Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2024; 14(7):2656-2674. doi:10.7150/thno.91456 This issue Cite
Research Paper
AXL-specific single domain antibodies show diagnostic potential and anti-tumor activity in Acute Myeloid Leukemia
Niels Vandewalle1, Hatice Satilmis1, Emma Verheye1,2,3, Rong Fan1, Yanmeng Wang1, Timo W.M. De Groof4, Jessica Bridoux4, Tessa Kerre5, Nathan De Beule6, Ann De Becker6, Elke De Bruyne1, Eline Menu1, Karin Vanderkerken1, Karine Breckpot7*, Nick Devoogdt4*, Kim De Veirman1,6* ![]()
1. Translational Oncology Research Center (TORC), team Hematology and Immunology (HEIM), Vrije Universiteit Brussel, Brussels, Belgium.
2. Laboratory of Dendritic Cell Biology and Cancer Immunotherapy, VIB Center for Inflammation Research, Brussels, Belgium.
3. Brussels Center of Immunology, Vrije Universiteit Brussel, Brussels, Belgium.
4. Laboratory of Molecular Imaging and Therapy (MITH), Vrije Universiteit Brussel, Brussels, Belgium.
5. Department of Hematology, Ghent University Hospital, Ghent, Belgium.
6. Translational Oncology Research Center (TORC), team Hematology and Immunology (HEIM), Universitair Ziekenhuis Brussel, Brussels, Belgium.
7. Translational Oncology Research Center (TORC), Laboratory for Molecular and Cellular Therapy (LMCT), Vrije Universiteit Brussel, Brussels, Belgium.
* Authors equally contributed.
Received 2023-10-24; Accepted 2024-3-16; Published 2024-4-15
Abstract

Rationale: AXL expression has been identified as a prognostic factor in acute myeloid leukemia (AML) and is detectable in approximately 50% of AML patients. In this study, we developed AXL-specific single domain antibodies (sdAbs), cross-reactive for both mouse and human AXL protein, to non-invasively image and treat AXL-expressing cancer cells.
Methods: AXL-specific sdAbs were induced by immunizing an alpaca with mouse and human AXL proteins. SdAbs were characterized using ELISA, flow cytometry, surface plasmon resonance and the AlphaFold2 software. A lead compound was selected and labeled with 99mTc for evaluation as a diagnostic tool in mouse models of human (THP-1 cells) or mouse (C1498 cells) AML using SPECT/CT imaging. For therapeutic purposes, the lead compound was fused to a mouse IgG2a-Fc tail and in vitro functionality tests were performed including viability, apoptosis and proliferation assays in human AML cell lines and primary patient samples. Using these in vitro models, its anti-tumor effect was evaluated as a single agent, and in combination with standard of care agents venetoclax or cytarabine.
Results: Based on its cell binding potential, cross-reactivity, nanomolar affinity and GAS6/AXL blocking capacity, we selected sdAb20 for further evaluation. Using SPECT/CT imaging, we observed tumor uptake of 99mTc-sdAb20 in mice with AXL-positive THP-1 or C1498 tumors. In THP-1 xenografts, an optimized protocol using pre-injection of cold sdAb20-Fc was required to maximize the tumor-to-background signal. Besides its diagnostic value, we observed a significant reduction in tumor cell proliferation and viability using sdAb20-Fc in vitro. Moreover, combining sdAb20-Fc and cytarabine synergistically induced apoptosis in human AML cell lines, while these effects were less clear when combined with venetoclax.
Conclusions: Because of their diagnostic potential, sdAbs could be used to screen patients eligible for AXL-targeted therapy and to follow-up AXL expression during treatment and disease progression. When fused to an Fc-domain, sdAbs acquire additional therapeutic properties that can lead to a multidrug approach for the treatment of AXL-positive cancer patients.
Keywords: single domain antibodies, AXL, nuclear imaging, therapy, acute myeloid leukemia
Introduction
Acute myeloid leukemia (AML) is a hematological malignancy characterized by the abnormal proliferation of immature myeloid cells and bone marrow failure [1]. The backbone therapy for AML patients consists of a combination of daunorubicin and cytarabine, also called the “7+3 induction regimen” [2, 3]. Unfortunately, because of increased treatment-related toxicity and unfavorable biological characteristics when compared to younger patients, the treatment of elderly AML patients (> 60 years) is more challenging. Therefore, their prognosis is poor with a 5-year overall survival of less than 20% [4]. Since AML is a highly heterogeneous disease, both clinically and genetically, there is an increased demand for patient-tailored approaches to diagnose and treat AML patients [5].
Receptor tyrosine kinase AXL, derived from the Greek word 'anexelecto' (uncontrollable), has been identified as a prognostic factor and therapeutic target in AML patients [6]. Binding of the ligand growth arrest-specific factor 6 (GAS6) to the AXL receptor was found to regulate various processes in cancer pathogenesis, including proliferation, survival, migration, stemness and chemoresistance [7, 8]. In normal tissues, the AXL receptor can be found on the surface of, among others, bone marrow stroma and myeloid cells, where activated AXL will efficiently dispose apoptotic material and reduce TLR-dependent inflammatory responses and natural killer cell activity. In cancer, AXL is expressed on tumor cells as well as adjacent immune cells (e.g., dendritic cells, macrophages), and is often considered as an inhibitor of the innate immune response [9].
Several AXL targeting compounds have been developed, including small molecule inhibitors (e.g., bemcentinib, dubermatinib) and human anti-AXL monoclonal antibodies (mAbs) (e.g., tilvestamab) [8]. Bemcentinib (also known as BGB324 or R428) is by far the most advanced anti-AXL therapeutic agent and was given FDA fast-track designation for the treatment of elderly patients with relapsed/refractory AML (NCT02488408). Since AXL is detectable in approximately 50% of the AML patients and appears to be highly predictive for response to AXL-targeted therapies (like bemcentinib) [6], monitoring AXL expression in cancer patients is highly recommended. While bone marrow biopsies are typically taken at diagnosis, repeated invasive biopsy procedures are considered painful and uncomfortable [10]. Moreover, a single-site biopsy may not provide a representative profile of the tumor heterogeneity as it is spatially and temporally constrained, and necessitates the development of a non-invasive tool to follow-up AXL expression during and/or after therapy [8, 11].
In this study, we developed alpaca-derived, small sized single domain antibodies (sdAbs, 12-15 kDa), cross-reactive for mouse and human AXL protein, and evaluated their potential in a diagnostic and therapeutic context in AML. SdAbs present unique advantages over conventional mAbs due to their small size, allowing them to target and bind obstructed antigen binding sites and penetrate tissues effectively. Moreover, the fast blood clearance of radiolabeled sdAbs allows an early acquisition of high-quality PET images and fast diagnosis of patients, with low radiation burden. Additionally, sdAbs exhibit remarkable stability, solubility, and resistance to proteases, making them promising candidates for drug development with lower immunogenicity and simplified production processes compared to conventional mAbs [12, 13].
Several anti-AXL sdAbs were characterized using ELISA, flow cytometry, surface plasmon resonance and the AlphaFold2 prediction software. sdAb20 was selected as a lead compound and was evaluated as a diagnostic tool in mice bearing human (THP-1) or mouse (C1498) AML cells. For therapeutic purposes, sdAb20 was fused to a mouse IgG2a-Fc tail (sdAb20-Fc) and in vitro functionality tests were performed, including viability, apoptosis and proliferation assays using human AML cell lines and primary AML patient samples. The anti-tumor effect of sdAb20-Fc was evaluated in monotherapy, and in a dual combination with standard-of-care agents venetoclax or cytarabine, on human AML cell lines.
Materials and Methods
Animal studies
CB17-SCID and C57BL/6-Ly5.1 mice of 6-8 weeks old were purchased from Charles River (France and Germany, respectively). For imaging and biodistribution experiments, we used both the immunodeficient THP-1 xenograft model and the immunocompetent syngeneic C1498 mouse model. For the THP-1 xenografts, CB17-SCID mice were inoculated subcutaneously in the right flank with five million THP-1 cells that were diluted 1:1 with Matrigel (Corning, New York, USA). Tumor inoculation was performed under 2.5% isoflurane anaesthesia and subcutaneous tumor growth was allowed to a size of 250 ± 50 mm³. For the syngeneic C1498 mouse model, C57BL/6-Ly5.1 mice were inoculated intravenously with one million lentivirally transduced AXLhigh C1498 cells under 2.5% isoflurane anaesthesia. C1498 mice were assessed daily for signs of leukemic disease (e.g., paralysis, weight loss). Prior to imaging, mice were anesthetized with 18.75 mg/kg of ketamine hydrochloride and 0.5 mg/kg of medetomidine hydrochloride. All animal experiments and housing conditions were approved by the Ethical Committee for Use of Laboratory Animals of the Vrije Universiteit Brussel and executed in accordance with the European guidelines for animal experimentation (License Agreement: LA 1230281, Files No.: 20-281-3, 21-281-2, 22-281-4, 23-281-1 and 23-281-4).
Single domain antibody isolation and selection
Anti-AXL sdAbs were generated as previously described [14]. In short, an alpaca (Vicugna pacos) was immunized with a mixture of 100 µg of recombinant mouse and 100 µg of recombinant human AXL proteins (R&D Systems, Oxon, UK; #154-AL, #854-AX) in Gerbu LQ 3000 adjuvant (GerbuBiotechnik GmbH, Heidelberg, Germany), at a rate of one injection per week for six weeks. Four days after the last immunization, 50 mL anti-coagulated blood was collected, lymphocytes were extracted with Leucosep (Greiner Bio-One, Kremsünster, Austria) by density centrifugation and total RNA was isolated and converted into cDNA by reverse transcription with oligo-dT primers. Gene fragments encoding the variable domain of camelid-specific heavy-chain-only antibodies were amplified via a two-step nested PCR and ligated into a pMECS phagemid vector. This vector was transformed into E. coli TG1 electrocompetent cells to generate a cloned sdAb-library of 108 transformants. These were then phage displayed using M13K07 helper phages, and selected by biopanning, as described [15]. After 3-4 consecutive rounds of in vitro selection on recombinant AXL protein-coated wells of Nunc Maxisorp flat bottom microtiter plates (Thermo Fisher scientific, Inc., Waltham, MA, USA), a total of 135 individual clones were selected and screened for binding on recombinant AXL proteins using enzyme-linked immunoassays (ELISA). In addition, their inserted DNA sequence was analyzed using the CLC Main Work Bench 8 software (Qiagen, Hilden, Germany), aligned according to the international ImMunoGeneTices (IMGT) database [16] and manually annotated.
Bacterial production, purification and quality control of anti-AXL sdAbs
Based on their sequence, 118 clones were further selected and subdivided into 11 families and from the 135 original clones, 14 were selected and re-cloned into pHEN6 plasmids (to encode a C-terminal hexa-histidine (His6) tag) for expression of the monomeric version of the selected sdAbs. For production, the pHEN6-sdAb plasmids were transformed into an E. coli WK6 strain. The sdAbs were obtained via periplasmic expression and subsequently purified by immobilized metal affinity chromatography (IMAC) and size exclusion chromatography (SEC), as previously described [14]. The concentration of the sdAbs was determined by spectrophotometry at 280 nm with NanodropTM using the theoretically calculated extinction coefficient, based on the amino acid sequence. The purity of the sdAbs was confirmed by a 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) under reducing conditions, followed by Coomassie blue staining. Besides anti-AXL sdAbs, an irrelevant non-targeting control sdAb (referred to as R3B23), specific for 5T2 M-proteins [17], was used for in vitro and in vivo experiments. As a last step, the periplasmic proteins were screened for cross-reactive specificity against human and mouse AXL proteins using flow cytometry on the human HCT-116 and the murine CT-26 cell lines.
Evaluation of affinity/kinetics using surface plasmon resonance
The affinity (KD-value) for both human and mouse AXL proteins of the purified sdAbs was determined using a Biacore-T200 device (GE Healthcare, Chicago, USA). The measurements were performed at 25°C using HEPES-Buffered Saline (HBS; 20 mM HEPES pH 7.4, 150 mM NaCl, 3.4 mM EDTA and 0.005% Tween 20) as running buffer. In short, recombinant AXL proteins were diluted and immobilized on a CM5 sensor chip Series S (GE-Healthcare) using amino coupling chemistry with 1-(3-(dimethylamino)propyl)-3-ethylcarbodi-imide (EDC) and N-hydroxy-succinimide (NHS). Remaining free EDC-NHS linkers were neutralized with 1 M ethanolamine-HCl pH 8.5. Seven serial dilutions were made of the purified sdAbs, after which the association (240 s) and dissociation (300 s) of each dilution to the immobilized recombinant AXL proteins were analyzed at a flow rate of 10 µL/min. After each cycle, the chip was regenerated twice for 30 s using 100 mM Glycine HCl pH 2.0 to completely remove any leftover sdAb. The response units from the flow channel on which recombinant AXL protein was immobilized were corrected by subtraction of the response units measured on a flow channel without protein. Additionally, the signal from the blank (HBS only) was subtracted from each measurement. The rate kinetic constants were determined by a mathematical fitting of a 1:1 binding model using the Biacore Evaluation software (GE Healthcare), and the kon/koff ratio was used to determine the equilibrium dissociation constant (KD).
99mTc-labeling of sdAbs, pinhole SPECT/CT imaging and image analysis
Radiolabeling of sdAb20 using 99mTc-tricarbonyl chemistry was performed as previously described [18]. In short, [99mTc(H2O)3(CO)3]+ was linked to the C-terminal His6-tag of the sdAb using the Isolink® labeling kit (Mallinckrodt Medical BV, Petten, The Netherlands). Next, unbound [99mTc(H2O)3(CO)3]+ was separated from the 99mTc-sdAb complex via gel filtration on a pre-equilibrated NAP-5 SEC column (Sephadex, GE Healthcare, Machelen, Belgium), followed by filtration through a 0.22 µm filter (Millex, Millipore, Haren, Belgium). Labeling efficiency and purity (> 95%) were assessed using instant thin-layer chromatography (iTLC-SG, Pall Corporation, Hoegaarden, Belgium). Mice bearing subcutaneous THP-1 tumors and C1498 mice reaching humane endpoints were intravenously injected with 100 µL 99mTc-labeled anti-AXL sdAb20 (5 µg) or the irrelevant 99mTc-labeled control sdAb R3B23 with on average 37.9 ± 18.3 MBq of injected activity. For the THP-1 xenograft model, mice were pre-inoculated with unlabeled sdAb20-Fc (90 µg) construct for a variety of timepoints (0 h/co-injection, 2, 4, 6, 8, 24, 48 and 72 h) to reduce aspecific uptake of 99mTc-labeled sdAb20. Pre-inoculation with sdAb20-Fc was not performed for imaging in the C1498 syngeneic AML model. One hour post-injection, pinhole SPECT-micro-CT imaging was performed using the MILabs VECTor/CT. SPECT images were generated using a rat SPECT-collimator (1.5 mm 75-pinhole general purpose collimator), in spiral mode, with six bed positions for whole-body imaging, 150 s per position. The CT-scan was set to 60 kV and 615 µA with a resolution of 80 µm. The total body scan duration was 15 min for SPECT and 139 s for CT. Images were reconstructed with 0.4 mm voxels with two subsets and seven iterations, without post-reconstruction filter. Images were analyzed using Medical Image Data Examiner (AMIDE) software and maximum intensity projections were generated using OsiriX Lite software (Pixmeo SARL, CH1233 Bernex, Switzerland).
DTPA-conjugation and 111In-labeling of Fc-conjugated sdAb constructs
All buffers were prepared metal free. SdAb-Fc constructs were buffer exchanged to 0.5 M Na2CO3 (pH 8.9-9.5 for R3B23-Fc; pH 9.5-9.9 for sdAb20-Fc) and concentrated to ± 4 mg/mL using Vivaspin 2 (MWCO 5000) (Sartorius), according to manufacturer's guidelines. For DTPA-conjugation, a 100-fold molar excess of NCS-DTPA (C22H28N4O10S·3HCL; S-2-(4-Isothiocyanatobenzyl)-diethylenetriamine pentaacetic acid; Macrocyclics) was added to the sdAb-Fc constructs and incubated for 2.5 h at room temperature with gentle shaking. The conjugated sdAb-Fc-DTPA complex was purified from excess NCS-DTPA via SEC (conjugation yield ~ 30%) on a Superdex 75 Increase 10/300GL column (GE Healthcare) with elution in 0.1 M NH4OAc (pH 7.0). The concentration of the conjugate was determined using a NanoDropTM 2000 spectrophotometer (Thermo Scientific), based on the new molecular weight and extinction coefficient of the sdAb-Fc-DTPA complexes.
For Indium-111 (111In)-labeling, 175-200 MBq of [111In]InCl3 (Curium Pharma, Petten, The Netherlands) was added to 75 µg DTPA-conjugated sdAb and 0.25 M NaOAc (pH 4.7) buffer, in a final volume of 500 µL. The reaction mixture was incubated for 30 min at room temperature, after which the radiotracer was purified via gel-filtration with a pre-equilibrated (5 mg/mL Vitamin C in 0.99% NaCl) NAP-5 column (GE Healthcare, Diegem, Belgium), according to manufacturer's protocol (decay-corrected radiochemical yield > 60%). Radiochemical purity (> 90%) was confirmed through iTLC-SG (Pall Corporation, Hoegaarden, Belgium), with 0.1 M sodium citrate (pH 5.0) as mobile phase, and via SEC on a Superdex 75 Increase 10/300 GL column (Cytiva) in 2x PBS at a flow rate of 0.8 mL/min. In addition, a radio-SDS-PAGE was performed, followed by Coomassie Blue staining, using a NovexTM WedgeWellTM 16% Tris-Glycerine Gel (Invitrogen) and a 1x Tris/Glycerine/SDS running buffer (Bio-Rad). The PageRuler™ Prestained Protein Ladder (Thermo ScientificTM) was used as a reference. Images were recorded with an Amersham Imager 680 and radioactive signals were detected with an autoradiograph Typhoon FLA 7000 Laser Scanner.
To confirm functionality, radio-ELISA was performed by coating Lockwell C8 Maxisorp plates (Thermo Scientific) with recombinant mouse antigen (R&D Systems, Oxon, UK; #854-AX). A serial dilution (300-11.1 nM) of the [111In]In-DTPA-sdAb-Fc constructs were added to the pre-coated plates, in the presence of PBS + 1% FBS. Plates were washed three times and binding was determined by assessing the radioactive counts with a γ-counter (Cobra Inspector 5003, Canberra, Packard, Illinois, IL, USA). Specific binding (KD) was calculated as the difference between total and non-specific binding.
Prior to in vivo injection, the radiolabeled tracers were passed through a 0.22 µm PVDF low protein binder filter (Millipore, Merck, Darmstadt, Germany), after which stability at room temperature was confirmed up to 72 h by SEC on a Superdex 75 increase 5/150 GL column in 2x PBS at a flow rate of 0.45 mL/min. Mice bearing subcutaneous THP-1 tumors were intravenously injected with 100 µL 111In-labeled anti-AXL sdAb20-Fc (5 µg) or the irrelevant 111In-labeled control sdAb R3B23 with on average 9.76 ± 2.34 MBq of injected activity.
Ex vivo biodistribution
After imaging, mice were sacrificed for an ex vivo biodistribution study. Several organs, tissues and tumors were isolated to evaluate the specific uptake of the tracer using a γ-counter (Cobra Inspector 5003, Canberra, Packard, Illinois, IL, USA). The tissue/organ/tumor uptake was corrected for decay and calculated as the percentage of injected activity per gram tissue (%IA/g). For the C1498 mouse model, a single cell suspension was made of various organs known for C1498 AML infiltration, including blood, bone marrow (BM), liver, lungs, spleen, and ovary. Therefore, organs were crushed and cell suspensions were filtered through a 70 µm nylon filter to obtain a single cell suspension. Erythrocytes were lysed using red blood cell lysis buffer (0.16 M NH4Cl, 0.17 M Tris, pH 7.2). Finally, cells were washed, centrifuged, counted (Countess™ Automated Cell Counter, Invitrogen) and prepared for flow cytometry staining and analysis.
Drugs and reagents
For in vitro experiments, anti-AXL sdAb20 and control sdAb R3B23 were reformatted into an Fc-construct (mIgG2a) and purchased from ImmunoPrecise Antibodies (Utrecht, Netherlands). The constructs were dissolved in PBS at a stock concentration of 3.71 mg/mL (sdAb20-Fc) and 3.11 mg/mL (R3B23-Fc). Three additional compounds were used during in vitro experiments: R428, cytarabine, and venetoclax. The small molecule AXL inhibitor R428 (BGB324, Bemcentinib) was obtained from SelleckChem (Munich, Germany) and dissolved in dimethylsulfoxide (DMSO; Sigma-Aldrich, St Louis, MO, USA) at a stock concentration of 10 mM. The chemotherapeutic agent cytarabine was purchased from Sigma (#147-94-4) and dissolved in sterile water at a stock concentration of 10 mM. Lastly, the BCL-2 inhibitor venetoclax (ABT-199) was obtained from SelleckChem (#1257044-40-8) and dissolved in DMSO at a stock concentration of 20 mM. All compounds were stored at -20°C.
Cell viability and apoptosis assay
50,000 cells were seeded in 500 µL serum-supplemented medium in a 24-well plate and were treated with R428, R3B23-Fc, or sdAb20-Fc constructs as a single treatment or in combination with cytarabine or venetoclax at increasing concentrations for 72 h. We treated the cells with 200 µg/mL R3B23-Fc construct and 10, 100, or 200 µg/mL of the sdAb20-Fc construct or R428. We selected the final working concentrations for cytarabine of 250 nM, 500 nM, and 1000 nM for the MOLM-13 cell line and 1, 2, and 4 µM for the THP-1 cell line. Lastly, final working concentrations for venetoclax were 100, 500, and 1000 nM for the MOLM-13 cell line and 15, 30, and 60 nM for the THP-1 cell line. Used concentrations were determined based on titration experiments, resulting in low, medium, and high effects observed in the cell lines. Cell viability was analyzed using the CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI, USA) and normalized to the untreated control sample. The remaining cell suspension was used to evaluate cell death (apoptosis) via AnnexinV-APC/7-AAD staining (BD Biosciences) and flow cytometry using the BD AccuriTM C6 Plus and FACSCanto flow cytometers (BD Biosciences). Data were processed using BD FACSDivaTM (BD Biosciences) and FlowJo 10.9® software (Tree Star; Inc., Ashland, OR, USA).
BrdU incorporation assay and cell cycle analysis
AML cell proliferation was assessed by the incorporation of 5-Bromo-2-deoxyUridine (BrdU) during DNA replication. Cells were cultured at a concentration of 500,000 cells/mL and treated with 200 µg/mL R3B23-Fc and increasing concentrations of sdAb20-Fc (10, 100, 200 µg/mL) for 48 h. Four hours prior to sample collection, 1 mg/mL BrdU (Sigma, #B5002) was added to the medium. Samples were washed and fixated/incubated for 10 min at 4°C with a 4% paraformaldehyde solution, followed by overnight incubation in PBS + 0.2% Tween (Sigma-Aldrich) at room temperature. Cells were then washed twice, stained for 30 min with 2 M HCl at room temperature and washed with FACS flow and a mixture of PBS + 0.5% Triton-X (Sigma-Aldrich) + 10% FCS (PTF), respectively. Thereafter, cells were stained with 3 µL of FITC-conjugated Anti-BrdU-Fluorescein Antibody (Sigma-Aldrich, #1120269001) in 50 µL of PBS + 0.5% Triton-X + 10% FCS and incubated in the dark for 30 min. After washing, cells were incubated with a Cell Cycle Analysis (CCA) staining solution (0.1% Triton-X + 1 mg/mL Na₃C₆H₅O₇ + 50 µg/mL Propidium Iodide + 100 µg/mL RNase A). The percentage of BrdU+ cells was detected via flow cytometry using the FACSCanto and LSRFortessa flow cytometers (BD Biosciences) and gathered data were processed using BD FACSDivaTM (BD Biosciences) and FlowJo 10.9® software (Tree Star; Inc., Ashland, OR, USA).
Isolation of whole mononuclear cells from bone marrow samples
Bone marrow samples were collected as part of routine diagnostic or evaluation procedures after patients' signed informed consent and in accordance with the Declaration of Helsinki and an institutional research board approval from the University Hospital of Brussels (BUN: 1432021000659, Table S3). Mononuclear cells were isolated using density gradient centrifugation with Lymphoprep™ (STEMCELL™ technologies, Grenoble, France) and stored in liquid nitrogen until further needed. Upon thawing, cells were first resuspended in Roswell Park Memorial Institute (RMPI)-1640 medium (Gibco; Thermo Fisher Scientific), supplemented with 20% fetal calf serum (FCS) (PAN Biotech, Aidenbach, Bayern, Germany), 100 U/mL penicillin, 100 µg/mL streptomycin, 2 mM L-glutamine (Lonza, Basel, Switzerland), 100 µg/mL DNase I (Merck, Belgium) and 10 mM MgCl2. After 30 min of incubation and 10 min of centrifugation at 400g, the cells were washed twice with RPMI-1640 medium with 20% FCS and then counted with Trypan Blue staining to determine the number of dead cells.
Statistical analysis
Statistical analyses were performed with GraphPad Prism 8.01 software (GraphPad Software Inc., La Jolla, CA, USA). All data are represented as mean ± standard deviation (SD). p-values were calculated using a one-sided Mann-Whitney U t-test (two groups) or a one-way ANOVA (three or more groups). p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***) and p ≤ 0.0001 (****) were considered statistically significant. Normality was confirmed using the Shapiro-Wilk and Kolmogorov-Smirnov tests.
Results
Development and in vitro characterization of cross-reactive AXL-targeting single domain antibodies
AXL has been shown to be overexpressed in different tumor types, including AML [6, 7, 19]. Since this protein is not only expressed by tumor cells, but also by various immune cells (e.g., macrophages, dendritic cells) [7, 20], evaluation in preclinical immunocompetent models is indispensable. To aid clinical translation, we immunized an alpaca with both mouse and human AXL proteins (homology ~88%, determined using the Ensembl Database), followed by four rounds of biopanning of the resulting phage-displayed immune sdAb library (Figure 1A). Upon sequencing, 118 unique anti-AXL sdAbs (subgrouped in 11 families based on CDR3 sequence similarities) were identified and sdAbs were classified as either mouse specific (n=82), human specific (n=7) or cross-reactive (n=29) (Figure 1B-C). To confirm this, periplasmatic extracts were screened by ELISA for binding to recombinant mouse and human AXL proteins (Figure 1C). Next, we evaluated the cell binding potential of sdAbs to AXL+ cancer cells (human HCT-116, murine CT26) using flow cytometry and sdAb affinity was determined using surface plasmon resonance (Figure 1C, Figure S1A,B). Based on all in vitro characteristics, five cross-reactive sdAbs were selected for amino acid sequence alignment (according to IMGT) (Figure 1D). From these five sdAbs, sdAb20 was selected as the lead compound for further in vitro and in vivo evaluation (Figure 1C-D), based on its properties, including low-nM affinity binding, cross-reactivity for mouse and human AXL, thermal stability (> 50°C; Figure S1C) and the ability to specifically block the interaction between GAS6 and AXL. Using AlphaFold2 and Pymol software, the 3D structure of sdAb20 (Figure 1E) and the extracellular domain of mouse and human AXL (Figure 1F), as well as the binding of sdAb20 (through its CDR1, CDR2 and CDR3 loops) to the AXL protein (Figure 1G-H, Figure S2A-D) was predicted. Interestingly, sdAb20 interacted with the GAS6 recognition region of the Ig-like C2-type 1 domain of AXL, possibly leading to blocking of the AXL-GAS6 interaction, and therefore suggesting that sdAb20 could also have a therapeutic effect.
Generation and initial characterization of AXL-specific single domain antibodies. (A) Schematic illustration of alpaca immunization strategy, sdAb library construction and phage display selection of AXL-specific sdAbs. mRNA from blood lymphocytes served as a template for sdAb-specific PCR reactions, and resultant purified fragments were cloned into phagemids. Subsequently, antigen-specific phages were identified from this phagemid library using ELISA, flow cytometry and surface plasmon resonance. Figure created with BioRender.com. (B) Initial screening of 135 selected clones allowed for further classification into negative, mouse-specific, human-specific and cross-reactive. (C) Based on their characteristics, five cross-reactive sdAbs were selected for further investigation. The table shows a summary of their most important features. (D) Amino acid sequences of the selected anti-AXL sdAbs (numbering accor-ding to IMGT) [14]. The CDR1, CDR2 and CDR3 regions are highlighted in cyan, green and magenta, respectively. (E-F) Structural analy-sis of the selected sdAb20 and the extracellular domain of both murine and human AXL using Alphafold2 and Pymol software. (G-H) Alpha-Fold2 prediction of the interaction between sdAb20 and AXL. CDR = complementarity-determining regions; EC = extracellular; FR = frame-work region.
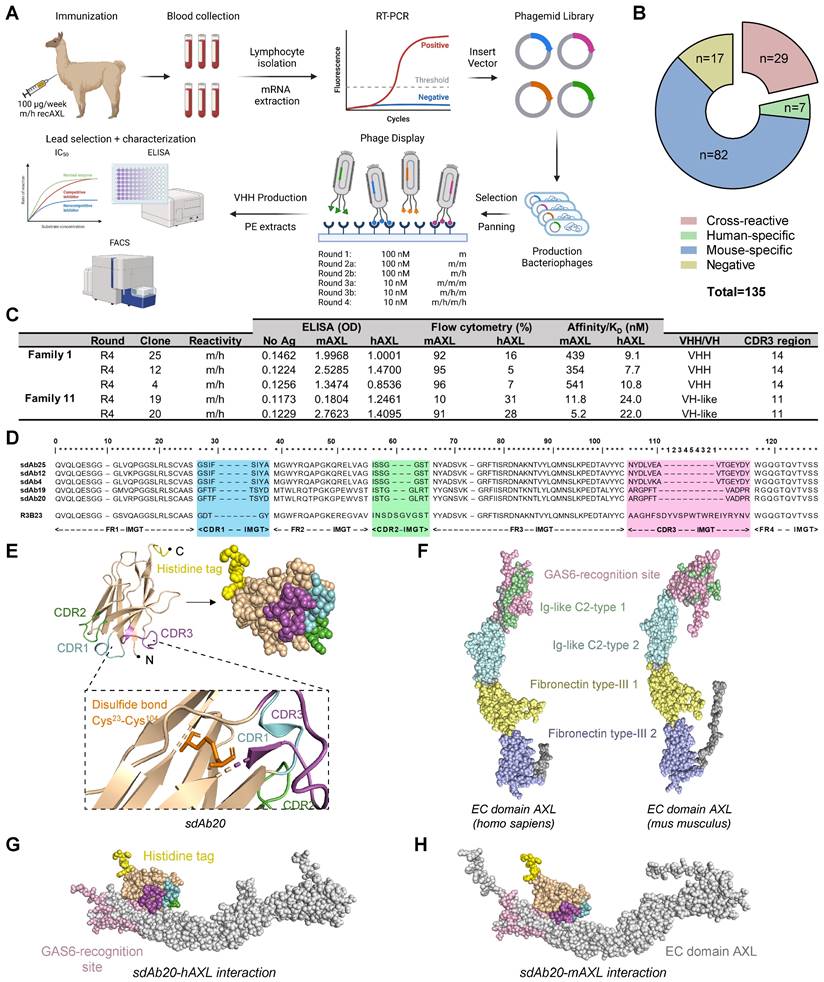
Binding potential of sdAb20 on human and murine AXL+ AML cell lines
Since AML is a commonly used model to evaluate AXL targeting therapeutics, we first assessed the expression of AXL in different human AML cell lines. Solid tumor cancer cells A549 (NSCLC) and HCT-116 (CRC) were included as positive controls. We observed a heterogeneous gene expression of AXL in AML cell lines, with the highest expression being detected in MV-4-11, MOLM-13, and THP-1 (Figure 2A). The highest protein expression of GAS6, AXL and phosphorylated (activated) AXL (P-AXL) was found in human THP-1 cells, while KG-1a cells were considered negative for AXL (Figure 2B). Since AXL expression is low in the murine C1498 AML cell line, we generated AXL-overexpressing C1498 cells (AXLhigh C1498) by lentiviral transduction and positive selection (Figure 2A,C; Figure S3A-C). This AML model was further used to assess the murine cell binding potential and for in vivo evaluation.
sdAb20 can specifically distinguish AXLhigh and AXLlow cancer cell lines. (A) Human (AXL) and murine (Axl) gene expression levels of Axl were measured using qRT-PCR after RNA extraction from human AML (KG-1a, OCI-AML3, MOLM-13, MV-4-11 and THP-1), human non-small cell lung carcinoma (A549), human colorectal carcinoma (HCT-116) and murine AML (AXLlow C1498 and AXLhigh C1498) cell lines. Human expression levels relative to OCI-AML3 cell line (n=5, ± SD). Murine expression levels relative to AXLlow C1498 cell line (n=5, ± SD). (B) Western blot analysis for protein expression of AXL, phosphorylated AXL (P-AXL) and GAS6 in extracts from several cancer cell lines (n=4). (C) Binding of sdAb20 to murine AML cell lines was assessed by flow cytometry and compared to an APC-coupled anti-AXL mAb. The anti-AXL mAb was compared to an isotype control and sdAb20 was compared to an irrelevant control sdAb (R3B23) (n=5, ± SD). (D-E) Flow cytometric assessment of AXL expression in several human cancer cell lines using an anti-AXL mAb and a corresponding isotype control (n=5, ± SD). (F-G) Flow cytometric analysis of the binding potential of sdAb20 to human cancer cell lines with a varying AXL expression. The irrelevant sdAb R3B23 served as a negative control (n=5, ± SD). (H) The specificity of the binding potential of sdAb20 to human (THP-1) and murine (C1498) AML cell lines was investigated by administering sdAb20 with increasing doses to two AML cell lines with a high AXL-expression (n=3, ± SD). p ≤ 0.05 (*), p ≤ 0.01 (**) and p ≤ 0.001 (***) were considered statistically significant. FSC = forward scatter, mAb = monoclonal antibody, MFI = median fluorescence intensity, ΔMFI = MFI (anti-AXL mAb) - MFI (isotype CTRL) or MFI (sdAb20) - MFI (CTRL sdAb).
Using flow cytometry, we compared the cell binding potential of sdAb20 to a commercially available mAb (Figure 2C-G). While the binding of the mAb to AXL+ AML cell lines resulted in a weak signal (Figure 2D-E), the MFI of the total population shifted significantly with sdAb20 compared to the control sdAb (R3B23) in AXL+ AML cells (Figure 2F-G). For solid tumor cell lines A549 and HCT-116, a clear AXL+ cell population could be distinguished using both the mAb and sdAb20. The binding of sdAb20 was also validated in the AXLhigh C1498 model (Figure 2C) and we observed a dose-dependent increase in mean fluorescence intensity in the THP-1 and AXLhigh C1498 cell lines (Figure 2H). These data illustrate the potential of sdAb20 to bind both murine and human AXL-expressing AML cells in a specific manner.
Specific tumor uptake of 99mTc-labeled sdAb20 in THP-1 xenografts
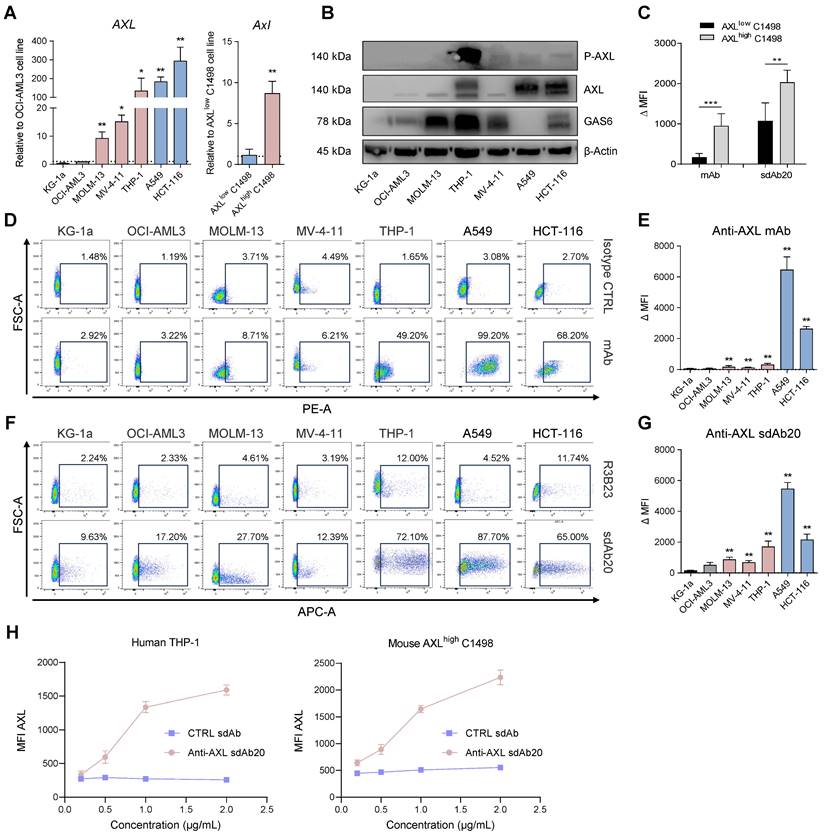
To validate the potential of sdAb20 as a diagnostic tracer in AML, we performed biodistribution studies in naive mice (Figure S1D) and in the subcutaneous THP-1 xenograft model (Figure 3A). The biodistribution of 99mTc-labeled sdAb20 in naive mice demonstrated an elevated uptake in especially lungs, liver and spleen (> 5 %IA/g) compared to radiolabeled R3B23 control sdAb (Figure S1D). Intravenous injection of 99mTc-labeled sdAb20 in tumor-bearing mice and ex vivo gamma counting of the organs demonstrated a significant uptake of sdAb20 within the tumor compared to control sdAb R3B23 (Figure 3B; p = 0.0040). However, the subcutaneous tumor uptake remained undetectable by SPECT/CT imaging. While CB17-SCID mice are deficient in B- and T-cells, myeloid cells such as macrophages remain present and are known to also express the AXL protein [20]. Therefore, sdAb20 was specifically taken up in organs like spleen, liver and lungs; resulting in a sink effect in these organs in tumor-bearing mice (Figure 3B-C). To reduce this background signal and increase tumor uptake, we further optimized our protocol using a pre-injection with cold (unlabeled) Fc-conjugated sdAb20. THP-1 xenografted mice were co-injected (0 h) or pre-treated with sdAb20-Fc for 2 h, 4 h, 6 h, 8 h, 24 h, 48 h and 72 h prior to 99mTc-labeled sdAb20 administration and SPECT/CT imaging (Figure 3A). Only pre-treatment for 24 h with cold sdAb20-Fc resulted in a significant reduction of the signal in the spleen and a significant increase in tumor uptake (Figure 3B-F; p = 0.0040), which could be clearly visualized using SPECT/CT imaging (Figure 3D). However, the tumor uptake of sdAb20 reduced after pre-treatment with sdAb20-Fc for 48 h and 72 h (Figure 3B-F).
sdAb20 can specifically recognize AXL-expressing cells in the human THP-1 AML xenograft mouse model. (A) Schematic overview of an optimized protocol to investigate the biodistribution profile of sdAb20. 21-24 days post tumor-inoculation, mice will first receive a pre-determined dose of unlabeled, cold sdAb20-Fc, after which they are injected with radiolabeled (99mTc) sdAb20 after 0-72 h. One hour later, mice will undergo SPECT/CT imaging and ex vivo biodistribution through organ collection. Figure created with BioRender.com. (B) The ex vivo biodistribution profile of 99mTc-R3B23, 99mTc-sdAb20 alone and 99mTc-sdAb20 after pre-treatment with sdAb20-Fc for 24 h. Results are presented as mean %IA/g ± SD. (C) Heatmap of accumulation patterns of 99mTc-sdAb20 in various organs at different timepoints upon pre-treatment with sdAb20-Fc. The colour key represents the mean value of tracer accumulation in organs (%IA/g, n=3 for each group, n=5 for 24 h group). (D) Reconstructed SPECT/CT images of THP-1 xenograft mice showing maximum intensity projection and transversal planes. One reconstructed image representing three individual mice. White dashed circles indicate THP-1 tumors. (E) Differences in lung, liver, spleen, bone and (F) tumor, relative to blood uptake. (G) Uptake of 111In-labeled sdAb20-Fc in lung, liver, spleen, bone and (H) tumor at different timepoints. Data were represented as organ-to-blood ratio. p ≤ 0.05 (*) and p ≤ 0.0001 (****) were considered statistically significant. IA = injected activity, ID/cc = injected dose per cubic centimeter.
To further clarify the observed effect, we analyzed the biodistribution profile of radiolabeled sdAb20-Fc and control R3B23-Fc sdAb. As the His6-tag of sdAb20 has been replaced with a functional mIgG2a-Fc region, and given the lower pharmacokinetics of sdAb20-Fc (evaluations up to 72 h), labeling with 99mTc (decay t1/2 = 6.0 hours) was no longer possible and had to be replaced by 111In (decay t1/2 = 2.8 days) instead. After DTPA-conjugation, sdAb20-Fc was radiolabeled with 111In (purity > 90%) and quality controls were performed, including radio-ELISA, radio-SDS-PAGE and a stability assay (radiochemical purity remained > 90% up to 72 h), to confirm the functionality of the compound (Figure S4A-E). Starting with a high uptake at 1 h post-injection in almost all organs, with spleen and blood having the highest %IA/g values (66.16 ± 6.51 and 9.80 ± 1.48 %IA/g, respectively), both sdAb-Fc constructs gradually decreased in circulation (Figure S5A-D). At 24 h post-injection, sdAb20-Fc primarily accumulated in the spleen (35.41 ± 3.73 %IA/g) and low uptake could be observed in liver (6.61 ± 0.69 %IA/g), bone (3.48 ± 0.52 %IA/g) and tumor (2.97 ± 0.35 %IA/g) (Figure 3G, Figure S5). At 48 h and 72 h, a significant increase in tumor-to-blood ratios could be observed for 111In-labeled sdAb20-Fc (12.99 ± 2.73 %IA/g and 23.86 ± 2.53 %IA/g, for 48 h and 72 h, respectively) (Figure 3H).
Taken together, these data illustrate that 24 h pre-treatment with sdAb20-Fc can reduce the uptake of 99mTc-labeled sdAb20 in the spleen while significantly increasing tumor uptake. On the other hand, the elevated tumor accumulation levels of sdAb20-Fc at later time points probably resulted in a reduced imaging signal of 99mTc-labeled sdAb20 using the 48 h and 72 h pre-treatment protocol.
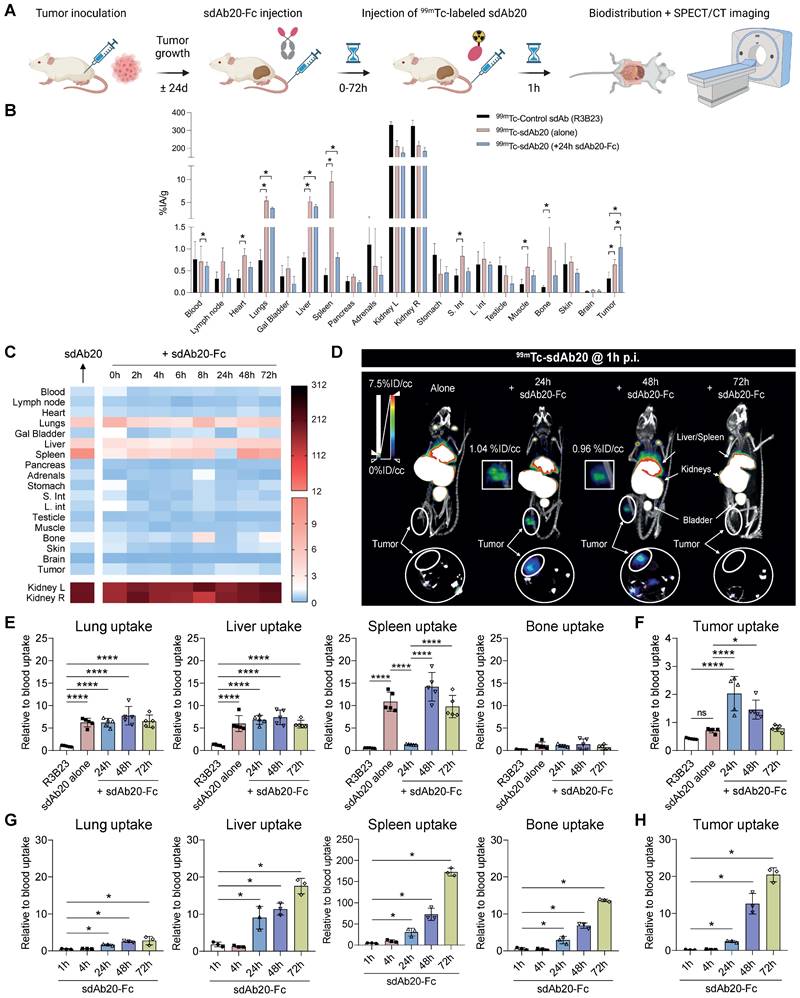
Specific uptake of 99mTc-labeled sdAb20 in naive and immunocompetent AXLhigh C1498 mice
Although the data in THP-1 xenografted mice already provided evidence for the use of sdAb20 as a diagnostic tool in AML, we aimed to further validate its potential using an immunocompetent syngeneic AML mouse model using the C1498 cell line. This murine AML cell line was isolated from C57BL/6 mice and is described to accumulate within the bone marrow, spleen, liver, lungs and ovaries upon intravenous injection [21]. However, as AXL expression in C1498 cells is low, we lentivirally modified this cell line to overexpress the AXL protein (Figure S3A-C). Naive mice and tumor-bearing mice (21 days post tumor inoculation) were intravenously injected with 99mTc-labeled sdAb20 or control sdAb R3B23, followed by SPECT/CT imaging after 1 h of biodistribution. In both naive and AXLhigh C1498 mice, we observed a significant uptake of sdAb20 in lungs (p < 0.0001), liver (p = 0.0035 and p < 0.0001), spleen (p < 0.0001), adrenals (p < 0.0001 and p = 0.0141), bone marrow (p < 0.0001 and p = 0.0003) and ovaries (p = 0.0054 and p < 0.0001) compared to R3B23 (Figure 4A). Interestingly, AXLhigh C1498 mice demonstrated a significant higher uptake in blood (p = 0.0249), spleen (p < 0.0001), liver (p < 0.0001), bone marrow (p = 0.0007) and ovary (p = 0.0008) compared to naive mice, in all organs that were found to be infiltrated with AXLhigh C1498 (CD45.2+) cells (Figure 4B-C). Despite the accumulation of tumor cells within the lungs of AXLhigh C1498 mice, no significant difference in ex vivo radioactivity could be found compared to naive mice (Figure 4B; p = 0.3992). Finally, SPECT/CT images illustrated a difference between the signal intensity of sdAb20 in AXLhigh C1498 mice compared to naive mice, confirming the potential of sdAb20 as a diagnostic tool to identify AXL+ tumors (Figure 4D).
99mTc-sdAb20 can specifically target and visualize AXL-expressing cells in the syngeneic murine C1498 AML mouse model. (A) Ex vivo γ-counting of isolated organs from naive and AXLhigh C1498 mice. Organs were isolated 90 min post-injection of radiolabeled (99mTc) sdAb20 or R3B23 control tracer (n=5). (B) Uptake of radiolabeled sdAb20 in various hematological organs of naive and AXLhigh C1498 tumor-bearing immunocompetent mice. Data were represented as organ-to-blood ratio. (C) Flow cytometric analysis of end-stage tumorload (CD45.2+ tumor cells) of different organs (n=8, ± SD). (D) Reconstructed SPECT/CT images of naive and AXLhigh C1498 mice, one hour post-injection of 99mTc-labeled anti-AXL sdAb20 and R3B23 control sdAb (n=8). Images show maximum intensity projections and transversal planes; and are reconstructed with OsiriX software. Representative intensities (%IA/g) are shown next to the images. Dashed circles represent ovaries. %ID/cc = injected dose per cubic centimeter. p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***) and p ≤ 0.0001 (****) were considered statistically significant.
Fc-coupled sdAb20 decreases cell viability and proliferation of human AXL positive AML cell lines in vitro
Since the binding of GAS6 to AXL has been found to promote cancer progression and sdAb20 was predicted to interact with the GAS6 recognition site, we further evaluated the GAS6/AXL blocking capacity of sdAb20 in vitro using a competition assay. Although sdAb20 could inhibit the binding of GAS6 to AXL (IC50 = 118.8 nM) (Figure 5C-D), we only observed a limited effect on AML cell viability or apoptosis (Figure S6A-B). To further improve the therapeutic capacity, sdAb20 was coupled to a mIgG2a Fc-domain, which has been described to offer superior anti-tumor activity to mIgG1 (Figure 5A-B) [22]. SdAb20-Fc was able to bind AXL+ MV-4-11, MOLM-13 and THP-1 AML cells (Figure 5E-F), however the shift in fluorescence signal (MFI) was less clear compared to the monovalent sdAb20. Interestingly, the Fc-fused sdAb20 construct resulted in an increased GAS6/AXL blocking capacity with an IC50 of 24.23 nM (Figure 5C,D).
sdAb20-Fc generation and characterization. (A) Graphical representation of size difference between a conventional Ab, a heavy-chain only Ab, a sdAb and a sdAb-Fc construct. Figure created with BioRender.com. (B) Summarizing table of the most important characteristics of R3B23-Fc and sdAb20-Fc, including the mIgG2a-sequence that was used to generate these constructs. (C-D) Comparison of IC50 values, for blocking of hAXL and hGAS6, of sdAb20 and sdAb20-Fc as determined via a competition assay using surface plasmon resonance. (E-F) Flow cytometric analysis of the binding potential of sdAb20-Fc to human cancer cell lines with a varying AXL expression. The irrelevant sdAb R3B23-Fc served as a negative control (n=4, ± SD). p ≤ 0.01 (**) was considered statistically significant. MFI = median fluorescence intensity.
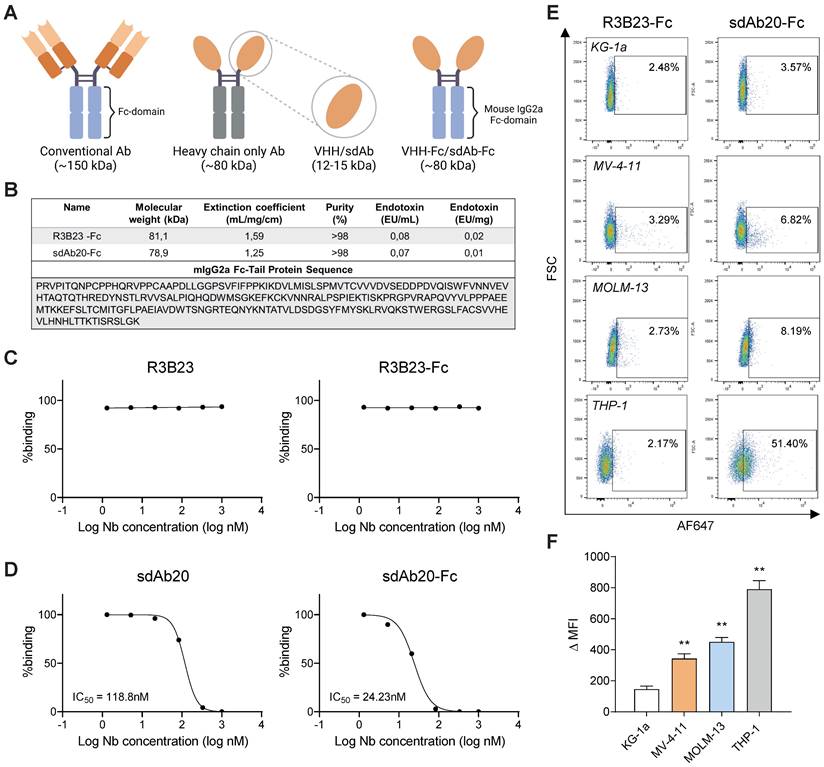
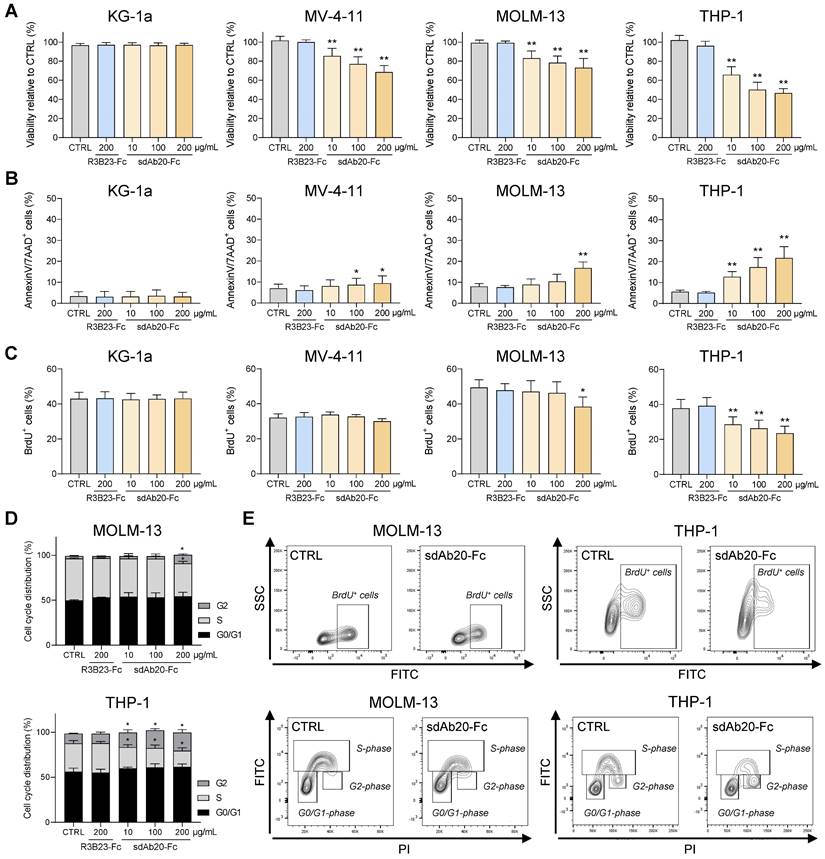
Next, we assessed the therapeutic effect of sdAb20-Fc in AXL-positive AML cell lines MV-4-11, MOLM-13 and THP-1. KG-1a was included as negative control. Treatment of MV-4-11, MOLM-13 and THP-1 with sdAb20-Fc resulted in a dose-dependent decrease in AML cell viability (Figure 6A) and increased apoptosis (Figure 6B) compared to R3B23-Fc, with the most pronounced effect in the THP-1 cell line. SdAb20-Fc treatment did not affect the KG-1a cell line. To further clarify the underlying mechanism of the observed anti-AML effect, we evaluated the effect of sdAb20-Fc in GAS6-/AXL+ (A549) and GAS6+/AXL+ (HCT-116) solid tumor cell lines. We selected A549 and HCT-116 since no AML cell lines were available with these characteristics. Anti-AXL sdAb20-Fc therapy significantly reduced the viability of the GAS6+ HCT-116 cell line, while no effect could be observed for the GAS6- A549 cell line (Figure S6C). Similar results were found on apoptosis (Figure S6D). These data indicate that the direct therapeutic effect of sdAb20-Fc on AXL+ tumors is mediated by blocking the interaction between GAS6 and AXL.
sdAb20-Fc has promising anti-AML effects as a single agent. (A-B) AML cell lines KG-1a, MV-4-11, MOLM-13 and THP-1 were treated with indicated concentrations of R3B23-Fc (200 μg/mL) or sdAb20-Fc (10, 100 and 200 μg/mL) for 72 h. The effect on cell viability (A) and apoptosis (B) was assessed via CellTiter-Glo assay and Annexin V/7-AAD staining, respectively (KG-1a: n=6; MV-4-11: n=6; MOLM-13: n=5; THP-1: n=6, ± SD). (C) MOLM-13 and THP-1 cells were treated with either R3B23-Fc or increasing concentrations of the sdAb20-Fc construct for 48 h. The number of proliferating cells was assessed using BrdU staining (THP-1: n=5; MOLM-13: n=3, ± SD). (D) Cell cycle analysis was performed on THP-1 and MOLM-13 cells using propidium iodide (PI) staining (THP-1: n=4; MOLM-13: n=3, ± SD). (E) Gating strategy used to determine the number of proliferating cells and to distinguish between the different cell cycle phases (G0/G1-phase, S-phase, G2-phase). p ≤ 0.05 (*) and p ≤ 0.01 (**) were considered statistically significant. CTRL = control.
As a positive control, we also tested the FDA-approved small molecule inhibitor R428 (bemcentinib). We observed a significant reduction in cell viability in all tested AML cell lines, but in contrast to sdAb20-Fc also in the AXL-negative KG-1a cell line (Figure S7A-B). Moreover, a direct comparison of the anti-AML effects of R428 and sdAb20-Fc at different molar concentrations (ranging from 10 nM to 10 µM) clearly demonstrated that R428 reduced the viability of both the KG-1a and THP-1 cell lines, regardless of their AXL expression, in a dose-dependent manner (Figure S7C-D). Surprisingly, more potent killing effects could be observed in the AXL- KG-1a cell line (IC50: 544.3 nM, Figure S7C) compared to the AXL+ THP-1 cell line (IC50: 1.2 µM, Figure S7D) after treatment with R428, indicating AXL-independent off-target effects. In contrast to these results, sdAb20-Fc did not have any effect on the viability of the AXL- KG-1a cell line, emphasizing its AXL-specific capacities. For the THP-1 cell line, we were able to replicate our previous findings, with an IC50 of 2.3 µM for sdAb20-Fc (Figure S7D).
Lastly, we also tested the effect of sdAb20-Fc on AML cell proliferation. SdAb20-Fc treatment resulted in a significant decrease in the percentage of BrdU-positive AML cells compared to R3B23-Fc treatment (Figure 6C,E), which was associated with a decrease in the S-phase and a significant increase in the G2-phase (Figure 6D-E).
Taken together, these data illustrate the direct anti-proliferative and pro-apoptotic effects of the Fc-fused sdAb20 construct by blocking the interaction between AXL and GAS6 in AML.
GAS6/AXL-targeting by sdAb20-Fc increases sensitivity towards standard-of-care agents in AML
The chemotherapeutic agent cytarabine and BCL-2 inhibitor venetoclax are commonly used drugs for the treatment of AML patients. In a first step, we evaluated the effect of both compounds on AXL expression in MOLM-13 and THP-1 cells using qRT-PCR and flow cytometry. Treatment with cytarabine for 48 h resulted in a significant increase of AXL expression in THP-1 and MOLM-13 cells, while this effect was absent with venetoclax (Figure S8A-D).
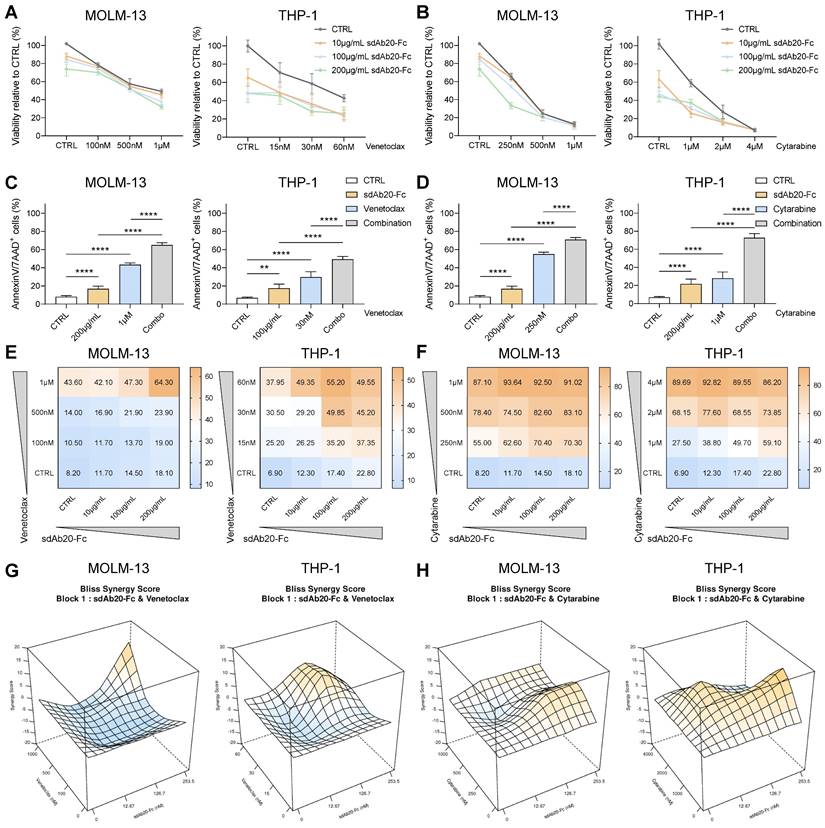
Next, we tested the anti-tumor effects of the combination of sdAb20-Fc with cytarabine or venetoclax in vitro. The combination of sdAb20-Fc with each agent could clearly reduce cell viability (Figure 7A-B) and significantly increase apoptosis (Figure 7C-D) at indicated concentrations in both THP-1 and MOLM-13 cells. Using the BLISS synergy method, we observed synergistic effects for the sdAb20-Fc/cytarabine combination and additive effects for the sdAb20-Fc/venetoclax combination (Figure 7E-H, Figure S9A-D) in the AML cell lines.
sdAb20-Fc has synergistic and additive effects when combined with standard-of-care agents cytarabine and venetoclax. (A-D) THP-1 and MOLM-13 cells were treated with increasing concentrations of sdAb20-Fc in combination with increasing concentrations of cytarabine or venetoclax for 72 h. The effect on cell viability (A-B) and induced apoptosis (C-D) was assessed by CellTiter-Glo viability assay and AnnexinV/7-AAD staining, respectively (MOLM-13: n=5, THP-1: n=6, ± SD). (E-F) Heatmaps display the percentage of apoptotic cells for combination treatment of sdAb20-Fc with venetoclax (E) and cytarabine (F). (G-H) Synergy of drug interactions was calculated using the BLISS synergy method. Output is generated in 3D format using the SynergyFinder Plus webtool. Data are represented as mean of five samples. p ≤ 0.01 (**) and p ≤ 0.0001 (****) were considered statistically significant. CTRL = control.
SdAb20-Fc reduces the cell viability of bone marrow-derived AXL-positive AML samples
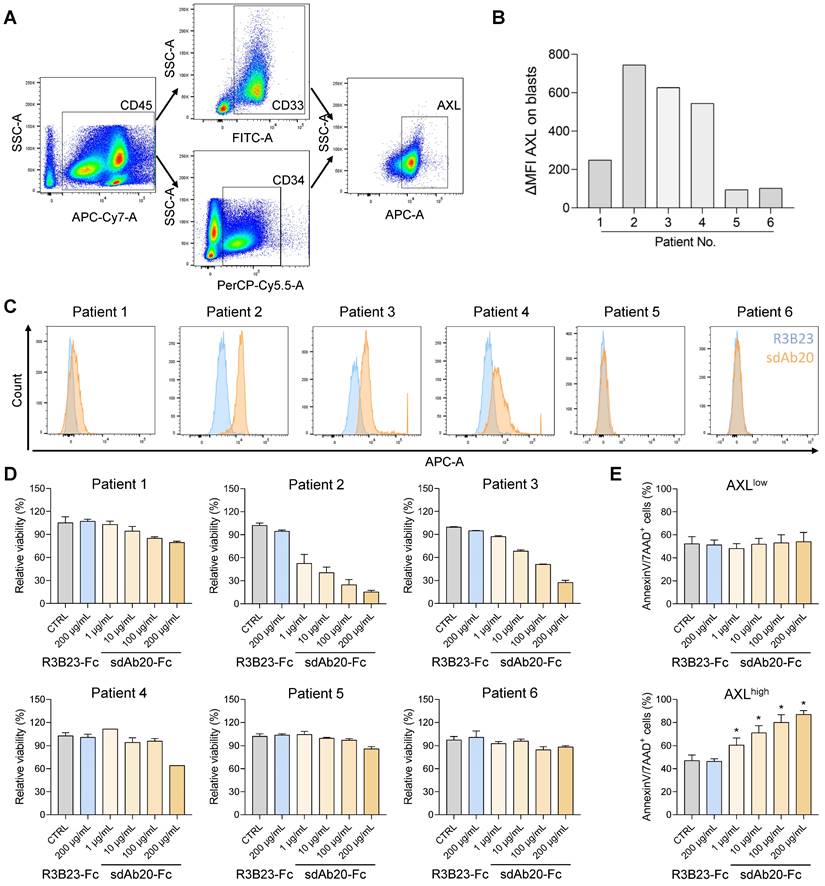
To further validate our findings, we also investigated the effect of sdAb20-Fc on primary bone marrow samples of AML patients at diagnosis or relapse. Using flow cytometry, the percentage of blasts (CD45/SSC), CD33/CD34 positivity and AXL expression (using sdAb20) were determined in six patient samples (Figure 8A-C, Table S3). Based on the median fluorescence intensity (MFI), the patient samples could be subdivided into an AXLlow (∆MFI < 300; patients 1, 5 and 6) and an AXLhigh (∆MFI > 300; patient 2, 3 and 4) group. Mononuclear cells were isolated from the primary bone marrow samples and incubated with sdAb20-Fc or the control R3B23-Fc construct. While sdAb20-Fc reduced the cell viability and increased apoptosis in AXLhigh samples, this effect could not be observed in the AXLlow group (Figure 8D-E). These data again confirm the AXL-specific anti-tumor effects of sdAb20-Fc in AML. Interestingly, the anti-tumor effect of sdAb20-Fc clearly correlated with the AXL-positivity within the blasts (Figure S10A-B).
sdAb20-Fc can specifically alter the viability and apoptosis of primary patient samples. (A) Gating strategy to determine the number of blasts (based on SSC-A/CD45-A), the percentage of CD33/CD34-positivity and the percentage of AXL-expression in various AML patients. (B-C) Flow cytometric assessment of AXL expression in CD33/CD34-positive cells of various primary samples using sdAb20 and the control sdAb R3B23. ΔMFI = MFI (sdAb20) - MFI (CTRL sdAb). (D-E) BMMCs of AML patients were treated with R3B23-Fc or increasing concentrations of sdAb20-Fc (10, 100 and 200 µg/mL). The effect on cell viability (D) was assessed using CellTiter-Glo assay (n=2/sample). Apoptosis (E) was measured using AnnexinV/7-AAD staining with flow cytometry. Patients were subdivided into AXLlow and AXLhigh, based on their AXL expression (n=3/group, ± SD). p ≤ 0.05 (*) was considered statistically significant. CTRL = control, BMMCs = bone marrow mononuclear cells.
Discussion
Although outcomes for young patients with AML have dramatically improved of the past 5 decades, this has not been the case for elderly patients [23]. Previous research already identified AXL as a prognostic marker and therapeutic target in AML [6]. In this study, we developed AXL-specific sdAbs and evaluated their value for personalized cancer treatment. Biodistribution studies demonstrated the tumor-specific uptake of sdAb20 in THP-1 xenografts and immunocompetent C1498 AML mice. Importantly, the signal in tumor-bearing mice was higher compared to the background signal in naive mice, illustrating its value as a diagnostic tracer in cancer patients. Therapeutically, sdAb20-Fc demonstrated significant anti-tumor effects by inhibiting cell proliferation and cell viability in human AML cell lines and primary patient samples. Moreover, besides its clear single agent anti-tumor effect, our data also demonstrated the therapeutic potential of AXL-specific sdAb20-Fc in combination with the standard-of-care agent cytarabine, which resulted in a synergistic effect.
Receptor tyrosine kinase AXL has been reported to be overexpressed in a variety of human cancers (e.g., lung cancer, breast cancer, colon cancer, melanoma, and pancreatic cancer) and often correlates with a poor prognosis [7, 19]. Importantly, the expression of AXL is highly heterogeneous (inter- and intra-patient variability) and is influenced by both the stage of the disease and the administered cancer treatments [6, 11]. Previous work by Wang and colleagues already demonstrated the potential of 64Cu-labeled anti-human AXL antibodies to visualize AXL+ tumors and monitor the effect of anti-AXL therapies in triple negative breast cancer (TNBC)-bearing nude mice [24]. However, since AXL is expressed in multiple immune cells (e.g., macrophages, natural killer cells) [20], the evaluation in immunocompetent mice and the use of murine AXL-targeting Abs is indispensable to realistically gauge the background signal in the tissue of interest. Moreover, because of their small size, rapid biodistribution, homogeneous tumor labeling, rapid renal clearance and low dosimetry, we preferred the development of sdAbs to generate high-contrast images in vivo early after administration [25-27]. We found that lead compound sdAb20 could better distinguish AXL+ from AXL- AML tumors compared to mAbs using flow cytometry. Moreover, the signal of 99mTc-labeled sdAb20 was clearly higher in tumor-bearing C1498 mice compared to naive mice, which was also visible on the generated SPECT/CT images. Co-injection of unlabeled sdAb20-Fc with 99mTc-labeled sdAb20 could significantly improve the tumor uptake and visualization in THP-1 xenografts by reducing the splenic background. This was further explained by assessing the biodistribution profile of 111In-labeled sdAb20-Fc, which demonstrated increased uptake in the spleen and low tumor accumulation after 24 h. A similar co-injection approach was previously reported as a useful method for PET imaging of 64Cu-labeled anti-PD-L1 antibodies in orthotopic pancreatic cancer models [28]. In the study of Movahedi et al., an excess of unlabeled, bivalent anti-MMR sdAbs were co-injected with monovalent anti-MMR sdAbs to reduce sdAb accumulation in extra-tumoral organs to background levels, without compromising tumor uptake [29].
It is important to emphasize that AXL is not only expressed by malignant cancer cells, but can also be located on the surface of various other cells, including immune cells. Therefore, we specifically developed cross-reactive sdAbs, targeting both murine and human AXL protein, allowing us to demonstrate that even in the presence of an immunocompetent environment and AXL+ non-AML cells, we were still able to distinguish the specific tumor uptake from the background levels using SPECT/CT imaging, fostering the clinical potential of sdAb20.
It is also important to mention that the biodistribution experiment illustrated a high kidney uptake of 99mTc-labeled sdAbs. This could be explained by their small size together with receptor-mediated endocytosis of these charged molecules in the kidney proximal tubuli cells with the number of polar residues in the C-terminal amino acid tag, which are the predominant reasons for sdAb-associated kidney retention [30]. The use of untagged sdAbs or the co-injection with plasma expander gelofusin are two known methods to significantly reduce the kidney uptake of radiolabeled sdAbs [30-32]. In comparison, the increased size of sdAb20-Fc (~ 80 kDa) resulted in a lower kidney uptake (6.48 ± 2.46 %IA/g) compared to sdAb20 (210.84 ± 64.25 %IA/g), related to the slower renal clearance. Furthermore, the use of 99mTc as a diagnostic tracer is particularly of interest for imaging in animal models (e.g. easy His6-tag based coupling, SPECT/CT imaging), but should be replaced and tested with a radionuclide that emits positrons, like Gallium-68, Fluor-18 or Zirconium-89, for PET imaging [33]. In patients, PET imaging is preferred based on its improved image quality and spatial resolution, higher diagnostic accuracy and lower patient dosimetry [34].
AXL's pleiotropic role in cell survival, drug resistance and immune suppression makes it a promising target for cancer therapy [8]. The past years, diverse AXL-targeting agents have been developed, including small molecule inhibitors (e.g., bemcentinib, bosutinib, gilteritinib, cabozantinib), anti-AXL mAbs (e.g., tilvestamab, YW327.6S2), antibody-drug conjugates (e.g., AXL-107-MMAE), nucleotide aptamers (e.g., GL21.T) and soluble receptors (e.g., AXL decoy receptor) [7, 8]. Although these AXL-targeted therapies appear promising, they often induce off-target effects through the inhibition of additional kinases. This may lead to subsequent unexpected toxicities and limit their clinical use [8]. In our study, the AXL inhibitor bemcentinib, that was included as a positive control, clearly demonstrated also AXL-independent anti-tumor effects in vitro. This aspect has previously been reported by Zdzalik-Bielecka et al. and was linked to bemcentinib-mediated targeting of the endo-lysosomal system and autophagy pathway, in an AXL-independent manner, in glioblastoma cells [35]. In a therapeutic context, sdAbs also possess desirable properties compared to mAbs, such as their low immunogenicity risk [13], modularity, and compact size, making them suitable for targeting recessed epitopes. Coupling of sdAb20 to an Fc tail increased its in vitro GAS6/AXL blocking capacity, while maintaining half the molecular weight of a conventional antibody. SdAb20-Fc treatment of AML cell lines and patient samples resulted in multiple anti-tumor effects in vitro, including reduced cell proliferation and viability, and increased apoptosis. Since GAS6/AXL interaction has been described to trigger various signaling pathways in cancer cells, including PI3K-AKT, NFκB, RAS-MEK-ERK, SRC-FAK and JAK-STAT [8]; it might be interesting to further elucidate the downstream mechanisms of action of sdAb20-Fc in AML.
Current clinical trials focus on the combination of AXL inhibitors with chemotherapeutic agents and immunotherapies in cancer [8, 36-38]. AXL inhibitors have been described to increase sensitivity towards standard-of-care agents venetoclax or cytarabine in AML [39, 40]. However, a direct comparison to identify the most promising combination therapy is missing. In concordance with previous studies, we observed increased anti-tumor effects combining sdAb20-Fc with either venetoclax or cytarabine. Interestingly, we found that cytarabine could induce AXL expression, and that the combination with sdAb20-Fc resulted in synergistic anti-AML effects. On the other hand, venetoclax had no effect on AXL expression and the combination with sdAb20-Fc resulted in additive effects in AML cell lines.
Altogether, this study describes the development and theragnostic potential of AXL-specific sdAbs in AML. Radiolabeled sdAb20-based imaging may be an accurate and non-invasive method to monitor AXL expression in cancer patients, paving the way for personalized treatment and increased effectiveness of AXL inhibitors. Our study also fosters the evaluation of sdAb20-Fc in combination with cytarabine in preclinical models for AML. Future PK/PD and toxicological studies are still required to determine the dose and safety profile of sdAb20 and sdAb20-Fc for first-in-human clinical trials.
Abbreviations
%IA/g: percentage injected activity per gram organ; 111In: indium-111; 2-ME: 2-mercapto-ethanol; 7-AAD: 7-aminoactinomycin D; 99mTc: technetium-99m; AF647: alexa fluor 647; AKT: protein kinase B; AMIDE: a medical image data examiner; AML: acute myeloid leukemia; ANOVA: analysis of variance; APC: allophycocyanin; ATCC: american type culture collection; AXL: anexelecto; BCA: bicinchronic acid; BCL-2: B-cell lyphoma 2; BM: bone marrow; BrdU: bromodeoxyuridine; BSA: bovine serum albumin; BV605: briljant violet 605; CCA: cell cycle arrest; CD33: cluster of differentiation 33; CDR: complementarity-determining region; CRC: colorectal cancer; CT: computed tomography; DMEM: dulbecco's modified eagle medium; DMSO: dimethylsulfoxide; ECL: enhanced chemiluminescent substrate; EDC: 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide; EDTA: ethylenediaminetetraacetic acid; ELISA: enzyme-linked immunosorbent assay; ERK: extracellular signal-regulated kinases; FACS: fluorescence-activated cell sorting; FAK: protein tyrosine kinase 2; FCS: forward scatter; FDA: food & drug administration; FITC: fluorescein isothiocyanate; GAS6: growth arrest specific factor 6; HA: hemagglutinin; HBS: hepes-buffered saline; IMAC: immobilized metal affinity chromatography; IMDM: iscove's modified dulbecco's medium; IMGT: ImMunoGeneTics; iTLC: instant thin-layer chromatography; JAK: janus kinase; kV: kilo volt; mAb: monoclonal antibody; MBq: mega becquerel; MEK: mitogen-activated protein kinase kinase; MFI: median fluorescence intensity; mIgG2a: mouse immunoglobuline G2a; NF-kB: nuclear factor-kappa B; NHS: n-hydroxy-succinimide; NSCLC: non-small cell lung carcinoma; PBS: phosphate-buffered saline; PCR: polymerase chain reaction; PD-L1: programmed death ligand 1; PE: phycoerythrin; PET: positron emission tomography; PI3K: phosphoinositide 3-kinase; PK/PD: pharmacokinetic/pharmacodynamic; qRT-PCR: quantitative reverse transcription polymerase chain reaction; RAS: rat sarcoma virus; RPMI: roswell park memorial institute; SCID: severe combined immunodeficiency; SD: standard deviation; sdAbs: single domain antibodies; SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis; SEC: size exclusion chromatography; SPECT: single-photon emission computed tomography; SRC: proto-oncogene tyrosine-protein kinase Src (Sarcoma); SSC: side scatter; STAT: signal transducer and activator of transcription; TNBC: triple-negative breast cancer; Tm: melting temperature.
Supplementary Material
Supplementary materials and methods, figures and tables.
Acknowledgements
The authors thank Carine Seynaeve, Charlotte Van De Walle, Elsy Vaeremans, Petra Rooman, Jan De Jonge and Kevin De Jonghe for excellent laboratory assistance. This work was supported by the Funds Catharina Weekers, Raymond Wuyts, Gezusters Loosveldt & Patrick Bouillon, managed by the King Baudouin Foundation; by the Brian D. Novis research grant by the International Myeloma Foundation; the Research Foundation Flanders (FWO) (G0A8522N, IOO1618N), Kom Op Tegen Kanker (KotK_VUB/2021/12786/1); Wetenschappelijk Fonds Willy Gepts of the UZ Brussel and by the VUB spearhead research programs. K.D.V. (12I0921N) and T.D.G. (12ZO723N) are postdoctoral fellows of FWO Vlaanderen. N.V. (1S66121N) is predoctoral fellow of FWO Vlaanderen.
Author contributions
Study design: K.D.V., K.B., N.D. and N.V.; Methodology and Investigation: N.V., H.S., E.V., R.F., Y.W., T.D.G.; J.B.; Resources: K.D.V., K.B., N.D.; Formal Analysis: N.V. and K.D.V.; Writing-Original Draft Preparation: N.V. and K.D.V.; Manuscript Editing: H.S., E.V., R.F., Y.W., T.D.G., J.B.; T.K., N.D.B., A.D.B., E.D.B., E.M., K.V., K.B., N.D.; Supervision: K.D.V., K.B., N.D. The author(s) read and approved the final manuscript.
Competing Interests
N.D. is co-founder and scientific consultant at Precirix NV and ABSCINT NV. All other authors declare no conflict of interest.
References
1. De Kouchkovsky I, Abdul-Hay M. 'Acute myeloid leukemia: a comprehensive review and 2016 update'. Blood Cancer J. 2016;6(7):e441
2. Jaramillo S, Schlenk RF. Update on current treatments for adult acute myeloid leukemia: to treat acute myeloid leukemia intensively or non-intensively?. That is the question. Haematologica. 2023;108(2):342-52
3. Bhansali RS, Pratz KW, Lai C. Recent advances in targeted therapies in acute myeloid leukemia. J Hematol Oncol. 2023;16(1):29
4. Cancer.Net. Leukemia - Acute Myeloid - AML: Statistics. American Society of Clinical Oncology (ASCO). 2023
5. Yu J, Jiang PYZ, Sun H, Zhang X, Jiang Z, Li Y. et al. Advances in targeted therapy for acute myeloid leukemia. Biomark Res. 2020;8:17
6. Ben-Batalla I, Schultze A, Wroblewski M, Erdmann R, Heuser M, Waizenegger JS. et al. Axl, a prognostic and therapeutic target in acute myeloid leukemia mediates paracrine crosstalk of leukemia cells with bone marrow stroma. Blood. 2013;122(14):2443-52
7. Yan S, Vandewalle N, De Beule N, Faict S, Maes K, De Bruyne E. et al. AXL Receptor Tyrosine Kinase as a Therapeutic Target in Hematological Malignancies: Focus on Multiple Myeloma. Cancers (Basel). 2019;11(11):1727
8. Zhu C, Wei Y, Wei X. AXL receptor tyrosine kinase as a promising anti-cancer approach: functions, molecular mechanisms and clinical applications. Mol Cancer. 2019;18(1):153
9. Gay CM, Balaji K, Byers LA. Giving AXL the axe: targeting AXL in human malignancy. Br J Cancer. 2017;116(4):415-23
10. Percival ME, Lai C, Estey E, Hourigan CS. Bone marrow evaluation for diagnosis and monitoring of acute myeloid leukemia. Blood Rev. 2017;31(4):185-92
11. Hong CC, Lay JD, Huang JS, Cheng AL, Tang JL, Lin MT. et al. Receptor tyrosine kinase AXL is induced by chemotherapy drugs and overexpression of AXL confers drug resistance in acute myeloid leukemia. Cancer Lett. 2008;268(2):314-24
12. Jin BK, Odongo S, Radwanska M, Magez S. Nanobodies: A Review of Generation, Diagnostics and Therapeutics. Int J Mol Sci. 2023;24(6):5994
13. Ackaert C, Smiejkowska N, Xavier C, Sterckx YGJ, Denies S, Stijlemans B. et al. Immunogenicity Risk Profile of Nanobodies. Front Immunol. 2021;12:632687
14. Vincke C, Gutierrez C, Wernery U, Devoogdt N, Hassanzadeh-Ghassabeh G, Muyldermans S. Generation of single domain antibody fragments derived from camelids and generation of manifold constructs. Methods Mol Biol. 2012;907:145-76
15. Romao E, Morales-Yanez F, Hu Y, Crauwels M, De Pauw P, Hassanzadeh GG. et al. Identification of Useful Nanobodies by Phage Display of Immune Single Domain Libraries Derived from Camelid Heavy Chain Antibodies. Curr Pharm Des. 2016;22(43):6500-18
16. Lefranc MP, Pommie C, Ruiz M, Giudicelli V, Foulquier E, Truong L. et al. IMGT unique numbering for immunoglobulin and T cell receptor variable domains and Ig superfamily V-like domains. Dev Comp Immunol. 2003;27(1):55-77
17. Lemaire M, D'Huyvetter M, Lahoutte T, Van Valckenborgh E, Menu E, De Bruyne E. et al. Imaging and radioimmunotherapy of multiple myeloma with anti-idiotypic Nanobodies. Leukemia. 2014;28(2):444-7
18. Xavier C, Devoogdt N, Hernot S, Vaneycken I, D'Huyvetter M, De Vos J. et al. Site-specific labeling of his-tagged Nanobodies with 99mTc: a practical guide. Methods Mol Biol. 2012;911:485-90
19. Wium M, Ajayi-Smith AF, Paccez JD, Zerbini LF. The Role of the Receptor Tyrosine Kinase Axl in Carcinogenesis and Development of Therapeutic Resistance: An Overview of Molecular Mechanisms and Future Applications. Cancers (Basel). 2021;13(7):1521
20. Son HY, Jeong HK. Immune Evasion Mechanism and AXL. Front Oncol. 2021;11:756225
21. Mopin A, Driss V, Brinster C. A Detailed Protocol for Characterizing the Murine C1498 Cell Line and its Associated Leukemia Mouse Model. Jove-J Vis Exp. 2016;116:54270
22. Vukovic N, Segues A, Huang S, Waterfall M, Sijts A, Zaiss DM. Mouse IgG2a Isotype Therapeutic Antibodies Elicit Superior Tumor Growth Control Compared with mIgG1 or mIgE. Cancer Res Commun. 2023;3(1):109-18
23. Sasaki K, Ravandi F, Kadia TM, DiNardo CD, Short NJ, Borthakur G. et al. De novo acute myeloid leukemia: A population-based study of outcome in the United States based on the Surveillance, Epidemiology, and End Results (SEER) database, 1980 to 2017. Cancer-Am Cancer Soc. 2021;127(12):2049-61
24. Wang W, Zhao J, Wen X, Lin CC, Li J, Huang Q. et al. MicroPET/CT Imaging of AXL Downregulation by HSP90 Inhibition in Triple-Negative Breast Cancer. Contrast Media Mol Imaging. 2017;2017:1686525
25. Harmand TJ, Islam A, Pishesha N, Ploegh HL. Nanobodies as in vivo, non-invasive, imaging agents. RSC Chem Biol. 2021;2(3):685-701
26. Yang EY, Shah K. Nanobodies: Next Generation of Cancer Diagnostics and Therapeutics. Front Oncol. 2020;10:1182
27. Berland L, Kim L, Abousaway O, Mines A, Mishra S, Clark L. et al. Nanobodies for Medical Imaging: About Ready for Prime Time? Biomolecules. 2021;11(5):637
28. Zhao J, Wen X, Li T, Shi S, Xiong C, Wang YA. et al. Concurrent Injection of Unlabeled Antibodies Allows Positron Emission Tomography Imaging of Programmed Cell Death Ligand 1 Expression in an Orthotopic Pancreatic Tumor Model. ACS Omega. 2020;5(15):8474-82
29. Movahedi K, Schoonooghe S, Laoui D, Houbracken I, Waelput W, Breckpot K. et al. Nanobody-based targeting of the macrophage mannose receptor for effective in vivo imaging of tumor-associated macrophages. Cancer Res. 2012;72(16):4165-77
30. D'Huyvetter M, Vincke C, Xavier C, Aerts A, Impens N, Baatout S. et al. Targeted radionuclide therapy with A 177Lu-labeled anti-HER2 nanobody. Theranostics. 2014;4(7):708-20
31. Wanner N, Eden T, Liaukouskaya N, Koch-Nolte F. Nanobodies: new avenue to treat kidney disease. Cell Tissue Res. 2021;385(2):445-56
32. Gainkam LO, Caveliers V, Devoogdt N, Vanhove C, Xavier C, Boerman O. et al. Localization, mechanism and reduction of renal retention of technetium-99m labeled epidermal growth factor receptor-specific nanobody in mice. Contrast Media Mol Imaging. 2011;6(2):85-92
33. Crisan G, Moldovean-Cioroianu NS, Timaru DG, Andries G, Cainap C, Chis V. Radiopharmaceuticals for PET and SPECT Imaging: A Literature Review over the Last Decade. Int J Mol Sci. 2022;23(9):5023
34. Bateman TM. Advantages and disadvantages of PET and SPECT in a busy clinical practice. J Nucl Cardiol. 2012;19(Supplement 1):S3-11
35. Zdzalik-Bielecka D, Kozik K, Poswiata A, Jastrzebski K, Jakubik M, Miaczynska M. Bemcentinib and Gilteritinib Inhibit Cell Growth and Impair the Endo-Lysosomal and Autophagy Systems in an AXL-Independent Manner. Mol Cancer Res. 2022;20(3):446-55
36. Sang YB, Kim JH, Kim CG, Hong MH, Kim HR, Cho BC. et al. The Development of AXL Inhibitors in Lung Cancer: Recent Progress and Challenges. Front Oncol. 2022;12:811247
37. Kennedy VE, Smith CC. FLT3 Mutations in Acute Myeloid Leukemia: Key Concepts and Emerging Controversies. Front Oncol. 2020;10:612880
38. Loges S, Sutamtewagul G, Heuser M, Chromik J, Kapp-Schwoerer S, Crugnola M. et al. Bemcentinib (Oral AXL Inhibitor) in Combination with Low-Dose Cytarabine Is Well Tolerated and Efficacious in Older Relapsed AML Patients.Updates from the Ongoing Phase II Trial (NCT02488408) and Preliminary Translational Results Indicating Bemcentinib Elicits Anti-AML Immune Responses. Blood. 2021;138(Supplement 1):3410
39. Niu X, Rothe K, Chen M, Grasedieck S, Li R, Nam SE. et al. Targeting AXL kinase sensitizes leukemic stem and progenitor cells to venetoclax treatment in acute myeloid leukemia. Blood. 2021;137(26):3641-55
40. Liu Y, Wei J, Liu J, Ma W, Duan Y, Liu D. Novel AXL-targeted agents overcome FLT3 inhibitor resistance in FLT3-ITD(+) acute myeloid leukemia cells. Oncol Lett. 2021;21(5):397
Author contact
![]() Corresponding author: Kim.De.Veirmanbe; Tel.: +324774535.
Corresponding author: Kim.De.Veirmanbe; Tel.: +324774535.