Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Experimental
Results and Discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(19):9652-9666. doi:10.7150/thno.64354 This issue Cite
Research Paper
Polyamino acid calcified nanohybrids induce immunogenic cell death for augmented chemotherapy and chemo-photodynamic synergistic therapy
Wei Qiu1, Mengyun Liang1, Yuan Gao1, Xuelian Yang1, Xingyao Zhang1, Xiaoli Zhang2 ![]() , Peng Xue1, Yuejun Kang1, Zhigang Xu1,3
, Peng Xue1, Yuejun Kang1, Zhigang Xu1,3 ![]()
1. Key Laboratory of Luminescence Analysis and Molecular Sensing (Southwest University), Ministry of Education, School of Materials and Energy and Chongqing Engineering Research Center for Micro-Nano Biomedical Materials and Devices, Southwest University, Chongqing 400715, P. R. China.
2. Pediatric Research Institute, Department of Hematology and Oncology, Shenzhen Children's Hospital, Shenzhen, Guangdong 518038, P. R. China.
3. Key Laboratory of Laser Technology and Optoelectronic Functional Materials of Hainan Province, College of Chemistry and Chemical Engineering, Hainan Normal University, Haikou 571158, P. R. China.
Received 2021-6-28; Accepted 2021-8-28; Published 2021-9-21
Abstract

Background: Monotherapy for cancer treatment is limited by unstable efficacy and uncontrollable toxic side effects, while the multifunctional nanoplatform with complex preparation process cannot avoid the potential toxicity of each functional component.
Methods: We exploited tumor-specific activated polyamino acid calcified nanoparticles (CHC NPs) as new-type oxidative stress amplification of anticancer drugs via building a safe and biodegradable multifunctional nanoplatform. Giving priority to chemotherapy, and synergizing chemodynamic therapy (CDT) with photodynamic therapy (PDT), this strategy was to achieve enhanced chemotherapy, simultaneously inducing immunogenic cell death and inhibiting tumor cell invasion.
Results: Based on amorphous calcium carbonate, pH-responsive nanocarrier was prepared with classical chemotherapeutic drug 10-hydroxycamplothecin (HCPT) and photosensitizer Chlorin e6 (Ce6) to realize multifunctional nanotheranostics.
Conclusion: Inventive calcified nanohybrids, where topoisomerase inhibited by HCPT to prevent DNA synthesis, the generation of •OH induced via Fenton reaction, along with a large amount of 1O2 produced by Ce6, might be a promising strategy for anti-tumor combination therapy in clinical translation.
Keywords: Calcified nanohybrids, multimodal nanotheranostics, chemotherapy, chemo-photodynamic therapy, immunogenic cell death
Introduction
Cancer has always examined the resilience of human beings. As cancer burden increases with years, malignancy continues to be one of the major threats to human life and health [1-3]. Aiming to seek accurate diagnosis and effective treatment remains the primary mission in nanomedicine researches, and the development of multimodal nanoplatforms to satisfy the demands of personalized treatment and significant efficacy [4, 5]. Constructed by assembling multiple components with unique advantages, multifunctional nanotheranostic platform is still subject to the complicated preparation process and poor repeatability [6]. Outmoded nanomedicine exhibits bad biodegradability and is prone to trigger long-term toxicity for patients, which impedes the transformation into clinical practice [7]. Exploitation for degradable and multifunctional nanomedicine with prominent biocompatibility and simple synthesis process has become an urgent need to achieve breakthroughs in cancer therapy. Calcium carbonate (CaCO3), as a typical mineral widely existing in organisms and nature, is expected to participate in the construction of multimodal nanoplatforms due to its biodegradation performance, excellent biocompatibility, and low cost, as well as great potentials for drug delivery, protein transportation and gene delivery [8-11]. The pH sensitivity of CaCO3 makes it one of the attractive drug candidates for responsive release in the tumor microenvironment to achieve controlled release without drug leakage [12, 13]. According to frontier researches, crystal calcium carbonates with micron size have trapped into some inherent limitations, which may hinder its application as a drug delivery system, even affect transfer efficiency [14]. Amorphous calcium carbonate breaks this impasse, and high solubility enables it to be better absorbed from the blood into cells, thus having greater absorbability and higher bioavailability [15, 16]. Additionally, amorphous calcium carbonate also possesses multiple functions such as microenvironmental pH regulated, local anti-inflammatory activity enhanced, immunity improved, and cancer cell metabolism influenced. Previous reports manifested that amorphous calcium carbonate-based nanodrug delivery platform could directionally release the cargo at the tumor site due to sensitive pH response [17]. Calcified nanosubstrate like this can be easy to carry various types of bioactive molecules without a fussy and complicated formation process, which indicates that it's potential to combine with diverse modifications or organic ligands. Introduction of PEGylation agent, like PEG-P(Glu) block copolymer is anticipated to further ameliorate biological function and biocompatibility of composite nanodrugs [18].
In addition to the development of various treatment methods and biocompatible nanomaterials, the improvement of tumor treatment effect is also inseparable from the discovery and application of clinical treatment mechanisms [19, 20]. At present, the therapeutic target of most clinical tumor administration is apoptosis-related pathway. 10-hydroxycamplothecin (HCPT), extracted from the unique plant of Camptotheca acuminata in China, is a classic and effective anti-tumor chemotherapy drug, and has been widely used in clinical practice in many countries. Compared with camptothecin, HCPT has better curative effect and less side effects in the treatment of malignancy. HCPT is also a semi-synthetic precursor compound of camptothecin anticancer drugs CPT-11 and TPT, permitted by FDA for clinical use. HCPT is a kind of natural alkaloid with broad spectrum, high efficiency and low toxicity, which participates in anti-tumor by inhibiting topoisomerase I (TOPO I) activity to interfere with the DNA replication of tumor cells [21, 22]. As a promising alternative therapy, chemodynamic therapy (CDT) produces reactive oxygen species (ROS) in specific tumor microenvironment, which is followed by Fenton reaction (2Fe3+ + H2O2 → 2Fe2+ + O2 + 2H+, Fe2++ H2O2 → Fe3+ + (OH)- + •OH), catalyzing the conversion of low-toxic H2O2 to highly toxic hydroxyl radical (•OH), and significantly elevating the ROS level in tumor cells [23-25]. Based on the reductive tumor microenvironment, the introduced ferric ions would be translated into ferrous ions to participate in Fenton reaction. Fenton reaction could play a better role under acidic condition, and ROS generated in TME is easily consumed by the high level of intracellular glutathione (GSH) [26]. Recently, phototherapy of tumor cells damaged by phototoxicity has attracted great attention in the field of tumor therapy owing to the presence of advantages such as less invasion, less adverse effects, strong temporal and spatial selectivity [27-29]. Photodynamic therapy (PDT) applies photosensitizers (PSs) for transforming laser into singlet oxygen 1O2 as substrate to injure tumor cells. As a commonly applied photosensitizer for clinical imaging and diagnose, Chlorin e6 (Ce6) is suitable for the exploitation of photodynamic therapy upon tumors thanks to high efficiency in producing 1O2 [30, 31]. However, like most photosensitizers so far, Ce6 is hydrophobic and tends to aggregate in aqueous solution, which has some difficulties in practical application. Properly speaking, the efficacy of single phototherapy is far away from our satisfactory goal in cancer nanomedicine [32-34]. Moreover, 1O2 produced by most photosensitizers (PSs) are heavily oxygen-dependent in PDT, and rapid depletion of molecular oxygen can further aggravate hypoxia [35]. Therefore, tumor hypoxia has become a major obstacle to PDT treatment. Clinical treatment-activated immunogenic cell death (ICD) can reinforce tumor immunotherapy by stimulating the autoimmune system through secreting associated signals. At present, therapeutic means like chemotherapy and phototherapy can induce ICD, with cytotoxic •OH and 1O2 involved in regulation ICD [36-38]. In consequence, increased intracellular ROS levels are highly proposed to amplify ICD and PDT could effectively arouse immunogenicity during treatment [39, 40].
Although chemotherapy is the main method of cancer treatment, both CDT based on ROS involvement in cell homeostasis and local treatment PDT as non-invasive therapy have also been widely investigated [41, 42]. With high level of H2O2 in cancer cells, CDT is specific to TME and thus minimizes adverse reaction on healthy tissue, compared with conventional PDT [43]. On the basis of the preliminary studies, chemotherapy agent (HCPT) and photosensitizer (Ce6) were incorporated into calcified nanohybrids (denoted as CHC NPs) to amplify oxidative stress. CHC NPs were fabricated through the complexation between drugs and amorphous calcium carbonate nanosubstrate, as well as tailored modification with PEG-P(Glu). The improved and optimized nanoparticle stability and drug release property, were likely to determine the efficacy of nanodrug in the body. As illustrated in Scheme 1, CDT utilized the Fenton reaction to produce •OH, while PDT produced 1O2 under laser irradiation to trigger tumor-associated apoptosis or necrosis. ROS derived from PDT caused mitochondrial dysfunction and accelerated the end of tumor cells during the process of immunogenic cell death (ICD). The efforts made by CDT and PDT were aimed at enhancing HCPT-based chemotherapy, making up for monotherapy shortcomings, and realizing the construction of multimode nanoplatform.
Schematic diagram about fabrication of CHC NPs and anticancer mechanism of multimodal therapies based on chemotherapy and CDT/PDT therapy.
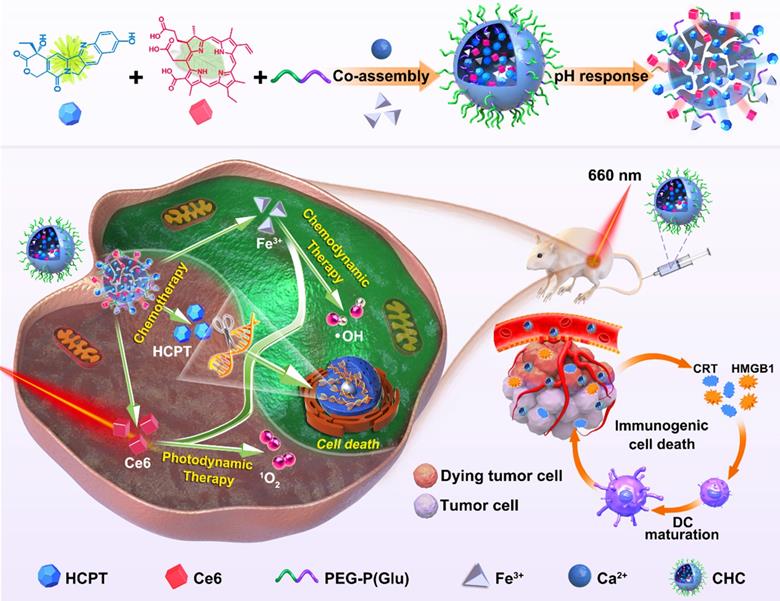
Experimental
Materials
10-Hydroxy camptothecin (HCPT, 98%) was purchased from Adamas-beta (China). Chlorin e6 (Ce6, 92%) was supplied by Yuanye Biotechnology Ltd (China). Calcium chloride anhydrous (CaCl2), iron (III) chloride hexahydrate (FeCl3∙6H2O), sodium carbonate (Na2CO3), tris-hydrochloride buffer and HEPES were acquired from Aladdin (China). Antibodies for chromatin-binding protein high mobility group B1 (HMGB1) and Calreticulin were provided by cell signaling technology (China). All other agents were supplied by Life Technologies (Carlsbad, USA) and used directly.
Preparation and characterization of CHC NPs
The formation of CHC nanohybrids referred to typical methods as previously reported with some modification [44]. Briefly, 1 mM Tris-HCl buffer (pH 7.6) containing 100 mM CaCl2 and 20 mM FeCl3 was dropwise added to 50 mM HEPES buffer (pH 7.1) according to the optimal solvent ratio of 1:2. The HEPES buffer encompassed 10 mM Na2CO3, HCPT and Ce6 (each 400 μg), as well as 5 mg PEG-P(Glu) constructed as formerly described [18, 45]. Further, the obtained mixture was continuously stirred at 25 °C for 24 h and centrifugated respectively at 3000 rpm and 13000 rpm for 5 min to remove excess ions, drugs and copolymers.
Cumulative drug release from CHC NPs
Due to the weak acidity in tumor microenvironment (TME), the release behavior of CHC NPs under different pH condition in vitro was studied by dialysis to simulate the real TME in vivo. Typically, 1.0 mL CHC was set in a 3.5 kDa dialysis bag and put into 15 mL buffer (pH 5.0, pH 6.5, pH 7.4, respectively). At different timepoints, 1 mL original buffer was collected and replaced by corresponding fresh buffer of consistent volume. Eventually, the concentration of Ce6 and HCPT released were determined by microplate reader (Tecan SPARK-10M).
Biosafety of CHC NPs
L929 culture
Biocompatibility of CHC NPs was initially evaluated by murine L929 fibroblasts (L929 cells). After adherence, cells were cultured with the medium including CHC NPs at Ce6 concentration of 1~100 µg/mL. Subsequent to incubation for 24 h, 100 µL MTT solution was contained into per well. The supernatant was discarded after 4 h reaction and filled with 150 µL DMSO. Then, optical absorption intensity was observed via microplate reader.
Hemolysis analysis
After whole blood was centrifuged at 3000 rpm, erythrocytes (RBC) were harvested and subsequently rinsed with PBS for three times. Afterwards, 0.3 mL of CHC NPs at different Ce6 concentration was introduced into the tube containing the diluent RBC for incubation for 2 h at 37 °C. Finally, the absorbance of supernatant was measured at 570 nm.
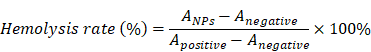
Blood routine test
6-7 weeks Balb/c mice were intravenously injected with PBS buffer, HCPT, Ce6 or CHC NPs dispersion (100 µL, 1 mg/mL). Next, blood specimen was collected from the eye socket at day 1, 7 and 14. Key blood indexes were measured by automatic hematology analyzer (NC-2600Vet, Mindary, China).
Cellular uptake and colocalization analysis
Initially, flow cytometry was applied to measure the phagocytosis of CT26 towards CHC NPs. In short, CT26 cells (7×104/well) were respectively exposed to medium containing CHC NPs (Ce6, 5 μg/mL) for 0.5 h, 1 h, 2 h, 4 h and 6 h. For colocalization assessment, after treated with CHC NPs (Ce6, 10 μg/mL) at various time points, CT26 cells were stained with Lyso-Tracker and Mito-Tracker reagents on behalf of lysosomes and mitochondria separately, finally recorded by confocal laser scanning microscope (CLSM 880, Carl Zeiss, Germany).
Cytotoxicity assessment in vitro
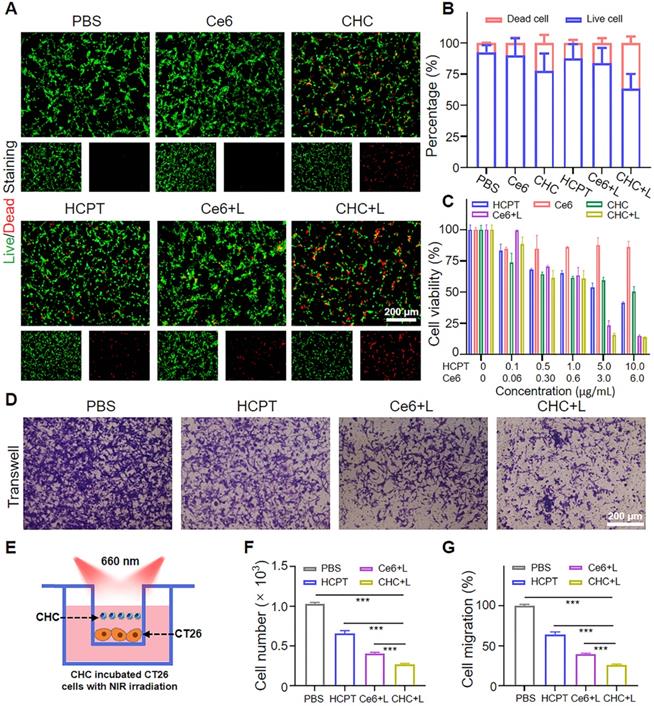
First of all, cytotoxicity of CHC NPs to CT26 cells was evaluated on the basis of standard MTT assay. CT26 cells in 96 well plates (1×104 per well) were cultured overnight prior to the addition of HCPT, Ce6 or CHC NPs for 24 h. Additionally, living/dead cell staining upon FDA/PI was carried out to direct observation of cell survival status and then quantified through Image J software. To analyze cell migration after different administration, CT26 cells in 12-well plate were incubated with HCPT, Ce6 and CHC NPs for 24 h. Subsequently, the cells (5×104 per well) treated as described above were transferred to upper chambers to visualize cell migration. Metastatic cells were viewed and imaged with the fluorescence microscope and calculated using Image J software. Cell migration rate (%) = Ntreatment / NPBS × 100%. (All laser tests were exposed to 660 nm irradiation after cultivation with drugs for 4 h).
ROS generation capacity
To monitor 1O2 generation in vitro, DPBF (1 mg/mL) was mixed with CHC NPs (Ce6: 5 μg/mL), irradiated by 660 nm laser every minute (180 mW/cm2). The UV absorption change of DPBF at 400 nm was determined at the corresponding time point above. MB (1 mg/mL) probe was applied to detect the production of •OH under various condition. Afterwards, intracellular ROS generation performance of various treatment towards CT26 was conducted. Specially, the cells were stained by DCFH-DA for 30 min with or without 660 nm laser irradiation for 3 min, while the nuclei were processed by Hoechst 33342. Furthermore, the generation of 1O2 was measured by SOSG, which could specifically react with 1O2 and produce green fluorescence that were observed upon CLSM. Besides, electron spin resonance (ESR) was utilized to validate the generation of 1O2 and •OH by CHC NPs in presence of H2O2, to illustrate the type of ROS generation in detail.
Immunofluorescence imaging of CHC NPs in DNA damage
Attributed to HCPT combination with topoisomerase to suppress the synthesis of DNA, this assay was exploited to evaluate the DNA damage of CHC NPs to tumor cells based on measuring the intensity of green fluorescence. Adherent CT26 cells were separately exposed to HCPT, Ce6 and CHC NPs at 37 °C for 4 h. After replacing with fresh medium, they were subjected to 660 nm laser irradiation or not and cultured for an additional 8 h. Through formaldehyde fixed for 20 min, Triton X-100 permeabilized for 20 min and BSA blocked for 2 h, the cells were incubated with the primary antibody γ-H2AX at 4 °C overnight for DNA damage detection. Replaced with the Alexa Fluor 488-secondary antibody, cells were finally visualized by CLSM. In addition, tumor sections in each group after treatment were used to further immunofluorescence detection to verify DNA damage in vivo.
Immunologic cell death (ICD) induced by CHC NPs
According to the performance of HMGB1 and CRT on the surface of tumor cells, immunofluorescence (IF) analysis was employed to estimate ICD induced by CHC NPs in vitro. Concretely, CT26 cells 5×104 were added into 12-well plates overnight and respectively treated by HCPT, Ce6 and CHC NPs for 24 h, with 660 nm irradiation or not. After rinsed with PBS, fixed, permeated and blocked in turns, then cultured with primary antibodies of anti-HMGB1 and anti-CRT at 4 °C overnight, cells were replaced with AF488-labeled secondary antibodies for 1 h and stained by DAPI for 10 min, finally tested by CLSM. In another aspect, solid tumors of each group were sliced into sections for HMGB1 and CRT immunofluorescence detection. By way of deparaffinized, dehydrated, heat induced epitope retrieval with citrate buffer (pH = 6.0) and closure by 10% goat serum in 37 °C, tumor sections were seperately incubated with primary and secondary antibodies, as same as the processs of intracellular detection of CRT and HMGB1 in vitro. All immunofluorescence staining slices were scanned by CLSM.
Penetration capacity of CHC NPs in CT26 MCSs
The manifestation of drug release behavior and vertical tumor penetration ability to CHC NPs were assessed by using CT26 multicellular spheroids (MCSs). On basis of previous reports [46], 1×103 CT26 cells were planted into 96-well plates containing 50 µL hot agarose and incubated for 3-5 days to constitute MCSs. After replaced by fresh medium including CHC NPs (Ce6, 15 µg/mL), CT26 MCSs were cultured for 2 h, 6 h and 12 h respectively. Lastly, the photographs of different depth were captured by CLSM.
NIR imaging and biodistribution
Balb/c mice inoculated with 4T1 cells were randomly divided into CHC group and Ce6 group (n = 3) to evaluate the biodistribution of CHC NPs. Once tumor volume arrived approximate 200 mm3, CHC NPs and free Ce6 at the equivalent Ce6 dosage were intravenously injected into mice. The mice NIR images were recorded by IVIS Lumina imaging system at different timepoints and the distribution of Ce6 in the tumor and organs were observed after 48 h.
Tumor suppression efficiency in vivo
CT26 cells were subcutaneously inoculated on 6-7 weeks Balb/c mice to establish CT26 xenograft models. Each animal process was executed obeying the administrative provisions of Southwest University ethics committee. Until tumor volume reached ~200 mm3, all mice were separated into 5 treatment groups at random (PBS, HCPT, CHC NPs, Ce6+L and CHC NPs+L in a single dose of 5.0 mg/kg of HCPT or 3 mg/kg of Ce6). Tumors were exposed to 660 nm laser (180 mW/cm2) after 24 h administration. After 3 times administration and NIR laser irradiation, the mice were sacrificed and tumor tissue sections and major organs were taken for H&E staining and further analyzed for Ki67 and TUNEL assay. Tumor volume was reckoned in accordance with the classic formula: length×width2 / 2.
Statistical analysis
All results are displayed as the mean ± standard deviation (SD). Statistical significance (*p < 0.05, **p < 0.01, ***p < 0.001) was performed by Student's t-test for two-group comparisons. Image J software was applied for various quantification.
Results and Discussion
Formation and characterization of CHC nanohybrids
Built upon various unique material capabilities, the carboxyl terminus of PEG-P(Glu) complexed with Ca2+ in the aqueous phase served as the core, and hydrophilic PEG chains formed the outside of CHC NPs upon hydrophilic and hydrophobic interactions, so that both hydrophobic chemotherapeutics HCPT and photosensitizer Ce6 were loaded inside of calcium carbonate nanoparticles (denoted CHC NPs) with stirring. Synergizing multiple modes of cancer therapy was successful, including HCPT-based chemotherapy to inhibit DNA replication, Fenton reaction for CDT and the mass production of 1O2 from PDT. HCPT and Ce6 following the optimal ratio were loaded into CHC NPs. The loading efficiency of HCPT in CHC nanohybrids was 8.6%, while to Ce6 was 5.1%, respectively, implying the high drug-loading capability of CHC NPs. The size of CHC NPs was 163.33 ± 15.45 nm or 185.53 ± 2.31 nm (polydispersity index, PDI = 0.155 ± 0.02) with approximately spherical morphology, respectively measured by transmission electron microscopy (TEM) and dynamic light scattering (DLS) (Figure 1A). The diameter and PDI of CHC nanoparticles were suitable so as to possess the excellent stability in PBS (pH 7.4) with or without 10% FBS for long-term storage over 2 weeks (Figure 1B and S1), and a small amount negative charge distributed on the surface of CHC might prolong circulation in the blood (Figure S2). Due to pH sensitivity of CaCO3, the diameter of CHC NPs in acid PBS (pH 5.0) was recorded by DLS in Figure S3, which further manifested that CHC was pH-responsive nanoparticle, promising to release drugs in response to acidic tumor microenvironment. The presence of HCPT and Ce6 embedded in CHC was confirmed by UV-vis shown in Figure 1C. As exhibited in Figure 1D-E and Figure S4, fluorescence spectra and FT-IR spectra further proved the successful preparation of CHC.
Physiochemical characterization of CHC NPs. (A) Hydrodynamic diameter and TEM image of CHC NPs. (B) Size, zeta potentials and polydispersity of CHC NPs dispersed in PBS during 2 weeks. (C) UV-vis absorption of Ce6, HCPT and CHC NPs. Fluorescence spectra of (D) Ce6 and (E) HCPT in CHC NPs. (F) XPS full survey spectrum of CHC NPs. (G) Core level XPS spectrum of Fe2p and (H) high-resolution Fe2p spectrum. (I) Quantitative analysis of Fe element in CHC NPs using ICP-MS.
The chemical valence of Ca and Fe elements were monitored by X-ray photoelectron spectroscopy (XPS) [47, 48], which showed the Ca(II) peak at 346.68 eV was exactly corresponding to the binding energy of Ca2p (Figure S5), Fe(III) at 713.68 eV and 726.48 eV along with Fe(II) at 710.53 eV and 723.29 eV were indexed to the binding energy of Fe2p (Figure 1F-H). As depicted in Figure 1I, the existence of Fe element in CHC NPs was further determined by inductively coupled plasma mass spectrometry (ICP-MS). To explore the decomposition of CHC NPs at different pH values, the release patterns of Ce6 and HCPT from CHC were simulated in weak acidic environment via dialysis. As described in Figure S6, CHC NPs revealed sustained slow release properties. More Ce6 and HCPT were dissociated from CHC NPs at the environment of pH = 5.0 or pH = 6.5, while only a few were separated from the normal condition of pH = 7.4, which suggested that CHC NPs were sensitive response to acidic tumor microenvironment, but no response to the normal.
CHC NPs as an oxidative stress amplifier
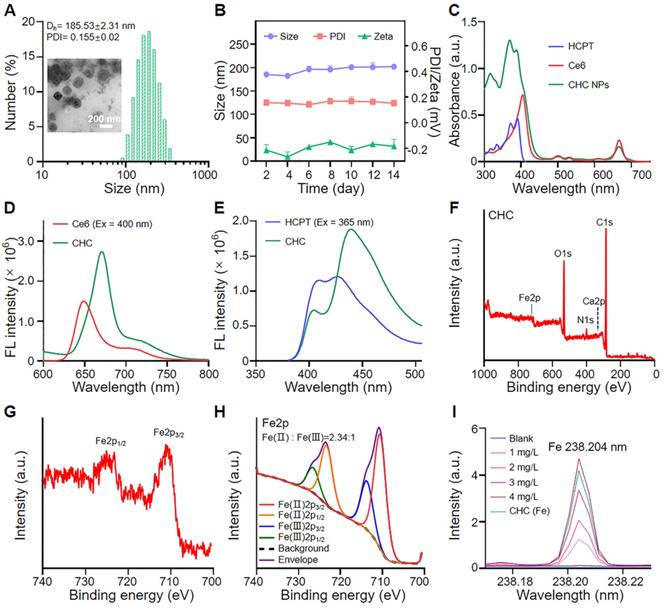
Methylene blue (MB) was employed to verify the generation of •OH, where both CHC NPs with or without drug could activate Fenton reaction to make MB oxidized and color changes occur (Figure 2A-B). The absorbance of MB at 665 nm reduced with irradiation time which was observed by ultraviolet spectrophotometer (Figure 2C). To prove 1O2 generation capacity of CHC NPs outside tumor cells upon 660 nm irradiation, the change of DPBF probe was recorded by UV-vis to confirm the large amount ROS yield (Figure 2D). Electron spin resonance (ESR) was employed to verify the existence of 1O2 or •OH instantaneously caught by 2,2,6,6-tetramethylpiperidine (TEMP) and 5,5-dimethyl-1-pyrroline N-oxide (DMPO), respectively. As demonstrated in Figure 2E and S7, the peak of •OH with intensity ratio of 1:2:1 was produced by CHC NPs in vitro with the addition of H2O2, while CHC+L group exhibited obvious characteristic peak with intensity ratio of 1:1:1 corresponding to the standard peak of 2,2,6,6-Tetramethyl-1-piperidinyloxy (TEMPO) in Figure 2F. The above results indicated that CHC NPs could significantly enhance oxidative stress by igniting the produce of •OH and 1O2 (Figure 2G).
Detection of ROS generation from CHC NPs. (A) Visual color variation of aqueous samples including MB. (B) •OH generation detected by MB probe upon different conditions. (C) UV-vis spectra of MB after reacting with CHC NPs upon 660 nm irradiation for various periods. (D) 1O2 emergence caught by DPBF probe subject to 660 nm irradiation for up to 6 min. ESR spectra of (E) DMPO/•OH and (F) TEMP/1O2 adducts in different solutions. (G) Illustration of drug release and robust radical generation.
Cytotoxicity of CHC NPs in vitro
FDA/PI staining was employed to observed the green living cells or red dead cells via fluorescence imaging to evaluate the cytotoxicity of various drugs visually (Figure 3A-B). Apart from that, MTT assay was applied to further manifest that CHC NPs represented the excellent cytotoxicity to CT26 tumor cells, where the cell viability was less than 15% treated with CHC+L (660 nm, 180 mW/cm2) at Ce6 of 6 μg/mL (Figure 3C). By virtue of Chou-Talalay method [49], the combination index (CI) was calculated to 0.635, which indicated that the co-delivery of HCPT and Ce6 enhanced the inhibition effifacy (CI < 1: synergistic effect; CI = 1: additive effect, CI > 1: antagonism effect). Due to synergized HCPT and Ce6, the consistance phenomenon emerged in Transwell assay (Figure 3D-E). As exhibited in Figure 3F and 3G, the migration capacities of CT26 cells were influenced by the treatment of PBS, HCPT, Ce6+L or CHC+L. Especially, CHC+L significantly inhibited the movement of CT26 cells from the upper to the lower chamber, corresponding migration rate as low as 26%.
Cytotoxicity evaluation of CHC NPs. (A) FDA (green)/PI (red) staining images of CT26 cells under various treatment for 12 h (Ce6 concentration: 10 µg/mL) and (B) quantification of living or dead cells area. (C) Viability of CT26 cells incubated in various groups. (D) CT26 cells migration influenced by PBS, HCPT, Ce6+L and CHC+L. (E) Illustration of transwell system experiment. The calculation of (F) cell number and (G) cell migration rate. Data denoted as mean ± SD, n = 3.
Oxidative stress amplification-enhanced CDT and PDT therapy fuel DNA damage-based chemotherapy
According to previous researches, irreversible cell damage could emerge under intense oxidative stress because of outrageous high ROS levels, which as a signal to initiate antitumor reaction (Figure 4A). DCFH-DA and SOSG probes were used to respectively detect the intracellular levels of ROS and 1O2, which could specifically react with them and both produce green fluorescence (Figure 4B). After incubation with CHC NPs for 6 h, NIR 660 nm laser irradiation was operated on CT26 cells for 3 min ahead of visualized by CLSM. Apparently, consistent green fluorescence of CHC+L group was located in the whole cells indicating the elevated ROS and 1O2 production, which different from other treatment (Figure 4C-D). The above facts implied that CHC NPs could induce the tumor-specific oxidative stress by producing various types of highly toxic reactive oxygen species along with drug toxicity for causing damage to tumor cells.
CHC NPs generate radicals and 1O2 upon 660 nm irradiation to push cell death. (A) Diagram of cellular endocytosis and enhanced DNA damage by producing ROS. (B) Detection of intracellular ROS and 1O2 generation in CT26 cells through various administration via CLSM and mean fluorescence intensity (MFI) of (C) DCFH-DA or (D) SOSG. (E) Flow cytometric profile of CHC NPs for different treatment time and (F) corresponding MFI. (G) The degree of DNA damage in different groups. Data signified as mean ± SD (n = 3).
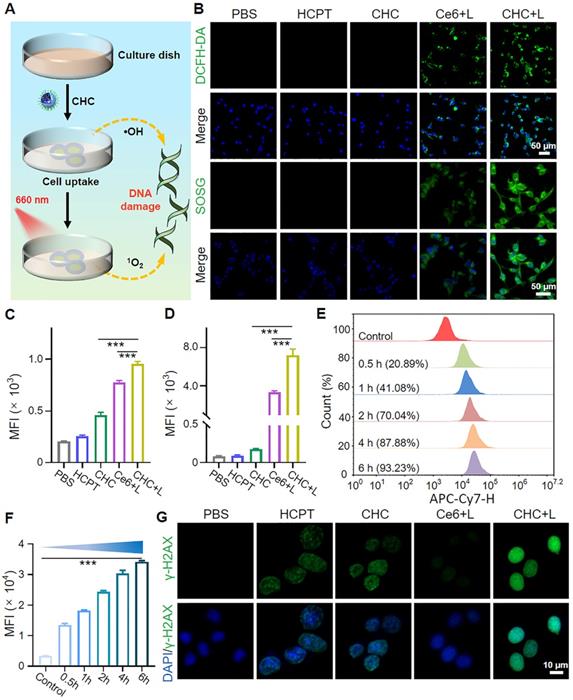
Owing to the suitable size and neutral potential, the cell uptake modes and intracellular distribution of CHC NPs in CT26 were made a thorough inquiry. CHC NPs chose endocytosis as a viable approach to enter the tumor cells, first nanoparticals attached to cell membrane and membrane invaginated to form vesicles, then CHC NPs-packaged vesicles detached from the membrane to further arrive at lysosomes and other organelles. Concretely, the time-dependent pinocytosis behavior of CHC NPs was recorded by flow cytometry (FCM), approximately 93.23% CHC NPs internalization throughout the whole CT26 cells at 6 h (Figure 4E-F). Colocalization assay and drug distribution were investigated by CLSM with Lyso-Tracker Green or Mito-Tracker Green dye, respectively (Figure S8 and S9). Among the above results indicated that quick uptake efficiency and particularity of CHC NPs made it easy to transport for exerting the combined therapeutic effects of HCPT and Ce6.
To investigate the mechanism of chemotherapeutic drug HCPT with DNA damage response, the DNA damage degree of CHC NPs or free HCPT has been compared in Figure 4G and S10. After incubated with drugs, the intensity of green fluorescence in CHC+L group was strongest in comparison with other treatment, while the group without drug treatment had no fluorescence at all. Since DNA has a certain self-repairing ability during a certain period of time, the strong DNA damage caused by HCPT, cooperated with the violent tumor-specific oxidative stress response induced by CDT and PDT has accelerated cell death.
CHC NPs induced ICD in vitro and in vivo
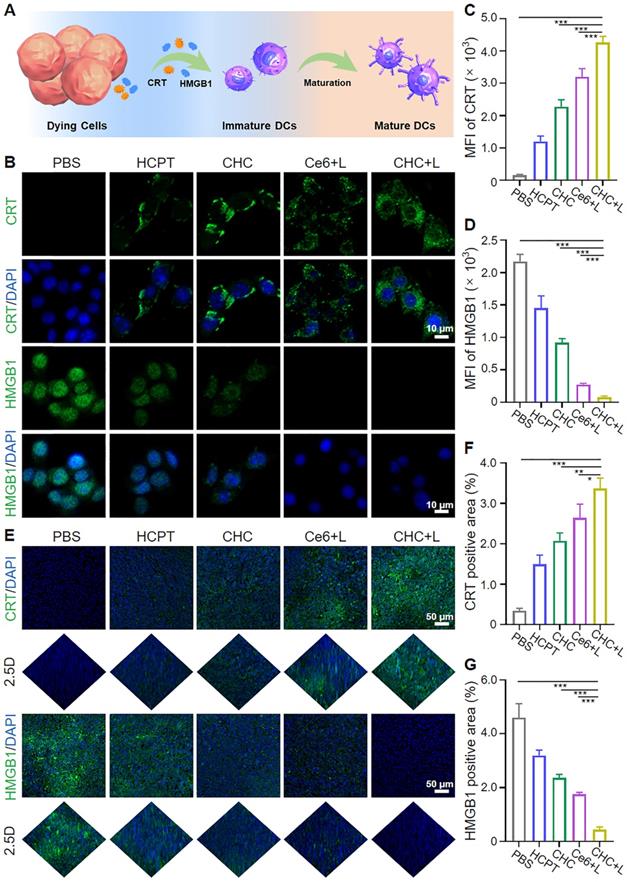
Photodynamic therapy (PDT) activating immunogenic cell death (ICD) to induce personalized tumor vaccine in situ has been verified to expose tumor-associated antigens and recruit immune cells to trigger tumor-specific immune response (Figure 5A). Whether CHC NPs can effectively trigger ICD was investigated in vitro by means of the major biochemical indicators, calreticulin (CRT) and high-mobility group box 1 (HMGB1). Transferring from rough endoplasmic reticulum (rER) to cytomembrane surface, CRT is involved in early stage of dendritic cell (DC) maturation induction, and subsequently HMGB1 spreads from the nucleus to the extracellular matrix for participating in the late ICD via binding antigen presenting cells (APCs) to speed up DC maturation. Expression of CRT on CT26 cell membrane and extracellular release of HMGB1 in vitro were observed and quantified with CLSM (Figure 5B-D). Since the upregulated level of ROS in tumor cells would accelerate CRT translocation and HMGB1 release, CHC NPs displayed superior ROS generation capacity stemming from chemodynamic effect of Fenton reaction and photodynamic effect of Ce6. CHC+L group appeared remarkably stronger CRT green fluorescence and weaker HMGB1 green fluorescence upon 660 nm irradiation. In addition to the evaluation of correlated biomarkers from damaged CT26 cells, tumor tissues under various treatments about CRT expression and HMGB1 migration were sequentially assessed by immunofluorescent staining (Figure 5E). As visualized in Figure 5F and 5G, CHC+L group resulted in the significant exposure of CRT from rER and the insignificant migration of HMGB1 from cell nucleus or cytoplasm to drive autoimmunity. Both in vitro and in vivo manifestations validated that CHC NPs could serve as optimal ICD-induction agent.
Immunogenic cell death induction of CHC NPs in vitro and vivo. (A) Schematic illustration of CHC NPs mediated activation of DCs maturation via CRT and HMGB1 secretion. (B) CLSM examination of CRT and HMGB1 exposure in CT26 cells with different treatments for 24 h and corresponding fluorescence intensity (C and D). (E) Immunofluorescence staining of CRT and HMGB1 release in CT26 tumor sections and corresponding quantification (F and G). Data displayed as mean ± SD, n = 3.
Biodistribution and penetration capacity of CHC NPs
After verifying the outstanding efficacy of CHC for ICD effect in vitro and in vivo, the overall assessment about the performance of drug delivery level from outside to inside was further carried out. Nanodrugs equipped with appropriate size are easier to exhibit EPR effects in comparison with small drug molecules. Using CT26 MCSs model simulated deep penetration of CHC NPs in tumor to estimate the permeability and therapeutic function of nanoparticles (Figure 6A). Chlorin e6 (Ce6), as an auto-fluorescent tracker upon 660 nm irradiation, was involved in fluorescence imaging to determine intracellular localization with the generation of red fluorescence, which agreed with vivid fluorescence emission of Ce6 at 670 nm visualized by fluorescence photometer (Figure 1D), proving the successful loading of Ce6 into CHC NPs. Besides, the blue HCPT fluorescence at different vertical layer was caught by CLSM Z-stack pattern after cultured with CHC NPs. The journey of CHC NPs started from the edge of MCSs at 2 h to nearly filled the entire MCSs at 12 h, was manifested through the change of fluorescence in detail (Figure S11). As described in Figure 6B, CLSM images of CT26 MCSs after 12 h treatment by CHC NPs were displayed in the form of individual channel to clearly distinct and ensure both Ce6 and HCPT encapsulation. The corresponding fluorescence intensity of HCPT and Ce6 at each vertical layer were shown in Figure 6C and 6D. Notably, both the strongest fluorescence intensity of HCPT and Ce6 were viewed at Z-axis distance of 80 µm under 12 h treatment with CHC NPs. Thus, the time-dependent permeation performance also demonstrated that CHC nanoparticles utilized EPR effect to effectively accumulate at tumor sites and gradually permeate.
Biodistribution and deep penetration of CHC NPs. (A) Estimate about penetration of CHC NPs via CT26 MCSs treated for 2 h, 6 h and 12 h. (B) Individual CLSM images of CT26 MCSs after 12 h treatment by CHC NPs and corresponding fluorescence intensity of (C) HCPT or (D) Ce6. (E) NIR fluorescence photographs of CT26 xenograft mice at various timepoints and (F) statistic comparison of fluorescence intensity in tumors. (G) NIR fluorescence images of major organs at 48 h and (H) quantification of fluorescence intensity in tumor and major organs. (I) Fluorescence imaging of the excised tumors after 48 h post-injection of Ce6 or CHC NPs and (J) mean fluorescence intensity. Data denoted as mean ± SD, n = 3.
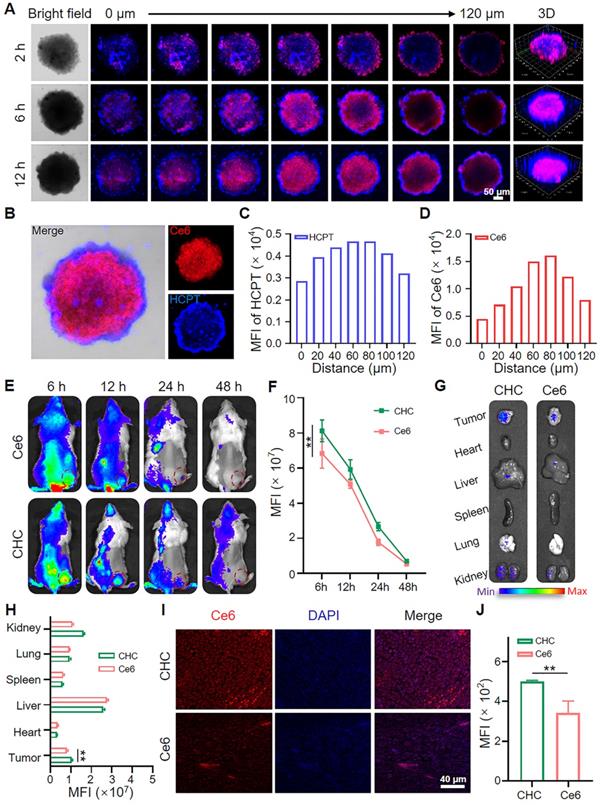
Meanwhile, biodistribution of CHC NPs in vivo was also analyzed on mice bearing CT26 tumor cells, taking advantage of Near-infrared imaging capability of Ce6 to endow fluorescent properties for CHC NPs. As depicted in Figure 6E, the fluorescence intensity of tumor site in CT26-bearing mice reduced with time, but the intensity of CHC always surpassed free Ce6, implying that CHC nanoparticles could be better enriched in tumor site and excreted from the body with the normal metabolism (Figure 6F). In terms of CHC NPs, there was no retention in major organs after 48 h and additional toxic side effects would not be formed (Figure 6G-H). Fluorescence imaging of tumor sections were applied to analyze the permeation and retention of CHC at 48 h, where CHC group still with obvious red fluorescence signal of Ce6 (Figure 6I and 6J). Therefore, CHC NPs exhibited remarkable penetration ability in vitro or in vivo, which could stay a balance of penetration and retention to elevate the therapeutic efficiency.
Antitumor effect of CHC NPs in vivo
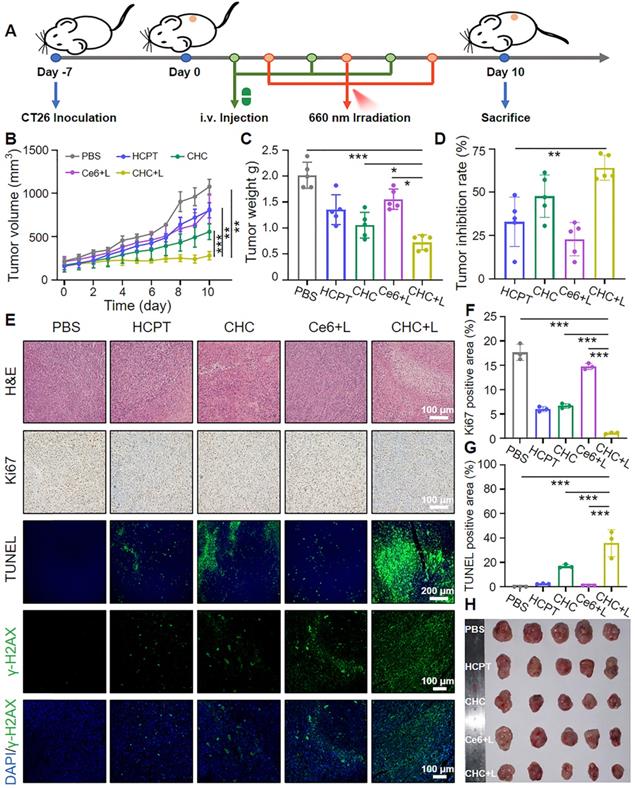
Given aforementioned fantastic treatment outcomes of CHC NPs in CT26 cells by utilizing oxidative lesions and ICD originated from CT/CDT/PDT, murine colon cancer CT26 model was constructed to further seek therapeutic efficacy on Balb/c mice. As present in Figure 7A, CT26 cells were subcutaneously inoculated to models, and CT26-bearing mice were grouped into five treatment types at random, respectively PBS, HCPT, CHC, Ce6+L, CHC+L. The treatment course included intravenous injection of drugs for three times, where groups of Ce6+L and CHC+L exposed to 660 nm laser (180 mW/cm2) after 24 h administration. The average tumor volume of mice in PBS group rose perpendicularly from ~218 mm3 to ~1078 mm3, but CHC+L treatment exhibited exceptional tumor inhibitory effect with significant reduction in tumor mass (Figure 7B). Groups of HCPT and Ce6+L had shown little benefit, however, HCPT and Ce6 loaded CHC NPs treatment was second only to CHC+L. It was mainly because that HCPT could significantly inhibit the genetic material DNA synthesis by suppressing the activity of DNA topoisomerase Ⅰ, but low toxicity compared to CPT. Further calculation of tumor weight and inhibition rate once again illustrated the advantages of mainly chemotherapy combined with CDT/PDT therapies in anti-tumor issues (Figure 7C and D). For tumor tissue sections through H&E staining, CHC+L treated group expressed evident tumor apoptosis/necrosis and the largest damaged area, CHC group appeared a little, while tumor cells of other treatment group were in good condition (Figure 7E). DNA damage was further validated in vivo by tumor sections after treatment and mean fluorescence intensity was shown in Figure S12. Ki67 immunohistochemical staining intuitively performed the least brown cell proliferation in CHC+L group, corresponding to the most intensive green fluorescence of apoptotic cells in CHC+L for TUNEL staining. The positive area quantification of Ki67 and TUNEL were displayed in Figure 7F and 7G in turns. During 10 days treatment, the body weight among the all groups had reasonable fluctuation and the digital photograph of the excised tumors from various administration groups as expected also witnessed the remarkable efficacy of CHC+L (Figure 7H and S13). Aforesaid results all confirmed the serious apoptosis induced by CHC+L group to emphasize the prominent therapy efficiency of CHC NPs. Beyond that, H&E sections of major organs in CHC+L group had no tumor metastasis by contrast with other treatment (Figure S14).
Tumor growth synergistic inhibition of CHC NPs. (A) Sketch map of dual-drug synergy therapy in CT26 xenograft mice. (B) Tumor growth curves and (C) tumor weight at different groups for 10 days. (D) Tumor inhibition rate after 10 days treatment. (E) H&E, Ki67, TUNEL and DNA damage staining of CT26 tumors at the end of treatment. Qualitative analysis of (F) Ki67 generating cells and (G) TUNEL apoptosis cells upon various treatments. (H) Digital photograph of the excised tumors from different groups.
Biosafety assessment
The primary prerequisite for CHC NPs to be transformed into clinical application in the future is excellent biological safety. Hemocompatibility is regarded as an important target to evaluate the effect of pharmaceutical preparations on blood stability by CHC NPs and erythrocyte cocultivation. We selected the concentration range of Ce6 in CHC from 0 to 200 μg/mL for hemolysis experiment, and all calculation results were under 2% (Figure S15A). The ultraviolet absorption peaks of hemoglobin appearing at 541 nm and 574 nm were measured by UV-Vis, where red blood corpuscles were severely damaged by Triton X-100 as the positive control group with exceptionally high absorbance, illustrating the prominent hemocompatibility and performance stability of CHC NPs for ensuring the normal blood circulation (Figure S15B). As for the biocompatibility of CHC NPs and murine L929 fibroblasts (L929 cells), MTT assay results showed the cell survival rate of L929 was above 89% at various concentration, indicating CHC as a superior biocompatible nanodrug (Figure S16). Furthermore, the biosafety of CHC NPs conduct in vivo was viewed as one of the vital contents. Undergoing intravenous injection of different reagents (PBS, HCPT, Ce6 and CHC NPs, respectively), the body weight of mice fluctuated within the normal range (Figure S17). The parameters of blood routine incorporating WBC, LYM, RBC, HGB, HCT, MCHC, RDW, PLT and MPV were investigated and no significant toxicity or inflammatory response in various treatment groups after 1 day, 7 days and 14 days (Figure S18). These analysis reports manifested that CHC NPs could suppress tumor growth via synergistic chemo-chemodynamic/photodynamic therapy with outstanding biosafety application.
Conclusion
This study launched an efficient and environmental-friendly process that calcium carbonate hybrid nanoparticles equipped with classic chemotherapy drug HCPT and photosensitizer Ce6 were successfully prepared, achieving chemotherapy-based, synergetic CDT/PDT therapy to trigger immunogenic death for eradicating tumors. The introduction of PEG-P(Glu) block copolymers greatly improved and optimized hydrophilicity and stabilization of CHC NPs, to facilitate the transfer of nanoparticles via the blood circulation and enrich at tumor sites through EPR effect. In addition, CHC NPs were manifested to trigger high ROS yield under CDT/PDT activation due to the internal stimulation of Fenton reaction to generate •OH and the production of 1O2 upon utilizing exogenous stimulation of 660 nm laser. Meanwhile, ICD was efficiently induced to promote antitumor immunogenicity for eliminating malignancy. Exploiting structure of calcium carbonate decomposed autonomously at low pH in tumor microenvironment, the proposed composite mode of chemotherapy and CDT/PDT for cancer therapy was found at the first time to inspire more related designs on the strength of responsive combination therapy, in order that promote the transformation of nanomedicine into clinic.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Nos. 51703187, 32071375), the Chongqing Talent Plan for Young Top Notch Talents (CQYC202005029, China) and Shenzhen Industry and Information Committee “Innovation Chain and Industry Chain” integration special support plan project (20180225103240819).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394-424
2. Rosenblum D, Joshi N, Tao W, Karp JM, Peer D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun. 2018;9:1410
3. Shi JJ, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: Progress, challenges and opportunities. Nat Rev Cancer. 2017;17:20-37
4. Chan WCW, Khademhosseini A, Parak W, Weiss PS. Cancer: Nanoscience and nanotechnology approaches. ACS Nano. 2017;11:4375-4376
5. Xue XD, Huang Y, Bo RN, Jia B, Wu H, Yuan Y. et al. Trojan horse nanotheranostics with dual transformability and multifunctionality for highly effective cancer treatment. Nat Commun. 2018;9:3653
6. Qi C, He J, Fu LH, He T, Blum NT, Yao XK. et al. Tumor-specific activatable nanocarriers with gas-generation and signal amplification capabilities for tumor theranostics. ACS Nano. 2021;15:1627-1639
7. Chen HM, Zhang WZ, Zhu GZ, Xie J, Chen XY. Rethinking cancer nanothe-ranostics. Nat Rev Mater. 2017;2:17024
8. Dong ZL, Feng LZ, Hao Y, Li QG, Chen MC, Yang ZJ. et al. Synthesis of CaCO3-based nanomedicine for enhanced sonodynamic therapy via amplification of tumor oxidative stress. Chem. 2020;6:1391-1407
9. Liu YJ, Yu BR, Dai XG, Zhao NN, Xu FJ. Biomineralized calcium carbonate nanohybrids for mild photothermal heating-enhanced gene therapy. Biomaterials. 2021;274:120885
10. Wan XY, Zhong H, Pan W, Li YH, Chen YY, Li N. et al. Programmed release of dihydroartemisinin for synergistic cancer therapy using a CaCO3 mineralized metal-organic framework. Angew Chem Int Ed. 2019;58:14134-14139
11. Zhu YJ, Yang ZJ, Dong ZL, Gong YM, Hao Y, Tian LL. et al. CaCO3-assisted preparation of pH-responsive immune-modulating nanoparticles for augmented chemo-immunotherapy. Nanomicro Lett. 2020;13:29
12. Liu YL, Pan YX, Cao W, Xia FF, Liu B, Niu JQ. et al. A tumor microenvironment responsive biodegradable CaCO3/MnO2-based nanoplatform for the enhanced photodynamic therapy and improved PD-L1 immunotherapy. Theranostics. 2019;9:6867-6884
13. Xu CY, Yan YF, Tan JC, Yang DH, Jia XJ, Wang L. et al. Biodegradable nanoparticles of polyacrylic acid-stabilized amorphous CaCO3 for tunable pH-responsive drug delivery and enhanced tumor inhibition. Adv Funct Mater. 2019;29:1808146
14. Ruan HT, Hu QY, Wen D, Chen Q, Chen GJ, Lu YF. et al. A dual-bioresponsive drug-delivery depot for combination of epigenetic modulation and immune checkpoint blockade. Adv Mater. 2019;31:e1806957
15. Xue CC, Li MH, Zhao Y. Zhou, J. Hu Y, Cai KY, et al. Tumor microenvironment-activatable Fe-doxorubicin preloaded amorphous CaCO3 nanoformulation triggers ferroptosis in target tumor cells. Sci Adv. 2020;6:eaax1346
16. Zheng P, Ding BB, Shi R, Jiang ZY, Xu WG, Li G. et al. A multichannel Ca2+ nanomodulator for multilevel mitochondrial destruction-mediated cancer therapy. Adv Mater. 2021;33:e2007426
17. Zheng P, Ding BB, Jiang ZY, Xu WG, Li G, Ding JX. et al. Ultrasound-augmented mitochondrial calcium ion overload by calcium nanomodulator to induce immunogenic cell death. Nano Lett. 2021;21:2088-2093
18. Nishiyama N, Okazaki S, Cabral H, Miyamoto M, Kato Y, Sugiyama Y. et al. Novel Cisplatin-incorporated polymeric micelles can eradicate solid tumors in mice. Cancer Res. 2003;63:8977-8983
19. Hu QY, Li HJ, Archibong E, Chen Q, Ruan HT, Ahn S. et al. Inhibition of post-surgery tumour recurrence via a hydrogel releasing CAR-T cells and anti-PDL1-conjugated platelets. Nat Biomed Eng. 2021 33903744
20. Li ML, Xia J, Tian RS, Wang JY, Fan JL, Du JJ. et al. Near-infrared light-initiated molecular superoxide radical generator: rejuvenating photodynamic therapy against hypoxic tumors. J Am Chem Soc. 2018;140:14851-14859
21. Ma XB, Zhang T, Qiu W, Liang MY, Gao Y, Xue P. et al. Bioresponsive prodrug nanogel-based polycondensate strategy deepens tumor penetration and potentiates oxidative stress. Chem Eng J. 2021;420:127657
22. Zhang LJ, Zhang X, Lu GH, Li F, Bao WE, Song C. et al. Cell membrane camouflaged hydrophobic drug nanoflake sandwiched with photosensitizer for orchestration of chemo-photothermal combination therapy. Small. 2019;15:e1805544
23. Ke WD, Li JJ, Mohammed F, Wang YH, Tou K, Liu XY. et al. Therapeutic polymersome nanoreactors with tumor-specific activable cascade reactions for cooperative cancer therapy. ACS Nano. 2019;13:2357-2369
24. Tang ZM, Liu YY, He MY, Bu WB. Chemodynamic therapy: Tumour micro-environment-mediated Fenton and Fenton-like reactions. Angew Chem Int Ed. 2019;58:946-956
25. Wang SQ, Yang LT, Cho HY, Dean Chueng ST, Zhang HP, Zhang QY. et al. Programmed degradation of a hierarchical nanoparticle with redox and light responsivity for self-activated photo-chemical enhanced chemodynamic therapy. Biomaterials. 2019;224:119498
26. Ranji-Burachaloo H, Gurr PA, Dunstan DE, Qiao GG. Cancer treatment through nanoparticle-facilitated fenton reaction. ACS Nano. 2018;12:11819-11837
27. Cheng K, Yang XQ, Zhang XS, Chen J, An J, Song YY. et al. High-security nanocluster for switching photodynamic combining photothermal and acid-induced drug compliance therapy guided by multimodal active-targeting imaging. Adv Funct Mater. 2018;28:1803118
28. Xiang HJ, Zhao LZ, Yu LD, Chen HZ, Wei CY, Chen Y. et al. Self-assembled organic nanomedicine enables ultrastable photo-to-heat converting theranostics in the second near-infrared biowindow. Nat Commun. 2021;12:218
29. Zhang WT, Li SH, Liu XN, Yang CY, Hu N, Dou LN. et al. Oxygen-generating MnO2 nanodots-anchored versatile nanoplatform for combined chemo-photodynamic therapy in hypoxic cancer. Adv Funct Mater. 2018;28:1706375
30. Shu M, Tang JJ, Chen LL, Zeng Q, Li C, Xiao ST. et al. Tumor microenvironment triple-responsive nanoparticles enable enhanced tumor penetration and synergetic chemo-photodynamic therapy. Biomaterials. 2021;268:120574
31. Feng LZ, Tao DL, Dong ZL, Chen Q, Chao Y, Liu Z. et al. Near-infrared light activation of quenched liposomal Ce6 for synergistic cancer phototherapy with effective skin protection. Biomaterials. 2017;127:13-24
32. Fan ZJ, Liu HX, Xue YH, Lin JY, Fu Y, Xia ZH. et al. Reversing cold tumors to hot: An immunoadjuvant-functionalized metal-organic framework for multimodal imaging-guided synergistic photo-immunotherapy. Bioact Mater. 2021;6:312-325
33. Ma TH, Zhang Q, Xuan QZ, Zhuang JF, Zhang W, Li H. et al. pH/Near-infrared light dual activated Ce6-doped silicon nanoparticles with tumor chemo-photodynamic synergistic therapy for improving efficiency of monotherapy. Chem Eng J. 2021;424:130536
34. Wen M, Ouyang J, Wei CW, Li H, Chen WS, Liu YN. Artificial enzyme catalyzed cascade reactions: antitumor immunotherapy reinforced by NIR-II light. Angew Chem Int Ed. 2019;58:17425-17432
35. Wan GY, Cheng YY, Song J, Chen Q, Chen BW, Liu YY. et al. Nucleus-targeting near-infrared nanoparticles based on TAT peptide-conjugated IR780 for photo-chemotherapy of breast cancer. Chem Eng J. 2020;380:122458
36. Gao T, Zhang ZP, Liang S, Fu SL, Mu WW, Guan L. et al. Reshaping antitumor immunity with chemo-photothermal integrated nanoplatform to augment checkpoint blockade-based cancer therapy. Adv Funct Mater. 2021;31:2100437
37. Martin JD, Cabral H, Stylianopoulos T, Jain RK. Improving cancer immuno-therapy using nanomedicines: Progress, opportunities and challenges. Nat Rev Clin Oncol. 2020;17:251-266
38. Yang AF, Dong X, Bai Y, Sheng SP, Zhang Y, Liu TJ. et al. Doxorubicin/CpG self-assembled nanoparticles prodrug and dendritic cells co-laden hydrogel for cancer chemo-assisted immunotherapy. Chem Eng J. 2021;416:129192
39. Li B, Hao GY, Sun B, Gu Z, Xu ZP. Engineering a therapy-induced “immunogenic cancer cell death” amplifier to boost systemic tumor elimination. Adv Funct Mater. 2020;30:1909745
40. Liu J, Ai XX, Cabral H, Liu JL, Huang Y, Mi P. Tumor hypoxia-activated combinatorial nanomedicine triggers systemic antitumor immunity to effectively eradicate advanced breast cancer. Biomaterials. 2021;273:120847
41. Jia D, Ma XB, Lu Y, Li XY, Hou SX, Gao Y. et al. ROS-responsive cyclodextrin nanoplatform for combined photodynamic therapy and chemotherapy of cancer. Chin Chem Lett. 2021;32:162-167
42. Tang ZM, Liu YY, Ni DL, Zhou JJ, Zhang M, Zhao PR. et al. Biodegradable nanoprodrugs: "Delivering" ROS to cancer cells for molecular dynamic therapy. Adv Mater. 2020;32:e1904011
43. Hou SX, Gao YE, Ma XB, Lu Y, Li XY, Cheng JQ. et al. Tumor microenvironment responsive biomimetic copper peroxide nanoreactors for drug delivery and enhanced chemodynamic therapy. Chem Eng J. 2021;416:129037
44. Chen Q, Wang C, Zhang XD, Chen GJ, Hu QY, Li HJ. et al. In situ sprayed bioresponsive immunotherapeutic gel for post-surgical cancer treatment. Nat Nanotechnol. 2019;14:89-97
45. Mi P, Kokuryo D, Cabral H, Wu HL, Terada Y, Saga T. et al. A pH-activatable nanoparticle with signal-amplification capabilities for non-invasive imaging of tumour malignancy. Nat Nanotechnol. 2016;11:724-30
46. Zhang T, Xiong HG, Ma XB, Gao Y, Xue P, Kang YJ. et al. Supramolecular Tadalafil nanovaccine for cancer immunotherapy by alleviating myeloid-derived suppressor cells and heightening immunogenicity. Small Methods. 2021;5:2100115
47. An SF, Zhang GH, Wang TW, Zhang WN, Li KY, Song CS. et al. High-density ultra-small clusters and single-atom Fe sites embedded in graphitic carbon nitride (g-C3N4) for highly efficient catalytic advanced oxidation processes. ACS Nano. 2018;12:9441-9450
48. Jiao L, Li JK, Richard LL, Sun Q, Stracensky T, Liu ES. et al. Chemical vapour deposition of Fe-N-C oxygen reduction catalysts with full utilization of dense Fe-N4 sites. Nat Mater. 2021 34112977
49. Xu ZG, Liu SY, Kang YJ, Wang MF. Glutathione- and pH-responsive nonporous silica prodrug nanoparticles for controlled release and cancer therapy. Nanoscale. 2015;7:5859-68
Author contact
![]() Corresponding authors: E-mail: zgxuedu.cn (Z. Xu); xlzhang82com (X. Zhang).
Corresponding authors: E-mail: zgxuedu.cn (Z. Xu); xlzhang82com (X. Zhang).