Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2022; 12(13):5761-5775. doi:10.7150/thno.72599 This issue Cite
Research Paper
SETD1A-SOX2 axis is involved in tamoxifen resistance in estrogen receptor α-positive breast cancer cells
Ming Li Jin1, Liu Yang2, Kwang Won Jeong1 ![]()
1. Gachon Research Institute of Pharmaceutical Sciences, College of Pharmacy, Gachon University, 191 Hambakmoero, Yeonsu-gu, Incheon, 21936, Republic of Korea
2. Collaborative Innovation Center for Chinese Medicine and Respiratory Diseases Co-Constructed by Henan Province & Education Ministry of P.R. China, Henan University of Chinese Medicine, Zhengzhou, 450046, China
Received 2022-3-4; Accepted 2022-7-2; Published 2022-7-18
Abstract

Rationale: Approximately 30-40% of estrogen receptor (ER)-positive breast cancer (BC) cases recur after tamoxifen therapy. Thus, additional studies on the mechanisms underlying tamoxifen resistance and more specific prognostic biomarkers are required. In this study, we investigated the role of the SET domain containing 1A (SETD1A), a histone H3-lysine 4 (H3K4) methyltransferase, in the development of tamoxifen resistance in BC.
Methods: The relationship between tamoxifen resistance and SETD1A protein level was investigated using resistant cell lines derived from the parent BC cells. Biochemical and molecular assays, such as RNA-sequencing, reverse transcription-quantitative polymerase chain reaction, chromatin-immunoprecipitation, and protein-binding assays, were used to identify the SETD1A target gene in tamoxifen-resistant BC cells. Additionally, the role of SETD1A in cancer stem cells (CSCs) was investigated using CSCs isolated from tamoxifen-resistant BC cells. Comprehensive transcriptome analysis and immunofluorescence staining using clinical datasets and tissue microarray were performed to determine the correlation between the expression of the SETD1A-SRY-box transcription factor 2 (SOX2) pair and recurrence in tamoxifen-treated patients with BC.
Results: SETD1A was expressed at higher levels in tamoxifen-resistant BC cells than in primary BC cells. Notably, SETD1A-depleted tamoxifen-resistant MCF-7 cells showed restored sensitivity to tamoxifen, whereas SETD1A overexpression in MCF-7 cells resulted in decreased sensitivity. SETD1A is recruited to the SOX2 gene via its interaction with SOX2, thereby enhancing the expression of SOX2 genes in tamoxifen-resistant BC cells. The growth of tamoxifen-resistant cells and CSCs was effectively suppressed by SETD1A knockdown. In addition, high levels of SETD1A and SOX2 were significantly correlated with a low survival rate in patients with ER-positive tamoxifen-resistant BC.
Conclusion: Our findings provide the first evidence of the critical role of the SETD1A-SOX2 axis in tamoxifen-resistant BC cells, implying that SETD1A may serve as a molecular target and prognostic indicator of a therapeutic response in patients with tamoxifen-resistant BC.
Keywords: breast cancer, tamoxifen resistance, SETD1A, SOX2, cancer stem cell
Introduction
Tamoxifen is a representative selective estrogen receptor modulator (SERM) that inhibits the proliferation of breast cancer (BC) cells by competitive antagonism of estrogen receptor (ER)-mediated transcription. After approval for use in patients with advanced BC, tamoxifen has been widely used as the gold standard for the treatment of BC at all stages [1]. However, approximately 30-40% of patients treated with tamoxifen for five years develop acquired resistance, which remains a major challenge in BC therapy. Early studies have shown that alterations in ER signaling by downregulation or mutation of ERα are potential mechanisms for acquired resistance [2-4]. Indeed, approximately 1% of all primary tumors carry ERα mutations [5], and approximately 15-20% of SERM-resistant BC arise from the loss of ER expression [6]. Several phenotypic changes besides ER have been reported in tamoxifen resistant (TamR) BC cells [6, 7]. Enhanced expression levels of epidermal growth factor receptor (EGFR), erb-b2 receptor tyrosine kinase 2, and insulin like growth factor 1 receptor as well as activation of the downstream signaling pathway components, extracellular signal-regulated kinase and phosphatidylinositol 3-kinase, are major changes that accompany tamoxifen resistance [8-10]. Moreover, reduced expression of p21 or p27 and overexpression of cyclin D1, cyclin E1, and MYC reduce estrogen sensitivity in vitro [11-14].
Recent studies have reported the association between tamoxifen resistance and enhanced expression levels of stem cell factors [15-17] For instance, SRY-box transcription factor 2 (SOX2) is aberrantly expressed in lung, brain, ovary, bone, colon, skin, and BC cells [18-24]. BC cells overexpressing SOX2 are highly proliferative, invasive, and tumorigenic [15, 25] and showed frequent resistance to chemotherapeutic drugs in clinical settings, resulting in poor prognosis in patients with BC [26]. However, the exact mechanism underlying the role of stem cell factors in endocrine resistance to ER-targeted SERM, such as tamoxifen, remains unclear.
Epigenetic regulators are considered one of the major drivers of anticancer drug resistance [7, 27]. For example, DNA hypermethylation of MLH1, Gas1, STA, C8orf4, LAMB3, TUBB, G0S2, and MCAM gene promoter regions has been shown to be involved in acquired resistance to cytotoxic chemotherapeutic drugs in several tumor cell line models [28-32]. In addition, inhibition of the histone H3K27 methyltransferase EZH2 in various cancers reduces the drug-resistant stem cell population [33-35]. Bromodomain inhibitor also increased the apoptosis of T-ALL cells resistant to γ-secretase inhibitor treatment [36]. Lung cancer cells that are resistant to tyrosine kinase inhibitors can be killed by histone deacetylase (HDAC) inhibitors [37]. SETD1A, a histone H3K4-specific methyltransferase, promotes the proliferation of metastatic castration-resistant prostate cancer by regulating the expression of forkhead box M1 and octamer-binding transcription factor 4 (OCT-4) [38]. In addition, our previous study demonstrated the involvement of SETD1A in the expression of genes characteristically expressed in TamR cancer cells (for example, EGFR) as well as ERα-mediated target gene expression [39].
Here, we investigated the mechanism underlying the involvement of SETD1A in the acquisition of tamoxifen resistance in ERα-positive BC cells. First, we measured the changes in SETD1A protein levels following tamoxifen resistance progression. Additionally, we investigated the role of SETD1A overexpression in the development of tamoxifen resistance. Further, we revealed that SETD1A regulates specific transcription factors required for maintaining the pluripotency of cancer stem cells in TamR BC cells. Our findings suggest that SETD1A plays a crucial role in the acquisition of tamoxifen resistance in ER-positive BC cells.
Materials and Methods
Cell culture, plasmids, and siRNAs
MCF-7 cells were cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). T47D cells were grown in Roswell Park Memorial Institute-1640 medium supplemented with 10% FBS. TamR cells were produced by 12-month treatment of MCF-7 and T47D cells with 4-hydroxytamoxifen (Tamoxifen, 5 μM). All cells were incubated at 37 °C with 5% CO2 in a humidified incubator. MCF-7 and T47D cells were obtained from the Korean Cell Line Bank (KCLB, Seoul, Korea). Full-length SOX2 expression plasmid was purchased from Addgene (Watertown, MA, USA). pcDNA3-flag-SETD1A was kindly provided by Professor David Skalnik (Purdue University, IN, USA). The sequences of siRNAs and shRNAs are listed in Table S1.
RNA-sequencing analysis
The total RNA purified from each group (n = 2) was processed to prepare an mRNA-seq library using the QuantSeq3 mRNA-seq Library Prep kit (Lexogen, Vienna, Austria). Each library was sequenced using an Illumina NextSeq500 instrument (Illumina, San Diego CA, USA). The original image data were converted into sequence data and stored in FASTQ format. Genes showing an absolute fold change (FC) of at least 1.5 between groups and P < 0.05 were considered to be differentially expressed. Gene set enrichment was measured via X2K analysis [40].
Analysis of nascent mRNA
Newly synthesized RNA was labeled and isolated using a Click-iT Nascent RNA Capture Kit (Invitrogen, Carlsbad, CA, USA). Nascent RNAs were labeled with 0.2 mM 5-ethynyl uridine (EU) for 16 h and harvested. RNA was isolated from the cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). A total of 1 μg of RNA was used for the click reaction, and 12 μL of Dynabeads (Invitrogen, Carlsbad, CA, USA) was used for 1 μg of biotinylated EU-RNA. Reverse transcription was performed using an iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA, USA). Quantitative polymerase chain reaction (qPCR) was performed using using LightCycler 480 II instrument (Roche, Indianapolis, IN, USA) with the primers listed in Table S2.
Tissue microarray and immunofluorescence staining
A human breast cancer tissue microarray (BRM961a) comprising normal breast tissue (n = 12) and invasive ductal carcinoma tissues (n = 47) was obtained from US Biomax Inc. (Rockville, MD, USA). Microarray slides were deparaffinized and incubated with anti-SETD1A (Bethyl Laboratories, Montgomery, AL, USA) and anti-SOX2 (Thermo Fisher Scientific, Waltham, MA, USA) primary antibodies. Anti-rabbit DyLight488 and anti-mouse DyLight594 (Vector Laboratories, Burlingame, CA, USA) antibodies were used as secondary antibodies (Table S3). The stained area was observed using a confocal microscope (Nikon, Tokyo, Japan) and the average integrated optical density (IOD) of SETD1A and SOX2 was measured at five randomly selected sites for each tissue sample using the ImageJ 1.8.0 software.
Protein-protein interaction assay
For in vitro binding assay, His-SETD1A (SET domain) was purified using Ni-NTA agarose beads. Glutathione S-transferase (GST)-fused SOX2 protein (a.a. 1-317, 1-204, 1-120, and 1-40) and SET domains of SETD1A (Win-SET, a.a. 1450-1707 and SET, a.a. 1538-1707) were expressed in BL21-DE3 and purified using glutathione-agarose beads. Full-length SETD1A was synthesized using the TNT T7 Quick kit (Promega, Madison, WI, USA). For the co-immunoprecipitation assay, cell extracts were prepared using a lysis buffer (20 mM Tris-Cl [pH 8.0], 150 mM NaCl, 1% NP-40, and 2 mM EDTA) [41]. The SETD1A antibody was immobilized on Protein A/G agarose beads (Santa Cruz Biotechnology, Dallas, TX, USA) and incubated overnight with cell extracts at 4 °C. The beads were washed with lysis buffer and analyzed via western blotting.
Mammosphere formation assay
MCF-7 or TamR cells (200 cells/well) were suspended in mammosphere medium (20 ng/mL epidermal growth factor, 10 ng/mL basic fibroblast growth factor, 5 μg/mL insulin, 0.4% bovine serum albumin, B27 supplement, and 500 mL DMEM) in a 96-well ultra-low attachment plate (Corning Inc., Corning, NY, USA). The cells were placed in an incubator at 37 °C and 5% CO2 for seven days. Mammosphere number was counted under a microscope using 4× and 10× magnification. The percentage of mammospheres formed was calculated by dividing the number of mammospheres formed by the number of single cells plated.
Data collection and analysis of the transcriptome datasets
The ROC Plotter platform (http://rocplt.org) [42] was used to evaluate the prognostic significance of SETD1A levels in tamoxifen-treated patients with BC. The data used to compare SETD1A and SOX2 mRNA levels in tamoxifen-treated patients with BC were obtained from microarray datasets (GSE9893). The R2 Genomics Analysis and Visualization platform (http://r2.amc.nl) was used to determine the correlation between SETD1A and SOX2 expression levels in BC datasets (GSE42568, GSE124648).
Statistical analysis
Statistical analyses of the data from cell viability, migration, qPCR, protein correlation analysis, and tumor growth assays in a mouse xenograft model were conducted using GraphPad Prism software v8.0.2 (La Jolla, CA, USA). Statistical significance was determined using an unpaired Student's t-test. Survival analysis was performed using BreastMark [43].
Results
SETD1A is overexpressed in tamoxifen-resistant BC cells
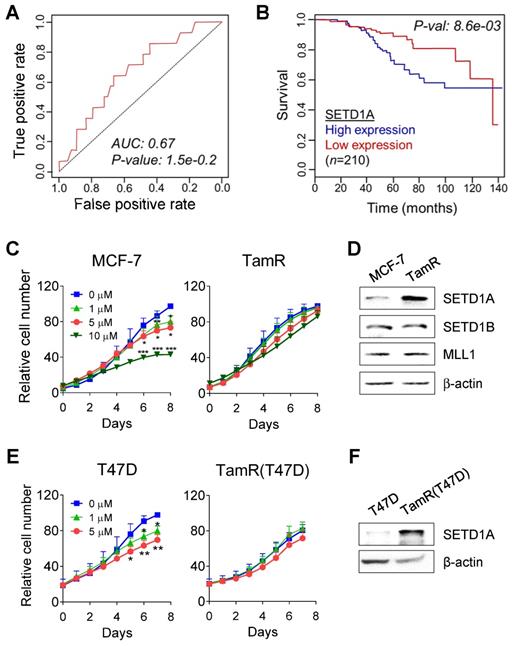
Our previous study confirmed that SETD1A mediates the survival of TamR cells [39]. Therefore, we performed an online receiver-operating characteristic (ROC) analysis [42] to detect whether the expression of SETD1A affected the prognosis of patients with BC treated with tamoxifen. During the first five years after tamoxifen therapy in ER-positive, HER2-negative, and nodal-positive luminal B patients with BC, high expression of SETD1A was associated with a high rate of relapse (AUC 0.67, P = 1.5E-02; Figure 1A). When the analysis was performed using a cohort of ER-positive and tamoxifen-treated patients with BC (n = 210) [42], a significant correlation was observed between the reduced overall survival and high SETD1A mRNA expression levels (P = 0.0086; Figure 1B). Next, we determined the expression levels of SETD1A in TamR cells (Figure 1C). SETD1A protein levels were significantly higher in TamR cells than in the parent cells (MCF-7). In contrast, the expression levels of SETD1B and MLL1, another family of H3K4 methylation enzymes, were not significantly affected by tamoxifen resistance (Figure 1D). SETD1A protein levels were higher in tamoxifen-resistant T47D (TamR T47D) cells than in the parent cells (T47D), another ER-positive BC cell line (Figure 1E-F). Collectively, our results suggest that SETD1A plays a potential role in tamoxifen resistance in BC.
SETD1A is overexpressed in tamoxifen-resistant breast cancer cells. (A) Results of SETD1A expression analysis that predict the poor outcome in patients with breast cancer treated with tamoxifen. Area under the curve (AUC), P-value, false-positive rate (FPR), and true-positive rate (TPR) were calculated for SETD1A gene signature. (B) Kaplan-Meier survival curves indicating the relationship between SETD1A mRNA expression levels and survival rate were generated using Breast Mark filtered cancer datasets (n = 210). (C, E) Parent breast cancer (MCF-7 and T47D) and their tamoxifen-resistant (TamR and TamR(T47D)) cells were treated with vehicle or tamoxifen. Cell proliferation was assessed for eight days after treatment using a live cell imaging system. Data are expressed as the mean ± S.D. (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001. (D, F) SETD1A expression levels in breast cancer and TamR cells were measured via western immunoblotting.
Role of SETD1A in tamoxifen resistance in BC cells
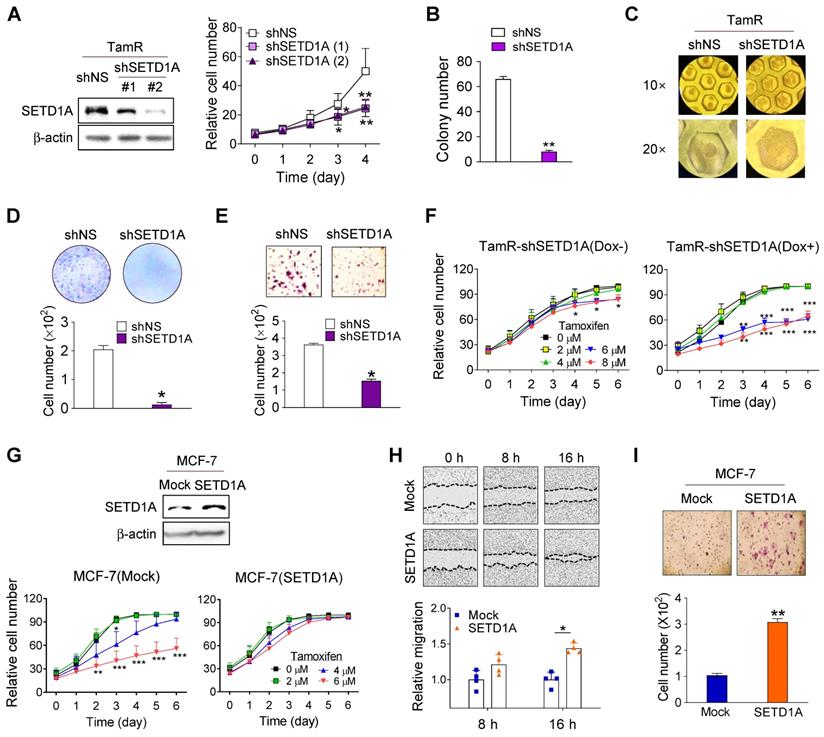
Based on the finding that SETD1A protein levels were increased in TamR cells, we sought to investigate the specific function of SETD1A in TamR cells. Similar to our previously published results [39], short hairpin RNA (shRNA) knockdown of SETD1A resulted in the significant downregulation of TamR cell proliferation (Figure 2A). In addition, SETD1A-depleted TamR cells displayed significantly lower colony and spheroid formation than the controls in both soft agar (Figure 2B) and 3D cultures that more accurately reflected the in vivo environment (Figure 2C). Moreover, SETD1A depletion markedly inhibited the migration and invasion of TamR cells (Figure 2D-E). To explore the biological effects of SETD1A on tamoxifen resistance, we investigated the effects of SETD1A knockdown in TamR cells or SETD1A overexpression in MCF-7 cells on sensitivity to tamoxifen. Compared with the control group, SETD1A-depleted TamR cells exhibited restored tamoxifen sensitivity (Figure 2F), whereas SETD1A overexpression resulted in a decreased sensitivity of MCF-7 cells to tamoxifen (Figure 2G). In addition, SETD1A overexpression in MCF-7 cells enhanced their migration and invasion abilities (Figure 2H-I). These data indicate that SETD1A plays an important role in the development of TamR BC cells.
Role of SETD1A in tamoxifen resistance in breast cancer cells. (A) Cell proliferation in short hairpin RNA (shRNA)-silenced tamoxifen-resistant MCF-7 cells (TamR) was analyzed using a live cell imaging system. Each value represents the mean ± S.D. (n = 6). (B) Soft agar assay of TamR cells transfected with non-specific shRNA (shNS) or shRNA targeting mRNA of SETD1A (shSETD1A). Cells were plated on soft agar and cultured for 30 days. (C) The 3D-culture of TamR cells transfected with shNS or shSETD1A. Microwell culture dish was used for shRNA-silenced TamR spheroid culture for 24 h. (D, E) Transwell migration (D) and invasion (E) assays of SETD1A-depleted TamR cells. Data are expressed as the mean ± S.D. (n = 3). (F) Effect of SETD1A depletion in TamR cells on tamoxifen sensitivity. Control TamR cells (Dox-, left panel) and Dox-induced SETD1A knockdown TamR cells (right panel) were grown in the presence of tamoxifen, and cell proliferation was analyzed using a live cell imaging system. (G) Effect of SETD1A overexpression in MCF-7 cells on tamoxifen sensitivity. MCF-7 cells stably overexpressing SETD1A or control (Mock) were grown in the presence of tamoxifen, and cell proliferation was analyzed using a live cell imaging system. Each value represents the mean ± S.D. (n = 9). (H) Cell migration assay of SETD1A-overexpressing MCF-7 cells. Motility of MCF-7 cells stably overexpressing SETD1A or control (Mock) cells was examined after 8 and 16 h. Representative cell migration images are shown (top panel), and cell migration was quantified by measuring the distance between the opposing cell boundaries (bottom panel). Data are expressed as the mean ± S.D. (n = 3). (I) Invasion assay of SETD1A-overexpressing MCF-7 cells. Data are presented as the mean ± S.D. (n = 4). *P < 0.05; ** P < 0.01; *** P < 0.001.
SETD1A regulates the transcription of SOX2 target genes in tamoxifen-resistant cells
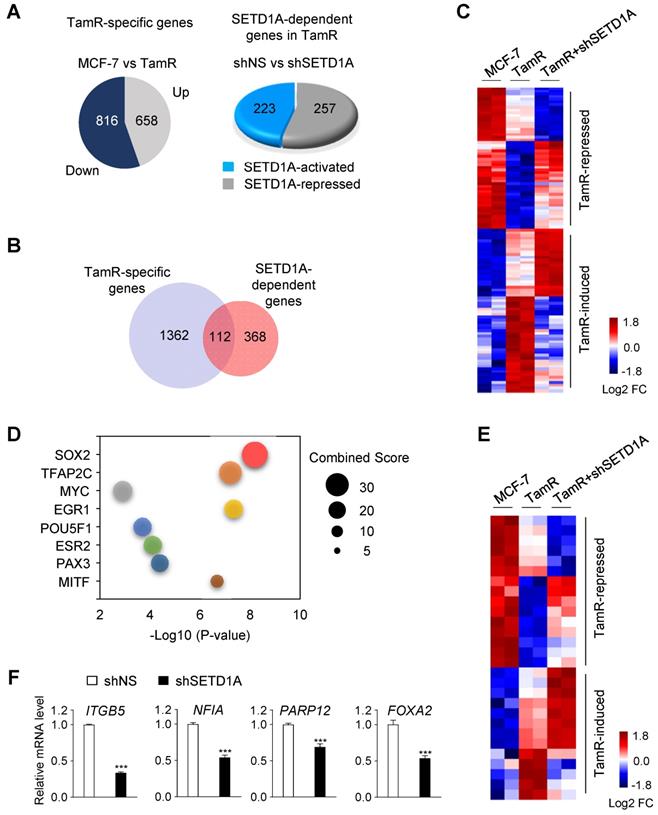
To investigate the mechanism of action of SETD1A in the development of tamoxifen resistance, a transcriptome analysis was conducted in MCF-7 and TamR cells. First, compared with MCF-7 cells, 1474 TamR-specific genes that were differentially expressed in TamR cells were identified (Figure 3A). Among these, 112 genes that were specifically affected by SETD1A knockdown were identified (Figure 3B-C). Next, we investigated the regulatory factors of these 112 genes using Expression2Kinase(X2K) [40]. SOX2 was predicted to be a potential transcriptional regulator of these genes (Figure 3D). Indeed, the expression levels of many known SOX2 target genes were altered in TamR cells, and most of these genes were affected by the depletion of SETD1A (Figure 3E). The effect of SETD1A on the expression levels of SOX2 target genes in TamR cells was validated via RT-qPCR (Figure 3F). Notably, SOX2 is a well-known prognostic biomarker for tamoxifen resistance [26]. Gene Ontology biological process analysis showed that the regulation of TamR-specific genes by SETD1A was enriched in tumor-associated pathways, such as apoptotic process, mitogen-activated protein kinase cascade, and cell migration (Figure S1). These pathway analyses support the hypothesis that SETD1A regulates the expression levels of SOX2 target genes in TamR BC cells.
Genome-wide analysis of SETD1A-dependent genes in MCF-7 and TamR cells. (A) TamR-specific genes were identified based on RNA-sequencing (RNA-seq) results of MCF-7 and TamR cells (|FC| ≥ 1.5, P < 0.05). SETD1A-dependent genes in TamR cells were identified by performing RNA-seq analysis after SETD1A depletion by shRNA in TamR cells. (B) Venn diagram summarizing the RNA-seq results for TamR-specific gene set and SETD1A-dependent genes in TamR cells. (C) Heatmap generated via RNA-seq analysis of MCF-7 and TamR cells expressing either shNS or shSETD1A showing differential expression of SETD1A-dependent genes among TamR-specific genes. (D) Bubble chart of top 8 transcription factors predicted using Expression2Kinases analysis. Y-axis represents the transcription factor, X-axis represents the P-value, and the size of bubble represents the Combined Score. (E) Effect of SETD1A depletion on the expression levels of SOX2 target genes among TamR-specific genes. (F) RT-qPCR validation of the effect of SETD1A knockdown on SOX2 target genes in TamR cells. Data are presented as the mean ± S.D. (n = 4). *** P < 0.001.
Correlation between SETD1A and SOX2 expression levels in tamoxifen-resistant cells
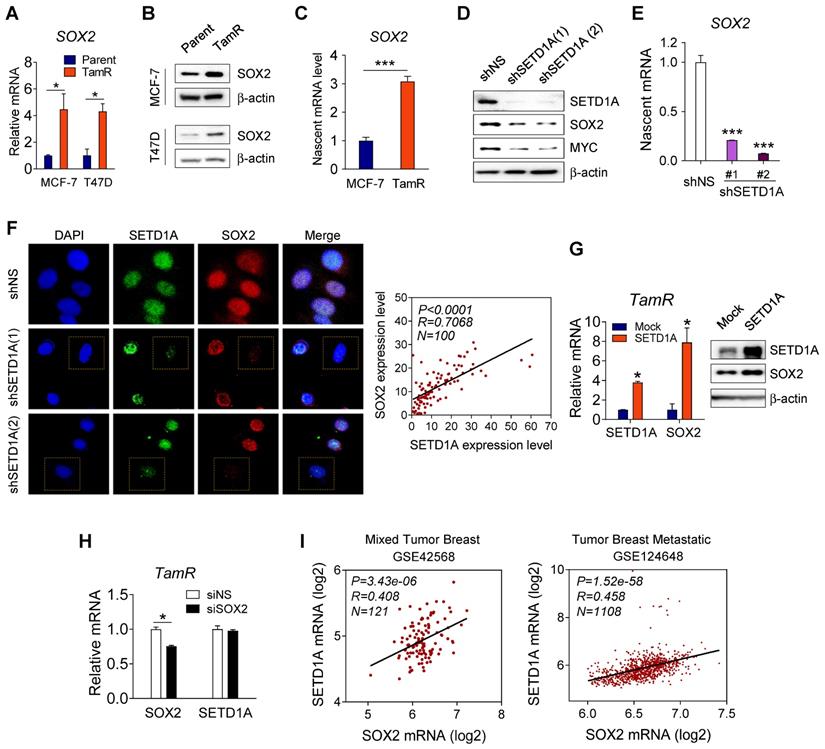
The finding that SOX2 regulates the expression of SOX2 target genes in TamR cells led us to investigate whether SETD1A regulated the expression of SOX2. First, we observed that compared to the parent cells, SOX2 mRNA and protein levels were significantly increased in TamR BC cells (TamR MCF-7 and TamR T47D) (Figure 4A-B). A similar result was observed for nascent mRNA level of SOX2 (Figure 4C). Next, we investigated whether SETD1A regulated the SOX2 gene transcription. Expression of a specific shRNA for SETD1A (shSETD1A) significantly reduced the SOX2 protein and nascent mRNA levels in TamR MCF-7 cells (Figure 4D-E). Immunofluorescence analysis of endogenous SETD1A and SOX2 showed that these proteins coexist in the nucleus. Specifically, the expression levels of SETD1A and SOX2 proteins were correlated in each cell; TamR cells exhibiting low levels of SETD1A exhibited low levels of SOX2 and vice versa (Figure 4F). In contrast, SETD1A overexpression in TamR and MCF-7 cells further increased SOX2 mRNA and protein levels (Figure 4G and Figure S2). However, downregulation of SOX2 did not affect the SETD1A mRNA levels (Figure 4H). Supporting the relevance of these findings, a positive correlation between SETD1A and SOX2 expression levels was observed in patients with BC (Figure 4I). Collectively, these results suggest that SETD1A acts as an upstream regulator of SOX2 transcription in TamR cells.
Correlation between SETD1A and SOX2 expression levels in tamoxifen-resistant cells. (A, B) Comparison of SETD1A expression levels in parent breast cancer cells and corresponding tamoxifen-resistant cells. Expression levels of SOX2 were measured via RT-qPCR and western immunoblotting in MCF-7 and T47D cells and tamoxifen-resistant cells (TamR MCF-7 and TamR T47D cells). Data are expressed as the mean ± S.D. (n = 3). (C) Analysis of nascent mRNA in MCF-7 and TamR cells. Newly synthesized RNA was labeled, and nascent SOX2 mRNA was analyzed via RT-qPCR. Levels of all nascent mRNAs were normalized to that of GAPDH. Data are expressed as the mean ± S.D. (n = 4). (D) Effect of SETD1A depletion using shRNA on SOX2 protein level in TamR cells. Extracts from the cells were analyzed for SETD1A, SOX2, MYC, and β-actin levels via western immunoblotting. (E) Analysis of nascent SOX2 mRNA levels in SETD1A-depleted TamR cells. Data are expressed as the mean ± S.D. (n = 3). (F) Immunofluorescence analysis of SETD1A (green) and SOX2 (red) in TamR cells transfected with shNS or shSETD1A. Plotted data show the intensity levels of SETD1A and SOX2 fluorescence (right panel). Pearson correlation analysis of SETD1A and SOX2 expression levels was conducted using GraphPad Software Prism v.8.0.2 (n = 100). (G) Effect of SETD1A overexpression on SOX2 levels in TamR cells measured via RT-qPCR and western immunoblotting. Data are expressed as the mean ± S.D. (n = 3). (H) Effect of SOX2 depletion on SETD1A expression levels in SOX2-depleted TamR cells measured via RT-qPCR. Data are presented as the mean ± S.D. (n = 4). (I) Correlation analysis of SETD1A and SOX2 expression levels in patients with breast cancer. Plotted data show log2 mRNA expression levels in GSE 42568 and GSE 124648 datasets. *P < 0.05; ** P < 0.01; *** P < 0.001.
SETD1A regulates the transcription of SOX2
To elucidate the basic mechanism by which SETD1A regulates SOX2 transcription, SETD1A recruitment to the SOX2 gene in MCF-7 and TamR cells was investigated. ChIP analysis showed that the promoter (-1kb) and super-enhancer regions of the SOX2 gene (-111 kb) were occupied by SETD1A, and SETD1A recruitment was higher in TamR cells than in MCF-7 cells (Figure 5A). ChIP assay using anti-H3K4me3 showed specific enrichment of H3K4me3 at the transcription start site (TSS) of SOX2 gene. Importantly, we observed that H3K4me3 was higher in the promoter region of the TSS of SOX2 gene in TamR cells than in MCF-7 cells (Figure 5B). These results are consistent with the finding of high recruitment of H3K4 methylation enzyme SETD1A to the SOX2 gene in TamR cells. The correlation between H3K4 methylation and chromatin accessibility is well documented. Consistently, chromatin accessibility of the promoter region of the SOX2 gene was increased in TamR cells (Figure 5C). However, SETD1A knockdown using shRNA reduced H3K4me3 in the promoter region (Figure 5D) and decreased chromatin accessibility of the promoter region (Figure 5E) of the SOX2 gene, eventually leading to reduced recruitment of RNA polymerase II (Pol II; Figure 5F).
SETD1A regulates the transcription of SOX2 gene. (A, B) SETD1A recruitment and histone H3K4me3 level at the SOX2 gene in parent breast cancer and corresponding tamoxifen-resistant cells. Chromatin immunoprecipitation (ChIP) assay was performed in MCF-7 and TamR cells. Quantification of the indicated region of SOX2 gene precipitated by anti-SETD1A or anti-H3K4me3 antibodies was performed via qPCR. Data are expressed as the mean ± S.D. (n = 3). (C) Chromatin accessibility at the SOX2 locus assessed by formaldehyde-assisted isolation of regulatory elements (FAIRE)-qPCR analysis using chromatin samples from MCF-7 and TamR cells. Data are normalized to non-crosslinked genomic DNA for each primer pair. Data are presented as the mean ± S.D. (n = 3). (D) Role of SETD1A in H3K4me3 methylation at the SOX2 locus. SETD1A levels in TamR cells were depleted using shRNA. Data are expressed as the mean ± S.D. (n = 3). (E) Effect of SETD1A on chromatin accessibility at SOX2 gene promoter. FAIRE-qPCR analysis was performed at the promoter region (-1 kb) of SOX2 gene in SETD1A-depleted TamR cells. Data are expressed as the mean ± S.D. (n = 3). (F) Effect of SETD1A on recruitment of RNA polymerase II to the SOX2 gene promoter region in TamR cells. Quantification of the indicated region of SOX2 gene precipitated by anti-SOX2 antibody was performed via qPCR. Data are expressed as the mean ± S.D. (n = 3) (G) Recruitment of SOX2 to the SOX2 gene in TamR cells. (H, I) Effect of SETD1A depletion on the recruitment of SOX2 and NANOG to the SOX2 gene in TamR cells. Data are presented as the mean ± S.D. (n = 3). (J) Effect of SOX2 depletion on the recruitment of SETD1A to the SOX2 promoter or enhancer region in TamR cells. (K) Restoration of tamoxifen resistance by SOX2 overexpression in SETD1A-depleted TamR cells. Sensitivity to tamoxifen was measured after transfection of the control or SOX2-overexpressing plasmids in SETD1A-depleted TamR cells (tamoxifen vs SOX2 + tamoxifen). Increase in SOX2 protein levels by the SOX2 expression plasmid was measured by western blotting (left panel). Cell proliferation was analyzed using a live cell imaging system (right panel). Data are expressed as the mean ± S.D. (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001.
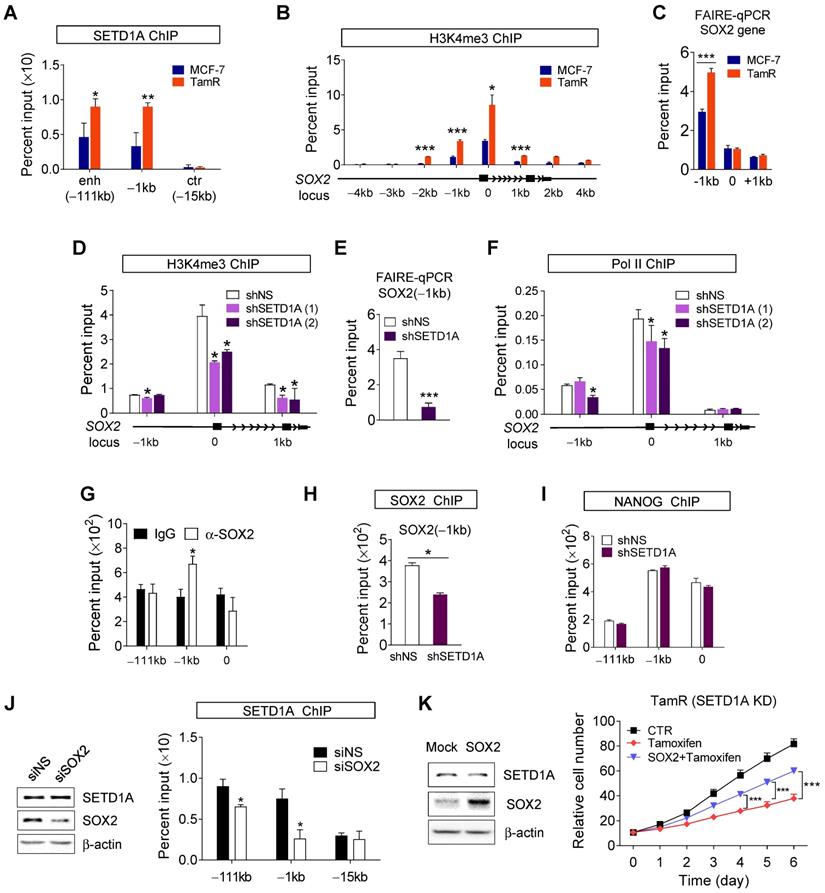
ChIP-seq analysis of immortalized multipotent otic progenitor cells revealed that SOX2 acts as a transcription factor that regulates the expression of SOX2 gene [44]. Similar results were observed in our study using TamR BC cells. SOX2 protein was recruited to the promoter region of SOX2 gene (Figure 5G). This recruitment was reduced by SETD1A knockdown, which may be due to a decrease in SOX2 protein levels after SETD1A knockdown (Figure 5H). In contrast, recruitment of NANOG, another transcription factor regulating SOX2 expression, to the SOX2 gene was not altered by SETD1A knockdown (Figure 5I). Depletion of SOX2 in TamR cells resulted in a reduction in SETD1A recruitment to the promoter and enhancer regions of the SOX2 gene without affecting SETD1A protein levels (Figure 5J). These results indicate that SETD1A binds to the promoter and enhancer regions of the SOX2 gene in SOX2-dependent manner to induce H3K4me3 at the promoter and TSS sites and to activate SOX2 transcription by maintaining the optimal chromatin structure required for transcription. To validate these results, we examined the role of SETD1A in the expression of two other SOX2 target genes, MYC and BMP7. Knockdown of SOX2 reduced mRNA of MYC in TamR cells (Figure S3A). SOX2 and SETD1A were recruited to the promoter and TSS regions of the MYC gene (Figure S3B-C). Trimethylation of histone H3K4 in MYC gene was SETD1A-dependent, and SETD1A regulated chromatin accessibility of MYC gene (Figure S3D-E). SETD1A protein was recruited to the MYC gene in a SOX2- dependent manner, and SETD1A knockdown reduced the nascent mRNA and protein levels of MYC in TamR cells (Figure S3F-G and Figure 4D). Additionally, SETD1A was recruited to the BMP7 gene, regulated H3K4 trimethylation and chromatin remodeling, eventually affecting the recruitment of Pol II to the BMP7 gene (Figure S4). These results indicate that SETD1A binds in SOX2-dependent manner to the promoter and enhancer regions of SOX2 target genes, including the SOX2 gene itself, to induce H3K4me3 at the promoter and TSS sites, and to activate SOX2 transcription by maintaining the optimal chromatin structure required for transcription. Consistently, overexpression of SOX2 in SETD1A knockdown TamR cells restored tamoxifen resistance (Figure 5K), indicating that SETD1A promoted tamoxifen resistance via SOX2 signaling.
SETD1A interacts with SOX2 via the Win motif
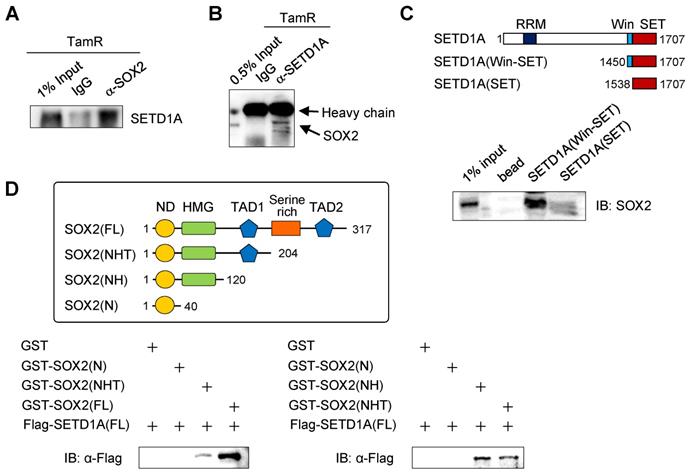
Our previous results suggest that SETD1A might associate with SOX2 at SOX2 target gene sites, including the SOX2 gene itself. In addition, the binding of SETD1A to the SOX2 target gene site is SOX2-dependent. Therefore, we investigated the direct interaction between SETD1A and SOX2 (Figure 6 and Figure S5). First, we observed that endogenous SETD1A was associated with SOX2 in TamR cells using a co-immunoprecipitation assay (Figure 6A-B). Next, we investigated whether SOX2 and SETD1A bind directly and identified their binding sites. In vitro binding assay revealed that the Win motif of SETD1A directly binds to SOX2 (Figure 6C). Previous studies have revealed direct binding of SOX2 and Ash2L subunits of the SET1/MLL complex [45], and it has been reported that the Win motif of SETD1A provides a binding site for various proteins, including WDR5 [46]. In this study, the Win motif played an important role in the interaction between SETD1A and SOX2 (Figure 6C). In addition, in vitro protein-binding experiments using various truncated forms of SOX2 have revealed that the HMG domain (a.a. 41-120) of SOX2 directly binds to SETD1A. The presence of the C-terminal region of SOX2 (serine-rich domain and TAD2, a.a. 205-317) further increased binding to SETD1A (Figure 6D).
Binding of SETD1A to SOX2 via the Win motif. (A, B) The interaction between SOX2 and SETD1A in TamR cells was determined by co-immunoprecipitation using anti-SOX2 or anti-SETD1A antibody followed by western immunoblotting of SETD1A or SOX2. (C) Direct binding assay using recombinant SET domain of SETD1A. Two types of His-tagged SET domains of SETD1A protein with (Win-SET) or without (SET) the Win motif were expressed in E. coli. After incubation of these bead-bound SET domains and TamR cell lysates immobilized in Ni-NTA beads, SOX2 protein bound to SETD1A was identified as an anti-SOX2 antibody. (D) Mapping study of SOX2 to identify SETD1A binding region. GST-tagged truncation mutants of SOX2 were expressed in E. coli and incubated with FLAG-tagged full-length SETD1A expressed using in vitro transcription and translation system. SETD1A bound to the SOX2 fragments was measured using the anti-FLAG antibody.
Role of SETD1A in tamoxifen resistance of cancer stem cells
SOX2, an embryonic stem cell marker, along with NANOG and OCT-4 is highly expressed in BC cells [47]. Therefore, we investigated mammosphere formation during the development of tamoxifen resistance. First, TamR cells formed significantly more mammospheres than control cells (Figure 7A), suggesting an increased self-renewal capacity. Second, among the stem cell factors reported to be involved in maintaining the self-renewal capacity of epithelial stem cells (ESCs), a significant difference between TamR and MCF-7 cells was observed only with respect to SOX2 expression (Figure 7B). However, in CSC isolated from TamR cells (TamR-CSC), the expression levels of all four stem cell factors, SOX2, OCT-4, NANOG, and KLF4, were significantly increased. However, SOX2 expression was most significantly increased (Figure 7C), consistent with the results of previous studies revealing the involvement of SOX2 in tamoxifen resistance [26]. This suggests that the increased expression of SOX2 by SETD1A plays an important role in the development of tamoxifen-induced resistance and stem/progenitor cell population formation in BC cells.
The role of SETD1A in cancer stem cells in TamR. (A) Mammosphere (MS) formation assay of MCF-7 and TamR cells. The efficiency of mammosphere formation was calculated by dividing the number of mammospheres formed by the number of cells plated. Data are expressed as the mean ± S.D. (n = 3). (B) Expression levels of SETD1A and stem cell factors, including SOX2, MYC, NANOG, OCT-4, and KLF4 in MCF-7 and TamR cells. Data are expressed as the mean ± S.D. (n = 3). (C) Increased expression levels of stem cell factors in cancer stem cells isolated from TamR cells (TamR-CSC) compared to that in TamR cells. Data are expressed as the mean ± S.D. (n = 3). (D) Venn diagram summarizing the RNA-seq results for cancer stem cell-specific genes (versus TamR) and SETD1A-dependent genes in cancer stem cells (|FC| ≥ 1.5, P < 0.05). (E) Bubble chart of top 5 transcription factors predicted using SETD1A-dependent genes in cancer stem cells Expression2Kinases suite analysis. Y-axis represents the transcription factor, X-axis represents the P-value, and the size of bubble represents the Combined Score. (F) Gene Ontology (GO) biological process analysis of SETD1A-dependent genes among cancer stem cell-specific genes. Statistically significant pathways (P < 0.05) are listed. (G) Mammosphere formation assay of SETD1A-depleted TamR cells. Data are expressed as the mean ± S.D. (n = 3). (H) Effect of SOX2 overexpression on tamoxifen-resistance in SETD1A-depleted TamR cells. The formation of mammosphere was measured after transfection of the control or SOX2-overexpressing plasmids in SETD1A-depleted TamR cells (mock vs SOX2). (I) SETD1A regulates the in vivo proliferation of TamR cells in the mouse xenograft model. TamR cells (shNS or shSETD1A) were injected subcutaneously into nude mice. Tumor volumes are shown as the mean ± S.D. (n = 10). ***P < 0.001. (J) The mRNA levels of SETD1A and SOX2 in ERα-positive and tamoxifen-treated patients with breast cancer (GSE9893). The 25th-75th percentiles are indicated by the closed box. (K) Representative image of SETD1A and SOX2 protein level in patients with breast cancer. Invasive: invasive ductal carcinoma; Normal: adjacent normal tissue. (L) Comparison of SETD1A and SOX2 protein level in normal breast tissue and invasive ductal carcinoma tissues. SETD1A and SOX2 protein expression levels were determined via immunofluorescence staining in human breast cancer tissue microarrays containing the normal breast tissues (n = 12) and invasive ductal carcinoma tissues (n = 47). SETD1A and SOX2 protein levels in individual tissue samples were presented as average integrated optical density (IOD). (M) Correlation between SOX2 and SETD1A protein levels in breast cancer tissue. Plotted data show the protein levels of SETD1A and SOX2 in normal breast tissues (n = 12) and invasive ductal carcinoma tissues (n = 47). Pearson correlation analysis of SETD1A and SOX2 expression levels was conducted using the GraphPad Software Prism V.8.0.2. (N) Kaplan-Meier overall survival curves show the prognostic ability of SETD1A and SOX2 signatures. The high-expressing group was defined as having both SETD1A and SOX2 levels higher than the median of all patients that participated in the study, while the remaining patients were in the low-expressing group.
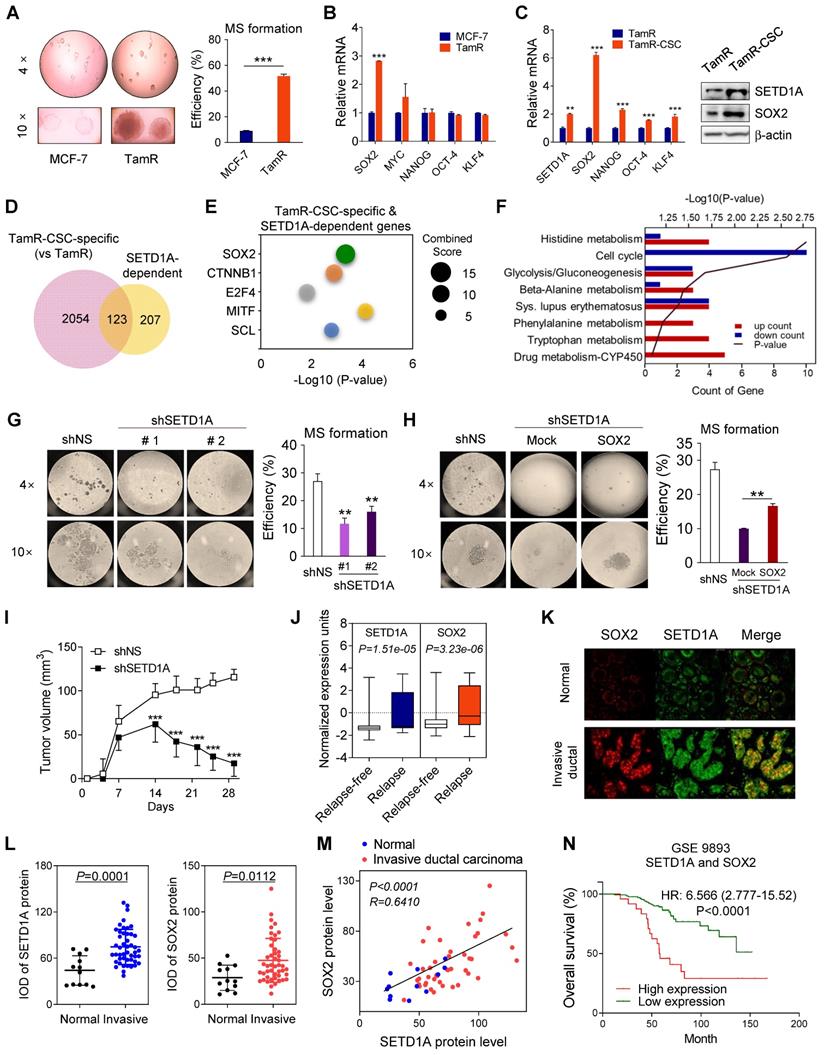
Our results show that SETD1A regulates the expression levels of SOX2 in TamR cells, suggesting that SETD1A may be involved in the self-renewal of BC stem cells (BCSCs). First, to investigate the genome-wide effects of SETD1A in cancer stem cell-specific gene expression, RNA-seq analysis was conducted in TamR cells and TamR-CSCs. Compared with TamR cells, 2177 genes were differentially expressed in TamR-CSCs; among them, 123 genes regulated by SETD1A were identified (Figure 7D and Figure S6). Next, by predicting the potential upstream regulators of these 123 genes, we confirmed that SOX2 was the most significant (P = 4.61E-04) protein among the upstream transcription factors of TamR-CSC-specific and SETD1A-dependent genes (Figure 7E). Indeed, the expression levels of many known SOX2 target genes were affected by the depletion of SETD1A in TamR-CSCs (Figure S6). Additionally, depletion of SETD1A resulted in the downregulation of all DNA replication and ubiquitination-related genes in TamR-CSCs, which participate in the maintenance of CSC stemness [48, 49] (Figure 7F). Next, we examined the role of SETD1A in the self-renewal of TamR-CSCs. Downregulation of SETD1A expression significantly reduced the mammosphere formation in TamR cells (Figure 7G). However, this effect was reversed by SOX2 overexpression (Figure 7H). Additionally, SETD1A-depleted TamR cells showed significantly decreased growth compared with the control group in a mouse xenograft model (Figure 7I). Finally, we determined the clinical relevance of SETD1A-SOX2 expression in the prognosis in TamR BC. In the transcriptome analysis of patients with ER-positive BC receiving tamoxifen, SETD1A and SOX2 transcriptional levels were significantly increased in tamoxifen-resistant patients than in the tamoxifen-sensitive patients (Figure 7J). Additionally, the protein levels of SETD1A and SOX2 were significantly higher in invasive ductal carcinoma tissues than in the normal breast tissues (Figure 7K-L). Importantly, a positive correlation between SETD1A and SOX2 protein levels was observed in patients with BC (Figure 7M), supporting our findings regarding the mRNA levels in TamR cells and patients with BC (Figure 4). Higher levels of both SOX2 and SETD1A were significantly associated with overall survival rates (Figure 7N). These results indicate that the BCSC-specific target gene regulation mechanism of the SETD1A-SOX2 axis plays an important role in the development of tamoxifen resistance and recurrence in patients via the formation and maintenance of BCSCs.
Discussion
Endocrine therapy can significantly reduce the mortality and recurrence rates by inhibiting ER signaling, thereby improving the survival rate in patients with ER-positive BC [50, 51]. However, more than 30% of ER-positive BC cases are intrinsically resistant to hormone therapy, and in a certain proportion of patients with BC, resistance to long-term endocrine therapy is inevitable (acquired resistance) [52, 53]. Currently, the treatment strategy for endocrine resistance involves the combination of hormone endocrine therapy with molecular targeting drugs such as those targeting mammalian target of rapamycin, cyclin-dependent kinase, or EGFR (www.clinicaltrials.gov) [54]. However, the identification of novel therapeutic targets or more specific biomarkers is necessary for the successful treatment of BC refractory to endocrine therapy. Recent studies have shown that epigenetic mechanisms regulate the growth of CSC-like cell subsets, resulting in the resistance to anticancer drugs [55]. HDACs required for maintaining CSCs, are found overexpressed in CSCs. The HDAC inhibitor, trichostatin A, was used to preferentially target CSCs [56]. Additionally, breast CSCs have a unique miRNA expression profile [57, 58]; for example, miRNA-200 targeting the CSC self-renewal factor BMI1 is downregulated in mammary CSCs [59].
Our study presents the clinical significance and mechanism of action of SETD1A, a histone H3K4-specific methyltransferase, in the development of tamoxifen resistance in BC. Data analysis of patients receiving tamoxifen monotherapy for more than five years revealed that SETD1A was strongly correlated with poor prognosis, and depletion of SETD1A in TamR BC cells led to alterations in the proliferation, migration, and invasion of these cells. We demonstrated that SETD1A plays an important role in TamR cell colonization and spheroid formation under both soft agar and 3D culture conditions. We also showed that SETD1A-depleted TamR cells exhibited significantly reduced tumor growth in a mouse xenograft model. Importantly, depletion of SETD1A in TamR cells restored their sensitivity to tamoxifen. Moreover, overexpression of SETD1A in MCF-7 cells led to the development of tamoxifen resistance, indicating that SETD1A is a key molecule that promotes tamoxifen resistance in BC cells. SETD1A directly binds to SOX2 and regulates the transcription of SOX2 target genes, including the SOX2 gene itself. As SOX2 is involved in maintaining stem cells, our results indicate that SETD1A plays an important role in the self-renewal of CSCs by regulating SOX2.
According to the cancer stem cell theory, a small number of CSCs may remain dormant following conventional cancer therapy, and during tumor remission, they regenerate to cause recurrent cancer, which has profound implications for cancer therapy. Similarly, the increase in SETD1A expression in tamoxifen-resistant cells seems to be the result of the dominant survival of resistant clones, CSCs, expressing high levels of SETD1A and SOX2 due to intra-tumor heterogeneity in primary BC. A previous report indicated that SOX2 was overexpressed in TamR cells, which conferred the stem cell-like and resistant phenotypes to BC cells [26]. SETD1A is involved in cell proliferation in various cancers, including lung cancer, colorectal cancer, hepatocellular carcinoma, and leukemia, and in the development of resistance to anticancer drugs [60-67].
Moreover, SETD1A is a key co-activator required for the transcription of OCT-4-mediated genes in ESCs [68]. Our study demonstrated the interaction of SETD1A with SOX2, and SETD1A overexpression during the development of tamoxifen resistance in ER-positive BC cells. SETD1A expression levels were higher in TamR-CSCs than in TamR cells grown in adherent cultures, and depletion of SETD1A significantly reduced the mammosphere formation capacity of TamR cells. SETD1A knockdown resulted in changes in the expression levels of several genes, including those associated with CSCs. These results suggest that SETD1A plays an important role in the self-renewal of CSCs in TamR BC. Further research is needed to determine whether the origin of TamR-CSCs in TamR cells is a product of de novo resistance or newly acquired mutations during tamoxifen treatment. If the mutation is newly acquired, BC cells may adapt to anticancer treatments via epigenetic modifications. A recent report that anticancer drug resistance can be caused not only by the survival of the fittest mutation, but also by the adaptation of tumor cells to the environment via non-genetic (epigenetic) heterogeneity further supports the above hypothesis [37, 69]. Collectively, our results suggest that SETD1A is a key regulator of SOX2 in BC cells and demonstrated the clinical significance of SETD1A in patients with BC that were treated with tamoxifen, indicating that SETD1A is a potential therapeutic target for the treatment of tamoxifen resistance.
Abbreviations
AUC: area under the curve; BC: breast cancer; ChIP: chromatin immunoprecipitation; CoIP: co-immunoprecipitation; CSC: cancer stem cell; ER: estrogen receptor; FAIRE: formaldehyde-assisted isolation of regulatory elements; GST: glutathione S-transferase; HMG: high mobility group; 4-HT: 4-hydroxytamoxifen; ITGB5: integrin subunit beta 5; KLF4: Kruppel-like factor 4; MLL1: mixed-lineage leukemia 1; NFIA: nuclear factor I A; OCT-4: octamer-binding transcription factor 4; Pol II: RNA polymerase II; SERM: selective estrogen receptor modulator; SETD1A: SET domain containing 1A; SETD1B: SET domain containing 1B; SOX2: SRY-box transcription factor 2; TamR: tamoxifen-resistant; TSS: transcription start site.
Supplementary Material
Supplementary methods, figures and tables.
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education to K.W.J. (2020R1A6A1A03043708 and 2021R1A2C1011132) and M.L.J. (2020R1I1A1A01051942).
Competing Interests
The authors have declared that no competing interest exists.
References
1. McDonnell DP. The Molecular Pharmacology of SERMs. Trends Endocrinol Metab. 1999;10:301-11
2. Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM. et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 2014;20:1757-67
3. Johnston SR, Saccani-Jotti G, Smith IE, Salter J, Newby J, Coppen M. et al. Changes in estrogen receptor, progesterone receptor, and pS2 expression in tamoxifen-resistant human breast cancer. Cancer Res. 1995;55:3331-8
4. Shi L, Dong B, Li Z, Lu Y, Ouyang T, Li J. et al. Expression of ER-{alpha}36, a novel variant of estrogen receptor {alpha}, and resistance to tamoxifen treatment in breast cancer. J Clin Oncol. 2009;27:3423-9
5. Roodi N, Bailey LR, Kao WY, Verrier CS, Yee CJ, Dupont WD. et al. Estrogen receptor gene analysis in estrogen receptor-positive and receptor-negative primary breast cancer. J Natl Cancer Inst. 1995;87:446-51
6. Fan W, Chang J, Fu P. Endocrine therapy resistance in breast cancer: current status, possible mechanisms and overcoming strategies. Future Med Chem. 2015;7:1511-9
7. Hanker AB, Sudhan DR, Arteaga CL. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell. 2020;37:496-513
8. McClelland RA, Barrow D, Madden TA, Dutkowski CM, Pamment J, Knowlden JM. et al. Enhanced epidermal growth factor receptor signaling in MCF7 breast cancer cells after long-term culture in the presence of the pure antiestrogen ICI 182,780 (Faslodex). Endocrinology. 2001;142:2776-88
9. Hutcheson IR, Knowlden JM, Madden TA, Barrow D, Gee JM, Wakeling AE. et al. Oestrogen receptor-mediated modulation of the EGFR/MAPK pathway in tamoxifen-resistant MCF-7 cells. Breast Cancer Res Treat. 2003;81:81-93
10. Miller TW, Pérez-Torres M, Narasanna A, Guix M, Stål O, Pérez-Tenorio G. et al. Loss of Phosphatase and Tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer. Cancer Res. 2009;69:4192-201
11. Prall OW, Rogan EM, Musgrove EA, Watts CK, Sutherland RL. c-Myc or cyclin D1 mimics estrogen effects on cyclin E-Cdk2 activation and cell cycle reentry. Mol Cell Biol. 1998;18:4499-508
12. Hui R, Finney GL, Carroll JS, Lee CS, Musgrove EA, Sutherland RL. Constitutive overexpression of cyclin D1 but not cyclin E confers acute resistance to antiestrogens in T-47D breast cancer cells. Cancer Res. 2002;62:6916-23
13. Cariou S, Donovan JC, Flanagan WM, Milic A, Bhattacharya N, Slingerland JM. Down-regulation of p21WAF1/CIP1 or p27Kip1 abrogates antiestrogen-mediated cell cycle arrest in human breast cancer cells. Proc Natl Acad Sci U S A. 2000;97:9042-6
14. Carroll JS, Prall OW, Musgrove EA, Sutherland RL. A pure estrogen antagonist inhibits cyclin E-Cdk2 activity in MCF-7 breast cancer cells and induces accumulation of p130-E2F4 complexes characteristic of quiescence. J Biol Chem. 2000;275:38221-9
15. Leis O, Eguiara A, Lopez-Arribillaga E, Alberdi MJ, Hernandez-Garcia S, Elorriaga K. et al. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene. 2012;31:1354-65
16. Gwak JM, Kim M, Kim HJ, Jang MH, Park SY. Expression of embryonal stem cell transcription factors in breast cancer: Oct4 as an indicator for poor clinical outcome and tamoxifen resistance. Oncotarget. 2017;8:36305-18
17. Yu F, Li J, Chen H, Fu J, Ray S, Huang S. et al. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. 2011;30:2161-72
18. Annovazzi L, Mellai M, Caldera V, Valente G, Schiffer D. SOX2 expression and amplification in gliomas and glioma cell lines. Cancer Genomics Proteomics. 2011;8:139-47
19. Basu-Roy U, Seo E, Ramanathapuram L, Rapp TB, Perry JA, Orkin SH. et al. Sox2 maintains self renewal of tumor-initiating cells in osteosarcomas. Oncogene. 2012;31:2270-82
20. Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS. et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet. 2012;44:1111-6
21. Zhang J, Chang DY, Mercado-Uribe I, Liu J. Sex-determining region Y-box 2 expression predicts poor prognosis in human ovarian carcinoma. Hum Pathol. 2012;43:1405-12
22. Neumann J, Bahr F, Horst D, Kriegl L, Engel J, Luque RM. et al. SOX2 expression correlates with lymph-node metastases and distant spread in right-sided colon cancer. BMC Cancer. 2011;11:518
23. Laga AC, Zhan Q, Weishaupt C, Ma J, Frank MH, Murphy GF. SOX2 and nestin expression in human melanoma: an immunohistochemical and experimental study. Exp Dermatol. 2011;20:339-45
24. Lengerke C, Fehm T, Kurth R, Neubauer H, Scheble V, Müller F. et al. Expression of the embryonic stem cell marker SOX2 in early-stage breast carcinoma. BMC Cancer. 2011;11:42
25. Chen Y, Shi L, Zhang L, Li R, Liang J, Yu W. et al. The molecular mechanism governing the oncogenic potential of SOX2 in breast cancer. J Biol Chem. 2008;283:17969-78
26. Piva M, Domenici G, Iriondo O, Rábano M, Simões BM, Comaills V. et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol Med. 2014;6:66-79
27. Yang L, Jin M, Jeong KW. Histone H3K4 Methyltransferases as Targets for Drug-Resistant Cancers. Biology. 2021;10:581
28. Zhang YW, Zheng Y, Wang JZ, Lu XX, Wang Z, Chen LB. et al. Integrated analysis of DNA methylation and mRNA expression profiling reveals candidate genes associated with cisplatin resistance in non-small cell lung cancer. Epigenetics. 2014;9:896-909
29. Plumb JA, Strathdee G, Sludden J, Kaye SB, Brown R. Reversal of drug resistance in human tumor xenografts by 2'-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer Res. 2000;60:6039-44
30. Chang X, Monitto CL, Demokan S, Kim MS, Chang SS, Zhong X. et al. Identification of hypermethylated genes associated with cisplatin resistance in human cancers. Cancer Res. 2010;70:2870-9
31. Strathdee G, MacKean MJ, Illand M, Brown R. A role for methylation of the hMLH1 promoter in loss of hMLH1 expression and drug resistance in ovarian cancer. Oncogene. 1999;18:2335-41
32. Gifford G, Paul J, Vasey PA, Kaye SB, Brown R. The acquisition of hMLH1 methylation in plasma DNA after chemotherapy predicts poor survival for ovarian cancer patients. Clin Cancer Res. 2004;10:4420-6
33. Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG. et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624-9
34. Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323-35
35. Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A. et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110:7922-7
36. Knoechel B, Roderick JE, Williamson KE, Zhu J, Lohr JG, Cotton MJ. et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet. 2014;46:364-70
37. Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S. et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69-80
38. Yang L, Jin M, Park SJ, Seo SY, Jeong KW. SETD1A Promotes Proliferation of Castration-Resistant Prostate Cancer Cells via FOXM1 Transcription. Cancers. 2020;12:1736
39. Jin ML, Kim YW, Jin HL, Kang H, Lee EK, Stallcup MR. et al. Aberrant expression of SETD1A promotes survival and migration of estrogen receptor alpha-positive breast cancer cells. Int J Cancer. 2018;143:2871-83
40. Clarke B, Daniel J, Kuleshov MV, Schilder BM, Torre D, Duffy ME. et al. eXpression2Kinases (X2K) Web: linking expression signatures to upstream cell signaling networks. Nucleic Acids Res. 2018;46:W171-W9
41. Jeong KW, Kim K, Situ AJ, Ulmer TS, An W, Stallcup MR. Recognition of enhancer element-specific histone methylation by TIP60 in transcriptional activation. Nat Struct Mol Biol. 2011;18:1358-65
42. Fekete JT, Gyorffy B. ROCplot.org: Validating predictive biomarkers of chemotherapy/hormonal therapy/anti-HER2 therapy using transcriptomic data of 3,104 breast cancer patients. Int J Cancer. 2019;145:3140-51
43. Madden SF, Clarke C, Gaule P, Aherne ST, O'Donovan N, Clynes M. et al. BreastMark: an integrated approach to mining publicly available transcriptomic datasets relating to breast cancer outcome. Breast Cancer Res. 2013;15:R52
44. Kwan KY, Shen J, Corey DP. C-MYC transcriptionally amplifies SOX2 target genes to regulate self-renewal in multipotent otic progenitor cells. Stem cell reports. 2015;4:47-60
45. Yang Z, Augustin J, Hu J, Jiang H. Physical Interactions and Functional Coordination between the Core Subunits of Set1/Mll Complexes and the Reprogramming Factors. PLoS One. 2015;10:e0145336
46. Dharmarajan V, Lee JH, Patel A, Skalnik DG, Cosgrove MS. Structural basis for WDR5 interaction (Win) motif recognition in human SET1 family histone methyltransferases. J Biol Chem. 2012;287:27275-89
47. Simoes BM, Piva M, Iriondo O, Comaills V, Lopez-Ruiz JA, Zabalza I. et al. Effects of estrogen on the proportion of stem cells in the breast. Breast Cancer Res Treat. 2011;129:23-35
48. Srivastava AK, Han C, Zhao R, Cui T, Dai Y, Mao C. et al. Enhanced expression of DNA polymerase eta contributes to cisplatin resistance of ovarian cancer stem cells. Proc Natl Acad Sci U S A. 2015;112:4411-6
49. Deng L, Meng T, Chen L, Wei W, Wang P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct Target Ther. 2020;5:11
50. Gradishar WJ, Anderson BO, Balassanian R, Blair SL, Burstein HJ, Cyr A. et al. NCCN Guidelines Insights Breast Cancer, Version 1.2016. J Natl Compr Canc Netw. 2015;13:1475-85
51. Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62:233-47
52. Badia E, Oliva J, Balaguer P, Cavaillès V. Tamoxifen resistance and epigenetic modifications in breast cancer cell lines. Curr Med Chem. 2007;14:3035-45
53. Clarke R, Liu MC, Bouker KB, Gu Z, Lee RY, Zhu Y. et al. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene. 2003;22:7316-39
54. Rasha F, Sharma M, Pruitt K. Mechanisms of endocrine therapy resistance in breast cancer. Mol Cell Endocrinol. 2021;532:111322
55. Abdel-Hafiz H. Epigenetic Mechanisms of Tamoxifen Resistance in Luminal Breast Cancer. Diseases. 2017;5:16
56. Witt AE, Lee C-W, Lee TI, Azzam DJ, Wang B, Caslini C. et al. Identification of a cancer stem cell-specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer. Oncogene. 2017;36:1707-20
57. Schwarzenbacher D, Balic M, Pichler M. The Role of MicroRNAs in Breast Cancer Stem Cells. Int J Mol Sci. 2013;14:14712-23
58. Gong C, Tan W, Chen K, You N, Zhu S, Liang G. et al. Prognostic Value of a BCSC-associated MicroRNA Signature in Hormone Receptor-Positive HER2-Negative Breast Cancer. EBioMedicine. 2016;11:199-209
59. Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D. et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009;138:592-603
60. Salz T, Li G, Kaye F, Zhou L, Qiu Y, Huang S. hSETD1A regulates Wnt target genes and controls tumor growth of colorectal cancer cells. Cancer Res. 2014;74:775-86
61. Wu J, Chai H, Shan H, Pan C, Xu X, Dong W. et al. Histone Methyltransferase SETD1A Induces Epithelial-Mesenchymal Transition to Promote Invasion and Metastasis Through Epigenetic Reprogramming of Snail in Gastric Cancer. Front Cell Dev Biol. 2021;9:657888
62. Wang R, Liu J, Li K, Yang G, Chen S, Wu J. et al. An SETD1A/Wnt/beta-catenin feedback loop promotes NSCLC development. J Exp Clin Cancer Res. 2021;40:318
63. Wu J, Chai H, Li F, Ren Q, Gu Y. SETD1A augments sorafenib primary resistance via activating YAP in hepatocellular carcinoma. Life Sci. 2020;260:118406
64. Wu J, Chai H, Xu X, Yu J, Gu Y. Histone methyltransferase SETD1A interacts with HIF1alpha to enhance glycolysis and promote cancer progression in gastric cancer. Mol Oncol. 2020;14:1397-409
65. Tajima K, Matsuda S, Yae T, Drapkin BJ, Morris R, Boukhali M. et al. SETD1A protects from senescence through regulation of the mitotic gene expression program. Nat Commun. 2019;10:2854
66. Du M, Gong P, Zhang Y, Liu Y, Liu X, Zhang F. et al. Histone methyltransferase SETD1A participates in lung cancer progression. Thorac Cancer. 2021;12:2247-57
67. Hoshii T, Cifani P, Feng Z, Huang CH, Koche R, Chen CW. et al. A Non-catalytic Function of SETD1A Regulates Cyclin K and the DNA Damage Response. Cell. 2018;172:1007-21 e17
68. Fang L, Zhang J, Zhang H, Yang X, Jin X, Zhang L. et al. H3K4 Methyltransferase Set1a Is A Key Oct4 Coactivator Essential for Generation of Oct4 Positive Inner Cell Mass. Stem Cells. 2016;34:565-80
69. Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014;54:716-27
Author contact
![]() Corresponding author: Kwang Won Jeong. Gachon Research Institute of Pharmaceutical Sciences, College of Pharmacy, Gachon University, 191 Hambakmoero, Yeonsu-gu, Incheon 21936, Republic of Korea. Tel.: +82 32 820 4925, Fax: +82 32 820 4829, E-mail: kwjeongac.kr
Corresponding author: Kwang Won Jeong. Gachon Research Institute of Pharmaceutical Sciences, College of Pharmacy, Gachon University, 191 Hambakmoero, Yeonsu-gu, Incheon 21936, Republic of Korea. Tel.: +82 32 820 4925, Fax: +82 32 820 4829, E-mail: kwjeongac.kr