Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2022; 12(9):4237-4249. doi:10.7150/thno.69035 This issue Cite
Research Paper
Extracellular vesicle-derived miR-144 as a novel mechanism for chronic intermittent hypoxia-induced endothelial dysfunction
Huina Zhang1 ![]() , Lu Peng1, Yifan Wang2, Wen Zhao1, Wayne Bond Lau3, Yajing Wang3, Yu Li1, Yunhui Du1, Linyi Li1, Yu Huang2, Shaoping Nie4, Yanwen Qin1, Xinliang Ma3, Yongxiang Wei5
, Lu Peng1, Yifan Wang2, Wen Zhao1, Wayne Bond Lau3, Yajing Wang3, Yu Li1, Yunhui Du1, Linyi Li1, Yu Huang2, Shaoping Nie4, Yanwen Qin1, Xinliang Ma3, Yongxiang Wei5
1. Beijing An Zhen Hospital, Capital Medical University, Beijing Institute of Heart Lung and Blood Vessel Disease, Beijing, 100029, China.
2. Department of Biomedical Scienecs, City University of Hong Kong, Hong Kong, 508057, China.
3. Department of Emergency Medicine, Thomas Jefferson University, Philadelphia, Pa, PA19107, USA.
4. Department of Emergency, Beijing An Zhen Hospital, Capital Medical University, Beijing, 100029, China.
5. Department of Otolaryngology, Head and Neck Surgery, Capital Institute of Pediatrics, Beijing, 100020, China.
Received 2021-11-14; Accepted 2022-4-18; Published 2022-5-16
Abstract

Rationale: Extracellular vesicles (EVs) play a significant role in cell-cell communication. However, whether and how extracellular vesicles are involved in chronic intermittent hypoxia-induced endothelial dysfunction is unknown.
Methods: Comparative transcriptomics analysis and miRNA screening were used to identify the possible pathways or target molecules mediating chronic intermittent hypoxia-induced endothelial function. Serum- or erythrocyte-derived EVs were isolated through ultracentrifugation plus filtration. After in vitro or in vivo treatment with EVs, aortic rings were treated with dihydroethidium staining for superoxidative anion measurement or mounted with wire myography to measure isometric forces. Immunoblotting and qPCR were used for evaluating the molecular mechanism mediating EV miR-144-induced endothelial function under intermittent hypoxia.
Results: We revealed a previously undefined importance of circulating extracellular vesicles in regulating endothelial function via delivery of miR-144 to endothelial cells, reducing nuclear factor erythroid 2-related factor 2 expression. Additionally, we identified that erythrocytes were the primary cellular source of miR-144-enriched serum-derived extracellular vesicles and that erythrocyte-derived extracellular vesicles were largely responsible for chronic intermittent hypoxia-impaired endothelial function. Furthermore, silencing of miR-144 by anti-miR-144 confirmed its essential role in endothelial dysfunction elicited by erythrocyte-derived extracellular vesicles from chronic intermittent hypoxia-exposed C57BL/6 mice.
Conclusion: The results expand the scope of blood-borne substances involved in vascular homeostasis and suggest that anti-miR-144-loaded extracellular vesicles may represent a promising therapeutic approach against obstructive sleep apnea or chronic intermittent hypoxia-associated endothelial dysfunction.
Keywords: Chronic intermittent hypoxia, endothelial dysfunction, extracellular vesicle, erythrocyte, miR144-Nrf2 Signaling
Introduction
Obstructive sleep apnea (OSA), hallmarked with chronic intermittent hypoxia (CIH) due to recurrent partial or complete pharyngeal collapse during sleep, is a highly prevalent chronic sleep disorder. OSA is identified as an independent risk factor for the development of systemic hypertension. Approximately 50% of OSA patients are hypertensive and an estimated 70-85% patients with resistant hypertensive patients have OSA [1]. However, the precise mechanisms underlying OSA-induced hypertension are only partially understood. Growing evidence has revealed that vascular endothelial dysfunction, characterized by impaired endothelium-dependent relaxations, is the earliest sign of vascular injury preceding the occurrence of clinically obvious cardiovascular complications in OSA [2]. Therefore, a holistic analysis of the molecular mechanism of endothelial dysfunction under CIH status will help to fully understand the pathogenesis of OSA-or CIH-associated hypertension.
Oxidative stress leads to endothelial dysfunction by promoting NO uncoupling. Anti-oxidative transcription factor NRF2 preserves endothelial function and prevents Ang II-induced hypertension [3]. It has been reported that CIH induced myocardial injury by inhibiting NRF2 protein expression [4], but whether NRF2 mediates CIH-related endothelial dysfunction and hypertension is still obscure.
The secretion of extracellular vesicles (EVs) into the blood or other body fluids is a universal cellular process that occurs in multicellular organisms. EVs, as a vital mediator of intercellular communication, perform multifaceted functions by delivering complex molecules to recipient cells, thus participating in the regulation of multiple physiological and pathological process [5]. Endothelial cells are the primary cell type known to take up EVs. Circulating EVs, which are in direct and constant contact with endothelial cells, can be absorbed by endothelial cells, thereby affecting the regulation of angiogenesis [6], vascular permeability [7], and vascular tone [8]. Previous literature documented that circulating exosomal miR-144-3p inhibited the mobilization of endothelial progenitor cells and then impaired neovascularization in diabetes-related myocardial infarction [9]. However, few studies concerned the effect and detail the mechanism of circulating EVs on CIH-related endothelial dysfunction and hypertension.
In this study, we examined the effects of serum-derived EVs as well as erythrocyte-derived EVs from CIH-exposed C57BL/6 mice on endothelial-dependent relaxation and elucidated the underlying mechanisms. This research will help to further understand OSA- or CIH-related endothelial dysfunction and hypertension from a new perspective.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. See Data Supplement for detailed methods.
Animal studies
Experiments were approved by Capital Medical University Animal Experimentation Ethics Committee and in compliance with the National Institutes of Health Guidelines on the Use of Laboratory Animals. C57BL/6 mice were treated under normoxia or chronic intermittent hypoxia as previous report [10]. Endothelial dependent dilation to acetylcholine was measured.
Molecular biology
EV was isolated from serum and red blood cells as previous reports [8, 11].
Statistics
Statistical analysis is summarized in the figure legends. In most cases, the results represent the mean ± standard error of the mean (SEM) of n separate experiments. Concentration-response curves were compared by two-way analysis of variance (two-way ANOVA) followed by Bonferroni post hoc test. Two-tailed Student's t-test was used when two groups were compared. One-way analysis of variance (ANOVA) was performed to determine whether there was a significant difference between more than two datasets, followed by Bonferroni's post hoc test. P < 0.05 indicates statistical difference between groups.
Results
Chronic intermittent hypoxia treatment induces endothelial dysfunction, promotes superoxide anion radical generation, and inhibits NRF2 expression
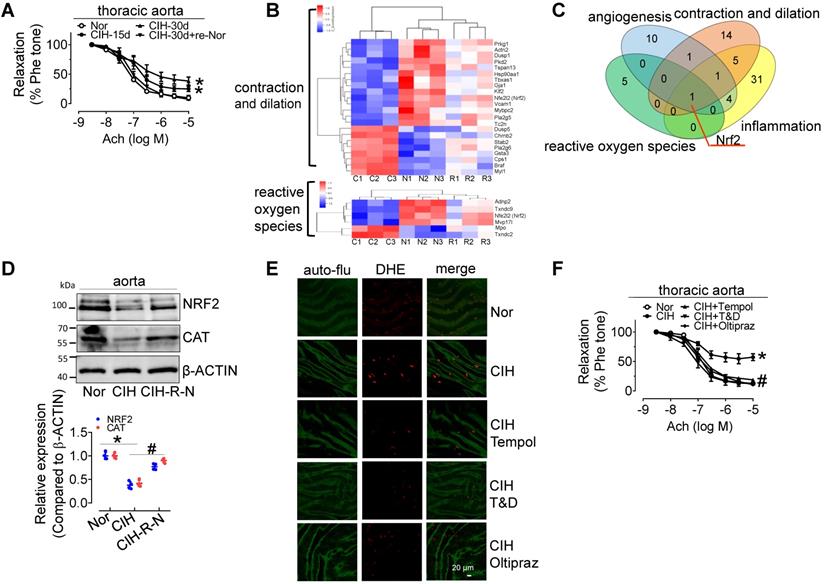
30-day CIH treatment severely attenuated endothelium-dependent relaxation (EDR) in C57BL/6 mouse thoracic aortas (mean relaxation from 92.1% to 56.4%) and carotid arteries (mean relaxation from 87.3% to 61.8%), and significantly elevated blood pressure, especially systolic blood pressure, but the effects were reversed by re-administering normal oxygen for 15 days after 30-day-CIH treatment (Figure 1A and Figure S1A-C). CIH treatment increased blood triglyceride levels, but did not significantly change glucose tolerance (Figure S1D-F). To determine the mechanism underlying CIH-induced endothelial dysfunction, microarray analysis was employed to identify differentially expressed genes in aortas from C57BL/6 mice exposed to normoxia (N), CIH (C), and normoxic conditions for 15 days after 30-day-CIH treatment (Re-Nor post-CIH; R). We identified 743 significantly differentially expressed coding genes (Table S1, https://data.mendeley.com/drafts/zbrmjpdv93) between CIH and normoxia mice, with a concomitant reverse tendency in the Re-Nor post-CIH group. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis demonstrated the cellular processing and functions of differentially expressed genes (Table S2-3). Based on the functional characterization, all differentially expressed genes to be associated with vessel constriction and dilatation, reactive oxygen species (ROS) generation, inflammation, and angiogenesis were manually categorized and presented with a heat map (Figure 1B and Figure S2A-B). Of note, a Venn diagram revealed only one gene, nuclear factor erythroid 2-related factor 2 (Nrf2), to be implicated in all four of the above-mentioned functions (Figure 1C). Consistently, Western-blotting results showed that the levels of NRF2 and its downstream target, CATALASE (CAT) were significantly reduced in the aorta from 30-day CIH-exposed C57BL/6 mice and restored in Re-Nor post-CIH group (Figure 1D). Meanwhile, 30-day CIH treatment increased the mRNA levels of NADPH oxidase (NOX) complex subunits, Nox2 and p47phox (Figure S2C). Correspondingly, increased superoxide production was clearly demonstrated in aortic endothelial cells from CIH-treated mice detected by en face fluorescence with dihydroethidium (DHE) staining, which was reversed by acute 30-min exposure to the ROS scavengers, Tempol (100 μM) or Tiron (1 mM) and DETCA (0.1 mM), as well as by oral treatment with NRF2 agonist, Oltipraz (0.5 g/kg daily in saline via gavage for 4 days before execution) (Figure 1E and Figure S2D). Furthermore, administration of Tempol, Tiron and DETCA, and Oltipraz reversed CIH-induced impairment of EDR in C57BL/6 thoracic aorta and carotid arteries (Figure 1F and Figure S2E).
CIH treatment impairs EDR, reduces NRF2 expression, and promotes superoxide anion production. (A) CIH treatment impaired EDR in mouse thoracic aortas in a time-dependent manner. (B) Some differentially expressed genes in aortas from C57BL/6 mice treated with normoxia (N), CIH (C), or Re-Nor post-CIH (R) were predicted to be associated with vessel contraction and dilation, and reactive oxygen species according to GO and KEGG analysis. (C) A Venn diagram illustrated overlap of genes with predicted functions related to angiogenesis (blue), reactive oxygen species (green), contraction and dilation (orange), and inflammation (yellow). (D) CIH-30d treatment reduced the expression of NRF2, and its downstream target, CAT, in C57BL/6 mouse aorta, whereas re-administering normoxia for 15 days after 30-day-CIH treatment restored the decreased expression of NRF2 and CAT induced by CIH. (E) CIH-30d treatment increased superoxide anion production in endothelial cells, as detected by en face fluorescence with dihydroethidium (DHE) dye (red), which was blocked by 30-min pretreatment with ROS scavengers, Tempol (100 µM), or Tiron (1 mM) and DETCA (0.1 mM) or NRF2 agonist, Oltipraz (0.5 g/kg in saline, gavage once per day for 4 days). Bar, 20 µm. Green autofluorescence (auto-flu), elastic fibers; Red DHE staining, ROS in endothelial cells. (F) 30-min pretreatment with ROS scavengers, Tempol (100 µM), Tiron (1 mM) and DETCA (0.1 mM), or 4-day treatment with NRF2 agonist, Oltipraz (0.5 g/kg), reversed CIH-induced endothelial dysfunction in thoracic aortas. Results are the mean ± SEM (n = 4). *P < 0.05 vs. Nor. #P < 0.05 vs. CIH (D and F). Two-way ANOVA (A and F) and two-tailed t test (D).
Serum EVs from CIH-treated C57BL/6 mice (CIH S-EVs) impair endothelial function, augment superoxide anion production, and decrease NRF2 expression
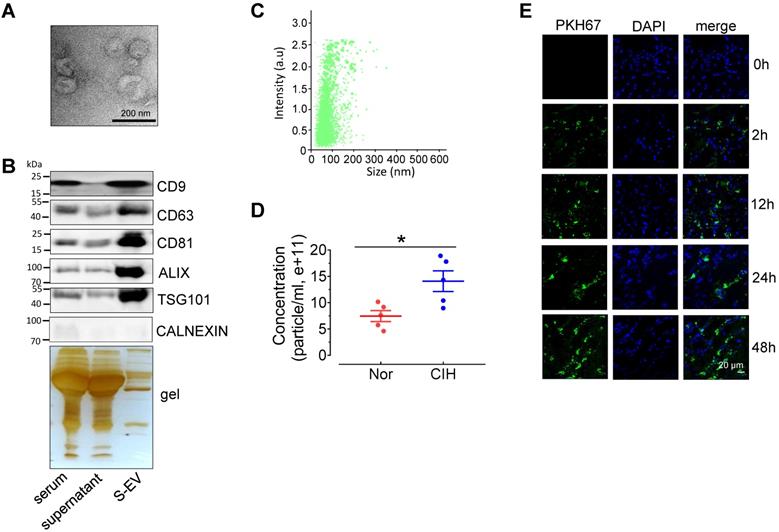
To investigate the effect of CIH S-EVs on EDRs, we first evaluated the characterization of S-EVs. Transmission electron microscopy showed that most S-EVs isolated from normal C57BL/6 mice presented classic disc-shaped vesicles with a diameter around 100nm (Figure 2A). The EV markers, especially exosome associated proteins including CD9, CD63, CD81, ALIX, and TSG101, were enriched in the EV fraction. The endoplasmic reticulum protein, Calnexin, was barely detectable in the EV fraction (Figure 2B). Nanoparticle tracking analysis revealed S-EVs with an average size 111.0 ± 57.2 nm yield by 300 μL serum (Figure 2C), while the amount CIH S-EVs was approximately 2-fold that of the normoxic group (Nor S-EVs) (Figure 2D). En face staining demonstrated that PKH67-labeled C57BL/6 mouse S-EVs were taken up by endothelial cells in a time-dependent fashion (Figure 2E).
Extracellular vesicles isolated from C57BL/6 mouse serum can be absorbed by aortic endothelial cells. (A) Electron microscopy image of whole-mount EVs purified from serum extracellular vesicles (S-EVs). Bar, 200 nm. (B) Enrichment of extracellular vesicle markers, CD63, CD81, CD9, ALIX, TSG101, and Calnexin in different serum protein fractions shown by Western blotting. Silver staining demonstrates protein loading and the protein profile of each sample. (C) S-EV size (111.0 ± 57.2 nm) was analyzed by NanoSight NS300. (D) The concentration of S-EVs was measured by NanoSight NS300. (E) Time-dependent uptake of S-EVs by mouse aortic endothelial cells assessed by en face staining. Nuclei were stained blue with DAPI. S-EVs were stained green by PKH67. Bar, 20 µm. Results are the mean ± SEM (n = 4-5). *P < 0.05 vs. Nor. Two-tailed t test (D).
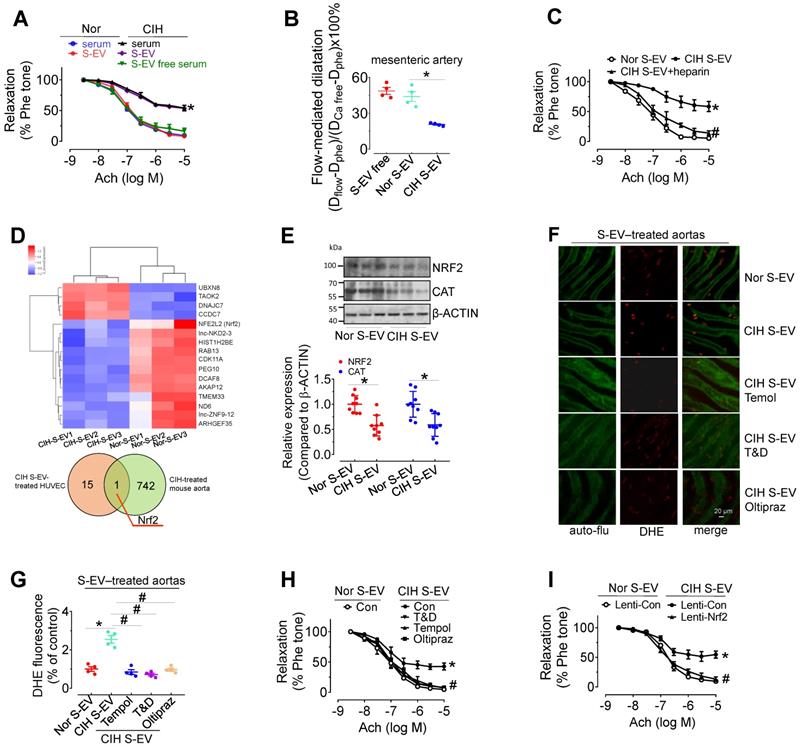
Ex vivo treatment for 48 h with CIH serum (serum from 1 mL of mouse blood made up to a final volume of 1 mL with serum-free DMEM) attenuated EDR in mouse aortas, whereas EV-free serum treatment did not produce such an inhibitory effect. Direct exposure to CIH S-EVs for 48 h attenuated EDR in C57BL/6 mouse thoracic aortas and decreased flow-mediated dilatation in C57BL/6 mouse mesenteric arteries (Figure 3A-B), but did not affect the endothelium-independent relaxation in mouse aortas (Figure S3A). This effect was more pronounced when aortas were treated with a higher S-EV concentration, or for a longer period (Figure S3B-C). Co-treatment with heparin (0.3 μg/mL, blocker of EV absorption) reversed CIH S-EV-impaired EDR in mouse aortas (Figure 3C). To further ascertain how CIH S-EVs exerts the adverse effect on EDR, transcriptome microarray analysis was used to identify differentially expressed genes in HUVECs treated with CIH S-EVs or Nor S-EVs. 19 significantly differentially expressed coding genes were identified between the two groups (Figure 3D, upper panel, and Table S4, https://data.mendeley.com/drafts/xk3krh5ffp). Among them, Nrf2 was the only differentially expressed gene common to both CIH S-EV-treated HUVECs and CIH-treated mouse aortas (Figure 3D, below panel). Further verification showed that CIH S-EV ex vivo treatment indeed decreased the protein expression of NRF2 and its target, CAT, in both murine endothelial cell line, H5V, (Figure S3D) and mouse aortas (Figure 3E). Furthermore, 48 h of CIH S-EV treatment increased superoxide anion production in H5V cells and in aortic endothelial cells, an effect blocked by 30-min pretreatment with ROS scavengers, Tempol or Tiron and DETCA, or the 48-h cotreatment of NRF2 agonist, Oltipraz (Figure 3F-G and Figure S3E-F). Whereas, the mRNA levels of NOX subunits or Xanthine dehydrogenase (Xdh) were not altered in CIH S-EV-treated H5V cells (Figure S3G), indicating that decreased NRF2 was the possible primary molecular responsible for CIH S-EV-induced superoxide anion overproduction in endothelial cells. Consistently, aorta EDR impaired by 48-h CIH S-EVs treatment was reversed by Tempol, Tiron and DETCA as well as NRF2 agonist Oltipraz or lentivirus-mediated NRF2 overexpression (Figure 3H-I and Figure S3H).
CIH S-EVs impair endothelial function, augment superoxide anion production, and decrease NRF2 expression in endothelial cells. (A) 48-h treatment with serum from CIH-treated mouse attenuated EDR in mouse aortas. This effect was absent after removal of EVs from the serum, while 48-h-treatment with CIH S-EVs isolated from 1 mL of blood significantly impaired EDR in mouse aortas. (B) Exposure (48 h) to CIH S-EVs reduced flow-mediated dilatation in C57BL/6 mouse mesenteric arteries. (C) Heparin (0.3 µg/mL, 48 h) ameliorated EDR induced by CIH S-EVs. (D) Transcriptome microarray analysis was used to identify differentially expressed genes in HUVECs treated with CIH S-EVs or Nor S-EVs (up panel). The Venn diagram showed the number of overlapping genes from CIH S-EV-treated HUVECs (orange) and CIH-treated mouse aortas (green) (below panel). (E) CIH S-EV reduced the expression of NRF2 and CAT in mouse aortas. (F-G) CIH S-EV-incubation increased superoxide anion production in aortic endothelial cells detected by DHE fluorescent dye, which was blocked by 30-min pretreatment with ROS scavengers, Tempol (100 µM), or Tiron (1 mM) and DETCA (0.1 mM), or NRF2 agonist, Oltipraz (100 µM, co-culture for 48 h). Bar, 50 µm. (H) 30-min pretreatment with ROS scavengers, Tempol (100 µM), or Tiron (1 mM) and DETCA (0.1 mM), or co-culture with NRF2 agonist, Oltipraz (100 µM) for 48 h reversed CIH S-EV-induced endothelial dysfunction in aortas. (I) CIH S-EV-attenuated EDR in mouse aortas was absent after Nrf2 overexpression mediated by lentivirus. Results are the means ± SEM (n = 4-9). *P < 0.05 vs. Nor S-EV. #P < 0.05 vs. CIH S-EV. Two-way ANOVA (A, C, H, I) and two-tailed t test (B, E, G).
miR-144 is elevated in CIH S-EVs
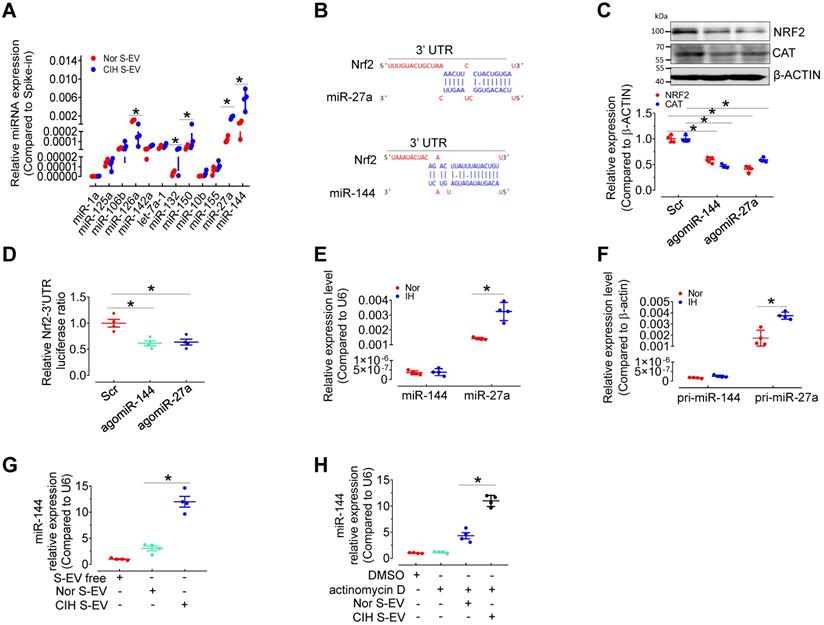
We have compared the S-EV protein profiles, but did not identify the possible differentially expressed S-EV protein participating in CIH-induced endothelial dysfunction [12]. To determine the mechanism by which CIH S-EVs decreased NRF2 and impaired endothelial function, we measured several EV-associated miRNAs related to oxidative stress or hypoxia signaling [13-19] in S-EVs from normoxia-and CIH-treated mice by qPCR. Levels of miR-126 were decreased while those of miR-132, miR-150, miR-27a, and miR-144 were increased in CIH S-EVs (Figure 4A). Previous reports [20] as well as miRNA target prediction by TargetScan indicated conserved binding sequences for miR-27a and miR-144a in the 3′-UTR of Nrf2 (Figure 4B). Therefore, we detected the expression regulation of NRF2 by miR-144 and miR-27a in H5V. As expected, overexpression of miR-144 and miR-27a using angomiR-144 and angomiR-27a reduced the protein levels of NRF2 and its target, CAT, in H5V (Figure 4C) and inhibited Nrf2-3'UTR-drived luciferase activity in 293A cells (Figure 4D). Furthermore, qPCR results demonstrated that mature miR-27a and its primary transcript pri-miR-27a (but not miR-144 or pri-miR-144), were induced in H5V after intermittent hypoxia (IH) treatment for 24 h (Figure 4E-F), suggesting that miR-27a, other than miR-144 was endogenously expressed in endothelial cells and responded to IH stimulus. miR-144 expression was almost undetectable in H5V cells, but CIH S-EV treatment significantly increased miR-144 abundance in H5V cells (Figure 4G). This effect persisted regardless of whether we inhibited the production of endogenous miRNAs with actinomycin D (10 µg/mL in DMSO) (Figure 4H), suggesting that the endothelial miR-144 signal was mainly from S-EVs. Hence, EV miR-144 was selected as our target molecule for further investigation.
miR-144 is increased in CIH S-EVs, and is delivered to endothelial cells by S-EVs. (A) Relative expression level of indicated miRNAs in CIH S-EV or Nor S-EV. (B) Diagram depicted the predicted binding sites of miR-27a and miR-144 on the 3'-UTR of Nrf2. (C) AgomiR-144 and agomiR-27a decreased the expression of NRF2 and CAT in H5V cells. The below panel showed the relative expression level of NRF2 and CAT. (D) The treatment of agomiR-144 plus co-transfection of Nrf2 3'UTR reporter plasmid led to a significant decrease of the luciferase activity. (E) The expression of miR-144 and miR-27a in S-EV-free medium-cultured H5V cells with 24-h normoxia or intermittent hypoxia (IH) treatment. (F) Pri-miR-27a and pri-miR-144 were measured by qPCR in EV-free medium-cultured H5V endothelial cells after normoxia or intermittent hypoxia-treatment for 24 h. (G-H) The expression of miR-144 in S-EV-free medium-cultured H5V cells with 24-h Nor S-EV or CIH S-EV treatment with (H) or without (G) actinomycin D (10 µg/mL in DMSO) cotreatment. Results are the mean ± SEM (n = 4). *P < 0.05 vs. Nor S-EV (A, G) or Nor S-EV plus actinomycin D or Scr (C, D) or Nor (E, F). One-way ANOVA (D, G, H) and two-tailed t test (A, C, E, F).
miR-144 is elevated in erythrocyte-derived extracellular vesicles from CIH-exposed mice (CIH E-EVs)
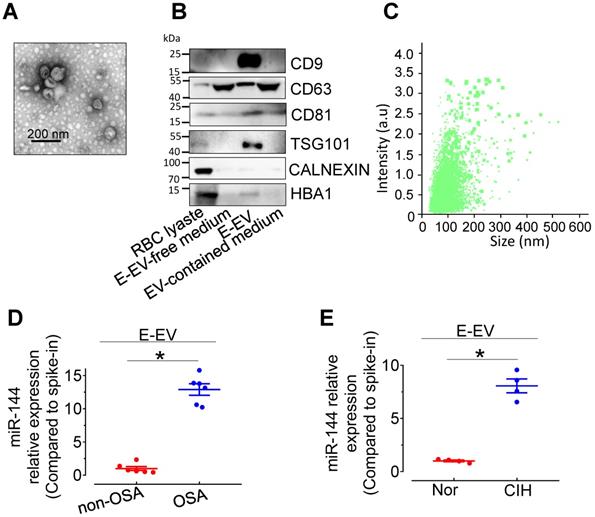
miR-144 is primarily expressed in erythrocytes and involved in erythroid differentiation [21]. To clarify the cellular origin of miR-144 in CIH S-EVs, erythrocyte-derived extracellular vesicles (E-EVs) were isolated from C57BL/6 mouse red blood cells by ultracentrifugation and visualized by transmission electron microscopy. E-EVs presented as classic disc shape (Figure 5A). Nanoparticle tracking analysis showed the average diameter of E-EVs was 115.0±50.9 nm (Figure 5B). The purity of the isolated E-EVs was determined by the relative abundance of E-EV-resident proteins, including CD9, CD81, and TSG101. The erythrocyte protein, HBA, was also observed in the E-EV fraction (Figure 5C). qPCR assay showed that miR-144 was markedly upregulated in E-EVs from both CIH-treated C57BL/6 mouse erythrocytes and OSA patient erythrocytes (Figure 5D-E and Figure S4A-B).
Characterization of isolated E-EVs and miR-144 expression in E-EVs. (A) E-EVs isolated from culture medium of mouse red blood cells were imaged by transmission electron microscopy. Bar, 200 nm. (B) E-EV size (111.0 ± 57.2 nm) was analyzed by NanoSight NS300. (C) Different fractions were processed for Western blotting with EV markers, CD9, CD63, CD81, and TSG101, endoplasmic reticulum marker, Calnexin, and erythrocyte marker, HBA. Silver staining revealed the protein loading amount and the protein profile of each sample. (D-E) qPCR assays determined the expression levels of miR-144 in E-EVs from OSA patients (D), CIH-exposed mice (E), and their control groups. Results are the means ± SEM (n = 4-6). *P < 0.05 vs. Nor E-EV or OSA E-EV. Two-tailed t test (D, E).
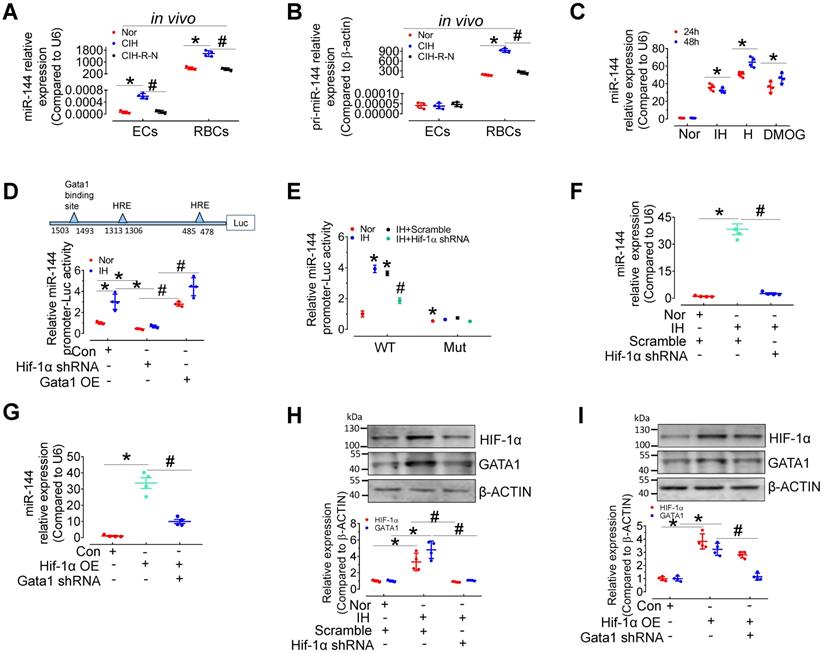
HIF-1α and GATA1 mediate upregulation of miR-144 in CIH E-EVs
To provide further evidence of EV-mediated cell-cell communication between erythrocytes and endothelial cells, we measured the levels of pri- and mature miR-144 in red blood cells and endothelial cells from C57BL/6 mice after normoxia, 30-day CIH, or normoxic conditions for 15 days after 30-day-CIH (CIH-R-N) treatment. qPCR results presented that the expression of miR-144 and pri-miR-144 was dominant in erythrocytes rather than in endothelial cells. Under CIH status, the expression of pri-miR-144 and miR-144 was significantly increased in erythrocytes and the tendency was reversed after returning to normoxia for 15 days; whereas, in aortic endothelial cells, only miR-144 was upregulated (Figure 6A-B), indicating that miR-144 in endothelial cells was most likely transferred exogenously from erythrocytes in vivo. We further studied the molecular regulation of miR-144 in erythrocytes under hypoxia status. miR-144 was upregulated in murine erythroleukemia (MEL) cells after continuous hypoxia, intermittent hypoxia, or DMOG (1 mM, HIF-1α stabilizer) treatment (Figure 6C). Transcriptional factor hypoxia inducible factor 1α (HIF-1α) plays a central role in hypoxia status. We predicted the localization of two hypoxia response elements (HREs) in the miR-144 promoter sequence and demonstrated that IH treatment increased miR-144 promoter activity, which was blocked by lentivirus-mediated knockdown of Hif-1α. Enhanced miR-144 promoter activity by Gata1 overexpression served as a positive control (Figure 6D). Furthermore, to evaluate the importance of GATA1 and HIF-1α in miR-144 promoter activity, we mutated GATA1 binding site on miR-144 promoter and investigated the effect of intermittent hypoxia and HIF-1α knockdown on the activity of the mutated miR-144 promoter. Results indicated that although intermittent hypoxia significantly induced miR-144 wild type promoter activity, the mutated miR-144 promoter was insensitive to IH stimulation as well as knockdown of Hif-1α (Figure 6E), suggesting that GATA1 plays a more critical role than HIF-1α in initiating miR-144 promoter activity. Accordingly, we observed that upregulation of miR-144 by intermittent hypoxia could be reversed by lentivirus-mediated silencing of Hif-1α (Figure 6F) and upregulation of miR-144 by HIF-1α overexpression was blocked by knockdown of Gata1 (Figure 6G). Meanwhile, intermittent hypoxia treatment and overexpression of Hif-1α increased GATA1 expression in MEL cells, which was blocked by Hif-1α knockdown (Figure 6H-I). These evidences indicated that miR-144 can be directly regulated by HIF-1α or indirectly regulated by HIF-1α/GATA1 pathway during hypoxia.
Expression and regulation of miR-144 in erythrocytes during intermittent hypoxia. (A-B) qPCR assay measured the expression level of pri-miRNA (A) and miR-144 (B) in aortic endothelial cells and red blood cells in C57BL/6 mice under normoxia, CIH, or readministering normoxia for 15 days after 30-day-CIH treatment (CIH-R-N). (C) The expression levels of miR-144 in MEL cells after continuous or intermittent hypoxia or DMOG (1mM in PBS, Hif-1α stabilizer) treatment for the indicated period. (D) Luciferase reporter gene assays demonstrated the activity of the miR-144 promoter in IH- or normoxia-exposed 293A cells after Hif-1α knockdown or Gata1 overexpression by lentivirus infection. (E) Luciferase reporter gene assays demonstrated the activity of the wild-type miR-144 promoter (WT) or GATA1 binding site mutant miR-144 promoter (Mut) in normoxia or IH-exposed 293A cells after lentivirus-mediated knockdown of Hif-1α. (F) The expression level of miR-144 in IH- or normoxia-treated MEL cells after silencing Hif-1α. (G) The level of miR-144 in MEL cells after Gata1 knockdown or Hif-1α overexpression. (H) The levels of HIF-1α and GATA1 in IH- or normoxia-treated MEL cells after lentiviral silencing of Hif-1α. (I) The levels of HIF-1α, and GATA1 in MEL cells after Gata1 knockdown or Hif-1α overexpression by lentivirus. Results are the mean ± SEM (n = 4). *P < 0.05 vs. Nor (A-C, E, F, H) or Con (D, G, I). #P < 0.05 vs. CIH (A, B) or Hif-1α shRNA (D) or IH Scramble (E, F, H) or Hif-1α OE (G, I). One-way ANOVA (C-I) and two-tailed t test (A, B).
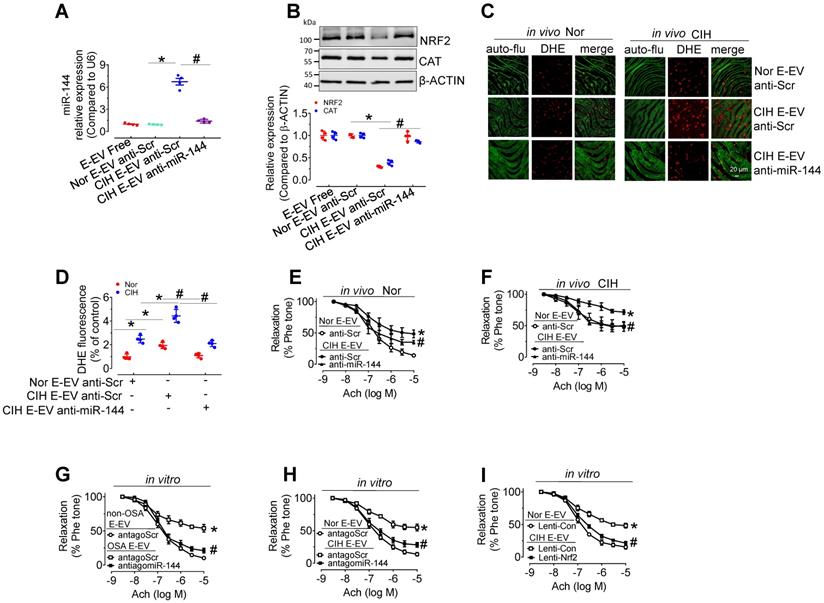
Anti-miR-144 blocked CIH E-EVs-induced NRF2 expression, superoxide anion production, and endothelial dysfunction
To further verify the endothelial effect of miR-144 delivery by CIH E-EVs, anti-miR-144 as well as anti-miR-144-loaded CIH E-EVs were utilized to treat aortas in vitro or treat CIH mice in vivo. As expected, anti-miR-144-loaded CIH E-EVs did not increase miR-144 levels in H5V cells after incubation for 48 h, while anti-scramble-loaded groups did (Figure 7A). Protein levels of NRF2 and CAT in H5V cells after 48-h treatment of anti-miR-144-loaded CIH E-EVs were maintained at the level observed in the control groups (Figure 7B). Consistently, in vivo CIH E-EV treatment by tail injection (one injection/3 day, for 12 days) potentiated superoxide anion production in endothelial cells. However, this effect was largely inhibited in anti-miR-144-loaded CIH E-EV treated groups, as shown by en face DHE staining (Figure 7C-D). Meanwhile, in vivo treatment of CIH E-EVs impaired EDR and increased systolic blood pressure in normoxia- and CIH-treated mice. These effects were reduced in groups treated with anti-miR-144-loaded CIH E-EVs (Figure 7E-F and Figure S4C-E). Similarly, antagomiR-144 significantly reversed endothelial dysfunction caused by E-EV from CIH mice or OSA patients (Figure 7G-H). Of note, lentivirus-mediated Nrf2 overexpression also effectively blocked CIH E-EV-impaired endothelial function (Figure 7I).
Anti-miR-144 reverses CIH E-EV-induced superoxide anion overproduction, NRF2 decrease, and endothelial dysfunction. (A) qPCR analysis verified the expression of miR-144 in H5V cells after the indicated E-EV treatment. (B) Protein levels of NRF2 and CAT in H5V cells treated with or without anti-miR-144-loaded CIH E-EVs. Below panel depicted the relative levels of NRF2 and CAT. (C) En face fluorescence images with DHE staining revealed the superoxide level in aortic endothelial cells after in vivo treatment with anti-Scr- or anti-miR-144-loaded CIH E-EVs. Bar, 20 µm. (D) Summary of the DHE fluorescence signal intensity. (E-F) Anti-miR-144-loaded CIH E-EVs restored CIH E-EV-induced endothelial dysfunction in aortas under normoxia (E) or CIH (F). (G-H) antagomiR-144 in vitro 48-h treatment improved OSA E-EV- (G) or CIH E-EV-(H) induced endothelial dysfunction. (I) Lentivirus-mediated overexpression of Nrf2 in mouse aortas reversed CIH E-EV-induced endothelial dysfunction. Results are the mean ± SEM (n = 4). *P < 0.05 vs. Nor E-EV anti-Scr (A, B, D-F) or non-OSA E-EV (G) or Nor E-EV antagoScr (H) or Nor E-EV Lenti-Con (I). #P < 0.05 vs. CIH E-EV anti-Scr (A, B, D-F) or OSA E-EV antagoScr (G) or CIH E-EV antagoScr (H) or CIH E-EV Lenti-Con (I). One-way ANOVA (A, B, D) and two-way ANOVA (E-I).
Discussion
This study highlights the role of S-EVs and E-EVs as blood-borne regulators of vascular function, and pinpoints erythrocyte-enriched miR-144 as a critical EV-derived miRNA that participates in CIH-induced endothelial dysfunction via inhibiting NRF2 expression.
OSA is a prevalent disease most frequently associated with secondary hypertension [1]. Endothelial dysfunction is the earliest vascular consequence of OSA or CIH and precedes hypertension onset [22, 23]. However, the underlying mechanism of OSA or CIH-induced endothelial dysfunction is not well characterized. Some reports propose that changes in vasoactive molecules, such as reduced NO bioavailability [23], excessive oxidative stress [24], increased levels of angiotensin II and endothelin-1 [25, 26], regulate endothelial function during the development of hypertension following CIH. Consistently, we confirmed that CIH treatment dramatically impaired EDR in the C57BL/6 mouse aorta and increased systolic blood pressure. To explore the mechanism more comprehensively and systematically, we performed global transcriptome analysis of aortas from CIH-treated mice. Among the 743 differentially expressed genes, Nrf2 was the only one commonly implicated in the four vascular-associated responses, namely, contraction and dilation, angiogenesis, ROS production, and inflammation. ROS regulates vascular homeostasis, and ROS overproduction-induced oxidative stress is a primary cause of vascular dysfunction by reducing NO availability [27]. Increased oxidative stress has been proposed to contribute to OSA or CIH-related endothelial dysfunction and hypertension. NRF2 is a key antioxidant transcriptional factor with a protective role in many free radical detoxification pathways related to aging, atherosclerosis, hypertension, ischemia, and other cardiovascular diseases [28, 29]. NRF2 binds to the antioxidant response element to transcriptionally activate downstream genes encoding Glutathione S-transferase, Aldehyde dehydrogenase, CAT, Heme oxygenase 1, and Thioredoxin [30]. However, most reports emphasize the role of increased NOX levels and activity in CIH-induced adverse vascular outcomes [24, 31], and few studies have linked NRF2 regulation to CIH-induced endothelial dysfunction, although some studies similarly revealed that CIH inhibited NRF2 expression [4, 32]. In the present study, we demonstrated that NRF2 downregulation in CIH-treated mouse aorta or in CIH-treated endothelial cells results in excess superoxide production and impaired endothelial function, an effect rescued by the NRF2 agonist, Oltipraz, or by the ROS scavengers, Tempol or Tiron and DETCA. This result confirms the importance of NRF2 in CIH-associated endothelial dysfunction induced by oxidative stress.
Circulating EVs are constantly in direct contact with and regulate the functions of endothelial cells under pathophysiological conditions [33]. EVs from OSA patient plasma impair endothelial adhesiveness and permeability [34]. Little attention has been given to the impact of EVs upon CIH-impaired EDR. Flow-mediated dilation was attenuated in mice after treatment with plasma EVs from OSA pediatric patients. This compromised vascular function was proposed to be caused by reduced eNOS expression and altered EV miR-630 [35]. Our group recently reported that EV derived from intermittent hypoxia-treated red blood cells impaired endothelial function through regulating eNOS phosphorylation and ET-1 expression [11]. Here, we demonstrate that EVs-derived from CIH S-EVs and CIH E-EVs profoundly impaired EDR in normal mouse aorta. Interestingly, NRF2 was differentially expressed in CIH S-EV-treated HUVECs and CIH-treated mouse aortas compared with their corresponding controls. CIH S-EVs attenuated NRF2 expression, which increased ROS production in endothelial cells without altering NOX signaling. This was validated by improved endothelial function in response to the NRF2 agonist, Oltipraz.
We previously reported that S-EVs mediate endothelial dysfunction in diabetes through delivery of arginase 1 [8]. We also compared the S-EV protein profiles between normoxic and CIH animal models, but did not identify the possible differentially expressed S-EV protein participating in CIH-induced endothelial dysfunction [12]. Therefore, we focused upon S-EV miRNAs through screening miRNAs related to oxidative stress or hypoxia signaling in S-EVs and found miR-144 and miR-27a was upregulated in CIH S-EVs. miR-144 and miR-27a are known to inhibit NRF2 expression by directly binding to the 3′-UTR of Nrf2 [20], and we confirmed this regulatory link in endothelial cells. Interestingly, unlike miR-27a, miR-144 is expressed at a very low endogenous level in endothelial cells. The expression of miR-144 or pri-miR-144 in endothelial cells did not change after intermittent hypoxia treatment, suggesting that the increased level of miR-144 in CIH S-EVs was likely to be derived from a non- endothelial origin. miR-144 is highly expressed in erythrocytes and is closely related to erythroid differentiation [21]. We confirmed high levels of miR-144 expression in erythrocytes and E-EVs following CIH and in OSA. miR-144 was reported to play versatile roles in maintaining homeostasis in the cardiovascular system. For instance, miR-144 significantly reduced infarct size in an acute ischemia reperfusion injury model and reduces left ventricular remodeling after myocardial infarction through promoting autophagy [36, 37], and protected against LPS-induced lung endothelial hyperpermeability [38]; but it also promoted atherosclerosis plaque formation [39], and mediated the 7-Ketocholesterol-induced endothelial dysfunction [40]. One research even found that circulating exosomal miR-144-3p impaired the mobilization ability of EPCs and then impaired ischemia-induced neovascularization [9]. To clarify the involvement of EV miR-144 in CIH E-EV-induced endothelial dysfunction, we demonstrated that CIH E-EV treatment increased miR-144 expression and reduced NRF2 expression, increased superoxide production in endothelial cells, attenuated aorta EDR, and increased systolic blood pressure. Furthermore, anti-miR-144-loaded CIH E-EVs effectively blocked miR-144 expression and increased the levels of NRF2 and CAT in endothelial cells. More importantly, functional studies revealed that anti-miR-144-loaded CIH E-EVs as well as antagomir-144 partially rescued EDR, reduced ROS, and decreased hypertension caused by CIH E-EVs, indicating a pivotal role of circulating EV miR-144 in these processes. Furthermore, we confirmed the protective role of NRF2 in CIH S-EV- or CIH E-EV-caused endothelial dysfunction through overexpression of Nrf2 by lentivirus.
The regulation of miR-144 in erythrocytes during hypoxia is not well documented in the literature. Sequencing analysis showed that miR-144 was upregulated after 16 h of hypoxic treatment in HUVECs [41]. Other papers have shown that miR-144 expression is significantly higher in cancer tissues that are often under hypoxia conditions compared to normal tissues [42, 43]. However, none of these articles addressed the mechanism of hypoxia-induced miR-144 upregulation. GATA1 is an important transcriptional factor that increases miR-144 expression during erythropoiesis [44]. Here, we predicated a hypoxia response element and a GATA1 binding site in the miR-144 promoter region and confirmed that intermittent hypoxia treatment promoted miR-144 expression in MEL cells via HIF-1α direct transcriptional regulation. Interestingly, we also demonstrated that both Hif-1α overexpression and CIH treatment upregulated GATA1 expression in MEL cells, indicating GATA1 to be a mediator participating in HIF-1α indirect regulation of miR-144.
The present study has several limitations. In this study, considering that OSA patients may have complex comorbidities and complex pathophysiological states, which will increase the heterogeneity of EVs and even lead to inconsistent results, we selected serum EVs or erythrocyte-derived EVs obtained under more consistent CIH conditions to treat aortic and endothelial cells to obtain more reproducible results. This result reflects the mechanism of EV-induced vascular impairment in CIH setting, however, this mode of treatment is less clinically relevant than EV of OSA patient origin. Meanwhile, given that many of the miRNAs in mouse EVs are not homologous to human ones, our treatment model may therefore miss other possible functional molecules. We provide clear evidence that miR-144 in erythrocyte-derived EVs is responsible for endothelial dysfunction induced by CIH, the major pathological alteration in OSA. However, it is not known whether the same or different EV miRNAs are involved in the endothelial dysfunction induced by other pathological features of OSA, such as fragmented sleep, sympathetic nerve activation, and increased negative chest pressure. Moreover, we cannot exclude the possible involvement of non-erythroid-derived miR-144, albeit to a lesser degree. Further investigation is warranted to address these problems. Meanwhile, considering that mature erythrocytes would not respond to any stimulus to induce the expression of miRNAs, in the current work, we used MEL to reveal the mechanism by which intermittent hypoxia promoted miR-144 expression, which maybe partially helped to understand the increased levels of miR-144 in mature erythrocytes under CIH status, however, the in vivo mechanism closer to the physiological state should be in future investigations. Due to technical constraints, the CIH paradigm used in the current study does not closely match that observed in clinical OSA status, new specific interfering tools and more rigorous studies are required to further explore the clinical relevance of S-EV or E-EV in the development of OSA-related vasculopathy. In addition, the present study cannot discount other possible signals, such as miR-27a, that are independent of E-EV miR-144/NRF2 might play a role in CIH-induced impairment of endothelial function, which requires future examination.
In summary, we systematically demonstrate that CIH S-EVs and CIH E-EVs can deliver functional miR-144 to endothelial cells, promoting superoxide anion production by reducing NRF2 expression, a critical pathogenic component of endothelial dysfunction and hypertension during CIH. Our study provides experimental evidence for the therapeutic potential of EV-loaded anti-miR-144 or antagomir-144 in treating OSA or CIH-associated vascular complications.
Abbreviations
EVs: extracellular vesicles; CIH: chronic intermittent hypoxia; DHE: dihydroethidium; OSA: obstructive sleep apnea; SEM: standard error of the mean; EDR: endothelium-dependent relaxation; FMD: For flow-mediated dilatation; GO: Gene ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; ROS: reactive oxygen species; NRF2: nuclear factor erythroid 2-related factor 2; NOX: NADPH oxidase; HIF-1α: hypoxia inducible factor 1α.
Supplementary Material
Supplementary materials and methods, figures, and tables.
Acknowledgements
This study was supported by the Beijing Natural Science Foundation (Grant No.7192031), the National Natural Science Foundation of China (Grant No. 81870335), and Hong Kong Research Grants Council (Grant No. SRFS2021-4S04, C4024-16W). We are grateful to: Dr. Wenxing Li (Shanghai OE Biotech. Co., Ltd. China) for bioinformatics assistance, Prof. Rui Chen (Prof. Rui Chen, Capital Medical University, China) for kindly providing us with Hif-1α knocking-down lentivirus and Ms. Yuqing Yang (Beijing ECHO Biotech. Co., Ltd. China) for transmission electron microscopy assistance.
Author Contributions
H.Z. conducted the experiments, analyzed the data, prepared the manuscript, and was responsible for all data, figures, and text. X.M. and Yongxiang W. designed the experiments, supported, and supervised the work. L.P., and Y.L. assisted with functional studies and data analysis. Yifan W., W.B.L., Yajing W. and Y.H. helped with manuscript revision; Y.D., L.L., Y.Q. and S.N. helped with sample collection.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Labarca G, Schmidt A, Dreyse J, Jorquera J, Enos D, Torres G. et al. Efficacy of continuous positive airway pressure (cpap) in patients with obstructive sleep apnea (osa) and resistant hypertension (rh): Systematic review and meta-analysis. Sleep Med Rev. 2021;58:101446
2. Mochol J, Gawrys J, Gajecki D, Szahidewicz-Krupska E, Martynowicz H, Doroszko A. Cardiovascular disorders triggered by obstructive sleep apnea-a focus on endothelium and blood components. Int J Mol Sci. 2021 22
3. Wang C, Luo Z, Carter G, Wellstein A, Jose PA, Tomlinson J. et al. Nrf2 prevents hypertension, increased adma, microvascular oxidative stress, and dysfunction in mice with two weeks of ang ii infusion. Am J Physiol Regul Integr Comp Physiol. 2018;314:R399-R406
4. Zhang H, Zhou L, Zhou Y, Wang L, Jiang W, Liu L. et al. Intermittent hypoxia aggravates non-alcoholic fatty liver disease via ripk3-dependent necroptosis-modulated nrf2/nfkappab signaling pathway. Life Sci. 2021;285:119963
5. Wortzel I, Dror S, Kenific CM, Lyden D. Exosome-mediated metastasis: Communication from a distance. Dev Cell. 2019;49:347-360
6. Hu Y, Rao SS, Wang ZX, Cao J, Tan YJ, Luo J. et al. Exosomes from human umbilical cord blood accelerate cutaneous wound healing through mir-21-3p-mediated promotion of angiogenesis and fibroblast function. Theranostics. 2018;8:169-184
7. Fang JH, Zhang ZJ, Shang LR, Luo YW, Lin YF, Yuan Y. et al. Hepatoma cell-secreted exosomal microrna-103 increases vascular permeability and promotes metastasis by targeting junction proteins. Hepatology. 2018;68:1459-1475
8. Zhang HN, Liu J, Qu D, Wang L, Wong CM, Lau CW. et al. Serum exosomes mediate delivery of arginase 1 as a novel mechanism for endothelial dysfunction in diabetes. P Natl Acad Sci USA. 2018;115:E6927-E6936
9. Liu Y, Xu J, Gu R, Li Z, Wang K, Qi Y. et al. Circulating exosomal mir-144-3p inhibits the mobilization of endothelial progenitor cells post myocardial infarction via regulating the mmp9 pathway. Aging (Albany NY). 2020;12:16294-16303
10. Du YH, Wei YX, Christopher T, Lopez B, Ma XL, Wang YJ. Mirna-mediated suppression of a cardioprotective cardiokine as a novel mechanism exacerbating post-mi remodeling by sleep breathing disorders. Faseb J. 2020 34
11. Peng L, Li Y, Li X, Du Y, Li L, Hu C. et al. Extracellular vesicles derived from intermittent hypoxia-treated red blood cells impair endothelial function through regulating enos phosphorylation and et-1 expression. Cardiovasc Drugs Ther. 2021;35:901-913
12. Zhang HN, Yang F, Guo YC, Wang L, Fang F, Wu H. et al. The contribution of chronic intermittent hypoxia to osahs: From the perspective of serum extracellular microvesicle proteins. Metabolism-Clinical and Experimental. 2018;85:97-108
13. Zhang J, Li S, Li L, Li M, Guo C, Yao J. et al. Exosome and exosomal microrna: Trafficking, sorting, and function. Genomics, proteomics & bioinformatics. 2015;13:17-24
14. Salimian J, Mirzaei H, Moridikia A, Harchegani AB, Sahebkar A, Salehi H. Chronic obstructive pulmonary disease: Micrornas and exosomes as new diagnostic and therapeutic biomarkers. Journal of research in medical sciences: the official journal of Isfahan University of Medical Sciences. 2018;23:27
15. Ong SG, Lee WH, Huang M, Dey D, Kodo K, Sanchez-Freire V. et al. Cross talk of combined gene and cell therapy in ischemic heart disease: Role of exosomal microrna transfer. Circulation. 2014;130:S60-69
16. Sangokoya C, Telen MJ, Chi JT. Microrna mir-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood. 2010;116:4338-4348
17. Zhou Y, Fang L, Yu Y, Niu J, Jiang L, Cao H. et al. Erythropoietin protects the tubular basement membrane by promoting the bone marrow to release extracellular vesicles containing tpa-targeting mir-144. American journal of physiology Renal physiology. 2016;310:F27-40
18. Zhao Y, Dong D, Reece EA, Wang AR, Yang P. Oxidative stress-induced mir-27a targets the redox gene nuclear factor erythroid 2-related factor 2 in diabetic embryopathy. American journal of obstetrics and gynecology. 2018;218:136 e131-136 e110
19. Saha B, Momen-Heravi F, Kodys K, Szabo G. Microrna cargo of extracellular vesicles from alcohol-exposed monocytes signals naive monocytes to differentiate into m2 macrophages. The Journal of biological chemistry. 2016;291:149-159
20. Cheng X, Ku CH, Siow RC. Regulation of the nrf2 antioxidant pathway by micrornas: New players in micromanaging redox homeostasis. Free radical biology & medicine. 2013;64:4-11
21. Zhang FL, Shen GM, Liu XL, Wang F, Zhao YZ, Zhang JW. Hypoxia-inducible factor 1-mediated human gata1 induction promotes erythroid differentiation under hypoxic conditions. Journal of cellular and molecular medicine. 2012;16:1889-1899
22. Korcarz CE, Benca R, Barnet JH, Stein JH. Treatment of obstructive sleep apnea in young and middle-aged adults: Effects of positive airway pressure and compliance on arterial stiffness, endothelial function, and cardiac hemodynamics. J Am Heart Assoc. 2016;5:e002930
23. Badran M, Abuyassin B, Golbidi S, Ayas N, Laher I. Uncoupling of vascular nitric oxide synthase caused by intermittent hypoxia. Oxid Med Cell Longev. 2016;2016:2354870
24. Nisbet RE, Graves AS, Kleinhenz DJ, Rupnow HL, Reed AL, Fan TH. et al. The role of nadph oxidase in chronic intermittent hypoxia-induced pulmonary hypertension in mice. Am J Respir Cell Mol Biol. 2009;40:601-609
25. Capone C, Faraco G, Coleman C, Young CN, Pickel VM, Anrather J. et al. Endothelin 1-dependent neurovascular dysfunction in chronic intermittent hypoxia. Hypertension. 2012;60:106-113
26. Morgan BJ, Schrimpf N, Rothman M, Mitzey A, Brownfield MS, Speth RC. et al. Effect of chronic intermittent hypoxia on angiotensin ii receptors in the central nervous system. Clin Exp Hypertens. 2018:1-7
27. Daiber A, Kroller-Schon S, Oelze M, Hahad O, Li H, Schulz R. et al. Oxidative stress and inflammation contribute to traffic noise-induced vascular and cerebral dysfunction via uncoupling of nitric oxide synthases. Redox Biol. 2020;34:101506
28. Satta S, Mahmoud AM, Wilkinson FL, Yvonne Alexander M, White SJ. The role of nrf2 in cardiovascular function and disease. Oxid Med Cell Longev. 2017;2017:9237263
29. Lopes RA, Neves KB, Tostes RC, Montezano AC, Touyz RM. Downregulation of nuclear factor erythroid 2-related factor and associated antioxidant genes contributes to redox-sensitive vascular dysfunction in hypertension. Hypertension. 2015;66:1240-1250
30. Chorley BN, Campbell MR, Wang X, Karaca M, Sambandan D, Bangura F. et al. Identification of novel nrf2-regulated genes by chip-seq: Influence on retinoid x receptor alpha. Nucleic Acids Res. 2012;40:7416-7429
31. Schulz R, Murzabekova G, Egemnazarov B, Kraut S, Eisele HJ, Dumitrascu R. et al. Arterial hypertension in a murine model of sleep apnea: Role of nadph oxidase 2. Journal of hypertension. 2014;32:300-305
32. Castro-Grattoni AL, Suarez-Giron M, Benitez I, Tecchia L, Torres M, Almendros I. et al. The effect of chronic intermittent hypoxia in cardiovascular gene expression is modulated by age in a mice model of sleep apnea. Sleep. 2021 44
33. Huber HJ, Holvoet P. Exosomes: Emerging roles in communication between blood cells and vascular tissues during atherosclerosis. Curr Opin Lipidol. 2015;26:412-419
34. Khalyfa A, Zhang C, Khalyfa AA, Foster GE, Beaudin AE, Andrade J. et al. Effect on intermittent hypoxia on plasma exosomal micro rna signature and endothelial function in healthy adults. Sleep. 2016;39:2077-2090
35. Khalyfa A, Kheirandish-Gozal L, Khalyfa AA, Philby MF, Alonso-Alvarez ML, Mohammadi M. et al. Circulating plasma extracellular microvesicle microrna cargo and endothelial dysfunction in children with obstructive sleep apnea. American journal of respiratory and critical care medicine. 2016;194:1116-1126
36. Li J, Rohailla S, Gelber N, Rutka J, Sabah N, Gladstone RA. et al. Microrna-144 is a circulating effector of remote ischemic preconditioning. Basic Res Cardiol. 2014;109:423
37. Li J, Cai SX, He Q, Zhang H, Friedberg D, Wang F. et al. Intravenous mir-144 reduces left ventricular remodeling after myocardial infarction. Basic Res Cardiol. 2018;113:36
38. Siddiqui MR, Akhtar S, Shahid M, Tauseef M, McDonough K, Shanley TP. Mir-144-mediated inhibition of rock1 protects against lps-induced lung endothelial hyperpermeability. Am J Respir Cell Mol Biol. 2019;61:257-265
39. Cheng J, Cheng A, Clifford BL, Wu X, Hedin U, Maegdefessel L. et al. Microrna-144 silencing protects against atherosclerosis in male, but not female mice. Arterioscler Thromb Vasc Biol. 2020;40:412-425
40. Fu X, Huang X, Li P, Chen W, Xia M. 7-ketocholesterol inhibits isocitrate dehydrogenase 2 expression and impairs endothelial function via microrna-144. Free radical biology & medicine. 2014;71:1-15
41. Janaszak-Jasiecka A, Siekierzycka A, Bartoszewska S, Serocki M, Dobrucki LW, Collawn JF. et al. Enos expression and no release during hypoxia is inhibited by mir-200b in human endothelial cells. Angiogenesis. 2018;21:711-724
42. Xiao W, Lou N, Ruan HL, Bao L, Xiong ZY, Yuan CF. et al. Mir-144-3p promotes cell proliferation, metastasis, sunitinib resistance in clear cell renal cell carcinoma by downregulating arid1a. Cell Physiol Biochem. 2017;43:2420-2433
43. Liu C, Su C, Chen YC, Li G. Mir-144-3p promotes the tumor growth and metastasis of papillary thyroid carcinoma by targeting paired box gene 8. Cancer Cell Int. 2018 18
44. Dore LC, Amigo JD, Dos Santos CO, Zhang Z, Gai X, Tobias JW. et al. A gata-1-regulated microrna locus essential for erythropoiesis. Proc Natl Acad Sci U S A. 2008;105:3333-3338
Author contact
![]() Corresponding author: Huina Zhang, Beijing An Zhen Hospital, Capital Medical University, Beijing Institute of Heart Lung and Blood Vessel Disease, No. 2 Anzhen Road, Beijing, 100029, China, Tel.: +86-10-64456509, E-mail: whinnerzhnccmu.edu.cn.
Corresponding author: Huina Zhang, Beijing An Zhen Hospital, Capital Medical University, Beijing Institute of Heart Lung and Blood Vessel Disease, No. 2 Anzhen Road, Beijing, 100029, China, Tel.: +86-10-64456509, E-mail: whinnerzhnccmu.edu.cn.