Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2023; 13(2):596-610. doi:10.7150/thno.78687 This issue Cite
Research Paper
Enhancer RNA promotes resistance to radiotherapy in bone-metastatic prostate cancer by m6A modification
Yu Zhao1# ![]() , Simeng Wen2#, Hang Li1#, Chun-Wu Pan3, Yulei Wei4, Ting Huang1, Zhaochen Li2, Yinhui Yang5, Saijun Fan1
, Simeng Wen2#, Hang Li1#, Chun-Wu Pan3, Yulei Wei4, Ting Huang1, Zhaochen Li2, Yinhui Yang5, Saijun Fan1 ![]() , Yingyi Zhang6
, Yingyi Zhang6 ![]()
1. Tianjin Key Laboratory of Radiation Medicine and Molecular Nuclear Medicine, Institute of Radiation Medicine, Chinese Academy of Medical Sciences and Peking Union Medical College, Tianjin, 300192, China
2. Department of Urology, The Second Hospital of Tianjin Medical University, Tianjin Medical University, Tianjin, 300211, China
3. Department of Urology, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, 200127, China
4. Department of Gynecology and Obstetrics, Tianjin First Central Hospital, Tianjin, 300192, China
5. Department of Urology, Shanghai Changhai Hospital, Naval Medical University, Shanghai, 200433, China
6. Department of Biochemistry and Molecular Biology, Mayo Clinic, Rochester, MN 55905, USA
#These authors contributed equally.
Received 2022-9-6; Accepted 2022-12-14; Published 2023-1-1
Abstract

Rationale: Prostate cancer metastasizes to the bone with the highest frequency and exhibits high resistance to 177Lu-prostate-specific membrane antigen (PSMA) radioligand therapy. Little is known about bone metastatic prostate cancer (mPCa) resistance to radiation.
Methods: We filtered the metastatic eRNA using RNA-seq, MeRIP-seq, RT-qPCR and bioinformation. Western blot, RT-qPCR, CLIP, co-IP and RNA pull-down assays were used for RNA/protein interaction, RNA or protein expression examination. MTS assay was used to determine cell viability in vitro, xenograft assay was used to examine the tumor growth in mice.
Results: In this study, we screened and identified bone-specific N6 adenosine methylation (m6A) on enhancer RNA (eRNA) that played a post-transcriptional functional role in bone mPCa and was correlated with radiotherapy (RT) resistance. Further data demonstrated that RNA-binding protein KHSRP recognized both m6A at eRNA and m6Am at 5'-UTR of mRNA to block RNA degradation from exoribonuclease XRN2. Depletion of the MLXIPe/KHSRP/PSMD9 regulatory complex inhibited tumor growth and RT sensitization of bone mPCa xenograft in vitro and in vivo.
Conclusions: Our findings indicate that a bone-specific m6A-modified eRNA plays a vital role in regulating mPCa progression and RT resistance and might be a novel specific predictor for cancer RT.
Keywords: Bone metastatic prostate cancer, m6A, Enhancer RNA, m6Am, Radiotherapy.
Introduction
Prostate cancer (PCa) is the most common malignancy in males worldwide and is the second-most common cause of cancer death [1, 2]. Metastasis is the leading cause of death in prostate cancer, with about 80% of metastasizing to bones, such as the hip, spine, and pelvis. Only 3% of patients with bone metastases survive for five years [1, 3-6]. Androgen deprivation therapy is the primary treatment for advanced prostate cancer; however castration-resistant prostate cancer (CRPC), especially metastatic prostate cancer (mPCa), remains a challenge for treatment [7]. Radiotherapy (RT) is another critical component of cancer management in over 50% of cancer patients [8, 9]. Even though RT shows benefits for high-risk localized advanced PCa, distant mPCa are RT resistant [7, 10, 11]. Recent clinical trials showed that androgen deprivation therapy in combination with radiation therapy for bone mPCa did not significantly differ in overall survival compared to androgen deprivation therapy alone [12]. The underlying molecular mechanism for RT resistance of bone mPCa cells remains unclear.
Among over 160 types of cellular RNA modifications, N6-adenosine methylation (m6A) is the most prevalent chemical modification of mRNAs in eukaryotes [13-15]. In eukaryotic cells, m6A is catalyzed by m6A methyltransferases METTL3 and METTL14 and demethylated by FTO and (ALKBH5) [16-18]. FTO was identified as the first RNA demethylase that catalyzes the reversal of m6A and 2-O-dimethyladenosine (m6Am) methylation on mRNAs [19]. The functional effects of mRNA m6A on cellular processes include RNA nuclear transport, secondary structure, splicing, stability and translation efficiency [20-29]. However, there is limited information on the m6A function in enhancer RNAs (eRNAs).
Tissue-specific or disease-specific gene expression is established primarily by the transcription complexes at enhancers [30], which are critical genomic elements that regulate transcription and are distant from gene promoters [31-33]. eRNAs are involved in the activation of gene expression and transcriptional regulation and act as regulators of adjacent elements and complexes in cis and trans modes [34-36]. Furthermore, eRNAs interact with cohesion complexes to facilitate promoter and enhancer looping [37]. Independent of their function in looping, eRNAs promote RNA polymerase-II (Pol-II) phosphorylation and elongation by activating the p-TEBb complex and removing the NELF complex [38, 39]. A recent study reported that eRNA facilitated transcriptional condensate formation through its m6A in Erα-related cells [40]. Due to the relevance of enhancers in cancer, immune diseases, and neurodegeneration, significant challenges need to be addressed in understanding the mechanisms of enhancers and designing new strategies for enhancer/eRNA targeting and novel therapeutic modalities in cancer medicine. However, the role and modification of eRNAs in cancer are not fully understood.
In this study, we identified a novel m6A function of eRNA that was involved in RT resistance in bone mPCa and demonstrated that eRNA m6A was associated with poor prognosis of patients with RT. Our data also showed that KHSRP recognized and interacted with m6A at MLXIPe eRNA and m6Am at 5'-UTR of mRNA to block RNA degradation by XRN2. Analysis of the regulatory complex involving eRNA/KHSRP/mRNA might provide new insights into the potential mechanism of RT resistance of bone metastatic prostate cancer. Thus, MLXIPe might be a potential predictive marker and therapeutic target for bone-mPCa.
Materials and Methods
Plasmids and antibodies
MLXIPe eRNA and KHSRP were generated by cloning the corresponding cDNAs into pcDNA3.1(+) vector with flag or myc tags designed in the primers. cDNA fragments were amplified by Phusion polymerase (NEB, USA) using Phusion High-Fidelity PCR Master Mix. The insert and deletion mutants were constructed using QuikChange II Site Directed-Mutagenesis Kit (Agilent, USA). Antibodies are shown in Table S4.
Clinical samples, cell lines, and patient-derived xenograft (PDX)
PCa primary tumors, bone metastases, and adjacent non-tumor tissues were obtained from patients at the Tianjin Second Hospital of Tianjin Medical University, Tianjin, between Jan 1, 2000 and Sep 1, 2020 in accordance with Tianjin Medical University Institutional Review Board approved protocols. Patient specimens were manually dissected using a sterile scalpel blade into 4 mm3 pieces and stored in a transport medium at 4 °C until xenograft preparation. Operative tumor tissue was mechanically disaggregated, triturated through a 1 mL syringe, mixed 1:1 with Growth Factor Reduced Matrigel (BD Bioscience, CA, USA) and 200 µL injected into the flank of Athymic Nude mice. Mice were monitored for tumor growth for at least nine months. At the time of surgery, host mice were supplemented with a subcutaneous 5 mm silastic testosterone implant to increase circulating testosterone levels [41]. Viable xenografts were propagated by direct serial transplantation for at least three generations before being considered an established xenograft line. Tumor tissue was cryopreserved at initial and subsequent early passages. Short-term explant cultures were obtained from a primary patient-derived bone metastatic prostate panel. PDXs were propagated in 6-8 weeks-old male NOD-SCID mice, and AI xenografts were propagated in SCID mice. Mice were housed in the Tianjin Medical University pathogen-free rodent facility. All procedures were approved by the Tianjin Medical University Institutional Animal Care and Use Committee [42]. LNCaP, C4-2B, and PC3 cells were purchased from American Type Culture Collection (ATCC) (Manassas, VA, USA). The C4-2 cell line was obtained from UroCorpoation. LNCaP, C4-2, C4-2B, and PDX-related primary cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) or charcoal-stripped, androgen-depleted FBS (Invitrogen) and 100 µg/ml penicillin-streptomycin-glutamine (Invitrogen) at 37ºC with 5% CO2. PC3 cells were cultured in a K-12 medium supplemented with 10% FBS or charcoal-stripped FBS. Cell authentication was performed using STR profiling, as previously described [43, 44].
Positron emission tomography (PET) image acquisition, reconstruction, and analysis
The median injected activity of the 68Ga-PSMA-11 (Glu-NH-CO-NH-Lys-(Ahx)-[68Ga]) ligand was 198 MBq (5.3 mCi) (range, 107.3-233.1 MBq [2.8-6.4 mCi]), and the median tracer uptake period was 1 h (range, 46-110 min). The images were acquired using a PET/CT scanner (Infinia Hawkeye4; GE) [45]. The PET image contained a whole-body scan (pelvis to vertex, 4 min/bed position depending on the patient weight), a dedicated pelvic scan after voiding (same acquisition time/bed position time as used for the whole body), and a dedicated scan of the lower extremities (pelvis to toes, 1 min/bed position for a total of 10 min). The PET images were reconstructed using random-event, attenuation, dead time, and scatter corrections. PET images were reconstructed with an iterative algorithm (ordered-subset expectation maximization) in an axial 168 × 168 matrix on the Biograph 64 TruePoint (2-dimensional, 2 iterations, 8 subsets) and in a 200 × 200 matrix on the Biograph mCT (3-dimensional, 2 iterations, 24 subsets, gaussian filter 5.0). The focal uptake of 68Ga-PSMA-11 above the background level and not related to the physiologic uptake or known pitfalls was considered the PSMA-positive signal [45, 46].
Mouse xenograft generation and tumor growth measurement
For tumor growth under ionizing radiation (IR), 6-week- old NSG male mice were injected with 5 × 106 cancer cells infected with lentivirus or shRNAs and/or expression vectors in 100 μl PBS with 100 μl of Matrigel matrix (BD Bioscience) in one side of flanks. After injection of tumor cells into mice, right flank tumors were radiated by 10 Gy X-ray beam for 14 days and monitored until they reached maximum tumor volumes of 1,000 mm3. Subsequently, tumor growth was measured with a caliper every 7 days.
Biotin-labeled RNA pull-down and Western blot analysis
RNAs were biotin-labeled during transcription using Biotin RNA Labeling Mix (Roche) and T7 polymerase (New England Biolabs). Various deletion mutations within this region were generated by mutagenesis using a KOD-Plus-Mutagenesis Kit (TOYOBO). P-90 primary cells cultured in androgen-depleted medium were lysed in modified binding buffer (150mM NaCl, 50mM Tris-HCl pH7.5, 0.1% SDS 1% Nonidet P-40 (NP-40), and 1% protease inhibitor cocktail). Cell lysates were incubated with biotin-labeled RNAs and streptavidin beads at 4ºC for 10 h. The beads were washed in the wash buffer (150 mM NaCl; 50 mM Tris; pH 7.5; 0.05% NP-40; 1mM MgCl2) at 4ºC six times. The samples were subjected to Western blot analyses as described previously [39]. Briefly, protein samples were denatured and subjected to SDS-polyacrylamide gel electrophoresis (SDS/PAGE) and transferred to nitrocellulose membranes (Bio-Rad). The membranes were immunoblotted with specific primary antibodies and horseradish peroxidase-conjugated secondary antibodies and visualized by SuperSignal West Pico Stable Peroxide Solution (Thermo Scientific). The antibodies are shown in Table S4. The original Western blotting data are shown in Figure S7.
Clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 system
The CRISPR-Cas9-generated assay was completed using the CRISPR-Cas9 tool kit (Santa Cruz). gRNAs were cloned in lentivirusV2 plasmid under the U6 promoter as previously described [42]. The gRNAs are shown in Table S4.
Cross-linking immunoprecipitation (CLIP)
Primary cells (6 x 106) were treated with 100 µM 4-thiouridine (4SU) for 8 h, washed with cold PBS, and irradiated once with 150 mJ/cm2 at 365 nm using a Stratalinker. Cells were lysed in the lysis buffer (100 mM NaCl, 50 mM Tris-HCl pH 7.5, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, protease inhibitor cocktail and RNase inhibitors) with protease inhibitors (1 mL) and transferred to 1.5 mL microtubes. The lysate was partially digested by 2.5 U/µL RNaseT1/A for 15 min at 22oC for iCLIP. RNA was immunoprecipitated with Flag, KHSRP, or XRN2 antibodies and protein A/G beads for 10 h at 4oC, washed 6 times, RNA was phosphorylated by T4 PNK and ligated between 5' and 3' ends by RNA T4 ligase. SDS-PAGE loading buffer was added, incubated at 65oC for 10 min, and the mixture was separated on the SDS-PAGE gel. The RNA-protein complexes were then transferred to a nitrocellulose membrane using a wet transfer apparatus (30V for 1 h). The membrane with the target protein was cut up, and the targeted membrane piece was incubated with proteinase K to de-crosslink, following which RNA was reverse transcribed into cDNA and subjected to real-time qPCR analysis (Yeasen) [42].
γ-H2AX staining
γ-H2AX staining in PDX primary cells was performed, as described previously [47]. Primary cells infected with empty vector (EV) or plasmids expressing MLXIPe were treated with 4Gy IR. At each time point after IR, the cells were harvested for immunofluorescence with antibodies for γ-H2AX. Data were calculated from six biological replicates.
Analysis of publicly available datasets
RNA-seq were deposited in the GEO. The GEO numbers are SRR21733498, SRR21733499, SRR21733500, SRR21733501, SRR21733502, and SRR21733503. m6A or m6Am related sequences (GSE63753, GSE92375) [48, 49] were obtained from our laboratory and publicly available RNA expression datasets in prostate cancer (GSE55032, GSE137209) [39, 50] using Gene Expression Omnibus (GEO).
Statistical analysis
Statistical analysis was performed by GraphPad Prism7 and R scripts. Animals were randomized for in vivo experiments, but randomization was not performed for all other experiments. Statistical analyses were performed using Student's t-test or two-way ANOVA for most comparisons. Experiments were carried out with three or more replicates unless otherwise stated. Non-parametric Kolmogorov-Smirnov (KS) test was used to evaluate the statistical significance of differential expression between primary prostate cancer and bone metastatic prostate cancer. All experiments were performed in biological triplicates unless otherwise specified. P < 0.05 was considered statistically significant.
Detailed methods are described in Supplementary Information.
Results
Identification of specific eRNA modification in bone mPCa
Compared to primary PCa, more than 30% of advanced prostate cancers were more RT-resistant [7, 8]. We analyzed patient survival with or without radiation to investigate the relationship between RT resistance and bone mPCa clinicopathologic characteristics. Consistent with clinical findings from other groups [7, 9], we found that PSMA radioligand therapy prolonged the survival time for patients with primary PCa and lymph node (LN) mPCa, but was limited for patients with bone mPCa in our cohort (Figure S1A). Since enhancers and eRNAs are tissue-specific and function-specific elements, we explored the specific eRNAs in bone mPCa and characterized the bone mPCa-related function of the epitranscriptome. We previously reported stage-related eRNAs in LNCaP (hormone hormone-naïve PCa cell line), C4-2 (CRPC cell line) and PC3 (bone-mPCa cell line) cells using RNA-seq [39, 50]. Among the 100 differentially upregulated enhancer transcripts identified by RNA-seq in CRPC cells, we found 43 eRNAs were highly expressed in bone mPCa PC3 cells (Figure 1A and Table S1). Oncogenic activity by genomic enhancers or eRNAs would be the specific targeting therapy in prostate cancer. A recent publication reported that m6A in eRNAs was involved in gene transcription in breast cancer cells [40]. Thus, we examined whether m6A was a specific modification at eRNA in oncogenic bone mPCa. We found 6 eRNAs with m6A signals by immunoprecipitating with two different m6A antibodies in combination with MeRIP-seq [48]. Subsequently, filtering the 6 eRNAs with intergenic peak selection and qPCR verification resulted in a unique m6A eRNA upregulated in bone-mPCa cells (Figure 1A-B). A meta-analysis of published ChIP-seq data from LNCaP and PC3 cells identified the region as an authentic enhancer, as evident by the enrichment of enhancer activator (BRD4), enhancer markers (H3K4me1 and H3K27ac) [30], and promoter marker (H3K4me3) (Figure 1B). Based on the naming rules for eRNAs, we labeled this eRNA as MLXIPe due to its proximity to MLXIP mRNA. Two putative m6A sites (#1: TTACA chr12 122502280-122502284; #2: GGACA chr12 122506423-122506427) were identified in MLXIPe by two independent m6A antibodies (Figure 1B).
m6A of MLXIPe was involved in metastasis of prostate cancer and radiotherapy. (A) Schematic of eRNA filtering analysis. Numbers of RNAs and methods were shown in the boxes. (B) Screen shots from UCSC genome browser showing signal profiles of eRNA and mRNA expression in LNCaP. C4-2, PC3; ChIP-seq in LNCaP and PC3 were shown as the references [39]. The enhancer regions were highlighted in yellow box. (C and D) Box and whisker plot showing eRNA expression and m6A signals upregulated in bone metastatic prostate cancer tissues. Analysis was of Tianjin Medical University data sets for levels of 12-m6A was based on the m6A-RIP and RT-PCR. n = 30 each group. P values were shown in the figures. (E and F) Kaplan-Meier survival analysis was of the Tianjin Medical University patient tissues cohort for the relationship between the levels of expression of MLXIPe (E) or #2-m6A of MLXIPe (F) and survival time in bone mPCa tissues. n = 30. P values were shown in the figures. (G) The PET-scan for metastatic prostate cancer before or after total 30 Gy 177Lu-prostate-specific membrane antigen (PSMA)-radioligand therapy-treatment in P-73 (MLXIPe m6A low) and P-90 (MLXIPe m6A high) patients.
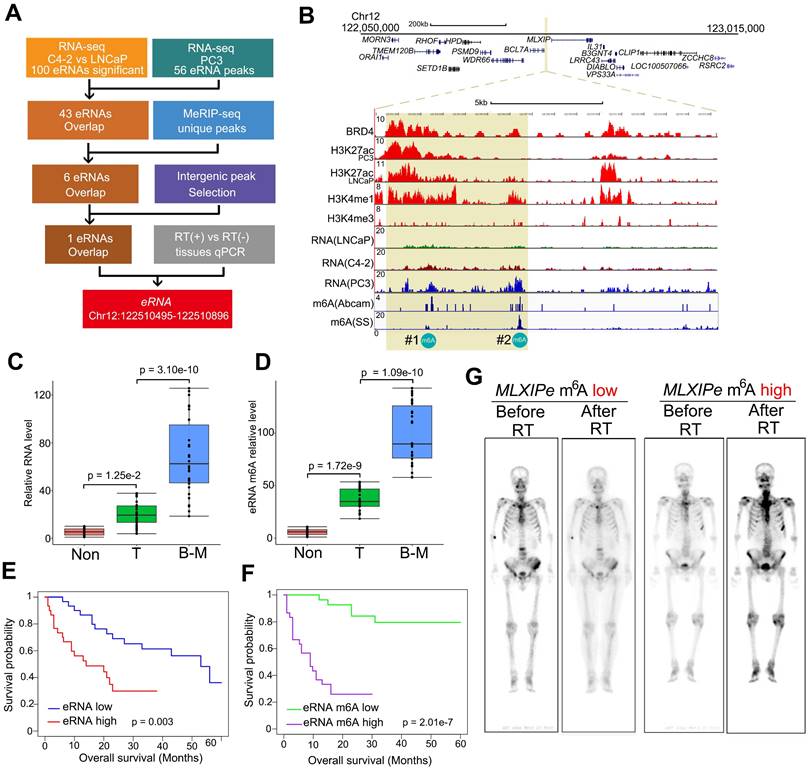
We harvested RNA immunoprecipitated by the m6A antibody from fresh samples of primary prostate cancer, adjacent normal prostate tissue, and bone metastatic cancer to demonstrate the functional role of m6A modifications in a clinicopathological context. Box plot analysis revealed that #2-m6A levels of MLXIPe were significantly higher in bone-mPCa samples than in primary PCa or adjacent prostate samples in the Tianjin Medical University Urology cohort (Figure 1C-D). However, there was no significant difference in the #1-m6A levels of MLXIPe between the metastatic prostate cancer, primary prostate cancer, and adjacent prostate tissue samples (Figure S1B). Next, we harvested RNAs from LNCaP, C4-2, PC3, and C4-2B cell lines and found that #2-m6A levels of MLXIPe were significantly higher in bone metastatic prostate cancer cell lines PC3 and C4-2B (Figure S1C-S1D), but there was no difference in the #1-m6A level of MLXIPe between all cell lines (Figure S1E). We further determined the survival significance of#2-m6A level of MLXIPe with clinical characteristics of PCa patients in the bone metastasis cohort. Kaplan-Meier survival analysis of the Tianjin Medical University cohort found that elevated RNA and m6A levels of MLXIPe were associated with shorter survival in PCa patients with bone metastasis (Figure 1E-F). We also examined the m6A levels of MLXIPe in patients with PMSA PET. PET analysis showed that in prostate cancer patient 73 (P-73)MLXIPe-m6Alow, distant bone metastatic tumor signal disappeared after RT, but stronger signals were detected on both sides of the spine and pelvis in P-90MLXIPe-m6Ahigh (Figure 1G), suggesting RT resistance in MLXIPe-m6Ahigh patients. Taken together, MLXIPe with m6A modification was specific for bone mPCa cells.
MLXIPe-induced RT resistance of mPCa via PSMD9 upregulation
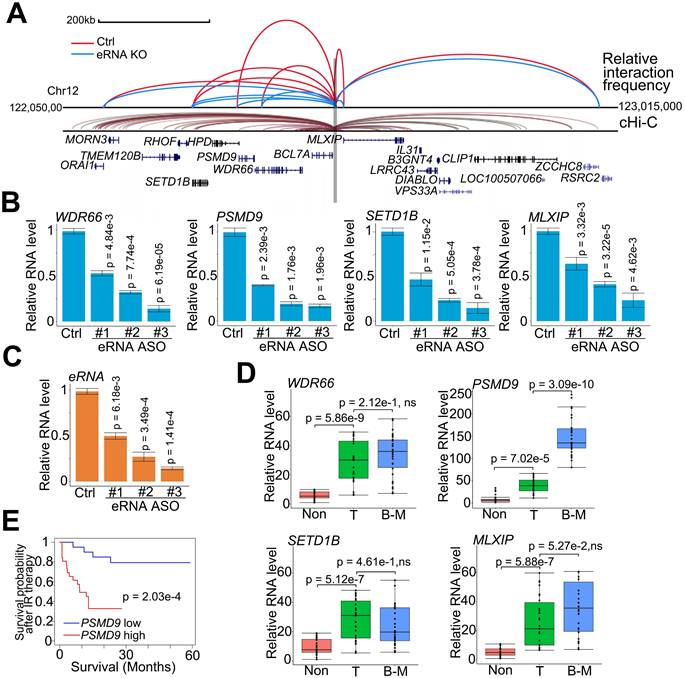
eRNAs transfer enhancer activity to one or several neighboring target promoters by increasing enhancer-promoter looping [37, 51]. To determine the downstream promoters regulated by MLXIPe's enhancer, we investigated the adjacent promoters using a strategy analogous to chromosome conformation capture (3C) and analyzed published data using the capture Hi-C (cHi-C) technique [52]. Enhancer-promoter looping by cHi-C identified 11 mRNAs around MLXIPe (Figure 2A). By excluding 4 mRNAs with no expression in prostate cancer cells by RNA-seq data (Figure S2A), we determined the spatial organization of 7 promoters by 3C assay in human bone-mPCa patient-derived xenograft (PDX) primary cell line-90 (P-90). 3C data showed that depletion of MLXIPe by antisense oligodeoxynucleotides (ASOs) decreased specific enhancer-promoter interaction between MLXIPe and WDR66 (also called CFAP251), PSMD9, SETD1B, and MLXIP loci (Figure 2A and S2B). After identifying the MLXIPe target genes by genomic DNA interactions, we investigated whether MLXIPe affected their transcription. We knocked down MLXIPe by ASOs to verify the eRNA function on the expression of the target. Reverse transcription qPCR and RNA-seq data using three independent ASOs showed that MLXIPe downregulated the expression of the four target genes in bone-mPCa primary cell line P-90 and PC3 cells (Figure 2B-C, S2C and Table S2). Interestingly, meta-data showed that the expression of PSMD9, but not the other 3 genes, was highly specific in bone marrow (Figure S2D). We also found that PSMD9 was a specific bone-mPCa-related gene, but WDR66, SETD1B, and MLXIP were oncogenic genes (Figure 2D). PSMD9 is related to 26S proteasome expression and could predict RT benefits in breast cancer [53, 54]. Higher levels of PSMD9 were associated with lower recurrence-free survival after RT (Figure 2E), which was consistent with the association of PSMD9 with disease-free survival in TCGA data (Figure S2E). Collectively, PSMD9, as an MLXIPe bone-specific target gene, was correlated with RT resistance and shorter survival in bone mPCa patients.
PSMD9 was the target gene regulated by MLXIPe and was involved in radiotherapy resistance. (A) Normalized three-dimensional DNA selection and ligation capture data from ORAI1 locus to RSRC2 locus in chromatin 12. Capture 4C data was analyzed from website http://3dgenome.fsm.northwestern.edu. (B and C) MLXIPe and mRNAs RNA expressions were measured by qRT-PCR in P-90 primary cells with three independent ASOs targeting MLXIPe. Data presented were means ± standard deviations (SD) of up to three independent replicates. P values were shown in the figures. (D) Box and whisker plot showing WDR66, PSMD9, SETD1B and MLXIP expressions elevated in prostate cancer tissues. Analysis was of Tianjin Medical University data cohort for levels of mRNAs were based on the PCR data.n = 30 each group. P values were shown in the figures. (E) Kaplan-Meier survival analysis was of the Tianjin Medical University cohort for the relationship between the levels of expression of PSMD9 RNA levels and survival time in bone mPCa tissues after radiotherapy. n = 26. P values were shown in the figures.
MLXIPe interacted with KHSRP through m6A
We explored the molecular mechanism of m6A at MLXIPe by generating CRISPR control (Ctrl) and MLXIPe #2-m6A site-deletion (-del) cell lines (Figure S3A) for unbiased tandem affinity purification by biotin-labeled probes targeting MLXIPe eRNA or PSMD9 mRNA. Subsequently, we performed mass spectrometry, showing that MLXIPe eRNA bound to multiple proteins, including the looping protein MED12 and KH domain proteins hnRNPK, IGF2BP2, or KHSRP in control (Ctrl) cells but not to KH domain proteins in MLXIPe -m6A-del cells (Figure 3A and Table S3). Similarly, PSMD9 mRNA bound to several proteins, including translation protein eIF4A1, eIF4G2, and the KH domain protein KHSRP in Ctrl cells, but not KH domain proteins in eRNA-m6A-del cells (Figure 3A and Table S3). KHSRP, an RNA binding protein involved in multiple cancers [55-58], was the common protein between both pull-down groups (Figure 3B). We deleted four domains (N-terminal, KH1/2, KH3/4, or C-terminal-deletion) in the KHSRP protein to confirm the mass spectrometry data (Figure 3C). The cross-linking immunoprecipitation (CLIP)-PCR assay showed that the KH1/2-deletion domain blocked the interaction between the KHSRP protein and PSMD9 mRNA (Figure 3D), and the interaction between KHSRP and MLXIPe was significantly inhibited by KH3/4-deletion domain (Figure 3E).
KHSRP bound to eRNA and mRNA to form a complex. (A) The top hits of MLXIPe and PSMD9 RNA pull-down proteins were identified by TAP-MS. The numbers of peptides were indicated in the list. (B) Venn diagram showing that proteins interacting by MLXIPe and PSMD9 RNA pull-down proteins overlapped in P-90 cells. (C) Upper, protein diagrams with hnRNP K homology (KH) motif were shown. Lower, western blot of Flag binding at the KHSRP fragments in P-90 cells transfected with Flag-KHSRP-WT, Flag-KHSRP-muts (N-terminal, KH1/2, KH3/4, C-terminal deletion). (D and E) CLIP-qPCR analysis of KHSRP binding at the MLXIPe and PSMD9 mRNA in P-90 cells with different primers targeting different regions. (F) RNA pull-down analysis of KHSRP binding at the MLXIPe RNA in P-90 cells transfected with WT, #1-m6Am and #2-m6A deletion using CRISPR system. (G) RNA pull-down analysis of KHSRP binding at the PSMD9 mRNA in P-90 cells with different probes targeting 5'UTR, CDS and 3'UTR regions.
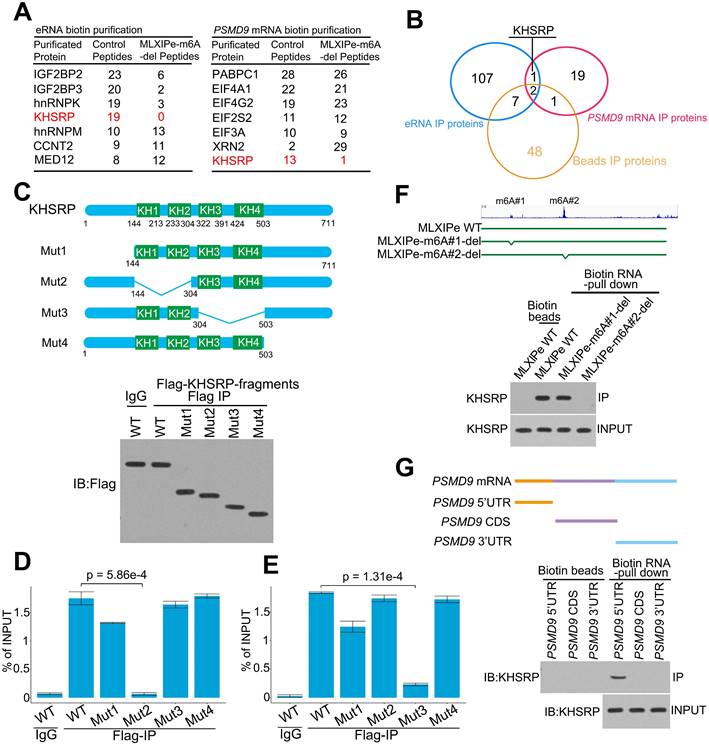
Next, we examined the effect of m6A modification on the interaction between KHSRP and MLXIPe and observed that #2-m6A site deletion blocked MLXIPe association with KHSRP, whereas #1-m6A site deletion failed to affect the interaction (Figure 3F and S3A). Furthermore, KHSRP bound to 5'UTR of PSMD9 mRNA but not to CDS and 3'UTR regions (Figure 3G). 5'UTR of mRNAs is methylated with N6,2'-O-dimethyladenosine (m6Am), which is also recognized by the m6A antibody [19]. It was important to characterize whether KHSRP bound to m6Am or m6A at 5'UTR of PSMD9 mRNA. For this, we analyzed the published m6Am-seq and performed epitranscriptome analysis to distinguish m6Am and m6A in cells through L-ascorbic acid changing Fe(II)/2 related affinity activation [49]. Thus, m6Am signals could be reduced by FTO treatment, but m6A signals were not changed [49], such as on the RBM48 locus (Figure S3B). The high-throughput sequencing data showed m6Am modification at the 5'UTR of PSMD9 and MLXIP mRNAs, suggesting that KHSRP bound to m6Am using its KH1/2 domain (Figure S3B). m6Am-seq showed an m6Am at the #1 site of MLXIPe (Figure S3B), explaining the reason the #1 m6Am-site of MLXIPe failed to interact with m6A reader IGF2BP2 or IGF2BP3 (Figure 3A and S3B). To further demonstrate that m6Am is necessary for KHSRP-PSMD9 interaction, we generated m6Am-deletion PSMD9 mRNA 5'UTR plasmids by QuickChange II Site Directed-Mutagenesis-kit. We found that m6Am deletion of PSMD9 mRNA 5'UTR fragment failed to bind KHSRP in PC3 cells (Figure S3C), suggesting that m6Am was necessary for the interaction between KHSRP and PSMD9 mRNA. The simultaneous interaction of KHSRP with eRNA and mRNA was examined by testing eRNA using PSMD9 pull-down assay and PSMD9 mRNA using eRNA pull-down assay with or without knocking down KHSRP. The data showed that KHSRP interacted with both eRNA and mRNA simultaneously (Figure S3D). Thus, KHSRP was an m6A and m6Am double reader that interacted with m6Am using the KH1/2 domain and with m6A using the KH3/4 domain (Figure 4A).
eRNA-KHSRP-mRNA complex protected PSMD9 mRNA against from XRN2. (A) A diagram showed eRNA-KHSRP-mRNA complex model. (B) A curve-figure showed PSMD9 mRNA half-life in P-90 cells. The cells were treated with Actinomycin D in time dependent manner. Data presented were means ± standard deviations (SD) of up to three independent replicates. Visible P values were shown in the figures. (C) Western blot (WB) and qPCR assay showed KHSRP knocking down (KD) in P-90 cells. Cells were transfected with sh-control and sh-KHSRPs, after 48 hrs the cells were harvested for WB or qPCR assay. Means and standard deviations (error bar) were determined from three replicates. P values were shown in the figures. (D) A curve-figure showed PSMD9 mRNA half-life in P-90 cells. The cells were transfected with sh-control and sh-KHSRPs and treated with Actinomycin D in time dependent manner. Data presented were means ± standard deviations (SD) of up to three independent replicates. P values were shown in the figures. (E) Western blot (WB) and qPCR assay showed PCIF1 knocking down in P-90 cells. Cells were transfected with si-control or si-PCIF1s and empty vector or MLXIPe RNA, after 48 hrs the cells were harvested for WB or qPCR assay. Means and standard deviations (error bar) were determined from three replicates. Visible P values were shown in the figures. (F) qPCR assay XRN2 knocking down (KD) in P-90 cells. Cells were transfected with si-control and si-XRN2s, after 48 hrs the cells were harvested for qPCR assay. Data presented were means ± standard deviations (SD) of up to three independent replicates. P values were shown in the figures. (G) CLIP-qPCR analysis of XRN2 binding at the PSMD9 mRNA in P-90 cells transfected with sh-control and sh-KHSRPs. (H) A curve-figure showed PSMD9 mRNA half-life in P-90 cells. The cells were transfected with si-control and si-XRN2s and treated with Actinomycin D in time dependent manner. Data presented were means ± standard deviations (SD) of up to three independent replicates. P values were shown in the figures. (I) A diagram showed how eRNA-KHSRP-mRNA complex protected mRNA against from degradation by XRN2.
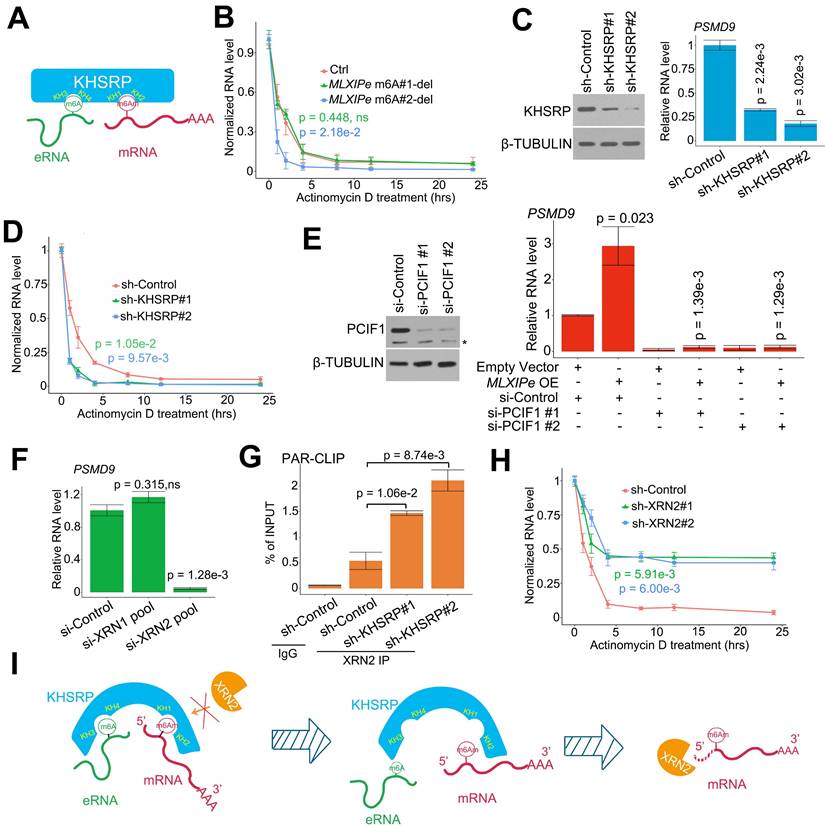
KHSRP stabilized mRNA by connecting it with eRNA
It has been reported that m6Am is involved in mRNA stability [19, 25, 28, 59]. We examined the half-life of RNA in P-90 cells to confirm the relevance m6Am in mRNA stability. The RNA half-life data indicated that #2 m6A deletion increased PSMD9 mRNA degradation, but #1-m6Am site deletion failed to complete it in P-90 and PC3 cells (Figure 4B and S4A). We analyzed the relationship among MLXIPe, PSMD9 mRNA and m6A levels in the clinical cohort. The data showed that MLXIPe m6A levels were significantly correlated with MLXIPe RNA and PSMD9 mRNA levels, suggesting that MLXIPe m6A modifications upregulated the stabilization of MLXIPe and PSMD9 mRNAs (Figure S4B). Meanwhile, KHSRP depletion with two independent shRNAs significantly downregulated PSMD9 RNA levels and enhanced PSMD9 mRNA degradation (Figures 4C-D). The m6Am writer is PCIF1 as reported [60]. PCIF1-KD impaired the PSMD9 RNA levels elevated by MLXIPe (Figure 4E). Importantly, RNA-seq data showed that elevated MLXIPe increased the 5'UTR signals in PC3 cells, whereas decreased MLXIPe exhibited lower 5'UTR signals in LNCaP cells (Figure S4C). The qPCR assay with paired primers targeting 5'UTR, CDS, and 3'UTR showed that high MLXIPe enhanced the ratios of 5'UTR/CDS and 5'UTR/3'UTR at PSMD9 and MLXIP loci (Figure S4C), suggesting that the degradation of PSMD9 mRNA might be from 5' to 3'. XRN1 and XRN2 are highly processive 5' > 3' exoribonucleases [61]. The Knockdown assay demonstrated that XRN2, but not XRN1, was the 5' > 3' exoribonuclease responsible for PSMD9 mRNA degradation. (Figure 4F). Depletion of KHSRP increased PSMD9 mRNA binding to XRN2 (Figure 4G). Furthermore, knocking down of XRN2 upregulated the stability of PSMD9 mRNA (Figure 4H and S4D). These data suggested that MLXIPe-KHSRP-PSMD9 blocked the XRN2-induced degradation of PSMD9 mRNA in bone mPCa cells (Figure 4I).
MLXIPe elevated levels of DNA repair proteins through m6A in prostate cancer cells
PSMD9 is an RT predicting marker related to RT resistance in breast cancer cells [53, 54]. In this study, we found that MLXIPe mediated the PSMD9 mRNA stability and upregulated its protein level via eRNA m6A (Figure 4). We selected four MLXIPe m6Ahigh and MLXIP m6Alow primary bone-mPCa PDX cells from our PDX cohort to determine whether MLXIPe m6A was the specific RT resistance marker in bone-mPCa. Different dose radiation curve data indicated RT resistance in MLXIPe m6Ahigh cells; however, MLXIPe m6Alow cells were killed in a dose-dependent manner by radiation (Figure 5A). PSMD9 belongs to 26S proteasome, a multi-catalytic proteinase complex for protein degradation [54]. Western blotting showed that ATM, DNA-PKcs, and p53 protein levels were upregulated in MLXIPe m6Ahigh cells, but the ATR expression was not affected (Figure 5B). We determined whether the functional role of MLXIPe m6A in mPCa RT resistance was through PSMD9, by overexpressing PSMD9 and/or knocking down KHSRP in mPCa primary cells, and performing MTS cell survival assay and protein expression by Western blotting. The data showed that PSMD9 depletion impaired cancer cell survival after RT (Figure S5A). Furthermore, we found that the knocking down of KHSRP downregulated ATM, DNA-PKcs, and p53 expression after RT. On the contrary, overexpression of PSMD9 rescued ATM, DNA-PKcs, and p53 protein expression in P-73 and P-90 cells following RT (Figure S5B).
MLXIPe elevated key proteins of DNA repair pathway. (A) Relative primary cell viabilities were completed by MTS assay. Bone-mPCa primary cells were radiated in a dose dependent manner, after radiation 72 hrs cells were treated with MTS for 2 hrs and measured. Data presented were means ± standard deviations (SD) of up to three independent replicates. P values were shown in the figures. (B) Expression of DNA repair related proteins were measured by western blot in MLXIPe m6A low and MLXIPe m6A high primary bone-mPCa cells. (C) Western blot and qPCR assay showed eRNA m6A knocking out (KO) in MLXIPe m6Alow P-73 cells. Cells were infected with empty vector, MLXIPe WT and MLXIPe m6A-del, after 48 hrs the cells were harvested for western blot or qPCR assay. Data presented were means ± standard deviations (SD) of up to three independent replicates. P values were shown in the figures. (D) Western blot and qPCR assay showed eRNA m6A and KHSRP knocking down in MLXIPe m6Alow P-73 cells. Cells were infected with empty vector or MLXIPe WT accompanying sh-control or sh-KHSRP, after 48 hrs the cells were harvested for western blot or qPCR assay. Data presented were means ± standard deviations (SD) of up to three independent replicates. P values were shown in the figures. (E) Western blot and qPCR assay showed eRNA knocking-down (KD) by ASO-mix and XRN2 knocking-down by shRNAs in MLXIPe m6Ahigh P-90 cells. Cells were infected with CRISPR empty vector or CRISPR targeting MLXIPe m6A site accompanying sh-control or sh-XRN2, after 48 hrs the cells were harvested for western blot or qPCR assay. Data presented were means ± standard deviations (SD) of up to three independent replicates. P values were shown in the figures.
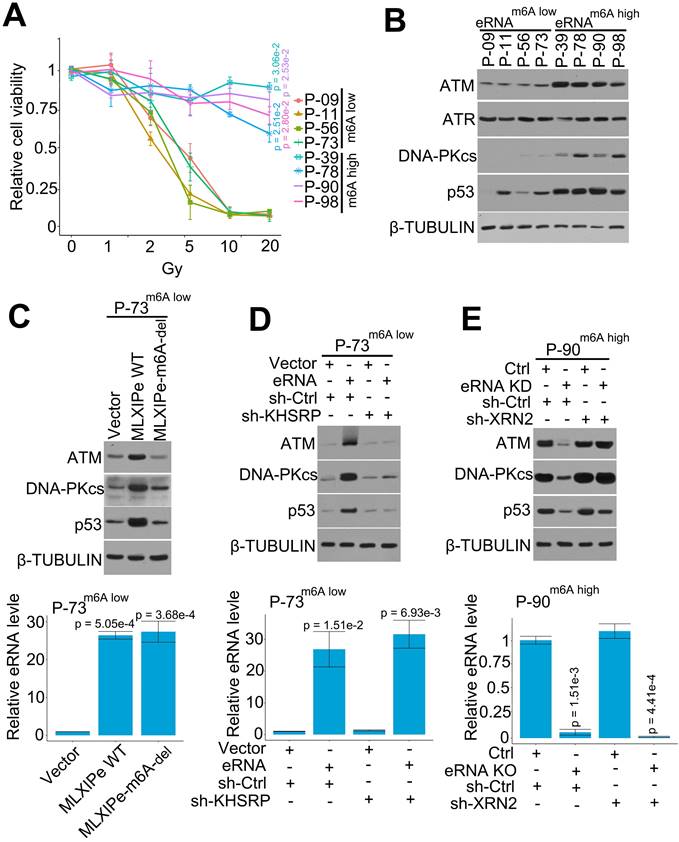
We deleted m6A in MLXIPe to further confirm m6A function in the DNA repair pathway. The expression data showed that MLXIPe WT but not MLXIPe-m6A deletion elevated ATM, DNA-PKcs, and p53 protein levels (Figure 5C). Also, MLXIPe WT and MLXIPe-m6A-deletion elevated DNA repair and decreased the number of γ-H2AX foci at 8 h after IR (Figure S5C). The function of the eRNA-KHSRP-mRNA complex in the DNA repair pathway was verified in MLXIPe m6Alow P-73 cells and MLXIPe m6Ahigh P-90 cells. The data showed that KHSRP-depletion inhibited the upregulation of ATM, DNA-PKcs, and p53 proteins by MLXIPe and impaired the ratio of γ-H2AX foci (8h/1h after IR) in MLXIPe m6Alow P-73 cells (Figure 5D and S5D). Meanwhile, XRN2 knock-down rescued ATM, DNA-PKcs, and p53 protein levels and enhanced the ratio of γ-H2AX foci (8h/1h after IR) in MLXIPe m6A-deleted cells (Figure 5E and S5E). These data indicated that MLXIPe elevated ATM, DNA-PKcs, and p53 protein levels through the m6A and KHSRP complex in bone-mPCa cells.
MLXIPe enhanced prostate PDX RT resistance through m6A and KHSRP
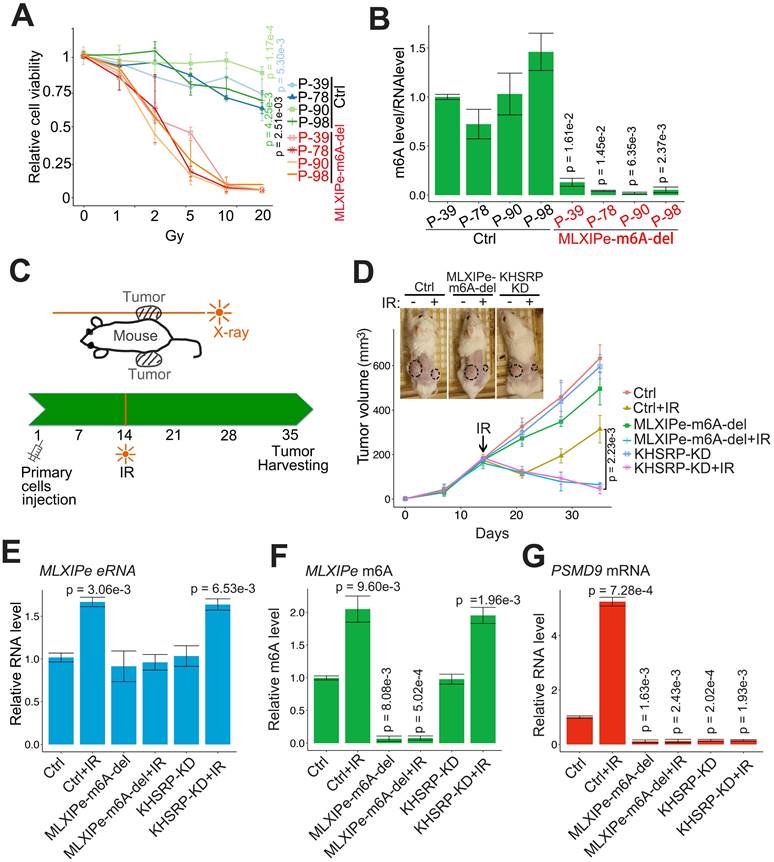
We tested the m6A function of MLXIPe in PDX cells by editing the m6A site in four MLXIPe m6Ahigh PDX cells. The data showed that the RT resistance was impaired and radiosensitivity was recovered in MLXIPe m6A sites-deleted (-del) mPCa cells (Figures 6A-B). Next, we determined the m6A function of MLXIPe in vivo in the xenograft model and examined the effect of m6A regulation of MLXIPe on prostate xenograft growth and radio resistance. When MLXIPe-m6A-del cells injected into the mice flank, the data showed that MLXIPe-m6A knock-out slightly decreased the growth of xenografts, whereas KHSRP-KD transfection did not affect xenograft growth (Figure 6C-D). Significantly, MLXIPe-m6A-del and KHSRP-KD groups recovered radiosensitivity after RT in 36 days, but not the Ctrl group (Figure 6D). The qPCR data showed that PSMD9 mRNA was associated with the RT resistance of xenografts (Figure 6E-G). Thus, our data indicated that m6A of MLXIPe or KHSRP elevated the mPCa RT resistance in the PDX model.
MLXIPe induced radiotherapy resistance in vivo. (A and B) Relative primary cell viabilities were completed by MTS assay. Bone-mPCa primary cells infected with CRISPR empty vector or CRISPR targeting MLXIPe m6A site were radiated in a dose dependent manner, after radiation 72 hrs cells were treated with MTS for 2 hrs and measured. The m6A status were measured by m6A-RIP. Data presented were means ± standard deviations (SD) of up to three independent replicates. P values were shown in the figures. (C and D) P-90 cells (3x106) transfected with lenti-virus shRNAs were injected into NSG mice flanks (n = 6 each group). The one-side xenografts of mice were subjected to X-ray radiation in 14 days. The tumor growth was observed every 7 days, and the data were shown in the bottom panel. P values were shown in the figures. Representative images out of 6 different mice were displayed in 35 day and then were harvest for RNAs and m6A measurements. (E) MLXIPe eRNA expressions were measured by qPCR in P-90 xenograft tissues. Data presented were means ± standard deviations (SD) of up to three independent replicates. P values were shown in the figures. (F) Effect of m6A of MLXIPe on growth of bone-mPCa xenografts. P-90 cells were injected into NSG mice flanks (n = 6 each group). After 35 days the xenografts were harvested for m6A-RIP. Data shown as means ± SD (n = 6). Statistical significance was determined by two-tail Student's t-test. P values were displayed in the figures. (G) PSMD9 mRNA expressions were measured by qPCR in P-90 xenograft tissues. Data presented were means ± standard deviations (SD) of up to three independent replicates. P values were shown in the figures.
Discussion
Metastatic prostate cancer is the principal cause of mortality in men. Bone is the most common target organ for prostate cancer metastasis in an overwhelming 80 % of patients with a 5-year survival rate of merely 3% [1, 3]. Androgen deprivation therapy is one of the main treatments for advanced prostate cancer, however, CRPC is a challenge for prostate cancer treatment by hormone therapy or drugs, especially for metastatic CRPC [62, 63]. RT is another critical component of cancer management in over 50% of cancer patients [9, 64]. Sometimes RT is the first treatment for cancers that have metastasized outside the prostate gland and into nearby tissues [7]. Furthermore, 177Lu- PSMA radioligand therapy (RLT) is a novel therapeutic option in patients with metastatic CRPC. RT or hormone therapy was considered an excellent independent therapy. However, combination therapy with RT exhibited an equal overall survival of metastatic CRPC (mCRPC) in recent clinical trials [8, 46, 64], suggesting that mCRPC is not only resistant to hormone therapy but also has the feature of being RT resistant. Therefore, we hypothesized that there is a common specific oncogenic factor in CRPC and mPCa.
In this study, we filtered the m6A eRNA in a hormone-naïve PCa cell line LNCaP, CRPC cell line C4-2, and bone-mPCa cell line PC3 by RNA-seq, MeRIP-seq, intergenic selection, and RT-qPCR, and identified a new eRNA MLXIPe with m6A as the candidate (Figure 1). We demonstrated four target genes of MLXIPe, including WDR66, SETD1B, PSMD9, and MLXIP (Figure 2). Among these, WDR66, SETD1B, and MLXIP were novel markers for cancer metastasis and involved in epithelial-mesenchymal transition (EMT) [65-67]. However, these three genes were not bone-specific (Figure 2 and S2). Importantly, PSMD9, the expression of was bone-specific, was involved in the RT resistance of bone mPCa, indicating that the MLXIPe-PSMD9 axis might be the key pathway mediating bone-mPCa RT resistance.
Despite the discovery of enhancers regulating gene transcription over 40 years, the enhancer-mediated post-transcription is not entirely understood. eRNA was demonstrated as the critical factor in the mechanism of enhancers regulating genes' expression [31, 37, 51]. Recently published finding reported that m6A at eRNA was involved into breast cancer oncogenesis [40]. It supplies two scientific questions whether m6A is at the disease-specific-expressed RNA (such as eRNA) in specific cancer cells and whether it is the functional modification in these cancer cells. To ask both questions, we detected the m6A status at eRNA in the prostate cancer. Here, unlike m6A abundant modification at mRNA, we found about 6% eRNA with m6A modification in prostate cancer cells, consistent with published data of about 18% eRNA marked with m6A in breast cancer cells [40]. Unlike the eRNA function in looping and transcription activation, we found a novel function of eRNA that protected XRN2 to maintain the stability of target mRNA at the post-transcriptional level. As shown in RNA-seq data (Figure 4 and S4), MLXIPelow cells exhibited a low ratio of 5'UTR/CDS and 5'UTR/3'UTR, suggesting that MLXIPe was involved in the stability of 5' end of mRNAs. KHSRP binding to PSMD9 mRNA 5'UTR significantly blocked the interaction of XRN2 with PSMD9 mRNA (Figure 4). These data provided new insight into eRNA post-transcriptional regulation and protection of the nascent mRNA stability from its 5' end.
m6A in mammals was identified as a highly abundant modification of mRNA among more than 160 chemical modifications [13-15]. Besides m6A, there is another reversible modification in eukaryotes known as m6Am. Unlike the m6A mainly located at 3'UTR, m6Am is precisely located at the first transcribed nucleotide and hence adjacent to the mRNA cap [19]. Recently, a limited number of writers, erasers, and readers of m6A were reported by many groups [18, 26, 28]. m6Am has been shown to be catalyzed by PCIF1, an interacting protein with RNA polymerase II [19, 49]. The only eraser of m6Am is FTO, which is also the same enzyme as m6A [49, 68]. However, the reader of m6Am remained unclear.
Using the published data of m6Am-seq [49], we distinguished m6Am and m6A in eRNAs and mRNAs (Figure S3). Consistent with our mass spec data in Figure 3A, #2 site m6A of MLXIPe bound to KHSRP, IGF2BP2, and IGF2BP3 of which IGF2BP2 and IGF2BP3 are m6A readers[69, 70], but #1 site of the putative m6A of MLXIPe failed to bind because it was an m6Am at eRNA as determined by m6Am-seq. Furthermore, MeRIP-seq (Figure 1B) showed that #1-site m6Am level was about 5 times lower than #2-site methylation level, suggesting that #1-site was a weaker eRNA methylation site than m6Am at the mRNA. Also, the RNA pull-down data displayed in Figure 3G showed that the KH1/2 domain of KHSRP bound to 5'UTR of PSMD9 mRNA, demonstrating that the KH1/2 domain of KHSRP was a potential reader of m6Am. Significantly, we found an m6Am and m6A double-recognized protein with different KH domains. The m6Am interaction mechanism was assisted by MLXIPe binding (Figure 3A). When eRNA-m6A was disassociated with the KHSRP KH3/4 domain, KHSRP did not bind to m6Am at PSMD9 mRNA (Figure 3A), suggesting that the eRNA-KHSRP interaction may change the KH1/2 structure and facilitate the interaction between KHSRP and m6Am at the PSMD9 mRNA. Functionally, the new complex of eRNA-KHSRP-mRNA protected the 5' end of PSMD9 mRNA from degradation by XRN2 in bone-mPCa cells. These data indicated that the new m6Am read complex played an RT-resistant role through PSMD9 in bone-mPCa cells.
In summary, our data provided new insights into mPCa RT resistance, eRNA modification, and the mechanism of the potential m6Am reader. We detected a bone-specific eRNA with m6A that facilitated the RT resistance function in bone metastatic prostate PDXs. KHSRP recognized and interacted with m6A at the eRNA and m6Am on 5'-UTR of mRNA to block RNA degradation from 5'-UTR by XRN2. The findings indicated that a bone-specific eRNA-m6A is vital in regulating mPCa growth and RT resistance and might be a novel target for cancer therapy (Figure 6). Our study highlights MLXIPe as a potential predictive biomarker and therapeutic target for bone-mPCa.
Availability of data and materials
All data that support the findings of this study are available from the corresponding authors upon reasonable request.
Abbreviations
CRPC: castration-resistant prostate cancer
CLIP: Cross-linking immunoprecipitation
eRNAs: Enhancer RNAs
mPCa: Metastatic prostate cancer
m6Am: N6,2'-O-dimethyladenosine
m6A: N6-methyladenosine
PCR: Polymerase chain reaction
qPCR: Quantitative real-time PCR
RT: Radiotherapy
RIP: RNA immunoprecipitation
5′ UTR: 5′-untranslated region
3′ UTR: 3′-untranslated region
Supplementary Material
Supplementary figures and methods.
Supplementary table 1.
Supplementary table 2.
Supplementary table 3.
Supplementary table 4.
Acknowledgements
We thank the patients and their families for their altruism in participating in research studies. We thank Dr. Yuanjie Niu for reagents, plasmids, and suggestions about PDXs.
Funding
This work was supported in part by grants from the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2021-RC310-010 to Y.Z.), the National Natural Science Foundation of China (81972654, 82273262 to S.W.; 81730086 to SJ.F.), the CAMS Innovation Fund for Medical Sciences (2021-I2M-1-042, to SJ.F.; 2021-I2M-1-060 to Y.Z.), Tianjin International Student Science and Technology Activities (20160014 to S.W.), the Mayo Clinic Center for Individualized Medicine (to YY.Z.).
Author information
These authors contributed equally: Yu Zhao, Simeng Wen and Hang Li.
Author contributions
YZ, HL, SMW, YW, CWP and TH performed the experiments. SMW, HL and YZ prepared all the figures and wrote the manuscript. SMW, ZCL, CWP, YHY and YLW collected the patients' samples. YZ, SJF and YYZ supervised the project. YZ, SW, YYZ and SJF provided fund for the whole project. All authors read and approved the final manuscript.
Ethics declarations
All experimental protocols were approved by the Ethics Committee of Tianjin Medical University and performed in accordance with the relevant guidelines and regulations (Ethical approval number: KY2019K036). Written informed consents were obtained from all patients. All animal experiments were approved by the Institutional Animal Care and Use Committee of Tianjin Medical University (Tianjin, China).
Competing Interests
The authors have declared that no competing interest exists.
References
1. van der Toom EE, Axelrod HD, de la Rosette JJ, de Reijke TM, Pienta KJ, Valkenburg KC. Prostate-specific markers to identify rare prostate cancer cells in liquid biopsies. Nat Rev Urol. 2019;16:7-22
2. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71:7-33
3. Yin JJ, Pollock CB, Kelly K. Mechanisms of cancer metastasis to the bone. Cell Res. 2005;15:57-62
4. Norgaard M, Jensen AO, Jacobsen JB, Cetin K, Fryzek JP, Sorensen HT. Skeletal related events, bone metastasis and survival of prostate cancer: a population based cohort study in Denmark (1999 to 2007). J Urol. 2010;184:162-7
5. Smith MR, Cook R, Lee KA, Nelson JB. Disease and host characteristics as predictors of time to first bone metastasis and death in men with progressive castration-resistant nonmetastatic prostate cancer. Cancer. 2011;117:2077-85
6. van der Toom EE, Verdone JE, Pienta KJ. Disseminated tumor cells and dormancy in prostate cancer metastasis. Curr Opin Biotechnol. 2016;40:9-15
7. Ali A, Parker CC, Clarke NW. Prostate radiotherapy in newly diagnosed metastatic prostate cancer. Curr Opin Urol. 2019;29:620-8
8. Wallis CJD, Saskin R, Choo R, Herschorn S, Kodama RT, Satkunasivam R. et al. Surgery Versus Radiotherapy for Clinically-localized Prostate Cancer: A Systematic Review and Meta-analysis. Eur Urol. 2016;70:21-30
9. Kamran SC, D'Amico AV. Radiation Therapy for Prostate Cancer. Hematol Oncol Clin North Am. 2020;34:45-69
10. Ali A, Hoyle AP, Parker CC, Brawley CD, Cook A, Amos C. et al. The Automated Bone Scan Index as a Predictor of Response to Prostate Radiotherapy in Men with Newly Diagnosed Metastatic Prostate Cancer: An Exploratory Analysis of STAMPEDE's "M1|RT Comparison". Eur Urol Oncol. 2020;3:412-9
11. Bolla M, Van Tienhoven G, Warde P, Dubois JB, Mirimanoff RO, Storme G. et al. External irradiation with or without long-term androgen suppression for prostate cancer with high metastatic risk: 10-year results of an EORTC randomised study. Lancet Oncol. 2010;11:1066-73
12. Boeve LMS, Hulshof M, Vis AN, Zwinderman AH, Twisk JWR, Witjes WPJ. et al. Effect on Survival of Androgen Deprivation Therapy Alone Compared to Androgen Deprivation Therapy Combined with Concurrent Radiation Therapy to the Prostate in Patients with Primary Bone Metastatic Prostate Cancer in a Prospective Randomised Clinical Trial: Data from the HORRAD Trial. Eur Urol. 2019;75:410-8
13. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S. et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201-6
14. Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell. 2012;149:1635-46
15. Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat Rev Genet. 2014;15:293-306
16. Fu Y, Jia G, Pang X, Wang RN, Wang X, Li CJ. et al. FTO-mediated formation of N6-hydroxymethyladenosine and N6-formyladenosine in mammalian RNA. Nat Commun. 2013;4:1798
17. Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y. et al. R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m(6)A/MYC/CEBPA Signaling. Cell. 2018;172:90-105 e23
18. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y. et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885-7
19. Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP. et al. Reversible methylation of m(6)Am in the 5' cap controls mRNA stability. Nature. 2017;541:371-5
20. Meyer KD, Jaffrey SR. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol. 2014;15:313-26
21. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D. et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117-20
22. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H. et al. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell. 2015;161:1388-99
23. Zhao X, Yang Y, Sun BF, Shi Y, Yang X, Xiao W. et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014;24:1403-19
24. Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. 2014;16:191-8
25. Wang T, Kong S, Tao M, Ju S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol Cancer. 2020;19:88
26. Nombela P, Miguel-Lopez B, Blanco S. The role of m(6)A, m(5)C and Psi RNA modifications in cancer: Novel therapeutic opportunities. Mol Cancer. 2021;20:18
27. Guo X, Li K, Jiang W, Hu Y, Xiao W, Huang Y. et al. RNA demethylase ALKBH5 prevents pancreatic cancer progression by posttranscriptional activation of PER1 in an m6A-YTHDF2-dependent manner. Mol Cancer. 2020;19:91
28. An Y, Duan H. The role of m6A RNA methylation in cancer metabolism. Mol Cancer. 2022;21:14
29. Shen S, Zhang R, Jiang Y, Li Y, Lin L, Liu Z. et al. Comprehensive analyses of m6A regulators and interactive coding and non-coding RNAs across 32 cancer types. Mol Cancer. 2021;20:67
30. Furlong EEM, Levine M. Developmental enhancers and chromosome topology. Science. 2018;361:1341-5
31. Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J. et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182-7
32. De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK. et al. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010;8:e1000384
33. Pefanis E, Wang J, Rothschild G, Lim J, Kazadi D, Sun J. et al. RNA exosome-regulated long non-coding RNA transcription controls super-enhancer activity. Cell. 2015;161:774-89
34. Li W, Notani D, Rosenfeld MG. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet. 2016;17:207-23
35. Skowronska-Krawczyk D, Ma Q, Schwartz M, Scully K, Li W, Liu Z. et al. Required enhancer-matrin-3 network interactions for a homeodomain transcription program. Nature. 2014;514:257-61
36. Hsieh CL, Fei T, Chen Y, Li T, Gao Y, Wang X. et al. Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proc Natl Acad Sci U S A. 2014;111:7319-24
37. Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY. et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature. 2013;498:516-20
38. Schaukowitch K, Joo JY, Liu X, Watts JK, Martinez C, Kim TK. Enhancer RNA facilitates NELF release from immediate early genes. Mol Cell. 2014;56:29-42
39. Zhao Y, Wang L, Ren S, Wang L, Blackburn PR, McNulty MS. et al. Activation of P-TEFb by Androgen Receptor-Regulated Enhancer RNAs in Castration-Resistant Prostate Cancer. Cell Rep. 2016;15:599-610
40. Lee JH, Wang R, Xiong F, Krakowiak J, Liao Z, Nguyen PT. et al. Enhancer RNA m6A methylation facilitates transcriptional condensate formation and gene activation. Mol Cell. 2021;81:3368-85 e9
41. Risbridger GP, Clark AK, Porter LH, Toivanen R, Bakshi A, Lister NL. et al. The MURAL collection of prostate cancer patient-derived xenografts enables discovery through preclinical models of uro-oncology. Nat Commun. 2021;12:5049
42. Wen S, Wei Y, Zen C, Xiong W, Niu Y, Zhao Y. Long non-coding RNA NEAT1 promotes bone metastasis of prostate cancer through N6-methyladenosine. Mol Cancer. 2020;19:171
43. Zhao Y, Peng J, Yang J, Zhang E, Huang L, Yang H. et al. Enhancing Prostate-Cancer-Specific MRI by Genetic Amplified Nanoparticle Tumor Homing. Adv Mater. 2019;31:e1900928
44. Shang Z, Yu J, Sun L, Tian J, Zhu S, Zhang B. et al. LncRNA PCAT1 activates AKT and NF-kappaB signaling in castration-resistant prostate cancer by regulating the PHLPP/FKBP51/IKKalpha complex. Nucleic Acids Res. 2019;47:4211-25
45. Calais J, Kishan AU, Cao M, Fendler WP, Eiber M, Herrmann K. et al. Potential Impact of (68)Ga-PSMA-11 PET/CT on the Planning of Definitive Radiation Therapy for Prostate Cancer. J Nucl Med. 2018;59:1714-21
46. Fendler WP, Rahbar K, Herrmann K, Kratochwil C, Eiber M. (177)Lu-PSMA Radioligand Therapy for Prostate Cancer. J Nucl Med. 2017;58:1196-200
47. Zhao F, Kim W, Gao H, Liu C, Zhang Y, Chen Y. et al. ASTE1 promotes shieldin-complex-mediated DNA repair by attenuating end resection. Nat Cell Biol. 2021;23:894-904
48. Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12:767-72
49. Sun H, Li K, Zhang X, Liu J, Zhang M, Meng H. et al. m(6)Am-seq reveals the dynamic m(6)Am methylation in the human transcriptome. Nat Commun. 2021;12:4778
50. Wu D, Yan Y, Wei T, Ye Z, Xiao Y, Pan Y. et al. An acetyl-histone vulnerability in PI3K/AKT inhibition-resistant cancers is targetable by both BET and HDAC inhibitors. Cell Rep. 2021;34:108744
51. Pan CW, Wen S, Chen L, Wei Y, Niu Y, Zhao Y. Functional roles of antisense enhancer RNA for promoting prostate cancer progression. Theranostics. 2021;11:1780-94
52. Wang X, Xu J, Zhang B, Hou Y, Song F, Lyu H. et al. Genome-wide detection of enhancer-hijacking events from chromatin interaction data in rearranged genomes. Nat Methods. 2021;18:661-8
53. Langlands FE, Dodwell D, Hanby AM, Horgan K, Millican-Slater RA, Speirs V. et al. PSMD9 expression predicts radiotherapy response in breast cancer. Mol Cancer. 2014;13:73
54. Koster F, Sauer L, Hoellen F, Ribbat-Idel J, Brautigam K, Rody A. et al. PSMD9 expression correlates with recurrence after radiotherapy in patients with cervical cancer. Oncol Lett. 2020;20:581-8
55. Bae JA, Bae WK, Kim SJ, Ko YS, Kim KY, Park SY. et al. A new KSRP-binding compound suppresses distant metastasis of colorectal cancer by targeting the oncogenic KITENIN complex. Mol Cancer. 2021;20:78
56. Huang J, Sachdeva M, Xu E, Robinson TJ, Luo L, Ma Y. et al. The Long Noncoding RNA NEAT1 Promotes Sarcoma Metastasis by Regulating RNA Splicing Pathways. Mol Cancer Res. 2020;18:1534-44
57. Du Y, Zhang JY, Gong LP, Feng ZY, Wang D, Pan YH. et al. Hypoxia-induced ebv-circLMP2A promotes angiogenesis in EBV-associated gastric carcinoma through the KHSRP/VHL/HIF1alpha/VEGFA pathway. Cancer Lett. 2022;526:259-72
58. Huang X, Liu X, Du B, Liu X, Xue M, Yan Q. et al. LncRNA LINC01305 promotes cervical cancer progression through KHSRP and exosome-mediated transfer. Aging (Albany NY). 2021;13:19230-42
59. Kramer K, Sachsenberg T, Beckmann BM, Qamar S, Boon KL, Hentze MW. et al. Photo-cross-linking and high-resolution mass spectrometry for assignment of RNA-binding sites in RNA-binding proteins. Nat Methods. 2014;11:1064-70
60. Sendinc E, Valle-Garcia D, Dhall A, Chen H, Henriques T, Navarrete-Perea J. et al. PCIF1 Catalyzes m6Am mRNA Methylation to Regulate Gene Expression. Mol Cell. 2019;75:620-30 e9
61. Li Y, Yamane D, Lemon SM. Dissecting the roles of the 5' exoribonucleases Xrn1 and Xrn2 in restricting hepatitis C virus replication. J Virol. 2015;89:4857-65
62. Slack FJ, Chinnaiyan AM. The Role of Non-coding RNAs in Oncology. Cell. 2019;179:1033-55
63. Zhao SG, Chen WS, Li H, Foye A, Zhang M, Sjostrom M. et al. The DNA methylation landscape of advanced prostate cancer. Nat Genet. 2020;52:778-89
64. Podder TK, Fredman ET, Ellis RJ. Advances in Radiotherapy for Prostate Cancer Treatment. Adv Exp Med Biol. 2018;1096:31-47
65. Wang Q, Ma C, Kemmner W. Wdr66 is a novel marker for risk stratification and involved in epithelial-mesenchymal transition of esophageal squamous cell carcinoma. BMC Cancer. 2013;13:137
66. Mejhert N, Kuruvilla L, Gabriel KR, Elliott SD, Guie MA, Wang H. et al. Partitioning of MLX-Family Transcription Factors to Lipid Droplets Regulates Metabolic Gene Expression. Mol Cell. 2020;77:1251-64 e9
67. Lindner P, Paul S, Eckstein M, Hampel C, Muenzner JK, Erlenbach-Wuensch K. et al. EMT transcription factor ZEB1 alters the epigenetic landscape of colorectal cancer cells. Cell Death Dis. 2020;11:147
68. Wei J, Liu F, Lu Z, Fei Q, Ai Y, He PC. et al. Differential m(6)A, m(6)Am, and m(1)A Demethylation Mediated by FTO in the Cell Nucleus and Cytoplasm. Mol Cell. 2018;71:973-85 e5
69. Muller S, Glass M, Singh AK, Haase J, Bley N, Fuchs T. et al. IGF2BP1 promotes SRF-dependent transcription in cancer in a m6A- and miRNA-dependent manner. Nucleic Acids Res. 2019;47:375-90
70. Huang H, Weng H, Sun W, Qin X, Shi H, Wu H. et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285-95
Author contact
![]() Corresponding authors: Yu Zhao, Ph.D. (zhaoyuac.cn), Saijun Fan, Ph.D. (fansaijunac.cn) and Yingyi Zhang, Ph.D. (zhang.yingyiedu).
Corresponding authors: Yu Zhao, Ph.D. (zhaoyuac.cn), Saijun Fan, Ph.D. (fansaijunac.cn) and Yingyi Zhang, Ph.D. (zhang.yingyiedu).