Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2022; 12(1):48-58. doi:10.7150/thno.65302 This issue Cite
Research Paper
Mucosal microbiome associates with progression to gastric cancer
Chin Wen Png1,2*, Wei Jie Jonathan Lee3,4,5*, Shijia Joy Chua3, Feng Zhu3, Gastric Consortium5, Khay Guan Yeoh3,4,5 ![]() , Yongliang Zhang1,2
, Yongliang Zhang1,2 ![]()
1. Department of Microbiology & Immunology, and NUSMED Immunology Translational Research Programme, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 117456, Singapore.
2. Immunology Programme, Life Science Institute, National University of Singapore, Singapore 117456, Singapore.
3. Department of Medicine, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 119228, Singapore.
4. Division of Gastroenterology and Hepatology, National University Health System, Singapore 119074, Singapore.
5. Singapore Gastric Cancer Consortium, Singapore 119074, Singapore.
*Co-first authors
Received 2021-7-24; Accepted 2021-9-9; Published 2022-1-1
Abstract

Background & Aims: Dysbiosis is associated with gastric cancer (GC) development. However, no longitudinal study was carried out to identify key bacteria that could predict for GC progression. Here, we aimed to investigate changes in bacterial metagenome prior to GC and develop a microbiome-based predictive model to accurately classify patients at risk of GC.
Methods: Bacterial 16S rDNA was sequenced from 89 gastric antral biopsies obtained from 43 participants. This study was nested in a prospective, longitudinal study, whereby study participants underwent screening gastroscopy, with further 1-2 yearly surveillance gastroscopies for at least 5 years. Putative bacterial taxonomic and functional features associated with GC carcinogenesis were identified by comparing between controls, patients with gastric intestinal metaplasia (IM) and patients with early gastric neoplasia (EGN).
Results: Patients with EGN had enrichment of Proteobacteria (in particular Proteus genus) and depletion of Bacteroidetes (in particular S24-7 family) in their gastric mucosa. Sequencing identified more patients with Helicobacter pylori compared to histopathological assessment, while H. pylori was also significantly enriched in EGN. Furthermore, a total of 261 functional features, attributing to 97 KEGG pathways were differentially abundant at baseline between patients who subsequent developed EGN (n = 13/39) and those who did not. At the same time, a constellation of six microbial taxonomic features present at baseline, provided the highest classifying power for subsequent EGN (AUC = 0.82).
Conclusion: Our study highlights early microbial changes associated with GC carcinogenesis, suggesting a potential role for prospective microbiome surveillance for GC.
Keywords: gastric cancer, microbiome, intestinal metaplasia, early gastric neoplasia, Helicobacter pylori
Introduction
Gastric cancer (GC) is one of the most prevalent malignant cancer worldwide and is amongst the top three leading cause of cancer-associated death globally [1]. Due to the late presentation of this disease, patients with early stages of GC are often asymptomatic [2]. Development of the most prevalent form of GC (i.e. intestinal subtype) follows a multistep process known as the Correa's cascade, progressing from precancerous gastritis, intestinal metaplasia (IM) to dysplasia.
Gastric carcinogenesis is the result of complex host-microbial interactions. Of which, Helicobacter pylori (H. pylori) infection remains as a major risk factor [3-5]. However, H. pylori infection alone does not explain all cases of GC. Only approximately < 5% of infected individuals developed GC. More importantly, eradication of H. pylori for GC resulted in mixed outcomes, with some studies demonstrating success in halting the progression of gastritis [6-8]. However, in most clinical trials, successful H. pylori eradication did not lead to reversal of neoplasia, or prevention of GC progression [9, 10].
Meanwhile, non-H. pylori microbiota was also found to play an important contributing role for development of GC. Hypergastrinaemic insulin-gastrin (INS-GAS) transgenic mice developed more severe GC in the presence of complex gastric microbiota compared to germ-free mice infected with H. pylori alone [11, 12]. Similarly from human microbiome studies, dysbiosis of non-H. pylori microbiota was associated with atrophic gastritis, IM and GC when compared to either healthy controls or patients with superficial gastritis [13-15].
However, there were no prior prospective studies, with repeated paired sampling of gastric mucosa, to investigate bacterial taxonomic and functional changes prior to gastric cancer. In this study, participants were part of a larger prospective longitudinal study [16], whereby participants underwent screening gastroscopy and subsequently 1-2 yearly surveillance gastroscopy. Gastric antral biopsies from these patients, at each visit, were profiled for the mucosal-associated bacterial composition to identify putative metagenomic taxonomic and functional changes prior to GC. Here, we report the differential gastric mucosal bacterial composition of participants across varying severity of IM, as well as of patients with early gastric neoplasia (EGN). Baseline bacterial features were also used to develop a predictive model to accurately classify patients at risk of EGN.
Methods
Participants and clinical data
This study was nested within the Singapore Gastric Cancer Epidemiology Program, GCEP [16, 17]. GCEP is a prospective multi-center longitudinal cohort, which recruited 2,980 Chinese subjects aged 50 or above, who had previous history of H. pylori infection and/or known premalignant gastric lesions such as atrophic gastritis and IM. All participants underwent gastroscopy by board-certified gastroenterologist and gastrointestinal surgeons using high quality white light endoscopy platforms with rapid escalation to high resolution image-enhanced endoscopy upon detection of abnormal pathology. The examination protocol was systematic with detailed photographic documentation. Participants who had never had an endoscopy or who had an endoscopy more than 12 months before the enrolment underwent a prospective baseline endoscopy. All tissues specimens were collected with ethics approval from the Singapore National Health Group Domain Specific Review Board (DSRB number: 2000/00329).
Prospective follow-up and definition of outcomes
After the baseline endoscopy, participants were scheduled for surveillance endoscopies 1-2 yearly. At each gastroscopy visit, 6 biopsies consisting of 2 each from the antrum, corpus, 1 from the incisura and 1 from the cardia were taken at each gastroscopy examination, and graded for chronic gastritis, gastric atrophy, intestinal metaplasia (IM), and dysplasia by two independent pathologists. Severity of gastritis and IM was scored using the updated Sydney System classification. Patients with focal, mild (< 30%) IM were classified as low-risk IM, whereas multi-focal, or moderate-severe IM (≥ 30%) were consider high-risk IM. The endpoint of GCEP was EGN, which included histological diagnosis of high-grade dysplasia, intramucosal carcinoma, and adenocarcinoma. Dysplasia was graded according to the revised Vienna classification and classification of carcinoma was done according to the WHO classification of tumours and the AJCC Cancer Staging Manual, Eight Edition [18, 19]. All participants were followed up annually, for at least 5 years, either at the clinic or via telephone for symptom review in the years that they were not scheduled for endoscopy surveillance. Participants who did not complete year 5 surveillance endoscopy were matched against the National Registry of Diseases Office for missed diagnoses of gastric cancer.
Sample collection and DNA extraction/QC
One gastric antral biopsy from each patient at each gastroscopy visit was obtained for bacterial 16S metagenomic analysis. DNA from snap-frozen mucosal biopsies were extracted with the Wizard™ Genomic DNA Purification kit (Promega, USA). Approximately 5 mg of antral gastric mucosal tissue was used. The samples were overlaid with 600 µl of Nuclei Lysis Buffer (50 mM Tris-HCl, pH 8.4, 0.5% SDS, 1 mM EDTA) and homogenized for 10 seconds. An aliquot of 3 µL of RNAase A Solution was added and incubated for 15 min at 37 °C. After which, 200 µl of Protein Precipitation Solution was added. DNA was further purified using the QIAamp DNA Mini kit (Qiagen, USA) before storage at -20 °C until required.
Bacterial 16S rRNA gene library preparation and sequencing
Bacterial 16S rRNA gene amplicon library preparation and sequencing was carried out according to Illumina “16S Metagenomic Sequencing Library Preparation” protocol. Briefly, bacterial 16S rDNA V3-V4 region was amplified using V3-V4 specific primers [20] with adapters and paired-end indices that allowed pooling and sequencing of PCR products. Amplicons were sequenced using MiSeq reagent kit V3 on Illumina MiSeq platform (Illumina, USA) that generated 2 x 300 bps paired-end reads. Demultiplex reads in FASTQ format (excluding reads that did not pass cluster filter) were used for data analysis as described below.
Data analysis
OTU construction and taxonomic assignment
OTU clustering and taxonomic profiling of 16 rRNA amplicon sequencing data was performed with the UPARSE pipeline (version 11) [21]. Briefly, paired-end reads from all datasets were first merged, filtered and de-replicated. For quality-filtering the UPARSE threshold of Emax = 1 was used, at which the most probable number of base errors per read is zero for filtered reads, and a truncation quality threshold of 15. Furthermore, reads were trimmed-filtered by a minimal length of 400 bp. OTUs were then sorted by size, singletons were discarded and OTUs were clustered at 97% similarity. Subsequently, the representative sequences for each cluster were mapped against the Greengenes 16S rDNA database (version 13.5) [22] to filter chimeras and obtain taxonomic assignment. This resulted in the generation of 644 OTUs, whereby 11% of the OTUs were classified at the species level, 59% genus, 87% family and 96% class. OTUs were further required to occur in at least 5% of the sample cohort, before use in differential abundance testing.
Statistical association analysis
Community microbial analysis were analysed using the R packages Phyloseq and Vegan [23, 24], whereby rarefaction was implemented to the depth at 90% of the minimum sample depth (3120 reads) of the dataset for diversity measurements. For prediction of functional gene content, raw OTU abundances were first normalised by their known/predicted 16S copy number prior to predicting metagenomic functional potential using the software PICRUSt2 [25], and reported as functional orthologs using the KEGG orthology database, whereby they were further agglomerated into higher-order KEGG modules and MetaCYC pathways. Gene set enrichment analysis of significant KOs (P < 0.05) were carried out using clusterProfiler R package [26]. For differential abundance testing of microbial features, DESeq2 [27] was used to normalised raw read counts whereby a case-control study design was adopted, with samples without IM/EGN as the control. Significant differentially abundant features were expressed as log2-fold change, and accounted for multiple testing through Benjamini-Hochberg correction. To construct a prediction model for subsequent gastric neoplasia OTUs abundances were converted to log10 relative abundances, then z-normalised, before passing through a 5-repeated 5-fold cross validation Lasso logistic regression using the SIAMCAT R package [28], and performance of the prediction model is presented as an AUROC with 95% confidence interval.
Results
Patient cohort and samples
Among the 43 participants who underwent a total of 89 gastroscopies, four were found to have EGN at baseline. Participants without EGN at baseline, were then stratified by stage of the Correa's cases, (a) superficial gastritis without IM (n = 17), focal, mild IM (i.e. low risk, n = 16), or moderate-severe, multifocal IM, (high risk, n = 6). One third (n = 13/39) of the participants at baseline subsequently developed EGN (Figure 1).
Schematic diagram showing clinical features of patients at baseline and at subsequent follow up. Each circle denotes a single biopsy sample from the respective patients, whereby they are coloured according to their corresponding histological diagnosis: No IM (grey), Low risk IM (yellow), High risk IM (orange) or EGN (red).
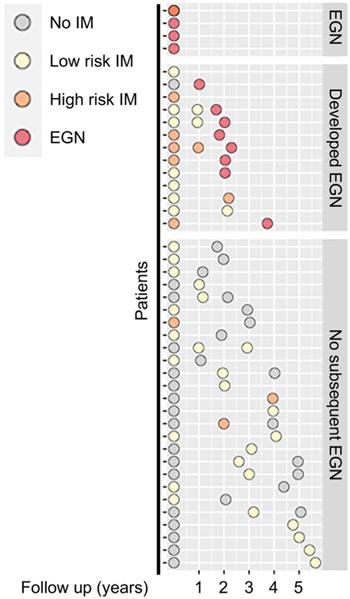
The baseline characteristics of the study participants, stratified by baseline histology, are detailed in Table 1. Majority of the patients had prior exposure to H. pylori (n = 31/43, 72.1%). However, none of the patients had H. pylori present upon histopathology review. There was a greater proportion of men with IM (77.3%, n = 17/22) and the mean age amongst patients with either high-risk or low-risk IM were significantly higher than patients with no IM (p = 0.01).
Clinical parameters of patients' samples and sequenced map reads output used in this study.
| No IM | Low Risk IM | High Risk IM | Dysplasia | P values | |
|---|---|---|---|---|---|
| N = 43 | 17 | 16 | 6 | 4 | - |
| Age (mean (SD)) | 54.82 (5.00) | 62.25 (7.49) | 64.33 (10.84) | 59.75 (5.74) | 0.011 |
| Sex = Male (%) | 6 (35.3) | 12 (75.0) | 5 (83.3) | 3 (75.0) | 0.055 |
| Clinical history of H. pylori infection = Yes (%) | 14 (82.4) | 9 (56.2) | 4 (66.7) | 4 (100.0) | 0.21 |
| IM grade (%) | < 0.001 | ||||
| Negative | 17 (100.0) | 0 (0.0) | 0 (0.0) | 2 (50.0) | - |
| Mild | 0 (0.0) | 3 (18.8) | 0 (0.0) | 0 (0.0) | - |
| Moderate | 0 (0.0) | 6 (37.5) | 1 (16.7) | 0 (0.0) | - |
| Marked | 0 (0.0) | 7 (43.8) | 5 (83.3) | 2 (50.0) | - |
| Multi focal IM (%) | 0 (NaN) | 2 (12.5) | 6 (100.0) | 2 (100.0) | NaN |
| Transition (%) | < 0.001 | ||||
| Baseline dysplasia | 0 (0.0) | 0 (0.0) | 0 (0.0) | 4 (100.0) | - |
| Developed EGN | 1 (5.9) | 7 (43.8) | 5 (83.3) | 0 (0.0) | - |
| No subsequent EGN | 16 (94.1) | 9 (56.2) | 1 (16.7) | 0 (0.0) | - |
| Map reads (mean (SD)) | 24246.41 (26665.40) | 17889.25 (13308.57) | 20217.00 (18521.55) | 20051.75 (19164.68) | 0.852 |
Differences in abundance of specific bacterial taxa in tissues from IM and EGN compared to no IM patients
To study the bacterial community changes associated with pre-GC stages (i.e. no IM, low risk IM, high risk IM, EGN), we assessed microbial diversity and richness of mucosal biopsy samples via the analysis of 16S rRNA gene hypervariable V3-4 regions. Overall, there were no significant differences in alpha and beta diversity across samples from different groups. Briefly, alpha diversity measurement based on various estimator including Chao1, ACE, Shannon, Simpson diversity index, did not show significant differences in species richness and phylogenetic diversity when comparing all 89 samples from across the different patient groups (Supplementary Figure 1A, 1B). Although bacterial profiles in samples from high risk IM and EGN group had larger variations compared to no IM and low risk IM (Supplementary Figure 1C), test of multivariant homogeneity of groups dispersions measured by the Bray-Curtis (P = 0.719) and ANOSIM based on Bray-Curtis (P = 0.209) did not demonstrate significant differences in beta diversity across the groups (Supplementary Figure 1D).
Associated microbial taxonomic in GC carcinogenesis
While changes in bacterial diversity were not identified, abundances of specific bacteria taxa in patients with IM or EGN compared to those with no IM were found to be significantly different (P < 0.05 and Benjamini-Hochberg adjusted P (Padj) < 0.1). Differentially abundant OTUs across the GC carcinogenesis stages were represented by enrichment of Proteobacteria and depletion of Bacteroidetes (Figure 2A). Among the Proteobacteria, abundance of OTUs from the genus Proteus were increased by 26 (log2) fold (Padj < 0.0001) in samples with EGN. In addition, H. pylori (species) was increased in EGN and high risk IM by ~41-fold (Padj < 0.05), whereas OTUs representing Phyllobacteriaceae and Enhydrobacter were increased in both EGN and low risk IM.
Differentially abundant bacterial OTUs in patients with IM and EGN compared to patients with no IM. (A) Bar chart shows the log2 fold change (X-axis) differences in abundance of top bacterial taxa in low risk IM (L-IM), high risk IM (H-IM) or patients with dysplasia (EGN) compared to No IM group based on Deseq2 univariant analysis. All comparisons P < 0.05, *denotes Benjamini-Hochberg adjusted P (Padj) < 0.1. (B) Box and whiskers plot showing increased Proteus bacteria in patients with dysplasia compared to No IM patients. ***P < 0.0007, FDR = 0.1, multivariant test (MaAsLin2).
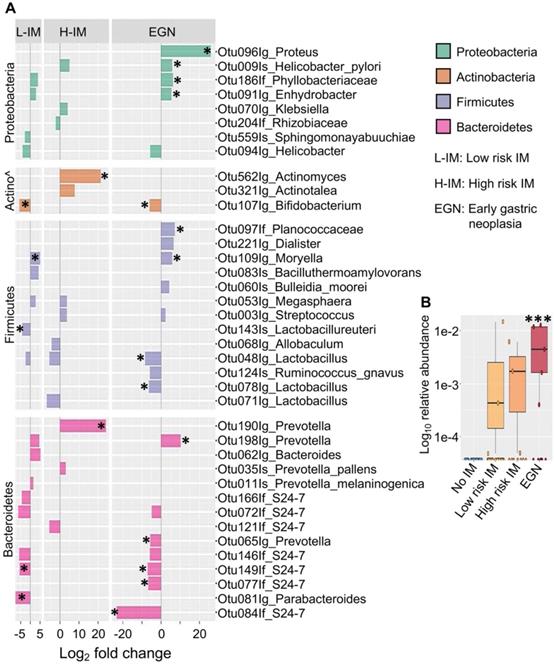
In addition, OTU representing Actinomyces (genus) that belongs to the Actinobacteria phylum was increased in high risk IM (Figure 2A). In contrast, Bifidobacteria (genus) were reduced in both EGN and low risk IM by 62-fold (Padj < 0.1). Changes in abundance of OTUs belonging to the Firmicutes phylum were also detected. These included Moryella (genus), which was increased in samples from EGN and low risk IM by ~55-fold (Padj = 0.07). Lactobacillus (Genus), which is a key lactic acid bacteria found in the gastric environment, was also significantly reduced by ~ 99 to 360-fold (Padj < 0.05) in EGN and low risk IM patients. Significant changes in the abundance of various OTUs belonging to the Prevotella genus and S24-7 group were also detected in EGN and low risk IM group. In particular, S24-7 group was reduced by 6.7 to 23 (log2) fold (Padj < 0.05) compared to no IM. Of note, S24-7 abundance was also found to be positively associated with IM reversal and negatively associated with IM progression (Supplementary Figure 2). Upon correction for other patient significant covariates such as age, gender, H. pylori exposure, tissue histological characteristics and repeated measurements, abundance of Proteus (P = 0.0007, Padj < 0.1, FDR = 0.1) remained as the only bacterial taxonomic feature that was significantly higher in samples with EGN compared to no IM (Figure 2B).
Specific gastric bacteria in baseline patient samples were predictive of EGN progression
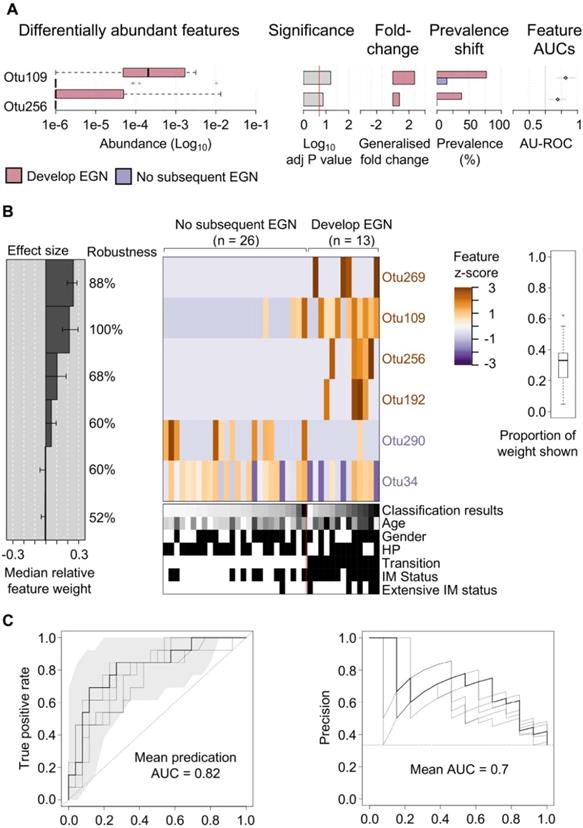
One third (n = 13/39) of study participants, without baseline EGN, were found to have EGN at the end of the 5-year study. Two microbial features demonstrated a statistically significant difference (FDR < 0.1) in the relative abundance at baseline between patients who did and did not develop EGN. Both OTU109 (Moryella genus) and OTU256 (Vibro genus) were significantly more abundant at baseline among patients who did develop EGN (Figure 3A). To evaluate if microbial taxonomic features would accurately predict GC risk, baseline microbial taxonomic features were analysed using feature-selecting machine-learning model (i.e. repeated 5-fold cross validation lasso regression). This model, which incorporated 6 taxonomic features, namely OTU109, OTU256 (described above), OTU269 (family: Comamonadaceae), OTU192 (genus: Paludibacter), OTU290 (genus: Agrobacterium), OTU34 (Order: Clostridiales) provided the highest classification power for progression to EGN, with the mean predication AUC at 0.82 for true positive and the mean AUC at 0.7 for precision recall (Figure 3B).
Specific gastric mucosal bacteria are predictive of disease progression to EGN. (A) Bar charts showing baseline OTUs (log10 relative abundance) that were significantly different between patients that progress to EGN compared to those who did not. (B) Model interpretation plots show the median relative feature weight (left barplot) of the top selected features, effect size, the robustness (percentages shown to the right of the barplot), and the feature z-scores across samples, ordered by group and classification score (right heatmap with annotations, black bar = positive status). HP denotes samples with prior clinical history of H. pylori infection. (C) Receiver operating characteristics (ROC) curve and precision recall curves showing the performance of the model shown in (B). SIAMCAT package was used for all analysis and calculations.
Microbial functional features associated with subsequent EGN
Gene content and predicated gene family abundance were inferred from the bacterial 16S rRNA gene sequencing data using PICRUSt2 [25]. In total, 261 differentially abundant KEGG orthologs (KOs) (P < 0.05) were identified and mapped to 97 KEGG pathways, 24 KEGG BRITE and 40 functional modules (Supplementary Data 1). Of these, the top differentially abundant KOs (P < 0.005) are shown in Figure 4A. These top KO entries were assigned to; (a) 19 KEGG pathways (most represented pathways included; “metabolic pathways” (38%), “galactose metabolism”, “phosphotransferase system” and “two-component system” (14% each); (b) 9 BRITE hierarchies (most represented BRITE list object included; “Enzymes” (71%), and “Transporters” (29%) and; (c) two functional modules including “M00704, tetracycline resistance” and “M00546, purine degradation” (Supplementary Data 1). Within the most represented metabolic pathways, key factors involved in galactosamine PTS system EIIB component (agaB, agaC, agaD) that are important for galactose metabolism were reduced in the progression group (Log2 fold change = -4.9, P = 0.003). Other metabolic pathway-related enzymes that were also reduced included levansucrase (sacB), arginine deiminase (arcA), CDP-paratose 2-epimerase (rfbE) and dTDP-L-rhamnose 4-epimerase (wbiB), which are involved in biosynthesis of secondary metabolites. On the other hand, taurine-2-oxoglutarate transaminase (toa) required for beta-alanine metabolism and FAD-dependent urate hydroxylase (HpxO), which catalyze the hydrolysis of uric acid for purine metabolism were increased in the progression group.
Gastric mucosal-associated microbiome functions by PICRUSt2 based on bacterial 16S rRNA gene sequences. Bar plots show top differentially abundant (A) KEGG orthology (KO) features (P < 0.005) and (D) MetaCyc pathways (P < 0.05) in baseline samples from patients that progress to EGN and patients that do not progress. *denote Benjamini-Hochberg Padj < 0.01 (DESeq2). (C) Dot plot showing top enrichment KEGG pathways that were either activated or suppressed in samples from patients that progress to EGN compared to patients that do not. “padj” and “counts” denote Benjamini-Hochberg adjusted P values and gene counts respectively. (D) Network plot showing KOs features and linkages associated with the enriched KEGG pathways.
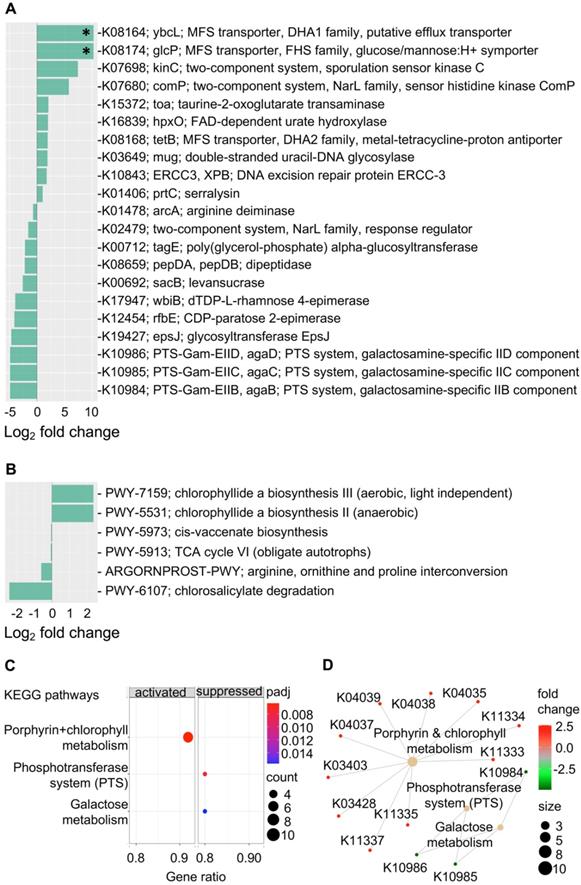
In addition, an increased abundance of KOs that is related to transcription machinery and DNA repair mechanisms were detected in the progression group. Functions represented in key KOs include the DNA excision repair protein ERCC-3, double-stranded uracil-DNA glycosylase, DNA repair protein RadD and DNA-directed RNA polymerase subunit beta-beta (Figure 4 & Supplementary Data 1). Functions related to protein kinases such as sporulation sensor kinase C and E, NarL family proteins including sensor histidine kinase ComP, nitrate/nitrite sensor histidine kinase NarQ and sensor histidine kinase NreB and sensor histidine kinase MalK from the CitB family protein were upregulated in baseline samples from patients that progressed to EGN. Of note, among the various top functional predications, increased cationic antimicrobial peptide (CAMP) resistance and vancomycin resistance were also identified in the progression group. Changes in these functions were represented by upregulation of serralysin, serralysin4-amino-4-deoxy-L-arabinose transferase (prtC), undecaprenyl phosphate-alpha-L-ara4N flippase subunit (arnF), undecaprenyl phosphate-alpha-L-ara4FN deformylase (arnD) and response regulator VanR (Figure 4 and Supplementary Data 1). At the same time, MFS transporters, namely ybcL and glcP were significantly increased (Log2 fold change = 10.3, P < 0.0001, Padj < 0.02) in the progression group.
Further KEGG pathway enrichment analysis of all 261 differentially abundant KOs showed that the interrelated phosphotransferase system (PTS) (enrichment score = -0.957, Padj = 0.006) and galactose metabolism (enrichment score = -0.904, Padj = 0.016) pathways were suppressed in the progression group, whereas porphyrin and chlorophyll metabolism were increased (enrichment score = 0.827, Padj = 0.006) (Figure 4C and 4D). In line with this, chlorophyllide, a biosynthesis pathways (PWY-7159, PWY-5531) that are important for the formation of chlorophyllide a, an intermediate for bacteriochlorophyll synthesis was overrepresented in the progression group (Figure 4B and Supplementary Data 2). Four other MetaCyc pathways that were overrepresented in the non-progression group included the (a) cis-vaccenate biosynthesis (PWY-5973) pathway required for the synthesis of the main unsaturated fatty acid cis-vaccenate; (b) TCA cycle VI (PWY-5913), a catabolic aerobic respiration reaction for the generation of energy; (c) arginine, ornithine and proline interconversion (ARGORNPROST-PWY) pathway that carries out fermentation of different forms of amino acids for growth based Stickland reactions and (d) the chlorosalicylate degradation (PWY-6107) pathway that is utilised by microbes that are able to chlorosalicylate as a carbon source for growth.
Discussion
Components of the microbiome, in particular H. pylori, contribute to the initiation and propagation of GC carcinogenesis. In this study, we characterised the gastric mucosal bacterial composition through 16S rRNA amplicon sequencing, demonstrating that the gastric mucosa was predominantly composed of phyla groups Proteobacteria and Firmicutes. This finding is consistent with other reports [29, 30]. In addition, H. pylori were also significantly enriched in patients with EGN. Although all patients included in this study were screened to be negative for H. pylori infection, DNA sequencing was still able to identified patients with H. pylori. This suggests that target sequencing to identify H. pylori could be applied clinically in addition to histological assessment to detect for H. pylori infection. Indeed, our group [17] and others have previously used PCR methods to detect H. pylori infection. However, most of such applications have been limited to stool analysis, with a high sensitivity of 93.8% [31]. Currently we are validating a point-of-care diagnostic for H. pylori infection using such PCR methods, by sampling the 23S rRNA to detect point mutations for additional predictive antibiotic (i.e. clarithromycin) resistance information [32], thereby providing patients with direct-based eradication therapies.
In this study, we also illustrated important non-H. pylori changes across different stages in the Correa's cascade of gastric carcinogenesis. Patients further along the progression of the Correa's cascade had an enrichment of metagenomic taxonomic features belonging to the Proteobacteria phyla, demonstrated through feature-based regression analysis, such as Proteus (genus) which is a potential gastrointestinal pathobiont shown to promote intestinal inflammation [33]. Other key Proteobacteria taxonomic features increased amongst patients with dysplastic lesions included Phyllobacteriaceae and Enhydrobacter. Changes in the abundance of these Proteobacteria were also previously reported to be significantly associated with GC development [14, 34].
Beneficial bacterial taxonomic features, such as lactic acid bacteria genera Lactobacillus and Bifidobacteria, were diminished in IM and EGN. Lactobacillus is often characterised as a transient probiotic bacteria from the oral cavity and is included in the formulation of many probiotics [35, 36]. Lactic acid bacteria have also been previously associated with CRC prevention by alleviating apoptosis and antioxidant DNA damages [37]. Apart from the above bacterial taxa, lower abundance of S24-7 (or Muribaculaceae) of the Bacteroidetes phylum was also detected in IM and EGN. Recently, S24-7 has been associated with increased ILC2 in the stomach, which in turn provides immune protection through the induction of IgA [38].
One third of patients in this study subsequently developed EGN, from which we identified a constellation of six bacterial taxonomic markers, at baseline, that accurately classify patients who would develop EGN (Figure 3). Significant predictors of EGN risk included microbial taxonomic features belonging to bacterial genera Moryella and Vibrio, which are also significantly increased at baseline amongst patients who would develop EGN. Moryella (genus) has been shown to exhibit specific co-occurrence with H. pylori in IM [15], which is consistent with our data that shows patients with either IM or EGN have both increased abundance of both H. pylori and Moryella (genus) (Figure 2). While, Vibrio (genus) is a known pathogen of the gastrointestinal tract [39], its association with GC pathogenesis has not been widely reported. When these two OTUs were implemented together with four other OTUs, the prediction model was able to identify patients who would progression to EGN with an accuracy of 82%. Unlike previous microbial biomarker discovery efforts for GC from cross-sectional case-control studies, we examined and compared prospectively the bacterial profiles of patients at baseline, before the development of EGN. Thus, we believe that our findings would more accurately represent putative metagenomic features associated with GC carcinogenesis. However agreements with external cohorts are required to further validate this panel of taxonomic features as a robust EGN progression biomarker.
We further investigated metagenomic functional changes associated with GC carcinogenesis. Features associated with dysregulation of nutrient metabolism were found to be significantly different between patients who developed EGN and those who did not. Patients who developed EGN had gastric microbial communities harbouring decreased abundances of agaB, agaC, agaD, sacB and rfbE, which represent reduced galactose, sucrose and starch metabolism (Figure 4A, 4C, 4D). This is in line with the observation of decreased mucin production in gastric IM [40], that could alter the nutritional content within the gastric mucosal, and in turn the composition of the gastric microbial community. In addition, patients that progressed to EGN had lower abundances of functional features that represent microbial pathways associated with arginine degradation. Arginine deprivation induced through bacterial arginine deiminase has been shown to supress growth of various tumour cell types [41-43]. Hence in patients that developed EGN, a baseline reduction in microbial arginine deiminase function may suggest possible increase in arginine availability for tumour cell growth.
Interestingly, prokaryote defence mechanism via increased antimicrobial function was also found to be increased in the progression group. This could result from increase in KOs that are representative of enzymes involved in antimicrobial resistance such as the KO for cationic antimicrobial peptide (CAMP) resistance. This may confer protection for specific pathogens or bacteria against gastric antimicrobial peptides [44] and in turn further alter gastric bacterial composition. Increased bacterial defence could also be achieved via increasing stress sensor functions conferred by proteins such as sensor histidine kinases that were found in the progression group [45]. This would allow bacteria to adapt to environmental changes such as those from host antitumour response. Further detail analysis identified six specific MetaCyc pathways that were differentially represented and highly related to the previous mentioned metabolic pathways. Of which, biosynthesis of cis-vaccenate which is regulated based on thermal condition of the microbes to control the fatty acid composition of membrane phospholipids [46] was reduced in the progression group. This may be related to the reduction of upstream metabolite in the host or overall changes in microbial composition that has reduced requirement for such processes. On the other hand, TCA cycle VI pathway was also under represented in the progression group. This may suggest a shift in bacterial population (i.e. reduced obligate autotrophs) that is less dependent on aerobic respiration for energy. Interestingly, the chlorophyllide a biosynthesis pathway was enriched in patients that progressed to EGN. Chlorophyllide is a key intermediate molecule during synthesis of chlorophyll and bacteriochlorophylls [47]. Several phototropic bacteria including Chloroflexi, Acidobacteria Heliobacteria, Chromatium, Ectothiorhodospira and Chloracidobacterium thermophilum carry out photosynthesis to produce bacteriochlorophylls as a form of energy [48, 49]. Of note, Chloroflexi was previously found to be increased in the gastric mucosal of GC patients compared to IM and gastritis [34], which suggests a shift in bacterial diversity in patients that progressed to EGN. Although 16S metagenomic functions were inferred, these results provide insights on the probable microbial pathways attributing to the progression to EGN patients.
Conclusion
Our study highlights early microbial changes associated with gastric carcinogenesis, whereby Proteobacteria microbes (e.g. Proteus genus) were enriched while Bacteroidetes microbes (e.g. S24-7 family) were depleted in gastric mucosal samples of patients with EGN. We identified a constellation of six microbial taxonomic features present at baseline, which provided the highest classifying power for subsequent EGN. This finding suggests a potential role for prospective microbiome monitoring for GC risk.
Abbreviations
GC: gastric cancer; IM: intestinal metaplasia; EGN: early gastric neoplasia; OTU: Operational Taxonomic Unit; KO: KEGG orthology; AUC: Area under the ROC Curve.
Supplementary Material
Supplementary figures.
Supplementary data 1.
Supplementary data 2.
Acknowledgements
This work was supported by grants from the National Research Foundation, Prime Minister's Office, Singapore, under its Campus of Research Excellence and Technological Enterprise (CREATE) program (R571-010-012-592 to Y. Z.), the Singapore National Medical Research Council (NMRC/OFIRG/0059/2017 to Y. Z.), the NUS Global Asia Institute (R571-000-043-133 to Y. Z.) and the National University Health System (NUHSRO/2020/113/T1/Seed-Mar/09 to Y. Z.). Furthermore, the Singapore Gastric Cancer Epidemiology and Molecular Genetics Program (GCEP) was supported by the National Research Foundation, Singapore, and Singapore Ministry of Health's National Medical Research Council under its Translational and Clinical Research (TCR) Flagship grant and Open Fund-Large Collaborative Grant (OF-LCG) (NMRC/TCR/009-NUHS/2013, NMRC/TCR/001-NUS/2007, MOH-OFLCG18May-0003), Biomedical Research Council (BMRC) grant (04/1/21/19/312), Singapore Cancer Syndicate (SCS) grant (SCS-GN0015), Singapore.
Data availability: The processed bacterial 16S rRNA gene V3-V4 amplicon data that supported the conclusions of this article is available from the National Center for Biotechnology Information GenBank accession numbers MZ263181 - MZ263772.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394-424
2. Necula L, Matei L, Dragu D, Neagu AI, Mambet C, Nedeianu S. et al. Recent advances in gastric cancer early diagnosis. World J Gastroenterol. 2019;25:2029-44
3. Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;1:1311-5
4. Warren JR, Marshall B. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet. 1983;1:1273-5
5. Chmiela M, Karwowska Z, Gonciarz W, Allushi B, Staczek P. Host pathogen interactions in Helicobacter pylori related gastric cancer. World J Gastroenterol. 2017;23:1521-40
6. Kuipers EJ, Nelis GF, Klinkenberg-Knol EC, Snel P, Goldfain D, Kolkman JJ. et al. Cure of Helicobacter pylori infection in patients with reflux oesophagitis treated with long term omeprazole reverses gastritis without exacerbation of reflux disease: results of a randomised controlled trial. Gut. 2004;53:12-20
7. Mera R, Fontham ET, Bravo LE, Bravo JC, Piazuelo MB, Camargo MC. et al. Long term follow up of patients treated for Helicobacter pylori infection. Gut. 2005;54:1536-40
8. Schenk BE, Kuipers EJ, Nelis GF, Bloemena E, Thijs JC, Snel P. et al. Effect of Helicobacter pylori eradication on chronic gastritis during omeprazole therapy. Gut. 2000;46:615-21
9. Han SJ, Kim SG, Lim JH, Choi JM, Oh S, Park JY. et al. Long-Term Effects of Helicobacter pylori Eradication on Metachronous Gastric Cancer Development. Gut Liver. 2018;12:133-41
10. Cheung KS, Leung WK. Risk of gastric cancer development after eradication of Helicobacter pylori. World J Gastrointest Oncol. 2018;10:115-23
11. Lertpiriyapong K, Whary MT, Muthupalani S, Lofgren JL, Gamazon ER, Feng Y. et al. Gastric colonisation with a restricted commensal microbiota replicates the promotion of neoplastic lesions by diverse intestinal microbiota in the Helicobacter pylori INS-GAS mouse model of gastric carcinogenesis. Gut. 2014;63:54-63
12. Lofgren JL, Whary MT, Ge Z, Muthupalani S, Taylor NS, Mobley M. et al. Lack of commensal flora in Helicobacter pylori-infected INS-GAS mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology. 2011;140:210-20
13. Yang I, Woltemate S, Piazuelo MB, Bravo LE, Yepez MC, Romero-Gallo J. et al. Different gastric microbiota compositions in two human populations with high and low gastric cancer risk in Colombia. Sci Rep. 2016;6:18594
14. Ferreira RM, Pereira-Marques J, Pinto-Ribeiro I, Costa JL, Carneiro F, Machado JC. et al. Gastric microbial community profiling reveals a dysbiotic cancer-associated microbiota. Gut. 2018;67:226-36
15. Coker OO, Dai Z, Nie Y, Zhao G, Cao L, Nakatsu G. et al. Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut. 2018;67:1024-32
16. Lee JWJ, Zhu F, Srivastava S, Tsao SK, Khor C, Ho KY. et al. Severity of gastric intestinal metaplasia predicts the risk of gastric cancer: a prospective multicentre cohort study (GCEP). Gut. 2021
17. Huang KK, Ramnarayanan K, Zhu F, Srivastava S, Xu C, Tan ALK. et al. Genomic and Epigenomic Profiling of High-Risk Intestinal Metaplasia Reveals Molecular Determinants of Progression to Gastric Cancer. Cancer Cell. 2018;33:137-50 e5
18. Dixon MF. Gastrointestinal epithelial neoplasia: Vienna revisited. Gut. 2002;51:130-1
19. Dixon MF, Genta RM, Yardley JH, Correa P. Classification and grading of gastritis. The updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am J Surg Pathol. 1996;20:1161-81
20. Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41:e1
21. Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996-8
22. DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069-72
23. McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217
24. Oksanen FJ, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D. et al. vegan: Community Ecology Package. R package version 2.4-4. https://CRANR-projectorg/package=vegan. 2017
25. Douglas GM, Maffei VJ, Zaneveld J, Yurgel SN, Brown JR, Taylor CM. et al. PICRUSt2: An improved and extensible approach for metagenome inference. bioRxiv. 2019: 672295.
26. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284-7
27. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550
28. Wirbel J, Zych K, Essex M, Karcher N, Kartal E, Salazar G. et al. Microbiome meta-analysis and cross-disease comparison enabled by the SIAMCAT machine learning toolbox. Genome Biol. 2021;22:93
29. Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F. et al. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci U S A. 2006;103:732-7
30. Liu X, Shao L, Liu X, Ji F, Mei Y, Cheng Y. et al. Alterations of gastric mucosal microbiota across different stomach microhabitats in a cohort of 276 patients with gastric cancer. EBioMedicine. 2019;40:336-48
31. Beckman E, Saracino I, Fiorini G, Clark C, Slepnev V, Patel D. et al. A Novel Stool PCR Test for Helicobacter pylori May Predict Clarithromycin Resistance and Eradication of Infection at a High Rate. J Clin Microbiol. 2017;55:2400-5
32. Chen D, Cunningham SA, Cole NC, Kohner PC, Mandrekar JN, Patel R. Phenotypic and Molecular Antimicrobial Susceptibility of Helicobacter pylori. Antimicrob Agents Chemother. 2017 61
33. Hamilton AL, Kamm MA, Ng SC, Morrison M. Proteus spp. as Putative Gastrointestinal Pathogens. Clin Microbiol Rev. 2018;31:e00085-17
34. Wang L, Xin Y, Zhou J, Tian Z, Liu C, Yu X. et al. Gastric Mucosa-Associated Microbial Signatures of Early Gastric Cancer. Front Microbiol. 2020;11:1548
35. Berg RD. The indigenous gastrointestinal microflora. Trends Microbiol. 1996;4:430-5
36. Sarao LK, Arora M. Probiotics, prebiotics, and microencapsulation: A review. Crit Rev Food Sci Nutr. 2017;57:344-71
37. Zhong L, Zhang X, Covasa M. Emerging roles of lactic acid bacteria in protection against colorectal cancer. World J Gastroenterol. 2014;20:7878-86
38. Satoh-Takayama N, Kato T, Motomura Y, Kageyama T, Taguchi-Atarashi N, Kinoshita-Daitoku R. et al. Bacteria-Induced Group 2 Innate Lymphoid Cells in the Stomach Provide Immune Protection through Induction of IgA. Immunity. 2020;52:635-49.e4
39. Janda JM, Newton AE, Bopp CA. Vibriosis. Clin Lab Med. 2015;35:273-88
40. Boltin D, Niv Y. Mucins in Gastric Cancer - An Update. J Gastrointest Dig Syst. 2013;3:15519
41. Al-Koussa H, El Mais N, Maalouf H, Abi-Habib R, El-Sibai M. Arginine deprivation: a potential therapeutic for cancer cell metastasis? A review. Cancer Cell Int. 2020;20:150
42. Liu J, Ma J, Wu Z, Li W, Zhang D, Han L. et al. Arginine deiminase augments the chemosensitivity of argininosuccinate synthetase-deficient pancreatic cancer cells to gemcitabine via inhibition of NF-kappaB signaling. BMC Cancer. 2014;14:686
43. Takaku H, Takase M, Abe S, Hayashi H, Miyazaki K. In vivo anti-tumor activity of arginine deiminase purified from Mycoplasma arginini. Int J Cancer. 1992;51:244-9
44. LaRock CN, Nizet V. Cationic antimicrobial peptide resistance mechanisms of streptococcal pathogens. Biochim Biophys Acta. 2015;1848:3047-54
45. Fabret C, Feher VA, Hoch JA. Two-component signal transduction in Bacillus subtilis: how one organism sees its world. J Bacteriol. 1999;181:1975-83
46. White SW, Zheng J, Zhang YM, Rock. The structural biology of type II fatty acid biosynthesis. Annu Rev Biochem. 2005;74:791-831
47. Bollivar DW. Recent advances in chlorophyll biosynthesis. Photosynth Res. 2006;90:173-94
48. Bryant DA, Costas AM, Maresca JA, Chew AG, Klatt CG, Bateson MM. et al. Candidatus Chloracidobacterium thermophilum: an aerobic phototrophic Acidobacterium. Science. 2007;317:523-6
49. Gisriel C, Sarrou I, Ferlez B, Golbeck JH, Redding KE, Fromme R. Structure of a symmetric photosynthetic reaction center-photosystem. Science. 2017;357:1021-5
Author contact
![]() Corresponding authors: Yongliang Zhang. Address: 28 Medical Drive. Centre for Life Sciences (CeLs), Life Science Institute, Immunology Programme, National University of Singapore, Singapore 117456, Singapore. Email: miczyedu.sg. Telephone: +65-6516-6407. Fax: +65-6873-3905. Khay Guan Yeoh. Address: Department of Medicine, Yong Loo Lin School of Medicine, National University of Singapore, 119228, Singapore. Email: mdcykgedu.sg. Telephone: +65-6772-3732. Fax: +65-6772-4361
Corresponding authors: Yongliang Zhang. Address: 28 Medical Drive. Centre for Life Sciences (CeLs), Life Science Institute, Immunology Programme, National University of Singapore, Singapore 117456, Singapore. Email: miczyedu.sg. Telephone: +65-6516-6407. Fax: +65-6873-3905. Khay Guan Yeoh. Address: Department of Medicine, Yong Loo Lin School of Medicine, National University of Singapore, 119228, Singapore. Email: mdcykgedu.sg. Telephone: +65-6772-3732. Fax: +65-6772-4361