Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Diabetes and cardiovascular...
Endothelial cell functions
Endothelial dysfunction (ED) in...
Current status of metformin in...
Pharmacological effects of...
Future directions
Conclusion
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(19):9376-9396. doi:10.7150/thno.64706 This issue Cite
Review
Metformin in cardiovascular diabetology: a focused review of its impact on endothelial function
Yu Ding1, Yongwen Zhou1,3, Ping Ling1,3, Xiaojun Feng2, Sihui Luo1, Xueying Zheng1, Peter J. Little4,5, Suowen Xu1 ![]() , Jianping Weng1,3
, Jianping Weng1,3 ![]()
1. Institute of Endocrine and Metabolic Diseases, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, China.
2. Department of Pharmacy, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, China.
3. Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, China.
4. Sunshine Coast Health Institute, University of the Sunshine Coast, Birtinya, QLD 4575, Australia.
5. School of Pharmacy, Pharmacy Australia Centre of Excellence, the University of Queensland, Woolloongabba, Queensland 4102, Australia.
Received 2021-7-8; Accepted 2021-8-30; Published 2021-9-9
Abstract

As a first-line treatment for diabetes, the insulin-sensitizing biguanide, metformin, regulates glucose levels and positively affects cardiovascular function in patients with diabetes and cardiovascular complications. Endothelial dysfunction (ED) represents the primary pathological change of multiple vascular diseases, because it causes decreased arterial plasticity, increased vascular resistance, reduced tissue perfusion and atherosclerosis. Caused by “biochemical injury”, ED is also an independent predictor of cardiovascular events. Accumulating evidence shows that metformin improves ED through liver kinase B1 (LKB1)/5'-adenosine monophosphat-activated protein kinase (AMPK) and AMPK-independent targets, including nuclear factor-kappa B (NF-κB), phosphatidylinositol 3 kinase-protein kinase B (PI3K-Akt), endothelial nitric oxide synthase (eNOS), sirtuin 1 (SIRT1), forkhead box O1 (FOXO1), krüppel-like factor 4 (KLF4) and krüppel-like factor 2 (KLF2). Evaluating the effects of metformin on endothelial cell functions would facilitate our understanding of the therapeutic potential of metformin in cardiovascular diabetology (including diabetes and its cardiovascular complications). This article reviews the physiological and pathological functions of endothelial cells and the intact endothelium, reviews the latest research of metformin in the treatment of diabetes and related cardiovascular complications, and focuses on the mechanism of action of metformin in regulating endothelial cell functions.
Keywords: Metformin, cardiovascular diabetology, endothelial function, diabetes, panvascular disease
Diabetes and cardiovascular disease - the dangerous liaison
Patients with diabetes suffer from a higher incidence and rate of mortality of cardiovascular disease (CVD) compared with nondiabetic subjects [1]. Diabetes per se is also a high-risk factor for CVD [2]. According to the disease statistic report from the International Diabetes Federation, the global prevalence of diabetes was 9.3% in 2019 estimated and is projected to reach 10.2% by 2030 [3]. Numerous studies have shown that there is a positive correlation between glucose levels and the incidence of CVD. It is reported that the incidence of heart failure increases by 15% when glycosylated hemoglobin increases by 1%, and the incidence of myocardial infarction decreases by 16% if anti-hyperglycemic drugs are used intensively [4, 5]. With multiple pathophysiological components, including hyperglycemia, insulin resistance (IR), hyperinsulinemia and hyperlipidemia, diabetes changes the vascular responsiveness to various vasoconstrictors and vasodilators and is a vital factor in the occurrence and development of the vascular complications of diabetes [5]. Hyperglycemia is the primary determinant factor of thrombosis in the coronary vessels through the modification of microbiota thrombus colonization in hyperglycemic patients with ST-segment elevation myocardial infarction (STEMI) [6], and via the direct modification of atherosclerotic plaque instability and rupture with consequent acute thrombosis (in STEMI) and worse prognosis [7, 8]. The overexpression of inflammatory and pro-thrombotic markers is linked to the hyperglycemic levels in the serum, the coronary thrombus [9] and the peri-coronary fat [10]. This is evidence of excessive inflammation and pro-thrombotic effects of hyperglycemia in patients with atherosclerotic plaque instability, rupture, acute thrombosis, and STEMI.
Various diabetes-related pathological changes vary with the size and location of blood vessels, leading to decreased arterial plasticity, increased vascular resistance, reduced tissue perfusion, and atherosclerosis [11, 12]. In the process of atherosclerotic plaque formation, circulating monocytes first adhere to the lesion area of the vascular wall, then infiltrate into the wall, and differentiate into macrophages. These macrophages take up modified lipids through phagocytosis and scavenger receptor mediated processes, and leading to the formation of foam cells, which represent the hallmark of arterial plaques [13-16]. Based on these pathological changes, the development and progression of atherosclerosis plays a critical role in the underlying pathological basis of most patients with stable coronary disease [17].
Ross and colleagues proposed the “endothelial cell injury” model of atherosclerosis (“response to injury”) sort of assuming that atherosclerosis results from physical damage to the endothelium [18]. It was subsequently found that biochemical molecules [19, 20], including turbulent blood flow and inflammatory cytokines/chemokines, actually serve as physiochemical injury. We propose the hypothesis that “biochemical injury” is the cause of endothelial dysfunction (ED). ED is related to a series of pathological events in diabetes and its complications [21]. In patients with stable coronary disease, with the increase of IR and carbohydrate intolerance, the functional coronary circulation abnormalities of endothelium-dependent vascular movement will gradually worsen [22]. Impaired NO-dependent vasodilatation is a predictor of future cardiac events and the development of coronary ED [23]. Moreover, coronary ED is as a cause of ischemic heart disease (IHD) linked worse prognosis in diabetic patients with stable coronary disease [24, 25]. This is evidenced in stable IHD patients during angiography of coronary vessels and invasive measurements of ED. It is noteworthy that the occurrence of ED and the development of atherosclerosis is very rapid in patients with diabetes in the absence of an effective pharmacological intervention [26]. ED is also linked to insulin resistant states including obesity, diabetes, and the metabolic dysfunction [27]. Hyperglycemia is the major causal factor in the development of ED in patients with diabetes. Hyperglycemia is also a main risk factor for CVD, particularly IHD alone and in combination with diabetes. Indeed, hyperglycemia is a risk factor for CVD/IHD independently from diabetes.
Diabetes can be classified into several categories, including type 1 diabetes (T1D), type 2 diabetes (T2D), gestational diabetes mellitus, and specific types of diabetes due to other causes. Accounting for 90 - 95% of all diabetes, T2D is generally due to a progressive loss of β-cell insulin secretion frequently on the background of IR [28]. The established relationship between T2D and CVD provides a therapeutic framework that anti-diabetic drugs may have beneficial effects on cardiovascular risk factors and help reduce the atherosclerotic process in diabetic patients. In addition to hypoglycemic benefits, whether anti-diabetic drugs can prevent or treat ED and improve atherosclerosis and CVD is an important research direction [29, 30]. When choosing pharmacological interventions for patients with the comorbidities of diabetes and CVD, the hypoglycemic effects and potential cardiovascular benefits should be considered simultaneously. These considerations are critical to our understanding of the potential role of antidiabetic medications in reducing the development and progression of atherosclerosis and CVD in patients both with and without diabetes.
Because the treatment of hyperglycemia changes many factors other than circulating glucose levels, clinical and experimental data support the hypothesis that factors other than hyperglycemia are the culprit [31]. As the first-line drug for most patients with T2D, metformin is indicated because of its benefits on glycemia management but also affects several other pathways/mechanisms including oxidative stress, inflammation, lipoprotein metabolism and thrombosis [32, 33]. For example, metformin provides a protective effect on cardiovascular function by reducing weight gain and serum-free insulin level, according to the report from UK Prospective Diabetes Study (UKPDS) [34]. Moreover, various studies have emphasized on the anti-inflammatory and antioxidant role of antidiabetic medications, with multiple mechanisms, which activation of 5'-adenosine monophosphat-activated protein kinase (AMPK) by metformin has had a key role in many of them [35-37]. Notably, the pleiotropic effects of antidiabetic medications have been seen for metformin and new classes of antidiabetic drugs and incretins. To date, metformin shows pleiotropic effects to reduce the peri-coronary inflammatory stress [10], including anti-thrombotic and anti-atherosclerotic properties, which are also played by incretins in STEMI [38] and non-STEMI [39] patients. Therefore, this evidences the protective effects played by incretins on the atherosclerotic plaque for patients with acute coronary syndrome, in the condition of acute coronary thrombosis [38] and multivessel disease with and ED [39]. For a detailed review of cardiovascular actions and molecular targets of glucagon-like peptide-1 receptor agonists (GLP-1RAs) [36] and sodium glucose cotransporter 2 inhibitors (SGLT2i) [37], please refer to our recent reviews. As mentioned above, ED can be an independent predictor of cardiovascular events and the first step toward coronary arteriosclerosis [40]. Therefore, evaluating the effects of metformin on improving ED would deepen our knowledge of the therapeutic potential of metformin. In this review, we aim to provide a comprehensive account of the physiological and pathological functions of endothelial cells and the intact endothelium, and summarize the latest research on metformin in diabetes and associated cardiovascular complications, with particular emphasis on the role of metformin on regulating endothelial cell functions.
Endothelial cell functions
There are three layers of blood vessels, and the innermost layer is the intima composed of a monolayer of endothelial cells and the underlying internal elastic lamella [41]. In the physiological state, the healthy endothelium serves diverse biological functions (Figure 1) and creates an adaptive life support system that extends to almost every tissue and organ [42].
Multi-faceted biological functions of endothelium. The healthy endothelium serves diverse biological functions in physiological state, including 1) serving as a physiological barrier and site of innate immunity; 2) regulation of vascular tone; 3) mediating mechanotransduction through mechanical sensors/mechanosensitive complexes; 4) keeping endothelial integrity by an effect on both endothelial injury and the capacity for endothelial repair; 5) keeping the balance of anticoagulant and procoagulant function; 6) regulating angiogenesis; 7) as an essential cell type for the metabolic and synthetic function.
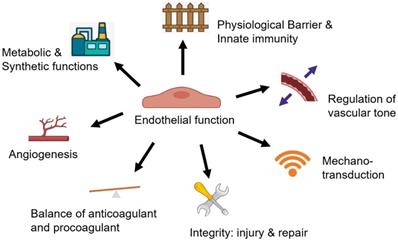
Among all the functions of the intact endothelium [43], the first is its role as a physiological semi-permeable barrier and an emerging component of innate immunity. As the barrier and site of innate immunity between blood and tissues, the endothelium actively regulates the exchange and transport of small and large molecules and defends against invading pathogens and infections [44, 45]. In addition, as a carbohydrate-rich layer lining the vascular endothelium [46], the glycocalyx acts as a barrier between blood, the endothelium and underlying tissues and plays an essential role in maintaining the barrier integrity and consequently homeostasis and quiescence of the vasculature [47].
A pivotal function of the endothelium is to regulate vascular tone through secreting substances that relax and constrict blood vessels. Nitric oxide (NO), a classical mediator of vasodilation, is produced by endothelial cells and glycocalyx. NO regulates the relaxation of vascular smooth muscle cells (VSMCs) mainly by stimulating soluble guanylyl cyclase to produce cyclic guanosine monophosphate [46, 48]. By reacting with the Cys thiol group and reversibly regulating S-nitrosylation, NO can play a role in relaxing blood vessels [49]. Endothelial nitric oxide synthase (eNOS) is the leading source of most vascular NO, and is also a critical enzyme to protect endothelium against vascular damage [50, 51]. Besides NO, vasodilators include other dissolved gasses such as hydrogen sulfide (H2S) and carbon monoxide (CO), which are also secreted by endothelial cells [52-54]. The endothelium also produces vasoconstrictor molecules, such as angiotensin II (AngII), endothelin 1 (ET-1), thromboxane A2 (TXA2), thrombin, and others [55]. The dynamic balance between vasoconstrictor and vasodilator mediators is an important foundation for endothelial cells to maintain homeostasis. On the other hand, the autonomic nervous system and various neurotransmitters play an essential role in the regulation of vascular tone [56]. The endothelial cells express a variety of receptors that specifically interact with neurotransmitters from the nerve terminal. Among various neurotransmitters, norepinephrine (NE), adenosine triphosphate (ATP) and neuropeptide Y (NPY) relax blood vessels, while acetylcholine (Ach) and calcitonin gene-related peptide (CGRP) act as vasodilators. The endothelial cells are triggered to produce NO and other vasoactive gases when the adrenergic endothelial receptors are activated [57].
The endothelium senses mechanical stress and mediates mechano-transduction through mechanosensors or mechanosensitive complexes [58]. Endothelial cells convert mechanical stress into intracellular biochemical signals, such as increased NO, H2S, and calcium concentration [59-61]. Thus, these responses of vascular mechano-transduction serve to prevent vascular cells from damage induced by mechanical injury [58]. Endothelial integrity, another primary function of the endothelium, is affected by the degree of injury and depends on the integrity of the endogenous repair ability of the vessels [62]. When an adjacent endothelial cell is damaged, the local mature endothelial cell can replicate to replace the lost and damaged cell. However, if endothelial cells only rely on local replication function, it is far from enough to resist external damage [63]. A recently study shows that the protection is driven by a transient activation of extracellular-signal-regulated kinase (ERK) in endothelial cells neighboring extruding cells, which inhibits caspase activation and prevents elimination of endothelial cells in clusters [64]. Endothelial progenitor cells (EPCs) in the circulation participate in another repairing mechanism for maintaining the integrity of the endothelium, which has been clearly explained [65]. Therefore, mechano-transduction and circulating EPCs may play an essential role in the pathogenesis of vascular disease by affecting endothelial injury and repair.
Maintaining the balance between anticoagulant and procoagulant status of the vessel wall and blood is a critical function of the endothelium. Although endothelial cells have close contact with platelets, there is no harmful reaction in the blood vessels of a healthy organism [66]. Plasminogen activator (PA) and plasminogen activator inhibitor (PAI) are synthesized and secreted by endothelial cells, providing anticoagulant and procoagulant regulatory mechanisms, respectively [67]. There are several mechanisms involved in the regulation process, such as enhanced plasminogen activation by tissue-PA (t-PA) or urokinase-PA (u-PA), reduced inhibition of plasmin by α2-antiplasmin or of PAs by PAIs, and enhanced conversion of single-chain u-PA to two chain u-PA [68].
Angiogenesis, depending on endothelial cell migration, is another basis function of the endothelium [69]. Endothelial cells regulate angiogenesis through producing several factors, including vascular endothelial growth factor (VEGF), NO, matrix metalloproteinases (MMPs), tissue inhibitors of metalloproteinases (TIMPs), and triggers [44, 70]. On this basis, endothelial cells have the ability to adjust their number and arrangement to meet local needs.
As a sizeable endocrine organ, endothelial cells have a metabolic (large source of energy form glycolysis) and synthetic function. In addition to vasoactive substances, endothelial cells can also secrete various mediators, thus affecting the whole-body cell function [44]. Endothelial cells are also processing factories of important metabolites, such as glucose, fatty acids, amino acids, lipids, ATP, acetyl coenzyme A, and lactate, and endothelial cells regulate the supply of nutrition and oxygen to all tissues of the body [44, 71]. The angiocrine and angiogenic function of endothelial cells underlie its importance in regulating tumor metastasis and neovascularization in states of myocardial infarction.
In summary, these physiological functions of the intact endothelium complement each other, and each role is crucial to maintaining the homeostasis of the vasculature.
Endothelial dysfunction (ED) in diabetes and its complications
Hyperglycemia and ED
ED occurs when the production and secretion of vasodilators and vasoconstrictors are out of balance, leading to impaired endothelium-dependent vasodilation. Hyperglycemia, as the defining clinical feature of diabetes mellitus, is known to cause ED through several mechanisms [72], including those involving advanced glycation end products (AGEs), hexosamine, glycocalyx reduction, protein kinase C (PKC), oxidative stress, proliferative dysfunction, ascorbic acid and aldose reductase [73-78]. Hyperglycemia induces changes in a variety of cellular signaling and metabolic pathways including altered hemodynamics due to decreased eNOS activity and increased ET-1 synthesis, increased oxidative stress and inflammation, basement membrane thickening caused by transforming growth factor-β (TGF-β) mediated increased synthesis of collagen IV and fibronectin, extracellular matrix changes, VEGF-induced damage to VSMC vascular permeability, cellular growth, inhibition of protein kinase B (PKB, also known as Akt) activation, angiogenesis, and decrease of Na+-K+-ATPase activity [79-81]. These changes can further lead to endothelial function injury. In addition, fluctuating plasma glucose levels in people with diabetes can also cause endothelial cell damage [82]. It is speculated that endoglin is involved in the regulation of ED, and oxidative stress may play a role in this regulation [83]. Through suppression of phosphatidylinositol 3 kinase (PI3K)-Akt signalling and activation of FOXO1 in cultured primary endothelial cells, hyperglycemia stimulated placenta growth factor (PlGF) secretion which is a pro-inflammatory angiogenic mediator [84]. Hyperglycemia drives ED via different mechanisms, including excessive oxidative stress, over activated inflammation and reduced NO bioavailability (see Figure 2). Mechanistically, metformin improves ED through liver kinase B1 (LKB1)/AMPK and AMPK-independent targets [85]. The mechnisms involved can interact with each other and amplify the influence, which may bring a vicious circle.
Role of endothelial dysfunction in diabetes and its cardiovascular complications. Hyperglycemia is the leading and common characteristic of diabetes and its cardiovascular complications, including insulin resistance and glucose fluctuation. Hyperglycemia drives endothelial dysfunction via different mechanisms, such as 1) Decreased bioavailability of NO and increased secretion of vasoconstrictors, e.g., ET-1 and Ang II; 2) Increased level of MGO/AGE/RAGE; 3) Enhanced inflammatory response, which is also affected by increased glucose fluctuation; 4) Enhanced oxidative stress; 5) Disruption of endothelial glycocalyx; 6) The expression of epigenetic, such as the level some miRNA, lncRNA. The role of endothelial dysfunction is reflected in the following aspects: 1) The vasodilation decreases and the vasoconstriction increases, and then the vascular homeostasis is broken; 2) Leukocytes adhesion and migration, and platelet aggregation increase; 3) The injury/apoptotic process of endothelial cells is increased; 4) The process of EndoMT is promoted; 5) The endothelial senescence is increased; 6) The permeability of endothelial cells increases. Abbreviations: AMPK, 5'-adenosine monophosphate-activated protein kinase; AGEs, advanced glycation end products; Ang II, angiotensin II; EndoMT, endothelial-to-mesenchymal transition; ET-1, endothelin 1; ICAM1, intercellular adhesion molecule 1; LncRNAs, long noncoding RNAs; MGO, methylglyoxal; MiRNAs, microRNAs; NF-κB, nuclear factor-KappaB; PARP, poly ADP-ribose polymerase; Akt, also known as PKB, protein kinase B; RAGE, receptor for AGEs; SIRT1, sirtuin 1; VCAM1, vascular cell adhesion molecule 1.
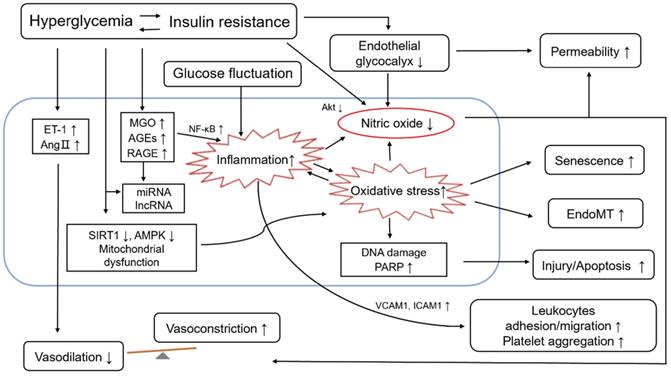
Previous studies have shown that hyperglycemia-induced oxidative stress could cause deoxyribonucleic acid (DNA) damage and induce endothelial cell senescence, injury, apoptosis, and endothelial-to-mesenchymal transition (EndoMT) [86-88]. Upon DNA damage, the nuclear enzyme poly ADP-ribose polymerase (PARP) is activated, promoting the production of adenosine diphosphate (ADP)-ribose polymers. Then, hyperglycemia-induced overproduction of superoxide also inhibits glyceraldehyde phosphate dehydrogenase (GAPDH) activity [89]. Furthermore, glycolytic intermediates produce to diacylglycerol, which can activate both classical and novel PKC pathways [81] involved in vascular hyperpermeability, cell growth, neovascularization and other vascular abnormalities of diabetes. There are at least 11 PKC isoforms, and the β- and δ-isoforms appear to be activated preferentially by the diabetic milieu [81]. High glucose-induced nuclear factor-kappaB (NF-κB) DNA binding activity may also induce a low-grade pro-inflammatory state and mediate this inhibition of cellular migration by regulating the NO levels in endothelial cells [90, 91]. In addition, the release of endothelial cell-derived vasoconstrictor factors and the disruption of the endothelial glycocalyx both increase in a high-glucose environment [92, 93]. The main precursor of AGEs, methylglyoxal (MGO), a reactive dicarbonyl generated is believed to play a critical role in ED. As presented in a previous study, after treatment with MGO for three months, it was observed that the formation of AGEs and the expression of monocyte chemoattractant protein 1 (MCP1) and the receptor for AGEs (RAGE) were increased compared to the matched control group [94], while the bioavailability of NO was decreased significantly concurrent with increased level of superoxide anion. Therefore, MGO scavengers have the potential to attenuate hyperglycemia-induced ED and MGO through ATP-sensitive potassium (KATP)/mitogen-activated protein kinase (MAPK) pathway activation [95].
Insulin resistance (IR) and ED
In diabetes mellitus, hyperinsulinemia and hyperglycemia are both reported to selectively impair receptor-dependent, endothelium-dependent relaxation [96]. The metabolic disorders of IR, hyperglycemia and excessive circulating free fatty acids cause vascular tissue oxidative stress, mild inflammation, platelet hyperactivity, and ED. In 1997, Pinkney et al. proposed that the IR syndrome was accompanied by many aspects of atherogenesis, which could be viewed as the different consequences of the effect of ED on different vascular beds [97]. In a rat model of IR, the endothelium-dependent relaxation is impaired due to the defect in NO/prostanoid-independent relaxation [98]. Associated with lipotoxicity, glucotoxicity, and inflammation, IR may damage the endothelial cell and thus trigger and accelerate atherosclerosis and CVD [30, 99-102].
Insulin activates the PI3K-Akt pathway, phosphorylates eNOS at Serine 1177 site, thereby promoting the synthesis of NO in endothelial cells under normal physiological conditions [103]. However, when the body is in a state of IR, the PI3K-Akt pathway is impaired, as well as the process of NO production. Alternatively, the MAPK pathway causes the synthesis of inflammatory markers, including PAI-1, intercellular adhesion molecule 1 (ICAM1), E-selectin, and vascular cell adhesion molecule 1 (VCAM1), thereby leading to ED [104]. Therefore, the pathogenic factors of ED induced by obesity, one of the main causes of IR, in the early stages will further impair the insulin signaling pathways of endothelial cells, resulting in reduced vasodilatation, abnormal capillary recruitment, and the transfer of insulin substrate to the target tissues [105]. Even after blood glucose levels have returned to normal, the known phenomenon of 'metabolic memory' persists and the harmful effects of hyperglycemia are perpetuated. In addition, molecular and vascular abnormalities involving NO persisted for several months even though the biochemical parameters of IR and ED were normalized, confirming the long-term effects of metabolic memory [106]. Increasing evidence shows that chronic inflammation of metabolic tissues plays a causal role in obesity-induced IR. However, it is still unclear how specific endothelial factors affect metabolic tissues [107].
Advanced glycation end products (AGEs) and ED
The reactive oxygen intermediates (ROI), the reactive chemical entities accelerating the formation of AGEs, activate multiple mechanisms that are potentially involved in the development of vascular complications, such as the activation of NF-κB, PKC and upregulation of the hexosamine pathway [108]. The binding of AGEs to RAGE increases intracellular enzymatic superoxide production [109] and induces the expression of heparanase in human umbilical vein endothelial cells (HUVECs), via activation of the forkhead box O4 (FOXO4) [110]. In a cohort study, independent of other cardiovascular risk factors, the elevated serum AGEs levels in patients with T2D was shown to be associated with ED [111]. AGEs impair endothelial-dependent vascular relaxation and the mechanism is primarily due to reduced bioavailability and activity of NO [112] and activation of p38 and ERK1/2 by reducing eNOS expression and increasing the level of reactive oxygen species (ROS) production [113]. Of note, serum AGEs do not follow the same time course even though glycated hemoglobin A1c and markers of endothelial function improve under insulin therapy [114], suggesting that there may be other factors affecting AGEs, in addition to hyperglycemia, such as oxidative stress.
More recently, mounting evidence has supported the contention that microRNAs (miRNAs) and long noncoding RNAs (lncRNAs) are associated with ED [115-118]. In this regard, AGEs induce downregulation of miR-200c and miR-200b, which leads to increased expression of target genes Rho-associated coiled-coil kinase (ROCK) and RhoA, respectively [119].
Oxidative stress, inflammation and ED
Oxidative stress is a state of imbalance between antioxidant systems and exposure to toxic ROS and related chemical species. It has been established that a low concentration of intracellular ROS is essential in maintaining antioxidant/redox homeostasis, vascular integrity and function. However, under pathological conditions such as occuring during the development of atherosclerosis, injury after ischemia-reperfusion and hypoxia, the ROS level is elevated in response to adverse stimuli [120]. ROS are produced by a variety of oxidase enzymes, including nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), lipoxygenase, xanthine oxidase, cyclooxygenase, glucose oxidase, eNOS uncoupling, and mitochondrial electron transport (cytochromeP450 monooxygenase) [121-128]. It follows that proteins and lipids are oxidized by excessive ROS (especially free radicals), which leads to the overexpression of redox genes, intracellular calcium overload and DNA fragmentation, and consequently damage to VSMCs, endothelial cells and myocardial cells [129]. In the early stage of atherosclerosis, the number of inflammatory cells (monocytes/macrophages/T-lymphocytes) and the process of arteriosclerosis are all associated with enhanced serum levels of inflammatory parameters [130]. As seen in chronic inflammation, the arteriosclerotic artery produces different adhesion molecules, cytokines, and growth factors [20, 131]. When endothelial cells are activated by inflammation, the increased expression of VCAM1, selectins and ICAM1 will promote monocyte adhesion to the vessel wall [132]. Many cytokines, including circulating inflammatory cytokines, tumor necrosis factor-α (TNF-α), ROS, oxidized low-density lipoprotein (oxLDL), and traditional risk factors can directly or indirectly induce abnormal functioning of endothelial cells, resulting in impaired vasodilation, increased endothelial permeability, increased leukocyte adhesion and thrombosis [133-135].
Over-inflammatory/oxidative stress causes ED in patients with and without diabetes [136, 137]. Indeed, in young women without IHD and low risk of CVD (“low risk subjects”), the over-inflammatory/oxidative stress may be the main determinant factors of ED and major adverse cardiovascular events (MACE) [137]. It is relevant to note that ED could be linked to other non-classical CVDs' risk factors as adipose tissue deposition and excess. This is observed in women [137] with different classes of breast fat density (lowest breast density) and increased risk of pre-diabetes and diabetes. On the other hand, it has been remarked in overweight patients with altered glycemia and IR, as those with pre-DM and without first clinical manifestation of CVD. Excess of abdominal adipose tissue in these overweight pre-diabetics patients with IR is a chronic source of inflammatory molecules that cause ED and reduction of cardiac performance and worse prognosis [138, 139].
In 2002, a study from Ceriello et al. [74] showed that oxidative stress was a common mediator of postprandial hyperglycemia and hypertriglyceridemia on endothelial function. Hyperglycemia increases intracellular ROS through the interrogation mechanism between cytosolic and mitochondrial ROS production [140], which once again shows that the decrease in NO production and the imbalance of increased ROS production may be related to the impaired endothelium-dependent vasodilation in patients with CVD. Conditions such as hyperlipidemia can also induce increased levels of AGEs, leading to increased ROS and oxLDL accumulation in diabetic animals [141]. Cytokines and oxLDL stimulate endothelial cell permeability, and NF-κB-dependent inflammatory gene expression and ROS appear to play a central role in mediating both responses [72]. Therefore, the vicious cycle among ED, oxidative stress and inflammation will lead to atherosclerosis in diabetes and associated conditions [128].
Endothelial-to-mesenchymal transition (EndoMT) and ED
EndoMT is an extremely complex pathophysiological process during which endothelial cells lose their specific markers and acquire the phenotypes of mesenchymal cells or myofibroblasts [142]. In hyperglycemia, it is deduced that high glucose levels induce EndoMT through ERK, Smad2/3, nucleotide-binding oligomerization domain-containing protein 2 (NOD2), ROCK1, and serum response factor in glomerular endothelial cells - this sequence results in increased expression of mesenchymal markers in different types of endothelial cells [143, 144]. Recently, evidence has suggested that EndoMT is a critical link in the interactions between ED and inflammation. Through this process, varieties of inflammatory mediators activate endothelial cells and transform them into mesenchymal-like cells. The factors implicated in this response are interleukin-1beta (IL-1β), TNF-α, NF-κB transcription factor, and endotoxins [145-147]. In addition to inflammation and hyperglycemia, hypoxia [148], dyslipidemia [149], miRNAs and lncRNAs [150] and other abnormalities are related to the induction of EndoMT. Notwithstanding these findings, the totalities of the mechanisms involved in EndoMT are not fully understood.
Current status of metformin in the amelioration of ED
Metformin is a biguanide derivative that exerts an anti-hyperglycemic effect with minimal risk of hypoglycemia. Metformin is currently the first choice for the majority of patients with newly diagnosed T2D requiring medical therapy. The primary status of metformin has arisen from the results of clinical evidence from the UKPDS [34]. The reduction of glucose production in liver cells and increased uptake in skeletal muscle are considered the primary processes mediating the hypoglycemic effect of metformin although multiple other actions have been suggested [151].
As for the efficacy of SGLT2i [37] and GLP-1RAs [36], numerous clinical studies have shown that metformin can improve vascular endothelial function and reduce CVD in patients with diabetes (see Table 1 [152-167]). A study reported in 2001, utilizing a moderate dose of metformin (500 mg twice daily) administered for 12 weeks and compared to placebo, showed improvement in Ach-stimulated flow (and IR) was observed among diet-treated T2D patients. In this study IR was the sole predictor of endothelium-dependent blood flow after metformin treatment [152]. Similar benefits were observed with longer-term and higher doses of metformin. Fifteen T2D patients took 1700 mg per day for three months, and t-PA, VCAM1 and ICAM1 levels were significantly reduced [155]. For obese patients who have previously received medical nutrition guidance and regular exercise, after taking metformin for 12 weeks (average dosage 1381 ± 85 mg/d), a decrease in PAI-1 and VEGF was observed, effects which were independent of favourable effects of metformin on body mass index (BMI) and glycemic control [156]. Among T2D patients with stable coronary heart disease, treatment with metformin affects VCAM1 and asymmetric dimethylarginine (ADMA) levels [166]. Still, it does not impair the endothelial healing of drug-eluting stents in these patients with or without insulin use [168]. In the long-term treatment (4.3 years) with metformin plus insulin, most of the inflammatory biomarkers, such as the von Willebrand factor (vWF), soluble VCAM1, t-PA, and ICAM1, were significantly reduced even after adjusting for baseline differences in age, sex, smoking and severity of previous CVD [161]. When comparing the effect with the other antihyperglycemic agents, at equivalent levels of glycemic control, metformin was more effective in reducing biomarkers reflecting inflammation and improving endothelial functions than repaglinide (an insulin secretagogue) [157], and only rosiglitazone (the insulin-sensitizing agent) improved endothelium-dependent vasodilatation and insulin sensitivity more than metformin [169]. In addition, with lipid-lowering agents, such as atorvastatin or fenofibrate, used in combination among T2D patients, metformin is demonstrated not only to prevent the glucose-induced impairment of endothelial functions partly but also potentiates lymphocyte-suppressing, endothelial protective and systemic anti-inflammatory effects of fenofibrate, superior to monotherapy [158, 170]. A recent randomized controlled trial showed that through distinct or complementary mechanisms of action on the vascular wall, metformin was able to improve functional capillary density during post-occlusive reactive hyperemia in obese newly diagnosed drug-naive women with T2DM [165].
Clinical evidence demonstrating the effect of metformin on endothelial function among diabetic patients
| Articles | Subjects | Intervention in metformin | Assessments | Conclusions |
|---|---|---|---|---|
| Mather, K. J (2001) [152] | Diet-treated T2D | 1000 mg/d, 12 weeks, N = 29 | ACh-stimulated flows:↑; Nitroprusside-stimulated flows: NS; Verapamil-stimulated flows: NS | IR was the sole predictor of endothelium-dependent blood flow following metformin treatment. |
| Abbasi F (2004) [153] | T2D | 1000-2000 mg/d, 12 weeks, N = 16 | sICAM-1↑; sVCAM-1: NS; ET-1: NS | Metformin, either as monotherapy or in combination with a sulfonylurea drug, led to a decrease in several CVD risk factors in patients with T2D. |
| De Jager J (2005) [154] | T2D treated with insulin | 850 mg/d, 16 weeks, N = 196 | vWf↓; sET-1↓; t-PA↓; PAI-1↓; s-ICAM-1: NS; s-VCAM-1↓ | An improvement of endothelial function with metformin in T2D treated with insulin, which was largely unrelated to changes in glycemic control. |
| Skrha J (2007) [155] | T2D | 1700 mg/d, 12 weeks, N = 15 | tPA↓; sVCAM-1↓; sICAM-1↓; Microcirculation by laser Doppler: NS | Metformin treatment promotes endothelium effects associated with a complex of metabolic changes in T2D. |
| Ersoy C (2008) [156] | Obese T2D | 1381 ± 85 mg/d, 12 weeks, N = 24 | PAI-1↑; VEGF↑ | A beneficial effect on VEGF and PAI-1 levels with metformin in obese T2D. |
| Lund SS (2008) [157] | Non-obese T2D without insulin | 2000 mg/d, 16 weeks, N = 83 | PAI-1↓; tPA↓; s ICAM-1↓; sVCAM-1↓; ET-1↓ | Metformin was more effective in reducing selected biomarkers reflecting inflammation and endothelial dysfunction compared with repaglinide despite similar glycemic levels between treatments. |
| Tousoulis D (2010) [158] | Newly diagnosed DM | 850 mg/d, 6 weeks, N = 15 | Resting FBF: NS; EDD: decrease in combination with atorvastatin | Combined with metformin and atorvastatin for 6 weeks partly prevented the glucose-induced impairment of EDD. |
| Fidan E (2011) [159] | T2D | 850-2550 mg/d, 12 weeks, N = 20 | PAI-1: NS; sICAM-1↓; ET-1: NS; Fibrinogen: NS | Metformin was effective in controlling inflammatory markers in addition to metabolic parameters. |
| Pitocco D (2013) [160] | uncomplicated T1D | 2550 mg/d, 6 months, N = 21 | FMD↑; NMD: NS | Metformin improved FMD and increased PGF2α in uncomplicated T1D. |
| de Jager J (2014) [161] | T2D treated with insulin | 850 mg/d, 4.3 years, N = 131 | vWf↓; sVCAM-1↓; s-ICAM-1↓; t-PA, PAI-1↓; ET-1: NS | Metformin is associated with improvement in some markers of endothelial function in T2D. |
| Kruszelnicka O (2015) [166] | T2D with stable CHD | Previous 1 year, N = 40 | sVCAM-1 ↓, ADMA ↑ | Metformin affects VCAM1 and ADMA levels among T2D patients with stable CHD. |
| Shigiyama F (2017) [162] | T2D treated with metformin | 750-1500 mg/d, 16 weeks, N = 54 | FMD: NS in alone; increase in combination with linagliptin | Among T2D patients with moderate hyperglycemia, metformin plus linagliptin induced both better glycemic control and improvement of endothelial function. |
| Kitao N (2017) [163] | T2D treated with metformin | 1000-1500 mg/d, 12weeks, N = 48 | FMD: NS | Combination of vildagliptin and metformin did not affect endothelial function but exert favorable effects on adipokine with T2D without advanced atherosclerosis. |
| Petrie JR (2017) [167] | T1D at increased risk for CVD | 2000 mg/d, 3 years, N = 219 | Progression of mean cIMT: NS Reactive hyperaemia index: NS | Metformin did not affect on endothelial function but might have a wider role in cardiovascular risk management. |
| Lunder, M (2018) [164] | T1D | 2000 mg/d, 12 weeks, N = 10 | Beta stiffness: NS in metformin alone; FMD↑ | Empagliflozin on top of metformin treatment significantly improved arterial stiffness compared to metformin in T1D. |
| Schiapaccassa, A (2019) [165] | Obesity T2DM women | 1700 mg/d, 30 days, N = 19 | Nutritive microvascular reactivity↑; Functional capillary density during post-occlusive reactive hyperemia↑ | Metformin was able to improve vascular function in obese newly diagnosed drug-naive T2DM women, through distinct or complementary mechanisms of action on the vascular wall. |
Abbreviations: Ach, acetylcholine; ADMA, asymmetric dimethylarginine; CHD, coronary heart disease; CVD, cardiovascular Disease; DM, diabetes mellitus; EDD, endothelium-dependent vasodilation; ET-1, endothelin-1; FBF, forearm blood flow; FMD, flow mediated dilation; IR, insulin resistance; NMD, nitroglycerin-mediated dilation; NS, non-significant; PAI-1, plasminogen activator inhibitor-1; PGF2α, prostaglandin F2α; s-ICAM-1, soluble intercellular adhesion molecule-1; s-VCAM-1, soluble vascular cell adhesion molecule-1; T1D, type 1 diabetes; T2D, type 2 diabetes; t-PA, tissue plasminogen activator; VEGF, vascular endothelial growth factor; vWf, von willebrand factor.
The traditional paradigms of T2D occurring only in adults and T1D only in children are no longer accurate, as both diseases occur in both age-groups. There is less data available about metformin affects on the endothelium among patients with T1D in whom increasing evidence suggests the deleterious effect of glycemic variability and fluctuations on CVD. The Reversing with Metformin Vascular Adverse Lesions (REMOVAL) trial, the largest and longest trial of metformin in T1D to date, has demonstrated that metformin does not reduce either the carotid intima-media thickness (IMT) or the reactive hyperemia index (RHI) after treatment with metformin for approximately three years [167]. However, among the uncomplicated subjects with T1D, metformin is shown to improve the flow mediated dilation (FMD) and the biomarker of oxidative stress (urinary 8-iso-prostaglandin F2α) after 6-month treatment, irrespective of its effects on glycemic control and BMI [160]. There was a cardioprotective effect of metformin for those without overt CVD through improving circulating EPCs, pro-angiogenic cells, and circulating endothelial cells count and function independently [171]. In addition, with the combination of metformin and the SGLT2i, empagliflozin, used in patients with T1D, arterial stiffness and endothelial function assessed by FMD and RHI were significantly improved in the combination therapy group compared to those on monotherapy [164].
Furthermore, metformin reduces the inflammatory biomarkers, such as vWF, sVCAM1, and sICAM1, among volunteers with impaired glucose tolerance [172]. Moreover, metformin may improve vascular function and decrease myocardial ischemia in nondiabetic women with chest pain and angiographically normal coronary arteries [173]. Among women with polycystic ovary syndrome (PCOS), those resulting from the critical pathophysiologic component contributing to hyperandrogenisms and the reproductive metabolic features with IR, conflicting results have been reported regarding the effects of metformin on endothelial function [174]. Metformin (850-1700 mg daily) therapy could reduce the baseline diameter of the brachial artery, FMD, IMT, and serum VEGFB after reactive hyperemia among women with PCOS, and even improve the FMD values, plasma ET-1 after 6-month treatment [175-179]. In one study which compared the characteristics among nonobese adolescents with PCOS and healthy age-matched volunteers, only the surrogate markers of cardiovascular risk such as blood pressure, levels of C-reactive protein (CRP), and PAI-1, whereas the deterioration of vascular structure and function has also been undetected even after 6-month treatment with metformin [180]. Metformin therapy reduces the high risk of cardiovascular events in pre-DM patients by reducing coronary ED with two-year therapy [181].
Pharmacological effects of metformin on endothelial function
Regulation of vascular tone
Impaired endothelium-dependent vasodilation is a common characteristic in arteries in both T1D and T2D. NO is the major vasodilator synthesized from its precursor L-arginine via eNOS [182]. Metformin (60 mg/kg/d) treatment improves endothelial functions in the T2D rat model (Goto-Kakizaki rats) and significantly improves NO bioavailability in rats [183]. Metformin (300 mg/kg/d) therapy does not effectively correct hyperglycemia; however, the reduction of vasodilation and total eNOS activity in a T1D rat model (streptozotocin, STZ-induced rats) can be corrected by metformin treatment [184]. In a 2006 study, AMPK was found to be necessary for metformin to enhance eNOS activation in vivo [35]. There is a positive correlation between the concentrations of metformin (50-500 mmol/L) and the levels of eNOS with serine-1179 (Ser1179) phosphorylation and heat shock protein 90 (HSP90), resulting in increased eNOS activation and NO bioactivity [35]. Henceforth, AMPK activation became one main recognised mechanisms of the vascular protective effects of metformin. Recently, the eNOS activating effect of metformin (250 mg/kg/d) on EPCs of STZ-induced diabetic mice has also been demonstrated [185].
In diabetes, part of the eNOS process produces superoxide anion instead of NO, and this is referred to as “eNOS uncoupling”. The decrease of endothelium-derived tetrahydrobiopterin (BH4, a vital cofactor for eNOS) promotes the uncoupling of eNOS [50, 51]. Metformin (300 mg/kg/d) treatment normalizes Ach-induced endothelial relaxation and increased GTP cyclohydrolase 1 (GCH1, the rate-limiting enzyme in BH4 biosynthesis) and BH4 levels [186] in both diabetic and wild type mice by slowing down the degradation of GCH1 via a post-translational mechanism [187]. Metformin (2 mmol/L, 200 mg/kg/d, respectively) may also inhibit NOX (p47-phox) via AMPK activation in HUVECs [188] and increase the phosphorylation of AMPKα and PARP1 in the aortas of spontaneously hypertensive rats [189]. Metformin (100-500 mg/kg/d) treatment increases NO bioavailability and attenuates high-sensitivity CRP (hCRP)-induced hypertension by activating of AMPK/peroxisome proliferator-activated receptor δ (PPARδ) pathway [190] and AMPK-eNOS phosphorylation pathway [191] in animal studies. Metformin (500 μM) also reduces p38 MAPK signal transduction through an AMPK dependent mechanism [192].
Metformin affects the expression of several vasoconstrictive molecules such as ET-1, AngII, and tissue factors to regulate vascular tone. In both clinical and basic research, metformin treatment reduces ET-1 levels in patients with PCOS (Diamanti-Kandarakis et al. 2001) and insulin-resistant human endothelial cells [193]. Metformin inhibits PKC membrane translocation and activity induced by AngII [194]. AngII stimulates vascular NOX and is activated through the AngII receptor AT1R-PKC pathway [195], and is closely related to vascular oxidative stress in ED under diabetic conditions [196]. In addition, by inhibiting vasoconstrictor prostanoids, metformin also improves the endothelial function of mesenteric arteries in a T2D rat model [197].
Inhibition of inflammation and leukocyte adhesion to endothelial cells
Metformin inhibits the pro-inflammatory changes induced by cytokines, which may be one mechanism for its vascular actions beyond the hypoglycemic effect. Like the AMPK activator AICAR, which can inhibit the increase in NF-κB reporter gene expression incubated with TNF-α in HUVECs [198], metformin (2-10 mM) is been found to inhibit inflammation through the AMPK-dependent IKK/IKBα/ NF-κB inhibitory pathway (Figure 3) [199]. Except for inhibition of NF-κB through blockade of the PI3K-Akt pathway in human saphenous vein endothelial cells [200], metformin also inhibits TNF-α induced production of Interleukin-6 (IL-6) [201] and several other inflammatory molecules responsible for monocyte adhesion to activated endothelial cells including VCAM1, MCP1, E-selectin, and ICAM1, by up-regulating B-cell lymphoma 6 (BCL6) and AMPK-induced phosphorylation of PARP1 at ser-177 [202], and the phosphorylation of histone deacetylase 5 (HDAC5) at serine 498 [203].
Protective effects of metformin against endothelial dysfunction and its molecular targets. Metformin improves endothelial dysfunction through the following mechanisms: 1) increasing NO production and inhibiting eNOS uncoupling, 2) inhibiting inflammation and leukocyte adhesion to endothelial cells, 3) inhibiting oxidative stress, 4) inhibiting endothelial cell senescence, 5) preventing endothelial cell death and apoptosis, 6) inhibition of EndoMT, 7) inhibition of endothelial permeability, 8) increasing differentiation of EPCs. These pharmacological effects of metformin were exerted through LKB1/AMPK and AMPK-independent targets. Abbreviations: AMPK, 5'-adenosine monophosphate-activated protein kinase; AGEs, advanced glycation end products; BCL6, B-cell lymphoma 6; BDNF, brain-derived neurotrophic factor; BH4, tetrahydrobiopterin; ChREBP, carbohydrate response element-binding protein; CREB, cyclic AMP response element binding; Ch25h, cholesterol-25-hydroxylase; DOT1L, Dot1-like protein; ER stress, endoplasmic reticulum stress; ERK, extracellular-signal-regulated kinase; eNOS, endothelial nitric oxide synthase; EPCs, endothelial progenitor cells; EndoMT, endothelial-to-mesenchymal transition; FOXO1, forkhead box O1; FOXO3, forkhead box O3; GCH1, Gtp cyclohydrolase 1; H3K79me3, histone H3 lysine 79 trimethylation; HSP90, heat shock protein 90; HDAC5-p, phosphorylation of histone deacetylase 5; HIF-1α, hypoxia inducible factor-1 alpha; ICAM1, intercellular adhesion molecule 1; IL-6, interleukin-6; LOX-1, lectin-like oxidized LDL receptor 1; KLF2, krüppel-like factor 2; KLF4, krüppel-like factor 4; LKB1, liver kinase B1; mTOR, mammalian target of rapamycin; MnSOD, manganese superoxide dismutase; MMPs, matrix metalloproteinases; mPTP, mitochondrial permeability transition pore; MAPK, mitogen-activated protein kinase; MCP1, monocyte chemoattractant protein 1; MiR, microRNA; NF-κB, nuclear factor-kappaB; NO, nitric oxide; NOX, NADPH oxidase; OGG1, 8-oxoguanine glycosylase 1; oxLDL, oxidized low-density lipoprotein; PPARδ, peroxisome proliferator-activated receptor δ; PGC1⍺, peroxisome proliferator-activated receptor-gamma coactivator 1⍺; PI3K, phosphatidylinositol 3 kinase; PARP1-p, phosphorylation of poly ADP-ribose polymerase 1; PFN1, profilin-1; PTEN, phosphatase and tensin homolog; ROS, reactive oxygen species; SIRT1, sirtuin 1; TRAF3IP2, TRAF3-interacting protein 2; TRX, thioredoxin; TXNIP, TRX-interacting protein; VCAM1, vascular cell adhesion molecule 1.
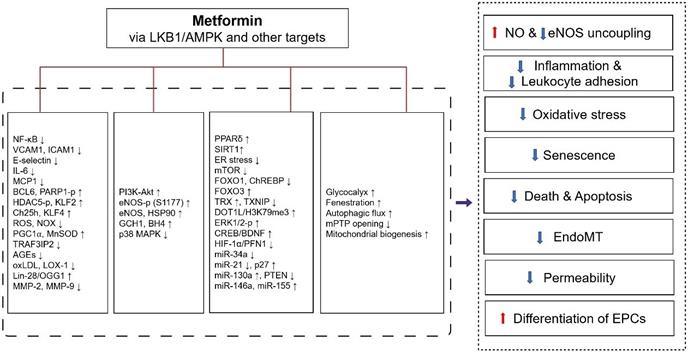
Treatment with metformin decreases retinal leukocyte adhesion [204], palmitic acid-induced monocyte adhesion [205], and reduces clinically relevant high levels of CRP-induced ED and hypertension [191]. Metformin (300-500 mg/kg/d) pretreatment also reduces ferrous chloride-induced thrombosis in carotid arteries [206]. Metformin can also reduces the uptake of oxLDL by endothelial cells and reduces subsequent inflammatory signals, thereby preventing macrophage adhesion and infiltration [207]. Additional mechanisms of metformin-stimulated autophagic flux [205] and krüppel-like factor 2 (KLF2) restoration [203, 208] partially contribute to the anti-inflammatory action of metformin in endothelial cells.
Inhibition of oxidative stress in endothelial cells
Metformin inhibits ROS production and reduces ROS levels in endothelial cells through multiple antioxidant mechanisms, including inhibiting NOX and stimulating catalase activity [209] and inhibiting lectin-like oxidized LDL receptor 1 (LOX-1) expression [210]. Other antioxidant mechanisms include reducing PKC membrane translocation and activity [194], increasing peroxisome proliferator-activated receptor-gamma coactivator 1α (PGC1α) [211] and thioredoxin (TRX) expression [212], and normalizing mitochondrial ROS production [211]. Some evidence suggests that PGC1α is an essential regulator of intracellular ROS levels. Metformin can induce PGC1α and manganese superoxide dismutase (MnSOD), resulting in inhibition of mitochondrial ROS [213] and increased TRX expression through activation of the AMPK-forkhead box O3 (FOXO3) pathway in human aortic endothelial cells (HAECs) [212]. In addition, metformin therapy inhibits endoplasmic reticulum stress (ER stress) and oxidative stress on activation of the AMPK/PPARδ pathway in obese diabetic mice [190]. ER stress plays a key role in progression of diabetes and development of complications, especially CVD [214]. While oxLDL is a product of chronic oxidative stress which generate pro-oxidant effects by inducing ROS generation, metformin (20 μM) decreases ROS levels by inhibiting LOX-1 expression and increasing Akt/eNOS levels [210].
Besides its effect on oxidative stress, metformin also reduces nitroxidative stress. The treatment of metformin (300 mg/kg/d) is observed to increase the production of NO by 37% and 57% in aortic and glomerular endothelial cells of experimental rats, respectively, while decreasing ONOO (-) (cytotoxic peroxynitrite) by 32% and 34%, compared with controlled animals [215]. Moreover, as the main DNA glycosylase, 8-oxoguanine glycosylase 1 (OGG1) can resist ROS and is involved in various vascular diseases. The level of OGG1 increases under metformin (0.5 mM) treatment through the AMPK/Lin-28/OGG1 pathway [216]. The anti-oxidative function of metformin (10 μM-1 mM) also relates to inhibition of TRAF3-interacting protein 2 (TRAF3IP2), which is a redox-sensitive cytoplasmic adapter protein functions upstream of IKK/NF-κB and c-Jun N-terminal kinase (JNK)/AP-1 (Activator protein-1) [217].
Inhibition of endothelial cell senescence
Endothelial cell senescence leads to ED and contributes to the progression of age-associated vascular disorders [218]. Endothelial cell senescence is characterized by cell cycle arrest and pro-inflammatory gene expression [219-221] and is driven by many factors, including high glucose, ROS, inflammatory cytokines, ionizing radiation, and telomere dysfunction [222, 223]. High glucose induces endothelial cell senescence by inhibiting sirtuin 1 (SIRT1) expression, which can be attenuated with metformin (50-250 μM) treatment by modulating the SIRT1 downstream targets forkhead box O1 (FOXO1) and p53/p21 [224, 225]. In ApoE-/- mice, metformin (50 mg/kg/d) therapy significantly reduces vascular aging and inhibits atherosclerotic plaque formation through AMPK activation leading to SIRT1/Dot1-like protein (DOT1L)/ histone H3 lysine 79 trimethylation (H3K79me3)-induced upregulation of sirtuin 3 (SIRT3) levels [226]. Metformin increases fenestrations in liver sinusoidal endothelial cells isolated form old and young mice [227]. Calcification is a vascular disease marker and a prognostic factor of CVD, which is related to the aging of the vascular system [228]. It is speculated that metformin, acting via AMPK dependent mechanisms, may be a potential target for the treatment of vascular calcification [229]. Indeed, clinical trials have been designed to assess the potential benefits of metformin as an anti-aging drug that enhances healthspan and extends lifespan [230]. The beneficial effects of metformin on aging and healthspan are primarily indirect via its effects on cellular metabolism and result from its anti-hyperglycemic action, enhancing insulin sensitivity, reduction of oxidative stress and protective effects on the endothelium and vascular function [230].
Inhibition of endothelial cell death and apoptosis
Metformin (100 μM) prevents endothelial cell death by inhibiting mitochondrial permeability transition pore (mPTP), which is a mitochondrial channel involved in cell death [231]. Metformin partly protects endothelial cells from dysfunction and apoptosis via reducing p38 MAPK phosphorylation [192], inhibiting the mammalian target of rapamycin (mTOR) pathway [232], promoting the PI3K-Akt and phospho-ERK 1/2 pathways [233], and down regulation of TRX-interacting protein (TXNIP) transcription by inactivating carbohydrate response element-binding protein (ChREBP) and FOXO1 [234] in endothelial cells. Metformin (0.01-1.0 mmol/L) also inhibits endothelial cell apoptosis via upregulation of VEGF receptors, fatty acid binding protein 4 (FABP4) [235] and the AMPK/ cyclic AMP response element binding (CREB)/brain-derived neurotrophic factor (BDNF) pathway [236]. Metformin (100 μmol/L or 10 mmol/L) can also inhibit mPTP (a sensitive channel) opening in three endothelial cell types, including HMEC-1, HUVECs, and bovine aortic endothelial cells (BAECs), from preventing hyperglycemia-induced cell death [231].
Inhibition of EndoMT
EndoMT is a driver event in the pathogenesis of atherosclerosis, diabetes associated myocardial remodeling, pulmonary hypertension and others. A study in 2018 explored the effects of different flow patterns on the EndoMT in endothelial cells, indicating that pharmacological activation of AMPK or SIRT1 could attenuate oscillatory shear stress (OS)-induced EndoMT [237]. The beneficial therapeutic effect of metformin on atherosclerosis may be mediated at least in part by inhibiting the EndoMT in the vasculature. The expression of Krüppel-like factor 4 (KLF4), a major transcription factor showing endothelial protection and maintenance of vascular homeostasis, and cholesterol-25-hydroxylase (Ch25h) decreases in high glucose level [238], while metformin (200 mg/kg/d) can increase the expression of KLF4 and Ch25h to suppress EndoMT [239].
Inhibition of the permeability of the endothelium
The increased permeability of the endothelium is accompanied by inflammation-mediated reversible cell rounding and inter-endothelial gap formation [240] and high glucose-activated PKC pathway [241]. Metformin (0.33 mg/ml, two weeks) treatment is associated with improved glycocalyx barrier properties and indicates reduced vascular permeability in mice [242]. Metformin (400 mg/kg/d, 100 mg/kg/d, respectively) administration is associated with reduced lung endothelial hyperpermeability and systemic inflammatory response in STZ-induced diabetic mice [243] and alleviates microvascular ED by suppressing hypoxia inducible factor-1 alpha (HIF-1α)/Profilin-1 (PFN1) signaling, which mediates endothelial cell permeability during diabetic retinopathy [244]. Moderate doses of metformin (0.1-1.0 mM) are shown to produce an increase in the trans-endothelial resistance of endothelial monolayers and enhance the vascular barrier integrity [245]. By activation of AMPK signaling pathway and regulation of oxidant/anti-oxidant balance, metformin abrogates the disruptive effect of polyphosphate which induce hyper-permeability and inflammatory responses through mTOR pathway in endothelial cells [246].
Increased differentiation of endothelial progenitor cells (EPCs)
By restoring damaged endothelial cells, EPCs represent new targets for treating vascular diseases [247, 248]. EPCs, originating from bone marrow [249] and circulating in peripheral blood, have the ability of endothelial differentiation and secretion of angiogenic growth factors and cytokines [250]. Metformin treatment significantly increases EPC differentiation in vitro (1 mM) [251] and in diabetic mice (250 mg/kg/d) [185]. As to mechanism, increased levels of AMPK and eNOS phosphorylation [185], light chain3 (LC3) expression and NO production, and decreased mTOR, p70 S6K, TGF-β expression [251] and MMP-2 and MMP-9 expression [252] are found in EPCs. In addition, the effects of metformin on improving EPC dysfunction are mediated by several miRNAs, such as miR‑130a/ phosphatase and tensin homolog (PTEN) [253] and miR-221/p27 [254].
Future directions
Epigenetic effects of metformin
MicroRNAs (miRNAs) are highly conserved, small, and noncoding RNAs involved in the post-transcriptional regulation of gene expression. MiRNAs participate in regulating endothelial function and dysfunction. Diabetes and hyperlipidemia-induced inflammatory responses upregulate the expression of connexins and Rho kinase by selective downregulation of miR-10a, miR-139b, miR-206, and miR-222 [116]. A study in 2006 found that metformin (50 μM), or the inhibition of miR-34a by treatment with an anti-miR-34a inhibitor, increased the expression of SIRT1 and attenuated the impairment in angiogenesis in high glucose-exposed mouse microvascular endothelial cells (MMECs) [255]. In metformin (0-10 mM) or miR-221 siRNA-treated EPCs, AMPK inhibition decreased the expression of p27 and AMPK-mediated autophagic activity [254]. In contrast, upregulation of miR‑130a [253], miR-146a and miR-155 [256] via the treatment of metformin (50 μM, 200 mg/kg/d) is observed in PA‑exposed EPCs and endothelium, respectively. Giuliani et al. [257] investigated the miRNA landscape in endothelial cells subject to replicative senescence after a long-term application with metformin (20 μM), and found the differential expression of 27 miRNAs (including miR-100-5p, -125b-5p, -654-3p, -217 and -216a-3p/5p).
Long noncoding RNAs (lncRNAs) represent a family of non-protein-coding transcripts (>200 nucleotides), which occupy the majority of the human genome [258], while circular RNAs (circRNAs) are a large category of noncoding RNAs widely expressed in eukaryotic cells [259]. Growing evidence suggests that lncRNAs [260-263] and circular RNAs [264-268] regulate endothelial functions, while the role of lncRNAs and circular RNAs in the protective effect of metformin on endothelial cells remains largely unclear.
Metformin on endothelial cell metabolism
Evidence has demonstrated the importance of endothelial cells in the maintenance of homeostasis across the entire vascular system. The initiation and progression of atherosclerosis may originate from the disorder of endothelium intracellular metabolism that can be detected at the earliest stages of developing the syndrome [269]. Endothelial cell metabolism is important for maintaining metabolic health, such as glucose, lipid and amino acid, as they have been most extensively studied [270]. The study identifies a metabolic program that promotes the acquisition of a quiescent endothelial state and emphasizes the role of metabolites as signaling molecules in endothelial cells [271]. The activity and migration of endothelial cells decrease when the enzymes related to glucose metabolisms, such as G6P dehydrogenase and transketolase, are inhibited [272]. Previous studies showed that metformin affects FABP4 mediated endothelial fatty acid metabolism [273, 274]. Endothelial cells internalize chylomicrons, which trigger lipid accumulation in aortic of lipoprotein lipase-deficient mice [275]. Recently, metformin (100 μM) is shown to reduce saturated fatty acid-induced lipid accumulation and inflammatory response by restoring autophagic flux in endothelial cells [205]. In addition, metformin (1-5 mM) takes part in regulation of hexose transport [276], reversing glucose starvation-induced ER stress [277] in endothelial cells. Metformin, GLP-1RAs, and SGLT2i are the only drugs screened in seven classes of anti-hyperglycemic drugs which reduce ER stress caused by pharmacological or hyperglycemic conditions in human coronary artery endothelial cells (HCAECs) [214]. In endothelial cells arginine, glutamine, leucine and valine play a role in the metabolism of endothelial cells [278-281].
Metformin on mitochondrial dynamics
Mitochondria are critical integrators that participate in signal transduction, energy production, ROS generation, and cell apoptosis. Recent studies have highlighted the importance of mitochondrial dynamics, especially mitochondrial fusion and fission in mitochondrial homeostasis [282]. In endothelial cells, more fragmented structures caused by fission are associated with mitochondrial dysfunction, contributing to ED with an unclear mechanism [283]. Mitochondrial division is triggered when Dynamin-related protein 1 (DRP1), a cytosolic guanosine-5'-triphosphatase, binds with fission 1 (FIS1) or mitochondrial fission factor (MFF) on mitochondria [282]. Metformin (300 mg/kg/d) is observed to suppress the progression of atherosclerosis through the inhibition of DRP1-mediated mitochondrial fission in STZ-induced diabetic ApoE-/- mice [284]. In addition to inhibiting mitochondrial division, promoting mitochondrial fusion may also benefit endothelial cell function and represent a potential therapeutic approach for diabetes patients with CVD, which is an area that needs further study. In terms of the efficacy of metformin, studies have also shown that metformin (0.01-2 mmol/L) can promote mitochondrial biogenesis by activating AMPK and inducing PGC1α in HUVECs [213].
Therapeutic potential of metformin in COVID-19 patients with diabetes
Coronavirus disease 19 (COVID-19), caused by the severe acute respiratory syndrome-coronavirus 2 (SARS-CoV-2), is associated with multiple factors, such as cytokine storm, hyperactive inflammatory and immune responses, and coagulation disorders [285]. Among COVID-19 patients with diabetes, the intensive care hospitalization rate and mortality rate are two to three times higher than that of patients without diabetes [286]. Diabetes has been identified as an independent risk factor for poor prognosis of COVID-19 [287, 288]. The underlying pathogenic links between COVID-19 and diabetes include effects on inflammation, glucose homeostasis, immune status changes and renin angiotensin aldosterone system (RAAS) activation [289]. Metformin treatment is associated with reduced mortality in COVID-19 patients with diabetes [290-292]. Patients who used metformin before hospitalization also showed lower severity parameters at admission [293]. During SARS-CoV-2 infection, in addition to controlling the glucose level, metformin may attenuate ED, inhibit viral entry and infection, resist oxidative stress, and change inflammatory and immune responses that would explain its ability to achieve cardiovascular protection in COVID-19 [294, 295]. Therefore, metformin may represent a drug candidate for treating COVID-19 and gain the advantage of fighting SARS-CoV-2 induced immune storm in COVID-19 patients with diabetes [296].
Metformin on other transcriptional factors
Recent data has shown that islet ED in db/db mice was evident compared with non-diabetic mice [297]. Metformin/empagliflozin treatment (6 weeks) could reduce the expression of dysfunction marker genes and increase insulin release in vivo [297]. It is speculated that metformin may improve the blood glucose level of T2D patients by enhancing the function of islet endothelial cells. Metformin (100 mg/kg/d) can also promote the development of pulmonary vessels and up regulate the expression of glioma-associated oncogene homolog 1 (Gli1) in pulmonary vascular endothelial cells of hyperoxia newborn mice [298].
Conclusion
Similar as SGLT2i and GLP-1RAs, numerous clinical studies have shown that metformin can improve vascular endothelial functions and reduce CVD in patients with diabetes. Mechanistically, metformin improves ED through LKB1/AMPK and AMPK-independent targets. In addition to several common mechanisms, including increasing NO production, inhibiting inflammation and oxidative stress, metformin can protect endothelial cell functions and the intact endothelium by reducing EndoMT and increasing the differentiation ability of EPCs. Based on the protective effects of metformin on blood vessels summarized in these basic and clinical studies, we suggest that metformin is also worthy of attention for potential treatments for CVD and other panvascular diseases in which ED plays a fundamental role.
Abbreviations
Ach: Acetylcholine; ADP: Adenosine Diphosphate; AMPK: 5'-Adenosine Monophosphate-activated Protein Kinase; AP-1: Activator Protein-1; ATP: Adenosine Triphosphate; AGEs: Advanced Glycation End Products; AngⅡ: AngiotensinⅡ; ADMA: Asymmetric Dimethylarginine; BCL6: B-Cell Lymphoma 6; BDNF: Brain-Derived Neurotrophic Factor; BH4: Tetrahydrobiopterin; BMI: Body Mass Index; BAECs: Bovine Aortic Endothelial Cells; CGRP: Calcitonin Gene-related Peptide; ChREBP: Carbohydrate Response Element-binding Protein; CO: Carbon Monoxide; CREB: Cyclic AMP Response Element Binding; CVD: Cardiovascular Disease; Ch25h: Cholesterol-25-Hydroxylase; CRP: C-Reactive Protein; DNA: Deoxyribonucleic Acid; DOT1L: Dot1-like Protein; ED: Endothelial Dysfunction; ER stress: Endoplasmic Reticulum Stress; ERK: Extracellular-signal-regulated Kinase; eNOS: Endothelial Nitric Oxide Synthase; EPCs: Endothelial Progenitor Cells; EndoMT: Endothelial-To-Mesenchymal Transition; ET-1: Endothelin 1; FABP4: Fatty Acid Binding Protein 4; FBF: Forearm Blood Flow; FMD: Flow Mediated Dilation; FOXO1: Forkhead Box O1; FOXO3: Forkhead Box O3; FOXO4: Forkhead Box O4; GAPDH: Glyceraldehyde Phosphate Dehydrogenase; Gli1: Glioma-associated Oncogene Homolog 1; GCH1: Gtp Cyclohydrolase 1; GLP-1RAs: Glucagon-like Peptide-1 Receptor Agonists; H3K79me3: Histone H3 Lysine 79 Trimethylation; HSP90: Heat Shock Protein 90; hCRP: High-Sensitivity CRP; HDAC5: Histone Deacetylase 5; HAECs: Human Aortic Endothelial Cells; HCAECs: Human Coronary Artery Endothelial Cells; HIF-1α: Hypoxia Inducible Factor-1 Alpha; HUVECs: Human Umbilical Vein Endothelial Cells; H2S: Hydrogen Sulfide; IR: Insulin Resistance; ICAM1: Intercellular Adhesion Molecule 1; IHD: Ischemic Heart Disease; IL-1β: Interleukin-1Beta; IL-6: Interleukin-6; IMT: Intima-Media Thickness; JNK: C-Jun N-terminal Kinase; KLF2: Krüppel-Like Factor 2; KLF4: Krüppel-Like Factor 4; LC3: Light Chain3; LKB1: Liver Kinase B1; LncRNAs: Long Noncoding RNAs; LOX-1: Lectin-like Oxidized LDL Receptor 1; MGO: Methylglyoxal; mTOR: Mammalian Target Of Rapamycin; MnSOD: Manganese Superoxide Dismutase; MMPs: Matrix Metalloproteinases; mPTP: Mitochondrial Permeability Transition Pore; MAPK: Mitogen-Activated Protein Kinase; MCP1: Monocyte Chemoattractant Protein 1; MiRNAs: MicroRNAs; MMECs: Mouse Microvascular Endothelial Cells; NADPH: Nicotinamide Adenine Dinucleotide Phosphate; NE: Norepinephrine; NF-κB: Nuclear Factor-KappaB; NMD: Nitroglycerin-Mediated Dilation; NO: Nitric Oxide; NOD2: Nucleotide-Binding Oligomerization Domain-Containing Protein 2; NOX: NADPH Oxidase; NPY: Neuropeptide Y; OGG1: 8-Oxoguanine Glycosylase 1; OS: Oscillatory Shear Stress; oxLDL: Oxidized Low-Density Lipoprotein; PFN1: Profilin-1; PPARδ: Peroxisome Proliferator-Activated Receptor δ; PGC1⍺: Peroxisome Proliferator-Activated Receptor-Gamma Coactivator 1α; PI3K: Phosphatidylinositol 3 Kinase; PA: Plasminogen Activator; PAI: Plasminogen Activator Inhibitor; PARP: Poly ADP-ribose Polymerase; PCOS: Polycystic Ovary Syndrome; PlGF: Placenta Growth Factor; PTEN: Phosphatase And Tensin Homolog; PKB, also known as Akt: Protein Kinase B; PKC: Protein Kinase C; RHI: Reactive Hyperemia Index; ROI: Reactive Oxygen Intermediates; ROS: Reactive Oxygen Species; RAGE: Receptor For AGEs; ROCK: Rho-Associated Coiled-Coil Kinase; SGLT2i: Sodium Glucose Cotransporter 2 inhibitors; SIRT1: Sirtuin 1; SIRT3: Sirtuin 3; STEMI: ST-segment Elevation Myocardial Infarction; STZ: Streptozotocin; TRAF3IP2: TRAF3-Interacting Protein 2; TRX: Thioredoxin; TXA2: Thromboxane A2; TIMPs: Tissue Inhibitors Of Metalloproteinases; t-PA: Tissue-PA; TGF-β: Transforming Growth Factor-Β; TXNIP: TRX-Interacting Protein; TNF-α: Tumor Necrosis Factor-Α; T1D: Type 1 Diabetes; T2D: Type 2 Diabetes; UKPDS: UK Prospective Diabetes Study; u-PA: Urokinase-PA; VCAM1: Vascular Cell Adhesion Molecule 1; VEGF: Vascular Endothelial Growth Factor; VSMCs: Vascular Smooth Muscle Cells; vWF: Von Willebrand Factor.
Acknowledgements
Funding
This study was supported by grants from National Natural Science Foundation of China (Grant No. 81941022, 81530025, 82070464) and Strategic Priority Research Program of Chinese Academy of Sciences (Grant No. XDB38010100). This work was also supported by Program for Innovative Research Team of The First Affiliated Hospital of USTC (CXGG02), Anhui Provincial Key Research and Development Program (Grant No. 202104j07020051), Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (Grant No. 2017BT01S131), Hefei Comprehensive National Science Center (Grant No. BJ9100000005), and Hefei Municipal Development and Reform Commission Emergency Funding for COVID-19 disease.
Author contributions
J.W. and S.X. conceived the project and guided the study. Y.D. wrote the manuscript. Y.Z., P.L., X.F., S.L., X.Z. and P.J.L. provided insightful comments on vascular biology and on the structure and revisions of the manuscript. All authors read and revised the manuscript and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gu K, Cowie CC, Harris MI. Diabetes and decline in heart disease mortality in US adults. JAMA. 1999;281(14):1291-7
2. Anna-Maria K, Dimitris T, Alexandros B, George L, Nikolaos P, Christodoulos S. Potential pathogenic inflammatory mechanisms of endothelial dysfunction induced by type 2 diabetes mellitus. Curr Pharm Des. 2011;17(37):4147-58
3. Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N. et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the international diabetes federation diabetes atlas, 9th edition. Diabetes Res Clin Pract. 2019;157:107843
4. Yamaguchi A, Ino T, Adachi H, Murata S, Kamio H, Okada M. et al. Left ventricular volume predicts postoperative course in patients with ischemic cardiomyopathy. Ann Thorac Surg. 1998;65(2):434-8
5. Stratton IM, Adler AI, Neil HAW, Matthews DR, Manley SE, Cull CA. et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. 2000;321:405-12
6. Sardu C, Consiglia Trotta M, Santella B, D'Onofrio N, Barbieri M, Rizzo MR. et al. Microbiota thrombus colonization may influence athero-thrombosis in hyperglycemic patients with ST segment elevation myocardialinfarction (STEMI). Marianella study. Diabetes Res Clin Pract. 2021;173:108670
7. D'Onofrio N, Sardu C, Paolisso P, Minicucci F, Gragnano F, Ferraraccio F. et al. MicroRNA-33 and SIRT1 influence the coronary thrombus burden in hyperglycemic STEMI patients. J Cell Physiol. 2020;235(2):1438-52
8. Sardu C, Barbieri M, Balestrieri ML, Siniscalchi M, Paolisso P, Calabrò P. et al. Thrombus aspiration in hyperglycemic ST-elevation myocardial infarction (STEMI) patients: clinical outcomes at 1-year follow-up. Cardiovasc Diabetol. 2018;17(1):152
9. Sardu C, D'Onofrio N, Mauro C, Balestrieri ML, Marfella R. Thrombus aspiration in hyperglycemic patients with high inflammation levels in coronary thrombus. J Am Coll Cardiol. 2019;73(4):530-31
10. Sardu C, D'Onofrio N, Torella M, Portoghese M, Loreni F, Mureddu S. et al. Pericoronary fat inflammation and major adverse cardiac events (MACE) in prediabetic patients with acute myocardial infarction: effects of metformin. Cardiovasc Diabetol. 2019;18(1):126
11. Love KM, Barrett EJ, Malin SK, Reusch JEB, Regensteiner JG, Liu Z. Diabetes pathogenesis and management: the endothelium comes of age. J Mol Cell Biol. 2021 doi: 10.1093/jmcb/mjab024. Online ahead of print
12. Maruhashi T, Higashi Y. Pathophysiological association between diabetes mellitus and endothelial dysfunction. Antioxidants (Basel). 2021;10(8):1306
13. Kaur R, Kaur M, Singh J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: molecular insights and therapeutic strategies. Cardiovasc Diabetol. 2018;17(1):121
14. Tian K, Ogura S, Little PJ, Xu SW, Sawamura T. Targeting LOX-1 in atherosclerosis and vasculopathy: current knowledge and future perspectives. Ann N Y Acad Sci. 2019;1443(1):34-53
15. Xu S, Ogura S, Chen J, Little PJ, Moss J, Liu P. LOX-1 in atherosclerosis: biological functions and pharmacological modifiers. Cell Mol Life Sci. 2013;70(16):2859-72
16. Wang D, Yang Y, Lei Y, Tzvetkov NT, Liu X, Yeung AWK. et al. Targeting foam cell formation in atherosclerosis: therapeutic potential of natural products. Pharmacol Rev. 2019;71(4):596-670
17. Frostegård J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013;11:117
18. Ross R, Glomset J, Harker L. Response to injury and atherogenesis. Am J Pathol. 1977;86(3):675-84
19. Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113(13):1708-14
20. Zhang C. The role of inflammatory cytokines in endothelial dysfunction. Basic Res Cardiol. 2008;103(5):398-406
21. Lunder M, Janić M, Šabovič M. Prevention of vascular complications in diabetes mellitus patients: focus on the arterial wall. Curr Vasc Pharmacol. 2019;17(1):6-15
22. Prior JO, Quiñones MJ, Hernandez-Pampaloni M, Facta AD, Schindler TH, Sayre JW. et al. Coronary circulatory dysfunction in insulin resistance, impaired glucose tolerance, and type 2 diabetes mellitus. Circulation. 2005;111(18):2291-8
23. Schächinger V, Britten MB, Zeiher AM. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation. 2000;101(16):1899-906
24. Suwaidi JA, Hamasaki S, Higano ST, Nishimura RA, Holmes DR Jr, Lerman A. Long-term follow-up of patients with mild coronary artery disease and endothelial dysfunction. Circulation. 2000;101(9):948-54
25. Sasso FC, Pafundi PC, Marfella R, Calabrò P, Piscione F, Furbatto F. et al. Adiponectin and insulin resistance are related to restenosis and overall new PCI in subjects with normal glucose tolerance: the prospective AIRE study. Cardiovasc Diabetol. 2019;18(1):24
26. Goraya TY, Leibson CL, Palumbo PJ, Weston SA, Killian JM, Pfeifer EA. et al. Coronary atherosclerosis in diabetes mellitus: a population-based autopsy study. J Am Coll Cardiol. 2002;40(5):946-53
27. Rask-Madsen C, King GL. Mechanisms of disease: endothelial dysfunction in insulin resistance and diabetes. Nat Clin Pract Endocrinol Metab. 2007;3(1):46-56
28. American Diabetes A. 2. Classification and diagnosis of diabetes: standards of medical care in diabetes-2021. Diabetes Care. 2021;44(Suppl 1):S15-S33
29. Yang SN, Burch ML, Tannock LR, Evanko S, Osman N, Little PJ. Transforming growth factor-β regulation of proteoglycan synthesis in vascular smooth muscle: contribution to lipid binding and accelerated atherosclerosis in diabetes. J Diabetes. 2010;2(4):233-42
30. Nigro J, Osman N, Dart AM, Little PJ. Insulin resistance and atherosclerosis. Endocr Rev. 2006;27(3):242-59
31. Eckel RH, Bornfeldt KE, Goldberg IJ. Cardiovascular disease in diabetes, beyond glucose. Cell Metab. 2021;33(8):1519-1545
32. Anabtawi A, Miles JM. Metformin: nonglycemic effects and potential novel indications. Endocr Pract. 2016;22(8):999-1007
33. Fujita Y, Inagaki N. Metformin: new preparations and nonglycemic benefits. Curr Diab Rep. 2017;17(1):5
34. UKPDS. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33).UK Prospective Diabetes Study (UKPDS) Group Lancet. 1998;352(9131):837-53
35. Davis BJ, Xie Z, Viollet B, Zou MH. Activation of the AMP-activated kinase by antidiabetes drug metformin stimulates nitric oxide synthesis in vivo by promoting the association of heat shock protein 90 and endothelial nitric oxide synthase. Diabetes. 2006;55(2):496-505
36. Ma X, Liu Z, Ilyas I, Little PJ, Kamato D, Sahebka A. et al. GLP-1 receptor agonists (GLP-1RAs): cardiovascular actions and therapeutic potential. Int J Biol Sci. 2021;17(8):2050-68
37. Liu Z, Ma X, Ilyas I, Zheng X, Luo S, Little PJ. et al. Impact of sodium glucose cotransporter 2 (SGLT2) inhibitors on atherosclerosis: from pharmacology to pre-clinical and clinical therapeutics. Theranostics. 2021;11(9):4502-15
38. Marfella R, Sardu C, Balestrieri ML, Siniscalchi M, Minicucci F, Signoriello G. et al. Effects of incretin treatment on cardiovascular outcomes in diabetic STEMI-patients with culprit obstructive and multivessel non obstructive-coronary-stenosis. Diabetol Metab Syndr. 2018;10:1
39. Marfella R, Sardu C, Calabrò P, Siniscalchi M, Minicucci F, Signoriello G. et al. Non-ST-elevation myocardial infarction outcomes in patients with type 2 diabetes with non-obstructive coronary artery stenosis: Effects of incretin treatment. Diabetes Obes Metab. 2018;20(3):723-29
40. Vanhoutte PM. Endothelial dysfunction: the first step toward coronary arteriosclerosis. Circ J. 2009;73(4):595-601
41. Tennant M, McGeachie JK. Blood vessel structure and function: a brief update on recent advances. Aust N Z J Surg. 1990;60(10):747-53
42. Stevens T, Garcia JG, Shasby DM, Bhattacharya J, Malik AB. Mechanisms regulating endothelial cell barrier function. Am J Physiol Lung Cell Mol Physiol. 2000;279(3):L419-22
43. Xu S, Ilyas I, Little PJ, Li H, Kamato D, Zheng X. et al. Endothelial dysfunction in atherosclerotic cardiovascular diseases and beyond: from mechanism to pharmacotherapies. Pharmacol Rev. 2021;73(3):924-67
44. Galley HF, Webster NR. Physiology of the endothelium. Br J Anaesth. 2004;93(1):105-13
45. Krüger-Genge A, Blocki A, Franke RP, Jung F. Vascular endothelial cell biology: an update. Int J Mol Sci. 2019;20(18):4411
46. Reitsma S, Slaaf DW, Vink H, van Zandvoort MA, oude Egbrink MG. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. 2007;454(3):345-59
47. Becker BF, Chappell D, Bruegger D, Annecke T, Jacob M. Therapeutic strategies targeting the endothelial glycocalyx: acute deficits, but great potential. Cardiovasc Res. 2010;87(2):300-10
48. Denninger JW, Marletta MA. Guanylate cyclase and the.NO/cGMP signaling pathway. Biochim Biophys Acta. 1999;1411(2-3):334-50
49. Stamler JS, Simon DI, Osborne JA, Mullins ME, Jaraki O, Michel T. et al. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci U S A. 1992;89(1):444-8
50. Laursen JB, Somers M, Kurz S, McCann L, Warnholtz A, Freeman BA. et al. Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation. 2001;103(9):1282-8
51. Yang YM, Huang A, Kaley G, Sun D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am J Physiol Heart Circ Physiol. 2009;297(5):H1829-36
52. Gheibi S, Jeddi S, Kashfi K, Ghasemi A. Regulation of vascular tone homeostasis by NO and H(2)S: Implications in hypertension. Biochem Pharmacol. 2018;149:42-59
53. Cirino G, Vellecco V, Bucci M. Nitric oxide and hydrogen sulfide: the gasotransmitter paradigm of the vascular system. Br J Pharmacol. 2017;174(22):4021-31
54. Citi V, Martelli A, Gorica E, Brogi S, Testai L, Calderone V. Role of hydrogen sulfide in endothelial dysfunction: pathophysiology and therapeutic approaches. J Adv Res. 2021;27:99-113
55. Cediel E, Vázquez-Cruz B, Navarro-Cid J, de las Heras N, Sanz-Rosa D, Cachofeiro V. et al. Role of endothelin-1 and thromboxane A2 in renal vasoconstriction induced by angiotensin II in diabetes and hypertension. Kidney Int Suppl. 2002(82):S2-7.
56. Sheng Y, Zhu L. The crosstalk between autonomic nervous system and blood vessels. Int J Physiol Pathophysiol Pharmacol. 2018;10(1):17-28
57. Conti V, Russomanno G, Corbi G, Izzo V, Vecchione C, Filippelli A. Adrenoreceptors and nitric oxide in the cardiovascular system. Front Physiol. 2013;4:321
58. Deng Q, Huo Y, Luo J. Endothelial mechanosensors: the gatekeepers of vascular homeostasis and adaptation under mechanical stress. Sci China Life Sci. 2014;57(8):755-62
59. Chatterjee S, Fisher AB. Mechanotransduction in the endothelium: role of membrane proteins and reactive oxygen species in sensing, transduction, and transmission of the signal with altered blood flow. Antioxid Redox Signal. 2014;20(6):899-913
60. Niu N, Xu S, Xu Y, Little PJ, Jin ZG. Targeting mechanosensitive transcription factors in atherosclerosis. Trends Pharmacol Sci. 2019;40(4):253-66
61. Krenning G, Barauna VG, Krieger JE, Harmsen MC, Moonen JR. Endothelial plasticity: shifting phenotypes through force feedback. Stem Cells Int. 2016;2016:9762959
62. Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation. 2007;115(10):1285-95
63. Op den Buijs J, Musters M, Verrips T, Post JA, Braam B, van Riel N. Mathematical modeling of vascular endothelial layer maintenance: the role of endothelial cell division, progenitor cell homing, and telomere shortening. Am J Physiol Heart Circ Physiol. 2004;287(6):H2651-8
64. Valon L, Davidović A, Levillayer F, Villars A, Chouly M, Cerqueira-Campos F. et al. Robustness of epithelial sealing is an emerging property of local ERK feedback driven by cell elimination. Dev Cell. 2021;56(12):1700-11.e8
65. Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T. et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275(5302):964-7
66. Ramasamy I. Inherited bleeding disorders: disorders of platelet adhesion and aggregation. Crit Rev Oncol Hematol. 2004;49(1):1-35
67. Lijnen HR, Collen D. Endothelium in hemostasis and thrombosis. Prog Cardiovasc Dis. 1997;39(4):343-50
68. Mackman N, Tilley RE, Key NS. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2007;27(8):1687-93
69. Lamalice L, Le Boeuf F, Huot J. Endothelial cell migration during angiogenesis. Circ Res. 2007;100(6):782-94
70. Golestani R, Jung JJ, Sadeghi MM. Molecular imaging of angiogenesis and vascular remodeling in cardiovascular pathology. J Clin Med. 2016;5(6):57
71. Li X, Sun X, Carmeliet P. Hallmarks of endothelial cell metabolism in health and disease. Cell Metab. 2019;30(3):414-33
72. Funk SD, Yurdagul A Jr, Orr AW. Hyperglycemia and endothelial dysfunction in atherosclerosis: lessons from type 1 diabetes. Int J Vasc Med. 2012;2012:569654
73. Price KD, Price CS, Reynolds RD. Hyperglycemia-induced ascorbic acid deficiency promotes endothelial dysfunction and the development of atherosclerosis. Atherosclerosis. 2001;158(1):1-12
74. Ceriello A, Taboga C, Tonutti L, Quagliaro L, Piconi L, Bais B. et al. Evidence for an independent and cumulative effect of postprandial hypertriglyceridemia and hyperglycemia on endothelial dysfunction and oxidative stress generation: effects of short- and long-term simvastatin treatment. Circulation. 2002;106(10):1211-8
75. Varma S, Lal BK, Zheng R, Breslin JW, Saito S, Pappas PJ. et al. Hyperglycemia alters PI3k and Akt signaling and leads to endothelial cell proliferative dysfunction. Am J Physiol Heart Circ Physiol. 2005;289(4):H1744-51
76. Nieuwdorp M, van Haeften TW, Gouverneur MC, Mooij HL, van Lieshout MH, Levi M. et al. Loss of endothelial glycocalyx during acute hyperglycemia coincides with endothelial dysfunction and coagulation activation in vivo. Diabetes. 2006;55(2):480-6
77. Hamilton SJ, Chew GT, Watts GF. Therapeutic regulation of endothelial dysfunction in type 2 diabetes mellitus. Diab Vasc Dis Res. 2007;4(2):89-102
78. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107(9):1058-70
79. Naruse K, Rask-Madsen C, Takahara N, Ha SW, Suzuma K, Way KJ. et al. Activation of vascular protein kinase C-beta inhibits Akt-dependent endothelial nitric oxide synthase function in obesity-associated insulin resistance. Diabetes. 2006;55(3):691-8
80. Chen S, Apostolova MD, Cherian MG, Chakrabarti S. Interaction of endothelin-1 with vasoactive factors in mediating glucose-induced increased permeability in endothelial cells. Lab Invest. 2000;80(8):1311-21
81. Meier M, King GL. Protein kinase C activation and its pharmacological inhibition in vascular disease. Vasc Med. 2000;5(3):173-85
82. Torimoto K, Okada Y, Mori H, Tanaka Y. Relationship between fluctuations in glucose levels measured by continuous glucose monitoring and vascular endothelial dysfunction in type 2 diabetes mellitus. Cardiovasc Diabetol. 2013;12:1
83. La Sala L, Pujadas G, De Nigris V, Canivell S, Novials A, Genovese S. et al. Oscillating glucose and constant high glucose induce endoglin expression in endothelial cells: the role of oxidative stress. Acta Diabetol. 2015;52(3):505-12
84. Sissaoui S, Egginton S, Ting L, Ahmed A, Hewett PW. Hyperglycaemia up-regulates placental growth factor (PlGF) expression and secretion in endothelial cells via suppression of PI3 kinase-Akt signalling and activation of FOXO1. Sci Rep. 2021;11(1):16344
85. Silva C, Rodrigues I, Andrade S, Costa R, Soares R. Metformin reduces vascular assembly in high glucose-treated human microvascular endothelial cells in an AMPK-independent manner. Cell J. 2021;23(2):174-83
86. Oeseburg H, Iusuf D, van der Harst P, van Gilst WH, Henning RH, Roks AJ. Bradykinin protects against oxidative stress-induced endothelial cell senescence. Hypertension. 2009;53(2):417-22
87. Aoki M, Nata T, Morishita R, Matsushita H, Nakagami H, Yamamoto K. et al. Endothelial apoptosis induced by oxidative stress through activation of NF-kappaB: antiapoptotic effect of antioxidant agents on endothelial cells. Hypertension. 2001;38(1):48-55
88. Thuan DTB, Zayed H, Eid AH, Abou-Saleh H, Nasrallah GK, Mangoni AA. et al. A potential link between oxidative stress and endothelial-to-mesenchymal transition in systemic sclerosis. Front Immunol. 2018;9:1985
89. Du XL, Edelstein D, Rossetti L, Fantus IG, Goldberg H, Ziyadeh F. et al. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc Natl Acad Sci U S A. 2000;97(22):12222-6
90. Hamuro M, Polan J, Natarajan M, Mohan S. High glucose induced nuclear factor kappa B mediated inhibition of endothelial cell migration. Atherosclerosis. 2002;162(2):277-87
91. Du XL, Edelstein D, Dimmeler S, Ju Q, Sui C, Brownlee M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J Clin Invest. 2001;108(9):1341-8
92. Guan Q, Liu W, Liu Y, Fan Y, Wang X, Yu C. et al. High glucose induces the release of endothelin-1 through the inhibition of hydrogen sulfide production in HUVECs. Int J Mol Med. 2015;35(3):810-4
93. Ushiyama A, Kataoka H, Iijima T. Glycocalyx and its involvement in clinical pathophysiologies. J Intensive Care. 2016;4(1):59
94. Sena CM, Matafome P, Crisóstomo J, Rodrigues L, Fernandes R, Pereira P. et al. Methylglyoxal promotes oxidative stress and endothelial dysfunction. Pharmacol Res. 2012;65(5):497-506
95. Dhar A, Dhar I, Desai KM, Wu L. Methylglyoxal scavengers attenuate endothelial dysfunction induced by methylglyoxal and high concentrations of glucose. Br J Pharmacol. 2010;161(8):1843-56
96. Pieper GM, Meier DA, Hager SR. Endothelial dysfunction in a model of hyperglycemia and hyperinsulinemia. Am J Physiol. 1995;269(3 Pt 2):H845-50
97. Pinkney JH, Stehouwer CD, Coppack SW, Yudkin JS. Endothelial dysfunction: cause of the insulin resistance syndrome. Diabetes. 1997;46(Suppl 2):S9-13
98. Miller AW, Hoenig ME, Ujhelyi MR. Mechanisms of impaired endothelial function associated with insulin resistance. J Cardiovasc Pharmacol Ther. 1998;3(2):125-34
99. McCarty MF. AMPK activation as a strategy for reversing the endothelial lipotoxicity underlying the increased vascular risk associated with insulin resistance syndrome. Med Hypotheses. 2005;64(6):1211-5
100. Imrie H, Abbas A, Kearney M. Insulin resistance, lipotoxicity and endothelial dysfunction. Biochim Biophys Acta. 2010;1801(3):320-6
101. Del Turco S, Gaggini M, Daniele G, Basta G, Folli F, Sicari R. et al. Insulin resistance and endothelial dysfunction: a mutual relationship in cardiometabolic risk. Curr Pharm Des. 2013;19(13):2420-31
102. Muniyappa R, Iantorno M, Quon MJ. An integrated view of insulin resistance and endothelial dysfunction. Endocrinol Metab Clin North Am. 2008;37(3):685-711 ix-x
103. Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399(6736):601-5
104. Montagnani M, Golovchenko I, Kim I, Koh GY, Goalstone ML, Mundhekar AN. et al. Inhibition of phosphatidylinositol 3-kinase enhances mitogenic actions of insulin in endothelial cells. J Biol Chem. 2002;277(3):1794-9
105. Prieto D, Contreras C, Sanchez A. Endothelial dysfunction, obesity and insulin resistance. Curr Vasc Pharmacol. 2014;12(3):412-26
106. Tallapragada DS, Karpe PA, Tikoo K. Long-lasting partnership between insulin resistance and endothelial dysfunction: role of metabolic memory. Br J Pharmacol. 2015;172(16):4012-23
107. Mao H, Li L, Fan Q, Angelini A, Saha PK, Wu H. et al. Loss of bone morphogenetic protein-binding endothelial regulator causes insulin resistance. Nat Commun. 2021;12(1):1927
108. Marx N, Walcher D, Ivanova N, Rautzenberg K, Jung A, Friedl R. et al. Thiazolidinediones reduce endothelial expression of receptors for advanced glycation end products. Diabetes. 2004;53(10):2662-8
109. Schmidt AM, Yan SD, Wautier JL, Stern D. Activation of receptor for advanced glycation end products: a mechanism for chronic vascular dysfunction in diabetic vasculopathy and atherosclerosis. Circ Res. 1999;84(5):489-97
110. An XF, Zhou L, Jiang PJ, Yan M, Huang YJ, Zhang SN. et al. Advanced glycation end-products induce heparanase expression in endothelial cells by the receptor for advanced glycation end products and through activation of the FOXO4 transcription factor. Mol Cell Biochem. 2011;354(1-2):47-55
111. Tan KC, Chow WS, Ai VH, Metz C, Bucala R, Lam KS. Advanced glycation end products and endothelial dysfunction in type 2 diabetes. Diabetes Care. 2002;25(6):1055-9
112. Rojas A, Romay S, Gonzalez D, Herrera B, Delgado R, Otero K. Regulation of endothelial nitric oxide synthase expression by albumin-derived advanced glycosylation end products. Circ Res. 2000;86(3):E50-4
113. Ren X, Ren L, Wei Q, Shao H, Chen L, Liu N. Advanced glycation end-products decreases expression of endothelial nitric oxide synthase through oxidative stress in human coronary artery endothelial cells. Cardiovasc Diabetol. 2017;16(1):52
114. Mentink CJ, Kilhovd BK, Rondas-Colbers GJ, Torjesen PA, Wolffenbuttel BH. Time course of specific AGEs during optimised glycaemic control in type 2 diabetes. Neth J Med. 2006;64(1):10-6
115. Silambarasan M, Tan JR, Karolina DS, Armugam A, Kaur C, Jeyaseelan K. MicroRNAs in hyperglycemia induced endothelial cell dysfunction. Int J Mol Sci. 2016;17(4):518
116. Li T, Yang GM, Zhu Y, Wu Y, Chen XY, Lan D. et al. Diabetes and hyperlipidemia induce dysfunction of VSMCs: contribution of the metabolic inflammation/miRNA pathway. Am J Physiol Endocrinol Metab. 2015;308(4):E257-69
117. Xu E, Hu X, Li X, Jin G, Zhuang L, Wang Q. et al. Analysis of long non-coding RNA expression profiles in high-glucose treated vascular endothelial cells. BMC Endocr Disord. 2020;20(1):107
118. Shao J, Zhang Y, Fan G, Xin Y, Yao Y. Transcriptome analysis identified a novel 3-LncRNA regulatory network of transthyretin attenuating glucose induced hRECs dysfunction in diabetic retinopathy. BMC Med Genomics. 2019;12(1):134
119. Wu XD, Liu WL, Zeng K, Lei HY, Zhang QG, Zhou SQ. et al. Advanced glycation end products activate the miRNA/RhoA/ROCK2 pathway in endothelial cells. Microcirculation. 2014;21(2):178-86
120. Rodrigo R, Gonzalez J, Paoletto F. The role of oxidative stress in the pathophysiology of hypertension. Hypertens Res. 2011;34(4):431-40
121. Higashi Y, Noma K, Yoshizumi M, Kihara Y. Endothelial function and oxidative stress in cardiovascular diseases. Circ J. 2009;73(3):411-8
122. Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87(10):840-4
123. Chan NN, Chan JC. Asymmetric dimethylarginine (ADMA): a potential link between endothelial dysfunction and cardiovascular diseases in insulin resistance syndrome? Diabetologia. 2002;45(12):1609-16
124. Incalza MA, D'Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul Pharmacol. 2018;100:1-19
125. Ray R, Shah AM. NADPH oxidase and endothelial cell function. Clin Sci (Lond). 2005;109(3):217-26
126. Rueckschloss U, Duerrschmidt N, Morawietz H. NADPH oxidase in endothelial cells: impact on atherosclerosis. Antioxid Redox Signal. 2003;5(2):171-80
127. Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552(Pt 2):335-44
128. Victor VM, Rocha M, Sola E, Banuls C, Garcia-Malpartida K, Hernandez-Mijares A. Oxidative stress, endothelial dysfunction and atherosclerosis. Curr Pharm Des. 2009;15(26):2988-3002
129. Pizzino G, Irrera N, Cucinotta M, Pallio G, Mannino F, Arcoraci V. et al. Oxidative stress: harms and benefits for human health. Oxid Med Cell Longev. 2017;2017:8416763
130. Sun H, Lu X, Wu S, Sun W. The effects of C-reactive protein, interleukin-6, and tumor necrosis factor-alpha in rat allograft adventitial inflammation and allograft arteriosclerosis. Transplant Proc. 2009;41(9):3909-12
131. Kume N, Cybulsky MI, Gimbrone MA Jr. Lysophosphatidylcholine, a component of atherogenic lipoproteins, induces mononuclear leukocyte adhesion molecules in cultured human and rabbit arterial endothelial cells. J Clin Invest. 1992;90(3):1138-44
132. Beekhuizen H, Verdegaal EM, Blokland I, van Furth R. Contribution of ICAM-1 and VCAM-1 to the morphological changes in monocytes bound to human venous endothelial cells stimulated with recombinant interleukin-4 (rIL-4) or rIL-1 alpha. Immunology. 1992;77(3):469-72
133. True AL, Rahman A, Malik AB. Activation of NF-kappaB induced by H(2)O(2) and TNF-alpha and its effects on ICAM-1 expression in endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2000;279(2):L302-11
134. Renkonen R, Mennander A, Ustinov J, Mattila P. Activation of protein kinase C is crucial in the regulation of ICAM-1 expression on endothelial cells by interferon-gamma. Int Immunol. 1990;2(8):719-24
135. Kim I, Moon SO, Kim SH, Kim HJ, Koh YS, Koh GY. Vascular endothelial growth factor expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin through nuclear factor-kappa B activation in endothelial cells. J Biol Chem. 2001;276(10):7614-20
136. Sardu C, Paolisso G, Marfella R. Impact of sex differences in incident and recurrent coronary events and all-cause mortality. J Am Coll Cardiol. 2021;77(6):829-30
137. Sardu C, Gatta G, Pieretti G, Viola L, Sacra C, Di Grezia G. et al. Pre-menopausal breast fat density might predict MACE during 10 years of follow-up: the BRECARD study. JACC Cardiovasc Imaging. 2021;14(2):426-38
138. D'Onofrio N, Pieretti G, Ciccarelli F, Gambardella A, Passariello N, Rizzo MR. et al. Abdominal fat SIRT6 expression and its relationship with inflammatory and metabolic pathways in pre-diabetic overweight patients. Int J Mol Sci. 2019;20(5):1153
139. Sardu C, Pieretti G, D'Onofrio N, Ciccarelli F, Paolisso P, Passavanti MB. et al. Inflammatory cytokines and SIRT1 levels in subcutaneous abdominal fat: relationship with cardiac performance in overweight pre-diabetics patients. Front Physiol. 2018;9:1030
140. Bhatt MP, Lim YC, Kim YM, Ha KS. C-peptide activates AMPKα and prevents ROS-mediated mitochondrial fission and endothelial apoptosis in diabetes. Diabetes. 2013;62(11):3851-62
141. Yu Y, Lyons TJ. A lethal tetrad in diabetes: hyperglycemia, dyslipidemia, oxidative stress, and endothelial dysfunction. Am J Med Sci. 2005;330(5):227-32
142. Piera-Velazquez S, Li Z, Jimenez SA. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am J Pathol. 2011;179(3):1074-80
143. Peng H, Li Y, Wang C, Zhang J, Chen Y, Chen W. et al. ROCK1 induces endothelial-to-mesenchymal transition in glomeruli to aggravate albuminuria in diabetic nephropathy. Sci Rep. 2016;6:20304
144. Shang J, Zhang Y, Jiang Y, Li Z, Duan Y, Wang L. et al. NOD2 promotes endothelial-to-mesenchymal transition of glomerular endothelial cells via MEK/ERK signaling pathway in diabetic nephropathy. Biochem Biophys Res Commun. 2017;484(2):435-41
145. Pérez L, Muñoz-Durango N, Riedel CA, Echeverría C, Kalergis AM, Cabello-Verrugio C. et al. Endothelial-to-mesenchymal transition: cytokine-mediated pathways that determine endothelial fibrosis under inflammatory conditions. Cytokine Growth Factor Rev. 2017;33:41-54
146. Good RB, Gilbane AJ, Trinder SL, Denton CP, Coghlan G, Abraham DJ. et al. Endothelial to mesenchymal transition contributes to endothelial dysfunction in pulmonary arterial hypertension. Am J Pathol. 2015;185(7):1850-8
147. Cho JG, Lee A, Chang W, Lee MS, Kim J. Endothelial to mesenchymal transition represents a key link in the interaction between inflammation and endothelial dysfunction. Front Immunol. 2018;9:294
148. Hu J, Zheng Z, Li X, Li B, Lai X, Li N. et al. Metformin attenuates hypoxia-induced endothelial cell injury by activating the AMP-activated protein kinase pathway. J Cardiovasc Pharmacol. 2021;77(6):862-74
149. Lin FY, Lin YW, Shih CM, Lin SJ, Tung YT, Li CY. et al. A novel relative high-density lipoprotein index to predict the structural changes in high-density lipoprotein and its ability to inhibit endothelial-mesenchymal transition. Int J Mol Sci. 2021;22(10):5210
150. Giordo R, Ahmed YMA, Allam H, Abusnana S, Pappalardo L, Nasrallah GK. et al. EndMT regulation by small RNAs in diabetes-associated fibrotic conditions: potential link with oxidative stress. Front Cell Dev Biol. 2021;9:683594
151. Wiernsperger NF, Bailey CJ. The antihyperglycaemic effect of metformin: therapeutic and cellular mechanisms. Drugs. 1999;58(Suppl 1):31-9 discussion 75-82
152. Mather KJ, Verma S, Anderson TJ. Improved endothelial function with metformin in type 2 diabetes mellitus. J Am Coll Cardiol. 2001;37(5):1344-50
153. Abbasi F, Chu JW, McLaughlin T, Lamendola C, Leary ET, Reaven GM. Effect of metformin treatment on multiple cardiovascular disease risk factors in patients with type 2 diabetes mellitus. Metabolism. 2004;53(2):159-64
154. De Jager J, Kooy A, Lehert P, Bets D, Wulffelé MG, Teerlink T. et al. Effects of short-term treatment with metformin on markers of endothelial function and inflammatory activity in type 2 diabetes mellitus: a randomized, placebo-controlled trial. J Intern Med. 2005;257(1):100-9
155. Skrha J, Prázný M, Hilgertová J, Kvasnicka J, Kalousová M, Zima T. Oxidative stress and endothelium influenced by metformin in type 2 diabetes mellitus. Eur J Clin Pharmacol. 2007;63(12):1107-14
156. Ersoy C, Kiyici S, Budak F, Oral B, Guclu M, Duran C. et al. The effect of metformin treatment on VEGF and PAI-1 levels in obese type 2 diabetic patients. Diabetes Res Clin Pract. 2008;81(1):56-60
157. Lund SS, Tarnow L, Stehouwer CD, Schalkwijk CG, Teerlink T, Gram J. et al. Impact of metformin versus repaglinide on non-glycaemic cardiovascular risk markers related to inflammation and endothelial dysfunction in non-obese patients with type 2 diabetes. Eur J Endocrinol. 2008;158(5):631-41
158. Tousoulis D, Koniari K, Antoniades C, Miliou A, Noutsou M, Nikolopoulou A. et al. Impact of 6 weeks of treatment with low-dose metformin and atorvastatin on glucose-induced changes of endothelial function in adults with newly diagnosed type 2 diabetes mellitus: A single-blind study. Clin Ther. 2010;32(10):1720-8
159. Fidan E, Onder Ersoz H, Yilmaz M, Yilmaz H, Kocak M, Karahan C. et al. The effects of rosiglitazone and metformin on inflammation and endothelial dysfunction in patients with type 2 diabetes mellitus. Acta Diabetol. 2011;48(4):297-302
160. Pitocco D, Zaccardi F, Tarzia P, Milo M, Scavone G, Rizzo P. et al. Metformin improves endothelial function in type 1 diabetic subjects: a pilot, placebo-controlled randomized study. Diabetes Obes Metab. 2013;15(5):427-31
161. de Jager J, Kooy A, Schalkwijk C, van der Kolk J, Lehert P, Bets D. et al. Long-term effects of metformin on endothelial function in type 2 diabetes: a randomized controlled trial. J Intern Med. 2014;275(1):59-70
162. Shigiyama F, Kumashiro N, Miyagi M, Iga R, Kobayashi Y, Kanda E. et al. Linagliptin improves endothelial function in patients with type 2 diabetes: A randomized study of linagliptin effectiveness on endothelial function. J Diabetes Investig. 2017;8(3):330-40
163. Kitao N, Miyoshi H, Furumoto T, Ono K, Nomoto H, Miya A. et al. The effects of vildagliptin compared with metformin on vascular endothelial function and metabolic parameters: a randomized, controlled trial (Sapporo Athero-Incretin Study 3). Cardiovasc Diabetol. 2017;16(1):125
164. Lunder M, Janić M, Japelj M, Juretič A, Janež A, Šabovič M. Empagliflozin on top of metformin treatment improves arterial function in patients with type 1 diabetes mellitus. Cardiovasc Diabetol. 2018;17(1):153
165. Schiapaccassa A, Maranhão PA, de Souza M, Panazzolo DG, Nogueira Neto JF, Bouskela E. et al. 30-days effects of vildagliptin on vascular function, plasma viscosity, inflammation, oxidative stress, and intestinal peptides on drug-naïve women with diabetes and obesity: a randomized head-to-head metformin-controlled study. Diabetol Metab Syndr. 2019;11:70
166. Kruszelnicka O, Chyrchel B, Golay A, Surdacki A. Differential associations of circulating asymmetric dimethylarginine and cell adhesion molecules with metformin use in patients with type 2 diabetes mellitus and stable coronary artery disease. Amino Acids. 2015;47(9):1951-9
167. Petrie JR, Chaturvedi N, Ford I, Brouwers M, Greenlaw N, Tillin T. et al. Cardiovascular and metabolic effects of metformin in patients with type 1 diabetes (REMOVAL): a double-blind, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2017;5(8):597-609
168. Cubero-Gallego H, Romaguera R, Gómez-Lara J, Gómez-Hospital JA, Sabaté M, Pinar E. et al. In vivo evaluation of the synergic effect of metformin and mTOR inhibitors on the endothelial healing of drug-eluting stents in diabetic patients. Rev Esp Cardiol (Engl Ed). 2018;71(11):917-25
169. Natali A, Baldeweg S, Toschi E, Capaldo B, Barbaro D, Gastaldelli A. et al. Vascular effects of improving metabolic control with metformin or rosiglitazone in type 2 diabetes. Diabetes Care. 2004;27(6):1349-57
170. Krysiak R, Gdula-Dymek A, Okopień B. Lymphocyte-suppressing, endothelial-protective and systemic anti-inflammatory effects of metformin in fenofibrate-treated patients with impaired glucose tolerance. Pharmacol Rep. 2013;65(2):429-34
171. Ahmed FW, Rider R, Glanville M, Narayanan K, Razvi S, Weaver JU. Metformin improves circulating endothelial cells and endothelial progenitor cells in type 1 diabetes: MERIT study. Cardiovasc Diabetol. 2016;15(1):116
172. Caballero AE, Delgado A, Aguilar-Salinas CA, Herrera AN, Castillo JL, Cabrera T. et al. The differential effects of metformin on markers of endothelial activation and inflammation in subjects with impaired glucose tolerance: a placebo-controlled, randomized clinical trial. J Clin Endocrinol Metab. 2004;89(8):3943-8
173. Jadhav S, Ferrell W, Greer IA, Petrie JR, Cobbe SM, Sattar N. Effects of metformin on microvascular function and exercise tolerance in women with angina and normal coronary arteries: a randomized, double-blind, placebo-controlled study. J Am Coll Cardiol. 2006;48(5):956-63
174. Heidari B, Lerman A, Lalia AZ, Lerman LO, Chang AY. Effect of metformin on microvascular endothelial function in polycystic ovary syndrome. Mayo Clin Proc. 2019;94(12):2455-66
175. Naka KK, Kalantaridou SN, Kravariti M, Bechlioulis A, Kazakos N, Calis KA. et al. Effect of the insulin sensitizers metformin and pioglitazone on endothelial function in young women with polycystic ovary syndrome: a prospective randomized study. Fertil Steril. 2011;95(1):203-9
176. Orio F Jr, Palomba S, Cascella T, De Simone B, Manguso F, Savastano S. et al. Improvement in endothelial structure and function after metformin treatment in young normal-weight women with polycystic ovary syndrome: results of a 6-month study. J Clin Endocrinol Metab. 2005;90(11):6072-6
177. Diamanti-Kandarakis E, Alexandraki K, Protogerou A, Piperi C, Papamichael C, Aessopos A. et al. Metformin administration improves endothelial function in women with polycystic ovary syndrome. Eur J Endocrinol. 2005;152(5):749-56
178. Romualdi D, Costantini B, Selvaggi L, Giuliani M, Cristello F, Macrì F. et al. Metformin improves endothelial function in normoinsulinemic PCOS patients: a new prospective. Hum Reprod. 2008;23(9):2127-33
179. Diamanti-Kandarakis E, Spina G, Kouli C, Migdalis I. Increased endothelin-1 levels in women with polycystic ovary syndrome and the beneficial effect of metformin therapy. J Clin Endocrinol Metab. 2001;86(10):4666-73
180. Fruzzetti F, Ghiadoni L, Virdis A, De Negri F, Perini D, Bucci F. et al. Adolescents with classical polycystic ovary syndrome have alterations in the surrogate markers of cardiovascular disease but not in the endothelial function. The possible benefits of metformin. J Pediatr Adolesc Gynecol. 2016;29(5):489-95
181. Sardu C, Paolisso P, Sacra C, Mauro C, Minicucci F, Portoghese M. et al. Effects of metformin therapy on coronary endothelial dysfunction in patients with prediabetes with stable angina and nonobstructive coronary artery stenosis: The CODYCE multicenter prospective study. Diabetes Care. 2019;42(10):1946-55
182. Nishida K, Harrison DG, Navas JP, Fisher AA, Dockery SP, Uematsu M. et al. Molecular cloning and characterization of the constitutive bovine aortic endothelial cell nitric oxide synthase. J Clin Invest. 1992;90(5):2092-6
183. Sena CM, Matafome P, Louro T, Nunes E, Fernandes R, Seiça RM. Metformin restores endothelial function in aorta of diabetic rats. Br J Pharmacol. 2011;163(2):424-37
184. Sartoretto JL, Melo GA, Carvalho MH, Nigro D, Passaglia RT, Scavone C. et al. Metformin treatment restores the altered microvascular reactivity in neonatal streptozotocin-induced diabetic rats increasing NOS activity, but not NOS expression. Life Sci. 2005;77(21):2676-89
185. Yu JW, Deng YP, Han X, Ren GF, Cai J, Jiang GJ. Metformin improves the angiogenic functions of endothelial progenitor cells via activating AMPK/eNOS pathway in diabetic mice. Cardiovasc Diabetol. 2016;15:88
186. Wang S, Xu J, Song P, Viollet B, Zou MH. In vivo activation of AMP-activated protein kinase attenuates diabetes-enhanced degradation of GTP cyclohydrolase I. Diabetes. 2009;58(8):1893-901
187. Kidokoro K, Satoh M, Channon KM, Yada T, Sasaki T, Kashihara N. Maintenance of endothelial guanosine triphosphate cyclohydrolase I ameliorates diabetic nephropathy. J Am Soc Nephrol. 2013;24(7):1139-50
188. An H, Wei R, Ke J, Yang J, Liu Y, Wang X. et al. Metformin attenuates fluctuating glucose-induced endothelial dysfunction through enhancing GTPCH1-mediated eNOS recoupling and inhibiting NADPH oxidase. J Diabetes Complications. 2016;30(6):1017-24
189. Shang F, Zhang J, Li Z, Zhang J, Yin Y, Wang Y. et al. Cardiovascular protective effect of metformin and telmisartan: reduction of PARP1 activity via the AMPK-PARP1 cascade. PLoS One. 2016;11(3):e0151845
190. Cheang WS, Tian XY, Wong WT, Lau CW, Lee SS, Chen ZY. et al. Metformin protects endothelial function in diet-induced obese mice by inhibition of endoplasmic reticulum stress through 5' adenosine monophosphate-activated protein kinase-peroxisome proliferator-activated receptor δ pathway. Arterioscler Thromb Vasc Biol. 2014;34(4):830-6
191. Cheng L, Wang L, Guo M, He J, Deng Y, Liu J. et al. Clinically relevant high levels of human C-reactive protein induces endothelial dysfunction and hypertension by inhibiting the AMPK-eNOS axis. Clin Sci (Lond). 2020;134(13):1805-19
192. Eriksson L, Nyström T. Activation of AMP-activated protein kinase by metformin protects human coronary artery endothelial cells against diabetic lipoapoptosis. Cardiovasc Diabetol. 2014;13:152
193. Chen H, Li J, Yang O, Kong J, Lin G. Effect of metformin on insulin-resistant endothelial cell function. Oncol Lett. 2015;9(3):1149-53
194. Mahrouf M, Ouslimani N, Peynet J, Djelidi R, Couturier M, Therond P. et al. Metformin reduces angiotensin-mediated intracellular production of reactive oxygen species in endothelial cells through the inhibition of protein kinase C. Biochem Pharmacol. 2006;72(2):176-83
195. Mollnau H, Wendt M, Szocs K, Lassegue B, Schulz E, Oelze M. et al. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ Res. 2002;90(4):E58-65
196. Booth G, Stalker TJ, Lefer AM, Scalia R. Mechanisms of amelioration of glucose-induced endothelial dysfunction following inhibition of protein kinase C in vivo. Diabetes. 2002;51(5):1556-64
197. Matsumoto T, Noguchi E, Ishida K, Kobayashi T, Yamada N, Kamata K. Metformin normalizes endothelial function by suppressing vasoconstrictor prostanoids in mesenteric arteries from OLETF rats, a model of type 2 diabetes. Am J Physiol Heart Circ Physiol. 2008;295(3):H1165-h76
198. Cacicedo JM, Yagihashi N, Keaney JF Jr, Ruderman NB, Ido Y. AMPK inhibits fatty acid-induced increases in NF-kappaB transactivation in cultured human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2004;324(4):1204-9
199. Hattori Y, Suzuki K, Hattori S, Kasai K. Metformin inhibits cytokine-induced nuclear factor kappaB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension. 2006;47(6):1183-8
200. Isoda K, Young JL, Zirlik A, MacFarlane LA, Tsuboi N, Gerdes N. et al. Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells. Arterioscler Thromb Vasc Biol. 2006;26(3):611-7
201. Huang NL, Chiang SH, Hsueh CH, Liang YJ, Chen YJ, Lai LP. Metformin inhibits TNF-alpha-induced IkappaB kinase phosphorylation, IkappaB-alpha degradation and IL-6 production in endothelial cells through PI3K-dependent AMPK phosphorylation. Int J Cardiol. 2009;134(2):169-75
202. Gongol B, Marin T, Peng IC, Woo B, Martin M, King S. et al. AMPKα2 exerts its anti-inflammatory effects through PARP-1 and Bcl-6. Proc Natl Acad Sci U S A. 2013;110(8):3161-6
203. Tian R, Li R, Liu Y, Liu J, Pan T, Zhang R. et al. Metformin ameliorates endotoxemia-induced endothelial pro-inflammatory responses via AMPK-dependent mediation of HDAC5 and KLF2. Biochim Biophys Acta Mol Basis Dis. 2019;1865(6):1701-12
204. Han J, Li Y, Liu X, Zhou T, Sun H, Edwards P. et al. Metformin suppresses retinal angiogenesis and inflammation in vitro and in vivo. PLoS One. 2018;13(3):e0193031
205. Kim HS, Ren G, Kim T, Bhatnagar S, Yang Q, Bahk YY. et al. Metformin reduces saturated fatty acid-induced lipid accumulation and inflammatory response by restoration of autophagic flux in endothelial cells. Sci Rep. 2020;10(1):13523
206. Ghatak SB, Dhamecha PS, Bhadada SV, Panchal SJ. Investigation of the potential effects of metformin on atherothrombotic risk factors in hyperlipidemic rats. Eur J Pharmacol. 2011;659(2-3):213-23
207. Ahmadi A, Panahi Y, Johnston TP, Sahebkar A. Antidiabetic drugs and oxidized low-density lipoprotein: A review of anti-atherosclerotic mechanisms. Pharmacol Res. 2021;172:105819
208. Wu H, Feng K, Zhang C, Zhang H, Zhang J, Hua Y. et al. Metformin attenuates atherosclerosis and plaque vulnerability by upregulating KLF2-mediated autophagy in apoE(-/-) mice. Biochem Biophys Res Commun. 2021;557:334-41
209. Gallo A, Ceolotto G, Pinton P, Iori E, Murphy E, Rutter GA. et al. Metformin prevents glucose-induced protein kinase C-beta2 activation in human umbilical vein endothelial cells through an antioxidant mechanism. Diabetes. 2005;54(4):1123-31
210. Hung CH, Chan SH, Chu PM, Lin HC, Tsai KL. Metformin regulates oxLDL-facilitated endothelial dysfunction by modulation of SIRT1 through repressing LOX-1-modulated oxidative signaling. Oncotarget. 2016;7(10):10773-87
211. Kim HJ, Park KG, Yoo EK, Kim YH, Kim YN, Kim HS. et al. Effects of PGC-1alpha on TNF-alpha-induced MCP-1 and VCAM-1 expression and NF-kappaB activation in human aortic smooth muscle and endothelial cells. Antioxid Redox Signal. 2007;9(3):301-7
212. Hou X, Song J, Li XN, Zhang L, Wang X, Chen L. et al. Metformin reduces intracellular reactive oxygen species levels by upregulating expression of the antioxidant thioredoxin via the AMPK-FOXO3 pathway. Biochem Biophys Res Commun. 2010;396(2):199-205
213. Kukidome D, Nishikawa T, Sonoda K, Imoto K, Fujisawa K, Yano M. et al. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes. 2006;55(1):120-7
214. Kapadia P, Bikkina P, Landicho MA, Parekh S, Haas MJ, Mooradian AD. Effect of anti-hyperglycemic drugs on endoplasmic reticulum (ER) stress in human coronary artery endothelial cells. Eur J Pharmacol. 2021;907:174249
215. Sambe T, Mason RP, Dawoud H, Bhatt DL, Malinski T. Metformin treatment decreases nitroxidative stress, restores nitric oxide bioavailability and endothelial function beyond glucose control. Biomed Pharmacother. 2018;98:149-56
216. Tao L, Fan X, Sun J, Zhang Z. Metformin prevented high glucose-induced endothelial reactive oxygen species via OGG1 in an AMPKα-Lin-28 dependent pathway. Life Sci. 2021;268:119015
217. Valente AJ, Irimpen AM, Siebenlist U, Chandrasekar B. OxLDL induces endothelial dysfunction and death via TRAF3IP2: inhibition by HDL3 and AMPK activators. Free Radic Biol Med. 2014;70:117-28
218. Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation. 2002;105(13):1541-4
219. Erusalimsky JD. Vascular endothelial senescence: from mechanisms to pathophysiology. J Appl Physiol (1985). 2009;106(1):326-32
220. Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192(4):547-56
221. Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res. 2012;111(2):245-59
222. Valenzuela MT, Nunez MI, Villalobos M, Siles E, McMillan TJ, Pedraza V. et al. A comparison of p53 and p16 expression in human tumor cells treated with hyperthermia or ionizing radiation. Int J Cancer. 1997;72(2):307-12
223. Vasto S, Candore G, Balistreri CR, Caruso M, Colonna-Romano G, Grimaldi MP. et al. Inflammatory networks in ageing, age-related diseases and longevity. Mech Ageing Dev. 2007;128(1):83-91
224. Arunachalam G, Samuel SM, Marei I, Ding H, Triggle CR. Metformin modulates hyperglycaemia-induced endothelial senescence and apoptosis through SIRT1. Br J Pharmacol. 2014;171(2):523-35
225. Zhang E, Guo Q, Gao H, Xu R, Teng S, Wu Y. Metformin and resveratrol inhibited high glucose-induced metabolic memory of endothelial senescence through SIRT1/p300/p53/p21 pathway. PLoS One. 2015;10(12):e0143814
226. Karnewar S, Neeli PK, Panuganti D, Kotagiri S, Mallappa S, Jain N. et al. Metformin regulates mitochondrial biogenesis and senescence through AMPK mediated H3K79 methylation: relevance in age-associated vascular dysfunction. Biochim Biophys Acta Mol Basis Dis. 2018;1864(4 Pt A):1115-28
227. Hunt NJ, Lockwood GP, Kang SWS, Pulpitel T, Clark X, Mao H. et al. The effects of metformin on age-related changes in the liver sinusoidal endothelial cell. J Gerontol A Biol Sci Med Sci. 2020;75(2):278-85
228. Radford NB, DeFina LF, Barlow CE, Lakoski SG, Leonard D, Paixao AR. et al. Progression of CAC score and risk of incident CVD. JACC Cardiovasc Imaging. 2016;9(12):1420-29
229. Lu Y, Yuan T, Min X, Yuan Z, Cai Z. AMPK: potential therapeutic target for vascular calcification. Front Cardiovasc Med. 2021;8:670222
230. Mohammed I, Hollenberg MD, Ding H, Triggle CR. A critical review of the evidence that metformin is a putative anti-aging drug that enhances healthspan and extends lifespan. Front Endocrinol (Lausanne). 2021;12:718942
231. Detaille D, Guigas B, Chauvin C, Batandier C, Fontaine E, Wiernsperger N. et al. Metformin prevents high-glucose-induced endothelial cell death through a mitochondrial permeability transition-dependent process. Diabetes. 2005;54(7):2179-87
232. Laouirem S, Sannier A, Norkowski E, Cauchy F, Doblas S, Rautou PE. et al. Endothelial fatty liver binding protein 4: a new targetable mediator in hepatocellular carcinoma related to metabolic syndrome. Oncogene. 2019;38(16):3033-46
233. Xu XX, Zhang SS, Lin HL, Lin Q, Shen LE, Ansong E. et al. Metformin promotes regeneration of the injured endometrium via inhibition of endoplasmic reticulum stress-induced apoptosis. Reprod Sci. 2019;26(4):560-68
234. Li X, Kover KL, Heruth DP, Watkins DJ, Moore WV, Jackson K. et al. New insight into metformin action: regulation of ChREBP and FOXO1 activities in endothelial cells. Mol Endocrinol. 2015;29(8):1184-94
235. Bakhashab S, Ahmed F, Schulten HJ, Ahmed FW, Glanville M, Al-Qahtani MH. et al. Proangiogenic effect of metformin in endothelial cells is via upregulation of VEGFR1/2 and their signaling under hyperglycemia-hypoxia. Int J Mol Sci. 2018;19(1):293
236. Han X, Wang B, Sun Y, Huang J, Wang X, Ma W. et al. Metformin modulates high glucose-incubated human umbilical vein endothelial cells proliferation and apoptosis through AMPK/CREB/BDNF pathway. Front Pharmacol. 2018;9:1266
237. Lai B, Li Z, He M, Wang Y, Chen L, Zhang J. et al. Atheroprone flow enhances the endothelial-to-mesenchymal transition. Am J Physiol Heart Circ Physiol. 2018;315(5):H1293-h303
238. Sharma N, Lu Y, Zhou G, Liao X, Kapil P, Anand P. et al. Myeloid kruppel-like factor 4 deficiency augments atherogenesis in ApoE-/- mice-brief report. Arterioscler Thromb Vasc Biol. 2012;32(12):2836-8
239. Yu B, Wu Y, Li Z. KLF4/Ch25h axis activated by metformin suppresses EndoMT in human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2020;522(4):838-44
240. Lum H, Malik AB. Mechanisms of increased endothelial permeability. Can J Physiol Pharmacol. 1996;74(7):787-800
241. Hempel A, Maasch C, Heintze U, Lindschau C, Dietz R, Luft FC. et al. High glucose concentrations increase endothelial cell permeability via activation of protein kinase C alpha. Circ Res. 1997;81(3):363-71
242. Eskens BJ, Zuurbier CJ, van Haare J, Vink H, van Teeffelen JW. Effects of two weeks of metformin treatment on whole-body glycocalyx barrier properties in db/db mice. Cardiovasc Diabetol. 2013;12:175
243. Yang Y, Dong R, Hu D, Chen Z, Fu M, Wang DW. et al. Liver kinase B1/AMP-activated protein kinase pathway activation attenuated the progression of endotoxemia in the diabetic mice. Cell Physiol Biochem. 2017;42(2):761-79
244. Ding H, Chen B, Lu Q, Wang J. Profilin-1 mediates microvascular endothelial dysfunction in diabetic retinopathy through HIF-1α-dependent pathway. Int J Clin Exp Pathol. 2018;11(3):1247-55
245. Uddin MA, Akhter MS, Kubra KT, Siejka A, Barabutis N. Metformin in acute respiratory distress syndrome: an opinion. Exp Gerontol. 2021;145:111197
246. Asgharzadeh F, Barneh F, Fakhraie M, Adel Barkhordar SL, Shabani M, Soleimani A. et al. Metformin inhibits polyphosphate-induced hyper-permeability and inflammation. Int Immunopharmacol. 2021;99:107937
247. Ii M, Nishimura H, Iwakura A, Wecker A, Eaton E, Asahara T. et al. Endothelial progenitor cells are rapidly recruited to myocardium and mediate protective effect of ischemic preconditioning via "imported" nitric oxide synthase activity. Circulation. 2005;111(9):1114-20
248. Modarai B, Burnand KG, Sawyer B, Smith A. Endothelial progenitor cells are recruited into resolving venous thrombi. Circulation. 2005;111(20):2645-53
249. Urbich C, Dimmeler S. Endothelial progenitor cells: characterization and role in vascular biology. Circ Res. 2004;95(4):343-53
250. Kamihata H, Matsubara H, Nishiue T, Fujiyama S, Tsutsumi Y, Ozono R. et al. Implantation of bone marrow mononuclear cells into ischemic myocardium enhances collateral perfusion and regional function via side supply of angioblasts, angiogenic ligands, and cytokines. Circulation. 2001;104(9):1046-52
251. Li WD, Du XL, Qian AM, Hu N, Kong LS, Wei S. et al. Metformin regulates differentiation of bone marrow-derived endothelial progenitor cells via multiple mechanisms. Biochem Biophys Res Commun. 2015;465(4):803-9
252. Li WD, Li NP, Song DD, Rong JJ, Qian AM, Li XQ. Metformin inhibits endothelial progenitor cell migration by decreasing matrix metalloproteinases, MMP-2 and MMP-9, via the AMPK/mTOR/autophagy pathway. Int J Mol Med. 2017;39(5):1262-68
253. Gu X, Wang XQ, Lin MJ, Liang H, Fan SY, Wang L. et al. Molecular interplay between microRNA-130a and PTEN in palmitic acid-mediated impaired function of endothelial progenitor cells: effects of metformin. Int J Mol Med. 2019;43(5):2187-98
254. Ni HZ, Liu Z, Sun LL, Zhou M, Liu C, Li WD. et al. Metformin inhibits angiogenesis of endothelial progenitor cells via miR-221-mediated p27 expression and autophagy. Future Med Chem. 2019;11(17):2263-72
255. Arunachalam G, Lakshmanan AP, Samuel SM, Triggle CR, Ding H. Molecular interplay between microRNA-34a and sirtuin1 in hyperglycemia-mediated impaired angiogenesis in endothelial cells: effects of metformin. J Pharmacol Exp Ther. 2016;356(2):314-23
256. Gou L, Liu G, Ma R, Regmi A, Zeng T, Zheng J. et al. High fat-induced inflammation in vascular endothelium can be improved by Abelmoschus esculentus and metformin via increasing the expressions of miR-146a and miR-155. Nutr Metab (Lond). 2020;17:35
257. Giuliani A, Londin E, Ferracin M, Mensà E, Prattichizzo F, Ramini D. et al. Long-term exposure of human endothelial cells to metformin modulates miRNAs and isomiRs. Sci Rep. 2020;10(1):21782
258. Xu S, Kamato D, Little PJ, Nakagawa S, Pelisek J, Jin ZG. Targeting epigenetics and non-coding RNAs in atherosclerosis: from mechanisms to therapeutics. Pharmacol Ther. 2019;196:15-43
259. Szabo L, Salzman J. Detecting circular RNAs: bioinformatic and experimental challenges. Nat Rev Genet. 2016;17(11):679-92
260. Josipovic I, Pflüger B, Fork C, Vasconez AE, Oo JA, Hitzel J. et al. Long noncoding RNA LISPR1 is required for S1P signaling and endothelial cell function. J Mol Cell Cardiol. 2018;116:57-68
261. Radhakrishnan R, Kowluru RA. Long noncoding RNA MALAT1 and regulation of the antioxidant defense system in diabetic retinopathy. Diabetes. 2021;70(1):227-39
262. Gong YP, Zhang YW, Su XQ, Gao HB. Inhibition of long noncoding RNA MALAT1 suppresses high glucose-induced apoptosis and inflammation in human umbilical vein endothelial cells by suppressing the NF-κB signaling pathway. Biochem Cell Biol. 2020;98(6):669-75
263. Leisegang MS, Fork C, Josipovic I, Richter FM, Preussner J, Hu J. et al. Long noncoding RNA MANTIS facilitates endothelial angiogenic function. Circulation. 2017;136(1):65-79
264. Dang RY, Liu FL, Li Y. Circular RNA hsa_circ_0010729 regulates vascular endothelial cell proliferation and apoptosis by targeting the miR-186/HIF-1α axis. Biochem Biophys Res Commun. 2017;490(2):104-10
265. Pan L, Lian W, Zhang X, Han S, Cao C, Li X. et al. Human circular RNA-0054633 regulates high glucose-induced vascular endothelial cell dysfunction through the microRNA-218/roundabout 1 and microRNA-218/heme oxygenase-1 axes. Int J Mol Med. 2018;42(1):597-606
266. Wen Y, Chun Y, Lian ZQ, Yong ZW, Lan YM, Huan L. et al. circRNA-0006896-miR1264-DNMT1 axis plays an important role in carotid plaque destabilization by regulating the behavior of endothelial cells in atherosclerosis. Mol Med Rep. 2021;23(5):311
267. Xu X, Wu Z, Qiu H, Wu J. Circular RNA circPHC3 promotes cell death and apoptosis in human BMECs after oxygen glucose deprivation via miR-455-5p/TRAF3 axis in vitro. Neuropsychiatr Dis Treat. 2021;17:147-56
268. Shan K, Liu C, Liu BH, Chen X, Dong R, Liu X. et al. Circular noncoding RNA HIPK3 mediates retinal vascular dysfunction in diabetes mellitus. Circulation. 2017;136(17):1629-42
269. McVeigh GE, Cohn JN. Endothelial dysfunction and the metabolic syndrome. Curr Diab Rep. 2003;3(1):87-92
270. Eelen G, de Zeeuw P, Treps L, Harjes U, Wong BW, Carmeliet P. Endothelial cell metabolism. Physiol Rev. 2018;98(1):3-58
271. Andrade J, Shi C, Costa ASH, Choi J, Kim J, Doddaballapur A. et al. Control of endothelial quiescence by FOXO-regulated metabolites. Nat Cell Biol. 2021;23(4):413-23
272. Zhang Z, Apse K, Pang J, Stanton RC. High glucose inhibits glucose-6-phosphate dehydrogenase via cAMP in aortic endothelial cells. J Biol Chem. 2000;275(51):40042-7
273. Song J, Ren P, Zhang L, Wang XL, Chen L, Shen YH. Metformin reduces lipid accumulation in macrophages by inhibiting FOXO1-mediated transcription of fatty acid-binding protein 4. Biochem Biophys Res Commun. 2010;393(1):89-94
274. Wang C, Liu F, Yuan Y, Wu J, Wang H, Zhang L. et al. Metformin suppresses lipid accumulation in skeletal muscle by promoting fatty acid oxidation. Clin Lab. 2014;60(6):887-96
275. Cabodevilla AG, Tang S, Lee S, Mullick AE, Aleman JO, Hussain MM. et al. Eruptive xanthoma model reveals endothelial cells internalize and metabolize chylomicrons, leading to extravascular triglyceride accumulation. J Clin Invest. 2021;131(12):e145800
276. Sasson S, Gorowits N, Joost HG, King GL, Cerasi E, Kaiser N. Regulation by metformin of the hexose transport system in vascular endothelial and smooth muscle cells. Br J Pharmacol. 1996;117(6):1318-24
277. Samuel SM, Ghosh S, Majeed Y, Arunachalam G, Emara MM, Ding H. et al. Metformin represses glucose starvation induced autophagic response in microvascular endothelial cells and promotes cell death. Biochem Pharmacol. 2017;132:118-32
278. Yang J, Chi Y, Burkhardt BR, Guan Y, Wolf BA. Leucine metabolism in regulation of insulin secretion from pancreatic beta cells. Nutr Rev. 2010;68(5):270-9
279. Jang C, Oh SF, Wada S, Rowe GC, Liu L, Chan MC. et al. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat Med. 2016;22(4):421-6
280. Yang Y, Wu Z, Meininger CJ, Wu G. L-Leucine and NO-mediated cardiovascular function. Amino Acids. 2015;47(3):435-47
281. Morris SM Jr. Recent advances in arginine metabolism: roles and regulation of the arginases. Br J Pharmacol. 2009;157(6):922-30
282. Kleele T, Rey T, Winter J, Zaganelli S, Mahecic D, Perreten Lambert H. et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature. 2021;593(7859):435-439
283. Miyao M, Cicalese S, Kawai T, Cooper HA, Boyer MJ, Elliott KJ. et al. Involvement of senescence and mitochondrial fission in endothelial cell pro-inflammatory phenotype induced by angiotensin II. Int J Mol Sci. 2020;21(9):3112
284. Wang Q, Zhang M, Torres G, Wu S, Ouyang C, Xie Z. et al. Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of Drp1-mediated mitochondrial fission. Diabetes. 2017;66(1):193-205
285. Morelli N, Rota E, Terracciano C, Immovilli P, Spallazzi M, Colombi D. et al. The baffling case of ischemic stroke disappearance from the casualty department in the COVID-19 era. Eur Neurol. 2020;83(2):213-15
286. Zhu L, She ZG, Cheng X, Qin JJ, Zhang XJ, Cai J. et al. Association of blood glucose control and outcomes in patients with COVID-19 and pre-existing type 2 diabetes. Cell Metab. 2020;31(6):1068-77 e3
287. Schlesinger S, Neuenschwander M, Lang A, Pafili K, Kuss O, Herder C. et al. Risk phenotypes of diabetes and association with COVID-19 severity and death: a living systematic review and meta-analysis. Diabetologia. 2021;64(7):1480-91
288. Boye KS, Tokar Erdemir E, Zimmerman N, Reddy A, Benneyworth BD, Dabora MC. et al. Risk factors associated with COVID-19 hospitalization and mortality: a large claims-based analysis among people with type 2 diabetes mellitus in the united states. Diabetes Ther. 2021:1-17
289. Lim S, Bae JH, Kwon HS, Nauck MA. COVID-19 and diabetes mellitus: from pathophysiology to clinical management. Nat Rev Endocrinol. 2021;17(1):11-30
290. Luo P, Qiu L, Liu Y, Liu XL, Zheng JL, Xue HY. et al. Metformin treatment was associated with decreased mortality in COVID-19 patients with diabetes in a retrospective analysis. Am J Trop Med Hyg. 2020;103(1):69-72
291. Hariyanto TI, Kurniawan A. Metformin use is associated with reduced mortality rate from coronavirus disease 2019 (COVID-19) infection. Obes Med. 2020;19:100290
292. Lalau JD, Al-Salameh A, Hadjadj S, Goronflot T, Wiernsperger N, Pichelin M. et al. Metformin use is associated with a reduced risk of mortality in patients with diabetes hospitalised for COVID-19. Diabetes Metab. 2020;47(5):101216
293. Tamura RE, Said SM, de Freitas LM, Rubio IGS. Outcome and death risk of diabetes patients with Covid-19 receiving pre-hospital and in-hospital metformin therapies. Diabetol Metab Syndr. 2021;13(1):76
294. Samuel SM, Varghese E, Busselberg D. Therapeutic potential of metformin in COVID-19: reasoning for its protective role. Trends Microbiol. 2021 doi: 10.1016/j.tim.2021.03.004
295. Varghese E, Samuel SM, Liskova A, Kubatka P, Büsselberg D. Diabetes and coronavirus (SARS-CoV-2): molecular mechanism of metformin intervention and the scientific basis of drug repurposing. PLoS Pathog. 2021;17(6):e1009634
296. Hua S, Yang Y, Zou D, Li J, Yan K, Xu Y. et al. COVID-19 and metabolic comorbidities: An update on emerging evidences for optimal therapies. Biomed Pharmacother. 2021;140:111685
297. Hogan MF, Hackney DJ, Aplin AC, Mundinger TO, Larmore MJ, Castillo JJ. et al. SGLT2-i improves markers of islet endothelial cell function in db/db diabetic mice. J Endocrinol. 2021;248(2):95-106
298. Xiang X, Wang L, Zhou L, Chen Y, Xia H. Metformin upregulates the expression of Gli1 in vascular endothelial cells in hyperoxia-exposed neonatal mice. Am J Transl Res. 2020;12(10):6092-106
Author contact
![]() Corresponding authors: Jianping Weng, MD, PhD, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, 230001, China. E-mail: wengjpedu.cn; Suowen Xu, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, 230001, China. E-mail: sxu1984edu.cn.
Corresponding authors: Jianping Weng, MD, PhD, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, 230001, China. E-mail: wengjpedu.cn; Suowen Xu, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, 230001, China. E-mail: sxu1984edu.cn.