Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
CBP/p300 protein structure and...
Modulation of CBP/p300-induced...
CBP/p300 are required for...
CBP/p300 as tumor suppressors
CBP/p300 induce oncogene...
Small molecule compound CBP/p300...
CBP/p300 inhibitors exert...
Conclusions and perspectives
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2022; 12(11):4935-4948. doi:10.7150/thno.73223 This issue Cite
Review
Histone acetyltransferases CBP/p300 in tumorigenesis and CBP/p300 inhibitors as promising novel anticancer agents
Qingjuan Chen1, Binhui Yang1, Xiaochen Liu1, Xu D. Zhang2,3 ![]() , Lirong Zhang4
, Lirong Zhang4 ![]() , Tao Liu3,5,6
, Tao Liu3,5,6 ![]()
1. Department of Oncology, 3201 Hospital of Xi'an Jiaotong University Health Science Center, Hanzhong, Shaanxi 723000, China.
2. School of Medicine and Public Health, Priority Research Centre for Cancer Research, University of Newcastle, Callaghan, Newcastle, NSW 2308, Australia.
3. Translational Research Institute, Henan Provincial People's Hospital, Academy of Medical Sciences, Zhengzhou University, Zhengzhou, China.
4. Department of Pharmacology, School of Basic Medical Sciences, Zhengzhou University, Zhengzhou, China.
5. Children's Cancer Institute Australia, Randwick, Sydney, NSW 2031, Australia.
6. School of Women's and Children's Health, University of New South Wales, Sydney, New South Wales, Australia.
Received 2022-3-24; Accepted 2022-5-23; Published 2022-6-21
Abstract

The histone acetyltransferases CBP and p300, often referred to as CBP/p300 due to their sequence homology and functional overlap and co-operation, are emerging as critical drivers of oncogenesis in the past several years. CBP/p300 induces histone H3 lysine 27 acetylation (H3K27ac) at target gene promoters, enhancers and super-enhancers, thereby activating gene transcription. While earlier studies indicate that CBP/p300 deletion/loss can promote tumorigenesis, CBP/p300 have more recently been shown to be over-expressed in cancer cells and drug-resistant cancer cells, activate oncogene transcription and induce cancer cell proliferation, survival, tumorigenesis, metastasis, immune evasion and drug-resistance. Small molecule CBP/p300 histone acetyltransferase inhibitors, bromodomain inhibitors, CBP/p300 and BET bromodomain dual inhibitors and p300 protein degraders have recently been discovered. The CBP/p300 inhibitors and degraders reduce H3K27ac, down-regulate oncogene transcription, induce cancer cell growth inhibition and cell death, activate immune response, overcome drug resistance and suppress tumor progression in vivo. In addition, CBP/p300 inhibitors enhance the anticancer efficacy of chemotherapy, radiotherapy and epigenetic anticancer agents, including BET bromodomain inhibitors; and the combination therapies exert substantial anticancer effects in mouse models of human cancers including drug-resistant cancers. Currently, two CBP/p300 inhibitors are under clinical evaluation in patients with advanced and drug-resistant solid tumors or hematological malignancies. In summary, CBP/p300 have recently been identified as critical tumorigenic drivers, and CBP/p300 inhibitors and protein degraders are emerging as promising novel anticancer agents for clinical translation.
Keywords: CBP/p300, gene transcription, tumorigenesis, small molecule inhibitors, cancer therapy
Introduction
Gene transcription is carried out by RNA polymerase (RNA Pol) enzymes, and RNA Pol II is responsible for the synthesis of mRNAs and non-coding RNAs [1]. To start gene transcription, RNA Pol II recognizes the promoter region of the target gene, synthesizes RNA from the DNA duplex, and then moves from the promoter to the gene body (elongation) until RNA synthesis completes at a termination signal [2, 3].
Transcriptional enhancers are short DNA elements which are characterized by acetylated histone H3 lysine 27 (H3K27ac) signals. Transcriptional enhancers bind RNA Pol II, transcription factors and co-regulators to enhance the transcription of neighboring genes, independent of their orientation, as enhancers can loop over long genomic distances to regulate promoter activity [4]. First reported in 2013, super-enhancers span tens of kilobases of DNA sequences, consist of clusters of enhancers, and selectively localize at cell identity gene and critical oncogene loci [5, 6]. Characterized by massive H3K27ac signals, super-enhancers are densely bound by master transcription factors, co-regulators and mediators [6-10].
The histone acetyltransferase CREB-binding protein (CBP) and its closely related p300 protein are often referred to as a single entity (CBP/p300), due to their considerable sequence homology and functional overlap and co-operation [11]. CBP/p300 are required for histone acetylation globally [11-14], at promoters [15], enhancers [12, 13] and super-enhancers [16]. Known as tumor suppressors in certain cancers more than a decade ago [17-19], CBP/p300 have recently been demonstrated as important regulators of enhancer- and super-enhancer-mediated transcriptional activation of critical oncogenes [13, 16, 20], and small molecule CBP/p300 inhibitors are emerging as efficacious anticancer agents [13, 20, 21].
CBP/p300 protein structure and histone acetyltransferase activity
CBP/P300 proteins have intrinsic histone acetyltransferase (HAT) activity due to their HAT domains and, like CBP/p300 associated factor (PCAF), induces the acetylation of histone H3 and H4 [14, 22-24]. The catalytic core of CBP/p300 proteins consists of the bromodomain, HAT, cysteine/histidine-rich region (CH2, comprising PHD and RING) and ZZ-type zinc finger (ZZ) domains (Figure 1A). The bromodomain, HAT and PHD domains establish a structural unit to which the RING and ZZ domains connect [25-27]. The catalytic core is preceded by the N-terminal nuclear receptor interaction domain (NRID), the transcriptional adaptor zinc-finger 1 (TAZ1) and the kinase-inducible CREB interaction region (KIX); and is succeeded by the second TAZ domain (TAZ2) and the interferon-binding domain (IBiD) [28-30] (Figure 1A). Identical mutations in the HAT domains of p300 and CBP reduce acetyltransferase activity to different extents, indicating minor differences between CBP and p300 proteins [31].
Modulation of CBP/p300-induced histone acetylation by histone reader, histone modification proteins and the ubiquitin hydrolase USP24. A. CBP/p300 proteins consist of nuclear receptor interaction domain (NRID), transcriptional-adaptor zinc-finger domain 1 (TAZ1, also known as CH1), kinase inducible domain of CREB interacting domain (KIX), bromodomain (Bromo), PHD finger, histone acetyltransferase domain (HAT) including the autoinhibitory loop (AIL), ZZ-type zinc finger domain (ZZ), TAZ2 and interferon-binding domain (IBiD). B. CBP/p300 interact with BRD4 to induce H3K27ac, and BRG1 is then recruited to acetylated histone sites to enhance H3K27ac and gene transcription. P300 also forms a complex with UTX and MLL4, driving H3K4 mono-methylation (Me1) which further augments H3K27ac and transcriptional activation. In addition, SMARCB1 and other SWI/SNF subunit proteins recruit p300 to distal enhancers, rather than promoters, to induce H3K27ac and gene transcription. C. USP24 decreases p300 protein ubiquitination and proteasome-mediated degradation, thereby increasing p300 protein expression and H3K27ac.
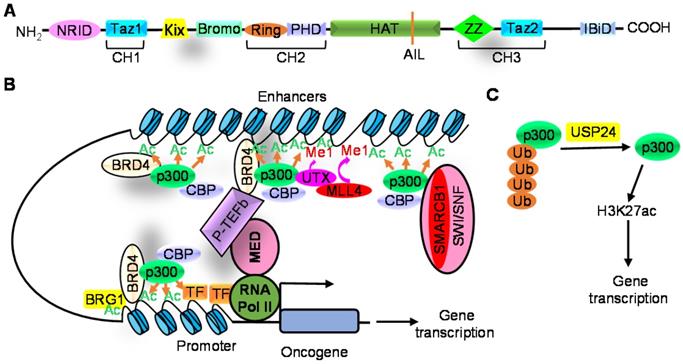
The bromodomain/PHD finger region of CBP/p300 can bind to acetylated HAT domain of CBP/p300 and magnify HAT activity. The binding of the bromodomain to the acetyl-lysine is required for H3 and H4 lysine acetylation [13, 26, 32], inhibition of the bromodomain leads to histone deacetylation [13], and deletion of the PHD finger reduces histone acetylation [33]. The ZZ domain recognizes histone H3 tail and promotes acetylation of histone H3K27 and H3K18 sites [26].
The RING finger of CBP/p300 suppresses HAT activity through blocking the HAT active site [25], and the autoinhibitory loop (AIL) of the HAT domain, when in a hypoacetylated form, also suppresses HAT activity [34-36]. Several lysine residues in the AIL are autoacetylated by the HAT domain, and autoacetylated AIL binds intramolecularly to the bromodomain and facilitates histone H3 acetylation [32, 37].
Interestingly, p300 has been found to form phase-separated condensates in the nucleus, in a HAT domain-dependent manner, and the p300 condensates bind to histone H3 tail but show reduced HAT activity [38]. The p300 condensates at repressed chromatin regions therefore function as a storage pool of p300 with reduced HAT activity [38].
Modulation of CBP/p300-induced histone acetylation
Histone acetylation and consequent transcriptional regulation by CBP/p300 is modulated by the histone “reader” BET bromodomain protein BRD4 [39], histone modification proteins [34, 40-42] and transcription factors [36]. CBP/p300 interacts with BRD4 to induce H3K27ac, and BRG1 is then recruited to acetylated histone sites [39]. In partnership with BRD4 and BRG1, CBP/p300 play an important role in inducing H3K27ac and the transcription of pluripotency genes, such as OCT4 and NANOG, in embryonic stem cells [39] (Figure 1B).
Drosophila Polycomb and its mammalian counterpart CBX proteins directly interact with un-acetylated CBP HAT domain at its autoinhibitory loop and suppresses HAT activity and H3K27ac at gene promoters and enhancers [34]. CBP autoacetylation inhibits Polycomb/CBX binding and augments H3K27ac [34]. The SET domains of Trithorax and Trithorax-related proteins show substantial histone H3K4 mono-methyltransferase activity. CBP, Trithorax and Trithorax-related proteins form complexes, and H3K4 mono-methylation enhances CBP-mediated H3K27ac at transcriptional enhancers [40].
P300 also forms an epigenetic protein complex with the H3K27 demethylase UTX and the H3K4 methyltransferase MLL4. UTX facilitates p300-modulated H3K27ac through recruiting p300 to enhancer and super-enhancer regions; p300, UTX and MLL4 form a feedforward regulatory loop that drives simultaneous H3K4 mono-methylation and H3K27ac on enhancers and super-enhancers to generate an active enhancer landscape; and MLL4-dependent H3K4 mono-methylation further augments CBP/p300-dependent H3K27ac and transcriptional activation [41, 42]. Through forming a protein complex with p300, the SMARCA4, SMARCB1 and SMARCC1 SWI/SNF subunit proteins recruit p300 to distal enhancers, rather than promoters, to induce H3K27ac and enhancer-associated gene transcription [43] (Figure 1B).
Transcription factors stimulate HAT activity, as CBP protein recruited by wild-type, but not dominant-negative mutant, transcription factors shows strong HAT activity [44]. The double homeodomain transcription factor DUX4 interacts with p300/CBP and recruits CBP/p300 to its target genes to induces H3K27ac and gene transcription [45]. Transcription factor ligands such as IRF3 and STAT1 bind to p300, and IRF3 and STAT1 dimerization renders p300 trans-autoacetylation in the HAT domain which augments p300 HAT activity [36].
CBP/p300 HAT activity is augmented by phosphorylation [46, 47], de-ubiquitination [15] and enhancer RNAs [48]. DYRK1A and mTORC1 induce p300 protein phosphorylation which blocks its RING domain from binding to its HAT domain, resulting in suppression of intra-molecular HAT activity inhibition [46, 47]. The ubiquitin hydrolase USP24 directly interacts with p300 protein, and reduces p300 protein ubiquitination and proteasome-mediated degradation, leading to p300 protein up-regulation and histone acetylation [15] (Figure 1C).While CBP is highly enriched at enhancers, enhancer RNAs bind to the RNA-binding region of the CBP HAT domain, resulting in improved substrate binding, magnified HAT activity and up-regulated transcription and expression of neighbouring protein-coding genes [48].
CBP/p300 are required for promoter-, typical enhancer- and super-enhancer-driven gene transcription
CBP/p300 can modulate gene transcription, directly through acetylating histone at gene promoters, typical enhancers and super-enhancers [39, 49-53] or indirectly through inducing the acetylation of transcription factors, such as p53, MYC and GATA-1 [54-56]. CBP/p300 are required for histone acetylation at BRD4-occupied sites and for BRD4 binding at gene promoters and enhancers [57] (Figure 1B). The recruitment of BRD4 by CBP/p300 in turn recruits and activates RNA Pol II and positive transcription elongation factor b (P-TEFb), leading to transcriptional initiation and the release of RNA Pol II from promoter-proximal-pause for elongation [16]. CBP/p300 can also recruit RNA Pol II, independent of BRD4, and stimulate transcription factor IID binding and pre-initiation complex assembly at gene promoters, enhancers and super-enhancers to activate gene transcription [16].
CBP/p300 bromodomain inhibition results in H3K27 deacetylation and reduction in enhancer-associated gene expression [13]. In comparison, CBP/p300 HAT inhibition down-regulates gene transcription, similar to that of CBP/p300 knockout, suggesting that CBP/p300 HAT activity is the main factor for CBP/p300-dependent gene transcription [58].
CBP/p300 play a key role in maintaining cell identity-specific gene expression and therefore cell identity [59]. In embryonic stem cells, the histone variant H3.3 is enriched at enhancers [60]. When phosphorylated, H3.3 stimulates p300 HAT activity to result in H3K27ac at gene enhancers and gene transcription [60]. Inhibition of CBP/p300 HAT activity induces global histone de-acetylation and prevents pluripotent stem cell formation, indicating that HAT activity is required for the function of a number of transcription factors and for reprogramming [59].
In genome- and proteome-wide analyses, CBP/p300 have been found to bind to and acetylate > 200 histone modification proteins, transcription factors and transcriptional co-activators, thereby regulating gene transcription [12]. For example, CBP/p300 bind to and acetylate the sequence-specific DNA-binding sites of p53, c-Myc and GATA-1 proteins to enhance their DNA-binding activity [54-56]. Acetylation of p53 by CBP/p300 leads to p53 protein conformational change and increased transcription of p53 target genes such as p21 and MDM2 [54, 61, 62], acetylation of GATA-1 protein by p300 stimulates GATA-1-mediated gene transcription [55], and acetylation of c-Myc protein at its carboxy-terminal by CBP augments c-Myc target gene transcription [56].
CBP/p300 as tumor suppressors
Reduction in CBP expression due to chromosomal rearrangement, chromosomal deletion or point mutation results in Rubenstein-Taybi Syndrome, and patients are prone to develop tumors [63]. In a mouse model of human myelodysplastic syndrome, deletion of EP300 significantly accelerates leukemogenesis. Mechanistically, EP300 deletion activates the mitogen-activated protein (MAP) kinase and the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathways, suppresses apoptosis and restores hematopoietic progenitor and stem cell self-renewal [64]. In chimeric mice, loss of CBP or p300 leads to up-regulation of NOTCH1, BMI1, MYC, CCNE and SKP2 oncogenes, as well as the development of thymic lymphoma and histiocytic sarcomas [17-19], further confirming the role of CBP/p300 deletion/loss in tumorigenesis.
Chromosomal rearrangements of the ZNF384 gene with CBP and EP300 genes result in CBP-ZNF384 and EP300-ZNF384 fusion genes and proteins [65]. The fusion proteins show loss of CBP/p300 HAT activity in a dominant-negative manner and significantly up-regulate CLCF1 and BTLA gene expression, leading to reduction in hematopoietic stem and progenitor cell differentiation and induction of malignant transformation [65].
CBP/p300 induce oncogene transcription, cancer cell proliferation, survival, tumorigenesis, metastasis and immune evasion
Transcription landscape is substantially reprogrammed during tumorigenesis. EP300 gene is amplified or gained in approximately 24% of human hepatocellular carcinoma tissues, and EP300 gene over-expression positively correlates with the gene copy number variations and poor patient prognosis [66]. P300 substantially reprograms super-enhancers during hepatocellular carcinoma tumorigenesis; stimulates over-expression of super-enhancer-associated oncogenes such as MYC, MYCN and CCND1; and induces hepatocellular carcinoma cell proliferation in vitro and tumor progression in vivo [66] (Table 1).
CBP/p300 induce oncogene transcription, cancer cell proliferation, survival, tumor initiation, tumor progression, metastasis and immune evasion
| Cancer type | Modulation of gene expression | Modulation of tumorigenesis | References |
|---|---|---|---|
| Liver cancer | EP300 gene is amplified/gained, reprograms super-enhancers, and up-regulates MYC and CCND1 | Hepatocellular carcinoma cell proliferation in vitro and tumor progression in mice | [66] |
| Prostate cancer | CBP and p300 bind to androgen receptor-binding sites, and activate MYC transcription | Prostate cancer cell proliferation and androgen deprivation therapy resistance | [67] |
| Melanoma | In melanoma cells, p300 increases MITF and FOXM1 gene transcription | Melanoma cell proliferation | [68] |
| Clear cell renal cell carcinoma | In clear cell renal cell carcinoma, VHL loss leads to p300 to MYC and ZNF395 gene super-enhancers, and MYC and ZNF395 over-expression | Clear cell renal cell carcinoma cell proliferation, survival, and colony formation in vitro and tumor progression in mice | [69] |
| Acute lymphoblastic leukemia | CBP induces super-enhancer formation at MYB binding sites, leading to TAL1 over-expression | T-cell acute lymphoblastic leukemia cell survival and leukemogenesis | [70] |
| Acute myeloid leukemia | CBP/p300 modulate the transcription of genes involved in DNA replication and repair, mitosis and cell cycle progression | Leukemia cell immortalization, cell proliferation, survival and leukemia initiation and maintenance in mice | [57, 71] |
| Chronic myeloid leukemia & lymphoma | P300 binds to the GATA1 and MYC gene super-enhancers and stimulate their over-expression | Chronic myeloid leukemia and lymphoma cell cycle progression and cell proliferation. | [72] |
| Diffuse large B-cell lymphoma | In diffuse large B-cell lymphoma with C-terminal truncated p300, the truncated p300 suppresses NF-κB and REL activity, reduces p53 | Diffuse large B-cell lymphoma cell proliferation | [37, 73] |
| CBP- deficient lung cancer | In CBP-deficient lung cancer cells, p300 up-regulates MYC expression | Lung cancer cell cycle progression, proliferation, survival and tumor progression in mice. | [74] |
| Non-small cell lung cancer | P300 up-regulates IL-6, increases mesenchymal markers and decreases epithelial markers | Non-small cell lung cancer cell migration, invasion and metastasis | [15, 75] |
| Immune cells | In T regulatory cells and myeloid-derived suppressor cells, CBP/p300 up-regulates the expression of STAT pathway genes, FOXP3 and GATA3 | Enhance T regulatory cell and myeloid-derived suppressor cell function and survival; and suppress cytotoxic T cell-driven immunity, lymphocyte activation and proliferation | [77-80] |
RNA sequencing data from primary and metastatic castration-resistant prostate cancer tissues show that CBP and p300 mRNAs are over-expressed in tumor than normal tissues, and that CBP and p300 expression positively correlates with androgen receptor expression, androgen receptor signature, and androgen deprivation therapy resistance [67]. In castration-resistant prostate cancer cells, CBP and p300 up-regulate androgen receptor signaling by binding to androgen receptor-binding sites at target gene promoters, activate the transcription of oncogenes such as MYC, and induce cancer cell proliferation [67] (Table 1).
In melanoma cells, 33 of the 250 genes down-regulated after p300 knockdown are target genes of the transcription factor MITF. P300 up-regulates MITF gene expression by inducing H3K27ac at the MITF gene promoter, resulting in the up-regulation of MITF target oncogenes, such as FOXM1, and melanoma cell proliferation [68]. In clear cell renal cell carcinoma, loss of the tumor suppressor VHL drives the recruitment of p300 to oncogene enhancers and super-enhancers. P300 thereby induces H3K27ac, MYC and ZNF395 oncogene over-expression, clear cell renal cell carcinoma cell proliferation, survival and colony formation in vitro, and tumor progression in mouse models [69] (Table 1).
In T-cell acute lymphoblastic leukemia patients, heterozygous somatic mutations introduce MYB binding motifs upstream of the TAL1 oncogene [70]. MYB binding results in the recruitment of CBP, leading to super-enhancer formation, TAL1 over-expression, cell survival and leukemogenesis [70] (Table 1). In acute myeloid leukemia, CBP/p300 modulate the transcription of genes involved in DNA replication, DNA repair, mitosis and cell cycle progression. CBP/p300 are therefore essential for acute myeloid leukemia cell proliferation, immortalization, leukemia initiation and maintenance [57, 71]. In chronic myeloid leukemia and lymphoma cells, the GATA1 and MYC gene loci are characterized by super-enhancers occupied by p300 [72]. CBP/p300 bromodomain inhibition blocks GATA1 and MYC oncogene expression and induces chronic myeloid leukemia and lymphoma cell cycle arrest and growth inhibition [72]. In diffuse large B-cell lymphoma cells with C-terminal truncated HAT domain-deficient p300, the truncated p300 suppresses NF-κB and REL activity, reduces p53 expression, and is required for lymphoma cell proliferation [37, 73] (Table 1).
In CBP-deficient lung cancer cells, genome-wide gene expression analysis identifies MYC as one of the top genes most significantly up-regulated by p300, and p300 is essential for CBP-deficient lung cancer cell proliferation and tumor progression in vivo [74]. In non-small cell lung cancer, p300 up-regulates the transcription of IL-6 in macrophages and cancer cells, increases the expression of mesenchymal markers and decreases the expression of epithelial markers. P300 thereby promotes cancer cell migration, invasion and metastasis [15, 75] (Table 1).
In myelodysplastic syndrome-derived acute myeloid leukemia cells, CBP/p300 promote the expression of ribosomal genes and CBP/p300 inhibition reduces global protein synthesis, leading to leukemia cell death [76].
The recruitment of T regulatory cells and myeloid-derived suppressor cells to tumor tissues is important for immune evasion. CBP/p300 induce H3K27 acetylation at the promoters and enhancers of genes critical for T regulatory cells and myeloid-derived suppressor cells, such as STAT pathway genes, FOXP3 and GATA3, and up-regulate their expression. CBP/p300 thereby augment T regulatory cell and myeloid-derived suppressor cell survival and function; suppress cytotoxic T cell-driven immunity, lymphocyte activation and proliferation; and promote tumor progression in vivo [77-80] (Table 1).
Small molecule compound CBP/p300 activators and inhibitors
CBP/p300 activators
Small molecule compound CBP/p300 activators and inhibitors have been discovered in the past two decades. CTPB, an amide derivative of anacardic acid, alters p300 protein structure, activates p300 HAT activity, and enhances HAT-dependent transcriptional activation [81]. The short-chain fatty acids butyrate and propionate have long been presumed to inhibit histone deacetylases, however, it has recently been shown that butyrate and propionate are converted to acetyl coenzyme A which catalyzes auto-acetylation of the autoinhibitory loop of p300, leading to HAT activation and histone acetylation [82]. The findings challenge many other publications demonstrating that butyrate acts mainly by inhibiting histone deacetylases [83, 84], and require independent validation.
CBP/p300 HAT inhibitors
Anacardic acid from cashew nutshell and Garcinol from garcinia are weak natural inhibitors of both p300 and PCAF HAT activity [81, 85]. The coenzyme A conjugate Lys-CoA is an effective p300 HAT inhibitor (half-maximal inhibitory concentration, IC50 of 0.5µM) and a much weaker inhibitor of PCAF (IC50 of 200µM) [86]. The isothiazolone-based PCAF and p300 HAT co-inhibitors CCT077791 and CCT077792 reduce histone acetylation and reduce colon cancer cell proliferation by 50% 72 hours after treatment with CCT077791 at 2-3 µM and CCT077792 at 0.4 µM [87]. Through compound screening and medicinal chemistry, “compound 12” was found to selectively suppress CBP/p300 HAT activity with an IC50 of 620 nM. “Compound 12” recapitulates p300 siRNA-mediated reduction in estrogen receptor target gene transcription in breast cancer cells [88].
Through in silico drug discovery pipelines, highly selective and potent drug-like small-molecule compound CBP/p300 HAT inhibitors have been discovered. The pyrazolone-containing p300 HAT inhibitor C646 suppresses p300-induced HAT activity with an inhibitory constant of 400 nM [89] (Table 2).
Small molecule CBP/p300 HAT inhibitors and their anticancer effects
| Compounds | Structures | HAT inhibition and anticancer effects | References |
|---|---|---|---|
| Lys-CoA | 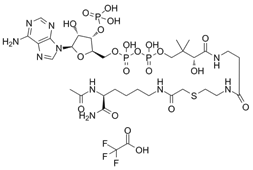 | Suppresses p300-mediated histone acetylation with an IC50 of 0.5µM. | [86] |
| CCT077791 | 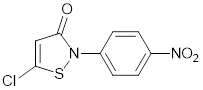 | Reduces histone H3 and H4 acetylation and induces colon cancer cell growth inhibition with an IC50 of 2-3µM. | [87] |
| CCT077792 | 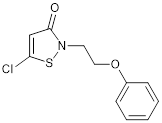 | Reduces histone H3 and H4 acetylation and induces colon cancer cell growth inhibition with an IC50 of 0.4 µM. | [87] |
| C646 | 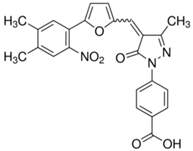 | Suppresses p300-induced HAT activity with an inhibitory constant of 400 nM, reduces oncogene expression, induces melanoma, non-small cell lung cancer and acute myeloid leukemia cell cycle arrest and apoptosis, and sensitizes lymphoma cells to EZH2 inhibitors. | [71, 74, 89, 106] |
| A-485 | 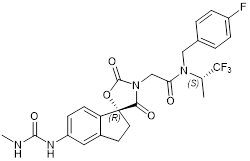 | Suppresses CBP (IC50 = 2.6 nM) and p300 (IC50 = 9.8 nM), reduces estrogen receptor, androgen receptor and MYC gene expression, and show anticancer effects against breast, prostate, leukaemia, myeloma, lymphoma and NUT midline carcinoma. | [67, 90, 97, 103] |
| B026 | 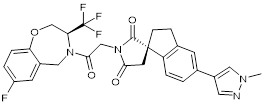 | Suppresses CBP (IC50 = 9.5nM) and p300 (IC50 = 1.8nM), reduces MYC transcription, and blocks acute myeloid leukemia cell proliferation in vitro and leukemia progression in vivo. | [91] |
| B029-2 | 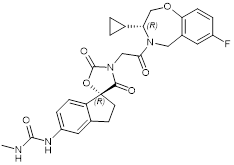 | Suppresses CBP (IC50 = 11nM) and p300 (IC50 = 0.5nM), decreases amino acid metabolism and nucleotide synthesis gene expression, and reduces liver cancer cell proliferation in vitro and tumor progression in vivo. | [92] |
Through virtual ligand screen, A-485 has been found to be a potent, selective and drug-like CBP and p300 HAT inhibitor [90]. A-485 binds to CBP/p300 catalytic active site, and reduces histone acetylation by CBP and p300 with IC50 of 2.6 nM and 9.8 nM respectively [90] (Table 2).
B026 and B029-2, which have similar chemical structure, are very efficient in suppressing CBP (IC50 of 9.5nM for B026 and 11nM for B029-2) and p300 (IC50 of 1.8nM for B026 and 0.5nM for B029-2) [91, 92] HAT activity (Table 2).
CBP/p300 bromodomain inhibitors
The CBP/p300 bromodomain inhibitors CPI703 and CPI644 exhibit good efficacy in inhibiting CBP bromodomain with IC50 values of 0.47 μM and 0.18 μM respectively and cellular EC50 of 2.1 μM and 0.53 μM respectively [93]. Treatment with CPI703 or CPI644 reduces H3K27ac and the transcription of a number of genes including FOXP3 in regulatory T cells, suppresses regulatory T cell differentiation and T helper 17 cell cytokine production [93]. The CBP/p300 bromodomain inhibitor CBP30 suppresses CBP (IC50 = 21 nM) and p300 (IC50 = 38 nM), and reduces IL-17A expression in immune cells and secretion by T helper 17 cells [94] (Table 3).
Small molecule CBP/p300 bromodomain inhibitors and their anticancer effects
| Compounds | Structures | Bromodomain inhibition and anticancer effects | References |
|---|---|---|---|
| CPI703 | 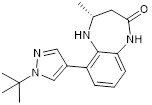 | Suppresses CBP with an IC50 of 0.47 μM and cellular EC50 of 2.1 μM, reduces FOXP3 transcription in regulatory T cells, and suppresses regulatory T cell differentiation and T helper 17 cell cytokine production. | [93] |
| CPI644 | 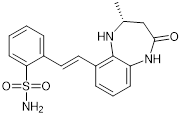 | Inhibits CBP bromodomain with an IC50 of 0.18 μM and cellular EC50 values of 0.53 μM, and reduces the percentage of FOXP3(+) cells in differentiating regulatory T cells. | [93] |
| CBP30 | 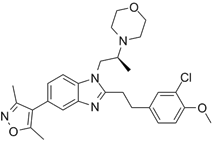 | Suppresses CBP (IC50 = 21 nM) and p300 (IC50 = 38 nM), reduces IL-17A expression in immune cells and secretion by T helper 17 cells, and inhibits MYC and androgen target gene expression and prostate cancer cell proliferation. | [72, 94] |
| I-CBP112 | 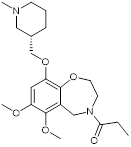 | Inhibits CBP/p300 with dissociation constants of 151 nM for CBP and 167 nM for p300 and IC50 of 142 nM for CBP and 625 nM for p300, reduces immune response and drug resistance genes, and inhibits leukemia cell differentiation and leukemia-initiating potential in mice. | [96, 104] |
| GNE-049 | 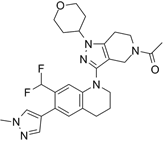 | Suppresses CBP (IC50 = 1.1 nM) and p300 (IC50 = 2.3 nM), and represses the expression of oncogenes, such as MYC (EC50 = 14 nM) and androgen receptor target gene transcription, and induce anticancer effects against castration-resistant prostate cancer in mice. | [97] |
| GNE-781 | 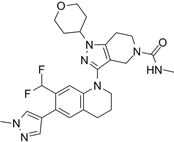 | Suppresses CBP (IC50 = 0.94 nM) and p300 (IC50 = 1.2 nM), reduces MYC and FOXP3 expression, decreases myeloid-derived suppressor and T regulatory cell function, augments tumor immune response, and suppresses colon and breast cancer in mice. | [20, 98] |
| CCS1477 | 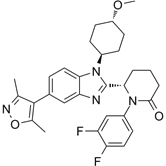 | Binds to CBP and p300 (dissociation constants of 1.7 nM for CBP and 1.3 nM for p300; IC50 =19 nM), reduces androgen receptor coactivator function and androgen target gene transcription, and induce anticancer effects against castration-resistant prostate cancer. Synergizes with azacytidine to induce leukemia cell death. | [67, 76] |
| NEO2734 | 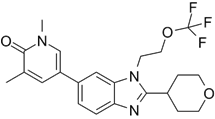 | Shows dissociation constants of 19 nM for CBP, 31 nM for p300 and 6 nM for BRD4, reduces the transcription of MYC target genes and genes involved in chemokine signalling and inflammation, and reduces leukemia, lymphoma and prostate cancer growth in vitro and in vivo. | [102, 108, 109] |
I-CBP112 is a selective CBP/p300 bromodomain ligand and inhibitor [95], and shows dissociation constant of 151 ± 6 nM for CBP and 167 ± 8 nM for p300, and IC50 of 142 nM for CBP and 625 nM for p300 [96] (Table 3).
The chemically synthesized CBP/p300 bromodomain inhibitor GNE-049 displays effective suppression of both CBP (IC50 = 1.1 nM) and p300 (IC50 = 2.3 nM), and exhibits strong efficacy in repressing the expression of oncogenes, such as MYC (EC50 = 14 nM), in leukaemia cells [97]. The structurally similar GNE-781 displays effective suppression of both CBP (IC50 = 0.94 nM) and p300 (IC50 = 1.2 nM), and strong efficacy in repressing the MYC oncogene (EC50 = 6.6 nM) in leukaemia cells [98] (Table 3).
CCS1477 is a potent, selective and orally bioavailable CBP/p300 bromodomain inhibitor. CCS1477 binds to CBP and p300 with dissociation constants of 1.7 nM and 1.3 nM respectively and an IC50 of 19 nM [67] (Table 3).
Proteolysis-targeted-chimaera (PROTAC) p300 protein degrader JQAD1
PROTACs are small molecule compounds that target proteins for E3 ligase-mediated ubiquitination and degradation [99]. JQAD1 binds to p300 protein and target it for ubiquitin proteasome-mediated degradation [100]. JQAD1 induces H3K27 deacetylation at core regulatory circuitry gene enhancers and reduces the transcription of critical oncogenes such as MYCN [100].
CBP/p300 and BET bromodomain dual inhibitors
Due to homology between CBP/p300 and BET bromodomains, CBP/p300 and BET bromodomain dual inhibitors, NEO2734 and NEO1132, have been designed [101]. NEO2734 binds both CBP/p300 and BET proteins with dissociation constants of 19 nM for CBP, 31 nM for p300 and 6 nM for BRD4. In comparison, NEO1132 displays dissociation constants of 61 nM for CBP, 80 nM for p300 and 63 nM for BRD4 [102]. NEO2734 downregulates the transcription of MYC target genes and genes involved in chemokine signalling and inflammation (Table 3) (Figure 2).
The CBP/p300 and BET bromodomain dual inhibitor NEO2734 suppresses H3K27 acetylation and oncogene transcription and expression. CBP/p300 induce H3K27 acetylation and recruit the BET bromodomain protein BRD4 at oncogene promoters, typical enhancers and super-enhancers, leading to oncogene transcriptional activation and over-expression. Treatment with NEO2734 blocks H3K27 acetylation and displaces BRD4 at oncogene promoters, typical enhancers and super-enhancers, leading to oncogene transcriptional suppression.
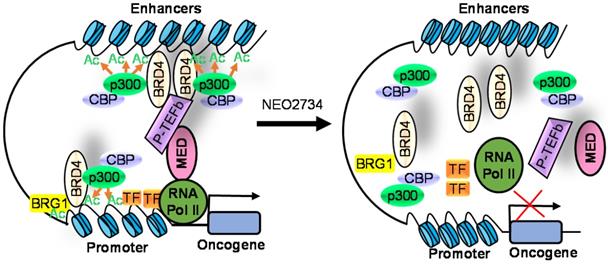
CBP/p300 inhibitors as novel anti-cancer agents
The CBP/p300 HAT inhibitor C646 down-regulates oncogene expression, reduces acute myeloid leukemia cell clonogenic potential, and induces acute myeloid leukemia cell cycle arrest, apoptosis and melanoma and non-small cell lung cancer cell growth arrest [71, 74, 89] (Table 2). Consistent with these findings, the CBP/p300 bromodomain inhibitor I-CBP112 substantially reduces leukemic cell colony formation, induces leukemia cell differentiation, and suppresses leukemia-initiating potential of acute myeloid leukemia cells in vitro and in mouse models [96] (Table 3).
The CBP/p300 HAT inhibitor A-485 shows potent anticancer activity against acute myeloid leukaemia, multiple myeloma and non-Hodgkin's lymphoma cells [90]. A-485 reduces H3K27ac at the enhancers of estrogen receptor α target genes, including MYC and CCND1, down-regulates their expression, and inhibits breast cancer cell proliferation. A-485 and the CBP/p300 bromodomain inhibitors CBP30, GNE-049 and CCS1477 reduce histone acetylation, suppress androgen receptor target gene transcription, and induce considerable anticancer effects against castration-resistant prostate cancer in mouse models [67, 72, 90, 97] (Tables 2 and 3).
In NUT midline carcinoma cells, A-485 reduces histone acetylation, disrupts BRD4-NUT binding, and down-regulates the expression of BRD4-NUT target genes including MYC and TP63. A-485 thereby strongly induces NUT midline carcinoma cell differentiation, cell cycle arrest and apoptosis [103] (Table 2).
The CBP/p300 HAT inhibitor B026 down-regulates MYC oncogene expression, and blocks acute myeloid leukemia cell proliferation in vitro and leukemia progression in vivo [91]. B029-2 decreases amino acid metabolism and nucleotide synthesis gene expression, reduces glycolytic function and nucleotide synthesis, and suppresses hepatocellular carcinoma cell proliferation, migration and invasion in vitro and tumor progression in vivo [92] (Table 2).
In human neuroblastoma tissues, high levels of p300 expression correlates with poor patient prognosis, independent of current prognostic markers [100]. The PROTAC compound JQAD1 selectively targets p300 protein for degradation, reduces the transcription of critical oncogenes such as MYCN, and induces neuroblastoma cell apoptosis in vitro and tumor growth inhibition in mouse models [100].
CBP/p300 inhibitors can also suppress tumor progression by activating immune response. Treatment with the CBP/p300 bromodomain inhibitor GNE-781 converts myeloid-derived suppressor cells from a suppressive to an inflammatory phenotype, decreases myeloid-derived suppressor cell differentiation and function, and impair FOXP3 expression and T regulatory cell function. GNE-781 therefore augments tumor immune response and suppresses tumor growth in mouse models of colon and breast cancers [20, 98] (Table 3).
CBP/p300 inhibitors exert synergistic anticancer effects with other anticancer agents
Synergistic anticancer effects between CBP/p300 inhibitors and chemotherapy, radiotherapy and epigenetic therapy agents
CBP/p300 inhibitors exert synergistic anticancer effects with chemotherapy, radiotherapy and epigenetic therapy agents. Treatment with I-CBP112 decreases the expression of a number of ATP-binding cassette transporter genes in breast, lung and hepatic cancer cell lines, leading to increased accumulation of chemotherapy agents including daunorubicin, doxorubicin and methotrexate inside the cancer cells. I-CBP112 thereby considerably sensitizes cancer cells to a wide range of chemotherapy agents [96, 104]. The CBP/p300 HAT inhibitor C646 sensitizes non-small cell lung cancer cells to radiotherapy by abrogating checkpoint maintenance and augmenting radiotherapy-induced mitotic catastrophe [105].
The DNA demethylation agent azacitidine is the best treatment for severe myelodysplastic syndromes, but patients eventually develop resistance. A loss of function shRNA screen identifies CBP as a top regulator of azacytidine resistance [76]. In myelodysplastic syndrome-derived acute myeloid leukemia cells, CBP/p300 promote the expression of genes important for protein synthesis, and the CBP/p300 HAT inhibitor A-485 and the CBP/p300 bromodomain inhibitor CCS1477 synergize with azacytidine to induce myelodysplastic syndrome-derived leukemia cell death by synergistically suppressing global protein synthesis [76].
EP300 gene transcription is up-regulated by EZH2 inhibitors, p300 drives oncogenic transcriptional reprograming and cancer cell resistance to EZH2 inhibitors, and C646 sensitizes diffuse large B-cell lymphoma cells to EZH2 inhibitors [106]. In addition, A-485 induces H3K27 deacetylation and exerts synergistic anticancer effects with the histone demethylase KDM6A inhibitor GSK-J4 against multiple myeloma [20].
Interestingly, CBP/p300 HAT inhibitors and bromodomain inhibitors exert synergistic anticancer effects with each other and with BET bromodomain inhibitors. A-485 and I-CBP112 co-operatively reduce CBP/p300 occupancy at chromatin and the expression of androgen-dependent oncogenes such as MYC and PSA, and synergistically suppress prostate cancer cell proliferation [107]. NUT midline carcinoma is characterized by NUT gene rearrangement with the BET bromodomain gene BRD4. Screening of an epigenetic compound library has identified the CBP/p300 HAT inhibitor A-485 and the BET bromodomain inhibitor JQ1 as the most effective in inducing NUT midline carcinoma cell growth inhibition and cell death [103]. A-485 suppresses p300-mediated histone acetylation and the transcription of key oncogenes such as MYC; and combination therapy with A-485 and JQ1 synergistically suppresses Wnt/β catenin pathway and c-Myc target gene expression, and synergistically induces NUT midline carcinoma cell apoptosis [103]. Similarly, I-CBP112 and the BET bromodomain inhibitor JQ1 synergistically induce leukemia cell apoptosis [96].
Anticancer efficacy of the CBP/p300 and BET bromodomain dual inhibitors
In a panel of 60 cancer cell lines from different cancers, NEO1132 and NEO2734 display anti-proliferative effects in a number of cancer cell lines, with the most potent efficacy in leukemia, lymphoma and prostate cancer cells [102]. NEO1132 and NEO2734 efficiently induce acute myeloid leukemia cell apoptosis, eliminates leukemic stem/progenitor cells, and enhance the anticancer effects of chemotherapies in mouse models of acute myeloid leukemia [108]. NEO2374 induces transcriptional changes in lymphoma cells, significantly suppresses lymphoma progression in mice, and is more effective than CBP/p300 inhibitors or BET bromodomain alone [102].
In castration-resistant prostate cancer cells, treatment with NEO1132 or NEO2734 reduces H3K27ac at androgen receptor binding site-gained cell lineage and cancer-promoting gene loci, down-regulates their expression, induces castration-resistant prostate cancer cell growth inhibition and cell death in vitro, and abrogates tumors in mouse models [109].
CBP/p300 inhibitors in clinical trials
Two CBP/p300 inhibitors are currently in clinical trials in cancer patients. CCS1477 is the first p300/CBP inhibitor in phase I/IIa clinical trials in patients with advanced solid tumours, such as MYC over-expressing small cell lung cancer, radiation-induced breast sarcoma and metastatic castrate-resistant prostate cancer (ClinicalTrials.gov Identifier: NCT03568656) [110]. CCS1477 has also been evaluated in an open-label Phase I/IIa study as monotherapy in patients with advanced haematological malignancies including acute myeloid leukemia, non-Hodgkin lymphoma and multiple myeloma (ClinicalTrials.gov Identifier NCT04068597).
While no published pre-clinical information is available, the orally active, potent and selective CBP/p300 bromodomain inhibitor FT-7051 has entered a multi-center, phase I, open-label clinical trial in patients with metastatic castration-resistant prostate cancer (ClinicalTrials.gov Identifier NCT04575766) [111]. Analysis of the first 5 enrolled patients has shown that FT-7051 causes minor side-effects including hyperglycemia [111].
Conclusions and perspectives
The histone acetyltransferase CBP/p300 contains HAT domain and bromodomain which are required for H3K27 acetylation. CBP/p300-mediated H3K27ac is augmented by the histone “reader” BRD4, histone modification proteins, such as CBX, Trithorax, Trithorax-related proteins, UTX, MLL4 and SWI/SNF subunit proteins, transcription factors such as DUX4, the ubiquitin hydrolase USP24 and enhancer RNAs. CBP/p300 catalyzes histone H3K27ac at gene promoters, enhancers and super-enhancers, leading to transcriptional initiation and productive elongation. While CBP/p300 deletion/loss was known to promote tumorigenesis more than a decade ago, CBP/p300 have recently been demonstrated to be over-expressed or activated in cancer cells, induce enhancer and super-enhancer activity, and up-regulate the expression of important oncogenes, such as MYC, MYCN, CCND1 and ZNF395. CBP/p300 therefore induce cancer cell proliferation, survival, migration, invasion, immune evasion, tumor initiation, progression and drug resistance in a number cancer types, such as leukemia, lymphoma, melanoma, hepatocellular carcinoma, lung and renal cancers.
It is currently not clear why CBP/p300 loss and over-expression/activity can both facilitate tumorigenesis. Possible explanations include: (i) CBP/p300 loss due to genetic deletion or rearrangement reduces the expression of tumor suppressor genes, the promoters of which are bound by CBP/p300; while CBP/p300 over-expression/activity leads to enhancer and super-enhancer formation at critical oncogene loci, leading to oncogene over-expression; (ii) cellular context and different expression levels of transcriptional co-factors alter CBP/p300 protein accessibility to oncogenic and tumor suppressive target gene loci; and (iii) CBP loss leads to over-activity of p300 and oncogene over-expression.
Originally regarded as a histone deacetylase inhibitor, the anticancer agent butyrate has now been confirmed to be CBP/p300 HAT activator. CBP/p300 HAT inhibitors and bromodomain inhibitors have been discovered through compound library screen, in silico compound screen and chemical synthesis. Among them, the CBP/p300 HAT inhibitors A-485, B026 and B029-2, and the bromodomain inhibitors GNE-049, GNE-781 and CCS1477 suppress CBP/p300 at < 10 nM. Other promising CBP/p300 targeting compounds include the PROTAC compound JQAD1 which induces p300 protein degradation, and the CBP/p300 and BET bromodomain dual inhibitor NEO2734. These CBP/p300 inhibitors and degrader reduce H3K27ac at oncogene promoters, enhancers and super-enhancer, down-regulate oncogene expression, induce cancer cell growth inhibition and apoptosis, suppress cancer cell migration, invasion, immune evasion and tumor progression. In addition, the CBP/p300 inhibitors exert synergistic anticancer effects with other anticancer agents such as chemotherapy, radiotherapy, EZH2 inhibitors and BET bromodomain inhibitors. Importantly, the CBP/p300 inhibitors CCS147 and FT-7051 are now in clinical trials in cancer patients.
While small molecule CBP/p300 HAT and bromodomain inhibitors have been the main focus of drug discovery research, the first PROTAC p300 protein degrader JQAD1 has recently been reported. Due to high homology between CBP and p300, current CBP/p300 HAT and bromodomain inhibitors suppress both of the enzymes with similar efficiency. In comparison, the PROTAC p300 degrader JQAD1 shows strong selectivity for p300 [100]. For clinical translation, CBP/p300 HAT and bromodomain inhibitors are expected to be preferred when both CBP and p300 are involved in tumorigenesis, such as prostate cancer with the over-expression of both CBP and p300 [67], while PROTAC CBP or p300 protein degraders are required when only CBP or p300 is the tumorigenic driver, such as in CBP-deficient but p300 active lung cancer [74] as well as CBP non-functional but p300 active MYCN gene-amplified neuroblastoma [100].
Future endeavours should focus on developing more potent and selective small-molecule CBP/p300 HAT inhibitors, CBP/p300 and BET bromodomain co-inhibitors, and PROTAC CBP or p300 selective protein degraders through structure-based virtual screen, laboratory-based screen of small molecule compound libraries and chemical synthesis. Their safety to normal cells, pharmacokinetics and anticancer efficacy should be investigated in vitro and in mouse models. The other anticancer agent which exerts the best synergistic anticancer effects with CBP/p300 inhibitors or protein degraders can then be identified through approved oncology drug library screen. Ultimately, ideal combination therapies with CBP/p300 inhibitors or degraders and other anticancer drugs are expected to be tested in patients with cancers characterized by CBP and/or p300 over-expression or over-activity, and potential side-effects from the CBP/p300 inhibitors, degraders or combination therapies will be identified.
Abbreviations
AIL: autoinhibitory loop; CBP: CREB-binding protein; CH2: cysteine/histidine-rich region; HAT: histone acetyltransferase; H3K27: histone H3 lysine 27; H3K27ac: histone H3 lysine 27 acetylation; IBiD: interferon-binding domain; IC50: half-maximal inhibitory concentration; Janus kinase: JAK; KIX: kinase inducible domain of CREB interacting domain; mitogen-activated protein: MAP; NRID: nuclear receptor interaction domain; PCAF: p300/CBP associated factor; PROTAC: proteolysis-targeted-chimaera; P-TEFb: positive transcription elongation factor b; RNA Pol: RNA polymerase; RNA Pol II: RNA polymerase II; signal transducer and activator of transcription: STAT; TAZ1: transcriptional-adaptor zinc-finger domain 1; TAZ2: transcriptional-adaptor zinc-finger domain 2; ZZ: ZZ-type zinc finger domain.
Acknowledgements
Children's Cancer Institute Australia is affiliated with UNSW Sydney and Sydney Children's Hospitals Network. The authors were supported by National Health & Medical Research Council Australia, National Natural Science Foundation of China and Chongqing Science and Health Joint Medical Research Project (2020MSXM007).
Author contributions
XDZ, LRZ and TL designed this review. QC, BY, XL, XDZ, LRZ and TL drafted and revised this manuscript, tables and figures. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Vannini A, Cramer P. Conservation between the RNA polymerase I, II, and III transcription initiation machineries. Mol Cell. 2012;45:439-46
2. Haberle V, Stark A. Eukaryotic core promoters and the functional basis of transcription initiation. Nat Rev Mol Cell Biol. 2018;19:621-37
3. Cramer P. Organization and regulation of gene transcription. Nature. 2019;573:45-54
4. Sur I, Taipale J. The role of enhancers in cancer. Nat Rev Cancer. 2016;16:483-93
5. Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR. et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320-34
6. Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA. et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934-47
7. Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH. et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307-19
8. Chen J, Nelson C, Wong M, Tee AE, Liu PY, La T. et al. Targeted Therapy of TERT-Rearranged Neuroblastoma with BET Bromodomain Inhibitor and Proteasome Inhibitor Combination Therapy. Clin Cancer Res. 2021;27:1438-51
9. Tee AE, Ciampa OC, Wong M, Fletcher JI, Kamili A, Chen J. et al. Combination therapy with the CDK7 inhibitor and the tyrosine kinase inhibitor exerts synergistic anticancer effects against MYCN-amplified neuroblastoma. Int J Cancer. 2020;147:1928-38
10. Wong M, Sun Y, Xi Z, Milazzo G, Poulos RC, Bartenhagen C. et al. JMJD6 is a tumorigenic factor and therapeutic target in neuroblastoma. Nat Commun. 2019;10:3319
11. Jin Q, Yu LR, Wang L, Zhang Z, Kasper LH, Lee JE. et al. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. Embo j. 2011;30:249-62
12. Weinert BT, Narita T, Satpathy S, Srinivasan B, Hansen BK, Schölz C. et al. Time-Resolved Analysis Reveals Rapid Dynamics and Broad Scope of the CBP/p300 Acetylome. Cell. 2018;174:231-44.e12
13. Raisner R, Kharbanda S, Jin L, Jeng E, Chan E, Merchant M. et al. Enhancer Activity Requires CBP/P300 Bromodomain-Dependent Histone H3K27 Acetylation. Cell Rep. 2018;24:1722-9
14. Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953-9
15. Wang YC, Wu YS, Hung CY, Wang SA, Young MJ, Hsu TI. et al. USP24 induces IL-6 in tumor-associated microenvironment by stabilizing p300 and β-TrCP and promotes cancer malignancy. Nat Commun. 2018;9:3996
16. Narita T, Ito S, Higashijima Y, Chu WK, Neumann K, Walter J. et al. Enhancers are activated by p300/CBP activity-dependent PIC assembly, RNAPII recruitment, and pause release. Mol Cell. 2021;81:2166-82.e6
17. Rebel VI, Kung AL, Tanner EA, Yang H, Bronson RT, Livingston DM. Distinct roles for CREB-binding protein and p300 in hematopoietic stem cell self-renewal. Proc Natl Acad Sci U S A. 2002;99:14789-94
18. Kung AL, Rebel VI, Bronson RT, Ch'ng LE, Sieff CA, Livingston DM. et al. Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP. Genes Dev. 2000;14:272-7
19. Kang-Decker N, Tong C, Boussouar F, Baker DJ, Xu W, Leontovich AA. et al. Loss of CBP causes T cell lymphomagenesis in synergy with p27Kip1 insufficiency. Cancer Cell. 2004;5:177-89
20. Hogg SJ, Motorna O, Cluse LA, Johanson TM, Coughlan HD, Raviram R. et al. Targeting histone acetylation dynamics and oncogenic transcription by catalytic P300/CBP inhibition. Mol Cell. 2021;81:2183-200.e13
21. Huang Y, Mouttet B, Warnatz HJ, Risch T, Rietmann F, Frommelt F. et al. The Leukemogenic TCF3-HLF Complex Rewires Enhancers Driving Cellular Identity and Self-Renewal Conferring EP300 Vulnerability. Cancer Cell. 2019;36:630-44.e9
22. Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641-3
23. Pasini D, Malatesta M, Jung HR, Walfridsson J, Willer A, Olsson L. et al. Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of Polycomb group target genes. Nucleic Acids Res. 2010;38:4958-69
24. Tie F, Banerjee R, Stratton CA, Prasad-Sinha J, Stepanik V, Zlobin A. et al. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development. 2009;136:3131-41
25. Delvecchio M, Gaucher J, Aguilar-Gurrieri C, Ortega E, Panne D. Structure of the p300 catalytic core and implications for chromatin targeting and HAT regulation. Nat Struct Mol Biol. 2013;20:1040-6
26. Zhang Y, Xue Y, Shi J, Ahn J, Mi W, Ali M. et al. The ZZ domain of p300 mediates specificity of the adjacent HAT domain for histone H3. Nat Struct Mol Biol. 2018;25:841-9
27. Kalkhoven E, Teunissen H, Houweling A, Verrijzer CP, Zantema A. The PHD type zinc finger is an integral part of the CBP acetyltransferase domain. Mol Cell Biol. 2002;22:1961-70
28. De Guzman RN, Liu HY, Martinez-Yamout M, Dyson HJ, Wright PE. Solution structure of the TAZ2 (CH3) domain of the transcriptional adaptor protein CBP. J Mol Biol. 2000;303:243-53
29. Kasper LH, Boussouar F, Ney PA, Jackson CW, Rehg J, van Deursen JM. et al. A transcription-factor-binding surface of coactivator p300 is required for haematopoiesis. Nature. 2002;419:738-43
30. Xu W, Chen H, Du K, Asahara H, Tini M, Emerson BM. et al. A transcriptional switch mediated by cofactor methylation. Science. 2001;294:2507-11
31. Bordoli L, Hüsser S, Lüthi U, Netsch M, Osmani H, Eckner R. Functional analysis of the p300 acetyltransferase domain: the PHD finger of p300 but not of CBP is dispensable for enzymatic activity. Nucleic Acids Res. 2001;29:4462-71
32. Park S, Stanfield RL, Martinez-Yamout MA, Dyson HJ, Wilson IA, Wright PE. Role of the CBP catalytic core in intramolecular SUMOylation and control of histone H3 acetylation. Proc Natl Acad Sci U S A. 2017;114:E5335-e42
33. Rack JGM, Lutter T, Kjæreng Bjerga GE, Guder C, Ehrhardt C, Värv S. et al. The PHD finger of p300 influences its ability to acetylate histone and non-histone targets. J Mol Biol. 2014;426:3960-72
34. Tie F, Banerjee R, Fu C, Stratton CA, Fang M, Harte PJ. Polycomb inhibits histone acetylation by CBP by binding directly to its catalytic domain. Proc Natl Acad Sci U S A. 2016;113:E744-53
35. Thompson PR, Wang D, Wang L, Fulco M, Pediconi N, Zhang D. et al. Regulation of the p300 HAT domain via a novel activation loop. Nat Struct Mol Biol. 2004;11:308-15
36. Ortega E, Rengachari S, Ibrahim Z, Hoghoughi N, Gaucher J, Holehouse AS. et al. Transcription factor dimerization activates the p300 acetyltransferase. Nature. 2018;562:538-44
37. Haery L, Lugo-Picó JG, Henry RA, Andrews AJ, Gilmore TD. Histone acetyltransferase-deficient p300 mutants in diffuse large B cell lymphoma have altered transcriptional regulatory activities and are required for optimal cell growth. Mol Cancer. 2014;13:29
38. Zhang Y, Brown K, Yu Y, Ibrahim Z, Zandian M, Xuan H. et al. Nuclear condensates of p300 formed though the structured catalytic core can act as a storage pool of p300 with reduced HAT activity. Nat Commun. 2021;12:4618
39. Wu T, Kamikawa YF, Donohoe ME. Brd4's Bromodomains Mediate Histone H3 Acetylation and Chromatin Remodeling in Pluripotent Cells through P300 and Brg1. Cell Rep. 2018;25:1756-71
40. Tie F, Banerjee R, Saiakhova AR, Howard B, Monteith KE, Scacheri PC. et al. Trithorax monomethylates histone H3K4 and interacts directly with CBP to promote H3K27 acetylation and antagonize Polycomb silencing. Development. 2014;141:1129-39
41. Wang SP, Tang Z, Chen CW, Shimada M, Koche RP, Wang LH. et al. A UTX-MLL4-p300 Transcriptional Regulatory Network Coordinately Shapes Active Enhancer Landscapes for Eliciting Transcription. Mol Cell. 2017;67:308-21.e6
42. Lai B, Lee JE, Jang Y, Wang L, Peng W, Ge K. MLL3/MLL4 are required for CBP/p300 binding on enhancers and super-enhancer formation in brown adipogenesis. Nucleic Acids Res. 2017;45:6388-403
43. Alver BH, Kim KH, Lu P, Wang X, Manchester HE, Wang W. et al. The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat Commun. 2017;8:14648
44. Soutoglou E, Viollet B, Vaxillaire M, Yaniv M, Pontoglio M, Talianidis I. Transcription factor-dependent regulation of CBP and P/CAF histone acetyltransferase activity. Embo j. 2001;20:1984-92
45. Choi SH, Gearhart MD, Cui Z, Bosnakovski D, Kim M, Schennum N. et al. DUX4 recruits p300/CBP through its C-terminus and induces global H3K27 acetylation changes. Nucleic Acids Res. 2016;44:5161-73
46. Wan W, You Z, Xu Y, Zhou L, Guan Z, Peng C. et al. mTORC1 Phosphorylates Acetyltransferase p300 to Regulate Autophagy and Lipogenesis. Mol Cell. 2017;68:323-35.e6
47. Li S, Xu C, Fu Y, Lei PJ, Yao Y, Yang W. et al. DYRK1A interacts with histone acetyl transferase p300 and CBP and localizes to enhancers. Nucleic Acids Res. 2018;46:11202-13
48. Bose DA, Donahue G, Reinberg D, Shiekhattar R, Bonasio R, Berger SL. RNA Binding to CBP Stimulates Histone Acetylation and Transcription. Cell. 2017;168:135-49.e22
49. An W, Palhan VB, Karymov MA, Leuba SH, Roeder RG. Selective requirements for histone H3 and H4 N termini in p300-dependent transcriptional activation from chromatin. Mol Cell. 2002;9:811-21
50. Sen P, Lan Y, Li CY, Sidoli S, Donahue G, Dou Z. et al. Histone Acetyltransferase p300 Induces De Novo Super-Enhancers to Drive Cellular Senescence. Mol Cell. 2019;73:684-98.e8
51. Martinez-Balbás MA, Bannister AJ, Martin K, Haus-Seuffert P, Meisterernst M, Kouzarides T. The acetyltransferase activity of CBP stimulates transcription. Embo j. 1998;17:2886-93
52. Kundu TK, Palhan VB, Wang Z, An W, Cole PA, Roeder RG. Activator-dependent transcription from chromatin in vitro involving targeted histone acetylation by p300. Mol Cell. 2000;6:551-61
53. Chan SH, Tang Y, Miao L, Darwich-Codore H, Vejnar CE, Beaudoin JD. et al. Brd4 and P300 Confer Transcriptional Competency during Zygotic Genome Activation. Dev Cell. 2019;49:867-81.e8
54. Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595-606
55. Boyes J, Byfield P, Nakatani Y, Ogryzko V. Regulation of activity of the transcription factor GATA-1 by acetylation. Nature. 1998;396:594-8
56. Vervoorts J, Lüscher-Firzlaff JM, Rottmann S, Lilischkis R, Walsemann G, Dohmann K. et al. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep. 2003;4:484-90
57. Roe JS, Mercan F, Rivera K, Pappin DJ, Vakoc CR. BET Bromodomain Inhibition Suppresses the Function of Hematopoietic Transcription Factors in Acute Myeloid Leukemia. Mol Cell. 2015;58:1028-39
58. Yokomizo C, Yamaguchi K, Itoh Y, Nishimura T, Umemura A, Minami M. et al. High expression of p300 in HCC predicts shortened overall survival in association with enhanced epithelial mesenchymal transition of HCC cells. Cancer Lett. 2011;310:140-7
59. Ebrahimi A, Sevinç K, Gürhan Sevinç G, Cribbs AP, Philpott M, Uyulur F. et al. Bromodomain inhibition of the coactivators CBP/EP300 facilitate cellular reprogramming. Nat Chem Biol. 2019;15:519-28
60. Martire S, Gogate AA, Whitmill A, Tafessu A, Nguyen J, Teng YC. et al. Phosphorylation of histone H3.3 at serine 31 promotes p300 activity and enhancer acetylation. Nat Genet. 2019;51:941-6
61. Tang Z, Chen WY, Shimada M, Nguyen UT, Kim J, Sun XJ. et al. SET1 and p300 act synergistically, through coupled histone modifications, in transcriptional activation by p53. Cell. 2013;154:297-310
62. Gu W, Shi XL, Roeder RG. Synergistic activation of transcription by CBP and p53. Nature. 1997;387:819-23
63. Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M. et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348-51
64. Cheng G, Liu F, Asai T, Lai F, Man N, Xu H. et al. Loss of p300 accelerates MDS-associated leukemogenesis. Leukemia. 2017;31:1382-90
65. Qian M, Zhang H, Kham SK, Liu S, Jiang C, Zhao X. et al. Whole-transcriptome sequencing identifies a distinct subtype of acute lymphoblastic leukemia with predominant genomic abnormalities of EP300 and CREBBP. Genome Res. 2017;27:185-95
66. Tsang FH, Law CT, Tang TC, Cheng CL, Chin DW, Tam WV. et al. Aberrant Super-Enhancer Landscape in Human Hepatocellular Carcinoma. Hepatology. 2019;69:2502-17
67. Welti J, Sharp A, Brooks N, Yuan W, McNair C, Chand SN. et al. Targeting the p300/CBP Axis in Lethal Prostate Cancer. Cancer Discov. 2021;11:1118-37
68. Kim E, Zucconi BE, Wu M, Nocco SE, Meyers DJ, McGee JS. et al. MITF Expression Predicts Therapeutic Vulnerability to p300 Inhibition in Human Melanoma. Cancer Res. 2019;79:2649-61
69. Yao X, Tan J, Lim KJ, Koh J, Ooi WF, Li Z. et al. VHL Deficiency Drives Enhancer Activation of Oncogenes in Clear Cell Renal Cell Carcinoma. Cancer Discov. 2017;7:1284-305
70. Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD. et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346:1373-7
71. Giotopoulos G, Chan WI, Horton SJ, Ruau D, Gallipoli P, Fowler A. et al. The epigenetic regulators CBP and p300 facilitate leukemogenesis and represent therapeutic targets in acute myeloid leukemia. Oncogene. 2016;35:279-89
72. Garcia-Carpizo V, Ruiz-Llorente S, Sarmentero J, Graña-Castro O, Pisano DG, Barrero MJ. CREBBP/EP300 bromodomains are critical to sustain the GATA1/MYC regulatory axis in proliferation. Epigenetics Chromatin. 2018;11:30
73. Haery L, Mussakhan S, Waxman DJ, Gilmore TD. Evidence for an oncogenic modifier role for mutant histone acetyltransferases in diffuse large B-cell lymphoma. Leuk Lymphoma. 2016;57:2661-71
74. Ogiwara H, Sasaki M, Mitachi T, Oike T, Higuchi S, Tominaga Y. et al. Targeting p300 Addiction in CBP-Deficient Cancers Causes Synthetic Lethality by Apoptotic Cell Death due to Abrogation of MYC Expression. Cancer Discov. 2016;6:430-45
75. Hou X, Gong R, Zhan J, Zhou T, Ma Y, Zhao Y. et al. p300 promotes proliferation, migration, and invasion via inducing epithelial-mesenchymal transition in non-small cell lung cancer cells. BMC Cancer. 2018;18:641
76. Diesch J, Le Pannérer MM, Winkler R, Casquero R, Muhar M, van der Garde M. et al. Inhibition of CBP synergizes with the RNA-dependent mechanisms of Azacitidine by limiting protein synthesis. Nat Commun. 2021;12:6060
77. Liu Y, Wang L, Han R, Beier UH, Akimova T, Bhatti T. et al. Two histone/protein acetyltransferases, CBP and p300, are indispensable for Foxp3+ T-regulatory cell development and function. Mol Cell Biol. 2014;34:3993-4007
78. de Almeida Nagata DE, Chiang EY, Jhunjhunwala S, Caplazi P, Arumugam V, Modrusan Z. et al. Regulation of Tumor-Associated Myeloid Cell Activity by CBP/EP300 Bromodomain Modulation of H3K27 Acetylation. Cell Rep. 2019;27:269-81.e4
79. Liu Y, Wang L, Predina J, Han R, Beier UH, Wang LC. et al. Inhibition of p300 impairs Foxp3⁺ T regulatory cell function and promotes antitumor immunity. Nat Med. 2013;19:1173-7
80. Xiao Y, Nagai Y, Deng G, Ohtani T, Zhu Z, Zhou Z. et al. Dynamic interactions between TIP60 and p300 regulate FOXP3 function through a structural switch defined by a single lysine on TIP60. Cell Rep. 2014;7:1471-80
81. Balasubramanyam K, Swaminathan V, Ranganathan A, Kundu TK. Small molecule modulators of histone acetyltransferase p300. J Biol Chem. 2003;278:19134-40
82. Thomas SP, Denu JM. Short-chain fatty acids activate acetyltransferase p300. Elife. 2021 10
83. Saito A, Yamashita T, Mariko Y, Nosaka Y, Tsuchiya K, Ando T. et al. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc Natl Acad Sci U S A. 1999;96:4592-7
84. Liang G, Taranova O, Xia K, Zhang Y. Butyrate promotes induced pluripotent stem cell generation. J Biol Chem. 2010;285:25516-21
85. Balasubramanyam K, Altaf M, Varier RA, Swaminathan V, Ravindran A, Sadhale PP. et al. Polyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. J Biol Chem. 2004;279:33716-26
86. Lau OD, Kundu TK, Soccio RE, Ait-Si-Ali S, Khalil EM, Vassilev A. et al. HATs off: selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Mol Cell. 2000;5:589-95
87. Stimson L, Rowlands MG, Newbatt YM, Smith NF, Raynaud FI, Rogers P. et al. Isothiazolones as inhibitors of PCAF and p300 histone acetyltransferase activity. Mol Cancer Ther. 2005;4:1521-32
88. Wu F, Hua Y, Kaochar S, Nie S, Lin YL, Yao Y. et al. Discovery, Structure-Activity Relationship, and Biological Activity of Histone-Competitive Inhibitors of Histone Acetyltransferases P300/CBP. J Med Chem. 2020;63:4716-31
89. Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA. et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem Biol. 2010;17:471-82
90. Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, Digiammarino EL. et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature. 2017;550:128-32
91. Yang Y, Zhang R, Li Z, Mei L, Wan S, Ding H. et al. Discovery of Highly Potent, Selective, and Orally Efficacious p300/CBP Histone Acetyltransferases Inhibitors. J Med Chem. 2020;63:1337-60
92. Cai LY, Chen SJ, Xiao SH, Sun QJ, Ding CH, Zheng BN. et al. Targeting p300/CBP Attenuates Hepatocellular Carcinoma Progression through Epigenetic Regulation of Metabolism. Cancer Res. 2021;81:860-72
93. Ghosh S, Taylor A, Chin M, Huang HR, Conery AR, Mertz JA. et al. Regulatory T Cell Modulation by CBP/EP300 Bromodomain Inhibition. J Biol Chem. 2016;291:13014-27
94. Hammitzsch A, Tallant C, Fedorov O, O'Mahony A, Brennan PE, Hay DA. et al. CBP30, a selective CBP/p300 bromodomain inhibitor, suppresses human Th17 responses. Proc Natl Acad Sci U S A. 2015;112:10768-73
95. Hay DA, Fedorov O, Martin S, Singleton DC, Tallant C, Wells C. et al. Discovery and optimization of small-molecule ligands for the CBP/p300 bromodomains. J Am Chem Soc. 2014;136:9308-19
96. Picaud S, Fedorov O, Thanasopoulou A, Leonards K, Jones K, Meier J. et al. Generation of a Selective Small Molecule Inhibitor of the CBP/p300 Bromodomain for Leukemia Therapy. Cancer Res. 2015;75:5106-19
97. Jin L, Garcia J, Chan E, de la Cruz C, Segal E, Merchant M. et al. Therapeutic Targeting of the CBP/p300 Bromodomain Blocks the Growth of Castration-Resistant Prostate Cancer. Cancer Res. 2017;77:5564-75
98. Romero FA, Murray J, Lai KW, Tsui V, Albrecht BK, An L. et al. GNE-781, A Highly Advanced Potent and Selective Bromodomain Inhibitor of Cyclic Adenosine Monophosphate Response Element Binding Protein, Binding Protein (CBP). J Med Chem. 2017;60:9162-83
99. Bondeson DP, Mares A, Smith IE, Ko E, Campos S, Miah AH. et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol. 2015;11:611-7
100. Durbin AD, Wang T, Wimalasena VK, Zimmerman MW, Li D, Dharia NV. et al. EP300 selectively controls the enhancer landscape of MYCN-amplified neuroblastoma. Cancer Discov. 2022;12:730-51
101. Giles F, Witcher M, Brown B. NEO2734: A novel potent oral dual BET and P300/CBP inhibitor. Ann Oncol. 2018 29(suppl 8)
102. Spriano F, Gaudio E, Cascione L, Tarantelli C, Melle F, Motta G. et al. Antitumor activity of the dual BET and CBP/EP300 inhibitor NEO2734. Blood Adv. 2020;4:4124-35
103. Zhang X, Zegar T, Lucas A, Morrison-Smith C, Knox T, French CA. et al. Therapeutic targeting of p300/CBP HAT domain for the treatment of NUT midline carcinoma. Oncogene. 2020;39:4770-9
104. Strachowska M, Gronkowska K, Michlewska S, Robaszkiewicz A. CBP/p300 Bromodomain Inhibitor-I-CBP112 Declines Transcription of the Key ABC Transporters and Sensitizes Cancer Cells to Chemotherapy Drugs. Cancers (Basel). 2021 13
105. Oike T, Komachi M, Ogiwara H, Amornwichet N, Saitoh Y, Torikai K. et al. C646, a selective small molecule inhibitor of histone acetyltransferase p300, radiosensitizes lung cancer cells by enhancing mitotic catastrophe. Radiother Oncol. 2014;111:222-7
106. Huang X, Yan J, Zhang M, Wang Y, Chen Y, Fu X. et al. Targeting Epigenetic Crosstalk as a Therapeutic Strategy for EZH2-Aberrant Solid Tumors. Cell. 2018;175:186-99.e19
107. Zucconi BE, Makofske JL, Meyers DJ, Hwang Y, Wu M, Kuroda MI. et al. Combination Targeting of the Bromodomain and Acetyltransferase Active Site of p300/CBP. Biochemistry. 2019;58:2133-43
108. van Gils N, Martiañez Canales T, Vermue E, Rutten A, Denkers F, van der Deure T. et al. The Novel Oral BET-CBP/p300 Dual Inhibitor NEO2734 Is Highly Effective in Eradicating Acute Myeloid Leukemia Blasts and Stem/Progenitor Cells. Hemasphere. 2021;5:e610
109. He Y, Wei T, Ye Z, Orme JJ, Lin D, Sheng H. et al. A noncanonical AR addiction drives enzalutamide resistance in prostate cancer. Nat Commun. 2021;12:1521
110. Crabb S, Plummer R, Greystoke A, Carter L, Pacey S, Walter H. et al. A phase I/IIa study to evaluate the safety and efficacy of CCS1477, a first in clinic inhibitor of p300/CBP, as monotherapy in patients with selected molecular alterations. Ann Oncol. 2021;32(Suppl 5):S617
111. Armstrong AJ, Gordon MS, Reimers MA, Hussain A, Patel VG, Lam ET. et al. Initial findings from an ongoing first-in-human phase 1 study of the CBP/p300 inhibitor FT-7051 in men with metastatic castration-resistant prostate cancer [abstract]. In: Proceedings of the AACR-NCI-EORTC Virtual International Conference on Molecular Targets and Cancer Therapeutics; 2021 Oct 7-10. Philadelphia (PA): AACR. Mol Cancer Ther. 2021;20(12 Suppl):Abstract nr P202
Author contact
![]() Corresponding authors: E-mail: xu.zhangedu.au (Xu D. Zhang), zhanglirongzzucom (Lirong Zhang); tliuunsw.edu.au (Tao Liu).
Corresponding authors: E-mail: xu.zhangedu.au (Xu D. Zhang), zhanglirongzzucom (Lirong Zhang); tliuunsw.edu.au (Tao Liu).