Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2022; 12(11):5086-5102. doi:10.7150/thno.74989 This issue Cite
Research Paper
UBR5 promotes tumor immune evasion through enhancing IFN-γ-induced PDL1 transcription in triple negative breast cancer
Bingbing Wu1, Mei Song2*, Qun Dong3*, Gang Xiang1, Jing Li3, Xiaojing Ma2 ![]() , Fang Wei1
, Fang Wei1 ![]()
1. Sheng Yushou Center of Cell Biology and Immunology, Joint International Research Laboratory of Metabolic & Developmental Sciences, School of Life Science and Biotechnology, Shanghai Jiao Tong University, Shanghai, China.
2. Department of Microbiology and Immunology, Weill Cornell Medicine, New York, New York.
3. Department of Bioinformatics and Biostatistics, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai, China
*Contribute equally to this work
Received 2022-5-11; Accepted 2022-6-7; Published 2022-7-4
Abstract

Background: The up-regulation of PD-L1 is recognized as an adaption of cancer cells to evade immune surveillance and attack. However, the intrinsic mechanisms of the induction of PD-L1 by interferon-γ (IFN-γ) in tumor microenvironment remain incompletely characterized. Ubiquitin ligase E3 component N-recognition protein 5 (UBR5) has a critical role in tumorigenesis of triple negative breast cancer (TNBC) by triggering specific immune responses to the tumor. Dual targeting of UBR5 and PD-L1 exhibited superior therapeutic benefits in a preclinical TNBC model in short term.
Methods: The regulation of UBR5 to PD-L1 upon IFN-γ stimulation was evaluated through in UBR5 deficiency, reconstitution or overexpression cell line models by quantitative PCR, immunohistochemistry and RNA-seq. The effects of PD-L1 regulation by UBR5 and double blockade of both genes were evaluated in mouse TNBC model. Luciferase reporter assay, chromatin immunoprecipitation-qPCR and bioinformatics analysis were performed to explore the transcription factors involved in the regulation of UBR5 to PD-L1.
Results: E3 ubiquitin ligase UBR5 plays a key role in IFN-γ-induced PDL1 transcription in TNBC in an E3 ubiquitination activity-independent manner. RNA-seq-based transcriptomic analyses reveal that UBR5 globally affects the genes in the IFN-γ-induced signaling pathway. Through its poly adenylate binding (PABC) domain, UBR5 enhances the transactivation of PDL1 by upregulating protein kinase RNA-activated (PKR), and PKR's downstream factors including signal transducers and activators of transcription 1 (STAT1) and interferon regulatory factor 1 (IRF1). Restoration of PD-L1 expression in UBR5-deficient tumor cells recoups their malignancy in vivo, whereas CRISPR/Cas9-mediated simultaneous abrogation of UBR5 and PD-L1 expression yields synergistic therapeutic benefits than either blockade alone, with a strong impact on the tumor microenvironment.
Conclusions: This study identifies a novel regulator of PDL1 transcription, elucidates the underlying molecular mechanisms and provides a strong rationale for combination cancer immunotherapies targeting UBR5 and PD-L1.
Keywords: UBR5, Interferon-γ, PD-L1, PKR/STAT1/IRF1, triple negative breast cancer
Introduction
Tumor cells can adapt immune regulatory signaling pathways to evade immune recognition and elimination. One of the mechanisms utilized by tumor cells is the upregulation of PD-L1, which has been identified as an indicator of poor prognosis in various tumor types, including breast cancer [1, 2]. It has been reported that the mRNA and protein levels of PD-L1 are elevated in triple negative breast cancer (TNBC) cells [3]. In breast cancers, higher expression level of PD-L1 is associated with larger tumor size, higher tumor grade, and increased positive lymph node number [4, 5]. The PD-1/PD-L1 interaction can cause T cells to enter a state of anergy/exhaustion, which manifests impaired active proliferation, cytokine production and cytotoxicity [6, 7]. Thus, targeting the PD-1/PD-L1 axis is a beneficial approach in the treatment of different cancers. Blockade of PD-1 or PD-L1 with monoclonal antibodies can reverse many of these phenomena and restore T cell function [8]. Recently, atezolizumab and pembrolizumab showed durable antitumor activity as first-line therapies for patients with PD-L1-positive TNBC by blocking the interaction between PD-1 and its ligand PD-L1 [9, 10]. However, the patient response rate was still lower than expected. Thus, a better understanding of PD-L1 regulation may help predict patient responses and improve treatment.
The expression of PD-L1 can be exogenously induced by various cytokines including interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), interleukins (ILs) and epidermal growth factor (EGF) through the Janus kinase (JAK)/signal transducer and activator of transcription 1 (STAT1)/interferon regulatory factor1 (IRF1), nuclear factor‑κB (NF-κB), phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) or JAK/STAT3 signaling pathways [11, 12]. Among these factors, IFN-γ in the tumor microenvironment (TME) affects both tumor and immune cells in both immunoactivating and immunosuppressive ways [13] which explains why that early approaches targeting IFN-γ in the TME largely failed to provide any clinical benefit [14-16]. IFN-γ-induced adaptive immune resistance highlights the importance of utilizing IFN-γ-mediated immunotherapies by simultaneously blocking the expression or activity of PD-L1 and other factors [13]. However, the intrinsic mechanism controlling IFN-γ-induced PDL1 transcription remains incompletely characterized.
Human ubiquitin protein ligase E3 component N-recognin 5 (UBR5) was originally identified in a screen for progestin-regulated genes in breast cancer cells [17]. UBR5, a member of a rare “homologous to E6-AP C-terminus” (HECT)-domain E3 ubiquitin ligase family [17], is highly conserved in metazoans and is essential for early embryonic development in mice [18, 19]. UBR5 is frequently amplified and overexpressed in many cancer types, especially in human breast cancer and ovarian cancer [20, 21]. Our previous work revealed a critical role of UBR5 in the aggression of a murine TNBC model [21]. Overexpression of UBR5 was shown to correlate with poor overall survival in breast cancer [22]. Two key functional domains, the HECT and poly adenylate binding C terminal (PABC) domains of UBR5, are well characterized. The HECT domain associates mainly with E3 ligase activity, and the PABC domain is thought to be a protein-protein interaction motif [23, 24] and may regulate ubiquitin transfer catalyzed by the HECT domain [25]. Frameshift mutations tend to occur in the PABC/HECT domain in tumors [22]. UBR5 has been reported to directly interact with various proteins implicated in a wide variety of cellular processes, including cell cycle, transcriptional and translational machinery, and DNA damage response. Known targets of UBR5 ligase activity include β-catenin [26], pregnane X receptor [27], and E6-AP [28].
We previously reported that UBR5-deficiency can facilitate the processing and presentation of tumor antigens by antigen-presenting cells to host T cells, triggering specific immune responses to the tumor [21]. Dual targeting of UBR5 and PD-L1 exhibited superior therapeutic benefits in a preclinical TNBC model in short term [29]. Here, we report for the first time that UBR5 globally regulates IFN-γ-mediated pathways and stimulated genes, particularly PD-L1 expression and uncover the underlying molecular mechanism. We also showed here that simultaneous abrogation of Ubr5 and Pdl1 expression has synergistic therapeutic benefits in long term.
Materials and Methods
Cell lines
Murine TNBC cell lines 4T1, and its derivative 4T1/GFP, 4T1/Ubr5-/-, human TNBC cell lines BT549, MDA-MB-231, ER+ breast carcinoma cell lines MCF7 and human embryonic kidney cell lines HEK293T were stored in Ma lab at SJTU. 4T1 cells were cultured with RPMI 1640 (Invitrogen) containing 10% fetal bovine serum (BI) and 100 μg/mL penicillin/streptomycin (Invitrogen). BT549, MDA-MB-231, MCF7 and HEK293T cells were maintained in DMEM (Invitrogen) supplemented with 10% FBS (BI) and 100 μg/mL penicillin/streptomycin (Invitrogen). All cell lines were incubated under an atmosphere of 5% CO2 at 37℃.
Mice and mouse tumor model
Wild-type (BALB/c) female mice (6-8 weeks old) were purchased from the Charles River Laboratories (Pinghu, China) and maintained in a pathogen-free facility, supplied with sterile food and water.
For 4T1 tumor model, 1×106 cells were injected into the 4th mammary fat pads of BALB/c mice. Tumor growth was measured every 3 days with a caliper and tumor volume was calculated as volume=1/2×length×width2. For lung metastasis experiment, 5×105 4T1 cells were suspended in 100 μL PBS and then intravenously injected into BALB/c mice through the tail vein. Twelve days later, mice were sacrificed and the lungs were collected and made single cell suspension to perform tumor cell metastasis assay in vitro with 6-thioguanine as described previously [21].
Plasmids and vectors
For the constructs used in luciferase reporter assay, mouse Pdl1 and human PDL1 promoters were amplified by PCR from genomic DNA isolated from 4T1 or BT549 cells using the primers listed in Table S1, then cloned into the multicloning site (MCS) of the pGL3 Basic Vector (Promega Corporation). Specific deletions of the putative binding sites were carried out by the protocol described elsewhere [11], with primers listed in Table S1. For UBR5 mutant UBR5-ΔPABC, 78 amino acids of the PABC domain were deleted using overlapping PCR. The mEif2ak2 cDNA were amplified by PCR from 4T1 cells cDNA pool using primers listed in Table S1. The RNA interference (RNAi) from a lentiviral vector were generated with specific short hairpin RNA (shRNA) expression for each gene. All shRNA sequences were listed in Table S2.
CRISPR/Cas9-mediated knockout
4T1 and its derivative cells were subjected to CRISPR/Cas9-mediated knockout of Pdl1 by transient transfection of lentiCRISPR v2 based vector carrying the guide sequences specific for PD-L1. Three guide sequences used per gene were listed in Table S2. Positive single-cell clones were screened using 4 μg/mL puromycin. Disruption was confirmed finally by western blot and FACS analysis.
Cell transfections and infections
For the reconstitution of human UBR5 or mouse Pdl1, 4T1/Ubr5-/- cells were transfected with hUBR5 and mPdl1 plasmids by Lipofectamine 2000 reagent (Invitrogen). Stable hUBR5 or mPdl1 reconstited-4T1/Ubr5-/- cells were obtained by transfecting plasmids pCDH-hUBR5 or pCDH-mPdl1 and selecting by puromycin. Western blot or FACS were used to confirm the efficiency of reconstitution. 4T1/GFP cells were transiently transfected with siRNA targeting JAK3, STAT1, STAT2, IRF1, and IRF7 individually by Lipofectamine 2000 reagent. siNC (non-target control) was used as the negative control. All siRNA were designed and purchased from GenePharm. Lentiviruses were produced by cotransfection of 293T cells with PSPA, pMD2G, and pGIPZ-dtTomato-shUBR5 or shEIF2AK2 with polyethyleneimine (PEI). Virus supernatants were collected at 24 h and 48 h post-transfection. MDA-MB-231 and BT549 cells were infected with shUBR5 containing lentivirus, then were selected with puromycin. 4T1 cells were infected with shEIF2AK2 containing lentivirus and treated with puromycin. A scrambled shRNA was used as the negative control (shNC).
Quantitative RT-PCR (RT-qPCR)
Total RNA was extracted with Trizol (Invitrogen) and cDNA was synthesized using the cDNA Synthesis Kit (Vazyme). RT-qPCR was performed with Hieff qPCR SYBR Green Master Mix (YEASEN). The cDNA was quantified using SYBR mRNA expression assays by CFX96 Touch Real-Time PCR Detection System (Bio-Rad). The primers sequence of target genes were listed in Table S3.
Western blot
Cells were lysed in RIPA buffer (Beyotime Biotechnology) on ice for 10 min. Cell lysates were centrifuged at 12,000 rpm for 15 min at 4℃, and supernatant was collected. Protein concentration was quantified by Beyotime protein assay (Beyotime Biotechnology, 5000006). Proteins were resolved on a 10% SDS PAGE gel and transferred to the NC membrane, blocked with 5% milk and probed for monoclonal antibodies against UBR5 (Santa Cruz, sc-515494), PD-L1 (Proteintech) #66248-1-lg, EIF2AK2 (Beyotime) #AF2125, STAT1 p84/91 (C-136) (Santa Cruz, sc-464), STAT1 (D1K9Y) Rabbit (Cell Signal Technology) mAb #14994, Phospho-STAT1 (Tyr701) Rabbit (Beyotime) #AF5935, IRF1 (E-4) (Santa Cruz, sc-514544) or anti-GAPDH (Proteintech).
Flow cytometry analysis
For IFN-γ stimulation and PD-L1 staining in cell lines, TNBC cell lines were seeded into 6-well plates on Day 1, targeting 70-80% of confluence on the day of surface staining. On Day 2, cells were treated with 10 ng/mL IFN-γ (mouse IFN-γ: Sino Biological #50709-MNAH; human IFN-γ: PEPROTECH #300-02) for 24 h. On Day 3, cells were trypsinized and stained with allophycocyanin (APC) labelled anti-PD-L1 antibodies (Biolegend) on ice for 30 min, then washed with staining buffer for three times. For single cell staining, cells were dissociated from tumors or lymph nodes, then stained with antibodies accordingly. Antibodies against CD4 (RM4-5), CD3 (17A2), CD8 (53-6.7), Foxp3 (150D), CD25 (PC61), Granzyme B (GB11), and IFN-γ (XMG1.2) were purchased from Biolegend. Antibodies against CD45 (30-F11), CD11c (HL3), and MHCⅡ (2G9) were purchased from eBioscience. Flow cytometry data were analyzed by FlowJo software.
Quantification of the micrometastases in lungs
To quantify micrometastases, mice were sacrificed twelve days after 5×105 4T1 cells injected into mice through i.v. Lungs were excised, minced, and digested with tissue dissociation buffer [0.25% collagenase IV (384 unit/mg, Worthington), 0.2% Dipase II (Roche), and 0.01% DNase I (Sigma) in PBS] with periodic votexing for 1 h in 37℃ water bath. Single cell suspension was washed and strained with 70 mm strainer, then plated in 60 μmol/L 6-thioguanine selection (serve as duplicates). After 1 to 2 weeks of selection, tumor colonies were stained with crystal violet for 10 min, rinsed with ultrapure water and dried overnight prior to counting.
RNA-seq analysis
Total RNA was isolated for RNA-seq analysis. Second generation of RNA sequencing was performed by Genomic Core Facilities at Weill Cornell Medicine. High-quality reads were aligned to the mouse reference genome (vM25) using Histat2. We next used the featureCounts function of the subread software package to count the number of reads that mapped to a reference gene and performed differential expression with DEseq2. The enrichment analyses were based on differential expressed genes (padj <0.05, |foldchange|>2) using clusterProfiler R package.
Bioinformatic analysis of TCGA database
The KEGG pathway analysis was used by R clusterProfiler. Based on the data of all cancer expression profiles (FPKM) of TCGA, Pearson Correlation was used to calculate pairwise expression correlations between UBR5 and ISGs.
Transient luciferase reporter assays
Cells were seeded in 6-well plates for 18 h and then transfected using Lipofectamine 2000 reagent with 0.5 µg plasmid each (0.05 µg Renilla DNA was used for normalization). Cells were then treated with or without 10 ng/mL IFN-γ. 24 h later, relative luciferase units (RLUs) were measured using the Dual-Luciferase Report Assay System and GloMax 96 Microplate Luminometer (Promega) according to the manufacturer's instructions. RLUs from firefly luciferase signal were normalized by RLUs from Renilla signal.
ChIP-qPCR
Formaldehyde-cross-linked chromatin was prepared from 1×107 WT, Ubr5-/- and hUBR5-reconstituted Ubr5-/- 4T1 cells, and chromatin immunoprecipitation (ChIP) was performed using the EZ ChIP Kit (#17-371) from Millipore according to the manufacturer's instructions. Normal Mouse immunoglobulin G (IgG, sc-2025), anti-STAT1 p84/91 (C-136) (sc-464) and anti-IRF1 (E-4) (sc-514544) antibodies were purchased from Santa Cruz. Normal rabbit IgG (#2729) was purchased from Cell Signal Technology. Anti-H3K27ac (ab4729) antibody was purchased from Abcam. Anti-H3K4me1 antibody (#39498) was purchased from Active Motif. Real-time qPCR was performed in a CFX96 real-time PCR system (Bio-Rad) using Hieff qPCR SYBR Green Master Mix (YEASEN) and the primers for the GAPDH and PD-L1 promoters were listed in Table S4.
Statistical analysis
All values were presented as mean ± SEM, and the Student t test was used to determine statistical differences between groups. Values of P < 0.05 were considered statistically significant. These analyses were carried out using the GraphPad Prism 6 for statistical software.
Results
UBR5 is required for IFN-γ-induced PDL1 gene expression
Tumor cells can respond to elevated IFN-γ levels in the tumor microenvironment by upregulating the expression of PD-L1 to evade immune surveillance [11]. Interestingly, we observed that the IFN-γ-mediated induction of PD-L1 in previous generated [29] 4T1/Ubr5-/- cells (Figure 1A) was weaker (~50%) than that in control 4T1/GFP cells at both the mRNA and protein levels (Figure 1A-B). To determine whether the same phenomenon occurs in vivo, 4T1/GFP and 4T1/Ubr5-/- cells were subcutaneously injected into the 4th mammary fat pads of BALB/c mice individually, and tumors were dissected at Day 28 after injection. Immunohistochemistry staining data (Figure 1C) showed that the PD-L1 levels were lower in 4T1/Ubr5-/- than in 4T1/GFP tumors.
UBR5 is required for IFN-γ-induced PDL1 gene expression. (A) Quantitative PCR analysis of the relative Pdl1 mRNA levels and western blot analysis of the UBR5 protein levels in WT and Ubr5-/- 4T1 cells with or without 10 ng/mL IFN-γ treatment for 24 h. (B) The relative expression levels of PD-L1 on the surfaces of WT and Ubr5-/- 4T1 cells with or without 10 ng/mL IFN-γ stimulation was evaluated by flow cytometry. (C) WT and Ubr5-/- 4T1 cells (1×106) were subcutaneously injected into the mammary pads to generate tumors in WT (BALB/c) mice. Immunohistochemical staining was performed with anti-UBR5 or anti-PD-L1 antibody in WT and Ubr5-/- 4T1 tumor sections. (D-F) Human TNBC BT549 and MDA-MB-231 cell lines with stable UBR5 knockdown were generated by infection with a lentivirus containing a control or UBR5-targeted shRNA sequence followed by puromycin selection. The mRNA (D) and protein levels of UBR5 (E) and PD-L1 (E and F) were measured after cells were treated with 10 ng/mL IFN-γ for 24 h.(G-I) BT549, MDA-MB-231 and MCF7 cells were transfected with an empty vector or UBR5 plasmids. 48 hours later, the cells were treated with 10 ng/mL IFN-γ for 24 h. The mRNA and protein levels of UBR5 (G) and PD-L1 (H and I) were measured by qPCR, western blot and FACS. All experiments were repeated at least three times, and the data are presented as the mean ± SEM. ns, no significance, **P < 0.01, ***P < 0.001, ****P < 0.0001.
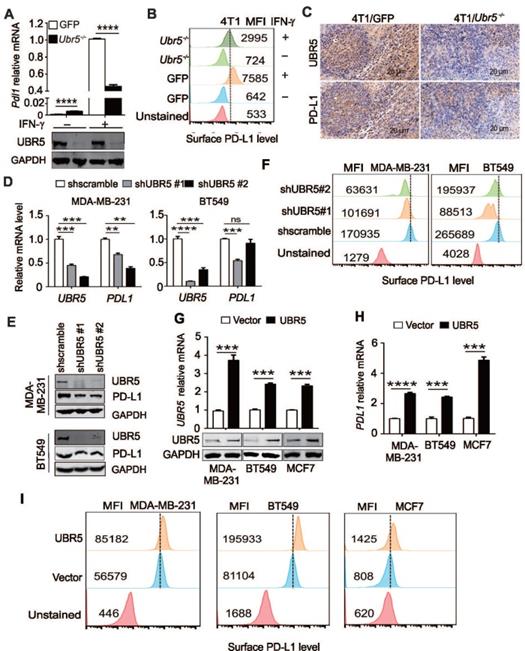
To confirm that the effect of UBR5 on IFN-γ-induced PD-L1 expression was not due to cell line specificity, we evaluated the expression level of PD-L1 in short hairpin RNA (shRNA)-mediated UBR5 knockdown MDA-MB-231 and BT549 cells. Upon IFN-γ stimulation, only 32-54% and 49-66% of PDL1 mRNA and protein were expressed in UBR5 knockdown MDA-MB-231 and BT549 cells compared with scramble control cells, which was consistent with the finding in 4T1 cells (Figure 1D-E). The PD-L1 surface protein levels in these cells were correlated with the mRNA levels (Figure 1F). Reciprocally, overexpression of UBR5 in BT549, MDA-MB-231 and MCF7 cell lines increased IFN-γ-induced PD-L1 expression at both the mRNA and surface protein levels (Figure 1G, H-I). Taken together, the findings indicate that there is a strong correlation between UBR5 and IFN-γ-stimulated PD-L1 expression.
Next, to explore whether the positive correlation between UBR5 and PD-L1 exists in other cancers beyond breast cancer, we evaluated the correlation between UBR5 and PD-L1 in TCGA database. TCGA-based analyses also highlight that the mRNA expression levels of UBR5 and PDL1 are highly correlated in many other cancer types, such as pancreatic adenocarcinoma (PAAD), thymoma (THYM), uveal melanoma (UVM), and prostate adenocarcinoma (PRAD) (Figure S1).
Restoration of PD-L1 expression in UBR5-deficient tumor regains malignancy
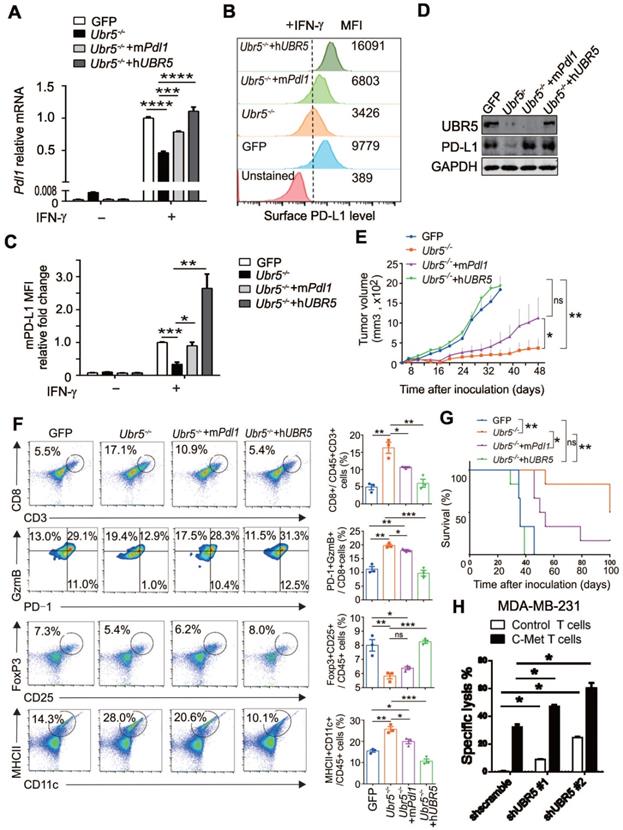
Given that UBR5 regulates IFN-γ-induced PD-L1 expression and that Ubr5-/- tumor growth is arrested from Day 10 onward [21], it is important to determine the role of PD-L1 in the impaired growth of Ubr5-/- tumors. We thus rescued PD-L1 in 4T1/Ubr5-/- cells with murine Pdl1 to levels similar to those in WT 4T1 cells (Figure 2A-C) without affecting the expression of UBR5 (Figure 2D). Upon inoculation in mice, the mPdl1-reconstituted 4T1/Ubr5-/- cells exhibited substantially enhanced growth compared with Ubr5-/- cells (Figure 2E). Analyses of tumor-infiltrating immune cells revealed that the number of CD8+ T cells and their cytolytic activity, manifested as increased granzyme B expression and decreased PD-1 expression, was strongly increased in Ubr5-/- tumors, which were subsequently reversed following the rescue of Pdl1 or Ubr5 expression (Figure S2, Figure 2F). The restoration also effected IFN-γ production of these CD8+ T cells (Figure S3), as well as the presence of CD25+ and FoxP3+ regulatory T cells (Figure S2, Figure 2F). Interestingly, the increased CD11c+ MHC Ⅱ+ mature dendritic cells (DCs) observed previously in mice bearing 4T1/Ubr5-/- tumor [21], was also lost when either mPdl1 or hUBR5 was reconstituted (Figure S2, Figure 2F). Accordingly, a significantly poorer prognosis was observed in the mPdl1-reconstituted 4T1/Ubr5-/- group than that of the Ubr5-/- group (Figure 2G). Given that the UBR5 deficiency decreased PD-L1 levels in MDA-MB-231 cells (Figure 1D-F), we tested the idea if this could enhance the function of effector T cells, using the c-Met specific human chimeric antigen receptor (CAR) T cells on the human TNBC cells. We found that, indeed, UBR5 deficiency in the target cells rendered them more susceptible to CAR T-mediated killing (Figure 2H, Figure S4).
Restoration of PD-L1 expression in UBR5-deficient tumors reinvigorates malignancy. (A-D) Ubr5-/- 4T1 cells were transfected with a plasmid encoding mPdl1, and then subjected to 4 μg/mL puromycin selection to generate stable mPdl1-reconstituted 4T1/Ubr5-/- cells. The mRNA and protein levels of PD-L1 and UBR5 protein levels in WT, Ubr5-/-, hUBR5- or mPdl1-reconstituted Ubr5-/- 4T1 cells were confirmed by RT-qPCR (A), FACS analysis (B) and western blot (D) after treatment with IFN-γ for 24 h, and the relative fold changes in PD-L1 based on the mean fluorescence intensity (MFI) are shown in (C). The data are presented as the mean ± SEM (error bar) from three replicates. *P< 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (E) A total of 1×106 WT, Ubr5-/-, hUBR5- or mPdl1-reconstituted Ubr5-/- 4T1 cells were subcutaneously injected into the mammary pads of WT (BALB/c) mice (n=6 mice per group). Tumor size was monitored every 3 days. (F) On Day 10 after tumor cells inoculation, flow cytometry analyzed the CD8+ T cells infiltration and GzmB+/PD-1-/CD8+ T cells in tumor tissue from mice bearing 4T1 WT, Ubr5-/-, hUBR5- or mPdl1-reconstituted Ubr5-/- tumor. Tumor-draining lymph nodes were analyzed by staining for CD25+, Foxp3+ Tregs and CD11c+, MHC Ⅱ+ DCs by flow cytometry. The results are presented as the mean ± SEM. ns, no significance, *P< 0.05, **P < 0.01, ***P < 0.001. n=3 mice per group. Mouse survival (G) were recorded daily. (H) T cells cytotoxicity difference toward MDA-MB-231 cells with different UBR5 expression levels. dt-Tomato Red stably expressed MDA-MB-231 cells (target cells) were mixed with CFSE labelled MCF7 cells (non-target cells) at a ratio of 1:1, and then cocultured for 18 h with either control or c-Met specific chimeric antigen receptor T cells at a ratio of 1:2 separately. Cells were harvested and analyzed by flow cytometry. The data are presented as the mean ± SEM (error bar) from three replicates. *P< 0.05.
Combined genetic targeting of Ubr5 and Pdl1 yields synergistic long term therapeutic effects
Next, we investigated whether combined abrogation of intrinsic UBR5 and PD-L1 expression has synergistic therapeutic benefits. GFP/Pdl1-/- and Ubr5-/-Pdl1-/- cell lines derived from the GFP and Ubr5-/- 4T1 cell lines were generated by CRISPR/Cas9 editing (Figure 3A-B). The cells were inoculated into the mammary pad of mice and the tumor growth was monitored over time (Figure 3C). The tumor growth of the Ubr5-/- group was dramatically reduced within 30 days, as we have reported previously [21], but gradually increased beyond 30 days. Tumor growth was considerably more arrested in the Ubr5-/-Pdl1-/- group compared with the other groups. Furthermore, the tumor did not recur in 83% of mice (4/6) in the Ubr5-/-Pdl1-/- group until Day 122. Markedly, no tumor recurrence was observed in these mice for more than 1 year. In contrast, no therapeutic benefit was observed in the GFP/Pdl1-/- group, which is consistent with the TNBC “cold tumor” theory [30].
Combined genetic targeting of Ubr5 and Pdl1 yields synergistic therapeutic effects. (A-B) 4T1/GFP/Pdl1-/- and 4T1/Ubr5-/-Pdl1-/- cells were generated by CRISPR/Cas9 editing. WT, Ubr5-/-, GFP/Pdl1-/- or Ubr5-/-Pdl1-/- 4T1 cells were treated with 10 ng/mL IFN-γ for 24 h, and then the UBR5 and PD-L1 protein levels were detected by western blot (A) and surface PD-L1 levels were measured by flow cytometry analysis (B). (C) A total of 1×106 WT, Ubr5-/-, GFP/Pdl1-/- or Ubr5-/-Pdl1-/- 4T1 cells were subcutaneously injected into the mammary pads to monitor tumor growth in WT (BALB/c) mice (n=6 mice per group). Tumor size was measured every 3 days. (D-G) WT, Ubr5-/-, GFP/Pdl1-/- or Ubr5-/-Pdl1-/- 4T1 cells (5×105) were injected via the tail vein into 7-week-old female BALB/c mice (n=5 mice per group). Twelve days later, the mice were sacrificed, and single-cell suspensions were obtained from lung tissue to perform clonogenic assays in order to evaluate lung metastasis. Images of 6-well plates in a representative experiment are shown in (D), and colonies were quantified (E). (F) On Day 10 after tumor cells inoculation, CD8+ T cells infiltration and GzmB+/PD-1-/CD8+ T cells in tumor tissue from mice bearing 4T1 WT, Ubr5-/-, hUBR5-or mPdl1-reconstituted Ubr5-/- tumor were analyzed by flow cytometry. Flow cytometry analyzed CD25+, Foxp3+ Tregs and CD11c+, MHC Ⅱ+ DCs in tumor-draining lymph nodes. n=3 mice per group. The survival curve is shown in (G). The results are presented as the mean ± SEM. ns, no significance, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
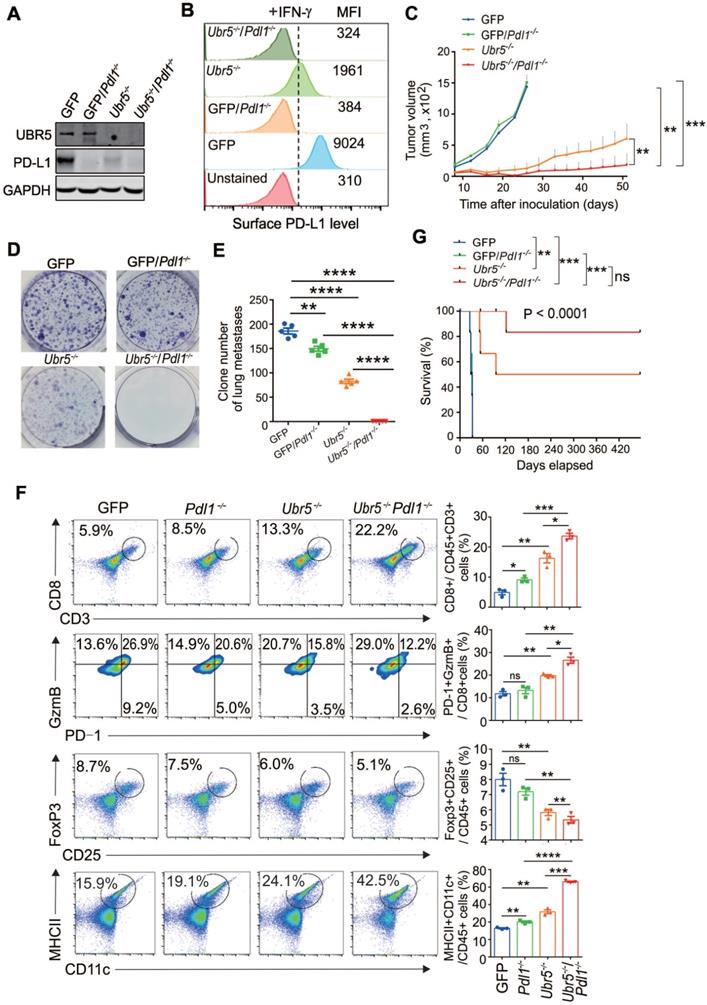
To evaluate the effect of dual targeting UBR5 and PD-L1 on the spontaneous lung metastasis of 4T1 tumors, 5×105 tumor cells were administrated i.v. to mice and lung metastasis was measured at Day 12 post injection using the 6-thioguanine clonogenicity assay (Figure 3D). We observed that the number of lung-colonizing Ubr5-/-Pdl1-/- tumor cells was significantly less than the number of colonizing Ubr5-/- tumor cells (Figure 3E). Notably, ~2 folds higher CD8+ T cell infiltration was observed in Ubr5-/-Pdl1-/- tumors than in Ubr5-/- tumors (Figure 3F). These infiltrating T cells were more active shown by an increased GzmB+/PD-1- effector T population and decreased regulatory T cells (Figure 3F) and more GzmB and IFN-γ production (Figure S5). Increased production of IFN-γ by CD4+ T cells were also observed in the tumors of mice bearing 4T1/Ubr5-/-Pdl1-/- tumor (Figure S5). Furthermore, CD11c+ MHC Ⅱ+ mature DCs in 4T1/Ubr5-/-Pdl1-/- tumor significantly increased (Figure 3F). Consequently, the Ubr5-/-Pdl1-/- bearing mice exhibited prolonged survival for up to 420 days (Figure 3G). These results demonstrate that simultaneous blockade of UBR5 and PD-L1 expression has synergistic therapeutic benefits with a strong impact on the infiltrating immune cells.
UBR5 globally regulates IFN-γ-mediated pathways and stimulated genes
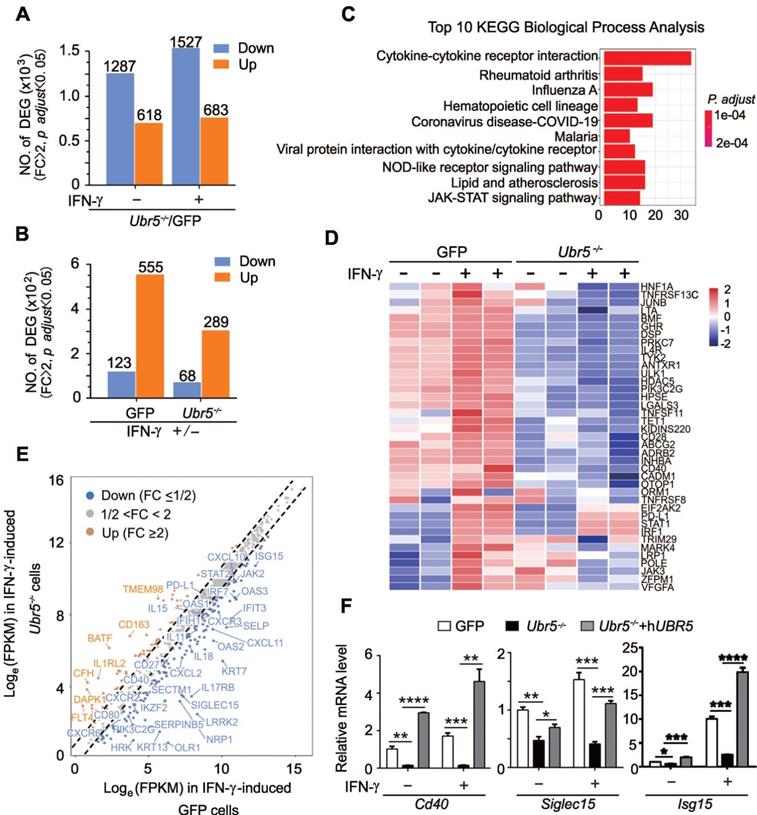
Since UBR5 could enhance IFN-γ-induced PD-L1 expression to promote tumor growth, it was of interest to explore whether there are other IFN-γ responsive factors affected by UBR5, we performed transcriptome profiling with RNA-sequencing (RNA-seq) in 4T1/GFP and 4T1/Ubr5-/- cells treated with or without IFN-γ. The RNA-seq data showed that there were more genes downregulated than upregulated in Ubr5-/- cells compared with GFP 4T1 cells no matter with or without IFN-γ stimulation (Figure 4A). A total of 555 genes were induced with IFN-γ in 4T1/GFP cells, while significantly fewer (289 genes) were induced in 4T1/Ubr5-/- cells (Figure 4B). In addition, more genes were downregulated in 4T1/GFP cells (123 genes in total) than in 4T1/Ubr5-/- cells (68 genes in total) (Figure 4B). RNA-seq data of IFN-γ-treated WT and Ubr5-/- 4T1 cells were further evaluated both by Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment and Gene Ontology (GO) functional annotation analysis. KEGG pathway enrichment analysis revealed that UBR5 was highly correlated with multiple signaling pathways including the JAK-STAT pathway and cytokine-receptor interactions, which suggests that UBR5 is involved in relatively broad regulation of the IFN-γ stimulation pathway (Figure 4C). The pathways of positive response to external stimulus and regulation of T cell activation/leukocyte proliferation ranked in the top 20 GO biological processes in the enrichment analysis (Figure S6). Interestingly, in selected interferon-stimulated gene (ISG) subsets, a set of genes respond to IFN-γ in GFP cells but completely not or only mildly responsive in Ubr5-/- cells (Figure 4D). Genes responding to IFN-γ differently in 4T1/Ubr5-/- cells, including the immune costimulatory and checkpoint genes Cd40 [31] and Siglec15 [32] significantly decreased in IFN-γ-induced 4T1/Ubr5-/- cells compared with IFN-γ-induced 4T1/GFP cells (Figure 4E), while expression of ISG resistance signature (ISG.RS) genes, such as ISG Isg15 [33], JAK2, OAS3, OAS1, IRF7, OAS2, IFIT3 and IFIH1 decreased (Figure 4E). These genes are predominantly expressed in cancer cells, albeit with variable expression [34]. Further analysis utilizing WT, Ubr5-/- and hUBR5-reconstituted Ubr5-/- 4T1 cells confirmed that the transcription of the Cd40, Siglec15 and Isg15 genes was affected by the expression of UBR5 (Figure 4F).
UBR5 globally regulates IFN-γ-mediated pathways and stimulated genes. (A) Numbers of differentially expressed genes (DEGs) in Ubr5-/- compared with GFP 4T1 cells with or without IFN-γ treatment. Fold change (FC) ≥ 2, p. adjust ˂ 0.05. (B) Numbers of DEGs in GFP 4T1 cells or Ubr5-/- 4T1 cells with IFN-γ treatment compared with that without IFN-γ treatment. Fold change (FC) ≥ 2, p. adjust ˂ 0.05. (C) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment of the top 10 biological process between IFN-γ-treated GFP and Ubr5-/- 4T1 cells. (D) Heatmap depicting the mRNA levels of selected ISGs in GFP and Ubr5-/- 4T1 cells with or without 10 ng/mL IFN-γ induction for 24 h. (E) Scatterplot diagram of genes responding to IFN-γ differently in Ubr5-/- cells compared to that in GFP cells. The grey dots represent unchanged expression (1/2<FC<2) in both cell lines. Upregulated ISGs with fold change (FC) values >2 in Ubr5-/- cells compared with GFP cells are shown with orange dots, while downregulated ISGs with fold change (FC) values <1/2 are shown with blue dots. (F) The mRNA levels of Cd40, Siglec15 and Isg15 were examined by qPCR analysis in WT, Ubr5-/- and hUBR5-reconstituted Ubr5-/- 4T1 cells treated with or without IFN-γ for 24 h. All experiments were repeated three times, and the data are presented as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
UBR5 is crucial for IFN-γ-mediated activation of STAT1 and IRF1 transcription
IFN-γ is generally considered the most prominent soluble inducer of PD-L1 and the JAK1/2-STAT1/3-IRF1 signaling axis has been shown to play a central role in the IFN-γ-mediated induction of PD-L1 [11, 35]. RNA-seq-based transcriptomic analyses suggested that the JAK-STAT signaling pathway (Figure 4C) played a role in the regulation of IFN-γ-stimulated PD-L1 by UBR5. The expression levels of JAK3, STAT2 and IRF7 were also significantly different between IFN-γ-treated GFP and Ubr5-/- 4T1 cells (Figure 4E). We then evaluated PD-L1-targeting transcription factors including JAK1, JAK2, JAK3, STAT1, STAT2, STAT3, IRF1, IRF7, and TYK2 (Figure S7), and found that the IFN-γ-stimulated mRNA and protein levels of IRF1 and STAT1 were decreased in UBR5-silenced 4T1 (Figure 5A-B), BT549 (Figure 5C-D) and MDA-MB-231 (Figure 5D) cells. Reciprocally, overexpression of UBR5 increased the mRNA (Figure 5E- F) and protein levels (Figure 5G) of STAT1 and IRF1 in different human breast cancer cell lines. The protein levels of pSTAT1 increased in UBR5 overexpression BT549 cells compared to the control cells (Figure S8). The phosphorylation level of STAT1 also markedly decreased in 4T1/Ubr5-/- cells (Figure 5B). Consistently, both the mRNA (Figure 5A) and protein (Figure 5B) levels of STAT1 and IRF1, as well as the levels of pSTAT1 were restored after hUBR5 was reconstituted in 4T1/Ubr5-/- cells. The mRNA levels of Jak1, Jak2, Stat3, Tyk2 altered in a similar pattern as those of Stat1 and Irf1 in GFP, Ubr5-/- and hUBR5-reconstituted Ubr5-/- 4T1 cells; however, Jak1 gene did not respond to IFN-γ treatment, and the mRNA levels of Jak2, Stat3 and Tyk2 increased less than 2 fold in IFN-γ-induced 4T1/GFP cells, which is inconsistent with Pdl1 mRNA level changes in 4T1 cells with or without IFN-γ stimulation (Figure S7). To further evaluate the roles of these genes in IFN-γ-induced PDL1 transcription in 4T1 cells, JAK3, STAT1, STAT2, IRF1, and IRF7 were silenced individually through siRNA (Figure S9). The expression of STAT1 and IRF1 transcripts, but not JAK3, STAT2 or IRF7 transcripts were required for IFN-γ-induced PDL1 transcription (Figure 5H, S9). These results suggest that upon IFN-γ-activation, UBR5 enhanced PDL1 transcription is mediated through STAT1 and IRF1.
UBR5 is crucial for IFN-γ-induced activation of STAT1 and IRF1 transcription. (A-B) The mRNA (A) and protein (B) levels of UBR5, STAT1, pSTAT1 and IRF1 were detected in WT, Ubr5-/- and hUBR5-reconstituted Ubr5-/- 4T1 cells with or without IFN-γ stimulation. (C-D) The mRNA (C) and protein levels (D) of STAT1 and IRF1were detected in IFN-γ-treated UBR5-knockdown BT549 (C and D) and MDA-MB-231 (D) stable cell lines. (E-G) BT549, MDA-MB-231 and MCF7 cells were transfected with either an empty vector or UBR5 plasmids. 24 hours later, the cells were treated with IFN-γ for 24 h. Then the mRNA and protein levels of STAT1 (E and G) and IRF1 (F and G) were measured by qPCR and western blot, respectively. (H) Surface PD-L1 levels were detected in IFN-γ-treated GFP, Ubr5-/- 4T1 cells and GFP 4T1 cells treated with siNC, siSTAT1 or siIRF1. GAPDH was used for normalization. (I) Luciferase reporter vectors containing either STAT1 or IRF1 promoter regions were cotransfected with an empty vector or UBR5 plasmids into the indicated cells. After 24 h, the transfected cells were treated with IFN-γ stimulation for 24 h, the cells were lysed to perform luciferase assay. The results are presented as the mean ± SEM from three individual experiments. *P < 0.05, **P < 0.01, ***P< 0.001, ****P < 0.0001. (J) The IRF1 and STAT1 binding sites in the mPdl1 promoter region were predicted using the ALGGEN website. (K) Summary of the results of a ChIP assay using anti-IRF1, STAT1, H3K4me1 and H3K27ac antibodies in WT, Ubr5-/- or hUBR5-reconstituted Ubr5-/- 4T1 cells after treatment with or without IFN-γ.
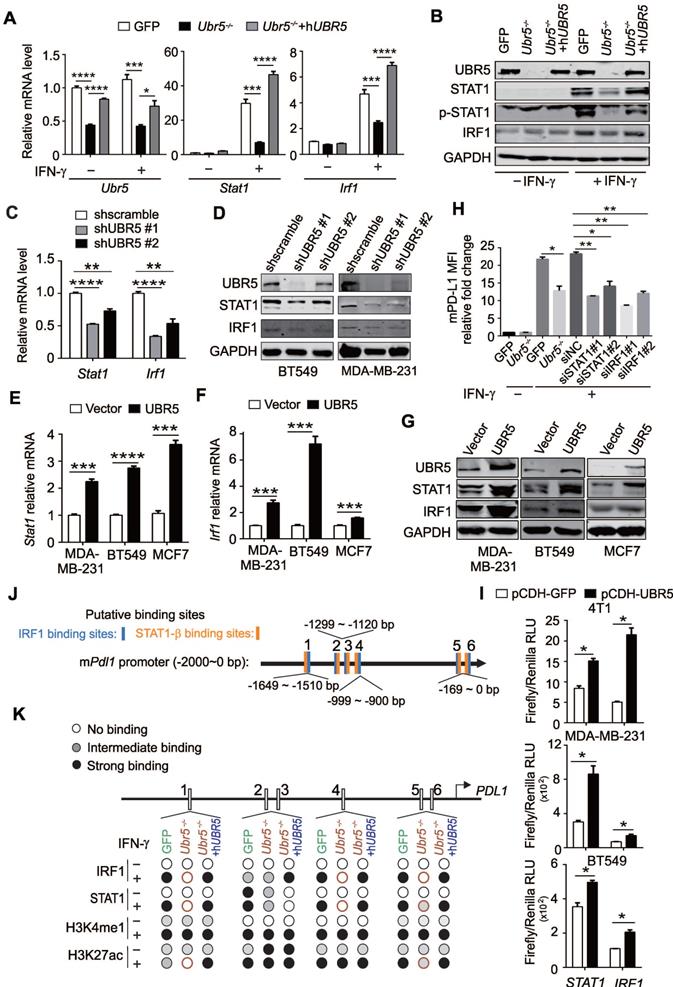
In eukaryotic cells, mRNA homeostasis is achieved through a balance between mRNA synthesis and degradation. Therefore, we sought to investigate whether UBR5 affects STAT1 and IRF1 mRNA by affecting its stability or transcription. 4T1 cells were treated with the transcriptional inhibitor actinomycin D and the mRNA levels of STAT1 and IRF1 were evaluated over time. The half-lives of both STAT1 and IRF1 mRNA showed no differences in WT, Ubr5-/- and hUBR5-reconstituted Ubr5-/- 4T1 cells (Figure S10). Next, we explored whether UBR5 affects the transcription of STAT1 and IRF1 via dual-luciferase reporter assay. Human STAT1 (from -972 to +884) and IRF1 (from -820 to +138) promoters and mouse Stat1 (from -2000 to +100) and Irf1 (from -2000 to +100) promoters were cloned into pGL3 plasmids separately. These reporter plasmids were transiently cotransfected with pCDH-UBR5/pCDH-eGFP plasmids into MDA-MB-231, BT549 or 4T1 cell lines separately to detect STAT1 and IRF1 transcription activity. The results showed that UBR5 indeed enhanced the transcriptional activity of the STAT1 and IRF1 promoters in BT549, MDA-MB-231 and 4T1 cells (Figure 5I). These data confirm that UBR5 plays an essential role in IFN-γ-induced activation of STAT1 and IRF1 transcription by enhancing synthesis rather than slowing degradation.
To further explore whether the effect of UBR5 on the transcription of PDL1 is mediated by the binding of STAT1-IRF1 in the PDL1 promoter region, we carried out chromatin immunoprecipitation (ChIP)-qPCR assays to analyse the enrichment of Stat1 and Irf1 in the mPdl1 promoter region. Six putative Stat1 binding sites and six putative Irf1 binding sites were predicted by the ALGGEN website (Figure 5J). Enhanced enrichment of Stat1 and Irf1 (in predicted binding sites 1 and 4 and predicted binding sites 5 and 6) in the mPdl1 promoter was observed in WT and hUBR5-reconstituted Ubr5-/- 4T1 cells compared to Ubr5-/- 4T1 cells, which further supported the idea that the UBR5-mediated enhancement of IFN-γ-induced PDL1 transcription is dependent on STAT1 and IRF1 (Figure 5K, Figure S11).
Increased post-translational histone modification, such as H3K4me1 and H3K27ac, have been reported to be responsible for the overexpression of PD-L1 as well as immune evasion in cancer [36, 37]. Although the status of H3K4me1 and H3K27ac differed when cells were stimulated with IFN-γ at sites 1, 2, 3, 4, 5, and 6, there were no differences among the 4T1 WT, Ubr5-/- and hUBR5-reconstituted Ubr5-/- cell lines (Figure S12). In addition, treatment with trichostatin A (TSA), a histone deacetylase (HDAC) inhibitor, and 5-aza-2'-deoxycytidine (5'-AZA-dC), a DNA methylation inhibitor did not change the expression of PD-L1 in 4T1 cell surface regardless of whether UBR5 was expressed (Figure S13). These results suggest that histone modifications such as H3K4me1 and H3K27ac are not involved in the regulation of PD-L1 expression by UBR5.
UBR5-mediated transactivation of PDL1 is independent of the E3 ligase activity
Given that UBR5 was found to be responsible for IFN-γ-induced STAT1 and IRF1 transcription, we further explored that which domain of UBR5 (Figure 6A) is critical to its regulation. The human PDL1 promoter was cloned and inserted it into a pGL3-based firefly luciferase reporter vector (Figure 6B), and transiently cotransfected the vector with plasmids encoding wild type UBR5, the HECT domain mutant C2768A or the PABC domain deletion (∆PABC) of UBR5 into MDA-MB-231 cells. The results showed that UBR5 increased the transcriptional activity of the PDL1 promoter (Figure 6C). The UBR5-ΔPABC mutant completely lost the IFN-γ-inducibility on the PDL1 promoter, suggesting that this domain is pivotal for regulation. PDL1 promoter constructs with site-directed mutations in the binding sites of IRF1 or STAT1/3 [11] were generated (Figure 6B), and transiently cotransfected into MDA-MB-231 cells with expression vector of UBR5 and the variants. Luciferase activity data indicated that the IRF1 and STAT1/3 binding sites were essential for the transcriptional activity of the PDL1 promoter by UBR5 upon IFN-γ stimulation (Figure 6C). Consistently, a highly analogous response was observed in IFN-γ-stimulated 4T1 cells on the mouse Pdl1 promoter (Figure 6D).
UBR5-mediated transactivation of PDL1 is independent of its E3 ubiquitin ligase. (A-D) MDA-MB-231 cells were cotransfected with WT, HECT-mutation (UBR5-C2768A) or PABC-deletion UBR5 (A) together with luciferase reporters containing the human PDL1 WT promoter or a promoter with 2 deletions, including the STAT1/3 or IRF1 binding sites (B), and then stimulated with IFN-γ for 24 h. Luciferase activity was measured in cell lysates of MDA-MB-231 (C) and 4T1 cells (D) by dual luciferase assay. (E-F) The protein levels of UBR5 (E) and PD-L1 (F) were measured in IFN-γ-treated GFP, Ubr5-/-, hUBR5-reconstituted Ubr5-/- 4T1 cells and Ubr5-/- 4T1 cells reconstituted with either hUBR5-C2768 or hUBR5-ΔPABC. The results are presented as the mean ± SEM from three individual experiments. ns, no significance, *P < 0.05, **P < 0.01, ***P < 0.001.
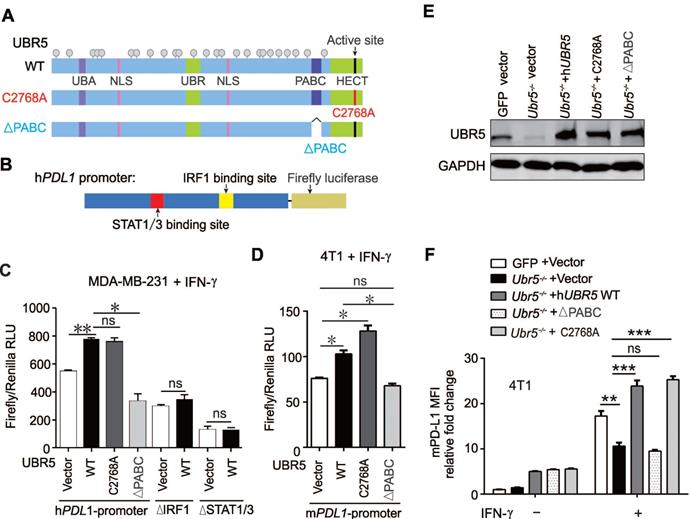
To further confirm that PABC is required for IFN-γ-induced PDL1 transcription, we transiently transfected plasmids encoding hUBR5, hUBR5-ΔPABC or hUBR5-C2768A respectively into 4T1/Ubr5-/- cells (Figure 6E) and performed flow cytometry assays to detect PD-L1 surface protein levels. Consistent with the dual-luciferase reporter assay results, the PABC domain, but not the E3 ligase catalytic site, was essential for the regulation of IFN-γ-induced PD-L1 expression (Figure 6F). Additionally, we corroborated the dispensability of UBR5 ubiquitin ligase activity by treating BC cells with MG132, an inhibitor of the 26S proteasome. The reduced surface PD-L1 level observed in UBR5-depleted cells did not result from increased protein degradation through the proteasome (Figure S14). Taken together, UBR5 promotes PD-L1 in a manner dependent on its PABC domain and independent of the E3 ligase activity.
UBR5 transactivation of STAT1 and PDL1 is mediated by PKR
Through RNA-seq (Figure 4D) and previous mass spectrometry (MS) analysis [29], we observed a significant downregulation of EIF2AK2 in RNA and protein levels, the gene that encodes protein kinase RNA-activated (PKR), in Ubr5-/- cells compared with WT 4T1 cells [29]. PKR can be activated by IFN-γ mRNA [38] and plays an important role in interferon and dsRNA‐signaling pathways by modulating the transcriptional function of STAT1 [39]. In addition, PKR can indirectly activate the modification of IRF1 and activation of its DNA-binding activity in a protein kinase-dependent manner [40]. Therefore, we speculated that UBR5 might regulate STAT1 and IRF1 through PKR. To test this possibility, we first evaluated Eif2ak2 mRNA levels in 4T1 WT, Ubr5-/-, hUBR5-reconstituted, hUBR5-ΔPABC and hUBR5-C2768A-reconstituted Ubr5-/- cells with or without IFN-γ treatment. Interestingly, the alterations in Eif2ak2 mRNA (Figure 7A) and protein (Figure 7B) levels were consistent with the patterns of Stat1, Irf1 and Pdl1 alterations with IFN-γ induction when the expression of UBR5 changed. Consistently, the Eif2ak2 mRNA (Figure 7C) and protein (Figure 7D) levels were decreased in BT549 cells after UBR5 was knocked down. The EIF2AK2 was expressed in response to IFN-γ in hUBR5-C2768A-reconstituted Ubr5-/- cells but not in hUBR5-ΔPABC-reconstituted Ubr5-/- cells (Figure 7A-B), which was consistent with our previous observation in Figure 6C-F that PD-L1 regulation by UBR5 is dependent on the PABC domain.
The transactivation of UBR5 to STAT1 and PDL1 is mediated by protein kinase RNA-activated. (A-B) The mRNA (A) and protein levels (B) of EIF2AK2 in WT, Ubr5-/-, hUBR5-reconstituted Ubr5-/- 4T1 cells and Ubr5-/- 4T1 cells reconstituted with either hUBR5-C2768A or hUBR5-ΔPABC were measured by qPCR and western blot analysis after cells were treated with or without IFN-γ. (C-D) The mRNA (C) and protein levels (D) levels of EIF2AK2 in WT and UBR5-knockdown BT549 cells were measured by qPCR and western blot analysis after cells were treated with IFN-γ for 24 h. (E-G) The mRNA (E) and protein levels of UBR5, STAT1, IRF1 and EIF2AK2 (F) and relative fold changes of cell-surface PD-L1 levels (G) in IFN-γ-treated WT, Ubr5-/-, and 4T1 cells with stable knockdown by shEif2ak2 #1 and shEif2ak2 #1. Shscramble served as the control. (H-J) The mRNA levels (H) of Eif2ak2, Ubr5, Pdl1, Stat1 and Irf1, the protein levels of UBR5, EIF2AK2, STAT1 and IRF1 (I) and the surface levels of PD-L1 (J) were measured in IFN-γ-treated 4T1 WT, Ubr5-/- and mEif2ak2-reconstituted Ubr5-/- cell lines. The results are presented as the mean ± SEM from three individual experiments. ns, no significance, *P< 0.05, **P < 0.01, ***P< 0.001, ****P < 0.0001. (K) The correlations of EIF2AK2 with UBR5, PDL1 and STAT1 mRNA levels were assessed in the TCGA BRCA database (1104 samples) by starBase database. (L) The summary correlations of EIF2AK2 with UBR5, PDL1, STAT1 and IRF1 mRNA levels (normalizated to GAPDH) were analysed in the TCGA BRCA database by starBase and GEPIA database respectively.
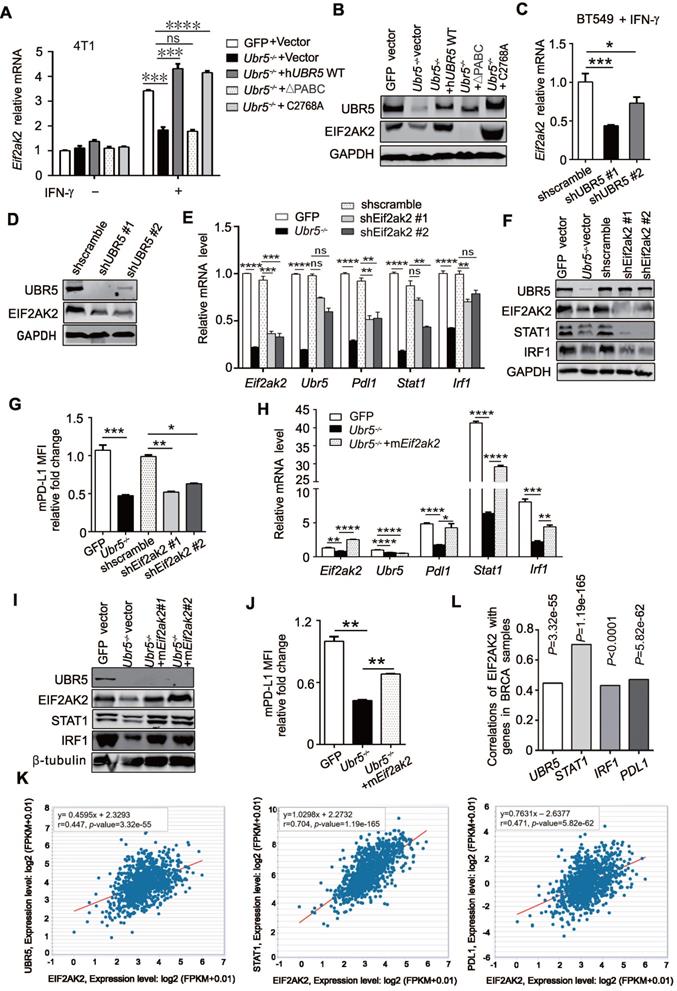
To further confirm that EIF2AK2 affects the mRNA levels of STAT1 and IRF1, we generated Eif2ak2-knockdown 4T1 cells via shRNA (Figure 7E). The mRNA levels of Pdl1, Stat1 and Irf1 (Figure 7E) and the protein levels of STAT1, IRF1 (Figure 7F) and PD-L1 (Figure 7G) decreased in 4T1 cells after Eif2ak2 knockdown. Conversely, the mRNA and protein levels of STAT1, IRF1 and PD-L1 were restored after we reconstituted mEif2ak2 expression in Ubr5-/- cells compared with GFP and Ubr5-/- 4T1 cells (Figure 7H-J). However, compared with WT cells, the cell surface protein level of PD-L1 restoration was not completely rescued in mEif2ak2-reconstituted cells, suggesting that additional mechanisms might be involved in the regulation of UBR5-mediated PD-L1 surface upregulation other than PKR. These data suggest that the transactivation of STAT1 and PDL1 by UBR5 is mediated by PKR.
To investigate whether our observations match the scenario in clinical human breast cancer samples, we performed bioinformatic analysis with data from TCGA breast cancer invasive carcinoma (BRCA) RNA-seq database by GEPIA and starBase database respectively. Moderate and above correlations between EIF2AK2 and UBR5, STAT1, IRF1 or PDL1 expression (Pearson correlation: R=0.447, R=0.704, R=0.43 and R=0.471, respectively) were observed (Figure 7K-L), which supports the idea that EIF2AK2 expression is highly positively correlated with those of UBR5, STAT1, IRF1 and PDL1.
PKR can mediate the activation of IFN-γ-stimulated STAT1 by regulating p38 MAPK and is required for efficient activation of JNK by IFN-γ [41]. To further explore whether UBR5 activates PKR-mediated STAT1 and PD-L1 through p38 or JNK, we used the PKR inhibitor oxindole/imidazole compound (C16) to treat GFP and Ubr5-/- 4T1 cells with or without IFN-γ stimulation. The results showed that there were no differences of the protein levels of STAT1, p38 and JNK (and their phosphorylated forms) with or without PKR inhibition in GFP and Ubr5-/- 4T1 cells in the presence of IFN-γ (Figure S15). Also, the surface levels of PD-L1 protein were not different between GFP cells and Ubr5-/- 4T1 cells treated with C16 (Figure S16), suggesting that PKR is involved in UBR5-mediated activation of STAT1 and PDL1 transcription in a kinase-independent manner. The kinase activity independent PKR was also observed in PKR-activated NF-κB signaling pathway [42, 43], as well as in broad mechanisms of inflammasome-mediated caspase-1 activation [44].
Since the PABC domain is required for the regulation of EIF2AK2 by UBR5 (Figure 7A-B) and plays a role in enhancing mRNA stability [45], we further explored whether UBR5 controlled Eif2ak2 by maintaining the latter's mRNA stability. IFN-γ-pretreated cells were treated with the transcriptional inhibitor actinomycin D for different times, and then the mRNA levels of Eif2ak2 were evaluated. The half-lives of Eif2ak2 mRNA were not different among 4T1 WT, Ubr5-/- and hUBR5-reconstituted cells (Figure S17). This result suggests that UBR5 regulates Eif2ak2 not by enhancing its mRNA stability, but by increasing the transcription of Eif2ak2.
Discussion
As PD-L1 is a ligand for the critical immune checkpoint molecule PD-1, the regulation of PD-L1 expression is complex and can occur at genetic, transcriptional and posttranscriptional levels. Here, we demonstrate that UBR5 is required for IFN-γ-induced PD-L1 expression in breast cancer, and that transactivation of PDL1 gene expression by UBR5 is mediated through the PKR/STAT1/IRF1 pathway. Furthermore, we show that PD-L1 regulation by UBR5 is dependent on the PABC domain but not the E3 ligase activity of UBR5. More significantly, the augmented expression of PD-L1 by UBR5 can enhance the latter's function in promoting tumor growth and metastasis. Lastly, combined blockade of UBR5 and PD-L1 leads to a synergistic therapeutic benefit in eradicating tumor growth and prolong the survival or even to a potential cure (Figure 3), although we need to more rigorously corroborate the synergism by other ways of targeting of UBR5 and PD-L1 than the genetic means.
However, since the UBR5 protein does not contain a DNA binding motif, it is still unclear how UBR5 regulates EIF2AK2, although there is evidence that UBR5 can enhance its transcription through the UBR5 PABC domain. Previous studies have implied that UBR5 can regulate gene transcription through several mechanisms involving three different stages: initiation of RNA transcription by transcription factors and their associated co-activators [46], RNA elongation, and RNA processing and nuclear export [47]. It seems from our observations that UBR5 does not affect EIF2AK2 mRNA stability, thus UBR5 may promote EIF2AK2 expression through other mechanisms at one or more of these three stages. UBR5 is overexpressed in many cancer types [20, 21], [48-54] and associated with poor prognosis in a variety of cancers [48-51, 55, 56]. EIF2AK2-encoded PKR also plays a critical role in tumorigenesis. High PKR expression is linked with prognosis [57-62] and tumor malignancy in multiple cancer types [63-65]. In addition, high PKR expression is related to genomic instability [66], reduced survival and shortened remission period [67]. We showed that UBR5 could influence the transcription of EIF2AK2. Interestingly, we observed significant co-expression between UBR5 and EIF2AK2 in the TCGA mRNA database (Figure 7K). It's worth noting that PKR may also be upregulated by IFN-γ indirectly by UBR5-induced changes in the tumor microenvironment and it is of interest to further delineate the role UBR5 in the regulation of PKR expression in vivo.
We demonstrate that UBR5 is broadly and critically involved in the IFN-γ signaling pathway and in the regulation of immune checkpoints (Figure 4). Furthermore, our previous study [21] showed that IFN-γ mRNA levels were elevated in 4T1/Ubr5-/- tumors compared to WT. As a cytokine, IFN-γ plays a dual role in tumor immune response. On the one hand, IFN-γ inhibits the occurrence of tumor and promotes the apoptosis of tumor by regulating immune responses and cell cycle, promoting cell apoptosis, and inhibiting angiogenesis [68, 69]. On the other hand, IFN-γ can promote immune escape of tumor cells by regulating the TME [70-79]. However, there is little research on the conversional mechanisms of IFN-γ between the two contrasting activities, which also illustrates that IFN-γ has great prospects in the field of tumor immunotherapy.
It is noted that UBR5 can upregulate PDL1 transcription to promote tumor immune evasion and that restoration of PD-L1 expression in UBR5-deficient tumor decreases T cell infiltration and restores malignancy, as was indirectly proven by the combination therapy targeting both UBR5 and PD-L1 in the TNBC mouse model. Therefore, dual targeting UBR5 and PD-L1 has better efficacy than single targeting for breast cancer, and potentially for other cancer types as well. These areas represent interesting directions for future work.
Together, our in vivo and in vitro results reveal for the first time, that UBR5 is a key player in the transcriptional regulation of IFN-γ-induced PDL1 and ISGs, and in directing cancer immune evasion. These findings provide strong evidence and rationale for targeting both UBR5 and PD-L1 as a novel approach to enhance the efficacy of immune checkpoint blockade-based therapy against breast cancer, particularly TNBC.
Abbreviations
ChIP: Chromatin immunoprecipitation; DCs: Dendritic Cells; EGF: Epidermal growth factor; GO: Gene Ontology; GrzmB: Granzyme B; HECT: homologous to E6-AP C-terminus; IFN-γ: Interferon-γ; ILs: Interleukins; IRF: Interferon regulatory factor; ISGs: IFN-γ-stimulated genes; JAK: Janus kinase; MFI: Mean fluorescence intensity; NF-κB: Nuclear factor-κB; PABC: Poly adenylate binding C terminal; PD-L1: Programmed cell death-ligand1; PD-1: Programmed cell death protein 1; PKR: Protein kinase RNA-activated; PI3K: Phosphatidylinositol 3-kinase; STAT: Signal transducer and activator of transcription; TCGA: The cancer genome atlas; TME: Tumor microenvironment; TNBC: Triple negative breast cancer; TNF-α: Tumor necrosis factors α; UBR5: E3 component N-recognin 5.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81772166 to FW; 81872353 and 31670913 to XM; 31871329 to JL). We are grateful to Drs. Qiliang Cai and Ming Yu for helpful suggestions and proofreading.
Author Contributions
Bingbing Wu: Methodology, Investigation, Formal analysis, Writing - Original Draft.
Mei Song: Methodology, Investigation, Formal analysis.
Qun Dong: Methodology, Investigation, Formal analysis.
Gang Xiang: Resources.
Jing Li: Validation, Funding acquisition, Validation.
Xiaojing Ma: Conceptualization, Data Curation, Writing - Review & Editing, Project administration, Funding acquisition.
Fang Wei: Conceptualization, Data Curation, Writing - Review & Editing, Project administration, Funding acquisition.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56-61
2. Boussiotis VA. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N Engl J Med. 2016;375:1767-78
3. Mittendorf EA, Philips AV, Meric-Bernstam F, Qiao N, Wu Y, Harrington S. et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res. 2014;2:361-70
4. Li F, Ren Y, Wang Z. Programmed death 1 Ligand 1 expression in breast cancer and its association with patients' clinical parameters. J Cancer Res Ther. 2018;14:150-4
5. Muenst S, Schaerli AR, Gao F, Däster S, Trella E, Droeser RA. et al. Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Breast Cancer Res Treat. 2014;146:15-24
6. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111-22
7. Chikuma S, Terawaki S, Hayashi T, Nabeshima R, Yoshida T, Shibayama S. et al. PD-1-mediated suppression of IL-2 production induces CD8+ T cell anergy in vivo. J Immunol. 2009;182:6682-9
8. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH. et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682-7
9. Adams S, Loi S, Toppmeyer D, Cescon DW, De Laurentiis M, Nanda R. et al. Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: cohort B of the phase II KEYNOTE-086 study. Ann Oncol. 2019;30:405-11
10. Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H. et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med. 2018;379:2108-21
11. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA. et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017;19:1189-201
12. Concha-Benavente F, Srivastava RM, Trivedi S, Lei Y, Chandran U, Seethala RR. et al. Identification of the Cell-Intrinsic and -Extrinsic Pathways Downstream of EGFR and IFNγ That Induce PD-L1 Expression in Head and Neck Cancer. Cancer Res. 2016;76:1031-43
13. Gocher AM, Workman CJ, Vignali DAA. Interferon-γ: teammate or opponent in the tumour microenvironment? Nat Rev Immunol. 2022;22:158-72
14. Dummer R, Eichmüller S, Gellrich S, Assaf C, Dreno B, Schiller M. et al. Phase II clinical trial of intratumoral application of TG1042 (adenovirus-interferon-gamma) in patients with advanced cutaneous T-cell lymphomas and multilesional cutaneous B-cell lymphomas. Mol Ther. 2010;18:1244-7
15. Gleave ME, Elhilali M, Fradet Y, Davis I, Venner P, Saad F. et al. Interferon gamma-1b compared with placebo in metastatic renal-cell carcinoma. Canadian Urologic Oncology Group. N Engl J Med. 1998;338:1265-71
16. Razaghi A, Owens L, Heimann K. Review of the recombinant human interferon gamma as an immunotherapeutic: Impacts of production platforms and glycosylation. J Biotechnol. 2016;240:48-60
17. Callaghan MJ, Russell AJ, Woollatt E, Sutherland GR, Sutherland RL, Watts CK. Identification of a human HECT family protein with homology to the Drosophila tumor suppressor gene hyperplastic discs. Oncogene. 1998;17:3479-91
18. Saunders DN, Hird SL, Withington SL, Dunwoodie SL, Henderson MJ, Biben C. et al. Edd, the murine hyperplastic disc gene, is essential for yolk sac vascularization and chorioallantoic fusion. Mol Cell Biol. 2004;24:7225-34
19. Kinsella E, Dora N, Mellis D, Lettice L, Deveney P, Hill R. et al. Use of a Conditional Ubr5 Mutant Allele to Investigate the Role of an N-End Rule Ubiquitin-Protein Ligase in Hedgehog Signalling and Embryonic Limb Development. PLoS One. 2016;11:e0157079
20. Clancy JL, Henderson MJ, Russell AJ, Anderson DW, Bova RJ, Campbell IG. et al. EDD, the human orthologue of the hyperplastic discs tumour suppressor gene, is amplified and overexpressed in cancer. Oncogene. 2003;22:5070-81
21. Liao L, Song M, Li X, Tang L, Zhang T, Zhang L. et al. E3 Ubiquitin Ligase UBR5 Drives the Growth and Metastasis of Triple-Negative Breast Cancer. Cancer Res. 2017;77:2090-101
22. Shearer RF, Iconomou M, Watts CK, Saunders DN. Functional Roles of the E3 Ubiquitin Ligase UBR5 in Cancer. Mol Cancer Res. 2015;13:1523-32
23. Kozlov G, De Crescenzo G, Lim NS, Siddiqui N, Fantus D, Kahvejian A. et al. Structural basis of ligand recognition by PABC, a highly specific peptide-binding domain found in poly(A)-binding protein and a HECT ubiquitin ligase. Embo j. 2004;23:272-81
24. Lim NS, Kozlov G, Chang TC, Groover O, Siddiqui N, Volpon L. et al. Comparative peptide binding studies of the PABC domains from the ubiquitin-protein isopeptide ligase HYD and poly(A)-binding protein. Implications for HYD function. J Biol Chem. 2006;281:14376-82
25. Muñoz-Escobar J, Matta-Camacho E, Kozlov G, Gehring K. The MLLE domain of the ubiquitin ligase UBR5 binds to its catalytic domain to regulate substrate binding. J Biol Chem. 2015;290:22841-50
26. Hay-Koren A, Caspi M, Zilberberg A, Rosin-Arbesfeld R. The EDD E3 ubiquitin ligase ubiquitinates and up-regulates beta-catenin. Mol Biol Cell. 2011;22:399-411
27. Ong SS, Goktug AN, Elias A, Wu J, Saunders D, Chen T. Stability of the human pregnane X receptor is regulated by E3 ligase UBR5 and serine/threonine kinase DYRK2. Biochem J. 2014;459:193-203
28. Tomaic V, Pim D, Thomas M, Massimi P, Myers MP, Banks L. Regulation of the human papillomavirus type 18 E6/E6AP ubiquitin ligase complex by the HECT domain-containing protein EDD. J Virol. 2011;85:3120-7
29. Song M, Wang C, Wang H, Zhang T, Li J, Benezra R. et al. Targeting ubiquitin protein ligase E3 component N-recognin 5 in cancer cells induces a CD8+ T cell mediated immune response. Oncoimmunology. 2020;9:1746148
30. Stanton SE, Adams S, Disis ML. Variation in the Incidence and Magnitude of Tumor-Infiltrating Lymphocytes in Breast Cancer Subtypes: A Systematic Review. JAMA Oncol. 2016;2:1354-60
31. Croft M, Benedict CA, Ware CF. Clinical targeting of the TNF and TNFR superfamilies. Nat Rev Drug Discov. 2013;12:147-68
32. Wang J, Sun J, Liu LN, Flies DB, Nie X, Toki M. et al. Siglec-15 as an immune suppressor and potential target for normalization cancer immunotherapy. Nat Med. 2019;25:656-66
33. Loeb KR, Haas AL. The interferon-inducible 15-kDa ubiquitin homolog conjugates to intracellular proteins. J Biol Chem. 1992;267:7806-13
34. Benci JL, Johnson LR, Choa R, Xu Y, Qiu J, Zhou Z. et al. Opposing Functions of Interferon Coordinate Adaptive and Innate Immune Responses to Cancer Immune Checkpoint Blockade. Cell. 2019;178:933-48.e14
35. Lee SJ, Jang BC, Lee SW, Yang YI, Suh SI, Park YM. et al. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7-H1 (CD274). FEBS Lett. 2006;580:755-62
36. Xiong W, Deng H, Huang C, Zen C, Jian C, Ye K. et al. MLL3 enhances the transcription of PD-L1 and regulates anti-tumor immunity. Biochim Biophys Acta Mol Basis Dis. 2019;1865:454-63
37. Xu Y, Wu Y, Zhang S, Ma P, Jin X, Wang Z. et al. A Tumor-Specific Super-Enhancer Drives Immune Evasion by Guiding Synchronous Expression of PD-L1 and PD-L2. Cell Rep. 2019;29:3435-47.e4
38. Ben-Asouli Y, Banai Y, Pel-Or Y, Shir A, Kaempfer R. Human interferon-gamma mRNA autoregulates its translation through a pseudoknot that activates the interferon-inducible protein kinase PKR. Cell. 2002;108:221-32
39. Wong AH, Tam NW, Yang YL, Cuddihy AR, Li S, Kirchhoff S. et al. Physical association between STAT1 and the interferon-inducible protein kinase PKR and implications for interferon and double-stranded RNA signaling pathways. Embo j. 1997;16:1291-304
40. Zamanian-Daryoush M, Der SD, Williams BR. Cell cycle regulation of the double stranded RNA activated protein kinase, PKR. Oncogene. 1999;18:315-26
41. Goh KC, deVeer MJ, Williams BR. The protein kinase PKR is required for p38 MAPK activation and the innate immune response to bacterial endotoxin. Embo j. 2000;19:4292-7
42. Bonnet MC, Daurat C, Ottone C, Meurs EF. The N-terminus of PKR is responsible for the activation of the NF-kappaB signaling pathway by interacting with the IKK complex. Cell Signal. 2006;18:1865-75
43. Bonnet MC, Weil R, Dam E, Hovanessian AG, Meurs EF. PKR stimulates NF-kappaB irrespective of its kinase function by interacting with the IkappaB kinase complex. Mol Cell Biol. 2000;20:4532-42
44. Hett EC, Slater LH, Mark KG, Kawate T, Monks BG, Stutz A. et al. Chemical genetics reveals a kinase-independent role for protein kinase R in pyroptosis. Nat Chem Biol. 2013;9:398-405
45. Yang R, Gaidamakov SA, Xie J, Lee J, Martino L, Kozlov G. et al. La-related protein 4 binds poly(A), interacts with the poly(A)-binding protein MLLE domain via a variant PAM2w motif, and can promote mRNA stability. Mol Cell Biol. 2011;31:542-56
46. Kwak J, Workman JL, Lee D. The proteasome and its regulatory roles in gene expression. Biochim Biophys Acta. 2011;1809:88-96
47. Weake VM, Workman JL. Histone ubiquitination: triggering gene activity. Mol Cell. 2008;29:653-63
48. O'Brien PM, Davies MJ, Scurry JP, Smith AN, Barton CA, Henderson MJ. et al. The E3 ubiquitin ligase EDD is an adverse prognostic factor for serous epithelial ovarian cancer and modulates cisplatin resistance in vitro. Br J Cancer. 2008;98:1085-93
49. Wang J, Zhao X, Jin L, Wu G, Yang Y. UBR5 Contributes to Colorectal Cancer Progression by Destabilizing the Tumor Suppressor ECRG4. Dig Dis Sci. 2017;62:2781-9
50. Zhang Z, Zheng X, Li J, Duan J, Cui L, Yang L. et al. Overexpression of UBR5 promotes tumor growth in gallbladder cancer via PTEN/PI3K/Akt signal pathway. J Cell Biochem. 2019;120:11517-24
51. Yang M, Jiang N, Cao QW, Ma MQ, Sun Q. The E3 ligase UBR5 regulates gastric cancer cell growth by destabilizing the tumor suppressor GKN1. Biochem Biophys Res Commun. 2016;478:1624-9
52. Ji SQ, Zhang YX, Yang BH. UBR5 promotes cell proliferation and inhibits apoptosis in colon cancer by destablizing P21. Pharmazie. 2017;72:408-13
53. Chen L, Yuan R, Wen C, Liu T, Feng Q, Deng X. et al. E3 ubiquitin ligase UBR5 promotes pancreatic cancer growth and aerobic glycolysis by downregulating FBP1 via destabilization of C/EBPα. Oncogene. 2021;40:262-76
54. Gu YF, Ge XP. UBR5 inhibits the radiosensitivity of non-small cell lung cancer cells via the activation of the PI3K/AKT pathway. J Investig Med. 2021;69:970-5
55. Wang K, Tang J, Liu X, Wang Y, Chen W, Zheng R. UBR5 regulates proliferation and radiosensitivity in human laryngeal carcinoma via the p38/MAPK signaling pathway. Oncol Rep. 2020;44:685-97
56. Bolt MJ, Stossi F, Callison AM, Mancini MG, Dandekar R, Mancini MA. Systems level-based RNAi screening by high content analysis identifies UBR5 as a regulator of estrogen receptor-α protein levels and activity. Oncogene. 2015;34:154-64
57. Guo C, Shao R, Correa AM, Behrens C, Johnson FM, Raso MG. et al. Prognostic significance of combinations of RNA-dependent protein kinase and EphA2 biomarkers for NSCLC. J Thorac Oncol. 2013;8:301-8
58. He Y, Correa AM, Raso MG, Hofstetter WL, Fang B, Behrens C. et al. The role of PKR/eIF2α signaling pathway in prognosis of non-small cell lung cancer. PLoS One. 2011;6:e24855
59. Pataer A, Raso MG, Correa AM, Behrens C, Tsuta K, Solis L. et al. Prognostic significance of RNA-dependent protein kinase on non-small cell lung cancer patients. Clin Cancer Res. 2010;16:5522-8
60. Kwon HC, Moon CH, Kim SH, Choi HJ, Lee HS, Roh MS. et al. Expression of double-stranded RNA-activated protein kinase (PKR) and its prognostic significance in lymph node negative rectal cancer. Jpn J Clin Oncol. 2005;35:545-50
61. Hii SI, Hardy L, Crough T, Payne EJ, Grimmett K, Gill D. et al. Loss of PKR activity in chronic lymphocytic leukemia. Int J Cancer. 2004;109:329-35
62. Shimada A, Shiota G, Miyata H, Kamahora T, Kawasaki H, Shiraki K. et al. Aberrant expression of double-stranded RNA-dependent protein kinase in hepatocytes of chronic hepatitis and differentiated hepatocellular carcinoma. Cancer Res. 1998;58:4434-8
63. Cheng X, Bennett RL, Liu X, Byrne M, Stratford May W. PKR negatively regulates leukemia progression in association with PP2A activation, Bcl-2 inhibition and increased apoptosis. Blood Cancer J. 2013;3:e144
64. Terada T, Maeta H, Endo K, Ohta T. Protein expression of double-stranded RNA-activated protein kinase in thyroid carcinomas: correlations with histologic types, pathologic parameters, and Ki-67 labeling. Hum Pathol. 2000;31:817-21
65. Roh MS, Kwak JY, Kim SJ, Lee HW, Kwon HC, Hwang TH. et al. Expression of double-stranded RNA-activated protein kinase in small-size peripheral adenocarcinoma of the lung. Pathol Int. 2005;55:688-93
66. Cheng X, Byrne M, Brown KD, Konopleva MY, Kornblau SM, Bennett RL. et al. PKR inhibits the DNA damage response, and is associated with poor survival in AML and accelerated leukemia in NHD13 mice. Blood. 2015;126:1585-94
67. Oshima M, Iwama A. Nuclear, not cytoplasmic, PKR maneuvers in AML. Blood. 2015;126:1523-4
68. Hao Q, Tang H. Interferon-γ and Smac mimetics synergize to induce apoptosis of lung cancer cells in a TNFα-independent manner. Cancer Cell Int. 2018;18:84
69. Briesemeister D, Sommermeyer D, Loddenkemper C, Loew R, Uckert W, Blankenstein T. et al. Tumor rejection by local interferon gamma induction in established tumors is associated with blood vessel destruction and necrosis. Int J Cancer. 2011;128:371-8
70. Kelly SA, Gschmeissner S, East N, Balkwill FR. Enhancement of metastatic potential by gamma-interferon. Cancer Res. 1991;51:4020-7
71. Chen HC, Chou AS, Liu YC, Hsieh CH, Kang CC, Pang ST. et al. Induction of metastatic cancer stem cells from the NK/LAK-resistant floating, but not adherent, subset of the UP-LN1 carcinoma cell line by IFN-γ. Lab Invest. 2011;91:1502-13
72. Lo UG, Pong RC, Yang D, Gandee L, Hernandez E, Dang A. et al. IFNγ-Induced IFIT5 Promotes Epithelial-to-Mesenchymal Transition in Prostate Cancer via miRNA Processing. Cancer Res. 2019;79:1098-112
73. Lane RS, Femel J, Breazeale AP, Loo CP, Thibault G, Kaempf A. et al. IFNγ-activated dermal lymphatic vessels inhibit cytotoxic T cells in melanoma and inflamed skin. J Exp Med. 2018;215:3057-74
74. Pai CS, Huang JT, Lu X, Simons DM, Park C, Chang A. et al. Clonal Deletion of Tumor-Specific T Cells by Interferon-γ Confers Therapeutic Resistance to Combination Immune Checkpoint Blockade. Immunity. 2019;50:477-92.e8
75. Mandai M, Hamanishi J, Abiko K, Matsumura N, Baba T, Konishi I. Dual Faces of IFNγ in Cancer Progression: A Role of PD-L1 Induction in the Determination of Pro- and Antitumor Immunity. Clin Cancer Res. 2016;22:2329-34
76. Zhang X, Zeng Y, Qu Q, Zhu J, Liu Z, Ning W. et al. PD-L1 induced by IFN-γ from tumor-associated macrophages via the JAK/STAT3 and PI3K/AKT signaling pathways promoted progression of lung cancer. Int J Clin Oncol. 2017;22:1026-33
77. Bellucci R, Martin A, Bommarito D, Wang K, Hansen SH, Freeman GJ. et al. Interferon-γ-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology. 2015;4:e1008824
78. Ramos-Casals M, Brahmer JR, Callahan MK, Flores-Chávez A, Keegan N, Khamashta MA. et al. Immune-related adverse events of checkpoint inhibitors. Nat Rev Dis Primers. 2020;6:38
79. Yang L, Li A, Lei Q, Zhang Y. Tumor-intrinsic signaling pathways: key roles in the regulation of the immunosuppressive tumor microenvironment. J Hematol Oncol. 2019;12:125
Author contact
![]() Corresponding author: Fang Wei, 800 Dongchuan Road, Minghang, Shanghai 200240, China. Phone: 86-21-34205287; Fax: 86-21-34205287; E-mail: fangweiedu.cn; Xiaojing Ma, xim2002cornell.edu
Corresponding author: Fang Wei, 800 Dongchuan Road, Minghang, Shanghai 200240, China. Phone: 86-21-34205287; Fax: 86-21-34205287; E-mail: fangweiedu.cn; Xiaojing Ma, xim2002cornell.edu