Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Genomic instability in AD
3. Telomere attrition in AD
4. Epigenetic alterations in AD
5. Loss of proteostasis
6. Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2023; 13(1):197-230. doi:10.7150/thno.79535 This issue Cite
Review
Role of primary aging hallmarks in Alzheimer´s disease
Jin Zhao, Jisen Huai ![]()
The Second Affiliated Hospital of Xinxiang Medical University (Henan Mental Hospital), Xinxiang, 453000, China.
Institute of Psychiatry and Neuroscience, Xinxiang Medical University, Xinxiang, 453003, China.
Received 2022-10-4; Accepted 2022-11-15; Published 2023-1-1
Abstract

Alzheimer's disease (AD) is the most common neurodegenerative disease, which severely threatens the health of the elderly and causes significant economic and social burdens. The causes of AD are complex and include heritable but mostly aging-related factors. The primary aging hallmarks include genomic instability, telomere wear, epigenetic changes, and loss of protein stability, which play a dominant role in the aging process. Although AD is closely associated with the aging process, the underlying mechanisms involved in AD pathogenesis have not been well characterized. This review summarizes the available literature about primary aging hallmarks and their roles in AD pathogenesis. By analyzing published literature, we attempted to uncover the possible mechanisms of aberrant epigenetic markers with related enzymes, transcription factors, and loss of proteostasis in AD. In particular, the importance of oxidative stress-induced DNA methylation and DNA methylation-directed histone modifications and proteostasis are highlighted. A molecular network of gene regulatory elements that undergoes a dynamic change with age may underlie age-dependent AD pathogenesis, and can be used as a new drug target to treat AD.
Keywords: Aging, Alzheimer's disease, Epigenetics, Molecular neurobiology, Neurodegeneration, Oxidative stress
1. Introduction
Dementia is an acquired loss of cognitive ability severe enough to interfere with daily living [1]. Alzheimer's disease (AD) is the most prevalent form of dementia, affecting more than 50 million individuals worldwide [2]. It is a progressive, amnestic, and fatal neurodegenerative disease characterized by the combined neuropathological burden of extracellular amyloid plaques and intracellular neurofibrillary tangles, two cornerstones of AD etiology [3-5]. These pathological changes often occur selectively in the limbic and neocortical brain regions, especially in the entorhinal cortex layer II (ECII), hippocampal CA1, and temporal and frontal lobes of the brain [6-8].
AD is a multifactorial disease with environmental (30%) and genetic (70%) causes. Environmental factors are usually associated with sporadic AD (SAD), while genetic factors are associated with familial AD (FAD) and SAD [9]. Interestingly, FAD and SAD differ in age of onset [10-12]. According to the age of onset, AD can be divided into two categories of early-onset AD (EOAD) and late-onset AD (LOAD) before or after the age of 65 [10]. In all AD cases, approximately 5% are EOAD and 95% are LOAD [11], indicating that most AD is caused by aging in concert with a complex interaction of genetic and environmental risk factors [13, 14].
AD, especially LOAD, is associated with aging and is characterized by selective neuronal vulnerability (SNV) [6, 7, 14, 15]. However, the relationship between aging and SNV and the molecular basis of AD are not completely understood which need to be urgently elucidated [6, 16-18]. Aging is the inevitable time-dependent decline in physiological organ integrity, leading to impaired function and increased vulnerability to death. It is characterized by nine tentative hallmarks grouped into three main categories: primary hallmarks (genomic instability, telomere attrition, epigenetic alterations, and loss of proteostasis), antagonistic hallmarks (deregulated nutrient sensing, altered mitochondrial function, and cellular senescence), and integrative hallmarks (stem cell exhaustion and altered intercellular communication) [19, 20]. To date, the role of each aging hallmark in AD development remains unclear. This article will focus on the primary aging hallmarks as these are interconnected with other aging characteristics and are at the base of the hierarchical order of aging features [19, 20], and have been shown to be related to AD [21, 22]. It is an attempt to improve our understanding of the pathological mechanisms of AD to find potential therapeutic approaches and diagnostic tools.
2. Genomic instability in AD
Recently, DNA damage has been shown to play a central role in aging and affects most aspects of the aging phenotype [23, 24], suggesting that DNA damage is a potentially unifying cause of aging [25]. DNA damage markers, including double-strand breaks (DSBs), have also been found in brain regions of AD patients [26-30], indicating that DNA damage may be an important pathological cause of AD, particularly in LOAD cases [21, 31, 32].
It is conceivable that easily damaged regions in AD may be more active in normal physiological processes, including oxidative stress, neuronal activation, and gene transcription, causing DNA damage in neurons [33-36]. However, to offset excessive DNA damage and prevent mutation, all organisms have evolved highly conserved DNA damage detection and repair mechanisms [37-41]. How the threshold of this homeostatic DNA damage and repair is disrupted has yet to be completely understood [42]. It has been proposed that the balance between DNA damage and repair processes is disrupted mainly by DNA repair becoming less efficient with age, finally causing genomic instability, and triggering cell death signalling cascades [23, 43, 44]. This is consistent with the idea that DNA damage accumulation during the lifetime in differentiated cells such as neurons is mainly attributed to insufficient DNA repair [45-47]. However, an age-dependent increase in DNA damaging factors such as reactive oxygen species (ROS) has also been reported, likely inducing more DNA damage [43, 48].
The human brain accounts for approximately 2% of our body mass, but it uses 20% of the total oxygen supply consumed by the whole body [49, 50]. It requires a continuous supply of energy in the form of ATP, produced by oxidative phosphorylation in mitochondria with ROS as the by-product [51]. Among the three main types of ROS, superoxide radicals (•O2-), hydrogen peroxide (H2O2), and the hydroxyl radical (•OH), •O2- is the proximal mitochondrial ROS, which can be converted to H2O2 by superoxide dismutase (SOD) and then to •OH by Fenton´s reaction of H2O2 with Fe2+ [52-54]. •OH is by far the most active form of ROS and is considered the main DNA damaging factor in neurons and can cause altered bases, abasic sites, and single- and double-strand breaks [55-57]. Excessive ROS can lead to approximately 100 different oxidative base damage and 2-deoxyribose modifications most likely to occur in neuronal cells [55, 58]. Oxidative DNA damage is notably related to human diseases [48, 59] and is the most significant DNA lesion affecting the progression of AD [60].
It has been shown that 8-oxo-2-deoxyguanosine (oxo8dG), a marker of oxidative DNA damage, accumulates with age in nuclear DNA in all tissues and strains of rodents and is associated with AD brain pathology [57, 61, 62]. However, the accumulation of oxyradical-associated DNA damage in different brain regions is not uniform [46, 63, 64]. The vulnerable areas in the brains of AD patients appear to be more sensitive to DNA damage insult and are also subjected to DNA repair deficiency with age. Although it is difficult to determine which of the two processes plays a more important role in AD development, DNA damage and repair deficiency contribute to the genomic instability, thereby causing systematic regional vulnerability of AD [33, 65, 66]. DNA damage may reduce the expression of selectively vulnerable genes involved in learning, memory, and neuronal survival, initiating a program of brain aging that starts early in adult life [33]. Genomic instability affects the expression of the genes linked to mitochondrial and metabolic dysfunction, altered proteostasis, and inflammation, which are centrally involved in aging, and supports the notion that DNA damage could be the root of aging and AD [23, 67]. Therefore, targeting DNA damage or other aging-related hallmarks provides a rationale for developing interventions to combat age-related diseases, including AD [25].
Neurons are post-mitotic cells vulnerable to oxidative damage comparable to dividing cells during aging [68]. However, neurons from different AD brain regions show significantly different vulnerability, are equipped with different antioxidants, and have different abilities to scavenge oxidized DNA groups, which play a decisive role in the SNV of AD [63, 69]. Therefore, the cell-autonomous mechanism is thought to be the primary driver responsible for initiating SNV in AD [16]. However, this notion is challenged by other findings. For example, although the regional basal levels of DNA damage inversely correlate with the regional capacity to remove oxo8dG from DNA [63], it has been reported that the age-related increase in oxo8dG in the nuclear DNA of aging mice is not due to a decrease in the ability to eliminate oxo8dG damage. Instead, the increase in oxo8dG levels seems to be caused by an age-related increase in the sensitivity of the tissues to oxidative stress [61, 70]. Intriguingly, the sensitivity of neurons to oxidative stress also depends on their surrounding environment. Neurons from different regions may have distinct surrounding cells and tissues, such as astrocytes, microglia, and vasculature, which can secrete other antioxidants or oxidants, thereby inhibiting or promoting ROS damage [71-78]. These observations suggest that the surrounding environment may play a dominant role in maintaining DNA integrity and the survival of neurons. Consistent with this hypothesis, a transplantation experiment showed that the lifespan of neurons was not limited by the maximum lifespan of the donor organism but was influenced when transplanted in a longer-living host [79]. Although both oxidative DNA damage and repair deficiency contribute to genomic instability and are involved in the neurodegenerative process of the AD brain [80, 81], oxidative DNA damage, which increases with age, appears to play a leading role. However, it still needs to be clarified whether neuronal DNA damage is caused mainly by autonomous or nonautonomous mechanisms and how the critical point for maintaining neuron redox balance is impaired and causes cell death and AD onset.
3. Telomere attrition in AD
The telomere is the protective end cap of the chromosome, a nucleoprotein complex composed of tandem TTAGGG DNA repeats and associated protein complexes known as shelterin [82, 83]. The shelterin complex consists of six associated proteins: telomeric repeat binding factor 1 and 2 (TRF1/2), TRF2 interacting protein (RAP1), TRF1-interacting nuclear factor 2 (TIN2), adrenocortical dysplasia protein homolog (TPP1), and protection of telomeres 1 (POT1) [83, 84]. The shelterin complex is believed to stabilize a lariat-like structure of the chromosomal end, the telomere loop (t-loop), to prevent telomeres from inadvertently activating DNA damage signaling and DSB repair pathways [85-87].
Telomeres shorten with age, and approximately 50 nucleotides are lost during each cell cycle [88]. Although neurons are post-mitotic cells, and their telomeres are not expected to be shortened through the division mechanism, they may still accumulate irreparable DNA damage, resulting in a senescent cell type or even apoptosis [89-92]. In contrast, neural stem cells (NSCs) are proliferative and affected by aging [93]. Telomere maintenance is crucial for NSC viability and self-renewal potential, while telomere shortening may cause cognitive impairment and psychiatric disorders [94, 95]. Nonneuronal brain cells such as NSCs and astrocytes divide more slowly than cells from other tissues, and the telomere attrition rate is slower than other cells [94]. Leukocyte telomere length (LTL) has been widely used as a surrogate marker for central nervous system telomere length (TL) [96], and an association between accelerated LTL shortening and the incidence of AD has been demonstrated [94, 97-99] despite several negative reports [100-102]. Since telomeres are the protective chromosomal end caps, their dysfunction may lead to genomic instability [103, 104]. For example, altered telomere chromatin structure has been linked to a defective DNA damage response [103]. Since genomic instability plays a key role in the pathogenesis of AD, it is not surprising that telomere attrition is associated with the incidence of AD. Deficits in telomere-associated enzymes such as telomerase and DNA-PKcs render neurons vulnerable to adverse conditions related to AD. In contrast, telomerase-increasing compounds protect hippocampal neurons from Aβ42 toxicity by enhancing the expression of neurotrophins and plasticity-related genes [105-107], confirming that telomere attrition may play an important role in AD pathogenesis.
Remarkably, the telomeres of hippocampal neurons become shorter in AD, and much evidence supports its correlation with elevated levels of oxidative stress [102, 108]. Interestingly, telomeres are not only favoured by oxidative attack due to their unique sequence but also less efficiently repaired when compared to non-telomeric damage [109-112]. Furthermore, genomic instability can adversely affect telomere integrity [113, 114]. For example, the DNA damage response can cause telomere erosion [113]. These results suggest that oxidative stress may cause SNV of AD through both non-telomeric and telomeric DNA damage in a synergistic manner and are consistent with the findings that oxidative stress plays a central role in the pathogenesis of AD [115, 116]. Furthermore, in mature human hippocampal neurons and activated microglia, telomerase reverse transcriptase (TERT) also plays a protective role against oxidative damage independent of its canonical function of telomere maintenance [117, 118]. In line with this finding, TERT protein is increased in mitochondria of AD hippocampal CA1 neurons compared to healthy controls [117], probably due to the compensatory effect of cells on oxidative stress in AD. However, overexpression of TERT or boosting its activity through compounds cannot prevent AD, although it has antioxidative and autophagy-promoting effects [89, 119, 120]. Collectively, telomeres form a special structure at the end of the chromosome to protect the integrity of the genome in canonical and noncanonical ways and are very important for the survival of neural cells. However, the telomere is very sensitive to oxidative stress and vulnerable to damage. Therefore, elevated free radical pressure may lead to telomere wear with increasing age, exacerbating the related genomic instability, and contributing to AD pathogenesis.
4. Epigenetic alterations in AD
Epigenetics focuses on the underlying mechanism of gene expression without changes in DNA sequences. The most studied epigenetic signatures are DNA methylation, histone modification, and noncoding RNAs (ncRNAs) [121]. Epigenetic mechanisms directly contribute to aging and aging-related diseases [122]. Recently, neuro-epigenetics has emerged as an important field that explores how reversible modifications can change gene expression to control behavior and cognitive abilities [123]. Accumulating findings have shown that epigenetic machinery plays a key role in maintaining genome integrity and regulating gene expression [124], and its dysfunction is closely related to AD pathogenesis [125].
4.1. DNA methylation
DNA methylation refers to the attachment of a methyl group to the DNA chain. While methylation is catalyzed by DNA methyltransferases (DNMTs), including DNMT1 (the primary maintenance DNA methyltransferase), DNMT3A, DNMT3B, and DNMT3L [126, 127], demethylation is mainly performed by ten-eleven translocation (TET) family enzymes [128, 129]. DNA methylation primarily occurs on the 5th carbon of a cytosine residue (5mC) at CpG and non-CpG (CpA, CpC and CpT) dinucleotides [130]. In vertebrates, CpG dinucleotides are predominantly methylated in all tissues, with approximately 80% of all CpG sites containing 5mC [131-133]. In contrast, non-CpG methylation (mCpHs, H = A/C/T) is usually restricted to specific cell types, such as pluripotent stem cells, oocytes, neurons, and glial cells, and plays a critical regulatory role in cognitive function [127, 134]. CpH methylation can be up to 25% of all CpH sites in adult mouse dentate neurons, and it is generated de novo during neuronal maturation and requires DNMT3A for active maintenance in post-mitotic neurons [132].
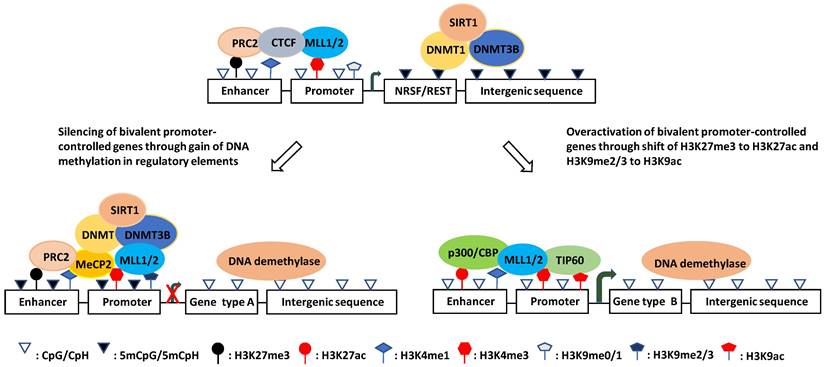
Intriguingly, although genome-wide CpGs are highly methylated, CpG islands (CGIs) are paradoxically unmethylated in most cases [127, 135, 136]. These CGIs are usually cis-regulatory elements, including more than 50% of mammalian gene promoters [133, 137-139]. They are high-intensity CpG promoters (HCPs) regulating housekeeping genes, developmental regulator genes, and a proportion of tissue-specific genes [140, 141]. By contrast, the remaining over 40% of human gene promoters are low-intensity CpG promoters (LCPs, also known as non-CGI promoters: NCPs), which are differentially methylated and are more prone to hypermethylation under stress [138, 142-144]. Recently, it was reported that methylated CpHs (mCpHs) and CpGs (mCpGs) in the brain increase with age [145-147]. Enhancers with hypermethylated CpHs are associated with genes functionally enriched in immune responses, and some of the genes are related to neuroinflammation and degeneration [145, 148, 149]. In general, DNA methylation in enhancers and promoters negatively regulates gene expression, whereas gene body methylation likely increases transcriptional activity (Fig. 1) [149-152]. However, some studies have shown that hypomethylation in the promoter region is associated with the downregulation of gene expression [153].
Schematic illustration of the hypothesized mechanisms of aberrant gene expression in AD. (left) Downregulation mechanism: DNMTs shift from non-CGI regions to CGI regulatory elements under oxidative stress and catalyze DNA methylation at the damage sites, which recruits MeCP2 and help DNMTs catalyze more DNA methylation thereby resulting in the target gene silencing. (Right) Upregulation mechanism: DNA damage repair induces co-occurence of H3K9ac and/or H3K27ac with H3K4me3, which prevents DNA methylation and represses H3K9me2 inhibitory effect on transcription to overactivate the genes.
Aberrant DNA methylation has been linked to many AD susceptibility genes, including amyloid precursor protein (APP), β- and γ-secretases [153-156], apolipoprotein E [157, 158], Triggering receptor expressed on myeloid cells 2 (TREM2) [159], hTERT [160], cAMP responsive element binding protein (CREB)-regulated transcription coactivator 1 (CRTC1) [161], brain-derived neurotrophic factor (BDNF) [162], thromboxane A2 receptor (TBXA2R, related to CREB activation), sorbin and SH3 domain-containing 3 (SORBS3, related to synapse formation), and spectrin beta 4 (SPTBN4, related to axon initial segment) [163]. Epigenome-wide association studies (EWAS) have uncovered more genes with altered DNA methylation levels in the hippocampus, entorhinal cortex, dorsolateral prefrontal cortex, and cerebellum of both early and late AD patients as compared with controls [164-167]. Interestingly, many of these genes were previously shown to be AD susceptibility genes or connected to the AD susceptibility network and participate in different pathological processes of neurons or nonneuronal cells, such as amyloid pathology (APP, ABCA7, SERPINF1 and 2), tau pathology (BIN1), inflammation (ANK1, RHBDF2, IL-1β, and IL-6), protein dyshomeostasis (RPL13 and HOXA3), calcium dyshomeostasis (S100B), and cellular skeleton defects (MAP2 and MCF2L) [164-167].
A genome-wide DNA methylation analysis of hippocampal or superior temporal gyrus samples from a cohort of AD patients and controls used Illumina 450K methylation arrays. The study showed that differentially methylated positions (DMPs) of AD patients were enriched in poised promoters (bivalent promoters) marked by H3K27me3 and H3K4me3, which are not generally maintained in committed neural cells but participate in neurodevelopment and neurogenesis (Fig. 1) [168, 169]. This is consistent with previous findings that epigenetic mechanisms are critical to adult hippocampal neurogenesis and are relevant to AD etiology [170-173]. It is conceivable that the impaired generation of neurons from NSCs will exacerbate the loss of neurons causing learning and memory deficits [174, 175].
Although the AD susceptible genes are regulated by DNA methylation, the mechanism for the aberrant methylation patterns is not fully understood. The primary point for DNA methylation in mammalian genomes is cytosine in CpG dinucleotides, which are highly enriched in CGIs, but paradoxically, CGIs are usually unmethylated [127]. It was proposed that CGI promoters (HCPs), which are transcriptionally active at totipotent stages of development, can act as origins of DNA replication; thus, these CGIs can exclude methylation due to occupancy with the molecules that initiate DNA replication [176], and the methylation-free form of the promoter is transmitted to all somatic cells [177]. Although most of these CGI promoters remain transcriptionally active (high expression HCPs), others become silenced by recruited polycomb repressive complex 2 (PRC2), which catalyzes H3K27me3 [178-180], or become poised with the coexistence of H3K27me3 and an activating histone marker H3K4me3 (poised HCPs) [181, 182]. Under stress conditions, the PRC2 catalytic subunit enhancer of zeste homolog 2 (EZH2) can recruit DNMTs, catalyzing de novo DNA methylation [183-185]. Critically, DNA methylation is enriched in poised promoters of AD susceptibility genes [168, 169]. Given that H3K4me3 protects against DNA methylation [186, 187], whereas H3K27me3 promotes DNA methylation, the ratio of H3K4me3/H3K27me3 appears to play a key role in determining the DNA methylation state at the CGI promoters. This is consistent with the finding that DNA methylation at these CGIs can be coordinated by H3K27me3 [188-190].
O´Hagan et al. found that under oxidative stress, DNMT1/DNMT3B, together with SIRT1, move from non-GC-rich regions to CGIs, forming a complex with two subunits of the PRC2 complex (EZH2 and EED2), and cause local DNA methylation [191]. Also, DNA methylation under chronic oxidative stress occurs only on low expression (poised HCPs) but not high expression CGI promoters (Fig. 1) [191]. The reason why highly expressed HCPs are protected from methylation is poorly understood. As mentioned previously, the molecules responsible for DNA replication might occupy the sites and exclude methylation of high-expression CGI promoters [176, 177]. It is also possible that proteins containing a CXXC zinc finger domain and its resultant H3K4me3 inhibit the activity of the de novo DNA methyltransferase DNMT3a, thus preventing methylation at these sites [144, 192]. In addition, HCPs with high expression contain high CpG intensity and are less occupied and can, therefore, produce more oxo8dG and 5hmC under oxidative stress. However, oxo8dG can induce DNA hypomethylation by inhibiting DNA methylation at nearby cytosine bases, and 5hmC can cause hypomethylation by activating DNA demethylation [193]. Therefore, so far, it is unclear which of these mechanisms plays a leading role in protecting highly expressed HCPs from methylation.
Notably, whilst the high-expression HCPs are protected from methylation, other mechanisms can still silence the genes. Either the specific oxo8dG base damage or the base excision repair (BER) pathway that repairs this type of damage can recruit members of the gene silencing complex to these promoters [191]. This is consistent with previous findings that oxo8dG is associated with the pathological process of the AD brain [57, 61, 62]. Additionally, 5hmC in the inferior parietal lobe, hippocampus, parahippocampal gyrus, and cerebellum of patients with mild cognitive impairment (MCI) and preclinical Alzheimer's disease (PCAD) has been shown to be significantly increased [194]. These findings may explain why housekeepers such as glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and β-actin (controlled by high expression CGI promoters) have a lower overall expression in AD cases than in controls [195]. Interestingly, many defective DNA repair genes in AD are not methylated in their promoters [196, 197], although DNA damage response factors, such as p53, poly(ADP-ribose) polymerase 1 (PARP-1) and Growth arrest and DNA damage-inducible alpha (GADD45a), have been shown to stimulate DNA methylation [198, 199]. Whether they are silenced by oxo8dG/BER-recruited silencing complexes is although likely but not known. In support of this notion, it has been reported that alteration of the expression of repair genes is controlled by aberrant transcriptional and epigenetic factors in addition to DNA methylation/demethylation and gene mutations [198].
In summary, DNA methylation is involved in both aging and AD. However, many de novo methylations of initially unmethylated sites are AD-specific and play an important role in AD pathogenesis. Although the detailed mechanism for aberrant DNA methylation patterns is unclear, there is evidence showing that oxidative DNA damage plays a key role. Since DNA methylation and damage response are closely related, it is tempting to propose that AD-specific methylation patterns derive from and reflect stronger oxidative damage, requiring extensive repair and therefore generating additional errors. Although the hypo- or unmethylated CGI promoters may become hypermethylated or remain unchanged under chronic oxidative stress, gene silencing complexes may form on both low- and high-expression HCPs. These complexes can coordinate different epigenetic markers and thereby determine gene transcription [191, 200-205]. Various DNA methylation states on promoters may form distinct patterns of other epigenetic modifications [206-208]. In line with this notion, it has been reported that histone modifications occurring on HCPs and LCPs are distinct and active LCPs require H3K4me3 and H3K79me1, while active HCPs require H3K27ac and H4K20me1 [209, 210]. In the next section, we summarize new findings of histone modifications and their interaction with DNA methylation.
4.2. Histone modification
Histone modification is another pivotal form of epigenetic regulation. Histones are covalently modified, altering the intrinsic chromatin structure, and regulating gene expression [211, 212]. In eukaryotes, the core structure of chromatin is a nucleosome, composed of a histone octamer formed by two copies of each of the four histones (H2A, H2B, H3, and H4) and 147 base pairs of DNA wrapped around the octamer [213]. Various posttranslational modifications occur at histones, the most common being methylation, acetylation, phosphorylation, and ubiquitination [121, 214]. Histone modification usually occurs on gene promoters and enhancers, with methylation and acetylation at positively charged lysine (K) and arginine (R) residues being the most prevalent [211, 215, 216]. Histone modification is dynamic and reversible and is controlled by both writers (e.g., histone acetyltransferases (HATs) or histone methyltransferases (HMTs)) and erasers (e.g., histone deacetylases (HDACs) or histone demethylases (HDMs)) required for proper in-printing and off-printing of genes [217]. The imbalance between writers and erasers, resulting in epigenetic marker disparity and transcription dysfunction, is associated with many brain disorders [218-221]. Accumulating evidence has shown that histone methylation and acetylation dysregulation is associated with AD pathology even in its early stage [222-226].
4.2.1. Histone acetylation variation in the AD brain
Dysfunction of HDACs (HDAC1, HDAC2, HDAC3, and HDAC6) has been reported to be involved in cognitive impairment, a debilitating feature of many neurodegenerative disorders, including AD [227-233]. At late AD stages, HDAC1 and HDAC2 were shown to be decreased in the prefrontal cortex (PFC) and hippocampus of AD patients [234, 235]. Lu et al. found that H3K9ac was significantly upregulated in early AD PFC neurons, whereas it was globally downregulated in normal aging PFC neurons [236]. In addition, Nativio et al. reported that H3K9ac was also enriched in the AD temporal lobe together with H3K27ac [237], but no information was available on the level of H3K9ac in the AD hippocampus. However, Tat-interacting 60 kDa protein (TIP60/KAT5, a HAT for expression of H3K9ac, H3K14ac, and H2AK5ac) was significantly reduced in both neurons and glial cells and largely absent from nuclei of neurons in the AD hippocampus [238], where TIP60 and GCN5/KAT2A were usually most strongly expressed [239-241], suggesting that H3K9ac and other acetylated histones catalyzed by TIP60, were decreased in the AD hippocampus. Since many cognition-related genes are regulated by H3K9ac [242], TIP60 reduction in the hippocampus might downregulate these cognition-related genes, which would be upregulated by increased H3K9ac in the PFC and temporal lobe. However, the genes involved in apoptosis (BAX and DAXX), Aβ production (presenilin-2), and neurotransmission (SST and CALB1) are also upregulated in the PFC and temporal lobe [236]. Thus, both H3K9ac downregulation in the hippocampus and upregulation in the PFC and temporal lobe of the AD brain might be detrimental.
An increase in genome-wide levels of H3K9ac and H3K27ac in a fly model of AD exacerbated Aβ42-driven neurodegeneration [237]. Enrichment of H3K9ac and H3K27ac on the angiopoietin-like protein 4 (ANGPTL4) gene promoter has also been shown to be involved in inflammation, vascular permeability, and metabolic dyshomeostasis [241, 243], and the expression of ANGPTL4 in brain glial cells was stimulated by hypoxia [244, 245], suggesting that enrichment of H3K9ac and/or H3K27ac in the brain may be a typical response to hypoxia [246]. However, in contrast to H3K9ac and H3K27ac, H3K18ac, H3K23ac and H4K16ac were significantly downregulated or lost in the temporal lobe of the AD brain [247, 248]. In AD, hippocampal CA1 H3K12ac was downregulated [249], although it was significantly elevated in peripheral monocytes [250]. Collectively, both upregulation and downregulation of these histone markers (H3K9ac, H3K27ac, and others) (Table 1) may cause cognition defects. Furthermore, H3K9ac upregulation in the PFC and temporal lobe and possibly the downregulation in the hippocampus plays a key role in AD pathogenesis, suggesting that H3K9ac might be a promising drug target for AD therapy.
Overview of the epigenetic signatures in AD, their distribution and related enzymes or factors
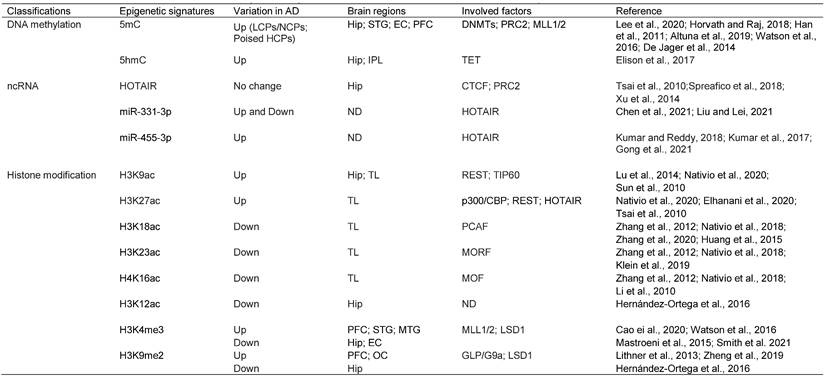
Abbreviations: 5mC: 5-Methylcytosine; 5hmC: 5-Hydroxymethylcytosine; HOTAIR: HOX antisense intergenic RNA; H3K9ac: Histone H3 lysine 9 acetylation; H3K27ac: Histone H3 lysine 27 acetylation; H3K18ac: Histone H3 lysine 18 acetylation; H3K23ac: Histone H3 lysine 23 acetylation; H4K16ac: Histone H4 lysine 16 acetylation; H3K12ac: Histone H3 lysine 12 acetylation; H3K4me3: Histone H3 lysine 4 trimethylation; H3K9me2: Histone H3 lysine 9 dimethylation; LCPs: low-intensity CpG promoters; NCPs: non-CGI promoters; HCPs: High-intensity CpG promoters; Hip: Hippocampus; STG: Superior temporal gyrus; EC: Entorhinal cortex; PFC: Prefrontal cortex; IPL: inferior parietal lobe; TL: temporal lobe; MTG: Medial temporal gyrus; OC: Occipital cortex; DNMT: DNA methyltransferase enzyme; PRC2: Polycomb repressive complex 2; MLL1/2: Mixed lineage leukemia1/2; TET: Ten-eleven translocation; CTCF: CCCTC-binding factor; REST: Repressor element 1 (RE1)-silencing transcription; TIP60: Tat-interacting protein of 60 kDa; p300/CBP: p300/CREB binding protein; PCAF: p300/CBP-associated factor; MORF: MOZ-related Factor; MOF: Males absent on the first; ND: Not determined; LSD1: Lysine-specific demethylase 1.
Of note, H3K9ac has been shown to co-localize with H3K14ac on the previously described poised promoters harbouring H3K4me3 and H3K27me3 [209, 251, 252]. The genes controlled by poised promoters are usually expressed at basal levels due to the coexistence of H3K27me3 and H3K4me3 [181]. Since H3K9ac is an active gene marker, and both H3K9ac and H3K14ac are involved in DNA damage repair [253, 254], their co-occurrence on poised promoters suggests that the expression level of these genes can be increased in response to DNA damage [181]. They are primed by H3K14ac, which is catalyzed by p300/CREB binding protein (CBP, a HAT that can also switch H3K27me3 to H3K27ac downstream of PHF8/KDM7B/JMJD-1.2), p300/CBP-associated factor (GCN5/PCAF) and/or MYST3, and then activated by H3K9ac, which is mainly catalyzed by GCN5/PCAF and/or TIP60 [251]. Consistent with this finding, H3K18ac, H3K23ac, and H4K16ac were found to be involved in the DNA damage response [255-257]. While H3K18ac enrichment promotes the transcriptional expression of nucleotide excision repair (NER)-related genes and inhibits DNA damage [255], H3K23ac is coupled to H3K14ac, which is known to facilitate DNA repair in a positioned nucleosome by stabilizing the binding of the chromatin remodeler [254, 256]. Also, H4K16ac plays an important role in DNA damage repair by modulating the recruitment of the DNA damage repair protein Mediator of DNA damage checkpoint 1 (MDC1) [257]. Critically, H3K18ac, H3K23ac, and H4K16ac occur mainly on promoters and/or enhancers [248, 256, 258-261] and may affect the presence of H3K9ac and H3K27ac and/or cooperate with them to determine whether the gene is activated or repressed in response to DNA damage. However, although it is known that H3K9ac is upregulated in the PFC and temporal lobe, while H3K18ac, H3K23ac, and H4K16ac are downregulated or lost in the temporal lobe of the AD brain [247, 248], information on these factors throughout the AD-affected brain regions is still needed.
Moreover, the expression of H3K9ac is normally inhibited by the level of repressor element 1 (RE1)-silencing transcription/neuron-restrictive silencer factor (REST/NRSF) [236]. REST is a gene-silencing transcription factor (a Krüppel type zinc finger protein), which can bind to thousands of sites in the human genome and repress a large array of coding and noncoding neuron-specific genes [262, 263] by interacting with many chromatin-modifying enzymes, including HDACs (e.g., HDAC1,2), HMTs (e.g., G9a), HDMs (e.g., lysine-specific demethylase 1 (LSD1/KDM1A)), and methyl CpG binding protein 2 (MeCP2) [263-265]. However, REST was found to be absent from the PFC and hippocampus only at late stages of AD [236] suggesting the existence of a mechanism compromising the inhibitory effect of REST on H3K9ac at the early stage of AD. Although the potential mechanism is currently unclear, it is conceivable that chromatin-modifying enzymes associated with REST play a role. This notion is supported by the observation that TIP60 and plant homeodomain finger protein 8 (PHF8/KDM7B/JMJD-1.2, an HDM for demethylation of H3K9me1/2 and H3K27me2) could form a REST-interacting complex and increase the local H3K9ac/H3K9me2 ratios [266-270]. Interestingly, TIP60 is also the core component of the DNA damage repair machinery and can acetylate ATM at damaged sites to regulate DNA repair signals [266].
In summary, histone acetylation markers, especially H3K9ac and H3K27ac, can become aberrant in AD (Table 1) and may lead to abnormal activation or overactivation of the affected genes, which may cause cell death. However, the aberrant expression of H3K9ac and H3K27ac in other regions of the AD brain is unclear, and the underlying mechanism that causes aberrant expression of H3K9ac is just beginning to be understood. Although DNA damage repair is involved and TIP60 may play an important role, details are still missing. For example, it is unclear whether TIP60 acts on histone modifications other than H3K9ac in response to DNA damage and the disparity between the levels of these markers is related to disease stages. Answering these questions will help develop a specific approach to downregulate H3K9ac for AD treatment.
4.2.2. Histone methylation variation in the AD brain
According to previous reports, H3K9me2 in the occipital cortex and PFC is significantly elevated at the late stage of AD [221, 271] and downregulated in the hippocampal CA1 (Table 1) [249]. H3K4me3, associated with memory formation [272], is also elevated in the AD PFC and superior and medial temporal gyrus but is downregulated in the AD entorhinal cortex and hippocampus (Table 1) [169, 273-275]. Interestingly, gene-repressive H3K9me2 and gene-activating H3K4me3 markers are concomitantly and globally upregulated in the AD PFC and downregulated in the AD hippocampus. This observation indicates that gene activation and inhibition are active in the AD PFC, reflecting the compensatory state of gene expression. However, in the AD hippocampus, both gene activation and inhibition are inactive, indicating the overall gene repression state. These results are consistent with the H3K9ac expression pattern in the AD brain; it is unclear whether they are induced in AD due to gradually decreasing REST levels. REST is a master organizer of enzymes involved in histone acetylation and histone methylation [263, 265], and its role in the imbalance of histone methylation has not been clarified.
Since REST is lost at the MCI and AD stages, it is conceivable that the enzymatic complexes organized by REST will be disrupted under these conditions. However, in neurons where REST is absent, REST corepressor (CoREST) is expressed at high levels and exists in complexes with HDAC1, HDAC2, and LSD1 [276, 277]. The LSD1-CoREST complex mediates H3K4me1/2 demethylation and is commonly associated with silencing gene expression [277, 278]. On the other hand, the CoREST complex is not recruited to its target loci without the REST scaffold [276], leading to an increase in H3K4me3 in the PFC of the AD brain due to a lack of demethylation catalyzed by LSD1 and its associated histone lysine demethylase 5A (KDM5A, an H3K4me3 demethylase) [279]. Also, PFC neurons depend critically on lysine methyltransferase 2A (KMT2A, also known as MLL1) and mildly on lysine methyltransferase 2B (KMT2B, also known as MLL2) to maintain H3K4me3 levels at a subset of genes with an essential role in cognition and emotion, for example, at the ARC immediate early gene, which is an important mediator of synaptic plasticity and an AD susceptibility gene [280, 281].
These data indicate that improved activities of KMT2A and 2B may contribute to an increase in H3K4me3 in the PFC of the AD brain. By contrast, it is difficult to define how H3K4me3 is reduced in the hippocampus of the AD brain without REST. In the hippocampus, the LSD1-CoREST complex can most likely bind to the loci, recruit KDM5A, and cause the ensuing demethylation of H3K4me3 [279, 282]. Thus, when REST is lost, the recruitment of the LSD1-CoREST complex and KDM5A to REST-binding loci is region dependent [283]. KMT2A/MLL1 and KMT2B/MLL2 have also been shown to mediate hippocampal H3K4 di- and trimethylation and are critical players in memory formation [284, 285]. Therefore, it cannot be excluded that KMT2A and 2B become deficient due to DNA damage/rearrangement or fail to be recruited to target genes via ncRNA Mistral in the AD hippocampus and cause H3K4me3 reduction [286-288]. Interestingly, LSD1 mediates the demethylation of histone H3K4me1/2 and H3K9me1/2, and its deficiency may lead to an increase in H3K9me2 in the AD PFC [289, 290]. However, other studies revealed that in AD PFC, GLP/G9a (EHMT1/2), a REST-interacting protein that catalyzes H3K9me0 to H3K9me1 and H3K9me1 to H3K9me2, is significantly elevated [221]. The H3K9me2 reduction in the hippocampus may be attributed to a region-dependent lack of GLP/G9a complex at the loci upon loss of REST; GLP/G9a complex is normally recruited to the loci by REST through chromodomain on Y-like (CDYL) and is required for memory consolidation [291-293]. It is noteworthy that there exist other related histone methyltransferases and demethylases for H3K4 and/or H3K9 modifications [289, 294-296], but their functional roles are unclear and need further investigation.
Notably, the same set of genes within different brain regions may be enriched in different markers. For example, in PFC, H3K4me3 is enriched at the ARC gene locus [280], whereas in the hippocampus, H3K9me2 is enriched at this locus [269]. The enrichment of H3K4me3 or H3K9me2 at the ARC locus in different brain regions reflects different activation or DNA repair states. However, H3K4me3 and H3K9me2 have also been reported to co-occur at the same locus, where PHF2 (an H3K9me2 demethylase) binds to H3K4me3 and recruits suppressor of variegation 39H1 (SUV39H1, an H3K9me2/3 methyltransferase) to coordinate H3K9me2/3 and H3K4me3 levels and thereby regulate their target gene expression [297]. PHF2 promotes the expression of memory-related genes by epigenetically reinforcing the Tyrosine kinase receptor B (TRKB)-CREB signaling pathway [298]. While the co-occurrence of H3K4me3 and H3K9me2 reflects the association of these two epigenetic markers, other factors can help determine whether the target gene is activated or inhibited. For example, H3K9ac, H3K4me3, and H3K9me2 concomitantly act on the same set of genes [299], suggesting that H3K9ac can disrupt the balance between H3K4me3 and H3K9me2 and cause activation of the target genes. Intriguingly, H3K9ac was enriched in cell death-promoting genes in AD, whereas H3K4me3 and H3K9me2 were enriched in cell protection and memory formation genes [236, 242]. Thus, the overall outcome of simultaneous enrichment of all three markers (H3K9ac, H3K4ac, and H3K9me2) may have a deleterious effect on cells because the overactivation of a beneficial gene can be toxic. For example, it has been shown that inhibition of GLP/G9a (H3K9me2) rescues synaptic and cognitive functions by stimulating glutamate receptor expression [221]; however, excessive glutamate receptor activity causes excitotoxicity and promotes cell death [300]. This is consistent with previous findings that REST is involved in repressing the expression of glutamate receptors and ARC in addition to synaptophysin and PSD-95 to maintain synaptic homeostasis [301], and H3K4me3, H3K9me2, and H3K9ac are coordinated by REST [221, 236, 280].
4.2.3. Cross-regulatory mechanism between histone methylation and acetylation
A chromatin-modifying complex containing PHF8 and TIP60 has been identified to be involved in the crosstalk between H3K9me2, H3K9acS10P, and H3K4me3 [267, 269]. Clinically, the mutation in PHF8 causes X-linked mental retardation (XLMR), while the mutation in TIP60 has been implicated in the pathogenesis of AD [269, 302]. Mechanistically, at REST-bound promoters, PHF8 can bind to H3K4me3 (also RNA polymerase II) and demethylate H3K9me1/2 to H3K9me0 [270], while TIP60 can bind to H3K9me3 and acetylate H3K9me0 to H3K9ac [266, 303]. In addition, the PHF8 target gene SMCX (JARID1C/KDM5C) can bind to H3K9me3 and demethylate H3K4me3 to H3K4me1/2 [265, 290]. Furthermore, Wang et al. found that H3K4me3 facilitates the acetylation of both H3K9 and H4K16 [304]. H3K4me3 and H4K16ac were shown to be specifically recognized by the Bromodomain and PHD finger transcription factor (BPTF, also known as FAC1) and constitute a unique trans-histone modification pattern in the human genome [305]. Besides, Katoh et al. found that FOXP3, an X-linked suppressor of autoimmune diseases, increases both H4K16ac and H3K4me3 at multiple FOXP3-activated genes by recruiting MOF (which interacts with MLL1) and displacing histone H3K4 demethylase PLU-1 (KDM5B or JARID1B) [306, 307]. It has been reported that BPTF is a subunit of the nucleosome remodeling factor (NURF) complex, which mediates ATP-dependent chromatin remodeling [308]. FOXP3 is a member of the forkhead transcription factor family, mainly expressed in a subset of CD4+ T cells and plays a suppressive role in the immune system [309]. Interestingly, both BPTF and FOXP3 are associated with AD. BPTF protein was found in Hirano bodies and swollen dendrites in the hippocampus of AD patients [310], whereas FOXP3 contributed to chronic neuroinflammation and disease escalation in AD [311]. Recently, Zhao et al. discovered that ectopic introduction of H3K27ac in the promoter region resulted in H3K4me3 enrichment around transcription start site (TSS) via Bromodomain-containing protein 2 (BRD2), while the presence of H3K4me3 at the promoter could not induce H3K27ac increase and failed to activate gene expression [312]. However, deposition of H3K4me3 via KMT2B/MLL2 could remove the repressive marker H3K27me3 and DNA methylation [313].
In summary, H3K4me3 appears to play a coordinating role among the transcriptionally permissive and repressive markers. It interacts directly with several proteins, including PHF8, FOXP3, and BPTF, which further interact with TIP60, MOF, and other subunits of the NURF complex to form an H3K4me3-centered interaction network. Interestingly, while H3K4me3 is required for the transcription of DNA repair genes [314, 315], it prevents DNA repair at the damage sites [316]. Thus, the coordination of PHF8, BPTF, and FOXP3 by H3K4me3 plays an important role in the early stage of AD, characterized by oxidative DNA damage [317]. TIP60 is the core component of the DNA damage repair machinery [266, 270]. In response to DNA damage, H3K9ac is downregulated in conjunction with the upregulation of H3K9me3 through KAP-1/HP1/SUV39H1. Subsequently, H3K9me3 activates TIP60, allowing the acetylation of H3K9 and ATM to prevent DNA damage [253, 303, 318]. In contrast, under hypoxic conditions, PHF8 sustains the level of H3K4me3 and downregulates H3K9me1/2 through KDM3A to prevent DNA damage [270, 319-321]. Interestingly, although PHF2 and G9a regulate the level of H3K9me2 in the opposite direction, both prevent DNA damage [322, 323], the discrepancy in these findings may be due to different cells used in the experiments [322, 323]. Also, H3K9me2 has been shown to be enriched at CGIs and selectively inhibit the expression of the genes involved in BER [324, 325]. Coincidently, H3K18ac and H3K27ac also play a role in BER in cooperation with uracil DNA glycosylase (UDG) and apurinic/apyrimidinic endonuclease 1 (APE1) [326, 327], and H3K27ac promotes oxidative stress-induced expression of noncoding RNA activated by DNA damage (NORAD), preserving genomic stability [328]. Moreover, KDM4, which demethylates H3K9me3 at promoters denoted by H3K4me3 [329], actively regulates the DNA damage response [330, 331]. Finally, MeCP2, the intermediator of DNA methylation and histone modifications, can concomitantly downregulate H3K9ac and upregulate H3K9me3 levels and is involved in the DNA damage response [332, 333]. Overall, histone acetylation (such as H3K9ac, H3K27ac and H3K18ac, and H4K16ac) and histone methylation (such as H3K4me3 and H3K9m2/3) are closely correlated with each other (Table 1) and form an internal regulatory network, which is involved in gene transcription and plays a role in DNA damage repair. Conversely, DNA damage and repair may disrupt the homeostatic state of the network. Therefore, maintaining homeostasis of the network and reorienting the expression profiles by interfering with these epigenetic markers may identify new AD drug candidates.
4.3. Noncoding RNAs (ncRNAs)
ncRNAs are a vast and diverse family of nonprotein-coding transcripts that are mainly divided into two groups: linear ncRNAs and circular ncRNAs. Linear ncRNAs can be further divided into two subgroups according to their lengths: small ncRNAs (sncRNAs, <200 nucleotides), including microRNAs (miRNAs, approximately 22 nucleotides), and long ncRNAs (lncRNAs, >200 nucleotides), including multiple natural antisense transcripts (NATs) [334, 335]. Approximately 80% of the human genome is transcribed as noncoding transcripts, whereas less than 2% encodes proteins [336, 337]. In the CNS, ncRNAs are particularly abundant. Approximately 70% of miRNAs and 40% of lncRNA genes are expressed in the brain [337]. Remarkably, these noncoding transcripts are predominantly located in the nucleus, suggesting that their major function is epigenetic regulation [336, 338, 339].
A large number of ncRNAs are expressed in the CNS with precise temporal and spatial patterns [340-342], and a complex mesh of multitasking ncRNAs (especially miRNAs and lncRNAs) is deregulated and associated with the core pathological phenotypes of AD [343-348]. Although the neurobiology of ncRNAs in the context of AD pathophysiology has been extensively reviewed [346, 347, 349], the underlying mechanisms remain incompletely understood due to their wide variety and complex nature. REST-related ncRNAs have recently been shown to play a key role in AD pathogenesis. REST is an epigenetic master regulator and a universal feature of normal aging in human cortical and hippocampal neurons, and its level is closely correlated with AD and longevity [236, 350]. Also, REST and ncRNAs cross-regulate each other [351, 352]. In this section, we review the current knowledge on the roles of REST-related ncRNAs in AD pathogenesis.
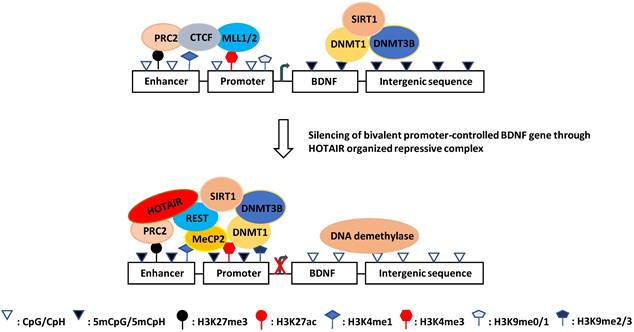
As mentioned above, REST binds to thousands of sites in the human genome and regulates a large array of coding and noncoding neuron-specific genes [262, 263]. It forms complexes with histone-modifying enzymes and other chromatin remodeling partners [353-356]. Especially, the REST complex can form a large complex with HOX antisense intergenic RNA (HOTAIR) and PRC2 [351]. HOTAIR is one of the most extensively studied lncRNAs in human cancer and various other diseases [357, 358]. It facilitates protein-protein interactions, thereby regulating many pathophysiological processes, including epigenetic reprogramming, protein degradation, miRNA sponging, NF-κB activation, inflammation, immune signaling, and DNA damage response [357-360]. HOTAIR also promotes the expression of DNA damage repair factors, including KU70, KU80, DNA-PKs, and ATM, by recruiting EZH2 to the target gene promoters and activates NF-κB by decreasing IκBα [361, 362]. PRC2 is present in almost all eukaryotic cells and is involved in the epigenetic regulation of more than 2000 genes on 10 chromosomes; it is also responsible for H3K27me3 modification of 5%-10% histones [363]. H3K27me3 and H3K4me3 often co-occur on bivalent promoters (poised HCPs) to keep the controlled genes expressed at basal levels. REST binding to the RE1 element increases the local level of H3K27me3 via PRC2 [263] and decreases the H3K4me3 level via SMCX [265, 290] for gene silencing. Interestingly, the loss of function of REST can induce H3K27ac [364]. Moreover, with aging, the level of H3K27me3 decreases while that of H3K4me3 increases, resulting in a progressive increase in gene expression [365, 366]. Since H3K27me3 and H3K4me3 are involved in the DNA damage response and are associated with AD [169, 314, 367], it is conceivable that HOTAIR may play a role in the DNA damage response and AD pathogenesis by coordinating the levels of H3K27ac, H3K27me3, H3K4me3 and other epigenetic markers on the bivalent promoters by recruiting REST and PRC2 [351].
Notably, REST is lost at the MCI and AD stages; therefore, it is unclear whether and how HOTAIR exerts its function at these AD stages [236, 368]. At the prodromal AD stage, HOTAIR may cooperate with REST and PRC2 in the DNA damage response and gene silencing across the genome. For example, under ischemic conditions, which cause oxidative stress/DNA damage and are thought to be a prelude to AD [369-371], miR-132 (a neuron-protective microRNA downregulated in AD) expression is selectively silenced in hippocampal CA1 neurons by REST binding to its promoter rich in H3K9ac and H3K4me2 (another bivalent promoter) [352, 372]. Interestingly, H3K27me3 is also rich in the promoter region of miR-132 [373]. In addition, polycomb group proteins (e.g., PRC2) are involved in ischemic tolerance [374]. Considering these findings, it is possible that HOTAIR represses the transcription of neuron-protective target genes at the prodromal stage of AD by organizing PRC2 and REST, facilitating AD onset. This regulatory mechanism at the prodromal stage of AD can be tissue (region)-specific, probably due to variation in oxidative stress. For example, REST is involved in the downregulation of BDNF [375]. However, forebrain ischemia induces downregulation of BDNF only selectively in hippocampal CA1 (like miR-132), but in CA3 and DG, the BDNF protein and other transcript versions are even upregulated [246]. Also, histone acetylation patterns of H3K27ac, H3K9ac, and H3K14ac on BDNF promoters are different in CA1 compared with CA3 and DG, including decreased H3K27ac, increased H3K9ac and H3K14ac in CA1, increased H3K9ac, H3K14a, and H3K27ac in CA3, and no significant change of H3K9ac, H3K14ac, and H3K27ac in DG [246].
Based on these observations, it appears that REST selectively represses its target genes in the hippocampus at the prodromal stage of AD by conjugating with HOTAIR in different hippocampal regions, where HOTAIR coordinates enzymatic complexes for specific chromatin remodeling. In keeping with this notion, many HOTAIR-organized molecules, including the REST-associated MeCP2-Sin3A-HDAC1 complex and PRC2-associated CBP-JMJD3 complex, have been observed at the BDNF promoter (Fig. 2) [181, 376-379]. Moreover, REST and PRC2 have been shown to be involved in the downregulation of BDNF [375, 380, 381].
Schematic illustration of the hypothesized mechanism of aberrant BDNF expression in AD. DNA methylation at the damage sites recruits MeCP2 and HOTAIR-PRC2-REST complex which prevents coupling of promoter and enhancer resulting in transcription silencing. This event may be involved in the onset of AD.
Finally, HOTAIR is one of the first described lncRNAs that can act in trans [382]. Recently, HOTAIR was found to act as a competing endogenous RNA (ceRNA) by sponging miR-331-3p in gastric cancer [383, 384]. Although it is unclear whether in the CNS, HOTAIR plays a similar role as in gastric cancer, two reports revealed that miR-331-3p is associated with AD [385, 386]. Moreover, it has been shown that HOTAIR negatively regulates the cyclin-dependent kinase 5 regulatory subunit 1 (CDK5R1) gene, which encodes p35, the main activator of CDK5. The active p35/CDK5 complex is involved in numerous aspects of brain development and function, and its deregulation is associated with AD onset and progression [387]. Recently, it was reported that HOTAIR negatively regulates the miR-455-3p/Nod-like receptor protein 1 (NLRP1) axis [388], whereas miR-455-3p has been shown to play a protective role against Aβ-induced toxicities and enhance cell survival and lifespan extension [389, 390].
In silico analysis for its target prediction showed the binding capacity of miR-455-3p with several AD-associated key genes, such as APP, Nerve growth factor (NGF), Ubiquitin specific peptidase 25 (USP25), p53 and DNA damage regulated 1 (PDRG1), Small mothers against decapentaplegic member 4 (SMAD4), Ubiquilin 1 (UBQLN1), SMAD family member 2 (SMAD2), Tumor protein p73 (TP73), Vesicle associated membrane protein 2 (VAMP2), HSPB1 Associated Protein 1 (HSPBAP1), and Neurexin 1 (NRXN1) [391, 392]. It is unclear whether the regulatory mechanisms of HOTAIR on CDKR5 and the miR-455-3p/NLRP1 axis are dependent on its role as a ceRNA or as a scaffold for REST and PRC2. However, miR-455-3p, a potential biomarker for MCI and AD, has been shown to be upregulated at the MCI and AD stages, suggesting that it is derepressed, probably due to the depletion of REST at these AD stages. Furthermore, PRC2, the other partner of HOTAIR, also decreases with age, potentially promoting LOAD development through the dysregulation of APP and PS1 [393]. PRC2 has also been found to bind promiscuous RNA to scan for target genes that have escaped repression [394, 395]. These results suggest that HOTAIR plays an important role in AD pathogenesis by coordinating REST and PRC2 as a scaffold and sponging miR-331-3p as ceRNA. Thus, HOTAIR may be another drug target for AD therapy (Table 1).
5. Loss of proteostasis
Proteins are important components of organisms and participate in all physiological functions. Thus, protein homeostasis must be tightly controlled to maintain fundamental biological processes and survival. However, as an organism ages, the proteome, like the genome, is easily disrupted and damaged [57, 396]. Genomic instability and loss of proteostasis are closely interrelated. DNA damage may reduce the expression of proteins [33], and altered protein expression can inversely affect the DNA structure [249]. Although genomic instability and loss of proteostasis are primary aging hallmarks, aging and its associated diseases are considered a snowballing phenotype of accumulated damaged or toxic proteins [22, 397]. In this context, at the early and advanced stages of AD, alteration of protein synthesis machinery, including nucleolar chaperones, ribosome proteins, and elongation factors, has been observed in the frontal cortex and hippocampus [249, 398]. Moreover, proteins related to many other biological processes are also aberrantly downregulated due to genomic instability in the AD brain [399-401].
5.1. Transcription factors in proteostasis
The regulation of proteostasis mainly includes protein production and degradation. For protein production, transcription factors (TFs) retrieve genetic information from the DNA to RNA together with other factors. Efficient transcription requires the initiation complex and the TFs that bind to the promoter sequence upstream of the TSS [402]. TFs are readers of epigenetic markers, which may regulate transcription by many incompletely understood mechanisms, including stabilization of the initiation complex, destabilization of the chromatin structure, and conformational change of the promoter domain [403, 404]. The binding of approximately 60% of TFs to their target genes is influenced by 5mC [207]. Distinct profile patterns of chromatin features are related to different TF binding events [405]. Conversely, transcription can shape the genome-wide methylome and histone acetylome patterns [148, 406]. Of particular interest, computational analysis of the importance of various DNA-intrinsic and chromatin-associated features of ENCODE data showed that only H3K27ac, H3K4me2, H3K4me3, and H3K9ac are more reliable predictors of TF occupancy [405], whereas position weight matrix (PWM)-scores are among the most important features only for two TFs, namely, REST and CCCTC-binding factor (CTCF) [407].
REST and CTCF are abundantly expressed in the brain and are critical for memory [408, 409]. Additionally, their binding sites are broadly distributed in the genome [207, 410-412], and their distribution patterns are markedly similar to each other when compared to those of other TFs [413, 414]. Furthermore, REST and CTCF are involved in DNA integrity maintenance [270, 415-419] and depletion of DNA methylation [148, 420]. In particular, REST and CTCF are critical for maintaining proteostasis [421, 422], and their defects are associated with AD pathogenesis [236, 423, 424]. In the following section, we review these two TFs and their mechanisms in AD pathogenesis.
5.2. REST
REST is a master transcriptional regulator that controls approximately 2000 target genes [425]. It is usually upregulated with aging in human cortical and hippocampal neurons, protecting them from oxidative stress and Aβ toxicity [236, 426]. However, in MCI and AD, REST is lost from the neuronal nucleus and appears in autophagosomes together with pathological misfolded proteins [236], suggesting that REST degradation is accelerated in AD. However, whether the production of REST protein is also problematic in AD has not been investigated.
5.2.1. DNMT1 and β-catenin in REST expression
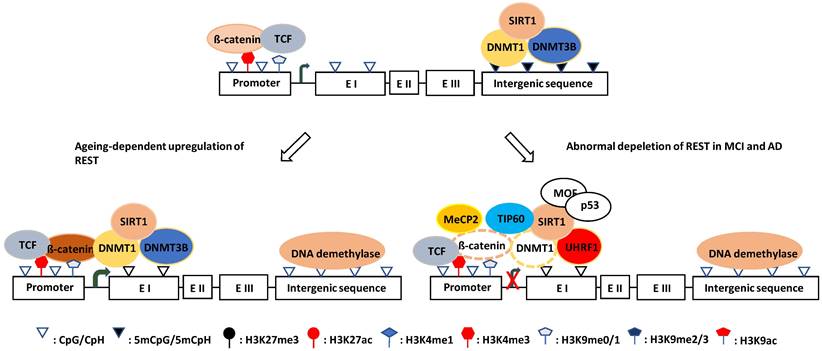
The canonical Wnt signaling pathway has been reported to regulate REST expression, and the β-catenin/TCF complex upregulates the REST gene transcription through a conserved element found in exon 1a [427]. Tomasoni et al. found that REST regulates its own expression through the REST-TSC2-ß-catenin signaling pathway in neural cell models [428]. Song et al. reported that DNMT1 and β-catenin mutually regulate each other, and DNMT1 upregulates β-catenin/TCF-driven transcription by forming a complex with β-catenin and LSD1, where LSD1 demethylates DNMT1 and stabilizes it [200, 429]. Moreover, Funato et al. demonstrated that oxidative stress could activate β-catenin/TCF through the redox-sensitive association between thioredoxin and dishevelled [430], while O´Hagan et al. showed that, under chronic oxidative stress, DNMT1 (also DNMT3B and SIRT1) shifts from non-GC-rich genes and chromosome regions to CGIs [191]. Oxidative stress can also promote DNMT1 enrichment at CGI promoters, which may then upregulate REST expression by forming a β-catenin stabilizing complex. This mechanism probably explains why the REST level increases with aging which is normally accompanied by chronic oxidative stress [431]. Notably, DNMT1 may exert its function in nonenzymatic and enzymatic ways during this process. On the one hand, it can stabilize β-catenin as described, and on the other hand, it can catalyze DNA methylation on exon 1a and/or the promoter of the REST gene (Fig. 3). Consistent with this, a significant portion of tissue-specific differentially methylated regions (T-DMRs) are positively correlated with the expression of transcriptional repressor genes [432].
Schematic illustration of the hypothesized mechanisms of REST expression in ageing and AD. (left) Upregulation mechanism: DNMT1 shifts from non-CGI region to CGI regulatory element under oxidative stress and forms a ß-catenin stabilizing complex to prevent DNA methylation and increase ß-catenin/TCF activity thereby increasing REST transcription. (Right) Downregulation mechanism: DNMT1 is degraded when acetylated by TIP60 and ubiquitinated by UHRF1. which can lead to ß-catenin destabilization. Whereas, MeCP2 can reduce availability of ß-catenin for binding to DNMT1. Both cases will downregulated REST transcription. MOF and p53 prevent SIRT1 expression, which can aggravate DNMT1 degradation.
5.2.2. UHRF1 in REST depletion
In contrast, the Wnt signaling pathway is defective [433, 434], and β-catenin is destabilized by phosphorylation in AD [435-437]. However, the upstream signaling that induces phosphorylation-dependent degradation of β-catenin is unknown. There are reports showing that hypermethylated in cancer 1 (HIC1, a zinc finger transcriptional repressor) can attenuate the Wnt signaling pathway by recruiting TCF-4 and β-catenin to the nuclear bodies [438], while SIRT1 can deacetylate β-catenin and decrease its affinity for TCF4, thereby reducing its activity [439]. It is unknown whether these two events initiate the degradation process of β-catenin. However, HIC1 is reduced with age by DNA methylation and H3K27me3 modification (trimethylated by PRC2 and demethylated by KDM6A/UTX and KDM6B/JMJD3) [440-442]. Additionally, in elderly and senile AD patients, the expression of SIRT1 (also SIRT3 and SIRT6) in the hippocampus and saliva was 1.5-4.9-fold reduced compared with age-matched healthy individuals [443]. Although DNA hypermethylation with increasing age can promote SIRT1 gene transcription through the downregulation of HIC1 [440], SIRT1 mRNA was degraded through the ROS-Chk2-HuR pathway in elderly and senile AD patients [444]. Therefore, it appears impossible that REST depletion is due to the direct action of HIC1 and SIRT1 on β-catenin, and its destabilization and degradation in AD need to be elucidated.
Oh et al. found that UHRF1 inhibits REST expression [445]. UHRF1 is an E3 ligase that can bind to H3K9me3 and interact with DNMT1 at the promoter region of the REST gene [445]. Most importantly, UHRF1 ubiquitinates DNMT1 and promotes its degradation when DNMT1 is acetylated by TIP60 [202, 203]. Considering the importance of DNMT1 for the stability and activity of β-catenin [200], it is conceivable that DNMT1 degradation may lead to β-catenin destabilization and inactivation, resulting in REST depletion. In line with this notion, the levels of SIRT1 in the hippocampus and parietal cortex of AD patients were reduced compared with controls [443, 446]. Because TIP60 is negatively regulated by SIRT1 [447-449], its reduction is expected to increase the TIP60 level, which can acetylate DNMT1 for degradation, which itself is a substrate of SIRT1 [201]. Thus, SIRT1 reduction combined with TIP60 upregulation aggravates DNMT1 acetylation and destabilization. However, this notion is challenged by the finding that in the early AD Drosophila brain, TIP60 is decreased while HDAC2 is increased before Aβ plaque formation [220]; it is probably caused due to the feedback regulation of TIP60 by UHRF1. While TIP60 interferes with the USP7‑UHRF1 association and induces UHRF1 degradation in an auto‑ubiquitination‑dependent manner [450], UHRF1 recruits and ubiquitinates TIP60 for degradation to prevent its overaction [451, 452]. Collectively, SIRT1 reduction may play a key role in REST depletion by destabilizing DNMT1 directly by deacetylating it or indirectly by stabilizing TIP60.
5.2.3. MeCP2 in REST depletion
MeCP2 is widely believed to be a transcriptional repressor involved in methylation-associated gene inactivation [453, 454], although it can also positively regulate gene expression [455, 456] and plays an important role in CNS development, spontaneous neurotransmission, and short-term synaptic plasticity [454]. Interestingly, MeCP2 is broadly expressed in mature human brain cells at all ages [457], binds to methylated cytosines (5mCpG and 5mCpA) with high affinity, and induces genome-wide histone deacetylation, especially on promoters, resulting in repression of many genes [457-459]. For example, MeCP2 represses many long genes (>100 kb) with neuronal functions [207, 460]. Abuhatzira et al. found that MeCP2 binds to the promoters of REST and CoREST genes despite their unmethylated state and is involved in the repression of their expression [380]. However, whether MeCP2 contributes to REST depletion in MCI and AD has not been studied. It usually binds to methylated CGIs and represses gene expression mainly through a histone deacetylase complex composed of mammalian switch-independent 3A (mSin3A), HDAC1, and HDAC2 [461, 462]. The complex deacetylates acetylated histones, which are associated with a target gene, then changes the chromatin structure and leads to the repression of gene expression, showing a direct causal relationship between DNA methylation-dependent transcriptional silencing and the modification of chromatin [332, 461, 462]. But at late AD stages, when REST is lost, HDAC1 and HDAC2 were decreased in the PFC and hippocampus of AD patients compared with controls [234, 235]. Thus, it seems highly unlikely that REST depletion in MCI and AD is caused by MeCP2-mSin3A-HDAC1/2-mediated deacetylation of acetylated histones associated with the REST gene promoter.
Kimura and Shiota discovered that MeCP2 could form a complex with DNMT1 through the same transcription repressor domain, which mediates interaction with mSin3A-HDAC1/2 [463]. It was proposed that DNA methylation in vivo is maintained by the MeCP2-DNMT1 complexes [463]. It is conceivable that the MeCP2-DNMT1 complex formation at the REST gene promoter interferes with the MeCP2-mSin3A-HDAC1/2 and the DNMT1-β-catenin complex formation and, therefore, may lead to MeCP2 acetylation and β-catenin destabilization/activity reduction, possibly inhibiting REST gene expression. It has been reported that SIRT1 reduction in AD increases acetylation of MeCP2, thereby preventing its binding to HDAC1 and SWI/SNF DNA helicase/ATPase (ATRX) and enabling its binding to DNMT1 [458, 463-465]. Interestingly, as competitors of DNMT1 binding, both MeCP2 and β-catenin regulate the expression of tau protein in an antagonistic manner to regulate the REST gene. While MeCP2 promotes tau expression and prevents REST expression, β-catenin does this in the opposite direction [466-468]. Moreover, MeCP2 and β-catenin interact with p300/CBP and are regulated by the p300/CBP-SIRT1 switch [469, 470]. Taken together, these observations suggest that MeCP2 contributes to REST gene silencing in MCI and AD by binding to DNMT1 and excluding β-catenin from the DNMT1-β-catenin complex and not by its interaction with the mSin3A-HDAC1/2 complex. This is consistent with a previous report that MeCP2 may mediate gene expression repression via a histone deacetylation-independent mechanism [471].
5.2.4. Signaling integration in REST depletion
As described above, REST expression is controlled by β-catenin, UHRF1, and MeCP2. While β-catenin promotes REST expression, MeCP2 and UHRF1 inhibit its expression [380, 427, 445]. Interestingly, all three molecules interact with DNMT1 [200, 202, 203, 463]. Given that the level of DNMT1 is positively correlated with the stability and activity of β-catenin, REST depletion in MCI and AD may be due to β-catenin deficiency induced by DNMT1 reduction or redistribution. In agreement with this notion, Wnt/β-catenin signaling is greatly suppressed in the AD brain, and its activation inhibits amyloid-β production and tau protein hyperphosphorylation in the brain [472]. It has been proposed that a sustained loss of function of Wnt/β-catenin signaling underlies the onset and progression of AD [473].
DNMT1, together with DNMT3B and SIRT1, shifts from non-GC-rich genes and chromosome regions to CGIs [191], promoting local DNA methylation and can then be degraded or redistributed through UHRF1 or MeCP2 [202, 203, 463]. In response to DNA damage, molecules including Males absent on the first (MOF/MYST1/KAT8), TIP60, and p53 are also recruited to the damage sites [257, 266, 474-477]. They are usually involved in DNA damage repair [257, 266, 475-477], but they may also regulate DNMT1. It has been shown that TIP60 accelerates DNMT1 degradation together with UHRF1 [202, 203]. In contrast, MOF and p53 have not been reported to be directly associated with DNMT1 degradation. However, MOF (and TIP60) can acetylate p53 at the DNA binding domain (K120), thereby inhibiting SIRT1 expression and promoting p300/CBP autoacetylation [478-480], which, in turn, can destabilize DNMT1 through TIP60 and MeCP2 [447, 465]. Besides, MOF is the major acetyltransferase for the expression of H4K16ac, which can recruit p53 via the MDC1-53BP1-BRCA1 complex to DNA damage foci [257, 481]. Thus, the MOF-p53 axis may induce REST depletion by downregulating SIRT1, which is decreased in AD [443]. Additionally, HDAC1, which can deacetylate p53 and thereby upregulate SIRT1, is decreased in AD [234, 235, 482]. Of particular importance, MOF and p53 are also AD risk factors [483, 484].
Notably, MOF and p53 are substrates of SIRT1 [448, 485-488], and p53 is also a substrate of HDAC1 [482]. SIRT1 is multifunctional and is an important player in the DNA damage response as a histone and non-histone deacetylase [489]. For example, SIRT1 interacts with the MYST domain of MOF via its catalytic domain and deacetylates autoacetylated MOF, the major acetyltransferase for the expression of H4K16ac [257, 490], while SIRT1 also deacetylates H4K16ac [491]. Although deacetylated MOF robustly binds to nucleosomes, becomes more active, and is more easily degraded, the acetylation of MOF decreases its binding ability and the global H4K16ac level [257, 448, 490]. Interestingly, normal aging leads to H4K16ac enrichment, but AD entails dramatic losses of H4K16ac in the proximity of genes linked to aging and AD, which are especially enriched in the REST binding motif [248]. Since SIRT1 is recruited to damaged CGIs together with DNMT1 and DNMT3B [191, 447, 448], it is possible that DNA damage-induced imbalance of the SIRT1/MOF ratio causes loss of H4K16ac at the gene regulatory elements, which may recruit REST and silence the gene. This mechanism may contribute to REST depletion because REST can regulate its own expression [428].
In summary, REST gene silencing is apparently related to the DNA damage response. An array of molecules, including DNMT1, TIP60, UHRF1, ß-catenin, MOF, p53, and SIRT1, play critical roles in the silencing process. These molecules may act in a cascade pathway or in an integrated platform and eventually cause β-catenin deficiency, resulting in REST depletion. Based on the above findings, REST depletion process appears to be as follows: (1) chronic oxidative stress causes DNA damage and DNA damage response, (2) continuous DNA damage and DNA damage response cause accumulation of DNMTs and DNA methylation at the regulatory elements, (3) MeCP2 binding to methylated CpGs and DNMT1 enhances DNA methylation and repels β-catenin from the β-catenin-DNMT1 complex, thereby inactivating β-catenin transcriptional activity, and (4) MeCP2-mediated gene silencing can be aggravated by TIP60/UHRF1-mediated DNMT1 degradation, MOF-mediated upregulation of p53 activity, and subsequent downregulation of SIRT1. Thus, MeCP2 plays a key role in REST depletion (Fig. 3). However, it is noteworthy that in addition to its multiple functions, MeCP2 also interacts with RNA binding fox-1 homolog (Rbfox) and the lncRNA Retinal non-coding RNA3 (RNCR3), affecting chromatin remodeling and mRNA splicing [492-494]. Whether and to what extent the changes in mRNA splicing caused by MeCP2 are related to REST loss are unknown. However, an altered RNA spliceosome was reported to be associated with the expression of APP and TRKB [495], which are closely related to REST depletion [380].
5.3. CTCF
CTCF is a multifunctional protein in genome regulation and gene expression [411] that can bind to tens of thousands of genomic sites relying on distinct pathways compared with REST [496, 497]. CTCF preferentially binds to unmethylated DNA sequences [496, 498, 499]; it can bind to DNA damage sites and activate a cascade reaction resulting in DNMT1 inactivation and DNA demethylation [500]. Moreover, CTCF interacts with histone modifiers, including HATs and HDACs, and is involved in histone modifications [501, 502]. In this respect, CTCF is especially required to implement both H3K27ac and H3K27me3 [503]. Collectively, CTCF is a master organizer of chromatin structure, which plays a key role in DNA damage repair and gene transcription, and its defect may rewire genome-wide chromatin accessibility and have serious implications [504]. Thus, the decline in CTCF with age may be an initial mechanism in AD pathogenesis [505, 506].
5.3.1. APP and Tau
It has previously been reported that CTCF is required for the expression of APP and PAX6 [423, 507, 508]. PAX6, in turn, directly regulates the transcription of GSK-3β, which further catalyzes amyloid-β-mediated tau phosphorylation [509, 510]. Since CTCF decreases with age, it does not seem to directly upregulate APP in AD [511, 512]. However, the decline in CTCF might indirectly interfere with APP expression. For example, SMAD3 and SMAD4 have been shown to be specifically associated with the CTCF-APP promoter binding beta (APBbeta) complex to promote the TGF-beta-induced expression of APP [513]. However, in another study, SMAD3 and 4 were shown to form nuclear complexes with SP1 in TGF-beta-mediated APP expression [514]. Since the CTCF binding site overlaps with that of SP1 [515], the decline in CTCF may enable the complex formation of SP1 with SMAD3 and 4 in regulating APP expression. Consistent with this notion, SP1 was upregulated in the cortex and hippocampus of the AD mouse brain, upregulating APP and tau [516, 517]. Also, HDAC1, which interacts with SP1 and represses its activity, was reduced in the PFC and hippocampus of AD patients [234, 235, 518]. REST, which interacts with and represses SP1, is also lost in MCI and AD [236, 519, 520].
5.3.2. NPAS4
Neuronal PAS domain protein 4 (NPAS4) is a brain-restricted and activity-induced TF for synaptic excitation/inhibition (E/I) homeostasis [521]. The expression of NPAS4 is synergistically repressed by REST and CTCF, with REST binding to the promoter and CTCF binding within intron I of the NPAS4 gene [521]. Accordingly, the decrease in CTCF or loss of REST can increase the level of NPAS4 due to reduction or loss of gene repression. Interestingly, as mentioned earlier, the decline in CTCF and loss of REST may increase APP level, whereas APP can also upregulate the NPAS4 expression [522, 523]. Thus, the decrease in CTCF and/or loss of REST may induce neuronal E/I imbalance by upregulating NPAS4 directly or indirectly via APP. Besides, APP can mediate tau phosphorylation, whereas APP-induced upregulation of NPAS4 can facilitate the autophagic clearance of tau [522]. Thus, CTCF appears critical for the homeostatic maintenance of the APP-NPAS4-tau axis, and its decline or loss with age may cause APP/tau dyshomeostasis and NPAS4-mediated E/I imbalance, which usually occurs in the early stage of AD [524].
5.3.3. PCDHs
Protoconherins (PCDHs) constitute the largest subgroup within the cadherin family of calcium-dependent adhesion molecules [525, 526]. There are three large protocadherin clusters (PCDHα, PCDHß, and PCDHγ) that are highly expressed in the brain [525, 526] and are involved in CNS development and establishment of cell diversity and appropriate neural circuits [525-530]. A decreased protocadherin expression was associated with severe dysregulation of dendritic morphology and arborization [531]. Thus, PCDHα is critical for learning and memory [532, 533].
CTCF functions as an architectural protein that can mediate inter- and intra-chromosomal interactions, playing an important role in chromatin loop formation and 3D DNA topology regulation [534, 535]. It is also the only well-defined mammalian insulator protein with properties to form long-range chromatin loops [536], which are often found between active promoters, enhancers, and CTCF binding sites [537]. However, the exact function of CTCF at a given genomic site is unpredictable [496] and determined by the associated TFs and the location of the binding site relative to the TSS, enhancer, and promoter of the gene [496]. Enhancers are the most salient noncoding DNA elements sprinkled throughout the genome that activate gene promoters and help orchestrate proper spatiotemporal gene expression [538, 539]. In the brain, enhancers accurately regulate intricate expression programs across different neuronal classes, providing incredible cellular and functional diversity [540]. Thus, enhancer dysregulation may induce different expression levels of their target genes and predispose or contribute to diseases [540].
Many studies have shown that CTCF is a master regulator of clustered PCDH genes and promotes their stochastic and combinatorial expression by mediating topological chromatin interactions between enhancers and promoters [531, 541-544]. Recently, Tang et al. found that REST directionally forms base-specific interactions with neuron-restrictive silencer elements (NRSEs) within distal enhancers and target gene promoters, preventing CTCF-mediated long-range enhancer-promoter interactions and leading to the downregulation of clustered PCDHα proteins [545]. Notably, CTCF binding to sites proximal to each promoter of PCDHα genes is demethylation dependent. Epigenetic methylation of CTCF binding sites (CBS) abrogates CTCF binding and abolishes its ability to bridge long-distance chromatin looping interactions between distal enhancers and their target promoters [546-548]. It is, therefore, conceivable that the loss of REST causes an increase in PCDHα, although the gained methylation at CBS and a decrease in CTCF may antagonize this effect, likely contributing to AD pathogenesis. This notion was supported by the observation that PCDHγC5 was increased in neuronal hyperexcitation conditions upon Aβ treatment and in APP/PS1 transgenic mice [549].
5.3.4. Arc and BDNF
In the brain, enhancers are necessary for genes that create cellular and functional diversity and activity-dependent gene expression associated with long-term potentiation (LTP) and homeostatic plasticity [540]. Likewise, CTCF-mediated enhancer-promoter interactions are involved in the regulation of basal [550, 551] and activity-regulated genes (e.g., Arc, Npas4, Fos, Rgs2, and Nr4a2) [540]. Interestingly, although these activity-regulated genes have variable physiological functions and have different enhancers controlling their transcription plasticity [540], their defects can similarly lead to AD [281, 523, 552-554].
The expression of BDNF and Arc is cell type- and stimulus-specific and is finely tuned [555-557]. It has been shown that the expression of BDNF and Arc is regulated by CTCF [558, 559]. However, Sams et al. found that CTCF is involved in the activity-dependent expression of BDNF and Arc but not under basal conditions [409]. As mentioned above, BDNF basal expression is controlled by poised promoters of H3K27me3 and H3K4me3 [181], whereas the basal expression of Arc is regulated by the PHF8-TIP60 complex, which specifically counteracts H3K9me2 to facilitate the formation of H3K9acS10P [267, 269]. Thus, the decline in CTCF with age may cause a decrease in BDNF and Arc by downregulating the coupling between the poised promoters and enhancers. In support of this notion, BDNF was found to be decreased in the hippocampus, parietal cortex, and temporal cortex of AD patients [560-562]. Additionally, Arc was reduced in the hippocampus of AD patients and most mouse AD models [563, 564] but was increased in some mouse AD models [565, 566]. Since BDNF and Arc are vital for activity-dependent plasticity and memory consolidation [567-571], and the expression of approximately 100 genes associated with the pathophysiology of AD depends on Arc [281], it is plausible that the decline in CTCF may contribute to the development of AD by downregulating BDNF and Arc.
5.3.5. MHC II
CTCF can act as a transcriptional activator or repressor and an insulator in a tissue-specific or conserved way [496]. Genome-wide studies support CTCF as a global insulator [572]. Insulators are classically defined by two experimental properties, enhancer blocking (EB) and barrier insulation, and CTCF is generally considered to function solely via the EB mechanism with no direct role in barrier insulation [414]. For example, CTCF binds to the XL9 enhancer element at the major histocompatibility complex class II (MHC-II) locus between human leukocyte antigen-DRB1 (HLA-DRB1) and HLA-DQA1 genes (driven by divergent promoters) and blocks their expression [573-575]. However, in many neuropathological disorders, including AD, the expression of MHC-II in microglia is markedly increased [576].
Microglia profoundly affect chronic inflammation in the brain and increase susceptibility to AD [577]. Nikolic et al. found that upon toll-like receptor (TLR) stimulation, Ctcf-deficient macrophages produced normal levels of proinflammatory cytokines IL-12 and IL-6 but manifested a strongly impaired capacity to produce tumor necrosis factor (TNF) and IL-10 and the IL-10 family members IL-19, IL-20, and IL-24 [578]. Furthermore, human genetic data point to a key role of microglia in the development and progression of AD pathology. Impaired microglial activities and altered microglial responses to β-amyloid are associated with increased AD risk. Activated microglia can also be harmful to neurons, mediate synapse loss by engulfing synapses, exacerbate tau pathology, and secrete inflammatory factors [579]. Thus, the decline in CTCF may contribute to AD pathogenesis by tuning macrophage function on MHC-II presentation and cytokine production.
5.3.6. H19 and IGF2
Another example of the EB function of CTCF is that it can bind to the maternally imprinted control region (ICR) of the H19/IGF2 locus with a group of downstream enhancers, thereby blocking IGF2 expression while activating the expression of H19 [580]. In parallel, on the paternal allele, IGF2 expression is activated, while H19 expression is silenced by binding of MeCP2-mSin3A-HDACs to ICR in a methylation-dependent manner [581, 582]. Furthermore, the decline in CTCF and/or biallelic methylation of ICR lead(s) to loss of imprinting (LOI) of IGF2 [583, 584], while the biallelic hypomethylation of ICR causes LOI of H19 [584]. Interestingly, IGF2 and H19 are associated with aging and AD [583, 585-587]. While a normal level of IGF2 correlates with memory performance, overexpression of IGF2 can induce cellular senescence [583, 588], which is linked to AD [589]. In contrast, H19 promotes neuroinflammation by driving HDAC1-dependent M1 microglial polarization [590, 591].
Intriguingly, the LOI of IGF2 has also been reported in some regions of normal adult and whole fetal brains [592], suggesting that IGF2 is overexpressed in these brain regions. However, there is evidence showing that the IGF2 level is decreased in the hippocampus of AD patients and a mouse model of this disease, and ectopic expression of IGF2 in the hippocampus of transgenic aged mice reduces Aβ plaques and reverses memory deficits [593]. Considering that LOAD is age-dependent, these findings may indicate that the LOI of IGF2 with age induces overexpression of IGF2, which, in turn, causes cellular senescence and subsequent IGF2 reduction. As such, the decline in CTCF may contribute to AD pathogenesis through the LOI of IGF2. In contrast, H19 expression was drastically reduced in the adult brain compared to the fetal brain and was detectable only in the pons and globus palludus [592]. However, Zhang et al. found that H19 knockdown promotes viability and inhibits oxidative stress in Aβ25-35-expressing PC12 cells by regulating the expression of miR-129 and high-mobility group box 1 (HMGB1), suggesting that H19 may be overexpressed in the AD brain [594]. H19 overexpression is caused by the biallelic hypomethylation of ICR in cancer [584]. There are no reports on whether and how biallelic hypomethylation of ICR occurs in the AD brain. However, oxo8dG and 5hmC in CGIs can induce DNA hypomethylation by inhibiting DNA methylation or activating the DNA demethylation processes [193]. Additionally, there is evidence showing that H19/IGF2 methylation states are potentially relevant to hippocampal structure and function across the life course [595, 596]. Taken together, the decline in CTCF may cooperate with the gained methylation on ICR at the initial stage of AD to induce LOI of IGF2, which then may cause cascade events resulting in LOI of H19.
5.3.7. Other proteins
CTCF mediates POLD1 (the catalytic subunit of DNA polymerase δ) transcription, and its decline with age has been shown to accelerate the progression of cell aging and cognitive impairment in AD [505, 597]. Also, a reduction in CTCF upregulates p16INK4a, which is involved in premature senescence [598]. CTCF shares DNA binding sites in post-mitotic cells with cohesin [496], a ring-shaped complex composed of four core subunits, Structural maintenance of chromosome 1 (SMC1), SMC3, RAD21, and stromal antigen 1 (STAG1), and several cohesin-associated proteins [599]. Cohesin is associated with DNA replication and sister chromatid cohesion during the S, G2, and M phases of the cell cycle. Also, it has intrinsic roles in maintaining the neuronal post-mitotic state, which is often disrupted by cohesin impairment and causes the aneuploidogenic phenotype of AD [600]. As with CTCF, cohesin decreases with age and is probably the reason for the aneuploidogenic phenotype of AD. CTCF co-occupies sites with Forkhead box A1 (FOXA1) and the estrogen receptor (ER) which are also associated with AD [601-603], suggesting that it facilitates their transcriptional activation [496]. CTCF also binds promiscuous RNA for genome organization [604]. These findings indicate that CTCF is a key TF that may regulate the expression of many AD-associated proteins by cooperating with diverse interaction partners (Table 2).
Overview of the AD-associated proteins/ncRNA regulated by CTCF and its co-factor
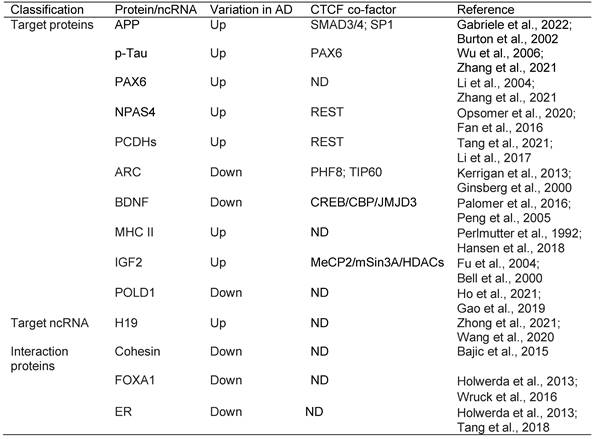
Abbreviations: APP: Amyloid precursor protein; p-Tau: Phosphorylated Tau; PAX6: Paired box 6; NPAS4: Neuronal PAS domain-containing protein 4; PCDHs: Protocadherins; ARC: Activity-regulated cytoskeletal; BDNF: Brain-derived neurotrophic factor; MHC II: Major Histocompatibility Complex Class II; IGF2: Insulin growth factor 2; POLD1: Polymerase delta; H19: Long non-coding RNA H19; FOXA1: Fork-head box protein A1; ER: Estrogen receptor; SMAD3/4: Small mother against decapentaplegic 3/4; SP1: Stimulatory protein 1; ND: Not determined; REST: Repressor element 1 (RE1)-silencing transcription; PHF8: PHF finger protein 8; TIP60: Tat-interacting protein of 60 kDa; CREB: cAMP response element binding protein; CBP: CREB binding protein; JMJD3: Jumonji domain-containing protein-3; MeCP2: Methyl-CpG-binding Protein 2; mSin3A: Mammalian switch-independent 3A; HDACs: Histone deacetylases.
In summary, REST and CTCF are a pair of TFs closely associated with many biological processes. Although they bind to different consensus sequences and have no reported physical interactions, they are spatially distributed near each other in the genome and have fine-tuned cooperation in maintaining genomic integrity and gene expression. REST homeostasis maintenance is dependent on DNMT1, whereas the homeostasis of REST and CTCF is required for decreasing DNA methylation. In response to DNA damage, they may play a compensatory role in the damage repair by keeping DNMT1 and its activity at the homeostatic level. However, in response to neuronal activity stimuli, REST and CTCF may play antagonistic or synergistic roles depending on their binding location and methylation state. Therefore, when REST and CTCF are absent or present at low levels, physiological processes associated with them may not function. Especially, DNA damage can be aggravated, and the neuronal E/I balance can be disrupted, causing cell death, and thereby contributing to AD pathogenesis.
6. Discussion
AD is the most prevalent form of dementia and is associated with many risk factors. This review highlights the primary hallmarks of aging in AD pathogenesis. Among the four primary aging features, three are related to genomic DNA, in agreement with the fact that the genome is a dynamic information and communication system sensitive to external and internal genotoxic stresses [605, 606]. In particular, ROS can accelerate genomic instability and telomere attrition [51, 80, 81, 607], thereby deregulating epigenetic modifications [191, 266, 270, 608] and protein expression profiles [609, 610].
AD is characterized by SNV [6, 16], which is positively correlated with oxidized DNA signatures [611, 612]. The accumulation of oxyradical-associated DNA damage in the hippocampus, midbrain, and caudate putamen is greater than in other regions [46, 63, 64], especially the hippocampus is vulnerable to damage at the early stages of AD [613]. Since reactive species are signaling molecules that reflect cellular activity, brain regions that are more active and produce high ROS levels are more easily damaged, although there are autonomous and nonautonomous antioxidants to protect them [61, 63, 70-78]. In agreement with above observations, DNA damage-associated DNA methylation and aberrant histone modifications in the AD hippocampus have been reported [168, 169, 236-238, 275]. DNA methylation and histone modifications are well-established biomarkers that reflect the biological age of any tissue and predict early disease risk and mortality [207, 614, 615].
Under chronic oxidative stress, HCPs with low expression (H3K27me3 and H3K4me3) become hypermethylated, while HCPs with high expression (H3K27ac and H4K20me1) remain unmethylated [191, 209]. De Jager et al. reported that DNA methylation at the strong promoters that drive fundamental cellular processes of neurons and glia in the healthy brain is not effectively altered in AD. Rather, methylation changes appear to primarily affect genomic regions that are weakly transcribed or inactive in the healthy older brain [164]. These observations are consistent with the finding that the most prominent alteration of DNA methylation in AD occurs mainly at hypomethylated or unmethylated promoters or enhancers [164], while the global variation of DNA methylation is not well defined [194, 616-619]. Interestingly, these hypomethylated or unmethylated promoters or enhancers are usually occupied by REST and CTCF in healthy cells [207, 496, 498, 620-623], which have been shown to protect against DNA methylation [148, 420, 624]. While CTCF protects against DNA methylation through a cascade reaction resulting in DNMT1 inactivation, it is unclear how REST performs this function. It likely prevents DNA methylation by recruiting KMT2B/MLL2, which catalyzes H3K4me3 [187, 313, 625]. Therefore, the reported aberrant DNA methylation in AD may be induced by the depletion of REST and a decrease in CTCF.
Although H3K4me3 prevents DNA methylation, it is widely accepted that DNA methylation can direct histone acetylation, especially at sites occupied by REST and PRC2 [332, 626, 627]. Interestingly, the reported histone acetylation markers in AD are associated with the DNA repair response and are most likely induced by DNA methylation. MeCP2 may play a critical role as it can bind to the mSin3A-HDAC1/2 complex via methylated CpG, stimulating genome-wide histone deacetylation [458, 459, 461, 462] and also regulates DNA methylation by binding to DNMT1 in a mutually exclusive manner [463]. However, in response to DNA damage, HATs, such as MOF and TIP60, can be recruited to the damaged sites by molecules other than MeCP2 and then interact with H3K9me3 [266, 628-630]. MOF specifically catalyzes H4K16ac [257, 631], whereas TIP60 can catalyze H3K9ac, H3K14ac, and H2AK5ac [238, 266, 475, 476]. Thus, DNA methylation-directed histone deacetylation and MOF-TIP60-mediated histone acetylation play antagonistic roles at the damaged sites. MeCP2 downregulates H3K9ac in the DNA damage response via HDAC1/2 [332, 333]; however, when MeCP2 or HDAC1/2 is decreased, the H3K9ac level is expected to increase. Indeed, MeCP2 was reduced in the AD hippocampus [632], and HDAC1 and HDAC2 were generally downregulated in the AD brain [234, 235]. This explains H3K9ac upregulation in the AD PFC and suggests that H3K9ac in the AD hippocampus may increase depending on MeCP2 and TIP60 levels.
H3K27ac is upregulated in parallel with H3K9ac in the AD brain. H3K27ac is catalyzed by p300/CBP and deacetylated by HDAC1/2 [229, 241, 633], whereas MeCP2 can recruit p300/CBP and HDAC1/2 (with different domains) to methylated CpGs [464, 634]. It is conceivable that the elevation of H3K27ac in AD is caused by loss of HDAC1/2 and local enrichment of p300/CBP. Given that MeCP2 binds to DNMT1 and regulates DNA methylation [463], the level of H3K27ac is correlated with DNA methylation. The finding supports the contention that p300/CBP and PRC2 are bound at unmethylated CGI loci occupied by REST and CTCF [178, 179, 184, 208, 363, 635, 636], which prevent DNA methylation and interact with PRC2 [637, 638]. CTCF was reported to be required for PRC2 stabilization and H3K27me3 expression [503], which in turn is needed for repressing HOTAIR production [639]. HOTAIR forms a scaffold with REST and PRC2 in a gene-repressive complex [351, 640]. Interestingly, HOTAIR also interacts with DNMT1 [641]. Therefore, CTCF and REST may play a critical role in DNA methylation-directed histone acetylation by cooperating with MeCP2, PRC2, p300/CBP, TIP60, and MOF.
It has been reported that DNA methylation can exclude H3K4me3, which is catalyzed by CXXC proteins, such as KMT2A/MLL1 and KMT2B/MLL2 [642-644]. On the contrary, DNA methylation can upregulate H3K9me2/3 concomitant with the downregulation of H3K9ac through the MeCP2-SUV39H1 complex [332, 645]. Although G9a also catalyzes H3K9me2, MeCP2 was reported to repress its activity [221, 646]. As previously mentioned, DNA methylation is increased in the PFC and hippocampus of AD patients, whereas H3K4me3 and H3K9me2 are significantly elevated in the PFC [221, 271, 273, 274] but downregulated in the hippocampus and entorhinal cortex [169, 249, 275]. MeCP2 is positively correlated with H3K9me2 and robustly expressed in the PFC but is downregulated in the hippocampus of AD patients [632, 647]. Therefore, it is understandable that H3K9me2 is increased in the PFC but is decreased in the hippocampus of AD. However, the reason for the H3K4me3 increase in the AD PFC is not known, it was expected to decrease as in the AD hippocampus due to aberrant DNA methylation. Notably, the H3K4me3 level is closely related to H3K9me2/3 through the PHF2-SUV39H1 complex [297]. MeCP2 can also interact with chromodomain helicase DNA binding protein 1 (CHD1) and poly (ADP-ribose) polymerase 1 (PARP1), which bind to H3K4me3 [648, 649]. These data suggest that the PHF2-SUV39H1 complex and MeCP2 can prevent H3K4me3 exclusion by DNA methylation and, therefore, create a bivalent intermediate state, especially under DNA damage conditions. Furthermore, H3K9me3 can inversely upregulate DNMT1-mediated DNA methylation [650], while H3K4me3 inhibits de novo DNA methylation [187, 651]. Thus, although DNA methylation may play a leading role in the regulation of histone methylation, there is a feedback loop and the crosstalk with H3K4me3, likely resulting in many intermediate states of these epigenetic markers. This notion is supported by the presence of many histone methylation markers, including H3K4me2, H3K9me2, H3K27me3, H3K36me2, H3K36me3, H3K79me2, H4K20me2, H4K20me3, and their related HMTs and HDMs at DNA damage sites [652].
In summary, age-dependent DNA damage, telomere attrition, and DNA repair processes can progressively change the overall pattern of DNA methylation and histone modifications [653-656]. A dynamic network that coordinates DNA methylation and histone modifications may form at CGI regulatory elements, which are more susceptible to oxidative stress. This network contains HOTAIR, REST, and PRC2 as core components, with HOTAIR scaffolding REST and PRC2 [361, 657, 658], REST scaffolding CoREST and MeCP2, and CoREST and MeCP2 scaffolding more epigenetic enzymes that interact with CTCF. Among them, only HOTAIR expression is not changed with age and sex [641], whereas CTCF, PRC2, and MeCP2 are decreased with age [393, 505, 632], and REST is increased with age but decreased in MCI and AD [236]. CoREST and its associated LSD1 and HDAC1/2 decrease with differentiation [659], but in neurons where REST is absent, CoREST is expressed at high levels [276, 277]. In addition, PRC1, which catalyzes the monoubiquitination of histone H2AK119 (H2AK119ub1) [393, 660, 661], was reduced and associated with AD [660, 661]. PRC1 may be another core component of the network due to its interaction with PRC2 and REST [637, 662].
Most of the components of the network exhibit age-related decrease in parallel with the accumulation of DNA damage and are closely associated with the overall upregulation of DNA methylation and dysregulation of histone modifications at the gene regulatory elements. Given that DNA methylation plays a dominant role in rebuilding the landscape of histone modifications and in REST depletion, it is tempting to propose that DNMTs, MeCP2, and SIRT1 may be potential drug candidates for AD therapy. HDAC inhibitors (HDACis), including that of SIRT1, have been tried as candidate drugs for AD therapy. Although HDACis have shown promising results in mouse AD models, their nonspecific nature could interfere with REST-mediated neuroprotective pathways that require histone deacetylation [236, 663]. This is consistent with the fact that SIRT1 plays a key role in the expression of REST and other proteins essential for the DNA damage response and cell survival. Because of the importance of SIRT1 in AD pathogenesis and its decrease with age, in the future, pharmacotherapy directed toward histone modifications could be employed to target the network of genes regulated by SIRT1 reduction [218, 277, 664].
Another approach would be to prevent the downregulation of REST, CTCF, and other related core components of the network, which play a key role in maintaining homeostasis of DNA methylation and histone modifications. To this end, we have further analyzed the related molecules interacting with REST and CTCF by bioinformatics methods. Figure 4 highlights the molecules which mediate the coupling of REST and CTCF. We found that the expression of REST, CTCF, and their interaction partners is positively correlated with H3K27ac and H3K4me3 (Fig. 5; Table S1). Thus, maintenance of the homeostasis of the two epigenetic markers, H3K27ac and H3K4me3, may play a key role in maintaining REST and CTCF levels, and therefore represent promising drug target candidates.
STRING network of REST and CTCF interaction proteins and REST-CTCF coupling through their interaction partners drawn using Cytoscape (v3.7.1). (A) REST Interaction partners; (B) CTCF interaction partners; (C) REST-CTCF coupling through their interaction partners. CHD8: Chromodomain Helicase DNA Binding Protein 8; CREBBP: CREB Binding Protein; CTDSP1: Carboxy-terminal Domain RNA Polymerase II Polypeptide A Small Phosphatase 1; DDX5: DEAD-Box Helicase 5; EP300: E1A Binding Protein P300; EED: Embyonic Ectoderm Development; FBXW7: F-Box And WD Repeat Domain Containing 7; HDAC2: Histone Deacetylase 2; HIST1H2BD: Histone Cluster 1 H2BD; HIST1H2BH: Histone Cluster 1 H2BH; HIST1H2BJ: HistoneCluster1 H2BJ; HTT: Huntingtin; KAT2A: Lysine Acetyltransferase 2A; KDM1A: Lysine Demethylase 1A: MBD2: Methyl-CpG Binding Domain Protein 2; MTA2: Metastasis Associated 1 Family member2; MYC: Myc Proto-Oncogene, BHLH Transcription factor; NANOG: Nanog Homeobox; NCOR1: Nuclear Receptor Corepressor 1; NIPBL: NIPBL Cohesion Loading factor; NPM1: Nucleophosmin 1; POU5F1: POU Class 5 Homeobox 1; RAD21: RAD21 Cohesion Complex Component; RB1: RB Transcriptional Corepressor 1; RBL2: RB Transcriptional Corepressor Like 2; RCOR1: REST Corepressor 1 (CoREST); SAP30: SIN3A Associated Protein 30; SIN3A: SIN3 Transcrition Regulatory Family Member A; SIN3B: SIN3 Transcrition Regulatory Family Member B; SKP2: S-Phase Kinase Associated Protein 2; SMC3: Structural Maintenance Of Chromosomes 3; SNAI1: Snail Family Transcriptional Repressor 1; STAG2: Stromal Antigen 2; SUZ12: SUZ12 Polycomb Repressive Complex 2 Subunit; TP53: Tumor Protein P53; YY1: YY1 Transcription Factor.
Schematic illustration of correlation between REST, CTCF and their interaction proteins and selected epigenetic markers. (A) Epigenetic markers of the genes related to neural and stem cells are recruited from http://dbtoolkit.cistrome.org, and the mode diagram was drawn using Goplot R package; (B) The mode diagram was drawn using Circlize R package.CHD8: Chromodomain Helicase DNA Binding Protein 8; CREBBP: CREB Binding Protein; CTDSP1: Carboxy-terminal Domain RNA Polymerase II Polypeptide A Small Phosphatase 1; DDX5: DEAD-Box Helicase 5; EP300: E1A Binding Protein P300; EED: Embyonic Ectoderm Development; FBXW7: F-Box And WD Repeat Domain Containing 7; HDAC2: Histone Deacetylase 2; HIST1H2BD: Histone Cluster 1 H2BD; HIST1H2BH: Histone Cluster 1 H2BH; HIST1H2BJ: HistoneCluster1 H2BJ; HTT: Huntingtin; KAT2A: Lysine Acetyltransferase 2A; KDM1A: Lysine Demethylase 1A: MBD2: Methyl-CpG Binding Domain Protein 2; MTA2: Metastasis Associated 1 Family member2; MYC: Myc Proto-Oncogene, BHLH Transcription factor; NANOG: Nanog Homeobox; NCOR1: Nuclear Receptor Corepressor 1; NIPBL: NIPBL Cohesion Loading factor; NPM1: Nucleophosmin 1; POU5F1: POU Class 5 Homeobox 1; RAD21: RAD21 Cohesion Complex Component; RB1: RB Transcriptional Corepressor 1; RBL2: RB Transcriptional Corepressor Like 2; RCOR1: REST Corepressor 1 (CoREST); SAP30: SIN3A Associated Protein 30; SIN3A: SIN3 Transcrition Regulatory Family Member A; SIN3B: SIN3 Transcrition Regulatory Family Member B; SKP2: S-Phase Kinase Associated Protein 2; SMC3: Structural Maintenance Of Chromosomes 3; SNAI1: Snail Family Transcriptional Repressor 1; STAG2: Stromal Antigen 2; SUZ12: SUZ12 Polycomb Repressive Complex 2 Subunit; TP53: Tumor Protein P53; YY1: YY1 Transcription Factor.
Supplementary Material
Supplementary table.
Acknowledgements
We thank all laboratory members for their comments and suggestions. We are grateful to Dr. Mingjie Chen from NewCore Biotech Co., Ltd. Shanghai, for helping us conduct bioinformatics analysis. We would like to extend our sincere gratitude to Professor Ruiling Zhang and Dr. Jia Tong for helpful discussions during the preparation of this manuscript and to Professor Zhongjian Zhang for his help. Due to space limitations, we regret not being able to cite all references in the field and apologize to investigators whose publications are not included in this review.
Funding
This work was supported entirely by the intramural program of the Henan Province Mental and Neurological Disease Dominant Discipline Construction Project of Xinxiang Medical University. The content of this manuscript is solely the responsibility of the authors and does not represent the official views of the funding agency.
Author Contributions
J. H. and J. Z. researched the references for this article. J. H. designed the study and wrote the first draft of the manuscript. J. Z. organized the references, contributed to the discussion of the content, and prepared tables and figures. Both authors contributed to revising the initial draft and reviewing, preparing, and approving the final draft of the manuscript for submission.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Arvanitakis Z, Shah RC, Bennett DA. Diagnosis and Management of Dementia: Review. JAMA. 2019;322:1589-99
2. Kumar A, Sidhu J, Goyal A, Tsao JW. Alzheimer Disease. StatPearls. Treasure Island (FL). 2022
3. Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S. et al. Alzheimer's disease. Lancet. 2016;388:505-17
4. Knopman DS, Petersen RC, Jack CR Jr. A brief history of “Alzheimer disease”: Multiple meanings separated by a common name. Neurology. 2019;92:1053-9
5. Kidd M. Paired helical filaments in electron microscopy of Alzheimer's disease. Nature. 1963;197:192-3
6. Roussarie JP, Yao V, Rodriguez-Rodriguez P, Oughtred R, Rust J, Plautz Z. et al. Selective Neuronal Vulnerability in Alzheimer's Disease: A Network-Based Analysis. Neuron. 2020;107:821-35 e12
7. Yu M, Engels MMA, Hillebrand A, van Straaten ECW, Gouw AA, Teunissen C. et al. Selective impairment of hippocampus and posterior hub areas in Alzheimer's disease: an MEG-based multiplex network study. Brain. 2017;140:1466-85
8. Lane CA, Hardy J, Schott JM. Alzheimer's disease. Eur J Neurol. 2018;25:59-70
9. Dorszewska J, Prendecki M, Oczkowska A, Dezor M, Kozubski W. Molecular Basis of Familial and Sporadic Alzheimer's Disease. Curr Alzheimer Res. 2016;13:952-63
10. Tellechea P, Pujol N, Esteve-Belloch P, Echeveste B, Garcia-Eulate MR, Arbizu J. et al. Early- and late-onset Alzheimer disease: Are they the same entity? Neurologia (Engl Ed). 2018;33:244-53
11. Bird TD. Alzheimer Disease Overview. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, et al. GeneReviews((R)). Seattle (WA). 1993
12. Sepulveda-Falla D, Chavez-Gutierrez L, Portelius E, Velez JI, Dujardin S, Barrera-Ocampo A. et al. A multifactorial model of pathology for age of onset heterogeneity in familial Alzheimer's disease. Acta Neuropathol. 2021;141:217-33
13. Blennow K, de Leon MJ, Zetterberg H. Alzheimer's disease. Lancet. 2006;368:387-403
14. Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL. et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15:565-81
15. Wagner J, Neher JJ. Killing ageing neurons, one cell at a time. Nat Neurosci. 2021;24:759-60
16. Wang ZT, Zhang C, Wang YJ, Dong Q, Tan L, Yu JT. Selective neuronal vulnerability in Alzheimer's disease. Ageing Res Rev. 2020;62:101114
17. Mrdjen D, Fox EJ, Bukhari SA, Montine KS, Bendall SC, Montine TJ. The basis of cellular and regional vulnerability in Alzheimer's disease. Acta Neuropathol. 2019;138:729-49
18. Dyer M, Phipps AJ, Mitew S, Taberlay PC, Woodhouse A. Age, but Not Amyloidosis, Induced Changes in Global Levels of Histone Modifications in Susceptible and Disease-Resistant Neurons in Alzheimer's Disease Model Mice. Front Aging Neurosci. 2019;11:68
19. Aunan JR, Watson MM, Hagland HR, Soreide K. Molecular and biological hallmarks of ageing. Br J Surg. 2016;103:e29-46
20. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194-217
21. Hou Y, Song H, Croteau DL, Akbari M, Bohr VA. Genome instability in Alzheimer disease. Mech Ageing Dev. 2017;161:83-94
22. Ainslie A, Huiting W, Barazzuol L, Bergink S. Genome instability and loss of protein homeostasis: converging paths to neurodegeneration? Open Biol. 2021;11:200296
23. Yousefzadeh M, Henpita C, Vyas R, Soto-Palma C, Robbins P, Niedernhofer L. DNA damage-how and why we age? Elife. 2021 10
24. Petr MA, Tulika T, Carmona-Marin LM, Scheibye-Knudsen M. Protecting the Aging Genome. Trends Cell Biol. 2020;30:117-32
25. Schumacher B, Pothof J, Vijg J, Hoeijmakers JHJ. The central role of DNA damage in the ageing process. Nature. 2021;592:695-703
26. Coppede F, Migliore L. DNA damage and repair in Alzheimer's disease. Curr Alzheimer Res. 2009;6:36-47
27. Wang J, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J Neurochem. 2006;96:825-32
28. Gabbita SP, Lovell MA, Markesbery WR. Increased nuclear DNA oxidation in the brain in Alzheimer's disease. J Neurochem. 1998;71:2034-40
29. Thadathil N, Delotterie DF, Xiao J, Hori R, McDonald MP, Khan MM. DNA Double-Strand Break Accumulation in Alzheimer's Disease: Evidence from Experimental Models and Postmortem Human Brains. Mol Neurobiol. 2021;58:118-31
30. Shanbhag NM, Evans MD, Mao W, Nana AL, Seeley WW, Adame A. et al. Early neuronal accumulation of DNA double strand breaks in Alzheimer's disease. Acta Neuropathol Commun. 2019;7:77
31. Lin X, Kapoor A, Gu Y, Chow MJ, Peng J, Zhao K. et al. Contributions of DNA Damage to Alzheimer's Disease. Int J Mol Sci. 2020 21
32. Zhu LS, Wang DQ, Cui K, Liu D, Zhu LQ. Emerging Perspectives on DNA Double-strand Breaks in Neurodegenerative Diseases. Curr Neuropharmacol. 2019;17:1146-57
33. Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J. et al. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883-91
34. Madabhushi R, Gao F, Pfenning AR, Pan L, Yamakawa S, Seo J. et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell. 2015;161:1592-605
35. Suberbielle E, Sanchez PE, Kravitz AV, Wang X, Ho K, Eilertson K. et al. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-beta. Nat Neurosci. 2013;16:613-21
36. Lodato MA, Woodworth MB, Lee S, Evrony GD, Mehta BK, Karger A. et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science. 2015;350:94-8
37. Volkova NV, Meier B, Gonzalez-Huici V, Bertolini S, Gonzalez S, Vohringer H. et al. Mutational signatures are jointly shaped by DNA damage and repair. Nat Commun. 2020;11:2169
38. Poetsch AR, Boulton SJ, Luscombe NM. Genomic landscape of oxidative DNA damage and repair reveals regioselective protection from mutagenesis. Genome Biol. 2018;19:215
39. Yang JL, Chen WY, Mukda S, Yang YR, Sun SF, Chen SD. Oxidative DNA damage is concurrently repaired by base excision repair (BER) and apyrimidinic endonuclease 1 (APE1)-initiated nonhomologous end joining (NHEJ) in cortical neurons. Neuropathol Appl Neurobiol. 2020;46:375-90
40. Yang JL, Chen WY, Chen YP, Kuo CY, Chen SD. Activation of GLP-1 Receptor Enhances Neuronal Base Excision Repair via PI3K-AKT-Induced Expression of Apurinic/Apyrimidinic Endonuclease 1. Theranostics. 2016;6:2015-27
41. Yang JL, Tadokoro T, Keijzers G, Mattson MP, Bohr VA. Neurons efficiently repair glutamate-induced oxidative DNA damage by a process involving CREB-mediated up-regulation of apurinic endonuclease 1. J Biol Chem. 2010;285:28191-9
42. Saxena S, Caroni P. Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron. 2011;71:35-48
43. Chen JH, Hales CN, Ozanne SE. DNA damage, cellular senescence and organismal ageing: causal or correlative? Nucleic Acids Res. 2007;35:7417-28
44. Maynard S, Fang EF, Scheibye-Knudsen M, Croteau DL, Bohr VA. DNA Damage, DNA Repair, Aging, and Neurodegeneration. Cold Spring Harb Perspect Med. 2015 5
45. Jeppesen DK, Bohr VA, Stevnsner T. DNA repair deficiency in neurodegeneration. Prog Neurobiol. 2011;94:166-200
46. Brasnjevic I, Hof PR, Steinbusch HW, Schmitz C. Accumulation of nuclear DNA damage or neuron loss: molecular basis for a new approach to understanding selective neuronal vulnerability in neurodegenerative diseases. DNA Repair (Amst). 2008;7:1087-97
47. Imam SZ, Karahalil B, Hogue BA, Souza-Pinto NC, Bohr VA. Mitochondrial and nuclear DNA-repair capacity of various brain regions in mouse is altered in an age-dependent manner. Neurobiol Aging. 2006;27:1129-36
48. Tonnies E, Trushina E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer's Disease. J Alzheimers Dis. 2017;57:1105-21
49. Vadas D, Kalichman L, Hadanny A, Efrati S. Hyperbaric Oxygen Environment Can Enhance Brain Activity and Multitasking Performance. Front Integr Neurosci. 2017;11:25
50. Shulman RG, Rothman DL, Behar KL, Hyder F. Energetic basis of brain activity: implications for neuroimaging. Trends Neurosci. 2004;27:489-95
51. Cunnane SC, Trushina E, Morland C, Prigione A, Casadesus G, Andrews ZB. et al. Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat Rev Drug Discov. 2020;19:609-33
52. Marlatt M, Lee HG, Perry G, Smith MA, Zhu X. Sources and mechanisms of cytoplasmic oxidative damage in Alzheimer's disease. Acta Neurobiol Exp (Wars). 2004;64:81-7
53. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1-13
54. Neyens E, Baeyens J. A review of classic Fenton's peroxidation as an advanced oxidation technique. J Hazard Mater. 2003;98:33-50
55. Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. 2017;58:235-63
56. Fishel ML, Vasko MR, Kelley MR. DNA repair in neurons: so if they don't divide what's to repair? Mutat Res. 2007;614:24-36
57. Markesbery WR, Lovell MA. DNA oxidation in Alzheimer's disease. Antioxid Redox Signal. 2006;8:2039-45
58. Whitaker AM, Schaich MA, Smith MR, Flynn TS, Freudenthal BD. Base excision repair of oxidative DNA damage: from mechanism to disease. Front Biosci (Landmark Ed). 2017;22:1493-522
59. Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634-58
60. Hou Y, Lautrup S, Cordonnier S, Wang Y, Croteau DL, Zavala E. et al. NAD(+) supplementation normalizes key Alzheimer's features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc Natl Acad Sci U S A. 2018;115:E1876-E85
61. Hamilton ML, Van Remmen H, Drake JA, Yang H, Guo ZM, Kewitt K. et al. Does oxidative damage to DNA increase with age? Proc Natl Acad Sci U S A. 2001;98:10469-74
62. Lovell MA, Markesbery WR. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer's disease. Nucleic Acids Res. 2007;35:7497-504
63. Cardozo-Pelaez F, Brooks PJ, Stedeford T, Song S, Sanchez-Ramos J. DNA damage, repair, and antioxidant systems in brain regions: a correlative study. Free Radic Biol Med. 2000;28:779-85
64. Rutten BP, Schmitz C, Gerlach OH, Oyen HM, de Mesquita EB, Steinbusch HW. et al. The aging brain: accumulation of DNA damage or neuron loss? Neurobiol Aging. 2007;28:91-8
65. Arendt T, Bruckner MK, Losche A. Regional mosaic genomic heterogeneity in the elderly and in Alzheimer's disease as a correlate of neuronal vulnerability. Acta Neuropathol. 2015;130:501-10
66. Unnikrishnan A, Raffoul JJ, Patel HV, Prychitko TM, Anyangwe N, Meira LB. et al. Oxidative stress alters base excision repair pathway and increases apoptotic response in apurinic/apyrimidinic endonuclease 1/redox factor-1 haploinsufficient mice. Free Radic Biol Med. 2009;46:1488-99
67. Fielder E, von Zglinicki T, Jurk D. The DNA Damage Response in Neurons: Die by Apoptosis or Survive in a Senescence-Like State? J Alzheimers Dis. 2017;60:S107-S31
68. Grimm A, Eckert A. Brain aging and neurodegeneration: from a mitochondrial point of view. J Neurochem. 2017;143:418-31
69. Ham S, Lee SV. Advances in transcriptome analysis of human brain aging. Exp Mol Med. 2020;52:1787-97
70. Lindqvist D, Mueller S, Mellon SH, Su Y, Epel ES, Reus VI. et al. Peripheral antioxidant markers are associated with total hippocampal and CA3/dentate gyrus volume in MDD and healthy controls-preliminary findings. Psychiatry Res. 2014;224:168-74
71. Tauffenberger A, Magistretti PJ. Reactive Oxygen Species: Beyond Their Reactive Behavior. Neurochem Res. 2021;46:77-87
72. Chen R, Lai UH, Zhu L, Singh A, Ahmed M, Forsyth NR. Reactive Oxygen Species Formation in the Brain at Different Oxygen Levels: The Role of Hypoxia Inducible Factors. Front Cell Dev Biol. 2018;6:132
73. Shaw K, Bell L, Boyd K, Grijseels DM, Clarke D, Bonnar O. et al. Neurovascular coupling and oxygenation are decreased in hippocampus compared to neocortex because of microvascular differences. Nat Commun. 2021;12:3190
74. Tarantini S, Tran CHT, Gordon GR, Ungvari Z, Csiszar A. Impaired neurovascular coupling in aging and Alzheimer's disease: Contribution of astrocyte dysfunction and endothelial impairment to cognitive decline. Exp Gerontol. 2017;94:52-8
75. Ledo A, Lourenco CF, Cadenas E, Barbosa RM, Laranjinha J. The bioactivity of neuronal-derived nitric oxide in aging and neurodegeneration: Switching signaling to degeneration. Free Radic Biol Med. 2021;162:500-13
76. Han K, Min J, Lee M, Kang BM, Park T, Hahn J. et al. Neurovascular Coupling under Chronic Stress Is Modified by Altered GABAergic Interneuron Activity. J Neurosci. 2019;39:10081-95
77. Burtscher J, Mallet RT, Burtscher M, Millet GP. Hypoxia and brain aging: Neurodegeneration or neuroprotection? Ageing Res Rev. 2021;68:101343
78. Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C. et al. A senescent cell bystander effect: senescence-induced senescence. Aging Cell. 2012;11:345-9
79. Magrassi L, Leto K, Rossi F. Lifespan of neurons is uncoupled from organismal lifespan. Proc Natl Acad Sci U S A. 2013;110:4374-9
80. Abolhassani N, Leon J, Sheng Z, Oka S, Hamasaki H, Iwaki T. et al. Molecular pathophysiology of impaired glucose metabolism, mitochondrial dysfunction, and oxidative DNA damage in Alzheimer's disease brain. Mech Ageing Dev. 2017;161:95-104
81. Sheng Z, Oka S, Tsuchimoto D, Abolhassani N, Nomaru H, Sakumi K. et al. 8-Oxoguanine causes neurodegeneration during MUTYH-mediated DNA base excision repair. J Clin Invest. 2012;122:4344-61
82. Smith EM, Pendlebury DF, Nandakumar J. Structural biology of telomeres and telomerase. Cell Mol Life Sci. 2020;77:61-79
83. de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100-10
84. Herrmann M, Pusceddu I, Marz W, Herrmann W. Telomere biology and age-related diseases. Clin Chem Lab Med. 2018;56:1210-22
85. de Lange T. Shelterin-Mediated Telomere Protection. Annu Rev Genet. 2018;52:223-47
86. Sfeir A, de Lange T. Removal of shelterin reveals the telomere end-protection problem. Science. 2012;336:593-7
87. Denchi EL, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007;448:1068-71
88. Zvereva MI, Shcherbakova DM, Dontsova OA. Telomerase: structure, functions, and activity regulation. Biochemistry (Mosc). 2010;75:1563-83
89. Saretzki G, Wan T. Telomerase in Brain: The New Kid on the Block and Its Role in Neurodegenerative Diseases. Biomedicines. 2021 9
90. Damjanovic AK, Yang Y, Glaser R, Kiecolt-Glaser JK, Nguyen H, Laskowski B. et al. Accelerated telomere erosion is associated with a declining immune function of caregivers of Alzheimer's disease patients. J Immunol. 2007;179:4249-54
91. Rossiello F, Herbig U, Longhese MP, Fumagalli M, d'Adda di Fagagna F. Irreparable telomeric DNA damage and persistent DDR signalling as a shared causative mechanism of cellular senescence and ageing. Curr Opin Genet Dev. 2014;26:89-95
92. Victorelli S, Passos JF. Telomeres and Cell Senescence - Size Matters Not. EBioMedicine. 2017;21:14-20
93. Zhang H, Li J, Ren J, Sun S, Ma S, Zhang W. et al. Single-nucleus transcriptomic landscape of primate hippocampal aging. Protein Cell. 2021;12:695-716
94. Anitha A, Thanseem I, Vasu MM, Viswambharan V, Poovathinal SA. Telomeres in neurological disorders. Adv Clin Chem. 2019;90:81-132
95. Palmos AB, Duarte RRR, Smeeth DM, Hedges EC, Nixon DF, Thuret S. et al. Telomere length and human hippocampal neurogenesis. Neuropsychopharmacology. 2020;45:2239-47
96. Daniali L, Benetos A, Susser E, Kark JD, Labat C, Kimura M. et al. Telomeres shorten at equivalent rates in somatic tissues of adults. Nat Commun. 2013;4:1597
97. Yu G, Lu L, Ma Z, Wu S. Genetically Predicted Telomere Length and Its Relationship with Alzheimer's Disease. Front Genet. 2021;12:595864
98. Fani L, Hilal S, Sedaghat S, Broer L, Licher S, Arp PP. et al. Telomere Length and the Risk of Alzheimer's Disease: The Rotterdam Study. J Alzheimers Dis. 2020;73:707-14
99. Zhan Y, Hagg S. Telomere Length Shortening in Alzheimer's Disease: Procedures for a Causal Investigation Using Single Nucleotide Polymorphisms in a Mendelian Randomization Study. Methods Mol Biol. 2018;1750:293-306
100. Zekry D, Herrmann FR, Irminger-Finger I, Ortolan L, Genet C, Vitale AM. et al. Telomere length is not predictive of dementia or MCI conversion in the oldest old. Neurobiol Aging. 2010;31:719-20
101. Rolyan H, Scheffold A, Heinrich A, Begus-Nahrmann Y, Langkopf BH, Holter SM. et al. Telomere shortening reduces Alzheimer's disease amyloid pathology in mice. Brain. 2011;134:2044-56
102. Levstek T, Kozjek E, Dolzan V, Trebusak Podkrajsek K. Telomere Attrition in Neurodegenerative Disorders. Front Cell Neurosci. 2020;14:219
103. Misri S, Pandita S, Kumar R, Pandita TK. Telomeres, histone code, and DNA damage response. Cytogenet Genome Res. 2008;122:297-307
104. Hande MP. DNA repair factors and telomere-chromosome integrity in mammalian cells. Cytogenet Genome Res. 2004;104:116-22
105. Baruch-Eliyahu N, Rud V, Braiman A, Priel E. Telomerase increasing compound protects hippocampal neurons from amyloid beta toxicity by enhancing the expression of neurotrophins and plasticity related genes. Sci Rep. 2019;9:18118
106. Liu MY, Nemes A, Zhou QG. The Emerging Roles for Telomerase in the Central Nervous System. Front Mol Neurosci. 2018;11:160
107. Zhang P, Dilley C, Mattson MP. DNA damage responses in neural cells: Focus on the telomere. Neuroscience. 2007;145:1439-48
108. Liu N, Xu J, Liu H, Zhang S, Li M, Zhou Y. et al. Hippocampal transcriptome-wide association study and neurobiological pathway analysis for Alzheimer's disease. PLoS Genet. 2021;17:e1009363
109. von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem Sci. 2002;27:339-44
110. Oikawa S, Kawanishi S. Site-specific DNA damage at GGG sequence by oxidative stress may accelerate telomere shortening. FEBS Lett. 1999;453:365-8
111. Kruk PA, Rampino NJ, Bohr VA. DNA damage and repair in telomeres: relation to aging. P roc Natl Acad Sci U S A. 1995;92:258-62
112. Fumagalli M, Rossiello F, Clerici M, Barozzi S, Cittaro D, Kaplunov JM. et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol. 2012;14:355-65
113. Tanaka H, Mendonca MS, Bradshaw PS, Hoelz DJ, Malkas LH, Meyn MS. et al. DNA damage-induced phosphorylation of the human telomere-associated protein TRF2. Proc Natl Acad Sci U S A. 2005;102:15539-44
114. d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T. et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194-8
115. Tobore TO. On the central role of mitochondria dysfunction and oxidative stress in Alzheimer's disease. Neurol Sci. 2019;40:1527-40
116. Huang TT, Leu D, Zou Y. Oxidative stress and redox regulation on hippocampal-dependent cognitive functions. Arch Biochem Biophys. 2015;576:2-7
117. Spilsbury A, Miwa S, Attems J, Saretzki G. The role of telomerase protein TERT in Alzheimer's disease and in tau-related pathology in vitro. J Neurosci. 2015;35:1659-74
118. Xiao Z, Zhang A, Lin J, Zheng Z, Shi X, Di W. et al. Telomerase: a target for therapeutic effects of curcumin and a curcumin derivative in Abeta1-42 insult in vitro. PLoS One. 2014;9:e101251
119. Miwa S, Czapiewski R, Wan T, Bell A, Hill KN, von Zglinicki T. et al. Decreased mTOR signalling reduces mitochondrial ROS in brain via accumulation of the telomerase protein TERT within mitochondria. Aging (Albany NY). 2016;8:2551-67
120. Wan T, Weir EJ, Johnson M, Korolchuk VI, Saretzki GC. Increased telomerase improves motor function and alpha-synuclein pathology in a transgenic mouse model of Parkinson's disease associated with enhanced autophagy. Prog Neurobiol. 2021;199:101953
121. Peixoto P, Cartron PF, Serandour AA, Hervouet E. From 1957 to Nowadays: A Brief History of Epigenetics. Int J Mol Sci. 2020 21
122. Saul D, Kosinsky RL. Epigenetics of Aging and Aging-Associated Diseases. Int J Mol Sci. 2021 22
123. Poon CH, Tse LSR, Lim LW. DNA methylation in the pathology of Alzheimer's disease: from gene to cognition. Ann N Y Acad Sci. 2020;1475:15-33
124. Kim JJ, Lee SY, Miller KM. Preserving genome integrity and function: the DNA damage response and histone modifications. Crit Rev Biochem Mol Biol. 2019;54:208-41
125. Tecalco-Cruz AC, Ramirez-Jarquin JO, Alvarez-Sanchez ME, Zepeda-Cervantes J. Epigenetic basis of Alzheimer disease. World J Biol Chem. 2020;11:62-75
126. Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. 2018;19:81-92
127. Jang HS, Shin WJ, Lee JE, Do JT. CpG and Non-CpG Methylation in Epigenetic Gene Regulation and Brain Function. Genes (Basel). 2017 8
128. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y. et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930-5
129. Matuleviciute R, Cunha PP, Johnson RS, Foskolou IP. Oxygen regulation of TET enzymes. FEBS J. 2021;288:7143-61
130. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484-92
131. Ehrlich M, Gama-Sosa MA, Huang LH, Midgett RM, Kuo KC, McCune RA. et al. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982;10:2709-21
132. Guo JU, Su Y, Shin JH, Shin J, Li H, Xie B. et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci. 2014;17:215-22
133. Angeloni A, Bogdanovic O. Sequence determinants, function, and evolution of CpG islands. Biochem Soc Trans. 2021;49:1109-19
134. de Mendoza A, Poppe D, Buckberry S, Pflueger J, Albertin CB, Daish T. et al. The emergence of the brain non-CpG methylation system in vertebrates. Nat Ecol Evol. 2021;5:369-78
135. Riggs AD, Pfeifer GP. X-chromosome inactivation and cell memory. Trends Genet. 1992;8:169-74
136. Razin A, Cedar H. DNA methylation and genomic imprinting. Cell. 1994;77:473-6
137. Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261-82
138. Mohn F, Schubeler D. Genetics and epigenetics: stability and plasticity during cellular differentiation. Trends Genet. 2009;25:129-36
139. Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010-22
140. Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A. 2006;103:1412-7
141. Zhu J, He F, Hu S, Yu J. On the nature of human housekeeping genes. Trends Genet. 2008;24:481-4
142. Han H, Cortez CC, Yang X, Nichols PW, Jones PA, Liang G. DNA methylation directly silences genes with non-CpG island promoters and establishes a nucleosome occupied promoter. Hum Mol Genet. 2011;20:4299-310
143. Fuso A, Lucarelli M. CpG and Non-CpG Methylation in the Diet-Epigenetics-Neurodegeneration Connection. Curr Nutr Rep. 2019;8:74-82
144. Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204-20
145. Lee JH, Saito Y, Park SJ, Nakai K. Existence and possible roles of independent non-CpG methylation in the mammalian brain. DNA Res. 2020 27
146. Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet. 2018;19:371-84
147. Ziller MJ, Gu H, Muller F, Donaghey J, Tsai LT, Kohlbacher O. et al. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500:477-81
148. Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Scholer A. et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490-5
149. Zhao X, Ji J, Wang S, Wang R, Yu Q, Li D. The regulatory pattern of target gene expression by aberrant enhancer methylation in glioblastoma. BMC Bioinformatics. 2021;22:420
150. Xiao FH, Wang HT, Kong QP. Dynamic DNA Methylation During Aging: A “Prophet” of Age-Related Outcomes. Front Genet. 2019;10:107
151. Greenberg MVC, Bourc'his D. The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol. 2019;20:590-607
152. Hahn MA, Wu X, Li AX, Hahn T, Pfeifer GP. Relationship between gene body DNA methylation and intragenic H3K9me3 and H3K36me3 chromatin marks. PLoS One. 2011;6:e18844
153. Wei X, Zhang L, Zeng Y. DNA methylation in Alzheimer's disease: In brain and peripheral blood. Mech Ageing Dev. 2020;191:111319
154. West RL, Lee JM, Maroun LE. Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer's disease patient. J Mol Neurosci. 1995;6:141-6
155. Fuso A, Seminara L, Cavallaro RA, D'Anselmi F, Scarpa S. S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Mol Cell Neurosci. 2005;28:195-204
156. Zawia NH, Lahiri DK, Cardozo-Pelaez F. Epigenetics, oxidative stress, and Alzheimer disease. Free Radic Biol Med. 2009;46:1241-9
157. Shao Y, Shaw M, Todd K, Khrestian M, D'Aleo G, Barnard PJ. et al. DNA methylation of TOMM40-APOE-APOC2 in Alzheimer's disease. J Hum Genet. 2018;63:459-71
158. Karlsson IK, Ploner A, Wang Y, Gatz M, Pedersen NL, Hagg S. Apolipoprotein E DNA methylation and late-life disease. Int J Epidemiol. 2018;47:899-907
159. Ozaki Y, Yoshino Y, Yamazaki K, Sao T, Mori Y, Ochi S. et al. DNA methylation changes at TREM2 intron 1 and TREM2 mRNA expression in patients with Alzheimer's disease. J Psychiatr Res. 2017;92:74-80
160. Silva PN, Gigek CO, Leal MF, Bertolucci PH, de Labio RW, Payao SL. et al. Promoter methylation analysis of SIRT3, SMARCA5, HTERT and CDH1 genes in aging and Alzheimer's disease. J Alzheimers Dis. 2008;13:173-6
161. Mendioroz M, Celarain N, Altuna M, Sanchez-Ruiz de Gordoa J, Zelaya MV, Roldan M. et al. CRTC1 gene is differentially methylated in the human hippocampus in Alzheimer's disease. Alzheimers Res Ther. 2016;8:15
162. Xie B, Xu Y, Liu Z, Liu W, Jiang L, Zhang R. et al. Elevation of Peripheral BDNF Promoter Methylation Predicts Conversion from Amnestic Mild Cognitive Impairment to Alzheimer's Disease: A 5-Year Longitudinal Study. J Alzheimers Dis. 2017;56:391-401
163. Sanchez-Mut JV, Aso E, Panayotis N, Lott I, Dierssen M, Rabano A. et al. DNA methylation map of mouse and human brain identifies target genes in Alzheimer's disease. Brain. 2013;136:3018-27
164. De Jager PL, Srivastava G, Lunnon K, Burgess J, Schalkwyk LC, Yu L. et al. Alzheimer's disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci. 2014;17:1156-63
165. Nicolia V, Cavallaro RA, Lopez-Gonzalez I, Maccarrone M, Scarpa S, Ferrer I. et al. DNA Methylation Profiles of Selected Pro-Inflammatory Cytokines in Alzheimer Disease. J Neuropathol Exp Neurol. 2017;76:27-31
166. Gasparoni G, Bultmann S, Lutsik P, Kraus TFJ, Sordon S, Vlcek J. et al. DNA methylation analysis on purified neurons and glia dissects age and Alzheimer's disease-specific changes in the human cortex. Epigenetics Chromatin. 2018;11:41
167. Semick SA, Bharadwaj RA, Collado-Torres L, Tao R, Shin JH, Deep-Soboslay A. et al. Integrated DNA methylation and gene expression profiling across multiple brain regions implicate novel genes in Alzheimer's disease. Acta Neuropathol. 2019;137:557-69
168. Altuna M, Urdanoz-Casado A, Sanchez-Ruiz de Gordoa J, Zelaya MV, Labarga A, Lepesant JMJ. et al. DNA methylation signature of human hippocampus in Alzheimer's disease is linked to neurogenesis. Clin Epigenetics. 2019;11:91
169. Watson CT, Roussos P, Garg P, Ho DJ, Azam N, Katsel PL. et al. Genome-wide DNA methylation profiling in the superior temporal gyrus reveals epigenetic signatures associated with Alzheimer's disease. Genome Med. 2016;8:5
170. Hsieh J, Eisch AJ. Epigenetics, hippocampal neurogenesis, and neuropsychiatric disorders: unraveling the genome to understand the mind. Neurobiol Dis. 2010;39:73-84
171. Ma DK, Marchetto MC, Guo JU, Ming GL, Gage FH, Song H. Epigenetic choreographers of neurogenesis in the adult mammalian brain. Nat Neurosci. 2010;13:1338-44
172. Fitzsimons CP, van Bodegraven E, Schouten M, Lardenoije R, Kompotis K, Kenis G. et al. Epigenetic regulation of adult neural stem cells: implications for Alzheimer's disease. Mol Neurodegener. 2014;9:25
173. Jia L, Li F, Wei C, Zhu M, Qu Q, Qin W. et al. Prediction of Alzheimer's disease using multi-variants from a Chinese genome-wide association study. Brain. 2021;144:924-37
174. Li X, Bao X, Wang R. Neurogenesis-based epigenetic therapeutics for Alzheimer's disease (Review). Mol Med Rep. 2016;14:1043-53
175. Moreno-Jimenez EP, Flor-Garcia M, Terreros-Roncal J, Rabano A, Cafini F, Pallas-Bazarra N. et al. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer's disease. Nat Med. 2019;25:554-60
176. Antequera F, Bird A. CpG islands as genomic footprints of promoters that are associated with replication origins. Curr Biol. 1999;9:R661-7
177. Antequera F. Structure, function and evolution of CpG island promoters. Cell Mol Life Sci. 2003;60:1647-58
178. Li H, Liefke R, Jiang J, Kurland JV, Tian W, Deng P. et al. Polycomb-like proteins link the PRC2 complex to CpG islands. Nature. 2017;549:287-91
179. Guo Y, Zhao S, Wang GG. Polycomb Gene Silencing Mechanisms: PRC2 Chromatin Targeting, H3K27me3 'Readout', and Phase Separation-Based Compaction. Trends Genet. 2021;37:547-65
180. von Schimmelmann M, Feinberg PA, Sullivan JM, Ku SM, Badimon A, Duff MK. et al. Polycomb repressive complex 2 (PRC2) silences genes responsible for neurodegeneration. Nat Neurosci. 2016;19:1321-30
181. Palomer E, Carretero J, Benvegnu S, Dotti CG, Martin MG. Neuronal activity controls Bdnf expression via Polycomb de-repression and CREB/CBP/JMJD3 activation in mature neurons. Nat Commun. 2016;7:11081
182. Lesch BJ, Page DC. Poised chromatin in the mammalian germ line. Development. 2014;141:3619-26
183. Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C. et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871-4
184. Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J. et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39:232-6
185. Zocher S, Overall RW, Berdugo-Vega G, Rund N, Karasinsky A, Adusumilli VS. et al. De novo DNA methylation controls neuronal maturation during adult hippocampal neurogenesis. EMBO J. 2021;40:e107100
186. Jia D, Jurkowska RZ, Zhang X, Jeltsch A, Cheng X. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature. 2007;449:248-51
187. Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z. et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714-7
188. Hagarman JA, Motley MP, Kristjansdottir K, Soloway PD. Coordinate regulation of DNA methylation and H3K27me3 in mouse embryonic stem cells. PLoS One. 2013;8:e53880
189. Cai Y, Zhang Y, Loh YP, Tng JQ, Lim MC, Cao Z. et al. H3K27me3-rich genomic regions can function as silencers to repress gene expression via chromatin interactions. Nat Commun. 2021;12:719
190. Maclary E, Hinten M, Harris C, Sethuraman S, Gayen S, Kalantry S. PRC2 represses transcribed genes on the imprinted inactive X chromosome in mice. Genome Biol. 2017;18:82
191. O'Hagan HM, Wang W, Sen S, Destefano Shields C, Lee SS, Zhang YW. et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell. 2011;20:606-19
192. Cao W, Guo J, Wen X, Miao L, Lin F, Xu G. et al. CXXC finger protein 1 is critical for T-cell intrathymic development through regulating H3K4 trimethylation. Nat Commun. 2016;7:11687
193. Wu Q, Ni X. ROS-mediated DNA methylation pattern alterations in carcinogenesis. Curr Drug Targets. 2015;16:13-9
194. Ellison EM, Abner EL, Lovell MA. Multiregional analysis of global 5-methylcytosine and 5-hydroxymethylcytosine throughout the progression of Alzheimer's disease. J Neurochem. 2017;140:383-94
195. Gebhardt FM, Scott HA, Dodd PR. Housekeepers for accurate transcript expression analysis in Alzheimer's disease autopsy brain tissue. Alzheimers Dement. 2010;6:465-74
196. Coppede F, Tannorella P, Stoccoro A, Chico L, Siciliano G, Bonuccelli U. et al. Methylation analysis of DNA repair genes in Alzheimer's disease. Mech Ageing Dev. 2017;161:105-11
197. Cuozzo C, Porcellini A, Angrisano T, Morano A, Lee B, Di Pardo A. et al. DNA damage, homology-directed repair, and DNA methylation. PLoS Genet. 2007;3:e110
198. Christmann M, Kaina B. Epigenetic regulation of DNA repair genes and implications for tumor therapy. Mutat Res Rev Mutat Res. 2019;780:15-28
199. Dhar GA, Saha S, Mitra P, Nag Chaudhuri R. DNA methylation and regulation of gene expression: Guardian of our health. Nucleus (Calcutta). 2021;64:259-70
200. Song J, Du Z, Ravasz M, Dong B, Wang Z, Ewing RM. A Protein Interaction between beta-Catenin and Dnmt1 Regulates Wnt Signaling and DNA Methylation in Colorectal Cancer Cells. Mol Cancer Res. 2015;13:969-81
201. Peng L, Yuan Z, Ling H, Fukasawa K, Robertson K, Olashaw N. et al. SIRT1 deacetylates the DNA methyltransferase 1 (DNMT1) protein and alters its activities. Mol Cell Biol. 2011;31:4720-34
202. Du Z, Song J, Wang Y, Zhao Y, Guda K, Yang S. et al. DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Sci Signal. 2010;3:ra80
203. Qin W, Leonhardt H, Spada F. Usp7 and Uhrf1 control ubiquitination and stability of the maintenance DNA methyltransferase Dnmt1. J Cell Biochem. 2011;112:439-44
204. Rountree MR, Bachman KE, Baylin SB. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat Genet. 2000;25:269-77
205. Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet. 2000;25:338-42
206. Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23-38
207. Luo C, Hajkova P, Ecker JR. Dynamic DNA methylation: In the right place at the right time. Science. 2018;361:1336-40
208. Schultz MD, He Y, Whitaker JW, Hariharan M, Mukamel EA, Leung D. et al. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature. 2015;523:212-6
209. Karlic R, Chung HR, Lasserre J, Vlahovicek K, Vingron M. Histone modification levels are predictive for gene expression. Proc Natl Acad Sci U S A. 2010;107:2926-31
210. Natsume-Kitatani Y, Mamitsuka H. Classification of Promoters Based on the Combination of Core Promoter Elements Exhibits Different Histone Modification Patterns. PLoS One. 2016;11:e0151917
211. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381-95
212. Henikoff S, Shilatifard A. Histone modification: cause or cog? Trends Genet. 2011;27:389-96
213. Campos EI, Reinberg D. Histones: annotating chromatin. Annu Rev Genet. 2009;43:559-99
214. Zhang M, Zhao J, Lv Y, Wang W, Feng C, Zou W. et al. Histone Variants and Histone Modifications in Neurogenesis. Trends Cell Biol. 2020;30:869-80
215. Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z. et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823-37
216. Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S. et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897-903
217. Tyagi SC, Stanisic D, Singh M. Epigenetic memory: gene writer, eraser and homocysteine. Mol Cell Biochem. 2021;476:507-12
218. Basavarajappa BS, Subbanna S. Histone Methylation Regulation in Neurodegenerative Disorders. Int J Mol Sci. 2021 22
219. Konsoula Z, Barile FA. Epigenetic histone acetylation and deacetylation mechanisms in experimental models of neurodegenerative disorders. J Pharmacol Toxicol Methods. 2012;66:215-20
220. Panikker P, Xu SJ, Zhang H, Sarthi J, Beaver M, Sheth A. et al. Restoring Tip60 HAT/HDAC2 Balance in the Neurodegenerative Brain Relieves Epigenetic Transcriptional Repression and Reinstates Cognition. J Neurosci. 2018;38:4569-83
221. Zheng Y, Liu A, Wang ZJ, Cao Q, Wang W, Lin L. et al. Inhibition of EHMT1/2 rescues synaptic and cognitive functions for Alzheimer's disease. Brain. 2019;142:787-807
222. Sharma VK, Mehta V, Singh TG. Alzheimer's Disorder: Epigenetic Connection and Associated Risk Factors. Curr Neuropharmacol. 2020;18:740-53
223. Stoccoro A, Coppede F. Role of epigenetics in Alzheimer's disease pathogenesis. Neurodegener Dis Manag. 2018;8:181-93
224. Wang BY, Zhong Y, Zhao Z, Miao Y. Epigenetic suppression of hippocampal BDNF mediates the memory deficiency induced by amyloid fibrils. Pharmacol Biochem Behav. 2014;126:83-9
225. Woldemichael BT, Bohacek J, Gapp K, Mansuy IM. Epigenetics of memory and plasticity. Prog Mol Biol Transl Sci. 2014;122:305-40
226. Narayan PJ, Lill C, Faull R, Curtis MA, Dragunow M. Increased acetyl and total histone levels in post-mortem Alzheimer's disease brain. Neurobiol Dis. 2015;74:281-94
227. Li Y, Stockton ME, Eisinger BE, Zhao Y, Miller JL, Bhuiyan I. et al. Reducing histone acetylation rescues cognitive deficits in a mouse model of Fragile X syndrome. Nat Commun. 2018;9:2494
228. Graff J, Rei D, Guan JS, Wang WY, Seo J, Hennig KM. et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature. 2012;483:222-6
229. Datta M, Staszewski O, Raschi E, Frosch M, Hagemeyer N, Tay TL. et al. Histone Deacetylases 1 and 2 Regulate Microglia Function during Development, Homeostasis, and Neurodegeneration in a Context-Dependent Manner. Immunity. 2018;48:514-29 e6
230. Zhu X, Wang S, Yu L, Jin J, Ye X, Liu Y. et al. HDAC3 negatively regulates spatial memory in a mouse model of Alzheimer's disease. Aging Cell. 2017;16:1073-82
231. Janczura KJ, Volmar CH, Sartor GC, Rao SJ, Ricciardi NR, Lambert G. et al. Inhibition of HDAC3 reverses Alzheimer's disease-related pathologies in vitro and in the 3xTg-AD mouse model. Proc Natl Acad Sci U S A. 2018;115:E11148-E57
232. Govindarajan N, Rao P, Burkhardt S, Sananbenesi F, Schluter OM, Bradke F. et al. Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer's disease. EMBO Mol Med. 2013;5:52-63
233. Zhang L, Sheng S, Qin C. The role of HDAC6 in Alzheimer's disease. J Alzheimers Dis. 2013;33:283-95
234. Schueller E, Paiva I, Blanc F, Wang XL, Cassel JC, Boutillier AL. et al. Dysregulation of histone acetylation pathways in hippocampus and frontal cortex of Alzheimer's disease patients. Eur Neuropsychopharmacol. 2020;33:101-16
235. Jung HG, Hwang YS, Park YH, Cho HY, Rengaraj D, Han JY. Role of Epigenetic Regulation by the REST/CoREST/HDAC Corepressor Complex of Moderate NANOG Expression in Chicken Primordial Germ Cells. Stem Cells Dev. 2018;27:1215-25
236. Lu T, Aron L, Zullo J, Pan Y, Kim H, Chen Y. et al. REST and stress resistance in ageing and Alzheimer's disease. Nature. 2014;507:448-54
237. Nativio R, Lan Y, Donahue G, Sidoli S, Berson A, Srinivasan AR. et al. An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer's disease. Nat Genet. 2020;52:1024-35
238. Li Z, Rasmussen LJ. TIP60 in aging and neurodegeneration. Ageing Res Rev. 2020;64:101195
239. Stilling RM, Ronicke R, Benito E, Urbanke H, Capece V, Burkhardt S. et al. K-Lysine acetyltransferase 2a regulates a hippocampal gene expression network linked to memory formation. EMBO J. 2014;33:1912-27
240. Urban I, Kerimoglu C, Sakib MS, Wang H, Benito E, Thaller C. et al. TIP60/KAT5 is required for neuronal viability in hippocampal CA1. Sci Rep. 2019;9:16173
241. Jin Q, Yu LR, Wang L, Zhang Z, Kasper LH, Lee JE. et al. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011;30:249-62
242. Mostafavi S, Gaiteri C, Sullivan SE, White CC, Tasaki S, Xu J. et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer's disease. Nat Neurosci. 2018;21:811-9
243. Fernandez-Hernando C, Suarez Y. ANGPTL4: a multifunctional protein involved in metabolism and vascular homeostasis. Curr Opin Hematol. 2020;27:206-13
244. Hata S, Nomura T, Iwasaki K, Sato R, Yamasaki M, Sato F. et al. Hypoxia-induced angiopoietin-like protein 4 as a clinical biomarker and treatment target for human prostate cancer. Oncol Rep. 2017;38:120-8
245. Vienberg SG, Kleinridders A, Suzuki R, Kahn CR. Differential effects of angiopoietin-like 4 in brain and muscle on regulation of lipoprotein lipase activity. Mol Metab. 2015;4:144-50
246. Li J, Yan D, Ma N, Chen J, Zhao X, Zhang Y. et al. Transient Forebrain Ischemia Induces Differential Bdnf Transcript Expression and Histone Acetylation Patterns in the Rat Hippocampus. J Mol Neurosci. 2020;70:568-75
247. Zhang K, Schrag M, Crofton A, Trivedi R, Vinters H, Kirsch W. Targeted proteomics for quantification of histone acetylation in Alzheimer's disease. Proteomics. 2012;12:1261-8
248. Nativio R, Donahue G, Berson A, Lan Y, Amlie-Wolf A, Tuzer F. et al. Dysregulation of the epigenetic landscape of normal aging in Alzheimer's disease. Nat Neurosci. 2018;21:497-505
249. Hernandez-Ortega K, Garcia-Esparcia P, Gil L, Lucas JJ, Ferrer I. Altered Machinery of Protein Synthesis in Alzheimer's: From the Nucleolus to the Ribosome. Brain Pathol. 2016;26:593-605
250. Plagg B, Ehrlich D, Kniewallner KM, Marksteiner J, Humpel C. Increased Acetylation of Histone H4 at Lysine 12 (H4K12) in Monocytes of Transgenic Alzheimer's Mice and in Human Patients. Curr Alzheimer Res. 2015;12:752-60
251. Karmodiya K, Krebs AR, Oulad-Abdelghani M, Kimura H, Tora L. H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genomics. 2012;13:424
252. Mas G, Blanco E, Ballare C, Sanso M, Spill YG, Hu D. et al. Promoter bivalency favors an open chromatin architecture in embryonic stem cells. Nat Genet. 2018;50:1452-62
253. Tjeertes JV, Miller KM, Jackson SP. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J. 2009;28:1878-89
254. Duan MR, Smerdon MJ. Histone H3 lysine 14 (H3K14) acetylation facilitates DNA repair in a positioned nucleosome by stabilizing the binding of the chromatin Remodeler RSC (Remodels Structure of Chromatin). J Biol Chem. 2014;289:8353-63
255. Zhang AL, Chen L, Ma L, Ding XJ, Tang SF, Zhang AH. et al. Role of H3K18ac-regulated nucleotide excision repair-related genes in arsenic-induced DNA damage and repair of HaCaT cells. Hum Exp Toxicol. 2020;39:1168-77
256. Klein BJ, Jang SM, Lachance C, Mi W, Lyu J, Sakuraba S. et al. Histone H3K23-specific acetylation by MORF is coupled to H3K14 acylation. Nat Commun. 2019;10:4724
257. Li X, Corsa CA, Pan PW, Wu L, Ferguson D, Yu X. et al. MOF and H4 K16 acetylation play important roles in DNA damage repair by modulating recruitment of DNA damage repair protein Mdc1. Mol Cell Biol. 2010;30:5335-47
258. Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ. et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107:21931-6
259. Wolfe JC, Mikheeva LA, Hagras H, Zabet NR. An explainable artificial intelligence approach for decoding the enhancer histone modifications code and identification of novel enhancers in Drosophila. Genome Biol. 2021;22:308
260. Berson A, Nativio R, Berger SL, Bonini NM. Epigenetic Regulation in Neurodegenerative Diseases. Trends Neurosci. 2018;41:587-98
261. Zhang S, Zhang H, Yu L. HMGA2 promotes glioma invasion and poor prognosis via a long-range chromatin interaction. Cancer Med. 2018
262. Rockowitz S, Zheng D. Significant expansion of the REST/NRSF cistrome in human versus mouse embryonic stem cells: potential implications for neural development. Nucleic Acids Res. 2015;43:5730-43
263. Zheng D, Zhao K, Mehler MF. Profiling RE1/REST-mediated histone modifications in the human genome. Genome Biol. 2009;10:R9
264. Noh KM, Hwang JY, Follenzi A, Athanasiadou R, Miyawaki T, Greally JM. et al. Repressor element-1 silencing transcription factor (REST)-dependent epigenetic remodeling is critical to ischemia-induced neuronal death. Proc Natl Acad Sci U S A. 2012;109:E962-71
265. Tahiliani M, Mei P, Fang R, Leonor T, Rutenberg M, Shimizu F. et al. The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature. 2007;447:601-5
266. Sun Y, Jiang X, Price BD. Tip60: connecting chromatin to DNA damage signaling. Cell Cycle. 2010;9:930-6
267. Zhu Z, Wang Y, Li X, Wang Y, Xu L, Wang X. et al. PHF8 is a histone H3K9me2 demethylase regulating rRNA synthesis. Cell Res. 2010;20:794-801
268. Kleine-Kohlbrecher D, Christensen J, Vandamme J, Abarrategui I, Bak M, Tommerup N. et al. A functional link between the histone demethylase PHF8 and the transcription factor ZNF711 in X-linked mental retardation. Mol Cell. 2010;38:165-78
269. Oey NE, Leung HW, Ezhilarasan R, Zhou L, Beuerman RW, VanDongen HM. et al. A Neuronal Activity-Dependent Dual Function Chromatin-Modifying Complex Regulates Arc Expression. eNeuro. 2015 2
270. Wang J, Lin X, Wang S, Wang C, Wang Q, Duan X. et al. PHF8 and REST/NRSF co-occupy gene promoters to regulate proximal gene expression. Sci Rep. 2014;4:5008
271. Lithner CU, Lacor PN, Zhao WQ, Mustafiz T, Klein WL, Sweatt JD. et al. Disruption of neocortical histone H3 homeostasis by soluble Abeta: implications for Alzheimer's disease. Neurobiol Aging. 2013;34:2081-90
272. Collins BE, Sweatt JD, Greer CB. Broad domains of histone 3 lysine 4 trimethylation are associated with transcriptional activation in CA1 neurons of the hippocampus during memory formation. Neurobiol Learn Mem. 2019;161:149-57
273. Cao Q, Wang W, Williams JB, Yang F, Wang ZJ, Yan Z. Targeting histone K4 trimethylation for treatment of cognitive and synaptic deficits in mouse models of Alzheimer's disease. Sci Adv. 2020 6
274. Smith AR, Smith RG, Macdonald R, Marzi SJ, Burrage J, Troakes C. et al. The histone modification H3K4me3 is altered at the ANK1 locus in Alzheimer's disease brain. Future Sci OA. 2021;7:FSO665
275. Mastroeni D, Delvaux E, Nolz J, Tan Y, Grover A, Oddo S. et al. Aberrant intracellular localization of H3k4me3 demonstrates an early epigenetic phenomenon in Alzheimer's disease. Neurobiol Aging. 2015;36:3121-9
276. Ballas N, Battaglioli E, Atouf F, Andres ME, Chenoweth J, Anderson ME. et al. Regulation of neuronal traits by a novel transcriptional complex. Neuron. 2001;31:353-65
277. Kalin JH, Wu M, Gomez AV, Song Y, Das J, Hayward D. et al. Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. Nat Commun. 2018;9:53
278. Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005;437:432-5
279. Liu Y, Zhou D, Qi D, Feng J, Liu Z, Hu Y. et al. Lysine-specific demethylase 1 cooperates with BRAF-histone deacetylase complex 80 to enhance HIV-1 Tat-mediated transactivation. Virus Genes. 2018;54:662-71
280. Jakovcevski M, Ruan H, Shen EY, Dincer A, Javidfar B, Ma Q. et al. Neuronal Kmt2a/Mll1 histone methyltransferase is essential for prefrontal synaptic plasticity and working memory. J Neurosci. 2015;35:5097-108
281. How-Wing Leung GWQF, Antonius M.J. VanDongen. Arc Regulates Transcription of Genes for Plasticity, Excitability and Alzheimer's Disease. bioRxiv. 2019
282. Pilotto S, Speranzini V, Tortorici M, Durand D, Fish A, Valente S. et al. Interplay among nucleosomal DNA, histone tails, and corepressor CoREST underlies LSD1-mediated H3 demethylation. Proc Natl Acad Sci U S A. 2015;112:2752-7
283. Ooi L, Wood IC. Chromatin crosstalk in development and disease: lessons from REST. Nat Rev Genet. 2007;8:544-54
284. Kerimoglu C, Agis-Balboa RC, Kranz A, Stilling R, Bahari-Javan S, Benito-Garagorri E. et al. Histone-methyltransferase MLL2 (KMT2B) is required for memory formation in mice. J Neurosci. 2013;33:3452-64
285. Kerimoglu C, Sakib MS, Jain G, Benito E, Burkhardt S, Capece V. et al. KMT2A and KMT2B Mediate Memory Function by Affecting Distinct Genomic Regions. Cell Rep. 2017;20:538-48
286. Nanya M, Sato M, Tanimoto K, Tozuka M, Mizutani S, Takagi M. Dysregulation of the DNA Damage Response and KMT2A Rearrangement in Fetal Liver Hematopoietic Cells. PLoS One. 2015;10:e0144540
287. El Chaer F, Keng M, Ballen KK. MLL-Rearranged Acute Lymphoblastic Leukemia. Curr Hematol Malig Rep. 2020;15:83-9
288. Bertani S, Sauer S, Bolotin E, Sauer F. The noncoding RNA Mistral activates Hoxa6 and Hoxa7 expression and stem cell differentiation by recruiting MLL1 to chromatin. Mol Cell. 2011;43:1040-6
289. Laurent B, Ruitu L, Murn J, Hempel K, Ferrao R, Xiang Y. et al. A specific LSD1/KDM1A isoform regulates neuronal differentiation through H3K9 demethylation. Mol Cell. 2015;57:957-70
290. Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH. et al. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128:1077-88
291. Mulligan P, Westbrook TF, Ottinger M, Pavlova N, Chang B, Macia E. et al. CDYL bridges REST and histone methyltransferases for gene repression and suppression of cellular transformation. Mol Cell. 2008;32:718-26
292. Liu F, Barsyte-Lovejoy D, Li F, Xiong Y, Korboukh V, Huang XP. et al. Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J Med Chem. 2013;56:8931-42
293. Gupta-Agarwal S, Franklin AV, Deramus T, Wheelock M, Davis RL, McMahon LL. et al. G9a/GLP histone lysine dimethyltransferase complex activity in the hippocampus and the entorhinal cortex is required for gene activation and silencing during memory consolidation. J Neurosci. 2012;32:5440-53
294. Shen E, Shulha H, Weng Z, Akbarian S. Regulation of histone H3K4 methylation in brain development and disease. Philos Trans R Soc Lond B Biol Sci. 2014 369
295. Collins BE, Greer CB, Coleman BC, Sweatt JD. Histone H3 lysine K4 methylation and its role in learning and memory. Epigenetics Chromatin. 2019;12:7
296. Ebata KT, Mesh K, Liu S, Bilenky M, Fekete A, Acker MG. et al. Vitamin C induces specific demethylation of H3K9me2 in mouse embryonic stem cells via Kdm3a/b. Epigenetics Chromatin. 2017;10:36
297. Shi G, Wu M, Fang L, Yu F, Cheng S, Li J. et al. PHD finger protein 2 (PHF2) represses ribosomal RNA gene transcription by antagonizing PHF finger protein 8 (PHF8) and recruiting methyltransferase SUV39H1. J Biol Chem. 2014;289:29691-700
298. Kim HJ, Hur SW, Park JB, Seo J, Shin JJ, Kim SY. et al. Histone demethylase PHF2 activates CREB and promotes memory consolidation. EMBO Rep. 2019;20:e45907
299. Song Y, Dagil L, Fairall L, Robertson N, Wu M, Ragan TJ. et al. Mechanism of Crosstalk between the LSD1 Demethylase and HDAC1 Deacetylase in the CoREST Complex. Cell Rep. 2020;30:2699-711 e8
300. Wang R, Reddy PH. Role of Glutamate and NMDA Receptors in Alzheimer's Disease. J Alzheimers Dis. 2017;57:1041-8
301. Xu C, Zhang M, Zu L, Zhang P, Sun L, Liu X. et al. Repressor element-1 silencing transcription factor regulates glutamate receptors and immediate early genes to affect synaptic plasticity. Aging (Albany NY). 2021;13:15569-79
302. Johnson AA, Sarthi J, Pirooznia SK, Reube W, Elefant F. Increasing Tip60 HAT levels rescues axonal transport defects and associated behavioral phenotypes in a Drosophila Alzheimer's disease model. J Neurosci. 2013;33:7535-47
303. Ayrapetov MK, Gursoy-Yuzugullu O, Xu C, Xu Y, Price BD. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc Natl Acad Sci U S A. 2014;111:9169-74
304. Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W. et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019-31
305. Ruthenburg AJ, Li H, Milne TA, Dewell S, McGinty RK, Yuen M. et al. Recognition of a mononucleosomal histone modification pattern by BPTF via multivalent interactions. Cell. 2011;145:692-706
306. Katoh H, Qin ZS, Liu R, Wang L, Li W, Li X. et al. FOXP3 orchestrates H4K16 acetylation and H3K4 trimethylation for activation of multiple genes by recruiting MOF and causing displacement of PLU-1. Mol Cell. 2011;44:770-84
307. Dou Y, Milne TA, Tackett AJ, Smith ER, Fukuda A, Wysocka J. et al. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell. 2005;121:873-85
308. Wysocka J, Swigut T, Xiao H, Milne TA, Kwon SY, Landry J. et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442:86-90
309. Kim CH. FOXP3 and its role in the immune system. Adv Exp Med Biol. 2009;665:17-29
310. Jordan-Sciutto K, Dragich J, Walcott D, Bowser R. The presence of FAC1 protein in Hirano bodies. Neuropathol Appl Neurobiol. 1998;24:359-66
311. Baruch K, Rosenzweig N, Kertser A, Deczkowska A, Sharif AM, Spinrad A. et al. Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer's disease pathology. Nat Commun. 2015;6:7967
312. Zhao W, Xu Y, Wang Y, Gao D, King J, Xu Y. et al. Investigating crosstalk between H3K27 acetylation and H3K4 trimethylation in CRISPR/dCas-based epigenome editing and gene activation. Sci Rep. 2021;11:15912
313. Douillet D, Sze CC, Ryan C, Piunti A, Shah AP, Ugarenko M. et al. Uncoupling histone H3K4 trimethylation from developmental gene expression via an equilibrium of COMPASS, Polycomb and DNA methylation. Nat Genet. 2020;52:615-25
314. Li J, Xing X, Zhang X, Liang B, He Z, Gao C. et al. Enhanced H3K4me3 modifications are involved in the transactivation of DNA damage responsive genes in workers exposed to low-level benzene. Environ Pollut. 2018;234:127-35
315. Pena PV, Hom RA, Hung T, Lin H, Kuo AJ, Wong RP. et al. Histone H3K4me3 binding is required for the DNA repair and apoptotic activities of ING1 tumor suppressor. J Mol Biol. 2008;380:303-12
316. Gong F, Clouaire T, Aguirrebengoa M, Legube G, Miller KM. Histone demethylase KDM5A regulates the ZMYND8-NuRD chromatin remodeler to promote DNA repair. J Cell Biol. 2017;216:1959-74
317. Hoekstra JG, Hipp MJ, Montine TJ, Kennedy SR. Mitochondrial DNA mutations increase in early stage Alzheimer disease and are inconsistent with oxidative damage. Ann Neurol. 2016;80:301-6
318. Meyer B, Fabbrizi MR, Raj S, Zobel CL, Hallahan DE, Sharma GG. Histone H3 Lysine 9 Acetylation Obstructs ATM Activation and Promotes Ionizing Radiation Sensitivity in Normal Stem Cells. Stem Cell Reports. 2016;7:1013-22
319. Maina PK, Shao P, Jia X, Liu Q, Umesalma S, Marin M. et al. Histone demethylase PHF8 regulates hypoxia signaling through HIF1alpha and H3K4me3. Biochim Biophys Acta Gene Regul Mech. 2017;1860:1002-12
320. Olcina M, Lecane PS, Hammond EM. Targeting hypoxic cells through the DNA damage response. Clin Cancer Res. 2010;16:5624-9
321. Baker M, Petasny M, Taqatqa N, Bentata M, Kay G, Engal E. et al. KDM3A regulates alternative splicing of cell-cycle genes following DNA damage. RNA. 2021;27:1353-62
322. Pappa S, Padilla N, Iacobucci S, Vicioso M, Alvarez de la Campa E, Navarro C. et al. PHF2 histone demethylase prevents DNA damage and genome instability by controlling cell cycle progression of neural progenitors. Proc Natl Acad Sci U S A. 2019;116:19464-73
323. Yang Q, Zhu Q, Lu X, Du Y, Cao L, Shen C. et al. G9a coordinates with the RPA complex to promote DNA damage repair and cell survival. Proc Natl Acad Sci U S A. 2017;114:E6054-E63
324. Schones DE, Chen X, Trac C, Setten R, Paddison PJ. G9a/GLP-dependent H3K9me2 patterning alters chromatin structure at CpG islands in hematopoietic progenitors. Epigenetics Chromatin. 2014;7:23
325. Ding X, Zhang A, Li C, Ma L, Tang S, Wang Q. et al. The role of H3K9me2-regulated base excision repair genes in the repair of DNA damage induced by arsenic in HaCaT cells and the effects of Ginkgo biloba extract intervention. Environ Toxicol. 2021;36:850-60
326. Banerjee DR, Deckard CE 3rd, Zeng Y, Sczepanski JT. Acetylation of the histone H3 tail domain regulates base excision repair on higher-order chromatin structures. Sci Rep. 2019;9:15972
327. Schick S, Fournier D, Thakurela S, Sahu SK, Garding A, Tiwari VK. Dynamics of chromatin accessibility and epigenetic state in response to UV damage. J Cell Sci. 2015;128:4380-94
328. Wang J, Sun Y, Zhang X, Cai H, Zhang C, Qu H. et al. Oxidative stress activates NORAD expression by H3K27ac and promotes oxaliplatin resistance in gastric cancer by enhancing autophagy flux via targeting the miR-433-3p. Cell Death Dis. 2021;12:90
329. Sankar A, Lerdrup M, Manaf A, Johansen JV, Gonzalez JM, Borup R. et al. KDM4A regulates the maternal-to-zygotic transition by protecting broad H3K4me3 domains from H3K9me3 invasion in oocytes. Nat Cell Biol. 2020;22:380-8
330. Lapidot M, Case AE, Weisberg EL, Meng C, Walker SR, Garg S. et al. Essential role of the histone lysine demethylase KDM4A in the biology of malignant pleural mesothelioma (MPM). Br J Cancer. 2021;125:582-92
331. Wu R, Wang Z, Zhang H, Gan H, Zhang Z. H3K9me3 demethylase Kdm4d facilitates the formation of pre-initiative complex and regulates DNA replication. Nucleic Acids Res. 2017;45:169-80
332. Thatcher KN, LaSalle JM. Dynamic changes in Histone H3 lysine 9 acetylation localization patterns during neuronal maturation require MeCP2. Epigenetics. 2006;1:24-31
333. Enikanolaiye A, Ruston J, Zeng R, Taylor C, Schrock M, Buchovecky CM. et al. Suppressor mutations in Mecp2-null mice implicate the DNA damage response in Rett syndrome pathology. Genome Res. 2020;30:540-52
334. Hombach S, Kretz M. Non-coding RNAs: Classification, Biology and Functioning. Adv Exp Med Biol. 2016;937:3-17
335. Zucchelli S, Fedele S, Vatta P, Calligaris R, Heutink P, Rizzu P. et al. Antisense Transcription in Loci Associated to Hereditary Neurodegenerative Diseases. Mol Neurobiol. 2019;56:5392-415
336. Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A. et al. Landscape of transcription in human cells. Nature. 2012;489:101-8
337. Salta E, De Strooper B. Noncoding RNAs in neurodegeneration. Nat Rev Neurosci. 2017;18:627-40
338. Mihailescu R. Gene expression regulation: lessons from noncoding RNAs. RNA. 2015;21:695-6
339. Gomes AQ, Nolasco S, Soares H. Non-coding RNAs: multi-tasking molecules in the cell. Int J Mol Sci. 2013;14:16010-39
340. Quan Z, Zheng D, Qing H. Regulatory Roles of Long Non-Coding RNAs in the Central Nervous System and Associated Neurodegenerative Diseases. Front Cell Neurosci. 2017;11:175
341. Zhao J, Yue D, Zhou Y, Jia L, Wang H, Guo M. et al. The Role of MicroRNAs in Abeta Deposition and Tau Phosphorylation in Alzheimer's Disease. Front Neurol. 2017;8:342
342. Ma N, Tie C, Yu B, Zhang W, Wan J. Identifying lncRNA-miRNA-mRNA networks to investigate Alzheimer's disease pathogenesis and therapy strategy. Aging (Albany NY). 2020;12:2897-920
343. Quinlan S, Kenny A, Medina M, Engel T, Jimenez-Mateos EM. MicroRNAs in Neurodegenerative Diseases. Int Rev Cell Mol Biol. 2017;334:309-43
344. Johnson R, Noble W, Tartaglia GG, Buckley NJ. Neurodegeneration as an RNA disorder. Prog Neurobiol. 2012;99:293-315
345. Moreno-Garcia L, Lopez-Royo T, Calvo AC, Toivonen JM, de la Torre M, Moreno-Martinez L. et al. Competing Endogenous RNA Networks as Biomarkers in Neurodegenerative Diseases. Int J Mol Sci. 2020 21
346. Idda ML, Munk R, Abdelmohsen K, Gorospe M. Noncoding RNAs in Alzheimer's disease. Wiley Interdiscip Rev RNA. 2018 9
347. Maoz R, Garfinkel BP, Soreq H. Alzheimer's Disease and ncRNAs. Adv Exp Med Biol. 2017;978:337-61
348. Salta E, De Strooper B. Non-coding RNAs with essential roles in neurodegenerative disorders. Lancet Neurol. 2012;11:189-200
349. Lauretti E, Dabrowski K, Pratico D. The neurobiology of non-coding RNAs and Alzheimer's disease pathogenesis: Pathways, mechanisms and translational opportunities. Ageing Res Rev. 2021;71:101425
350. Zullo JM, Drake D, Aron L, O'Hern P, Dhamne SC, Davidsohn N. et al. Regulation of lifespan by neural excitation and REST. Nature. 2019;574:359-64
351. Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F. et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689-93
352. Hwang JY, Kaneko N, Noh KM, Pontarelli F, Zukin RS. The gene silencing transcription factor REST represses miR-132 expression in hippocampal neurons destined to die. J Mol Biol. 2014;426:3454-66
353. Lepagnol-Bestel AM, Zvara A, Maussion G, Quignon F, Ngimbous B, Ramoz N. et al. DYRK1A interacts with the REST/NRSF-SWI/SNF chromatin remodelling complex to deregulate gene clusters involved in the neuronal phenotypic traits of Down syndrome. Hum Mol Genet. 2009;18:1405-14
354. Epping MT, Lunardi A, Nachmani D, Castillo-Martin M, Thin TH, Cordon-Cardo C. et al. TSPYL2 is an essential component of the REST/NRSF transcriptional complex for TGFbeta signaling activation. Cell Death Differ. 2015;22:1353-62
355. Tsang KH, Lai SK, Li Q, Yung WH, Liu H, Mak PH. et al. The nucleosome assembly protein TSPYL2 regulates the expression of NMDA receptor subunits GluN2A and GluN2B. Sci Rep. 2014;4:3654
356. Li TY, Sleiman MB, Li H, Gao AW, Mottis A, Bachmann AM. et al. The transcriptional coactivator CBP/p300 is an evolutionarily conserved node that promotes longevity in response to mitochondrial stress. Nat Aging. 2021;1:165-78
357. Rajagopal T, Talluri S, Akshaya RL, Dunna NR. HOTAIR LncRNA: A novel oncogenic propellant in human cancer. Clin Chim Acta. 2020;503:1-18
358. Price RL, Bhan A, Mandal SS. HOTAIR beyond repression: In protein degradation, inflammation, DNA damage response, and cell signaling. DNA Repair (Amst). 2021;105:103141
359. Cheng S, Zhang Y, Chen S, Zhou Y. LncRNA HOTAIR Participates in Microglia Activation and Inflammatory Factor Release by Regulating the Ubiquitination of MYD88 in Traumatic Brain Injury. J Mol Neurosci. 2021;71:169-77
360. Zhang Q, Huang XM, Liao JX, Dong YK, Zhu JL, He CC. et al. LncRNA HOTAIR Promotes Neuronal Damage Through Facilitating NLRP3 Mediated-Pyroptosis Activation in Parkinson's Disease via Regulation of miR-326/ELAVL1 Axis. Cell Mol Neurobiol. 2021;41:1773-86
361. Qian L, Fei Q, Zhang H, Qiu M, Zhang B, Wang Q. et al. lncRNA HOTAIR Promotes DNA Repair and Radioresistance of Breast Cancer via EZH2. DNA Cell Biol. 2020
362. Ozes AR, Miller DF, Ozes ON, Fang F, Liu Y, Matei D. et al. NF-kappaB-HOTAIR axis links DNA damage response, chemoresistance and cellular senescence in ovarian cancer. Oncogene. 2016;35:5350-61
363. Kouznetsova VL, Tchekanov A, Li X, Yan X, Tsigelny IF. Polycomb repressive 2 complex-Molecular mechanisms of function. Protein Sci. 2019;28:1387-99
364. Elhanani O, Salame TM, Sobel J, Leshkowitz D, Povodovski L, Vaknin I. et al. REST Inhibits Direct Reprogramming of Pancreatic Exocrine to Endocrine Cells by Preventing PDX1-Mediated Activation of Endocrine Genes. Cell Rep. 2020;31:107591
365. Jin C, Li J, Green CD, Yu X, Tang X, Han D. et al. Histone demethylase UTX-1 regulates C. elegans life span by targeting the insulin/IGF-1 signaling pathway. Cell Metab. 2011;14:161-72
366. Han Y, Han D, Yan Z, Boyd-Kirkup JD, Green CD, Khaitovich P. et al. Stress-associated H3K4 methylation accumulates during postnatal development and aging of rhesus macaque brain. Aging Cell. 2012;11:1055-64
367. Basenko EY, Sasaki T, Ji L, Prybol CJ, Burckhardt RM, Schmitz RJ. et al. Genome-wide redistribution of H3K27me3 is linked to genotoxic stress and defective growth. Proc Natl Acad Sci U S A. 2015;112:E6339-48
368. Meyer K, Feldman HM, Lu T, Drake D, Lim ET, Ling KH. et al. REST and Neural Gene Network Dysregulation in iPSC Models of Alzheimer's Disease. Cell Rep. 2019;26:1112-27 e9
369. Li P, Stetler RA, Leak RK, Shi Y, Li Y, Yu W. et al. Oxidative stress and DNA damage after cerebral ischemia: Potential therapeutic targets to repair the genome and improve stroke recovery. Neuropharmacology. 2018;134:208-17
370. Pluta R, Januszewski S, Czuczwar SJ. Brain Ischemia as a Prelude to Alzheimer's Disease. Front Aging Neurosci. 2021;13:636653
371. Sweeney MD, Kisler K, Montagne A, Toga AW, Zlokovic BV. The role of brain vasculature in neurodegenerative disorders. Nat Neurosci. 2018;21:1318-31
372. Cong L, Cong Y, Feng N, Liang W, Wu Y. Up-regulated microRNA-132 reduces the cognition-damaging effect of sevoflurane on Alzheimer's disease rats by inhibiting FOXA1. Genomics. 2021;113:3644-52
373. Lin L, Wang Z, Jin H, Shi H, Lu Z, Qi Z. MiR-212/132 is epigenetically downregulated by SOX4/EZH2-H3K27me3 feedback loop in ovarian cancer cells. Tumour Biol. 2016
374. Stapels M, Piper C, Yang T, Li M, Stowell C, Xiong ZG. et al. Polycomb group proteins as epigenetic mediators of neuroprotection in ischemic tolerance. Sci Signal. 2010;3:ra15
375. Tabuchi A, Yamada T, Sasagawa S, Naruse Y, Mori N, Tsuda M. REST4-mediated modulation of REST/NRSF-silencing function during BDNF gene promoter activation. Biochem Biophys Res Commun. 2002;290:415-20
376. KhorshidAhmad T, Acosta C, Cortes C, Lakowski TM, Gangadaran S, Namaka M. Transcriptional Regulation of Brain-Derived Neurotrophic Factor (BDNF) by Methyl CpG Binding Protein 2 (MeCP2): a Novel Mechanism for Re-Myelination and/or Myelin Repair Involved in the Treatment of Multiple Sclerosis (MS). Mol Neurobiol. 2016;53:1092-107
377. Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC. et al. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885-9
378. Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y. et al. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890-3
379. Im HI, Hollander JA, Bali P, Kenny PJ. MeCP2 controls BDNF expression and cocaine intake through homeostatic interactions with microRNA-212. Nat Neurosci. 2010;13:1120-7
380. Abuhatzira L, Makedonski K, Kaufman Y, Razin A, Shemer R. MeCP2 deficiency in the brain decreases BDNF levels by REST/CoREST-mediated repression and increases TRKB production. Epigenetics. 2007;2:214-22
381. Ma KH, Duong P, Moran JJ, Junaidi N, Svaren J. Polycomb repression regulates Schwann cell proliferation and axon regeneration after nerve injury. Glia. 2018;66:2487-502
382. Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA. et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311-23
383. Liu XH, Sun M, Nie FQ, Ge YB, Zhang EB, Yin DD. et al. Lnc RNA HOTAIR functions as a competing endogenous RNA to regulate HER2 expression by sponging miR-331-3p in gastric cancer. Mol Cancer. 2014;13:92
384. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353-8
385. Chen ML, Hong CG, Yue T, Li HM, Duan R, Hu WB. et al. Inhibition of miR-331-3p and miR-9-5p ameliorates Alzheimer's disease by enhancing autophagy. Theranostics. 2021;11:2395-409
386. Liu Q, Lei C. Neuroprotective effects of miR-331-3p through improved cell viability and inflammatory marker expression: Correlation of serum miR-331-3p levels with diagnosis and severity of Alzheimer's disease. Exp Gerontol. 2021;144:111187
387. Spreafico M, Grillo B, Rusconi F, Battaglioli E, Venturin M. Multiple Layers of CDK5R1 Regulation in Alzheimer's Disease Implicate Long Non-Coding RNAs. Int J Mol Sci. 2018 19
388. Gong H, Wan X, Zhang Y, Liang S. Downregulation of HOTAIR reduces neuronal pyroptosis by targeting miR-455-3p/NLRP1 axis in propofol-treated neurons in vitro. Neurochem Res. 2021;46:1141-50
389. Kumar S, Reddy PH. A New Discovery of MicroRNA-455-3p in Alzheimer's Disease. J Alzheimers Dis. 2019;72:S117-S30
390. Kumar S, Reddy AP, Yin X, Reddy PH. Novel MicroRNA-455-3p and its protective effects against abnormal APP processing and amyloid beta toxicity in Alzheimer's disease. Biochim Biophys Acta Mol Basis Dis. 2019;1865:2428-40
391. Kumar S, Reddy PH. MicroRNA-455-3p as a Potential Biomarker for Alzheimer's Disease: An Update. Front Aging Neurosci. 2018;10:41
392. Kumar S, Vijayan M, Reddy PH. MicroRNA-455-3p as a potential peripheral biomarker for Alzheimer's disease. Hum Mol Genet. 2017;26:3808-22
393. Jung M, Pfeifer GP. Aging and DNA methylation. BMC Biol. 2015;13:7
394. Davidovich C, Zheng L, Goodrich KJ, Cech TR. Promiscuous RNA binding by Polycomb repressive complex 2. Nat Struct Mol Biol. 2013;20:1250-7
395. Wang X, Goodrich KJ, Gooding AR, Naeem H, Archer S, Paucek RD. et al. Targeting of Polycomb Repressive Complex 2 to RNA by Short Repeats of Consecutive Guanines. Mol Cell. 2017;65:1056-67 e5
396. Sin O, Nollen EA. Regulation of protein homeostasis in neurodegenerative diseases: the role of coding and non-coding genes. Cell Mol Life Sci. 2015;72:4027-47
397. Krisko A, Radman M. Protein damage, ageing and age-related diseases. Open Biol. 2019;9:180249
398. Garcia-Esparcia P, Sideris-Lampretsas G, Hernandez-Ortega K, Grau-Rivera O, Sklaviadis T, Gelpi E. et al. Altered mechanisms of protein synthesis in frontal cortex in Alzheimer disease and a mouse model. Am J Neurodegener Dis. 2017;6:15-25
399. Wang X, Michaelis ML, Michaelis EK. Functional genomics of brain aging and Alzheimer's disease: focus on selective neuronal vulnerability. Curr Genomics. 2010;11:618-33
400. Blalock EM, Buechel HM, Popovic J, Geddes JW, Landfield PW. Microarray analyses of laser-captured hippocampus reveal distinct gray and white matter signatures associated with incipient Alzheimer's disease. J Chem Neuroanat. 2011;42:118-26
401. Wang M, Roussos P, McKenzie A, Zhou X, Kajiwara Y, Brennand KJ. et al. Integrative network analysis of nineteen brain regions identifies molecular signatures and networks underlying selective regional vulnerability to Alzheimer's disease. Genome Med. 2016;8:104
402. Pugh BF, Tjian R. Diverse transcriptional functions of the multisubunit eukaryotic TFIID complex. J Biol Chem. 1992;267:679-82
403. Hughes TR, Lambert SA. Transcription factors read epigenetics. Science. 2017;356:489-90
404. Pabo CO, Sauer RT. Transcription factors: structural families and principles of DNA recognition. Annu Rev Biochem. 1992;61:1053-95
405. Liu L, Zhao W, Zhou X. Modeling co-occupancy of transcription factors using chromatin features. Nucleic Acids Res. 2016;44:e49
406. Martin BJE, Brind'Amour J, Kuzmin A, Jensen KN, Liu ZC, Lorincz M. et al. Transcription shapes genome-wide histone acetylation patterns. Nat Commun. 2021;12:210
407. Kumar S, Bucher P. Predicting transcription factor site occupancy using DNA sequence intrinsic and cell-type specific chromatin features. BMC Bioinformatics. 2016;17(Suppl 1):4
408. Tsai LH, Madabhushi R. Alzheimer's disease: A protective factor for the ageing brain. Nature. 2014;507:439-40
409. Sams DS, Nardone S, Getselter D, Raz D, Tal M, Rayi PR. et al. Neuronal CTCF Is Necessary for Basal and Experience-Dependent Gene Regulation, Memory Formation, and Genomic Structure of BDNF and Arc. Cell Rep. 2016;17:2418-30
410. Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497-502
411. Kim S, Yu NK, Kaang BK. CTCF as a multifunctional protein in genome regulation and gene expression. Exp Mol Med. 2015;47:e166
412. Wang H, Maurano MT, Qu H, Varley KE, Gertz J, Pauli F. et al. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 2012;22:1680-8
413. Jothi R, Cuddapah S, Barski A, Cui K, Zhao K. Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 2008;36:5221-31
414. Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137:1194-211
415. Nechiporuk T, McGann J, Mullendorff K, Hsieh J, Wurst W, Floss T. et al. The REST remodeling complex protects genomic integrity during embryonic neurogenesis. Elife. 2016;5:e09584
416. Kwon JH, Shin JH, Kim ES, Lee N, Park JY, Koo BS. et al. REST-dependent expression of TRF2 renders non-neuronal cancer cells resistant to DNA damage during oxidative stress. Int J Cancer. 2013;132:832-42
417. Tanwar VS, Jose CC, Cuddapah S. Role of CTCF in DNA damage response. Mutat Res Rev Mutat Res. 2019;780:61-8
418. Ray Chaudhuri A, Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol. 2017;18:610-21
419. Hilmi K, Jangal M, Marques M, Zhao T, Saad A, Zhang C. et al. CTCF facilitates DNA double-strand break repair by enhancing homologous recombination repair. Sci Adv. 2017;3:e1601898
420. Guastafierro T, Cecchinelli B, Zampieri M, Reale A, Riggio G, Sthandier O. et al. CCCTC-binding factor activates PARP-1 affecting DNA methylation machinery. J Biol Chem. 2008;283:21873-80
421. Rocchi A, Carminati E, De Fusco A, Kowalska JA, Floss T, Benfenati F. REST/NRSF deficiency impairs autophagy and leads to cellular senescence in neurons. Aging Cell. 2021;20:e13471
422. Torrano V, Navascues J, Docquier F, Zhang R, Burke LJ, Chernukhin I. et al. Targeting of CTCF to the nucleolus inhibits nucleolar transcription through a poly(ADP-ribosyl)ation-dependent mechanism. J Cell Sci. 2006;119:1746-59
423. Vostrov AA, Quitschke WW. The zinc finger protein CTCF binds to the APBbeta domain of the amyloid beta-protein precursor promoter. Evidence for a role in transcriptional activation. J Biol Chem. 1997;272:33353-9
424. Choi DI, Kim M, Kim S, Yu NK, Kwak C, Seo H. et al. Conditional knock out of transcription factor CTCF in excitatory neurons induces cognitive deficiency. Mol Brain. 2021;14:1
425. Hwang JY, Zukin RS. REST, a master transcriptional regulator in neurodegenerative disease. Curr Opin Neurobiol. 2018;48:193-200
426. Cavadas MA, Mesnieres M, Crifo B, Manresa MC, Selfridge AC, Keogh CE. et al. REST is a hypoxia-responsive transcriptional repressor. Sci Rep. 2016;6:31355
427. Nishihara S, Tsuda L, Ogura T. The canonical Wnt pathway directly regulates NRSF/REST expression in chick spinal cord. Biochem Biophys Res Commun. 2003;311:55-63
428. Tomasoni R, Negrini S, Fiordaliso S, Klajn A, Tkatch T, Mondino A. et al. A signaling loop of REST, TSC2 and beta-catenin governs proliferation and function of PC12 neural cells. J Cell Sci. 2011;124:3174-86
429. Pradhan S, Chin HG, Esteve PO, Jacobsen SE. SET7/9 mediated methylation of non-histone proteins in mammalian cells. Epigenetics. 2009;4:383-7
430. Funato Y, Michiue T, Asashima M, Miki H. The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt-beta-catenin signalling through dishevelled. Nat Cell Biol. 2006;8:501-8
431. Zhao Y, Zhu M, Yu Y, Qiu L, Zhang Y, He L. et al. Brain REST/NRSF Is Not Only a Silent Repressor but Also an Active Protector. Mol Neurobiol. 2017;54:541-50
432. Wan J, Oliver VF, Wang G, Zhu H, Zack DJ, Merbs SL. et al. Characterization of tissue-specific differential DNA methylation suggests distinct modes of positive and negative gene expression regulation. BMC Genomics. 2015;16:49
433. Inestrosa NC, Varela-Nallar L. Wnt signaling in the nervous system and in Alzheimer's disease. J Mol Cell Biol. 2014;6:64-74
434. Wan W, Xia S, Kalionis B, Liu L, Li Y. The role of Wnt signaling in the development of Alzheimer's disease: a potential therapeutic target? Biomed Res Int. 2014;2014:301575
435. He P, Shen Y. Interruption of beta-catenin signaling reduces neurogenesis in Alzheimer's disease. J Neurosci. 2009;29:6545-57
436. Inestrosa NC, Varela-Nallar L, Grabowski CP, Colombres M. Synaptotoxicity in Alzheimer's disease: the Wnt signaling pathway as a molecular target. IUBMB Life. 2007;59:316-21
437. Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y. et al. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837-47
438. Valenta T, Lukas J, Doubravska L, Fafilek B, Korinek V. HIC1 attenuates Wnt signaling by recruitment of TCF-4 and beta-catenin to the nuclear bodies. EMBO J. 2006;25:2326-37
439. Levy L, Wei Y, Labalette C, Wu Y, Renard CA, Buendia MA. et al. Acetylation of beta-catenin by p300 regulates beta-catenin-Tcf4 interaction. Mol Cell Biol. 2004;24:3404-14
440. Chen WY, Wang DH, Yen RC, Luo J, Gu W, Baylin SB. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell. 2005;123:437-48
441. Svedlund J, Koskinen Edblom S, Marquez VE, Akerstrom G, Bjorklund P, Westin G. Hypermethylated in cancer 1 (HIC1), a tumor suppressor gene epigenetically deregulated in hyperparathyroid tumors by histone H3 lysine modification. J Clin Endocrinol Metab. 2012;97:E1307-15
442. Xiang Y, Zhu Z, Han G, Lin H, Xu L, Chen CD. JMJD3 is a histone H3K27 demethylase. Cell Res. 2007;17:850-7
443. Pukhalskaia AE, Dyatlova AS, Linkova NS, Kozlov KL, Kvetnaia TV, Koroleva MV. et al. Sirtuins as Possible Predictors of Aging and Alzheimer's Disease Development: Verification in the Hippocampus and Saliva. Bull Exp Biol Med. 2020;169:821-4
444. Abdelmohsen K, Pullmann R Jr, Lal A, Kim HH, Galban S, Yang X. et al. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol Cell. 2007;25:543-57
445. Oh YM, Mahar M, Ewan EE, Leahy KM, Zhao G, Cavalli V. Epigenetic regulator UHRF1 inactivates REST and growth suppressor gene expression via DNA methylation to promote axon regeneration. Proc Natl Acad Sci U S A. 2018;115:E12417-E26
446. Julien C, Tremblay C, Emond V, Lebbadi M, Salem N Jr, Bennett DA. et al. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J Neuropathol Exp Neurol. 2009;68:48-58
447. Yamagata K, Kitabayashi I. Sirt1 physically interacts with Tip60 and negatively regulates Tip60-mediated acetylation of H2AX. Biochem Biophys Res Commun. 2009;390:1355-60
448. Peng L, Ling H, Yuan Z, Fang B, Bloom G, Fukasawa K. et al. SIRT1 negatively regulates the activities, functions, and protein levels of hMOF and TIP60. Mol Cell Biol. 2012;32:2823-36
449. Yang C, Wu J, Zheng YG. Function of the active site lysine autoacetylation in Tip60 catalysis. PLoS One. 2012;7:e32886
450. Ahmad T, Ashraf W, Ibrahim A, Zaayter L, Muller CD, Hamiche A. et al. TIP60 governs the autoubiquitination of UHRF1 through USP7 dissociation from the UHRF1/USP7 complex. Int J Oncol. 2021 59
451. Achour M, Fuhrmann G, Alhosin M, Ronde P, Chataigneau T, Mousli M. et al. UHRF1 recruits the histone acetyltransferase Tip60 and controls its expression and activity. Biochem Biophys Res Commun. 2009;390:523-8
452. Dai C, Shi D, Gu W. Negative regulation of the acetyltransferase TIP60-p53 interplay by UHRF1 (ubiquitin-like with PHD and RING finger domains 1). J Biol Chem. 2013;288:19581-92
453. Meehan RR, Lewis JD, Bird AP. Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res. 1992;20:5085-92
454. Na ES, Monteggia LM. The role of MeCP2 in CNS development and function. Horm Behav. 2011;59:364-8
455. Maphis NM, Jiang S, Binder J, Wright C, Gopalan B, Lamb BT. et al. Whole Genome Expression Analysis in a Mouse Model of Tauopathy Identifies MECP2 as a Possible Regulator of Tau Pathology. Front Mol Neurosci. 2017;10:69
456. Wittrahm R, Takalo M, Marttinen M, Kuulasmaa T, Makinen P, Kemppainen S. et al. MECP2 Increases the Pro-Inflammatory Response of Microglial Cells and Phosphorylation at Serine 423 Regulates Neuronal Gene Expression upon Neuroinflammation. Cells. 2021 10
457. Pejhan S, Siu VM, Ang LC, Del Bigio MR, Rastegar M. Differential brain region-specific expression of MeCP2 and BDNF in Rett Syndrome patients: a distinct grey-white matter variation. Neuropathol Appl Neurobiol. 2020;46:735-50
458. Ho KL, McNae IW, Schmiedeberg L, Klose RJ, Bird AP, Walkinshaw MD. MeCP2 binding to DNA depends upon hydration at methyl-CpG. Mol Cell. 2008;29:525-31
459. Lilja T, Wallenborg K, Bjorkman K, Albage M, Eriksson M, Lagercrantz H. et al. Novel alterations in the epigenetic signature of MeCP2-targeted promoters in lymphocytes of Rett syndrome patients. Epigenetics. 2013;8:246-51
460. Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND. et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905
461. Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN. et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386-9
462. Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N. et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187-91
463. Kimura H, Shiota K. Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J Biol Chem. 2003;278:4806-12
464. Zocchi L, Sassone-Corsi P. SIRT1-mediated deacetylation of MeCP2 contributes to BDNF expression. Epigenetics. 2012;7:695-700
465. Pandey S, Simmons GE Jr, Malyarchuk S, Calhoun TN, Pruitt K. A novel MeCP2 acetylation site regulates interaction with ATRX and HDAC1. Genes Cancer. 2015;6:408-21
466. Ramanujam PL, Mehrotra S, Kumar RP, Verma S, Deshpande G, Mishra RK. et al. Global chromatin organizer SATB1 acts as a context-dependent regulator of the Wnt/Wg target genes. Sci Rep. 2021;11:3385
467. Zhang M, Poplawski M, Yen K, Cheng H, Bloss E, Zhu X. et al. Role of CBP and SATB-1 in aging, dietary restriction, and insulin-like signaling. PLoS Biol. 2009;7:e1000245
468. Chatterjee S, Cassel R, Schneider-Anthony A, Merienne K, Cosquer B, Tzeplaeff L. et al. Reinstating plasticity and memory in a tauopathy mouse model with an acetyltransferase activator. EMBO Mol Med. 2018 10
469. Yang T, Fu M, Pestell R, Sauve AA. SIRT1 and endocrine signaling. Trends Endocrinol Metab. 2006;17:186-91
470. Simic P, Zainabadi K, Bell E, Sykes DB, Saez B, Lotinun S. et al. SIRT1 regulates differentiation of mesenchymal stem cells by deacetylating beta-catenin. EMBO Mol Med. 2013;5:430-40
471. Yu F, Thiesen J, Stratling WH. Histone deacetylase-independent transcriptional repression by methyl-CpG-binding protein 2. Nucleic Acids Res. 2000;28:2201-6
472. Jia L, Pina-Crespo J, Li Y. Restoring Wnt/beta-catenin signaling is a promising therapeutic strategy for Alzheimer's disease. Mol Brain. 2019;12:104
473. De Ferrari GV, Avila ME, Medina MA, Perez-Palma E, Bustos BI, Alarcon MA. Wnt/beta-catenin signaling in Alzheimer's disease. CNS Neurol Disord Drug Targets. 2014;13:745-54
474. Gupta A, Sharma GG, Young CS, Agarwal M, Smith ER, Paull TT. et al. Involvement of human MOF in ATM function. Mol Cell Biol. 2005;25:5292-305
475. Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol Cell. 2017;66:801-17
476. Tang M, Li Z, Zhang C, Lu X, Tu B, Cao Z. et al. SIRT7-mediated ATM deacetylation is essential for its deactivation and DNA damage repair. Sci Adv. 2019;5:eaav1118
477. Lakin ND, Jackson SP. Regulation of p53 in response to DNA damage. Oncogene. 1999;18:7644-55
478. Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS. et al. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24:841-51
479. Yuan F, Liu L, Lei Y, Tang P. p53 inhibits the upregulation of sirtuin 1 expression induced by c-Myc. Oncol Lett. 2017;14:4396-402
480. Kaypee S, Sahadevan SA, Patil S, Ghosh P, Roy NS, Roy S. et al. Mutant and Wild-Type Tumor Suppressor p53 Induces p300 Autoacetylation. iScience. 2018;4:260-72
481. Ekblad CM, Friedler A, Veprintsev D, Weinberg RL, Itzhaki LS. Comparison of BRCT domains of BRCA1 and 53BP1: a biophysical analysis. Protein Sci. 2004;13:617-25
482. Yang H, Yan B, Liao D, Huang S, Qiu Y. Acetylation of HDAC1 and degradation of SIRT1 form a positive feedback loop to regulate p53 acetylation during heat-shock stress. Cell Death Dis. 2015;6:e1747
483. Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S. et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. 2019;51:404-13
484. Kitamura Y, Shimohama S, Kamoshima W, Matsuoka Y, Nomura Y, Taniguchi T. Changes of p53 in the brains of patients with Alzheimer's disease. Biochem Biophys Res Commun. 1997;232:418-21
485. Solomon JM, Pasupuleti R, Xu L, McDonagh T, Curtis R, DiStefano PS. et al. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol Cell Biol. 2006;26:28-38
486. Liu T, Ma X, Ouyang T, Chen H, Lin J, Liu J. et al. SIRT1 reverses senescence via enhancing autophagy and attenuates oxidative stress-induced apoptosis through promoting p53 degradation. Int J Biol Macromol. 2018;117:225-34
487. Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK. et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149-59
488. Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A. et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137-48
489. Alves-Fernandes DK, Jasiulionis MG. The Role of SIRT1 on DNA Damage Response and Epigenetic Alterations in Cancer. Int J Mol Sci. 2019 20
490. Lu L, Li L, Lv X, Wu XS, Liu DP, Liang CC. Modulations of hMOF autoacetylation by SIRT1 regulate hMOF recruitment and activities on the chromatin. Cell Res. 2011;21:1182-95
491. Vaquero A, Scher M, Erdjument-Bromage H, Tempst P, Serrano L, Reinberg D. SIRT1 regulates the histone methyl-transferase SUV39H1 during heterochromatin formation. Nature. 2007;450:440-4
492. Jiang Y, Fu X, Zhang Y, Wang SF, Zhu H, Wang WK. et al. Rett syndrome linked to defects in forming the MeCP2/Rbfox/LASR complex in mouse models. Nat Commun. 2021;12:5767
493. Damianov A, Ying Y, Lin CH, Lee JA, Tran D, Vashisht AA. et al. Rbfox Proteins Regulate Splicing as Part of a Large Multiprotein Complex LASR. Cell. 2016;165:606-19
494. Maxwell SS, Pelka GJ, Tam PP, El-Osta A. Chromatin context and ncRNA highlight targets of MeCP2 in brain. RNA Biol. 2013;10:1741-57
495. Wong J. Altered expression of RNA splicing proteins in Alzheimer's disease patients: evidence from two microarray studies. Dement Geriatr Cogn Dis Extra. 2013;3:74-85
496. Holwerda SJ, de Laat W. CTCF: the protein, the binding partners, the binding sites and their chromatin loops. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120369
497. Barisic D, Stadler MB, Iurlaro M, Schubeler D. Mammalian ISWI and SWI/SNF selectively mediate binding of distinct transcription factors. Nature. 2019;569:136-40
498. Prickett AR, Barkas N, McCole RB, Hughes S, Amante SM, Schulz R. et al. Genome-wide and parental allele-specific analysis of CTCF and cohesin DNA binding in mouse brain reveals a tissue-specific binding pattern and an association with imprinted differentially methylated regions. Genome Res. 2013;23:1624-35
499. Maurano MT, Wang H, John S, Shafer A, Canfield T, Lee K. et al. Role of DNA Methylation in Modulating Transcription Factor Occupancy. Cell Rep. 2015;12:1184-95
500. Zampieri M, Guastafierro T, Calabrese R, Ciccarone F, Bacalini MG, Reale A. et al. ADP-ribose polymers localized on Ctcf-Parp1-Dnmt1 complex prevent methylation of Ctcf target sites. Biochem J. 2012;441:645-52
501. Ohlsson R, Renkawitz R, Lobanenkov V. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet. 2001;17:520-7
502. Yang J, Corces VG. Chromatin insulators: a role in nuclear organization and gene expression. Adv Cancer Res. 2011;110:43-76
503. Wang G, Xia B, Zhou M, Lv J, Zhao D, Li Y. et al. MACMIC Reveals A Dual Role of CTCF in Epigenetic Regulation of Cell Identity Genes. Genomics Proteomics Bioinformatics. 2021;19:140-53
504. Xu B, Wang H, Wright S, Hyle J, Zhang Y, Shao Y. et al. Acute depletion of CTCF rewires genome-wide chromatin accessibility. Genome Biol. 2021;22:244
505. Hou Y, Song Q, Gao S, Zhang X, Wang Y, Liu J. et al. CTCF Mediates Replicative Senescence Through POLD1. Front Cell Dev Biol. 2021;9:618586
506. Wang Y, Zhang X, Song Q, Hou Y, Liu J, Sun Y. et al. Characterization of the chromatin accessibility in an Alzheimer's disease (AD) mouse model. Alzheimers Res Ther. 2020;12:29
507. Wu D, Li T, Lu Z, Dai W, Xu M, Lu L. Effect of CTCF-binding motif on regulation of PAX6 transcription. Invest Ophthalmol Vis Sci. 2006;47:2422-9
508. Li T, Lu Z, Lu L. Regulation of eye development by transcription control of CCCTC binding factor (CTCF). J Biol Chem. 2004;279:27575-83
509. Zhang Y, Zhang Y, Aman Y, Ng CT, Chau WH, Zhang Z. et al. Amyloid-beta toxicity modulates tau phosphorylation through the PAX6 signalling pathway. Brain. 2021;144:2759-70
510. Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D'Avanzo C. et al. A three-dimensional human neural cell culture model of Alzheimer's disease. Nature. 2014;515:274-8
511. Gabriele RMC, Abel E, Fox NC, Wray S, Arber C. Knockdown of Amyloid Precursor Protein: Biological Consequences and Clinical Opportunities. Front Neurosci. 2022;16:835645
512. Menendez-Gonzalez M, Perez-Pinera P, Martinez-Rivera M, Calatayud MT, Blazquez Menes B. APP processing and the APP-KPI domain involvement in the amyloid cascade. Neurodegener Dis. 2005;2:277-83
513. Burton T, Liang B, Dibrov A, Amara F. Transforming growth factor-beta-induced transcription of the Alzheimer beta-amyloid precursor protein gene involves interaction between the CTCF-complex and Smads. Biochem Biophys Res Commun. 2002;295:713-23
514. Docagne F, Gabriel C, Lebeurrier N, Lesne S, Hommet Y, Plawinski L. et al. Sp1 and Smad transcription factors co-operate to mediate TGF-beta-dependent activation of amyloid-beta precursor protein gene transcription. Biochem J. 2004;383:393-9
515. Kim M, Li D, Cui Y, Mueller K, Chears WC, DeJong J. Regulatory factor interactions and somatic silencing of the germ cell-specific ALF gene. J Biol Chem. 2006;281:34288-98
516. Citron BA, Dennis JS, Zeitlin RS, Echeverria V. Transcription factor Sp1 dysregulation in Alzheimer's disease. J Neurosci Res. 2008;86:2499-504
517. Santpere G, Nieto M, Puig B, Ferrer I. Abnormal Sp1 transcription factor expression in Alzheimer disease and tauopathies. Neurosci Lett. 2006;397:30-4
518. Doetzlhofer A, Rotheneder H, Lagger G, Koranda M, Kurtev V, Brosch G. et al. Histone deacetylase 1 can repress transcription by binding to Sp1. Mol Cell Biol. 1999;19:5504-11
519. Paonessa F, Latifi S, Scarongella H, Cesca F, Benfenati F. Specificity protein 1 (Sp1)-dependent activation of the synapsin I gene (SYN1) is modulated by RE1-silencing transcription factor (REST) and 5'-cytosine-phosphoguanine (CpG) methylation. J Biol Chem. 2013;288:3227-39
520. Plaisance V, Niederhauser G, Azzouz F, Lenain V, Haefliger JA, Waeber G. et al. The repressor element silencing transcription factor (REST)-mediated transcriptional repression requires the inhibition of Sp1. J Biol Chem. 2005;280:401-7
521. Bersten DC, Wright JA, McCarthy PJ, Whitelaw ML. Regulation of the neuronal transcription factor NPAS4 by REST and microRNAs. Biochim Biophys Acta. 2014;1839:13-24
522. Fan W, Long Y, Lai Y, Wang X, Chen G, Zhu B. NPAS4 Facilitates the Autophagic Clearance of Endogenous Tau in Rat Cortical Neurons. J Mol Neurosci. 2016;58:401-10
523. Opsomer R, Contino S, Perrin F, Gualdani R, Tasiaux B, Doyen P. et al. Amyloid Precursor Protein (APP) Controls the Expression of the Transcriptional Activator Neuronal PAS Domain Protein 4 (NPAS4) and Synaptic GABA Release. eNeuro. 2020 7
524. Vico Varela E, Etter G, Williams S. Excitatory-inhibitory imbalance in Alzheimer's disease and therapeutic significance. Neurobiol Dis. 2019;127:605-15
525. Frank M, Kemler R. Protocadherins. Curr Opin Cell Biol. 2002;14:557-62
526. Wu Q, Jia Z. Wiring the Brain by Clustered Protocadherin Neural Codes. Neurosci Bull. 2021;37:117-31
527. Monahan K, Rudnick ND, Kehayova PD, Pauli F, Newberry KM, Myers RM. et al. Role of CCCTC binding factor (CTCF) and cohesin in the generation of single-cell diversity of protocadherin-alpha gene expression. Proc Natl Acad Sci U S A. 2012;109:9125-30
528. Hasegawa S, Kobayashi H, Kumagai M, Nishimaru H, Tarusawa E, Kanda H. et al. Clustered Protocadherins Are Required for Building Functional Neural Circuits. Front Mol Neurosci. 2017;10:114
529. Lefebvre JL, Kostadinov D, Chen WV, Maniatis T, Sanes JR. Protocadherins mediate dendritic self-avoidance in the mammalian nervous system. Nature. 2012;488:517-21
530. Ing-Esteves S, Kostadinov D, Marocha J, Sing AD, Joseph KS, Laboulaye MA. et al. Combinatorial Effects of Alpha- and Gamma-Protocadherins on Neuronal Survival and Dendritic Self-Avoidance. J Neurosci. 2018;38:2713-29
531. Hirayama T, Tarusawa E, Yoshimura Y, Galjart N, Yagi T. CTCF is required for neural development and stochastic expression of clustered Pcdh genes in neurons. Cell Rep. 2012;2:345-57
532. Fukuda E, Hamada S, Hasegawa S, Katori S, Sanbo M, Miyakawa T. et al. Down-regulation of protocadherin-alpha A isoforms in mice changes contextual fear conditioning and spatial working memory. Eur J Neurosci. 2008;28:1362-76
533. Yamagishi T, Yoshitake K, Kamatani D, Watanabe K, Tsukano H, Hishida R. et al. Molecular diversity of clustered protocadherin-alpha required for sensory integration and short-term memory in mice. Sci Rep. 2018;8:9616
534. Ong CT, Corces VG. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet. 2014;15:234-46
535. Splinter E, Heath H, Kooren J, Palstra RJ, Klous P, Grosveld F. et al. CTCF mediates long-range chromatin looping and local histone modification in the beta-globin locus. Genes Dev. 2006;20:2349-54
536. Zlatanova J, Caiafa P. CCCTC-binding factor: to loop or to bridge. Cell Mol Life Sci. 2009;66:1647-60
537. Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY. et al. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013;503:290-4
538. Gray JM, Kim TK, West AE, Nord AS, Markenscoff-Papadimitriou E, Lomvardas S. Genomic Views of Transcriptional Enhancers: Essential Determinants of Cellular Identity and Activity-Dependent Responses in the CNS. J Neurosci. 2015;35:13819-26
539. Lewis MW, Li S, Franco HL. Transcriptional control by enhancers and enhancer RNAs. Transcription. 2019;10:171-86
540. Carullo NVN, Day JJ. Genomic Enhancers in Brain Health and Disease. Genes (Basel). 2019 10
541. Golan-Mashiach M, Grunspan M, Emmanuel R, Gibbs-Bar L, Dikstein R, Shapiro E. Identification of CTCF as a master regulator of the clustered protocadherin genes. Nucleic Acids Res. 2012;40:3378-91
542. Guo Y, Monahan K, Wu H, Gertz J, Varley KE, Li W. et al. CTCF/cohesin-mediated DNA looping is required for protocadherin alpha promoter choice. Proc Natl Acad Sci U S A. 2012;109:21081-6
543. Guo Y, Xu Q, Canzio D, Shou J, Li J, Gorkin DU. et al. CRISPR Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function. Cell. 2015;162:900-10
544. Jia Z, Li J, Ge X, Wu Y, Guo Y, Wu Q. Tandem CTCF sites function as insulators to balance spatial chromatin contacts and topological enhancer-promoter selection. Genome Biol. 2020;21:75
545. Tang Y, Jia Z, Xu H, Da LT, Wu Q. Mechanism of REST/NRSF regulation of clustered protocadherin alpha genes. Nucleic Acids Res. 2021;49:4506-21
546. Hashimoto H, Wang D, Horton JR, Zhang X, Corces VG, Cheng X. Structural Basis for the Versatile and Methylation-Dependent Binding of CTCF to DNA. Mol Cell. 2017;66:711-20 e3
547. Canzio D, Nwakeze CL, Horta A, Rajkumar SM, Coffey EL, Duffy EE. et al. Antisense lncRNA Transcription Mediates DNA Demethylation to Drive Stochastic Protocadherin alpha Promoter Choice. Cell. 2019;177:639-53 e15
548. Yin M, Wang J, Wang M, Li X, Zhang M, Wu Q. et al. Molecular mechanism of directional CTCF recognition of a diverse range of genomic sites. Cell Res. 2017;27:1365-77
549. Li Y, Chen Z, Gao Y, Pan G, Zheng H, Zhang Y. et al. Synaptic Adhesion Molecule Pcdh-gammaC5 Mediates Synaptic Dysfunction in Alzheimer's Disease. J Neurosci. 2017;37:9259-68
550. Ren G, Jin W, Cui K, Rodrigez J, Hu G, Zhang Z. et al. CTCF-Mediated Enhancer-Promoter Interaction Is a Critical Regulator of Cell-to-Cell Variation of Gene Expression. Mol Cell. 2017;67:1049-58 e6
551. Eldholm V, Haugen A, Zienolddiny S. CTCF mediates the TERT enhancer-promoter interactions in lung cancer cells: identification of a novel enhancer region involved in the regulation of TERT gene. Int J Cancer. 2014;134:2305-13
552. Moon M, Jung ES, Jeon SG, Cha MY, Jang Y, Kim W. et al. Nurr1 (NR4A2) regulates Alzheimer's disease-related pathogenesis and cognitive function in the 5XFAD mouse model. Aging Cell. 2019;18:e12866
553. Hadar A, Milanesi E, Squassina A, Niola P, Chillotti C, Pasmanik-Chor M. et al. RGS2 expression predicts amyloid-beta sensitivity, MCI and Alzheimer's disease: genome-wide transcriptomic profiling and bioinformatics data mining. Transl Psychiatry. 2016;6:e909
554. Bonnycastle LL, Yu CE, Wijsman EM, Orr HT, Patterson D, Clancy KP. et al. The c-fos gene and early-onset familial Alzheimer's disease. Neurosci Lett. 1993;160:33-6
555. Tuvikene J, Esvald EE, Rahni A, Uustalu K, Zhuravskaya A, Avarlaid A. et al. Intronic enhancer region governs transcript-specific Bdnf expression in rodent neurons. Elife. 2021 10
556. Schaukowitch K, Joo JY, Liu X, Watts JK, Martinez C, Kim TK. Enhancer RNA facilitates NELF release from immediate early genes. Mol Cell. 2014;56:29-42
557. Kelly MP, Deadwyler SA. Experience-dependent regulation of the immediate-early gene arc differs across brain regions. J Neurosci. 2003;23:6443-51
558. Chang J, Zhang B, Heath H, Galjart N, Wang X, Milbrandt J. Nicotinamide adenine dinucleotide (NAD)-regulated DNA methylation alters CCCTC-binding factor (CTCF)/cohesin binding and transcription at the BDNF locus. Proc Natl Acad Sci U S A. 2010;107:21836-41
559. Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J. et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182-7
560. Phillips HS, Hains JM, Armanini M, Laramee GR, Johnson SA, Winslow JW. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer's disease. Neuron. 1991;7:695-702
561. Peng S, Wuu J, Mufson EJ, Fahnestock M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer's disease. J Neurochem. 2005;93:1412-21
562. Lee J, Fukumoto H, Orne J, Klucken J, Raju S, Vanderburg CR. et al. Decreased levels of BDNF protein in Alzheimer temporal cortex are independent of BDNF polymorphisms. Exp Neurol. 2005;194:91-6
563. Ginsberg SD, Hemby SE, Lee VM, Eberwine JH, Trojanowski JQ. Expression profile of transcripts in Alzheimer's disease tangle-bearing CA1 neurons. Ann Neurol. 2000;48:77-87
564. Kerrigan TL, Randall AD. A new player in the "synaptopathy" of Alzheimer's disease - arc/arg 3.1. Front Neurol. 2013;4:9
565. Grinevich V, Kolleker A, Eliava M, Takada N, Takuma H, Fukazawa Y. et al. Fluorescent Arc/Arg3.1 indicator mice: a versatile tool to study brain activity changes in vitro and in vivo. J Neurosci Methods. 2009;184:25-36
566. Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL. et al. Synaptic targeting by Alzheimer's-related amyloid beta oligomers. J Neurosci. 2004;24:10191-200
567. Karpova NN. Role of BDNF epigenetics in activity-dependent neuronal plasticity. Neuropharmacology. 2014 76 Pt C: 709-18
568. Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem. 2003;10:86-98
569. Plath N, Ohana O, Dammermann B, Errington ML, Schmitz D, Gross C. et al. Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron. 2006;52:437-44
570. Bramham CR, Alme MN, Bittins M, Kuipers SD, Nair RR, Pai B. et al. The Arc of synaptic memory. Exp Brain Res. 2010;200:125-40
571. Kaushik M, Kaushik P, Parvez S. Memory related molecular signatures: The pivots for memory consolidation and Alzheimer's related memory decline. Ageing Res Rev. 2022;76:101577
572. Xie X, Mikkelsen TS, Gnirke A, Lindblad-Toh K, Kellis M, Lander ES. Systematic discovery of regulatory motifs in conserved regions of the human genome, including thousands of CTCF insulator sites. Proc Natl Acad Sci U S A. 2007;104:7145-50
573. Majumder P, Gomez JA, Boss JM. The human major histocompatibility complex class II HLA-DRB1 and HLA-DQA1 genes are separated by a CTCF-binding enhancer-blocking element. J Biol Chem. 2006;281:18435-43
574. Majumder P, Gomez JA, Chadwick BP, Boss JM. The insulator factor CTCF controls MHC class II gene expression and is required for the formation of long-distance chromatin interactions. J Exp Med. 2008;205:785-98
575. Majumder P, Boss JM. CTCF controls expression and chromatin architecture of the human major histocompatibility complex class II locus. Mol Cell Biol. 2010;30:4211-23
576. Perlmutter LS, Scott SA, Barron E, Chui HC. MHC class II-positive microglia in human brain: association with Alzheimer lesions. J Neurosci Res. 1992;33:549-58
577. Edler MK, Mhatre-Winters I, Richardson JR. Microglia in Aging and Alzheimer's Disease: A Comparative Species Review. Cells. 2021 10
578. Nikolic T, Movita D, Lambers ME, Ribeiro de Almeida C, Biesta P, Kreefft K. et al. The DNA-binding factor Ctcf critically controls gene expression in macrophages. Cell Mol Immunol. 2014;11:58-70
579. Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer's disease. J Cell Biol. 2018;217:459-72
580. Kurukuti S, Tiwari VK, Tavoosidana G, Pugacheva E, Murrell A, Zhao Z. et al. CTCF binding at the H19 imprinting control region mediates maternally inherited higher-order chromatin conformation to restrict enhancer access to Igf2. Proc Natl Acad Sci U S A. 2006;103:10684-9
581. Bell AC, Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature. 2000;405:482-5
582. Drewell RA, Goddard CJ, Thomas JO, Surani MA. Methylation-dependent silencing at the H19 imprinting control region by MeCP2. Nucleic Acids Res. 2002;30:1139-44
583. Fu VX, Schwarze SR, Kenowski ML, Leblanc S, Svaren J, Jarrard DF. A loss of insulin-like growth factor-2 imprinting is modulated by CCCTC-binding factor down-regulation at senescence in human epithelial cells. J Biol Chem. 2004;279:52218-26
584. Ulaner GA, Vu TH, Li T, Hu JF, Yao XM, Yang Y. et al. Loss of imprinting of IGF2 and H19 in osteosarcoma is accompanied by reciprocal methylation changes of a CTCF-binding site. Hum Mol Genet. 2003;12:535-49
585. Mellott TJ, Pender SM, Burke RM, Langley EA, Blusztajn JK. IGF2 ameliorates amyloidosis, increases cholinergic marker expression and raises BMP9 and neurotrophin levels in the hippocampus of the APPswePS1dE9 Alzheimer's disease model mice. PLoS One. 2014;9:e94287
586. Zhong L, Liu P, Fan J, Luo Y. Long non-coding RNA H19: Physiological functions and involvements in central nervous system disorders. Neurochem Int. 2021;148:105072
587. Wang B, Suen CW, Ma H, Wang Y, Kong L, Qin D. et al. The Roles of H19 in Regulating Inflammation and Aging. Front Immunol. 2020;11:579687
588. Pardo M, Cheng Y, Sitbon YH, Lowell JA, Grieco SF, Worthen RJ. et al. Insulin growth factor 2 (IGF2) as an emergent target in psychiatric and neurological disorders. Review. Neurosci Res. 2019;149:1-13
589. Lyons CE, Bartolomucci A. Stress and Alzheimer's disease: A senescence link? Neurosci Biobehav Rev. 2020;115:285-98
590. Wang J, Zhao H, Fan Z, Li G, Ma Q, Tao Z. et al. Long Noncoding RNA H19 Promotes Neuroinflammation in Ischemic Stroke by Driving Histone Deacetylase 1-Dependent M1 Microglial Polarization. Stroke. 2017;48:2211-21
591. Wang J, Cao B, Han D, Sun M, Feng J. Long Non-coding RNA H19 Induces Cerebral Ischemia Reperfusion Injury via Activation of Autophagy. Aging Dis. 2017;8:71-84
592. Pham NV, Nguyen MT, Hu JF, Vu TH, Hoffman AR. Dissociation of IGF2 and H19 imprinting in human brain. Brain Res. 1998;810:1-8
593. Pascual-Lucas M, Viana da Silva S, Di Scala M, Garcia-Barroso C, Gonzalez-Aseguinolaza G, Mulle C. et al. Insulin-like growth factor 2 reverses memory and synaptic deficits in APP transgenic mice. EMBO Mol Med. 2014;6:1246-62
594. Zhang YY, Bao HL, Dong LX, Liu Y, Zhang GW, An FM. Silenced lncRNA H19 and up-regulated microRNA-129 accelerates viability and restrains apoptosis of PC12 cells induced by Abeta25-35 in a cellular model of Alzheimer's disease. Cell Cycle. 2021;20:112-25
595. Lorgen-Ritchie M, Murray AD, Staff R, Ferguson-Smith AC, Richards M, Horgan GW. et al. Imprinting methylation predicts hippocampal volumes and hyperintensities and the change with age in later life. Sci Rep. 2021;11:943
596. Paulina Troncoso-Escudero RLV. Insulin-like Growth Factor 2: Beyond its Role in Hippocampal-dependent Memory. Journal of Cellular Immunology. 2021;3:46-52
597. Gao S, Zhang X, Song Q, Liu J, Ji X, Wang P. POLD1 deficiency is involved in cognitive function impairment in AD patients and SAMP8 mice. Biomed Pharmacother. 2019;114:108833
598. Hirosue A, Ishihara K, Tokunaga K, Watanabe T, Saitoh N, Nakamoto M. et al. Quantitative assessment of higher-order chromatin structure of the INK4/ARF locus in human senescent cells. Aging Cell. 2012;11:553-6
599. Peters JM, Tedeschi A, Schmitz J. The cohesin complex and its roles in chromosome biology. Genes Dev. 2008;22:3089-114
600. Bajic V, Spremo-Potparevic B, Zivkovic L, Isenovic ER, Arendt T. Cohesion and the aneuploid phenotype in Alzheimer's disease: A tale of genome instability. Neurosci Biobehav Rev. 2015;55:365-74
601. Wruck W, Schroter F, Adjaye J. Meta-Analysis of Transcriptome Data Related to Hippocampus Biopsies and iPSC-Derived Neuronal Cells from Alzheimer's Disease Patients Reveals an Association with FOXA1 and FOXA2 Gene Regulatory Networks. J Alzheimers Dis. 2016;50:1065-82
602. Domanskyi A, Alter H, Vogt MA, Gass P, Vinnikov IA. Transcription factors Foxa1 and Foxa2 are required for adult dopamine neurons maintenance. Front Cell Neurosci. 2014;8:275
603. Tang Y, Min Z, Xiang XJ, Liu L, Ma YL, Zhu BL. et al. Estrogen-related receptor alpha is involved in Alzheimer's disease-like pathology. Exp Neurol. 2018;305:89-96
604. Saldana-Meyer R, Rodriguez-Hernaez J, Escobar T, Nishana M, Jacome-Lopez K, Nora EP. et al. RNA Interactions Are Essential for CTCF-Mediated Genome Organization. Mol Cell. 2019;76:412-22 e5
605. Aguilera A, Garcia-Muse T. Causes of genome instability. Annu Rev Genet. 2013;47:1-32
606. Murnane JP. Telomeres and chromosome instability. DNA Repair (Amst). 2006;5:1082-92
607. Coluzzi E, Colamartino M, Cozzi R, Leone S, Meneghini C, O'Callaghan N. et al. Oxidative stress induces persistent telomeric DNA damage responsible for nuclear morphology change in mammalian cells. PLoS One. 2014;9:e110963
608. O'Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 2008;4:e1000155
609. Potashkin JA, Meredith GE. The role of oxidative stress in the dysregulation of gene expression and protein metabolism in neurodegenerative disease. Antioxid Redox Signal. 2006;8:144-51
610. Kyng KJ, May A, Stevnsner T, Becker KG, Kolvra S, Bohr VA. Gene expression responses to DNA damage are altered in human aging and in Werner Syndrome. Oncogene. 2005;24:5026-42
611. Cobley JN, Fiorello ML, Bailey DM. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018;15:490-503
612. Popa-Wagner A, Mitran S, Sivanesan S, Chang E, Buga AM. ROS and brain diseases: the good, the bad, and the ugly. Oxid Med Cell Longev. 2013;2013:963520
613. Mu Y, Gage FH. Adult hippocampal neurogenesis and its role in Alzheimer's disease. Mol Neurodegener. 2011;6:85
614. Salameh Y, Bejaoui Y, El Hajj N. DNA Methylation Biomarkers in Aging and Age-Related Diseases. Front Genet. 2020;11:171
615. Hashimoto H, Vertino PM, Cheng X. Molecular coupling of DNA methylation and histone methylation. Epigenomics. 2010;2:657-69
616. Lashley T, Gami P, Valizadeh N, Li A, Revesz T, Balazs R. Alterations in global DNA methylation and hydroxymethylation are not detected in Alzheimer's disease. Neuropathol Appl Neurobiol. 2015;41:497-506
617. Chouliaras L, Mastroeni D, Delvaux E, Grover A, Kenis G, Hof PR. et al. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer's disease patients. Neurobiol Aging. 2013;34:2091-9
618. Yokoyama AS, Rutledge JC, Medici V. DNA methylation alterations in Alzheimer's disease. Environ Epigenet. 2017;3:dvx008
619. Bradley-Whitman MA, Lovell MA. Epigenetic changes in the progression of Alzheimer's disease. Mech Ageing Dev. 2013;134:486-95
620. Yoshizaki K, Kimura R, Kobayashi H, Oki S, Kikkawa T, Mai L. et al. Paternal age affects offspring via an epigenetic mechanism involving REST/NRSF. EMBO Rep. 2021;22:e51524
621. Han Y, Franzen J, Stiehl T, Gobs M, Kuo CC, Nikolic M. et al. New targeted approaches for epigenetic age predictions. BMC Biol. 2020;18:71
622. Wang Y, Karlsson R, Lampa E, Zhang Q, Hedman AK, Almgren M. et al. Epigenetic influences on aging: a longitudinal genome-wide methylation study in old Swedish twins. Epigenetics. 2018;13:975-87
623. Yuan T, Jiao Y, de Jong S, Ophoff RA, Beck S, Teschendorff AE. An integrative multi-scale analysis of the dynamic DNA methylation landscape in aging. PLoS Genet. 2015;11:e1004996
624. Marchal C, Miotto B. Emerging concept in DNA methylation: role of transcription factors in shaping DNA methylation patterns. J Cell Physiol. 2015;230:743-51
625. McGann JC, Oyer JA, Garg S, Yao H, Liu J, Feng X. et al. Polycomb- and REST-associated histone deacetylases are independent pathways toward a mature neuronal phenotype. Elife. 2014;3:e04235
626. Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295-304
627. Eden S, Hashimshony T, Keshet I, Cedar H, Thorne AW. DNA methylation models histone acetylation. Nature. 1998;394:842
628. Singh DK, Pandita RK, Singh M, Chakraborty S, Hambarde S, Ramnarain D. et al. MOF Suppresses Replication Stress and Contributes to Resolution of Stalled Replication Forks. Mol Cell Biol. 2018 38
629. Squatrito M, Gorrini C, Amati B. Tip60 in DNA damage response and growth control: many tricks in one HAT. Trends Cell Biol. 2006;16:433-42
630. Sun Y, Jiang X, Xu Y, Ayrapetov MK, Moreau LA, Whetstine JR. et al. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol. 2009;11:1376-82
631. Mujoo K, Hunt CR, Horikoshi N, Pandita TK. A multifaceted role for MOF histone modifying factor in genome maintenance. Mech Ageing Dev. 2017;161:177-80
632. Huang JL, Zhang F, Su M, Li J, Yi W, Hou LX. et al. MeCP2 prevents age-associated cognitive decline via restoring synaptic plasticity in a senescence-accelerated mouse model. Aging Cell. 2021;20:e13451
633. Tie F, Banerjee R, Stratton CA, Prasad-Sinha J, Stepanik V, Zlobin A. et al. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development. 2009;136:3131-41
634. Lai B, Lee JE, Jang Y, Wang L, Peng W, Ge K. MLL3/MLL4 are required for CBP/p300 binding on enhancers and super-enhancer formation in brown adipogenesis. Nucleic Acids Res. 2017;45:6388-403
635. Boija A, Mahat DB, Zare A, Holmqvist PH, Philip P, Meyers DJ. et al. CBP Regulates Recruitment and Release of Promoter-Proximal RNA Polymerase II. Mol Cell. 2017;68:491-503 e5
636. Narita T, Ito S, Higashijima Y, Chu WK, Neumann K, Walter J. et al. Enhancers are activated by p300/CBP activity-dependent PIC assembly, RNAPII recruitment, and pause release. Mol Cell. 2021;81:2166-82 e6
637. Dietrich N, Lerdrup M, Landt E, Agrawal-Singh S, Bak M, Tommerup N. et al. REST-mediated recruitment of polycomb repressor complexes in mammalian cells. PLoS Genet. 2012;8:e1002494
638. Wang J, Wang J, Yang L, Zhao C, Wu LN, Xu L. et al. CTCF-mediated chromatin looping in EGR2 regulation and SUZ12 recruitment critical for peripheral myelination and repair. Nat Commun. 2020;11:4133
639. Xu M, Zhao GN, Lv X, Liu G, Wang LY, Hao DL. et al. CTCF controls HOXA cluster silencing and mediates PRC2-repressive higher-order chromatin structure in NT2/D1 cells. Mol Cell Biol. 2014;34:3867-79
640. Jarroux J, Foretek D, Bertrand C, Gabriel M, Szachnowski U, Saci Z. et al. HOTAIR lncRNA promotes epithelial-mesenchymal transition by redistributing LSD1 at regulatory chromatin regions. EMBO Rep. 2021;22:e50193
641. Song H, Chen L, Liu W, Xu X, Zhou Y, Zhu J. et al. Depleting long noncoding RNA HOTAIR attenuates chronic myelocytic leukemia progression by binding to DNA methyltransferase 1 and inhibiting PTEN gene promoter methylation. Cell Death Dis. 2021;12:440
642. Okitsu CY, Hsieh CL. DNA methylation dictates histone H3K4 methylation. Mol Cell Biol. 2007;27:2746-57
643. Sharifi-Zarchi A, Gerovska D, Adachi K, Totonchi M, Pezeshk H, Taft RJ. et al. DNA methylation regulates discrimination of enhancers from promoters through a H3K4me1-H3K4me3 seesaw mechanism. BMC Genomics. 2017;18:964
644. Bogdanovic O, Long SW, van Heeringen SJ, Brinkman AB, Gomez-Skarmeta JL, Stunnenberg HG. et al. Temporal uncoupling of the DNA methylome and transcriptional repression during embryogenesis. Genome Res. 2011;21:1313-27
645. Bian E, Chen X, Xu Y, Ji X, Cheng M, Wang H. et al. A central role for MeCP2 in the epigenetic repression of miR-200c during epithelial-to-mesenchymal transition of glioma. J Exp Clin Cancer Res. 2019;38:366
646. Zhang Z, Tao W, Hou YY, Wang W, Kenny PJ, Pan ZZ. MeCP2 repression of G9a in regulation of pain and morphine reward. J Neurosci. 2014;34:9076-87
647. Akbarian S, Chen RZ, Gribnau J, Rasmussen TP, Fong H, Jaenisch R. et al. Expression pattern of the Rett syndrome gene MeCP2 in primate prefrontal cortex. Neurobiol Dis. 2001;8:784-91
648. Cheng TL, Chen J, Wan H, Tang B, Tian W, Liao L. et al. Regulation of mRNA splicing by MeCP2 via epigenetic modifications in the brain. Sci Rep. 2017;7:42790
649. Harikumar A, Meshorer E. Chromatin remodeling and bivalent histone modifications in embryonic stem cells. EMBO Rep. 2015;16:1609-19
650. Ren W, Fan H, Grimm SA, Guo Y, Kim JJ, Yin J. et al. Direct readout of heterochromatic H3K9me3 regulates DNMT1-mediated maintenance DNA methylation. Proc Natl Acad Sci U S A. 2020;117:18439-47
651. Balasubramanian D, Akhtar-Zaidi B, Song L, Bartels CF, Veigl M, Beard L. et al. H3K4me3 inversely correlates with DNA methylation at a large class of non-CpG-island-containing start sites. Genome Med. 2012;4:47
652. Gong F, Miller KM. Histone methylation and the DNA damage response. Mutat Res Rev Mutat Res. 2019;780:37-47
653. Niu Y, DesMarais TL, Tong Z, Yao Y, Costa M. Oxidative stress alters global histone modification and DNA methylation. Free Radic Biol Med. 2015;82:22-8
654. Klein CB, Costa M. DNA methylation, heterochromatin and epigenetic carcinogens. Mutat Res. 1997;386:163-80
655. Hernando-Herraez I, Garcia-Perez R, Sharp AJ, Marques-Bonet T. DNA Methylation: Insights into Human Evolution. PLoS Genet. 2015;11:e1005661
656. Law PP, Holland ML. DNA methylation at the crossroads of gene and environment interactions. Essays Biochem. 2019;63:717-26
657. Wu L, Murat P, Matak-Vinkovic D, Murrell A, Balasubramanian S. Binding interactions between long noncoding RNA HOTAIR and PRC2 proteins. Biochemistry. 2013;52:9519-27
658. Portoso M, Ragazzini R, Brencic Z, Moiani A, Michaud A, Vassilev I. et al. PRC2 is dispensable for HOTAIR-mediated transcriptional repression. EMBO J. 2017;36:981-94
659. Saez JE, Gomez AV, Barrios AP, Parada GE, Galdames L, Gonzalez M. et al. Decreased Expression of CoREST1 and CoREST2 Together with LSD1 and HDAC1/2 during Neuronal Differentiation. PLoS One. 2015;10:e0131760
660. Flamier A, El Hajjar J, Adjaye J, Fernandes KJ, Abdouh M, Bernier G. Modeling Late-Onset Sporadic Alzheimer's Disease through BMI1 Deficiency. Cell Rep. 2018;23:2653-66
661. Hogan R, Flamier A, Nardini E, Bernier G. The Role of BMI1 in Late-Onset Sporadic Alzheimer's Disease. Genes (Basel). 2020 11
662. Morey L, Helin K. Polycomb group protein-mediated repression of transcription. Trends Biochem Sci. 2010;35:323-32
663. Xu K, Dai XL, Huang HC, Jiang ZF. Targeting HDACs: a promising therapy for Alzheimer's disease. Oxid Med Cell Longev. 2011;2011:143269
664. Chan AM, Fletcher S. Shifting the paradigm in treating multi-factorial diseases: polypharmacological co-inhibitors of HDAC6. RSC Med Chem. 2021;12:178-96
Author contact
![]() Corresponding author: E-mail: jisen.huaide.
Corresponding author: E-mail: jisen.huaide.