Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
KRAS-induced macropinocytosis
Robustness of macropinocytosis...
The difference in...
Drug delivery systems exploiting...
Opportunities that synergize...
Limitations in the current...
Off-target risks of exploiting...
Pitfalls in our current...
Concluding remarks
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2022; 12(3):1321-1332. doi:10.7150/thno.67889 This issue Cite
Review
Exploiting macropinocytosis for drug delivery into KRAS mutant cancer
Huiqin Liu, Feng Qian ![]()
School of Pharmaceutical Sciences, Beijing Advanced Innovation Center for Structural Biology, and Key Laboratory of Bioorganic Phosphorus Chemistry & Chemical Biology (Ministry of Education), Tsinghua University, Beijing 100084, P.R. China.
Received 2021-10-9; Accepted 2021-12-9; Published 2022-1-1
Abstract

KRAS mutations are one of the most common gene mutations linked to cancer, presenting in approximately 25% of all tumors, especially pancreatic, lung, and colorectal cancers. Mutant KRAS has long been considered an undruggable target, stalling progress in direct KRAS targeting for many years, while targeted drug delivery into KRAS mutant cells utilizing their transformed metabolic behavior might present an alternative opportunity. Macropinocytosis, a nonselective, fluid-phase, endocytic route, was found to be upregulated as a metabolic feature in KRAS-driven tumors and plays a critical role in nutrient acquisition from extracellular fluids. With the observation that a variety of drug delivery systems could be internalized by KRAS mutant cancer cells through macropinocytosis, exploiting macropinocytosis for intracellular delivery of therapeutics into KRAS mutant tumor cells is emerging as a new drug delivery expedition. In this article, we summarized cancer biology studies that examined KRAS mutation-induced macropinocytosis, reviewed recent studies exploiting macropinocytosis enhancement for KRAS mutant cancer cell-selective drug delivery, and discussed the potential opportunities, challenges and pitfalls of this strategy.
Keywords: Macropinocytosis, KRAS, drug delivery, pancreatic cancer
Introduction
Active transportation of extracellular substances into cells is one of the basic activities of living cells and is required to implement many important biological functions, such as signal transduction, nutrient absorption, and antigen uptake [1]. The major endocytic routes, including macropinocytosis, clathrin-mediated endocytosis, caveolae-mediated endocytosis, and clathrin- or caveolae-independent endocytosis, are characterized by diverse regulation mechanisms and endocytic vesicles with different sizes and biological natures [1]. Among them, macropinocytosis is a receptor-independent, actin-dependent, nonselective endocytic pathway utilized by cells to “drink” fluids and solutes from the external milieu, which was defined based on morphological observation in the early days [2]. As early as 1931, Warren Lewis first described this process morphologically. Upon growth factor stimulation, waving sheet-like extensions of the plasma membrane were observed in rat macrophages, and evolved into heterogeneous phase-bright organelles with a diameter greater than 0.2 μm [3]. To distinguish this pathway from endocytic processes involving smaller vesicles, Lewis named this activity pinocytosis (later renamed macropinocytosis), and the vesicles generated in this pathway macropinosome. Later, macropinocytosis, but not other receptor-mediated endocytosis pathways, was found to be selectively inhibited by amiloride, a Na+/H+ exchangers (NHE) inhibitor [4]. The functional association between NHE remained unknown for many years until Koivusalo et al. addressed mechanisms mediating NHE inhibition of EGF-induced macropinocytosis in 2010 [5]. They confirmed that pH maintenance, rather than NHE activity itself or the associated Na+ gain, is required for macropinocytosis in A431 cells. Briefly, upon NHE inhibition, H+ extrusion was impaired, resulting in decreased cytosolic pH, which exclusively impaired the recruitment and activation of Rac1 and Cdc42 to membrane ruffles. Subsequently, suppression with amiloride and its analog 5-(NEthyl-N-isopropyl) amiloride (EIPA) has been widely used to identify macropinocytosis [6]. To quantify the intensity of macropinocytosis in certain cell types, detecting the uptake of fluorescence-labelled agents, such as dextran, a known macropinocytosis marker, is the most common and usually also the only option [7].
The formation, trafficking and maturation of macropinosomes through the endocytic system and the regulatory mechanisms responsible for these dynamic processes have been comprehensively reviewed in several other excellent reviews [6,8,9,10] and thus will not be repeated here. Briefly, with the activation of small GTPases (such as Rac1) and the cell division control protein 42 homolog (Cdc42), the macropinocytosis process begins. Actin-rich, sheet-like membrane protrusions or ruffles are produced and further form circular cups. Enclosing these cups into macropinosomes depends on the production of phosphatidylinositol (3,4,5)-triphosphate (PIP3) and the inactivation of Rac1. The newly generated macropinosomes can fuse with the plasma membrane and release their contents back to the extracellular space or be transported to the lysosome for degradation.
Macropinocytosis is considered to be a conserved endocytic pathway shared by all vertebrate cells, while the regulation and functions of macropinocytosis in different cells could differ, as reviewed elsewhere [2,11]. A molecular-level understanding of macropinocytosis and its signalling networks has gradually been unveiled [12,13]. Generally, macropinocytosis in normal vertebrate cells is induced in response to growth factor stimulation (EGF, PDGF, etc.); however, some special cell types, such as innate immune cells and KRAS-transformed cancer cells, have intrinsic constitutive macropinocytosis apart from the induced forms [14]. In the sight of immunology, macropinocytosis of dendritic cells and macrophages have been most studied, which is a major pathway for the capture of antigens [11]. In KRAS-transformed cancer cells, enhanced macropinocytosis was recognized as a metabolic adaptation under nutrient stress conditions, supporting the survival and proliferation of aggressive tumors by scavenging extracellular proteins, lipids and cell debris [12,15,16].
KRAS is an oncogene protein with the capability of controlling diverse cellular functions, including macropinocytosis [17,12]. Wild-type KRAS signalling is well controlled by switching between the active GTP-bound state and inactive GDP-bound state, stimulated by upstream EGFR activation and deactivated by guanine-nucleotide exchange factors [18]. KRAS mutations, most of which (84%) occur at G12, resulting in single amino acid substitutions that activate the oncoprotein by hindering its ability to hydrolyze GTP, were detected in various solid tumors, such as lung cancer (~25% of cases), colorectal cancer (~35% of cases), and pancreatic cancer (~95% of cases), affecting an estimated 147,000 patients per year in the US [19,20]. Although sotorasib (LumakrasTM), an inhibitor specifically targeting glycine to cysteine (G12C) missense mutations has been approved by US FDA, to date, there are arduous challenges to develop molecular targeted therapies against other KRAS mutations due to the special protein structure and biochemical properties of KRAS, making the strategy of targeting drug delivery into KRAS mutant cancer cells an attractive alternate [21].
The supposition that KRAS mutant tumor cells have constitutive and enhanced macropinocytosis drives an interesting question: could such enhanced macropinocytosis be distinctive enough to act as a “phenotypic target” for KRAS mutant cancer, where targeted therapeutics are urgently needed? Apparently, the addiction and dependence of KRAS mutant tumor cells on macropinocytosis lay the theoretical basis for obtaining therapeutic benefits by inhibiting macropinocytosis. On the other hand, instead of blocking macropinocytosis directly, some scientists are attempting to utilize this process to obtain drug delivery benefits. It has been reported that various drug delivery systems, including albumin, micelles, liposomes, exosomes, etc., could be actively internalized by KRAS mutant cancer cells through macropinocytosis, suggesting that macropinocytosis regulation in the tumor setting can be harnessed for the delivery of anticancer therapeutics. In this article, we focused on constitutive macropinocytosis in KRAS mutant cancer cells and discussed the significance of this biological process from a drug delivery perspective. We analyzed the specificity and robustness of this phenomenon as a drug target candidate, summarized recent efforts in exploiting macropinocytosis for drug delivery into KRAS mutant cancer, and discussed the opportunities, challenges and pitfalls of this strategy.
KRAS-induced macropinocytosis
One of the distinctive features that define macropinocytosis is the fact that it can be stimulated by growth factors, including EGF. KRAS is downstream of the EGFR signalling pathway, could be activated by either growth factor stimulation or oncogenic mutation, leading to the stimulation of different signal transduction pathways, including Rac, Cdc42, etc., that are necessary for macropinocytosis. The mechanistic underpinnings of oncogenic KRAS-induced macropinocytosis have been reviewed elsewhere [12]. Here, we focus on the literature and studies that define the correlation and causality between oncogenic KRAS and macropinocytosis.
There are three closely related RAS isoforms: HRAS, KRAS, and NRAS in mammalian cells playing a central role in the regulation of cell growth and differentiation. Oncogenic HRAS protein (rather than gene)-induced macropinocytosis was reported as early as 1986 by Bar-Sagi and Feramisco, when RAS oncogenes were considered to be one of the contributing events of certain types of human cancers [22]. To determine the cell activity directly triggered by the RAS protein, human HRAS protein was transformed into quiescent rat embryonic fibroblasts (REF-52) via microinjection. After injection, cell surface ruffles were induced within 30 mins to 1 hour, as revealed by scanning electron microscopy, and fluid-phase pinocytosis was significantly increased, as measured by uptake of fluorescein-conjugated dextran. Both the proto-oncogenic HRAS protein and oncogenic HRAS protein can induce rapid enhancement of cell membrane ruffles and pinocytosis. The difference is that the effect of oncogenic protein is relatively long-acting for more than 15 hours after injection, while the effect of proto-oncogenic protein is short-lived, limited to an interval of 3 hours after injection. Dose dependence of the stimulation of pinocytosis by the HRAS oncogene protein was also demonstrated in this study. The abilities of the HRAS and KRAS isoforms to induce membrane ruffling and macropinocytosis was compared in a follow-up study by the Bar-Sagi group [23]. The average surface area of KRASG12V-induced membrane ruffles and the number of KRASG12V-induced pinocytic vesicles per cell were found to be ~2-fold greater than those of the HRASG12V groups, suggesting a stronger capability of macropinocytosis induction by KRASG12V, since the expression levels and cellular distribution of the two were similar. To the best of our knowledge, there have not been published researches regarding NRAS related macropinocytosis.
Robustness of macropinocytosis in KRAS mutant cancer cells
It is worth first commenting on the robustness of elevated macropinocytosis of KRAS mutant cancer cells because any conclusion that is true but only true under an extremely narrow set of conditions is therapeutically intractable regardless of effect size [24]. In other words, we need to confirm that elevated macropinocytosis of KRAS mutant cancer cells could be reproduced consistently over a range of experimental conditions (for example, by investigating multiple cell lines or patient tumor samples in the setting of different KRAS mutant alleles, reproducing the findings in independent studies).
KRAS mutation is an oncogenic driver in solid tumors, including but not limited to pancreatic cancer, where KRAS mutation-induced macropinocytosis has been studied relatively earlier and more comprehensively. A study published in 2013 by Commisso and colleagues demonstrated that macropinocytosis of proteins in RAS-transformed cells acting as an amino acid supply route [16]. Briefly, robust levels of macropinocytosis have been demonstrated in PDAC cell lines that harbor oncogenic KRAS mutations, observed in tumors of KPC mice (LSL-KRASG12D; TP53R172H/+; Pdx1-Cre; a transgenic spontaneous pancreatic cancer mouse model), and meaningfully, detected in human PDAC tumor samples regardless of some intratumoral variability. The necessity and sufficiency of KRAS mutation for macropinocytosis induction have been verified using classical in vitro experimental systems by KRAS knockdown in cell lines with KRAS mutation and transforming exogenous KRAS mutation into normal cells like NIH3T3. The macropinocytosis levels in these experiments were detected under similar experimental settings, that is, cells or tissue samples were incubated in a serum-free medium containing 1-2 mg/mL fluorescently labelled dextran for 30 minutes and analyzed by fluorescence microscopy. We have assessed whether this biological phenomenon was robust and persistent enough to be exploited for drug delivery in a previous study, wherein we compared the uptake kinetics of FITC-dextran (~70 kDa) by KRAS-mutant (mt) and KRAS wild-type (wt) cells across wide ranges of concentrations and times using fluorescence-activated cell sorting (FACS) rather than microscope [25]. Although the methods and experimental conditions are different, the trends are similar: MIA PaCa-2, which is a human pancreatic cell line with mutant KRAS, demonstrated obviously elevated macropinocytosis compared to the KRAS wt pancreatic cancer cell line BxPC-3. Across a period of 0.5-8 h and a dextran concentration range from 0.2-5 mg/mL, elevated extents and kinetics of macropinocytosis were also observed in KRASG12V transformed BxPC-3 cells compared to BxPC-3 control cells.
KRAS mutations are a cluster containing different versions, and several KRAS mutant subtypes related to the effect of macropinocytosis have been studied, including G12C, G12D, G12V, and G12R. Simply put, the first three subtypes have shown the capability to induce macropinocytosis in different studies, with the exception of G12R. MIA PaCa-2, a human pancreatic cancer cell line with a homozygous KRASG12C allele, displayed appreciably higher macropinocytosis levels as measured by 70 kDa dextran uptake compared to another pancreatic cell line with wild-type KRAS (i.e. BxPC-3) [16,25]. Macropinosomes were detected in pancreatic intraepithelial neoplasia lesions derived from KPC mice harboring KRASG12D, while pancreases collected from wild-type mice were found to be negative for macropinocytosis signals. Oncogenic KRASGV12 was also proven to be sufficient to stimulate robust EIPA-sensitive TMR-dextran uptake in mouse NIH 3T3 cells and BxPC-3 cells [16,25]. In agreement with these findings, Aaron Hobbs et al. found that transient siRNA suppression of mutant KRAS reduced macropinocytosis in KRASG12D, KRASG12V, and KRASG12C mutant cell lines in a separate study [20]. The surprising discovery of this research is that atypical KRAS mutation G12R, which is relatively rare (~1% in lung and colorectal cancers, ~20% in PDAC), has unique properties in driving macropinocytosis. Suppression of KRAS did not reduce macropinocytosis in any of the seven KRASG12R mutant pancreatic cancer cell lines investigated in their study, and KRASG12R uniquely failed to stimulate macropinocytosis in three model systems (rat intestinal epithelial cells RIE-1, mouse fibroblasts NIH/3T3 and hTERT-immortalized human pancreatic duct-derived epithelial cells), similar to KRASG12D or KRASG12V [20]. In terms of mechanism, KRASG12R is impaired in PI3K-AKT activation, which is required for macropinocytosis in KRASG12R-mutant PDAC, due to structural perturbations and thus defective for interaction with p110α PI3K, a key downstream effector of KRAS.
The pervasiveness of macropinocytosis in various malignant tumors has been summarized by Commisso and colleagues [26]. Since their initial study was published in 2013, which focused on KRAS-mutated pancreatic cancer, macropinocytosis has been recognized as a prevalent metabolic feature of other solid tumors, including lung, prostate, bladder and colon cancer, demonstrated by accumulating studies employing different in vitro systems as well as in vivo animal models. For example, Qian and colleagues explored the role of macropinocytosis in A549, a human non-small cell lung cancer (NSCLC) cell line that is addicted to oncogenic KRAS. Relying on integrin avβ3 and its modulator galectin-3, A549 NSCLC cells exhibited high levels of macropinocytosis to sustain their cellular fitness [27]. Nevertheless, it has become apparent that the driving forces for cancer-associated macropinocytosis may be more diversified than expected since independent studies revealed that macropinocytosis could be enhanced by KRAS, HRAS, EGFR activation [28], galectin-3 [27], loss of PTEN [29], Src activation [30], etc.
The difference in macropinocytosis in KRAS mt cancer cell vs. KRAS wt cells
Is there a therapeutic window for macropinocytosis-based treatments against KRAS mutant cancer? A major challenge for such a treatment strategy is that macropinocytosis is a universal cellular process that exists in both KRAS mt cancer cells and normal cells. Therefore, a sufficiently distinctive macropinocytosis activity between cancer and normal cells is the significant determinant of the rationale for this strategy from the biological perspective, and quantitative analysis of the levels of macropinocytosis in KRAS mt tumor cells and KRAS wt normal cells can help to determine to what extent this KRAS-enhanced macropinocytosis can be harnessed for the delivery of anti-cancer therapeutics.
To date, the majority of the literature reports macropinocytosis levels measured by the method reported by Commisso et al., that is, incubating the cell or tissue sample in serum-free medium containing 1-2 mg/mL fluorescently labelled dextran for 30 minutes and counting the number of fluorescent spots per cell using a fluorescence microscope. Although elevated macropinocytosis of KRAS mt tumor cells has been repeatedly verified by different laboratories, the levels are largely variable, and there is currently no conclusive answer to the exact extent of macropinocytosis enhancement by mutant KRAS. Two reasons could have caused such complications. First, different experimental settings (timepoints, with/without serum depletion), analytical methods (macroscope or FACS) and different model systems (cell lines or tissues) could provide different results, and the difference could span two orders of magnitude (Table 1). Second, most of these studies used immortalized cells with wt KRAS as a control, such as NIH3T3, BxPC-3, and MCF-7, none of which are truly normal human cells.
Relative macropinocytosis activity of KRAS mt cancer cells compared to KRAS wt cells
| References | Macropinocytosis assay | Sample system | Relative levels |
|---|---|---|---|
| Commisso et al., 2013 [16] | TMR-dextran was added to the serum-free medium at a final concentration of 1 mg/mL for 30 min at 37 °C. Total fluorescent particle area per cell was determined from at least five fields using the ImageJ. | MIA PaCa-2 (KRASG12C) compared to BxPC-3 cells | 8-fold |
| KRASG12V NIH 3T3 cells compared to untransformed control cells | 4-fold | ||
| Qian et al., 2014 [27] | Macropinocytosis assays were performed as described previously. [16] | A549 cells (KRASG12S) vs. MCF7 breast cancer cells | ~100 fold |
| Kamphorst et al., 2015 [31] | Freshly acquired human tumor specimens (n=5) were incubated with high molecular weight TMR-dextran (1-2 mg/mL, at 37 °C for 20-30 minutes) and intracellular uptake of TMR-dextran was assessed by fluorescence microscopy. | CK19-positive tumor cells vs. normal adjacent tissue | An accurate quantitative comparison is not provided |
| Kamerkar et al. 2017. [32] | Macropinocytosis assays were performed as described previously. [16] | PANC-1(KRASG12D) and BxPC-3 | ~9 fold |
| Aaron Hobbs et al., 2020 [20] | Macropinocytosis assays were performed as described previously. [16] | KRASG12D-transformed RIE-1 cells compared to untransformed control cells | >40 fold |
| KRASG12V-transformed RIE-1 cells compared to untransformed control cells | ~40 fold | ||
| Aubert et al., 2020 [33] | Cells were incubated in serum-free media containing 0.5 mg/mL of Lysine-fixable TMR-Dextran (10 kDa) for 30 min at 37 °C. | Isogenic intestinal epithelial cell model (IEC-6) stably expressing KRASG12V compared to untransformed control cells | ~3 fold |
| Liu et al., 2019 [25] | Cells were seeded into 12 wells plates (5 × 105 cells/well) and pulsed for 0, 1, 2, 4, or 8 h with 0.2-5 mg/mL 70 kDa FITC-dextran at 37 °C in complete growth medium. | BxPC-3 stably expressing KRASG12V compared to BxPC-3 control cells | 1-3 fold |
We noticed that, in most of these studies, macropinocytosis levels in KRAS mutant cells and control cells were determined at a single time point, i.e., 30 min, with a few exceptions. The macropinocytosis flux of KRAS mutant cancer cells may be underestimated since induction macropinocytosis of oncogenic RAS protein was found to last for more than 15 hours, while endogenously expressed mutant proteins are expected to induce persistent macropinocytosis [22]. It may provide extra useful information if the total amount of macropinocytosis flux across a certain time frame was calculated.
Drug delivery systems exploiting macropinocytosis of KRAS mutant cancer cells
With these observations, exploiting macropinocytosis for intracellular drug delivery into KRAS mutant cancer is emerging as a recent exploration. Based on the uptake inhibition induced by EIPA (a Na+/H+ exchange blocker used as a specific inhibitor of macropinocytosis), a variety of delivery systems have been identified, of which macropinocytosis is the dominant internalization mechanism in KRAS mt cancer cells. As previously reviewed by Desai and colleagues, lipids, polymers, peptides, extracellular vesicles, and inorganic nanoparticles have been reported to be internalized by KRAS mt cancer cells through macropinocytosis [34].
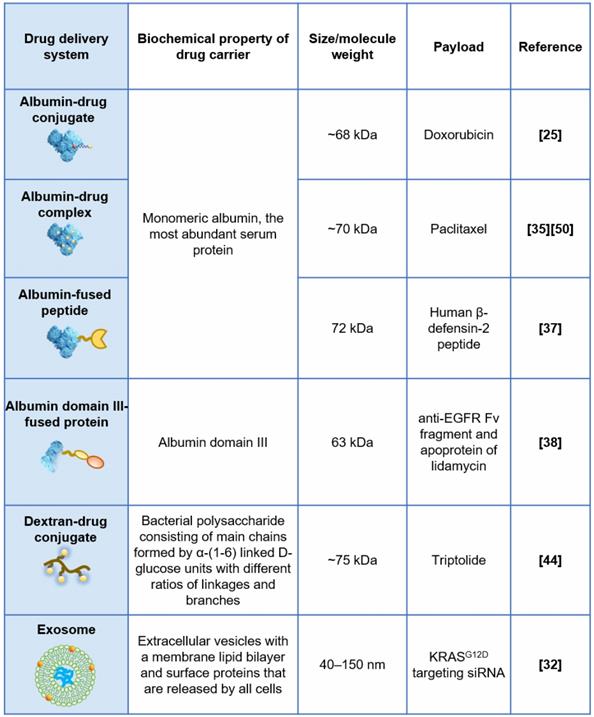
Human serum albumin (HSA), the most abundant serum protein in the human body, has attracted much attention since it was reported that pancreatic cancer cells with KRAS mutations can take up extracellular albumin through macropinocytosis and degrade it into amino acids, serving as an important nutrient source to fuel their metabolic addiction. Based on this finding, we covalently conjugated the model drug doxorubicin to native albumin to imitate the endocytosis and intracellular trafficking of albumin to expand the therapeutic windows of the cytotoxic payload in KRAS mt PDAC utilizing enhanced macropinocytosis [25]. We observed that doxorubicin-albumin conjugates exhibited EIPA-sensitive macropinocytosis in KRASG12V-overexpressing BxPC-3 cells and MIA PaCa-2 cells with an inherent KRAS mutation but not in BxPC-3 control cells, in a similar manner as native albumin.
AbraxaneTM is a nanoparticle albumin-bound formulation of paclitaxel (referred to as nab-PTX), that is currently used as the first-line treatment for advanced pancreatic cancer in combination with gemcitabine. Macropinocytotic uptake of nab-PTX by KP1.9 cells that are derived from the tumor of lung adenocarcinoma mouse model and express KRASG12D, was supported by EIPA inhibition and punctate subcellular colocalization of nab-PTX with fluorescent dextran [35]. Consistent with previous reports regarding native albumin, transient KRASG12D overexpression in KRAS wt BxPC-3 cells and inducible KRASG12D expression in tumor cells derived from a genetically engineered mouse model of pancreatic cancer, both enhanced nab-PTX uptake by ≥8-fold. In addition to monomeric albumin, cross-linked albumin nanoparticles also demonstrated enhanced macropinocytotic uptake in cells with mutant RAS compared to RAS wild-type control cells, as reported by Liu et al. [36].
Albumin-based drug carriers have also been used to deliver protein therapeutics. Du and colleagues utilized HSA to deliver human β-defensin-2 (DF), a small cationic peptide that has antibacterial and antiviral activity and tumor cell cytotoxicity against MIA PaCa-2 cells and xenograft tumors [37]. Based on the observation of fluorescence colocalization with dextran and the response to EIPA inhibition, the authors concluded that the internalization of DF-HSA in MIAPaCa-2 is apparently mediated by macropinocytosis and that there exists a differential pattern of intensity between KRAS mt MIAPaCa-2 cells and wt BxPC-3 cells. Wang et al. constructed the recombinant protein Fv-LDP-D3 by fusion domain III of HSA with an Fv fragment of an anti-EGFR antibody and the apoprotein of the antitumor antibiotic lidamycin (LDP) [38]. The authors demonstrated that, with EGFR-targeting and macropinocytosis-intensifying bifunctional attributes, Fv-LDP-D3 has shown the remarkable potential of tumor imaging and prominent tumor inhibition in AsPC-1 (human pancreatic cancer cells with a homozygous KRASG12D mutation)-derived xenografts. The uptake mechanism of Fv-LDP-D3 in MIA PaCa-2 and AsPC-1 cells was identified by EIPA inhibition assay and colocalization of fluorescent macropinocytosis marker (TMR-dextran) and FITC-labeled recombinant proteins.
Exosomes are nanosized extracellular vesicles (40-150 nm) with a membrane lipid bilayer. They could be released by all cells and have been used to deliver siRNA to specifically target KRASG12D in pancreatic tumors by Kamerkar and colleagues [32]. They found that exosome uptake but not liposome uptake in PANC-1 cells (PDAC cell line with KRASG12D) was reduced upon EIPA inhibition, suggesting that exosomes enter PANC-1 cells via macropinocytosis. Inspired by the application of exosomes in KRAS mutant pancreatic cancer treatment, Deng and colleagues constructed an exosome-mimicking membrane hybrid nanoplatform by fusing PEGylated lipids with the DC2.4 cell membrane for the benefits of easier generation and better loading efficiency [39]. The uptake of these exosome-mimicking nanoparticles in KRAS mt PANC-1 cells was significantly higher than that in BxPC-3 cells and was significantly decreased in response to EIPA inhibition, suggesting that this biomimetic system also imitates the uptake pattern of exosomes.
Dextran, a bacterial polysaccharide constructed by α-(1-6)-linked D-glucose units with different ratios of linkages and branches, has been used in drug delivery in the past to achieve solubilization or systemic PK modification before it became a popular “macropinocytosis marker” [40,41]. Based on the hypothesis that coupling anti-cancer drugs to high-molecular-weight carriers may optimize drug disposition and improve tumor inhibition efficacy, a doxorubicin-dextran (70 kDa) conjugate was synthesized via Schiff's base reaction, exhibited better antitumor activity and optimized biodistribution compared to the free drug in tumor models and was evaluated in a clinical phase I trial for patients with refractory solid tumors in 1993 [42,43]. Recently, we also confirmed the feasibility of selectively targeting KRAS mutant pancreatic cancer with 70 kDa dextran-drug conjugates via both in vitro cellular studies and in vivo tumor model assessment [44].
In summary, a variety of drug delivery systems with different physical and chemical properties have been reported to enter KRAS mutant cancer cells through macropinocytosis (Figure 1). Among them, albumin-based systems have been studied intensively. Despite the fact that most of the published studies confirmed that KRAS enhanced uptake and improved efficacy both in vitro and/or in vivo, the clinical significance of such a drug delivery strategy has yet to be validated.
Drug delivery systems exploiting macropinocytosis of KRAS mutant cancer cells.
Opportunities that synergize with macropinocytosis to further optimize drug delivery
The entry of complex delivery systems into KRAS tumor cells is only the first step to achieve effective drug delivery, followed by the process of intracellular transport, dissociation and release of the drug payload. Taking the albumin-drug conjugate as an example, after being encapsulated in the macropinosome, the protein and linker need to be degraded in acidic lysosomes to release the drug, and then the free drug reaches the site of function to induce corresponding pharmacological effects. In this dynamic intracellular process, a complex biological signalling network and microenvironment within the involved organelles could contribute to the final pharmacological effects. Consequently, distinct properties of KRAS mutant cancer cells compared to normal cells in these processes could also act in synergy with enhanced macropinocytosis in the pursuit of an ultimate goal for KRAS-targeted drug delivery. For example, the unique properties of lysosomes (pH, composition, abundance and degradation activity of lysosomal hydrolase, etc.) of KRAS mutant tumor cells, if any, could guide us in designing conditional active drug delivery systems. Unfortunately, it appears that there were few conclusive studies in this area. Although enhanced albumin degradation was observed in KRAS mutant tumor cells, as revealed by DQ-BSA (a probe that displays fluorescence after degradation) [16], it is difficult to precisely quantify the contribution of the intrinsic differences in lysosomal function due to the enhanced upstream albumin uptake, as well as the dynamic interaction between macropinosomes and lysosomes.
The neonatal Fc receptor (FcRn), which is responsible for albumin recycling by binding with it in a pH-sensitive manner, shields albumin from lysosomal degradation to achieve a long half-life (~19 days). We previously observed that in pancreatic cancer cells, albumin recycling was decreased while lysosomal catabolism of albumin was increased upon the reduction of FcRn expression, thus sensitized KRAS mutant PDAC to an albumin-conjugated drug but not to an unconjugated drug [25]. Although the intriguing connection between KRAS genotype and FcRn expression level remains blurred, the two critical regulators of albumin metabolism have demonstrated synergistic impacts on the sensitivity of pancreatic cancer to albumin-conjugated drugs both in vitro and in vivo. These results imply an enlarged therapeutic window of albumin-conjugated drugs against PDAC which harbors KRAS mutation and also lacks FcRn expression.
Li and colleagues found that in KRAS mt cells, the macropinocytosis of nab-PTX can be further enhanced therapeutically thus further improving the efficacy of the drug [35]. Among 13 tested compounds, six hits were found, including an IGF1R inhibitor AXL1717, which showed the ability to enhance nab-PTX uptake in two independent KRAS mt cancer cell lines [35]. In vitro, pretreatment with AXL1717 or another IGF1R inhibitor linsitinib decreased the IC50 of nab-PTX but not solvent-based PTX in PDAC cells with inducible KRASG12D by roughly >5-10-fold. In vivo, simultaneous dosing of AXL1717 and nab-PTX also notably improved tumor growth inhibition efficacy and extended the survival of allograft animal models. These observations led to the conclusion that an IGF1R-targeted kinase inhibitor enhances nab-PTX macropinocytotic uptake and efficacy, suggesting the potential of combination therapy.
Limitations in the current qualitative and quantitative methods for macropinocytosis detection
EIPA is the most commonly used tool reagent to qualitatively test whether cell entry of a given drug delivery system is macropinocytosis-dependent. However, to be precise, EIPA is a selective inhibitor of Na+/H+ exchangers other than a specific macropinocytosis inhibitor, which could indirectly inhibit macropinocytosis by adjusting the cytosolic pH and affect the recruitment and activation of Rac1 and Cdc42 to membrane ruffles. According to other reports as well as our own experiences, treatment with 50-75 μM EIPA for 30 mins could induce nonspecific endocytosis-independent effects and even unignorable cell viability in some sensitive cells.
Therefore, it would be quite helpful to conduct further studies aim to identify the specific regulators to macropinocytosis and develop corresponding inhibitors with precise working mechanisms. In addition to the mechanistic underpinnings of oncogenic KRAS-induced macropinocytosis that have been reviewed elsewhere [12], new mechanistic links between RAS signals and macropinocytosis induction have been gradually revealed [24]. Recently, Yao and colleagues revealed that syndecan 1 (SDC1) is a critical mediator for oncogenic RAS-induced macropinocytosis in PDAC using in vivo proteomic surfaceome screening [25]. Lin et al. attempted to identify novel macropinocytosis inhibitors using an FDA-approved drug library. Among 640 tested compounds, imipramine, which is used for depression treatment, was found to be able to potently inhibit macropinocytosis without exerting cytotoxic effects or inhibiting other endocytic pathways in cancer cells, as well as in dendritic cells and macrophages, whereas the mechanism has not yet been explored [45]. However, whether these newly discovered regulatory proteins (such as SDC1) or compounds have sufficient specificity to detect macropinocytosis still needs further research. At present, it is worthwhile to combine multiple methods (i.e., morphological definition, response to amiloride inhibition and growth factor stimulation) to define macropinocytosis.
The most commonly reported macropinocytosis quantification method is the one established by Commisso et al., briefly, first incubate the cells or tissue samples of interest in serum-free medium containing 1-2 mg/mL fluorescently labelled dextran (70 kDa) for 30 minutes, after washing and fixation, then analyze the cells attached on glass slides by fluorescent microscope. Sometimes, FACS is also used as an auxiliary verification method. Jin et al. established a real-time live cell surface imaging method to observe the dynamic macropinocytosis process using three-dimensional-structured illumination microscopy, providing a new approach for qualitative observation. They acquired real-time morphological data of internalized structures (numbers, depth, size of macropinocytic cups) on the surface of MIA PaCa-2 cells during macropinocytosis [46].
In addition, the experimental approaches for the analysis of macropinocytosis in individual patients are also quite limited, which might be a bottleneck in clinical translation for macropinocytosis-exploiting therapeutic strategies. Although some ex vivo studies have investigated several freshly acquired human tumor tissues, information regarding the levels and heterogenicity of macropinocytosis in the tumors of patients has not been reported. Moreover, most of the patients with metastatic tumors are not suitable for surgery, limiting the application of the ex vivo analytical methods that require isolated tumor tissues. A quantitative, real-time, in vivo evaluation method, would be highly desired. On the other hand, identifying reliable biomarkers for elevated macropinocytosis that can be easily detected will be of great value but also faces daunting challenges.
Off-target risks of exploiting macropinocytosis for targeted drug delivery
Off-target risks are critical for the efficacy and safety of macropinocytosis-based drug delivery strategies. Non-specific uptake in healthy tissues such as the liver and spleen, facilitated by a network of phagocytic cells (referred to as the reticular endothelial system, or mononuclear phagocyte system), is an important off-target concern for nanomedicines including macropinocytosis-exploiting drug delivery systems discussed earlier. The effect of physicochemical and surface properties on in vivo fate of drug nanocarriers has been intensively reviewed elsewhere [47]. For macropinocytosis-based drug delivery strategies, further off-target risk caused by the heterogeneity of macropinocytosis at tissue and cellular levels would be an extra consideration. For instance, beyond RAS-transformed tumor cells, some non-transformed mammalian cells were also reported to have constitutive macropinocytosis, as summarized in Figure 2.
The regulation and functions of macropinocytosis in different cell types.
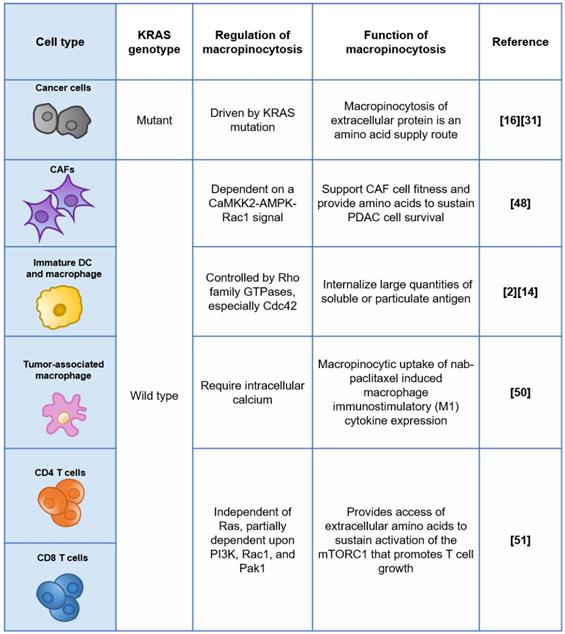
Within the tumor microenvironment (TME) of PDAC, KRAS mutant tumor cells are not the only cell category displaying elevated macropinocytosis, while other non-cancerous cells, such as KRAS wild-type cancer-associated fibroblasts (CAFs), which occupy the majority of the tissue volume, demonstrate similar behavior [48]. Zhang and colleagues found that stromal CAFs marked by α-SMA exhibited robust levels of macropinocytosis within KPC-derived orthotopic PDAC tumors, while PDGFR-expressing fibroblasts residing in normal murine pancreas did not display appreciable macropinocytosis. Mechanistically, macropinocytosis in CAFs is not reliant on EGFR signalling and instead is dependent on a CaMKK2-AMPK-Rac1 signal that is potentiated by elevated cytosolic Ca2+ [48]. Their study highlighted the functional role of macropinocytosis in the tumor stroma to support CAF cell fitness and provide amino acids to sustain PDAC cell survival, suggesting that CAFs could also be targeted by macropinocytosis-aiding drug carriers, and the net effect is unpredictable due to the complicated and contradictory role of CAFs.
Importantly, some immune cells have the capability of constitutive macropinocytosis. For example, immature dendritic cells (DCs) but not mature DCs have a very robust process of constitutive macropinocytosis, which is controlled by the Rho GTPase Cdc42 and Rac-dependent remodelling of the actin cytoskeleton [49]. Cullis and colleagues reported that nab-PTX is internalized by macrophages via macropinocytosis predominantly and is capable to drive M1 macrophage polarization in vitro and in vivo [50]. This study, of course, revealed a previously unappreciated mechanism of action of nab-PTX but also suggested that nab-PTX may induce off-target drug delivery into macrophages with active macropinocytosis. More recently, the active uptake of macropinocytosis probes (70 kDa dextran or albumin) was observed in both murine and human CD4+ and CD8+ T cells, and the T cells activated upon CD3/28 monoclonal antibody stimulation increased the extent of probe uptake [51]. Mechanistically, macropinocytosis could promote T cell growth by providing access to extracellular amino acids that are essential for the sustained activation of the mechanistic target of rapamycin complex 1 (mTORC1), even under amino acid replete conditions, as reported [51]. With the great breakthrough of immunotherapy in the treatment of cancer, we now have an unprecedented realization of the great significance and energy of the immune system in resisting tumors. A well-designed delivery system that enters KRAS mutant cancer cells through macropinocytosis and kills them efficiently while sparing immune cells will be much more promising.
Pitfalls in our current understanding of macropinocytosis
Last but not least, there are some widely conceived beliefs in this field, which have not yet been rigorously proven. First, macropinocytosis is a nonselective, bulk uptake process. Second, 70 kDa dextran is a marker of macropinocytosis. These two notions are contradictory: with the belief that macropinocytosis is a bulk uptake process without substrate selectivity, we are puzzled facing this question: when all the options are listed on the menu, do gourmets with KRAS mutations take on all these substrates, such as various components of the TME including extracellular proteins, drug delivery systems with different physical and chemical properties and different drug encapsulation mechanisms etc., with the same mechanisms and kinetics yet without any preference?
Intuitively, the answer will be “not very likely”. There are a few pieces of evidence worth discussing. First, the size range of the substances that enter KRAS mt cancer cells via macropinocytosis was not the maximum value of the theoretical volume of the macropinosome structures. Jin et al. observed that single-walled 2 μm carbon nanotubes and 420 nm SiO2 nanoparticles could not be internalized into MIA PaCa-2 cells, although their sizes were covered in the size range of macropinosomes [46]. Second, fluorescent dextran with different molecular weights has distinct endocytic pathways. Li et al. assessed the dependence of basal uptake of dextran with distinct molecular weights on macropinocytosis by using the Na+/H+ exchange-inhibitor amiloride and found that macropinocytosis dominates the fluid uptake of 70 kDa dextran, but not 10 kDa dextran, into HeLa cells [52]. The uptake of 70 kDa dextran was found to be inhibited to a much larger extent (up to 76%) than that of 10 kDa dextran (32%). Interestingly, although their molecular weights differ by 7 times, their hydrodynamic sizes differ by only 2-3 times, and the mechanism of how this difference affects macropinocytosis is still unknown.
These studies demonstrated that the physical and chemical properties of the substrate, size and molecular weight may all affect its internalization by macropinocytosis mechanism. At this stage, all we have seen is merely the tip of the iceberg regarding the science of macropinocytosis biology, and the presumption that it is a “non-selective” endocytosis process might just be an oversimplified understanding.
Concluding remarks
Multiple independent studies have demonstrated that tumor cells with endogenous KRAS mutations have enhanced macropinocytosis, which is oncogenic KRAS-dependent. These cells take in extracellular proteins through macropinocytosis and degrade them into amino acids, providing sources for the biosynthesis of tumor cells. In addition to extracellular proteins, many drug carriers can also enter KRAS mutant tumor cells through macropinocytosis, encouraging continuous attempts exploiting macropinocytosis to deliver drugs into tumor cells with KRAS mutations, and promising results (elevated uptake and improved efficacy) have been observed in vitro and in vivo. However, we still have some unknowns and risks: the limitation of specific chemical and genetic interference tools makes it tricky to identify macropinocytosis; the lack of real-time, quantitative methods for macropinocytosis level detection in vivo is an obvious blockage in translational medicine research; persistent macropinocytosis of macrophages and dendritic cells suggests the risk of off-target effects, etc. Thinking about these tough questions and seeking solutions by well-designed studies will truly help us understand macropinocytosis and thus guide us in the design of drug delivery systems, paving the way for the clinical application of this drug delivery strategy in the future.
Abbreviations
NHE: Na+/H+ exchanger; PDAC: pancreatic duct adenocarcinoma; TME: tumor microenvironment; CAFs: cancer-associated fibroblasts; KPC: Pdx1-Cre, LSL-KrasG12D/+, LSL-p53R172H/+; α-SMA: α-smooth muscle actin; EIPA: 5-(NEthyl-N-isopropyl) amiloride; Cdc42: cell division control protein 42; FACS: fluorescence-activated cell sorting; wt: wild type; mt: mutant; HAS: human serum albumin; nab-PTX: albumin-bound formulation of paclitaxel; NSCLC: non-small cell lung cancer; FcRn: neonatal Fc receptor; DCs: dendritic cells; mTORC1: mechanistic target of rapamycin complex 1.
Acknowledgements
Funding Sources
F.Q. received funding support from the China National Nature Science Foundation (project number 82073769) and Beijing Advanced Innovation Center for Structural Biology for drug delivery research targeting pancreatic cancer.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Schmid SL, Conner SD. Regulated portals of entry into the cell. Nature. 2003;422:37-44
2. Lin XP, Mintern JD, Gleeson PA. Macropinocytosis in different cell types: Similarities and differences. Membranes (Basel). 2020;10(8):1-21
3. Lewis W. Pinocyosis. Bull JOHNS HOPKINS Hosp. 1931;49:17-27
4. West MA, Bretscher MS, Watts C. Distinct endocytotic pathways in epidermal growth factor-stimulated human carcinoma A431 cells. J Cell Biol. 1989;109(6):2731-2739
5. Koivusalo M, Welch C, Hayashi H, Scott CC, Kim M, Alexander T. et al. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J Cell Biol. 2010;188(4):547-563
6. Kerr MC, Teasdale RD. Defining macropinocytosis. Traffic. 2009;10(4):364-371
7. Commisso C, Flinn RJ, Bar-Sagi D. Determining the macropinocytic index of cells through a quantitative image-based assay. Nat Protoc. 2014;9(1):182-192
8. Amyere M, Mettlen M, Van Der Smissen P, Platek A, Payrastre B, Veithen A. et al. Origin, originality, functions, subversions and molecular signalling of macropinocytosis. Int J Med Microbiol. 2001;291(6-7):487-494
9. Buckley CM, King JS. Drinking problems: mechanisms of macropinosome formation and maturation. FEBS J. 2017;284(22):3778-3790
10. Egami Y, Taguchi T, Maekawa M, Arai H, Araki N. Small GTPases and phosphoinositides in the regulatory mechanisms of macropinosome formation and maturation: Gtpases and phosphoinositides in macropinocytosis. Front Physiol. 2014;5:1-11
11. Lim JP, Gleeson PA. Macropinocytosis: An endocytic pathway for internalising large gulps. Immunol Cell Biol. 2011;89(8):836-843
12. Recouvreux MV, Commisso C. Macropinocytosis: A metabolic adaptation to nutrient stress in cancer. Front Endocrinol (Lausanne). 2017;8:1-7
13. Kay RR. Macropinocytosis: Biology and mechanisms. Cells Dev. 2021(April):203713. doi:10.1016/j.cdev.2021.203713
14. Stow JL, Hung Y, Wall AA. Macropinocytosis: Insights from immunology and cancer. Curr Opin Cell Biol. 2020;65:131-140
15. Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014;39(2):91-100
16. Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S. et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497(7451):633-637
17. Waters AM, Der CJ. KRAS: The critical driver and therapeutic target for pancreatic cancer. Cold Spring Harb Perspect Med. 2018;8(9):1-18
18. Mccormick F. KRAS as a Therapeutic Target. Clin Cancer Res. 2015;21(8):1797-1801
19. Kim D, Xue JY, Lito P. Targeting KRAS(G12C): From Inhibitory Mechanism to Modulation of Antitumor Effects in Patients. Cell. 2020;183(4):850-859
20. Hobbs GA, Baker NM, Miermont AM, Thurman RD, Pierobon M, Tran TH. et al. Atypical KRASG12R mutant is impaired in PI3K signaling and macropinocytosis in pancreatic cancer. Cancer Discov. 2020;10(1):104-123
21. McCormick F. Progress in targeting RAS with small molecule drugs. Biochem J. 2019;476(2):365-374
22. Bar-Sagi D, Feramisco JR. Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science. 1986;233(4768):1061-1068
23. Walsh AB, Bar-Sagi D. Differential Activation of the Rac Pathway by H-Ras and K-Ras. J Biol Chem. 2001;276(19):15609-15615
24. Kaelin WG. Common pitfalls in preclinical cancer target validation. Nat Rev Cancer. 2017;17(7):441-450
25. Liu H, Sun M, Liu Z, Kong C, Kong W, Ye J. et al. KRAS-enhanced macropinocytosis and reduced FcRn-mediated recycling sensitize pancreatic cancer to albumin-conjugated drugs. J Control Release. 2019;296:40-53
26. Commisso C. The pervasiveness of macropinocytosis in oncological malignancies. Philos Trans R Soc B Biol Sci. 2019 374(1765)
27. Qian Y, Wang X, Liu Y, Li Y, Colvin RA, Tong L. et al. Extracellular ATP is internalized by macropinocytosis and induces intracellular ATP increase and drug resistance in cancer cells. Cancer Lett. 2014;351(2):242-251
28. Lee SW, Zhang Y, Jung M, Cruz N, Alas B, Commisso C. EGFR-Pak Signaling Selectively Regulates Glutamine Deprivation-Induced Macropinocytosis. Dev Cell. 2019;50(3):381-392.e5
29. Kim SM, Nguyen TT, Ravi A, Kubiniok P, Finicle BT, Jayashankar V. et al. PTEN deficiency and AMPK activation promote nutrient scavenging and anabolism in prostate cancer cells. Cancer Discov. 2018;8(7):866-883
30. Tumbarello DA, Turner CE. Hic-5 Contributes to Transformation Through a RhoA / ROCK-dependent Pathway. J Cell Physiol. 2006;211(3):736-747
31. Nofal M, Drebin JA, Miller G, Grabocka E, Rabinowitz JD, Commisso C. et al. Human Pancreatic Cancer Tumors Are Nutrient Poor and Tumor Cells Actively Scavenge Extracellular Protein. Cancer Res. 2015;75(3):544-553
32. Kamerkar S, Lebleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA. et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546(7659):498-503
33. Aubert L, Nandagopal N, Steinhart Z, Lavoie G, Nourreddine S, Berman J. et al. Copper bioavailability is a KRAS-specific vulnerability in colorectal cancer. Nat Commun. 2020;11(1):1-15
34. Desai AS, Hunter MR, Kapustin AN. Using macropinocytosis for intracellular delivery of therapeutic nucleic acids to tumour cells. Philos Trans R Soc B Biol Sci. 2019 374(1765)
35. Li R, Ng TSC, Wang SJ, Prytyskach M, Rodell CB, Mikula H. et al. Therapeutically reprogrammed nutrient signalling enhances nanoparticulate albumin bound drug uptake and efficacy in KRAS-mutant cancer. Nat Nanotechnol. 2021;16:830-839
36. Liu X, Ghosh D. Intracellular nanoparticle delivery by oncogenic KRAS-mediated macropinocytosis. Int J Nanomedicine. 2019;14:6589-6600
37. Du Y, Shang B yang, Sheng W jin, Zhang S hua, Li Y. A recombinantly tailored β-defensin that displays intensive macropinocytosis-mediated uptake exerting potent efficacy against K-Ras mutant pancreatic cancer. Oncotarget. 2016Sep6;7(36):58418-58434
38. Wang X, Sheng W, Wang Y, Li L, Li Y, Zhang S. et al. A Macropinocytosis-Intensifying Albumin Domain-Based scFv Antibody and Its Conjugate Directed against K-Ras Mutant Pancreatic Cancer. Mol Pharm. 2018;15(6):2403-2412
39. Deng L, Zhang H, Zhang Y, Luo S, Du Z, Lin Q. et al. An exosome-mimiking membrane hybrid nanoplatform for targeted treatment towards Kras-mutant pancreatic carcinoma. Biomater Sci. 2021Aug21;9(16):5599-5611
40. Varshosaz J. Dextran conjugates in drug delivery. Expert Opin Drug Deliv. 2012;9(5):509-523
41. Chen F, Huang G, Huang H. Preparation and application of dextran and its derivatives as carriers. Int J Biol Macromol. 2020;145:827-834
42. Kikukawa A, Munechika K, Ueda Y, Yamanouchi K, Yokoyama K, Tsukagoshi S. Tissue concentration of adriamycin and adriamycin-oxidized dextran conjugate in tumor bearing rats and mice. Drug Deliv Syst. 1990;5(4):255-260
43. Danhauser-Riedl S, Hausmann E, Schick HD, Bender R, Dietzfelbinger H, Rastetter J. et al. Phase I clinical and pharmacokinetic trial of dextran conjugated doxorubicin (AD-70, DOX-OXD). Invest New Drugs. 1993;11(2-3):187-195
44. Fang Yuan, Mengnan Sun(co-first author), Zhengsheng Liu, Huiqin Liu, Weijian Kong, Rui Wang. et al. Macropinocytic dextran facilitates KRAS-targeted delivery while reducing drug-induced tumor immunity depletion in pancreatic cancer. Theranostics. Published online. 2022 doi:10.7150/thno.65299
45. Lin HP, Singla B, Ghoshal P, Faulkner JL, Cherian-Shaw M, O'Connor PM. et al. Identification of novel macropinocytosis inhibitors using a rational screen of Food and Drug Administration-approved drugs. Br J Pharmacol. 2018;175(18):3640-3655
46. Jin J, Shen Y, Zhang B, Deng R, Huang D, Lu T. et al. In situ exploration of characteristics of macropinocytosis and size range of internalized substances in cells by 3D-structured illumination microscopy. Int J Nanomedicine. 2018;13:5321-5333
47. Zhao Z, Ukidve A, Krishnan V, Mitragotri S. Effect of physicochemical and surface properties on in vivo fate of drug nanocarriers. Adv Drug Deliv Rev. 2019;143:3-21
48. Zhang Y, Recouvreux MV, Jung M, Galenkamp KMO, Li Y, Zagnitko O. et al. Macropinocytosis in Cancer-Associated Fibroblasts Is Dependent on CaMKK2/ARHGEF2 Signaling and Functions to Support Tumor and Stromal Cell Fitness. Cancer Discov. 2021Jul;11(7):1808-1825
49. Garrett WS, Chen LM, Kroschewski R, Ebersold M, Turley S, Trombetta S. et al. Developmental control of endocytosis in dendritic cells by Cdc42. Cell. 2000;102(3):325-334
50. Cullis J, Siolas D, Avanzi A, Barui S, Maitra A, Bar-Sagi D. Macropinocytosis of Nab-paclitaxel drives macrophage activation in pancreatic cancer. Cancer Immunol Res. 2017;5(3):182-190
51. Charpentier JC, Chen D, Lapinski PE, Turner J, Grigorova I, Swanson JA. et al. Macropinocytosis drives T cell growth by sustaining the activation of mTORC1. Nat Commun. 2020;11(1):1-9
52. Li L, Wan T, Wan M, Liu B, Cheng R, Zhang R. The effect of the size of fluorescent dextran on its endocytic pathway. Cell Biol Int. 2015;39(5):531-539
Author contact
![]() Corresponding author: Feng Qian, E-mail: qianfengedu.cn.
Corresponding author: Feng Qian, E-mail: qianfengedu.cn.