Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
1. Overview of OMVs
2. Engineered OMV for GI tumor...
3. Advantages of OMVs in GI...
4. Summary and future...
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2024; 14(2):761-787. doi:10.7150/thno.85917 This issue Cite
Review
Engineered bacterial outer membrane vesicles: a versatile bacteria-based weapon against gastrointestinal tumors
Keshuang Zheng1,2,3*, Yongpu Feng1,2,3*, Lei Li4*, Fanyang Kong1,2,3 ![]() , Jie Gao5
, Jie Gao5 ![]() , Xiangyu Kong1,2,3,5
, Xiangyu Kong1,2,3,5 ![]()
1. National Key Laboratory of Immunology and Inflammation, Naval Medical University, Shanghai, 200433, China.
2. Institute of Neuroscience, Key Laboratory of Molecular Neurobiology of the Ministry of Education and the Collaborative Innovation Center for Brain Science, Naval Medical University, Shanghai, 200433, China.
3. Department of Gastroenterology, Changhai Hospital, Naval Medical University, Shanghai, China.
4. Digestive Endoscopy Center, Shanghai Tenth People's Hospital, Shanghai, China.
5. Changhai Clinical Research Unit, Changhai Hospital, Naval Medical University, Shanghai, China.
* Co-first authors.
Received 2023-5-6; Accepted 2023-10-1; Published 2024-1-1
Abstract

Outer membrane vesicles (OMVs) are nanoscale lipid bilayer structures released by gram-negative bacteria. They share membrane composition and properties with their originating cells, making them adept at traversing cellular barriers. These OMVs have demonstrated exceptional membrane stability, immunogenicity, safety, penetration, and tumor-targeting properties, which have been leveraged in developing vaccines and drug delivery systems. Recent research efforts have focused on engineering OMVs to increase production yield, reduce cytotoxicity, and improve the safety and efficacy of treatment. Notably, gastrointestinal (GI) tumors have proven resistant to several traditional oncological treatment strategies, including chemotherapy, radiotherapy, and targeted therapy. Although immune checkpoint inhibitors have demonstrated efficacy in some patients, their usage as monotherapy remains limited by tumor heterogeneity and individual variability. The immunogenic and modifiable nature of OMVs makes them an ideal design platform for the individualized treatment of GI tumors. OMV-based therapy enables combination therapy and optimization of anti-tumor effects. This review comprehensively summarizes recent advances in OMV engineering for GI tumor therapy and discusses the challenges in the clinical translation of emerging OMV-based anti-tumor therapies.
Keywords: outer membrane vesicles, gastrointestinal tumors, genetic engineering, cargo delivery, tumor vaccine
Introduction
Gastrointestinal (GI) tumors, comprising pancreatic ductal adenocarcinoma (PDAC), hepatocellular carcinomas (HCC), esophageal cancer, gastric cancer, and colorectal cancer (CRC), and so on, constitute a substantial proportion of both total cancer cases and cancer-related deaths, accounting for 26% and 35% respectively, on a global scale [1]. In recent times, immunotherapy has manifested marked advantages and efficacy in treating malignancies, in addition to conventional therapies like surgery, radiation, chemotherapy, and targeted therapy. Presently, the most commonly employed immunotherapeutic agents primarily comprise checkpoint inhibitor monoclonal antibodies directed against programmed cell death 1 (PD-1) or its ligand, PD-L1, and chimeric antigen receptor T-cell (CAR-T) therapies, with other agents currently undergoing clinical trials [2]. Despite the significant advancements in immunotherapy, its effectiveness and safety remain subject to reservations, thereby limiting its clinical utility in treating certain GI tumors [3], such as PDAC. Statistically, more than 99% of patients with PDAC are unresponsive to monotherapy with any of the currently approved immunotherapeutic agents by the U.S. Food and Drug Administration (FDA)-approved immunotherapeutic agents, except for a small fraction of patients with high microsatellite instability. The overall survival rate at 5 years in patients with PDAC in the U.S. is only around 9% [4]. While immune checkpoint inhibitor-based therapy has displayed encouraging results in treating CRC, only a minute fraction of CRC patients with defective mismatch repair and high microsatellite instability have demonstrated any tangible benefits from it [5]. Immunotherapy has showcased suboptimal outcomes in treating advanced CRC and HCC, with individual variability also affecting the therapy's efficacy. Consequently, there exists a pressing need for novel immunotherapeutic approaches such as improved drug delivery methods, as well as the identification and presentation of neoantigens, to treat GI tumors effectively.
Recent research has shed light on the potential of bacteria in the realm of cancer therapy. Bacteria exhibit inherent motility in that they colonize tumors preferentially and modulate the tumor immuno-microenvironment, thereby impacting the efficacy of immunotherapy [1]. The utilization of bacteria themselves for the treatment of solid tumors dates back to the mid-19th century. However, the toxicity of bacteria presents a challenge in managing unnecessary cytotoxicity and mortality during treatment. Extracellular vesicles are a heterogeneous group of cell-derived lipid-bound structures, which involved in multiple physiological and pathological processes. Bacterial membrane vesicles, a form of extracellular vesicles secreted by bacteria, play a vital role in various biological functions of bacteria, including bacterial pathogenesis, interspecies communication and competition, oxidative stress, biofilm formation, and, significantly, regulation of the tumor microenvironment (TME) of GI tumors [6]. Generally, extracellular vesicles from gram-negative bacteria are defined as outer membrane vesicles (OMVs), while those from gram-positive bacteria and eukaryotic vesicles are defined as membrane vesicles and exosomes, respectively [7]. The OMVs could be observed at any stage of gram-negative bacteria [8]. The GI tract, which is a vast reservoir of gram-negative bacteria, is a significant source of various OMVs in humans. Previous studies have demonstrated that OMVs from GI bacteria could enter intestinal epithelium sufficiently through various mechanisms to exert pathogenic or non-pathogenic effects [9]. Given the impact on local immunity of the intestine and systematic immunity efficacy, OMVs are suitable for oral administration and have natural advantages in the treatment of GI tumors. Several studies have utilized OMVs to deliver drugs with some therapeutic efficacy in experimental animal models. OMVs possess several advantages over traditional drug delivery carriers, including greater drug-loading space and stability, higher biocompatibility, appropriate immunogenicity, and lower cytotoxicity. Despite the intrinsic immune adjuvant properties, the natural anti-tumor effects of them alone are limited. Recently, engineering modifications of OMVs have been developed to endow extracellular vesicles with new properties through genetic engineering or physicochemical methods, aimed at improving the yield, safety, and targeting capability of OMVs during drug delivery. Notably, the promising strategies combined with nanotechnology are able to evoke potent tumor-specific immune responses, inhibiting tumor growth and metastasis (Figure 1). In conclusion, engineering strategies provide great opportunities for improving the efficacy of tumor treatment.
Summary of engineered OMV-based anti-tumor treatment. Immunotherapy is important in the field of OMV-based cancer therapy. In addition to immunotherapy, OMVs have been applied in combination with chemotherapy, gene therapy, and photothermal therapy to amplify anti-tumor efficacy. Engineered OMVs improved tumor immunogenicity by 1) delivering exogenous immunogenic antigens that mainly target lymph nodes to promote DC maturation and 2) inducing immunogenic cell death, to release endogenous immunogenic agents to promote DC maturation. Both strategies could promote T-cell priming and clonal expansion of T cells, leading to the suppression of both orthotopic and distal tumors.
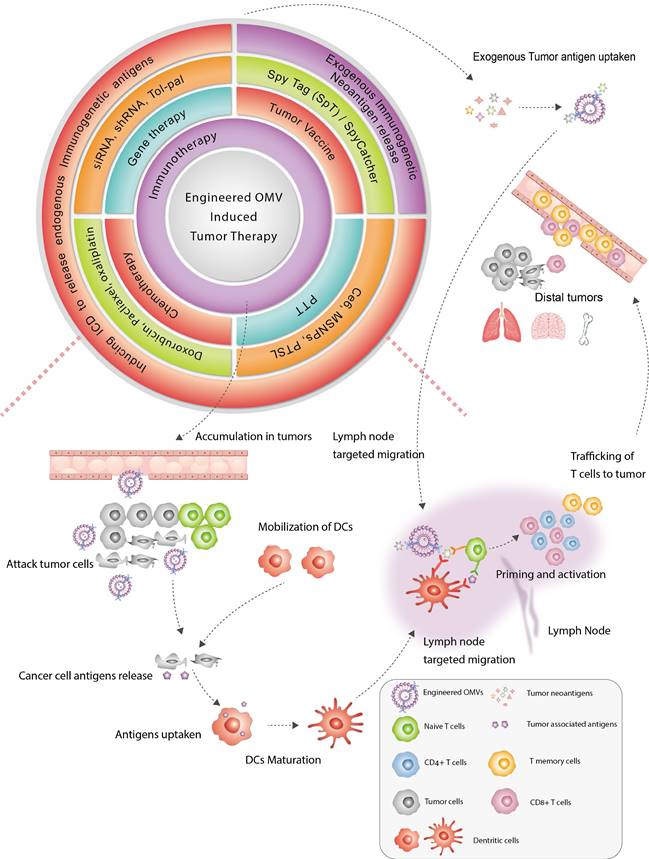
As an emerging class of immune-oncology therapy, tumor vaccines attack tumor cells via specific cytotoxic T-cells lymphocytes (CTLs), which are activated by tumor antigens. Oral vaccines should elicit robust anti-tumor immune responses since the intestine acts as the largest immune organ, containing about 70% of the immune cells of the body. OMVs derived from commensal bacteria assemble natural epithelial penetration and good oral tolerability, as well as the underlying roles in modulating the gut microbiota, mucosal adaptive immune responses, and the physicochemical barrier in assisting anti-tumor therapy. Increasingly researchers have spotlighted the potential of using gut microbiota-derived OMVs as an alternative to artificial nano-materials, for safer and more effective oral GI tumor vaccine. For instance, some studies have demonstrated that oral administration of Akkermansia muciniphila (Akk) OMV enhances immune checkpoint blockade (ICB) therapy against GI tumors by maintaining intestinal homeostasis, reprogramming the TME, as well as promoting CTL-related immune response [10,11].
Over the past decade, some reviews have reported progress in the application of engineered OMVs in anti-tumor drug delivery and immunotherapy [12-15]. However, to the best of our knowledge, few reviews have specifically focused on the potential application of engineered OMVs in the treatment of GI tumors. Based on the literature, our review covered common engineering strategies including genetic modification, drug-loading, surface modification, as well as biomimetic nanoparticles (NPs). Moreover, the advantages and disadvantages of engineered OMVs for the treatment of GI tumors have also been discussed. Finally, the challenges associated with these emerging OMVs platform-mediated anti-tumor therapies and their clinical translation are mentioned, which will help to better understand the current advance and future research directions in this field.
1. Overview of OMVs
1.1 Structure and biogenesis of OMVs
OMVs are non-replicative spherical nanovesicles ranging from 20-250nm, which consist of a typical phospholipid bilayer naturally derived by Gram-negative bacteria. The vesicles inherited components from bacterial membranes, presenting excellent intrinsic immunostimulatory properties. Besides some physiological processes including intracellular and extracellular communication, quorum sensing, and horizontal gene transfer, OMVs are ascribed to many biological functions such as ligand recognition, and biological targeting. Further, OMVs have been shown to be highly stable even upon elevated temperatures and several harsh environmental conditions [16]. The outer membrane of OMVs is composed of phospholipids, lipopolysaccharides (LPS), and outer membrane proteins (OMPs). The stability of the membrane structure is co-maintained by the covalent crosslinking between OMPs and lipoproteins (Lpps), as well as the non-covalent crosslinking between outer membrane pore proteins and the peptidoglycan (PG) layer that located in the periplasmic space. The membrane proteins of OMVs include OMPs, soluble periplasmic proteins, and the Tolerance peptidoglycan-associated lipoprotein (Tol-Pal) complex, which consists of TolA, TolB, TolQ, TolR, and Pal. Tol-Pal complexes are partially anchored to the inner membrane of OMVs, cross the periplasmic space, and are connected to the PG layer and the outer membrane of OMVs by noncovalent bonds. The absence of any component of Tol-Pal and any factor that destroys the cross-linkage between PG and Lpp or OMP may change the stability of the OMVs membrane and thus trigger OMVs vesiculation [17]. Although there are no definite conclusions regarding the biogenesis of OMVs, several biogenesis pathways have been reported. The basic principles for membrane curvature induction include (1) Selective local protein and cargo protein crowding in the periplasm or outer membrane. (2) Insertion of the Pseudomonas quinolone sequence (PQS) increases the surface area of the outer leaflet relative to the inner leaflet by a wedging effect [18]. (3) Specific enrichment of different fatty acids and LPS in some areas. (4) Phospholipids accumulation in the outer membrane in the absence of the VacJ/Yrb ATP-binding cassette (ABC) transport system. All models depend on an initial decoupling of the outer membrane by breakage of the Lpp crosslink loss, even this decoupling alone is sufficient to induce membrane vesiculation.
1.2 Preparation of OMVs
High OMV quality and purity are required in bringing OMV into the clinical setting. To meet this requirement, several methods have been developed to isolate and purify OMVs. Density- or size-based isolation is the most widely used method for OMV preparation that includes ultracentrifugation, ultrafiltration, tangential flow filtration, etc. These methods are usually simple in procedures but get OMV in limited purity. Another method is affinity-based OMV isolation, which collects OMVs according to special ligands presenting on the surface, such as antibodies, aptamers, and resin. These techniques isolate OMV in high purity from the culture broth. Nevertheless, the process of affinity isolation is time-consuming and results in product loss.
To meet the requirement for OMV basic and clinical research, Liu et al. have summarized a set of preparation protocols for the vast majority of bacteria, which has been used for E. coli Nissle 1917 and Akk OMV separation successfully [19]. Firstly, the culture medium containing bacteria and their debris is usually removed by low-speed centrifugation and a 0.22μm sterile filter. Secondly, the small molecule proteins are eliminated by ultrafiltration (100 kDa). Further, the OMVs are purified by ultracentrifugation as well as iodixanol gradient centrifugation. Finally, OMVs should be characterized by transmission electron microscopy, nanoparticle tracking analysis, or western blotting, if necessary [20]. These steps mentioned above achieve sufficient yield and purity in most cases. However, combining different techniques is imperative in OMVs isolation in complex media, such as biofluids [20]. Different methods could hamper the repeatability and reproducibility of outcomes between researchers. To promote in-depth studies of OMVs and their clinical translation, we dire need standardized guidelines that take cost, efficiency, and quality into consideration at the same time [20].
1.3 Internalization of OMVs
In GI tumors, OMVs have been reported to interact directly with epithelial cells at mucosal surfaces, immune cells as well and other host cells including endothelial cells, platelets, osteoblasts, and synovial cells. Several mechanisms have been put forward for OMV uptake into host cells, which can be roughly classified into 2 types. The first one is receptor-mediated OMV internalization. OMVs can bind to certain receptors, such as clathrin, caveolin, or lipid raft, and then activate receptor-induced intracellular signaling in recipient cells. OMVs of Porphyromonas gingivalis and Helicobacter pylori (H. pylori) utilized clathrin-mediated endocytosis to gain entry into GI epithelial cells [21]. Later, Kaparakis et al. declared that H. pylori OMVs enter GI tumor cells via both clathrin and caveolin-mediated endocytosis, with a preference for dynamin-dependent and caveolin-mediated endocytosis [22]. Subsequently, O'Donoghue et al. reported that H. influenzae, M. catarrhalis, V cholerae, as well as Enterotoxigenic Escherichia Coli (ETEC) OMVs, invade GI epithelial cells mainly via caveolin-mediated endocytosis [14,22]. Another mechanism of OMV internalization is direct fusion to host cell membranes. OMV fusion to host cell lipid rafts induces actin remolding to allow OMV soluble cargo to diffuse directly into the host cytoplasm. Lipid rafts-mediated entry has been observed in V cholerae, P. Aeruginosa, C. jejuni, A. baumannii, and P. gingivalis etc. [21]. Membrane fusion is used by L. pneumophila, P. Aeruginosa, and A. actinomycetemcomitans OMVs. As a special type of receptor-independent OMV Internalization, macropinocytosis is described as the inward folding by some of the cell surface ruffles to fuse with the basal membrane, which has been observed in OMVs from Shigella flexneri internalized by the human epithelial cells and fibroblasts [23]. This is also the mechanism proposed for P. aeruginosa OMVs' interaction with the airway epithelial cells [24]. As described above, the pathway employed is organism-, or even strain-dependent. Moreover, the size of the OMV population is relevant when studying endocytic routes. Clathrin-mediated endocytosis generally allows the internalization of larger cargo than clathrin-independent routes, while the macropinocytosis allows internalization of endocytic vesicles up to 1 um in diameter [25].
1.4 Biological characteristics of OMVs
Based on previous studies, OMVs showed a more rigid drug package as well as larger drug loading space when compared to normal liposomes [26]. Further, OMVs exhibit high environmental stability at higher ambient temperatures and a wide range of pH values. Consequently, OMVs safeguard their payload from enzymatic degradation during long-distance drug delivery in vivo without obvious leakage in systemic circulation. Alves et al. successfully packaged the enzyme phosphotriesterase (PTE) into the lumen of E. coli-derived OMVs and observed enhanced stability of OMVs-encapsulated PTE relative to free PTE after numerous freeze-thaw cycles [26]. Their subsequent study showed that E. coli-derived OMV-encapsulated PTE protects enzyme activity in harsh environments, such as heating and freeze-drying [27]. In addition, OMVs derived from Salmonellae and Shigella contain adhesins that enable themselves to be recognized and endocytosed by the GI tract cells without any targeting ligands assembled [25]. These studies efficiently underscore the remarkable features of OMVs as a platform for anti-tumor treatment.
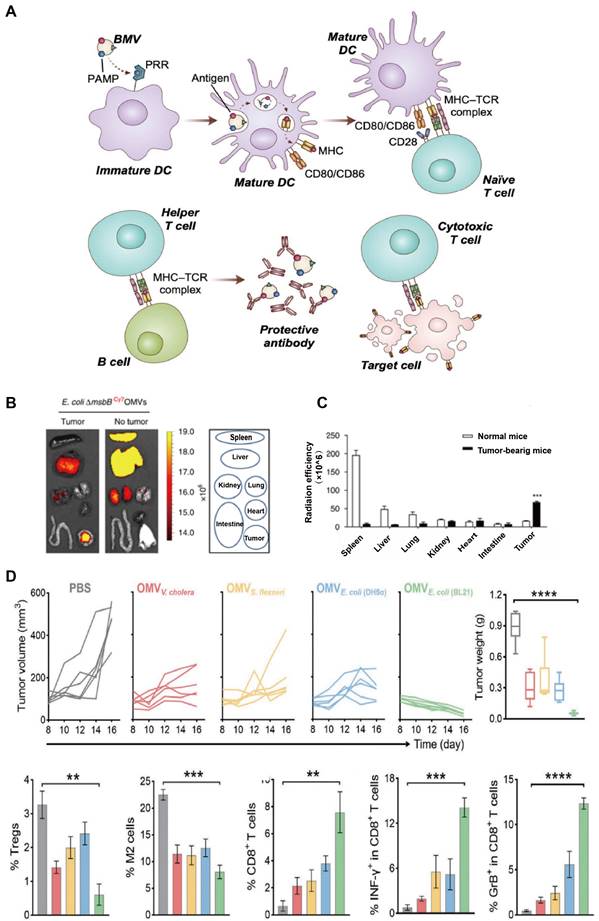
Moreover, immunogenecity is the most prominent property of OMV in its application in tumor treatment. OMV can be identified and phagocytosed by antigens to antigen-presenting cells (APCs) more easily than other nanostructures (e.g., liposomes). The LPS presenting on the OMVs surface can stimulate different pattern recognition receptors (PRRs), producing proinflammatory cytokines which are crucial for dendritic cell (DC) recruitment and maturation. Additionally, the spherical nanoscale structure of OMV stimulates APCs to present tumor antigen, thus provoking both antigen-specific B-cell and CTL (Figure 2A). Some studies have examined the interaction between OMVs and DCs. OMVs from Streptococcus were observed rapidly taken up by the DC with increased TNF-α releasing [28]. In another research, the intraperitoneal injection of Salmonellae-derived OMVs into mice elicited an increase in the expression of CTLs along with a high level of TNF-α in the spleen [29]. All of the above evidence indicates the potential of OMVs in enhancing antigen presentation and immune responses in GI tumors (Table 1).
OMVs-induced immunomodulatory effects
| Parental Bacteria | Immunomodulatory effects | Target cells | Reference |
|---|---|---|---|
| Helicobacter pylori | Dose-dependent IL-8 release | Gastric epithelial cells | [30] |
| Escherichia coli | Increased TLR-4 and IL-8 production | A498 and T-24 cells | [31] |
| Helicobacter pylori | Increased pro-inflammatory signal (NOD-1) | HEK 293 cells | [32] |
| Neisseria meningitidis | Increased IL-1b, IL-6, IL-8, IL-10, IL-12p40, TNF-α | Macrophages and monocytes | [33] |
| Salmonella | Increased TNF and NO | Mouse macrophages | [29] |
| Salmonella | Increased expression of CD86 and MHC-II. Increased release of TNF and IL-12 | Dendritic cells | [29] |
| Streptococcus | The rapid uptake of MVs into DC2.4 cell lines. Increased release of TNF-α | Dendritic cells | [28] |
The safety, immunomodulatory and anti-tumor effects of OMVs. A) OMVs containing various PAMPs that promote DC maturation following interaction with PRRs. Mature DCs elicit the proliferation and activation of antigen-specific CD4+ T and CD8+ T cells in lymph nodes. Adapted with permission from [34], copyright 2022 Advanced Drug Delivery Reviews. B) In vivo fluorescence images of CT26 tumor-bearing mice and tumor-free mice post i.v, injection of OMV@Cy7. The circles indicate different organs. Ex vivo imaging showing the distribution of OMV@Cy7 in the major organs and tumor sites at 24 hours post injection. C) Quantitative analysis of the Cy7 fluorescence intensity in the spleen, liver, heart, kidney, lung, intestine, and tumor. D) The CT26 mice were intravenous injected with OMVs three times every other day. Individual tumor growth kinetics after treatment with different types of OMVs and their tumor weights, as well as percentage of Tregs in tumor tissues were recorded. Adapted with permission from [35], copyright 2020 Advanced Materials.
2. Engineered OMV for GI tumor therapy
Compared with live or attenuated bacteria, native OMVs are considered safer since they cannot replicate autonomously in vivo [36]. Furthermore, nano-sized bacterial native OMVs can penetrate various cellular barriers and evade clearance by the immune system more easily than the bacteria. Nevertheless, emerging evidence reflected shortcomings of native OMV in tumor therapy recently. Kim et al. labeled OMVs with Cyanine7 (Cy7) and detected different levels of OMVs aggregation in the GI tract, liver, and heart, even though the highest signal was detected in tumor sites 12 hours after intravenous injection (Figure 2B-C). Native OMVs may exhibit off-target effects, thereby leading to unpredictable effects on sites other than tumor tissues. Further, the native OMV-induced anti-tumor effect based on IFN-α was generally limited to tumor enlargement inhibition rather than tumor regression [35] (Figure 2D). To enhance the efficacy and reliability of OMVs in the treatment of GI tumors, there is a need to optimize their targeting ability and immunogenicity while also enhancing their safety and controllability through engineering approaches.
It has been found that genetic modification and physicochemical methods, also known as the engineering of OMVs, hold promise for overcoming these problems. Commonly used methods for modification include (i) Genetic engineering of source bacteria to obtain 'customized' OMVs. (ii) Loading of therapeutic drugs through electroporation, extrusion, ultrasonication, or coincubation. (iii) Combination of OMVs with nanocarriers to form biomimetic NPs. (iv) Surface modification of OMVs using physical or chemical methods to reduce adverse effects. Modification of OMVs often requires multiple approaches due to tumor heterogeneity and organism complexity. The three main engineering methods (i-iii) are categorized and discussed below, while surface modification of OMVs is introduced in each case in this article. Additionally, the progress of several primary cancer therapies using engineered OMVs such as tumor vaccines and photothermal therapy (PTT) will be discussed.
2.1 Genetic Modification of OMVs
Despite the demonstrated anti-tumor effects of OMVs in certain studies, natural OMVs typically contain an excessive amount of immunogenic substances, potentially leading to severe systemic inflammatory response syndrome upon intravenous administration [37]. Moreover, the insufficient yield of OMVs poses a significant challenge to their clinical development and application. As such, there is a pressing need to explore and refine techniques for processing and modifying OMVs to address these issues.
2.1.1 Optimization of the targeting ability of OMVs
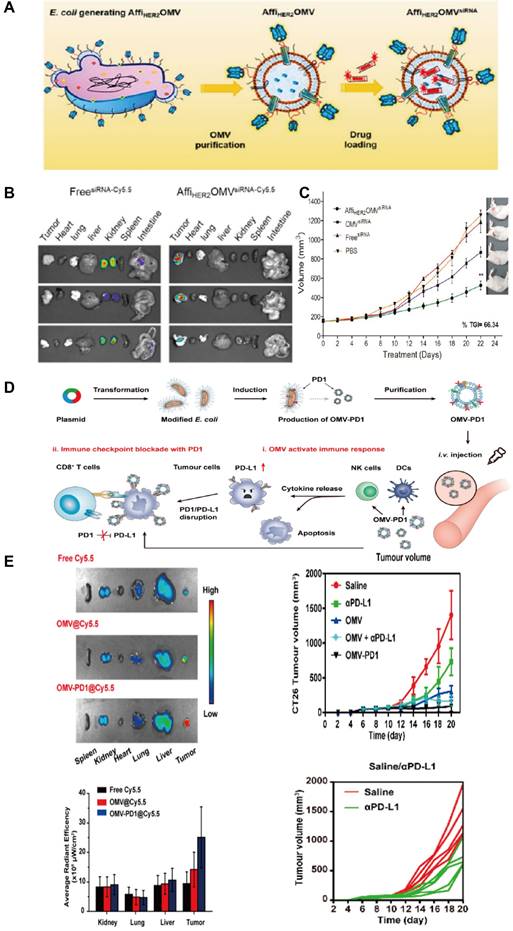
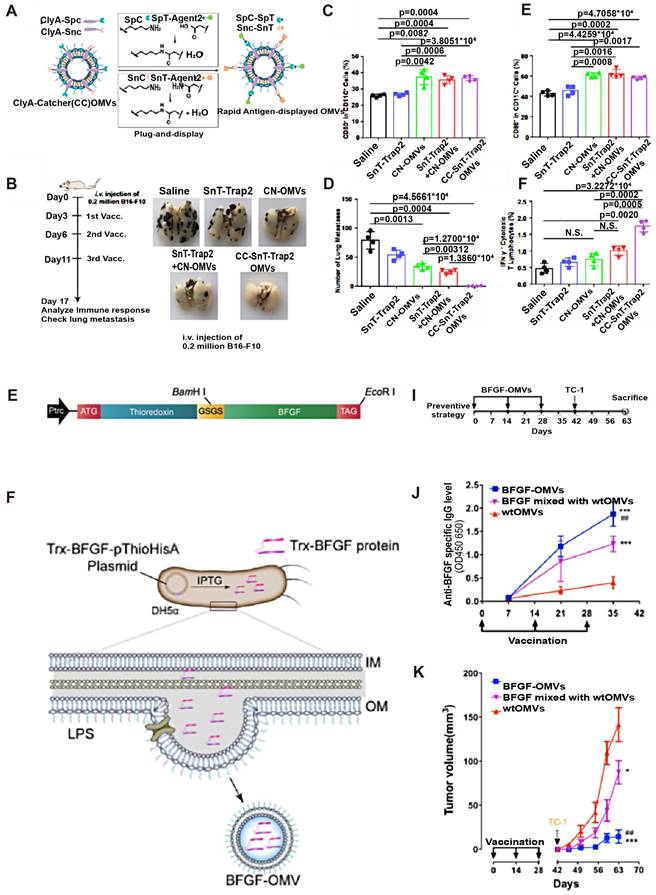
Most of the research on tumor-targeting optimization adopts genetic modification. Human epidermal growth factor receptor 2(HER2) is overexpressed in some GI tumors (e.g. Gastric cancer) and plays an important role in tumor cell growth, survival, and differentiation, thus becoming a popular target for cancer diagnosis and treatment [38]. Gujrati et al. fused HER2 ligands with Cytolysin A (ClyA), commonly expressed on E. coli OMVs, by genetic modification (Figure 3C). It was observed that engineered E. coli OMVs-specific accumulated in HER2-overexpressing tumor tissues after tail vein injection in an HCC mouse model [39] (Figure 3A). This was the initial in vivo study to use engineered OMVs for the purpose of optimizing the targeting ability of OMVs. In addition to molecular targeting, Li et al. combined engineered OMVs with ICB in CRC therapy and obtained OMV-PD1 [40] (Figure 3E-F) by fusing the PD-1 coding region with the ClyA coding region of E. coli. As expected, a large number of immune cell infiltrates was observed in tumor tissues because of the OMV-PD1 induced PD-L1 blocked on the surface of tumor cells, which suggests that genetically engineered OMV-PD1 has reversed the immunosuppressive TME in CRC besides specific target. Another key target in ICB is the CD47 which is abnormally expressed on tumor cells that bind to signal-regulatory protein alpha (SIRPα) of macrophages, and responsible for evading phagocytosis. Feng et al. modified OMV with CD47 antibodies to block the CD47-SIRPα binding efficiently. Additionally, the OMV-CD47nb were coated with the PEG/Se layer, which enabled the OMV-CD47nb with X-ray-controlled tumor targeting [41]. In another study that focused on targeted transportation of tumor APCs, Cheng et al. constructed a 'tumor antigen plug-and-display procedure' aiming to modify multiple tumor target antigens on the surface of OMVs (Figure 4A-F). On a SpyTag (SpT)/SpyCatcher (SpC) capture system, the SpT protein tag is used to label various tumor antigens, whereas SpC is an antigen catcher that binds to the OMVs surface protein ClyA. SpT with bound tumor antigens can bind to SpC via peptide bonds, thereby concentrating tumor antigens bound by SpT on the surface of engineered OMVs (Figure 4A). Subsequently, engineered OMVs carrying tumor antigens activate DCs in the TME and are redirected into peritumoral lymph nodes owing to their superior nanoscale size and immunogenicity. Ultimately, these OMVs induce a systemic, memory-based anti-tumor immune response in vivo [42]. The anti-tumor efficiency of the OMV-based antigen capture system has been confirmed in mouse models, which is presented in Figure 4B-F. Collectively, genetic engineering enhances the tumor-targeting ability of OMVs and allows their integration with immunotherapeutic strategies, such as ICB therapy and tumor vaccines, to elicit a broader anti-tumor-immune response. Engineered OMVs, which act as tumor antigen-presentation systems, could avoid the loss of tumor antigens and exert potent tumor-killing effects.
Anti-tumor effects of engineered OMVs with gene modifications. A) The OMV was purified after vesiculation from the parent bacteria that inserted with ClyA-Affibody and further loaded with siRNA using an electroporation method. B) For optical imaging, HCC-bearing mice were given a single injection of Cy5.5-labeled AffiHER2OMVsiRNA‑Cy5.5, which shows the accumulation of OMVs in the whole body after systematic administration. C) The engineered OMVs exerted the strongest anti-tumor effects compared to all controls. Adapted with permission from [43], copyright 2013 ACS Nano. D) OMV-PD1 were obtained by engineering E. coli to stably express the PD1 ectodomain fused with the ClyA. OMV-PD1 accumulation at the tumor site increases the infiltration of immune cells. At the same time, the PD1 ectodomain on the OMV-PD1 and protects CD8+ T cells, which can then attack tumor cells. E) Tumor tissues and major vital organs (lung, liver, kidney) were analyzed separately. The images show tumor-specific retention and accumulation of engineered OMV delivery system. Engineered OMVs exerted the strongest anti-tumor effects compared to all controls. Adapted with permission from [44], copyright 2020 ACS Nano.
Anti-tumor effects of engineered OMVs with optimized immunogenicity. A) A versatile OMV-based vaccine platform can rapidly and simultaneously display multiple tumor antigens and consequently elicit synergistic anti-tumor immune responses for personalized tumor vaccines. B, D) Engineered OMV vaccination inhibits tumor metastasis. The percentage of CD80+(C) or CD86+ (E) cells in CD11c cells was determined by flow cytometry. F) The percentage of IFN-γ cells in the CD3+CD8+ T-cell subpopulation is shown. Adapted with permission from [28], copyright 2021 Nature Communication. G) Schematic diagram of gene recombination of Trx and the whole BFGF molecule. H) Schematic diagram of the principle of production of BFGF-OMVs. Trx guided BFGF to the periplasmic space. The fusion protein was coated during the process of OMV formation in bacteria. I) Schematic diagram of the procedures for the tumor preventive experiment with BFGF-OMVs J) Detection of the level of specific anti-BFGF autoantibodies in mouse serum using ELISA. K) Continuous measurements of tumor volume in mice. BFGF-OMVs significantly reduced tumor volume. Adapted with permission from [36]. Copyright 2020, Acta Biomaterial.
2.1.2 Optimization of the immunogenicity of OMVs
Native OMVs caused mild to moderate immune responses, most of which were not directed against the heterologous expressed antigens necessarily [45]. To improve the tumor-targeting immune response, Zhang et al. engineered the factor H binding protein (fHbp) of E. coli, which increased both fHbp and antigen yields per OMV, thus improving the antibody responses against fHbp and target antigen. Likewise, Scatters et al. modified the surface of OMVs to express numerous ovalbumin (OVA) fragments by injecting plasmids containing OVA DNA sequences into Salmonella typhi. OVA fragments contain two antigenic epitopes capable of binding to MHC-I and MHC-II. After activation of DCs by engineered OMVs owing to their immunogenicity, the OVA fragments activated CD4+ T cells and CD8+ T cells, which in turn induced a persistent immune response [46].
The interaction of the OMV with the immune system can be considerably tuned. It has been proved that using monoclonal antibodies to block tumor angiogenesis is an effective Anti-tumor method. However, it is very expensive, time-consuming, and even easily induces drug resistance. Following the use of anti-vascular endothelial growth factor (VEGF) monoclonal antibodies such as bevacizumab, compensatory basic fibroblast growth factor (BFGF) expression promotes angiogenesis and resistance to targeted therapy. To address this issue, Huang et al. constructed a recombinant plasmid containing the bfgf gene transfected with E. coli (Figure 4G and H), thus resulting in spontaneous production of anti-BFGF immunoglobulins in CRC-bearing mice (Figure 4I-K) [47].
2.1.3 Optimization of the yield of OMVs
OMVs have exhibited immense potential in the field of biomedical applications. However, the current production levels of OMVs are insufficient to meet the demand for their clinical translation. In response to this challenge, Lloubès et al. investigated to elucidate the role of bacterial membrane structural integrity in the production of OMVs. Through gene screening and analysis, the researchers discovered that mutations, either individually or combined, in certain genes from the Tol-Pal protein family led to an increase in OMV production [17]. The Tol-Pal family, composed of five interacting proteins forming a cytoplasmic periplasmic space membrane protein complex, interacts with OMPs and Lpps in the bacterial outer membrane. Mutations in any of the five Tol-Pal genes lead to defects in the bacterial outer membrane, which result in elevated production of OMVs and the release of periplasmic proteins [17]. Reimer et al. also reported a noteworthy increase in OMV production upon the deletion or mutation of the tolA, tolR, and tolB genes in E. coli., which are keys in maintaining membrane stability [48]. In addition, the deletion of RmpM, a member of the Lpps family and encoding an outer membrane lipoprotein covalently cross-linked with PG layer, loosens the outer membrane and promotes OMVs release [49]. Waterbeemd et al. verified this principle through the knockout of the RmpM gene in Neisseria meningitides, which resulted in a significant increase in OMV production [37]. According to McBroom et al., in addition to interventions on genes related to membrane integrity, affecting bacterial membrane stress pathways can also enhance OMV production. Specifically, they found a 100-fold increase in OMV production upon knocking out the E. coli σE stress pathway-related genes degS and degP, without any associated membrane integrity defects [50]. Other identified genes such as the nlpI and the ompR were similarly linked to the cell envelope, affecting outer membrane protein expression and localization. The waaG and the ponB were related to LPS and PG biosynthesis and the deep and the rseA affected the σE envelope stress response pathway [51]. It is conceivable that certain mutations that reliably force hyper-vesiculation might have unexpected benefits for different OMV applications. These findings underscore the role of gene modification in altering the production of OMVs via various pathways, thus enabling the clinical translation of engineered OMVs (Table 2).
Genetic Modifications to Regulate OMVs Production
| Parental Bacteria | Genetic modification | Description | Relative vesicular production | Reference |
|---|---|---|---|---|
| Escherichia coli | ΔdegP, ΔdegS, ΔrseA | Cpx, or σE pathways-deregulated membrane stress responses | Improved yield | [52,53] |
| ΔnlpI | Deregulated anchored lipoprotein | Improved yield | [53] | |
| ΔompC, ΔompR, ΔompF | Deregulated outer membrane porin | Improved yield | [53] | |
| Δpnp | Deregulated polynucleotide phosphorylase | Improved yield | [53] | |
| ΔponB | Peptidoglycan synthesis | Improved yield | [53] | |
| ΔrmpM | Outer membrane integrity | Improved yield | [37] | |
| ΔtatC | Inner membrane secretion apparatus | Improved yield | [53] | |
| ΔtolA, ΔtolQ, ΔtolR, ΔtolB | Outer membrane integrity and periplasmic protein | Improved yield | [48,53] | |
| ΔwaaG/rfaG | ClyA biosynthesis; Glucosyl transferase | Improved yield | [53] | |
| ΔwzxE, Δ wecF | Inner membrane integrity | Improved yield | [53] | |
| ΔyieM | Unclarified | Improved yield | [53] | |
| ΔypjM | Unclarified | Improved yield | [53] | |
| ΔglnA | Glutamine synthetase | Decreased yield | [53] | |
| ΔlysS/herC | Lysyl tRNA synthetase | Decreased yield | [53] | |
| ΔnlpA | Outer membrane lipoprotein | Decreased yield | [53] | |
| ΔpepP | Proline aminopeptidase | Decreased yield | [53] | |
| Acinetobacter baumannii | ΔBfmS | Unclarified | Improved yield | [50] |
| ΔAbOmpA | Outer membrane lipoprotein | Improved yield | [54] | |
| Campylobacter jejuni | ΔmlaA | Lipid accumulation in the outer membrane | Improved yield | [55] |
| Pseudomonas aeruginosa | ΔoprI, ΔoprF | Outer membrane lipoprotein | Improved yield | [56] |
| Serratia marcescens | ΔwecG, ΔwecD | Decreased accumulation of lipid I at the inner membrane | Improved yield | [57] |
| Bacillus subtilis | ΔxhlAB/ΔxlyA | Unclarified | No effect | [58] |
| ΔytCDEF | Unclarified | No effect | [58] | |
| Streptococcus mutants | Δsfp | Phosphopantetheinyl transferase | Improved yield | [59] |
| Mycobacterium tuberculosis | ΔpstA1 | Increased outer membrane lipoprotein | Improved yield | [60,61] |
| ΔvirR | Unclarified | Improved yield | [60] | |
| Staphylococcus aureus | Insert Psmα | Membrane integrity | Decreased yield | [62] |
| ΔtagO, Δpbp4 | Decreased PG cross-linking | Improved yield | [62] | |
| Listeria monocytogenes | ΔsigB | Membrane integrity | Improved yield | [63] |
| Shigella sonnei | ΔtolR | Inner membrane integrity | Improved yield | [64] |
| Vibrio cholera | ΔvacJ, ΔyrbE | Lipid accumulation in the outer membrane | Improved yield | [65] |
Δ means gene deletion
A main shortcoming of genetic modulation is that one mutation may not work widely in all Gram-negative bacteria. Emerging evidence shows that exploring environmental triggers for OMV release could be a convenient way to enhance production. For example, Gerritzen et al. found that up-regulated dissolved oxygen tension in cysteine depleted cultivations resulted in N. meningitidis OMV double production. The increased yield may be caused by a reduced resistance to oxidative stress due to cysteine limitation [66]. However, cysteine-depletion also leads to bacteria growth arrest and even accumulation of undesired components, like DNA, and ammonium that hinder further purification of OMV [67]. Besides, Waterbeemd et al. found that sterile equipment provides better stability and higher yield OMVs because the filtration step removes most constituents that might inhibit growth [68]. Recently, Gerritzen et al. combined sulfur source depletion with high dissolved oxygen and reached OMVs continuous production, which has been used clinically in the production of vaccines [69].
2.1.4 Optimization of the biosafety of OMVs
One of the main considerations of OMVs application is safety. Traditional approaches principally use deoxycholate or sodium dodecyl sulfate to produce less toxic OMVs, such as OMV-based N. meningitides vaccines. While effective, the use of detergent partially decreased both Toll-like receptors (TLR) 4 and TLR2 activation induced by OMVs. Hence, novel strategies are required to manage the toxicity of OMVs while preserving their immune adjuvant effects. The mechanism of different OMVs initiates PRR signaling is highly heterogeneous. S Robbana-Barnat et al. have declared that both tri-acylated lipoprotein and bi-acylated lipoproteins can stimulate TLR2 responses, followed by TLR2 dimerization with TLR1 and TLR6 separately. Flagellin stimulates TLR5 responses, unmethylated bacterial CpG DNA stimulates TLR9 responses, and bacterial ribosomal RNA stimulates TLR13 responses. Moreover, one of the most important TLR for pathogen-associated molecular patterns (PAMPs) recognition is TLR4, which detects the lipid A of LPS [70].
A previous study reported that the use of Neisseria meningitides OMVs incorporating Hexa-acylated LPS as vaccines led to a severe, even lethal, inflammatory, while OMVs with the lpxL1 gene deletion displayed significantly reduced toxicity. This could be ascribed to the production of Penta-acylated lipid A resulting from the altered biosynthetic pathway of LPS after the knockdown of LpxL1 or LpxL2 [71]. Likewise, Xue et al. modified lipid A in E. coli, Shigella, and Salmonellae via the inactivation of genes encoding late acyltransferases (e.g., de-acylase LpxR, lpxM or PagL), which causes lipid A structural rearrangements with decreased ligand affinity for the TLR4/MD-2 complex and subsequently reducing the inflammatory response [12]. Alterations in the number of acyl chains and phosphate groups are commonly used methods, more studies on the regulation of OMVs virulence through the genetic modulation of bacteria are summarized in Table 3.
Genetic modification to regulate OMVs toxicity
| Parental Bacteria | Genetic modulation | Effect on Lipid A | Outcome | Reference |
|---|---|---|---|---|
| Escherichia coli | Δlpx, ΔlpxE, ΔLpxF | Monophosphorylated lipid A | Decreased toxicity | [72] |
| Salmonella typhimurium | ΔmsbB | Pentaacylated lipid A | Decreased toxicity | [73] |
| E. coli K-12 W3110, Salmonella typhimurium | Insert LpxR | Pentaacylated lipid A | Decreased toxicity | [74] |
| Neisseria meningitidis | Insert pagL | Pentaacylated lipid A | Decreased toxicity | [75] |
| E. coli K-12 W3110 | ΔmsbB, ΔpagP | Pentaacylated lipid A | Decreased toxicity | [76] |
| Escherichia coli | Insert lpxO | Pentaacylated lipid A | Decreased toxicity | [77] |
| Helicobacter pylori | Insert Hp0021 | Monophosphorylated lipid A | Decreased toxicity | [78] |
| Helicobacter pylori | ΔLpxE, ΔLpxF | Monophosphorylated lipid A | Decreased toxicity | [79] |
Δ means gene deletion
In fact, other explorations except for the genetic modification in reducing excessive OMV immunogenicity have also been undertaken. Qing et al. encapsulated OMVs with a highly biocompatible pH-sensitive shell of calcium phosphate, which overcame the severe systemic inflammation observed in naked OMVs after intravenous injection [35]. Zheng et al. came up with bacterium-mimicking vectors by rearranging the PAMPs, which displayed optimized anti-tumor therapeutic and prophylactic effects [80]. Further, LPS-neutralizing peptides could be used to reduce strong inflammatory responses induced by OMVs. Unfortunately, there is no universal peptide that can be applied to all OMVs. Selecting OMVs from commensal bacteria with lower immunogenicity or designing OMV-based oral formulations tend to be promising approaches, especially in the context of treating GI tumors. Oral administration prevents endotoxins from entering the circulatory system, at the same time, it can be in direct contact with the GI tumor. To sum up, although a series of trials evaluating the biosafety of OMV have been performed in animal models, there is still a lack of clinical research in humans. Moreover, determining the optimal safety and effectiveness tradeoff of OMVs remains one of the most challenging problems.
2.2 Engineered OMV-mediated delivery of anti-tumor drugs
OMVs were initially known to transport various biomolecules, such as nucleic acid, virulence factors, as well as proteins between cells. One of the core functions of OMVs better than other transport systems is their ability to carry several types of biomolecules simultaneously and prevent them from lytic enzymes over long distances transportation. This property of OMVs aroused the interest of OMVs involved in drug delivery systems in scholars. Initial studies focus on the use of OMV for drug delivery dating back to 1975. Since then, a steady stream of research has energized the field of OMV-mediated delivery of anti-tumor drugs [81].
2.2.1 Delivery of chemotherapeutic agents by engineered OMVs
Chemotherapy has been the first therapeutic choice for most patients with GI tumors during the last decades. However, non-specific accumulation of chemotherapeutic agents in healthy tissues, instead of their targeted distribution, has diminished their application value and led to numerous side effects, such as cardiotoxicity, peripheral neurotoxicity, bone marrow suppression, hair loss, nausea, and vomiting [82]. The intrinsic characteristics of certain chemotherapeutic drugs, like the high lipid solubility of paclitaxel and the facile degradation of camptothecin, also posed challenges to OMVs as drug carriers. Thus, researchers attempted to modify OMV to construct more stable and efficient drug delivery systems to improve the comprehensive effect of chemotherapy.
The E. coli strain Nissle 1917 (EcN), one of the gut probiotics, prefers to proliferate at the interface between the necrotic and hypoxic regions of hosts, which enables them natural tumor-targeting potential. The derivates of EcN, OMVs, were also proved to deliver Anti-tumor drugs to the tumor hypoxic areas, thereby reducing the tumor burden of CRC mice [83]. A comparative study showed that the chemotherapeutic drug Doxorubicin (DOX) loaded in OMVs exhibited better therapeutic response than that in liposomes or DOX alone in CRC. OMV-based drug delivery increased the half-life of drugs, decreased the clearance rate, and enhanced the bioavailability of the loaded drug [84]. The additional anti-tumor response may be attributed to the immunogenicity-induced aggregation of macrophages and significantly elevated cytokine levels in tumor tissues.
Paclitaxel (PTX), primarily targets cellular microtubule proteins to inhibit the cell cycle and has been widely used in the treatment of GI tumors. When PTX is synergistically administered with Salmonella typhimurium OMVs, the anti-tumor effect is further enhanced in CRC and HCC patients. Besides inhibiting angiogenesis by downregulating VEGF expression, the OMV-enveloping drug increased tumor cell apoptosis and autophagy, and increased Natural killer cells (NK cells) infiltration in tumor tissues [85]. The OMV-based platform enhances the efficacy of chemotherapy and minimizes its off-target and adverse effects. Solomon et al. utilized OMVs incorporated with PTX and attached bispecific antibodies (BsAb) to the OMV surface. When first used in clinical trials, the PTX-encapsulated OMVs were shown to be safe and well-tolerated in patients with advanced gastric, esophageal cancer, CRC, and PDAC. A significant secretion of the anti-tumor-associated cytokines (e.g., TNF-α) was also observed in the serum of patients four hours after intravenous administration, which could partially explain the anti-tumor effect of the engineered OMVs [86]. In addition to PTX, other traditional chemotherapeutic agents commonly used in the treatment of digestive system tumors, including hydrophilic (e.g., irinotecan), hydrophobic (e.g., cisplatin, carboplatin), or amphiphilic (e.g., DOX, vinblastine, and 5-fluorouracil) drugs, have been successfully incorporated into engineered OMVs. Engineered OMVs have been reported to transport chemotherapeutic drugs in a manner that greatly reduces the concentration of required chemotherapeutic agents and the accumulation of chemotherapeutic agents in normal tissues [87].
Engineered OMVs also offer a promising platform for the delivery of more potent and cytotoxic drugs that cannot be used as conventional chemotherapeutic agents. PNU-159682, a metabolite of Anthracyclines and a DNA topoisomerase I inhibitor, overcomes multiple resistance mechanisms in tumor cells. However, it is about 2000-fold more toxic than the conventional drug Adriamycin, which precludes its clinical application [88]. Sagnella et al. developed a drug delivery system incorporating PNU-159682 into S. Typhimurium OMVs, which were administered to mice with CRC. Engineered OMVs were modified with EGFR ligands on their surface in this study to improve their tumor-targeting properties. The new system exhibited significant anti-tumor effects without any apparent adverse effects. A schematic of the proposed mechanism of engineered OMV-induced activation of the immune system has been presented in Figure 5A. Anti-tumor related cytokines have significantly increased in the TME in Ep-EDV-682-treated CRC mice groups, which are known to promote both DC maturation and NK cell activation thus triggering a cascade releasing of cytokine production, resulting in a stronger cytolytic function. Notably, this engineered OMVs was further administrated in patients with advanced PDAC. CA19-9, and CRP levels were significantly decreased in the peripheral blood after the 12th dose, and no significant adverse reactions were observed during treatment [89]. This is one of the few engineered bacterial OMVs that have been used in GI tumor patients with therapeutic effects.
Anti-tumor effects of engineered OMVs with optimized immunogenicity. A-B) Schematic of the proposed mechanism of engineered OMVs reduced tumor volume with increasing concentrations of TNF-ɑ (a), IL-12p40 (b), and IL-6 (c), as well as growth inhibition in CT26 tumors. Adapted with permission from [96], copyright 2020 Cancer Cell. C) Drug resistant Caco-2/MDR1 xenografts were treated with shMDR1-loaded OMVs followed by irinotecan-loaded OMVs on days shown below the x-axis, which resulted in significant antitumor effects. D) Kaplan-Meier survival analysis showing 100% survival only in the mice receiving the sequential treatment of shMDR1-loaded, EGFR presenting OMVs and irinotecan-loaded EGFROMVs. Reproduced with permission from reference. Adapted with permission from [97], copyright 2009 Nature Biotechnology.
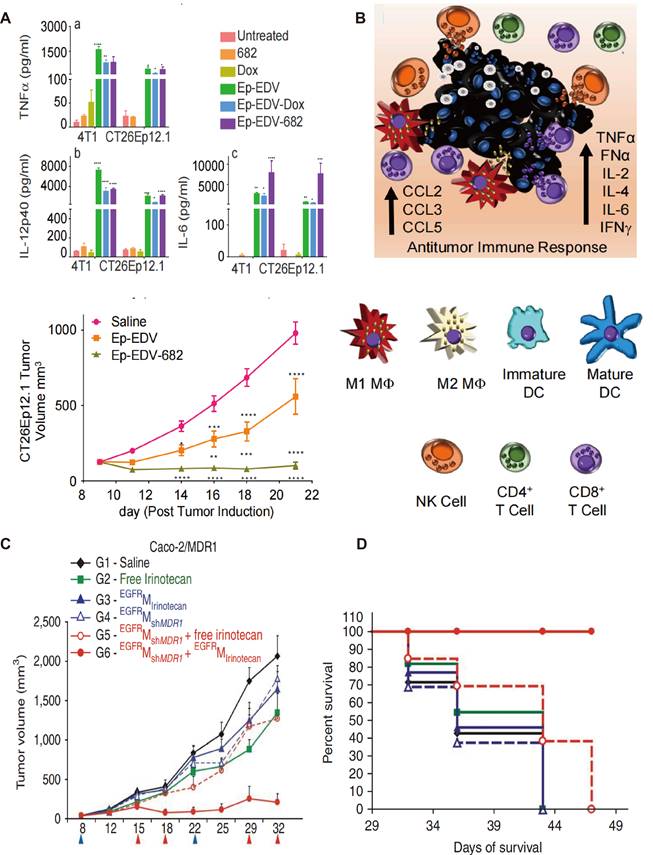
Engineered OMVs as carriers of chemotherapeutic drugs have higher drug loading capacity compared with traditional nanocarriers, which largely improved the efficiency of drug delivery. As reported, one OMV can encapsulate 10 million drug molecules at most. PTX shows a favorable binding affinity with OMVs compared to liposomes, because of the hydrogen bonds between PTX and DD-transpeptidase, the teichoic acid of the bacterial OMVs. Similarly, the quantitation of DOX packaging in OMVs showed approximately 100-fold higher than liposomes under the same co-incubation conditions [90]. Except for internal drug loading, a small number of drugs can be genetically engineered onto the surface of OMVs. As the study reported by Ren et al., Polybia-mastoparan I, which exhibits preferential toxicity to tumor cells while having little damage to nontumorigenic cells, has been engineered onto OMVs surface with enhanced anti-tumor immune responses [91].
2.2.2 Delivery of siRNAs by engineered OMVs
Short interfering RNA (siRNA), short hairpin RNA (shRNA), or micro RNA (miRNA) are subsets of double-stranded RNAs that effectively silence the expression of target genes at specific sequences, a process referred to as RNA interference (RNAi) in molecular biology [92]. In cancer therapy, these RNAs have emerged as a powerful tool to inhibit GI tumor progression by downregulating pro-oncogenes, such as Nuf2, Rap80, Hif-1α, and Vegfa [93]. However, the clinical use of siRNAs is limited due to their poor membrane permeability and stability, making them hardly cross the biofilm barrier and easily degraded by serum nucleases. Jivrajani et al. packed shRNAs targeting Vegfa in OMVs surface-modified with folic acid to treat GI tumor Xenograft mice. The engineered OMVs silenced the mRNA and protein expression of Vegfa in tumor tissues, and inhibited neovascularization to block the supply of oxygen and nutrients to tumors, eventually leading to tumor stabilization and regression [94]. The combination of engineered OMVs containing therapeutic siRNAs and cytotoxic drugs may improve treatment efficacy and enhance susceptibility to chemotherapeutic agents. Jenner et al. demonstrated that the engineered OMVs delivering specific siRNAs against multidrug resistance genes (e.g., Plk1) led to the elimination of previously chemoresistant tumors in an animal model of CRC [95]. The positive results showed the potential of OMV as a platform for siRNA-based combination therapy. Combination therapy involving lower concentrations of chemotherapeutic drugs, siRNAs, and antibodies delivered by engineered OMVs offers a promising strategy to overcome the limitations of conventional systemic therapy. As Figure 5B presented, OMVs surface was modified in another study with EGFR-specific ligands and internally loaded with siRNAs against cell cycle-associated proteins, which have produced significant tumor growth inhibition in colon cancer models. Further, engineered OMV-based dual sequential treatment of shRNA and irinotecan reversed the chemoresistance and acquired improved survival benefit in GI tumor patients [36].
Currently, electroporation is a prevalent technique for introducing siRNAs into OMVs. Electroporation applies short, high-voltage pulses to induce the formation of transient pores in the cell membrane, creating a temporary state of permeability that enables the uptake of drugs, fluorochrome compounds, and large molecules such as nucleotides. Following the completion of the electroporation process, the phospholipid membrane undergoes structural reorganization without any permanent damage [98]. Electroporation was first used to modify bacterial OMVs in 2014 [39], which inspired the following studies to use this technique to introduce nucleotide cargoes into OMVs. Guo et al. add the specific siRNAs into OMV with electroporation, which targets the Redd1 metabolic pathway in tumor-associated macrophages. This novel approach significantly increased the infiltration of M2-type macrophages in tumor tissues and the levels of anti-tumor-associated cytokines, ultimately improving the survival rate of tumor-bearing mice [99]. It is worth noting that different bacteria and drug payloads require precise regulation of electroporation parameters to avoid destabilizing the OMVs membrane [100]. Besides internal drug loading, drugs can also be inserted into the membrane depending on lipophilicity or hydrophobicity.
2.3 Engineered OMV-based biomimetic NPs for GI tumors
Nanomaterials have emerged as important candidates for biomedical applications owing to their unique characteristics, such as naturally optical, electrical, magnetic, and electrochemical properties [101]. However, the high cost of certain NPs (e.g., gold) limitations in mass production, complex process of design and synthesis, and unproven biocompatibility and safety have restricted the clinical translation of NP-based drug delivery systems. OMV can be a great ally for improving NPs selective cell penetration, it can also prevent the immune clearance of NPs in clinics due to its good natural properties [90]. The optimized pharmacokinetic properties of OMV-NPs have popularized their application in biomedical fields. The ain techniques of engineering OMVs with classical NPs and their applications in GI tumor treatment are described below.
2.3.1 Gold NPs
Gold NPs have been widely used throughout the medical field because of their excellent stability. Gold NPs could improve the magnetic resonance imaging (MRI) contrast sensitivity of esophageal and gastric cancers without any apparent side effects, thereby significantly enhancing the tumor detection rate [86]. Gold NPs offer a stable physical environment that facilitates electron transfer between electrode surfaces in contact with them and preserves protein bioactivity when bound to proteins. Additionally, they directly couple and interact with a broad range of molecules such as proteins, drugs, antibodies, enzymes, nucleic acids (DNA or RNA), and fluorescent dyes. Thus, they are considered versatile platforms for drug loading and targeted delivery. Gold NPs have also been utilized as targeted labeling agents for GI tumor tissues [102]. In addition to their application in diagnostics, gold NPs are a good platform for drug loading and targeted delivery. Given their tuneable and unique optical properties, gold NPs have attracted widespread interest as mediators of noninvasive radiofrequency ablation and photothermal therapy (PTT) for improving treatment efficacy in GI tumors. As previous literature reported, gold NP-gemcitabine complexes can reverse chemotherapy resistance in PDAC and significantly improve patient survival [88]. Their tunable and unique optical properties have attracted widespread interest in their use as mediators of non-invasive radiofrequency ablation and PTT in GI tumor therapy [103].
Recent studies have explored the use of gold NPs combined with biological materials, such as cell membranes, bacterial extracellular vesicles, and antibodies, to further functionalize gold NPs for optimized efficacy. Piao et al. used erythrocyte membranes to encapsulate gold NPs and found them to prolong circulation time and tumor targeting in both PTT and chemotherapy [104]. Recently, engineered OMVs have been shown to be more promising biomimetic materials for NP modification, as they are inexpensive, easily editable, and biocompatible. Gao et al. used E. coli OMVs encapsulated with gold NPs to form biomimetic NPs with a size of 30 nm and used them as tumor vaccine adjuvants. These biomimetic NPs triggered T-cell responses in vivo by inducing the production of INF-γ and IL-17. In addition, the biomimetic NPs enhanced the activation and maturation of DCs in lymph nodes and generated humoral immune responses with higher affinity [105]. OMV was also utilized to establish a platform to combine NP with another therapeutic agent. In a preclinical study, the E. coli OMV-based biomimetic Gold NPs were cleverly constructed for synergetic PTT and immunotherapy. The modified NPs enhance macrophage chemotaxis by upregulating TNF-α expression and induce the accumulation of reactive oxygen species (ROS) after low-dose laser irradiation, resulting in strong anti-tumor effects and improved survival of tumor-bearing mice [106].
2.3.2 Mesoporous silica NPs
Mesoporous silica nanoparticles (MSNPs) have garnered significant attention as potential drug delivery vehicles for the treatment of GI disorders, particularly inflammatory bowel disease (IBD), due to their desirable characteristics such as controlled particle size, high specific surface area, low toxicity, hemocompatibility, and ease of surface modification [107]. MSNPs achieve site-specific and controlled release of guest drugs from pore channels with external triggering motifs. Whereas, MSNPs may be destabilized in saline buffers to form aggregation, thus leading to the physically adsorbed drugs' premature release. To improve the drug-loading stability as well as the tumor-targeting ability of MSNPs, researchers explored OMV to wrap MSNPs for immune evasion. Covering of OMV improved MSNP biocompatibility, which can be explained by phagocytosis and delivery of OMV by neutrophils [108]. In a study on CRC, MSNPs that loaded with DOX internal were also surface-decorated with E. coli OMVs. OMVs as biological outer membranes help the drug delivery system adsorb on the intestinal mucosa to target tumor cells, which is responsible for not only improved therapeutic effect but also reduces the leakage of DOX. In another study, Shi et al. treated CRC cells with 5-Fluorouracil (5-FU) loaded MSNPs, which were encapsulated in engineered E. coli OMVs [109]. This delivery system profits from the high drug-loading capacity of MSNPs as well as the intestinal absorption ability of OMV via a specific Hyaluronic Acid (HA) receptor. The modified carriers enabled specific targeting of the tumor site and provided a viable strategy to improve the oral bioavailability of 5-FU, leading to effective tumor growth inhibition while minimizing side effects [110].
2.3.3 Ferrosoferric oxide -Manganese dioxide (Fe3O4-MnO2) NPs
It has been reported. that systemically or locally administered iron oxide nanoparticles inhibited cancer growth by inducing a pro-inflammatory immune response with M1 macrophage polarization besides ferroptosis [111]. Nevertheless, iron oxide nanoparticles triggered ROS-induced tumor damage may be resisted by the intracellular redox balancing mechanisms, limiting the therapeutic efficacy of ferrotherapy alone. Assisting materials therefore need to be applied to overcome the role of this endogenous balance. MnO2 is a widespread multifunctional therapeutic agent for relieving tumor hypoxia and improving tumor treatment efficacy. The structure of MnO2 includes Nanospheres, nanosheets, as well as hollow nanoparticles. Liu et al. deposited MnO2 with Fe3O4 in the envelopes of OMVs from E. coli. Taking Fe3O4 as PTA, they utilize E. coli OMVs as peroxidase carriers to relieve tumor hypoxia, thus Mn2+ activates ICD effects through glutathione (GSH)-consumed ferroptosis as well as the generation of ROS. The immune-stimulated OMVs hitchhike on the circulating neutrophils, resulting in improved tumor targeting. Finally, the engineered OMV produces stronger anti-tumor immune responses and enhances the therapeutic effect [112].
2.4 Engineered OMV-mediated photothermal therapy for GI tumors
PTT involves the use of photosensitizers (PSs) with high efficiency of photothermal conversion to convert light energy into heat energy to kill cancer cells. This approach induces the release of tumor antigens from heat-damaged cells, leading to the activation of tumor-specific CTLs, which mediate anti-tumor immune responses. Compared with traditional therapeutic modalities for GI tumors, PTT is relatively non-invasive and less toxic. However, the heterogeneity of tumor antigens, the complexity of laser dosimetry, and the potential damage of tumor surrounding tissue damage constrain its broad application [113]. It is therefore of great importance to come up with combinatorial therapeutic methods to improve the PTT efficacy. A study on CRC showed that Salmonella-derived OMVs systemic administration induced tumor site inflammation, then causing erythrocyte leakage, which increased the absorbance of the near-infrared (NIR) laser in tumor tissues. After low-dose laser irradiation, neither tumor recurrence nor metastasis was observed besides significant tumor regression. Compared to conventional PTT, the OMVs-combined strategy reduces the required doses of PSs and NIR and enhances tumor-targeting and killing activities (Figure 6A). Zhuang et al. found that intravenous injection of Salmonella typhimurium OMVs significantly increased NIR optical absorption of GI tumors without adding any exogenous PSs [114]. To take advantage of the membrane stability of OMVs, some biologically active molecules, such as enzymes, can be loaded into OMVs to inhibit tumor growth after laser irradiation. Engineered OMVs have demonstrated the potential to achieve precise targeting of tumors by exploiting the host immune response. Zhang et al. assembled hydrophobic PSs Ce6 with catalase (CAT) and loaded it into bacterial OMVs surface-modified with PD-L1. Relative to CAT-Ce6 alone, that modified with bacterial OMVs resulted in higher intratumor drug concentration and smaller tumor volume after laser irradiation [115].
Anti-tumor effects of engineered OMV-based PTT. A) Intravenous injection of engineered OMVs could lead to extravasation of red blood cells in the tumor, further enabling effective photothermal ablation of the tumor by the NIR laser. B) IR thermal images and the corresponding time-dependent tumor temperature changes of CT26 tumor-bearing mice under NIR laser irradiation post injection of PBS or OMVs. C)Body weights of CT26 tumor-bearing mice post different treatments as indicated. Adapted with permission from [120], copyright 2021, Biomaterials. D) Schematic of the membrane derived from OMV and CC fusion and the resulting engineered OMV-CC camouflaged HPDA NPs to produce HPDA@[OMV-CC] NPs. E) Synergistic Photothermal/Immunotherapy of engineered OMVs and the relative tumor volume and long-term survival of mice after different treatments. Reproduced with permission from reference. Adapted with permission from [116], copyright 2020, Applied Materials.
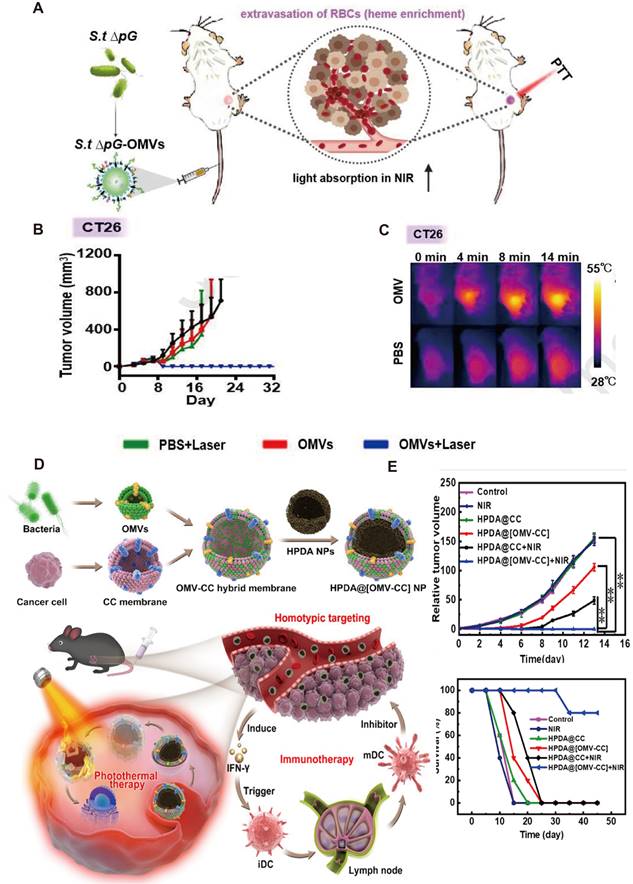
The recent emergence of membrane fusion approaches facilitated PTT development. On the one hand, the incorporation of tumor cell membranes into OMVs prevents the loss of tumor antigens. On the other hand, the introduction of OMVs enhances the recognition of tumor membrane antigens by the immune system. Wang et al. fused bacterial OMVs with tumor cell membranes to obtain OMV-CC hybrid membranes and successfully coated them on hollow polydopamine NPs (Figure 6B). Combined with OMVs, polydopamine NP-mediated PTT activated immune responses in tissues of tumor and resulted in enhanced anti-tumor effects. On intravenous administration, these biomimetic NPs can homogeneously target tumor tissues and activate immune responses by rapidly stimulating DC maturation in lymph nodes. The anti-tumor immune response and PTT mutually enhance the therapeutic effects to completely eradicate tumors without causing obvious adverse effects [116]. Synthetic OMV that integrates immunogenicity, cargo encapsulation, and photothermal conversion effect combined with other therapeutic methods has been raised to improve the therapeutic efficacy of PTT. For example, Zhai et al. constructed fusion bio-nanocarrier liposomes by fusing OMVs with photothermal-sensitive liposomes (PTSLs). High expression of CD38 on T cells in TME is associated with the immunosuppression of tumors. The use of PTSLs to deliver CD38-targeting siRNAs and PD-1 monoclonal antibodies produced significant anti-tumor effects in CRC and HCC. They also observed CD8+ T-cell infiltration at both primary and metastatic tumor sites and increased levels of anti-tumor-associated cytokines [117].
As mentioned above, the gold NPs and Fe3O4-MnO2 have shown promise as PSs in the treatment of various GI cancers, including esophageal, gastric, biliary tract, and pancreatic cancer. However, it has been uncovered that only 0.7% of these PSs successfully target the tumor site since the rapid clearance by the mononuclear phagocytic system in animal models. OMVs were utilized to coat PS, thereby in situ hitchhike circulating neutrophils for PSs' tumor targeting transportation. Through this work, neutrophil-mediated intratumor accumulation of PSs was improved by 300 to 600% which could be attributed to OMVs-caused inflammatory chemotaxis, and immune evasion by cell membrane camouflage [118]. In another research on CRC mice, OMVs were encapsulated into nano-long micelles that loaded with PSs to harness the immunogenicity of OMVs, which attracted neutrophils for phagocytosis [119]. Upon subsequent irradiation of tumor tissue with infrared light, an inflammatory microenvironment was induced, further enhancing the tumor-targeting and killing efficacy of PTT. Li et al. presented a promising OMV-based vaccine that facilitates immune-mediated tumor clearance after PTT in situ. The E. coli-derived OMVs were surface-modified with maleimide (Mal) groups for tumor neoantigens capture. At the same time, the 1-methyl-tryptophan (1-MT) was loaded into the OMV-Mal to reverse the tumor immunosuppression. The anti-tumor effect of PTT combined with 1-MT@OMV-Mal was confirmed both in primary and distant lesions in murine CRC models [119]. To sum up, the application of engineered OMVs in PTT warrants further investigation in future research.
2.5 Engineered OMV-mediated tumor vaccines for GI tumor
Many tumor vaccines have been proven to be therapeutically efficient in animals, whereas only a few have been evaluated in clinical trials, which suggests that numerous problems remain to be overcome. One of the major limitations is identifying sufficiently immunostimulating tumor-specific antigens [121]. Li et al. modified the E. coli ClyA sequence with the addition of the binding protein L7Ae and listeriolysin O (LLO), a protein known for promoting lysosomal escape. Followed by co-incubation, a therapeutic tumor vaccine that presents tumor-specific antigen mRNA was developed using OMVs as a vector (Figure 7B). Compared to free mRNA or liposome-based vaccines, the OMV-based mRNA vaccine notably facilitated DCs maturation, enhanced phagocytosis, and presentation of tumor antigens, and elicited stronger cellular immune responses, ultimately leading to the reduction of CRC tumor as shown as shown in Figure 7B. Remarkably, the vaccine demonstrated an anti-tumor immunological memory effect that persisted even 6 months after vaccination [122]. Another limitation is finding a representative neoantigen that is co-expressed by all tumors is almost impossible, because of the heterogeneity of tumors [121]. Zhao et al. evaluated the efficacy of two anti-tumor vaccines based on engineered OMVs. The first vaccine employed a 'tumor antigen plug-and-display procedure' in which protein tags were used to label tumor antigens, with the tags binding to OMVs expressing the 'catcher' for the efficient presentation of multiple tumor antigens on the surface of OMVs. The second vaccine was developed through a membrane fusion technique, where the fusion of tumor cell membranes and bacterial protoplasmic membranes onto poly lactic-co-glycolic acid (PLGA) NPs produced an immunogenic whole-tumor vaccine. Both vaccines elicited potent anti-tumor immune responses and demonstrated considerable tumor-killing activity in a mouse model of CRC. However, the former vaccine is insufficient in low mutation burden tumors, while the latter needs enough tumor tissue for vaccine synthesis, which can be obtained only through invasive procedures [123].
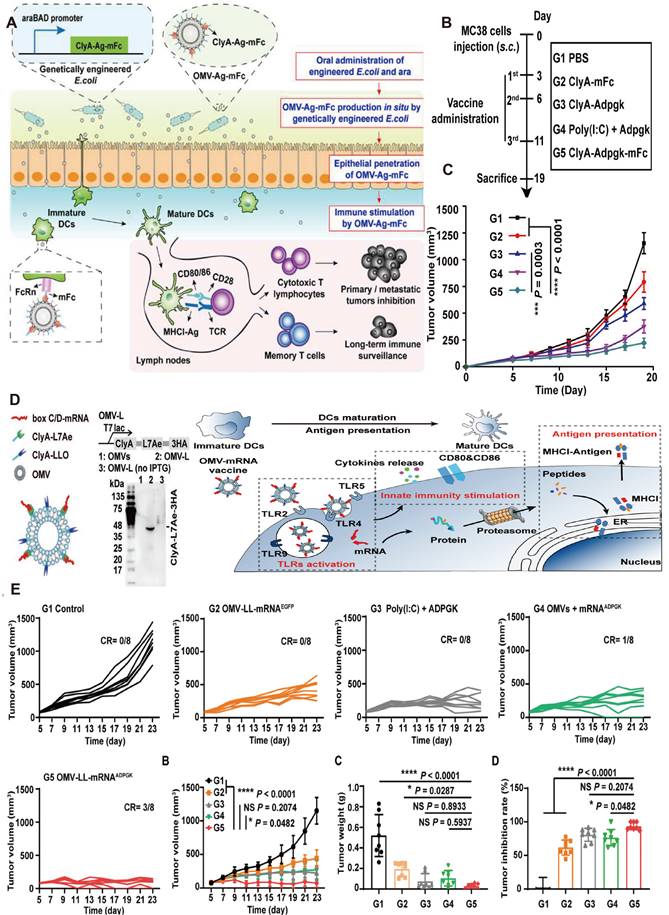
Engineered OMV-based oral tumor vaccine. A) Engineered E. coli were obtained by transformation with a plasmid expressing ClyA fused with a tumor antigen and Fc fragment of mouse IgG (ClyA-Ag-mFc). An arabinose-inducible promoter was introduced to control the fusion protein expression. OMV-Ag-mFc effectively penetrate the intestinal epithelial barriers, and are recognized and taken up by DCs in lamina propria. B) The schema showing the timeline of model construction and oral vaccination, and the tumor volumes were recorded every two days. Adapted with permission from [126], copyright 2022 Nature Biomedical Engineering. C) Schematic of the engineering OMV strategies. The RNA binding protein L7Ae and endosomal escape-promoting protein LLO were fused to the C-terminal of the ClyA surface protein on OMVs. D) The OMV-based mRNA vaccine triggering TLR activation, innate immunity stimulation, and antigen presentation. E) Growth curves of MC38 tumors bearing mice, growth curves of the average volumes, and the tumor weight of excised MC38 tumors have been recorded. Adapted with permission from [128], copyright 2022 Applied Materials.
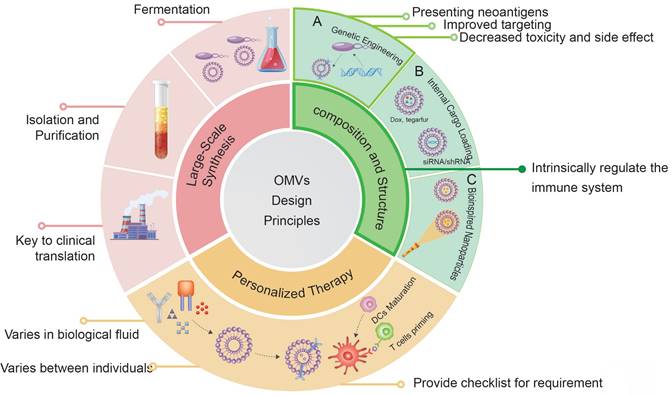
Design principles for engineered OMVs.
Future design of OMVs for advanced therapeutic applications.
Additionally, tumor vaccines can be limited by the tumor burden-associated suppression of the systemic immune response [124]. Surgical excision is contraindicated for advanced GI tumors due to its inability to improve survival and quality of life for patients. Research by Ma et al. proved that the debulking surgery for primary tumors before the vaccine could lead to a more satisfactory therapeutic effect in pancreatic cancer, hepatic cancer, and colon cancer mouse models. They explained that primary tumor resection has reversed the tumor-induced suppression of immune responses thus further preventing metastasis [124]. Similarly, efferocytosis has been concluded as one of the mechanisms that contribute to tumor immunosuppression. Zhuang et al. engineered the OMVs for the efferocytosis blockade, tumor antigens capture, and presentation, consequently boosting the OMV-based vaccine efficiency [125].
The immune potency of the OMVs vaccine is influenced by the administration' s ways. Given that vaccines are injected intramuscularly or subcutaneously, OMVs-aroused anti-tumor response is generally restricted by limited lymph nodes and immune cells. Yue et al. recently devised an oral anti-tumor vaccine that can be controlled in vitro and relies on the capability of OMVs to traverse the intestinal mucosal barrier and interact with immune cells in the lamina propria, especially DCs. The oral administration of engineered bacteria and the expression inducer Arabinose (Ara) resulted in the production of OMVs expressing tumor antigens (OMV-Ag-mFc) within the intestine (Figure 7A). As Figure 7 showcased, OMVs-induced anti-tumor vaccine with oral administration has triggered anti-tumor immune responses and significantly suppressed tumor growth in murine models of CRC [54]. Compared with subcutaneous or intramuscular tumor vaccines, oral administration has superior safety and patient compliance as well as reduced healthcare expenses.
Toxicity is generally considered one of the biggest hindrances to the clinical application of OMV-based vaccines. Several strategies have been addressed recently. First of all, developing new ways for vaccine administration, like oral administration. Without direct entry into blood circulation, oral delivery systems avoid cytokine storm induction, tissue extravasation, and reticuloendothelial system clearance, especially fit for the treatment of gastroenterological conditions, like GI tumors. Yue et al. used luciferase labeling to visualize the biodistribution and pharmacokinetics of OMVs. The results indicated that the bioluminescent signals are hardly detected in tissues other than those in the GI tract, which proved the biosafety of OMV-based oral vaccine (Accumulation in the cecum at 2 hours after administration, and gradually moved to the colon within 12 hours) [126]. The epithelial cell penetration and immune activation of oral vaccines have also been well-proved. To prevent unexpected immune tolerance, Yue et al. also introduced an Ara-inducible promoter to guarantee the tumor antigen expression. Selection of certain strains, like probiotics or commensal bacteria seems to be another choice to avoid unwanted cytokine storm.
The AKK is a recognized intestinal probiotic. The protective effects produced by oral administration of AKK OMV on the gut barrier and commensal microbiota homeostasis were explored in the background of several GI tumor treatments. For instance, Shi et al. combined IL-2-based immunotherapy with Akk OMV, by which enhancing the anti-tumor immune response and tumor clearance [127]. OMV derived from Akk was proven to enhance the immune activity of CTLs in CRC mice, whereas administration of Akk itself has no obvious influence on intestinal barrier integrity and gut homeostasis [10]. In addition, ensuring dimensional uniformity with dynamic light scattering, as well as avoiding the interference of impurities during preparation are two other ways to reduce side effects. Apart from the above measures, specific elimination of LPS in OMVs by genetic engineering as well as membrane fusion with tumor cells are also feasible options [19]. Overall, immunogenicity is the major source of OMV vaccine-induced adverse events and immunogenicity control is a double-edged sword because it may inhibit the anti-tumor effect. Investigations on methods to identify an ideal balance between low toxicity and high immunogenicity of OMV are still currently required.
3. Advantages of OMVs in GI Tumor Treatment
Different from other types of tumors, the GI mucosa possesses a large, orally accessible, and highly immunologically active interface, and thus represents attraction for tumor vaccine oral administration. Some studies have proved that mucosal immunizations are usually more effective than other parenteral routes, like subcutaneous or intramuscular injection in developing protective immunity to mucosal in GI tumors. GI tumors remain highly aggressive cancers, despite progress in therapy. Apart from conventional approaches, immunotherapy has become one of the primary treatment options for GI tumors especially those in advanced stages [129].
The intestine is considered the first immune organ of the human body, which is rich in both immune cells and mucosa-associated lymphoid tissues. High levels of T cells can be activated by mucosal immunostimulation in both the mucosal compartment and the related mesenteric lymph nodes [130]. Then governing GI and the mucosal tumors through integrins-induced lymphocyte homing as well as proper cytotoxic activation within the tumor site, thus maximizing the efficiency of oral tumor vaccine. Karaki et al. found that only mucosal vaccination elicited CD8+ T cells expressing mucosal integrins (CD49a and CD103), which is crucial for intratumoral CD8+ T cell infiltration and tumor elimination [131].
The environment of the GI tract provided various natural triggers of OMV-based therapeutic systems like special pH, pressure, and temperature. Thus, therapeutic cargo can be released at target sites mediated by the specific bacteria or enzyme in the intended biological environments. Besides, OMV is superior to other drug carriers in protecting cargoes from harsh GI environment like gastric acids or lipases secreted by the pancreas and bile salts [132]. Some probiotic OMVs were confirmed to penetrate intestinal physiochemical barriers, interact with specific epithelial cells, and even manipulate the gut microbiome proving alleviated primary resistance to PD-1 blockade in tumor immunotherapy [10].
4. Summary and future perspectives
Engineered OMV represents a promising nanoscale drug delivery platform due to its superior biosafety and capability to improve drug uptake and delivery efficiency. They can stabilize the transport of chemotherapeutic drugs, mitigate damage to normal tissues, and reduce drug leakage. Moreover, OMVs protect bioactive molecules (e.g., enzymes, small-molecular proteins, and siRNAs) from degradation, which enhances the stability and functional completeness of the cargo. As membrane structures, OMVs can be readily processed and modified to enhance their existing properties or confer additional functions, such as targeting tumor cells, reversing TME immunosuppression, and activating an effective anti-tumor immune response. OMVs combined with organic or inorganic synthetic nanomaterials to achieve complementary advantages, which promotes the functionalization of OMV that improves the efficiency of therapy, and reduces the burden of purely synthetic nanomaterials mass production. It has been confirmed that the disturbed gut microbiota as well as their metabolites strongly affects the occurrence and the progression of GI tumor. Thus, the combination of OMV-regulated gut microecology with other types of therapy is a powerful strategy. In addition, oral administrated OMV contact with GI tumor is direct, without LPS entry into the bloodstream, and avoids the OMVs clearance by the reticuloendothelial system. This review has summarized the recent advances related to the application of engineered OMVs in the treatment of GI tumors. Approaches for engineering OMVs include genetic modification, internal drug loading, surface modification, and combination with NPs, etc. (Table 4). Owing to their unique advantages, engineered OMVs have garnered substantial attention from researchers, and numerous encouraging preclinical studies have been reported on their use in the past few years [133].
Engineered OMV-based tumor therapy
| Type of engineering | Engineering strategy | Effect of modification | Effect of Engineered OMV-based tumor therapy | Reference |
|---|---|---|---|---|
| Genetic Engineering | ∆msbB | Higher production yield OMVs with impaired lipidA | NK cells and T cells accumulate and produce IFN-γ and CXCL10 in the tumor | [134] |
| ∆msbB and fused ClyA with HA tags | Impaired lipidA, expression of the ClyA-mPD1E-3HA protein on OMVs | OMV-PD1 blocks the PD1/PD-L1 inhibitory axis, high levels of IFN-γ, IL-6, and TNF-α, NK cells, and CTL cell accumulation | [40] | |
| ∆msbB and fused ClyA with HER2 affibody | OMVs expressed ClyA-Affibody recombinant protein | AffiHER2 OMVs were uptaken selectively by the tumor tissue | [135] | |
| ∆relA and ∆spot | OMVs with lower toxicity | Inflammatory and extravasation of RBCs in the TME, greatly enhanced tumor NIR absorbance | [120] | |
| Fused a plasmid expressing ClyA with a tumor antigen and Fc fragment of mouse IgG (ClyA-Ag-mFc) | Controllable production of OMVs loaded with tumor antigen (OMV-Ag-mFc) in the intestine | Inhibited the tumor metastatic and offering long-term protection | [54] | |
| Insert the gene fragments SpC or SnC into the plasmid | Engineered OMVs can display multiple tumor antigens | CD3+CD8+ T cells, CD3+CD4+ T cells, CD11b+Ly6G+ activated neutrophils, and CD11c+ DCs are all significantly elevated in the MC38 tumor microenvironment. | [136] | |
| Cargo Loading | OMVs packed PNU-159682 by coincubation | OMVs loaded with 682 stably and modified with BsAb | M1 macrophage polarization, NK cell activation, CD8+ T cells generation | [96] |
| OMVs capsulated MSN and 5-FU by high-pressure coextrusion | OMVs-MSN-5-FU | 5-FU was delivered with tumor-targeting and reduced liver and spleen damage | [110] | |
| Tegafur was loaded into OMVs by high-pressure coextrusion | With immune modulation, lower drug leakage decreased side effects | A robust anti-tumor immune Memory generated | [42] | |
| Hybrid membrane | OMVs and cancer cell membrane fusion by sonicating and extruding | Engineered OMVs present melanoma-specific antigens with high stability and low cytotoxicity | Engineered OMVs vaccination increases the CTLs level and CD4+ population, and suppresses tumorigenesis | [137] |
| Fused OMVs with the cancer cell membrane by sonicating | Engineered OMVs present various tumor-specific antigens | The mice inoculated with CT26 tumor cells show complete tumor elimination and a tumor inhibition rate of 100% | [136] | |
| The PTSLs incorporated with Cypate fused with OMVs by coextrude | Cascaded double-target OMVs were constructed with the ability to carry the loaded cargos to the tumor tissues and target the T cells, respectively | Increased infiltration and cytotoxicity of T cells, improve the therapeutic effects of the PD-1 antibody | [138] | |
| Surface modification | The DSPE-PEG-CA-PTX was escorted into OMVs by coincubation | Sequential pH-triggered prerelease of paclitaxel | Have satisfactory inhibition of tumor progression and metastasis | [99] |
| OMVs fused with DSPE-PEG-RGD through the coextrusion | Improved blood circulation and tumor-targeting ability, decreased immunogenicity | A robust anti-tumor immune Memory generated | [42] |
Δ means gene deletion
The intrinsic immunostimulatory property is the basis of engineered OMV in anti-tumor combinational therapies (Table 5). Nevertheless, OMV membrane substances cause excessive immune activation is still one of the most pressing concerns before clinical application. Developing multiple routes of administration or selecting OMVs derived from human commensal bacteria may become a new breakthrough. Cancer vaccines are primarily designed to stimulate tumor-specific immune responses through the delivery of tumor-associated antigens. OMVs, serving as immune adjuvants and tumor antigen carriers, represent promising candidates for tumor vaccine delivery. It is worth mentioning that suppressed tumor recurrence and metastasis have been observed in several OMV vaccine-mediated tumor treatments. It can be attributed to the long-term immune memory effects caused by the interaction of OMV-presented tumor-associated antigens tumor associated antigen with immune cells.
Engineered OMVs involved in GI cancer
| Parental Bacteria | Engineered strategy | Cancer type | Therapeutic strategy | Reference |
|---|---|---|---|---|
| E. Coli W3110 | Gene engineering (PD-L1) | Colorectal Neoplasms | Immunotherapy | [40] |
| E. Coli W3110 | Gene engineering (Decreased toxicity) | Colorectal Neoplasms | Immunotherapy | [134] |
| E. coli Nissle1917 | Surface modification with ClyA (ClyA)-hyaluronidase (Hy) | Pancreatic and Colorectal Neoplasms | Immunotherapy | [139] |
| E. Coli DH5α | Hybrid membrane with cancer cells | Colorectal Neoplasms | Immunotherapy | [140] |
| Salmonella Typhimurium | Drug loading | Colorectal and Liver Neoplasms | Immunotherapy and chemotherapy | [141] |
| Lactobacillus rhamnosus GG | Gene engineering (Decreased toxicity) | Liver Neoplasms | Immunotherapy | [142] |
| E. coli DH5α | OMV-loaded micromotors | Colorectal Neoplasms | Immunotherapy | [143] |
| E. coli BL21 (DE3) | Drug loading | Colorectal Neoplasms | Immunotherapy and chemotherapy | [144] |
| Salmonella Typhimurium | Gene engineering (Decreased toxicity) | Colorectal Neoplasms | Immunotherapy and PTT | [120] |
| S. Typhimurium minCDE- strain | Drug loading | Colorectal Neoplasms | Immunotherapy and chemotherapy | [89] |
| S. Typhimurium minCDE- strain | Gene engineering (EGFR) and siRNA loading | Colorectal Neoplasms | Immunotherapy and Gene therapy | [97] |
| Escherichia coli | OMVs-MSNs-5-FU mesoporous silica | Colorectal Neoplasms | Immunotherapy and chemotherapy | [110] |
| Salmonella | Membrane fused with liposomes and siRNA loading | Colorectal Neoplasms | Immunotherapy and Gene therapy and PTT | [138] |
| Escherichia coli | Genetic engineering (fused the SpC with ClyA) | Colorectal Neoplasms | Immunotherapy and surgery | [124] |
| E. coli BL21 (DE3) | Gene engineering (CD47nb) and surface modification (PEG/Se) | Colorectal Neoplasm | Immunotherapy and radiotherapy | [41] |
| Escherichia coli MG1655 | Drug loaded (UNC2025) and surface modification (Mal) | Colorectal Neoplasm | Immunotherapy and targeted therapy | [114] |
| E. Coli | Drug-loaded (IDO inhibitor) and surface modification (Mal) | Colorectal Neoplasm | Immunotherapy and PTT | [145] |
Although there are many advancements in engineered OMVs in the treatment of GI tumors, several obstacles must be overcome before their clinical translation. Apart from the potential immune-related adverse effects, significant problems including the relatively intricate and time-consuming isolation and purification procedures, as well as the unclear composition and mechanisms of OMVs are yet unsolved. Another challenge in the large-scale production of OMVs is the variability of batches due to the overexpression or knockout of specific genes, which affects the size, yield, and immunogenicity of OMVs. Thus, it is crucial to establish a standardized approach for the preparation of OMVs. To sum up, OMVs provide an excellent platform for integrating various therapeutic strategies for GI tumors. By engineering OMVs, personalized treatment strategies for GI tumors can be developed. Since engineered OMVs provide a platform to combine different therapeutic methods, optimizing the combination approach to achieve the most synergistic anti-tumor effect is a promising avenue for future research efforts.
Abbreviations
OMVs: outer membrane vesicles; PDAC: pancreatic ductal adenocarcinoma; HCC: hepatocellular carcinomas; CRC: colorectal cancer; CAR-T: chimeric antigen receptor T-cell; GI: gastrointestinal; PD-1: programmed cell death 1; PD-L1: programmed cell death ligand 1; FDA: Food and Drug Administration; TME: tumor microenvironment; CTLs: cytotoxic T-cell lymphocytes; Akk: Akkermansia muciniphila; ICB: Immune checkpoint blockade; NPs: nanoparticles; LPS: lipopolysaccharides; OMPs: outer membrane proteins; Lpps: lipoproteins; PG: peptidoglycan; Tol-Pal: tolerance peptidoglycan-associated lipoprotein; PQS: pseudomonas quinolone sequence; ABC: ATP-binding cassette; E. coli: Escherichia Coli; H. pylori: Helicobacter pylori; ETEC: enterotoxigenic Escherichia Coli; PTE: phosphotriesterase; PRRs: pattern recognition receptors; TLR: Toll-like receptors; DCs: dendritic cells; APCs: antigen-presenting cells; MHC-I/II: major histocompatibility complex I/II; Cy7: Cyanine7; PTT: photothermal therapy; HER2: human epidermal growth factor receptor 2; ClyA: Cytolysin A; SIRPα: signal-regulatory protein alpha; SpT/SpC: SpyTag /SpyCatcher; fHbp: factor H binding protein; OVA: ovalbumin; VEGF: vascular endothelial growth factor; BFGF: basic fibroblast growth factor; PAMPs: pathogen-associated molecular patterns; EcN: E. coli strain Nissle 1917; NK cells: natural killer cells; BsAb: bispecific antibodies; siRNAs: short interfering RNAs; shRNAs: short hairpin RNAs; miRNAs: micro RNAs; RNAi: RNA interference; ROS: reactive oxygen species; MSNPs: mesoporous silica nanoparticles; IBD: inflammatory bowel disease; HA: hyaluronic acid; 5-FU: 5-Fluorouracil; Fe3O4-MnO2: Ferrosoferric oxide-Manganese dioxide; GSH: glutathione; PSs: photosensitizers; NIR: near-infrared; CAT: catalase; PTSLs: photothermal-sensitive liposomes; Mal: maleimide; 1-MT: 1-methyl-tryptophan; LLO: listeriolysin O; PLGA: poly lactic-co-glycolic acid.
Acknowledgements
This work was supported in part by grant 82172572 (LL), grant 82072760 (XYK) and grant 82372932 (FYK) from the National Natural Science Foundation of China; National Key R&D Program of China No. 2019YFC1315900 and 2019YFC1315802, and The Innovation Platform for Academicians of Hainan Province (YSPTZX202029).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Meng C, Bai C, Brown TD, Hood LE, Tian Q. Human gut microbiota and gastrointestinal cancer. Genomics Proteomics Bioinformatics. 2018;16:33-49
2. Eshkiki ZS, Agah S, Tabaeian SP, Sedaghat M, Dana F, Talebi A. et al. Neoantigens and their clinical applications in human gastrointestinal cancers. World J Surg Oncol. 2022;20:321
3. Bear AS, Vonderheide RH, O'Hara MH. Challenges and opportunities for pancreatic cancer immunotherapy. Cancer Cell. 2020;38:788-802
4. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7-30
5. Tan S, Li D, Zhu X. Cancer immunotherapy: pros, cons and beyond. Biomed Pharmacother. 2020;124:109821
6. Villageliu DN, Samuelson DR. The role of bacterial membrane vesicles in human health and disease. Front Microbiol. 2022;13:828704
7. Schwechheimer C, Kuehn MJ. Outer-membrane vesicles from gram-negative bacteria: biogenesis and functions. Nat Rev Microbiol. 2015;13:605-19
8. Amatya SB, Salmi S, Kainulainen V, Karihtala P, Reunanen J. Bacterial extracellular vesicles in gastrointestinal tract cancer: an unexplored territory. Cancers (Basel). 2021;13:5450
9. Kesty NC, Mason KM, Reedy M, Miller SE, Kuehn MJ. Enterotoxigenic escherichia coli vesicles target toxin delivery into mammalian cells. EMBO J. 2004;23:4538-49
10. Wang X, Lin S, Wang L, Cao Z, Zhang M, Zhang Y. et al. Versatility of bacterial outer membrane vesicles in regulating intestinal homeostasis. Sci Adv. 2023;9:eade5079
11. Jiang Y, Xu Y, Zheng C, Ye L, Jiang P, Malik S. et al. Acetyltransferase from akkermansia muciniphila blunts colorectal tumourigenesis by reprogramming tumour microenvironment. Gut. 2023;72:1308-18
12. Xue K, Wang L, Liu J. Bacterial outer membrane vesicles and their functionalization as vehicles for bioimaging, diagnosis and therapy. Mater Adv. 2022;3:7185-97
13. Long Q, Zheng P, Zheng X, Li W, Hua L, Yang Z. et al. Engineered bacterial membrane vesicles are promising carriers for vaccine design and tumor immunotherapy. Adv Drug Deliv Rev. 2022;186:114321
14. Zhang Y, Fang Z, Li R, Huang X, Liu Q. Design of outer membrane vesicles as cancer vaccines: a new toolkit for cancer therapy. Cancers (Basel). 2019;11:1314
15. Wang S, Guo J, Bai Y, Sun C, Wu Y, Liu Z. et al. Bacterial outer membrane vesicles as a candidate tumor vaccine platform. Front Immunol. 2022;13:987419
16. Juodeikis R, Carding SR. Outer membrane vesicles: biogenesis, functions, and issues. Microbiol Mol Biol Rev. 2022;86:e0003222
17. Lloubès R, Cascales E, Walburger A, Bouveret E, Lazdunski C, Bernadac A. et al. The tol-pal proteins of the escherichia coli cell envelope: an energized system required for outer membrane integrity? Res Microbiol. 2001;152:523-9
18. Schertzer JW, Whiteley M. A bilayer-couple model of bacterial outer membrane vesicle biogenesis. mBio. 2012;3:e00297-11
19. Liu H, Zhang H, Han Y, Hu Y, Geng Z, Su J. Bacterial extracellular vesicles-based therapeutic strategies for bone and soft tissue tumors therapy. Theranostics. 2022;12:6576-94
20. Xie J, Li Q, Haesebrouck F, Van Hoecke L, Vandenbroucke RE. The tremendous biomedical potential of bacterial extracellular vesicles. Trends Biotechnol. 2022;40:1173-94
21. O'Donoghue EJ, Krachler AM. Mechanisms of outer membrane vesicle entry into host cells. Cell Microbiol. 2016;18:1508-17
22. Kaparakis-Liaskos M, Ferrero RL. Immune modulation by bacterial outer membrane vesicles. Nat Rev Immunol. 2015;15:375-87
23. Weiner A, Mellouk N, Lopez-Montero N, Chang Y-Y, Souque C, Schmitt C. et al. Macropinosomes are key players in early shigella invasion and vacuolar escape in epithelial cells. PLoS Pathog. 2016;12:e1005602
24. Bomberger JM, MacEachran DP, Coutermarsh BA, Ye S, O'Toole GA, Stanton BA. Long-distance delivery of bacterial virulence factors by pseudomonas aeruginosa outer membrane vesicles. PLoS Pathog. 2009;5:e1000382
25. Torchilin VP. Passive and active drug targeting: drug delivery to tumors as an example. Handb Exp Pharmacol. 2010:3-53
26. Alves NJ, Turner KB, Daniele MA, Oh E, Medintz IL, Walper SA. Bacterial nanobioreactors-directing enzyme packaging into bacterial outer membrane vesicles. ACS Appl Mater Interfaces. 2015;7:24963-72
27. Alves NJ, Turner KB, Medintz IL, Walper SA. Protecting enzymatic function through directed packaging into bacterial outer membrane vesicles. Sci Rep. 2016;6:24866
28. Mehanny M, Koch M, Lehr C-M, Fuhrmann G. Streptococcal extracellular membrane vesicles are rapidly internalized by immune cells and alter their cytokine release. Front Immunol. 2020;11:80
29. Alaniz RC, Deatherage BL, Lara JC, Cookson BT. Membrane vesicles are immunogenic facsimiles of salmonella typhimurium that potently activate dendritic cells, prime b and t cell responses, and stimulate protective immunity in vivo. J Immunol. 2007;179:7692-701
30. Ismail S, Hampton MB, Keenan JI. Helicobacter pylori outer membrane vesicles modulate proliferation and interleukin-8 production by gastric epithelial cells. Infect Immun. 2003;71:5670-5
31. Söderblom T, Oxhamre C, Wai SN, Uhlén P, Aperia A, Uhlin BE. et al. Effects of the escherichia coli toxin cytolysin a on mucosal immunostimulation via epithelial ca2+ signalling and toll-like receptor 4. Cell Microbiol. 2005;7:779-88
32. Kaparakis M, Turnbull L, Carneiro L, Firth S, Coleman HA, Parkington HC. et al. Bacterial membrane vesicles deliver peptidoglycan to nod1 in epithelial cells. Cell Microbiol. 2010;12:372-85
33. Tavano R, Franzoso S, Cecchini P, Cartocci E, Oriente F, Aricò B. et al. The membrane expression of neisseria meningitidis adhesin a (nada) increases the proimmune effects of menb omvs on human macrophages, compared with nada- omvs, without further stimulating their proinflammatory activity on circulating monocytes. J Leukoc Biol. 2009;86:143-53
34. Krishnan N, Kubiatowicz LJ, Holay M, Zhou J, Fang RH, Zhang L. Bacterial membrane vesicles for vaccine applications. Adv Drug Deliv Rev. 2022;185:114294
35. Qing S, Lyu C, Zhu L, Pan C, Wang S, Li F. et al. Biomineralized bacterial outer membrane vesicles potentiate safe and efficient tumor microenvironment reprogramming for anticancer therapy. Adv Mater. 2020;32:e2002085
36. Yang Y, Liu Q, Li P, Luo H, Wang H, Kong Q. Role of bacterial minicells in cancer therapy. Chin J Biotechnol. 2019;35:998-1008
37. van de Waterbeemd B, Streefland M, van der Ley P, Zomer B, van Dijken H, Martens D. et al. Improved omv vaccine against neisseria meningitidis using genetically engineered strains and a detergent-free purification process. Vaccine. 2010;28:4810-6
38. Yarden Y, Sliwkowski MX. Untangling the erbb signalling network. Nat Rev Mol Cell Biol. 2001;2:127-37
39. Gujrati V, Kim S, Kim S-H, Min JJ, Choy HE, Kim SC. et al. Bioengineered bacterial outer membrane vesicles as cell-specific drug-delivery vehicles for cancer therapy. ACS Nano. 2014;8:1525-37
40. Li Y, Zhao R, Cheng K, Zhang K, Wang Y, Zhang Y. et al. Bacterial outer membrane vesicles presenting programmed death 1 for improved cancer immunotherapy via immune activation and checkpoint inhibition. ACS Nano. 2020 33232124
41. Feng Q, Ma X, Cheng K, Liu G, Li Y, Yue Y. et al. Engineered bacterial outer membrane vesicles as controllable two-way adaptors to activate macrophage phagocytosis for improved tumor immunotherapy. Adv Mater. 2022;34:e2206200
42. Cheng K, Zhao R, Li Y, Qi Y, Wang Y, Zhang Y. et al. Bioengineered bacteria-derived outer membrane vesicles as a versatile antigen display platform for tumor vaccination via plug-and-display technology. Nat Commun. 2021;12:2041
43. Gujrati V, Prakash J, Malekzadeh-Najafabadi J, Stiel A, Klemm U, Mettenleiter G. et al. Bioengineered bacterial vesicles as biological nano-heaters for optoacoustic imaging. Nat Commun. 2019;10:1114
44. Li Y, Zhao R, Cheng K, Zhang K, Wang Y, Zhang Y. et al. Bacterial outer membrane vesicles presenting programmed death 1 for improved cancer immunotherapy via immune activation and checkpoint inhibition. ACS Nano. 2020.
45. Rosenthal JA, Huang C-Jr, Doody AM, Leung T, Mineta K, Feng DD. et al. Mechanistic insight into the th1-biased immune response to recombinant subunit vaccines delivered by probiotic bacteria-derived outer membrane vesicles. PLoS One. 2014;9:e112802
46. Schetters STT, Jong WSP, Horrevorts SK, Kruijssen LJW, Engels S, Stolk D. et al. Outer membrane vesicles engineered to express membrane-bound antigen program dendritic cells for cross-presentation to cd8+ t cells. Acta Biomater. 2019;91:248-57
47. Huang W, Shu C, Hua L, Zhao Y, Xie H, Qi J. et al. Modified bacterial outer membrane vesicles induce autoantibodies for tumor therapy. Acta Biomater. 2020;108:300-12
48. Reimer SL, Beniac DR, Hiebert SL, Booth TF, Chong PM, Westmacott GR. et al. Comparative analysis of outer membrane vesicle isolation methods with an escherichia coli tola mutant reveals a hypervesiculating phenotype with outer-inner membrane vesicle content. Front Microbiol. 2021;12:628801
49. Arigita C, Jiskoot W, Westdijk J, van Ingen C, Hennink WE, Crommelin DJA. et al. Stability of mono- and trivalent meningococcal outer membrane vesicle vaccines. Vaccine. 2004;22:629-42
50. Kim SY, Kim MH, Kim SI, Son JH, Kim S, Lee YC. et al. The sensor kinase bfms controls production of outer membrane vesicles in acinetobacter baumannii. BMC Microbiol. 2019;19:301
51. Qiao L, Rao Y, Zhu K, Rao X, Zhou R. Engineered remolding and application of bacterial membrane vesicles. Front Microbiol. 2021;12:729369
52. McBroom AJ, Kuehn MJ. Release of outer membrane vesicles by gram-negative bacteria is a novel envelope stress response. Mol Microbiol. 2007;63:545-58
53. McBroom AJ, Johnson AP, Vemulapalli S, Kuehn MJ. Outer membrane vesicle production by escherichia coli is independent of membrane instability. J Bacteriol. 2006;188:5385-92
54. Yue Y, Xu J, Li Y, Cheng K, Feng Q, Ma X. et al. Antigen-bearing outer membrane vesicles as tumour vaccines produced in situ by ingested genetically engineered bacteria. Nat Biomed Eng. 2022;6:898-909
55. Davies C, Taylor AJ, Elmi A, Winter J, Liaw J, Grabowska AD. et al. Sodium taurocholate stimulates campylobacter jejuni outer membrane vesicle production via down-regulation of the maintenance of lipid asymmetry pathway. Front Cell Infect Microbiol. 2019;9:177
56. Wessel AK, Liew J, Kwon T, Marcotte EM, Whiteley M. Role of pseudomonas aeruginosa peptidoglycan-associated outer membrane proteins in vesicle formation. J Bacteriol. 2013;195:213-9
57. McMahon KJ, Castelli ME, Vescovi EG, Feldman MF. Biogenesis of outer membrane vesicles in serratia marcescens is thermoregulated and can be induced by activation of the rcs phosphorelay system. J Bacteriol. 2012;194:3241-9
58. Rivera J, Cordero RJB, Nakouzi AS, Frases S, Nicola A, Casadevall A. Bacillus anthracis produces membrane-derived vesicles containing biologically active toxins. Proc Natl Acad Sci U S A. 2010;107:19002-7
59. Wen ZT, Jorgensen AN, Huang X, Ellepola K, Chapman L, Wu H. et al. Multiple factors are involved in regulation of extracellular membrane vesicle biogenesis in streptococcus mutans. Mol Oral Microbiol. 2021;36:12-24
60. Rath P, Huang C, Wang T, Wang T, Li H, Prados-Rosales R. et al. Genetic regulation of vesiculogenesis and immunomodulation in mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2013;110:E4790-7
61. White DW, Elliott SR, Odean E, Bemis LT, Tischler AD. Mycobacterium tuberculosis pst/senx3-regx3 regulates membrane vesicle production independently of esx-5 activity. mBio. 2018;9:e00778-18
62. Wang X, Thompson CD, Weidenmaier C, Lee JC. Release of staphylococcus aureus extracellular vesicles and their application as a vaccine platform. Nat Commun. 2018;9:1379
63. Lee JH, Choi C-W, Lee T, Kim SI, Lee J-C, Shin J-H. Transcription factor σb plays an important role in the production of extracellular membrane-derived vesicles in listeria monocytogenes. PLoS One. 2013;8:e73196
64. Berlanda Scorza F, Colucci AM, Maggiore L, Sanzone S, Rossi O, Ferlenghi I. et al. High yield production process for shigella outer membrane particles. PLoS One. 2012;7:e35616
65. Roier S, Zingl FG, Cakar F, Durakovic S, Kohl P, Eichmann TO. et al. A novel mechanism for the biogenesis of outer membrane vesicles in gram-negative bacteria. Nat Commun. 2016;7:10515
66. Gerritzen MJH, Stangowez L, van de Waterbeemd B, Martens DE, Wijffels RH, Stork M. Continuous production of neisseria meningitidis outer membrane vesicles. Appl Microbiol Biotechnol. 2019;103:9401-10
67. Van De Waterbeemd B, Zomer G, Van Den IJssel J, Van Keulen L, Eppink MH, Van Der Ley P. et al. Cysteine depletion causes oxidative stress and triggers outer membrane vesicle release by neisseria meningitidis; implications for vaccine development. PLoS One. 2013;8:e54314
68. van de Waterbeemd B, Streefland M, van Keulen L, van den Ijssel J, de Haan A, Eppink MH. et al. Identification and optimization of critical process parameters for the production of nomv vaccine against neisseria meningitidis. Vaccine. 2012;30:3683-90
69. Gerritzen MJH, Maas RHW, van den Ijssel J, van Keulen L, Martens DE, Wijffels RH. et al. High dissolved oxygen tension triggers outer membrane vesicle formation by neisseria meningitidis. Microb Cell Fact. 2018;17:157
70. Robbana-Barnat S, Lafarge-Frayssinet C, Cohen H, Neish GA, Frayssinet C. Immunosuppressive properties of deoxynivalenol. Toxicology. 1988;48:155-66
71. van der Ley P, Steeghs L, Hamstra HJ, ten Hove J, Zomer B, van Alphen L. Modification of lipid a biosynthesis in neisseria meningitidis lpxl mutants: influence on lipopolysaccharide structure, toxicity, and adjuvant activity. Infect Immun. 2001;69:5981-90
72. Mancini F, Rossi O, Necchi F, Micoli F. OMV vaccines and the role of tlr agonists in immune response. Int J Mol Sci. 2020;21:E4416
73. Lee S-R, Kim S-H, Jeong K-J, Kim K-S, Kim Y-H, Kim S-J. et al. Multi-immunogenic outer membrane vesicles derived from an msbb-deficient salmonella enterica serovar typhimurium mutant. J Microbiol Biotechnol. 2009;19:1271-9
74. Reynolds CM, Ribeiro AA, McGrath SC, Cotter RJ, Raetz CRH, Trent MS. An outer membrane enzyme encoded by salmonella typhimurium lpxr that removes the 3'-acyloxyacyl moiety of lipid a. J Biol Chem. 2006;281:21974-87
75. Arenas J, van Dijken H, Kuipers B, Hamstra HJ, Tommassen J, van der Ley P. Coincorporation of lpxl1 and pagl mutant lipopolysaccharides into liposomes with neisseria meningitidis opacity protein: influence on endotoxic and adjuvant activity. Clin Vaccine Immunol. 2010;17:487-95
76. Lee DH, Kim S-H, Kang W, Choi YS, Lee S-H, Lee S-R. et al. Adjuvant effect of bacterial outer membrane vesicles with penta-acylated lipopolysaccharide on antigen-specific t cell priming. Vaccine. 2011;29:8293-301
77. Needham BD, Carroll SM, Giles DK, Georgiou G, Whiteley M, Trent MS. Modulating the innate immune response by combinatorial engineering of endotoxin. Proc Natl Acad Sci U S A. 2013;110:1464-9
78. Tran AX, Karbarz MJ, Wang X, Raetz CRH, McGrath SC, Cotter RJ. et al. Periplasmic cleavage and modification of the 1-phosphate group of helicobacter pylori lipid a. J Biol Chem. 2004;279:55780-91
79. González MF, Díaz P, Sandoval-Bórquez A, Herrera D, Quest AFG. Helicobacter pylori outer membrane vesicles and extracellular vesicles from helicobacter pylori-infected cells in gastric disease development. Int J Mol Sci. 2021;22:4823
80. Zhou J, Ma S, Zhang Y, He Y, Mao H, Yang J. et al. Bacterium-mimicking sequentially targeted therapeutic nanocomplexes based on o-carboxymethyl chitosan and their cooperative therapy by dual-modality light manipulation. Carbohydr Polym. 2021;264:118030
81. Aytar Çelik P, Erdogan-Gover K, Barut D, Enuh BM, Amasya G, Sengel-Türk CT. et al. Bacterial membrane vesicles as smart drug delivery and carrier systems: a new nanosystems tool for current anticancer and antimicrobial therapy. Pharmaceutics. 2023;15:1052
82. Aghajanzadeh M, Zamani M, Rajabi Kouchi F, Eixenberger J, Shirini D, Estrada D. et al. Synergic antitumor effect of photodynamic therapy and chemotherapy mediated by nano drug delivery systems. Pharmaceutics. 2022;14:322
83. Behrouzi A, Mazaheri H, Falsafi S, Tavassol ZH, Moshiri A, Siadat SD. Intestinal effect of the probiotic escherichia coli strain nissle 1917 and its omv. J Diabetes Metab Disord. 2020;19:597-604
84. Kuerban K, Gao X, Zhang H, Liu J, Dong M, Wu L. et al. Doxorubicin-loaded bacterial outer-membrane vesicles exert enhanced anti-tumor efficacy in non-small-cell lung cancer. Acta Pharm Sin B. 2020;10:1534-48
85. Aly RG, El-Enbaawy MI, Abd El-Rahman SS, Ata NS. Antineoplastic activity of salmonella typhimurium outer membrane nanovesicles. Exp Cell Res. 2021;399:112423
86. Solomon BJ, Desai J, Rosenthal M, McArthur GA, Pattison ST, Pattison SL. et al. A first-time-in-human phase i clinical trial of bispecific antibody-targeted, paclitaxel-packaged bacterial minicells. PLOS One. 2015;10:e0144559
87. Naskar A, Cho H, Lee S, Kim K-S. Biomimetic nanoparticles coated with bacterial outer membrane vesicles as a new-generation platform for biomedical applications. Pharmaceutics. 2021;13:1887
88. Brahmbhatt H, MacDiarmid JA. Bacterial minicells to the rescue: cyto-immunotherapy for the treatment of late stage cancers with minimal to no toxicity. Microb Biotechnol. 2021;15:91-4
89. Sagnella SM, Trieu J, Brahmbhatt H, MacDiarmid JA, MacMillan A, Whan RM. et al. Targeted doxorubicin-loaded bacterially derived nano-cells for the treatment of neuroblastoma. Mol Cancer Ther. 2018;17:1012-23
90. Ali MK, Liu Q, Liang K, Li P, Kong Q. Bacteria-derived minicells for cancer therapy. Cancer Lett. 2020;491:11-21
91. Ren C, Li Y, Cong Z, Li Z, Xie L, Wu S. Bioengineered bacterial outer membrane vesicles encapsulated polybia-mastoparan i fusion peptide as a promising nanoplatform for bladder cancer immune-modulatory chemotherapy. Front Immunol. 2023;14:1129771
92. Xin Y, Huang M, Guo WW, Huang Q, Zhang LZ, Jiang G. Nano-based delivery of rnai in cancer therapy. Mol Cancer. 2017;16:134
93. Mirzaei S, Gholami MH, Ang HL, Hashemi F, Zarrabi A, Zabolian A. et al. Pre-clinical and clinical applications of small interfering rnas (sirna) and co-delivery systems for pancreatic cancer therapy. Cells. 2021;10:3348
94. Jivrajani M, Nivsarkar M. Minicell-based targeted delivery of shrna to cancer cells: an experimental protocol. Methods Mol Biol. 2019;1974:111-39
95. MacDiarmid JA, Amaro-Mugridge NB, Madrid-Weiss J, Sedliarou I, Wetzel S, Kochar K. et al. Sequential treatment of drug-resistant tumors with targeted minicells containing sirna or a cytotoxic drug. Nat Biotechnol. 2009;27:643-51
96. Sagnella SM, Yang L, Stubbs GE, Boslem E, Martino-Echarri E, Smolarczyk K. et al. Cyto-immuno-therapy for cancer: a pathway elicited by tumor-targeted, cytotoxic drug-packaged bacterially derived nanocells. Cancer Cell. 2020;37:354-370.e7
97. Zanella I, König E, Tomasi M, Gagliardi A, Frattini L, Fantappiè L. et al. Proteome-minimized outer membrane vesicles from escherichia coli as a generalized vaccine platform. J Extracell Vesicles. 2021;10:e12066
98. Kumar P, Nagarajan A, Uchil PD. Electroporation. Cold Spring Harb Protoc. 2019;2019:31262965
99. Guo Q, Li X, Zhou W, Chu Y, Chen Q, Zhang Y. et al. Sequentially triggered bacterial outer membrane vesicles for macrophage metabolism modulation and tumor metastasis suppression. ACS Nano. 2021 34382768
100. Zong R, Ruan H, Liu C, Fan S, Li J. Bacteria and bacterial components as natural bio-nanocarriers for drug and gene delivery systems in cancer therapy. Pharmaceutics. 2023;15:2490
101. Volovat S-R, Ursulescu CL, Moisii LG, Volovat C, Boboc D, Scripcariu D. et al. The landscape of nanovectors for modulation in cancer immunotherapy. Pharmaceutics. 2022;14:397
102. Jahangirian H, Kalantari K, Izadiyan Z, Rafiee-Moghaddam R, Shameli K, Webster TJ. A review of small molecules and drug delivery applications using gold and iron nanoparticles. Int J Nanomedicine. 2019;14:1633-57
103. Connor DM, Broome A-M. Gold nanoparticles for the delivery of cancer therapeutics. Adv Cancer Res. 2018;139:163-84
104. Piao J-G, Wang L, Gao F, You Y-Z, Xiong Y, Yang L. Erythrocyte membrane is an alternative coating to polyethylene glycol for prolonging the circulation lifetime of gold nanocages for photothermal therapy. ACS Nano. 2014;8:10414-25
105. Naskar A, Cho H, Lee S, Kim K. Biomimetic nanoparticles coated with bacterial outer membrane vesicles as a new-generation platform for biomedical applications. Pharmaceutics. 2021;13:1887
106. Chen M-H, Liu T-Y, Chen Y-C, Chen M-H. Combining augmented radiotherapy and immunotherapy through a nano-gold and bacterial outer-membrane vesicle complex for the treatment of glioblastoma. Nanomaterials (Basel). 2021;11:1661
107. Zhang W, Liu M, Liu A, Zhai G. Advances in functionalized mesoporous silica nanoparticles for tumor targeted drug delivery and theranostics. Curr Pharm Des. 2017;23:3367-82
108. Su J, Sun H, Meng Q, Zhang P, Yin Q, Li Y. Enhanced blood suspensibility and laser-activated tumor-specific drug release of theranostic mesoporous silica nanoparticles by functionalizing with erythrocyte membranes. Theranostics. 2017;7:523-37
109. Wang Z, Shi J, Pan H, Liu M, Sang Y, Ai J. et al. Membrane-cloaked polydopamine modified mesoporous silica nanoparticles for cancer therapy. Nanotechnology. 2022;33:345101
110. Shi J, Ma Z, Pan H, Liu Y, Chu Y, Wang J. et al. Biofilm-encapsulated nano drug delivery system for the treatment of colon cancer. J Microencapsul. 2020;37:481-91
111. Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A. et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol. 2016;11:986-94
112. Liu XZ, Wen ZJ, Li YM, Sun WR, Hu XQ, Zhu JZ. et al. Bioengineered bacterial membrane vesicles with multifunctional nanoparticles as a versatile platform for cancer immunotherapy. ACS Appl Mater Interfaces. 2023;15:3744-59
113. Yan J, Wang C, Jiang X, Wei Y, Wang Q, Cui K. et al. Application of phototherapeutic-based nanoparticles in colorectal cancer. Int J Biol Sci. 2021;17:1361-81
114. Zhuang Q, Xu J, Deng D, Chao T, Li J, Zhang R. et al. Bacteria-derived membrane vesicles to advance targeted photothermal tumor ablation. Biomaterials. 2021;268:120550
115. Zhang J, Li Z, Liu L, Li L, Zhang L, Wang Y. et al. Self-assembly catalase nanocomplex conveyed by bacterial vesicles for oxygenated photodynamic therapy and tumor immunotherapy. Int J Nanomedicine. 2022;17:1971-85
116. Wang D, Liu C, You S, Zhang K, Li M, Cao Y. et al. Bacterial vesicle-cancer cell hybrid membrane-coated nanoparticles for tumor specific immune activation and photothermal therapy. ACS Appl Mater Interfaces. 2020;12:41138-47
117. Zhai Y, Ma Y, Pang B, Zhang J, Li Y, Rui Y. et al. A cascade targeting strategy based on modified bacterial vesicles for enhancing cancer immunotherapy. J Nanobiotechnology. 2021;19:434
118. Bourzac K. News feature: cancer nanomedicine, reengineered. Proc Natl Acad Sci U S A. 2016;113:12600-3
119. Li M, Li S, Zhou H, Tang X, Wu Y, Jiang W. et al. Chemotaxis-driven delivery of nano-pathogenoids for complete eradication of tumors post-phototherapy. Nat Commun. 2020;11:1126
120. Li M, Zhou H, Yang C, Wu Y, Zhou X, Liu H. et al. Bacterial outer membrane vesicles as a platform for biomedical applications: an update. J Control Release. 2020;323:253-68
121. Li Y, Li S, Jiang Z, Tan K, Meng Y, Zhang D. et al. Targeting lymph node delivery with nanovaccines for cancer immunotherapy: recent advances and future directions. J Nanobiotechnology. 2023;21:212
122. Li Y, Ma X, Yue Y, Zhang K, Cheng K, Feng Q. et al. Rapid surface display of mrna antigens by bacteria-derived outer membrane vesicles for a personalized tumor vaccine. Adv Mater. 2022;34:e2109984
123. Zhao X, Zhao R, Nie G. Nanocarriers based on bacterial membrane materials for cancer vaccine delivery. Nat Protoc. 2022;17:2240-74
124. Ma N, Chen Z, Liu G, Yue Y, Li Y, Cheng K. et al. Normalizing the immune macroenvironment via debulking surgery to strengthen tumor nanovaccine efficacy and eliminate metastasis. ACS Nano. 2023;17:437-52
125. Zhuang W-R, Wang Y, Nie W, Lei Y, Liang C, He J. et al. Bacterial outer membrane vesicle based versatile nanosystem boosts the efferocytosis blockade triggered tumor-specific immunity. Nat Commun. 2023;14:1675
126. Carvalho AL, Fonseca S, Miquel-Clopés A, Cross K, Kok K-S, Wegmann U. et al. Bioengineering commensal bacteria-derived outer membrane vesicles for delivery of biologics to the gastrointestinal and respiratory tract. J Extracell Vesicles. 2019;8:1632100
127. Shi L, Sheng J, Chen G, Zhu P, Shi C, Li B. et al. Combining il-2-based immunotherapy with commensal probiotics produces enhanced antitumor immune response and tumor clearance. J Immunother Cancer. 2020;8:e000973
128. Jiang J, Huang Y, Zeng Z, Zhao C. Harnessing engineered immune cells and bacteria as drug carriers for cancer immunotherapy. ACS Nano. 2023 36598956
129. Shalapour S, Karin M. Cruel to be kind: epithelial, microbial, and immune cell interactions in gastrointestinal cancers. Annu Rev Immunol. 2020;38:649-71
130. Allaire JM, Crowley SM, Law HT, Chang S-Y, Ko H-J, Vallance BA. The intestinal epithelium: central coordinator of mucosal immunity. Trends Immunol. 2019;40:174
131. Karaki S, Blanc C, Tran T, Galy-Fauroux I, Mougel A, Dransart E. et al. CXCR6 deficiency impairs cancer vaccine efficacy and cd8 + resident memory t-cell recruitment in head and neck and lung tumors. J Immunother Cancer. 2021;9:e001948
132. Ensign LM, Cone R, Hanes J. Oral drug delivery with polymeric nanoparticles: the gastrointestinal mucus barriers. Adv Drug Deliv Rev. 2012;64:557-70
133. Wang Y, Li Y, Bo L, Zhou E, Chen Y, Naranmandakh S. et al. Progress of linking gut microbiota and musculoskeletal health: casualty, mechanisms, and translational values. Gut Microbes. 2023;15:2263207
134. Kim OY, Park HT, Dinh NTH, Choi SJ, Lee J, Kim JH. et al. Bacterial outer membrane vesicles suppress tumor by interferon-γ-mediated antitumor response. Nat Commun. 2017;8:626
135. MacDiarmid JA, Mugridge NB, Weiss JC, Phillips L, Burn AL, Paulin RP. et al. Bacterially derived 400 nm particles for encapsulation and cancer cell targeting of chemotherapeutics. Cancer Cell. 2007;11:431-45
136. Zhou M, Tang Y, Xu W, Hao X, Li Y, Huang S. et al. Bacteria-based immunotherapy for cancer: a systematic review of preclinical studies. Front Immunol. 2023;14:1140463
137. Chen Q, Huang G, Wu W, Wang J, Hu J, Mao J. et al. A hybrid eukaryotic-prokaryotic nanoplatform with photothermal modality for enhanced antitumor vaccination. Adv Mater. 2020;32:e1908185
138. Liu Y, Niu L, Li N, Wang Y, Liu M, Su X. et al. Bacterial-mediated tumor therapy: old treatment in a new context. Adv Sci (Weinh). 2023;10:e2205641
139. Thomas SC, Madaan T, Kamble NS, Siddiqui NA, Pauletti GM, Kotagiri N. Engineered bacteria enhance immunotherapy and targeted therapy through stromal remodeling of tumors. Adv Healthc Mater. 2022;11:e2101487
140. Chen L, Qin H, Zhao R, Zhao X, Lin L, Chen Y. et al. Bacterial cytoplasmic membranes synergistically enhance the antitumor activity of autologous cancer vaccines. Sci Transl Med. 2021;13:eabc2816
141. Aly RGO, El-Enbaawy MIH, Abd El-Rahman SS, Ata NS. Antineoplastic activity of salmonella typhimurium outer membrane nanovesicles. Exp Cell Res. 2021;399:112423
142. Behzadi E, Mahmoodzadeh Hosseini H, Imani Fooladi AA. The inhibitory impacts of lactobacillus rhamnosus gg-derived extracellular vesicles on the growth of hepatic cancer cells. Microb Pathog. 2017;110:1-6
143. Zhou J, Karshalev E, Mundaca-Uribe R, Esteban-Fernández de Ávila B, Krishnan N, Xiao C. et al. Physical disruption of solid tumors by immunostimulatory microrobots enhances antitumor immunity. Adv Mater. 2021;33:2103505
144. Hua L, Yang Z, Li W, Zhang Q, Ren Z, Ye C. et al. A novel immunomodulator delivery platform based on bacterial biomimetic vesicles for enhanced antitumor immunity. Adv Mater. 2021;33:e2103923
145. Li Y, Zhang K, Wu Y, Yue Y, Cheng K, Feng Q. et al. Antigen capture and immune modulation by bacterial outer membrane vesicles as in situ vaccine for cancer immunotherapy post-photothermal therapy. Small. 2022;18:e2107461
Author contact
![]() Corresponding authors: Xiangyu Kong, Jie Gao and Fanyang Kong, Email: xiangyukong185com, gaojiehighcleacom, and kfy8195288com.
Corresponding authors: Xiangyu Kong, Jie Gao and Fanyang Kong, Email: xiangyukong185com, gaojiehighcleacom, and kfy8195288com.