Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2024; 14(2):622-639. doi:10.7150/thno.88759 This issue Cite
Research Paper
Pharmacological inhibition of MYC to mitigate chemoresistance in preclinical models of squamous cell carcinoma
Shuo Liu#, Zhen Qin#, Yaqing Mao, Ning Wang, Wenbo Zhang, Yujia Wang, Yiwen Chen, Lingfei Jia ![]() , Xin Peng
, Xin Peng ![]()
Department of Oral and Maxillofacial Surgery, Peking University School and Hospital of Stomatology, Beijing 100081, China. National Center of Stomatology & National Clinical Research Center for Oral Diseases & National Engineering Laboratory for Digital and Material Technology of Stomatology, Beijing 100081, China.
#Shuo Liu and Zhen Qin contributed equally to this work.
Received 2023-8-2; Accepted 2023-12-3; Published 2024-1-1
Abstract

Rationale: Cisplatin-based chemotherapy is the first-line treatment for late-stage head and neck squamous cell carcinoma (HNSCC). However, resistance to cisplatin has become a major obstacle for effective therapy. Cancer stem cells (CSCs) are critical for tumor initiation, growth, metastasis, and chemoresistance. How to effectively eliminate CSCs and overcome chemoresistance remains a key challenge. Herein, we confirmed that MYC plays critical roles in chemoresistance, and explored targeting MYC to overcome cisplatin resistance in preclinical models.
Methods: The roles of MYC in HNSCC cisplatin resistance and cancer stemness were tested in vitro and in vivo. The combined therapeutic efficiency of MYC targeting using the small molecule MYC inhibitor MYCi975 and cisplatin was assessed in a 4‑nitroquinoline 1-oxide-induced model and in a patient-derived xenograft model.
Results: MYC was highly-expressed in cisplatin-resistant HNSCC. Targeting MYC using MYCi975 eliminated CSCs, prevented metastasis, and overcame cisplatin resistance. MYCi975 also induced tumor cell-intrinsic immune responses, and promoted CD8+ T cell infiltration. Mechanistically, MYCi975 induced the DNA damage response and activated the cGAS-STING-IRF3 signaling pathway to increase CD8+ T cell-recruiting chemokines.
Conclusions: Our findings suggested that targeting MYC might eliminate CSCs, prevent metastasis, and activate antitumor immunity to overcome cisplatin resistance in HNSCC.
Keywords: Cisplatin resistance, Cancer stem cells, MYC, Head and neck squamous cell carcinoma, DNA damage response
Introduction
Head and neck squamous cell carcinoma (HNSCC), developing from the oral cavity, oropharynx, larynx, and hypopharynx, is a highly malignant tumor with poor prognosis, which frequently metastasizes to cervical lymph nodes [1]. Current treatment, including surgical resection, radiation therapy, chemotherapy, or their combinations, represent the classical options for managing HNSCC [2, 3]. However, the 5-year survival rate for patients with HNSCC has not increased over past decades because of lymph node metastasis, local recurrence, and the high incidence of therapeutic resistance [4-6]. Therefore, identifying the cellular and molecular mechanisms of recurrence and developing novel effective therapies is urgently required.
At present, cisplatin-based chemotherapy remains the most effective first-line agent to treat recurrent and metastatic disease [7]. Although HNSCC responds initially to chemotherapy, many patients rapidly develop chemoresistance and eventually experience recurrence. Thus, overcoming cisplatin resistance remains a critical goal for anticancer therapy and considerable efforts have been undertaken to solve this problem throughout the past three decades [8]. The complex mechanisms of cisplatin resistance involve cancer stem cells (CSCs), autophagy, epithelial-mesenchymal transition, drug efflux, and metabolic reprogramming [8-10]. CSCs are critical for tumor initiation and growth, and might be associated with metastasis and recurrence. Growing evidence suggests that CSCs are responsible for cancer therapy resistance and relapse or recurrence [11-14]. A number of studies have focused on the inhibition of regulatory pathways that are critical for the stemness and tumorigenic potential of CSCs [15-18]. Recently, using in vivo lineage tracing, we showed that CSCs have vital functions in chemotherapy resistance, recurrence, and metastasis of HNSCC [18, 19]. However, little is known about how cancer stemness is regulated molecularly and epigenetically.
In this study, we identified that MYC, which plays critical roles in tumorigenesis and therapeutic resistance [20], was highly expressed in a cisplatin-resistant patient-derived xenograft (PDX) model of HNSCC and regulates cancer stemness. Targeting MYC using a small molecule inhibitor (MYCi975) not only eliminated CSCs, prevented metastasis, and overcame cisplatin resistance, but also promoted CD8+ T cell intratumor infiltration in HNSCC treated with cisplatin. Our preclinical studies provide an important foundation to develop a new clinical trial for cisplatin-based combination therapy using MYC inhibitors.
Methods
Human HNSCC samples
The use of human HNSCC samples in this study was approved by the Ethics Committee of Peking University School and Hospital of Stomatology (PKUSSIRB-2012010). Fresh human HNSCC primary tissues were obtained from Peking University Hospital of Stomatology and subcutaneously inoculated into the flanks of 6-8 weeks old Nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice, which lack functional T cells and B cells, and show lymphopenia and hypogammaglobulinemia, but have a normal hematopoietic microenvironment.
Mice
Bmi1CreER (JAX: 010531) and RosatdTomato (JAX: 007909) mouse strains were crossbred to generate Bmi1CreER; RosatdTomato mice. K14CreER (NM-KI-190024) and Mycflox/flox (NM-CKO-210184) mouse strains were crossbred to generate K14CreER; Mycflox/flox. Bmi1CreER and RosatdTomato mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). K14CreER and Mycflox/flox were purchased from Shanghai Model Organisms Center, Inc. (Shanghai, China). The NOD-SCID mice, and BALB/c-nude female mice were purchased from Beijing Sibeifu Biotechnology Co. Ltd (Beijing, China). All the above mice were housed under specific-pathogen-free (SPF) conditions. All animal studies were performed in compliance with the regulations and the Peking University institutional animal care guidelines.
HNSCC PDX Model
Subcutaneous inoculation of human HNSCC primary tissues into the flanks of 6 - 8-week-old NOD-SCID mice was used to generate the PDX models. The patients used to generate the PDX models did not receive cisplatin treatment. Cisplatin‑resistant PDX models were established as described previously [21]. Briefly, 10 independent PDX models were given cisplatin (3 mg/kg) for 4-6 cycles. Cisplatin (Selleck, Houston, TX, USA; Cat# S1166) was dissolved in saline. We observed a spectrum of responses: Eight out of ten models showed a broad index of partial responses, ranging from 55% to 80% tumor growth inhibition, while two showed 30-40% tumor growth inhibition, without any dose-limiting toxicity, as measured by animal weights. To ensure that tumors progressing through multiple cycles of chemotherapy would develop intrinsic chemoresistance, the tumors of two cases of PDX model that showed the lowest response to cisplatin therapy were disaggregated, re-implanted into a second generation of mice, and selected again through 4-6 cycles of cisplatin therapy. Following this, we established two cisplatin‑resistant PDX models.
MYCi975 (MedMol, Shanghai, China; Cat# S89011) was dissolved in 5% dimethyl sulfoxide (DMSO) in corn oil. For the cisplatin-resistant PDX mice, two weeks after tumor implantation, the mice were divided into four groups and given: 1) control vehicle; 2) cisplatin (3 mg/kg body weight once a week); 3) MYCi975 (100 mg/kg, every 2 days); and 4) MYCi975 plus cisplatin for 6 weeks. Thereafter, the mice were sacrificed and the tumor samples were dissected and isolated immediately. The tumor volume was determined using the volume formula for an ellipsoid: 1/2 × D × d2, where D is the longer diameter and d is the shorter diameter.
Subcutaneous dorsal cell line-derived xenograft model
Cisplatin-resistant HN6-R cells (5 × 106) transfected by shMYC or shCtrl and Matrigel were injected into the subcutaneous dorsal region of BALB/c-nude mice. One week after injection, the mice were divided into difference groups, and given vehicle or cisplatin (3 mg/kg body weight once a week) for 4 weeks. Thereafter, the mice were sacrificed and the tumor samples were dissected and isolated immediately. The tumor volume was determined using the volume formula for an ellipsoid: 1/2 × D × d2, where D is the longer diameter and d is the shorter diameter.
4-nitroquinoline 1-oxide mouse model of HNSCC, treatment and histology
For the induction of HNSCC, six-week-old mice were treated with drinking water containing 50 µg/mL 4-nitroquinoline 1-oxide (4NQO) (Santa Cruz Biotechnology, Santa Cruz, CA, USA; Cat# 256815) for 16 weeks for tumor formation and lymph node metastasis and then given normal drinking water. The mice were divided randomly into different groups at 22 weeks. For lineage tracing and Myc knockout studies, mice were intraperitoneally injected with tamoxifen (9 mg per 40 g body weight; Sigma-Aldrich, St. Louis, MO, USA; Cat# T5648).
For treatment, the tumor-bearing mice were divided randomly into four groups and given: 1) control vehicle; 2) cisplatin (3 mg/kg body weight once a week); 3) MYCi975 (100 mg/kg, every 2 days); and 4) MYCi975 plus cisplatin for 4 weeks. After the mice were sacrificed, their cervical lymph nodes and tongues were harvested immediately, and the lesion surface areas were measured. For histological analysis and immunostaining, longitudinally cut tongues (dorsal/ventral) and intact lymph nodes were fixed overnight in 10% buffered formalin and embedded in paraffin. Tissue blocks were cut into 10-16 sections at 4 μm thickness and stained with hematoxylin and eosin (H&E). The SCC number was counted and areas were measured as described previously [15]. HNSCC invasiveness was scored based on the following criteria: showing signs of normal or epithelial dysplasia appearance (grade 1); distinct invasion, unclear basement membrane, and diffuse infiltration into the superficial portion of the muscle layer (grade 2); and loss of the basement membrane, extensive invasion into the deep muscle layer (grade 3). To assess lymph node metastasis, the sections of cervical lymph nodes were immunostained using anti‑pan-cytokeratin (PCK) antibodies (Santa Cruz Biotechnology, Cat# sc-8018, 1:200), which specifically detected epithelial tumor cells in lymph nodes.
To test MYCi975 and cisplatin tolerability in vivo, the major organs were collected for histopathological analysis and blood was also collected for routine blood and biochemical index analysis.
Cell culture, MYC knockdown, and drug treatment
Human HNSCC cell lines, HN6 and SCC15, were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). These cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum. Cisplatin-resistant HN6-R and SCC15-R cell lines were established using a gradient increase in the cisplatin dose and were maintained in 3 μM cisplatin, as described previously [15]. The IC50 and EC50 of the parental and resistant cells were calculated. All the cells were tested and were not contaminated with mycoplasmas.
The lentiviruses used for MYC knockdown (KD) were purchased from Integrated Biotech Solutions Co., Ltd (Shanghai, China). Briefly, the scramble control or the specific MYC KD (shMYC, a short hairpin RNA)) lentiviral plasmid were cotransfected into HEK293T cells together with two helper plasmids psPAX2 (Addgene, Watertown, MA, USA Cat# 12260) and pMD2.G (Addgene, Cat# 12259). Viral supernatants were harvested for cell infection at 72 h after transfection. Cells were infected with lentiviruses in the presence of polybrene (Sigma-Aldrich, Shanghai, China, Cat# H9268), selected with puromycin (Sigma-Aldrich, Cat# P9620), and expanded before being used for subsequent assays.
For drug administration, cisplatin was dissolved in saline and MYCi975 was dissolved in DMSO, and were added to the culture medium when the cells reached 70-80% confluence.
TUNEL experiment
To detect the apoptosis in HNSCC cells, TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) positivity was assessed using a TUNEL apoptosis detection kit (Solarbio, Beijing, China; Cat# T2196) according to the manufacturer's instructions. Images were then captured under a fluorescence microscope (Olympus, Tokyo, Japan).
Cell proliferation assay
Cell proliferation was measured in vitro using the Cell Counting Kit-8 (CCK-8) assay (Beyotime; Jiangsu, China; Cat# C0037). Briefly, 3 × 103 cells per well were added into 96-well plates with DMEM containing 10% FBS. Every 24 h, 100 μl of CCK-8 solution (1:10 dilution) was added to each well, and the cultures were incubated for 1 h at 37 °C. Color development was quantified photometrically at 450 nm using an ELx808 absorbance microplate reader (BioTeK Instruments, Winooski, VT, USA).
CSCs isolation and flow cytometry
To isolate CSCs from HNSCC PDXs, mice were euthanized and tumors were dissected and isolated. Tumors were chopped into small pieces using scalpels in petri dishes on ice, and then dissociated into a single cell suspension using a Tumor Dissociation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany; Cat# 130-095-929). Then, the single cell suspension was stained using an ALDHEFLUOR assay kit (STEMCELL Technologies, Vancouver, Canada; Cat# 01700) to label the alcohol dehydrogenase (ALDH)high populations. The single cell suspension was incubated with anti-epithelial cell adhesion molecule (EpCAM)-phycoerythrin (PE) antibodies (Miltenyi Biotec, Cat# 130-111-116, 1:100) for 30 min at room temperature and then sorted by fluorescence activated cell sorting (FACS). The results were analyzed using FlowJo software (Treestar Inc., Ashland, OR, USA).
Tumorsphere formation assay
For the tumorsphere formation assay, FACS-sorted cells were seeded in ultra-low attachment plates and cultured in serum-free DMEM/F12 (Thermo Fisher Scientific, Waltham, MA, USA; Cat# 11330-032) supplemented with 1% B27 supplement (Thermo Fisher Scientific, Cat# 17504044), 1% N2 supplement (Thermo Fisher Scientific, Cat# 17502048), antibiotics (streptomycin and penicillin, 100 mg/mL; Thermo Fisher Scientific, Cat# 15140122), human recombinant epidermal growth factor (EGF; 20 ng/mL; R&D Systems, Minneapolis, MN, USA; Cat# 236-EG-01M), and human recombinant basic fibroblast growth factor (bFGF; 10 ng/mL; R&D Systems, Cat# 233-FB-025/CF), in an incubator at 37 °C with a 5% CO2 atmosphere.
Extreme Limiting-dilution assay and the orthotopic HNSCC model
For in vivo extreme limiting-dilution transplantation in nude mice, HN6-derived ALDHhigh CSC-like cells or EpCAM+ ALDHhigh CSCs were mixed with Matrigel and subcutaneously injected into nude mice. Then, the mice were divided into two groups, and given vehicle or MYCi975 (100 mg/kg, every 2 days). The Extreme Limiting Dilution Analysis software was used to analyze the data, as described previously [22].
For the orthotopic nude mouse model of HNSCC, EpCAM+ ALDHhigh CSCs from HNSCC PDX were injected into the tongues of nude mice. One week after injection, the mice were divided into four groups and given: 1) control vehicle; 2) cisplatin (3 mg/kg body weight once a week); 3) MYCi975 (100 mg/kg, every 2 days); and 4) MYCi975 plus cisplatin for 4 weeks. After the mice were sacrificed, their cervical lymph nodes and tongues were harvested immediately. Then, the tumor volume was computed using the formula for an ellipsoid: 1/2 × D × d2, where D is the longer diameter and d is the shorter diameter.
Immunostaining
Immunohistochemical staining was performed on 4 μm-thick sections from 10% buffered formalin fixed, paraffin-embedded samples. Sections were incubated with the following primary antibodies at 4 °C overnight: anti-MYC (Cell Signaling Technology, Danvers, MA, USA; Cat# 18583, 1:100) and anti-PCK (Santa Cruz Biotechnology, Cat# sc-8018, 1:200). Then, the samples were stained with horseradish peroxidase-labelled secondary antibody (ZSGB-BIO, Beijing, China; Cat# PV-6001) for 1 h at room temperature. The signals were detected using 3,3′-Diaminobenzidine (DAB) as a chromogen (ZSGB-BIO, Cat# ZLI-9018), followed by counterstaining using hematoxylin. All images were acquired under an optical microscope (Olympus).
For immunofluorescent staining, sections were stained with the following primary antibodies: anti-CD8α (Cell Signaling Technology, Cat# 98941, 1:200), anti-PCK (Abcam, Cat# ab9377, 1:200), anti-phospho-Histone H2A.X (Cell Signaling Technology, Cat# 9718, 1:400), and anti-phosphorylated interferon regulatory factor 3 (p‑IRF3) (Cell Signaling Technology; Cat# 37829; 1:400) overnight at 4 °C. The immunocomplexes were detected and visualized using related secondary antibodies conjugated with cyanine (Cy)2 or Cy3 (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Sections were then counterstained with 4'6'‑diamidino‑2‑phenilindole (DAPI, Solarbio, Cat# C0060) and mounted with Antifade Reagents (ZSGB-BIO, Cat# ZLI-9557) for imaging and analysis.
For the quantification of MYC+, CD8+, and Tomato+ cells, we followed previously described methods [19]. At least three sections from each HNSCC lesion were immunostained and analyzed. Tumor cells (> 150) and MYC+, CD8+, and Tomato+ cells in these tumor cell areas were counted manually in each section. The percentage of MYC+, CD8+, and Tomato+ cells were calculated by dividing those cells with tumor cells and were averaged from the sections.
Quantitative real-time reverse transcription PCR (qRT-PCR)
Total RNA was prepared using the TRIzol reagent (Thermo Fisher Scientific, Cat# 15596026), and 500 ng of RNA was reverse transcribed to cDNA using a reverse transcription kit (Takara, Dalian, China; Cat# RR036A). The cDNA was then quantified using a SYBRGreen kit (Roche, Basel, Switzerland; Cat# 04913914001). GAPDH (encoding glyceraldehyde-3-phosphate dehydrogenase) was detected as the internal control. The qRT-PCR primers are listed in Table S1.
Western blot
Cells were lysed using radioimmunoprecipitation assay (RIPA) buffer (Solarbio, Cat# R0010) and added with a cocktail of protease inhibitors and phosphatase inhibitors (Huaxingbio, Chaohu, China; Cat# HX1864). Total proteins (30 μg) were separated using SDS polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred onto polyvinylidene fluoride membranes. The membranes were blocked with 5% skim milk for 1 h and then incubated with primary antibodies. The primary antibodies used in this study were: anti-MYC (Cell Signaling Technology; Cat# 18583; 1:1000), anti-B lymphoma Mo-MLV insertion region 1 homolog (BMI1) (Cell Signaling Technology; Cat# 6964; 1:1000), anti-SRY-box transcription factor 2 (SOX2) (Cell Signaling Technology; Cat# 14962; 1:1000), anti‑ALDH1 (Cell Signaling Technology; Cat# 54135; 1:1000), anti-phospho-Histone H2A.X (Cell Signaling Technology; Cat# 9718; 1:1000), anti-p-IRF3 (Cell Signaling Technology; Cat# 37829; 1:1000), anti-GAPDH (ZSGB-BIO; Cat# TA-08; 1:1000), and anti-Histone H3 (ABclonal, Wuhan, China; Cat# A2348; 1:1000). The membranes were then stained with the appropriate secondary antibodies. The signals were detected using a Clarity Western ECL kit (Thermo Fisher Scientific; Cat# 34577).
Cytosolic dsDNA Staining
Following treatment, HN6 and SCC15 cells were incubated with culture medium containing PicoGreen (a dsDNA stain, 200-fold dilution, Thermo Fisher Scientific; Cat# P11496) and MitoTracker (mitochondrial dsDNA stain, 500 nM, Thermo Fisher Scientific; Cat# M7512). At 1 h after incubation, the cells were fixed using 4% paraformaldehyde for 10 min. Cells were then washed three times with phosphate‑buffered saline (PBS) and stained with DAPI (ZSGB-BIO; Cat# ZLI-9557). Staining was imaged and assessed using a Leica SP5X laser scanning confocal microscope (Leica, Wetzlar, Germany).
Comet assays
Single cell gel electrophoresis (SCGE) comet assays were performed using an SCGE assay Kit (Enzo Life Sciences, Farmingdale, NY, USA; Cat# ADI-900-166). Following treatment, the cells were mixed with low melting point agarose at a volume ratio of 1:50, and 75 μL aliquots were loaded onto pre-warmed slides. The slides were incubated in pre-chilled lysis solution for 1 h and then in pre-chilled alkaline solution for 30 min. Electrophoresis was run at 25 V in Tris/Borate/EDTA (TBE) buffer for 15 min. Comets were stained using CYGREEN dye (Enzo Life Sciences) for 30 min and imaged. At least 100 individual cells per sample were evaluated in duplicate using the CASP Version 1.2.2 analysis tool (СASPlab, Wroclaw, Poland).
Statistical analysis
SPSS 21.0 (IBM Corp., Armonk, NY, USA) was used for the statistical analysis. All in vitro experiments were repeated at least two times, and the in vivo experiments were repeated at least once. Student's t test was used to analyze the data between two groups. The differences among multiple groups were evaluated using one-way analysis of variance (ANOVA). A χ2 test and Cochran-Armitage test were used to analyze the HNSCC invasion data. P < 0.05 was considered statistically significant.
Results
MYC is high expressed in cisplatin-resistant HNSCC models
To explore the role of CSCs in cisplatin-resistant, we established cisplatin‑resistant HN6 (HN6-R) and cisplatin-resistant SCC15 (SCC15-R) cells. The IC50 and EC50 of the resistant cells were increased than those of the parental cells (Figure 1A, 1B, 1C, 1D, 1E, 1F,1G and 1H). Compared with that of the parental cells, the proliferation of HN6-R and SCC15-R cells was similar under cisplatin (3 µM) treatment (Figure S1A and S1B). Furthermore, TUNEL assays showed that HN6-R and SCC15-R cells were resistant to cisplatin‑induced apoptosis (Figure S1C, S1D, S1E and S1F). Previous studies have successfully used ALDH activity to isolate CSCs from human HNSCC cell lines and primary tumor tissues [15, 17, 18, 23]. Using an ALDEFLUOR kit, we detected that ALDHhigh CSC‑like cells were enriched among HN6-R and SCC15-R cells (Figure 1I and 1J).
MYC is highly expressed in cisplatin-resistant HNSCC models. (A-B) CCK-8 assay analysis of the IC50 of cisplatin in parental and cisplatin-resistant HN6 cells. (C-D) CCK-8 assay analysis of the EC50 of cisplatin in parental and cisplatin-resistant HN6 cells. (E-F) CCK-8 assay analysis of the IC50 of cisplatin in parental and cisplatin-resistant SCC15 cells. (G-H) CCK-8 assay analysis of the EC50 of cisplatin in parental and cisplatin-resistant SCC15 cells. (I-J) Representative FACS plots of ALDHhigh cells in HN6 and SCC15 cells with or without cisplatin resistance. Means ± SD are shown. **P < 0.01 using an unpaired Student's t test. A specific inhibitor of ALDH, diethylaminobenzaldehyde (DEAB), was used to control for background fluorescence. (K) Schematic diagrams of acquired cisplatin resistance PDX model generation. (L-M) qRT-PCR analysis of HNSCC CSC-characteristic genes in two paired PDX models. Means ± SD are shown. *P < 0.05 and **P < 0.01 using an unpaired Student's t test. (N) Western blot analysis of MYC protein levels in HN6 and SCC15 cells with or without cisplatin resistance. GAPDH was used as the internal control.
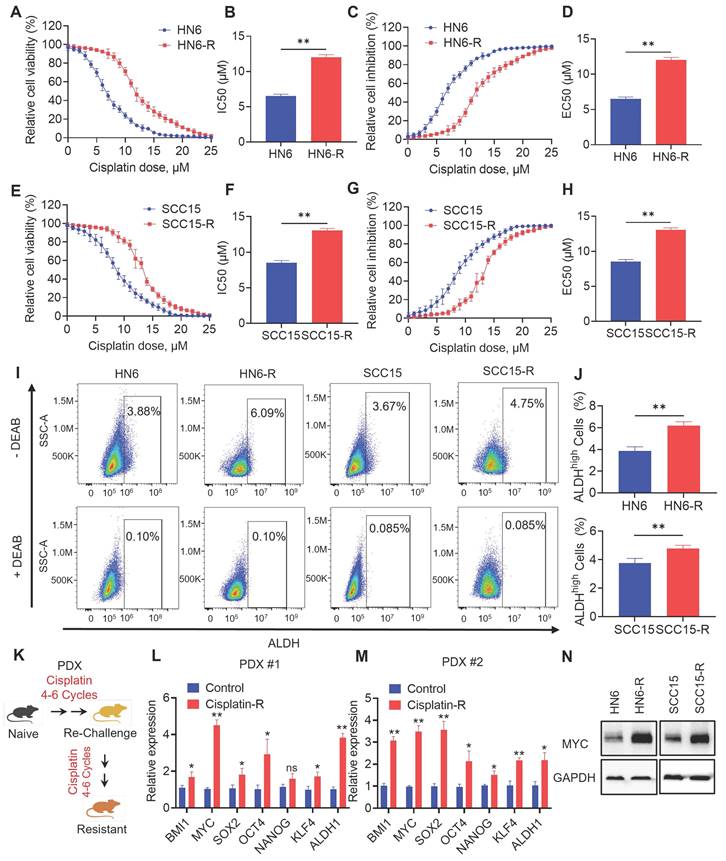
To simulate acquired cisplatin resistance in vivo, we adopted an analogous approach of repeated chemotherapy cycles in human PDX models of HNSCC to select populations of tumors that were resistant to cisplatin (Figure 1K). We established two PDX models with different genotypes for resistance to cisplatin, as previously described [21]. Then, we focused on analyzing potential cancer stemness markers of acquired cisplatin resistance. The mRNA expression of CSC-related genes, including BMI1, MYC, SOX2, OCT4, NANOG, KLF4, and ALDH1 were analyzed in two paired PDX models with or without cisplatin resistance. qRT-PCR showed that MYC was one of the mostly high expressed genes in cisplatin resistant PDX models (Figure 1L and 1M). The MYC protein level was also increased in HN6-R and SCC15-R compared with that in the controls (Figure 1N). Thus, we chose MYC for further investigation.
MYC inhibition reduces the stemness of HNSCC cells
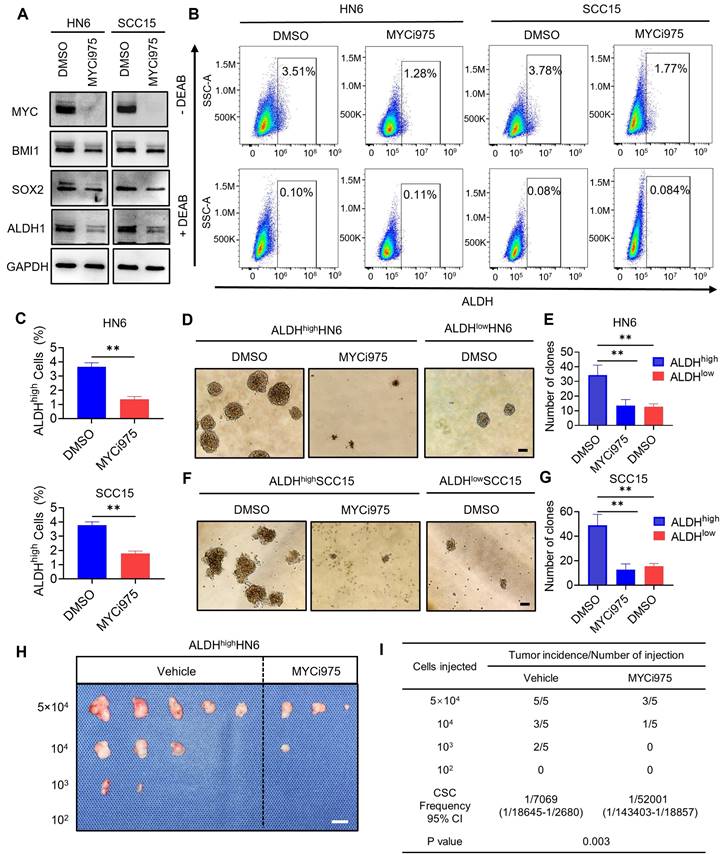
To investigate whether MYC-targeted therapy could eliminate CSCs in HNSCC, we took advantage of the specific inhibitor of MYC, MYCi975, which has been shown to effectively inhibit MYC [24-26]. Inhibition of MYC by MYCi975 in HN6 and SCC15 parental and cisplatin-resistant cells was confirmed by Western blot (Figure 2A and S2A). We also found that levels of CSC-related proteins, BMI1, SOX2, and ALDH1, were reduced under MYCi975 treatment. MYC KD using an shRNA produced similar results (Figure S3A). FACS analysis showed that MYCi975 or shMYC treatment reduced the proportion of ALDHhigh CSC-like cells among HN6 and SCC15 cells (Figure 2B, 2C, S3B, and S3C). Tumorsphere formation assays showed that MYCi975 inhibited the size and number of the ALDHhigh CSC-like cell spheres (Figure 2D, 2E, 2F and 2G). Moreover, MYC KD using an shRNA in ALDHhigh CSC‑like cells led to similar results (Figure S3D, S3E, S3F and S3G). In addition, in vivo limiting dilution tumorigenicity assays showed that MYCi975 inhibited the tumorigenic potential of ALDHhigh HN6 cells in nude mice (Figure 2H and 2I). To further confirm whether MYC controls the tumorigenic potential of CSCs, we isolated CSCs from the HNSCC PDX models using EpCAM+ALDHhigh markers, as described previously [15] (Figure S4A). In vivo limiting dilution tumorigenicity assays showed that MYCi975 inhibited the tumorigenic potential of EpCAM+ALDHhigh CSCs in nude mice (Figure S4B and S4C). These findings suggested that MYC regulates the pro-tumorigenic potential and self-renewal of CSCs.
MYCi975 suppresses cancer stemness in HNSCC cells. (A) Western blot analysis showing the inhibition of MYC, BMI1, SOX2, and ALDH1 after MYCi975 treatment. GAPDH was used as the internal control. (B-C) Representative FACS plots of ALDHhigh cells in HN6 and SCC15 cells after MYCi975 treatment. Means ± SD are shown. **P < 0.01 using an unpaired Student's t test. A specific inhibitor of ALDH, diethylaminobenzaldehyde (DEAB), was used to control for background fluorescence. (D-G) Images and numbers of tumorspheres formed by ALDHhigh and ALDHlow HN6 and SCC15 cells, with or without MYCi975 treatment. Scale bar: 100 μm. Means ± SD are shown. **P < 0.01 using an unpaired Student's t test. (H-I) Extreme limiting dilution analysis (ELDA) of ALDHhigh CSC‑like cells from HN6 cells treated with or without MYCi975 in vivo (n = 5). The frequency of allograft formation is displayed for each cell dose injected. ELDA software was used to analyze the data. Scale bar: 1 cm.
MYC inhibition suppresses the tumorigenic potential and metastasis of CSCs, thereby overcoming cisplatin resistance of HNSCC
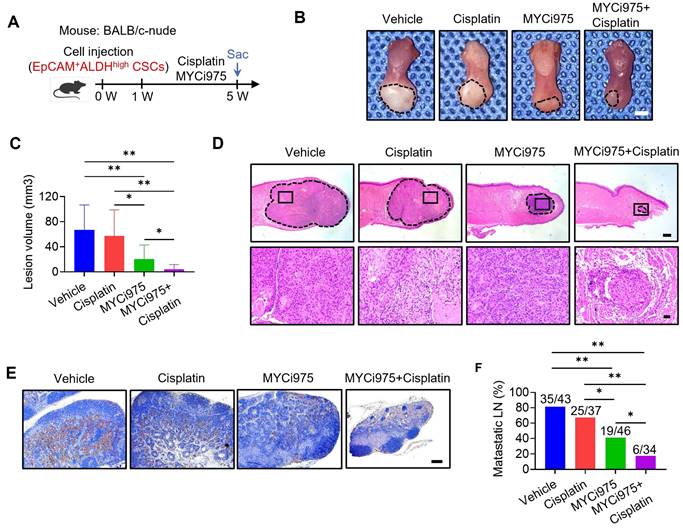
HNSCC tumor cells, most likely CSCs, frequently metastasize to, and grow in, cervical lymph nodes. To explore whether MYC inhibition suppresses CSCs and their progeny's ability to develop HNSCC and metastasize, we isolated EpCAM+ALDHhigh CSCs from the HNSCC PDX model and inoculated them into mice tongues to established an orthotopic HNSCC model. Mice were treated with control vehicle, cisplatin, MYCi975, or MYCi975 plus cisplatin for 4 weeks (Figure 3A). The reduction in MYC level induced by MYCi975 in tumors was confirmed by immunostaining (Figure S5A and S5B) and Western blot (Figure S5C). Treatment with MYCi975 alone reduced the orthotopic tumor growth, whereas cisplatin did not show inhibition compared with the control vehicle group. The addition of MYCi975 to cisplatin further enhanced the inhibitory effects compared with MYCi975 alone (Figure 3B, 3C and 3D). Cervical lymph node metastasis is a key prognostic factor for patients with HNSCC. To accurately detect whether the treatment inhibited HNSCC lymph node metastasis, the cervical lymph nodes of mice were immunostained with anti-PCK antibodies, which revealed that MYCi975 alone, or MYCi975 plus cisplatin, significantly reduced lymph node metastasis, whereas cisplatin alone could not suppress lymph node metastasis compared with the control vehicle group (Figure 3E and 3F). These findings suggested that CSCs were resistant to cisplatin, and MYC inhibition suppressed CSCs and lymph node metastasis of HNSCC.
MYCi975 suppresses the tumorigenic potential and metastasis of CSCs, and overcomes cisplatin resistance of HNSCC. (A) Schematic diagram of the time points of MYCi975 and cisplatin treatment and sacrifice (Sac) of mice injected with EpCAM+ALDHhigh CSCs from HNSCC PDX tissue. (B) Representative image of nude mice orthotopic tumors in the tongue from the mice injected with EpCAM+ALDHhigh CSCs in the different treatment groups. The lesion areas are circled using a white dotted line. Scale bar, 2 mm. (C) Quantification of the tumor volume of nude mice in the different treatment groups. The values are means ± SDs from the pool of two independent experiments. n = 10. *P < 0.05 and **P < 0.01 using one-way ANOVA. (D) Representative H&E staining of nude mice orthotopic tumors. Scale bar, 500 μm. Enlarged images are shown in the lower panels. Scale bar, 50 μm. (E) Immunostaining of metastatic cells in cervical lymph nodes using anti-PCK antibodies. Scale bar, 200 μm. (F) Percentage of metastatic lymph nodes from mice. The number of metastatic lymph nodes in each group is indicated in the figure. *P < 0.05 and **P < 0.01 using a χ2 test.
To further confirm these results, the cisplatin-resistant PDX model was treated with control vehicle, cisplatin, MYCi975, or MYCi975 plus cisplatin for 4 weeks. The reduction in the MYC level induced by MYCi975 in tumors was confirmed by Western blot (Figure 4A). The cisplatin-resistant PDX model showed no response to cisplatin treatment, whereas MYCi975 alone or MYCi975 plus cisplatin significantly inhibited tumor growth in terms of volume and weight (Figure 4B, 4C and 4D), suggesting that MYC inhibition overcame cisplatin resistance of HNSCC in vivo.
MYCi975 overcomes cisplatin resistance of HNSCC. (A) Western blot showing the reduction in MYC levels in PDX-R tumor tissues after MYCi975 treatment. GAPDH was used as the internal control. (B) Representative image of tumor samples harvested from cisplatin-resistant PDX mice. (C) Tumor volume growth curve of cisplatin-resistant PDX mice. *P < 0.05 and **P < 0.01 using one-way ANOVA. (D) Tumor weights of cisplatin-resistant PDX mice after 6 weeks of treatment. *P < 0.05 and **P < 0.01 using one-way ANOVA. (E) Representative image of tumor samples harvested from nude mice with injection of HN6-R cells. (F) Tumor volume growth curve of subcutaneous tumor models in nude mice with injection of HN6-R cells. *P < 0.05 and **P < 0.01 using one-way ANOVA. (G) Tumor weights of subcutaneous tumor models in nude mice with injection of HN6-R cells after 4 weeks of treatment. *P < 0.05 and **P < 0.01 using one-way ANOVA.
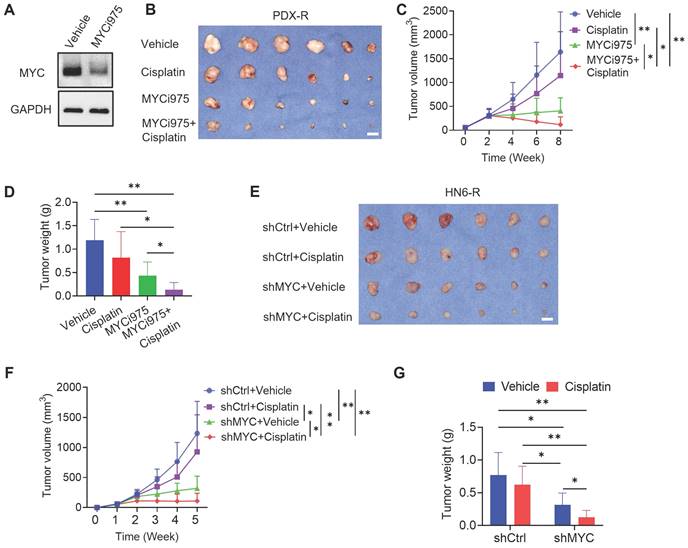
Meanwhile, to investigate whether MYC KD could overcome cisplatin resistance of HNSCC, we used shMYC to knockdown MYC in HN6-R cells. HN6-R cells transfected by shMYC or shCtrl were injected into the subcutaneous dorsal region of nude mice. One week after injection, the mice were divided in to difference groups. Mice injected with HN6-R cells transfected by shCtrl showed no response to cisplatin. shMYC-mediated KD of MYC significantly reduced the weight and volume of HN6-R-derived tumors in nude mice compared with that achieved by shCtrl and overcome cisplatin resistance (Figure 4E, 4F and 4G).
MYC inhibition plus cisplatin eliminates CSCs in vivo, potently inhibiting HNSCC
Previously, we established a 4NQO-induced HNSCC Bmi1CreER; RosatdTomato mouse model, which allows us to perform the lineage tracing of CSCs in an in vivo unperturbed environment using tamoxifen-induced Cre-mediated recombination [19]. To explore whether targeting MYC could suppress CSCs in vivo and augmented cisplatin therapy, we treated Bmi1CreER; RosatdTomato mice with 4NQO in their drinking water for 16 weeks and then replaced it with normal drinking water.
At 22 weeks from the initial 4NQO treatment, tumor-bearing mice were treated with cisplatin, MYCi975, MYCi975 plus cisplatin, or control vehicle. A single dose of tamoxifen was administered 1 day before sacrificing the mice to label Tomato+ BMI1+ CSCs (Figure 5A). The reduction in MYC levels by MYCi975 in tumors was confirmed using immunostaining (Figure S6A and S6B). MYCi975 plus cisplatin significantly reduced more lesion surface areas compared with cisplatin or MYCi975 alone (Figure 5B and 5C). Histological analysis indicated that MYCi975 plus cisplatin significantly reduced HNSCC numbers, areas, and invasiveness compared with MYCi975 or cisplatin alone (Figure 5D, 5E, 5F and 5G). Moreover, the metastatic status of isolated cervical lymph nodes was compared between the groups. The results showed that MYCi975 plus cisplatin effectively eliminated the majority of lymph node metastasis of HNSCC, as determined by anti-PCK immunostaining (Figure 5H and 5I).
MYCi975 plus cisplatin eliminates CSCs in vivo, potently inhibits HNSCC. (A) Schematic diagrams showing the treatment and lineage tracing of primary HNSCC in Bmi1CreER; RosatdTomato mice. Tamoxifen (Tam) was administered 1 day before sacrificing (Sac) the mice to label BMI1+ CSCs. (B) Representative image of tongue visible lesions in the different treatment groups. Black dashed lines denote the lesion areas. Scale bar, 2 mm. (C) Quantification of the HNSCC lesion area from mice treated as indicated. The values are means ± SDs from the pool of two independent experiments. n = 13. (D) Representative H&E staining of HNSCC from mice treated as indicated. Scale bar, 200 μm. Enlarged images are shown in the lower panels. Scale bar, 40 μm. (E-F) Quantification of HNSCC tumor number and area from mice treated as indicated. The values are means ± SDs from the pool of two independent experiments. n = 13. *P < 0.05 and **P < 0.01 using one-way ANOVA. (G) Quantification of HNSCC invasion grades from mice treated as indicated. The data were pooled from two independent experiments. n = 13. *P < 0.05 and **P < 0.01 using the Cochran-Armitage test. (H) Immunostaining of metastatic cells in cervical lymph nodes using anti-PCK antibodies. Scale bar, 200 μm. (I) Percentage of metastatic lymph nodes from mice treated as indicated. The number of metastatic lymph nodes in each group is indicated in the figure. Data were pooled from two independent experiments. *P < 0.05 and **P < 0.01 using a χ2 test. (J) Representative images of Tomato+ BMI1+ CSCs in HNSCC from mice treated as indicated. The white dashed lines denote the tumor-stromal junction. Scale bar, 20 μm. (K) Quantification of the percentage of Tomato+ cells in HNSCC from mice treated as indicated. The values are means ± SDs from the pool of two independent experiments. n = 13. *P < 0.05 and **P < 0.01 using one-way ANOVA. (L) Immunostaining images for CD8+ T cells from mice treated as indicated. Scale bar, 10 μm. (M) Quantification of CD8+ T cells in HNSCC from mice treated as indicated. The values are means ± SDs from the pool of two independent experiments. n = 13. *P < 0.05 and **P < 0.01 using one-way ANOVA.
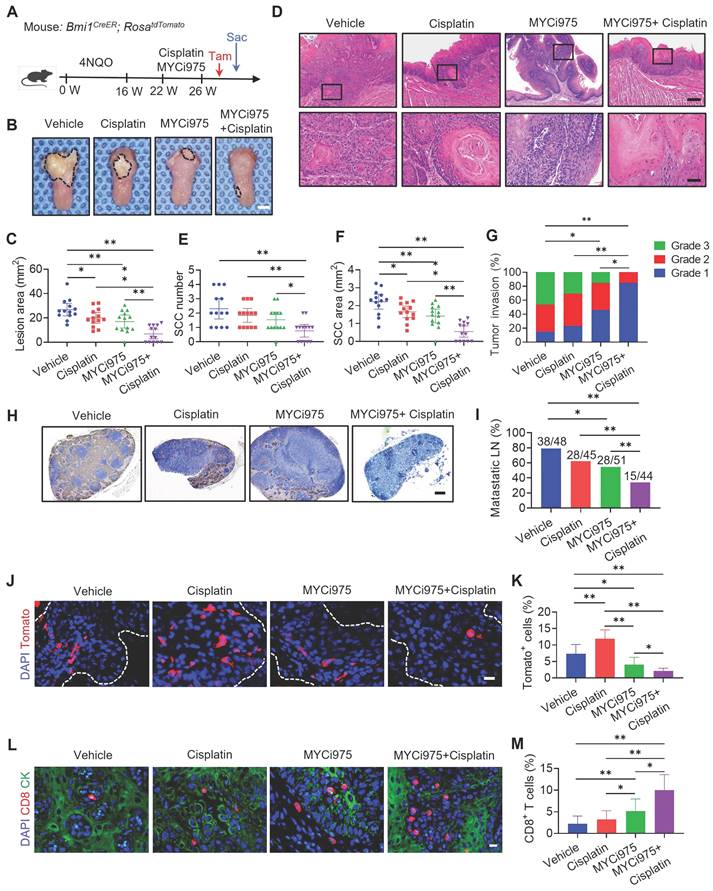
MYCi975 plus cisplatin efficiently inhibited HNSCC; therefore, we used in vivo labeling of BMI1+ CSCs to determine whether the combination could efficiently eliminate BMI1+ CSCs, which play a critical role in HNSCC chemoresistance and relapse [15]. Consistent with previous studies [15, 19], cisplatin significantly enriched BMI1+ CSCs in HNSCC. In sharp contrast to cisplatin, in vivo labeling showed that MYCi975 reduced BMI1+ CSCs. MYCi975 plus cisplatin further enhanced CSC elimination compared with MYCi975 alone (Figure 5J and 5K).
We then investigated the effect of the combination treatment on antitumor T cell immunity. Immunostaining showed that MYCi975 significantly increased CD8+ T cell infiltration, which was further significantly increased by MYCi975 plus cisplatin treatment (Figure 5L and 5M).
To determine whether secondary cisplatin resistance in 4NQO-induced HNSCC responds to MYCi975, we set up a new experiment to establish 4NQO-induced HNSCC with secondary cisplatin resistance as previously described [15, 19]. Administration of cisplatin for 4 weeks resulted in tumor stasis. Then, we extended our observation for additional 4 weeks. The results showed that cisplatin treatment discontinuation led to re-initiation of HNSCC growth. Next, mice with recurrent HNSCC were treated with cisplatin, MYCi975 or control vehicle for another 4 weeks (Figure S7A). Treatment with MYCi975 reduced the lesion surface areas, whereas cisplatin did not show inhibition compared with the control vehicle in recurrent HNSCC (Figure S7B and S7C). Histological analysis found that MYCi975 significantly reduced HNSCC numbers, areas, and invasiveness, while no such inhibitory effect was observed under cisplatin treatment (Figure S7D, S7E, S7F and S7G). Furthermore, immunostaining with anti-PCK antibodies revealed that MYCi975 inhibited lymph node metastasis effectively rather than cisplatin (Figure S7H and S7I). In vivo labeling of BMI1+ CSCs revealed that MYCi975 efficiently eliminated BMI1+ CSCs in recurrent HNSCC, while BMI1+ CSCs remained in recurrent HNSCC treated with cisplatin (Figure S7J and S7K). These results demonstrated that recurrent HNSCC after cisplatin therapy was resistant to further cisplatin treatment, while MYCi975 had a significant impact on recurrent and cisplatin secondary resistance HNSCC.
Toxicology studies were performed using C57BL/6J mice treated with MYCi975 or cisplatin. Histopathological analysis revealed that no tissue damage occurred in major organs (including the heart, liver, lung, and kidney) after MYCi975 or cisplatin administration (Figure S6C). In addition, the evaluation of routine blood and blood biochemical indicators also suggested that MYCi975 or cisplatin were well tolerated by the mice (Figures S6D and S6E).
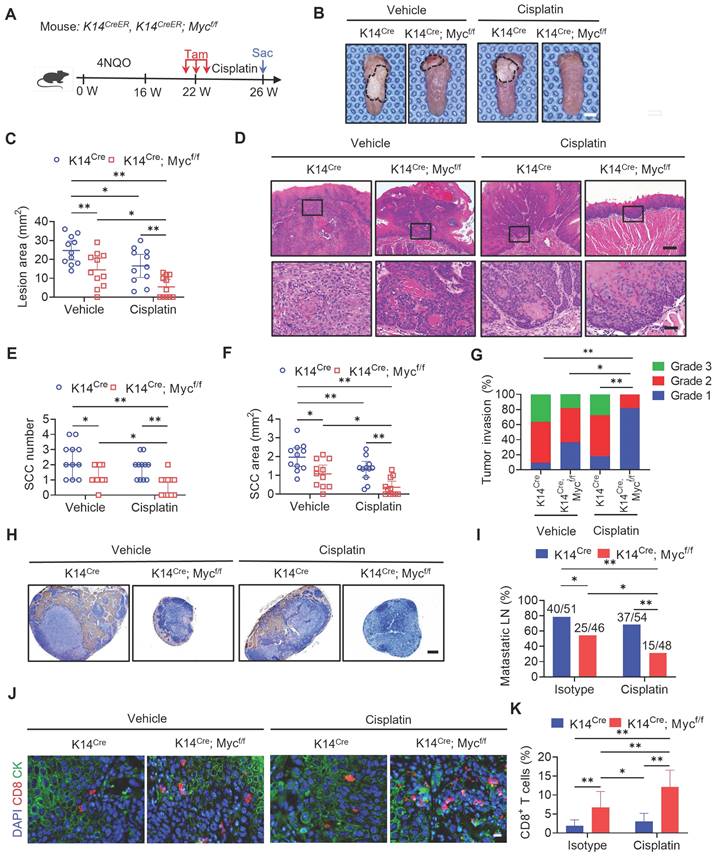
MYC knockout with cisplatin suppress HNSCC growth and metastasis
To further investigate whether targeting tumor cell-intrinsic MYC augmented cisplatin therapy, we crossed Mycflox/flox (Mycf/f) mice with keratin 14-Cre/ERT2 mice (K14CreER) to generate K14CreER; Mycflox/flox mice, in which epithelial Myc can be inducibly deleted by tamoxifen treatment. Three successive applications of tamoxifen were applied to both K14CreER; Mycflox/flox and the control K14CreER mice at 22 weeks after the initial 4NQO treatment (Figure 6A). Myc knockout (Myc KO) in mouse HNSCC was confirmed by immunostaining (Figure S8A and S8B). Treatment with cisplatin or Myc KO alone reduced the lesion areas; whereas, the addition of cisplatin to Myc KO further enhanced the inhibitory effects on the lesion areas compared with either single treatment (Figure 6B and 6C). Histological analysis revealed that Myc KO or cisplatin alone could reduce the numbers, areas, and invasive grades of HNSCC compared with those in the control vehicle group. Myc KO plus cisplatin significantly inhibited the numbers, areas, and invasive grades of HNSCC compared with Myc KO or cisplatin alone (Figure 6D, 6E, 6F and 6G). Moreover, Myc KO plus cisplatin also exhibited superior inhibitory effects on lymph node metastasis compared with Myc KO alone (Figure 6H and 6I). Immunostaining revealed that Myc KO alone, but not cisplatin, could induce CD8+ T cell infiltration in HNSCC, which was further increased in HNSCC treated with Myc KO plus cisplatin (Figure 6J and 6K).
Epithelial Deletion of MYC Collaborates with Cisplatin to Suppress HNSCC Growth and Metastasis. (A) Experimental design for MYC knockout in tumor cells and Cisplatin treatment in vivo. Three administrations of Tam were given to tumor-bearing mice. Mice were randomly divided into four experimental groups (n = 11 per group from 2 independent experiments): K14Cre with control vehicle, K14Cre with cisplatin, K14Cre; Mycf/f with control vehicle, and K14Cre; Mycf/f with cisplatin. (B) Representative image of tongue visible lesions. Black dashed lines denote the lesion areas. Scale bar, 2 mm. (C) Quantification of the HNSCC lesion area from mice treated as indicated. The values are means ± SDs from the pool of two independent experiments. n = 11. (D) Representative H&E staining of HNSCC from mice treated as indicated. Scale bar, 200 μm. Enlarged images are shown in the lower panels. Scale bar, 40 μm. (E-F) Quantification of HNSCC tumor number and area from mice treated as indicated. The values are means ± SDs from the pool of two independent experiments. n = 11. *P < 0.05 and **P < 0.01 using one-way ANOVA. (G) Quantification of HNSCC invasion grades from mice treated as indicated. The data were pooled from two independent experiments. n = 11. *P < 0.05 and **P < 0.01 using the Cochran-Armitage test. (H) Immunostaining of metastatic cells in cervical lymph nodes using anti-PCK antibodies. Scale bar, 200 μm. (I) Percentage of metastatic lymph nodes from mice treated as indicated. The number of metastatic lymph nodes in each group is indicated in the figure. Data were pooled from two independent experiments. *P < 0.05 and **P < 0.01 using a χ2 test. (J) Immunostaining images for CD8+ T cells from mice treated as indicated. Scale bar, 10 μm. (K) Quantification of CD8+ T cells in HNSCC from mice treated as indicated. The values are means ± SDs from the pool of two independent experiments. n = 11. *P < 0.05 and **P < 0.01 using one-way ANOVA.
MYC inhibition plus cisplatin facilitates cellular DNA damage and increases the tumor cell-intrinsic immune response
Previous studies showed that MYC is associated with the DNA damage response and repair regulation [27-29]. Our recent study showed that MYCi975 induced the DNA damage response [26]. We hypothesized that MYCi975 plus cisplatin would enhance the DNA damage response. To test this hypothesis, we used Western blot and immunostaining to confirm that the level of p-H2A.X, a specific marker for DNA damage, was significantly increased in cells treated with MYCi975 plus cisplatin compared with that in cells treated with cisplatin or MYCi975 alone (Figure 7A, 7B, 7C and 7D). Furthermore, p-H2A.X was also increased in MYCi975-treated 4NQO induced‑HNSCC (Figure S9A and S9B). Comet assays showed that MYCi975 plus cisplatin treatment significantly increased the comet tail moment compared with that obtained using cisplatin or MYCi975 alone in HN6 and SCC15 cells (Figure 7E, 7F, S9C and S9D), which confirmed that damaged cellular DNA fragments were separated from intact DNA. DNA damage in the nucleus could bring about the cytosolic accumulation of DNA fragments. We stained live HNSCC cells with PicoGreen, a dsDNA-specific vital dye. It was reported that PicoGreen also stains mitochondrial DNA [30]; therefore, mitochondrial DNA was visualized using MitoTracker staining to exclude its interference during the quantification of cytosolic dsDNA. Upon cisplatin or MYCi975 treatment, multiple PicoGreen staining areas were observed in the cytoplasm of HN6 and SCC15 cells, which did not overlap with the MitoTracker staining, indicating that MYCi975 induced the accumulation of cytosolic dsDNA. In addition, MYCi975 plus cisplatin enhanced the accumulation of cytosolic dsDNA (Figures 7G, 7H, S9E and S9F).
MYCi975 plus cisplatin induces DNA damage and activates the cGAS‑STING-IRF3 signaling pathway. (A) Western blot analysis of p-H2A.X in HN6 and SCC15 cell under different conditions as indicated. H3 was used as the internal control. (B-D) Immunostaining and quantification of p-H2A.X (red) in HN6 and SCC15 cells under different conditions as indicated. Nuclei were stained using DAPI (blue). Means ± SD are shown from three independent experiments. Scale bar, 50 μm. **P < 0.01 using one-way ANOVA. (E-F) Representative images and quantification of DNA Comet assays in HN6 cells treated under different conditions as indicated. More than 100 cells were analyzed per group. Means ± SD are shown. Scale bar, 100 μm. ** P < 0.01 using one‑way ANOVA. (G-H) Confocal microscopy images showing cytosolic DNA accumulation and quantification in HN6 cells under different conditions as indicated. Double‑stranded DNA (dsDNA) was stained using PicoGreen (green). Mitochondria and nuclei were stained using MitoTracker (Red) and DAPI (blue), respectively. The white arrows indicate cytosolic dsDNA. Scale bar, 10 μm. More than 100 cells were analyzed per group. Means ± SD are shown. ** P < 0.01 using one-way ANOVA. (I) Western blot analysis of the phosphorylation of IRF3 (p-IRF3) in HN6 and SCC15 cells under different conditions as indicated. GAPDH was used as the internal control. (J‑L) Immunostaining and quantification of p-IRF3 (green) in HN6 and SCC15 cells under different conditions as indicated. Nuclei are stained using DAPI (blue). Means ± SD are shown from three independent experiments. Scale bar, 50 μm. *P < 0.05 and ** P < 0.01 using one-way ANOVA.
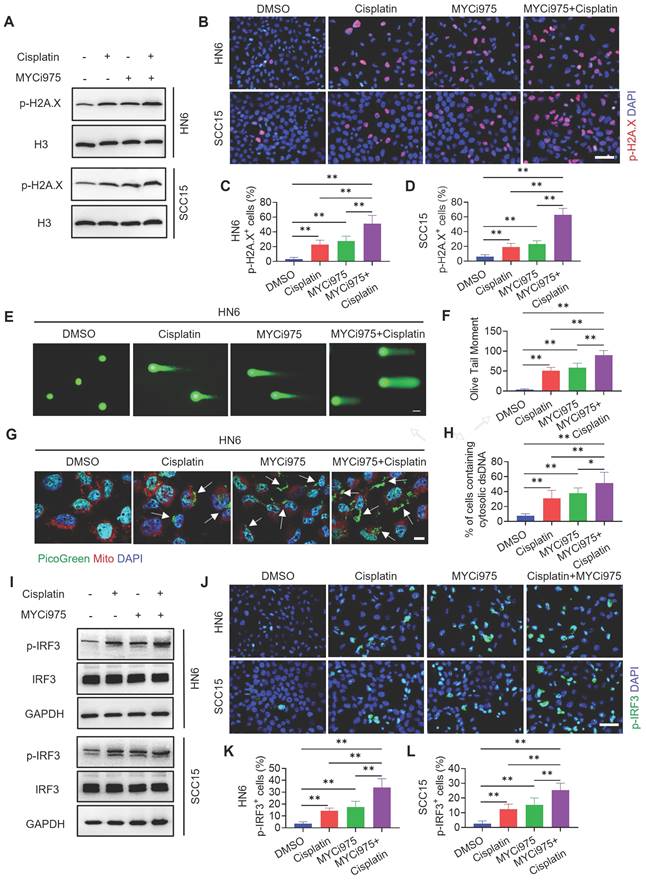
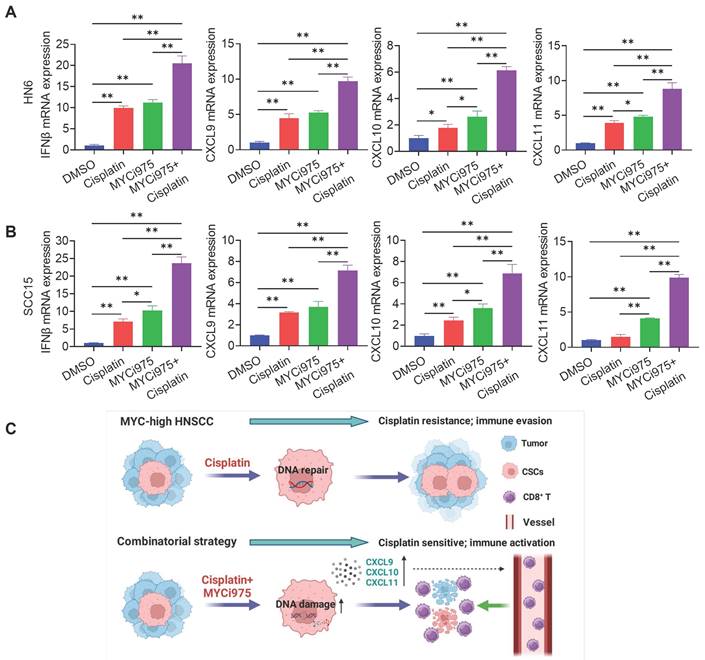
DNA damage can activate a tumor immune response [19, 31-33]. The accumulation of cytosolic DNA can activate the cyclic guanosine monophosphate (GMP)-AMP synthase-stimulator of interferon (IFN) gene (cGAS-STING) signaling axis by the sequential phosphorylation of IRF3. Then, p-IRF3 dimerizes and translocates to the nucleus to trigger the expression of IFN and IFN-regulated chemokines [34], including C-X-C motif chemokine ligand (CXCL)9, CXCL10, and CXCL11[35]. Western blot (Figure 7I) and immunostaining (Figure 7J, 7K and 7L) analysis showed that MYCi975 plus cisplatin enhanced the levels of p-IRF3 in HN6 and SCC15 cells. Consequently, MYCi975 plus cisplatin increased the mRNA expression levels of IFNB, CXCL9, CXCL10, and CXCL11 in HN6 and SCC15 cells (Figure 8A and 8B). We also observed that the p-IRF3 level was significantly increased in 4NQO induced-HNSCC upon MYCi975 plus cisplatin treatment, confirming that MYCi975 plus cisplatin enhanced the activation the cGAS-STING-IRF3 pathway in vivo. (Figures S9G and S9H).
MYCi975 plus cisplatin induces the expression of CD8+ T cell‑attracting chemokines in HNSCC cells. (A) The mRNA expression levels of IFNB, CXCL9, CXCL10, and CXCL11 in HN6 cells induced under different conditions as indicated. Means ± SD are shown. *P < 0.05 and **P < 0.01 using one-way ANOVA. (B) The mRNA expression levels of IFNB, CXCL9, CXCL10, and CXCL11 in SCC15 cells induced under different conditions as indicated. Means ± SD are shown. *P < 0.05 and **P < 0.01 using one-way ANOVA. (C) A diagram of the mechanism by which MYCi975 plus cisplatin eliminate CSCs and activate antitumor immunity to overcome cisplatin resistance.
Discussion
CSCs have been demonstrated to play a critical role in tumor development, progression, and metastasis [19, 36]. Traditional chemotherapies, such as cisplatin, are effective at abolishing actively proliferating tumor cells, but are usually not effective to eradicate CSCs, which leads to CSC enrichment and tumor recurrence [15, 16, 36]. In this study, we demonstrated that MYC inhibition could overcome cisplatin resistance in HNSCC. The combination treatment of MYCi975 plus cisplatin not only potently inhibited HNSCC invasive growth, but also significantly reduced lymph node metastasis. Mechanistically, while MYC inhibition destroyed CSCs, it also augmented the ability of cisplatin to kill non-stem tumor cells in HNSCC by enhancing the DNA damage response, which activated antitumor immunity by recruiting CD8+ T cells through chemokines (Figures 8C). Importantly, we showed that MYC plays a critical role in CSC tumorigenic potential and the survival of HNSCC, highlighting that MYC might be a new therapeutic target for the effective elimination of human HNSCC CSCs.
Cisplatin remains the most effective first-line agent for recurrent and metastatic HNSCC [7]. Although cisplatin improves important organ preservation and enhances the quality of life of patients with HNSCC [37], patients rapidly develop chemoresistance and are able to regrow and regenerate tumors locally and/or at distant sites. The overall survival of patients with head and neck cancer has not improved significantly for the last 40 years [1], despite the extensive use of platinum-based therapies for the last three decades. Based on the CSC hypothesis, CSCs might be associated with tumor development, metastasis, recurrence, and chemotherapy resistance because of their self-renewing capacity [38]. Understanding the epigenetic mechanisms that regulate the cancer stemness will help to develop new targeting strategies to eliminate the CSCs.
Our results showed that MYC was highly expressed in cisplatin-resistant PDX models. Growing evidence indicates that MYC regulates tumorigenesis through a variety of mechanisms, and is functionally involved in up to 70% of all human cancers [20]. Multiple studies demonstrated that MYC plays an important role in tumorigenesis, and is regarded as an attractive target for cancer therapy [39-45]. Recently, we discovered that MYC was associated with lymph node metastasis and poor prognosis in HNSCC [26]. In the present study, in vitro data showed that MYC inhibition suppressed tumorsphere formation and the tumorigenic potential of CSCs in HNSCC, indicating that MYC plays a critical role in CSC tumorigenic potential. Furthermore, our orthotopic tumor model suggested that MYCi975 could effectively inhibit CSCs and their progeny's ability to develop HNSCC in vivo. MYCi975 not only inhibited MYC, but also disrupted well-documented CSC-related proteins, BMI1, SOX2, and ALDH1. Previous studies identified that BMI1+ CSCs were resistant to cisplatin treatment in HNSCC [15, 19, 46], and SOX2 is another important transcription factor regulating CSC properties [47]. Using in vivo lineage tracing, we identified that cisplatin enriched BMI1+ CSCs, while MYCi975 could effectively eliminate cisplatin‑enriched CSCs. These findings indicated that targeting MYC is a good strategy to suppresses cancer stemness and MYCi975 might be an effective therapeutic approach to overcome drug resistance in cancer therapy.
In our study, other CSC-associated genes also raise in the cisplatin-resistant PDX models. Our previous study showed that the BMI1 inhibitor could effectively inhibit HNSCC growth, and prevent metastasis and relapse [19]. However, we also observed that MYC was elevated after BMI1 inhibition in another study. Thus, the use of BMI1 inhibitors is not appropriate for the treatment of cisplatin resistance HNSCC with MYC elevation. For other CSC-associated proteins, such as SOX2, OCT4, and KLF4, there are still no appropriate drugs to use. For NANOG and ALDH1 inhibitors, such as Amcasertib and Disulfiram [48, 49], our preliminary data showed that their anti-tumor effect in HNSCC was not as good as MYCi975.
HNSCC frequently metastasizes to cervical lymph nodes, and the poor prognosis of patients with HNSCC is closely associated with lymph node metastasis [50, 51]. A previous study suggested that CSCs might be associated with tumor metastasis [15]. In this study, we demonstrated that MYCi975 effectively inhibited tumor growth and metastasis by eliminating CSCs. Although cisplatin could reduce the tumor volume, it could not eliminate CSCs and suppress metastasis [52]. Using orthotopic tumor and 4NQO-induced mouse models, we identified that cisplatin alone could not suppress lymph node metastasis of HNSCC, but MYCi975 effectively suppressed lymph node metastasis and this suppression was enhanced using the combination therapy.
Furthermore, we found that MYCi975 could overcome cisplatin resistance and demonstrated that MYCi975 combined with cisplatin potently inhibited HNSCC. A previous study provided evidence that cisplatin treatment mainly killed proliferating cells, rather than CSCs [15]. Although the use of the MYC inhibitor impaired HNSCC growth and metastasis, it was not as effective as the combination therapy. We speculated that there are several potential reasons. Although MYCi975 could ablate CSCs with high MYC expression, the tumor cells with low MYC expression were able to proliferate and sustain the tumor bulk until they were worn out. It was also possible that non-stem tumor cells might have evolved and dedifferentiated into CSCs in the tumor microenvironment. Targeting MYC might promote tumor cells to dedifferentiate into other different CSCs. In fact, a previous study suggested that cytokines or growth factors could activate oncogenic signaling pathways that might be able to reprogram or dedifferentiate non-stem tumor cells into CSCs [53].
Immune surveillance is critical to prevent tumor development and progression. HNSCC tumor cells, most likely CSCs, likely have some unique mechanisms to evade immune surveillance [54]. Recently, we showed that MYC inhibition leads to cellular apoptosis and the DNA damage response [26]. In this study, while MYCi975 suppressed CSCs in HNSCC, it also activated antitumor immunity by recruiting CD8+ T cells through the cGAS-STING signaling pathway. Moreover, we demonstrated that the combination therapy enriched this effect. Initially, we showed that the extent of DNA damage was increased by the combination therapy, compared with that induced by MYCi975 or cisplatin alone, using p-H2A.X and comet assays. Then, consistent with DNA damage, we confirmed the accumulation of cytosolic dsDNA, which subsequently activated the phosphorylation of IRF3 to induce the expression of type I IFN chemokines (CXCL9, CXCL10, and CXCL11) and activated CD8+ T cell recruitment. This also explained the reason for the effective treatment by MYCi975 and cisplatin combination therapy. Previous studies demonstrated that MYC inhibition modulates the tumor immune microenvironment and enhances anti-PD1 immunotherapy [24]. The close relationship between cGAS-STING-IRF3 pathway and immunotherapy was also well documented [19, 31, 34], suggesting that MYCi975 could be also used to enhance the sensitivity of HNSCC to immunotherapy in the future.
Conclusions
The results demonstrated that MYCi975 could eliminate CSCs, prevent metastasis, activate the intrinsic immune responses, and overcome cisplatin resistance in HNSCC. We propose MYCi975 combined with cisplatin as a potential therapeutic option for clinical trials to treat HNSCC.
Abbreviations
HNSCC: head and neck squamous cell carcinoma; CSC: cancer stem cell; PDX: patient-derived xenograft; qRT-PCR: quantitative real-time reverse transcription PCR; SD: standard deviation; TUNEL: terminal deoxynulceotidyl transferase nick-end-labeling; 4NQO: 4-nitroquinoline 1-oxide; MYC: MYC proto-oncogene, BHLH transcription factor; ALDH: alcohol dehydrogenase; BMI-1: B Lymphoma Mo-MLV insertion region 1 homolog; CCK8: cell counting kit 8; DAPI: 4, 6-diamidino-2-phenylindole; FACS: fluorescence activated cell sorting; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; KLF4: Kruppel like factor 4; OCT4: octamer-binding protein 4; SOX2: SRY-box transcription factor 2; CXCL: C-X-C motif chemokine ligand; IFN: interferon; IRF3: interferon regulatory factor 3; cGAS-STING: cyclic guanosine monophosphate-adenosine monophosphate synthase-stimulator of interferon gene.
Supplementary Material
Supplementary figures.
Acknowledgements
Funding
This study was financially supported by grants from the Natural Science Foundation of Beijing [grant number 7222221]; the National Natural Science Foundation of China [grant number 82272706]; and the Research Foundation of Peking University School and Hospital of Stomatology [grant number PKUSS20220111].
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73:17-48
2. De Felice F, Musio D, Terenzi V, Valentini V, Cassoni A, Tombolini M. et al. Treatment improvement and better patient care: which is the most important one in oral cavity cancer? Radiat Oncol. 2014;9:263
3. Machiels JP, Rene Leemans C, Golusinski W, Grau C, Licitra L, Gregoire V. et al. Squamous cell carcinoma of the oral cavity, larynx, oropharynx and hypopharynx: EHNS-ESMO-ESTRO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2020;31:1462-1475
4. Chi AC, Day TA, Neville BW. Oral cavity and oropharyngeal squamous cell carcinoma-an update. CA Cancer J Clin. 2015;65:401-21
5. Hedberg ML, Goh G, Chiosea SI, Bauman JE, Freilino ML, Zeng Y. et al. Genetic landscape of metastatic and recurrent head and neck squamous cell carcinoma. J Clin Invest. 2016;126:1606
6. Lee YS, Johnson DE, Grandis JR. An update: emerging drugs to treat squamous cell carcinomas of the head and neck. Expert Opin Emerg Drugs. 2018;23:283-299
7. Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S. et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359:1116-1127
8. Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O. et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869-83
9. Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265-79
10. Tyagi A, Kaushal K, Chandrasekaran AP, Sarodaya N, Das S, Park CH. et al. CRISPR/Cas9-based genome-wide screening for deubiquitinase subfamily identifies USP1 regulating MAST1-driven cisplatin-resistance in cancer cells. Theranostics. 2022;12:5949-5970
11. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983-8
12. Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M. et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature. 2014;511:246-50
13. Brooks MD, Burness ML, Wicha MS. Therapeutic implications of cellular heterogeneity and plasticity in breast cancer. Cell Stem Cell. 2015;17:260-71
14. Burtness B, Harrington KJ, Greil R, Soulieres D, Tahara M, de Castro G. et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet. 2019;394:1915-1928
15. Chen D, Wu M, Li Y, Chang I, Yuan Q, Ekimyan-Salvo M. et al. Targeting BMI1(+) cancer stem cells overcomes chemoresistance and inhibits metastases in squamous cell carcinoma. Cell Stem Cell. 2017;20:621-634 e6
16. Li J, Yu B, Deng P, Cheng YD, Yu YX, Kevork K. et al. KDM3 epigenetically controls tumorigenic potentials of human colorectal cancer stem cells through Wnt/beta-catenin signalling. Nat Commun. 2017;8:15146
17. Dong JQ, Li J, Li Y, Ma ZK, Yu YX, Wang CY. Transcriptional super-enhancers control cancer stemness and metastasis genes in squamous cell carcinoma. Nat Commun. 2021;12:3974
18. Qin Z, Zhang WB, Liu S, Wang YJ, Peng X, Jia LF. PVT1 inhibition stimulates anti-tumor immunity, prevents metastasis, and depletes cancer stem cells in squamous cell carcinoma. Cell Death & Disease. 2023;14:187
19. Jia L, Zhang W, Wang CY. BMI1 inhibition eliminates residual cancer stem cells after PD1 blockade and activates antitumor immunity to prevent metastasis and relapse. Cell Stem Cell. 2020;27:238-253
20. Dang CV. MYC on the path to cancer. Cell. 2012;149:22-35
21. Gardner EE, Lok BH, Schneeberger VE, Desmeules P, Miles LA, Arnold PK. et al. Chemosensitive relapse in small cell lung cancer proceeds through an EZH2-SLFN11 axis. Cancer Cell. 2017;31:286-299
22. Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70-8
23. Wang C, Li Y, Jia L, Kim JK, Li J, Deng P. et al. CD276 expression enables squamous cell carcinoma stem cells to evade immune surveillance. Cell Stem Cell. 2021;28:1597-1613
24. Han H, Jain AD, Truica MI, Izquierdo-Ferrer J, Anker JF, Lysy B. et al. Small-molecule MYC inhibitors suppress tumor growth and enhance immunotherapy. Cancer Cell. 2019;36:483-497
25. Holmes AG, Parker JB, Sagar V, Truica MI, Soni PN, Han H. et al. A MYC inhibitor selectively alters the MYC and MAX cistromes and modulates the epigenomic landscape to regulate target gene expression. Sci Adv. 2022;8:eabh3635
26. Liu S, Qin Z, Mao Y, Zhang W, Wang Y, Jia L. et al. Therapeutic targeting of MYC in head and neck squamous cell carcinoma. Oncoimmunology. 2022;11:2130583
27. Yu H, Wu S, Liu S, Li X, Gai Y, Lin H. et al. Venetoclax enhances DNA damage induced by XPO1 inhibitors: A novel mechanism underlying the synergistic antileukaemic effect in acute myeloid leukaemia. J Cell Mol Med. 2022;26:2646-2657
28. Lu HR, Meng LH, Huang M, Zhu H, Miao ZH, Ding J. DNA damage, c-myc suppression and apoptosis induced by the novel topoisomerase II inhibitor, salvicine, in human breast cancer MCF-7 cells. Cancer Chemother Pharmacol. 2005;55:286-94
29. Veo B, Danis E, Pierce A, Wang D, Fosmire S, Sullivan KD. et al. Transcriptional control of DNA repair networks by CDK7 regulates sensitivity to radiation in MYC-driven medulloblastoma. Cell Rep. 2021;35:109013
30. Ashley N, Harris D, Poulton J. Detection of mitochondrial DNA depletion in living human cells using PicoGreen staining. Exp Cell Res. 2005;303:432-46
31. Zhang WC, Liu W, Jia LF, Chen DM, Chang I, Lake M. et al. Targeting KDM4A epigenetically activates tumor-cell-intrinsic immunity by inducing DNA replication stress. Mol Cell. 2021;81:2148-2165
32. Ghosh M, Saha S, Bettke J, Nagar R, Parrales A, Iwakuma T. et al. Mutant p53 suppresses innate immune signaling to promote tumorigenesis. Cancer Cell. 2021;39:494-508 e5
33. Sen T, Rodriguez BL, Chen LM, Della Corte CM, Morikawa N, Fujimoto J. et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 2019;9:646-661
34. Wu JX, Sun LJ, Chen X, Du FH, Shi HP, Chen C. et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826-30
35. Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. 2018;215:1287-1299
36. Prager BC, Xie Q, Bao SD, Rich JN. Cancer stem cells: the architects of the tumor ecosystem. Cell Stem Cell. 2019;24:41-53
37. Marur S, Forastiere AA. Update on role of chemotherapy in head and neck squamous cell cancer. Indian J Surg Oncol. 2010;1:85-95
38. Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17:313-9
39. Zhu L, Chen Z, Zang H, Fan S, Gu J, Zhang G. et al. Targeting c-Myc to overcome acquired resistance of EGFR mutant NSCLC cells to the third-generation EGFR tyrosine kinase inhibitor, Osimertinib. Cancer Res. 2021;81:4822-4834
40. Ala M. Target c-Myc to treat pancreatic cancer. Cancer Biol Ther. 2022;23:34-50
41. Zhou Y, Gao X, Yuan M, Yang B, He Q, Cao J. Targeting Myc interacting proteins as a winding path in cancer therapy. Front Pharmacol. 2021;12:748852
42. Foley SA, Casten A, Kavanagh E, Synnott NC, Crown J, Larsson LG. et al. MYC as a therapeutic target for the treatment of triple-negative breast cancer. J Clin Oncol. 2019;37:e12550-e12550
43. Ghaffarnia R, Nasrollahzadeh A, Bashash D, Nasrollahzadeh N, Mousavi SA, Ghaffari SH. Inhibition of c-Myc using 10058-F4 induces anti-tumor effects in ovarian cancer cells via regulation of FOXO target genes. Eur J Pharmacol. 2021;908:174345
44. Wang HC, Zhang SS, Zhang Y, Jia JY, Wang JX, Liu XQ. et al. TAZ is indispensable for c-MYC-induced hepatocarcinogenesis. J Hepatol. 2022;76:123-134
45. Robinson AM, Rathore R, Redlich NJ, Adkins DR, VanArsdale T, Van Tine BA. et al. Cisplatin exposure causes c-Myc-dependent resistance to CDK4/6 inhibition in HPV-negative head and neck squamous cell carcinoma. Cell Death Dis. 2019;10:867
46. Nor C, Zhang ZC, Warner K, Bernardi L, Visioli F, Helman JI. et al. Cisplatin induces Bmi-1 and enhances the stem cell fraction in head and neck cancer. Neoplasia. 2014;16:137-146
47. Watanabe H, Ma QP, Peng SY, Adelmant G, Swain D, Song WY. et al. SOX2 and p63 colocalize at genetic loci in squamous cell carcinomas. J Clin Invest. 2014;124:1636-45
48. Chang Y, Zhang J, Huo X, Qu X, Xia C, Huang K. et al. Substrate rigidity dictates colorectal tumorigenic cell stemness and metastasis via CRAD-dependent mechanotransduction. Cell Rep. 2022;38:110390
49. Peng Z, Tan X, Xie L, Li Z, Zhou S, Li Y. PKR deficiency delays vascular aging via inhibiting GSDMD-mediated endothelial cell hyperactivation. iScience. 2023;26:105909
50. Eskander A, Givi B, Gullane PJ, Irish J, Brown D, Gilbert RW. et al. Outcome predictors in squamous cell carcinoma of the maxillary alveolus and hard palate. Laryngoscope. 2013;123:2453-8
51. Koerdt S, Rockl J, Rommel N, Mucke T, Wolff KD, Kesting MR. Lymph node management in the treatment of oral cancer: Analysis of a standardized approach. J Craniomaxillofac Surg. 2016;44:1737-1742
52. Iglesias-Bartolome R, Gutkind JS. Unleashing immunotherapy by targeting cancer stem cells. Cell Stem Cell. 2020;27:187-189
53. Ding XM, Pan HY, Li J, Zhong Q, Chen XH, Dry SM. et al. Epigenetic activation of AP1promotes squamous cell carcinoma metastasis. Sci Signal. 2013;6:273
54. Jia L, Wang Y, Wang CY. circFAT1 promotes cancer stemness and immune evasion by promoting STAT3 activation. Adv Sci (Weinh). 2021;8:2003376
Author contact
![]() Corresponding authors: Xin Peng, MD, DDS, 22 Zhongguancun South Avenue, Beijing 100081, China. Tel: (8610)82195210; Fax: (8610)62173402; E-mail: pxpengxinedu.cn. Lingfei Jia, PhD, 22 Zhongguancun South Avenue, Beijing 100081, China. E-mail: jialingfei1984com.
Corresponding authors: Xin Peng, MD, DDS, 22 Zhongguancun South Avenue, Beijing 100081, China. Tel: (8610)82195210; Fax: (8610)62173402; E-mail: pxpengxinedu.cn. Lingfei Jia, PhD, 22 Zhongguancun South Avenue, Beijing 100081, China. E-mail: jialingfei1984com.