Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2022; 12(13):5820-5823. doi:10.7150/thno.76902 This issue Cite
Editorial
Multiple functions of MLKL in liver fibrosis, from necroptosis to hepatic stellate cell activation
Valeria Pistorio1,2,3, Chantal Housset2,3,4, Jérémie Gautheron2,3, ![]()
1. Department of Chemical Sciences, University of Naples Federico II, Naples, Italy
2. Sorbonne Université, Inserm, Centre de Recherche Saint-Antoine (CRSA), Paris, France
3. Institute of Cardiometabolism and Nutrition (ICAN), Paris, France
4. Assistance Publique-Hôpitaux de Paris (AP-HP), Saint-Antoine Hospital, Department of Hepatology, Reference Center for Inflammatory Biliary Diseases and Autoimmune Hepatitis (CMR MIVB-H), Paris, France
Received 2022-7-7; Accepted 2022-7-9; Published 2022-7-25

Hepatic fibrosis is the typical result of an exuberant wound healing reaction occurring after repeated or prolonged tissue injury [1]. It is characterized by either an accumulation of extracellular matrix and its defective degradation, or both. Alanine aminotransaminase (ALT) and aspartate aminotransferase (AST) are indicators of hepatocellular injury [2]. They are increased in virtually all chronic liver diseases, including non-alcoholic steatohepatitis (NASH), viral hepatitis, autoimmune disorders, and cholestatic liver diseases [1, 2]. Hepatocellular injury induces an activation of Kupffer cells (KCs), the resident macrophages of the liver and a major source of inflammatory mediators, including cytokines, nitric oxide, chemokines, or lysosomal and proteolytic enzymes, which in turn exacerbate cytotoxicity [3]. Thus, KCs play a pivotal role in hepatocellular injury [3]. They promote the activation of hepatic stellate cells (HSCs) and their switch from a quiescent, retinoic acid storing phenotype to a myofibroblast-like phenotype, resulting in increased production of collagens types I and III [4]. This excessive deposition of extracellular matrix in the subendothelial space of Disse disrupts the normal architecture of the hepatic lobule, and can lead to cirrhosis, liver failure, and portal hypertension, requiring liver transplantation [4]. Hepatocyte cell death is now considered as the primary trigger for the persistent leukocyte infiltration and inflammation that fuels the fibrogenic process [5]. Therefore, understanding and ultimately being able to control hepatic cell death in chronic liver injury is of paramount importance.
In recent years, it has become clear that programmed cell death was not restricted to apoptosis, but comprised other forms of regulated cell death [6]. Necroptosis is one of them, combining the molecular machinery of the extrinsic apoptotic pathways with an execution similar to necrosis [6, 7]. Unlike apoptosis, which requires the activation of aspartate-specific proteases known as caspases, necroptosis is first driven by the activation of the receptor-interacting protein kinase (RIPK) 1 and 3, followed by the activation of the pseudo kinase mixed lineage kinase domain-like (MLKL) [7]. Phosphorylation of MLKL leads to its oligomerization. Oligomerized MLKL then binds to and disrupts the plasma membrane releasing cellular components including the damage-associated molecular patterns (DAMPs), which exacerbate the inflammatory process [7]. Necroptosis has emerged as a novel mode of cell death in various chronic liver diseases such as non-alcoholic steatohepatitis (NASH) [6, 8-11]. Deficiency of Mlkl alleviates hepatic insulin resistance and glucose intolerance [12] and has a protective effect on NASH induced by high fat, fructose, and cholesterol diet (FFC) through inhibition of hepatocyte autophagy in hepatocytes [13]. Pharmacological inhibition of necroptosis reduces hepatic inflammation and fibrosis in various murine models of liver diseases [12, 14]. Although necroptosis was shown to play an important role in a number of liver diseases, the function of MLKL in liver fibrosis is still unclear. Moreover, there is growing evidence to indicate that MLKL function is not restricted to necroptosis but can also serve as a regulator of many diseases via non-necroptotic functions [15].
In the study from the group of Xin Xie published in the previous issues of Theranostics [16], the authors examined the role of MLKL in CCl4- and bile duct ligation (BDL)-induced liver injury and fibrosis (Figure 1). They showed that MLKL content positively correlated with a number of fibrotic markers in liver samples from both patients with and animal models of liver fibrosis. Mlkl deficiency in mice significantly reduced CCl4- and BDL-induced liver injury and fibrosis. Considering that hepatocyte injury is the main trigger of liver fibrosis, an adeno-associated virus (AAV) type 8 carrying Mlkl shRNA was used to specifically knock down Mlkl in hepatocytes. The authors demonstrated that AAV8-mediated specific knock down of Mlkl in hepatocytes remarkably alleviated CCl4-induced liver injury. The authors also showed that fibrosis reduction was not only due to the reduction in hepatocyte necroptosis but also to a reduction of HSCs activation, suggesting that targeting MLKL may be an effective way to treat liver fibrosis by acting both on the initiation (i.e., death of hepatocytes) and progression (i.e., HSC activation) stages of fibrogenesis.
MLKL contributes to liver inflammation and fibrosis in CCL4- and BDL-induced cytotoxicity. Chronic hepatocyte injury causes hepatocyte necroptosis, which relies on MLKL. The release of DAMPs activates KCs, which secrete pro-inflammatory cytokines and thus enhance hepatocyte injury. Inflammation drives HSCs trans-differentiation into myofibroblasts (activated HSCs) that are ultimately responsible for the excessive synthesis, deposition and remodeling of extracellular matrix proteins in fibrosis. MLKL has multidirectional functions by promoting cell death in hepatocytes but also by favoring activation of HSCs possibly by transcriptional regulation. Abbreviations: DAMPs: damage-associated patterns; HSCs: Hepatic stellate cells; KCs: Kupffer cells; MLKL: Mixed lineage kinase domain-like protein.
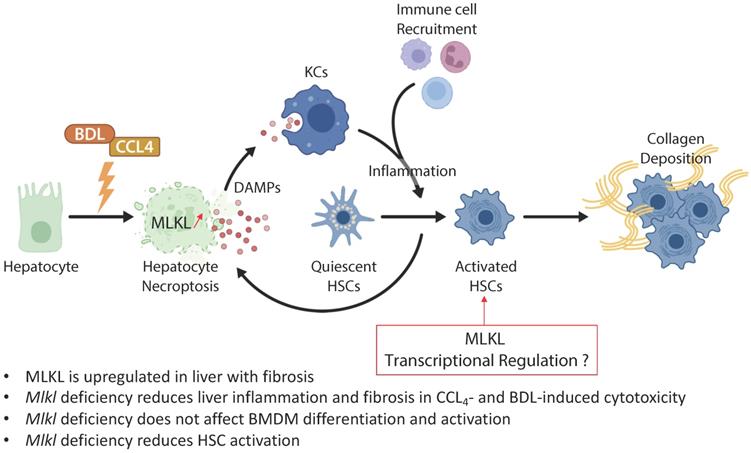
Given the assumption that hepatocyte death is the key trigger of liver disease progression towards fibrosis, and the privileged role of MLKL in regulating necroptosis, it is not surprising that Mlkl deficiency protects mice from CCl4- and BDL-induced liver injury and fibrosis. However, Mlkl deficiency also reduced the activation of HSCs in vitro, possibly via the regulation of TGFβ/Smad 2/3 signaling pathway. It has been demonstrated that fully activated HSCs are resistant to apoptosis and can survive to prolonged serum deprivation, exposure to Fas ligand, NGF, TNF‐α, doxorubicin, etoposide, and oxidative stress mediators such as hydrogen peroxide, superoxide anion, and 4‐hydroxynonenal [17, 18]. This resistance has been attributed to the antiapoptotic gene Bcl-2, which was overexpressed in HSCs in areas localized near fibrotic septa [18]. Therefore, it would be interesting to evaluate if HSCs are also resistant to necroptosis. Moreover, since apoptosis and necroptosis are interconnected with each other but generally do not co-exist [19], it is possible that the acquired resistance of HSCs to apoptosis has facilitated the activation of pro-fibrogenic signaling pathways controlled by MLKL, especially if necroptosis was inhibited. The use of a conditional lecithin-retinol acyltransferase (Lrat)-cre/loxP based knockdown approach of MLKL in HSCs will be necessary to confirm these assumptions [20].
Cytoplasmic MLKL is translocated into the plasma membrane for necroptosis induction. However, preceding necroptosis induction, MLKL is also located to the nucleus [21]. Three-dimensional analysis of immunocytochemistry has revealed that MLKL is located within the nucleus without being associated to the nuclear membrane [21]. Noticeably, the necroptotic function of MLKL is independent of its translocation, and pharmacological inhibition of necroptosis by necrosulfonamide (NSA) has no effect on its nuclear translocation [21]. Therefore, these data suggest that MLKL may have a necroptotic-independent function in regulating transcriptional activities. From this point of view, a recent study has revealed that MLKL interacts with RNA-binding motif protein 6 (RBM6) to promote the expression of several adhesion molecules including intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin [22]. Moreover, MLKL deficiency compromises the invasion of the nasopharyngeal carcinoma cells by reverting epithelial-mesenchymal transition (EMT) [23]. Given that MLKL appears to regulate adhesion molecules and EMT in cancer cells, we may hypothesize that MLKL exerts its necroptotic-independent function through transcriptional regulation by promoting EMT-like mechanisms [24] and thus HSCs trans-differentiation into scar-forming myofibroblasts.
While activation of MLKL has been reported to induce macrophage necroptosis, and targeting macrophage necroptosis may have therapeutic and diagnostic value in atherosclerosis [25], the data of Ren Guo et al. indicate that Mlkl deficiency does not influence bone marrow-derived macrophages (BMDM) polarization and cytokines production. Previous reports exploring the role of necroptosis in cerebral ischemia have shown that defective necroptosis induced polarization of macrophages/microglia toward M2 phenotype, but had not impact on polarization of naïve blood macrophages [26]. Taken together, these results indicate that MLKL may have cell type-specific functions, not only related to cell fate but also to its embryonic origin.
Next to its role as an effector of necroptosis, MLKL also participates in the assembly of the NLRP3 (NOD-like receptor family, pyrin domain containing 3) inflammasome [27]. MLKL-induced NLRP3 inflammasome formation and IL-1β cleavage occur before cell lysis. Furthermore, necroptotic activation of NLRP3, but not necroptotic cell death alone, is necessary for the activation of NF-κB in healthy bystander cells, suggesting that NLRP3 inflammasome activity is a driving force of inflammation in MLKL-dependent diseases [27]. Therefore, it would be useful to evaluate the contribution of MLKL-dependent inflammasome in the progression of liver fibrosis.
The evidence presented by Ren Guo et al. contributes to a better understanding of the role of MLKL in the development of liver fibrosis. It clearly confirmed that MLKL does not only serve as an effector of necroptosis but exhibits non-necroptotic functions that should be considered in future drug therapy. Nevertheless, further studies are needed to evaluate the differential contribution of MLKL in non-parenchymal cells vs. hepatocytes and whether pharmacological inhibition can hold promise as a therapy for liver fibrosis.
Acknowledgements
Funding
JG is supported by Société Francophone du Diabète (R19114DD), Fondation pour la Recherche Médicale (ARF20170938613 & EQU202003010517) and the Agence nationale de la recherche (ANR-21-CE17-0002-01).
Consent for publication
All authors have read and agreed to the published version of the manuscript.
Author contributions
All authors wrote the commentary.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209-218
2. Limdi JK, Hyde GM. Evaluation of abnormal liver function tests. Postgrad Med J. 2003;79:307-312
3. Kolios G, Valatas V, Kouroumalis E. Role of Kupffer cells in the pathogenesis of liver disease. World J Gastroenterol. 2006;12:7413-7420
4. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397-411
5. Shojaie L, Iorga A, Dara L. Cell Death in Liver Diseases: A Review. Int J Mol Sci. 2020 21
6. Gautheron J, Gores GJ, Rodrigues CMP. Lytic cell death in metabolic liver disease. J Hepatol. 2020;73:394-408
7. Choi ME, Price DR, Ryter SW, Choi AMK. Necroptosis: a crucial pathogenic mediator of human disease. JCI Insight. 2019 4
8. Afonso MB, Rodrigues PM, Mateus-Pinheiro M, Simao AL, Gaspar MM, Majdi A. et al. RIPK3 acts as a lipid metabolism regulator contributing to inflammation and carcinogenesis in non-alcoholic fatty liver disease. Gut. 2021;70:2359-2372
9. Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C. et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol Med. 2014;6:1062-1074
10. Afonso MB, Rodrigues PM, Carvalho T, Caridade M, Borralho P, Cortez-Pinto H. et al. Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin Sci (Lond). 2015;129:721-739
11. Gautheron J, Vucur M, Luedde T. Necroptosis in Nonalcoholic Steatohepatitis. Cell Mol Gastroenterol Hepatol. 2015;1:264-265
12. Xu H, Du X, Liu G, Huang S, Du W, Zou S. et al. The pseudokinase MLKL regulates hepatic insulin sensitivity independently of inflammation. Mol Metab. 2019;23:14-23
13. Wu X, Poulsen KL, Sanz-Garcia C, Huang E, McMullen MR, Roychowdhury S. et al. MLKL-dependent signaling regulates autophagic flux in a murine model of non-alcohol-associated fatty liver and steatohepatitis. J Hepatol. 2020;73:616-627
14. Majdi A, Aoudjehane L, Ratziu V, Islam T, Afonso MB, Conti F. et al. Inhibition of receptor-interacting protein kinase 1 improves experimental non-alcoholic fatty liver disease. J Hepatol. 2020;72:627-635
15. Zhan C, Huang M, Yang X, Hou J. MLKL: Functions beyond serving as the Executioner of Necroptosis. Theranostics. 2021;11:4759-4769
16. Guo R, Jia X, Ding Z, Wang G, Jiang M, Li B, Chen S, Xia B, Zhang Q, Liu J, Zheng R, Gao Z, Xie X. Loss of MLKL ameliorates liver fibrosis by inhibiting hepatocyte necroptosis and hepatic stellate cell activation. Theranostics. 2022;12(11):5220-5236 doi:10.7150/thno.71400
17. Kawada N. Human hepatic stellate cells are resistant to apoptosis: implications for human fibrogenic liver disease. Gut. 2006;55:1073-1074
18. Novo E, Marra F, Zamara E, Valfre di Bonzo L, Monitillo L, Cannito S. et al. Overexpression of Bcl-2 by activated human hepatic stellate cells: resistance to apoptosis as a mechanism of progressive hepatic fibrogenesis in humans. Gut. 2006;55:1174-1182
19. Vanden Berghe T, Kaiser WJ, Bertrand MJ, Vandenabeele P. Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol Cell Oncol. 2015;2:e975093
20. Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH. et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823
21. Yoon S, Bogdanov K, Kovalenko A, Wallach D. Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ. 2016;23:253-260
22. Dai J, Zhang C, Guo L, He H, Jiang K, Huang Y. et al. A necroptotic-independent function of MLKL in regulating endothelial cell adhesion molecule expression. Cell Death Dis. 2020;11:282
23. Dong Y, Sun Y, Huang Y, Fang X, Sun P, Dwarakanath B. et al. Depletion of MLKL inhibits invasion of radioresistant nasopharyngeal carcinoma cells by suppressing epithelial-mesenchymal transition. Ann Transl Med. 2019;7:741
24. Zhao YL, Zhu RT, Sun YL. Epithelial-mesenchymal transition in liver fibrosis. Biomed Rep. 2016;4:269-274
25. Karunakaran D, Geoffrion M, Wei L, Gan W, Richards L, Shangari P. et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci Adv. 2016;2:e1600224
26. Yang J, Zhao Y, Zhang L, Fan H, Qi C, Zhang K. et al. RIPK3/MLKL-Mediated Neuronal Necroptosis Modulates the M1/M2 Polarization of Microglia/Macrophages in the Ischemic Cortex. Cereb Cortex. 2018;28:2622-2635
27. Conos SA, Chen KW, De Nardo D, Hara H, Whitehead L, Nunez G. et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc Natl Acad Sci U S A. 2017;114:E961-E969
Author contact
![]() Corresponding author: Dr. Jérémie Gautheron, Centre de Recherche Saint-Antoine (CRSA), Sorbonne Université, 27 rue Chaligny 75571 Paris Cedex 12, France. Email: jeremie.gautheronfr.
Corresponding author: Dr. Jérémie Gautheron, Centre de Recherche Saint-Antoine (CRSA), Sorbonne Université, 27 rue Chaligny 75571 Paris Cedex 12, France. Email: jeremie.gautheronfr.