Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2021; 11(18):9022-9037. doi:10.7150/thno.60350 This issue Cite
Review
Targeting of promising transmembrane proteins for diagnosis and treatment of pancreatic ductal adenocarcinoma
Vida Mashayekhi1*, Orsola Mocellin1*, Marcel H.A.M. Fens2, Gerard C. Krijger3, Lodewijk A.A. Brosens4, Sabrina Oliveira1,2 ![]()
1. Cell Biology, Neurobiology and Biophysics, Department of Biology, Faculty of Science, Utrecht University, 3584 CH Utrecht, the Netherlands.
2. Pharmaceutics, Department of Pharmaceutical Sciences, Faculty of Science, Utrecht University, 3584 CG Utrecht, the Netherlands.
3. Department of Radiology and Nuclear Medicine, University Medical Center Utrecht, Faculty of Medicine, Utrecht University, 3584 CX Utrecht, the Netherlands.
4. Department of Pathology, University Medical Center Utrecht, Faculty of Medicine, Utrecht University, 3584 CX Utrecht, the Netherlands.
*These authors contributed equally to this work.
Received 2021-3-11; Accepted 2021-7-12; Published 2021-8-25
Abstract

Pancreatic ductal adenocarcinoma (PDAC) is one of the most fatal types of cancer due to the relatively late diagnosis and the limited therapeutic options. Current treatment regimens mainly comprise the cytotoxic agents gemcitabine and FOLFIRINOX. These compounds have shown limited efficacy and severe side effects, highlighting the necessity for earlier detection and the development of more effective, and better-tolerated treatments. Although targeted therapies are promising for the treatment of several types of cancer, identification of suitable targets for early diagnosis and targeted therapy of PDAC is challenging. Interestingly, several transmembrane proteins are overexpressed in PDAC cells that show low expression in healthy pancreas and may therefore serve as potential targets for treatment and/or diagnostic purposes. In this review we describe the 11 most promising transmembrane proteins, carefully selected after a thorough literature search. Favorable features and potential applications of each target, as well as the results of the preclinical and clinical studies conducted in the past ten years, are discussed in detail.
Keywords: PDAC, transmembrane proteins, targeted therapy, diagnosis
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the fourth cause of cancer-related deaths in the world [1]. Although several oncogenic mutations such as KRAS, CDKN2 A, TP53, SMAD4, GNAS, and RNF43 have been identified, the pathophysiology and genetic profile of PDAC are complex and heterogeneous [1,2]. In addition to gene mutations, overexpression of several transmembrane proteins such as growth factor receptors, mucins, tight junction and adhesion proteins, which are crucial for the activation of oncogenic pathways, have been reported in PDAC [2-4]. Moreover, the dense stroma and extracellular matrix surrounding the tumor cells create a hypovascular and hypoxic microenvironment that not only supports proliferation and immune evasion but also interferes with drug delivery, adding to the complexity of this disease [1].
The incidence of PDAC is increasing, while current treatments have not significantly improved the life expectancy over the last few decades, with less than 10% of PDAC patients reaching a 5-year overall survival [5]. This high mortality rate is mostly due to the fact that up to 80% of PDAC cases are diagnosed in advanced stages of the disease, limiting the treatment options [6]. Early-stage symptoms such as indigestion, abdominal pain and nausea, are generally unspecific and, in some cases, patients are asymptomatic until the later stages, allowing the disease to escalate unnoticed. Moreover, even after surgical resection, PDAC frequently recurs and shows limited response to current chemotherapeutic agents [6]. Therefore, there is a tremendous need for more effective diagnostic and therapeutic options.
A considerable number of preclinical studies have investigated potential novel therapeutic options for PDAC, but only few reached the clinic, showing limited efficacy and an increase in survival by only a few months [6]. Currently approved treatment options include surgery, chemotherapy and more recently targeted therapies, namely tyrosine kinase inhibitors such as erlotinib. In general, targeted therapies are meant to block specific target molecules involved in the growth and progression of cancer [7,8]. Surgery in combination with adjuvant therapy, such as chemoradiotherapy, is usually the recommended option for resectable PDAC. In spite of the initial effects seen after treatment, relapse is very common [9]. Moreover, up to 85% of PDAC cases are non-resectable due to presence of distant metastases and/or locally advanced disease [9,10]. For these patients, chemotherapy with gemcitabine or FOLFIRINOX is usually recommended as single agent or in combination therapies. However, these agents have shown limited efficacy and often come with severe side effects that have a substantial impact on patients' quality of life.
Many studies focused on the identification and targeting of PDAC-specific proteins that can potentially serve as targets for novel therapies with higher efficacy and fewer adverse effects [11]. A variety of targeted therapies, namely monoclonal antibodies and antibody-drug conjugates are currently under clinical investigation for treatment of PDAC. These therapies are usually administered in combination with gemcitabine. For instance, erlotinib, a tyrosine kinase inhibitor, in combination with gemcitabine has been shown to increase survival compared to gemcitabine alone [11,12]. Alternatively, immunotherapy has also been utilized for the treatment of PDAC, as recently reviewed [13]. Different types of immunotherapy have been investigated, such as the use of patients' own chimeric antigen receptor (CAR) T cells directed against cancer cells, cancer vaccines, monoclonal antibodies specifically targeting immune components, and conjugates of these with drugs (also known as antibody-drug conjugates) [4,6].
Antibody-targeted photodynamic therapy (PDT), known as photoimmunotherapy (PIT), has also been investigated preclinically for the treatment of PDAC [14-16]. This approach makes use of photosensitizers (PS) that are specifically delivered to the tumor tissue by means of monoclonal antibodies or other targeting moieties. Activation of PS at the tumor site after illumination results in reactive oxygen species production which subsequently induces tumor cell death [14,15]. While targeted PDT and PIT strategies have not yet reached clinical trials in PDAC, preclinical studies have so far shown promising results, highlighting the potential of this therapeutic strategy [15,16].
Another type of targeted therapy is receptor-targeted radionuclide therapy (RTRT), in which radionuclides conjugated to peptides, antibodies or antibody fragments deliver radiation specifically into the tumor [17]. RTRT has shown to achieve significant improvements both in diagnostics and therapeutic efficacy in several clinical studies, especially in patients with neuroendocrine tumors and prostate cancer [17-19]. By using different radionuclides for diagnostics and therapeutics, the attractive “theranostic” approach can be used for patient identification, selection and treatment. Nonetheless, few clinical studies have investigated such approaches for treatment of PDAC, with variations both in inclusion criteria and results, indicating that additional studies are required to further explore the full potential of this approach [17].
Based on an extensive literature review and specific criteria used for selection, we propose the 11 most promising proteins that may serve as targets for PDAC diagnostics and/or therapies. In particular, these targets can be explored for delivery of cytotoxic compounds, namely radionuclides and drugs, conjugated to targeting moieties that specifically associate with these proteins on tumor cells. This information can guide further preclinical studies and may contribute to the management of patients with PDAC in the future.
Search strategy and target selection
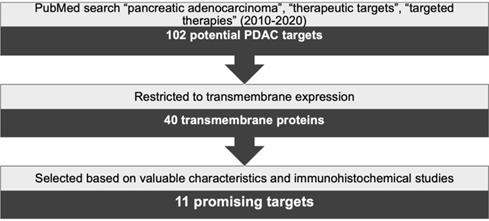
A thorough search was conducted using PubMed and combinations of the keywords “pancreatic adenocarcinoma”, “therapeutic targets” and “targeted therapies”. Results were limited to reviews published between 2010 and 2020 yielding 102 possible target proteins. Next, this number was reduced by selecting the targets showing transmembrane expression, resulting in 40 proteins. In the subsequent steps, a literature research was implemented to establish, for each target, the frequency and level of expression in PDAC, its expression in healthy tissues, and the results from preclinical in vivo or clinical studies. In addition, immunohistochemical expression of these targets in PDAC was assessed based on published studies in consultation with an experienced pathologist (LAAB). Protein expression was verified to be solely or mostly confined to cancer cells and not to the stroma, while also being present in the majority of the cancer cells. Based on this strategy, schematically represented in Figure 1, 11 targets were selected as the most promising targets to be explored for PDAC diagnosis and/or therapies. The characteristics of these selected transmembrane proteins are summarized in Table 1.
Schematic representation of target selection for the diagnosis and treatment of PDAC.
Summary of the characteristics of the 11 selected transmembrane proteins described in this review for diagnosis and/or treatment of PDAC
| Target | Molecular weight (kDa) | Ligands | Protein type | Role | Reference |
|---|---|---|---|---|---|
| ASPH | ~86 | No ligand | α-ketoglutarate-dependent dioxygenase | Proliferation, migration, invasion, hydroxylation of EGF-like domain containing proteins | [9] |
| EGFR | ~134 | EGF, TGFA, HBEGF, BTC, AREG, EREG, EPGN | Tyrosine kinase receptor | Proliferation, metastasis, drug resistance, survival | [11,20,21] |
| HER2 | ~185 | No ligand | Tyrosine kinase receptor | Proliferation, migration, invasion, survival | [22-24] |
| PDGFRβ | ~90 | PDGF | Tyrosine kinase receptor | proliferation, migration, invasion, angiogenesis, survival | [25-27] |
| GPRC5A | ~40 | No ligand | G protein-coupled receptor | Proliferation, invasion, metastasis, chemoresistance | [28-30] |
| Claudin 4 | ~22 | CPE | Adhesion protein | Tumor progression, invasion, chemoresistance | [31-34] |
| Integrin αvβ6 | ~114 | Fibronectin,Vitronectin, Tenascin-C, TGF-β1 | Adhesion glycoprotein | Proliferation, metastasis, invasion, immune evasion | [35,36] |
| Mesothelin | ~40 | No ligand | GPI-anchored surface protein | Proliferation, metastasis, angiogenesis, chemoresistance, activation of matrix metalloproteinases by interaction with MUC16 | [37-39] |
| MUC4 | 550-590 | No ligand | Highly glycosylated tight junction protein | Proliferation, metastasis, immune evasion, chemoresistance | [40-42] |
| MUC13 | ~175 | No ligand | Highly glycosylated protein | Activation of pro-oncogenic proteins, metastasis, down regulation of p53 | [43-46] |
| MUC16 | 3000-5000 | No ligand | Highly glycosylated protein | Proliferation, activation of matrix metalloproteinases by interaction with mesothelin, metastasis, immune evasion | [47-49] |
ASPH: Aspartate-β-hydroxylase; EGFR: epidermal growth factor receptor; EGF: epidermal growth factor; TGFA: transforming growth factor-alpha; HBEGF: heparin-binding EGF-like growth factor; BTC: betacellulin; AREG: amphiregulin; EREG: epiregulin; EPGN: epigen; HER2: human epidermal growth factor receptor 2; PDGFRβ: platelet derived growth factor receptor beta; PDGF: platelet derived growth factor; GPRC5A: G protein-coupled receptor family C member 5 group A; CPE: Clostridium perfringens enterotoxin; TGF-β1: transforming growth factor; HER2: human epidermal growth factor receptor 2; GPI: glycosylphosphatidylinositol.
Based on the described strategy, the most promising transmembrane proteins can be grouped into four categories based on their function, as illustrated in Figure 2:
Representative immunohistochemical staining pattern of the selected transmembrane proteins in patient-derived PDAC samples. The scheme presents transmembrane proteins grouped based on their function, i.e. enzymatic activity, signal transduction, cell adhesion or cell-cell recognition. The images were adapted from peer-reviewed papers or Protein Atlas (www.proteinatlas.org/). Enzymatic activity: ASPH [50] EGFR [51], HER2 [52], PDGFRβ [25] (copyright 2016 Springer Nature); Signal transduction: GPRC5A [53]; Cell adhesion: Claudin 4 [54], Integrin αvβ6 [55] (copyright 2019 John Wiley and Sons), Mesothelin [56]; Cell-cell recognition: Mucin 4 [57], Mucin 13 [58] (copyright 2015 Spandidos Publications), Mucin 16 [59]. ASPH: ASPH: Aspartate-β-hydroxylase; EGFR: epidermal growth factor receptor; GPRC5A: G protein-coupled receptor family member 5 group A; HER2; human epidermal growth factor receptor 2; PDGFRβ: platelet derived growth factor receptor beta.
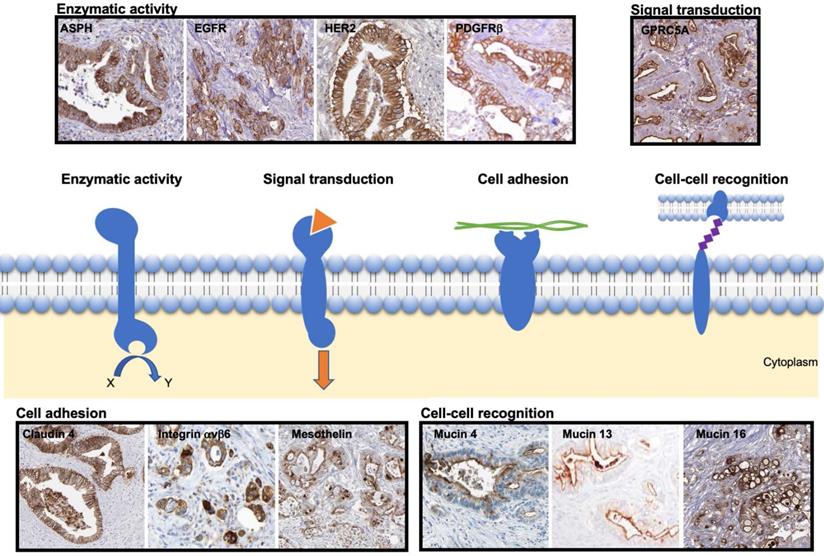
- Enzymatic activity: ASPH, EGFR, HER2 and PDGFRβ;
- Signal transduction: GPRC5A;
- Cell adhesion: Claudin 4, Integrin αvβ6, Mesothelin;
- Cell-cell recognition: Mucins (4, 13 and 16).
In the next sections, for each of these transmembrane proteins, details about their expression in cancer and normal tissue are provided. Representative immunochemistry staining patterns of these proteins are shown in Figure 2. In addition, encouraging preclinical studies, and, when available, clinical studies are described in the following sections. An overview of the transmembrane proteins described in this review is summarized in Table 2.
Selected transmembrane proteins described for the diagnosis or treatment of PDAC
| Target | Expression in PDAC | Percentage of expression | Stage of disease | Normal tissue expression | Reference |
|---|---|---|---|---|---|
| ASPH | Tumor cells | 97% | Lesions<4 | Placental trophoblasts | [9,10,60] |
| EGFR | Tumor cells, endothelial cells of the microenvironment | 30-90% | Lesion-4 | Gastrointestinal tract, pancreas, skin, brain, vasculature, heart, lungs, kidneys, liver, reproductive organs | [4,61-64] |
| HER2 | Tumor cells | 0-82% | 1-4 | Adrenal gland, liver, skin, brain, heart | [65-68] |
| PDGFRβ | Tumor cells, stroma | ~100% positive, 49% strong | 1<4 | Reproductive tissues, kidneys | [25,26,69-71] |
| GPRC5A | Tumor cells | 82% | 1<4 | Lung (high), intestines, kidneys, thyroid, bladder (low) | [28-30,72] |
| Claudin 4 | Tumor cells | 70-97% | Lesions-4 | Lungs, stomach, liver, intestines, kidneys, pancreas | [73-76] |
| Integrin αvβ6 | Tumor cells (mainly), fibroblasts, vessels (rarely) | ~90% | Lesions<4 | esophagus, stomach, intestines, liver | [35,36,77-79] |
| Mesothelin | Tumor cells | 100% positive, 80-85% strong | Lesions-4 | Pleura, pericardium and peritoneum | [37,80-83] |
| MUC4 | Tumor cells | 70-95% | Lesions<4 | Airway epithelia, body fluids | [42,84-86] |
| MUC13 | Tumor cells | 95% | Lesions<4 | Trachea, stomach, intestines, kidneys | [43-45] |
| MUC16 | Tumor cells | 82% | 1<4 | Airway epithelia, female reproductive tissues, cornea, conjunctiva | [41,87,88] |
ASPH: Aspartate-β-hydroxylase; EGFR: epidermal growth factor receptor; GPRC5A: G protein-coupled receptor family C member 5 group A; HER2: human epidermal growth factor receptor 2; PDGFRβ: platelet derived growth factor receptor beta.
Stage of disease indicates in which phase overexpression of transmembrane proteins is observed. In some targets the expression remains steady over the disease progression, i.e. from lesion to stage 4 (lesion-4) or from stage 1 to stage 4 (1-4), while increasing in other targets i.e. from lesion to stage 4 (lesion<4) or from stage 1 to stage 4 (1<4).
Transmembrane proteins with enzymatic activity
ASPH
Aspartate-β-hydroxylase (ASPH) is a type II transmembrane protein which belongs to the α-ketoglutarate-dependent dioxygenase family. This receptor consists of a cytoplasmic domain, a transmembrane domain, a luminal region, and a C-terminal catalytic domain. ASPH has an important role in embryonic development where it promotes organ formation and growth but shows virtually no expression in healthy adult tissues, mostly limited to the placenta [9].
Expression of ASPH is aberrantly increased in PDAC and also other cancers such as hepatocellular carcinoma and prostate cancer, and has been reported to contribute to proliferation, migration, invasion, recurrence, and tumor invasiveness [9,89,90]. Although the exact tumor-promoting mechanisms of ASPH have not been elucidated yet, it has been hypothesized that this protein might be involved in several tumorigenic pathways, including the Notch pathway. This will subsequently result in PDAC progression, somatic mutations of mitochondrial DNA, as well as inhibition of natural killer cells function, thus protecting tumor cells from immune surveillance [9]. To our knowledge, expression of ASPH in the tumor microenvironment has not been reported so far.
In two independent studies, ASPH upregulation has been reported in 97% of 101 [10] and of 104 [60] patient-derived PDAC samples, while its expression was not detected in normal pancreas. Moreover, the upregulation of this receptor has been shown in precursor lesions and increases during neoplastic progression, indicating that ASPH could be a promising diagnostic and therapeutic target for PDAC from the early stages of the disease [60]. So far, no ASPH-targeted agents have been assessed in clinical trials and only few have shown promising results in preclinical studies [91,92]. In a PDAC patient-derived xenograft murine model, the ASPH-targeting antibody-drug conjugate SNS-622-DM1 inhibited tumor growth, likely by inducing cell cycle arrest and promoting apoptosis. In the same model, SNS-622-DM1 could also inhibit lung metastasis while being well-tolerated [10]. In another study, the efficacy of MO-I-1182, a small molecule targeting the enzymatic activity of ASPH, was investigated in a patient-derived murine xenograft model for PDAC in both primary tumors and metastases. MO-I-1182 effectively and specifically targeted ASPH activity, thereby inhibiting PDAC growth, progression and suppression of metastases to the lungs [60].
EGFR
The epidermal growth factor receptor (EGFR) is a transmembrane glycoprotein which belongs to the ErbB family of receptor tyrosine kinases. This receptor contains an extracellular-binding domain, a transmembrane region and a cytoplasmic region [4]. EGFR is activated through binding of a variety of growth factors, such as epidermal growth factor, transforming growth factor-α, amphiregulin, and epigen. Upon ligand binding, EGFR undergoes conformational changes that lead to the dimerization and phosphorylation of the cytoplasmic domain, which induces a signaling cascade of intracellular proteins that ultimately contribute to cell survival, growth, differentiation, and proliferation in healthy tissues [61].
EGFR overexpression has been reported in various types of cancer, including in up to 90% of PDAC cases. Overexpression has been reported in late stage precursor lesions and PDAC, and it is significantly increased in tumor tissues compared to healthy pancreatic tissue [4,62]. In PDAC, EGFR is involved in the activation of pathways that contribute to tumor proliferation, metastasis, malignant transformation, survival, and drug resistance [11,20]. RAS/RAF/MEK/ERK and RAS/PI3K/PTEN/Akt/mTOR are the most common activated pathways in PDAC, not only due to the EGFR overexpression, but also as a result of KRAS mutation [4,20]. Although KRAS mutation was shown to inhibit the efficacy of the EGFR-targeted therapies in non-small cell lung cancer, controversial results were obtained in PDAC. Therefore, further research is required to confirm the effect of KRAS in EGFR targeted therapies [93,94]. In addition, EGFR is expressed on endothelial cells of the tumor microenvironment, where it contributes to angiogenesis, proliferation and metastasis through stimulation of vascular endothelial and fibroblast growth factors, and interleukin-8 [4,63].
Because of the long-standing history of research on EGFR and its important role in PDAC, this receptor has been investigated extensively for targeted therapies. Most studied agents are the anti-EGFR targeted monoclonal antibody cetuximab and kinase inhibitors such as erlotinib [4,11,12]. However, these treatments failed to improve PDAC therapy substantially. Therefore, several other EGFR targeted agents that have shown promising results in preclinical and early clinical stages, and particularly in combination therapies, are currently under investigation [4,95,96]. For example, Li and colleagues explored the anti-EGFR monoclonal antibody RC68 conjugated to the cytotoxin monomethyl auristatin E (MMAE) in a murine xenograft model. In this study, the RC68-MMAE conjugate successfully inhibited tumor growth and even induced tumor regression, while being well tolerated [96]. Recently, EGFR was also targeted for photodynamic therapy using cetuximab photoimmuno-nanoconjugates in vivo [16]. The nanoconjugates were shown to have high efficacy and good tolerability, providing a promising strategy for the treatment of PDAC. EGFR-targeted agents have also been investigated in combination with radiotherapy due to the role of EGFR overexpression in radiotherapy resistance. Combinations of cetuximab and radiotherapy have shown promising results both in terms of efficacy and tolerability in preclinical and early stages of clinical studies [4,95]. The combination of EGFR-targeted therapies with cytotoxic agents is also extensively investigated, mostly with gemcitabine, both agents in combination with tyrosine kinase inhibitors (NCT00075686, NCT00040183), or the use of antibody-drug conjugates to achieve selective and specific delivery of cytotoxic agents to the tumor cells [4,20]. An overview of the approved clinical trials targeting EGFR is summarized in Table 3.
Summary of the identified clinical trials targeting the transmembrane proteins for the diagnosis and treatment of PDAC
| Target | Trial number | Phase | Intervention | # of patients |
|---|---|---|---|---|
| EGFR | NCT04137536 | 1 | Anti-EGFR-BATs | 25 |
| NCT00243854 | 1 | Neoadjuvant hypofractioned radiotherapy + gemcitabine + erlotinib | 8 | |
| NCT00536614 | 2 | Gemcitabine/Cisplatin +/- Cetuximab | 86 | |
| NCT03269526 | 1/2 | Anti-CD3 x anti-EGFR-BATs | 22 | |
| NCT02620865 | 1/2 | Anti-CD3 x anti-EGFR-BATs + aldesleukin + sargramostim | 2 | |
| NCT00042939 | 2 | Cetuximab + irinotecan + docetaxel | 94 | |
| NCT00424827 | 2 | Cetuximab + gemcitabine + 5-FU + external beam radiotherapy | 11 | |
| NCT01077986 | 1/2 | Cetuximab + capecitabine + everolimus | 35 | |
| NCT01420874 | 1 | Anti-EGFR-BATs + FOLFOX6 | 30 | |
| NCT00040183 | 3 | Erlotinib + gemcitabine | 569 | |
| NCT00862524 | 1/2 | ARRY-334543 + gemcitabine | 20 | |
| NCT00234416 | 1/2 | Gefitinib + gemcitabine | 45 | |
| NCT01505413 | 2 | Erlotinib + gemcitabine + oxaliplatin | 33 | |
| NCT01210911 | 2 | Erlotinib + gemcitabine +/- metformin | 120 | |
| NCT02451553 | 1 | Afatinib + capecitabine | 48 | |
| NCT00260364 | 1/2 | Gemcitabine + capecitabine + erlotinib + bevacizumab | 44 | |
| NCT01693419 | 2 | Erlotinib + gemcitabine + S-1 | 37 | |
| NCT04464967 | 1/2 | SNK01 + trastuzumab; SNK01 + cetuximab | 154 | |
| NCT00026338 | 3 | Gemcitabine +/- erlotinib | 569 | |
| NCT04429542 | 1 | BCA101 +/- pembrolizumab | 292 | |
| NCT00225784 | 2 | Cetuximab + gemcitabine + radiotherapy | 37 | |
| NCT00480584 | 1 | Erlotinib + gemcitabine + capecitabine | 20 | |
| NCT00397384 | 1 | Cetuximab + erlotinib | 43 | |
| NCT00383149 | 2 | Cetuximab + ixabepilone | 58 | |
| NCT00622674 | 1 | Cetuximab + bortezomib | 37 | |
| NCT03319459 | 1 | FATE-NK100; FATE-NK100 + cetuximab; FATE-NK100 + trastuzumab | 100 | |
| NCT00962312 | 2 | Lapatinib + capecitabine | 9 | |
| NCT00075686 | 3 | Gemcitabine +/- cetuximab | 766 | |
| NCT01222689 | 2 | Erlotinib + selumetinib | 46 | |
| NCT03989115 | 1/2 | RMC-4630 + Osimertinib; RMC-4630 + cometinib | 168 | |
| NCT03878524 | 1 | Several drug combinations including afatinib,dacomitinib, erlotinib, imatinib, trastuzumab | 40 | |
| NCT02465060 | 2 | Several drug combinations including afatinib, dacomitinib, erlotinib, imatinib, trastuzumab | 6452 | |
| HER2 | NCT00862524 | 1/2 | ARRY-334543 + gemcitabine | 20 |
| NCT04464967 | 1/2 | SNK01 + trastuzumab or SNK01 + cetuximab | 154 | |
| NCT02999672 | 2 | Trastuzumab | 20 | |
| NCT02451553 | 1 | Afatinib + capecitabine | 48 | |
| NCT03602079 | 1/2 | A166 | 82 | |
| NCT04482309 | 2 | Trastuzumab deruxtecan | 280 | |
| NCT02912949 | 1/2 | Zenocutuzumab | 250 | |
| NCT03586869 | 1/2 | ETBX-021 + several other drugs | 173 | |
| NCT00005926 | 2 | Herceptin(trastuzumab) + gemcitabine + radiotherapy | 50 | |
| NCT00034281 | 1 | TAX-165 | 16 | |
| NCT00004074 | 1 | Trastuzumab + IL-12 | 15 | |
| NCT01384253 | 1 | ²¹²Pb-TCMC-Trastuzumab + trastuzumab | 18 | |
| NCT00962312 | 2 | Lapatinib + capecitabine | 9 | |
| NCT03319459 | 1 | FATE-NK100; FATE-NK100 + cetuximab; FATE-NK100 + trastuzumab | 100 | |
| NCT04319757 | 1 | ACE1702 (anti-HER2 oNK cells) + cyclophosphamide + fludarabine | 24 | |
| NCT03425773 | 1 | BVAC-B | 8 | |
| NCT02465060 | 2 | Several drug combinations including afatinib, dacomitinib, erlotinib, imatinib, trastuzumab | 6452 | |
| Integrin αvβ6 | NCT02683824 | 1 | [18F]-R01-MG-F2 for PET/CT and PET/MRI | 25 |
| NCT04285996 | NA* | [18F]FBA-A20FMDV2 for PET | 12 | |
| NCT03023722 | 2 | Anetumab ravtansine | 18 | |
| NCT03638193 | NA* | CART-meso cells | 10 | |
| NCT01897415 | 1 | Mesothelin targeted CAR-T | 16 | |
| NCT03816358 | 1/2 | Anetumab ravtansine + nivolumab; +ipilimumab + nivolumab; + gemcitabine + nivlumab | 64 | |
| NCT03122106 | 1 | Personalized neoantigen DNA vaccine | 10 | |
| Mesothelin | NCT00066651 | 1 | SS1(dsFv)-PE38 immunotoxin | 15 |
| NCT00006981 | 1 | SS1(dsFv)-PE38 immunotoxin | 30 | |
| NCT00325494 | 1 | MORAb-009(amatuximab) | 24 | |
| NCT03956056 | 1 | Neoantigen peptide vaccine + poly ICLC | 15 | |
| NCT01521325 | 1 | Amatuximab | 6 | |
| NCT03323944 | 1 | huCART-meso cells | 18 | |
| NCT03102320 | 1 | Antumab ravtansine +/- gemcitabine/cisplatin | 173 | |
| NCT04034238 | 1 | LMB-100 + tofacitinib | 45 | |
| MUC16 | NCT01959672 | 2 | Oregovomab + gemcitabine + leucovorin + 5-FU + nelfinavir + radiotherapy | 11 |
| PDGFRβ | NCT03766295 | 3 | Masitinib + gemcitabine; gemcitabine | 377 |
| NCT00789633 | 3 | Masitinib + gemcitabine; gemcitabine | 353 | |
| NCT00967603 | 2 | Sunitinib | 56 | |
| NCT00673504 | 2 | Sunitinib + gemcitabine; gemcitabine | 105 | |
| NCT00397787 | 2 | Sunitinib | 64 | |
| NCT03878524 | 1 | Several drug combinations including sunitinib, dacomitinib, erlotinib, imatinib, trastuzumab | 40 | |
| NCT02465060 | 2 | Several drug combinations including sunitinib, dacomitinib, erlotinib, imatinib, trastuzumab | 6452 | |
| NCT00462553 | 1 | Sunitinib + gemcitabine | 37 | |
| NCT00703638 | 1 | Sorafenib + cisplatin + pemetrexed | 16 | |
| NCT00837876 | 2 | Sorafenib + erlotinib | 37 | |
| NCT00789763 | 1 | Sorafenib + gemcitabine + radiotherapy | 12 | |
| NCT00696696 | 2 | Sorafenib + erlotinib + gemcitabine | 45 | |
| NCT00114244 | 2 | Sorafenib; sorafenib + gemcitabine | 52 | |
| NCT00375310 | 1 | Sorafenib + gemcitabine + radiotherapy | 27 | |
| NCT00095966 | 2 | Sorafenib + gemcitabine | 35 | |
| NCT02349867 | 1 | Sorafenib + gemcitabine + vorinostat + radiotherapy | 23 | |
| NCT00981162 | 1 | Sorafenib + everolimus | 12 | |
| NCT01497392 | 1 | Dovitinib + gemcitabine + capecitabine | 26 |
*NA: Not Applicable;
EGFR: epidermal growth factor receptor; HER2: human epidermal growth factor receptor 2; PDGFRβ: platelet derived growth factor receptor beta.
While the therapeutic potential of EGFR-targeted therapies is thoroughly studied, it is also under investigation for imaging purposes. For instance, EGFR and vascular endothelial growth factor were simultaneously targeted for fluorescence imaging using a bispecific fusion protein in an orthotopic PDAC murine model, showing promising results [97]. Another study using monoclonal antibody fragments for microPET/CT imaging was also able to visualize pancreatic tumors in a xenograft mouse model [98].
HER2
HER2 is a member of the ErbB family of receptor tyrosine kinases which consists of four domains in the extracellular region, a single membrane-spanning domain and an intracellular region comprising a juxtamembrane domain, a tyrosine kinase domain, and a C-terminal tail. Although HER2 has no natural ligand, this protein is involved in several cell growth signaling pathways through heterodimerization with other ErbB receptors [22].
While the exact role of HER2 in PDAC has not yet been confirmed, activation of this receptor has been correlated with the cell proliferation, migration and survival [23]. HER2 is thought to exert its activity through activation of several tumorigenic intracellular pathways, such as PI3K/Akt/mTOR and RAS/RAF/MEK/ERK1/2 to enhance tumor cell growth, proliferation and invasion [22]. Moreover, HER2 was shown to interact with other proteins which are overexpressed in PDAC, including EGFR, MUC4 and MUC13. These interactions can result in the activation of the tumorigenic pathways MAPK, JNK and STAT, leading to the tumor proliferation and metastasis [42]. Several studies have reported a correlation between the expression of HER2 with the development of metastasis and poor survival, as well as tumor differentiation [61,99,100]. To our knowledge, the expression and role of HER2 in the PDAC microenvironment has not been explicitly reported.
Low levels of HER2 expression are present in various normal tissues and not healthy pancreas [52,101]. In contrast, overexpression of this receptor has been observed on the cell membranes of 0-82% of PDAC samples. Nonetheless, the correlation of clinicopathological features of PDAC showing significant HER2 expression is unclear, and contradictory results have been reported in literature [65].
In addition to PDAC, HER2 is one of the main receptors overexpressed in breast cancer, making it a well-characterized target. Several therapeutic agents such as monoclonal antibodies trastuzumab and zenocutuzumab, have been developed and tested in clinical trials for PDAC treatment. Moreover, the efficacy of tyrosine kinase inhibitors has also been assessed in the clinical studies for several types of cancer, including PDAC, showing contradictory results with a number of studies reporting little to no efficacy, while others indicated more favorable outcomes. For instance, trastuzumab has not yet displayed improvement over chemotherapy in clinical studies [11,23]. On the other hand, HER2-specific CAR T cell therapy in a phase I trial has shown promising results [13]. Cancer vaccines such as ETBX-021 are also assessed in clinical trials, though results for PDAC have not yet been published (NCT03586869). An overview of the clinical trials targeting HER2 is summarized in Table 3.
PDGFRβ
Platelet-derived growth factor receptor beta (PDGFRβ) is one of the two isoforms of PDGFR and, like EGFR and HER2, it belongs to the receptor tyrosine kinase family. The structure of PDGFRβ presents an extracellular part consisting of five immunoglobulin-like domains, a transmembrane domain, and, intracellularly, a juxtamembrane domain, a tyrosine kinase domain, and a C-terminus [102]. Like other receptor tyrosine kinases, PDGFRβ requires dimerization to be activated upon ligand binding. This is achieved either through homo- or heterodimerization, the latter of which can involve other receptors, such as EGFR or integrins, leading to the activation of various signaling pathways [102]. Under healthy conditions, PDGFRβ is mainly involved in blood vessel formation during embryonic development, while in adults it displays roles in wound healing and the control of interstitial fluid pressure [102].
In PDAC, PDGFRβ levels were detected on the cell membrane of most or all of the patient-derived samples, with nearly 50% showing particularly high expression [25,70]. On the other hand, expression in healthy pancreatic tissue was not detected, making this receptor suitable as a potential target in PDAC [70,71]. The potential of this target is further supported by its role in disease progression, as studies have indicated that PDGFRβ contributes to several tumorigenic processes and is particularly involved in cell invasion and metastasis. In addition, PDGFRβ is consistently expressed in the tumor microenvironment, where it promotes angiogenesis via the tumor stroma, further contributing to the tumor progression and metastasis [26]. The metastasis-promoting activity of PDGFRβ might also be related to mutant p53. In fact, mutant p53 is thought to induce PDGFRβ expression, enhancing metastatic potential and thus correlating with poor prognosis [25,26]. In fact, the angiogenic effects of PDGFRβ were shown to negatively affect radiation treatment efficacy, and it is suggested that this receptor could influence transport of therapeutic agents to the tumor site by regulating interstitial fluid pressure [103].
Until now, PDGFRβ-targeted therapies that have reached the stage of clinical trials are mainly small molecule tyrosine kinase inhibitors (Table 3). These inhibitors have shown promising results in preclinical studies, where therapeutic agents such as masitinib and sunitinib could suppress tumor growth and increase PDAC sensitivity to radiation [104,105]. Although clinical trials on neuroendocrine pancreatic tumors showed positive results, the effects on PDAC were not encouraging [106]. The reason for these discrepancies has not been fully elucidated but might be partially caused by interstitial fluid pressure. Ongoing clinical trials might provide further insights on the effectiveness of tyrosine kinase inhibitors (NCT03766295, NCT02349867). To our knowledge, no other PDGFRβ-targeted therapies are currently under investigation in PDAC than tyrosine kinase inhibitors.
Signal transduction transmembrane protein
GPRC5A
G protein-coupled receptor family C member 5, group A (GPRC5A), also known as retinoic acid-inducible 3 (RAI3), is a transmembrane protein which is part of the large protein superfamily of G protein coupled receptors [72]. The structure of GPRC5A is characterized by seven membrane-spanning domains connected by three extracellular and three intracellular loops, with an intracellular C-terminus and a short extracellular N-terminus [29]. The exact role of GPRC5A, both in cancer and normal conditions, has not been fully elucidated, though it is thought to be related to cell cycle progression and involvement of this protein in several signaling pathways [30]. In particular, GPRC5A is involved in the activation of proteins and signaling pathways often related to tumor development, such as NF-κB, STAT, and FAK/Src. However, studies have reported both tumor-promoting and tumor-suppressing activity of GPRC5A in these pathways, indicating that the role of this receptor is possibly tumor dependent [29].
In healthy conditions, GPRC5A is predominantly expressed in lung tissue, with minor expression in intestines, kidneys, thyroid, bladder, and low to non-detectable expression in pancreas [28-30]. However, in PDAC the expression of GPRC5A is significantly increased, showing strong membranous expression in up to 82% of 376 patient-derived samples [28]. GPRC5A expression in PDAC appears to be mostly limited to the cancer cells and not present in the surrounding microenvironment [107]. Of note, while GPRC5A expression was shown to be consistently upregulated in certain types of cancer, including PDAC, downregulation was observed in others, such as non-small cell lung cancer and hepatocellular carcinoma [29]. In PDAC, GPRC5A is involved in tumor proliferation, invasion and metastasis [28,29]. The potential comprehensive roles of this protein in several aspects of cancer development, plus its frequent overexpression in PDAC, make GPRC5A a potentially promising therapeutic and diagnostic target. In addition, GPRC5A also plays a role in the development of chemoresistance, thus making this protein a potential target to increase the efficacy of the chemotherapeutic agents [28,29]. To our knowledge, so far no therapeutic or diagnostic agents targeting GPRC5A have been investigated in clinical trials. This could be due to the lack of detailed insight on the role of this protein in PDAC, indicating further studies are needed to explore the potential of this target in PDAC for therapeutic and diagnostic applications.
Cell adhesion transmembrane proteins
Claudin 4
Claudin 4 is one of the 27 members of the claudin family of transmembrane proteins, composed of four transmembrane domains and two extracellular loops. Claudins have functional and structural activity in the composition of the tight junction barrier in normal epithelium [3,73,74]. As part of the tight junction barrier, claudins have several roles in normal cell functioning, including the maintenance of cell polarity and adhesion, as well as controlling of traffic of substances between cells [31]. Several studies have shown that claudin expression is dysregulated in various types of cancer, where it contributes to tumor progression and invasion [3,73]. This is likely due to the loss of cell adhesion and polarity caused by tight junction barrier disruption, which is one of the hallmarks of tumorigenesis [75,108].
Overexpression of claudin 4 has been reported in a variety of cancers, including PDAC, with high and diffuse expression detected in 70-97% of patient samples [73,74]. Claudin 4 expression was shown to be restricted to the tumor cells and not present in the surrounding stroma [109]. Importantly, overexpression of claudin 4 in PDAC has been found not only in primary tumors and metastases but also in precursor lesions, making it a potential target for early diagnosis and treatment [75]. Moreover, the role of claudin 4 in the development of chemoresistance by inhibiting access of therapeutic agents to tumor cells has been reported in several studies [31-33]. Therefore, agents able to interfere with the function of this protein may improve therapeutic efficacy of chemotherapeutic regimens, reduce the side effects, and improve patients' quality of life.
Two main approaches have been described for targeting of claudin 4. One strategy makes use of Clostridium perfringens enterotoxin (CPE). The C-terminal fragment of CPE (C-CPE) is one of the ligands of claudin 4, and therefore C-CPE-fused proteins and CPE could be utilized to target claudin 4. However, this strategy has revealed some drawbacks, particularly the immunogenicity of CPE and C-CPE-fused proteins, and poor selectivity due to the possible interaction of CPE with other members of the claudin family [108]. The second approach relies on the use of monoclonal antibodies to target the extracellular domains of claudin 4. However, development of anti-claudin targeted antibodies has shown to be challenging. This is mainly due to the fact that claudins have small extracellular domains (~18-50 amino acids) and high inter-species similarity, causing low immunogenicity. Nonetheless, a number of monoclonal antibodies against human claudin 4 have been developed that show both anti-tumor effects and induction of chemosensitivity in vivo [31,108]. Sasaki and co-workers investigated the effects of anti-claudin 4 antibody 4D3 in combination with FOLFIRINOX in a PDAC subcutaneous murine tumor model. The combination of the 4D3 antibody and FOLFIRINOX showed significant anti-tumor effects while the adverse events were significantly reduced due to the lower FOLFIRINOX dose. The increased activity seen for co-administered agents has suggested the induction of chemosensitivity by anti-claudin 4 agents in addition to its anti-tumor effects [31].
As an early-stage marker, claudin 4 has also been investigated for imaging and diagnosis. Several studies have explored application of CPE and monoclonal antibodies for the imaging of PDAC in vivo, showing promising results [75,110,111]. To our knowledge, claudin 4 has not been investigated yet in any clinical trials for the treatment or diagnosis of pancreatic cancer [91,92].
Integrin αvβ6
Integrin αvβ6 is a heterodimeric cell surface receptor with two subunits, α and β, both being a type I transmembrane glycoprotein. Each subunit comprises a short cytoplasmic domain, a transmembrane domain, and a large extracellular region projecting from the membrane [112]. Integrins are mainly involved in cell adhesion, cell-extracellular matrix interactions, as well as intracellular and cell-cell signaling [36,112,113].
Aberrant expression of several integrins has been implicated in various cancers, with both inhibitory and stimulatory functions in tumor proliferation, metastasis and invasion [35]. Dysregulation of integrin αvβ6 has been shown in numerous cancers, including PDAC, where its expression is progressively upregulated during neoplastic progression from precursor lesions to invasive adenocarcinoma and metastasis. Several studies have reported integrin αvβ6 upregulation, with diffuse membranous and cytoplasmic expression in nearly 90% of PDAC samples, whereas healthy pancreas and other healthy epithelia show low or undetectable expression [35,77,78]. Expression of this protein was reported to be mostly restricted to the tumor cells, though a few samples showed limited expression in the surrounding stroma and blood vessels [36].
The role of integrin αvβ6 in PDAC has not yet been completely elucidated, although it is partly involved in activation of transforming growth factor-β (TGF-β) [36,113]. The growth suppressing effect of TGF-β has negligible effect on tumor cells, while healthy cells are greatly affected. This subsequently results in growth inhibition and proliferation of healthy cells at the tumor site, thereby facilitating tumor proliferation invasive growth [113]. Moreover, increased levels of TGF-β possibly contribute to other tumor-promoting processes, such as epithelial-mesenchymal transition, metastasis, and immune evasion [113].
Despite the significance of integrin αvβ6 in PDAC, therapeutic agents targeting this protein have not yet reached clinical trials. However, a number of preclinical studies have shown promising results. Recently, the anti-integrin αvβ6-targeted antibody 264RAD was tested in combination with gemcitabine in a human xenograft model of PDAC in mice [55]. In this study, combining 246RAD with gemcitabine showed significant tumor growth reduction compared to control and monotherapy, with three subjects achieving full recovery. These effects were likely the result of reduced tumor cell proliferation, growth signaling, and also increased apoptosis. Another study investigated the potential of integrin αvβ6-targeted for photodynamic therapy in vivo [15]. The study assessed the targeted near-infrared agent Dye-SA-B-HK (IRDye700-Streptavidin-biotin-HK), in a subcutaneous mouse model of PDAC. The results showed that the treatment was well-tolerated and had significant anti-tumor effects. The authors proposed targeted PDT as a potential strategy for the treatment of PDAC, although clinical studies are required to substantiate these findings.
As opposed to the therapeutic agents, imaging strategies targeting this integrin have been more thoroughly investigated and some are currently in clinical trials. Several of these studies include the integrin αvβ6-targeting radiotracers 18F-αvβ6-binding-peptide (NCT03164486) and [18F]-R01-MG-F2 (NCT02683824); although, results have not yet been published. An overview of the clinical trials targeting αvβ6 is given in Table 3.
Mesothelin
Mesothelin is a glycoprotein normally found on the surface of mesothelial cells in the pleura, pericardium and peritoneum [81,82]. It is a glycosylphosphatidylinositol-anchored membrane protein formed from a precursor which is further processed into the soluble serum mesothelin relative peptide (SMRP) [37,80]. The role of mesothelin in healthy cells has not been identified yet, although it has been suggested that mesothelin might have a role in cell adhesion [38,114]. Preclinical studies have shown that this protein is not essential for normal functioning and development [38,114]. In PDAC, expression of mesothelin was reported in up to 100 of patient-derived samples and overexpression was found in 80-85% cases, while no expression was observed in healthy pancreatic tissue [37,80]. Expression of mesothelin has been shown to be limited to the tumor cells, which could be clearly distinguished from the mesothelin-negative surrounding stroma [80].
In several types of cancer, aberrant expression of mesothelin has been shown to play a role in angiogenesis, apoptosis resistance as well as promoting tumor proliferation, invasiveness and metastasis [37]. The mechanisms by which mesothelin exerts its tumorigenic effects have been thoroughly investigated [37]. Mesothelin is involved in a variety of tumor-promoting pathways such as STAT3, NFκB, ERK, and PI3K/Akt. Moreover, it has been shown to influence the expression of PDAC-related proteins, such as BAX and BCL-2. Activation of Akt/PI3K/NFκB and the MEK1/2 pathways lead to apoptosis resistance, which further contributes to the tumor survival [37]. In addition, mesothelin plays a role in metastasis through binding to MUC16, leading to enhanced matrix metalloproteinase-7 activity thereby promoting angiogenesis in metastatic tissues [37]. Finally, mesothelin is involved in epithelial-mesenchymal transition, a process known to contribute to tumor invasiveness [37]. A correlation between mesothelin and chemoresistance has also been proposed, but the underlying mechanisms are unclear [37].
Given its involvement in tumorigenic processes and its marginal expression in healthy tissue, mesothelin has been extensively investigated as a therapeutic target for cancer. Currently, the main focus for mesothelin-targeted therapies is the use of anti-mesothelin antibodies for the development of immunotoxins and antibody-drug conjugates. Some of these treatments have shown promising results in vivo and have advanced to early phase clinical trials [37,91,115]. The antibody-drug conjugate anetumab ravtansine was tested in 148 adult patients with advanced, metastatic or recurrent solid tumors in a phase I clinical study. Anetumab ravtansine is composed of an anti-mesothelin monoclonal antibody conjugated to the tubulin inhibitor DM4. The phase I study showed an adequate safety profile with manageable side effects, and preliminary data indicated that anetumab ravtansine showed promising anti-tumor effects in mesothelin-expressing tumors [116]. Monoclonal antibodies against mesothelin have also been radiolabeled with thorium-227 (BAY2287411) for radioimmunotherapy. BAY2287411 exhibited anti-tumor effects in vivo and is currently studied for PDAC and other solid tumors in a phase I clinical trial (NCT03507452) [37]. CAR-T cell therapy and vaccines directed against mesothelin have also shown promising results in preclinical studies and are presently undergoing clinical trials (e.g. NCT03497819, NCT03122106) [37,91,115].
In addition to therapy, mesothelin is being investigated as a target for imaging of PDAC, although studies have not reached clinical trials yet. A number of imaging strategies have been tested in vivo, including a technetium-99m labeled anti-mesothelin antibody for SPECT-CT imaging and a nanoprobe modified with an anti-mesothelin antibody [117,118]. Results have so far been promising, suggesting that mesothelin-targeted agents could be suitable tools for visualization of PDAC. An overview of the clinical trials targeting mesothelin is summarized in Table 3.
Cell-cell recognition transmembrane proteins
Mucins
Mucins are a family of 21 high-molecular-weight glycoproteins expressed on different types of epithelial tissues, whose functions vary from protecting mucous membrane barriers to contributing to cellular regeneration, differentiation, adhesion and signaling [40,41]. Mucins comprise membrane-bound MUC1, MUC3, MUC4, MUC11-13, MUC15-17, MUC20-21 and, secreted MUC2, MUC5AC, MUC5B, MUC6, MUC7, MUC19. The focus of this review is solely on the membrane-bound mucins. The structure of mucins is characterized by the presence of tandem repeat regions, to which several glycans are linked. In addition, these proteins contain various functional domains and a transmembrane domain is also found in membrane-bound mucins [40,41].
Aberrant expression of several mucins has been reported in pancreatic cancer, increasing steadily through neoplastic progression [40]. These proteins are thought to have numerous functions in the pathophysiology of PDAC. In fact, dysregulation of mucin expression can lead to changes in the mucin mesh found on the cell surface, which might affect drug absorption and subsequently cause chemoresistance and immune evasion [40,41]. In addition, aberrant expression of certain mucins possibly leads to enhanced pro-tumorigenic signaling, and also their role in cell adhesion contributes to tumor invasion and metastasis [41]. Due to the important role of mucins from the very early stages of PDAC, several members are considered as potential diagnostic and therapeutic targets. The membrane bound mucins, MUC4, MUC13 and MUC16 have shown most promising properties and therefore are discussed in more detail.
MUC4
MUC4 is one of the most frequently overexpressed mucins in PDAC, and in recent years several studies have been focusing on its potential as a therapeutic target. MUC4 is a glycoprotein composed of two subunits, α and β. The extracellular domain forms subunit MUC4α, which contains a tandem repeat region of variable length, a nidogen-like and an adhesion associated-domain in MUC4 and other proteins functional domains. Whereas subunit MUC4β comprises a cytoplasmic tail, a transmembrane domain and, extracellularly, one Von Willebrand factor type D domain, and three EGF-like domains [42].
MUC4 expression in healthy tissues is mainly seen in epithelial cells of the airways and in secretory fluids such as saliva, breast milk and tears [42,84]. While MUC4 in normal pancreatic tissue has been consistently reported to be undetectable, expression has been observed in precursor lesions and increases during neoplastic progression, with positive expression in 70 to nearly 95% of PDAC cases [85,86].
The role of MUC4 in PDAC has been shown to be fairly extensive, with involvement of this mucin with several other proteins, promoting tumorigenic processes. One of the main interactions is with growth factor receptors, particularly HER2 and HER3, via the EGF-like domains on the extracellular domain of MUC4. With this interaction, MUC4 mediates HER2 and HER3 activation, thus enhancing signaling cascades such as MAPK, JNK, STAT-1, promoting tumor proliferation and metastasis [40-42]. MUC4 was also recognized to play a role in E- and N-cadherin signaling, further enhancing proliferation and contributing to epithelial-mesenchymal transition [42]. Moreover, studies have indicated MUC4's contribution to immune evasion by numerous mechanisms, such as epitope masking and apoptosis of MUC4 specific T-cells. Finally, MUC4 appears to play a role in gemcitabine chemoresistance, by inhibiting gemcitabine-induced apoptosis and increasing the level of transporters that remove gemcitabine from the tumor cells [42]. MUC4 is also known to be involved in the interaction between the tumor cells and surrounding environment [42]. However, to our knowledge studies have not focused on detecting its expression in the tumor microenvironment.
With such an important role in PDAC, MUC4 is being evaluated as a potential therapeutic target, especially in the field of immunotherapy. In fact, like other cancer-associated mucins, MUC4 not only is overexpressed in PDAC but also displays aberrant glycosylation patterns which is absent in healthy tissues, making it a potential target for cancer vaccines [42,119]. While no MUC4-targeted vaccines have been studied in clinical trials, a few have been evaluated in vivo, with promising results [120,121]. Recently, Liu et al. developed a MUC4 nanovaccine by encapsulating a recombinant MUC4β subunit in nanoparticles. To assess antibody response, they immunized mice using the nanovaccine and analyzed the serum, showing the presence of MUC4β-specific antibodies, which were not detected in the control group [120]. The potent immune response induced by this and the other MUC4 vaccines in vivo suggests that this protein could have a significant role as a target for immunotherapy in PDAC.
In addition to the therapeutic potential, the frequent overexpression and aberrant glycosylation of MUC4 makes it a potential target for imaging. To this end, a recent study investigated both the therapeutic and imaging potential of MUC4 and two other targets using single-chain antibodies conjugated to magnetic iron oxide nanoparticles, in a subcutaneous mouse model of PDAC. The conjugate successfully acted as an MRI negative contrast agent and also showed anti-tumor effects in vivo [121].
MUC13
MUC13 is less known compared to the other mucin family members, but the studies published reveal this protein as a potential target in PDAC. MUC13 presents a short transmembrane domain, a cytoplasmic domain and a large extracellular portion composed of a tandem repeat domain, three EGF-like domains and a sea urchin sperm protein, enterokinase and agrin (SEA) domain [43].
MUC13 is normally expressed in the healthy trachea, stomach, intestines and kidneys, while expression in normal pancreas is virtually absent [43,45]. However, MUC13 expression is highly upregulated in PDAC. In fact, immunohistochemical studies of over 200 patient-derived samples confirmed membrane, cytoplasmic and nuclear localization of MUC13 in nearly 95% of the PDAC cases. Moreover, MUC13 expression was detected in all 29 precursor lesions, with increasing levels following the disease progression [45]. The expression of MUC13 in PDAC tumor microenvironment has not been reported yet.
Similar to MUC4, MUC13 has been shown to interact with HER2, likely through binding of HER2 with the EGF-like functional domains of MUC13. In vivo studies have reported elevated expression levels of HER2 in MUC13 overexpressing tumors, indicating that this mucin might affect the expression and activation of HER2 [43,44]. Activation of HER2 would subsequently lead to downstream signaling of oncogenic pathways PI3K/Akt and MAPK, as described in the HER2 section of this review. In addition, MUC13 is suspected to play a role in the expression of other oncogenic proteins, such as PAK1 and S100A4 which play role in tumor migration, invasion and metastasis. Furthermore, MUC13 overexpression was correlated with an increase in lymph node metastasis in vivo, supporting that MUC13 likely plays a role in these processes [43-45]. In addition to its role in the activation of pro-oncogenic proteins, MUC13 overexpression has been linked to downregulation of the tumor suppressing protein p53 [43]. Overall, these findings point to a significant role of MUC13 in PDAC tumorigenesis [43-45].
With such a frequent overexpression of MUC13 and an extensive role in the pathophysiology of PDAC, this protein is a potentially interesting target for both diagnostic and therapeutic purposes. Nonetheless, MUC13 has not been gaining as much attention as other members of this family and, to our knowledge, no MUC13 targeting-based treatments have been tested in preclinical or clinical studies.
MUC16
MUC16, also known as carbohydrate antigen (CA) 125, is often used as a serum diagnostic and prognostic biomarker for PDAC and other cancers, in combination with other well-known markers such as CA19-9 and carcinoembryonic antigen (CEA). MUC16 is the largest membrane-associated mucin, constituted by a short intracellular tail, a transmembrane domain, and a large extracellular region containing several tandem repeats, multiple SEA functional groups and a heavily glycosylated N-terminal domain [122,123].
MUC16 is normally found on epithelial cells of the respiratory tract, female reproductive tissues, cornea and conjunctiva, while overexpression has been observed in various types of cancer. In PDAC, expression of MUC16 has been reported in over 80% of 200 cases, whereas it is absent in the vast majority of normal pancreatic samples (112/115) [87]. In immunohistochemical analysis of patient-derived tissues, the expression of MUC16 is mostly limited to the cancer cells and not to the surrounding stroma [88]. Although MUC16 expression is shown to increase as PDAC advances, levels in precursor lesions were found to be relatively low, suggesting this mucin might not be the best target for the early diagnosis [87,88]. Nonetheless, MUC16 expression was shown to be strongly correlated with short patient survival, with particularly high expression at metastatic sites, indicating that this target plays a significant role in PDAC pathophysiology [40,87].
The role of MUC16 in PDAC involves the interaction of this mucin with several proteins. One of the most reported interactions is with mesothelin, which activates matrix metalloproteinases, resulting in increased cell motility and invasion [47]. MUC16 is also thought to be involved in cell invasion and metastasis, with a potential role of the aberrant glycans at its surface in MUC16-galectin-3 mediated metastasis, and also through interaction of MUC16 with focal adhesin kinase (FAK). The latter possibly can cause changes in the tumor cell adhesion and invasion, as a result of FAK-mediated Akt and ERK/MAPK signaling [47]. MUC16 upregulation was also linked to increased cell proliferation and tumor size, indicating that this mucin might induce cell proliferation, possibly through interaction with FAK [47]. Moreover, MUC16 might play a role in immune evasion, as its expression has been correlated with upregulation of immunosuppressive regulatory T cells [48]. Furthermore, MUC16 mutation has been correlated with extremely long survival and selective loss of high quality and MUC16 neoantigenic clones on metastatic progression has been found, suggesting neoantigen immunoediting [124].
As an established biomarker in PDAC, MUC16 has potential as diagnostic and therapeutic target. Of the developed treatments, however, only the monoclonal antibody oregovomab was evaluated in clinical trials, also in combination with chemo- and radiotherapy in a phase II study (NCT01959672). As expression of MUC16 has been linked to T-cell specific immunosuppression, one of the goals of this study was to evaluate the effects of oregovomab on the immune response. The study reported the development of MUC16-specific CD8 T-lymphocytes in some of the patients [48]. However, these results are not conclusive as the study was limited by a small sample size, and therefore further studies are needed to confirm these findings.
MUC16 is currently, being investigated in preclinical studies for imaging to aid in the removal of PDAC tumor during surgery. For instance, the fluorescent MUC16-targeted antibody probe AR9.6-IRDye800 was recently tested in an orthotopic xenograft mouse model. The probe was able to achieve tumor visualization with low background, indicating the potential of MUC16 for imaging [48]. An overview of the clinical trials targeting MUC16 is summarized in Table 3.
Discussion
PDAC is a complex disease with high mortality rate. The lack of effective treatment is due to high percentage of late-stage diagnosis, as well as biological aggressiveness and therapeutic resistance. Therefore, it is imperative to develop strategies for early diagnosis of PDAC and more effective and better-tolerated therapeutic options. While many targets have been investigated, only few have shown encouraging results. In this review we discussed a number of transmembrane proteins as promising targets in PDAC, based on studies published in the last 10 years. The selection of the targets was based on favorable features, namely high expression in a large percentage of PDAC cases, low expression in healthy tissues, as well as promising preclinical or clinical results.
Transmembrane proteins containing extracellular domains, can be excellent targets for the delivery of the therapeutic agents and (early) imaging. Various examples, especially in nuclear medicine, are proven successful for other clinical indications. In PDAC the expression of many transmembrane proteins is significantly increased, making them viable candidates for the development of specific imaging strategies and effective treatments with low side effects. Importantly, large variation between patients in terms of the protein expression level has been reported, resulting in inconsistent patterns of protein expression. Among the large number of proteins overexpressed in PDAC, only a few have shown favorable characteristics for effective targeting and imaging with promising preclinical and clinical results. Due to the large patient-to-patient variability, it is important to focus on targets that are overexpressed in the majority of PDAC patients. The majority of the targets described in this review have shown overexpression in a large percentage of cases, although for some targets there is large variation and expression in PDAC has not been as extensively studied for all targets.
Mesothelin and PDGFRβ have shown de novo expression in up to 100% of cases, showing strong expression in 80-85% and 49% of the cases, respectively. Importantly, mesothelin and PDGFRβ are not expressed in normal pancreatic tissue and therefore can be ideal targets for therapy and imaging. In particular for PDGFRβ, which has also been described to be expressed by tumor stroma, further studies are needed to assess the consequence of this for diagnostic or therapeutic purposes. To our understanding, even though ASPH, GPRC5A, HER2, integrin αvβ6 and mucins are expressed to some extend in normal tissues, their expression in normal pancreatic tissue has not been reported.
Expressions of EGFR and HER2 have shown a large variation in PDAC patients. In fact, while some studies have reported EGFR expression in nearly 90% of cases, others have reported much lower percentage (30%) [4,62]. Similarly, HER2 expression in PDAC varies from 0-82% [65]. The possible explanation for the differences might be related to the different scoring methods or interpatient variation, making it difficult to draw firm conclusions about the actual percentage of expression in PDAC. Thus, patient eligibility for EGFR or HER2 targeted therapies may need to be determined in a more personalized manner, for instance by assessing EGFR or HER2 expression by immunohistochemistry (IHC) on biopsies.
In addition to the therapeutic potential of the targets, overexpression of these proteins can be exploited for development of novel imaging strategies for (early) diagnosis and follow-up of PDAC. Early PDAC diagnosis has proven to be challenging, contributing to poor survival of patients. Among the targets discussed, currently only integrin αvβ6 is assessed for imaging purposes in clinical trials (NCT03164486, NCT02683824). However, other targets such as claudin 4, EGFR, mesothelin, MUC4 and MUC16 have shown promising results for PDAC imaging in vivo [48,75,97,98,110,111,117,118,121]. Although this has not yet been confirmed, proteins such as ASPH and PDGFRβ that are not detectable in healthy pancreas could also be potential targets for PDAC imaging.
Besides overexpression of the targets on tumor cells compared to healthy pancreas, absence of expression in other healthy tissues is also important for therapy. Therefore, identification of targets with high expression in PDAC and low or preferably no expression in healthy tissues is essential for the development of effective treatment with low side effects. In this regard, mesothelin and ASPH have shown the most promising characteristics. The expression of mesothelin is limited to mesothelial cells in the pleura, pericardium and peritoneum, and also according to the preclinical studies, mesothelin does not play a role in normal cell processes and development [38,81,82,114]. Similarly, ASPH is not expressed in healthy pancreatic tissue nor is it in many healthy tissues. Importantly, ASPH is an oncofetal protein which remains inactive after the embryonic development, further encouraging targeting of this protein in PDAC.
In spite of the expression of EGFR, claudin 4, GPRC5A, HER2, integrin αvβ6 and mucins in several normal tissues, the expression level is significantly higher in PDAC compared to other tissues, although the exact difference in expression levels has not been reported [61,74,125]. Nonetheless, lower expression in normal tissues would likely reduce side effects compared to the effects on tumor cells. To further reduce the side effects on the normal tissues, therapeutic agents could be applied locally at the tumor site. Last but not least, the elevated interstitial fluid pressure particularly present in PDAC appears to hinder efficient drug delivery. Combination of targeted therapies with local ablation therapies, such as radiofrequency (RFA), microwave (MWA), cryoablation (CA), and irreversible electroporation, could help to overcome this.
In conclusion, the development of effective treatments has been challenging due to the complexity of PDAC, and novel diagnostic and therapeutic strategies are needed. Many clinical studies have been conducted and more trials are still ongoing investigating the potential of therapeutic agents as monotherapy or in combination. Transmembrane proteins described in this review could serve as potential targets for diagnosis, and also to improve treatment efficacy and quality of patients' life.
Acknowledgements
A grant was provided by AAA, a Novartis company, solely for the research part of this project.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Aguirre AJ, Collisson EA. Advances in the genetics and biology of pancreatic cancer. Cancer J (United States). 2017;23:315-20
2. Aier I, Semwal R, Sharma A, Varadwaj PK. A systematic assessment of statistics, risk factors, and underlying features involved in pancreatic cancer. Cancer Epidemiol. 2019;58:104-10
3. Kyuno D. Targeting tight junctions during epithelial to mesenchymal transition in human pancreatic cancer. World J Gastroenterol. 2014;20:10813
4. Grapa CM, Mocan T, Gonciar D, Zdrehus C, Mosteanu O, Pop T. et al. Epidermal Growth Factor Receptor and Its Role in Pancreatic Cancer Treatment Mediated by Nanoparticles. Int J Nanomedicine. 2019 Volume 14: 9693-706
5. Wang Y, Wang H, Zhou L, Lu J, Jiang B, Liu C. et al. Photodynamic therapy of pancreatic cancer: Where have we come from and where are we going? Photodiagnosis Photodyn Ther. 2020;31:101876
6. Lai E, Puzzoni M, Ziranu P, Pretta A, Impera V, Mariani S. et al. New therapeutic targets in pancreatic cancer. Cancer Treat Rev. 2019;81:101926
7. Carrington C. Oral targeted therapy for cancer. Aust Prescr. 2015;38:171-6
8. Gilad Y, Gellerman G, Lonard DM, O'Malley BW. Drug Combination in Cancer Treatment - From Cocktails to Conjugated Combinations. Cancers (Basel). 2021;13:669
9. Hou G, Xu B, Bi Y, Wu C, Ru B, Sun B. et al. Recent advances in research on aspartate β-hydroxylase ( ASPH ) in pancreatic cancer : A brief update. 2018; 297-304.
10. Nagaoka K, Bai X, Ogawa K, Dong X, Zhang S, Zhou Y. et al. Anti-tumor activity of antibody drug conjugate targeting aspartate-β-hydroxylase in pancreatic ductal adenocarcinoma. Cancer Lett. 2019;449:87-98
11. Mukherjee I, Powell B, Parianos M, Downs D, Ross SB. Available technologies and clinical applications of targeted chemotherapy in pancreatic cancer. Cancer Genet. 2016;209:582-91
12. Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin A V. et al. Pancreatic cancer. Nat Rev Dis Prim. 2016;2:16022
13. Schizas D, Charalampakis N, Kole C, Economopoulou P, Koustas E, Gkotsis E. et al. Immunotherapy for pancreatic cancer: A 2020 update. Cancer Treat Rev. 2020;86:102016
14. Nishimura T, Mitsunaga M, Sawada R, Saruta M, Kobayashi H, Matsumoto N. et al. Photoimmunotherapy targeting biliary-pancreatic cancer with humanized anti-TROP2 antibody. Cancer Med. 2019;8:7781-92
15. Gao D, Gao L, Zhang C, Liu H, Jia B, Zhu Z. et al. A near-infrared phthalocyanine dye-labeled agent for integrin αvβ6-targeted theranostics of pancreatic cancer. Biomaterials. 2015;53:229-38
16. Obaid G, Bano S, Mallidi S, Broekgaarden M, Kuriakose J, Silber Z. et al. Impacting Pancreatic Cancer Therapy in Heterotypic in vitro Organoids and in vivo Tumors with Specificity-Tuned, NIR-Activable Photoimmunonanoconjugates: Towards Conquering Desmoplasia? Nano Lett. 2019;19:7573-87
17. Hull A, Li Y, Bartholomeusz D, Hsieh W, Allen B, Bezak E. Radioimmunotherapy of Pancreatic Ductal Adenocarcinoma: A Review of the Current Status of Literature. Cancers (Basel). 2020;12:481
18. Strosberg J, El-Haddad G, Wolin E, Hendifar A, Yao J, Chasen B. et al. Phase 3 Trial of 177 Lu-Dotatate for Midgut Neuroendocrine Tumors. N Engl J Med. 2017;376:125-35
19. Hofman MS, Emmett L, Sandhu S, Iravani A, Joshua AM, Goh JC. et al. [177Lu]Lu-PSMA-617 versus cabazitaxel in patients with metastatic castration-resistant prostate cancer (TheraP): a randomised, open-label, phase 2 trial. Lancet. 2021
20. Fitzgerald TL, Lertpiriyapong K, Cocco L, Martelli AM, Libra M, Candido S. et al. Roles of EGFR and KRAS and their downstream signaling pathways in pancreatic cancer and pancreatic cancer stem cells. Adv Biol Regul. 2015;59:65-81
21. Singh B, Carpenter G, Coffey RJ. EGF receptor ligands: recent advances. F1000Research. 2016;5:2270
22. Omar N, Yan B, Salto-Tellez M. HER2: An emerging biomarker in non-breast and non-gastric cancers. Pathogenesis. 2015;2:1-9
23. Harder J, Ihorst G, Heinemann V, Hofheinz R, Moehler M, Buechler P. et al. Multicentre phase II trial of trastuzumab and capecitabine in patients with HER2 overexpressing metastatic pancreatic cancer. Br J Cancer. 2012;106:1033-8
24. Lv Q, Meng Z, Yu Y, Jiang F, Guan D, Liang C. et al. Molecular Mechanisms and Translational Therapies for Human Epidermal Receptor 2 Positive Breast Cancer. Int J Mol Sci. 2016;17:2095
25. Kurahara H, Maemura K, Mataki Y, Sakoda M, Shinchi H, Natsugoe S. Impact of p53 and PDGFR-β Expression on Metastasis and Prognosis of Patients with Pancreatic Cancer. World J Surg. 2016;40:1977-84
26. Weissmueller S, Manchado E, Saborowski M, Morris JP, Wagenblast E, Davis CA. et al. Mutant p53 Drives Pancreatic Cancer Metastasis through Cell-Autonomous PDGF Receptor β Signaling. Cell. 2014;157:382-94
27. Chen P-H, Chen X, He X. Platelet-derived growth factors and their receptors: Structural and functional perspectives. Biochim Biophys Acta - Proteins Proteomics. 2013;1834:2176-86
28. Jahny E, Yang H, Liu B, Jahnke B, Lademann F, Ru P. et al. The G Protein-Coupled Receptor RAI3 Is an Independent Prognostic Factor for Pancreatic Cancer Survival and Regulates Proliferation via STAT3 Phosphorylation. 2017; 1-15.
29. Jiang X, Xu X, Wu M, Guan Z, Su X, Chen S. et al. GPRC5A: An Emerging Biomarker in Human Cancer. Biomed Res Int. 2018;2018:1-11
30. Zhou H, Rigoutsos I. The emerging roles of GPRC5A in diseases. Oncoscience. 2014;1:765-76
31. Sasaki T, Fujiwara-Tani R, Kishi S, Mori S, Luo Y, Ohmori H. et al. Targeting claudin-4 enhances chemosensitivity of pancreatic ductal carcinomas. Cancer Med. 2019;8:6700-8
32. Fujiwara-Tani R, Sasaki T, Luo Y, Goto K, Kawahara I, Nishiguchi Y. et al. Anti-claudin-4 extracellular domain antibody enhances the antitumoral effects of chemotherapeutic and antibody drugs in colorectal cancer. Oncotarget. 2018;9:37367-78
33. Nishiguchi Y, Fujiwara-Tani R, Sasaki T, Luo Y, Ohmori H, Kishi S. et al. Targeting claudin-4 enhances CDDP-chemosensitivity in gastric cancer. Oncotarget. 2019;10:2189-202
34. Mosley M, Knight J, Neesse A, Michl P, Iezzi M, Kersemans V. et al. Claudin-4 SPECT Imaging Allows Detection of Aplastic Lesions in a Mouse Model of Breast Cancer. J Nucl Med. 2015;56:745-51
35. Desgrosellier JS, Cheresh DA. Integrins in cancer: Biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9-22
36. Sipos B, Hahn D, Carceller A, Piulats J, Hedderich J, Kalthoff H. et al. Immunohistochemical screening for beta6-integrin subunit expression in adenocarcinomas using a novel monoclonal antibody reveals strong up-regulation in pancreatic ductal adenocarcinomas in vivo and in vitro. Histopathology. 2004;45:226-36
37. Montemagno C, Cassim S, Pouyssegur J, Broisat A, Pagès G. From Malignant Progression to Therapeutic Targeting: Current Insights of Mesothelin in Pancreatic Ductal Adenocarcinoma. Int J Mol Sci. 2020;21:4067
38. Hassan R. Mesothelin: A New Target for Immunotherapy. Clin Cancer Res. 2004;10:3937-42
39. Tang Z, Qian M, Ho M. The Role of Mesothelin in Tumor Progression and Targeted Therapy. Anticancer Agents Med Chem. 2013;13:276-80
40. Wang S, You L, Dai M, Zhao Y. Mucins in pancreatic cancer: A well-established but promising family for diagnosis, prognosis and therapy. J Cell Mol Med. 2020;24:10279-89
41. Kaur S, Kumar S, Momi N, Sasson AR, Batra SK. Mucins in pancreatic cancer and its microenvironment. Nat Rev Gastroenterol Hepatol. 2013;10:607-20
42. Gautam SK, Kumar S, Cannon A, Hall B, Bhatia R, Nasser MW. et al. MUC4 mucin- a therapeutic target for pancreatic ductal adenocarcinoma. Expert Opin Ther Targets. 2017;21:657-69
43. Chauhan SC, Ebeling MC, Maher DM, Koch MD, Watanabe A, Aburatani H. et al. MUC13 mucin augments pancreatic tumorigenesis. Mol Cancer Ther. 2012;11:24-33
44. Khan S, Ebeling MC, Zaman MS, Sikander M, Yallapu MM, Chauhan N. et al. MicroRNA-145 targets MUC13 and suppresses growth and invasion of pancreatic cancer. Oncotarget. 2014;5:7599-609
45. Khan S, Zafar N, Khan SS, Setua S, Behrman SW, Stiles ZE. et al. Clinical significance of MUC13 in pancreatic ductal adenocarcinoma. HPB. 2018;20:563-72
46. Maher DM, Gupta BK, Nagata S, Jaggi M, Chauhan SC. Mucin 13: Structure, Function, and Potential Roles in Cancer Pathogenesis. Mol Cancer Res. 2011;9:531-7
47. Muniyan S, Haridas D, Chugh S, Rachagani S, Lakshmanan I, Gupta S. et al. MUC16 contributes to the metastasis of pancreatic ductal adenocarcinoma through focal adhesion mediated signaling mechanism. Genes Cancer. 2016;7:110-24
48. Lin C, Verma V, Lazenby A, Ly QP, Berim LD, Schwarz JK. et al. Phase I/II Trial of Neoadjuvant Oregovomab-based Chemoimmunotherapy Followed by Stereotactic Body Radiotherapy and Nelfinavir For Locally Advanced Pancreatic Adenocarcinoma. Am J Clin Oncol. 2019;42:755-60
49. Aithal A, Rauth S, Kshirsagar P, Shah A, Lakshmanan I, Junker WM. et al. MUC16 as a novel target for cancer therapy. Expert Opin Ther Targets. 2018;22:675-86
50. The Human Protein Atlas - ASPH. https://www.proteinatlas.org/ENSG00000198363-ASPH/pathology/pancreatic+cancer
51. The Human Protein Atlas - EGFR. https://www.proteinatlas.org/ENSG00000146648-EGFR/pathology/pancreatic+cancer
52. The Human Protein Atlas - ERBB2. https://www.proteinatlas.org/ENSG00000141736-ERBB2
53. The Human Protein Atlas - GPRC5A. https://www.proteinatlas.org/ENSG00000013588-GPRC5A
54. The Human Protein Atlas - CLDN4. https://www.proteinatlas.org/ENSG00000189143 CLDN4/pathology/pancreatic+cancer
55. Reader CS, Vallath S, Steele CW, Haider S, Brentnall A, Desai A. et al. The integrin αvβ6 drives pancreatic cancer through diverse mechanisms and represents an effective target for therapy. J Pathol. 2019;249:332-42
56. The Human Protein Atlas - MSLN. https://www.proteinatlas.org/ENSG00000102854-MSLN/pathology/pancreatic+cancer
57. The Human Protein Atlas - MUC4. https://www.proteinatlas.org/ENSG00000145113-MUC4/pathology/pancreatic+cancer#ihc
58. NISHII Y, YAMAGUCHI M, KIMURA Y, HASEGAWA T, ABURATANI H, UCHIDA H. et al. A newly developed anti-Mucin 13 monoclonal antibody targets pancreatic ductal adenocarcinoma cells. Int J Oncol. 2015;46:1781-7
59. The Human Protein Atlas - MUC16. https://www.proteinatlas.org/ENSG00000181143 MUC16/pathology/pancreatic+cancer
60. Ogawa K, Lin Q, Li L, Bai X, Chen X, Chen H. et al. Aspartate β-hydroxylase promotes pancreatic ductal adenocarcinoma metastasis through activation of SRC signaling pathway. J Hematol Oncol. 2019;12:144
61. Ioannou N. Expression pattern and targeting of HER family members and IGF-IR in pancreatic cancer. Front Biosci. 2012;17:2698
62. Park SJ, Gu MJ, Lee DS, Yun SS, Kim HJ, Choi JH. EGFR expression in pancreatic intraepithelial neoplasia and ductal adenocarcinoma. 2015; 8: 8298-304.
63. De Luca A, Carotenuto A, Rachiglio A, Gallo M, Maiello MR, Aldinucci D. et al. The role of the EGFR signaling in tumor microenvironment. J Cell Physiol. 2008;214:559-67
64. Chen J, Zeng F, Forrester SJ, Eguchi S, Zhang M-Z, Harris RC. Expression and Function of the Epidermal Growth Factor Receptor in Physiology and Disease. Physiol Rev. 2016;96:1025-69
65. Saxby AJ, Nielsen A, Scarlett CJ, Clarkson A, Morey A, Gill A. et al. Assessment of HER-2 Status in Pancreatic Adenocarcinoma. Am J Surg Pathol. 2005;29:1125-34
66. Gurzu S, Bara T, Molnar C, Bara T, Butiurca V, Beres H. et al. The epithelial-mesenchymal transition induces aggressivity of mucinous cystic neoplasm of the pancreas with neuroendocrine component: An immunohistochemistry study. Pathol - Res Pract. 2019;215:82-9
67. Li Q, Zhang L, Li X, Yan H, Yang L, Li Y. et al. The prognostic significance of human epidermal growth factor receptor family protein expression in operable pancreatic cancer. BMC Cancer. 2016;16:910
68. Gerson JN, Skariah S, Denlinger CS, Astsaturov I. Perspectives of HER2-targeting in gastric and esophageal cancer. Expert Opin Investig Drugs. 2017;26:531-40
69. Yuzawa S, Kano MR, Einama T, Nishihara H. PDGFRβ expression in tumor stroma of pancreatic adenocarcinoma as a reliable prognostic marker. Med Oncol. 2012;29:2824-30
70. Inoue K, Ohtsuka H, Tachikawa M, Motoi F, Shijo M, Douchi D. et al. MK2461, a Multitargeted Kinase Inhibitor, Suppresses the Progression of Pancreatic Cancer by Disrupting the Interaction Between Pancreatic Cancer Cells and Stellate Cells. Pancreas. 2017;46:557-66
71. The Human Protein Atlas - PDGFRB. https://www.proteinatlas.org/ENSG00000113721-PDGFRB/tissue
72. Liu B, Yang H, Pilarsky C, Weber G. The Effect of GPRC5a on the Proliferation, Migration Ability, Chemotherapy Resistance, and Phosphorylation of GSK-3β in Pancreatic Cancer. Int J Mol Sci. 2018;19:1870
73. Borka K, Kaliszky P, Szabó E, Lotz G, Kupcsulik P, Schaff Z. et al. Claudin expression in pancreatic endocrine tumors as compared with ductal adenocarcinomas. Virchows Arch. 2007;450:549-57
74. Nichols LS, Ashfaq R, Iacobuzio-donahue CA. Claudin 4 Protein Expression in Primary and Metastatic Pancreatic Cancer Support for Use as a Therapeutic Target. 2004; 226-30.
75. Baguña Torres J, Mosley M, Koustoulidou S, Hopkins S, Knapp S, Chaikuad A. et al. Radiolabeled cCPE Peptides for SPECT Imaging of Claudin-4 Overexpression in Pancreatic Cancer. J Nucl Med. 2020 jnumed.120.243113
76. Hou J. Paracellular Channel in Organ System. In: The Paracellular Channel. Elsevier. 2019:93-141
77. de Geus SWLL, Boogerd LSFF, Swijnenburg R-JJ, Mieog JSD, Tummers WSFJFJ, Prevoo HAJMJM. et al. Selecting Tumor-Specific Molecular Targets in Pancreatic Adenocarcinoma: Paving the Way for Image-Guided Pancreatic Surgery. Mol Imaging Biol. 2016;18:807-19
78. Steiger K, Schlitter A-M, Weichert W, Esposito I, Wester H-J, Notni J. Perspective of α v β 6 -Integrin Imaging for Clinical Management of Pancreatic Carcinoma and Its Precursor Lesions. Mol Imaging. 2017;16:153601211770938
79. Li Z, Lin P, Gao C, Peng C, Liu S, Gao H. et al. Integrin β6 acts as an unfavorable prognostic indicator and promotes cellular malignant behaviors via ERK-ETS1 pathway in pancreatic ductal adenocarcinoma (PDAC). Tumor Biol. 2016;37:5117-31
80. Argani P, Iacobuzio-Donahue C, Rosty C, Goggins M, Wilentz RE, Murugesan SR. et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: Identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin Cancer Res. 2001;7:3862-8
81. Hagerty BL, Pegna GJ, Xu J, Tai C-H, Alewine C. Mesothelin-Targeted Recombinant Immunotoxins for Solid Tumors. Biomolecules. 2020;10:973
82. Einama T, Kamachi H, Nishihara H, Homma S, Kanno H, Takahashi K. et al. Co-Expression of Mesothelin and CA125 Correlates With Unfavorable Patient Outcome in Pancreatic Ductal Adenocarcinoma. Pancreas. 2011;40:1276-82
83. Le K, Wang J, Zhang T, Guo Y, Chang H, Wang S. et al. Overexpression of Mesothelin in Pancreatic Ductal Adenocarcinoma (PDAC). Int J Med Sci. 2020;17:422-7
84. Moulshree Kohli, Animesh Kumar Shivam, Puneet Ahuja JD. Mucin-4: A novel marker for oral cancer. J Oral Maxillofac Pathol. 2019;23:49-53
85. Seshacharyulu P, Ponnusamy MP, Rachagani S, Lakshmanan I, Haridas D, Yan Y. et al. Targeting EGF-receptor(s) - STAT1 axis attenuates tumor growth and metastasis through downregulation of MUC4 mucin in human pancreatic cancer. Oncotarget. 2015;6:5164-81
86. Urey C, Andersson B, Ansari D, Sasor A, Said-Hilmersson K, Nilsson J. et al. Low MUC4 expression is associated with survival benefit in patients with resectable pancreatic cancer receiving adjuvant gemcitabine. Scand J Gastroenterol. 2017;52:595-600
87. Streppel MM, Vincent A, Mukherjee R, Campbell NR, Chen S, Konstantopoulos K. et al. Mucin 16 ( cancer antigen 125 ) expression in human tissues and cell lines and correlation with clinical outcome in adenocarcinomas of the pancreas, esophagus, stomach. Hum Pathol. 2012;43:1755-63
88. Haridas D, Chakraborty S, Ponnusamy MP, Lakshmanan I, Rachagani S, Cruz E. et al. Pathobiological Implications of MUC16 Expression in Pancreatic Cancer. Srivastava RK, Ed. PLoS One. 2011;6:e26839
89. Zou Q, Hou Y, Wang H, Wang K, Xing X, Xia Y. et al. Hydroxylase Activity of ASPH Promotes Hepatocellular Carcinoma Metastasis Through Epithelial-to-Mesenchymal Transition Pathway. EBioMedicine. 2018;31:287-98
90. Barboro P, Benelli R, Tosetti F, Costa D, Capaia M, Astigiano S. et al. Aspartate β-hydroxylase targeting in castration-resistant prostate cancer modulates the NOTCH/HIF1α/GSK3β crosstalk. Carcinogenesis. 2020;41:1246-52
91. ClinicalTrials.gov. https://clinicaltrials.gov
92. National Cancer Institute. https://www.cancer.gov/
93. Renouf DJ, Tang PA, Hedley D, Chen E, Kamel-Reid S, Tsao MS. et al. A phase II study of erlotinib in gemcitabine refractory advanced pancreatic cancer. Eur J Cancer. 2014;50:1909-15
94. Wang JP, Wu C-Y, Yeh Y-C, Shyr Y-M, Wu Y-Y, Kuo C-Y. et al. Erlotinib is effective in pancreatic cancer with epidermal growth factor receptor mutations: a randomized, open-label, prospective trial. Oncotarget. 2015;6:18162-73
95. Cohenuram M, Saif MW. Epidermal growth factor receptor inhibition strategies in pancreatic cancer: past, present and the future. J Pancreas. 2007;8:4-15
96. Li Z, Wang M, Yao X, Luo W, Qu Y, Yu D. et al. Development of a Novel EGFR-Targeting Antibody-Drug Conjugate for Pancreatic Cancer Therapy. Target Oncol. 2019;14:93-105
97. Wang Q, Yan H, Wang Z, Li Z, Li D, Li Z. et al. Construction of a novel bispecific fusion protein to enhance targeting for pancreatic cancer imaging. Biomaterials. 2020;255:120161
98. Boyle AJ, Cao P-J, Hedley DW, Sidhu SS, Winnik MA, Reilly RM. MicroPET/CT imaging of patient-derived pancreatic cancer xenografts implanted subcutaneously or orthotopically in NOD-scid mice using 64Cu-NOTA-panitumumab F(ab')2 fragments. Nucl Med Biol. 2015;42:71-7
99. Yamanaka Y, Friess H, Kobrin MS, Büchler M, Kunz J, Beger HG. et al. Overexpression of HER2/neu oncogene in human pancreatic carcinoma. Hum Pathol. 1993;24:1127-34
100. Dugan MC, Dergham ST, Kucway R, Singh K, Biernat L, Du W. et al. HER-2/neu Expression in Pancreatic Adenocarcinoma. Pancreas. 1997;14:229-36
101. Shibata W, Kinoshita H, Hikiba Y, Sato T, Ishii Y, Sue S. et al. Overexpression of HER2 in the pancreas promotes development of intraductal papillary mucinous neoplasms in mice. Sci Rep. 2018;8:6150
102. Heldin C-H, Lennartsson J. Structural and Functional Properties of Platelet-Derived Growth Factor and Stem Cell Factor Receptors. Cold Spring Harb Perspect Biol. 2013;5:a009100-a009100
103. Askoxylakis V, Marr A, Altmann A, Markert A, Mier W, Debus J. et al. Peptide-Based Targeting of the Platelet-Derived Growth Factor Receptor Beta. Mol Imaging Biol. 2013;15:212-21
104. Marech I, Patruno R, Zizzo N, Gadaleta C, Introna M, Zito AF. et al. Masitinib (AB1010), from canine tumor model to human clinical development: Where we are? Crit Rev Oncol Hematol. 2014;91:98-111
105. Cuneo KC, Geng L, Fu A, Orton D, Hallahan DE, Chakravarthy AB. SU11248 (Sunitinib) Sensitizes Pancreatic Cancer to the Cytotoxic Effects of Ionizing Radiation. Int J Radiat Oncol. 2008;71:873-9
106. Martínez-Bosch N, Guerrero PE, Moreno M, José A, Iglesias M, Munné-Collado J. et al. The pancreatic niche inhibits the effectiveness of sunitinib treatment of pancreatic cancer. Oncotarget. 2016;7:48265-79
107. Zhou H, Telonis AG, Jing Y, Xia NL, Biederman L, Jimbo M. et al. GPRC5A is a potential oncogene in pancreatic ductal adenocarcinoma cells that is upregulated by gemcitabine with help from HuR. Cell Death Dis. 2016;7:e2294-e2294
108. Hashimoto Y, Kawahigashi Y, Hata T, Li X, Watari A, Tada M. et al. Efficacy and safety evaluation of claudin-4-targeted antitumor therapy using a human and mouse cross-reactive monoclonal antibody. Pharmacol Res Perspect. 2016;4:e00266
109. Neesse A, Griesmann H, Gress TM, Michl P. Claudin-4 as therapeutic target in cancer. Arch Biochem Biophys. 2012;524:64-70
110. Torres JB, Knight JC, Mosley MJ, Kersemans V, Koustoulidou S, Allen D. et al. Imaging of Claudin-4 in Pancreatic Ductal Adenocarcinoma Using a Radiolabelled Anti-Claudin-4 Monoclonal Antibody. Mol Imaging Biol. 2018;20:292-9
111. Neesse A, Hahnenkamp A, Griesmann H, Buchholz M, Hahn SA, Maghnouj A. et al. Claudin-4-targeted optical imaging detects pancreatic cancer and its precursor lesions. Gut. 2013;62:1034-43
112. Alberts B, Johnson A, Lewis J. Molecular Biology of the Cell. 4th ed. New York: Garland Science. 2002
113. Quigley NG, Tomassi S, Saverio Di Leva F, Di Maro S, Richter F, Steiger K. et al. Click-Chemistry (CuAAC) Trimerization of an α v β 6 Integrin Targeting Ga-68-Peptide: Enhanced Contrast for in-Vivo PET Imaging of Human Lung Adenocarcinoma Xenografts. ChemBioChem. 2020 cbic.202000200
114. Bera TK, Pastan I. Mesothelin Is Not Required for Normal Mouse Development or Reproduction. Mol Cell Biol. 2000;20:2902-6
115. Nichetti F, Marra A, Corti F, Guidi A, Raimondi A, Prinzi N. et al. The Role of Mesothelin as a Diagnostic and Therapeutic Target in Pancreatic Ductal Adenocarcinoma: A Comprehensive Review. Target Oncol. 2018;13:333-51
116. Hassan R, Blumenschein GR, Moore KN, Santin AD, Kindler HL, Nemunaitis JJ. et al. First-in-Human, Multicenter, Phase I Dose-Escalation and Expansion Study of Anti-Mesothelin Antibody-Drug Conjugate Anetumab Ravtansine in Advanced or Metastatic Solid Tumors. J Clin Oncol. 2020;38:1824-35
117. Montemagno Cassim, Trichanh Savary, Pouyssegur Pagès. et al. 99mTc-A1 as a Novel Imaging Agent Targeting Mesothelin-Expressing Pancreatic Ductal Adenocarcinoma. Cancers (Basel). 2019;11:1531
118. Shao C, Liu F, Le W, Mei T, Wang T, Chen L. et al. In vitro and in vivo targeting imaging of pancreatic cancer using a Fe3O4@SiO2 nanoprobe modified with anti-mesothelin antibody. Int J Nanomedicine. 2016 2195
119. Gautam SK, Kumar S, Dam V, Ghersi D, Jain M, Batra SK. MUCIN-4 (MUC4) is a novel tumor antigen in pancreatic cancer immunotherapy. Semin Immunol. 2020;47:101391
120. Liu L, Kshirsagar P, Christiansen J, Gautam SK, Aithal A, Gulati M. et al. Polyanhydride nanoparticles stabilize pancreatic cancer antigen MUC4β. J Biomed Mater Res Part A. 2020
121. Cai H, Palitzsch B, Hartmann S, Stergiou N, Kunz H, Schmitt E. et al. Antibody Induction Directed against the Tumor-Associated MUC4 Glycoprotein. ChemBioChem. 2015;16:959-67
122. Kufe DW. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer. 2009;9:874-85
123. O'Brien TJ, Beard JB, Underwood LJ, Dennis RA, Santin AD, York L. The CA 125 Gene: An Extracellular Superstructure Dominated by Repeat Sequences. Tumor Biol. 2001;22:348-66
124. Balachandran VP, Łuksza M, Zhao JN, Makarov V, Moral JA, Remark R. et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017;551:512-6
125. Michl P, Buchholz M, Rolke M, Kunsch S, Löhr M, McClane B. et al. Claudin-4: A new target for pancreatic cancer treatment using Clostridium perfringens enterotoxin. Gastroenterology. 2001;121:678-84
Author contact
![]() Corresponding author: E-mail: s.oliveiranl; Tel.: +31-63-410-3460.
Corresponding author: E-mail: s.oliveiranl; Tel.: +31-63-410-3460.