Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
The production and downstream...
The role of PGE2 on organ repair...
PGE2-based therapeutic...
Conclusions and future...
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(18):8836-8854. doi:10.7150/thno.63396 This issue Cite
Review
Role of prostaglandin E2 in tissue repair and regeneration
Hui Cheng1,2,3, Haoyan Huang1,2, Zhikun Guo4, Ying Chang3 ![]() , Zongjin Li1,2,3,4,5
, Zongjin Li1,2,3,4,5 ![]()
1. Nankai University School of Medicine, Tianjin, China.
2. The Key Laboratory of Bioactive Materials, Ministry of Education, Nankai University, the College of Life Sciences, Tianjin, China.
3. Tianjin Key Laboratory of Human Development and Reproductive Regulation, Nankai University Affiliated Hospital of Obstetrics and Gynecology, Tianjin, China.
4. Henan Key Laboratory of Medical Tissue Regeneration, Xinxiang Medical University, Xinxiang, China.
5. State Key Laboratory of Kidney Diseases, Chinese PLA General Hospital, Beijing, China.
Received 2021-6-1; Accepted 2021-8-5; Published 2021-8-13
Abstract

Tissue regeneration following injury from disease or medical treatment still represents a challenge in regeneration medicine. Prostaglandin E2 (PGE2), which involves diverse physiological processes via E-type prostanoid (EP) receptor family, favors the regeneration of various organ systems following injury for its capabilities such as activation of endogenous stem cells, immune regulation, and angiogenesis. Understanding how PGE2 modulates tissue regeneration and then exploring how to elevate the regenerative efficiency of PGE2 will provide key insights into the tissue repair and regeneration processes by PGE2. In this review, we summarized the application of PGE2 to guide the regeneration of different tissues, including skin, heart, liver, kidney, intestine, bone, skeletal muscle, and hematopoietic stem cell regeneration. Moreover, we introduced PGE2-based therapeutic strategies to accelerate the recovery of impaired tissue or organs, including 15-hydroxyprostaglandin dehydrogenase (15-PGDH) inhibitors boosting endogenous PGE2 levels and biomaterial scaffolds to control PGE2 release.
Keywords: Prostaglandin E2, Stem cell, Tissue repair, Regeneration, Inflammation
Introduction
Effective tissue repair and regeneration after injury is critical for function maintenance and survival of all living organisms, which attract researchers to identify common regulators in tissue repair and regeneration [1]. In recent years, increasing evidences have defined that prostaglandin E2 (PGE2), a lipid signaling molecule involved in pain and inflammation, might potentiate tissue regeneration and repair following injury in diverse organ systems [2-4]. The utilization of nonsteroidal anti-inflammatory drugs (NSAIDs) reducing PGE2 levels in patients usually cause side effects on the tissue repair process [5-7]. Thus, further understanding and exploring the universal and unique mechanism of PGE2 in the process of organ repair might contribute to the development of the field of regenerative medicine. Unexpectedly, PGE2 has a faster turnover rate in vivo due to degradation by 15-hydroxyprostaglandin dehydrogenase (15-PGDH), converting it into an inactivated biomolecule. In recent years, research on 15-PGDH inhibitors in the field of regeneration has gradually increased, while biomaterials delivery system delivery regulating PGE2 release has improved the regeneration efficacy of PGE2. Here, we summarize recent researches about PGE2 instructing tissue repair and regeneration in different organ systems including skin, heart, liver, kidney, gut, bone, skeletal muscle, and hematopoietic system. Then, we describe the advancement of therapeutic strategies for enhancing tissue regeneration with PGE2.
The production and downstream signal of endogenous PGE2
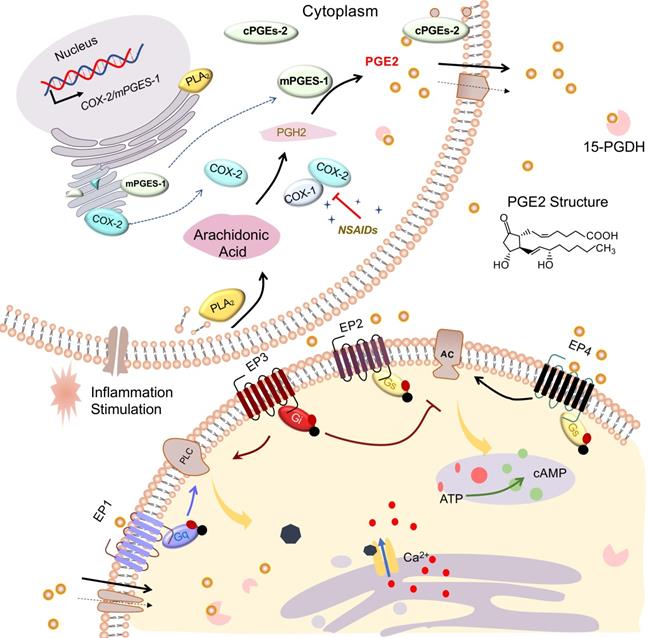
PGE2, a mediator of many physiological and pathological functions [8], can be produced by nearly all cell types of the body, such as epithelia, fibroblasts, and especially infiltrating inflammatory cells. The production of PGE2 increases significantly in damaged tissue [9]. Under diverse stimulation such as inflammation, PGE2 is generated from arachidonic acid (AA) that is liberated from membrane phospholipids catalyzed by phospholipase A2 (PLA2). Then the AAs are oxygenated to form Prostaglandin H2 (PGH2) by cyclooxygenase (COX), which is then transformed into PGE2 via terminal PGE2 synthases (PGES).
In detail, AA is immediately converted at the luminal side of nuclear and endoplasmic reticulum (ER)-membranes into the intermediate PGH2 by COX and converted into different prostaglandins by cell- and tissue-specific prostaglandin synthases (PGS). Constitutively expressed COX-1 is responsible for catalyzing the production of prostaglandins, which involve in several physiological functions. Inducible COX-2 is a rate-limiting enzyme for the synthesis of PGE2 and has been reported to be highly induced by inflammatory mediators following injury [9]. Then, cytosolic prostaglandin E synthases (cPGES) and microsomal prostaglandin E synthases1/2 (mPGES-1/2) are responsible for the production of PGE2 from PGH2. mPGES-1 is mainly coupled with COX-2 to increase the production of PGE2, especially under the stimulation of inflammatory factors. However, mPGES-2 and cPGES expression are constitutive rather than regulatory. To perform further function, the synthesized PGE2 may be actively transported by the prostaglandin efflux transporter MRP4 (multidrug resistance protein 4) in addition to exit from the cell by simple diffusion [10]. NSAIDs, widely used drugs in patients requiring pain reduction and inflammation control, not only decrease PGE2 production by suppressing COX activity but also inhibit MRP4-regulated PGE2 release [10, 11].
Subsequently, massively synthesized PGE2 will exert its diverse and complex biologic effects by binding to different downstream prostaglandin E receptors EP1, EP2, EP3, and EP4 that are located on the cell membrane or organelle membrane [12, 13]. EP receptors belong to a large family of seven transmembrane domain receptors coupled to specific G proteins that activate different second messenger signaling pathways. The final output of PGE2 signaling depends on the expression of each EP receptor and the strength of each EP signal [14]. EP1 (couple to Gq) and EP3 (couple to Gi) mediate PGE2-induced intracellular calcium mobilization. The EP2 and EP4 receptors coupled to Gs activate adenylate cyclase (AC) and increase cAMP production, whereas the EP3 receptor inhibits cAMP signaling. EP receptors, selective agonists, or antagonists were used to amplify or antagonize PGE2 signal effects [15-18]. It's reported that solute carrier organic anion transporter family member 2A1 (SLCO2A1), known as prostaglandin transporter (PGT), is responsible for transporting extracellular PGE2 into the cell and performs a function in the intracellular activity of PGE2 [13, 19]. However, PGE2 influx mediated by SLCO2A1 is rapidly metabolized into inactivated 15-keto PGE2 by 15-PGDH [19, 20]. Therefore, inhibition 15-PGDH may be an effective way to elevate PGE2 levels and enhance its downstream signal effects [21, 22]. Finally, we summarize the overall process of PGE2 from synthesis to activating downstream signaling in Figure 1.
PGE2 synthesis and signaling pathways. Arachidonic acid (AA) is liberated from membrane phospholipids by catalysis of phospholipase A2 (PLA2), subsequently converted into PGH2 via COXs including COX-1 and COX-2. NSAIDs can restrain COX activity. Synthesized by PGE synthase (cPGES, mPGES-1, and mPGES-2), PGE2 exerts its effects by binding to G protein-coupled receptors EP1-EP4. Both activated EP1 (coupled to Gq) and EP3 (coupled with Gi) could increase intracellular Ca2+ via phospholipase C (PLC). Activated EP3 receptors could inhibit cAMP production via adenylate cyclase (AC). Activated EP2 or EP4 (both coupled to Gs) could elevate cAMP production via AC. Arrowheads refer to stimulation, whereas blunt ends indicate inhibition.
The role of PGE2 on organ repair and regeneration
PGE2 and cutaneous wound healing
Non-healing cutaneous wounds caused by surgery, diabetes, or extreme temperature, have always been a severe spirit and financial burden for patients with abnormal wound healing [23, 24]. Normal wound healing processes comprise several overlapping phases including hemostasis, inflammatory response, proliferation, and tissue remodeling. Among them, the proliferation stage includes re-epithelialization, granulation tissue formation, and angiogenesis. If one or more stages of the wound healing process are interrupted or dysregulated, the healing rate will be delayed. In the cutaneous wound model, the expression of COX-2/PGE2/EP4 rises shortly after damage [25, 26]. Elevated PGE2 level accelerates the cutaneous wound healing process [25, 27]. Even though COX-1 is expressed constitutively, it still benefits PGE2 production and prevents wound healing disorders [28].
PGE2 displays great promise for the therapy of excisional skin wounds as it participates in different pathological repair processes with its function of anti-inflammatory, promoting angiogenesis, especially preventing scar formation [2]. To effectively keep the concentration of PGE2, we incorporated PGE2 into chitosan hydrogel to treat injured sites in a murine model of cutaneous wound healing [2]. Consistent with previous reports, PGE2 not only accelerates the healing rate but also remodels the skin structure in injured sites with new hair follicles and sebaceous glands. Furthermore, PGE2 hydrogel displays obvious anti-inflammatory and pro-angiogenesis effects via inducing macrophage polarization from the M1 phenotype to M2 phenotype at injured sites. More importantly, PGE2 can reduce pathological scar formation caused by the deposition of excessive extracellular matrix (ECM) secreted by myofibroblasts because PGE2 hydrogel markedly reduces the infiltration of myofibroblasts. Besides, enhanced expression of anti-fibrotic genes and decreased collagen deposition was observed after PGE2 hydrogel treatment. Inhibiting the TGF‐β1/SMAD pathway by PGE2 might be relevant to its ability to restrain collagen synthesis in dermal fibroblasts [29]. Similarly, previous data have also shown that PGE2 affects the migration and contraction of human fetal dermal fibroblasts, preventing fibrotic processes during wound healing [30, 31]. In excessive wound scarring, the content of PGE2 reduces. To improve PGE2 levels, Hoon Cho et al. designed a series of 15-PGDH inhibitors [32, 33]. Then, the 15-PGDH inhibitor (TD88) was investigated for its potential healing ability in vivo and in vitro [33]. As a result, a significant decrease in scar formation was observed after TD88 treatment following improved reepithelization [33].
Several investigators have identified PGE2 as an angiogenesis promotor in the healing of chronic cutaneous wounds induced by diabetes diseases, in which dermal endothelial cells are seriously impaired in neovascularization, resulting in peripheral ischemia and delayed healing process [31, 34, 35]. Regulating the activity of PGT to elevate the content of PGE2 in the circulation is essential for accelerating diabetes-associated wound closure [35, 36]. COX-2/PGE2 pathway is increased in murine and human diabetic monocytes/macrophages. Unsuitable PGE2 activity might maintain the inflammatory phenotype of wound macrophages, which is not conducive to the repair of diabetic wounds [37]. The duality of PGE2 and the complexity of the body require us to explore more to find the better therapeutic effect.
PGE2 in myocardial injury and repair
Occlusion of the coronary artery results in myocardial ischemia that is a common clinical symptom characterized by low pH values, low oxygen, and high extracellular potassium concentration. However, myocardial reperfusion supplying oxygen and nutrients results in a series of abrupt biochemical and metabolic changes within the myocardium. Myocardial ischemia/reperfusion (I/R) injuries induce arrhythmias, myocardial stunning, microvascular dysfunction, and even myocyte death [38]. Therefore, it's necessary to develop effective cardioprotective strategies and agents against myocardial I/R injury to improve myocardial function and to diminish the risk of cardiovascular events. In the heart with acute myocardial infarction, the production of PGE2 increases significantly in fibroblasts, myocardial cells, and vascular endothelial cells [39, 40]. An accumulating body of evidence indicates that both exogenous and endogenous PGE2 could exert cardiac protection function against ischemia reperfusion injury [41].
Failure to effectively stimulate the proliferation of cardiomyocytes is still the main obstacle to adult heart regeneration. Cardiac stem cells can be observed not only in the infarcted area and but also in the peri-infarcted area of the injured myocardium after injury. PGE2 is an important lipid molecule that activates endogenous stem/progenitor cells for myocardium regeneration after infarction [42-44]. Patrick C H Hsieh's team indicated that COX-2/PGE2/EP2 signaling promotes cardiac stem/progenitor cell differentiation into cardiomyocytes after infarction in young mice [43]. Surprisingly, PGE2 also rescues the cardiomyocyte regeneration function in aged mice [43]. In a study after that, Patrick C H Hsieh's team emphasized the importance of the PGE2 signaling pathway in myocardial regeneration once again [45]. In zebrafish heart resection injury models, injury-induced PGE2 signaling might drive cardiomyocyte proliferation during the regeneration process [42]. Moreover, by taking advantage of the natural infarct homing ability of platelet membrane and the overexpression of EP in infarcted myocardium, a novel biomimetic therapeutic nanoparticle with a platelet membrane shell conjugated with PGE2 and a core containing regeneration factors were designed to target injured heart and achieve delivery of the factors in the core. Consequently, increased cycling of cardiomyocytes, activated endogenous stem/progenitor cells, and angiogenesis was observed [44].
Both COX and mPGES-1 that determine the generation of PGE2 are required for cardiac structure repair and functional recovery following myocardial I/R injury. They are mutually irreplaceable in repair function [46, 47]. There is increased cardiovascular disease risk and worse repairability, particularly in patients utilizing high-dose NSAIDs [48]. Likewise, the same negative symptoms occur in animal models with genetic deletion of COX-1 or COX-2 [46, 48, 49]. For example, selective deletion of COX-2 in cardiomyocytes in mice can result in induced arrhythmia and reduced cardiac function. In contrast, cardiac-specific COX-2 overexpressing or constitutive expression transgene mice had small infarct size and better functional recovery in I/R injury models compared with wild type (WT) mice [41, 50]. mPGES-1, the dominant synthetic enzyme for PGE2 production in vivo, is being considered as a new therapeutic target. Deletion of mPGES-1 in bone marrow-derived leukocytes results in impaired left ventricular (LV) remodeling such as impaired LV systolic and diastolic, leukocyte infiltration, and higher mortality after acute myocardial infarction [40]. Consistently, in a myocardial infarction model, global deletion of mPGES-1can increase infarct size, reduce fractional shortening and ejection fraction [46] as well as impair microvascular perfusion via further enhancing myeloperoxidase levels and limiting leukocyte-endothelial cells interactions with EP4 receptor. These results indicate that the PGE2 signaling pathway is essential for the repair of myocardial ischemia-reperfusion and the utilization of related inhibitors such as NSAIDs should be cautious.
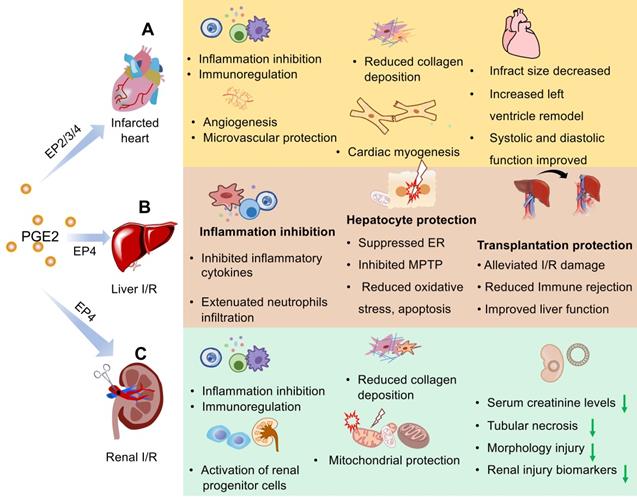
The expression levels of EP receptors may affect the role of PGE2 in the repair of myocardial I/R injury [43, 44]. Among EP receptors, EP4 is the most abundantly expressed and there are more studies on EP4 in the myocardial I/R model [51-55]. It's reported that overexpression of EP4 improves cardiac function after I/R injury through the reduction in adverse cardiac remodeling, inhibition of inflammation cytokine/chemokine production, and attenuated collagen deposition [51, 52] while some specific EP4 receptors agonists uncover the same protective function [51, 53, 56]. As expected, cardiac function was worse after myocardial injury with various EP4 receptors and genetic deletion approaches [45, 54]. Some researchers report that despite the expression of EP4 subtypes in hearts, selective stimulation of the EP2/3 receptor also displays cardio-protection against I/R injury. As we mentioned, EP2 mediates cardiomyocyte regeneration of PGE2 after myocardial ischemia via regulating macrophage activation such as migration towards the injured myocardium and phenotypic transformation from M1 to M2 in the injured myocardium [43, 45]. According to previous research, cardiac specific overexpression of the EP3 receptor could mitigate ischemic contracture, creatine kinase, and lactate dehydrogenase release and improve left ventricular remodeling post- myocardial I/R injury [57]. Targeted overexpression of EP3 receptors in macrophages can also facilitate cardiac healing by enhancing angiogenesis via vascular endothelial growth factor (VEGF) secretion [58]. To sum up, The EP receptor is an important target for the treatment of myocardial infarction. The EP receptor agonist can mimic PGE2 to deactivate downstream signaling pathways and should be considered as a good strategy to treat MI patients (Figure 2A).
Schematic representation of the protective effects of PGE2 in I/R injury of heart, liver, and kidney. I/R injury that includes initial simple ischemia and subsequent reperfusion results in impaired endothelial cell barrier function, activation of cell death programs, and transcriptional reprogramming associated with inflammation and immune reaction. By binding to members of the EP family, PGE2 can decrease the inflammation response and maintain immune homeostasis in three organs with I/R injury. PGE2 can also promote angiogenesis, protect microcirculation against reoxygenation injury, and alleviate fibrosis caused by I/R injury. Apoptosis or necrosis of endothelial or epithelial cells and some parenchymal cells is prevented by PGE2. In different organs, PGE2 plays different roles. (A) In the myocardial infarction model, PGE2 reduces infarct size, promotes left ventricle remodeling as well as systolic and diastolic function. PGE2 also offers the benefit of cardiomyocyte regeneration. In hepatic (B) or renal (C) transplantation-induced I/R injury, PGE2 contributes substantially to the success rate and survival rate. Furthermore, PGE2 exerts remarkable effects on functional protection in organs with I/R injury. The downward green arrow indicates the inhibition effects by PGE2. ER, Endoplasmic reticulum stress; MPTP, mitochondrial permeability transition pore.
PGE2 in hepatic injury and repair
Liver I/R injury is a serious complication that occurs in liver surgery, liver transplantation, and trauma. The insult induces not only damage of hepatocytes and liver sinusoidal endothelial cells but also destroys liver function and a cascade of dysfunction of other organs. Metabolic dysregulation, oxygen lack, and ATP depletion occur in hepatocytes during ischemia, leading to multifactorial hepatocyte death. Metabolically stressed hepatocytes release damage-associated molecular patterns, resulting in the induction of the innate immune response during subsequent reperfusion, while severe oxidative stress and necro apoptosis are induced because of reoxygenation. Hepatic I/R injury can be categorized into warm I/R and cold storage reperfusion injury. Warm hepatic I/R injury regularly occurs during liver resection as several procedures require intermittent total vascular occlusion and the ischemia time is relatively short. However, it's substantially longer in liver transplantation, with both cold ischemia up to 12 hours during preservation and warm ischemia during implantation, leading to increased reperfusion injury. In addition, drug-induced liver injury represents multiple reactions after exposure to any man-made or naturally occurring compound, and drug-induced hepatotoxicity is a frequent cause of liver injury [59].
Over the past 30 years, the protective role of PGE2 in liver I/R has aroused the interest of scientists [60-64]. During hepatic injury, the activity of AA, PLA2, COX-2, mPGES-1, or PGE2 receptors EPs were significantly increased following I/R injury [65-68]. In other words, increased hydrolysis of AA (a sort of unsaturated fatty acid) via PLA2 triggers the activity of COX and leads to increased PGE2 levels and relative downstream signals. Endogenous PGE2 is produced by many cells in the liver, mainly by hepatocytes [69], Kupffer cells [60, 61], and endothelial.
PGE2 can expedite the repair of liver ischemia-reperfusion injury by reducing liver inflammation, fibrosis, necrosis, etc. The inhibition of arachidonic acid may mediate aggravated inflammation in liver I/R injury [68]. Compared with WT mice, hepatocyte-specific COX-2 transgenic mice with liver I/R injury have less hepatocyte necrosis, reduced recruitment and infiltration of neutrophils, decreased expression levels of proinflammatory factors, attenuated oxidative stress injury, and endoplasmic reticulum stress [66]. It's demonstrated that COX-2 expression in hepatocytes restricts the activation of hepatic stellate cells, attenuating liver fibrosis in mice, reminding us that PGE2 may play a key role in alleviating hepatic fibrosis mediated by I/R [70]. Gradually increasing PGE2 molecular secreted by activated Kupffer cells in the late period of hepatic I/R injury, it was observed to serve as a mediator to regulate and control local TNF-α release that induces inflammation reaction and organ dysregulation [60].
In response to PGE2, the role of each PGE2 receptor in liver I/R is responsible for signal transmission. All four receptors were expressed in both the naive and ischemic liver, whereas there are more experimental data about EP4 on liver I/R among all EP receptors. This may be due to injured organ-dependent or cell type-dependent. Notably, the application of EP4 agonists may contribute greatly to liver function protection in murine models of hepatic I/R injury [67, 71]. It was also documented that upregulated hepatic EP4 expression could alleviate liver I/R injury by inhibiting mitochondrial permeability transition pore opening via activation of the ERK1/2-GSK3β pathway as well as ameliorating several proinflammatory cytokines, chemokines, and adhesion molecules in the early phase of reperfusion [71, 72].
However, some research results evoke an interesting paradox that deletion or inhibition of mPGES-1 or EP receptors facilitates liver repair following partial hepatic I/R injury through the accumulation of anti-inflammatory macrophages [73]. In summary, there is more evidence that PGE2 is beneficial and the reverse results may be due to different ischemia reperfusion times, distinct model animals with different pathological backgrounds, or diverse treatment methods such as local or global I/R, warm or cold reperfusion.
In recent years, the role of PGE2 in liver transplantation has attracted the attention of scientists. Recipients who are measured with higher PGE2 levels in plasma may have a better graft effect [66, 74] and donors with ischemia preconditioning to elevate PGE2 levels may be protective against liver transplantation related injury [61, 66]. Consistent with this, researchers making use of antibiotics for pretreatment in orthotopic liver transplantation recipients (human and mice), detected increased serum PGE2 levels and hepatic EP4 expression that suppress ER stress, enhance the autophagy pathway and improve hepatocellular function, resulting in alleviated liver transplant damage [74]. PGE2 also seems to play key roles in protection against acute liver failure [75], acute graft rejection following liver transplantation [76], and other liver disorders [77].
Liver removal from a donor is also a major operation, and its trauma and complications are inevitable. Previous evidence suggests that the utilization of COX-2 antagonists accelerates liver regeneration [78, 79]. Recently, researchers revealed that the liver regeneration rate and liver weight markedly increased after resection of two-thirds size in both 15-PGDH knockout mice and SW033291-treated mice compared with the control group [80]. Likewise, higher levels of cyclic AMP also were observed after operation in the regenerating liver [80]. Collectively, these results indicate that PGE2 has great potential in liver regeneration and is a chemical molecular promising for clinical application (Figure 2B).
PGE2 has also emerged as a positive regulator of drug-induced liver injury. Acetaminophen (APAP), one of the NSAIDs, and carbon tetrachloride are the two most common drugs inducing acute liver failure. In zebrafish models with APAP-induced liver injury, PGE2 has been identified as a potential therapeutic agent to decrease APAP-associated toxicity via enhancing Wnt activity [81]. According to reports, sPLA2, overly secreted by the liver, is a leading mediator of the progression of hepatic injury initiated by APAP and CCl4 due to the lack of COX-2 [82, 83]. Furthermore, liver injury is distinctly higher in COX-2 knockout mice compared with the WT mice, emphasizing the unique contribution of PGE2 to drug-induced liver injury once again [82, 83]. Celecoxib, inhibiting the activity of COX-2, aggravates liver fibrosis in CCl4-treated mice compared with the control group [84]. Along with COX-2, COX-1 also protects the liver against apoptosis, oxidative stress, and inflammatory response in the CCL4-induced liver injury model [85]. Lipopolysaccharide (LPS), an endotoxin composed of lipids and polysaccharides, often is utilized as an inducer of inflammation. PGE2/EP4 has been benefited to alleviate LPS-induced hepatic TNF-α expression and injury [86]. In contrast, another study showed that 15-PGDH converting PGE2 to 15-keto-PGE2 may have the advantage of preventing LPS/Galactosamine induced acute liver inflammation and injury, which may be related to PPAR-γ signaling restraining inflammation [87]. Taken together, these data remind us of the complexity of the body's repair mechanism and the diversified function of PGE2 under different injury backgrounds.
PGE2 and renal I/R injury
Renal I/R injury, arising from shock or kidney transplantation, is one of the leading causes of acute kidney injury. During kidney I/R injury, initial ischemia triggers alterations in tubular endothelial structure and function, significantly leading to the overall kidney injury. The microcirculation is subsequently compromised by further vascular perfusion and oxygenation, while hypoperfusion still persists in the outer medulla of the kidney [88]. There is increased vascular permeability, interstitial edema, and endothelial cell injury. Chemokines and cytokines together with other factors promote the inflammatory response leading to activation of the innate immune system as well as the adaptive immune system. If the inflammatory reaction continues within the graft tissue, progressive interstitial fibrosis will develop, whichimpacts long-term graft outcome [89, 90]. Moreover, the number of peritubular capillaries is reduced after I/R injury leading to chronic hypoxia, further promoting fibrosis.
PGE2, produced by all renal cells, is the most abundant prostaglandin and plays important physiological and pathological roles in the kidney (Figure 2C). For instance, it participates in modulating vasopressin on osmotic water reabsorption and regulating body water metabolism [91-93]. Tubular water and sodium transport, glomerular filtration, and vascular resistance are also regulated by PGE2 via its four EP receptors [91, 94, 95].
More recently, there is growing evidence regarding the pronounced effects of PGE2 on ameliorating renal I/R injury mainly via a variety of anti-oxidation, antiapoptotic, and inflammation inhibition effects. Paricalcitol treatment can prevent renal I/R with upregulated COX-2/PGE2/EP4 pathway [96, 97], reflecting the favorable role of PGE2 and EP4 in I/R injury [96, 97]. EP4 agonist CAY10598 can also inhibit alterations of mitochondrial membrane potential, cytochrome C release, and cell apoptosis, as well as the energy imbalance induced by renal I/R injury [98]. Excessive mitochondrial autophagy is also blocked by CAY10598 via activating the cAMP/PKA signaling pathway [98]. Additionally, the expression of PGE2 may play direct or indirect roles in the immune enhancement of the damaged kidney. Previous studies reported that mesenchymal stem cells (MSCs) partially mediated Treg differentiation by the secretion of PGE2. IL-17A pretreatment could enhance the expression of COX-2/PGE2 in MSCs to increase the Treg percentage, resulting in the improved therapeutic efficacy of MSCs on renal I/R injury [99]. PGE2 also exerts antifibrotic function in acute renal injury models [100]. In case of the metabolism of endogenous renal PGE2, SW033291, an inhibitor of 15-hydoxyprostaglandin dehydrogenase, was administered prior to I/R injury. In addition to reduced inflammation, it was also observed that decreased injury scores, tubular apoptosis, and biomarkers of renal injury such as blood urea nitrogen, creatinine, and neutrophil gelatinase-associated lipocalin [22].
However, several data show that the synthesis of PGE2 exerts adverse effects on the repair of renal I/R. Renal fibrosis is triggered by I/R injury, which no treatment can halt or regress this process. The blockade of COX-1 and COX-2 could decrease the development of fibrosis and renal fibrosis besides their anti-inflammatory effects [101]. In addition, the utilization of some COX inhibitors did reduce renal I/R injury in a dose-dependent manner [102-105]. Netrin-1 suppresses the infiltration of neutrophils and macrophages by inhibiting their COX-2 expression and PGE2 production through IKB-α mediated inhibition of NF-kB in renal I/R mice [106]. Moreover, it's reported that EP4 mediates the inflammatory response and ischemia reperfusion injury [106]. PGE2 also reduces the expression of the proximal tubular organic anion transporters Oat1 and Oat3, exacerbating renal I/R injury [107, 108]. The inconsistency between the various studies may be due to different experimental strategies such as different animal species and I/R models with different duration of ischemia or reperfusion time as well as the selection of different inhibitors.
PGE2 in intestinal injury
PGE2 is proved that it can promote the regeneration of epithelial crypts and keep epithelial homeostasis in the face of injury induced by dextran sulfate sodium (DSS) [80], chemotherapy [109], radiation [109], colonic surgery [110], or ischemia-reperfusion [111]. Both 15-PGDH knockout and 15-PGDH inhibitor (SW033291) treatment increased PGE2 levels and protected mice against DSS reduced colitis, leading to faster recovery of weight, colon length, and colitis histology scores [80]. Moreover, the potential of EP4 agonists on healing and regeneration of the bowel has attracted much attention in recent years [112-114]. PGE2 is conducive to wound healing of intestinal epithelial cells. A study shows that oxytocin, a drug interacting with its receptor that expresses in intestine crypt epithelial cells, also prevents intestine injury by evoking pulsatile PGE2 release [109]. Under homeostasis conditions, the PGE2 signaling pathway is essential to intestinal stem cell proliferation, such as Lgr5+ stem cells [115], and induces stem cell differentiation towards enterocytes [116]. In the face of intestinal injury, high local PGE2 levels can induce differentiation of intestinal epithelial stem cells to wound-associated epithelial (WAE) cells instead of enterocytes through EP4 and then the WAE cells migrate to cover and seal the wound bed to reestablish the epithelial barrier [116, 117].
Some signaling pathways are involved in the repair of PGE2 in intestinal injury models. YAP activity is also essential for intestinal regeneration after injury caused by DSS or radiation [118, 119]. However, it is worth noting that colon cancers are highly correlated with enhanced YAP and PGE2 status [119]. Another study indicated that COX-1/PGE2/EP4 also upregulates the β-arrestin1 mediated Akt signaling pathway to prevent mucosal injury in ulcerative colitis [120].
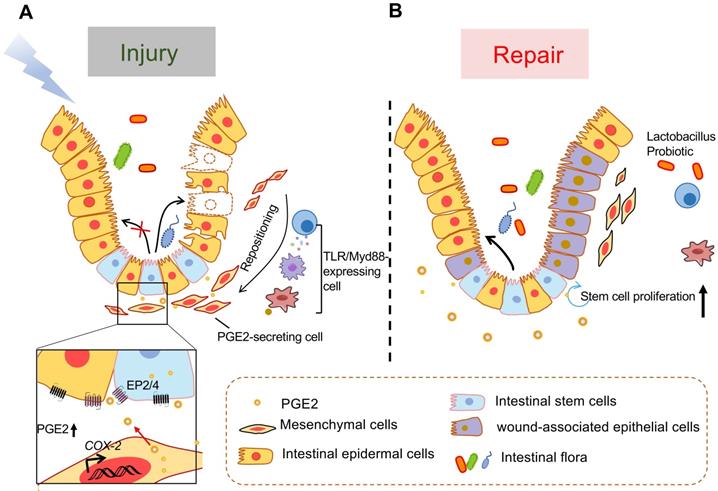
The crosstalk between PGE2-secreting mesenchymal cells around the crypt and intestinal epithelial cells is critical for wound repair (Figure 3). In the steady state, the PGE2-producing cells include both intestinal epithelial cells and MSCs that are known as (myo)fibroblasts [121-124]. MSCs are mostly present in the lamina propria of the upper and middle regions of the rectal crypt. In response to injury, they migrate to the base of the crypt to produce PGE2 depending on TLR/Myd88 expression in myeloid cells such as macrophages [115, 123, 125, 126]. Both intestinal epithelial cells and lamina propria macrophages also express COX-2 in a TLR4 and MyD88-dependent manner [127]. As we all know, TLRs, pattern recognition receptors expressed by immune or nonimmune cells, send signals in response to pathogen-related molecular patterns. Some substances, such as hyaluronic acid [115, 128] and Lactobacillus probiotics [122-124] have been reported that they prime the epithelial stem cell niche to protect epithelial stem cells by triggering a multicellular, adaptive immune signaling cascade involving macrophages and PGE2 secreting MSCs. Besides, exogenous adipose-derived MSCs also have a therapeutic effect on intestinal I/R injury, repairing via activating the COX-2-PGE2 signaling axis [111]. It's reported that fibroblast growth factor 9 and insulin-like growth factor 2 binding protein 1 are important factors regulating COX-2 expression in colonic MSCs [121, 129].
Model of TLR/MyD88-dependent repositioning of PGE2-producing cells and of intestinal epithelial cell formation in response to mucosal injury. (A) PGE2-secreting mesenchymal cells that mostly exist in the upper and middle portions of the rectal crypts in the steady state, migrate to the bottom of the crypt and occupy a position near the stem cell niche following intestinal injury. This migration depends on MyD88 expression by immune cells such as macrophages that are stimulated by TLR recognition of microbial products after barrier disruption. The PGE2 secreted by these newly located cells acts on intestinal stem cells or intestinal progenitor cells through EP2 or EP4 to promote their differentiation into wound-related epidermal cells. (B) During tissue repair, PGE2/EP4 promotes the proliferation of intestinal stem cells and the formation of intestinal epithelial cells.
PGE2 and bone fracture
Bone includes the periosteum, sclerotin, and bone marrow. Among them, the periosteum is made up of fibrous connective tissue and has abundant nerves and blood vessels, which are essential for bone regeneration, sensation, and nutrition. Periosteum can be divided into inner and outer layers. Osteoclasts and osteoblasts in the inner layer of bone are responsible for the absorption and formation of bone tissue, respectively. In addition, skeletal muscle is the tissue that controls the movement of the bone. It's usually located next to the the bone and connected to bone by tendons. In response to injury, bone tissue has an extraordinary ability of self-regeneration and healing. However, large bone defects, complex fractures, or muscle damage are still major challenges facing the medical community. PGE2 is an important regulator of bone metabolism and has an anabolic effect on the repairment and regeneration of skeletal muscle tissue.
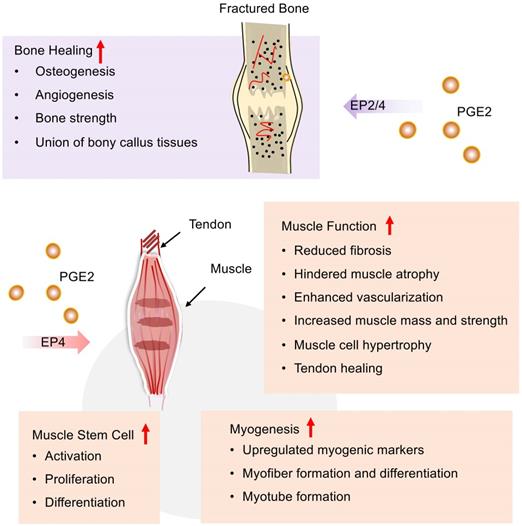
COX-2/PGE2/EP4 signaling pathway contributes to bone fracture healing and repair (Figure 4). In the early inflammatory stage of fracture repair, PGE2 is mainly produced by osteoblasts [130] and found at fracture sites, while COX-2 expression regulates key subsequent events, including cartilage formation, bone formation, and remodeling. For example, PGE2 can help the new bone formation and an increase of bone mass by stimulating MSC differentiation into an osteoblastic cell line, mostly of bone marrow origin [131]. Retroviral-based gene therapy with COX-2 promotes the union of bony callus tissues and accelerates fracture healing in the rat [132]. The absence or inhibition of COX-2 results in impaired periosteal endochondral bone formation and marked reduction of osteogenesis and angiogenesis [133]. Among all PGE2 receptors, EP4 receptors play a major role in fracture repair. Periosteal injection of EP4 agonists could markedly improve the impaired periosteal endochondral bone repair [133]. EP4 agonists might help in decreasing sternal necrosis in high-risk patients or permit wider application of bilateral internal thoracic arteries in coronary artery bypass surgery, even in patients with diabetes [134]. Furthermore, EP4 receptor deficiency delays fracture healing by interfering with intramembranous and cartilaginous ossification in mice [135]. Additionally, an EP2 agonist has also been found to increase bone formation and strength in a rat model of femoral fracture [136]. According to previous reviews, we know that COX-2/PGE2 may mediate osteoinductive communication between inflammatory macrophages and bone marrow mesenchymal stem cells, contributing to fracture healing [137].
PGE2 potentiates the repair of bone and skeletal muscle after injury. PGE2 can not only accelerate the fracture healing process via EP2/4 but also promote the repair of muscles and tendons following injury by EP4. In the process of muscle repair, PGE2 can improve muscle function, regulate muscle-derived stem cells, as well as facilitate muscle formation.
PGE2 and skeletal muscle
PGE2 participates in skeletal muscle repairment during all phases of healing including inflammation, regeneration, and fibrosis (Figure 4). COX-2/PGE2 pathway, which is mostly induced in pathological situations, is important in promoting skeletal muscle regeneration and reducing fibrosis formation [138]. For example, COX-2-/-mice exhibits decreased skeletal muscle regeneration post-laceration injury to the tibialis anterior relative to WT mice [139]. Both COX-1 and COX-2 contribute to lipid mediator synthesis production during myogenesis in vitro, which is most likely important for skeletal muscle myogenesis, repairment, and regeneration. In addition, AA supplementation stimulates PGE2 release and skeletal muscle cell hypertrophy via a COX-2-dependent pathway in vitro. Interestingly, a recent study suggests that PGE2 signaling may combat muscle atrophy and provides new insights into sarcopenia. Recently, A. R. Palla et al. revealed that elevated 15-PGDH is observed in aged tissues including skeletal muscle, leading to reduced PGE2 production. Furthermore, they uncovered that rejuvenated PGE2 levels via inhibition of 15-PGDH could augment mitochondrial function, resulting in increased muscle mass and strength [3].
Muscle-derived stem cells (MDSCs), also known as satellite cells, are crucial to muscle regeneration in response to injury [140] (Figure 4). PGE2 is a crucial and positive regulator of MDSCs that are known as the building blocks of muscle regeneration. PGE2 could directly target MDSCs via the EP4 receptor and results in either endogenous or transplanted MDSCs expansion, which robustly augments muscle regeneration and strength [141]. Similarly, the COX-2/PGE2 signaling pathway can also stimulate transplanted MDSCs to differentiate into chondrocytes, osteoblasts, and osteocytes [142]. Muscle-derived stem cell MDSCs incubated with SW033291 (15-PGDH inhibitor) can elevate the production of PGE2. In vitro, SW033291 enhanced myogenic differentiation and myotube formation via upregulating a series of myogenic markers with an activated PI3K/Akt pathway involved. Additionally, SW033291 incorporating the compound with MDSCs in fibrin gel significantly facilitates myofiber formation within the defect region with mild immune response, less fibrosis, and sufficient vascularization [143]. Together with these findings, PGE2 can facilitate the proliferation and differentiation of MDSCs. Combined use of PGE2 and MDSC might be a nice therapy strategy to boost muscle repair after injury.
Tendons are responsible for the attachment and fixation of muscles, and the PGE2 pathway also involves the tendon healing process (Figure 4). In a rotator cuff tear model, Atorvastatin is known as a lipid-lowering drug, which can enhance tendon healing by stimulating tenocyte proliferation, migration, and adhesion via increased COX2/PGE2/EP4 signaling pathway [144]. Post-operative adhesions forming between the tendon and the surrounding soft tissue complicate the repair process. Systemic EP4 antagonism leads to increased adhesion formation and matrix deposition, which counts with flexor tendon healing [145]. Furthermore, the specific function of EP4 may be dependent on cell type [146].
PGE2 and hematopoiesis
PGE2 regulates hematopoietic stem/progenitor cell (HSC/HSPC) homeostasis [147, 148] and modulates hematopoietic potential [149]. It's known that PGE2 enhances HSC homing, engraftment, survival, and expansion [148, 150, 151]. Researchers also reported that EP4 is a key receptor involving PGE2-mediated positive regulation of HSC/HSPC [152, 153]. PGE2 can not only directly regulate HSC but also can stimulate the differentiation of bone marrow mesenchymal progenitor cells into hematopoietic progenitor cells via EP4 receptors in murine [152]. The PGE2/Wnt interaction and cAMP/PKA signaling axis involves the regulation of PGE2 on hematopoietic stem and progenitor proliferation [78, 149].
Lots of studies confirm the key role of PGE2 for hematologic reconstitution after bone marrow transplant (BMT) in detail (Figure 5). Importantly, 15-PGDH is becoming an attractive therapeutic target for potentiating HSC transplantation because 15-PGDH inhibitor provides a well-tolerated strategy to therapeutically target multiple HSC niches, promote hematopoietic regeneration, and improve the clinical outcomes of BMT [80, 154-156]. In addition to neutrophils, 15-PGDH knockout or SW033291 treated, mice have more cell number of two specific bone marrow cell populations, which is enriched for bone marrow stem cells. Moreover, SW033291 treatment not only enhances the expression of CXCL12 and SCF in the hematopoietic niche for better supporting and homing of transplanted HSCs but also accelerates the recovery of platelets, and red cells after BMT. Consistently, SW033291 also enhances the generation of both myeloid and erythroid colonies in vitro [80]. Subsequently, a second-generation 15PGDH inhibitor (SW209415), has been further proven to facilitate BMT recovery regardless of age, transplantation dose, or granulocyte colony stimulating factors [155]. Overall, these results indicate that 15-PGDH inhibitors may have the unique advantage to increase PGE2 at physiological levels or directly at local sites, thereby avoiding extremely high concentrations caused by exogenous provision. Cautiously, overproduction of PGE2 post-BMT may impair host defense against pathogens, leading to the occurrence of infectious diseases, mainly including lung injury [157, 158]. At this time, blocking PGE2 synthesis with NSAIDS or related EP receptor antagonists should be considered.
PGE2 accelerates blood reconstitution after HSC transplantation. PGE2 can target multiple HSC niches, therapeutically, promote hematopoietic regeneration, and improve the clinical outcome of hematopoietic stem cell transplantation. In addition, PGE2 also facilitates the homing of transplanted HSCs and accelerates the recovery from HSCs transplantation or radiation damage.
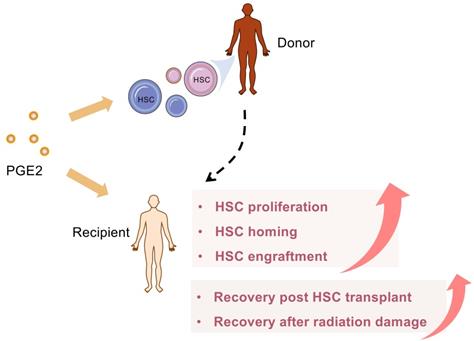
Umbilical cord blood (UCB) is a valuable source of HSC and is used for allogeneic transplantation when a suitable adult donor is not available. However, due to the low HSC content, the delay of hematopoietic and immunological recovery is still a problem to be overcome. From preclinical and clinical data, the coculture of PGE2 and cord blood stem cells is considered a cheap, safe, and practical method that can increase the homing and implantation potential of UCB stem cells [154, 159]. In addition, PGE2 exhibits a strong immune regulation function. PGE2 can not only enhance the immune tolerance of UCB stem cells after transplantation but also regulate the immune reconstitution after transplantation of UCB stem cells [160, 161].
PGE2-based therapeutic strategies for tissue regeneration
One of the limitations for using PGE2 as signal molecules is its rapid conversion under physiological conditions. As PGE2 is a key mediator of tissue regeneration and repair following injury, augmenting its effects and preventing its degradation at damaged sites are being searched as potential therapeutic strategies for a number of injured organs.
Application of 15-PGDH inhibitors
As we mentioned earlier, PGE2 molecular is degraded into 15-keto-PGE2 in cells by 15-PGDH [19]. Therefore, 15-PGDH is a negative regulator of tissue repair and regeneration in multiple organs. Scientists have been committed to the research of 15-PGDH inhibitors. Hoon Cho et al. synthesis a series of thiazolidinedione analogs [32, 162, 163] or derivatives [33, 164] to prevent the degradation of PGE2. Consequently, each of these inhibitors shows promising inhibition effects on 15-PGDH and accelerates scratch wound healing in vitro. However, only TD88 has been applied in vivo skin damage model and obtained positive results [33]. It was not until the discovery of an efficient small molecule 15-PGDH inhibitor (SW033291) by Sanford D. Markowitz's team that a lot of studies were widely carried out in vivo [80, 165]. There is no doubt that filtering such small molecule inhibitors that have a short half-life and safe pharmacological characteristics, is an outstanding job with certain application potential and clinical value.
Researchers have successively clarified the repair capabilities of SW033291 in different injury models including liver resection [80], I/R renal injury [22], intestinal injury [80], bone marrow transplantation [80, 155, 156], bone marrow failure [166], muscle defect [143], or muscle aging [3], which we have mentioned above. In fact, SW033291 was also explored in aspects of cervical ripening, drug-induced kidney injury, and pulmonary fibrosis. First, PGE2 is a cervical ripening agent, but about a quarter of full-term pregnant women who use PGE2 tablets fail to obtain a mature cervix. Furthermore, PGE2 treatment might lead to uterine hyperstimulation and abnormal fetal heart rhythms. Interestingly, a combination treatment of PGE2 and SW033291 increases the success rate of PGE2 alone-induced cervical ripening in mice [167], which should be considered in labor resulting from an unfavorable perinatal environment. Besides, since PGE2 induces vasodilation of intrarenal arteries and alleviates acute kidney injury to a certain degree, researchers also, respectively, applied SW033291 to treat contrast-induced [168] and LPS-induced [169] acute kidney injury in addition to I/R induced kidney injury [22]. Consequently, SW033291 blocks intrarenal vasoconstriction as well as renal tubular cytotoxicity in contrast-induced acute kidney ischemia injury [168], while increasing the survival rate and ameliorating injury via preventing apoptosis, oxidative stress, and facilitating autophagy in LPS-induced kidney injury models [169]. Additionally, a large number of reports show the protective effects of PGE2 in the bleomycin model. Studies demonstrated that suppressing PGE2 degradation with systemic administrated SW033291 shows antifibrotic effects in bleomycin-induced pulmonary fibrosis mice and human tissues [170, 171]. The antifibrotic effects are specifically manifested in reduced alveolar epithelial cell apoptosis, decreased fibroblast proliferation, and diminished pulmonary fibrocyte accumulation in mice [170]. Likewise, inhibitions of collagen secretion were disclosed in mice and end-stage human lung slices with bleomycin-induced fibrosis [170]. Moreover, according to further investigation by Sanford D. Markowitz's team, alveolar macrophages, mast cells, as well as endothelial cells may be the key target cells 15-PGDH inhibitor therapy in murine pulmonary fibrosis models [171].
On the one hand, even though SW033291 has high 15PGDH inhibition efficiency and superior repair-promoting function, it still requires further optimization to adapt to different injury backgrounds without altering the potency inhibiting 15PGDH. For instance, it's better to utilize intravenous administration drugs with high aqueous solubility such as in hematopoietic stem cell transplantation patients. Sanford D. Markowitz's team developed a second-generation 15-PGDH inhibitor, SW209415, with 10,000-fold more solubility compared with SW033291 [155], facilitating recovery from hematopoietic stem cell transplantation. On the other hand, the application of 15-PGDH inhibitors is not just limited to regenerative medicine. For example, a study reports that reduced PGE2 levels in patients lead to anaphylaxis via resulting in mast cell hyperresponsiveness. Then the 15-PGDH inhibitor elevating PGE2 levels decreases the severity of the disease in murine models with passive systemic anaphylaxis [12]. Of note, the utilization of 15-PGDH inhibitors might be positively correlated with the risk of cancer and maybe should not be considered in this special context [172-174].
PGE2 delivery systems
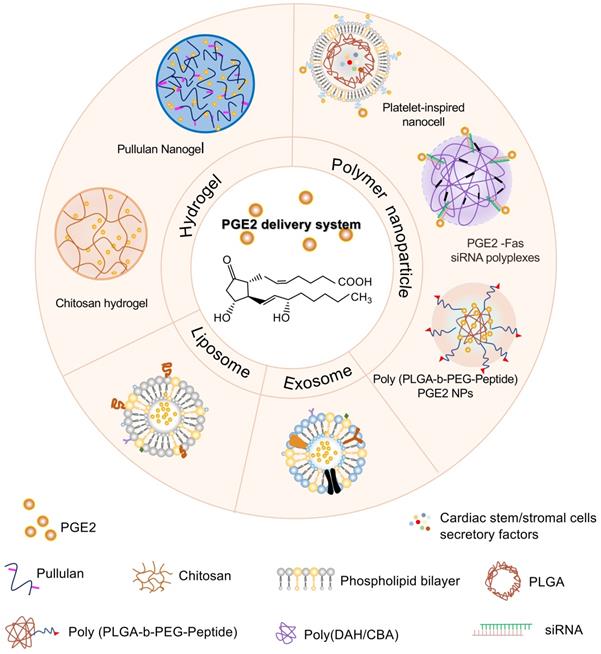
How to deliver exogenous PGE2 to the sites of action is worthy of exploration besides increasing endogenous PGE2 levels. Direct administration of PGE2 seems not to be ideal because it has a very rapid turnover rate in vivo and is eliminated from tissues or circulation even in some successful cases. Currently, several biological materials delivery systems have already been designed and synthesized to control PGE2 release, which increases the treatment efficacy of PGE2 and has potential applications in regeneration medicine. Those delivery vehicles include injectable hydrogels, liposomes or extracellular vesicles, and polymeric nanoparticles (Figure 6).
Different PGE2 biomaterial delivery systems. PGE2 delivery system is mainly divided into three categories including injectable hydrogels, liposomes or exosomes, and polymeric nanoparticles. Different hydrogel scaffolds containing PGE2 such as chitosan and pullulan hydrogel, contribute to the sustained release of PGE2. Exosomes and liposomes formed by the phospholipid bilayers encapsulate PGE2 within the hydrophilic core. Polymeric nanoparticles loading PGE2 are composed of natural or synthetic polymers such as poly (DAH/CBA) and PLGA. Platelet-inspired nanocell consists of an outer platelet-derived phospholipid bilayer binding PGE2 and inner PLGA nanoparticles encapsulated with cardiac stem/stromal cell secretory factors. PGE2 conjugation with siRNA to silence the apoptosis-related Fas genes is condensed with a reducible degradable cationic copolymer (poly (DAH/CBA)). Consistent with the platelet-derived nanocells, PGE2 molecules are exposed on the surface of the copolymer for targeting. The self-assembly of PLGA-b-PEG copolymer linked to an endothelial-targeted peptide forms poly PLGA-PEG-Peptide polymer encapsulating PGE2 in the core. PLGA, poly lactic-co-glycolic acid; poly (DAH/CBA), poly (1,6-diaminohexane, and cystamine bisacrylamide); PLGA-b-PEG, a poly (lactide-coglycolide acid)-b-poly (ethylene glycol) (PLGA-b-PEG) copolymer.
Injectable hydrogel
Hydrogel is a cross-linked network of hydrophilic polymers, capable of holding plenty of water but remaining insoluble and keeping their three-dimensional structure [23, 175]. In recent years, injectable hydrogel has raised increasing attention to their application as reservoir systems for effector molecule delivery so that biodegradability and biosorption can be ensured after appropriate design [4, 176, 177]. Pharmacokinetic studies have also revealed that effector molecules encapsulated within the hydrogel matrix could prolong the life of PGE2 in the circulation or wound surrounding tissues [178-180].
The application of the PGE2 sustained-release system may originate from Prepidil (an intracervical PGE2 gel) and Cervidil (a controlled-release hydrogel pessary) used as a cervical ripening agent [181]. Our previous study revealed that incorporating PGE2 with chitosan hydrogel to prolong the release of PGE2 contributes to improved cutaneous repair and regeneration [2]. PGE2 hydrogel significantly improves wound healing and ameliorates inflammation by promoting the polarization of M2 macrophages. Moreover, several nanogels based on cholesterol-bearing pullulan polysaccharides (CHP) encapsulating PGE2 exert positive effects on enhanced bone formation [182], while free PGE2 do not increase the thickness of the calvariae. Similarly, a biodegradable temperature-sensitive hydrogel permitting controlled release of PGE2 also plays beneficial effects on post-infarction ventricular function via preserving the immune privilege of implanted allogeneic MSCs that contribute to cardiac ischemia reperfusion repairment [183].
Liposomes or extracellular vesicles
Liposomes are spherical lipid bilayer vesicles formulated with an outer lipid bilayer that consists of natural, synthetic, or modified lipid species and an internal aqueous core [184]. Therefore, these structures can carry hydrophilic molecules in the core and hydrophobic ones between the bilayers. The lipophilic character of the liposomes makes it possible to be an efficient delivery system to protect the drugs against chemical or enzymatic degradation. T. Minko et al. demonstrated that ex vivo liposome containing PGE2 delivered locally to the pulmonary by inhalation was an effective therapeutic strategy to prevent idiopathic, pulmonary fibrosis through limiting fibroblast proliferation, activation, migration, collagen secretion, and/or myofibroblast differentiation [185]. Furthermore, extracellular vehicles (EVs) also have a phospholipid bilayer structure, which act as intermediaries between the membrane and cytoplasmic proteins, lipids, and RNA [24, 186, 187]. It's known that most EVs will accumulate in the liver after intravenous injection [188]. Moreover, a study showed that EVs-like nanoparticles loading PGE2 that derive from the intestine mucus could be transferred to the liver to maintain liver NK T cell homeostasis [189]. Therefore, EVs carrying PGE2 have potentially valuable therapeutic capabilities for a series of liver injuries. Another possibility is that engineering EVs with targeting functions may deliver PGE2 to the injured sites accurately [190]. PGE2 stored in liposomes or EVs might not directly act on EP receptors at the intracellular lipid membrane surface because of endocytosis. In conclusion, EVs, as a next-generation drug delivery platform, have extensive application prospects [191].
Polymeric nanoparticles
Polymeric nanoparticles are composed of many materials and components, mainly including natural or synthetic polymers so that satisfying characteristics such as good biocompatibility, broad structure variety, and noticeable bio-imitative are available. Polymeric nanoparticles can transport the drug to targeted tissues or organs and boost delivery efficiency via adjusting the chemical or physical characteristics of the polymer matrix [192, 193].
As we have known, EP receptors have a higher level of expression in myocardial cells after myocardial infarction, and the apoptosis-related genes, Fas and Fas ligands, are overexpressed in apoptotic cardiomyocytes. PGE2-Fas siRNA synthesized via the conjugation between PGE2 and Fas siRNA molecules. Subsequently, PGE2-siRNA conjugation is tightly condensed with reducible and degradable cationic copolymers synthesized by Michael-type polyaddition of 1,6-diaminohexane and cystamine bisacrylamide (poly (DAH/CBA)) to form nanosize polyplexes. This kind of polymeric nanoparticles is intracellularly degraded and releases siRNA after PGE2 receptor-mediated endocytosis, which inhibits cardiomyocyte apoptosis significantly after myocardial infarction [194]. Recently, scientists designed a platelet-inspired nano cell (PINC) consisting of a core and a platelet membrane shell conjugated with PGE2. The core is composed of resident cardiac stem/stromal cell secretome-loaded PLGA nanoparticles. Experimental results proved that the PINC linking PGE2 increases cycling cardiomyocytes, activates endogenous stem/progenitor cells, and promotes functional recovery more effectively compared to the groups without PGE2, which makes use of the dual features of PGE2 including targeting to cardiovascular cells and facilitating endogenous repair [44].
For PGE2 stimulated angiogenesis, Robert Langer et al. utilized a poly (lactide-coglycolide acid) - b - poly (ethylene glycol) (PLGA-b-PEG) copolymer to encapsulate 16,16-dimethyl PGE2 (a stable PGE2 analog) or another angiogenic stimulator to form vasculature-targeted polypeptide-nanoparticle, providing us with new avenues in exploring the therapeutic potential of PGE2 [195].
The future of PGE2-based treatment strategies
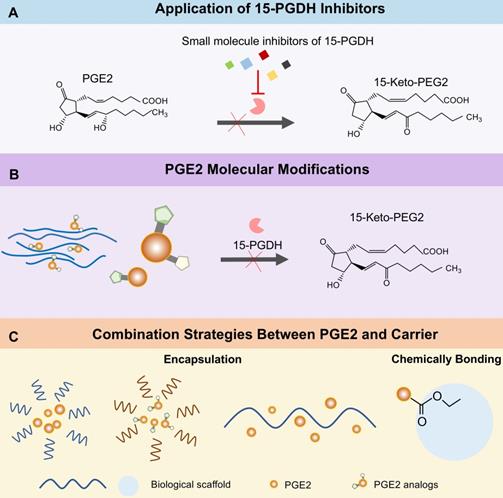
In recent years, 15-PGDH inhibitors have acquired convincing data in extensive basic research and revealed potential in clinical regeneration therapy. Intracellular PGE2 metabolized by 15-PGDH leads to the suppression of the downstream signals, which can be reversed by 15-PGDH inhibitors (Figure 7A). The development of 15-PGDH inhibitors with different mechanisms of action and stable drug characteristics will be able to provide different treatment options to patients with different types of injuries. Rodeo Therapeutics Corporation focuses on the exploration of small molecule therapies to promote regeneration and repair of diverse tissues. Its first oral 15-PGDH modulator, RTX-1688, was designed to treat intestinal diseases and this preclinical project has broad application potential. In addition, an alternative strategy to enhance the self-stability of PGE2 by chemical modification [195], such as adding or replacing some functional groups, is also promising for PGE2 based therapy. PGE2 analogs with spatial structures different from PGE2, are not subordinated to the degradation of 15-PGDH (Figure 7B). For example, previous experiments have discovered that 15-methyl PGE2, or 16, 16-dimethyl PGE2 are more stable than PGE2 when administered intravenously, orally and intrajejunal [196-198]. Besides, PGE2 analogs have been widely used in clinical practice for cervical ripening and labor induction [199]. Another strategy available is the incorporation of engineered biomatrices for PGE2 control release by different approaches, such as encapsulation and chemical bonding (Figure 7C). The limitations of current polymer drug delivery systems indicate that once drug molecules are encapsulated, they are susceptible to diffuse out of the nanocarrier. Hence, chemically bonding PGE2 to multifunctional biomaterials through stimulation-sensitive linkers, it may open new vistas in the design of smart and controllable release systems. How to accurately locate PGE2 delivery materials to damaged organs/sites in the body is a key issue that needs to be overcome, which is highly demanded for the development of optimized PGE2 delivery systems.
Therapeutic strategies for enhancing tissue regeneration of PGE2. (A) Inhibiting the activity via several small molecular compounds of 15-PGDH prevents PGE2 from degradation. (B) Modifying the PGE2 molecule to change its spatial structure or dehydrogenation site can resist the metabolism of 15-PGDH without changing the activity of PGE2. (C) Strategies for controlling release of PGE2 from biomaterials.
Conclusions and future perspectives
A broad range of studies in vitro and in vivo suggests that the PGE2 signaling pathway could protect different organs against injury from inflammation, oxidative stress, or fibrosis. Collectively, prior and current studies have been exploring efficient 15-PGDH inhibitors and have confirmed SW033291 as an ideal one that potentiates repair and regeneration of diverse organ systems following injury with increased PGE2 production. In addition, biomaterial delivery strategies have been proven effective, which reminds us that we should pay close attention to the potential combination therapies between PGE2 and the delivery system. In summary, the content presented in this review not only provides the mechanisms by which PGE2 works against injury in different organs, but also investigates the application of PGE2-based therapeutic strategies. Extensive exploration is necessary to further uncover the therapeutic potential of PGE2 in regeneration areas.
Abbreviations
AA: arachidonic acid; AC: adenylate cyclase; APAP: acetaminophen; ATP: adenosine triphosphate; cAMP: cyclic adenosine monophosphate; BMT: bone marrow transplant; CHP: cholesterol-bearing pullulan polysaccharides; CD206: cluster of differentiation 206; COX: cyclooxygenase; DAH/CBA: 1,6-diaminohexane/cystamine bisacrylamide; DSS: dextran sulfate sodium; ECM: extracellular matrix; EP: E-type prostanoid; ER: endoplasmic reticulum; ERK1/2: extracellular signal-regulated kinase1/2; EVs: extracellular vehicles; GSK3β: glycogen synthase kinase 3β; HSC/HSPC: hematopoietic stem/progenitor cell; I/R: ischemia/reperfusion; LPS: lipopolysaccharide; LV: left ventricle; MDSCs: muscle-derived stem cells; MI/R: myocardial ischemia reperfusion; MRP4: multidrug resistance protein 4; MSCs: mesenchymal stem cells; NSAIDs: nonsteroidal anti-inflammatory drugs; 15-PGDH: 15-hydroxyprostaglandin dehydrogenase; PGE2: prostaglandin E2; PGES: prostaglandin E synthase; cPGES: cytosolic prostaglandin E synthases; mPGES-1/2: microsomal prostaglandin E synthases1/2; PGH2: prostaglandin H2; PGS: prostaglandin synthase; PGT: prostaglandin transporter; PI3K: phosphoinositide 3-kinases; PKA: protein kinase A; PLA2: phospholipase A2; sPLA2: secretory phospholipase A2; PINC: platelet-inspired nano cell; PLGA-b-PEG: poly (lactide-coglycolide acid) - b - poly (ethylene glycol); PPAR- γ: peroxisome proliferators-activated receptors-γ; RELM-α: resistin-like molecule-α; SLCO2A1: solute carrier organic anion transporter family member 2A1; SMAD: drosophila mothers against decapentaplegic protein; TGF-β: transforming growth factor-β; TLR: toll-like receptor; TNF-α: tumor necrosis factor-α; UCB: umbilical cord blood; VEGF: vascular endothelial growth factor; WAE: wound-associated epithelial; WT: wild type.
Acknowledgements
This research was partially supported by National Key R&D Program of China (2017YFA0103200), National Natural Science Foundation of China (U2004126).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Galliot B, Crescenzi M, Jacinto A, Tajbakhsh S. Trends in tissue repair and regeneration. Development. 2017;144:357-64
2. Zhang S, Liu Y, Zhang X, Zhu D, Qi X, Cao X. et al. Prostaglandin E(2) hydrogel improves cutaneous wound healing via M2 macrophages polarization. Theranostics. 2018;8:5348-61
3. Palla AR, Ravichandran M, Wang YX, Alexandrova L, Yang AV, Kraft P. et al. Inhibition of prostaglandin-degrading enzyme 15-PGDH rejuvenates aged muscle mass and strength. Science. 2021 371
4. Cao X, Duan L, Hou H, Liu Y, Chen S, Zhang S. et al. IGF-1C hydrogel improves the therapeutic effects of MSCs on colitis in mice through PGE2-mediated M2 macrophage polarization. Theranostics. 2020;10:7697-709
5. Lisowska B, Kosson D, Domaracka K. Positives and negatives of nonsteroidal anti-inflammatory drugs in bone healing: the effects of these drugs on bone repair. Drug Des Devel Ther. 2018;12:1809-14
6. Geusens P, Emans PJ, de Jong JJ, van den Bergh J. NSAIDs and fracture healing. Curr Opin Rheumatol. 2013;25:524-31
7. Baker M, Perazella MA. NSAIDs in CKD: Are They Safe? Am J Kidney Dis. 2020;76:546-57
8. Kondeti V, Al-Azzam N, Duah E, Thodeti CK, Boyce JA, Paruchuri S. Leukotriene D4 and prostaglandin E2 signals synergize and potentiate vascular inflammation in a mast cell-dependent manner through cysteinyl leukotriene receptor 1 and E-prostanoid receptor 3. J Allergy Clin Immunol. 2016;137:289-98
9. Xiao CY, Yuhki K, Hara A, Fujino T, Kuriyama S, Yamada T. et al. Prostaglandin E2 protects the heart from ischemia-reperfusion injury via its receptor subtype EP4. Circulation. 2004;109:2462-8
10. Reid G, Wielinga P, Zelcer N, van der Heijden I, Kuil A, de Haas M. et al. The human multidrug resistance protein MRP4 functions as a prostaglandin efflux transporter and is inhibited by nonsteroidal antiinflammatory drugs. Proc Natl Acad Sci U S A. 2003;100:9244-9
11. Machado GC, Abdel-Shaheed C, Underwood M, Day RO. Non-steroidal anti-inflammatory drugs (NSAIDs) for musculoskeletal pain. BMJ. 2021;372:n104
12. Rastogi S, Willmes DM, Nassiri M, Babina M, Worm M. PGE(2) deficiency predisposes to anaphylaxis by causing mast cell hyperresponsiveness. J Allergy Clin Immunol. 2020;146:1387-96.e13
13. Konger RL, Billings SD, Thompson AB, Morimiya A, Ladenson JH, Landt Y. et al. Immunolocalization of low-affinity prostaglandin E receptors, EP and EP, in adult human epidermis. J Invest Dermatol. 2005;124:965-70
14. Santilli F, Boccatonda A, Davì G, Cipollone F. The Coxib case: Are EP receptors really guilty? Atherosclerosis. 2016;249:164-73
15. Bradbury P, Rumzhum NN, Ammit AJ. EP(2) and EP(4) receptor antagonists: Impact on cytokine production and β(2) -adrenergic receptor desensitization in human airway smooth muscle. J Cell Physiol. 2019;234:11070-7
16. Sharif NA, Klimko PG. Prostaglandin FP receptor antagonists: discovery, pharmacological characterization and therapeutic utility. Br J Pharmacol. 2019;176:1059-78
17. Toyoda Y, Morimoto K, Suno R, Horita S, Yamashita K, Hirata K. et al. Ligand binding to human prostaglandin E receptor EP(4) at the lipid-bilayer interface. Nat Chem Biol. 2019;15:18-26
18. Wang M, Wang Y, Xie T, Zhan P, Zou J, Nie X. et al. Prostaglandin E(2)/EP(2) receptor signalling pathway promotes diabetic retinopathy in a rat model of diabetes. Diabetologia. 2019;62:335-48
19. Nakanishi T, Nakamura Y, Umeno J. Recent advances in studies of SLCO2A1 as a key regulator of the delivery of prostaglandins to their sites of action. Pharmacol Ther. 2021;223:107803
20. Guda K, Fink SP, Milne GL, Molyneaux N, Ravi L, Lewis SM. et al. Inactivating mutation in the prostaglandin transporter gene, SLCO2A1, associated with familial digital clubbing, colon neoplasia, and NSAID resistance. Cancer Prev Res (Phila). 2014;7:805-12
21. FitzGerald GA. Bringing PGE(2) in from the cold. Science. 2015;348:1208-9
22. Kim HJ, Kim SH, Kim M, Baik H, Park SJ, Kang MS. et al. Inhibition of 15-PGDH prevents ischemic renal injury by the PGE(2)/EP(4) signaling pathway mediating vasodilation, increased renal blood flow, and increased adenosine/A(2A) receptors. Am J Physiol Renal Physiol. 2020;319:F1054-66
23. Wang C, Li G, Cui K, Chai Z, Huang Z, Liu Y. et al. Sulfated glycosaminoglycans in decellularized placenta matrix as critical regulators for cutaneous wound healing. Acta Biomater. 2021;122:199-210
24. Zhao X, Liu Y, Jia P, Cheng H, Wang C, Chen S. et al. Chitosan hydrogel-loaded MSC-derived extracellular vesicles promote skin rejuvenation by ameliorating the senescence of dermal fibroblasts. Stem Cell Res Ther. 2021;12:196
25. Futagami A, Ishizaki M, Fukuda Y, Kawana S, Yamanaka N. Wound healing involves induction of cyclooxygenase-2 expression in rat skin. Lab Invest. 2002;82:1503-13
26. Honma Y, Arai I, Hashimoto Y, Futaki N, Sugimoto M, Tanaka M. et al. Prostaglandin D2 and prostaglandin E2 accelerate the recovery of cutaneous barrier disruption induced by mechanical scratching in mice. Eur J Pharmacol. 2005;518:56-62
27. Fairweather M, Heit YI, Buie J, Rosenberg LM, Briggs A, Orgill DP. et al. Celecoxib inhibits early cutaneous wound healing. J Surg Res. 2015;194:717-24
28. Kämpfer H, Bräutigam L, Geisslinger G, Pfeilschifter J, Frank S. Cyclooxygenase-1-coupled prostaglandin biosynthesis constitutes an essential prerequisite for skin repair. J Invest Dermatol. 2003;120:880-90
29. Zhao J, Shu B, Chen L, Tang J, Zhang L, Xie J. et al. Prostaglandin E2 inhibits collagen synthesis in dermal fibroblasts and prevents hypertrophic scar formation in vivo. Exp Dermatol. 2016;25:604-10
30. Sandulache VC, Parekh A, Li-Korotky HS, Dohar JE, Hebda PA. Prostaglandin E2 differentially modulates human fetal and adult dermal fibroblast migration and contraction: implication for wound healing. Wound Repair Regen. 2006;14:633-43
31. Kämpfer H, Schmidt R, Geisslinger G, Pfeilschifter J, Frank S. Wound inflammation in diabetic ob/ob mice: functional coupling of prostaglandin biosynthesis to cyclooxygenase-1 activity in diabetes-impaired wound healing. Diabetes. 2005;54:1543-51
32. Piao YL, Seo SY, Lim SC, Cho H. Wound healing effects of new 15-hydroxyprostaglandin dehydrogenase inhibitors. Prostaglandins Leukot Essent Fatty Acids. 2014;91:325-32
33. Seo SY, Han SI, Bae CS, Cho H, Lim SC. Effect of 15-hydroxyprostaglandin dehydrogenase inhibitor on wound healing. Prostaglandins Leukot Essent Fatty Acids. 2015;97:35-41
34. Syeda MM, Jing X, Mirza RH, Yu H, Sellers RS, Chi Y. Prostaglandin transporter modulates wound healing in diabetes by regulating prostaglandin-induced angiogenesis. Am J Pathol. 2012;181:334-46
35. Liu Z, Benard O, Syeda MM, Schuster VL, Chi Y. Inhibition of Prostaglandin Transporter (PGT) Promotes Perfusion and Vascularization and Accelerates Wound Healing in Non-Diabetic and Diabetic Rats. PLoS One. 2015;10:e0133615
36. Lu Y, Ding X, Qi F, Ru Y, Kuai L, Chen S. et al. Quyu Shengji Formula Facilitates Diabetic Wound Healing via Inhibiting the Expression of Prostaglandin Transporter. Evid Based Complement Alternat Med. 2021;2021:8849935
37. Davis FM, Tsoi LC, Wasikowski R, denDekker A, Joshi A, Wilke C. et al. Epigenetic regulation of the PGE2 pathway modulates macrophage phenotype in normal and pathologic wound repair. JCI Insight. 2020 5
38. Medina de Chazal H, Del Buono MG, Keyser-Marcus L, Ma L, Moeller FG, Berrocal D. et al. Stress Cardiomyopathy Diagnosis and Treatment: JACC State-of-the-Art Review. J Am Coll Cardiol. 2018;72:1955-71
39. Sugiyama A, Okada M, Yamawaki H. Pathophysiological roles of canstatin on myofibroblasts after myocardial infarction in rats. Eur J Pharmacol. 2017;807:32-43
40. Degousee N, Simpson J, Fazel S, Scholich K, Angoulvant D, Angioni C. et al. Lack of microsomal prostaglandin E(2) synthase-1 in bone marrow-derived myeloid cells impairs left ventricular function and increases mortality after acute myocardial infarction. Circulation. 2012;125:2904-13
41. Guo Y, Nong Y, Tukaye DN, Rokosh G, Du J, Zhu X. et al. Inducible cardiac-specific overexpression of cyclooxygenase-2 (COX-2) confers resistance to ischemia/reperfusion injury. Basic Res Cardiol. 2019;114:32
42. FitzSimons M, Beauchemin M, Smith AM, Stroh EG, Kelpsch DJ, Lamb MC. et al. Cardiac injury modulates critical components of prostaglandin E(2) signaling during zebrafish heart regeneration. Sci Rep. 2020;10:3095
43. Hsueh YC, Wu JM, Yu CK, Wu KK, Hsieh PC. Prostaglandin E₂ promotes post-infarction cardiomyocyte replenishment by endogenous stem cells. EMBO Mol Med. 2014;6:496-503
44. Su T, Huang K, Ma H, Liang H, Dinh PU, Chen J. et al. Platelet-Inspired Nanocells for Targeted Heart Repair After Ischemia/Reperfusion Injury. Adv Funct Mater. 2019;29:1803567
45. Wu JMF, Cheng YY, Tang TWH, Shih C, Chen JH, Hsieh PCH. Prostaglandin E(2) Receptor 2 Modulates Macrophage Activity for Cardiac Repair. J Am Heart Assoc. 2018;7:e009216
46. Zhu L, Xu C, Huo X, Hao H, Wan Q, Chen H. et al. The cyclooxygenase-1/mPGES-1/endothelial prostaglandin EP4 receptor pathway constrains myocardial ischemia-reperfusion injury. Nat Commun. 2019;10:1888
47. Bolli R, Shinmura K, Tang XL, Kodani E, Xuan YT, Guo Y. et al. Discovery of a new function of cyclooxygenase (COX)-2: COX-2 is a cardioprotective protein that alleviates ischemia/reperfusion injury and mediates the late phase of preconditioning. Cardiovasc Res. 2002;55:506-19
48. Adams JA, Uryash A, Lopez JR. Cyclooxygenase inhibition prior to ventricular fibrillation induced ischemia reperfusion injury impairs survival and outcomes. Med Hypotheses. 2020;135:109485
49. Timmers L, Sluijter JP, Verlaan CW, Steendijk P, Cramer MJ, Emons M. et al. Cyclooxygenase-2 inhibition increases mortality, enhances left ventricular remodeling, and impairs systolic function after myocardial infarction in the pig. Circulation. 2007;115:326-32
50. Inserte J, Molla B, Aguilar R, Través PG, Barba I, Martín-Sanz P. et al. Constitutive COX-2 activity in cardiomyocytes confers permanent cardioprotection Constitutive COX-2 expression and cardioprotection. J Mol Cell Cardiol. 2009;46:160-8
51. Bryson TD, Ross J, Peterson E, Harding P. Prostaglandin E(2) and an EP4 receptor agonist inhibit LPS-Induced monocyte chemotactic protein 5 production and secretion in mouse cardiac fibroblasts via Akt and NF-κB signaling. Prostaglandins Other Lipid Mediat. 2019;144:106349
52. Bryson TD, Gu X, Khalil RM, Khan S, Zhu L, Xu J. et al. Overexpression of prostaglandin E2 EP4 receptor improves cardiac function after myocardial infarction. J Mol Cell Cardiol. 2018;118:1-12
53. Hishikari K, Suzuki J, Ogawa M, Isobe K, Takahashi T, Onishi M. et al. Pharmacological activation of the prostaglandin E2 receptor EP4 improves cardiac function after myocardial ischaemia/reperfusion injury. Cardiovasc Res. 2009;81:123-32
54. Qian JY, Harding P, Liu Y, Shesely E, Yang XP, LaPointe MC. Reduced cardiac remodeling and function in cardiac-specific EP4 receptor knockout mice with myocardial infarction. Hypertension. 2008;51:560-6
55. Pang L, Cai Y, Tang EH, Irwin MG, Ma H, Xia Z. Prostaglandin E Receptor Subtype 4 Signaling in the Heart: Role in Ischemia/Reperfusion Injury and Cardiac Hypertrophy. J Diabetes Res. 2016;2016:1324347
56. Maruyama T, Kuwabe SI, Kawanaka Y, Shiraishi T, Shinagawa Y, Sakata K. et al. Design and synthesis of a selective EP4-receptor agonist. Part 4: practical synthesis and biological evaluation of a novel highly selective EP4-receptor agonist. Bioorg Med Chem. 2002;10:2103-10
57. Martin M, Meyer-Kirchrath J, Kaber G, Jacoby C, Flögel U, Schrader J. et al. Cardiospecific overexpression of the prostaglandin EP3 receptor attenuates ischemia-induced myocardial injury. Circulation. 2005;112:400-6
58. Tang J, Shen Y, Chen G, Wan Q, Wang K, Zhang J. et al. Activation of E-prostanoid 3 receptor in macrophages facilitates cardiac healing after myocardial infarction. Nat Commun. 2017;8:14656
59. McGill MR, Jaeschke H. Animal models of drug-induced liver injury. Biochim Biophys Acta Mol Basis Dis. 2019;1865:1031-9
60. Wanner GA, Müller P, Ertel W, Busch CJ, Menger MD, Messmer K. Differential effect of cyclooxygenase metabolites on proinflammatory cytokine release by Kupffer cells after liver ischemia and reperfusion. Am J Surg. 1998;175:146-51
61. Arai M, Peng XX, Currin RT, Thurman RG, Lemasters JJ. Protection of sinusoidal endothelial cells against storage/reperfusion injury by prostaglandin E2 derived from Kupffer cells. Transplantation. 1999;68:440-5
62. Alvarez-Lopez A, de Hemptinne B, Hoebeke Y, Lambotte L. Prostaglandin E2 increases the tolerance of the rat liver to warm ischemia in absence of splanchnic congestion. Transplant Proc. 1987;19:4105-9
63. Hossain MA, Wakabayashi H, Izuishi K, Okano K, Yachida S, Maeta H. The role of prostaglandins in liver ischemia-reperfusion injury. Curr Pharm Des. 2006;12:2935-51
64. Nilsson B, Delbro D, Wallin M, Friman S. Protective effect of nitric oxide and prostaglandin E(2) in ischemia/reperfusion injury of the liver. Transplant Proc. 2001;33:2518-20
65. Kirac E, Özcan F, Tuzcu H, Elpek GO, Aslan M. Analysis of polyunsaturated fatty acids and the omega-6 inflammatory pathway in hepatic ischemia/re-perfusion injury. Mol Med Rep. 2015;12:4149-56
66. Motiño O, Francés DE, Casanova N, Fuertes-Agudo M, Cucarella C, Flores JM. et al. Protective Role of Hepatocyte Cyclooxygenase-2 Expression Against Liver Ischemia-Reperfusion Injury in Mice. Hepatology. 2019;70:650-65
67. Kuzumoto Y, Sho M, Ikeda N, Mizuno T, Hamada K, Akashi S. et al. Role of EP4 prostaglandin E2 receptor in the ischemic liver. Transplant Proc. 2005;37:422-4
68. Aslan M, Özcan F, Tuzcu H, Kıraç E, Elpek GO. Inhibition of neutral sphingomyelinase decreases arachidonic acid mediated inflammation in liver ischemia-reperfusion injury. Int J Clin Exp Pathol. 2014;7:7814-23
69. Masaki N, Ohta Y, Shirataki H, Ogata I, Hayashi S, Yamada S. et al. Hepatocyte membrane stabilization by prostaglandins E1 and E2: favorable effects on rat liver injury. Gastroenterology. 1992;102:572-6
70. Brea R, Motiño O, Francés D, García-Monzón C, Vargas J, Fernández-Velasco M. et al. PGE(2) induces apoptosis of hepatic stellate cells and attenuates liver fibrosis in mice by downregulating miR-23a-5p and miR-28a-5p. Biochim Biophys Acta Mol Basis Dis. 2018;1864:325-37
71. Kuzumoto Y, Sho M, Ikeda N, Hamada K, Mizuno T, Akashi S. et al. Significance and therapeutic potential of prostaglandin E2 receptor in hepatic ischemia/reperfusion injury in mice. Hepatology. 2005;42:608-17
72. Cai LL, Xu HT, Wang QL, Zhang YQ, Chen W, Zheng DY. et al. EP4 activation ameliorates liver ischemia/reperfusion injury via ERK1/2-GSK3β-dependent MPTP inhibition. Int J Mol Med. 2020;45:1825-37
73. Nishizawa N, Ito Y, Eshima K, Ohkubo H, Kojo K, Inoue T. et al. Inhibition of microsomal prostaglandin E synthase-1 facilitates liver repair after hepatic injury in mice. J Hepatol. 2018;69:110-20
74. Nakamura K, Kageyama S, Ito T, Hirao H, Kadono K, Aziz A. et al. Antibiotic pretreatment alleviates liver transplant damage in mice and humans. J Clin Invest. 2019;129:3420-34
75. Liu Y, Ren H, Wang J, Yang F, Li J, Zhou Y. et al. Prostaglandin E2 secreted by mesenchymal stem cells protects against acute liver failure via enhancing hepatocyte proliferation. Faseb j. 2019;33:2514-25
76. You Y, Zhang J, Gong J, Chen Y, Li Y, Yang K. et al. Mesenchymal stromal cell-dependent reprogramming of Kupffer cells is mediated by TNF-α and PGE2 and is crucial for liver transplant tolerance. Immunol Res. 2015;62:292-305
77. Mayoral R, Mollá B, Flores JM, Boscá L, Casado M, Martín-Sanz P. Constitutive expression of cyclo-oxygenase 2 transgene in hepatocytes protects against liver injury. Biochem J. 2008;416:337-46
78. Goessling W, North TE, Loewer S, Lord AM, Lee S, Stoick-Cooper CL. et al. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell. 2009;136:1136-47
79. Rudnick DA, Perlmutter DH, Muglia LJ. Prostaglandins are required for CREB activation and cellular proliferation during liver regeneration. Proc Natl Acad Sci U S A. 2001;98:8885-90
80. Zhang Y, Desai A, Yang SY, Bae KB, Antczak MI, Fink SP. et al. Inhibition of the prostaglandin-degrading enzyme 15-PGDH potentiates tissue regeneration. Science. 2015;348:aaa2340
81. North TE, Babu IR, Vedder LM, Lord AM, Wishnok JS, Tannenbaum SR. et al. PGE2-regulated wnt signaling and N-acetylcysteine are synergistically hepatoprotective in zebrafish acetaminophen injury. Proc Natl Acad Sci U S A. 2010;107:17315-20
82. Bhave VS, Donthamsetty S, Latendresse JR, Muskhelishvili L, Mehendale HM. Secretory phospholipase A2 mediates progression of acute liver injury in the absence of sufficient cyclooxygenase-2. Toxicol Appl Pharmacol. 2008;228:225-38
83. Bhave VS, Donthamsetty S, Latendresse JR, Cunningham ML, Mehendale HM. Secretory phospholipase A₂-mediated progression of hepatotoxicity initiated by acetaminophen is exacerbated in the absence of hepatic COX-2. Toxicol Appl Pharmacol. 2011;251:173-80
84. Harris TR, Kodani S, Rand AA, Yang J, Imai DM, Hwang SH. et al. Celecoxib Does Not Protect against Fibrosis and Inflammation in a Carbon Tetrachloride-Induced Model of Liver Injury. Mol Pharmacol. 2018;94:834-41
85. Xiao J, Liong EC, Huang H, On Tse W, Lau KS, Pan J. et al. Cyclooxygenase-1 serves a vital hepato-protective function in chemically induced acute liver injury. Toxicol Sci. 2015;143:430-40
86. Jin P, Chen Y, Lv L, Yang J, Lu H, Li L. Lactobacillus fermentum ZYL0401 Attenuates Lipopolysaccharide-Induced Hepatic TNF-α Expression and Liver Injury via an IL-10- and PGE2-EP4-Dependent Mechanism. PLoS One. 2015;10:e0126520
87. Yao L, Chen W, Song K, Han C, Gandhi CR, Lim K. et al. 15-hydroxyprostaglandin dehydrogenase (15-PGDH) prevents lipopolysaccharide (LPS)-induced acute liver injury. PLoS One. 2017;12:e0176106
88. Zuk A, Bonventre JV. Acute Kidney Injury. Annu Rev Med. 2016;67:293-307
89. Liu Y, Cui J, Wang H, Hezam K, Zhao X, Huang H. et al. Enhanced therapeutic effects of MSC-derived extracellular vesicles with an injectable collagen matrix for experimental acute kidney injury treatment. Stem Cell Res Ther. 2020;11:161
90. Vuiblet V, Fere M, Gobinet C, Birembaut P, Piot O, Rieu P. Renal Graft Fibrosis and Inflammation Quantification by an Automated Fourier-Transform Infrared Imaging Technique. J Am Soc Nephrol. 2016;27:2382-91
91. Li Y, Wei Y, Zheng F, Guan Y, Zhang X. Prostaglandin E2 in the Regulation of Water Transport in Renal Collecting Ducts. Int J Mol Sci. 2017;18:2539
92. Sonnenburg WK, Smith WL. Regulation of cyclic AMP metabolism in rabbit cortical collecting tubule cells by prostaglandins. J Biol Chem. 1988;263:6155-60
93. Hébert RL, Jacobson HR, Breyer MD. PGE2 inhibits AVP-induced water flow in cortical collecting ducts by protein kinase C activation. Am J Physiol. 1990;259:F318-25
94. Nørregaard R, Kwon TH, Frøkiær J. Physiology and pathophysiology of cyclooxygenase-2 and prostaglandin E2 in the kidney. Kidney Res Clin Pract. 2015;34:194-200
95. Breyer MD, Breyer RM. Prostaglandin E receptors and the kidney. Am J Physiol Renal Physiol. 2000;279:F12-23
96. Hong YA, Yang KJ, Jung SY, Park KC, Choi H, Oh JM. et al. Paricalcitol Pretreatment Attenuates Renal Ischemia-Reperfusion Injury via Prostaglandin E(2) Receptor EP4 Pathway. Oxid Med Cell Longev. 2017;2017:5031926
97. Hwang HS, Yang KJ, Park KC, Choi HS, Kim SH, Hong SY. et al. Pretreatment with paricalcitol attenuates inflammation in ischemia-reperfusion injury via the up-regulation of cyclooxygenase-2 and prostaglandin E2. Nephrol Dial Transplant. 2013;28:1156-66
98. Ding C, Han F, Xiang H, Wang Y, Dou M, Xia X. et al. Role of prostaglandin E2 receptor 4 in the modulation of apoptosis and mitophagy during ischemia/reperfusion injury in the kidney. Mol Med Rep. 2019;20:3337-46
99. Bai M, Zhang L, Fu B, Bai J, Zhang Y, Cai G. et al. IL-17A improves the efficacy of mesenchymal stem cells in ischemic-reperfusion renal injury by increasing Treg percentages by the COX-2/PGE2 pathway. Kidney Int. 2018;93:814-25
100. Higashi AY, Aronow BJ, Dressler GR. Expression Profiling of Fibroblasts in Chronic and Acute Disease Models Reveals Novel Pathways in Kidney Fibrosis. J Am Soc Nephrol. 2019;30:80-94
101. Feitoza CQ, Gonçalves GM, Semedo P, Cenedeze MA, Pinheiro HS, Beraldo FC. et al. Inhibition of COX 1 and 2 prior to renal ischemia/reperfusion injury decreases the development of fibrosis. Mol Med. 2008;14:724-30
102. Zhu SH, Zhou LJ, Jiang H, Chen RJ, Lin C, Feng S. et al. Protective effect of indomethacin in renal ischemia-reperfusion injury in mice. J Zhejiang Univ Sci B. 2014;15:735-42
103. Feitoza CQ, Câmara NO, Pinheiro HS, Gonçalves GM, Cenedeze MA, Pacheco-Silva A. et al. Cyclooxygenase 1 and/or 2 blockade ameliorates the renal tissue damage triggered by ischemia and reperfusion injury. Int Immunopharmacol. 2005;5:79-84
104. Feitoza CQ, Sanders H, Cenedeze M, Câmara NO, Pacheco-Silva A. Pretreatment with indomethacin protects from acute renal failure following ischemia-reperfusion injury. Transplant Proc. 2002;34:2979-80
105. Jia Z, Wang N, Aoyagi T, Wang H, Liu H, Yang T. Amelioration of cisplatin nephrotoxicity by genetic or pharmacologic blockade of prostaglandin synthesis. Kidney Int. 2011;79:77-88
106. Ranganathan PV, Jayakumar C, Mohamed R, Dong Z, Ramesh G. Netrin-1 regulates the inflammatory response of neutrophils and macrophages, and suppresses ischemic acute kidney injury by inhibiting COX-2-mediated PGE2 production. Kidney Int. 2013;83:1087-98
107. Schneider R, Meusel M, Betz B, Held C, Möller-Ehrlich K, Büttner-Herold M. et al. Oat1/3 restoration protects against renal damage after ischemic AKI. Am J Physiol Renal Physiol. 2015;308:F198-208
108. Schneider R, Meusel M, Renker S, Bauer C, Holzinger H, Roeder M. et al. Low-dose indomethacin after ischemic acute kidney injury prevents downregulation of Oat1/3 and improves renal outcome. Am J Physiol Renal Physiol. 2009;297:F1614-21
109. Chen D, Zhao J, Wang H, An N, Zhou Y, Fan J. et al. Oxytocin evokes a pulsatile PGE2 release from ileum mucosa and is required for repair of intestinal epithelium after injury. Sci Rep. 2015;5:11731
110. Reisinger KW, Schellekens DH, Bosmans JW, Boonen B, Hulsewé KW, Sastrowijoto P. et al. Cyclooxygenase-2 Is Essential for Colorectal Anastomotic Healing. Ann Surg. 2017;265:547-54
111. Liu L, He YR, Liu SJ, Hu L, Liang LC, Liu DL. et al. Enhanced Effect of IL-1β-Activated Adipose-Derived MSCs (ADMSCs) on Repair of Intestinal Ischemia-Reperfusion Injury via COX-2-PGE(2) Signaling. Stem Cells Int. 2020;2020:2803747
112. Hosono K, Kojo K, Narumiya S, Majima M, Ito Y. Prostaglandin E receptor EP4 stimulates lymphangiogenesis to promote mucosal healing during DSS-induced colitis. Biomed Pharmacother. 2020;128:110264
113. Watanabe Y, Murata T, Amakawa M, Miyake Y, Handa T, Konishi K. et al. KAG-308, a newly-identified EP4-selective agonist shows efficacy for treating ulcerative colitis and can bring about lower risk of colorectal carcinogenesis by oral administration. Eur J Pharmacol. 2015;754:179-89
114. Jiang GL, Nieves A, Im WB, Old DW, Dinh DT, Wheeler L. The prevention of colitis by E Prostanoid receptor 4 agonist through enhancement of epithelium survival and regeneration. J Pharmacol Exp Ther. 2007;320:22-8
115. Riehl TE, Alvarado D, Ee X, Ciorba MA, Stenson WF. Hyaluronic acid promotes Lgr5(+) stem cell proliferation and crypt fission through TLR4 and PGE(2) transactivation of EGFR. Am J Physiol Gastrointest Liver Physiol. 2020;319:G63-g73
116. Miyoshi H, VanDussen KL, Malvin NP, Ryu SH, Wang Y, Sonnek NM. et al. Prostaglandin E2 promotes intestinal repair through an adaptive cellular response of the epithelium. Embo j. 2017;36:5-24
117. Jain U, Lai CW, Xiong S, Goodwin VM, Lu Q, Muegge BD. et al. Temporal Regulation of the Bacterial Metabolite Deoxycholate during Colonic Repair Is Critical for Crypt Regeneration. Cell Host Microbe. 2018;24:353-63.e5
118. Hong AW, Meng Z, Guan KL. The Hippo pathway in intestinal regeneration and disease. Nat Rev Gastroenterol Hepatol. 2016;13:324-37
119. Kim HB, Kim M, Park YS, Park I, Kim T, Yang SY. et al. Prostaglandin E(2) Activates YAP and a Positive-Signaling Loop to Promote Colon Regeneration After Colitis but Also Carcinogenesis in Mice. Gastroenterology. 2017;152:616-30
120. Peng X, Li J, Tan S, Xu M, Tao J, Jiang J. et al. COX-1/PGE(2)/EP4 alleviates mucosal injury by upregulating β-arr1-mediated Akt signaling in colitis. Sci Rep. 2017;7:1055
121. Manieri NA, Drylewicz MR, Miyoshi H, Stappenbeck TS. Igf2bp1 is required for full induction of Ptgs2 mRNA in colonic mesenchymal stem cells in mice. Gastroenterology. 2012;143:110-21.e10
122. Ciorba MA, Riehl TE, Rao MS, Moon C, Ee X, Nava GM. et al. Lactobacillus probiotic protects intestinal epithelium from radiation injury in a TLR-2/cyclo-oxygenase-2-dependent manner. Gut. 2012;61:829-38
123. Riehl TE, Alvarado D, Ee X, Zuckerman A, Foster L, Kapoor V. et al. Lactobacillus rhamnosus GG protects the intestinal epithelium from radiation injury through release of lipoteichoic acid, macrophage activation and the migration of mesenchymal stem cells. Gut. 2019;68:1003-13
124. Uribe G, Villéger R, Bressollier P, Dillard RN, Worthley DL, Wang TC. et al. Lactobacillus rhamnosus GG increases cyclooxygenase-2 expression and prostaglandin E2 secretion in colonic myofibroblasts via a MyD88-dependent mechanism during homeostasis. Cell Microbiol. 2018;20:e12871
125. Brown SL, Riehl TE, Walker MR, Geske MJ, Doherty JM, Stenson WF. et al. Myd88-dependent positioning of Ptgs2-expressing stromal cells maintains colonic epithelial proliferation during injury. J Clin Invest. 2007;117:258-69
126. Viola MF, Boeckxstaens G. Intestinal resident macrophages: Multitaskers of the gut. Neurogastroenterol Motil. 2020;32:e13843
127. Fukata M, Chen A, Klepper A, Krishnareddy S, Vamadevan AS, Thomas LS. et al. Cox-2 is regulated by Toll-like receptor-4 (TLR4) signaling: Role in proliferation and apoptosis in the intestine. Gastroenterology. 2006;131:862-77
128. Zheng L, Riehl TE, Stenson WF. Regulation of colonic epithelial repair in mice by Toll-like receptors and hyaluronic acid. Gastroenterology. 2009;137:2041-51
129. Walker MR, Brown SL, Riehl TE, Stenson WF, Stappenbeck TS. Growth factor regulation of prostaglandin-endoperoxide synthase 2 (Ptgs2) expression in colonic mesenchymal stem cells. J Biol Chem. 2010;285:5026-39
130. Ozturk AM, Cila E, Kanatli U, Isik I, Senkoylu A, Uzunok D. et al. Treatment of segmental bone defects in rats by the stimulation of bone marrow osteo-progenitor cells with prostaglandin E2. Int Orthop. 2005;29:73-7
131. Zhang X, Schwarz EM, Young DA, Puzas JE, Rosier RN, O'Keefe RJ. Cyclooxygenase-2 regulates mesenchymal cell differentiation into the osteoblast lineage and is critically involved in bone repair. J Clin Invest. 2002;109:1405-15
132. Rundle CH, Strong DD, Chen ST, Linkhart TA, Sheng MH, Wergedal JE. et al. Retroviral-based gene therapy with cyclooxygenase-2 promotes the union of bony callus tissues and accelerates fracture healing in the rat. J Gene Med. 2008;10:229-41
133. Xie C, Liang B, Xue M, Lin AS, Loiselle A, Schwarz EM. et al. Rescue of impaired fracture healing in COX-2-/- mice via activation of prostaglandin E2 receptor subtype 4. Am J Pathol. 2009;175:772-85
134. Marui A, Hirose K, Maruyama T, Arai Y, Huang Y, Doi K. et al. Prostaglandin E2 EP4 receptor-selective agonist facilitates sternal healing after harvesting bilateral internal thoracic arteries in diabetic rats. J Thorac Cardiovasc Surg. 2006;131:587-93
135. Li M, Healy DR, Li Y, Simmons HA, Crawford DT, Ke HZ. et al. Osteopenia and impaired fracture healing in aged EP4 receptor knockout mice. Bone. 2005;37:46-54
136. Li M, Ke HZ, Qi H, Healy DR, Li Y, Crawford DT. et al. A novel, non-prostanoid EP2 receptor-selective prostaglandin E2 agonist stimulates local bone formation and enhances fracture healing. J Bone Miner Res. 2003;18:2033-42
137. Pajarinen J, Lin T, Gibon E, Kohno Y, Maruyama M, Nathan K. et al. Mesenchymal stem cell-macrophage crosstalk and bone healing. Biomaterials. 2019;196:80-9
138. Shen W, Li Y, Zhu J, Schwendener R, Huard J. Interaction between macrophages, TGF-beta1, and the COX-2 pathway during the inflammatory phase of skeletal muscle healing after injury. J Cell Physiol. 2008;214:405-12
139. Shen W, Prisk V, Li Y, Foster W, Huard J. Inhibited skeletal muscle healing in cyclooxygenase-2 gene-deficient mice: the role of PGE2 and PGF2alpha. J Appl Physiol (1985). 2006;101:1215-21
140. Relaix F, Bencze M, Borok MJ, Der Vartanian A, Gattazzo F, Mademtzoglou D. et al. Perspectives on skeletal muscle stem cells. Nat Commun. 2021;12:692
141. Ho ATV, Palla AR, Blake MR, Yucel ND, Wang YX, Magnusson KEG. et al. Prostaglandin E2 is essential for efficacious skeletal muscle stem-cell function, augmenting regeneration and strength. Proc Natl Acad Sci U S A. 2017;114:6675-84
142. Gao X, Usas A, Proto JD, Lu A, Cummins JH, Proctor A. et al. Role of donor and host cells in muscle-derived stem cell-mediated bone repair: differentiation vs. paracrine effects. Faseb j. 2014;28:3792-809
143. Dong Y, Li Y, Zhang C, Chen H, Liu L, Chen S. Effects of SW033291 on the myogenesis of muscle-derived stem cells and muscle regeneration. Stem Cell Res Ther. 2020;11:76
144. Dolkart O, Liron T, Chechik O, Somjen D, Brosh T, Maman E. et al. Statins enhance rotator cuff healing by stimulating the COX2/PGE2/EP4 pathway: an in vivo and in vitro study. Am J Sports Med. 2014;42:2869-76
145. Geary MB, Orner CA, Bawany F, Awad HA, Hammert WC, O'Keefe RJ. et al. Systemic EP4 Inhibition Increases Adhesion Formation in a Murine Model of Flexor Tendon Repair. PLoS One. 2015;10:e0136351
146. Ackerman JE, Best KT, O'Keefe RJ, Loiselle AE. Deletion of EP4 in S100a4-lineage cells reduces scar tissue formation during early but not later stages of tendon healing. Sci Rep. 2017;7:8658
147. Hoggatt J, Mohammad KS, Singh P, Hoggatt AF, Chitteti BR, Speth JM. et al. Differential stem- and progenitor-cell trafficking by prostaglandin E2. Nature. 2013;495:365-9
148. North TE, Goessling W, Walkley CR, Lengerke C, Kopani KR, Lord AM. et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature. 2007;447:1007-11
149. Diaz MF, Li N, Lee HJ, Adamo L, Evans SM, Willey HE. et al. Biomechanical forces promote blood development through prostaglandin E2 and the cAMP-PKA signaling axis. J Exp Med. 2015;212:665-80
150. Hoggatt J, Singh P, Sampath J, Pelus LM. Prostaglandin E2 enhances hematopoietic stem cell homing, survival, and proliferation. Blood. 2009;113:5444-55
151. Ben Nasr M, D'Addio F, Malvandi AM, Faravelli S, Castillo-Leon E, Usuelli V. et al. Prostaglandin E2 Stimulates the Expansion of Regulatory Hematopoietic Stem and Progenitor Cells in Type 1 Diabetes. Front Immunol. 2018;9:1387
152. Ikushima YM, Arai F, Hosokawa K, Toyama H, Takubo K, Furuyashiki T. et al. Prostaglandin E(2) regulates murine hematopoietic stem/progenitor cells directly via EP4 receptor and indirectly through mesenchymal progenitor cells. Blood. 2013;121:1995-2007
153. Patterson AM, Wu T, Chua HL, Sampson CH, Fisher A, Singh P. et al. Optimizing and Profiling Prostaglandin E2 as a Medical Countermeasure for the Hematopoietic Acute Radiation Syndrome. Radiat Res. 2021;195:115-27
154. Cutler C, Multani P, Robbins D, Kim HT, Le T, Hoggatt J. et al. Prostaglandin-modulated umbilical cord blood hematopoietic stem cell transplantation. Blood. 2013;122:3074-81
155. Desai A, Zhang Y, Park Y, Dawson DM, Larusch GA, Kasturi L. et al. A second-generation 15-PGDH inhibitor promotes bone marrow transplant recovery independently of age, transplant dose and granulocyte colony-stimulating factor support. Haematologica. 2018;103:1054-64
156. Smith JN, Dawson DM, Christo KF, Jogasuria AP, Cameron MJ, Antczak MI. et al. 15-PGDH inhibition activates the splenic niche to promote hematopoietic regeneration. JCI Insight. 2021;6:e143658
157. Domingo-Gonzalez R, Katz S, Serezani CH, Moore TA, Levine AM, Moore BB. Prostaglandin E2-induced changes in alveolar macrophage scavenger receptor profiles differentially alter phagocytosis of Pseudomonas aeruginosa and Staphylococcus aureus post-bone marrow transplant. J Immunol. 2013;190:5809-17
158. Martínez-Colón GJ, Taylor QM, Wilke CA, Podsiad AB, Moore BB. Elevated prostaglandin E(2) post-bone marrow transplant mediates interleukin-1β-related lung injury. Mucosal Immunol. 2018;11:319-32
159. Goessling W, Allen RS, Guan X, Jin P, Uchida N, Dovey M. et al. Prostaglandin E2 enhances human cord blood stem cell xenotransplants and shows long-term safety in preclinical nonhuman primate transplant models. Cell Stem Cell. 2011;8:445-58
160. Li L, Kim HT, Nellore A, Patsoukis N, Petkova V, McDonough S. et al. Prostaglandin E2 promotes survival of naive UCB T cells via the Wnt/β-catenin pathway and alters immune reconstitution after UCBT. Blood Cancer J. 2014;4:e178
161. Kim SK, Yun CH, Han SH. Dendritic Cells Differentiated from Human Umbilical Cord Blood-Derived Monocytes Exhibit Tolerogenic Characteristics. Stem Cells Dev. 2015;24:2796-807
162. Wu Y, Karna S, Choi CH, Tong M, Tai HH, Na DH. et al. Synthesis and biological evaluation of novel thiazolidinedione analogues as 15-hydroxyprostaglandin dehydrogenase inhibitors. J Med Chem. 2011;54:5260-4
163. Sun KH, Karna S, Moon YS, Cho H, Choi CH. The wound-healing effect of 7,3',4'-trimethoxyflavone through increased levels of prostaglandin E(2) by 15-hydroxyprostaglandin dehydrogenase inhibition. Biotechnol Lett. 2017;39:1575-82
164. Choi D, Piao YL, Wu Y, Cho H. Control of the intracellular levels of prostaglandin E₂ through inhibition of the 15-hydroxyprostaglandin dehydrogenase for wound healing. Bioorg Med Chem. 2013;21:4477-84
165. Antczak MI, Zhang Y, Wang C, Doran J, Naidoo J, Voruganti S. et al. Inhibitors of 15-Prostaglandin Dehydrogenase To Potentiate Tissue Repair. J Med Chem. 2017;60:3979-4001
166. Smith JNP, Otegbeye F, Jogasuria AP, Christo KF, Antczak MI, Ready JM. et al. Inhibition of 15-PGDH Protects Mice from Immune-Mediated Bone Marrow Failure. Biol Blood Marrow Transplant. 2020;26:1552-6
167. Kishore AH, Liang H, Kanchwala M, Xing C, Ganesh T, Akgul Y. et al. Prostaglandin dehydrogenase is a target for successful induction of cervical ripening. Proc Natl Acad Sci U S A. 2017;114:E6427-e36
168. Kim BW, Kim HJ, Kim SH, Baik HJ, Kang MS, Kim DH. et al. 15-Hydroxyprostaglandin dehydrogenase inhibitor prevents contrast-induced acute kidney injury. Ren Fail. 2021;43:168-79
169. Miao S, Lv C, Liu Y, Zhao J, Li T, Wang C. et al. Pharmacologic Blockade of 15-PGDH Protects Against Acute Renal Injury Induced by LPS in Mice. Front Physiol. 2020;11:138
170. Bärnthaler T, Theiler A, Zabini D, Trautmann S, Stacher-Priehse E, Lanz I. et al. Inhibiting eicosanoid degradation exerts antifibrotic effects in a pulmonary fibrosis mouse model and human tissue. J Allergy Clin Immunol. 2020;145:818-33.e11
171. Smith JNP, Witkin MD, Jogasuria AP, Christo KF, Raffay TM, Markowitz SD. et al. Therapeutic targeting of 15-PGDH in murine pulmonary fibrosis. Sci Rep. 2020;10:11657
172. Arima K, Ohmuraya M, Miyake K, Koiwa M, Uchihara T, Izumi D. et al. Inhibition of 15-PGDH causes Kras-driven tumor expansion through prostaglandin E2-ALDH1 signaling in the pancreas. Oncogene. 2019;38:1211-24
173. Smartt HJ, Greenhough A, Ordóñez-Morán P, Talero E, Cherry CA, Wallam CA. et al. β-catenin represses expression of the tumour suppressor 15-prostaglandin dehydrogenase in the normal intestinal epithelium and colorectal tumour cells. Gut. 2012;61:1306-14
174. Ogino S, Jhun I, Mata DA, Soong TR, Hamada T, Liu L. et al. Integration of pharmacology, molecular pathology, and population data science to support precision gastrointestinal oncology. NPJ Precis Oncol. 2017;1:40
175. Li Q, Cui J, Huang H, Yue Z, Chang Y, Li N. et al. IGF-1C domain-modified chitosan hydrogel accelerates cutaneous wound healing by promoting angiogenesis. Future Med Chem. 2020;12:1239-51
176. Zhao X, Cui K, Li Z. The role of biomaterials in stem cell-based regenerative medicine. Future Med Chem. 2019;11:1777-90
177. Zhang S, Nie Y, Tao H, Li Z. Hydrogel-Based Strategies for Stem Cell Therapy. In: Thakur VK, Thakur MK, editors. Hydrogels: Springer Singapore. 2018 p. 87-112
178. Vermonden T, Censi R, Hennink WE. Hydrogels for protein delivery. Chem Rev. 2012;112:2853-88
179. Soni KS, Desale SS, Bronich TK. Nanogels: An overview of properties, biomedical applications and obstacles to clinical translation. J Control Release. 2016;240:109-26
180. Okamoto T, Saito T, Tabata Y, Uemoto S. Immunological tolerance in a mouse model of immune-mediated liver injury induced by 16,16 dimethyl PGE2 and PGE2-containing nanoscale hydrogels. Biomaterials. 2011;32:4925-35
181. Shirley M. Dinoprostone Vaginal Insert: A Review in Cervical Ripening. Drugs. 2018;78:1615-24
182. Kato N, Hasegawa U, Morimoto N, Saita Y, Nakashima K, Ezura Y. et al. Nanogel-based delivery system enhances PGE2 effects on bone formation. J Cell Biochem. 2007;101:1063-70
183. Dhingra S, Li P, Huang XP, Guo J, Wu J, Mihic A. et al. Preserving prostaglandin E2 level prevents rejection of implanted allogeneic mesenchymal stem cells and restores postinfarction ventricular function. Circulation. 2013;128:S69-78
184. Wang L, Su W, Liu Z, Zhou M, Chen S, Chen Y. et al. CD44 antibody-targeted liposomal nanoparticles for molecular imaging and therapy of hepatocellular carcinoma. Biomaterials. 2012;33:5107-14
185. Ivanova V, Garbuzenko OB, Reuhl KR, Reimer DC, Pozharov VP, Minko T. Inhalation treatment of pulmonary fibrosis by liposomal prostaglandin E2. Eur J Pharm Biopharm. 2013;84:335-44
186. Li H, Huang H, Chen X, Chen S, Yu L, Wang C. et al. The delivery of hsa-miR-11401 by extracellular vesicles can relieve doxorubicin-induced mesenchymal stem cell apoptosis. Stem Cell Res Ther. 2021;12:77
187. Zhang K, Chen S, Sun H, Wang L, Li H, Zhao J. et al. In vivo two-photon microscopy reveals the contribution of Sox9+ cell to kidney regeneration in a mouse model with extracellular vesicle treatment. J Biol Chem. 2020;295:12203-13
188. Cao H, Cheng Y, Gao H, Zhuang J, Zhang W, Bian Q. et al. In vivo Tracking of Mesenchymal Stem Cell-Derived Extracellular Vesicles Improving Mitochondrial Function in Renal Ischemia-Reperfusion Injury. ACS Nano. 2020;14:4014-26
189. Deng ZB, Zhuang X, Ju S, Xiang X, Mu J, Liu Y. et al. Exosome-like nanoparticles from intestinal mucosal cells carry prostaglandin E2 and suppress activation of liver NKT cells. J Immunol. 2013;190:3579-89
190. Liang Y, Duan L, Lu J, Xia J. Engineering exosomes for targeted drug delivery. Theranostics. 2021;11:3183-95
191. Herrmann IK, Wood MJA, Fuhrmann G. Extracellular vesicles as a next-generation drug delivery platform. Nat Nanotechnol. 2021;16:748-59
192. Banik BL, Fattahi P, Brown JL. Polymeric nanoparticles: the future of nanomedicine. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2016;8:271-99
193. El-Say KM, El-Sawy HS. Polymeric nanoparticles: Promising platform for drug delivery. Int J Pharm. 2017;528:675-91
194. Kim SH, Jeong JH, Ou M, Yockman JW, Kim SW, Bull DA. Cardiomyocyte-targeted siRNA delivery by prostaglandin E(2)-Fas siRNA polyplexes formulated with reducible poly(amido amine) for preventing cardiomyocyte apoptosis. Biomaterials. 2008;29:4439-46
195. Xue Y, Xu X, Zhang XQ, Farokhzad OC, Langer R. Preventing diet-induced obesity in mice by adipose tissue transformation and angiogenesis using targeted nanoparticles. Proc Natl Acad Sci U S A. 2016;113:5552-7
196. Porter RL, Georger MA, Bromberg O, McGrath KE, Frisch BJ, Becker MW. et al. Prostaglandin E2 increases hematopoietic stem cell survival and accelerates hematopoietic recovery after radiation injury. Stem Cells. 2013;31:372-83
197. Patterson AM, Liu L, Sampson CH, Plett PA, Li H, Singh P. et al. A Single Radioprotective Dose of Prostaglandin E(2) Blocks Irradiation-Induced Apoptotic Signaling and Early Cycling of Hematopoietic Stem Cells. Stem Cell Reports. 2020;15:358-73
198. Robert A, Schultz JR, Nezamis JE, Lancaster C. Gastric antisecretory and antiulcer properties of PGE2, 15-methyl PGE2, and 16, 16-dimethyl PGE2. Intravenous, oral and intrajejunal administration. Gastroenterology. 1976;70:359-70
199. Fischer DP, Griffiths AL, Lui S, Sabar UJ, Farrar D, O'Donovan PJ. et al. Distribution and Function of Prostaglandin E(2) Receptors in Mouse Uterus: Translational Value for Human Reproduction. J Pharmacol Exp Ther. 2020;373:381-90
Author contact
![]() Corresponding authors: Zongjin Li, E-mail: zongjinliedu.cn; or Ying Chang, E-mail: changying4470com.
Corresponding authors: Zongjin Li, E-mail: zongjinliedu.cn; or Ying Chang, E-mail: changying4470com.