Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Single-cell isolation
Single-cell DNA methylation...
Single-cell multi-omics...
Biological applications of...
Perspectives
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2023; 13(8):2439-2454. doi:10.7150/thno.82582 This issue Cite
Review
Technologies and applications of single-cell DNA methylation sequencing
Fang Liu1,2,3, Yunfei Wang4, Hongcang Gu1,2,3 ![]() , Xiaoxue Wang5
, Xiaoxue Wang5 ![]()
1. Anhui Province Key Laboratory of Medical Physics and Technology, Institute of Health and Medical Technology, Hefei Institutes of Physical Science, Chinese Academy of Sciences, Hefei, 230031, China.
2. University of Science and Technology of China, Hefei, 230026, China.
3. Hefei Cancer Hospital, Chinese Academy of Sciences, Hefei, 230031, China.
4. Zhejiang ShengTing Biotech. Ltd, Hangzhou, 310000, China.
5. Department of Hematology, the First Hospital of China Medical University, Shenyang, 110001, China.
Received 2023-1-11; Accepted 2023-4-9; Published 2023-4-23
Abstract

DNA methylation is the most stable epigenetic modification. In mammals, it usually occurs at the cytosine of CpG dinucleotides. DNA methylation is essential for many physiological and pathological processes. Aberrant DNA methylation has been observed in human diseases, particularly cancer. Notably, conventional DNA methylation profiling technologies require a large amount of DNA, often from a heterogeneous cell population, and provide an average methylation level of many cells. It is often not realistic to collect sufficient numbers of cells, such as rare cells and circulating tumor cells in peripheral blood, for bulk sequencing assays. It is therefore essential to develop sequencing technologies that can accurately profile DNA methylation using small numbers of cells or even single cells. Excitingly, many single-cell DNA methylation sequencing and single-cell omics sequencing technologies have been developed, and applications of these methods have greatly expanded our understanding of the molecular mechanism of DNA methylation. Here, we summaries single-cell DNA methylation and multi-omics sequencing methods, delineate their applications in biomedical sciences, discuss technical challenges, and present our perspective on future research directions.
Keywords: DNA methylation, single-cell sequencing, single-cell multi-omics sequencing
Introduction
DNA methylation refers to the phenomenon in which a methyl group (CH3) from S-adenosylmethionine is transferred to the C-5 position of cytosine by DNA methyltransferases (DNMTs) [1, 2]. DNA methylation is the most stable epigenetic modification. Another type of DNA methylation in mammals occurs at the N-6 position of adenine, although its functions are still under extensive investigation [3, 4]. The 5-methylcytosine (5mC) is the dominant type of DNA modification, accounting for approximately 1% of the human genome [5]. It occurs almost exclusively in the form of 5'-3' cytosine-phosphate-guanine (CpG) dinucleotides, and approximately 70-80% of CpGs are methylated in mammals [6, 7]. CpGs are not randomly distributed across the genome, but exhibit widely scattered and locally clustered distributions [8]. The CpG-rich regions, where the C+G content exceeds 50% and the observed to expected CpG ratio is equal to or greater than 0.6, are called CpG islands (CGIs). CGIs are typically 300-3,000 bp in length and overlap with 60% of human gene promoters and almost 100% of housekeeping gene promoters [9-12]. Like CGIs, most CpG-rich regions show low levels of methylation, whereas CpG-poor regions are generally hypermethylated in mammals [13, 14]. Remarkably, gene regulatory elements, including enhancers and transcription-factor binding sites, exhibit dynamic DNA methylation across tissues and cell types [15].
DNA methylation plays a critical role at the molecular, biological, and pathological levels [6, 13, 14, 16]. Promoter hypermethylation is often associated with gene silencing and has been frequently observed in cancer [17]. The repressive role of 5mC at gene promoters can be caused by directly preventing transcription factors (TFs) from binding to the corresponding elements, thereby blocking gene transcription. Alternatively, 5mC attracts methyl-CpG-binding domain (MBD) proteins to attach to promoter regions, consequently blocking TF binding to regulatory elements [14, 18]. However, DNA methylation in the gene bodies shows complicated correlations with transcription: most low and highly-expressed genes exhibit low levels of methylation in the gene bodies, whereas moderately expressed genes show the highest levels of methylation [19]. DNA methylation is also associated with increased levels of C-to-T mutations [8, 15, 20]. Repetitive elements, which comprise more than 55% of the human genome, consist mainly of retrotransposons and are the primary targets of DNA methylation [21]. Loss of DNA methylation in repetitive elements contributes to genome instability and global hypomethylation, which are considered hallmarks of cancer [14, 16, 17, 22, 23]. Notably, the mammalian genome undergoes two waves of global demethylation and remethylation during development. In the first wave, primordial germ cells undergo genome-wide DNA demethylation, forming identical hermaphroditic epigenomes and ultimately developing new sex-specific epigenetic modifications [24, 25]. The second wave occurs immediately after fertilization, initially erasing the methylation profiles of the gametes and later rewriting the methylation markers of the embryos [26]. Mammalian development is also characterized by X chromosome inactivation (XCI) and genomic imprinting, resulting in monoallelic gene expression [27-29].
The biochemical processes of methylation and demethylation are beginning to be elucidated. De novo methylation is catalyzed by DNMT3A, 3B, and 3L, while the methylation profiles of dividing cells are maintained by DNMT1 [26]. On the other hand, demethylation is the result of an active enzymatic process and passive replication-dependent dilution. Specifically, 5mC is first converted to 5-hydroxymethylcytosine (5hmC), then to 5-formylcytosine (5fC), and finally to 5-carboxylcytosine (5caC) under the catalysis of ten-eleven translocation enzymes (TET1, 2, 3) [30-33]. These oxidized derivatives of 5mC are diluted during DNA replication. Alternatively, 5fC and 5caC are actively removed by thymine DNA glycosylase (TDG) and subsequently replaced by cytosine through base excision repair (BER) [34]. The dynamic nature of DNA methylation is attracting scientists to develop epigenetic drugs. Among them, DNA methyltransferase inhibitors, 5-azacytidine (Azacytidine, Vidaza®) and 5-aza-2'-deoxycytidine (Decitabine, Dacogen®), are the most successful epigenetic drugs. These drugs have been approved by the FDA and are widely used to treat patients with myelodysplastic syndrome (MDS) [35, 36].
DNA methylation analysis is essential to dissect its role in the development and human diseases, such as cancer. Methods for DNA methylation detection, including the principles and applications, have been described in the literature [8, 37-42]. Next-generation sequencing (NGS)-based assays combined with the sodium bisulfite treatment are widely adopted due to their high reproducibility and accuracy at the single base level [37]. In particular, whole-genome bisulfite sequencing (WGBS) is considered as the gold standard; however, the high sequencing costs and a significant amount of non-CpG reads make it less efficient for DNA methylation detection [8, 38]. Reduced representation bisulfite sequencing (RRBS) enriches CpG-rich fragments using restriction endonucleases, such as MspI (C|CGG) and HaeIII (GG|CC), and gel-based size selection. It therefore significantly reduces sequencing costs while still covering most CpG islands and promoters, a good representation of other genomic features, including enhancers and CpG island shores [43-45]. Conventional NGS-based methods, such as WGBS and RRBS, require a large amount of DNA, mainly measure the average DNA methylation level of many cells, and cannot identify the methylation status of individual cells [37]. Yet, cellular heterogeneity is a pervasive phenomenon in multicellular organisms, suggesting that the NGS-based measurements may not reflect the actual methylation status. The accessibility of certain rare cells, such as embryonic stem cells in early development and cancer stem cells, also limits the application of bulk sequencing methods [46]. Therefore, DNA methylation profiling at the single-cell level is essential, and many such technologies have been reported recently [47].
In this review, we summarize single-cell sequencing methods for the assessment of DNA methylation alone or in combination with other omics, outline the applications, and present with our perspective on these technologies.
Single-cell isolation
Isolating intact individual cells is crucial for single-cell sequencing, and various methods have been documented and summarized in Table 1 [48]. The limiting dilution method is characterized by low cost and low throughput (Figure 1A). Micromanipulators utilize automated pipetting under microscopic observation, allowing operators to isolate single cells efficiently and accurately (Figure 1B). Laser capture microdissection (LCM) also allows targeted cell collection under microscopic visualization. The difference is that LCM focuses on isolating single cells from stained tissue and is, therefore, able to collect single cells with specific histological characteristics (Figure 1C).
Advantages and disadvantages of single-cell isolation methods
| Isolation methods | Advantages | Disadvantages |
|---|---|---|
| Random limiting dilution | Easy to operate, no special equipment required | Less efficient |
| Mouth pipetting | Low cost and virtually no cell damage | Difficult to operate |
| Micromanipulation | Cost-effective and accurate single cell access | Difficult to operate |
| Laser microdissection | Retention of spatiotemporal information | Low throughput |
| Microfluidic devices | High-throughput cell sorting based on cell surface markers | Expensive consumables |
| Flow cytometry | High throughput, cell sorting using antibodies and fluorescent markers | Requires large number of cells, high cost, harmful to cells |
Schematic of single-cell isolation technologies. (A) Random limiting dilution; (B) Micromanipulation; (C) Laser capture microdissection; (D) Fluorescence activated cell sorting; (E) Microfluidic devices based on hydrodynamic cell traps; (F) Microwell-based microfluidics; (G) Droplet-based microfluidics.
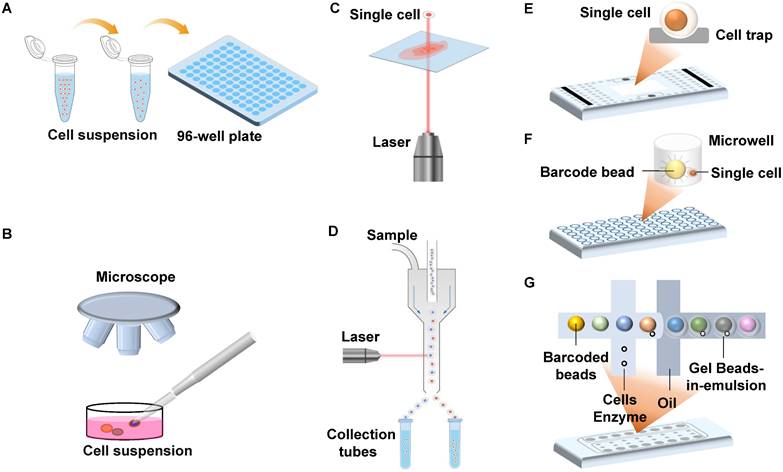
Many single-cell isolation platforms use flow cytometry or microfluidic devices to automatically sort single cells with high throughput. The most common platform is the fluorescence-activated cell sorting (FACS) system (Figure 1D), and one of its main advantages is the throughput, which enables the isolation of hundreds of single cells within 30 minutes [49]. Secondly, the platform can sort cells according to their functional properties using fluorescent staining, thus targeting individual cells of interest [50]. Most commercial single-cell sorting platforms are based on microfluidic technology, such as the C1™ Single-Cell Auto Prep System (Figure 1E), the BD Rhapsody™ Single-Cell Analysis System (Figure 1F), and the 10× Chromium Single Cell Gene Expression Solution (Figure 1G) [51, 52]. Commercial platforms can simultaneously perform cell sorting and barcode each cell, improving sequencing throughput and reducing sequencing costs.
Single-cell DNA methylation sequencing
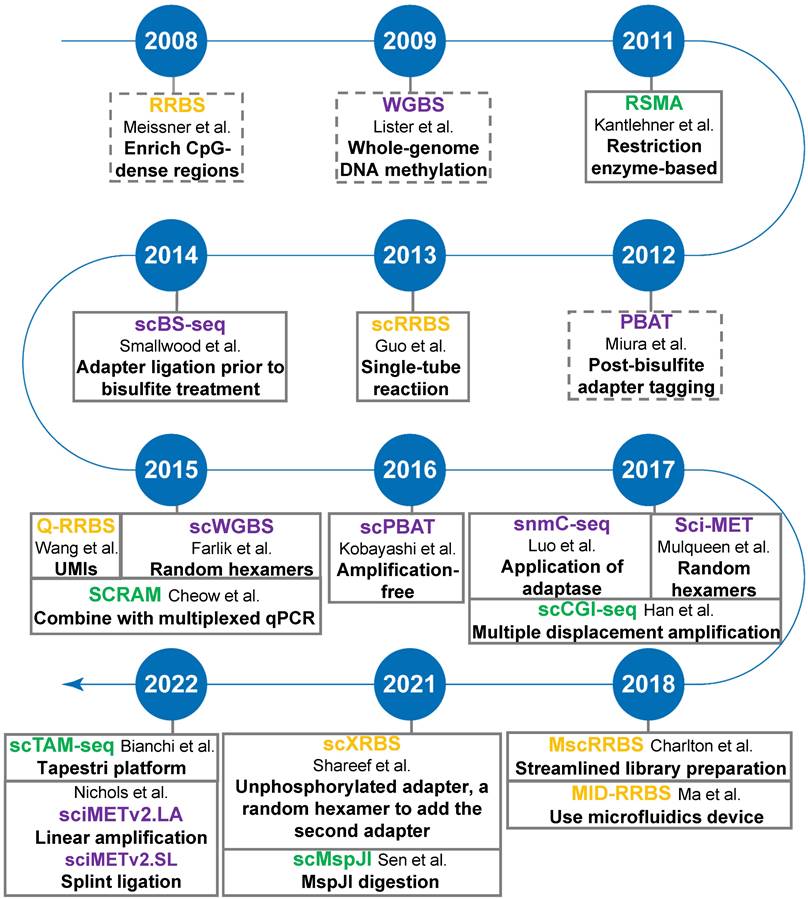
The advent of single-cell isolation technologies and the optimization of methylation sequencing technologies are accelerating the development of single-cell DNA methylation sequencing. Most, if not all, single-cell sequencing methods are based on the corresponding bulk-based assays. Here we focus mainly on the most common types of single-cell DNA methylation sequencing technologies based on either restriction digestion (including methylation-insensitive and methylation-sensitive restriction endonucleases) or post-bisulfite adapter tagging (PBAT) (Figure 2).
Timeline of single-cell DNA methylation sequencing technologies. Yellow-highlighting: RRBS-based single-cell DNA methylation methods; Green-highlighting: MSRE-based single-cell DNA methylation methods; Purple-highlighting: PBAT-based single-cell DNA methylation methods. Dotted line square: conventional bulk sequencing assays, including RRBS, WGBS, and PBAT.
Restriction digestion-based DNA methylation profiling methods
Methods in this category rely on restriction endonucleases that recognize and cleave double-stranded DNA at specific sites. Combined with DNA size selection, these techniques allow analysis of the methylation status of targeted CpG sites with reduced sequencing costs. Assays based on methylation-insensitive endonucleases typically require the treatment of adapter-equipped DNA with sodium bisulfite, which converts unmethylated cytosine (C) to uracil and leaves methylated C unchanged. Therefore, unmethylated and methylated Cs can be accurately inferred from sequencing analysis [43]. Conversely, the methylation-sensitive methods bypass the sodium bisulfite treatment. It only profiles unmethylated CpGs at cleavage sites, while the corresponding CpGs missed in the sequencing data are inferred as methylated [53]. The characteristics of the two types of methods are summarized in Table 2.
Single-cell DNA methylation profiling method based on restriction endonuclease digestion
| Method | Strategies | Coverage per cell* | Throughput | Advantages | Disadvantages | Applications | Ref |
|---|---|---|---|---|---|---|---|
| scRRBS | One-tube enzymatic reactions | ∼1.0M CpGs (mouse, mean coverage) | 10s of cells | High promoter/CGI coverage | Sequence bias due to two rounds of PCR application | DNAm dynamics in development and disease | [57] |
| Q-RRBS | Unique molecular identifiers | 0.5-1M CpGs (human) | 1-100 cells | Eliminate PCR-derived duplication; detect allele-specific methylation | Low CpGs coverage | DNAm dynamics in development and disease | [58] |
| MscRRBS | Inline barcode, one-well enzymatic reactions | ∼0.9M CpGs (human, mean coverage) | 384 cells | High-throughput and easy set-up with automation | Low CpGs coverage | DNAm dynamics in development and disease | [62] |
| scXRBS | Unphosphorylated adapter, a random hexamer to add the second adapter | up to 3.4M CpGs (human) | 96 cells | Extended CpG coverage for regulatory elements | Complicated library preparation | CNV and DNAm changes across single cells | [63] |
| MID-RRBS | Reactions in a microfluidic device | 35k-231K CpGs (mouse) | 96 cells | Efficient bisulfite conversion with increased DNA recovery | Low CpG coverage for single cells; requires non-commercial instrumentation | Cell type-specific epigenetic drug screening and drug-methylome interaction studies | [60] |
| RSMA | Reactions in a multi-well PCR slide | 10-20 CpGs (human) | 48 cells | Cost-effective | Unable to confirm heterozygous methylation | CpG methylation status in many single cells | [66] |
| SCRAM | PCR using two forward primers and one reverse primer | up to 24 CpGs (mouse) | 48 cells | High reliability and accuracy | Not suitable for genome-wide screening DNAm | Targeted CpG methylation changes | [67] |
| scCGI-seq | Two rounds of MSRE digestion, MDA | ∼21K CpGIs (human) | 10-100 cells | High and consistent CpGI coverage | Low CpGs coverage, low-throughput | DNAm heterogeneity in development, differentiation and cancer | [68] |
| scMspJI-seq | Enrich methylated CpGs with MspJI | 212-977K CpGs (mouse) | 384 cells | Cost-effective | Difficult to map allele-specific DNA methylation | Strand-specific DNAm; investigation of mechanisms regulating demethylation dynamics | [70] |
| scTAM-seq | Tapestri platform; gene-specific primers | 650 CpGs (human) | up to 10k cells | High-throughput; automation | Low CpG coverage | Targeted CpG methylation changes in development and disease | [69] |
Notes: *Description in parentheses indicates sample source for assay development. DNAm--DNA methylation; MDA--Multiple Displacement Amplification.
Single-cell methylation sequencing method based on methylation-insensitive restriction enzyme
RRBS is the first NGS-based method for DNA methylation profiling [54]. It relies on methylation-insensitive endonucleases, such as MspI (C|CGG), and size selection to cleave and enrich CpG-dense DNA fragments [55, 56]. Several groups have published modified RRBS protocols by streamlining library processing, barcoding library DNA fragments to remove duplicates, or reducing genomic DNA inputs from microgram to picogram (single cell) levels [57-59]. For example, Q-RRBS introduces 6-bp barcodes to the 5'- and 3'-ends of library DNA inserts, guaranteeing 4,096 adapter combinations and thus attempting to eliminate PCR-related duplicates [58]. Microfluidic diffusion-based RRBS (MID-RRBS) uses a microfluidic device that allows DNA bisulfite treatment and subsequent purification steps to be performed in tiny chambers (240 or 480 nl). The modification minimizes DNA loss and allows profiling of DNA methylation changes with nanograms of DNA input or even with DNA from single cells [60]. However, the method only captures about 35-231K CpGs in the mouse genome.
To generate single-cell RRBS (scRRBS) libraries, Guo and colleagues minimized the library DNA loss by performing five consecutive reactions from cell lysis, MspI digestion, end-repair, A-tailing to adapter ligation and the bisulfite conversion in one tube (Figure 3A). After two rounds of PCR enrichment, the amplified scRRBS libraries were pair-ended sequenced and the sequencing data indicated that scRRBS was capable of covering up to 1.5 million CpGs [57, 61]. Using scRRBS to profile mouse sperm, oocytes, and zygotes reveals fine demethylation landscapes after fertilization [57]. However, scRRBS can only process a limited number of single cells manually. The multiplexed single-cell RRBS (MscRRBS) is performed in a 96- or 384-well PCR plate and can be processed automatically [59, 62]. By prefixing each cell with an inline barcode, dozens of adapter-equipped libraries can be pooled, dramatically reducing the subsequent workload and archiving coverage of up to 2 million unique CpGs for single human cells [62].
Schematic comparison of single-cell DNA methylation profiling methods: sc-RRBS (A), scBS-seq (B), and RSMA (C).
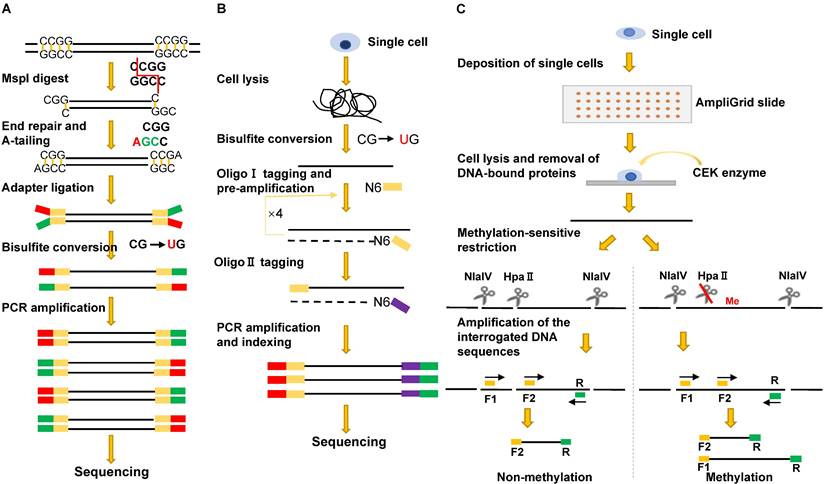
Extended-representation bisulfite sequencing (XRBS) deliberately uses Illumina adapters with unphosphorylated bottom strands. After the sodium bisulfite treatment, the converted DNA fragments have only a 5'-terminal adapter, and the 3'-terminal adapter sequences are introduced using random hexamer-tagged PCR primers. As a result, XRBS captures more CpG sites within two MspI cleavage sites [63]. The single-cell XRBS (scXRBS) also barcodes each DNA sample prior to bisulfite conversion and PCR amplification. The modifications allow each scXRBS library to cover up to 3.43 million CpGs with less than 2 million reads and can identify PCR duplicates [63].
Single-cell methylation sequencing method based on methylation-sensitive restriction enzymes (MSREs)
MSREs are a group of restriction endonucleases that cannot cleave DNA if their recognition sites contain methylated cytosines. Interestingly, some have isoschizomers with identical recognition sequences but are insensitive to methylation. MSRE-based assays can use multiple enzymes to extend genomic coverage, and the missed CpG sites in the enzyme binding sites are methylated and inferred from the sequencing analysis [64]. In contrast, Methyl-seq, which uses paired methylation-sensitive and methylation-insensitive isoschizomers MspI and HapII, can directly identify the methylation status of CpGs in their binding sites [65]; however, this strategy is unsuitable for single-cell sequencing.
The first MSRE-based single-cell DNA methylation assay was described in 2011, and the method, restriction enzyme-based single-cell methylation assay (RSMA) (Figure 3C), can only detect a limited number of CpGs [66]. The sequential reactions, including single-cell lysis, methylation-sensitive restriction digestion, and PCR amplification, are all performed on an AmpliGrid slide containing 48 microreactors for water-in-oil emulsions. The enzyme cleavage sites are located between the two forward primers so that the CpG methylation of the cleavage sites can be inferred either from the size of the PCR products or by pyrosequencing of the PCR product pool [66]. Subsequently, Cheow et al. developed another MSRE-based single-cell methylation method called single-cell restriction analysis of methylation (SCRAM) by combining MSRE digestion and multiplex PCR amplification [67]. The method applies a microfluidic qPCR chip and can detect the DNA methylation levels of 24 loci in up to 48 cells per assay. SCRAM is cost-effective but detects far fewer CpG sites than NGS-based single-cell DNA methylation assays. The method cannot distinguish between heterozygous methylated alleles and homozygous methylated alleles either. The single-cell CpG island methylation assay (scCGI-seq) is based on one round of MSRE digestion followed by multiplexed displacement amplification (MDA) and a second round of MSRE digestion [68]. The method enables genome-wide measurement of CGI methylation levels from single cells (covering 76% of CGIs in the human genome). Although the coverage of CpG sites is lower than that of scWGBS, scCGI-seq shows good reproducibility across multiple single cells.
Single-cell targeted analysis of the methylome (scTAM-seq) is another MSRE-based sequencing technology [69]. It can detect 650 CpG sites in up to 10,000 cells simultaneously. The assay uses a commercial microfluidic droplet device, the Mission Bio Tapestri platform, to mix individual cell lysate with barcoded beads tagged to gene-specific primers. Following methylation-sensitive restriction digestion and targeted PCR application in a thermal cycler, only targeted and methylated CpGs within the enzyme binding sites are amplified and sequenced. The application of scTAM-seq reveals the dynamic methylation status during B-cell differentiation in peripheral blood and bone marrow [69]. Despite low coverage, scTAM-seq achieves an excellent high throughput and low false-positive rates of less than 0.2% [49, 69].
In contrast to MSRE-based assays, which generally detect symmetric DNA methylation on both the plus and minus strands, single-cell MspJⅠ-dependent sequencing (scMspJI-seq) is designed to assess strand-specific 5mC [70]. The modification-dependent endonuclease MspJI targets mCNNR sites and cleaves downstream genomic DNA at approximately 9-13 bp. After the incorporation of double-stranded adapters containing T7 promoter, Illumina adapter, and unique molecular identifier sequences, DNA libraries are generated by in vitro transcription and PCR application of transcribed RNAs. Thus, scMspJI-seq specifically enriches methylated sites and has been used to study the dynamics of DNA demethylation in early development [70].
PBAT-based single-cell WGBS
In conventional bisulfite-based sequencing methods, fragmented DNA is typically tagged by methylated adapters prior to bisulfite conversion. PBAT implements an initial bisulfite treatment protocol and then uses random primers to amplify bisulfite-converted DNA fragments, allowing more DNA fragments to be subsequently amplified and sequenced [71]. Single-cell WGBS methods based on PBAT are summarized in Table 3.
Single-cell DNA methylation profiling methods based on PBAT
| Method | Strategies | Coverage per cell* | Throughput | Advantages | Disadvantages | Applications | Ref |
|---|---|---|---|---|---|---|---|
| scBS-seq | Two rounds of random priming | ∼3.7 M CpGs (mouse, mean coverage) | 96 cells | Genome-wide CpG coverage | Difficult to detect allele-specific methylation | DNAme in rare cells and heterogeneous populations | [72] |
| scWGBS | Integration of sequencing adapters using tagged random hexamers and terminal tagging | ∼1.0 M CpGs (mouse, mean coverage) | 96 cells | Fast, cost-effective | Low library complexity | DNAm dynamics in development and disease | [74] |
| scPBAT | Targeting DNAm at repetitive elements | 0.7%-6.6% (mouse) | 1-100 cells | Low sequencing costs and low PCR bias | Low mappability ofsequencing reads | Role of intergenic DNAm in mammals and other vertebrates | [75] |
| snmC-seq | Adapter sequence incorporation using random primers and adaptase | 22.2 ± 5.7% (mouse) | 384 cells | Improved mappability | High levels of adapter dimer sequences | Role of DNAm in disease, drug screening and cognition | [78] |
| snmC-seq2 | Adapter sequence incorporation using degenerate random primers and adaptase | 30.8 ± 7.5% (mouse) | 384 cells | Increased throughput, reduced artificial reads and improved library complexity | Adapter dimer sequences of about 10% | Role of DNAm in disease, drug screening and cognition | [79] |
| sci-MET | Transposase tagmentation, linear amplification, indexed PCR enrichment | 0.05-7% (human) | 96 cells | High-throughput potential and improved mappability | Low coverage, custom sequencing recipe & primers | DNAm alterations in cancer and neurological disorders | [76] |
| sciMETv2.LA | Indexed tagmentation, streamlined linear amplification, and indexed PCR amplification | ∼2.2 M CpGs (human, mean coverage) | 96 cells | High-coverage | DNA libraries with short-inserts, read pairs not fully overlapped | DNAm alterations in cancer and neurological disorders | [77] |
| sciMETv2.SL | Indexed tagmentation, splint ligation to add the TruSeq I5 adapter, and indexed PCR amplification | ∼0.3 M CpGs (human, mean coverage) | 96 cells | Low cost and less processing time | Low-coverage | DNAm alterations in cancer and neurological disorders | [77] |
Notes: *Description in parentheses indicates the sample source for assay development. DNAm--DNA methylation; MDA--Multiple displacement amplification.
Single-cell bisulfite sequencing (scBS-seq) is the first PBAT-based genome-wide methylation sequencing method in which bisulfite-treated DNA is subjected to two cycles of random primer extension [72, 73] (Figure 3B). Two critical steps, direct bisulfite treatment of the single-cell lysate and amplification of converted DNA before the purification of synthesized first-strand DNA, minimize DNA loss. In addition, the use of modified random hexamers eliminates the need to trim artificial bases introduced during conventional library preparation. On average, scBS-seq can detect 3.4 million CpGs per single cell. However, the method often fails to detect methylation differences for some alleles due to allele dropout caused by bisulfite conversion and enrichment-induced bias [49, 72].
The scWGBS method developed in the Bock laboratory primarily uses a commercial product, the EpiGnome™ Methyl-Seq Kit (Epicenter, EGMK81312), to generate sequencing libraries [74]. Bisulfite-converted genomic DNA is first transcribed using tagged random hexamer primers, and then the 3′-terminal ends of the newly synthesized DNA strands are linked to a second specific sequence tag. scWGBS does not undergo pre-amplification, reducing reagent costs, processing time, and amplification bias [74]. However, excessive PCR amplification to introduce Illumina-compatible sequencing adapters and generate library DNA negatively impacts library complexity, resulting in a relatively low coverage of approximately 1.4 million CpGs per cell.
Single-cell PBAT (scPBAT) uses a pair of primers, the Bio-PEA2-W4N4 primer (5'-biotin-ACACTCTTTCCCTACACGACGCTCTTCCGATCTWWWWNNNN-3') and the PE-index-W4N4 primer (5'-CAAGCAGAAGACGGCATACGAGATXXXXXXXXXGTAAAACGACGGCCAGCAGGAAACAGCTATGACWWWWNNNN-3') for the first- and second-strand DNA synthesis sequentially. It is noteworthy that scPBAT does not undergo PCR-based amplification and is tailored for methylation analysis of repetitive regions [75].
Some single-cell methylation libraries start from single-cell lysates, while others utilize single-cell nuclei instead, such as single-cell combinatorial indexing for methylation analysis (sci-MET), sciMETv2 linear amplification (sciMETv2.LA), sciMETv2 splint ligation (sciMETv2.SL), and single-nucleus methylcytosine sequencing (snmC-seq) [76-79]. In the sci-MET assay, each nucleus is indexed by transposase tagmentation in a 96-well plate prior to pooling for bisulfite treatment, linear amplification of the bisulfite-converted DNA, and sequential PCR enrichment of the library pools. The sci-MET covers a low percentage of CpGs per cell (0.05-7.0%), but is capable of sequencing DNA methylation for thousands of cells and achieving high alignment rates of 60-76% [76]. The optimized versions of sci-MET, sciMETv2.LA and sciMETv2.SL, achieve better tagmentation efficiency and increased coverage per cell, averaging 2.2 million and 325K unique CpGs, respectively [77]. The improvement benefits from using methylated indexed tagmentation adapters and updated nucleosome disruption technology. The two sciMETv2 methods can identify cell subtypes in the human brain.
Both snmC-seq and snmC-seq2 rely on barcoded random primers to amplify bisulfite-converted DNA and on the adaptase (Swift Biosciences) to tag a short oligo tail at the 3'-terminal of synthesized DNA. Sequencing libraries are generated by PCR using a pair of custom indexing primers containing Illumina P7 and P5 sequences, respectively. Several modifications, including the use of a different degenerate random primer (RP-H9, H=A, T, C) and the deactivation of free random primers and dNTP, dramatically improve the library qualities of snmC-seq2 compared to snmC-seq, such as better mapping rates (64.7±2.6% vs. 52.4±4% for the mouse genome), fewer artifactual reads (6.1±5.2%) and improved library complexity (30.8±7.5% vs. 22.2±5.7) [79].
Single-cell multi-omics sequencing
The emergence of single-cell genomic, epigenomic, and transcriptomic sequencing methods motivates scientists to explore technologies for parallel single-cell multi-omics profiling. Remarkably, single-cell multi-omics sequencing technologies have been reported and are summarized (Table 4). Applications of these technologies have greatly improved our understanding of cellular and molecular heterogeneities and the internal correlations within multi-omics in development and human disease.
Summary of single-cell multi-omics sequencing technologies
| Name | Omics | Methodology | Ref |
|---|---|---|---|
| Smart-RRBS | Transcriptome, methylome | Smart-seq2, MscRRBS | [80] |
| scMT-seq | Smart-seq2, scRRBS | [82] | |
| scM&T-seq | Smart-seq2, scBS-seq | [81] | |
| scTrio-seq | Transcriptome, methylome, CNVs | Smart-seq, scRRBS | [83] |
| scNMT-seq | Transcriptome, methylome, chromatin accessibility | Smart-seq2, NOMe-seq | [92] |
| iscCOOL-seq | scCOOL-seq, NOMe-seq | [87] | |
| scNOMeRe-seq | scNOMe-seq, MATQ-seq | [93] | |
| scGEM | Methylome, transcriptome | SCRAM | [89] |
| epi-gSCAR | Methylome, genetic variants | MSRE | [91] |
| scNOMe-seq | Methylome, chromatin accessibility | NOMe-Seq | [85] |
| scCOOL-seq | Methylome, chromatin accessibility, ploidy | COOL-seq | [86] |
Single-cell transcriptome and methylome sequencing allow the simultaneous assessment of gene expression and DNA methylation variation and the investigation of their correlation. Methodologically, most single-cell transcriptome and methylome sequencing assays have been developed by combining two types of single-cell sequencing methods. For example, Smart-RRBS combines Smart-seq2 and Msc-RRBS, scMT-seq is derived from scRRBS and Smart-seq2, and scM&T-seq is based on Smart-seq2 and scBS-seq [80-82]. Single-cell triple omics sequencing (scTrio-seq) combines scRRBS and scRNA-seq, and the third layer of omics, copy number variation (CNV), is deduced from the scRRBS data [83].
The critical step in parallel RNA and DNA methylation sequencing is isolating DNA and mRNA from the same cell properly. Two methods are commonly used for this purpose. One is to completely lyse single-cells and then separate mRNA from DNA using oligo-dT-coated magnetic beads. The second method is to gently lyse the cell membrane to release the cytoplasm and mRNAs, then transfer the cytoplasm and mRNAs to a separate tube, leaving the genomic DNA in the nucleus for further processing. Both scM&T-seq and Smart-RRBS take advantage of oligo-dT-coated magnetic beads for DNA and RNA separation, while scMT-seq and scTrio-seq benefit from the separation of intact nuclei and the cytoplasm for subsequent processing [80-82, 84].
Nucleosome-free regions (NFRs) or accessible chromatin regions often overlap with transcriptional regulatory elements. Methods capable of simultaneously assessing the chromosomal accessibility and DNA methylation include single-cell nucleosome occupancy and methylation (scNOMe-seq) [85], single-cell chromatin overall omic-scale landscape sequencing (scCOOL-seq) [86], and improved scCOOL-seq (iscCOOL-seq) [87]. scNOMe-seq relies on the GpC methyltransferase, M.CviPl, to catalyze the cytosine methylation of GpCs in NFRs. After bisulfite conversion of the M.CviPl-treated DNA and sequencing analysis, NFRs and endogenous DNA methylation are inferred according to the methylation patterns of regular CpGs and naturally unmethylated cytosines at GpC sites [85]. The method is also developed from the bulk type NOMe-seq [88]. By spiking in a certain amount of lambda DNA as an internal control, scCOOL-seq allows the profiling of individual cell ploidy [86]. In addition, iscCOOL-seq offers a better mapping rate, 74.55% vs. 22.01%, compared to scCOOL-seq - the improvement benefits from the optimized protocol for constructing single-cell PBAT-based methylation libraries [87].
The single-cell multiple omics assay for genotype, gene expression, and methylation profiling (sc-GEM) combines the single-cell restriction analysis of methylation (SCARM) technique with NGS-based single-cell genotyping. Targeted-gene transcripts are assessed by qPCR. Most of the experimental procedures are performed on the Fluidigm C1 single-cell auto-prep system [89, 90]. In particular, the methylation analysis is based on the digestion of an MSRE, HpaII (5'-C|CGG-3'), followed by qPCR amplification on the Fluidigm array. The assay covers a limited number of genes and gene transcripts; however, by performing the test on the Fluidigm instrument, hundreds of single cells can be analyzed simultaneously [89]. Another assay that can measure DNA methylation and identify genetic variants is epi-gSCAR (epigenomics and genomics of single cells analyzed by restriction) [91]. The method is based on Hhal, an MSRE that recognizes 5'-GCG|C-3' and can significantly enrich for CGIs and transcription start sites (TSSs). Sequencing analysis of epi-gSCAR libraries can detect up to half a million CpG sites and 1.2 million single-nucleotide variants (SNVs) [91].
Single-cell nucleosome, methylation, and transcription sequencing (scNMT-seq) can concurrently evaluate chromatin accessibility, DNA methylation, and gene transcription by applying M.CviPI to label the open chromatin regions. The method also uses oligo-dT-coated magnetic beads to precipitate mRNAs for RNA-seq library construction, leaving M.CviPI-treated DNA in the lysate for methylation analysis [92]. scNMT-seq can detect methylation changes in approximately half of the mouse promoters, three-quarters of gene bodies, and one-quarter of enhancers. Similarly, scNOMeRe-seq integrates scNOMe-seq and multiple annealing and dC-tailing-based quantitative single-cell RNA sequencing (MATQ-seq) to profile chromatin accessibility, DNA methylation, and gene transcription of the same cell [93, 94]. Unlike scNMT-seq, in which single cells are FACS sorted, scNOMeRe-seq is based on manually picking single cells and transferring the cytoplasm to another tube for MATQ-seq, leaving the nuclei for the GpC methylase treatment followed by scBS-seq [77]. scNOMeRe-seq can detect 3.49 million CpGs per single cell and more than 1000 gene transcripts for 94.8% of single cells [78].
Biological applications of single-cell DNA methylation sequencing
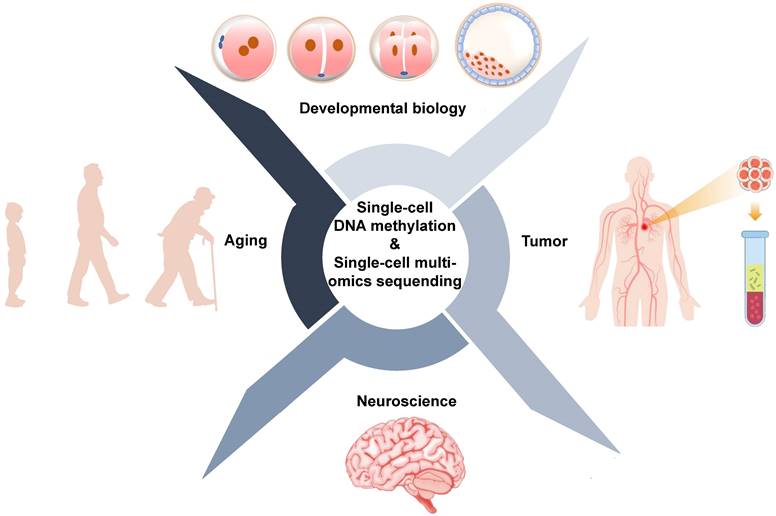
Conventional sequencing approaches require thousands to millions of cells and provide average changes at the genetic, epigenetic, and transcriptional levels. However, bulk sequencing technologies cannot reveal what is happening in rare cells or subpopulations of cells. Single-cell sequencing technologies provide tools to precisely profile DNA methylation and other omics for individual cells. Applications of single-cell DNA methylation and single-cell multi-omics sequencing are primarily focused on the development and human disease, particularly cancer [95] (Figure 4, Table 5).
Biological applications of single-cell DNA methylation sequencing and single-cell multi-omics sequencing.
Biological applications of single-cell DNA methylation sequencing
| Application | Method | Conclusion | Ref |
|---|---|---|---|
| Developmental biology | scBS-seq, Smart-seq2 | Dynamic DNAm during preimplantation with global demethylation and localized remethylation. | [97] |
| scCOOL-seq | Major changes in chromatin state and DNA methylation do not occur simultaneously after fertilization. | [98] | |
| scRRBS | Genic regions demethylated faster than intergenic regions in early mouse embryo development. | [57] | |
| scM&T-seq | Methylation patterns of distal regulatory regions correlate with gene expression. | [81] | |
| scNOMeRe-seq | DNAm remodeling is essential for reconstructing genetic lineages in early embryos. | [93] | |
| scTrio-seq2 | Genome remethylation in primitive endoderm cells is slower than in epiblast and trophectoderm cells. | [99] | |
| Tumor | scTrio-seq | Identification of cancer cell subpopulations and cellular heterogeneity within a subpopulation. | [83] |
| scTrio-seq2 | DNAm variation between primary and metastatic colorectal tumors reflects different sublineage composition. | [111] | |
| scRRBS | Abnormal DNAm in gliomas is associated with early genetic changes, and accumulated genetic variation is due to altered cellular states and environmental stress. | [112] | |
| Msc-RRBS, Smart-RRBS | Illustration of the lineage history of CLL and its evolution under pharmacological treatment. | [59] | |
| MARS-seq | Epigenetic memory diversifies the genetic subclonal structure of cancer cells. | [114] | |
| scCOOL-seq | Detection of enriched DNA demethylation in heterochromatin regions in pancreatic ductal adenocarcinoma (PDAC) and identification of two candidate biomarkers for the diagnosis of PDAC. | [115] | |
| scBS-seq | Classification of tumor origin using DNAm landscapes of CTCs. | [117] | |
| scWGBS | Hypomethylation of CTC clusters associated with poor prognosis in breast cancer. | [118] | |
| Neuroscience | snmC-seq | Establishment of a comprehensive DNAm atlas of mammalian neurons, demonstration of the essential role of epigenetic diversity in neuronal development. | [78] |
| snmC-seq2 | Creation of a sophisticated DNAm atlas of the mouse brain. | [124] | |
| Aging | sc-DNAm | DNAm as an epigenetic clock for age estimation in mammals. | [128] |
| scM&T-seq | Aging is associated with a global increase in transcription and methylome heterogeneity. | [130] |
Application of single-cell DNA methylation sequencing in developmental biology
Mammalian life begins at fertilization, where both paternal and maternal genomes undergo global demethylation, reaching its lowest level at the blastocyst stage [14, 96]. Using single-cell PBAT-based WGBS, Zhu and colleagues showed that local remethylation is interspersed with global demethylation. The authors further showed that methylation levels decrease more rapidly in the paternal genome, resulting in the paternal genome having consistently lower methylation levels from the two-cell stage to the blastocyst stage [97]. The same group further profiled DNA methylation and chromosome accessibility of early human embryos using scCOOL-seq. The results indicate that the chromatin of the paternal genome tends to be more open compared to the maternal genome shortly after fertilization up to the 4-cell stage [98].
Mouse is the most commonly used model animal to study early mammalian development. Single-cell DNA methylation analysis of the paternal and maternal genomes in mouse zygotes shows that the demethylation process of the genic region is faster than that of the intergenic regions [57]. Simultaneous profiling of the methylome and transcriptome of mouse embryonic stem cells by scM&T reveals novel correlations between the methylation patterns of regulatory elements and the expression of pluripotent genes [81]. Using scNOMeRe-seq, Wang et al. mapped the chromatin accessibility, detected DNA methylome variation, and profiled the transcriptomes of the mouse preimplantation embryos at the single-cell level. The authors also constructed genetic lineages from zygotes to the 8-cell stage and demonstrated that asymmetric cleavage may result from the transcriptional heterogeneity of blastomeres [93].
After blastocyst implantation, DNMT3A and 3B catalyze de novo methylation of the genome [14, 96]. Single-cell triple omics sequencing reveals that the genome remethylation of the primitive endoderm (PrE) cells is slower than that of the epiblast and trophectoderm cells, despite the fact that PrE and epiblast are both derived from the inner cell mass [99].
Application of single-cell DNA methylation sequencing analysis in tumors
Extensive studies show that epigenetic abnormalities are closely associated with the development and evolution of cancer [100-102]. Genome-wide hypomethylation and focal hypermethylation, particularly at the promoters of tumor suppressor gene, have been implicated as hallmarks of cancer [103-105]. Although observations based on 'bulk' DNA methylation analysis are likely valid, the superiority of single-cell sequencing analysis for cancer studies is evident. First, solid tumor tissues contain many cell types, including cancer cells, fibroblasts, endothelial cells, and infiltrating immune cells and nerves [106]. Therefore, bulk sequencing may not faithfully reflect the genetic and epigenetic status of tumor cells. Second, different subclones may coexist within the same tumor, and epigenetic plasticity permits cancer cells to alter their cellular state in response to microenvironmental and therapeutic stimuli [107]. Both directly contribute to the complexity of tumor heterogeneity. Finally, the accessible tumor cells may be limited, such as circulating tumor cells (CTCs) in the peripheral blood of cancer patients.
Cellular heterogeneity is closely associated with cancer development, evolution, and response to treatment. Many studies have used single-cell DNA methylation sequencing to investigate cellular heterogeneity in cancers, such as colorectal cancer, breast cancer, liver cancer, and chronic lymphocytic lymphoma (CLL) [59, 83, 108-110]. One study evaluates genetic, epigenetic, and transcriptional abnormalities in colorectal cancer using scTric-seq2 to analyze single cells derived from primary, lymphatic, and metastatic tissues [111]. The study identifies significant differences in overall methylation levels between genetic sublineages but less variation within a sublineage. Interestingly, the demethylation patterns of cancer cells are comparable across all ten patients [111]. An independent study investigates 914 single-cell methylomes, 55,284 single-cell transcriptomes, and bulk multi-omics sequences from 11 glioma patients with or without isocitrate dehydrogenase (IDH) gene mutation [112]. The study suggests that aberrant methylation is associated with early genetic alterations and that accumulated genetic alterations are related to altered cellular states and environmental stresses.
Understanding tumor heterogeneities and clonal evolutionary trajectories could help scientists elucidate the underlying mechanisms and develop specific targeted drugs. Using scTrio-seq, Hou et al. reported two subpopulations based on the CNV, methylation, and transcriptional profiles of 25 single cells isolated from the liver tissue of one patient with hepatocellular carcinoma [83]. The authors also found cellular heterogeneity within the subpopulations. Single-cell sequencing analysis not only sheds new light on solid tumor research but also provides mechanistic insight into chronic lymphocytic lymphoma (CLL). By applying Msc-RRBS to B cells from CLL patients and healthy donors, Gaiti and colleagues constructed the lineage tree and showed different branching patterns and lengths in the two cell populations [59]. Further analysis of the B cells using Smart-RRBS identified an ibrutinib-related bias in the methylation-based lineage tree, demonstrating how the therapeutic intervention affects the clonal evolutionary trajectory of CLL patients. Moreover, the upregulation of multiple Toll-like receptor (TLR) signalling pathway genes in ibrutinib-treated patients suggests a new direction for the development of targeted therapy [59].
Single-cell multi-omics sequencing technology is able to identify differentially expressed and differentially methylated genes in colorectal cancer, which can be used as biomarkers to guide targeted therapy for patients [113]. In one single-cell multi-omics study, DNA methylation is linked to the clonal stability of colorectal cancer cells and is strongly associated with cancer progression [114]. By simultaneously profiling the methylome, chromatin accessibility, and transcriptome, Fan et al. showed that hypermethylation is common in heterochromatin regions in the genome of patients with pancreatic ductal adenocarcinoma. In contrast, hypomethylation is typical in euchromatin regions. The authors also identified two biomarkers, ZNF667 and ZNF667-AS1, and showed that expression of these biomarkers is associated with a better prognosis [115].
Circulating tumor cells (CTCs) are cancer cells shed from primary or metastatic tumors into the peripheral blood. CTCs are rare, and often fewer than ten cells can be isolated from 10 ml of peripheral blood [116]. However, CTCs carry intact genetic, epigenetic, and transcriptional characteristics of tumor cells, making them ideal for studying tumor biology and monitoring tumor development and evolution. It is, therefore, possible to trace the cancer tissue of origin. The hypothesis was tested by applying scBS-seq to CTCs from six cancer types, and the investigation revealed tumor heterogeneities and an evolutionary pathway during cancer metastasis. The tumor tissue origin was also successfully identified based on the methylation landscapes of CTCs [117]. In addition, a systemic evaluation of the DNA methylation patterns of single CTCs and clustered CTCs reveals hypomethylation of binding sites for stemness- and proliferation-associated transcription factors (TFs), particularly in clustered cells [118]. The study demonstrates that an FDA-approved compound, a Na+/K+ ATPase inhibitor, disrupts CTC clustering, alters DNA methylation at TF-binding sites, and inhibits metastasis [118]. Another research using targeted bisulfite sequencing for three-EMT (epithelial-to-mesenchymal transition) genes tested 159 single CTCs from breast or prostate cancer patients. The study concluded that the methylation profiles of CTCs mirror those of epithelial-like cells and that CTCs have different methylation levels [119].
Single-cell DNA methylation sequencing in neuroscience and aging
Applications of single-cell DNA methylation sequencing and single-cell multi-omics sequencing technologies have also been extended to other research areas, such as neuroscience and aging. DNA methylation in neurons exhibits a unique feature, with a significant amount of methylated cytosine at CpH sites (H=A/T/C) in post-mitotic human and mouse neurons [120-122]. Notably, both CpG and non-CpG methylation are essential for neuronal development in the brain [120, 121, 123]. Single-cell methylation analysis of >6000 mouse and human frontal cortex neurons classifies these cells into 16 mouse and 21 human subpopulations, and both CpG and non-CpG methylation show cell-type-oriented landscapes [78]. In a parallel study, Liu and coworkers generated a brain DNA methylation atlas using 103,982 nuclei from 45 mouse brain regions. Single-cell methylation analysis reveals 161 subpopulations with distinct spatial locations and projection targets [124]. The integration of single-cell DNA methylation and chromatin accessibility datasets ultimately provides an epigenetic atlas for interpreting gene-enhancer interactions and understanding the 3D structure of neurons throughout the mouse cerebrum [124, 125].
A hallmark change of aging is genome-wide DNA hypomethylation [126]. Accordingly, DNA methylation-based biomarkers have been evaluated for predicting age and are considered the most promising of six distinct age estimators [127]. Recently developed pan-tissue epigenetic clocks can accurately estimate age using virtually any tissue from any mammalian species, suggesting that highly conserved DNA methylation patterns exist across mammals [128]. Gaiti and collaborators created a molecular clock based on the single-cell methylation dataset of a CLL patient. The authors predicted the subclonal divergence in the evaluation path and showed that the ancestral clone had evolved 2,180 ± 219 days, suggesting that the molecular clock could guide the treatment of CLL patients [59]. Another hallmark change is increased epigenetic or transcriptional heterogeneity during aging [129]. However, conventional bulk sequencing assays are unable to detect cell-to-cell variability. One study exploits the joint profiling of the single-cell transcriptome and single-cell methylome of mouse muscle stem cells. The assay reveals aged stem cells with increased transcriptional heterogeneity and localized DNA methylation changes, suggesting epigenetic drafting during aging [130]. Likewise, single-cell DNA methylation analysis of young and old mouse livers shows that mouse liver DNA methylation levels are highly variable, with an epivariation rate of 3.3%. Furthermore, DNA methylation heterogeneity is associated with genomic characteristics [131].
Perspectives
Over the last two decades, DNA methylation profiling technologies have changed dramatically from Sanger sequencing-based low-throughput to NGS-based high-throughput, from bulk DNA/RNA inputs to requiring only single cells [47]. Many single-cell DNA methylation sequencing technologies are currently available with varying coverage and mapping rates. However, improved CpG coverage often comes at the cost of reduced reproducibility. Different sequencing technologies can jointly provide comprehensive and accurate interpretations of genetic, epigenetic, and transcriptional changes. As illustrated earlier, most single-cell DNA methylation sequencing methods are based on bisulfite treatment, which causes significant DNA degradation and limits library complexity [132, 133].
Conversely, TET-assisted pyridine borane sequencing (TAPS) is based on TET oxidation of 5mC and 5hmC to 5caC, followed by pyridine borane reduction of 5caC to dihydrouracil. Enzyme-based bisulfite conversion is milder and generally does not cause DNA damage. TAPS can effectively identify modified cytosines with better mapping rates and uniform coverage [134]. However, the method requires a large amount of DNA input, and TAPS-based single-cell assays are not yet available as we draft the manuscript. In addition, most single-cell DNA methylation methods cannot process large numbers of cells, although many of them have improved throughput, such as sci-MET and Smart-RRBS [76, 80]. The development of efficient and high throughput assays is needed to analyze millions of CpG sites in hundreds or even thousands of single cells at a time in the future.
Sequencing costs have fallen dramatically over the last two decades, but profiling genetic and epigenetic changes at the single-cell level remains a challenge for many academic laboratories. One critical reason is that single-cell-based assays typically require sequencing hundreds or even thousands of single cells to obtain a comprehensive population-level picture [59, 112, 135]. A prototype sequencer from Ultima Genomics (Ultima), which adopts the mostly natural sequencing-by-synthesis (mnSBS) chemistry, can sequence the human genome with sufficient coverage at a cost of $100 [136, 137]. The new sequencer significantly reduces the sequencing cost and sheds new light on single-cell sequencing. However, whether it can be used to profile the methylome requires further investigation.
Single-cell DNA methylation sequencing has been widely used to profile rare cells and investigate cellular heterogeneity. CTCs preserve tumor genetic and epigenetic information well and are excellent candidates for cancer prognosis and diagnosis [138-140]. It is foreseeable that single-cell methylation sequencing and site-specific methylation assays will be incorporated into clinical testing. In addition to cancer, many publications report aberrant DNA methylation in other diseases, such as cardiovascular disease (CVD) [141, 142]. One study investigates whether the prevalence of CVD is associated with the global genomic DNA methylation levels in peripheral blood leukocytes (PBL) in a cohort of 286 Singaporean Chinese [143]. The study shows that increased DNA methylation is positively associated with the prevalence of CVD. In a recent case-control study involving thousands of participants, Fernandez-Sanles et al. identified 34 CpGs associated with acute myocardial infarction and four strongly correlated with coronary heart disease (CHD) and CVD [144]. However, how DNA methylation contributes to the development of CVD is still not fully understood [141]. The above single-cell-based assays will provide tools to dissect the molecular mechanism of CVD and identify biomarkers for diagnosis and prognosis of the disease.
Finally, the role of DNA methylation in gene regulation is complex [5, 23, 145]. For example, increased DNA methylation at promoter regions is generally thought to be anti-correlated with gene expression [17, 54, 146]. The relationships between gene expression and gene body methylation appear to be cell type dependent, being positively correlated in embryonic stem cells and negatively correlated in neurons [8, 14, 121, 147]. Furthermore, single-cell multi-omics sequencing shows that only a small percentage of promoter methylation levels are negatively associated with gene expression [81, 92, 148]. Similarly, significant correlations are only observed for a few gene bodies [81, 82]. The application of single-cell DNA methylation and single-cell multi-omics sequencing technologies across different cell types will help to elucidate the precise function of DNA in gene regulation in the coming years.
Acknowledgements
We thank all members of the Gu and Wang Labs for their comments and suggestions on the manuscript. Tong Zhang from Zhejiang ShengTing Biotech. Ltd helped with the figures. XW is supported by the National Natural Science Foundation of China (No.81900153) and the Natural Science Foundation of Liaoning Province (No.2022-YGJC-62).
Author contribution
FL drafted the manuscript. HG and XW reviewed and edited the draft. YW contributed to the figure design. All authors have read and approved the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6-21
2. Shen L, Song CX, He C, Zhang Y. Mechanism and function of oxidative reversal of DNA and RNA methylation. Annu Rev Biochem. 2014;83:585-614
3. Li VS, Tang MS, Kohn H. The effect of C(5) cytosine methylation at CpG sequences on mitomycin-DNA bonding profiles. Bioorgan Med Chem. 2001;9:863-73
4. Meyer KD, Jaffrey SR. Expanding the diversity of DNA base modifications with N-6-methyldeoxyadenosine. Genome Biol. 2016;17:5
5. Moore LD, Le T, Fan GP. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23-38
6. Jaenisch R, Bird A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33:245-54
7. Hernando-Herraez I, Garcia-Perez R, Sharp AJ, Marques-Bonet T. DNA methylation: Insights into human evolution. Plos Genet. 2015;11:e1005661
8. Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J. et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315-22
9. Wang Y, Leung FCC. An evaluation of new criteria for CpG islands in the human genome as gene markers. Bioinformatics. 2004;20:1170-7
10. Aissani B, Bernardi G. CpG islands, genes and isochores in the genomes of vertebrates. Gene. 1991;106:185-95
11. Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A. 2002;99:3740-5
12. Fatemi M, Pao MM, Jeong S, Gal-Yam EN, Egger G, Weisenberger DJ. et al. Footprinting of mammalian promoters: use of a CpG DNA methyltransferase revealing nucleosome positions at a single molecule level. Nucleic Acids Res. 2005;33:e176
13. Parry A, Rulands S, Reik W. Active turnover of DNA methylation during cell fate decisions. Nat Rev Genet. 2021;22:59-66
14. Greenberg MVC, Bourc'his D. The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol. 2019;20:590-607
15. Ziller MJ, Gu H, Muller F, Donaghey J, Tsai LT, Kohlbacher O. et al. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500:477-81
16. Jones PA. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484-92
17. Ehrlich M. DNA methylation in cancer: Too much, but also too little. Oncogene. 2002;21:5400-13
18. Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S. et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017;356:eaaj2239
19. Jjingo D, Conley AB, Yi SV, Lunyak VV, Jordan IK. On the presence and role of human gene-body DNA methylation. Oncotarget. 2012;3:462-74
20. Holliday R, Grigg GW. DNA methylation and mutation. Mutat Res. 1993;285:61-7
21. Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J. et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860-921
22. Zheng Y, Joyce BT, Liu L, Zhang Z, Kibbe WA, Zhang W. et al. Prediction of genome-wide DNA methylation in repetitive elements. Nucleic Acids Res. 2017;45:8697-711
23. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415-28
24. Guibert S, Forne T, Weber M. Global profiling of DNA methylation erasure in mouse primordial germ cells. Genome Res. 2012;22:633-41
25. Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W. et al. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev. 2002;117:15-23
26. Smith ZD, Meissner A. DNA methylation: Roles in mammalian development. Nat Rev Genet. 2013;14:204-20
27. Li L, Li L, Li QQ, Liu XX, Ma XY, Yong J. et al. Dissecting the epigenomic dynamics of human fetal germ cell development at single-cell resolution. Cell Res. 2021;31:463-77
28. Mohandas T, Sparkes RS, Shapiro LJ. Reactivation of an inactive human X-chromosome - evidence for X inactivation by DNA methylation. Science. 1981;211:393-6
29. Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362-5
30. He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q. et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303-7
31. Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA. et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300-3
32. Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129-33
33. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y. et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930-5
34. Wu H, Zhang Y. Reversing DNA methylation: Mechanisms, genomics, and biological functions. Cell. 2014;156:45-68
35. Li KH, Yang JX, Han XC. Lidocaine sensitizes the cytotoxicity of cisplatin in breast cancer cells via up-regulation of RAR beta 2 and RASSF1A demethylation. Int J Mol Sci. 2014;15:23519-36
36. Flotho C, Sommer S, Lubbert M. DNA-hypomethylating agents as epigenetic therapy before and after allogeneic hematopoietic stem cell transplantation in myelodysplastic syndromes and juvenile myelomonocytic leukemia. Semin Cancer Biol. 2018;51:68-79
37. Singer BD. A practical guide to the measurement and analysis of DNA methylation. Am J Respir Cell Mol Biol. 2019;61:417-28
38. Cokus SJ, Feng SH, Zhang XY, Chen ZG, Merriman B, Haudenschild CD. et al. Shotgun bisulphite sequencing of the arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452:215-19
39. Fouse SD, Nagarajan RP, Costello JF. Genome-scale DNA methylation analysis. Epigenomics. 2010;2:105-17
40. Khulan B, Thompson RF, Ye K, Fazzari MJ, Suzuki M, Stasiek E. et al. Comparative isoschizomer profiling of cytosine methylation: The HELP assay. Genome Res. 2006;16:1046-55
41. Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL. et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37:853-62
42. Rauch TA, Zhong XY, Wu XW, Wang M, Kernstine KH, Wang ZD. et al. High-resolution mapping of DNA hypermethylation and hypomethylation in lung cancer. Proc Natl Acad Sci U S A. 2008;105:252-7
43. Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33:5868-77
44. Smith ZD, Gu HC, Bock C, Gnirke A, Meissner A. High-throughput bisulfite sequencing in mammalian genomes. Methods. 2009;48:226-32
45. Harris RA, Wang T, Coarfa C, Nagarajan RP, Hong CB, Downey SL. et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol. 2010;28:1097-105
46. Rothova MM, Nielsen AV, Proks M, Wong YF, Riveiro AR, Linneberg-Agerholm M. et al. Identification of the central intermediate in the extra-embryonic to embryonic endoderm transition through single-cell transcriptomics. Nat Cell Biol. 2022;24:833-44
47. Karemaker ID, Vermeulen M. Single-cell DNA methylation profiling: Technologies and biological applications. Trends Biotechnol. 2018;36:952-65
48. Gross A, Schoendube J, Zimmermann S, Steeb M, Zengerle R, Koltay P. Technologies for single-cell isolation. Int J Mol Sci. 2015;16:16897-919
49. Zhang X, Wei X, Wei YJ, Chen ML, Wang JH. The up-to-date strategies for the isolation and manipulation of single cells. Talanta. 2020;218:121147
50. Muller S, Nebe-von-Caron G. Functional single-cell analyses: Flow cytometry and cell sorting of microbial populations and communities. FEMS Microbiol Rev. 2010;34:554-87
51. Shields CW, Reyes CD, Lopez GP. Microfluidic cell sorting: A review of the advances in the separation of cells from debulking to rare cell isolation. Lab Chip. 2015;15:1230-49
52. Roman GT, Chen YL, Viberg P, Culbertson AH, Culbertson CT. Single-cell manipulation and analysis using microfluidic devices. Anal Bioanal Chem. 2007;387:9-12
53. Zilberman D, Henikoff S. Genome-wide analysis of DNA methylation patterns. Development. 2007;134:3959-65
54. Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A. et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766-70
55. Wang JW, Xia YD, Li LL, Gong DS, Yao Y, Luo HJ. et al. Double restriction-enzyme digestion improves the coverage and accuracy of genome-wide CpG methylation profiling by reduced representation bisulfite sequencing. Bmc Genomics. 2013;14:11
56. Chatterjee A, Ozaki Y, Stockwell PA, Horsfield JA, Morison IM, Nakagawa S. Mapping the zebrafish brain methylome using reduced representation bisulfite sequencing. Epigenetics. 2013;8:979-89
57. Guo H, Zhu P, Wu X, Li X, Wen L, Tang F. Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Res. 2013;23:2126-35
58. Wang KL, Li XF, Dong SS, Liang JL, Mao FB, Zeng C. et al. Q-RRBS: a quantitative reduced representation bisulfite sequencing method for single-cell methylome analyses. Epigenetics. 2015;10:775-83
59. Gaiti F, Chaligne R, Gu H, Brand RM, Kothen-Hill S, Schulman RC. et al. Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia. Nature. 2019;569:576-80
60. Ma S, Revenga MD, Sun ZX, Sun C, Murphy TW, Xie HH. et al. Cell-type-specific brain methylomes profiled via ultralow-input microfluidics. Nat Biomed Eng. 2018;2:183-94
61. Guo HS, Zhu P, Guo F, Li XL, Wu XL, Fan XY. et al. Profiling DNA methylome landscapes of mammalian cells with single-cell reduced-representation bisulfite sequencing. Nat Protoc. 2015;10:645-59
62. Charlton J, Downing TL, Smith ZD, Gu H, Clement K, Pop R. et al. Global delay in nascent strand DNA methylation. Nat Struct Mol Biol. 2018;25:327-332
63. Shareef SJ, Bevill SM, Raman AT, Aryee MJ, van Galen P, Hovestadt V. et al. Extended-representation bisulfite sequencing of gene regulatory elements in multiplexed samples and single cells. Nat Biotechnol. 2021;39:1086-94
64. Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD. et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253-7
65. Brunner AL, Johnson DS, Kim SW, Valouev A, Reddy TE, Neff NF. et al. Distinct DNA methylation patterns characterize differentiated human embryonic stem cells and developing human fetal liver. Genome Res. 2009;19:1044-56
66. Kantlehner M, Kirchner R, Hartmann P, Ellwart JW, Alunni-Fabbroni M, Schumacher A. A high-throughput DNA methylation analysis of a single cell. Nucleic Acids Res. 2011;39:e44
67. Cheow LF, Quake SR, Burkholder WF, Messerschmidt DM. Multiplexed locus-specific analysis of DNA methylation in single cells. Nat Protoc. 2015;10:619-31
68. Han L, Wu HJ, Zhu HY, Kim KY, Marjani SL, Riester M. et al. Bisulfite-independent analysis of CpG island methylation enables genome-scale stratification of single cells. Nucleic Acids Res. 2017;45:e77
69. Bianchi A, Scherer M, Zaurin R, Quililan K, Velten L, Beekman R. scTAM-seq enables targeted high-confidence analysis of DNA methylation in single cells. Genome Biol. 2022;23:229
70. Sen M, Mooijman D, Chialastri A, Boisset JC, Popovic M, Heindryckx B. et al. Strand-specific single-cell methylomics reveals distinct modes of DNA demethylation dynamics during early mammalian development. Nat Commun. 2021;12:1286
71. Miura F, Enomoto Y, Dairiki R, Ito T. Amplification-free whole-genome bisulfite sequencing by post-bisulfite adaptor tagging. Nucleic Acids Res. 2012;40:e136
72. Smallwood SA, Lee HJ, Angermueller C, Krueger F, Saadeh H, Peet J. et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods. 2014;11:817-20
73. Clark SJ, Smallwood SA, Lee HJ, Krueger F, Reik W, Kelsey G. Genome-wide base-resolution mapping of DNA methylation in single cells using single-cell bisulfite sequencing (scBS-seq). Nat Protoc. 2017;12:534-47
74. Farlik M, Sheffield NC, Nuzzo A, Datlinger P, Schonegger A, Klughammer J. et al. Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Reports. 2015;10:1386-97
75. Kobayashi H, Koike T, Sakashita A, Tanaka K, Kumamoto S, Kono T. Repetitive DNA methylome analysis by small-scale and single-cell shotgun bisulfite sequencing. Genes Cells. 2016;21:1209-22
76. Mulqueen RM, Pokholok D, Norberg SJ, Torkenczy KA, Fields AJ, Sun D. et al. Highly scalable generation of DNA methylation profiles in single cells. Nat Biotechnol. 2018;36:428-31
77. Nichols RV, O'Connell BL, Mulqueen RM, Thomas J, Woodfin AR, Acharya S. et al. High-throughput robust single-cell DNA methylation profiling with sciMETv2. Nat Commun. 2022;13:7627
78. Luo C, Keown CL, Kurihara L, Zhou J, He Y, Li J. et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science. 2017;357:600-4
79. Luo C, Rivkin A, Zhou J, Sandoval JP, Kurihara L, Lucero J. et al. Robust single-cell DNA methylome profiling with snmC-seq2. Nat Commun. 2018;9:3824
80. Gu H, Raman AT, Wang X, Gaiti F, Chaligne R, Mohammad AW. et al. Smart-RRBS for single-cell methylome and transcriptome analysis. Nat Protoc. 2021;16:4004-30
81. Angermueller C, Clark SJ, Lee HJ, Macaulay IC, Teng MJ, Hu TX. et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat Methods. 2016;13:229-232
82. Hu Y, Huang K, An Q, Du G, Hu G, Xue J. et al. Simultaneous profiling of transcriptome and DNA methylome from a single cell. Genome Biol. 2016;17:88
83. Hou Y, Guo HH, Cao C, Li XL, Hu BQ, Zhu P. et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016;26:304-19
84. Hu Y, An Q, Guo Y, Zhong J, Fan S, Rao P. et al. Simultaneous profiling of mRNA transcriptome and DNA methylome from a single cell. Methods Mol Biol. 2019;1979:363-77
85. Pott S. Simultaneous measurement of chromatin accessibility, DNA methylation, and nucleosome phasing in single cells. Elife. 2017;6:23203
86. Guo F, Li L, Li J, Wu X, Hu B, Zhu P. et al. Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Res. 2017;27:967-88
87. Gu C, Liu S, Wu Q, Zhang L, Guo F. Integrative single-cell analysis of transcriptome, DNA methylome and chromatin accessibility in mouse oocytes. Cell Res. 2019;29:110-23
88. Kelly TK, Liu Y, Lay FD, Liang G, Berman BP, Jones PA. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 2012;22:2497-506
89. Cheow LF, Courtois ET, Tan Y, Viswanathan R, Xing Q, Tan RZ. et al. Single-cell multimodal profiling reveals cellular epigenetic heterogeneity. Nat Methods. 2016;13:833-6
90. Lorthongpanich C, Cheow LF, Balu S, Quake SR, Knowles BB, Burkholder WF. et al. Single-cell DNA-methylation analysis reveals epigenetic chimerism in preimplantation embryos. Science. 2013;341:1110-2
91. Niemoller C, Wehrle J, Riba J, Claus R, Renz N, Rhein J. et al. Bisulfite-free epigenomics and genomics of single cells through methylation-sensitive restriction. Commun Biol. 2021;4:153
92. Clark SJ, Argelaguet R, Kapourani CA, Stubbs TM, Lee HJ, Alda-Catalinas C. et al. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat Commun. 2018;9:781
93. Wang Y, Yuan P, Yan ZQ, Yang M, Huo Y, Nie YL. et al. Single-cell multiomics sequencing reveals the functional regulatory landscape of early embryos. Nat Commun. 2021;12:1247
94. Sheng K, Cao W, Niu Y, Deng Q, Zong C. Effective detection of variation in single-cell transcriptomes using MATQ-seq. Nat Methods. 2017;14:267-70
95. Mehrmohamadi M, Sepehri MH, Nazer N, Norouzi MR. A comparative overview of epigenomic profiling methods. Front Cell Dev Biol. 2021;9:714687
96. Guo HS, Zhu P, Yan LY, Li R, Hu BQ, Lian Y. et al. The DNA methylation landscape of human early embryos. Nature. 2014;511:606-10
97. Zhu P, Guo HS, Ren YX, Hou Y, Dong J, Li R. et al. Single-cell DNA methylome sequencing of human preimplantation embryos. Nat Genet. 2018;50:12-9
98. Li L, Guo F, Gao Y, Ren YX, Yuan P, Yan LY. et al. Single-cell multi-omics sequencing of human early embryos. Nat Cell Biol. 2018;20:847-58
99. Zhou F, Wang R, Yuan P, Ren YX, Mao YU, Li R. et al. Reconstituting the transcriptome and DNA methylome landscapes of human implantation. Nature. 2019;572:660-4
100. Gan YL, Li N, Xin YC, Zou GB. TriPCE: A novel tri-clustering algorithm for identifying pan-cancer epigenetic patterns. Front Genet. 2020;10:1298
101. Lee JE, Kim MY. Cancer epigenetics: Past, present and future. Semin Cancer Biol. 2022;83:4-14
102. Jeong HM, Kwon MJ, Shin YK. Overexpression of cancer-associated genes via epigenetic derepression mechanisms in gynecologic cancer. Front Oncol. 2014;4:12
103. Li W, Chen BF. Aberrant DNA methylation in human cancers. J Huazhong Univ Sci Technolog Med Sci. 2013;33:798-804
104. Mehdipour P, Murphy T, De Carvalho DD. The role of DNA-demethylating agents in cancer therapy. Pharmacol Ther. 2020;205:107416
105. Wang Z, Yin J, Zhou W, Bai J, Xie Y, Xu K. et al. Complex impact of DNA methylation on transcriptional dysregulation across 22 human cancer types. Nucleic Acids Res. 2020;48:2287-302
106. Giraldo NA, Sanchez-Salas R, Peske JD, Vano Y, Becht E, Petitprez F. et al. The clinical role of the TME in solid cancer. Br J Cancer. 2019;120:45-53
107. Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science. 2017;357:eaal2380
108. Yan X, Xie Y, Yang F, Hua Y, Zeng T, Sun C. et al. Comprehensive description of the current breast cancer microenvironment advancements via single-cell analysis. J Exp Clin Cancer Res. 2021;40:142
109. Roerink SF, Sasaki N, Lee-Six H, Young MD, Alexandrov LB, Behjati S. et al. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature. 2018;556:457-62
110. Li QL, Xue X, Li WZ, Wang Q, Han L, Brunson T. et al. Heterogeneous DNA methylation status in same-cell subpopulations of ovarian cancer tissues. Tumour Biol. 2017;39:1010428317701650
111. Bian SH, Hou Y, Zhou X, Li XL, Yong J, Wang YC. et al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science. 2018;362:1060-3
112. Johnson KC, Anderson KJ, Courtois ET, Gujar AD, Barthel FP, Varn FS. et al. Single-cell multimodal glioma analyses identify epigenetic regulators of cellular plasticity and environmental stress response. Nat Genet. 2021;53:1456-68
113. Sun X, Guo Y, Zhang Y, Zhao P, Wang Z, Wei Z. et al. Colon cancer-related genes identification and function study based on single-cell multi-omics integration. Front Cell Dev Biol. 2021;9:789587
114. Meir Z, Mukamel Z, Chomsky E, Lifshitz A, Tanay A. Single-cell analysis of clonal maintenance of transcriptional and epigenetic states in cancer cells. Nat Genet. 2020;52:709-18
115. Fan X, Lu P, Wang H, Bian S, Wu X, Zhang Y. et al. Integrated single-cell multiomics analysis reveals novel candidate markers for prognosis in human pancreatic ductal adenocarcinoma. Cell Discov. 2022;8:13
116. Batth IS, Mitra A, Manier S, Ghobrial IM, Menter D, Kopetz S. et al. Circulating tumor markers: harmonizing the yin and yang of CTCs and ctDNA for precision medicine. Ann Oncol. 2017;28:468-77
117. Chen HY, Su Z, Li RY, Zhang N, Guo H, Bai F. Single-cell DNA methylome analysis of circulating tumor cells. Chinese J Cancer Res. 2021;33:391-404
118. Gkountela S, Castro-Giner F, Szczerba BM, Vetter M, Landin J, Scherrer R. et al. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell. 2019;176:98-112 e14
119. Pixberg CF, Raba K, Muller F, Behrens B, Honisch E, Niederacher D. et al. Analysis of DNA methylation in single circulating tumor cells. Oncogene. 2017;36:3223-31
120. Xie W, Barr CL, Kim A, Yue F, Lee AY, Eubanks J. et al. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell. 2012;148:816-31
121. Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND. et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905
122. Guo JU, Su Y, Shin JH, Shin J, Li H, Xie B. et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci. 2014;17:215-22
123. Cerrizuela S, Kaya O, Kremer LPM, Sarvari A, Ellinger T, Straub J. et al. High-throughput scNMT protocol for multiomics profiling of single cells from mouse brain and pancreatic organoids. STAR Protoc. 2022;3:101555
124. Liu H, Zhou J, Tian W, Luo C, Bartlett A, Aldridge A. et al. DNA methylation atlas of the mouse brain at single-cell resolution. Nature. 2021;598:120-8
125. Li YE, Preissl S, Hou X, Zhang Z, Zhang K, Qiu Y. et al. An atlas of gene regulatory elements in adult mouse cerebrum. Nature. 2021;598:129-36
126. Gonzalo S. Epigenetic alterations in aging. J Appl Physiol. 2010;109:586-97
127. Jylhava J, Pedersen NL, Hagg S. Biological age predictors. EBioMedicine. 2017;21:29-36
128. Trapp A, Kerepesi C, Gladyshev VN. Profiling epigenetic age in single cells. Nat Aging. 2021;1:1189-201
129. Veitia RA, Govindaraju DR, Bottani S, Birchler JA. Aging: Somatic mutations, epigenetic drift and gene dosage imbalance. Trends Cell Biol. 2017;27:299-310
130. Hernando-Herraez I, Evano B, Stubbs T, Commere PH, Jan Bonder M, Clark S. et al. Ageing affects DNA methylation drift and transcriptional cell-to-cell variability in mouse muscle stem cells. Nat Commun. 2019;10:4361
131. Gravina S, Dong X, Yu B, Vijg J. Single-cell genome-wide bisulfite sequencing uncovers extensive heterogeneity in the mouse liver methylome. Genome Biol. 2016;17:150
132. Raiber EA, Beraldi D, Martinez Cuesta S, McInroy GR, Kingsbury Z, Becq J. et al. Base resolution maps reveal the importance of 5-hydroxymethylcytosine in a human glioblastoma. NPJ Genom Med. 2017;2:6
133. Olova N, Krueger F, Andrews S, Oxley D, Berrens RV, Branco MR. et al. Comparison of whole-genome bisulfite sequencing library preparation strategies identifies sources of biases affecting DNA methylation data. Genome Biol. 2018;19:33
134. Liu YB, Siejka-Zielinska P, Velikova G, Bi Y, Yuan F, Tomkova M. et al. Bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution. Nat Biotechnol. 2019;37:424-9
135. Chen S, Teichmann SA. Completing the cancer jigsaw puzzle with single-cell multiomics. Nat Cancer. 2021;2:1260-2
136. Pennisi E. Upstart DNA sequencers could be a 'game changer'. Science. 2022;376:1257-8
137. Simmons SK, Lithwick-Yanai G, Adiconis X, Oberstrass F, Iremadze N, Geiger-Schuller K. et al. Mostly natural sequencing-by-synthesis for scRNA-seq using Ultima sequencing. Nat Biotechnol. 2023;41:204-11
138. Ozimski LL, Gremmelspacher D, Aceto N. A fatal affair: Circulating tumor cell relationships that shape metastasis. iScience. 2021;24:103073
139. Kim TM, Yoo JS, Moon HW, Hur KJ, Choi JB, Hong SH. et al. Distinct mutation profiles between primary bladder cancer and circulating tumor cells warrant the use of circulating tumors cells as cellular resource for mutation follow-up. BMC Cancer. 2020;20:1203
140. Herath S, Rad HS, Radfar P, Ladwa R, Warkiani M, O'Byrne K. et al. The role of circulating biomarkers in lung cancer. Front Oncol. 2022;11:801269
141. Shi Y, Zhang H, Huang S, Yin L, Wang F, Luo P. et al. Epigenetic regulation in cardiovascular disease: Mechanisms and advances in clinical trials. Signal Transduct Target Ther. 2022;7:200
142. Zhong J, Agha G, Baccarelli AA. The role of DNA methylation in cardiovascular risk and disease: Methodological aspects, study design, and data analysis for epidemiological studies. Circ Res. 2016;118:119-31
143. Kim M, Long TI, Arakawa K, Wang R, Yu MC, Laird PW. DNA methylation as a biomarker for cardiovascular disease risk. PLoS One. 2010;5:e9692
144. Fernandez-Sanles A, Sayols-Baixeras S, Subirana I, Senti M, Perez-Fernandez S, de Castro Moura M. et al. DNA methylation biomarkers of myocardial infarction and cardiovascular disease. Clin Epigenetics. 2021;13:86
145. Uzun Y, Wu H, Tan K. Predictive modeling of single-cell DNA methylome data enhances integration with transcriptome data. Genome Res. 2021;31:101-9
146. Mikkelsen TS, Hanna J, Zhang X, Ku M, Wernig M, Schorderet P. et al. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49-55
147. Lee DS, Luo C, Zhou J, Chandran S, Rivkin A, Bartlett A. et al. Simultaneous profiling of 3D genome structure and DNA methylation in single human cells. Nat Methods. 2019;16:999-1006
148. Argelaguet R, Clark SJ, Mohammed H, Stapel LC, Krueger C, Kapourani CA. et al. Multi-omics profiling of mouse gastrulation at single-cell resolution. Nature. 2019;576:487-91
Author contact
![]() Corresponding authors: gu_hongcangac.cn; xx-wang119com
Corresponding authors: gu_hongcangac.cn; xx-wang119com