Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Building blocks for the design...
Mechanisms of action of...
Challenges and Perspectives
Concluding remarks
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2023; 13(3):1028-1041. doi:10.7150/thno.81494 This issue Cite
Review
When three is not a crowd: trispecific antibodies for enhanced cancer immunotherapy
Antonio Tapia-Galisteo1,2,3, Marta Compte4, Luis Álvarez-Vallina1,2,3 ![]() , Laura Sanz5
, Laura Sanz5 ![]()
1. Immuno-oncology and Immunotherapy Group, Biomedical Research Institute Hospital 12 de Octubre, Madrid, Spain.
2. Cancer Immunotherapy Unit (UNICA), Hospital Universitario 12 de Octubre, Madrid, Spain.
3. H12O-CNIO Cancer Immunotherapy Clinical Research Unit, Spanish National Cancer Research Centre (CNIO), Madrid, Spain.
4. Department of Antibody Engineering, Leadartis S.L., Madrid, Spain.
5. Molecular Immunology Unit, Biomedical Research Institute Hospital Universitario Puerta de Hierro Majadahonda, Madrid, Spain.
Received 2022-12-3; Accepted 2022-12-31; Published 2023-1-22
Abstract

Despite the clinical success of the first bispecific antibody approved by the FDA against B cell malignancies (blinatumomab), many obstacles remain such as dosing, treatment resistance, and modest efficacy in solid tumors. To overcome these limitations, considerable efforts have been dedicated to the development of multispecific antibodies, opening up new avenues to address both the complex biology of cancer and the onset of anti-tumoral immune responses. Simultaneous targeting of two tumor-associated antigens is presumed to enhance cancer cell selectivity and reduce immune escape. Co-engagement of CD3, along with agonists of co-stimulatory molecules or antagonists of co-inhibitory immune checkpoint receptors in a single molecule, may revert T cell exhaustion. Similarly, targeting of two activating receptors in NK cells may improve their cytotoxic potency. And these are only examples of the potential of antibody-based molecular entities engaging three (or more) relevant targets. From the perspective of health care costs, multispecific antibodies are appealing, since a similar (or superior) therapeutic effect could be obtained with a single therapeutic agent as with a combination of different monoclonal antibodies. Despite challenges in production, multispecific antibodies are endowed with unprecedented properties, which may render them more potent biologics for cancer therapy.
Keywords: antibody engineering, cancer immunotherapy, trispecific antibody, T cell engager, NK cell engager
Introduction
In 2021, the 100th antibody-based product was approved by the FDA [1]. Approximately half of the antibodies (Ab) in the market are indicated for the treatment of oncological patients, confirming the key role of antibodies in cancer drug discovery. Most of them are conventional monospecific bivalent IgG, but seven are bispecific antibodies (BsAb), four of them approved in 2022, and another two are currently in review. As of March 2019, the commercial clinical pipeline included over 85 BsAb and ~86% were being tested in patients with cancer [2]. These data reflect the growing interest in the field of multispecific antibodies.
The first BsAb was described by Nisonoff et al. in early 1960s, based on the combination of binding fragments from two different polyclonal sera through mild re-oxidation [3]. The advent of monoclonal Ab (mAb) technology allowed the development of BsAb obtained from quadromas (hybrid-hybridomas) [4,5] or based on the chemical conjugation of Fab fragments obtained after pepsinization of hybridoma-derived mAb [6,7]. However, it was the recombinant DNA technology along with antibody engineering which prompted the eclosion of a thriving field, with a plethora of different formats [8], resembling or not native IgG. In fact, the first BsAb approved by the FDA for clinical use in 2014, blinatumomab, lacks an Fc region. As an evolution of BsAb, trispecific antibodies (TsAb) have experienced a sudden interest in the last few years. However, the proof of concept was provided more than 30 years ago, using conjugation of three Fab to obtain a molecule able to engage effector T cells with two different arms, and tumor cells with a third one [9,10]. As of Nov 2022, around 20 molecular entities with three (or even four) specificities are in early phase clinical trials for the treatment of hematological malignancies or solid tumors. This review outlines the fundamental strategies used for the generation of multispecific Ab, describes recent advances and emerging applications and provides future perspectives for this field.
Building blocks for the design of multispecific antibodies
As previously described for BsAb [8,11], TsAb can be divided in IgG-like formats (with a Fc region) and non-IgG-like formats, (without a Fc region), according to their similarity with the conventional IgG structure (Figure 1). Non-IgG-like formats consists of antibody-derived building blocks fused via flexible linkers, the simplest arrangement being 'beads-on-string'. From higher to smaller molecular weight, these binding domains can be antigen-binding fragments (Fab), single-chain variable fragments (scFv) or single domain antibodies (sdAb). A comprehensive review on the building blocks used to tailor the design of multispecific Ab can be found in Wu et al. [12]; here, we will only address some basic concepts.
Schematic representation of some formats of multispecific antibodies with (A) or without (B) Fc domain. Numbers in brackets are the references to the original papers.
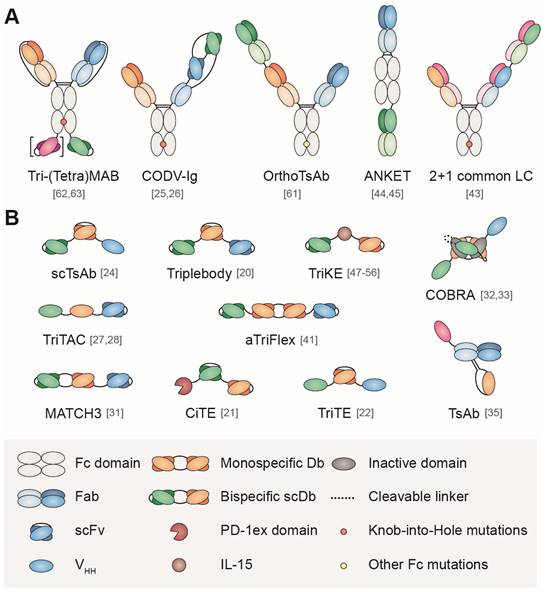
Fab fragments (50 kDa), comprise the full-length light (L) chain (VL and CL domains) and VH and CH1 domains of the heavy (H) chain from a conventional IgG, connected by an interchain disulfide bond. On the other hand, scFv are moieties of 25-30 kDa, which contain only VH and VL domains joined by a flexible polypeptide linker, usually (GGGGS)3. Finally, sdAb (12-15 kDa) consist of a unique variable domain derived from heavy-chain only antibodies present in camelids (named VHH) or cartilaginous fish (named VNAR). Obviously, the non-human nature of sdAb fragments makes them potentially immunogenic, although humanization approaches have been developed to mitigate this problem. Coding sequences of variable domains can be obtained from IgG with known specificities or by selection in vitro of large Ab libraries using phage, bacterial or yeast display. Of note, scFv and sdAb are easily produced as soluble, properly folded and stable recombinant proteins in bacteria, yeast and mammalian systems, with suitable ranges of affinities [13,14].
Multimerization of these building blocks enables the modification of antibody size, shape, and valence to optimize half-life and tumor penetration. For instance, the tandem scFv or (scFv)2 BsAb format, consists of two scFv fused in a single polypeptide chain by a five-amino acid linker [14]. Similarly, the assembly into a single Fc-less polypeptidic chain of three building blocks with different specificities allows the design of a variety of non-IgG-like TsAb formats, avoiding Fc-associated adverse effects in some contexts. In general, their smaller size in comparison with conventional IgG enhances tumor penetration, although at the cost of faster clearance [15].
On the other hand, IgG-like formats comprise molecules that partially or completely resemble natural antibodies, including a Fc region (functional or not). Designing Fc-based TsAb facilities dimerization, purification and promotes stability and extended half-life. Most of IgG-like formats consist of asymmetric chains, which include modifications in CH3 domains (such as the knob into hole -KIH- technology) in order to enforce the heterodimerization ot two different heavy chains. The correct pairing of light chains is a challenge that is addressed with different strategies. However, a percentage of non-functional molecules or undesirable homodimers can still cause impurities. BsAb in IgG format may gain a third specificity by appending an antibody fragment to the N- or C-terminal ends of heavy or light chains. However, the large molecular weight of IgG-like TsAb formats hampers tumor penetration, although guarantee long serum half-lifes.
Mechanisms of action of trispecific antibodies for cancer treatment
The main application field of TsAb remains oncology, with at least one of the three specificities intended to bind T or NK cells, and at least one targeting a tumor-associated antigen (TAA) in most designs [16] (Table 1).
Tri- and tetraspecific antibodies in preclinical development
| Name | Format | Specificity | Model | Ref |
|---|---|---|---|---|
| T cell-redirecting | ||||
| TsAb | F(ab)3 | CD2/CD3/CD37 | LNH | Tutt et al. [9] |
| Tribody | Fab-(scFv)2 | BCL1/hPLAP/CD3 | Mouse lymphoma | Schoonjans et al. [17] |
| A-2019 | scFv-Fab-scFv | CD19/CD3/CD20 | B-ALL | Wang et al. [18] |
| TsAb | scFv-Fab-Nb | CD19/CD22/CD3 | B-ALL | Zhao et al. [19] |
| triplebody | (scFv)3 | CD33/CD3/CD19 | MLL | Roskopf et al. [20] |
| CiTE | PD-1ex-scFv-scFv | PD-L1/CD3/CD33 | AML | Herrmann et al. [21] |
| TsAb (TriTE) | VHH-scFv-VHH | EpCAM/CD3/EGFR | CRC | Tapia-Galisteo et al. [22] |
| TsAb | F(ab)3 | TA/CD3/CD28 | Melanoma | Jung et al. [10] |
| TR3H (scTsAb) | scFv-VH-VH | OCA/CD3/CD28 | Ovarian cancer | Liu et al. [23] |
| scTsAb | scFv-scFv-VH | CEA/CD3/CD28 | CRC | Wang et al. [24] |
| CD38 TsAb | Fab-Fc-CODV-Fab | CD38/CD3/CD28 | MM, NHL | Wu et al. [25] |
| HER2 TsAb | Fab-Fc-CODV-Fab | HER2/CD3/CD28 | HER2+ solid tumors | Seung, et al. [26] |
| TriTAC | VHH-VHH-scFV | PSMA/HSA/CD3 | Prostate cancer | Austin et al. [27] |
| TriTAC | VHH-VHH-scFV | MSLN/HSA/CD3 | MSLN+ tumors | Molloy et al. [28] |
| LiTCo-Albu | VHH-scFv-Albumin | EGFR/CD137/HSA | CRC | Hangiu et al. [29] |
| CB213 (humabody) | (Human VH)4 | LAG3/LAG3/PD1/HSA | CRC | Edwards et al. [30] |
| MATCH3 (TsAb) | Db-scFc | CD137/PDL-1/HSA | NSCLC | Warmuth et al. [31] |
| COBRA | VHH-scFc-VHH | EGFR/CD3/HSA | CRC | Panchal et al. [32]; Dettling et al. [33] |
| MATCH4 (TtsAb) | scFv-Db-scFv | IL-23R/IL-5R/TNFa/CD3 | N/A | Egan et al. [34] |
| TsAb | VHH-Fab-VHH | HER/VEGFR2/CD3 | Breast, prostate cancer | Liu et al. [35] |
| NK cell-redirecting | ||||
| triplebody | ULBP2-scFv-scFv | NKG2D/CD19/CD33 | MLL | Vyas et al. [36] |
| triplebody | (scFv)3 | CD33/CD16/CD19 | MLL | Schubert et al. [37] |
| triplebody (sctb) | (scFv)3 | CD123/CD16/CD33 | AML | Kugler et al. [38] |
| triplebody SPM-2 | (scFv)3 | CD33/CD16/CD123 | AML | Braciak et al. [39] |
| TriKE | (scFv)3 | CD16/CD22/CD19 | B-ALL, B-CLL, AML | Gleason et al. [40] |
| ATriFlex | scFv-diabody-scFv | BCMA/CD200/CD16A | MM | Gantke et al. [41] |
| SEEDbody | IgG-like VHH-based | EGFR/HER2/NKG2D | Breast cancer | Pekar et al. [42] |
| TsAb | Bs IgG-Fab | EGFR/CD16a/PD-L1 | Epidermoid carcinoma | Bogen et al. [43] |
| ANKET | Fab-Fc-Fab | NKp46/CD16/CD19-CD20-EGFR | LNH | Gauthier et al. [44] |
| ANKET | Fab/Fc/Fab | NKp46-NKp30/CD16/ CD19-CD20 | B-ALL | Colomar et al. [45] |
| ANKET4 | Fab/Fc/Fab | NKp46/CD16/CD20/IL-2v | B-ALL | Demaria et al. [46] |
| TriKE (GTB-3550) | scFv/IL-15/scFv | CD16/IL-15/CD33 | AML | Vallera et al. [47]; Felices et al. [48] |
| TriKE | scFv/IL-15/scFv | CD16/IL-15/EpCAM | Various carcinomas | Schmohl et al. [49] |
| TriKE | scFv/IL-15/scFv | CD16/IL-15/CD133 | Cancer stem cells | Schmohl et al. [50] |
| TriKE | scFv/IL-15/scFv | CD16/IL-15/CD19 | B-CLL | Felices et al. [51] |
| TriKE | VHH/IL-15/scFv | CD16/IL-15/B7H3 | Ovarian cancer | Vallera et al. [52] |
| TriKE | VHH/IL-15/scFV | CD16/IL-15/HER2 | Ovarian cancer | Vallera et al. [53] |
| TriKE | VHH/IL-15/scFV | CD16/IL-15/CLEC12A | AML | Arvindam et al. [54] |
| TriKE | VHH/IL-15/scFV | CD16/IL-15/STEM8 | NSCLC | Kaminski et al. [55] |
| TriKE (GTB-3650) | VHH/IL-15/scFV | CD16/IL-15/CD33 | AML, MDS | Felices et al. [56] |
| HLE-nano-BiKE | (VHH)3 | CD38/CD16/HSA | MM | Hambach et al. [57] |
| DuoBody (DB)-VHH (TtsAb) | bs IgG-(VHH)2 | HER2/cMET/EGFR-IL6R-NKG2D | Breast cancer | Yanakieva et al. [58] |
| TetraKE (TtsAb) | scFv/IL-15/scFv/scFv | CD16/IL-5 /EpCAM/ CD133 | CRC | Schmohl et al. [59] |
| Oher MOA | ||||
| TAC | F(ab)3 | EGFR/HER2/CD64 | Breast cancer | Somasundaram et al. [60] |
| OrthoTsAb | Fab-Fab-Fab; bs IgG-Fab | PD-1/CTLA-4 /CD137;HER-2/EGFR/cMet | N/A | Wu et al. [61] |
| TriMAb | Bs IgG-scFv | EGFR/IGF1R/cMet-HER3 | Pancreas cancer | Castoldi et al. [62] |
| TetraMab | Bs IgG-(scFv)2 | EGFR/IGF1R/cMet/HER3 | NSCLC | Castoldi et al. [63] |
| GB263T | enhanced Fc/NA | EGFR/cMET/cMET* | NSCLC | Du et al. [64] |
| TsAb | IgG-(scDb)2 | EphA2/EphB4/EphA4 | Prostate cancer | Dimasi et al. [65] |
| mAb2, Fab-exchanged TsAb | Fab-Fcab-Fab | EGFR/VEGF/Her2 | Epidermoid carcinoma | Natale et al. [66] |
| TsAb | scFv/scFv/Fab | HER2/FAP/mPEG | Breast cancer | Chen et al. [67] |
*Binds two different cMet epitopes.
Immune cell engagers (ICE)
Immune cell engagers are molecules able to redirect immune effector cells (regardless of their antigen specificity) against cancer cells with the aim of triggering efficient tumor cell killing, acting as a bridge between immune cells and target cells [68]. Catumaxomab and blinatumomab were the first ICE approved for clinical use, both with a bispecific format and including an anti-CD3 moiety for T cell redirection. Substitution of anti-CD3 binding domain with an anti-CD16 one allows the recruitment of NK cells. In both cases, the addition of a third binding domain endows ICE with new and appealing properties, including increased specificity for tumors, or enhanced immune-cell activation.
T cells engagers (TCE)
Dual targeting of tumor cells by T cells
Beyond increasing the avidity and size to overcome fast clearance in small antibody fragment-based constructs, the incorporation of a second in-cis binding antibody moiety targeting a different TAA expressed on the same tumor cells offers additional advantages. For example, dual TAA-targeting may contribute to prevent tumor escape by antigen loss caused by selective pressure in comparison to conventional single TAA-targeting TCE and may help to overcome antigenic heterogeneity.
In a pioneering study, Schoonjans et al. described in 2000 a TsAb format, which they refer to as tribody, constituted by the fusion of two scFv against different TAA (BCL1 and hPLAP) to an anti-CD3 Fab, rendering an intermediate sized construct with a molecular weight around 100 kDa [17]. Moreover, they showed concurrent binding to the three cognate antigens.
Subsequently, dual TAA-targeting TsAb have been designed for leukemic cell killing, as an evolution of the CD19/CD3 BiTE (Bispecific T-cell Engager) blinatumomab, a CD3-based tandem scFv. This BsAb has shown remarkable efficacy in patients with B-ALL, although the durability of the response is limited, and the relapse rate is ~50% after 12-18 months. Interestingly, around 30% of blinatumomab-relapsed cases are characterized by CD19 negative leukemic cells [69]. To potentially overcome CD19 antigen escape, a second anti-CD20 scFv was added to a CD19/CD3 Fab-scFv BsAb, although TsAb efficacy in vitro was inferior to that of the BsAb [18]. In another work, a CD19/CD22/CD3 TsAb was designed by fusing anti-CD19 scFv and anti-CD22 sdAb to a CD3 binding Fab [19]. In this case, the TsAb was superior inducing T-cell specific cytotoxicity and cytokine production in vitro against CD19+ and/or CD22+ tumor cells and demonstrated significantly enhanced antitumor efficacy in a patient-derived xenograft (PDX) model of B-ALL, compared with the corresponding BsAb alone or in combination. Remarkably, leukemia relapse was observed in all animals, independently of the initial response, except in the group treated with the TsAb.
Leukemia cells with mixed B/myeloid origin are found in a small percentage and usually have poor prognosis. To deal with this condition, a CD33/CD3/CD19 TsAb, termed triplebody, was designed. Triplebodies consist of three scFv in tandem fused in a single polypeptide chain, with a molecular weight around 80-90 kDa. The two distal binding domains recognize different TAA, while the central domain recruits effector T cells. The CD33/CD3/CD19 triplebody showed efficient lysis of different ALL and AML cell lines, with enhanced selectivity for CD19+ CD33+ double-positive cells over CD19+ with comparable antigen density [20].
Fine-tuning affinity in TAA-binding domains can help to further increase tumor selectivity. Another TsAb designed for AML treatment, named CiTE (Checkpoint Inhibitory T cell-Engaging) Ab, combined T cell redirection to CD33 on AML cells with PD-L1 blockade on the same cells [21]. In this study, a PD-L1/CD3/CD33 triplebody structurally similar to the referred above was compared to a trifunctional construct where the high-affinity anti-PD-L1 scFv had been substituted by the low-affinity PD-1 extracellular domain. In such a construct, PD-L1 binding was dependent on CD33 targeting, restricting checkpoint blockade to the surface of leukemic cells. Consequently, the CiTE only induced lysis of CD33+, PDL1+ cells in vitro, whereas PD-L1+ non-AML cells were spared. Moreover, the CiTE construct achieved complete AML eradication in a PDX model without immune-related adverse effects.
In another work, a TsAb for dual targeting in solid tumors, termed trispecific T-cell engager (TriTE), was designed. This construct consists of a CD3-specific scFv flanked by an anti-epidermal growth factor receptor (EGFR) sdAb and an anti-epithelial cell (EpCAM) sdAb [22]. The TriTE enabled the specific cytolysis of EGFR- and/or EpCAM-expressing colorectal cancer cells. Bispecific bivalent targeting of double-positive HCT116 cells by TriTE improved in vitro potency up to 100-fold compared to single-positive cells and significantly prolonged survival in vivo compared to control BsAb. To avoid systemic toxicity, it has been proposed that a dual-TAA targeting BsAb should preferentially bind to malignant cells rather than normal cells if the affinity of the individual binding domains are sufficiently low as to require the presence of both target antigens (through the avidity effect) for efficient binding [70]. According to this principle, the TriTE antibody was less efficient at killing single-positive target cells than the corresponding BsAb controls, leading to potentially enhanced tumor specificity.
Recently, Liu and coworkers reported a thorough strategy to determine the most suitable fusion site of two VHH (anti-HER2 and anti-VEGFR2) to an anti-CD3 Fab for optimal antigen recognition. The resulting TsAb induced not only a more durable growth inhibition of double-positive tumors compared with the combination of corresponding BsAb, but also overcame tumor heterogeneity and elicited tumor regression in mice coimplanted with HER2+ or VEGFR2+ tumors in each flank. Moreover, the TsAb-based treatment inhibited tumor growth in a PC3 tumor model resistant to the approved mAb trastuzumab (anti-HER2) and ramucirumab (anti-VEGFR2). These results further supported the rationale that TAA dual targeting can improve the therapeutic index of TsAb [35].
Targeting of two T cell costimulatory and/or checkpoint receptors
Antigen-independent stimulation of T cells with canonical bivalent anti-CD3 mAb was demonstrated decades ago. On the other hand, monovalent CD3 binding, as it occurs with TCE comprising a single CD3-targeting domain, can induce T cell activation only upon crosslinking to TAA on targets cells. However, this must be accompanied by co-stimulatory signals for optimal activation and T cell proliferation. Furthermore, activated T cells, in turn, express inhibitory checkpoint receptors to control the immune response. Hence, the design of trispecific TCE including agonists of co-stimulatory molecules or antagonists of checkpoint receptors, along with the anti-CD3 and anti-TAA moieties, constitutes an appealing strategy. Such molecules, comprising an agonist of the co-stimulatory receptor CD28 (TAA/CD3/CD28), were expressed as single-chain polypeptides (in scFv-sdAb-sdAb or scFv-scFv-sdAb formats) and characterized in vitro for the killing of ovarian [23] or colon [24] cancer cells, respectively.
However, it was the study by Wu et al. [25], building on previous work by this group on a TsAb anti-HIV [71], that brought this modality of TsAb into the spotlight [72]. Using the Cross-Over Dual Variable (CODV) antibody format, different combinations of three binding domains (CD38/CD3/CD28) were mutated to assess the individual contributions of each specificity. To minimize the risk of cytokine release syndrome (CRS), the authors used the non-complement fixing IgG4 isotype, and introduced previously described mutations (F234A, L235A) into the Fc region to abrogate binding to Fc receptors. The presence of the anti-CD28 domain on the TsAb enhanced T cell activation and killing of different multiple myeloma (MM) cell lines in vitro, far above the potency of daratumumab (the anti-CD38 mAb approved for the treatment of MM). Similarly, the TsAb was superior in a humanized mouse model of MM, even at the lowest dose tested.
The use of CD28 agonists is associated to the risk of severe side effects due to cytokine release since the 2006 TeGenero clinical trial [73]. Importantly, maximum MM cell killing in vitro was observed at CD38/CD3/CD28 TsAb concentrations far below serum levels well tolerated in non-human primates (NHP). The authors argue that monovalent CD28 binding by the TsAb is less prone to induce cytokine release than the bivalent anti-CD28 IgG used in the above-mentioned trial. This TsAb, coined SAR442257, is currently in a phase I clinical trial with MM patients which eventually will dilucidate if safety concerns are justified [74] (Table 2).
Tri-and tetraspecific antibodies in clinical trials as of Nov 2022
| Drug name | Specificity | Indication | Clinical trial | Phase | Sponsor |
|---|---|---|---|---|---|
| SAR442257 (CODV-Fab) | CD38/CD3/CD28 | MM, NHL | NCT04401020 | 1 | Sanofi |
| SAR443216 (CODV-Fab) | HER2/CD3/CD28 | HER2+ solid tumors | NCT05013554 | 1 | Sanofi |
| SAR443579 (ANKET) | CD123/CD16/NKp46 | AML, MDS | NCT05086315 | 1/2 | Sanofi |
| CB307 (Humabody) | PSMA/CD137/HSA | PSMA+ tumors | NCT04839991 | 1 | Crescendo Biologics |
| HPN217 (TriTAC) | BCMA/HSA/CD3 | MM | NCT04184050 | 1 | Harpoon Therapeutics |
| HPN328 (TriTAC) | DLL3/HSA/CD3 | SCLC | NCT04471727 | 1/2 | Harpoon Therapeutics |
| HPN424 (TriTAC) | PSMA/HSA/CD3 | Prostate cancer | NCT03577028 | 1/2 | Harpoon Therapeutics |
| HPN536 (TriTAC) | MSLN/HSA/CD3 | MSLN+ tumors | NCT03872206 | 1/2 | Harpoon Therapeutics |
| MP0317 (DARPin) | CD40/FAP/HSA | Advanced Solid Tumors | NCT05098405 | 1 | Molecular Partners |
| MP0310 (DARPin) | CD137/FAP/HSA | Advanced Solid Tumors | NCT04049903 | 1 | Molecular Partners |
| GTB-3550 (TriKE) | CD16/IL-15/CD33 | AML | NCT03214666* | 1/2 | GT Biopharma |
| DF1001 (TriNKET) | HER2/CD16/NKG2D | HER2+ solid tumors | NCT04143711 | 1/2 | Dragonfly Therapeutics |
| GB263T | EGFR/cMET/cMET* | NSCLC | NCT05332574 | 1/2 | Genor Biopharma |
| NM21-1480 (scMATCH3) | PDL-1/CD137/HSA | NSCLC | NCT04442126 | 1/2 | Numab Therapeutics |
| GNC-035 | CD3/CD137/PD-L1/ROR1 | Breast cancer | NCT05160545 | 1 | Sichuan Baili/Systimmune |
| GNC-038 | CD3/CD137/PD-L1/CD19 | NHL | NCT04606433 | 1 | Sichuan Baili/Systimmune |
| GNC-039 | CD3/CD137/PD-L1/EGFRvIII | Glioma | NCT04794972 | 1 | Sichuan Baili/Systimmune |
| TAK-186 | EGFR/CD3/HSA | CCR, NSCLC, SCCHN | NCT04844073 | 1/2 | Takeda |
*Terminated, due to development of the second-generation TriKE GTB-3650.
A second TsAb HER2/CD3/CD28, based on the same platform, has recently joined it in clinical trials. In a humanized breast cancer model, this TsAb mediated effective tumor regression, even in low HER2-expressing tumors, suggesting a potential benefit to breast cancer patients who are not candidates for current anti-HER2 therapies [26]. Although CD8 T cells are widely accepted to be responsible for tumor cell elimination, an intriguing finding of this work was the main role of CD4 cells in the reported therapeutic effect.
TCE engineered for half-life extension
A third subtype of trispecific TCE comprises binding domains against a single TAA and a single T-cell activating receptor, along with a moiety for increased serum half-life. TsAb based on small Ab fragments, such as scFv and sdAb, usually exhibit molecular weights below the renal filtration cut-off, despite their multimeric nature. In fact, the 55-kDa BiTE blinatumomab has a serum half-life of only 2 hours and, therefore, continuous intravenous infusion is required [75].
The inclusion of albumin-binding sdAb is a widely used half-life extension strategy for small biotherapeutics. Albumin has a serum half-life of 3 weeks because of its size and FcRn-mediated recycling, properties shared with canonical IgG antibodies [76]. Half-life extension of antibody fragments by fusion to an albumin-binding sdAb has been demonstrated in different species in preclinical studies [77]. The great advantage of extended serum persistence is a more even drug concentration, less frequent dosing, and the potential to decrease doses without compromising therapeutical efficacy. Not surprisingly, a considerable proportion of TsAb currently in clinical trials have been half-life extended (HLE) using this strategy.
For example, the single-chain TriTAC (Trispecific T-cell Activating Construct) format, with a molecular weight of approximately 53 kDa, comprises a N-terminal humanized anti-TAA sdAb and a C-terminal humanized anti-CD3 scFv, separated by an anti-albumin sdAb which provides TriTAC with extended serum half-life. The serum levels of the anti-PSMA TriTAC HPN424 revealed a half-life of about 3.4 days in NHP [27] and up to 4.7 days for the anti-mesothelin TriTAC HPN536 [28]. As of November 2022, four TriTAC are in early phase clinical trials: the anti-BCMA (B cell maturation antigen) HPN217 for MM, the anti-DLL3 HPN328 for small cell lung cancer (SCLC), HPN424 for prostate cancer and HPN536 for mesothelin-positive solid tumors. In March 2022, the FDA granted Orphan Drug Designation to HPN328 for the treatment of patients with SCLC.
The MATCH3 (Multispecific Antibody-based Therapeutics by Cognate Heterodimerization) technology allows the assembly of three binding domains by fusion of a single-chain diabody to a scFv (scDb-scFv). NM21-1480 (PD-L1/CD137/HSA) combines in such a construct the ability to target CD137-mediated T cell costimulation to PD-L1 overexpressed in the tumor microenvironment (avoiding liver toxicity which hampers anti-CD137 IgG) and simultaneously blocking the PD-1/PD-L1 axis. Combination of HSA binding with a molecular weight around 80 kD endows these molecules with a suggested plasma half-life in humans of up to 2 weeks [31]. NM21-1480 is being tested in Phase 1-2 clinical trial with non-small cell lung cancer (NSCLC) patients.
A similar strategy is exploited by the Humabody CB307, with three fully human sdAb targeting PSMA, CD137 and HSA (molecular weight <50 kDa), currently in phase I trial for the treatment of advanced PSMA+ tumors. CB307 functions as a CD137 agonist selectively in the presence of PSMA+ cancer cells, thereby enabling tumor-specific T cell activation and decreasing the risk of systemic side effects. Based on the same technology, CB213 is a tetravalent trispecific Humabody engaging simultaneously PD1 and LAG3 on T cells and HSA. CB213 showed superior activity compared to an anti-PD1 mAb to induce ex vivo proliferation of T cells from NSCLC patients and to suppress tumor growth in vivo. In addition, NHP pharmacokinetics suggested a potential weekly clinical administration, which is a favorable regimen considering the small size of these molecules [30].
However, non-covalent HSA binding by sdAb is susceptible to competition and displacement by endogenous ligands and other albumin binding drugs. In contrast, the genetic fusion of drugs to recombinant HSA offers the opportunity to control half-life by the inclusion of albumin sequences with different FcRn binding affinities [78]. Recently, a new class of tumor-specific CD137 agonists was generated, named light T cell costimulatory (LiTCo) antibodies, composed of an anti-EGFR sdAb and a CD137-specific scFv. Genetic fusion to a human albumin sequence engineered for high affinity FcRn binding (LiTCo-Albu) demonstrated a prolonged circulatory half-life of 30 hours and in vivo tumor inhibition [29].
NK cells engagers (NKCE)
Multispecific molecules targeting one (or two) activating NK cell receptors and a TAA constitute another type of ICE, developed to enhance tumor cell recognition and killing by NK cells [79]. Among activating NK cell receptors, a hierarchy of CD16 > NKp46 > NKG2D has been established on the basis of their capability to trigger resting NK cells [80]. Most of the constructs that address CD16 (FcγRIIIa) on NK cells contain a CD16-specific scFv moiety instead of an IgG Fc domain, the natural ligand for CD16, to avoid side effects associated with the Fc domain [81]. Moreover, previous works have demonstrated that directly engaging CD16 on NK cells with a scFv can be more potent than CD16 engagement of Fc domains [82] and even have better safety profiles [74].
Dual targeting of tumor cells by NK cells
As in TCE, concurrent binding to two different TAA has also been implemented in NKCE antibodies to increase their therapeutic window. Addition of an extra anti-CD19 scFv to a BsAb CD19/CD16 rendered a construct with a molecular weight around 90 kDa termed single-chain Fv triplebody [83]. Incorporation of the second tumor-binding scFv led to increased avidity for CD19+ leukemia cells, promoted NK-mediated antibody-dependent cellular cytotoxicity (ADCC), and improved serum half-life. Dual targeting of AML cells was then been pursued with a TsAb CD33/CD16/CD123, which induced significantly stronger NK lysis of primary leukemic cells than the trivalent BsAb CD123/CD16/CD123 (38). Humanized versions of the three original murine scFv were used to produce the clinical candidate CD33/CD16/CD123 triplebody SPM-2 [39]. Additional triplebodies have been reported targeting simultaneously CD33 or HLA-DR and CD19, and recruiting NK cells via an anti-CD16 moiety [37,84] or using a NKG2D ligand [36]. A CD16/CD22/CD19 Ts Ab also based on a tandem scFv format was generated for the NK-mediated elimination of different types of leukemic cells [40].
Another CD16-directed trispecific, but tetravalent antibody format, termed aTriFlex, has been generated for the redirection of NK cell cytotoxicity to two multiple myeloma antigens. This format consists of an anti-CD16 monospecific diabody with two scFv in both ends, one anti-BCMA and another anti-CD200. Dual-targeting of multiple myeloma cells resulted in enhanced selectivity and 17-fold increase in potency [41].
IgG-like TsAb in this section include a sdAb-based SEEDbody, generated using the Strand-Exchanged Engineered Domains (SEED) heterodimerization technology, by replacement of the VH and VL regions of a conventional antibody by two different sdAb domains and grafting a third sdAb onto the hinge region of the second heavy chain [42]. As a proof of principle, an EGFR/HER2/NKG2D TsAb, which simultaneously bound each antigen, was obtained. Another TsAb was generated with the common light chain technology for the simultaneous targeting of EGFR, CD16 and PD-L1. This TsAb was based on a BsAb EGFR/CD16 IgG with an anti-PD-L1 arm N-terminally fused to the anti-CD16 Fab. The Fc-based approach was chosen to endow the molecule with prolonged half-life, mediated by the elevated molecular size (around 200 kDa) and FcRn-mediated recycling. From the mechanistical point of view, it should be noted that the PD-1/PD-L1 axis is an immune checkpoint for not only T cells but also for NK cells, and EGF signaling can induce PD-L1 upregulation. Simultaneous targeting of EGFR and PD-L1 on the same tumor cell increased TsAb avidity and tumor specificity, along with checkpoint inhibition, which translated into a more potent ADCC effect in vitro compared to the parental BsAb [43].
Dual Targeting of NK cell activation receptors
Full activation of NK cells to induce anti-tumor immunity has been shown to require the co-engagement of different cell-surface receptors. Based on this consideration, a series of molecules which bind a TAA and trigger two NK cell activation receptors have been designed.
The Antibody-based NK cell Engager Technology (ANKET) platform has generated a range of promising NKCE targeting different tumor antigens. In a pioneering work, Gauthier et al. reported the characterization of a trifunctional NKCE consisting of two Fab antibody fragments targeting NKp46 and a TAA (CD19, CD20 or EGFR), separated by an Fc domain to promote ADCC via CD16 [44]. Thereby, this format cannot be strictly considered as trispecific since it lacks an anti-CD16 moiety. These trifunctional molecules were effective against several tumor types in vitro, with no off-target cytotoxicity. In Raji B lymphoma models, mice treated with Fc-silenced NKCE or trifunctional NKCE controlled disease better than the group treated with the mAb rituximab, but the trifunctional construct was significantly more potent and outperformed a mixture of reagents activating NKp46 and CD16 separately. In a related work, trifunctional NKCE were designed targeting CD19 or CD20 and triggering either NKp46 or NKp30, aimed to the treatment of pediatric B-ALL [45]. In vitro experiments demonstrated efficient NK cell-mediated killing by all NKCE of leukemia cell lines and primary blasts, including the NK cell-resistant MHH-CALL-4. Moreover, a clinical study using an anti-NKp46-based ANKET targeting CD123 (IPH6101/SAR443579) is ongoing in patients with AML (NCT05086315). Another trifunctional ANKET (IPH6401/SAR'514), directed against BCMA, has recently been selected for clinical development.
A different platform for the development of multifunctional NKCE has been named TriNKET (Trispecific NKCE Therapies). DF1001 is one of several agents belonging to this platform, which targets HER2 and coengages CD16 and NKG2D [85]. The safety and efficacy of this HER2-targeting TriNKET are being investigated in a Phase 1/2 trial in patients with various advanced-stage HER2+ solid tumors, alone or in combination with immune checkpoint inhibitors.
IL-15-based trifunctional NK cells engagers
TriKEs (Trispecific Killer cell Engagers) are not strictly speaking TsAb, since they bind a TAA with one arm and CD16 with the other, while their third moiety is IL-15, which activates the recruited NK cells via interaction with the corresponding receptor. Nevertheless, since they are antibody-based, trifunctional molecules, they will be considered here, as proposed by Elsiathy et al. [16].
The first IL-15-based TriKE reported in the literature was an evolution of a previously characterized BiKE (Bispecific Killer cell Engager) based on a tandem scFv CD33/CD16 [47]. When compared with the BiKE, the TriKE induced superior NK cell cytotoxicity, degranulation, and cytokine production against the CD33+ AML cell line HL-60 and increased NK survival and proliferation in vitro. In a HL-60 mouse tumor model, the TriKE also showed enhanced antitumor activity relative to BiKE. Interestingly, TriKE-treated animals had substantial numbers of circulating human NK cells three weeks after, when they were hardly detectable in untreated or BiKE-treated animals. A phase 1 clinical trial (NCT03214666) with this TriKE, named GTB-3550, demonstrated absence of severe side effects, expansion of NK cells and clinical effect in patients with AML, with significant reduction in the numbers of leukemic blasts in the bone marrow [48]. However, it was terminated due to development of a second-generation TriKE. Other three first generation TriKE were generated against EpCAM [49], with activity in breast, prostate, ovarian and head and neck carcinomas; CD133, for the elimination of cancer stem cells [50]; and CD19, for the treatment of CLL [51].
Second generation TriKE were based on a humanized anti-CD16 sdAb, since the camelid CD16-engaging moiety provided further potency than the corresponding scFv [56]. A CD33-targeting second-generation TriKE induced more potent NK cell proliferation than its first-generation counterpart and showed enhanced tumor control in preclinical mouse models. This TriKE, coined GTB-3650, is the first clinical candidate being developed using this technology. Similarly, other second generation TriKE prototypes contain moieties anti B7-H3 for ovarian and lung carcinoma [52]; anti-CLEC12A, for AML [54]; and anti-HER2, for breast and ovarian carcinoma [53].
Very recently, it has been reported a second-generation TriKE with a novel mechanism of action, directed against the stroma-associated antigen TEM8 (Tumor Endothelial Marker 8) [55]. TEM8 is expressed on tumor and tumor stromal cells (endothelial cells, fibroblasts and pericytes) in the tumor microenvironment (TME) of different types of cancers. This TriKE selectively promoted NK cell degranulation, cytokine secretion and NK cell-mediated inhibition of tumor growth and tumor angiogenesis in A549 lung cancer xenograft models, along with increased numbers of tumor-infiltrating NK cells.
Targeting multiple receptors in tumor cells or immune effector cells
In this section, we will review TsAb which do not bridge tumor and effector cells, as they recognize in-cis antigens expressed on the surface of the same cell.
Multitargeting of tumor cells
Small molecule EGFR tyrosine kinase inhibitors are used for the treatment of NSCLC driven by EGFR mutations, but resistance frequently appears due to different mechanisms such as cMET amplification. The BsAb amivantamab, which recognizes EGFR and cMet and blocks their respective signalling pathways, was approved in 2021 for the treatment of NSCLC with EGFR exon 20 insertion mutations. The incorporation of a third binding domain to an in-cis targeting TsAb could potentially increase its efficacy. Indeed, a HER2/EGFR/cMet TsAb has been obtained using Fab interface designs to induce correct chain pairing, denoted OrthoTsAb. TsAb were designed with varied geometries, and overall, every construct demonstrated strong trispecific binding although their therapeutic potential was not addressed [61]. However, in the configuration with the anti-cMet Fab at the C-terminus of the TsAb, binding to this antigen was considerably impaired, suggesting steric inhibition of epitope recognition. Interestingly, the positioning effect on the binding affinity of the anti-EGFR VHH fused in the C-terminal end of TsAb has been previously described by different groups [22,27]. Modification of linker length and/or composition could be a strategy to improve the kinetic on-rates for binding moieties in unfavorable positions.
Another example of multiple tumor targeting is the IgG-like TriMAb directed against EGFR, IGF1R and cMet or HER3 [62]. The TsAb inhibited ligand-dependent receptor phosphorylation similarly to the parental antibodies. However, in a proliferation assay comparing TriMAb activity with individual antibodies or combinations, a superior growth inhibitory effect of the EGFR/IGF1R/HER3 TriMAb was observed. In addition, CD16 binding to the Fc domain of TriMAb was not impaired by scFv fusions at the C-terminus of the heavy chains.
A different approach is under the design of GB263T, an IgG-like TsAb recognizing EGFR and two different cMET epitopes, with enhanced ADCC function. In vitro, the TsAb blocked phosphorylation of both receptors and inhibited the proliferation of NSLC cells with different EGFR mutations. Significant in vivo anti-tumoral efficacy of GB263T was shown in several tumor models of EGFR exon 20 insertion, EGFR C797S mutation, cMET amplification, and cMET exon 14 skipping mutation. Currently, GB263T is under clinical testing in a phase 1 study (NCT05332574) [64].
On the other hand, ephrin (Eph) receptors constitute another family of tyrosine kinase receptors less explored as therapeutic targets, with EphA2, EphA4, and EphB4 found overexpressed in different types of carcinoma [65]. In the design of a TsAb targeting these three receptors, the anti-EphB4 and anti-EphA4 variable domains in a diabody configuration were linked to the C-terminal end of the heavy chains of a full-length anti-EphA2 IgG, rendering a bulky molecule around 250 kDa. Concurrent binding of the TsAb to the three antigens was demonstrated using BIAcore, although it did not translate into enhanced antitumor activity over parental antibodies.
An interesting approach could be the use of TsAb-based immunoconjugates for tumor delivery of drugs, toxins or radioisotopes, potentially increasing selectivity and therefore their therapeutic window.
Multitargeting of effector cells
Immunomodulatory mAb include immune checkpoint inhibitors and agonists of costimulatory receptors. Given the nonredundant signaling of the respective pathways, it is conceivable that combinations of agonist and antagonist antibodies could synergize to obtain higher response rates [86,87]. And doing so with a single molecule may offer additional benefits, although concerns about systemic unspecific T cell activation could rise since these constructs lack an anti-TAA moiety to redirect them. In the same work by Wu et al. the generation of a TsAb combining antagonists of PD-1 and CTLA-4 with a CD137 agonist was reported, although it was not functionally characterized [61].
Other mechanisms of action
Combinations of three different binding moieties in a single molecule offer a wide variety of new therapeutic options. A good example is the TsAb HER2/FAP/mPEG (scFv/scFv/Fab format) for conversion of mPEG (methoxypolyethylene glycol)-coated liposomes containing doxorubicin to immunoliposomes directed towards HER2+ breast cancer cells and tumor-associated fibroblasts (TAF) expressing FAP (fibroblast activation protein) [67]. Notably, the TsAb-decorated liposomes inhibited the growth of tumors containing TAFs (but not TAF-free tumors) more efficiently than liposomes modified with the control BsAb HER2/mPEG or FAP/mPEG.
Challenges and Perspectives
Beyond three different specificities
If antibody engineering allows the assembly in a single functional molecule of three different binding domains, adding a fourth one would not be a cumbersome task. Indeed, tetraspecific Ab (TtsAb) are not a future perspective any longer, but a reality. So real that some of them have reached clinical testing.
One of the most interesting platforms for this purpose is named CrossMAb. This technology is based on the crossover of VH and VL or CH1 and CL domains in one Fab arm of a Bs IgG to promote the correct assembling of light chains, along with the KIH technology to enforce the heterodimerization of heavy chains [88]. The 4-in-one CrossMab targeting EGFR, HER2, HER3 and VEGF [89] showed superior antitumor activity in different cancer models in vivo relative to combinations of BsAb, and they did so at lower antibody concentrations enabled by higher-avidity binding of tumor cells.
Another format amenable to TtsAb is coined MATCH4, which can be applied to produce homogeneous and stable antibody-derived molecules [34]. This concept is based on a core of 2 split variable domain pairs, with each chain containing either 2 VL domains or 2 VH domains in tandem, thereby driving heterodimerization of both chains. Additional scFv appended to the N-terminal end of both chains (or the N-terminus of one chain and the C-terminus of another) provide the two extra specificities. In a proof-of-concept study, tetraspecific heterodimers were generated in this combination with specificities for human TNFα, CD3, IL-5R and IL-23R. Antigen affinities of the binding domains in the MATCH formats were similar to those of the corresponding scFvs and single chain diabodies (scDbs), and the tetraspecific molecules were able to bind all target antigens simultaneously, irrespective of the order in which they encounter antigen.
The Tetramab concept is based on the combination of single-chain Fab and Fv fragments in an IgG antibody format enhanced by KIH technology [63]. A TsAb targeting HER3, cMet, HER1 and IGF1R was designed with the potential to avoid compensation mechanisms between different RTK signaling pathways. This construct was able to bind the cognate antigens with affinities in the nM range, comparable to the parental monospecific antibodies, and showed inhibition of RTK activation in pancreatic, mammary and lung tumor cells in presence of the corresponding growth factors. Importantly, the TetraMab showed improved tumor growth inhibition over individual monospecific or BsAb in co-cultures of different NSCLC cell lines.
Tetraspecific NKCEs also come in different formats. The DuoBody (DB)-VHH incorporates VHH with different specificities attached to the heavy chain C-terminal ends of a bispecific, Fc-silent IgG [58]. One or two VHH against EGFR, IL6R or NKG2D were appended to a HER2/cMET BsAb. Tri-and tetraspecific DB-VHH demonstrated binding to their cognate antigens and the NKG2D constructs showed promising results of NK cell-mediated cytotoxicity in vitro, although not significant differences were observed among them.
An evolution of TriKE format is TetraKE, which has been described to simultaneously engage EpCAM and CD133 for targeting carcinoma cells and cancer stem cells, along with a CD16-engaging moiety and IL-15 for NK cell expansion [59]. However, NK cytotoxic activity against EpCAM+, CD133+ Caco2 cells promoted by the TetraKE was similar to that of the EpCAM/CD16 BiKE.
Furthermore, a new type of multifunctional molecule based on the ANKET technology (named ANKET4) incorporates an IL-2 variant, binding IL-2 receptor β and γ chains, to NKp46 and CD16 co-engagement and targeting of a single TAA. ANKET4 induced NK cell proliferation and accumulation at the TME and had a higher anti-tumor efficacy than approved therapeutic antibodies targeting the same TAA. In NHP, a CD20-directed ANKET4 resulted in sustained CD20+ B-cell depletion with minimal systemic cytokine release and no clinical sign of toxicity [46].
On the other hand, the TriTECM platform enables the fusion of trispecific TCE to an anti-IL6R scFv antibody in order to mitigate the CRS mainly promoted by IL6. It is well known that on-target T cell activation is associated with release of cytokines that can potentially result in CRS, being this one of the major safety concerns intrinsic to the mode of action of TCE. Several EGFR/PD-L1/CD3/IL6R TriTECM candidates were generated by fusing one or two anti-IL6R scFv to the TsAb and ensuring the heterodimerization using KiH technology. In both cases, TriTECM antibodies inhibited simultaneously and dose-dependently EGFR and IL6R signaling pathways and triggered efficiently T cell activation and cytotoxicity against EGFR+ cells. In addition, TriTECM-mediated antitumor T cell activity was attenuated in comparison with EGFR/PD-L1/CD3 TsAb variants, indicating TriTECM potential as TCE with cytokine release modulator activity [90].
A search in clinicaltrials.gov as of November 2022 renders at least three tetraspecific TCE: GNC-038 (CD3/41BB/PD-L1/CD19), being tested in phase 1 trials with patients with NHL (NCT04606433) and DLBCL (NCT05192486); GNC-035 (CD3/41BB/PD-L1/ROR1) in patients with breast cancer; and GNC-039 (CD3/41BB/PD-L1/EGFRvIII) in glioma (NCT05160545).
Nonimmunoglobulin-trispecific binding proteins
An alternative to Ab-based constructs is the use of non-immunoglobulin scaffolds as building blocks [91]. Among the most advanced scaffold protein platforms are anticalins, affibodies and DARPins. DARPins are based on naturally occurring ankyrin repeat domains, which constitute a scaffold amenable to the generation of multispecific proteins [92]. Two trispecific DARPins are in clinical trials: MP0310 (CD137/FAP/HSA) and MP0317 (CD40/FAP/HSA). The first is intended to localize CD137 agonistic activity to FAP+ tumor-associated macrophages (TAM) to preferentially activate T cells in the TME. The second aims to selectively activate CD40 bearing B-cells, dendritic cells, and macrophages in the presence of FAP. In both cases, the objective is to avoid toxicity associated with systemic activation. Interestingly, suppressive TAM were repolarized by MP0317 to an anti-tumoral phenotype, restoring T cell responses [93,94].
Conditionally active trispecific T-cell engagers
Another interesting approach to widen the therapeutic window of TsAb is the use of conditionally active TCE, administered as inactive prodrugs, and designed for prolonged release (to avoid CRS) or tumor-restricted activation (to avoid on target-off tumor side effects) [95]. One example of the first option is the TriTAC-XR platform, which become slowly activated in systemic circulation, reducing the maximum drug concentration and prolonging dosing intervals, similar to subcutaneous dosing [96]. The prodrug was obtained by the incorporation of a peptide mask which binds to the anti-CD3 moiety and a protease-cleavable linker to the N-terminus of a TriTAC. Active TCE is released after cleavage of the linker by systemic proteases. In NHP, TriTAC-XR showed significantly reduced cytokine production while maintaining comparable pharmacodynamic properties as a non-masked TCE.
Other strategies include proteolytic cleavage sites in the TCE design to take advantage of the enhanced protease activity in the TME, as the ProTriTAC platform. An alternative to the masking peptide has been named COBRA (COnditional Bispecific Redirected Activation) where anti-CD3 VH and VL domains are forced to pair with inactive VL and VH domains in a diabody-like structure. Anti-EGFR and anti-HSA sdAb were added to the N- and C-terminal ends in a two-chain or single chain configuration [32]. MMP-9 release of the inactive domains allowed assembly of active anti-CD3 VH and VL domains on the surface of tumor cells. Complete regressions in colorectal or head and neck squamous cell carcinoma xenograft models were observed after treatment with the clinical COBRA candidate TAK-186, but not with the construct bearing non-cleavable linkers. Furthermore, TAK-186-mediated antitumoral effect was found to be dependent on the EGFR expression levels in tumors [33]. Currently, TAK-186 is the first and only trispecific conditionally active TCE in clinical trials in patients with EGFR+ solid tumors (NCT04844073).
Another EGFR/CD3/HSA trispecific construct named tumor-activated T cell engager (TRACTr) [97] contains two protease cleavable masks that inhibit both EGFR binding on target cells and CD3 engagement on T cells. Both cleavable JANX008 TRACTr and the non-masked construct induced complete eradication of HCT116 colorectal (CRC) tumors in mice, although the TRACTr exhibited enhanced safety and pharmacokinetic properties relative to the EGFR-TCE in NHP.
Finally, TwoGATE (Two component Guided Antibody Tumor Engager) platform consists of two HLE BsAb each targeting a different TAA and forming an active anti-CD3 moiety only when both bind to a cancer cell. Each of the CD3-targeting split paratopes are associated with an inactivating domain cleaved by tumor-specific proteases in the TME.
In all approaches described above, the HLE domain (usually, an anti-HSA antibody fragment) is lost upon proteolytic activation, reducing the risk of systemic side effects if the active form of the TCE is released from the tumor.
Concluding remarks
The development of complex therapeutics such as TsAb and TtsAb offers important challenges in terms of manufacturability. Some constructs may suffer from compromised stability, resulting in unfolding and aggregation. In addition, production of multichain multispecifics may be hampered by non-desired by-products arising due to incorrect assembly. Good manufacturing practice-compliant processes should be optimized to solve these problems, although in some cases production yields for TsAb were found to be better than for BsAb [74].
Many of the studies commented in this review are only proof-of-concept, focusing mainly on the design, expression, and preliminary functional characterization of TsAb, but a portion of them offer compelling in vivo data, and overall, they reveal the growing interest in the TsAb field that has blossomed in recent years. Indeed, multispecific antibodies are expanding the toolbox of antitumor reagents given their improved ability to target the multifactorial nature of cancer.
Specially interesting will be the demonstration of the TsAb therapeutic effect in non-hematological cancers compared to leukemias or lymphomas, currently a challenge for more advanced clinical trial-stage strategies such as BsAb and CAR-T cells, hampered by the lack of suitable single target antigens, intratumoral heterogeneity and the hostile tumor microenvironment promoting exclusion and/or exhaustion of recruited effector cells. It can be expected that TsAb, in their variety of flavors, will be better equipped to deal with these hurdles.
Ongoing early-stage clinical trials, most of them in patients with solid tumors, will provide relevant information in the next future on the added value of TsAb compared to the plethora of immunotherapies in development.
Abbreviations
B-ALL: B acute lymphoid leukemia; AML: acute myeloid leukemia; ANKET: antibody-based NK cell engager therapeutics; BCMA: B-cell maturation antigen; BiTE: bispecific T-cell engager; BsAb: bispecific antibody; B-CLL: B chronic lymphoid leukemia; CiTE: checkpoint inhibitory T cell-engaging; CODV: cross-over dual variable format; CRC: colorectal cancer; CRS: cytokine release syndrome; Db: diabody; EGFR: epidermal growth factor receptor; EpCAM: epithelial cell adhesion molecule; Fab: antigen-binding fragment; FAP: fibroblast activation protein; Fcab: antigen-binding Fc fragment; HAS: human serum albumin; mAb: monoclonal antibody; MATCH: multispecific antibody-based therapeutics by cognate heterodimerization; MDS: myelodysplastic syndrome; MLL: mixed lineage leukemia; mPEG: methoxypolyethylene glicol; MM: multiple myeloma; MSLN: mesothelin; NHL: non-Hodgkin lymphoma; NSCLC: non-small cell lung cancer; OCA: ovarian cancer antigen; PD-1ex: PD-1 extracellular domain; PSMA: prostate-specific membrane antigen; scDb: single chain diabody; scFab: single chain Fab; scFv: single-chain variable fragment; sctb: single chain triplebody; scTsAb: single chain trispecific antibody; SCLC: small cell lung cáncer; sdAb: single domain antibody; TAA: tumor-associated antigen; TAC: tsAb conjugate; TetraKE: tetraspecific killer engager; TriKE: trispecific killer engager; TriNKET: trispecific, NK cell engager therapies; TriTE: trispecific T-cell engager; TsAb: trispecific antibody; ULBP2: natural ligand for human NKG2D receptor.
Acknowledgements
This study was funded by grants from Instituto de Salud Carlos III (PI19/00132), partially supported by the European Regional Development Fund (ERDF) to L.S., and by grants from the Spanish Ministry of Science and Innovation (PID2020-117323RB-100 and PDC2021-121711-100), the Carlos III Health Institute (DTS20/00089), the CRIS Cancer Foundation (FCRIS-2021-0090), the Spanish Association Against Cancer (PROYE19084ALVA), the Fundación ''La Caixa'' (HR21-00761 project IL7R_LungCan) and the Fundación de Investigación Biomédica 12 de Octubre Programa Investiga (2022-0082) to L.A-V. A.T-G. was supported by a predoctoral fellowship from Comunidad Autónoma de Madrid (PEJD-2018-PRE/BMD-8314).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mullard A. FDA approves 100th monoclonal antibody product. Nat Rev Drug Discov. 2021;20:491-5
2. Labrijn AF, Janmaat ML, Reichert JM, Parren PWHI. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov. 2019;18:585-608
3. Nisonoff A, Rivers MM. Recombination of a mixture of univalent antibody fragments of different specificity. Arch Biochem Biophys. 1961;93:460-2
4. Lindhofer H, Mocikat R, Steipe B, Thierfelder S. Preferential species-restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a single-step purification of bispecific antibodies. J Immunol. 1995;155:219-25
5. Milstein C, Cuello AC. Hybrid hybridomas and the production of bi-specific monoclonal antibodies. Immunol Today. 1984;5:299-304
6. Brennan M, Davison PF, Paulus H. Preparation of bispecific antibodies by chemical recombination of monoclonal immunoglobulin G1 fragments. Science. 1985;229:81-3
7. Glennie MJ, McBride HM, Worth AT, Stevenson GT. Preparation and performance of bispecific F(ab' gamma)2 antibody containing thioether-linked Fab' gamma fragments. J Immunol. 1987;139:2367-75
8. Brinkmann U, Kontermann RE. The making of bispecific antibodies. mAbs. 2017;9:182-212
9. Tutt A, Stevenson GT, Glennie MJ. Trispecific F(ab')3 derivatives that use cooperative signaling via the TCR/CD3 complex and CD2 to activate and redirect resting cytotoxic T cells. J Immunol. 1991;147:60-9
10. Jung G, Freimann U, Von Marschall Z, Reisfeld RA, Wilmanns W. Target cell-induced T cell activation with bi- and trispecific antibody fragments. Eur J Immunol. 1991;21:2431-5
11. Jin S, Sun Y, Liang X, Gu X, Ning J, Xu Y. et al. Emerging new therapeutic antibody derivatives for cancer treatment. Signal Transduct Target Ther. 2022;7:39
12. Wu X, Demarest SJ. Building blocks for bispecific and trispecific antibodies. Methods. 2019;154:3-9
13. Els Conrath K, Lauwereys M, Wyns L, Muyldermans S. Camel single-domain antibodies as modular building units in bispecific and bivalent antibody constructs. J Biol Chem. 2001;276:7346-50
14. Nuñez-Prado N, Compte M, Harwood S, Álvarez-Méndez A, Lykkemark S, Sanz L. et al. The coming of age of engineered multivalent antibodies. Drug Discov Today. 2015;20:588-94
15. Xenaki KT, Oliveira S, van Bergen En Henegouwen PMP. Antibody or antibody fragments: implications for molecular imaging and targeted therapy of solid tumors. Front Immunol. 2017;8:1287
16. Elshiaty M, Schindler H, Christopoulos P. Principles and current clinical landscape of multispecific antibodies against cancer. Int J Mol Sci. 2021;22:5632
17. Schoonjans R, Willems A, Schoonooghe S, Leoen J, Grooten J, Mertens N. A new model for intermediate molecular weight recombinant bispecific and trispecific antibodies by efficient heterodimerization of single chain variable domains through fusion to a Fab-chain. Biomol Eng. 2001;17:193-202
18. Wang S, Peng L, Xu W, Zhou Y, Zhu Z, Kong Y. et al. Preclinical characterization and comparison between CD3/CD19 bispecific and novel CD3/CD19/CD20 trispecific antibodies against B-cell acute lymphoblastic leukemia: targeted immunotherapy for acute lymphoblastic leukemia. Front Med. 2022;16:139-49
19. Zhao L, Li S, Wei X, Qi X, Liu D, Liu L. et al. A novel CD19/CD22/CD3 trispecific antibody enhances therapeutic efficacy and overcomes immune escape against B-ALL. Blood. 2022;140:1790-802
20. Roskopf CC, Braciak TA, Fenn NC, Kobold S, Fey GH, Hopfner K-P. et al. Dual-targeting triplebody 33-3-19 mediates selective lysis of biphenotypic CD19+ CD33+ leukemia cells. Oncotarget. 2016;7:22579-89
21. Herrmann M, Krupka C, Deiser K, Brauchle B, Marcinek A, Ogrinc Wagner A. et al. Bifunctional PD-1 × αCD3 × αCD33 fusion protein reverses adaptive immune escape in acute myeloid leukemia. Blood. 2018;132:2484-94
22. Tapia-Galisteo A, Sánchez Rodríguez Í, Aguilar-Sopeña O, Harwood SL, Narbona J, Ferreras Gutierrez M. et al. Trispecific T-cell engagers for dual tumor-targeting of colorectal cancer. Oncoimmunology. 2022;11:2034355
23. Liu J, Zhao Q, Zhao B, Cheng J, Wang X, Song L. et al. A new format of single chain tri-specific antibody with diminished molecular size efficiently induces ovarian tumor cell killing. Biotechnol Lett. 2005;27:1821-7
24. Wang X-B, Zhao B-F, Zhao Q, Piao J-H, Liu J, Lin Q. et al. A new recombinant single chain trispecific antibody recruits T lymphocytes to kill CEA (carcinoma embryonic antigen) positive tumor cells in vitro efficiently. J Biochem. 2004;135:555-65
25. Wu L, Seung E, Xu L, Rao E, Lord DM, Wei RR. et al. Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nat Cancer. 2020;1:86-98
26. Seung E, Xing Z, Wu L, Rao E, Cortez-Retamozo V, Ospina B. et al. A trispecific antibody targeting HER2 and T cells inhibits breast cancer growth via CD4 cells. Nature. 2022;603:328-34
27. Austin RJ, Lemon BD, Aaron WH, Barath M, Culp PA, DuBridge RB. et al. TriTACs, a Novel Class of T-Cell-Engaging Protein Constructs Designed for the Treatment of Solid Tumors. Mol Cancer Ther. 2021;20:109-20
28. Molloy ME, Austin RJ, Lemon BD, Aaron WH, Ganti V, Jones A. et al. Preclinical Characterization of HPN536, a Trispecific, T-Cell-Activating Protein Construct for the Treatment of Mesothelin-Expressing Solid Tumors. Clin Cancer Res. 2021;27:1452-62
29. Hangiu O, Compte M, Dinesen A, Navarro R, Tapia-Galisteo A, Mandrup OA. et al. Tumor targeted 4-1BB agonist antibody-albumin fusions with high affinity to FcRn induce anti-tumor immunity without toxicity. iScience. 2022;25:104958
30. Edwards CJ, Sette A, Cox C, Di Fiore B, Wyre C, Sydoruk D. et al. The multi-specific VH-based Humabody CB213 co-targets PD1 and LAG3 on T cells to promote anti-tumour activity. Br J Cancer. 2022;126:1168-77
31. Warmuth S, Gunde T, Snell D, Brock M, Weinert C, Simonin A. et al. Engineering of a trispecific tumor-targeted immunotherapy incorporating 4-1BB co-stimulation and PD-L1 blockade. OncoImmunology. 2021;10:2004661
32. Panchal A, Seto P, Wall R, Hillier BJ, Zhu Y, Krakow J. et al. COBRATM: a highly potent conditionally active T cell engager engineered for the treatment of solid tumors. mAbs. 2020;12:1792130
33. Dettling DE, Kwok E, Quach L, Datt A, Degenhardt JD, Panchal A. et al. Regression of EGFR positive established solid tumors in mice with the conditionally active T cell engager TAK-186. J Immunother Cancer. 2022;10:e004336
34. Egan TJ, Diem D, Weldon R, Neumann T, Meyer S, Urech DM. Novel multispecific heterodimeric antibody format allowing modular assembly of variable domain fragments. mAbs. 2017;9:68-84
35. Liu D, Qi X, Wei X, Zhao L, Wang X, Li S. et al. A Novel Her2/VEGFR2/CD3 trispecific antibody with an optimal structural design showed improved T-cell-redirecting antitumor efficacy. Theranostics. 2022;12:7788-7803
36. Vyas M, Schneider A-C, Shatnyeva O, Reiners KS, Tawadros S, Kloess S. et al. Mono- and dual-targeting triplebodies activate natural killer cells and have anti-tumor activity in vitro and in vivo against chronic lymphocytic leukemia. Oncoimmunology. 2016;5:e1211220
37. Schubert I, Kellner C, Stein C, Kügler M, Schwenkert M, Saul D. et al. A single-chain triplebody with specificity for CD19 and CD33 mediates effective lysis of mixed lineage leukemia cells by dual targeting. mAbs. 2011;3:21-30
38. Kügler M, Stein C, Kellner C, Mentz K, Saul D, Schwenkert M. et al. A recombinant trispecific single-chain Fv derivative directed against CD123 and CD33 mediates effective elimination of acute myeloid leukaemia cells by dual targeting. Br J Haematol. 2010;150:574-86
39. Braciak TA, Roskopf CC, Wildenhain S, Fenn NC, Schiller CB, Schubert IA. et al. Dual-targeting triplebody 33-16-123 (SPM-2) mediates effective redirected lysis of primary blasts from patients with a broad range of AML subtypes in combination with natural killer cells. Oncoimmunology. 2018;7:e1472195
40. Gleason MK, Verneris MR, Todhunter DA, Zhang B, McCullar V, Zhou SX. et al. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol Cancer Ther. 2012;11:2674-84
41. Gantke T, Weichel M, Herbrecht C, Reusch U, Ellwanger K, Fucek I. et al. Trispecific antibodies for CD16A-directed NK cell engagement and dual-targeting of tumor cells. Protein Eng Des Sel. 2017;30:673-84
42. Pekar L, Busch M, Valldorf B, Hinz SC, Toleikis L, Krah S. et al. Biophysical and biochemical characterization of a VHH-based IgG-like bi- and trispecific antibody platform. mAbs. 2020;12:1812210
43. Bogen JP, Carrara SC, Fiebig D, Grzeschik J, Hock B, Kolmar H. Design of a Trispecific Checkpoint Inhibitor and Natural Killer Cell Engager Based on a 2 + 1 Common Light Chain Antibody Architecture. Front Immunol. 2021;12:669496
44. Gauthier L, Morel A, Anceriz N, Rossi B, Blanchard-Alvarez A, Grondin G. et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell. 2019;177:1701-1713
45. Colomar-Carando N, Gauthier L, Merli P, Loiacono F, Canevali P, Falco M. et al. Exploiting Natural Killer Cell Engagers to Control Pediatric B-cell Precursor Acute Lymphoblastic Leukemia. Cancer Immunol Res. 2022;10:291-302
46. Demaria O, Vivier E, Vetizou M, Alvarez AB, Habif G, Bonnafous C. et al. 851 Harnessing innate immunity in cancer therapies: the example of natural killer cell engagers. J Immunother Cancer. 2021;9:A892-A892
47. Vallera DA, Felices M, McElmurry R, McCullar V, Zhou X, Schmohl JU. et al. IL15 Trispecific Killer Engagers (TriKE) Make natural killer cells specific to CD33+ targets while also inducing persistence, in vivo expansion, and enhanced function. Clin Cancer Res. 2016;22:3440-50
48. Felices M, Warlick E, Juckett M, Weisdorf D, Vallera D, Miller S. et al. 444 GTB-3550 tri-specific killer engager TriKETM drives NK cells expansion and cytotoxicity in acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) patients. J Immunother Cancer. 2021;9:doi 10.1136/jitc-2021-SITC2021.444
49. Schmohl JU, Felices M, Taras E, Miller JS, Vallera DA. Enhanced ADCC and NK cell activation of an anticarcinoma bispecific antibody by genetic insertion of a modified IL-15 cross-linker. Mol Ther. 2016;24:1312-22
50. Schmohl JU, Felices M, Oh F, Lenvik AJ, Lebeau AM, Panyam J. et al. Engineering of anti-CD133 trispecific molecule capable of inducing NK expansion and driving antibody-dependent cell-mediated cytotoxicity. Cancer Res Treat. 2017;49:1140-52
51. Felices M, Kodal B, Hinderlie P, Kaminski MF, Cooley S, Weisdorf DJ. et al. Novel CD19-targeted TriKE restores NK cell function and proliferative capacity in CLL. Blood Adv. 2019;3:897-907
52. Vallera DA, Ferrone S, Kodal B, Hinderlie P, Bendzick L, Ettestad B. et al. NK-cell-mediated targeting of various solid tumors using a B7-H3 tri-specific killer engager in vitro and in vivo. Cancers. 2020;12:2659
53. Vallera DA, Oh F, Kodal B, Hinderlie P, Geller MA, Miller JS. et al. A HER2 tri-specific NK cell engager mediates efficient targeting of human ovarian cancer. Cancers. 2021;13:3994
54. Arvindam US, van Hauten PMM, Schirm D, Schaap N, Hobo W, Blazar BR. et al. A trispecific killer engager molecule against CLEC12A effectively induces NK-cell mediated killing of AML cells. Leukemia. 2021;35:1586-96
55. Kaminski MF, Bendzick L, Hopps R, Kauffman M, Kodal B, Soignier Y. et al. TEM8 Tri-specific Killer Engager binds both tumor and tumor stroma to specifically engage natural killer cell anti-tumor activity. J Immunother Cancer. 2022;10:e004725
56. Felices M, Lenvik TR, Kodal B, Lenvik AJ, Hinderlie P, Bendzick LE. et al. Potent cytolytic activity and specific IL15 delivery in a second-generation trispecific killer engager. Cancer Immunol Res. 2020;8:1139-49
57. Hambach J, Fumey W, Stähler T, Gebhardt AJ, Adam G, Weisel K. et al. Half-life extended nanobody-based CD38-specific bispecific killer cell engagers induce killing of multiple myeloma cells. Front Immunol. 2022;13:838406
58. Yanakieva D, Pekar L, Evers A, Fleischer M, Keller S, Mueller-Pompalla D. et al. Beyond bispecificity: controlled Fab arm exchange for the generation of antibodies with multiple specificities. mAbs. 2022;14:2018960
59. Schmohl JU, Felices M, Todhunter D, Taras E, Miller JS, Vallera DA. Tetraspecific scFv construct provides NK cell mediated ADCC and self-sustaining stimuli via insertion of IL-15 as a cross-linker. Oncotarget. 2016;7:73830-44
60. Somasundaram C, Sundarapandiyan K, Keler T, Deo YM, Graziano RF. Development of a trispecific antibody conjugate that directs two distinct tumor-associated antigens to CD64 on myeloid effector cells. Hum Antibodies. 1999;9:47-54
61. Wu X, Yuan R, Bacica M, Demarest SJ. Generation of orthogonal Fab-based trispecific antibody formats. Protein Eng Des Sel. 2018;31:249-56
62. Castoldi R, Jucknischke U, Pradel LP, Arnold E, Klein C, Scheiblich S. et al. Molecular characterization of novel trispecific ErbB-cMet-IGF1R antibodies and their antigen-binding properties. Protein Eng Des Sel. 2012;25:551-9
63. Castoldi R, Schanzer J, Panke C, Jucknischke U, Neubert NJ, Croasdale R. et al. TetraMabs: simultaneous targeting of four oncogenic receptor tyrosine kinases for tumor growth inhibition in heterogeneous tumor cell populations. Protein Eng Des Sel. 2016;29:467-75
64. Du Q, Zeng W, Yang X, Liang L, Li X, Li Y. et al. Abstract LB538: Characterization of GB263T, a tri-specific antibody against EGFR/cMET/cMET for NSCLC. Cancer Res. 2022;82:LB538
65. Dimasi N, Fleming R, Hay C, Woods R, Xu L, Wu H. et al. Development of a trispecific antibody designed to simultaneously and efficiently target three different antigens on tumor cells. Mol Pharm. 2015;12:3490-501
66. Natale V, Stadlmayr G, Benedetti F, Stadlbauer K, Rüker F, Wozniak-Knopp G. Trispecific antibodies produced from mAb2 pairs by controlled Fab-arm exchange. Biol Chem. 2022;403:509-23
67. Chen M, Sheu M-T, Cheng T-L, Roffler SR, Lin S-Y, Chen Y-J. et al. A novel anti-tumor/anti-tumor-associated fibroblast/anti-mPEG tri-specific antibody to maximize the efficacy of mPEGylated nanomedicines against fibroblast-rich solid tumor. Biomater Sci. 2021;10:202-15
68. Fucà G, Spagnoletti A, Ambrosini M, de Braud F, Di Nicola M. Immune cell engagers in solid tumors: promises and challenges of the next generation immunotherapy. ESMO Open. 2021;6:100046
69. Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J. et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest. 2016;126:3814-26
70. Mazor Y, Sachsenmeier KF, Yang C, Hansen A, Filderman J, Mulgrew K. et al. Enhanced tumor-targeting selectivity by modulating bispecific antibody binding affinity and format valence. Sci Rep. 2017;7:40098
71. Xu L, Pegu A, Rao E, Doria-Rose N, Beninga J, McKee K. et al. Trispecific broadly neutralizing HIV antibodies mediate potent SHIV protection in macaques. Science. 2017;358:85-90
72. Garfall AL, June CH. Trispecific antibodies offer a third way forward for anticancer immunotherapy. Nature. 2019;575:450-1
73. Hünig T. The rise and fall of the CD28 superagonist TGN1412 and its return as TAB08: a personal account. FEBS J. 2016;283:3325-34
74. Mullard A. Trispecific antibodies take to the clinic. Nat Rev Drug Discov. 2020;19:657-8
75. Viardot A, Bargou R. Bispecific antibodies in haematological malignancies. Cancer Treat Rev. 2018;65:87-95
76. Andersen JT, Dalhus B, Viuff D, Ravn BT, Gunnarsen KS, Plumridge A. et al. Extending serum half-life of albumin by engineering neonatal Fc receptor (FcRn) binding. J Biol Chem. 2014;289:13492-502
77. Hoefman S, Ottevaere I, Baumeister J, Sargentini-Maier ML. Pre-Clinical Intravenous Serum Pharmacokinetics of Albumin Binding and Non-Half-Life Extended Nanobodies®. Antibodies. 2015;4:141-56
78. Mandrup OA, Ong SC, Lykkemark S, Dinesen A, Rudnik-Jansen I, Dagnæs-Hansen NF. et al. Programmable half-life and anti-tumour effects of bispecific T-cell engager-albumin fusions with tuned FcRn affinity. Commun Biol. 2021;4:310
79. Demaria O, Gauthier L, Debroas G, Vivier E. Natural killer cell engagers in cancer immunotherapy: Next generation of immuno-oncology treatments. Eur J Immunol. 2021;51:1934-42
80. Bryceson YT, March ME, Ljunggren H-G, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood. 2006;107:159-66
81. Vyas M, Koehl U, Hallek M, von Strandmann EP. Natural ligands and antibody-based fusion proteins: harnessing the immune system against cancer. Trends Mol Med. 2014;20:72-82
82. Johnson S, Burke S, Huang L, Gorlatov S, Li H, Wang W. et al. Effector cell recruitment with novel Fv-based dual-affinity re-targeting protein leads to potent tumor cytolysis and in vivo B-cell depletion. J Mol Biol. 2010;399:436-49
83. Kellner C, Bruenke J, Stieglmaier J, Schwemmlein M, Schwenkert M, Singer H. et al. A novel CD19-directed recombinant bispecific antibody derivative with enhanced immune effector functions for human leukemic cells. J Immunother. 2008;31:871-84
84. Schubert I, Kellner C, Stein C, Kügler M, Schwenkert M, Saul D. et al. A recombinant triplebody with specificity for CD19 and HLA-DR mediates preferential binding to antigen double-positive cells by dual-targeting. mAbs. 2012;4:45-56
85. Myers JA, Miller JS. Exploring the NK cell platform for cancer immunotherapy. Nat Rev Clin Oncol. 2021;18:85-100
86. Etxeberria I, Glez-Vaz J, Teijeira Á, Melero I. New emerging targets in cancer immunotherapy: CD137/4-1BB costimulatory axis. ESMO Open. 2020;4:e000733
87. Blanco B, Domínguez-Alonso C, Alvarez-Vallina L. Bispecific Immunomodulatory Antibodies for Cancer Immunotherapy. Clin Cancer Res. 2021;27:5457-64
88. Klein C, Schaefer W, Regula JT. The use of CrossMAb technology for the generation of bi- and multispecific antibodies. mAbs. 2016;8:1010-20
89. Hu S, Fu W, Xu W, Yang Y, Cruz M, Berezov SD. et al. Four-in-one antibodies have superior cancer inhibitory activity against EGFR, HER2, HER3, and VEGF through disruption of HER/MET crosstalk. Cancer Res. 2015;75:159-70
90. Carrara SC, Harwardt J, Grzeschik J, Hock B, Kolmar H. TriTECM: A tetrafunctional T-cell engaging antibody with built-in risk mitigation of cytokine release syndrome. Front Immunol. 2022;13:1051875
91. Hober S, Lindbo S, Nilvebrant J. Bispecific applications of non-immunoglobulin scaffold binders. Methods. 2019;154:143-52
92. Stumpp MT, Dawson KM, Binz HK. Beyond antibodies: The DARPin® drug platform. BioDrugs. 2020;34:423-33
93. Jeger S, Schönborn-Kellenberger O, Winter HD, Steeghs N, Gort E, Gomez-Roca C. et al. 170TiP A phase I study to characterize the safety and tolerability of MP0317, a tumor targeting FAP dependent CD40 agonist DARPin®, in patients with relapsed/refractory solid tumors. Ann Oncol. 2021;32:S1456-7
94. Ioannou K, Ragusa S, Roquette J, Florescu A, Gachechiladze M, Müller E. et al. MP0317, a CD40xFAP targeting multi-specific DARPin® therapeutic, drives immune activation and reverts myeloid-mediated T-cell suppression in vitro and ex vivo. Cancer Res. 2021;81:Abstract 1733
95. Baeuerle PA, Wesche H. T-cell-engaging antibodies for the treatment of solid tumors: challenges and opportunities. Curr Opin Oncol. 2022;34:552-8
96. Kwant K, Rocha S, Stephenson K, Dayao M, Thothathri S, Banzon R. et al. 867 TriTAC-XR is an extended-release T cell engager platform designed to minimize cytokine release syndrome by reducing Cmax in systemic circulation. J Immunother Cancer. 2021;9:A908
97. Chen TT. Conditionally active T cell engagers for the treatment of solid tumors: rationale and clinical development. Expert Opin Biol Ther. 2022;22:955-63
Author contact
![]() Corresponding authors: Luis Álvarez-Vallina, E-mail: lalvarezvcnio.es; Laura Sanz, E-mail: lsalcobermadrid.org.
Corresponding authors: Luis Álvarez-Vallina, E-mail: lalvarezvcnio.es; Laura Sanz, E-mail: lsalcobermadrid.org.