, traditional Chinese medicine (TCM) is characterized by multiple targets, multiple pathways, and holistic regulation, thereby resulting in greater complexity. Notably, ST can capture the interactions of TCM between cells and tissues, assisting in revealing the intricate mechanisms of TCM action and the patterns of multipathway regulation, making it a powerful tool for revealing new pathways of TCM. To elucidate the mechanism by which the traditional Chinese medicine formula Shexiang Baoxin Pill (SBS) protects the heart, researchers have combined ST with single-nucleus RNA sequencing to assess the effects of SBS on myocardial cell types and subpopulation states [127]. They discovered that SBS could increase the gene expression of Nppb and Npr3 in the infarct area of the heart, regulating vascular generation mediated by endocardial cells. However, this study did not further validate this pathway. Despite the enormous potential of ST in traditional Chinese medicine, its current application in TCM research remains limited.
Additionally, in terms of validating drug pathways, ST has the ability to capture characteristic spatial differences following drug action. Through the combined application of single-cell transcriptomics and ST, it was observed that the interaction between tumor cells and immune cells (particularly the increased interaction between CD8+ T cells and tumor cells) was strengthened after NP137 treatment, further confirming the inhibitory effect of NP137 treatment on Netrin-1 and subsequently reducing EMT in tumors [128].
4.2.2 Panoramic and dynamic depiction of the drug action network
ST offers an unprecedented opportunity to gain a more detailed view of drug action regions at the molecular level within tissues or cells, enabling the construction of dynamic panoramic drug action networks and offering novel insights into drug efficacy assessment. In a previous study of prostatic hyperplasia, a large population of differentiating cells was identified to facilitate prostate regeneration using scRNA-seq [129]. Nevertheless, this study did not explore the impact of intercellular communication and the microenvironment on the status of prostate cells. With the application of ST in heterogeneous response studies to drug treatments, the molecular characteristics associated with the heterogeneous response to 5-alpha reductase inhibitor (5ARI) therapy for benign prostatic hyperplasia have been identified [130]. This study revealed a transition from prostate epithelial cells to club-like cells following 5ARI therapy and revealed that this transition was strongly associated with the degree of glandular atrophy. ST also provides a promising tool for identifying spatially relevant regions of drug action within the tumor microenvironment that have biological and therapeutic implications [131]. In the first study that mapped spatially resolved gene expression in Sonic hedgehog (SHH) patient-derived orthotopic xenograft (PDOX) medulloblastoma, a reduction in cellular diversity within the tumor following treatment with the CDK4/6 inhibitor palbociclib was revealed [132]. Interestingly, despite palbociclib treatment, cells at the tumor-microenvironment interface continue to proliferate, suggesting that cells specific to transcriptional states or spatial positions play a critical role in the response to palbociclib therapy. This finding elucidates the reason behind the recurrence observed in 80% of SHH PDOX tumors following cessation of palbociclib treatment in earlier studies [133]. A clear example of the use of ST analysis in assessing the potential effects of natural active compounds is the assessment of the potential effects of 47 ROIs in I/R-induced liver injury, which revealed that celastrol mitigated the infiltration of macrophages induced by I/R in specific injury zones and partially restored the characteristics of certain hepatic nonparenchymal cells affected by I/R [117]. Accurate drug targeting enhances the efficacy of medications. Through the integration of snRNA-seq and ST, researchers revealed the precise localization of prostaglandin E2 receptors, revealing that the site at which prostaglandin E2 is injected during uterine fibroid resection surgery should be at the junction of pseudocysts and smooth muscle tumors, leading to a greater reduction in bleeding [134].
ST has a unique advantage in deciphering intricate interaction networks between drugs in combination therapies and between drugs and the organism. Combination therapy has been effectively employed in cancer treatment to reduce drug resistance and enhance therapeutic efficacy for patients. However, numerous unanswered questions persist regarding the mechanisms underlying the improved drug efficacy achieved through combination therapy. In the field of combination therapy with immune checkpoint inhibitors (ICIs), identifying the tumor immune interactions that contribute to the response to ICIs remains a pivotal objective. A recent study examined the spatial transcriptomic profiles of T cells, stromal cells, and leukemia cells within the treatment sites of acute myeloid leukemia (R-AML) patients receiving pembrolizumab and decitabine combination ICI therapy and elucidated the impact of pembrolizumab and decitabine combination therapy on the tumor immune microenvironment [135]. Identifying new predictive biomarkers for tumor resistance in specific regions will aid in the exploration of new combination therapy approaches to enhance therapeutic efficacy. During the investigation of intraductal papillary mucinous neoplasms (IPMN), an analysis revealed an increase in mucin O-glycosylation pathways throughout IPMN progression and revealed that tantalate can target GCNT3 for mucin regulation and that combining tantalates with chemotherapy enhances immune infiltration, leading to improved therapeutic efficacy [136]. Due to its capacity to construct more comprehensive and accurate dynamic maps, there is increasing interest in the application of spatiotemporal transcriptomics in drug action research. However, their current application in pharmacological research remains relatively limited.
4.2.3 Pharmacological model construction
Organoids are three-dimensional cell clusters constructed with multiple cell types through the induction and self-assembly of human tissues, stem cells, or pluripotent stem cells in vitro [137]. Within each individual organoid, diverse cell types resembling natural human organs are present. The remarkable similarity in both function and structure to those of native human organs makes organoids a robust platform for studying human disease mechanisms and drug screening [138]. Cell culture systems are mainly classified into 2D and 3D culture systems [139]. Compared with traditional 2D cell models, organoids exhibit exceptional biological similarity [140]. Additionally, compared to animal models, organoids offer reduced culture expenses and significant advantages in high-throughput screening [141]. Furthermore, they can faithfully recapitulate human organ development characteristics and do not exhibit species differences [142,143]. Consequently, organoids have found extensive applications in diverse fields, including disease modelling [144], drug screening [145], drug evaluation [146], toxicity assessment [147], and cancer research [148]. In September 2022, the U.S. Food and Drug Administration (FDA) issued new guidelines, urging the reduction of animal testing and the adoption of novel technologies as alternatives whenever possible, with organoids having emerged as a high-quality substitute for preclinical animal experiments [138,149]. To determine the heterogeneity of organoids, single-cell sequencing has been employed in organoid research. Single-cell sequencing allows researchers to determine transcriptional characteristics and identify cell types within organoids that closely resemble those found in actual organs [150-152]. Nevertheless, these methods lack the ability to provide precise spatial transcriptional information [153]. ST, in contrast, offers the ability to decipher the spatial distribution and transcriptional characteristics of various cell types within organoids [154]. Currently, ST primarily provides assistance in three key aspects within the realm of organoids: 1). Understanding the spatial composition of cell types within organoids; 2). The spatial expression profiles of the organoid transcriptome were explored; 3). Investigating the intercellular interactions within organoids. However, the application of spatial transcriptomes in organoid research remains unpopular.
The formation of organoids closely mimics the characteristics and cellular composition of real organs, thus recapitulating the self-organizing processes of organ development. Therefore, studying human organ development through organoids in vitro offers a higher level of fidelity compared to that of model organisms such as mice or cell-based models [155]. From another perspective, gaining an understanding of organ development patterns in human embryos is also advantageous for the cultivation of organoids [156]. The application of scRNA-seq in developmental biology enables the comprehensive analysis of tissue transcriptomes at single-cell resolution across various stages of development [157,158]. However, current organoid analysis approaches still have certain limitations, and addressing how to depict the organoid development process more faithfully remains a challenge. For example, due to the nonfixed composition of organoids and the heterogeneity both between and within organoids, the need for integrating multiple technologies for high-throughput analysis in dynamic organ development models remains unmet [159]. In the study of neurological disorders, the human brain possesses unique characteristics distinct from those of the brains of other animals [160]. Cerebral organoids emulate the tissue architecture and multilineage differentiation seen in the human brain, offering a new window into investigating human brain development [161]. Single-cell transcriptomic analysis throughout the entire human brain organoid development process, from the pluripotency to neuroectoderm and neural epithelial stages, enables the understanding of the cellular composition of organoids and the reconstruction of differentiation trajectories [162]. Similarly, comparing the transcriptomes of human cortical organoids and fetal brains can validate the authenticity of organoid models [163]. However, previous single-cell transcriptomics approaches have not been able to capture the dynamic spatial changes in the transcriptome during developmental processes. In lineage tracing of human cerebral organoids, cellular populations in different regions of the brain may be associated with clonal spatial arrangements, while ST serve as a bridge connecting molecular states, cellular lineages, and spatial positional information [164]. Spatial transcriptomics has also been widely used in retinal organoid development. Retinal organoids are complex organoids containing a rich population of rod and cone cells, and they are capable of recapitulating the human retina's developmental process from the optic vesicle to the optic cup [165,166]. The development of retinal organoids is strongly correlated with retinal differentiation programs and genes related to development, both in terms of their chemical composition and cellular makeup [167]. The integration of ST with single-cell transcriptomics and temporal transcriptomics has enabled the precise reconstruction of the process of retinal neuron differentiation and localization during retinal organoid development [168]. Through multiple smFISH methods, researchers obtained spatial transcriptomic information and inferred the gene regulatory networks underlying retinal organoid development [169].
In addition, the study of organ development to achieve higher-fidelity construction of organoid models remains a research direction worth exploring. Legnini et al. devised a method for spatiotemporally programming organoids to establish unique gene expression domains, validated this approach using ST, and revealed the role of the SHH pathway in an organoid model of human neurodevelopment [170]. In the study of kidney development, spatial and temporal transcriptomic analysis of human embryonic kidneys has been employed to elucidate intercellular signalling during organoid development and the key factors that regulate kidney development, thus facilitating the cultivation of kidney organoids [156].
4.3 Precision medicine
Due to variations in genetics, physiology, biochemistry, and other factors among different individuals, patients exhibit diverse responses to the same disease and the same drug treatment [171,172]. Some individuals might be highly sensitive to a particular medication, while others may show insensitivity or even severe side effects to the same drug [173]. Therefore, there is a need to determine a better treatment approach based on individual characteristics to enhance treatment effectiveness and reduce adverse reactions. ST has emerged as a powerful tool for precision medicine, facilitating precise diagnosis and targeted therapies [174]. The precise spatial localization capability of ST enables accurate examination of biomarker variations subsequent to drug administration across various patients, aiding in patient stratification for predicting individual responses to treatment. In a study of the tumor immune microenvironment, scRNA-seq and ST datasets from various patients and tumor types were analysed, leading to the establishment of a tumor immune atlas for precision therapy [175]. The comprehensive single-cell atlas of the spatial location of gastric cancer delineates high-resolution maps between patients with various subtypes of the same gastric cancer and across different subtypes, identifying key factors predicting adverse reactions related to prognosis [176]. The combined therapy of antiangiogenic therapy and immune checkpoint inhibition is effective against HCC but lacks universal efficacy across all patients, with no identified biomarkers to differentiate responders from non-responders [177,178]. An ST-analysis of tumor samples from patients with HCC treated with cabozantinib and nivolumab revealed significant crosstalk between cancer cells and the tumor microenvironment, which plays a pivotal role in tumor response and drug resistance, and identified biomarkers capable of predicting patient responses, offering new avenues for precise classification and therapeutic strategies [179]. With a combination of scRNA-seq, RNA-Mutect, and ST, circulating lncRNAs associated with chemotherapy resistance in plasma have been identified as novel predictive circulating biomarkers for the progression of lung adenocarcinoma [180]. To predict the chemotherapy response in advanced hepatobiliary cancer patients and elucidate the molecular mechanisms underlying drug resistance, researchers have analysed the transcriptomic characteristics of rapid progressors (RP) and long-term survivors (LS), proposing the RPLS metric as a novel indicator of the effect of chemotherapy [181].
ST can precisely identify and locate therapeutic targets suitable for individual patients and validate the effectiveness of these targets. A close relationship exists between senescent cells and malignant tumors [182]. Senescent cells, on the one hand, express age-related genes that inhibit tumors, while on the other hand, age-related genes facilitate tumor growth, invasion, and metastasis [183]. In a recent study, three distinct tumor aging microenvironment (AME) regulatory patterns and 36 AME regulatory factors were identified through ST analysis of the AME in prostate cancer, providing potential targets for personalized therapies [184]. In patient tissue biopsies, ST has the potential to provide precise insights into the molecular mechanisms underlying disease progression. A study in precision medicine related to the kidneys analysed the gene expression characteristics of a biopsy tissue region from a diabetic patient, discovering the molecular mechanisms of neovascularization formation in the glomeruli, thereby introducing novel personalized diagnostic methods for renal biopsies [178]. ST analysis of biopsy material from primary central nervous system lymphomas revealed the spatial heterogeneity of malignant B-cell clusters and revealed the colocalization of T-cell exhaustion markers with malignant cells, demonstrating the potential of ST in personalized therapy [185]. In summary, analysing ST information aids in the development of personalized drugs or treatment strategies tailored to individual pathological features (Figure 5).
Spatial transcriptomics in personalized medicine. ST identifies biomarkers of drug response by analysing the spatial transcriptome information of different drug-responsive populations. The spatial transcriptome was used to analyse the spatial expression information of patient biomarkers to achieve personalized medicine.
5. Challenges and prospects
5.1 Technological limitations and advancements
At the technological level, current spatial transcriptomic techniques are still in their early stages. Despite ongoing technological advancements, there are still limitations in spatial resolution compared to single-cell transcriptomic techniques [186]. ST still cannot provide sufficiently precise information for certain subcellular structures within cells or areas with small intercellular distances. To address this issue, researchers have developed a series of approaches for integrating single-cell transcriptomic and spatial transcriptomic data [187]. Additionally, spatial transcriptomic resolution can be enhanced through algorithms such as the BayesSpace algorithm, which is based on a fully Bayesian statistical approach [188]. This algorithm utilizes spatial neighborhood information to improve the resolution of spatial transcriptomic data to the level and models the low-dimensional representation of gene expression to achieve spatial clustering, significantly enhancing the utilization of spatial information. Resolution can also be improved by optimizing the technology for capturing transcriptomic information. The Visium HD platform recently introduced by 10X Genomics, for example, has reduced the diameter of spots from 55 micrometers to an edge length of 2 micrometers and eliminated gaps between spots. This significant enhancement enables gap-free and bias-free ST at single-cell resolution.
Furthermore, current mainstream spatial transcriptomic techniques are mostly based on two-dimensional tissue sections, which can obtain two-dimensional spatial transcriptomic information but still lack adequate capability for acquiring transcriptional information in three-dimensional space. An R language toolkit called STUtility has been developed, which can automatically perform data conversion, align multiple tissue slices, and regionally annotate continuous two-dimensional slice data, ultimately creating a final three-dimensional model of tissues from processed sequencing and image data [189]. However, this method still relies on computer algorithms for three-dimensional model construction and still faces challenges related to ensuring slice quality and discrepancies between constructed three-dimensional models and actual scenarios. Addressing this issue requires revolutionary technology to develop methods that can directly capture three-dimensional spatial transcriptomic information. Imaging-based spatial transcriptomic methods can directly observe three-dimensional tissues and theoretically hold more potential and advantages in three-dimensional ST. For example, as mentioned earlier, STARmap analyses three-dimensional transcriptomic data using hydrogel and tissue-clearing technologies [46]. The key to this approach lies in enhancing imaging techniques to observe more comprehensive positional signals and decoding them to construct comprehensive three-dimensional transcriptomic maps.
The current mainstream platform for ST is the commercialized 10X Genomic Visium Spatial Gene Expression platform. However, due to the high cost, operational complexity, and requirement for specialized equipment, its widespread application and promotion in drug research and personalized therapy are restricted. Encouragingly, ST, as a trending technology in recent years, has continued to evolve with the emergence of new methodologies [190]. It is believed that in the foreseeable future, there will be more affordable and user-friendly solutions to lower the threshold of this technology.
5.2 Integration of ST with AI
With the rapid advancement of ST, a substantial volume of spatial transcriptomic data has accumulated. However, the current challenge lies in improving the efficiency of data acquisition and extracting valuable knowledge from this extensive dataset. Consequently, there is an urgent need for data standardization and the establishment of databases to facilitate the sharing of spatial transcriptomic data and streamline the data acquisition process for researchers. Additionally, the application of artificial intelligence (AI), such as machine learning (ML) and deep learning (DL) methodologies, can assist in uncovering potential drug targets and drug effects from the vast spatial transcriptomic dataset, maximizing the exploration of valuable spatial transcriptomic data resources. With respect to database establishment, researchers have already developed several comprehensive spatial transcriptomic databases [191-194], and future databases will integrate more analytical tools to achieve a one-stop solution for data collection and analysis.
AI technologies contribute to the computational analysis of spatial transcriptomic data, offering limited possibilities for the advancement of ST. DeepST is a DL-based ensemble algorithm that can accurately identify regions with similarities in gene expression and tissue morphology [195]. The utilization of AI for the analysis of ST data contributes to enhanced resolution. By way of illustration, the SCS method employs a transformer architecture to more accurately and efficiently segment individual cells from high-resolution ST images and even enables the analysis of heterogeneity within individual cells [196]. A recent study introduced SpatialScope, a method employing deep generative models to integrate single-cell transcriptomic and spatial transcriptomic data [197]. This approach not only enhances the single-cell resolution of sequence-based spatial transcriptomic data but also infers complete transcriptomic expression levels based on image-based spatial transcriptomic data. Artificial intelligence ingeniously provides a novel approach to address the challenges in constructing three-dimensional transcriptomic maps traditionally associated with ST. STitch3D innovatively combines probability models with deep neural networks to simultaneously analyse multiple consecutive spatial transcriptomic slices, thereby reconstructing three-dimensional tissue structures [198]. A series of AI tools have been developed, including STAGATE for identifying spatial substructures in biological tissues [199], STAligner for integrating and analysing multislice spatial transcriptomic data [195], and STAMarker for spatial domain recognition and identification of spatially variable genes [200]. This series of tools integrates artificial intelligence models to improve the efficiency and accuracy of computing and analysing spatial transcriptomic data.
5.3 Spatial multi-omics and spatiotemporal transcriptomics
Integrating ST with spatial metabolomics, spatial proteomics, and similar technologies facilitates the simultaneous acquisition of spatial distribution information for gene expression, metabolites, and proteins [201,202]. The combined analysis of single-cell transcriptomics, ST, and spatial proteomics has been employed to elucidate multicellular mechanisms underlying the progressive onset of Alzheimer's disease and their spatial relationships with stages of neurodegenerative changes [203]. The integration of ST with spatial metabolomics has enabled the construction of comprehensive spatial expression maps for genes and metabolites in complex human brains affected by injury [204].
Moreover, spatial-temporal transcriptomics technology enables the acquisition of dynamic and comprehensive information but has been extensively applied in developmental biology, but its potential in drug research remains largely unexplored [205-207].
5.4 Potential application of ST
Although ST has been applied in discovering disease targets, predicting drug action pathways, and delineating spatial maps, its potential in the field of drug development extends far beyond these aspects. Furthermore, despite the limited application of ST in natural medicine, its ability to construct comprehensive post-drug transcriptomic change maps holds promise as a robust tool for evaluating the effects of complex medications, predicting drug interactions, and potential pathway assessments. Hence, the field of natural medicine research, particularly in traditional Chinese medicine pharmacology, stands as a highly promising area for the application of ST. Pharmacological models are advancing toward building models that closely mimic the human body. ST provides a spatial viewpoint for comprehending the cellular composition and molecular features of human organs, facilitating the advancement of more precise preclinical models. However, due to limitations in resolution and sample processing techniques, the current application of ST in organoids remains relatively limited.
The ST is burgeoning and holds vast potential opportunities in the field of drug development, despite facing challenges such as data processing and technical limitations. In the future, advancements such as high-resolution ST, the integration of AI technologies with ST, three-dimensional ST, spatiotemporal transcriptomics, spatial multi-omics technologies, and human spatial mapping will greatly expand the application prospects of ST (Figure 6). These developments will offer more comprehensive insights for drug development, hastening the drug discovery process, and facilitating personalized therapy.
Rapidly advancing spatial transcriptomics technology and its application prospects. The integration of spatial transcriptome technology with other emerging technologies promotes understanding of disease and the construction of human spatial maps.
6. Conclusion
Overall, ST, as an emerging omics technology, holds immense potential in the field of drug discovery and development. With higher spatial resolution, ST effectively reveals gene expression at specific locations within tissue structures and their interaction relationships. This information is crucial for enhancing our understanding of diseases and discovering new therapeutic targets and drugs. Additionally, ST aids in establishing improved pharmacological research models, such as organoid models. Moreover, analysing drug pathways from a spatial resolution perspective can aid in more accurate prediction and validation of drug effects. The construction of dynamic panoramic networks of drug action further deepens our understanding of drug effects, particularly in combined therapies. Analysing the interaction of drugs within critical areas of a patient's body contributes to advancing personalized treatments, accelerating the discovery of new biomarkers, and providing more efficient treatment plans for patients. The distinct advantages in unraveling spatial information within complex tissues offered by ST, thereby providing crucial support for researching the distribution and mechanisms of action of complex drugs like traditional Chinese medicine. However, the current application of ST in drug development remains limited and faces certain challenges. Encouragingly, with the increasing maturity of high-throughput sequencing technologies, new opportunities are emerging, and ST may significantly alter the landscape of drug development.
Acknowledgements
This work was supported by the China Academy of Chinese Medical Sciences Innovation Fund (CI2021A00610), the National Natural Science Foundation of China (82274224), the Fundamental Research Funds for the Central Public Welfare Research Institutes of China (JJPY2022023, ZZ-YQ-082-C1), and the Beijing Nova Program (20230484243).
Author contributions
Junxian Cao: Conceptualization, Methodology, Writing - Original Draft, Visualization, Literature Review. Caifeng Li: Conceptualization, Literature Review, Writing - Original Draft, Methodology. Zhao Cui: Literature Review, Visualization, Methodology. Shiwen Deng: Visualization, Literature Review, Writing - Original Draft. Tong Lei: Methodology, Literature Review. Wei Liu: Conceptualization, Writing - review & editing & polishing. Hongjun Yang: Conceptualization, Writing - review & editing. Peng Chen: Conceptualization, Writing - review & editing, Supervision, Funding acquisition. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ståhl PL, Salmén F, Vickovic S. et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353:78-82
2. Strell C, Hilscher MM, Laxman N. et al. Placing RNA in context and space - methods for spatially resolved transcriptomics. FEBS J. 2019;286:1468-81
3. Zhou Y, Jia E, Pan M, Zhao X, Ge Q. Encoding Method of Single-cell Spatial Transcriptomics Sequencing. Int J Biol Sci. 2020;16:2663-74
4. Tian L, Chen F, Macosko EZ. The expanding vistas of spatial transcriptomics. Nat Biotechnol. 2023;41:773-82
5. Rao A, Barkley D, França GS, Yanai I. Exploring tissue architecture using spatial transcriptomics. Nature. 2021;596:211-20
6. Van De Sande B, Lee JS, Mutasa-Gottgens E. et al. Applications of single-cell RNA sequencing in drug discovery and development. Nat Rev Drug Discov. 2023;22:496-520
7. Chu X-Y, Quan Y, Zhang H-Y. Human accelerated genome regions with value in medical genetics and drug discovery. Drug Discov Today. 2020;25:821-7
8. Dias MH, Kitano ES, Zelanis A, Iwai LK. Proteomics and drug discovery in cancer. Drug Discov Today. 2016;21:264-77
9. Shyur L-F, Yang N-S. Metabolomics for phytomedicine research and drug development. Curr Opin Chem Biol. 2008;12:66-71
10. Anderson AC. Spatial transcriptomics. Cancer Cell. 2022;40:895-900
11. Choe K, Pak U, Pang Y, Hao W, Yang X. Advances and Challenges in Spatial Transcriptomics for Developmental Biology. Biomolecules. 2023;13:156
12. Shan Y, Zhang Q, Guo W. et al. TIST: Transcriptome and Histopathological Image Integrative Analysis for Spatial Transcriptomics. Genomics Proteomics Bioinformatics. 2022;20:974-88
13. Wu D, Liu X, Zhang J, Li L, Wang X. Significance of single-cell and spatial transcriptomes in cell biology and toxicology. Cell Biol Toxicol. 2021;37:1-5
14. Marx V. Method of the Year 2020: spatially resolved transcriptomics. Nat Methods. 2021;18:1-1
15. Maynard KR, Collado-Torres L, Weber LM. et al. Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nat Neurosci. 2021;24:425-36
16. Rodriques SG, Stickels RR, Goeva A. et al. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science. 2019;363:1463-7
17. Gunderson KL, Kruglyak S, Graige MS. et al. Decoding Randomly Ordered DNA Arrays. Genome Res. 2004;14:870-7
18. Saunders A, Macosko EZ, Wysoker A. et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell. 2018;174:1015-1030.e16
19. Stickels RR, Murray E, Kumar P. et al. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat Biotechnol. 2021;39:313-9
20. Vickovic S, Eraslan G, Salmén F. et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat Methods. 2019;16:987-90
21. Liao J, Lu X, Shao X, Zhu L, Fan X. Uncovering an organ's molecular architecture at single-cell resolution by spatially resolved transcriptomics. Trends Biotechnol. 2021;39:43-58
22. Chen H, Li D, Bar-Joseph Z. SCS: cell segmentation for high-resolution spatial transcriptomics. Nat Methods. 2023;20:1237-43
23. Han S, Li J, Ting AY. Proximity labeling: spatially resolved proteomic mapping for neurobiology. Curr Opin Neurobiol. 2018;50:17-23
24. Fazal FM, Han S, Parker KR. et al. Atlas of Subcellular RNA Localization Revealed by APEX-Seq. Cell. 2019;178:473-490.e26
25. Cho C-S, Xi J, Si Y. et al. Microscopic examination of spatial transcriptome using Seq-Scope. Cell. 2021;184:3559-3572.e22
26. Bentley DR, Balasubramanian S, Swerdlow HP. et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53-9
27. Liu Y, Yang M, Deng Y. et al. High-spatial-resolution multi-omics sequencing via deterministic barcoding in tissue. Cell. 2020;183:1665-1681.e18
28. Fu X, Sun L, Dong R. et al. Polony gels enable amplifiable DNA stamping and spatial transcriptomics of chronic pain. Cell. 2022;185:4621-4633.e17
29. Chen A, Liao S, Cheng M. et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell. 2022;185:1777-1792.e21
30. Natarajan KN, Miao Z, Jiang M. et al. Comparative analysis of sequencing technologies for single-cell transcriptomics. Genome Biol. 2019;20:70
31. Zugazagoitia J, Gupta S, Liu Y. et al. Biomarkers Associated with Beneficial PD-1 Checkpoint Blockade in Non-Small Cell Lung Cancer (NSCLC) Identified Using High-Plex Digital Spatial Profiling. Clin Cancer Res. 2020;26:4360-8
32. Singer RH, Ward DC. Actin gene expression visualized in chicken muscle tissue culture by using in situ hybridization with a biotinated nucleotide analog. Proc Natl Acad Sci U S A. 1982;79:7331-5
33. Gall JG, Pardue ML. Formation and detection of RNA-DNA hybrid molecules in cytological preparations. Proc Natl Acad Sci U S A. 1969;63:378-83
34. Femino A, Fay F, Fogarty K, Singer R. Visualization of single RNA transcripts in situ. Science. 1998:585-90
35. Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods. 2008;5:877-9
36. Shah S, Takei Y, Zhou W. et al. Dynamics and spatial genomics of the nascent transcriptome by intron seqFISH. Cell. 2018;174:363-376.e16
37. Codeluppi S, Borm LE, Zeisel A. et al. Spatial organization of the somatosensory cortex revealed by osmFISH. Nat Methods. 2018;15:932-5
38. Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science. 2015;348:aaa6090
39. Wang G, Moffitt JR, Zhuang X. Multiplexed imaging of high-density libraries of RNAs with MERFISH and expansion microscopy. Sci Rep. 2018;8:4847
40. Weinstein JA, Regev A, Zhang F. DNA microscopy: Optics-free spatio-genetic imaging by a stand-alone chemical reaction. Cell. 2019;178:229-241.e16
41. Borm LE, Mossi Albiach A, Mannens CCA. et al. Scalable in situ single-cell profiling by electrophoretic capture of mRNA using EEL FISH. Nat Biotechnol. 2023;41:222-31
42. Weibrecht I, Lundin E, Kiflemariam S. et al. In situ detection of individual mRNA molecules and protein complexes or post-translational modifications using padlock probes combined with the in situ proximity ligation assay. Nat Protoc. 2013;8:355-72
43. Ke R, Mignardi M, Pacureanu A. et al. In situ sequencing for RNA analysis in preserved tissue and cells. Nat Methods. 2013;10:857-60
44. Gyllborg D, Langseth CM, Qian X. et al. Hybridization-based in situ sequencing (HybISS) for spatially resolved transcriptomics in human and mouse brain tissue. Nucleic Acids Res. 2020;48:e112-e112
45. Lee JH, Daugharthy ER, Scheiman J. et al. Highly Multiplexed Subcellular RNA Sequencing in Situ. Science. 2014;343:1360-3
46. Wang X, Allen WE, Wright MA. et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science. 2018;361:eaat5691
47. Chen X, Sun Y-C, Church GM, Lee JH, Zador AM. Efficient in situ barcode sequencing using padlock probe-based BaristaSeq. Nucleic Acids Res. 2018;46:e22-e22
48. Chen X, Sun Y-C, Zhan H. et al. High-Throughput Mapping of Long-Range Neuronal Projection Using In Situ Sequencing. Cell. 2019;179:772-786.e19
49. Zhu J, Wang Y, Chang WY. et al. Mapping cellular interactions from spatially resolved transcriptomics data. Cold Spring Harbor Laboratory. 2023
50. Salas SM, Czarnewski P, Kuemmerle LB. et al. Optimizing Xenium In Situ data utility by quality assessment and best practice analysis workflows. Cold Spring Harbor Laboratory. 2023
51. Derry JMJ, Burns C, Frazier JP. et al. Trackable intratumor microdosing and spatial profiling provide early insights into activity of investigational agents in the intact tumor microenvironment. Clin Cancer Res Off J Am Assoc Cancer Res. 2023;29:3813-25
52. Nichterwitz S, Benitez JA, Hoogstraaten R, Deng Q, Hedlund E. LCM-Seq: A method for spatial transcriptomic profiling using laser capture microdissection coupled with PolyA-Based RNA sequencing. Methods Mol Biol Clifton NJ. 2018;1649:95-110
53. Peng G, Suo S, Cui G. et al. Molecular architecture of lineage allocation and tissue organization in early mouse embryo. Nature. 2019;572:528-32
54. Kruse F, Junker JP, van Oudenaarden A, Bakkers J. Tomo-seq: A method to obtain genome-wide expression data with spatial resolution. Methods Cell Biol. 2016;135:299-307
55. Casasent AK, Schalck A, Gao R. et al. Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing. Cell. 2018;172:205-217.e12
56. Lovatt D, Ruble BK, Lee J. et al. Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue. Nat Methods. 2014;11:190-6
57. Yang L, von Trentini D, Kim H, Sul J-Y, Eberwine JH, Dmochowski IJ. Photoactivatable circular caged oligonucleotides for transcriptome In Vivo analysis (TIVA). ChemPhotoChem. 2021;5:940-6
58. Wang T, Shen P, He Y, Zhang Y, Liu J. Spatial transcriptome uncovers rich coordination of metabolism in E. coli K12 biofilm. Nat Chem Biol. 2023:1-11
59. Hassan MI. Multi-omics approaches to therapeutic target identification. Brief Funct Genomics. 2023;22:75
60. Konda P, Garinet S, Van Allen EM, Viswanathan SR. Genome-guided discovery of cancer therapeutic targets. Cell Rep. 2023;42:112978
61. Zhang X, Ruan C, Wang Y. et al. Integrated protein solubility shift assays for comprehensive drug target identification on a proteome-wide scale. Anal Chem. 2023;95:13779-87
62. Duan Y-Y, Ke X, Wu H. et al. Multi-tissue transcriptome-wide association study reveals susceptibility genes and drug targets for insulin resistance-relevant phenotypes. Diabetes Obes Metab. 2024;26:135-47
63. Park H-E, Jo SH, Lee RH. et al. Spatial transcriptomics: Technical aspects of recent developments and their applications in neuroscience and cancer research. Adv Sci Weinh Baden-Wurtt Ger. 2023;10:e2206939
64. Wang W-J, Chu L-X, He L-Y. et al. Spatial transcriptomics: recent developments and insights in respiratory research. Mil Med Res. 2023;10:38
65. Rajachandran S, Zhang X, Cao Q. et al. Dissecting the spermatogonial stem cell niche using spatial transcriptomics. Cell Rep. 2023;42:112737
66. Zhao S, Wang Q, Ni K. et al. Combining single-cell sequencing and spatial transcriptome sequencing to identify exosome-related features of glioblastoma and constructing a prognostic model to identify BARD1 as a potential therapeutic target for GBM patients. Front Immunol. 2023;14:1263329
67. Causer A, Tan X, Lu X. et al. Deep spatial-omics analysis of Head & Neck carcinomas provides alternative therapeutic targets and rationale for treatment failure. NPJ Precis Oncol. 2023;7:89
68. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15:81-94
69. Marusyk A, Janiszewska M, Polyak K. Intratumor heterogeneity: The rosetta stone of therapy resistance. Cancer Cell. 2020;37:471-84
70. Bao Z, Wang Y, Wang Q. et al. Intratumor heterogeneity, microenvironment, and mechanisms of drug resistance in glioma recurrence and evolution. Front Med. 2021;15:551-61
71. Skarne N, D'Souza R, Harris L. et al. P02.12.B UNDERSTANDING THE ROLES OF EPHRIN A5 IN GLIOBLASTOMA AND ITS POTENTIAL AS A THERAPEUTIC TARGET. Neuro-Oncol. 2023;25:ii32-ii32
72. Offenhäuser C, Al-Ejeh F, Puttick S. et al. EphA3 pay-loaded antibody therapeutics for the treatment of glioblastoma. Cancers. 2018;10:519
73. Ji AL, Rubin AJ, Thrane K. et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell. 2020;182:497-514.e22
74. Kilmister EJ, Koh SP, Weth FR, Gray C, Tan ST. Cancer metastasis and treatment resistance: Mechanistic insights and therapeutic targeting of cancer stem cells and the tumor microenvironment. Biomedicines. 2022;10:2988
75. Zhang Q, Abdo R, Iosef C. et al. The spatial transcriptomic landscape of non-small cell lung cancer brain metastasis. Nat Commun. 2022;13:5983
76. Kim N, Kim HK, Lee K. et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat Commun. 2020;11:2285
77. Hwang WL, Jagadeesh KA, Guo JA. et al. Single-nucleus and spatial transcriptome profiling of pancreatic cancer identifies multicellular dynamics associated with neoadjuvant treatment. Nat Genet. 2022;54:1178-91
78. Maniatis S, Äijö T, Vickovic S. et al. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science. 2019;364:89-93
79. Chen W-T, Lu A, Craessaerts K. et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer's Disease. Cell. 2020;182:976-991.e19
80. Kaufmann M, Schaupp A-L, Sun R. et al. Identification of early neurodegenerative pathways in progressive multiple sclerosis. Nat Neurosci. 2022;25:944-55
81. Jia E, Sheng Y, Shi H. et al. Spatial Transcriptome Profiling of Mouse Hippocampal Single Cell Microzone in Parkinson's Disease. Int J Mol Sci. 2023;24:1810
82. Navarro JF, Croteau DL, Jurek A. et al. Spatial Transcriptomics Reveals Genes Associated with Dysregulated Mitochondrial Functions and Stress Signaling in Alzheimer Disease. iScience. 2020;23:101556
83. Chen S, Chang Y, Li L. et al. Spatial transcriptomics of human middle temporal gyrus reveals layer-specific gene expression in early Alzheimer's disease. Alzheimers Amp Dement. 2021 17
84. Wu J, Li Y, Huang Y. et al. Integrating spatial and single-nucleus transcriptomic data elucidates microglial-specific responses in female cynomolgus macaques with depressive-like behaviors. Nat Neurosci. 2023;26:1352-64
85. Batiuk MY, Tyler T, Dragicevic K. et al. Upper cortical layer-driven network impairment in schizophrenia. Sci Adv. 2022;8:eabn8367
86. Zhang Z, Li Y, Jiang S, Shi F-D, Shi K, Jin W-N. Targeting CCL5 signaling attenuates neuroinflammation after seizure. CNS Neurosci Ther. 2023;29:317-30
87. Mantri M, Hinchman MM, McKellar DW. et al. Spatiotemporal transcriptomics reveals pathogenesis of viral myocarditis. Nat Cardiovasc Res. 2022;1:946-60
88. Yamada S, Ko T, Hatsuse S. et al. Spatiotemporal transcriptome analysis reveals critical roles for mechano-sensing genes at the border zone in remodeling after myocardial infarction. Nat Cardiovasc Res. 2022;1:1072-83
89. Long X, Yuan X, Du J. Single-cell and spatial transcriptomics: Advances in heart development and disease applications. Comput Struct Biotechnol J. 2023;21:2717-31
90. Boogerd CJ, Lacraz GPA, Vértesy Á. et al. Spatial transcriptomics unveils ZBTB11 as a regulator of cardiomyocyte degeneration in arrhythmogenic cardiomyopathy. Cardiovasc Res. 2023;119:477-91
91. Asp M, Giacomello S, Larsson L. et al. A spatiotemporal organ-wide gene expression and cell atlas of the developing human heart. Cell. 2019;179:1647-1660.e19
92. Kuppe C, Ramirez Flores RO, Li Z. et al. Spatial multi-omic map of human myocardial infarction. Nature. 2022;608:766-77
93. Lacraz GPA, Junker JP, Gladka MM. et al. Tomo-Seq Identifies SOX9 as a Key Regulator of Cardiac Fibrosis During Ischemic Injury. Circulation. 2017;136:1396-409
94. Ko T, Nomura S, Yamada S. et al. Cardiac fibroblasts regulate the development of heart failure via Htra3-TGF-β-IGFBP7 axis. Nat Commun. 2022;13:3275
95. Porritt RA, Zemmour D, Abe M. et al. NLRP3 inflammasome mediates immune-stromal interactions in vasculitis. Circ Res. 2021;129:e183-200
96. Sun J, Singh P, Shami A. et al. Spatial Transcriptional Mapping Reveals Site-Specific Pathways Underlying Human Atherosclerotic Plaque Rupture. J Am Coll Cardiol. 2023;81:2213-27
97. Cross AR, de Andrea CE, Villalba-Esparza M. et al. Spatial transcriptomic characterization of COVID-19 pneumonitis identifies immune circuits related to tissue injury. JCI Insight. 2023;8:e157837
98. Downes DJ, Cross AR, Hua P. et al. Identification of LZTFL1 as a candidate effector gene at a COVID-19 risk locus. Nat Genet. 2021;53:1606-15
99. Kasmani MY, Topchyan P, Brown AK. et al. A spatial sequencing atlas of age-induced changes in the lung during influenza infection. Nat Commun. 2023;14:6597
100. Ma F, Hughes TK, Teles RMB. et al. Single cell and spatial transcriptomics defines the cellular architecture of the antimicrobial response network in human leprosy granulomas. Cold Spring Harbor Laboratory. 2020
101. Janosevic D, Myslinski J, McCarthy TW. et al. The orchestrated cellular and molecular responses of the kidney to endotoxin define a precise sepsis timeline. eLife. 2021;10:e62270
102. Airs PM, Vaccaro K, Gallo KJ. et al. Spatial transcriptomics reveals antiparasitic targets associated with essential behaviors in the human parasite Brugia malayi. PLoS Pathog. 2022;18:e1010399
103. Dixon EE, Wu H, Muto Y, Wilson PC, Humphreys BD. Spatially resolved transcriptomic analysis of acute kidney injury in a female murine model. J Am Soc Nephrol JASN. 2022;33:279-89
104. Kayhan M, Vouillamoz J, Rodriguez DG. et al. Intrinsic TGF-β signaling attenuates proximal tubule mitochondrial injury and inflammation in chronic kidney disease. Nat Commun. 2023;14:3236
105. Kuppe C, Ibrahim MM, Kranz J. et al. Decoding myofibroblast origins in human kidney fibrosis. Nature. 2021;589:281-6
106. Canela VH, Bowen WS, Ferreira RM. et al. A spatially anchored transcriptomic atlas of the human kidney papilla identifies significant immune injury in patients with stone disease. Nat Commun. 2023;14:4140
107. Chen D, Shao M, Song Y. et al. Single-cell RNA-seq with spatial transcriptomics to create an atlas of human diabetic kidney disease. FASEB J Off Publ Fed Am Soc Exp Biol. 2023;37:e22938
108. Salem F, Perin L, Sedrakyan S. et al. The spatially resolved transcriptional profile of acute T cell-mediated rejection in a kidney allograft. Kidney Int. 2022;101:131-6
109. Lin P, Yan X, Jing S. et al. Single-cell and spatially resolved transcriptomics for liver biology. Hepatol Baltim Md. 2023 Publish Ahead of Print
110. Wu R, Guo W, Qiu X. et al. Comprehensive analysis of spatial architecture in primary liver cancer. Sci Adv. 2021;7:eabg3750
111. Hildebrandt F, Andersson A, Saarenpää S. et al. Spatial Transcriptomics to define transcriptional patterns of zonation and structural components in the mouse liver. Nat Commun. 2021 12
112. Umbaugh DS, Ramachandran A, Jaeschke H. Spatial reconstruction of the early hepatic transcriptomic landscape after an acetaminophen overdose using single-cell RNA-Sequencing. Toxicol Sci Off J Soc Toxicol. 2021;182:327-45
113. Nault R, Saha S, Bhattacharya S, Sinha S, Maiti T, Zacharewski T. Single-cell transcriptomics shows dose-dependent disruption of hepatic zonation by TCDD in mice. Toxicol Sci Off J Soc Toxicol. 2023;191:135-48
114. Zhang G, Ji P, Xia P. et al. Identification and targeting of cancer-associated fibroblast signature genes for prognosis and therapy in Cutaneous melanoma. Comput Biol Med. 2023;167:107597
115. Yamasaki J, Hirata Y, Otsuki Y. et al. MEK inhibition suppresses metastatic progression of KRAS-mutated gastric cancer. Cancer Sci. 2022;113:916-25
116. Arnold K, Xu Y, Liao Y-E, Cooley BC, Pawlinski R, Liu J. Synthetic anticoagulant heparan sulfate attenuates liver ischemia reperfusion injury. Sci Rep. 2020;10:17187
117. Xin J, Yang T, Wu X. et al. Spatial transcriptomics analysis of zone-dependent hepatic ischemia-reperfusion injury murine model. Commun Biol. 2023;6:194
118. Lin H-Y, Wu H-J, Chu P-Y. Multi-omics and experimental analysis unveil theragnostic value and immunological roles of inner membrane mitochondrial protein (IMMT) in breast cancer. J Transl Med. 2023;21:189
119. Arora R, Cao C, Kumar M. et al. Spatial transcriptomics reveals distinct and conserved tumor core and edge architectures that predict survival and targeted therapy response. Nat Commun. 2023;14:5029
120. Bergen V, Lange M, Peidli S, Wolf FA, Theis FJ. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol. 2020;38:1408-14
121. Qiu X, Zhang Y, Martin-Rufino JD. et al. Mapping transcriptomic vector fields of single cells. Cell. 2022;185:690-711.e45
122. Wen C-Y, Hsiao J-H, Tzeng Y-DT. et al. Single-cell landscape and spatial transcriptomic analysis reveals macrophage infiltration and glycolytic metabolism in kidney renal clear cell carcinoma. Stress Aging Brain. 2023;15:11298-312
123. Lee J, Kim Y, Jin S. et al. Q-omics: Smart software for assisting oncology and cancer research. Mol Cells. 2021;44:843-50
124. Huang C, Wu Z, Wang D. et al. Myelin-associated oligodendrocytic basic protein-dependent myelin repair confers the long-lasting antidepressant effect of ketamine. Mol Psychiatry. 2023 10.1038/s41380-023-02288-5
125. Lee A, Floyd K, Wu S. et al. BCG vaccination stimulates integrated organ immunity by feedback of the adaptive immune response to imprint prolonged innate antiviral resistance. Nat Immunol. 2024;25:41-53
126. Ballester Roig MN, Leduc T, Dufort-Gervais J, Maghmoul Y, Tastet O, Mongrain V. Probing pathways by which rhynchophylline modifies sleep using spatial transcriptomics. Biol Direct. 2023;18:21
127. Lin W, Chen X, Wang D. et al. Single-nucleus ribonucleic acid-sequencing and spatial transcriptomics reveal the cardioprotection of Shexiang Baoxin Pill (SBP) in mice with myocardial ischemia-reperfusion injury. Front Pharmacol. 2023;14:1173649
128. Cassier PA, Navaridas R, Bellina M. et al. Netrin-1 blockade inhibits tumour growth and EMT features in endometrial cancer. Nature. 2023;620:409-16
129. Karthaus WR, Hofree M, Choi D. et al. Regenerative potential of prostate luminal cells revealed by single-cell analysis. Science. 2020;368:497-505
130. Joseph DB, Henry GH, Malewska A. et al. 5-Alpha reductase inhibitors induce a prostate luminal to club cell transition in human benign prostatic hyperplasia. J Pathol. 2022;256:427-41
131. Gouin KH 3rd, Ing N, Plummer JT. et al. An N-Cadherin 2 expressing epithelial cell subpopulation predicts response to surgery, chemotherapy and immunotherapy in bladder cancer. Nat Commun. 2021;12:4906
132. Vo T, Balderson B, Jones K. et al. Spatial transcriptomic analysis of Sonic hedgehog medulloblastoma identifies that the loss of heterogeneity and promotion of differentiation underlies the response to CDK4/6 inhibition. Genome Med. 2023;15:29
133. Cook Sangar ML, Genovesi LA, Nakamoto MW. et al. Inhibition of CDK4/6 by palbociclib significantly extends survival in medulloblastoma patient-derived xenograft mouse models. Clin Cancer Res Off J Am Assoc Cancer Res. 2017;23:5802-13
134. Wang J, Xu P, Zou G. et al. Integrating Spatial Transcriptomics and Single-nucleus RNA Sequencing Reveals the Potential Therapeutic Strategies for Uterine Leiomyoma. Int J Biol Sci. 2023;19:2515-30
135. Zhao C, Wong-Rolle A, Divakar P, Calvo K, Hourigan C. 250 Spatial-transcriptomic analysis of tumor-immune microenvironment in AML patients receiving pembrolizumab and decitabine. J Immunother Cancer. 2021;9:A270-A270
136. Agostini A, Guerriero I, Piro G. et al. Talniflumate abrogates mucin immune suppressive barrier improving efficacy of gemcitabine and nab-paclitaxel treatment in pancreatic cancer. J Transl Med. 2023 21
137. Simian M, Bissell MJ. Organoids: A historical perspective of thinking in three dimensions. J Cell Biol. 2017;216:31-40
138. Li M, Izpisua Belmonte JC. Organoids — Preclinical Models of Human Disease. N Engl J Med. 2019;380:569-79
139. Ikari R, Mukaisho K, Kageyama S. et al. Differences in the Central Energy Metabolism of Cancer Cells between Conventional 2D and Novel 3D Culture Systems. Int J Mol Sci. 2021;22:1805
140. Clevers H. Modeling Development and Disease with Organoids. Cell. 2016;165:1586-97
141. Liu J, Li T, Li R. et al. Hepatic Organoid-Based High-Content Imaging Boosts Evaluation of Stereoisomerism-Dependent Hepatotoxicity of Stilbenes in Herbal Medicines. Front Pharmacol. 2022;13:862830
142. Lancaster MA, Knoblich JA. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science. 2014;345:1247125
143. Dong Q, Su X, Li X. et al. In vitro construction of lung cancer organoids by 3D bioprinting for drug evaluation. Colloids Surf Physicochem Eng Asp. 2023;666:131288
144. Adult human kidney organoids. cellular origin and disease modeling. Nat Genet. 2022;54:1593-4
145. Mao Y, Wang W, Yang J. et al. Drug repurposing screening and mechanism analysis based on human colorectal cancer organoids. Protein Cell. 2024;15:285-304
146. Deng S, Li C, Cao J. et al. Organ-on-a-chip meets artificial intelligence in drug evaluation. Theranostics. 2023;13:4526-58
147. Brooks A, Liang X, Zhang Y. et al. Liver organoid as a 3D in vitro model for drug validation and toxicity assessment. Pharmacol Res. 2021;169:105608
148. Yan HHN, Chan AS, Lai FP-L, Leung SY. Organoid cultures for cancer modeling. Cell Stem Cell. 2023;30:917-37
149. Hockney S, Parker J, Turner JE. et al. Next generation organoid engineering to replace animals in cancer drug testing. Biochem Pharmacol. 2023;213:115586
150. Sachs N, de Ligt J, Kopper O. et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell. 2018;172:373-386.e10
151. Min J, Vega PN, Engevik AC. et al. Heterogeneity and dynamics of active Kras-induced dysplastic lineages from mouse corpus stomach. Nat Commun. 2019;10:5549
152. Kim S-C, Park JW, Seo H-Y. et al. Multifocal organoid capturing of colon cancer reveals pervasive intratumoral heterogenous drug responses. Adv Sci Weinh Baden-Wurtt Ger. 2022;9:e2103360
153. Moncada R, Barkley D, Wagner F. et al. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat Biotechnol. 2020;38:333-42
154. Piwecka M. Single-cell and spatial transcriptomics: deciphering brain complexity in health and disease. Nat Rev Neurol. 2023 19
155. Kim J, Koo B-K, Knoblich JA. Human organoids: model systems for human biology and medicine. Nat Rev Mol Cell Biol. 2020;21:571-84
156. Wu H, Liu F, Shangguan Y. et al. Integrating spatial transcriptomics with single-cell transcriptomics reveals a spatiotemporal gene landscape of the human developing kidney. Cell Biosci. 2022;12:80
157. Cai C, Yue Y, Yue B. Single-cell RNA sequencing in skeletal muscle developmental biology. Biomed Pharmacother Biomedecine Pharmacother. 2023;162:114631
158. Barrozo ER, Aagaard KM. Human placental biology at single-cell resolution: a contemporaneous review. BJOG Int J Obstet Gynaecol. 2022;129:208-20
159. Hao Y, Hao S, Andersen-Nissen E. et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573-3587.e29
160. Lancaster MA, Renner M, Martin C-A. et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373-9
161. Giandomenico SL, Sutcliffe M, Lancaster MA. Generation and long-term culture of advanced cerebral organoids for studying later stages of neural development. Nat Protoc. 2021;16:579-602
162. Kanton S, Boyle MJ, He Z. et al. Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature. 2019;574:418-22
163. Tanaka Y, Cakir B, Xiang Y, Sullivan GJ, Park I-H. Synthetic Analyses of Single-Cell Transcriptomes from Multiple Brain Organoids and Fetal Brain. Cell Rep. 2020;30:1682-1689.e3
164. He Z, Maynard A, Jain A. et al. Lineage recording in human cerebral organoids. Nat Methods. 2022;19:90-9
165. O'Hara-Wright M, Gonzalez-Cordero A. Retinal organoids: a window into human retinal development. Development. 2020;147:dev189746
166. Kim S, Lowe A, Dharmat R. et al. Generation, transcriptome profiling, and functional validation of cone-rich human retinal organoids. Proc Natl Acad Sci. 2019;116:10824-33
167. Kim HJ, O'Hara-Wright M, Kim D. et al. Comprehensive characterization of fetal and mature retinal cell identity to assess the fidelity of retinal organoids. Stem Cell Rep. 2023;18:175-89
168. Dorgau B, Collin J, Rozanska A. et al. Deciphering the spatiotemporal transcriptional and chromatin accessibility of human retinal organoid development at the single-cell level. iScience. 2024;27:109397
169. Wahle P, Brancati G, Harmel C. et al. Multimodal spatiotemporal phenotyping of human retinal organoid development. Nat Biotechnol. 2023;41:1765-75
170. Legnini I, Emmenegger L, Zappulo A. et al. Spatiotemporal, optogenetic control of gene expression in organoids. Nat Methods. 2023;20:1544-52
171. de Joode K, Heersche N, Basak EA. et al. Review - The impact of pharmacogenetics on the outcome of immune checkpoint inhibitors. Cancer Treat Rev. 2023;122:102662
172. Back DJ, Breckenridge AM, Crawford FE, MacIver M, Orme ML, Rowe PH. Interindividual variation and drug interactions with hormonal steroid contraceptives. Drugs. 1981;21:46-61
173. Skånland SS, Henrik B, Cremaschi A. et al. Ex vivo drug sensitivity screens identify personalized treatment options for CLL patients. Blood. 2019;134:5446-5446
174. Park YM, Lin D-C. Moving closer towards a comprehensive view of tumor biology and microarchitecture using spatial transcriptomics. Nat Commun. 2023;14:7017
175. Nieto P, Elosua-Bayes M, Trincado JL. et al. A single-cell tumor immune atlas for precision oncology. Genome Res. 2021;31:1913-26
176. Kumar V, Ramnarayanan K, Sundar R. et al. Single-Cell Atlas of Lineage States, Tumor Microenvironment, and Subtype-Specific Expression Programs in Gastric Cancer. Cancer Discov. 2022;12:670-91
177. Aldea M, Andre F, Marabelle A, Dogan S, Barlesi F, Soria J-C. Overcoming resistance to tumor-targeted and immune-targeted therapies. Cancer Discov. 2021;11:874-99
178. Zhu AX, Abbas AR, de Galarreta MR. et al. Molecular correlates of clinical response and resistance to atezolizumab in combination with bevacizumab in advanced hepatocellular carcinoma. Nat Med. 2022;28:1599-611
179. Zhang S, Yuan L, Danilova L. et al. Spatial transcriptomics analysis of neoadjuvant cabozantinib and nivolumab in advanced hepatocellular carcinoma identifies independent mechanisms of resistance and recurrence. Genome Med. 2023;15:72
180. Lin S, He C, Song L. et al. Exosomal lncCRLA is predictive for the evolvement and development of lung adenocarcinoma. Cancer Lett. 2023;582:216588
181. O'Rourke CJ, Salati M, Rae C. et al. Molecular portraits of patients with intrahepatic cholangiocarcinoma who diverge as rapid progressors or long survivors on chemotherapy. Gut. 2024;73:496-508
182. Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513-22
183. Lee S, Schmitt CA. The dynamic nature of senescence in cancer. Nat Cell Biol. 2019;21:94-101
184. Gui C, Wei J, Mo C. et al. Therapeutic implications for localized prostate cancer by multiomics analyses of the ageing microenvironment landscape. Int J Biol Sci. 2023;19:3951-69
185. Heming M, Haessner S, Wolbert J. et al. Intratumor heterogeneity and T cell exhaustion in primary CNS lymphoma. Genome Med. 2022;14:109
186. Shen X, Zhao Y, Wang Z, Shi Q. Recent advances in high-throughput single-cell transcriptomics and spatial transcriptomics. Lab Chip. 2022;22:4774-91
187. Longo SK, Guo MG, Ji AL, Khavari PA. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat Rev Genet. 2021;22:627-44
188. Zhao E, Stone MR, Ren X. et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat Biotechnol. 2021;39:1375-84
189. Bergenstråhle J, Larsson L, Lundeberg J. Seamless integration of image and molecular analysis for spatial transcriptomics workflows. BMC Genomics. 2020;21:482
190. Rao A. Exploring tissue architecture using spatial transcriptomics
191. Zheng Y, Chen Y, Ding X, Wong KH, Cheung E. Aquila: a spatial omics database and analysis platform. Nucleic Acids Res. 2023;51:D827-34
192. Wang G, Wu S, Xiong Z, Qu H, Fang X, Bao Y. CROST: a comprehensive repository of spatial transcriptomics. Nucleic Acids Res. 2024;52:D882-90
193. Xu Z, Wang W, Yang T. et al. STOmicsDB: a comprehensive database for spatial transcriptomics data sharing, analysis and visualization. Nucleic Acids Res. 2024;52:D1053-61
194. Fan Z, Chen R, Chen X. SpatialDB: a database for spatially resolved transcriptomes. Nucleic Acids Res. 2020;48:D233-7
195. Xu C, Jin X, Wei S. et al. DeepST: identifying spatial domains in spatial transcriptomics by deep learning. Nucleic Acids Res. 2022;50:e131
196. Li R, Chen X, Yang X. Navigating the landscapes of spatial transcriptomics: How computational methods guide the way. WIREs RNA. 2024 15
197. Sun D, Liu Z, Li T, Wu Q, Wang C. STRIDE: accurately decomposing and integrating spatial transcriptomics using single-cell RNA sequencing. Nucleic Acids Res. 2022;50:e42
198. Wang G, Zhao J, Yan Y, Wang Y, Wu AR, Yang C. Construction of a 3D whole organism spatial atlas by joint modelling of multiple slices with deep neural networks. Nat Mach Intell. 2023;5:1200-13
199. Dong K, Zhang S. Deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder. Nat Commun. 2022;13:1739
200. Zhang C, Dong K, Aihara K, Chen L, Zhang S. STAMarker: determining spatial domain-specific variable genes with saliency maps in deep learning. Nucleic Acids Res. 2023;51:e103
201. Ben-Chetrit N, Niu X, Swett AD. et al. Integration of whole transcriptome spatial profiling with protein markers. Nat Biotechnol. 2023;41:788-93
202. Vicari M, Mirzazadeh R, Nilsson A. et al. Spatial multimodal analysis of transcriptomes and metabolomes in tissues. Nat Biotechnol. 2023 10.1038/s41587-023-01937-y
203. Qian Z, Qin J, Lai Y, Zhang C, Zhang X. Large-scale integration of single-cell RNA-seq data reveals astrocyte diversity and transcriptomic modules across six central nervous system disorders. Biomolecules. 2023;13:692
204. Zheng P, Zhang N, Ren D, Yu C, Zhao B, Zhang Y. Integrated spatial transcriptome and metabolism study reveals metabolic heterogeneity in human injured brain. Cell Rep Med. 2023;4:101057
205. Li Y, Li Z, Wang C. et al. Spatiotemporal transcriptome atlas reveals the regional specification of the developing human brain. Cell. 2023;186:5892-5909.e22
206. Gao S, Shi Q, Zhang Y. et al. Identification of HSC/MPP expansion units in fetal liver by single-cell spatiotemporal transcriptomics. Cell Res. 2022;32:38-53
207. Piña JO, Raju R, Roth DM. et al. Multimodal spatiotemporal transcriptomic resolution of embryonic palate osteogenesis. Nat Commun. 2023;14:5687
Author contact
![]() Corresponding authors: Hongjun Yang (hongjun0420sina.com); Peng Chen (chenpengac.cn); Wei Liu (lwlancerlllcom).
Corresponding authors: Hongjun Yang (hongjun0420sina.com); Peng Chen (chenpengac.cn); Wei Liu (lwlancerlllcom).