Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2021; 11(17):8535-8549. doi:10.7150/thno.61452 This issue Cite
Research Paper
Neuronal-driven glioma growth requires Gαi1 and Gαi3
Yin Wang1#, Yuan-yuan Liu2#, Min-bin Chen3#, Kai-Wen Cheng1, Li-na Qi1, Zhi-qing Zhang1, Ya Peng4, Ke-ran Li5 ![]() , Fang Liu6
, Fang Liu6 ![]() , Gang Chen7
, Gang Chen7 ![]() , Cong Cao1,8
, Cong Cao1,8 ![]()
1. Jiangsu Key Laboratory of Neuropsychiatric Diseases and Institute of Neuroscience, Soochow University, Suzhou, China.
2. Clinical research & lab center, Affiliated Kunshan Hospital of Jiangsu University, Kunshan, China.
3. Department of Radiotherapy and Oncology, Affiliated Kunshan Hospital of Jiangsu University, Kunshan, China.
4. Department of Neurosurgery, The Third Affiliated Hospital of Soochow University, Changzhou, China.
5. The Fourth School of Clinical Medicine, The Affiliated Eye Hospital, Nanjing Medical University, Nanjing, China.
6. Department of Neurosurgery, Nanjing Medical University Affiliated Changzhou NO.2 People's Hospital, Changzhou, China.
7. Department of Neurosurgery, the First Affiliated Hospital of Soochow University, Suzhou, China.
8. The Central Lab, North District, The Affiliated Suzhou Hospital of Nanjing Medical University, Suzhou Municipal Hospital, Suzhou, China.
# Co-first authors.
Received 2021-4-10; Accepted 2021-7-8; Published 2021-7-25
Abstract

Neuroligin-3 (NLGN3) is necessary and sufficient to promote glioma cell growth. The recruitment of Gαi1/3 to the ligand-activated receptor tyrosine kinases (RTKs) is essential for mediating oncogenic signaling.
Methods: Various genetic strategies were utilized to examine the requirement of Gαi1/3 in NLGN3-driven glioma cell growth.
Results: NLGN3-induced Akt-mTORC1 and Erk activation was inhibited by decreasing Gαi1/3 expression. In contrast ectopic Gαi1/3 overexpression enhanced NLGN3-induced signaling. In glioma cells, NLGN3-induced cell growth, proliferation and migration were attenuated by Gαi1/3 depletion with shRNA, but facilitated with Gαi1/3 overexpression. Significantly, Gαi1/3 silencing inhibited orthotopic growth of patient-derived glioma xenografts in mouse brain, whereas forced Gαi1/3-overexpression in primary glioma xenografts significantly enhanced growth. The growth of brain-metastatic human lung cancer cells in mouse brain was largely inhibited with Gαi1/3 silencing. It was however expedited with ectopic Gαi1/3 overexpression. In human glioma Gαi3 upregulation was detected, correlating with poor prognosis.
Conclusion: Gαi1/3 mediation of NLGN3-induced signaling is essential for neuronal-driven glioma growth.
Keywords: Neuron-glioma communication, NLGN3, Gαi1/3, Signaling
Introduction
Glioma is the most common type of brain tumor of either astrocytic or oligodendroglial origin [1, 2]. High-grade gliomas, such as glioblastoma (GBM) and anaplastic astrocytoma, cause human mortality both in children and adults [3, 4]. Dysregulation of multiple signaling cascades and complex signaling crosstalk are vital in the tumorigenesis and progression of human glioma [5-9]. These cascades include phosphoinositide 3-kinase (PI3K)-Akt-mammalian target of rapamycin (mTOR) [10], p53, retinoblastoma (Rb), epidermal growth factor receptor (EGFR) and Wnt/β-catenin [11] as well as signal transducer and activator of transcription (STAT), neurotrophic tyrosine receptor kinase (NTRK), fibroblast growth factor receptor (FGFR) and many others [5, 6]. It is therefore extremely important to identify the dominant and key driving signaling molecules of glioma in order to develop efficient targeted therapies [5, 6, 12, 13].
Recent studies have revealed that neuronal activity and neuron-glioma circuits are vital for the malignancy of human glioma, and that the neuronal activity could be a key determinant for glioma tumorigenesis and progression [14-16]. Studies highlighted a crucial role of neuroligin-3 (NLGN3) in the mechanism of glioma progression [17-20]. NLGN3, a synaptic protein cleaved and secreted in an activity-dependent manner, was identified as the primary factor responsible for glioma progression [17-20]. Xenografts derived from high-grade primary human glioma cells failed to grow in a NLGN3-deficient brain [18], demonstrating that NLGN3 is required for glioma cell growth. Furthermore, inhibition of ADAM10 (A Disintegrin And Metalloproteinase 10) blocked activity-dependent NLGN3 secretion to inhibit orthotopic growth of primary glioma xenografts [19].
NLGN3 secreted from neurons was found to induce phosphorylation and activation of several key receptor tyrosine kinases (RTKs) on glioma cells to promote glioma progression [17-19]. Additionally, NLGN3-activated glioma cells produce feedforward expression of NLGN3 to further increase glioma cell growth and proliferation [17-19]. Recent studies found that NLGN3 induces the expression of synaptic genes required for neuron-glioma synapse formation, as well as genes essential for glioma proliferation and invasiveness [14, 15]. These studies identify NLGN3 as a key neuronal-derived factor for promoting activity-dependent glioma growth [17-19].
Gαi proteins, the inhibitory α subunits of G proteins (heterotrimeric guanine nucleotide-binding proteins), have three primary family subunits, Gαi1, Gαi2 and Gαi3 [21]. Through binding to GPCRs (G protein-coupled receptors) Gαi proteins and the βγ complexes can suppress adenylate cyclase (AC) to deplete cyclic AMP (cAMP) levels [21]. Gαi proteins are inhibited by pertussis toxin. Glider et al., showed that pertussis toxin potently inhibited migration and invasion of high grade glioma cells [22]. In addition, pertussis toxin and temozolomide co-treatment resulted in better anti-glioma efficiency in a rat intracerebral glioma model [23]. Moreover, a D2 dopamine receptor agonist stimulated mitogenesis of C6 glioma cells through activation of pertussis toxin-sensitive Gαi proteins [24].
Alternatively, our studies show that Gαi1 and Gαi3 proteins also play a key role in mediating the signal transduction of several RTKs, including EGFR (epidermal growth factor receptor) [25], fibroblast growth factor receptor (FGFR) [26], keratinocyte growth factor receptor (KGFR) [27], brain derived neurotrophic factor (BDNF) receptor TrkB [28], and vascular endothelial growth factor receptor 2 (VEGFR2) [29]. Specifically, the recruitment of either Gαi1 or Gαi3 subunit to these ligand-activated RTKs was found to be essential for mediating PI3K-Akt-mTORC1 and Erk-MAPK signaling [25-29]. As neuron-secreted NLGN3 activates RTKs to promote glioma progression, we explored the potential function of Gαi1 and Gαi3 in NLGN3-induced signaling and glioma cell progression.
Methods
Ethics. Protocols of this study were approved by the Ethics Committee of Soochow University.
Reagents. From Gibco (Shanghai, China) cell culture reagents were obtained. The antibodies were described early [26]. NLGN3, pertussis toxin (PTX), LY294002 and PD98059 were purchased from Sigma-Aldrich (St. Louis, Mo). All primers, sequences and viral constructs were provided by Shanghai Genechem Co. (Shanghai, China), unless otherwise mentioned.
Cell lines. U251MG glioma cell line was obtained from the Cell Bank of Shanghai Biological Institution (Shanghai, China), cultivated as described. The cell line has previously been tested and authenticated by the Cell Bank of Shanghai Biological Institution. Wild-type (WT), Gαi1 and Gαi3 double knockout (DKO), Gαi1, Gαi2 or Gαi3 single knockout (SKO) mouse embryonic fibroblasts (MEFs), as well as WT and Gab1 KO MEFs, were cultivated as previously described [25, 27-30]. Cells were starved in 0.5% FBS medium overnight plus 30 min warm PBS for signaling analyses.
Human glioma tissues. According to principles of the Declaration of Helsinki, this study was approved by the Ethics Committee of Soochow University. Human tissues, including the glioma tissues and surrounding normal brain tissues, were described previously [26]. Tissues were obtained from The First, Second, Third Affiliated and Children Hospitals of Soochow University. All participates provided written-informed consent.
Primary culture of human cancer cells. The source and culturing of primary human glioma cells were previously described [26]. From a lung cancer patient with brain metastases (female, 50 years old, brain metastatic lung papillary adenocarcinoma, CK7+, CK20—, Villin—, TTF-1+, CDX2—, PAX8—, CD10—, CAIX—, PR—, EMA+, GFAP—), surgical removed brain metastatic cancer tissues were washed, minced into small pieces, and digested with Collagenase I and DNase (Sigma-Aldrich). Single-cell suspensions were then pelleted and washed. Fibroblasts, blood vessel cells, immune cells and other non-cancerous cells were abandoned. Purified brain-metastatic human lung cancer cells (bmLCs) were ex vivo cultured in the described medium [31]. The study protocols were reviewed and approved by the Ethics Committee of Soochow University, and conformed to the guidelines of Helsinki declaration. The informed consent was obtained from all subjects before their participation.
Quantitative real-time reverse transcriptase polymerase chain reaction (qPCR). As described, qPCR was carried out by an ABI 7600 Prism equipment through utilizing a SYBR Green PCR kit. Gαi3 and NLGN3 mRNA levels were quantified by a ΔΔCt protocol [32], using GAPDH as the internal control [26]. Primers are listed in Table 1.
Primers and sgRNA sequences utilized in the study.
| Genes | qPCR primers |
|---|---|
| GAPDH Forward | 5'-GTCGTGTGAACGGATTTG-3' |
| GAPDH Reverse | 5'-AAGATGGTGATGGGCTTCC-3' |
| NLGN3 Forward | 5'- GTCTGGTTCACTGCCAACTTGG -3' |
| NLGN3 Reverse | 5'- CCGTCATTATCCGCTAAGTCCTC-3' |
| GNAI3 Forward | 5'- CACTTCACCTGTGCCACAGACA-3' |
| GNAI3 Reverse | 5'- GTCTGGTCTCAACACTCCACAC-3' |
| sgRNA | Target DNA Sequence |
| Murine GNAI3 sgRNA | GGCTCGTATGATTGCAATGA |
| Murine GNAI1 sgRNA | CCATCATTAGAGCCATGGGG |
Western blotting and co-immunoprecipitation. Protocols for Western blotting and co-immunoprecipitation (co-IP), as well as data quantification, have been extensively described previously [25, 28]. For all Western blotting assays, each lane was loaded with exact same amount of quantified protein lysates (30-40 μg in each treatment). Same set of lysate samples were run in parallel (“sister”) gels to test different proteins when necessary. Expression of indicated proteins was quantified via ImageJ software, with results normalized to the equal loadings.
Endosome fractions. Cells with the applied treatments were harvested and re-suspended in the hypotonic swelling buffer [33], and lysed with 30 strokes in a Dounce homogenizer using a tight pestle, and swelling was stopped by the addition of two fold homogenization buffer [33]. Lysates were centrifuged to obtain the post-nuclear supernatants, and centrifuged [33]. The resulting supernatants were centrifuged, and the pellet solubilized in the homogenization buffer [33]. Insoluble particles were removed by short centrifugation and the supernatant loaded onto a 5-20% continuous OptiprepTM (Sigma-Aldrich), poured using homogenization buffer. The gradient was further centrifuged at 60,000 g for 24h, with total 10 endosomal fractions collected, and proteins precipitated with 12% TCA for 1h. Fractions were centrifuged at 12,000 g for 1h. The protein pellets, combining all ten endosomal fractions, were dissolved in SDS-sample buffer for analysis by Western blotting.
Gαi1/3 shRNA. Glioma cells or bmLCs were seeded into six-well plates at 50-60% confluence, treated with Gαi1 shRNA lentiviral particles (sc-105382-V) (Santa Cruz, CA) and Gαi3 shRNA lentiviral particles [29]. After 24h, cells were further cultured in puromycin (1.0 μg/mL)-containing complete medium for 12-14 days. Gαi1/3 knockdown (over 95% efficiency) in stable cells was confirmed by Western blotting. shRNA-mediated knockdown of Gαi1 and Gαi3 in MEFs was reported previously [27-29]. Gab1 shRNA lentiviral particles, for both human and mouse, were also obtained from Santa Cruz Biotech.
CRISPR/Cas9 knockout of Gαi1 and Gαi3. The lentiviral CRISPR/Cas-9 Gαi1 KO construct and the lentiviral CRISPR/Cas-9 Gαi3 KO construct were provided by Shanghai Genechem (Shanghai, China), transfected into MEFs, and selected with puromycin. Gαi1/3 knockout was confirmed by Western blotting. Control cells were treated with the empty vector with nonsense sgRNA (Santa Cruz Biotech). The sgRNA sequences used for Gαi1 and Gαi3 KO are listed in Table 1.
Gαi1/3 overexpression. The recombinant adenovirus containing full-length Gαi1 (“Ad-Gαi1”, human or mouse) and Gαi3 (“Ad-Gαi3”, human or mouse) were described earlier [29]. Virus was filtered, enriched and added to cultured MEFs, glioma cells or bmLCs. Stable cells were established following selection by puromycin, and Gαi1/3 overexpression confirmed by Western blotting.
Glioma cell functional assays. Cell growth, proliferation, CCK-8 viability and EdU staining assays [26, 34, 35] and cell migration by the “Transwell” assays [29, 36, 37], were carried out using previously described protocols.
The orthotopic primary-derived xenograft assay. For intracranial tumor implantation, primary human glioma cells or bmLCs (5 × 105 cells of each mouse in 200 µL of Matrigel gel/10% FBS medium, with different genetic treatments) were implanted using the previously described coordinates [26, 38]. Magnetic resonance imaging (MRI) was carried out to visualize tumor xenografts. On the day when the first mouse in any group exhibited symptoms (severe fever, vomiting, or greater than 15% body weight loss), all groups were sacrificed and tumors isolated through surgery. Tumor volumes were measured by the formula: π/6 × larger diameter × (smaller diameter)2. Immunohistochemistry (IHC) was performed using the previously described procedures [26]. All animal procedures were approved by Soochow University Ethics Review Board.
Statistical analysis. The in vitro experiments were replicated three times or more, and data expressed as means ± standard deviation (SD). To examine statistical differences among different groups one-way ANOVA was carried out with multiple comparisons performed by post hoc Bonferroni test (SPSS 18.0). P values < 0.05 were considered statistically significant. A two-tailed unpaired t test (Excel 2007) was applied to examine significance between two treatment groups.
Results
Gαi1 and Gαi3 double knockout inhibits NLGN3-induced Akt, Erk and mTORC1 activation in mouse embryonic fibroblasts (MEFs)
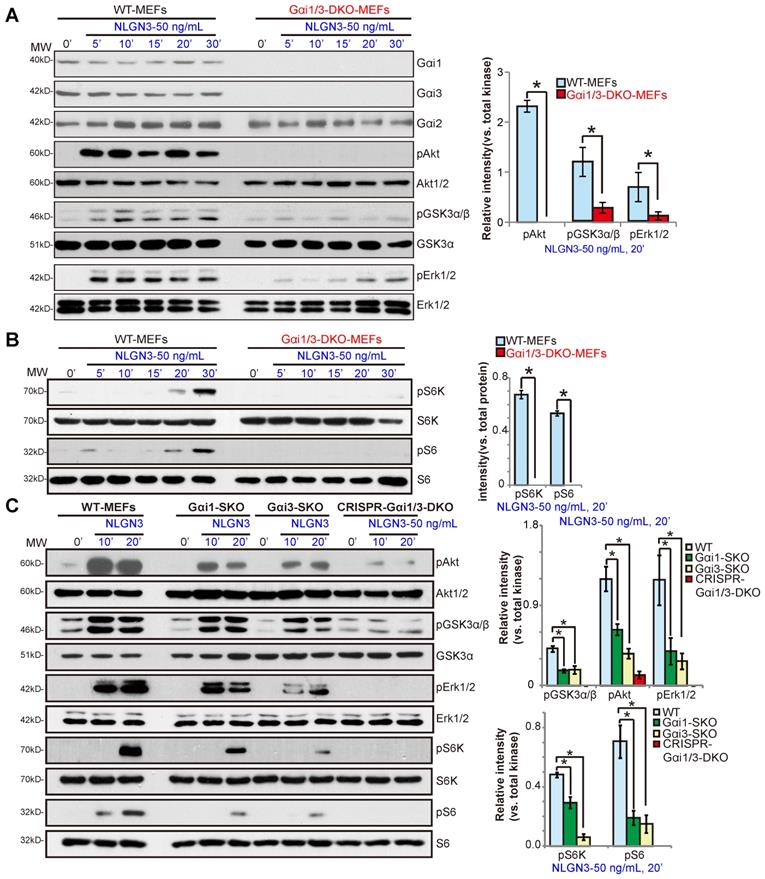
To test whether Gαi proteins are required for NLGN3-induced signal transduction, wild-type (WT) and Gαi1 and Gαi3 double knockout (Gαi1/3 DKO) MEFs [28, 29] were treated with NLGN3 (50 ng/mL) for 5-30 min. Western blotting results demonstrated that NLGN3 significantly increased phosphorylation of Akt (Ser-473) and GSK3α/β (Ser21/9) in WT MEFs, whereas phosphorylation was impaired in Gαi1/3 DKO MEFs (Figure 1A). Furthermore, NLGN3-induced Erk activation, tested by p-Erk1/2 at Thr202/Tyr204, was also attenuated in the DKO MEFs (Figure 1A). In WT MEFs NLGN3 activated mTORC1 signaling by inducing phosphorylation of p70S6K1(“S6K”, Thr389) and S6 (Ser-235/236), which was abolished in Gαi1/3 DKO MEFs (Figure 1B). Levels of total Akt, Erk1/2, S6K and S6 were equivalent between WT and DKO MEFs (Figure 1A-B). These results show that NLGN3-induced Akt, Erk and mTORC1 activation was significantly inhibited in Gαi1/3 DKO MEFs.
In Gαi1 or Gαi3 single knockout (SKO) MEFs, NLGN3-induced phosphorylation of Akt, Erk1/2, S6K and S6 was partially inhibited (P < 0.05 vs. WT MEFs, Figure 1C), indicating that both Gαi1 and Gαi3 can be utilized for NLGN3-induced signaling. To control for possible epigenetic modifications in the knockout MEFs, the CRISPR/Cas9 gene-editing method was applied to knockout both Gαi1 and Gαi3 in WT MEFs. As shown, NLGN3-induced Akt, Erk and mTORC1 activation was almost completely blocked in Gαi1/3 KO MEFS (“CRISPR-Gαi1/3-DKO”) (Figure 1C).
Gαi1 and Gαi3 are required for NLGN3-induced Akt, Erk and mTORC1 activation in MEFs
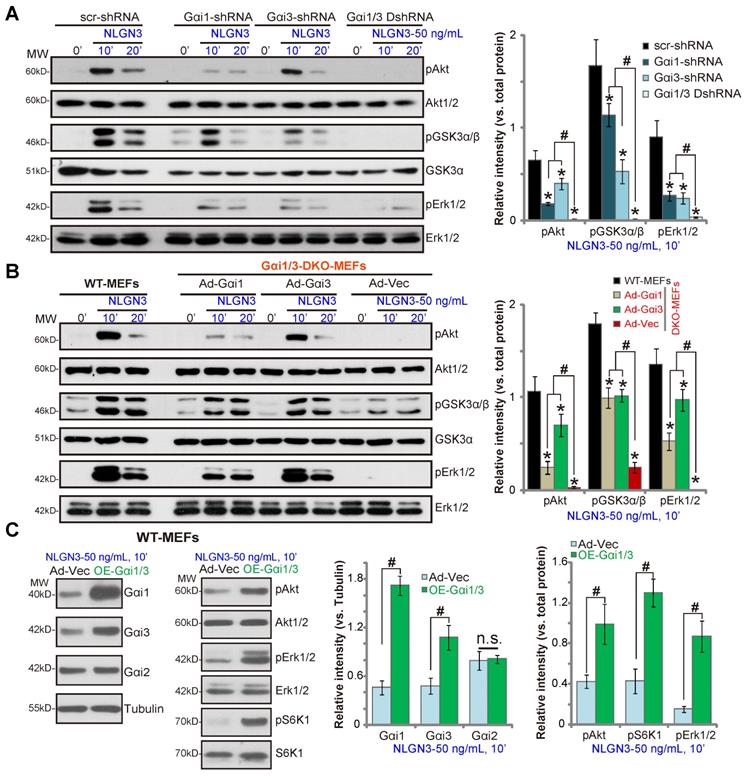
To further confirm that Gαi1/3 are required for NLGN3-induced signaling the shRNA method was utilized. As described previously [29], Gαi1 shRNA lentiviral particles and/or Gαi3 shRNA lentiviral particles were added to WT MEFs to silence Gαi1 and/or Gαi3. As shown, shRNA-mediated single knockdown of Gαi1 or Gαi3 resulted in partial inhibition of Akt, GSK3α/β and Erk1/2 phosphorylation in response to NLGN3 (P < 0.05 vs. MEFs with control shRNA, Figure 2A). Significantly, Gαi1 and Gαi3 double knockdown by targeted shRNAs (“Gαi1/3 DshRNA”) led to further inhibition of NLGN3-induced signaling in MEFs (Figure 2A).
To determine whether exogenously expressed Gαi proteins can rescue signaling, Gαi proteins were re-expressed in Gαi1/3 DKO MEF cells by infection with an adenovirus Gαi1 construct (“Ad-Gαi1”, no Tag) [28, 29] or adenovirus Gαi3 construct (“Ad-Gαi3”, no Tag) [28, 29]. As shown, Ad-Gαi1 or Ad-Gαi3 restored NLGN3-induced Akt, GSK3α/β and Erk1/2 in DKO MEFs (Figure 2B), further demonstrating that Gαi1 and Gαi3 are required for NLGN3-induced signaling. Conversely overexpression of Ad-Gαi1 (adenovirus-packed Gαi1) and Ad-Gαi3 in WT MEFs significantly increased NLGN3-induced signaling. Stable Gαi1 and Gαi3 overexpressing MEFs (“OE-Gαi1/3”) were established. Protein levels of Gαi1 and Gαi3 (but not Gαi2) were significantly increased in OE-Gαi1/3 MEFs (P < 0.05 vs. vector control MEFs, Figure 2C), where NLGN3-induced Akt, Erk1/2 and S6K phosphorylation were significantly increased (P < 0.05 vs. vector control MEFs, Figure 2C). Therefore, Gαi1/3 overexpression in WT MEFs enhanced NLGN3-induced downstream signaling activation.
Gαi1 and Gαi3 are essential for NLGN3 signaling in glioma cells
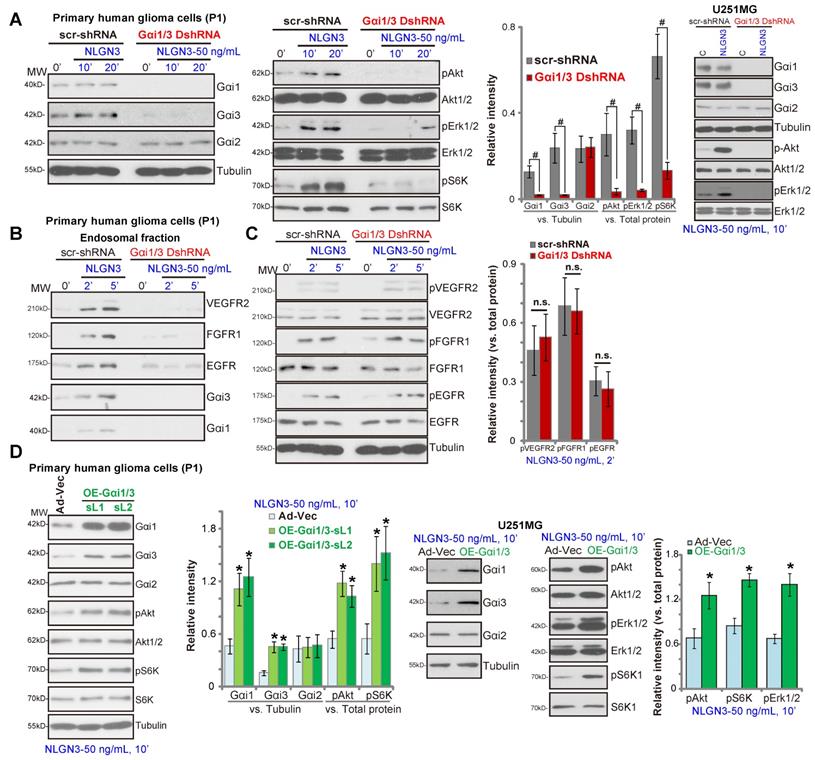
To silence Gαi1 and Gαi3 in glioma cells, primary human glioma cells (“P1”) were utilized. The pathology of P1 patients (female, 69) was: WHO grade IV, GFAP+, S100+, IDH1—, Nestin+, Olig2+, CD34+, Ki67+, p53-mutant, SOX10+, ATRX+, H3K27M— and MGMT—. Cells were infected with Gαi1 shRNA lentivirus plus Gαi3 shRNA lentivirus [29]. Following puromycin selection, stable glioma cells were established with depleted Gαi1 and Gαi3 (“Gαi1/3 DshRNA”, Figure 3A). In control P1 glioma cells (expressing a scramble nonsense shRNA/“scr-shRNA”) NLGN3 potently induced phosphorylation of Akt, Erk1/2 and S6K, whereas signaling was almost blocked in P1 glioma cells transduced with Gαi1/3 DshRNA (Figure 3A). In U251MG glioma cells, shRNA-mediated silencing of Gαi1/3 (“Gαi1/3 DshRNA”) similarly inhibited NLGN3-induced Akt and Erk1/2 phosphorylation (Figure 3A).
Gαi1/3 association is essential for RTK endocytosis and endosomal translocation, a key step for downstream signal activation [28, 29]. In response to NLGN3, multiple RTKs (VEGFR2, EGFR and FGFR1) as well as Gαi1 and Gαi3 were enriched in the endosomal fraction in P1 glioma cells (Figure 3B), but not in Gαi1/3-silenced cells (Figure 3B). RTKs expression and NLGN3-induced RTKs phosphorylation were not affected by Gαi1/3 silencing (Figure 3C). These results indicated that Gαi1/3 are required for NLGN3-induced RTK endocytosis and downstream signaling transduction.
Gαi1 and Gαi3 double knockout largely inhibits NLGN3-induced Akt, Erk and mTORC1 activation in MEFs. Wild-type (WT), Gαi1 and Gαi3 double knockout (DKO) (A-B), Gαi1, or Gαi3 single knockout (SKO) (C) mouse embryonic fibroblasts (MEFs), or WT MEFs with Gαi1 plus Gαi3 CRISPR/Cas9 KO constructs (“CRISPR-Gαi1/3-DKO”, C), were treated with NLGN3 (50 ng/mL) for applied time, tested by Western blotting of listed proteins in total cell lysates. “MW” stands for molecular weight (Same for all Figures). Data were expressed as mean ± standard deviation (SD, same for all Figures). Quantifications were from five replicate blot data. *P < 0.05.
Gαi1 and Gαi3 are required for NLGN3-induced Akt, Erk and mTORC1 activation in MEFs. WT MEFs with the scramble control shRNA (“scr-shRNA”), the lentiviral Gαi1 shRNA, the lentiviral Gαi3 shRNA, or both (“Gαi1/3 DshRNA”), were treated with NLGN3 (50 ng/mL) for applied time periods, and tested by Western blotting of listed proteins in total cell lysates (A). Gαi1/3 DKO MEFs (B) or WT MEFs (C) were transfected with the adenovirus Gαi1 construct (“Ad-Gαi1”), the adenovirus Gαi3 construct (“Ad-Gαi3”) or the empty vector (“Ad-Vec”), treated with NLGN3 (50 ng/mL) for applied time periods, and tested of listed proteins. Quantifications were from five replicate blot data. *P < 0.05 vs. “scr-shRNA” (A) or WT MEFs (B). #P < 0.05. “n.s.” stands for P > 0.05 (no statistical differences, C).
Conversely ectopic overexpression of Gαi1 and Gαi3 in P1 glioma cells significantly increased NLGN3-induced phosphorylation of Akt and S6K (Figure 3D), without affecting their expression (Figure 3D). For these experiments, Ad-Gαi1 (no tag) and Ad-Gαi3 (no tag) were transduced into P1 glioma cells, establishing Gαi1 plus Gαi3-overexpressed cells (“OE-Gαi1/3”, two stable cell lines, sL1/sL2) (Figure 3D). Similarly, ectopic overexpression of Gαi1 and Gαi3 in U251MG cells (“OE-Gαi1/3”) enhanced NLGN3-induced Akt, S6K1 and Erk1/2 phosphorylation (Figure 3D). Together, these results show that Gαi1 and Gαi3 are essential proteins for NLGN3 signaling in human glioma cells.
Gab1 is a key adaptor protein for NLGN3 signaling in human glioma cells
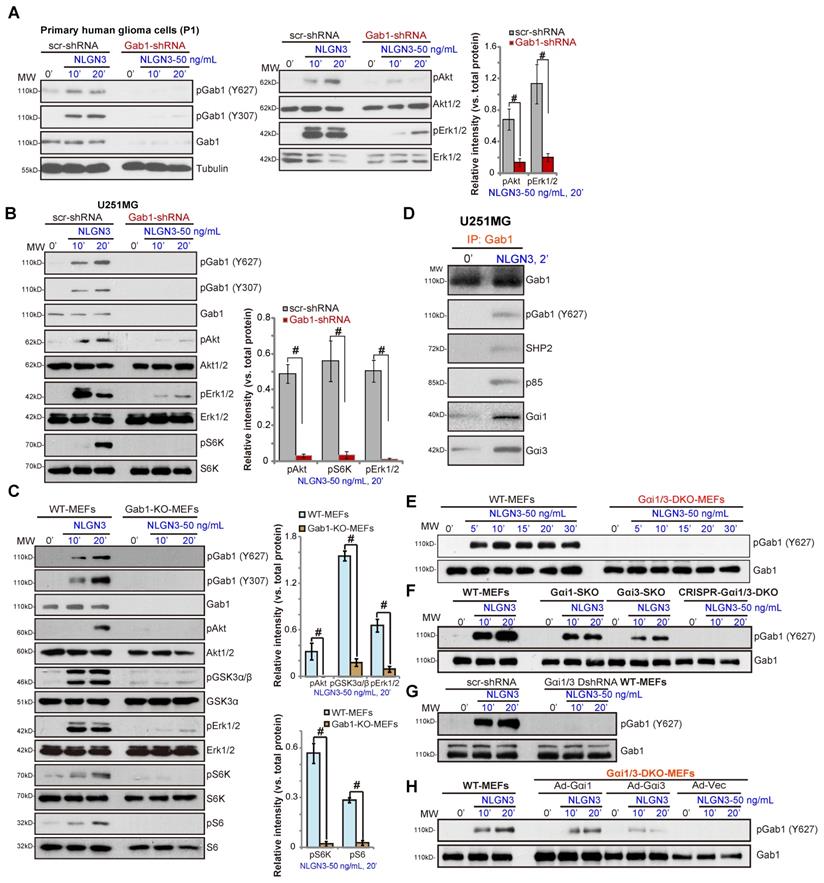
Gab1 is a key Gαi1/3 adaptor protein required for RTK signal transduction [25, 27-29]. We tested whether Gab1 is a required adaptor protein in NLGN3 signaling. NLGN3 was found to induce Gab1 activation, indicated by increased phosphorylation at Tyr-627 and Tyr-307 [39] in P1 primary human glioma cells (Figure 4A) and U251MG cells (Figure 4B). Significantly, shRNA-mediated stable knockdown of Gab1 inhibited NLGN3-induced Akt, Erk1/2 and S6K phosphorylation in U251 and primary glioma cells (Figure 4A and B).
In MEFs Gab1 KO abolished NLGN3 signaling by blocking downstream Akt-mTORC1 and Erk1/2 activation (Figure 4C). Co-IP results in U251MG cells, Figure 4D, demonstrated that, in response to NLGN3, Gab1 associated with Gαi1, Gαi3, SHP2 and p85. As Gab1-p85 association mediates PI3K-Akt activation and Gab1-SHP2 association is vital for Erk-MAPK cascade activation [39], we propose that Gab1 is a key adaptor protein for NLGN3 signaling in human glioma cells.
Importantly, NLGN3-induced Gab1 activation (p-Tyr at 627) was completely blocked in Gαi1/3 DKO MEFs (Figure 4E) and in CRISPR-Cas9-induced Gαi1/3 DKO MEFs (Figure 4F), being partially inhibited with Gαi1 or Gαi3 SKO (Figure 4F). Additionally, shRNA-induced Gαi1/3 double knockdown also attenuated NLGN3-induced Gab1 activation in MEFs (Figure 4G). Moreover, in DKO MEFs, re-expression of Gαi1 or Gαi3 restored NLGN3-induced Gab1 activation (Figure 4H). These results demonstrate that Gab1, lying downstream of Gαi1/3, mediates NLGN3-induced Akt-mTORC1 and Erk activation.
Gab1 is a key adaptor protein of NLGN3 signaling. The primary human glioma cells (“P1”) (A) or U251MG cells (B), with Gab1 shRNA or the scramble control shRNA (“scr-shRNA”), were treated with or without NLGN3 (50 ng/mL) for applied time periods, listed proteins in total cell lysates were tested by Western blotting. Wild-type (WT) and Gab1 knockout (Gab1-KO) MEFs were treated with NLGN3 (50 ng/mL) for applied time, tested by Western blotting of listed proteins in total cell lysates (C). U251MG cells were treated with NLGN3 (50 ng/mL) for 5 min, association of Gαi1/3-Gab1-SHP2-p85 was tested by co-IP assays (D). WT, Gαi1/3 DKO (E), Gαi1 or Gαi3 SKO (F), as well as WT MEFs with Gαi1 plus Gαi3 CRISPR/Cas9 DKO constructs (F) or Gαi1 shRNA plus Gαi3 shRNA (“Gαi1/3 DshRNA”) (G) were treated with NLGN3 (50 ng/mL) for applied time, total- and p-Gab1 were tested. Gαi1/3 DKO MEFs were transfected with the adenovirus Gαi1 construct (“Ad-Gαi1”), the adenovirus Gαi3 construct (“Ad-Gαi3”) or the empty vector (“Ad-Vec”), treated with NLGN3 (50 ng/mL) for applied time periods, total- and p-Gab1 were tested (H). Quantifications were from five replicate blot data. #P < 0.05.
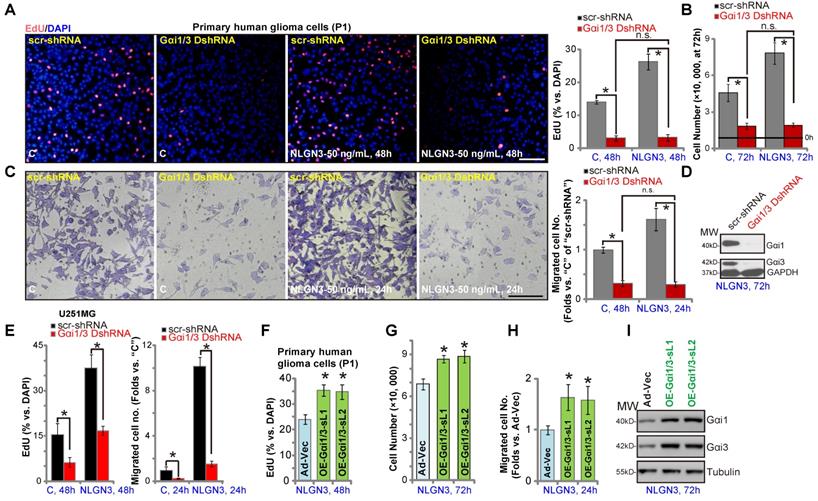
Gαi1 and Gαi3 mediates NLGN3-induced glioma cell progression in vitro
We next examined the role of Gαi1/3 proteins in glioma cell function. NLGN3 treatment promoted the growth of P1 primary glioma cells (with “scr-shRNA”), evidenced by increased EdU incorporation (Figure 5A, at 48h) and cell number (Figure 5B, at 72h). Significantly, NLGN3-induced effects were completely blocked by Gαi1 and Gαi3 shRNA (“Gαi1/3 DshRNA”) (Figure 5A and B). Notably, basal glioma cell growth (by complete medium, no NLGN3) was also inhibited by Gαi1/3 DshRNA (Figure 5A and B). Moreover, NLGN3 significantly induced glioma cell migration, evidenced by an increased number of migrated cells in “Transwell” tests (Figure 5C), which was reversed by Gαi1/3 DshRNA (Figure 5C). To confirm the efficiency of the applied shRNA, we showed that Gαi1 and Gαi3 proteins were silenced in Gαi1/3 DshRNA-expressing P1 glioma cells (with NLGN3 treatment, at 72h) (Figure 5D). In U251MG cells, NLGN3-induced cell proliferation (EdU-positive nuclei ratio, Figure 5D) and migration (“Transwell” cell number, Figure 5E) were significantly inhibited by Gαi1/3 DshRNA, which also inhibited medium-induced basal cell proliferation and migration (Figure 5E). These results demonstrated that Gαi1/3 silencing inhibited NGLN3-induced cell growth, proliferation and migration in vitro.
Conversely, ectopic overexpression of Gαi1 and Gαi3 (“OE-Gαi1/3”, see Figure 3) in P1 glioma cells promoted NGLN3-induced cell proliferation (increased EdU incorporation, Figure 5F), growth (Figure 5G), and migration (“Transwell” assay, Figure 5H). Western blotting results, Figure 5I, confirmed Gαi1/3 overexpression in OE-Gαi1/3 glioma cells (72h after NLGN3 treatment). Notably, basal glioma cell proliferation (by complete medium, no NLGN3) and migration were enhanced with OE-Gαi1/3 (Figure S1A-B). Akt and Erk1/2 phosphorylation was augmented as well (Figure S1C).
Gαi1 and Gαi3 are essential for NLGN3 signaling in glioma cells. The primary human glioma cells (“P1”) or U251MG cells, with Gαi1 shRNA plus Gαi3 shRNA (“Gαi1/3 DshRNA”) or the scramble control shRNA (“scr-shRNA”), were treated with NLGN3 (50 ng/mL) for applied time periods, listed proteins in total cell lysates or endosomal fractions were tested by Western blotting assays (A-C). P1 glioma cells or U251MG cells were transfected with the adenovirus Gαi1 construct plus the adenovirus Gαi3 construct (“OE-Gαi1/3”, two lines: “sL1/sL2”) or the empty vector (“Ad-Vec”), treated with NLGN3 (50 ng/mL) for 10 min, and tested by Western blotting assays of listed proteins (D). Quantifications were from five replicate blot data. #P < 0.05 (A).* P < 0.05 vs. “Ad-Vec” cells (D). “n.s.” stands for P > 0.05 (no statistical differences, C).
Gαi1 and Gαi3 mediate NLGN3-induced glioma cell progression in vitro. The P1 primary glioma cells or U251MG cells, with Gαi1 shRNA plus Gαi3 shRNA (“Gαi1/3 DshRNA”) or the scramble control shRNA (“scr-shRNA”), were treated with or without NLGN3 (50 ng/mL) for applied time periods, cell proliferation (A and E), growth (B), and migration (C and E) were examined by the assays mentioned in the text, with Gαi1 and Gαi3 expression tested as well (D, for P1 glioma cells). P1 glioma cells, with the adenovirus Gαi1 construct plus the adenovirus Gαi3 construct (“OE-Gαi1/3”, two lines: “sL1/sL2”) (F-I), were treated with or without NLGN3 (50 ng/mL) for applied time period, cell proliferation (F), growth (G), migration (H) and Gαi1/3 expression (I) were tested, with results quantified. For the EdU staining assay, ten random views were included to calculate EdU/DAPI ratios. For the “Transwell” assays, ten random views of each condition were included to calculate the average number of migrated cells. For all functional assays exact same number of viable cells of different genetic treatments were initially seeded into each well/dish (at 0h). “C” stands for medium control. Blotting data was repeated five times (D and I). *P < 0.05 (A-E). *P < 0.05 vs. “Ad-Vec” cells (F-H). “n.s.” stands for non-statistical difference (A-C). Scale bar=100 μm (A and C).
Gαi1 and Gαi3 are required for orthotopic growth of primary glioma xenografts in mouse brain
We performed orthotopic primary human glioma xenograft experiments using a previously-described protocol [26]. P1 glioma cells, derived from a GBM patient (see MRI diagnosis in Figure 6A), were intracranially (using the described parameters [38]) injected into brains of nude mice. At experimental day-26, when the first mouse in the control group exhibited obvious symptoms, all the mice were sacrificed and tumors isolated [26]. MRI imaging results confirmed that the growth of orthotopic primary glioma xenografts (Figure 6B, yellow boxes) was significantly inhibited when expressing Gαi1 shRNA plus Gαi3 shRNA (“Gαi1/3 DshRNA”). Gαi1 and Gαi3 double silencing resulted in an 80% reduction of orthotopic glioma xenograft growth (Figure 6C), and was significantly more effective than Gαi1 single silencing (50-60% inhibition, see our previous study [26]). Mouse body weights were equivalent between the Gαi1/3 DshRNA and control shRNA groups (Figure 6D). Gαi1/3 protein expression levels were significantly decreased in orthotopic primary xenografts with Gαi1/3 DshRNA, and levels of p-Akt, p-S6K and p-Erk1/2 inhibited (Figure 6E). Immunohistochemistry (IHC) results, shown in Figure 6F, further confirmed reductions in p-Akt and p-S6 in Gαi1/3-silenced orthotopic tumors (vs. control tumors). Therefore Gαi1/3 silencing significantly inhibited orthotopic growth of primary glioma xenografts.
Gαi1 and Gαi3 are required for orthotopic growth of primary glioma xenografts in mouse brain. Brain MRI images of a primary glioma patient providing primary human glioma cells (“P1” cells, A). The exact same amount of glioma cells (5 × 105 cells of each mouse), expressing Gαi1 shRNA plus Gαi3 shRNA (“Gαi1/3 DshRNA”), scramble non-sense shRNA (“scr-shRNA”), the adenovirus Gαi1 plus Gαi3 constructs (“OE-Gαi1/3”), or the empty vector (“Ad-Vec”), were intracranially injected to brains of nude mice (5-6 week old), after 26/20 days, representative MRI images of orthotopic glioma xenografts were presented (B and G); Animals were decapitated and tumors were isolated by surgery, tumor volumes (C and H) and mice body weights (D and I) were recorded. Tumor tissue lysates were tested by Western blot assay of listed proteins (E and J). Immunohistochemistry (IHC) images of p-Akt and p-S6K were shown (F). Western blotting quantifications were from five replicate blot data. IHC experiments were repeated in three pairs of tissues. # P < 0.05. “n.s.” stands for non-statistical difference (D and I). Scale bar=100 μm (F).
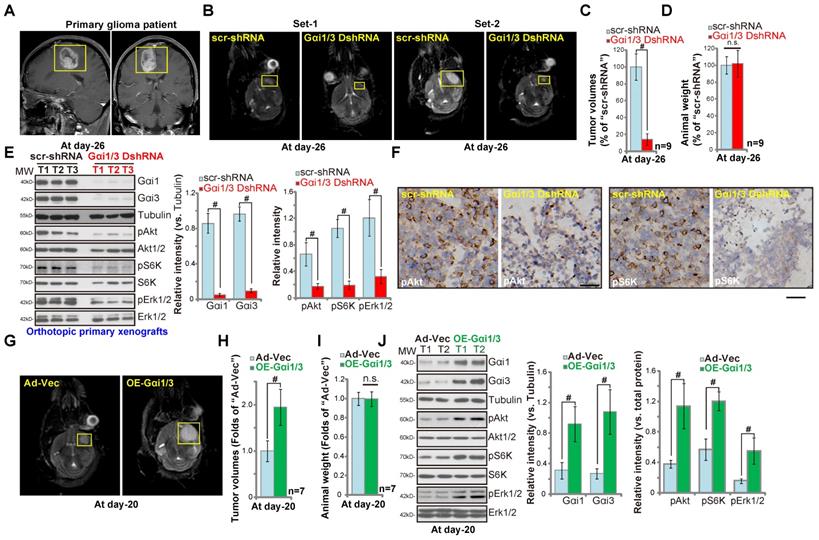
In contrast, ectopic Gαi1/3 overexpression promoted orthotopic growth of primary glioma xenografts. Primary glioma cells transduced with Ad-Gαi1 plus Ad-Gαi3 (“OE-Gαi1/3”) or the vector control (“Ad-Vec”) were injected into the brains of the nude mice. At experimental day-20, when the first mouse in the OE-Gαi1/3 group exhibited obvious symptoms, all mice were sacrificed and the tumors isolated. MRI images showed that Gαi1/3-overexpressing primary glioma xenografts grew faster than the control tumors (with Ad-Vec) (Figure 6G), presenting with higher tumor volumes (Figure 6H). Mouse body weights were unchanged (Figure 6I). Western blotting of tumor lysates confirmed Gαi1/3 overexpression in OE-Gαi1/3-tumor tissues, where levels of p-Akt, p-S6K and p-Erk1/2 increased (Figure 6J). Taken together, Gαi1 and Gαi3 are required for orthotopic primary glioma xenograft growth in mouse brain.
Orthotopic growth of brain-metastatic human lung cancer cells requires Gαi1 and Gαi3
Next we tested whether Gαi1 and Gαi3 were also required for the growth of metastatic brain tumor. Brain-metastatic lung cancer cells, bmLCs, from a lung cancer patient with brain metastasis (see the MRI image of brain metastatic tumor, Figure 7A) were obtained and primary-cultured. The bmLCs were infected with the lentivirus encoding Gαi1 shRNA plus Gαi3 shRNA (“Gαi1/3 DshRNA”) or scramble control shRNA (“scr-shRNA”), and stable bmLCs established after puromycin selection. In ex vivo cultured scr-shRNA control bmLCs, NLGN3 (50 ng, 10 min) increased phosphorylation of Akt, S6K and Erk1/2 (Figure S2). Gαi1 and Gαi3 double silencing by Gαi1/3 DshRNA potently inhibited NLGN3-induced signaling (Figure S2) in bmLCs.
Thereafter, cells were intracranially injected into brains of nude mice. At experimental day-25, when the first mouse in the control group exhibited obvious symptoms, all the mice were sacrificed. MRI image results confirmed that orthotopic bmLCs xenografts bearing Gαi1/3 DshRNA grew significantly slower than control bmLCs xenografts with scr-shRNA (Figure 7B). Gαi1 and Gαi3 double shRNA led to over 80% reduction of orthotopic bmLCs xenograft growth (Figure 7C). The mice body weights were indifferent between the two groups (Figure 7D). Next, signaling proteins in orthotopic bmLCs xenografts were examined. Western blotting assay results, Figure 7E, confirmed Gαi1 and Gαi3 silencing in bmLCs xenografts with Gαi1/3 DshRNA, while Gαi2 expression was unchanged (Figure 7E). Importantly, phosphorylations of Akt, S6K and Erk1/2 were largely decreased in Gαi1/3 DshRNA-expressing orthotopic bmLCs xenografts (Figure 7E). IHC images further confirmed decreased Akt-S6K phosphorylations in orthotopic bmLCs xenografts with Gαi1/3 DshRNA (Figure 7F). These results showed that silencing of Gαi1 and Gαi3 inhibited orthotopic growth of bmLCs in mouse brain.
Orthotopic growth of brain-metastatic human lung cancer cells requires Gαi1 and Gαi3. Brain MRI images of a primary lung cancer patient with brain metastasis (A) providing the brain-metastatic human lung cancer cells (“bmLCs”). bmLCs (5 × 105 cells of each mouse), expressing Gαi1 shRNA plus Gαi3 shRNA (“Gαi1/3 DshRNA”), scramble non-sense shRNA (“scr-shRNA”), the adenovirus Gαi1 plus Gαi3 constructs (“OE-Gαi1/3”), or the empty vector (“Ad-Vec”), were intracranially injected to brains of nude mice (5-6 week old); After 25/17 days, representative MRI images of orthotopic bmLCs xenografts were presented (B and G); Animals were decapitated and tumors were isolated by surgery, tumor volumes (C and H) and mice body weights (D and I) were recorded. The orthotopic bmLCs xenograft tissue lysates were tested by Western blot assay of listed proteins (E and J). Immunohistochemistry (IHC) images of p-Akt and p-S6K were shown (F). Western blotting quantifications were from five replicate blot data. IHC experiments were repeated in three pairs of tissues.# P < 0.05. “n.s.” stands for non-statistical difference (D and I). Scale bar=100 μm (F).
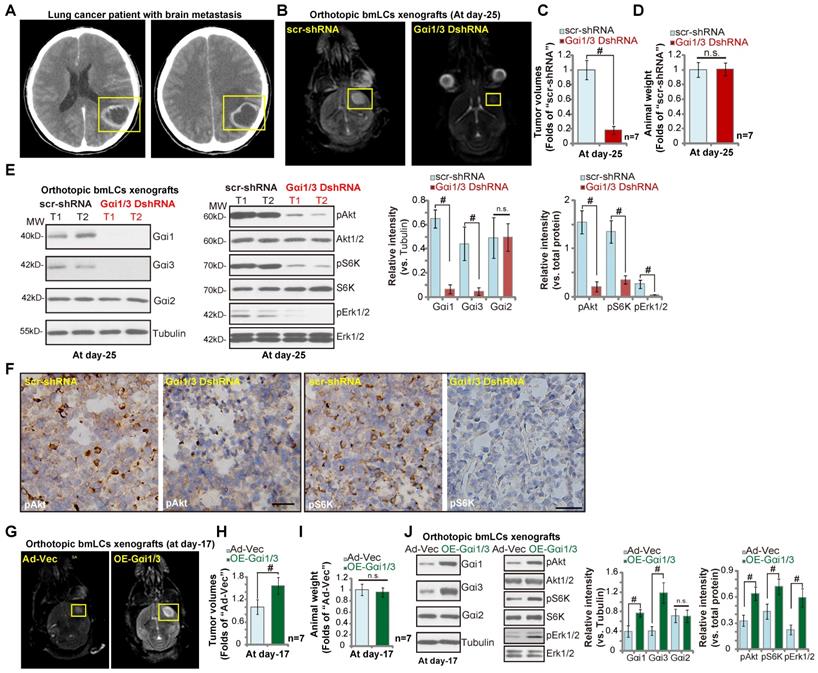
To further support out hypothesis, primary bmLCs, stably transduced with Ad-Gαi1 plus Ad-Gαi3 (“OE-Gαi1/3”), as well as the control cells with the vector control (“Ad-Vec”), were injected into the brains of the nude mice. At day-17, the first mice in the OE-Gαi1/3 group exhibited obvious neurological symptoms. MRI images showed that the orthotopic bmLCs xenografts with OE-Gαi1/3 were larger than the control xenografts (Figure 7G). Tumor volumes of OE-Gαi1/3 bmLCs xenografts were significantly higher than those of Ad-Vec xenografts (Figure 7H). The mice body weights were indifferent (Figure 7I). Western blotting assaying of orthotopic bmLCs xenograft tissues confirmed Gαi1/3 overexpression as well as increased phosphorylations of Akt-S6K1 and Erk1/2 in OE-Gαi1/3 xenograft tissues (Figure 7J). Together, these results show the orthotopic growth of bmLCs requires Gαi1 and Gαi3.
Gαi3 upregulation in human glioma tissues
We have previously shown that Gαi1 mRNA and protein levels are upregulated in human glioma, correlating with high tumor grade [26]. To examine Gαi3 expression in human glioma, we first consulted the TCGA database to examine RNA-Seq data. As shown, in human glioma tissues Gαi3 mRNA expression is significantly upregulated [P <0.05 vs. normal tissues] (Figure 8A). The overall survival of the low grade glioma (LGG) patients with Gαi3-high gliomas is significantly lower than those with Gαi3-high gliomas (P < 0.001, Figure 8B). There is however no significant difference in the overall survival of Gαi3-low GBM patients and Gαi3-high GBM patients (Figure 8B). This could be due to the extremely low overall survival of GBM patients (Figure 8B). When combining all glioma data, we showed that the average survival of Gαi3-high glioma patients is significantly lower than those with Gαi3-low gliomas (P < 0.001, Figure 8B). These results indicate that Gαi3 upregulation in human glioma is correlated with a poor overall survival.
To confirm the significance of the bioinformatics observations, we examined Gαi3 expression in human glioma specimens (“T”) and surrounding normal brain (“N”) tissues, from sixteen (16) low-grade (Grade I-II) tumors and another sixteen high-grade (Grade III-IV) tumors [26]. qPCR results, Figure 8C, demonstrated that Gαi3 mRNA levels are significantly upregulated in human glioma tissue (vs. normal brain tissue, Figure 8C). In late-stage gliomas (Grade III-IV, n=16) Gαi3 mRNA is more highly upregulated than in early-stage gliomas (Grade I-II, n=16) (Figure 8C). In the late-stage gliomas (Grade III-IV, n=16) Gαi3 mRNA is highly upregulated than that in early-stage gliomas (Grade I-II, n=16) (Figure 8C). Western blotting quantification results confirmed Gαi3 protein upregulation in human glioma tissues (Figure 8D), being more significant in the high-grade tumors (Figure 8D).
Gαi3 upregulation in human glioma tissues. TCGA database shows Gαi3 and NLGN3 expression (RNASeq, RSEM) in glioma tissues and normal brain tissues (A and F). Kaplan Meier Survival analyses of Gαi3-low and Gαi3-high glioma (LGG and GBM) patients (B). Human glioma tissues (“T”, 16 high-grade and 16 low-grade) and the paired normal brain tissues (“N”) were homogenized and dissolved in the tissue lysis buffer, Gαi3 mRNA and protein levels were tested by qPCR (C) and Western blotting (D), respectively, with results quantified. NLGN3 mRNA was tested by qPCR (E). * P < 0.05 vs.“N” group. #P < 0.05.
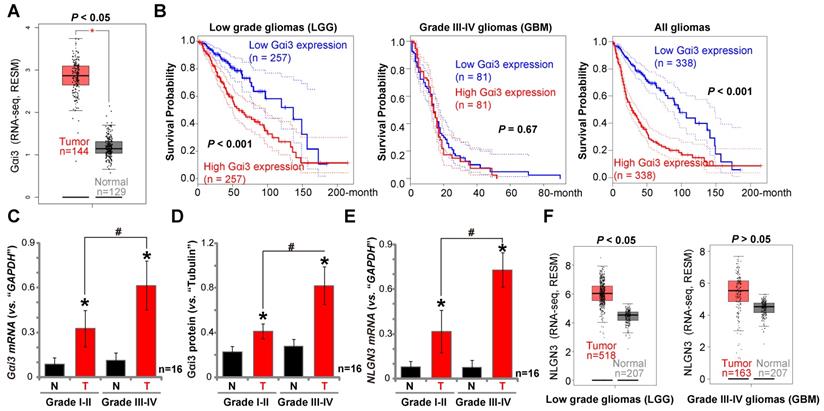
In line with previous studies [18, 19, 40], NLGN3 mRNA levels were increased in human glioma tissues (Figure 8E), and levels were higher in the late-stage gliomas (Figure 8E). TCGA cohorts showed that NLGN3 transcripts are higher in glioma tissues when compared to normal brain tissues (Figure 8F), being significant in LGG tissues (Figure 8F). Therefore, Gαi3, similar to Gαi1 [26], is upregulated in human glioma tissues, correlating with poor survival, high tumor grade and NLGN3 upregulation.
Discussion
GPCR components, including GPR56 [41], GPR55 [42] and many others, are dysregulated in human glioma and in other brain tumors. cAMP contents and the adenyl cyclase (AC) activity were decreased in brain tumors [43]. He et al., showed that Gαs subunit was downregulated in medulloblastoma, functioning as a tumor suppressor by blocking Sonic hedgehog signaling [44]. Lelievre et al., indicated that AC-mediated cAMP regulation plays an important cooperating role in the genesis of medulloblastoma [45]. Importantly, Gαi-coupled GPCRs, including cannabinoid receptors [46], CXCR4 [47, 48], dopamine D2 receptors [24] and melatonin receptor II (MTII) [49], were reported to be important for glioma progression. Pharmacological inhibition or genetic silencing of the Gαi-coupled GPCRs has potent anti-tumor effects [46-49]. Similarly, Urotensin II-activated Urotensin II receptor coupled to both Gαi/o and Gα13 to activate PI3K-PIP3-GEF-Rac-Cdc42 signaling cascade [50]. Le Joncour et al., reported that Urotensin II and its receptor were overexpressed in human glioma, important for angiogenesis of glioma [51]. Consistently, CXCR4-mediated Gαi activation and cAMP suppression is stimulated by the Sonic hedgehog pathway [52].
Our results show that Gαi1 and Gαi3 are essential players in NLGN3 signaling. In MEFs, Gαi1/3 DKO blocked NLGN3-induced Akt-mTORC1 and Erk-MAPK activation, with Gαi1 or Gαi3 SKO resulting in partial inhibition. Furthermore, in WT MEFs shRNA-mediated silencing of Gαi1 and Gαi3 attenuated Akt-mTORC1 and Erk-MAPK activation by NLGN3. Ectopic Gαi1/3 expression restored NLGN3-induced Akt-mTORC1 and Erk-MAPK activation in Gαi1/3 DKO MEFs, and enhanced NLGN3-induced signaling in WT MEFs. Similarly in established (U251MG) or primary human glioma cells, NLGN3-induced Akt-mTORC1 and Erk-MAPK activation was inhibited by Gαi1/3 shRNA, but enhanced with ectopic Gαi1/3 overexpression. These results show that Gαi1 and Gαi3 are required for NLGN3-induced mitogenic signal transduction.
Sustained and constitutive activation of multiple key RTKs is a primary driving force for glioma cell progression [53, 54], causing hyperactivation of downstream mitogenic pathways, including PI3K-Akt-mTOR and Erk-MAPK [54, 55]. There are reports of GPCR-RTKs crosstalk in glioma cells. For example, Huang et al., found that activation of the formylpeptide receptor induced trans-activation of EGFR via Gαi proteins, exacerbating malignant behaviors in glioblastoma cells [56]. Ghosh et al., found that Gαi-GIV (a novel non-receptor GEF for Gαi [57]) complex directly associated with and activated EGFR to promote cancer cell migration and proliferation [57].
Studies have shown that NLGN3 induces phosphorylations of multiple RTKs as well as downstream PI3K-Akt-mTOR and Erk-MAPK cascades, responsible for glioma cell growth and proliferation [19]. We found that Gαi1/3 proteins associate with activated RTKs (VEGFR and TrkB ([28, 29]) and are required for their endocytosis and downstream signaling. Here in glioma cells, Gαi1/3 silencing, by targeted shRNAs, blocked NLGN3-induced RTKs endosomal translocation and subsequent Gab1 activation, without affecting RTK phosphorylation and expression. Importantly, Gab1 silencing inhibited NLGN3-induced downstream signaling activation in glioma cells. These results suggest that Gαi1 and Gαi3 mediate NLGN3-induced signal transduction by promoting RTK endocytosis and adaptor protein Gab1 activation.
Functional studies demonstrate that Gαi1/3 are essential in mediating NLGN3-induced glioma cell progression in vitro and orthotopic primary glioma xenograft growth in vivo.NLGN3-stimulated glioma cell growth, proliferation and migration were largely inhibited by Gαi1/3 silencing, but enhanced by ectopic Gαi1/3 overexpression. Glioma xenografts are unable to grow in NLGN3 KO brain suggesting an essential dependency on NLGN3 for orthotopic glioma growth [28, 29]. Significantly, our results show that the growth of orthotopic primary glioma xenografts was largely inhibited with Gαi1/3 silencing. Conversely the Gαi1/3-overexpressed primary glioma xenografts grew significantly faster than the control tumors. We concluded that possibly by mediating NLGN3-induced signaling transduction, Gαi1/3 are essential for glioma cell progression in vitro and in vivo.
One important finding of this study is that Gαi1 and Gαi3 are required for the orthotopic growth of bmLCs in mouse brain. One possibility is that neuron-secreted NLGN3 is also important for orthotopic growth of bmLCs. Indeed, ex vivo cultured bmLCs were responsible to NLGN3. NLGN3 activated Akt-mTOR and Erk1/2 signalings, and promoted bmLCs proliferation and migration in vitro. Such actions were almost abolished with Gαi1/3 DshRNA. Importantly, orthotopic growth of bmLCs in mouse brain was largely inhibited with Gαi1/3 DshRNA, but augmented with ectopic Gαi1/3 overexpression. In addition, Akt-mTOR and Erk1/2 activation in orthotopic bmLCs xenograft tissues was inhibited with Gαi1/3 DshRNA, but enhanced with Gαi1/3 overexpression. Other mitogens, including the BDNF and VEGF, could also be important contributors for the growth of brain-metastatic cancer cells [18]. Interestingly, we have previously shown that BDNF- and VEGF-induced mitogenic signalings rely on Gαi1/3 [28, 29]. This could also demonstrate the essential role of Gαi1/3 in mediating orthotopic growth of bmLCs in mouse brain.
NLGN3 is a crucial factor for neuron-glioma synapse formation and neuronal activity-dependent glioma progression [14, 15, 20]. Interestingly, Hanahan and his colleagues have shown that brain metastatic breast-cancer cells can form a specialized type of pseudo-tripartite synapse with glutamatergic neurons [58]. This will allow cancer cells to take up glutamate and activate GluN2B-mediated NMDAR (N-methyl-D-aspartate receptor) signaling, required for metastatic cancer cell growth [58]. Thus, another possibility is that bmLCs might form synapses with neurons to activate glutamate signaling, and neuron-secreted NLGN3 could be essential for the process. The underlying mechanisms warrant further characterizations.
NLGN3 expression levels negatively correlate with overall survival of glioblastoma patients [18, 19]. Furthermore high NLGN3 expression is associated with glioma recurrence [40]. Our current and previous results [26] have demonstrated that Gαi1 and Gαi3 levels are significantly upregulated in human glioma tissues, and their upregulation is correlated with poor patients survival, high tumor grade and NLGN3 upregulation. Gliomas, especially the high grade GBM, are composed of a pathologically heterogeneous mixture of cells exhibiting different cellular and nuclear polymorphism [59-61]. Therefore, Gαi1/3 and NLGN3 upregulation in glioma tissues might be derived from both glioma cells and possible other cells in the tumor bulk (immune cells, endothelial cells and cancer stem cells, etc). Although the underlying mechanisms of Gαi1/3-mediated NLGN3 signaling and glioma cell progression need further characterization, the results of the present and previous studies [26] strongly indicate that Gαi1 and Gαi3 could be valuable therapeutic targets for treating human glioma.
Conclusions
Gαi1/3 mediation of NLGN3-induced signaling is essential for glioma growth in vitro and in vivo.
Acknowledgements
This work was generously supported by grants from the National Natural Science Foundation of China (81571282, 81771457, 81922025, 81773192, 81472786, 81802511, 81974388, 82072712, and 81700859); A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions; the Natural Science Foundation of Jiangsu Province (BK20170060). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7-30
2. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7-30
3. Reardon DA, Wen PY. Glioma in 2014: unravelling tumour heterogeneity-implications for therapy. Nat Rev Clin Oncol. 2015;12:69-70
4. Wen PY, Reardon DA. Neuro-oncology in 2015: Progress in glioma diagnosis, classification and treatment. Nat Rev Neurol. 2016;12:69-70
5. Kwiatkowska A, Symons M. Signaling Determinants of Glioma Cell Invasion. Adv Exp Med Biol. 2020;1202:129-49
6. Le Rhun E, Preusser M, Roth P, Reardon DA, van den Bent M, Wen P. et al. Molecular targeted therapy of glioblastoma. Cancer Treat Rev. 2019;80:101896
7. Wang L, Yang C, Wang Q, Liu Q, Wang Y, Zhou J. et al. Homotrimer cavin1 interacts with caveolin1 to facilitate tumor growth and activate microglia through extracellular vesicles in glioma. Theranostics. 2020;10:6674-94
8. Ran D, Zhou J, Chai Z, Li J, Xie C, Mao J. et al. All-stage precisional glioma targeted therapy enabled by a well-designed D-peptide. Theranostics. 2020;10:4073-87
9. Han B, Meng X, Wu P, Li Z, Li S, Zhang Y. et al. ATRX/EZH2 complex epigenetically regulates FADD/PARP1 axis, contributing to TMZ resistance in glioma. Theranostics. 2020;10:3351-65
10. Cheng CK, Fan QW, Weiss WA. PI3K signaling in glioma-animal models and therapeutic challenges. Brain Pathol. 2009;19:112-20
11. Zhang K, Zhang J, Han L, Pu P, Kang C. Wnt/beta-catenin signaling in glioma. J Neuroimmune Pharmacol. 2012;7:740-9
12. Wang H, Jiang D, Li W, Xiang X, Zhao J, Yu B. et al. Evaluation of serum extracellular vesicles as noninvasive diagnostic markers of glioma. Theranostics. 2019;9:5347-58
13. Huang W, Zhong Z, Luo C, Xiao Y, Li L, Zhang X. et al. The miR-26a/AP-2alpha/Nanog signaling axis mediates stem cell self-renewal and temozolomide resistance in glioma. Theranostics. 2019;9:5497-516
14. Venkatesh HS, Morishita W, Geraghty AC, Silverbush D, Gillespie SM, Arzt M. et al. Electrical and synaptic integration of glioma into neural circuits. Nature. 2019;573:539-45
15. Venkataramani V, Tanev DI, Strahle C, Studier-Fischer A, Fankhauser L, Kessler T. et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature. 2019;573:532-8
16. Gillespie S, Monje M. An active role for neurons in glioma progression: making sense of Scherer's structures. Neuro Oncol. 2018;20:1292-9
17. Thompson EG, Sontheimer H. A frightening thought: Neuronal activity enhances tumor growth. Cell Res. 2015;25:891-2
18. Venkatesh HS, Johung TB, Caretti V, Noll A, Tang Y, Nagaraja S. et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell. 2015;161:803-16
19. Venkatesh HS, Tam LT, Woo PJ, Lennon J, Nagaraja S, Gillespie SM. et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature. 2017;549:533-7
20. Johung T, Monje M. Neuronal activity in the glioma microenvironment. Curr Opin Neurobiol. 2017;47:156-61
21. Fan H, Li P, Zingarelli B, Borg K, Halushka PV, Birnbaumer L. et al. Heterotrimeric Galpha(i) proteins are regulated by lipopolysaccharide and are anti-inflammatory in endotoxemia and polymicrobial sepsis. Biochim Biophys Acta. 2011;1813:466-72
22. Gilder AS, Wang L, Natali L, Karimi-Mostowfi N, Brifault C, Gonias SL. Pertussis Toxin Is a Robust and Selective Inhibitor of High Grade Glioma Cell Migration and Invasion. PLoS One. 2016;11:e0168418
23. Magana-Maldonado R, Manoutcharian K, Hernandez-Pedro NY, Rangel-Lopez E, Perez-De la Cruz V, Rodriguez-Balderas C. et al. Concomitant treatment with pertussis toxin plus temozolomide increases the survival of rats bearing intracerebral RG2 glioma. J Cancer Res Clin Oncol. 2014;140:291-301
24. Luo Y, Kokkonen GC, Wang X, Neve KA, Roth GS. D2 dopamine receptors stimulate mitogenesis through pertussis toxin-sensitive G proteins and Ras-involved ERK and SAP/JNK pathways in rat C6-D2L glioma cells. J Neurochem. 1998;71:980-90
25. Cao C, Huang X, Han Y, Wan Y, Birnbaumer L, Feng GS. et al. Galpha(i1) and Galpha(i3) are required for epidermal growth factor-mediated activation of the Akt-mTORC1 pathway. Sci Signal. 2009;2:ra17
26. Liu YY, Chen MB, Cheng L, Zhang ZQ, Yu ZQ, Jiang Q. et al. microRNA-200a downregulation in human glioma leads to Galphai1 over-expression, Akt activation, and cell proliferation. Oncogene. 2018;37:2890-902
27. Zhang YM, Zhang ZQ, Liu YY, Zhou X, Shi XH, Jiang Q. et al. Requirement of Galphai1/3-Gab1 signaling complex for keratinocyte growth factor-induced PI3K-AKT-mTORC1 activation. J Invest Dermatol. 2015;135:181-91
28. Marshall J, Zhou XZ, Chen G, Yang SQ, Li Y, Wang Y. et al. Antidepression action of BDNF requires and is mimicked by Galphai1/3 expression in the hippocampus. Proc Natl Acad Sci U S A. 2018;115:E3549-E58
29. Sun J, Huang W, Yang SF, Zhang XP, Yu Q, Zhang ZQ. et al. Galphai1 and Galphai3mediate VEGF-induced VEGFR2 endocytosis, signaling and angiogenesis. Theranostics. 2018;8:4695-709
30. Bai JY, Li Y, Xue GH, Li KR, Zheng YF, Zhang ZQ. et al. Requirement of Galphai1 and Galphai3 in interleukin-4-induced signaling, macrophage M2 polarization and allergic asthma response. Theranostics. 2021;11:4894-909
31. Minjie S, Defei H, Zhimin H, Weiding W, Yuhua Z. Targeting pancreatic cancer cells by a novel hydroxamate-based histone deacetylase (HDAC) inhibitor ST-3595. Tumour Biol. 2015;36:9015-22
32. Schmittgen TD, Zakrajsek BA. Effect of experimental treatment on housekeeping gene expression: validation by real-time, quantitative RT-PCR. J Biochem Biophys Methods. 2000;46:69-81
33. Gschweitl M, Ulbricht A, Barnes CA, Enchev RI, Stoffel-Studer I, Meyer-Schaller N. et al. A SPOPL/Cullin-3 ubiquitin ligase complex regulates endocytic trafficking by targeting EPS15 at endosomes. Elife. 2016;5:e13841
34. Lv Y, Si M, Chen N, Li Y, Ma X, Yang H. et al. TBX2 over-expression promotes nasopharyngeal cancer cell proliferation and invasion. Oncotarget. 2017;8:52699-707
35. Shao NY, Wang DX, Wang Y, Li Y, Zhang ZQ, Jiang Q. et al. MicroRNA-29a-3p Downregulation Causes Gab1 Upregulation to Promote Glioma Cell Proliferation. Cell Physiol Biochem. 2018;48:450-60
36. Wang SS, Lv Y, Xu XC, Zuo Y, Song Y, Wu GP. et al. Triptonide inhibits human nasopharyngeal carcinoma cell growth via disrupting Lnc-RNA THOR-IGF2BP1 signaling. Cancer Lett. 2019;443:13-24
37. Cao C, Sun Y, Healey S, Bi Z, Hu G, Wan S. et al. EGFR-mediated expression of aquaporin-3 is involved in human skin fibroblast migration. Biochem J. 2006;400:225-34
38. Agnihotri S, Gajadhar AS, Ternamian C, Gorlia T, Diefes KL, Mischel PS. et al. Alkylpurine-DNA-N-glycosylase confers resistance to temozolomide in xenograft models of glioblastoma multiforme and is associated with poor survival in patients. J Clin Invest. 2012;122:253-66
39. Wang W, Xu S, Yin M, Jin ZG. Essential roles of Gab1 tyrosine phosphorylation in growth factor-mediated signaling and angiogenesis. Int J Cardiol. 2015;181:180-4
40. Liu R, Qin XP, Zhuang Y, Zhang Y, Liao HB, Tang JC. et al. Glioblastoma recurrence correlates with NLGN3 levels. Cancer Med. 2018;7:2848-59
41. Shashidhar S, Lorente G, Nagavarapu U, Nelson A, Kuo J, Cummins J. et al. GPR56 is a GPCR that is overexpressed in gliomas and functions in tumor cell adhesion. Oncogene. 2005;24:1673-82
42. Andradas C, Caffarel MM, Perez-Gomez E, Salazar M, Lorente M, Velasco G. et al. The orphan G protein-coupled receptor GPR55 promotes cancer cell proliferation via ERK. Oncogene. 2011;30:245-52
43. Furman MA, Shulman K. Cyclic AMP and adenyl cyclase in brain tumors. J Neurosurg. 1977;46:477-83
44. He X, Zhang L, Chen Y, Remke M, Shih D, Lu F. et al. The G protein alpha subunit Galphas is a tumor suppressor in Sonic hedgehog-driven medulloblastoma. Nat Med. 2014;20:1035-42
45. Lelievre V, Seksenyan A, Nobuta H, Yong WH, Chhith S, Niewiadomski P. et al. Disruption of the PACAP gene promotes medulloblastoma in ptc1 mutant mice. Dev Biol. 2008;313:359-70
46. Calatozzolo C, Salmaggi A, Pollo B, Sciacca FL, Lorenzetti M, Franzini A. et al. Expression of cannabinoid receptors and neurotrophins in human gliomas. Neurol Sci. 2007;28:304-10
47. Ehtesham M, Winston JA, Kabos P, Thompson RC. CXCR4 expression mediates glioma cell invasiveness. Oncogene. 2006;25:2801-6
48. Zhou Y, Larsen PH, Hao C, Yong VW. CXCR4 is a major chemokine receptor on glioma cells and mediates their survival. J Biol Chem. 2002;277:49481-7
49. Kinker GS, Ostrowski LH, Ribeiro PAC, Chanoch R, Muxel SM, Tirosh I. et al. MT1 and MT2 melatonin receptors play opposite roles in brain cancer progression. J Mol Med (Berl). 2021;99:289-301
50. Lecointre C, Desrues L, Joubert JE, Perzo N, Guichet PO, Le Joncour V. et al. Signaling switch of the urotensin II vasosactive peptide GPCR: prototypic chemotaxic mechanism in glioma. Oncogene. 2015;34:5080-94
51. Le Joncour V, Guichet PO, Dembele KP, Mutel A, Campisi D, Perzo N. et al. Targeting the Urotensin II/UT G Protein-Coupled Receptor to Counteract Angiogenesis and Mesenchymal Hypoxia/Necrosis in Glioblastoma. Front Cell Dev Biol. 2021;9:652544
52. Sengupta R, Dubuc A, Ward S, Yang L, Northcott P, Woerner BM. et al. CXCR4 activation defines a new subgroup of Sonic hedgehog-driven medulloblastoma. Cancer Res. 2012;72:122-32
53. Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R. et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318:287-90
54. Snuderl M, Fazlollahi L, Le LP, Nitta M, Zhelyazkova BH, Davidson CJ. et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell. 2011;20:810-7
55. Lo HW. EGFR-targeted therapy in malignant glioma: novel aspects and mechanisms of drug resistance. Curr Mol Pharmacol. 2010;3:37-52
56. Huang J, Hu J, Bian X, Chen K, Gong W, Dunlop NM. et al. Transactivation of the epidermal growth factor receptor by formylpeptide receptor exacerbates the malignant behavior of human glioblastoma cells. Cancer Res. 2007;67:5906-13
57. Ghosh P, Beas AO, Bornheimer SJ, Garcia-Marcos M, Forry EP, Johannson C. et al. A G{alpha}i-GIV molecular complex binds epidermal growth factor receptor and determines whether cells migrate or proliferate. Mol Biol Cell. 2010;21:2338-54
58. Zeng Q, Michael IP, Zhang P, Saghafinia S, Knott G, Jiao W. et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature. 2019;573:526-31
59. Ellis HP, Greenslade M, Powell B, Spiteri I, Sottoriva A, Kurian KM. Current Challenges in Glioblastoma: Intratumour Heterogeneity, Residual Disease, and Models to Predict Disease Recurrence. Front Oncol. 2015;5:251
60. Bonavia R, Inda MM, Cavenee WK, Furnari FB. Heterogeneity maintenance in glioblastoma: a social network. Cancer Res. 2011;71:4055-60
61. Little SE, Popov S, Jury A, Bax DA, Doey L, Al-Sarraj S. et al. Receptor tyrosine kinase genes amplified in glioblastoma exhibit a mutual exclusivity in variable proportions reflective of individual tumor heterogeneity. Cancer Res. 2012;72:1614-20
Author contact
![]() Corresponding authors: Prof. Ke-ran Li, The Fourth School of Clinical Medicine, The Affiliated Eye Hospital, Nanjing Medical University,. 138 Han-zhong Road, Nanjing, China, 210029. Tel/Fax: +86-025-86677699. E-mail: kathykeran860327com. Prof. Fang Liu, Department of Neurosurgery, Nanjing Medical University Affiliated Changzhou NO.2 People's Hospital, Changzhou, 213003, Jiangsu, China. Tel/Fax:+86-0519-86557524. Email: czdoctorliucom. Dr. Gang Chen, Professor. Department of Neurosurgery, The First Affiliated Hospital of Soochow University, Suzhou 215006, China. Tel:+86 512 65223637; Fax: +86-512-65223638. E-mail address: nju_neurosurgerycom. Prof. Cong Cao (primary contact), Jiangsu Key Laboratory of Neuropsychiatric Diseases and Institute of Neuroscience, Soochow University, 199 Ren-ai Road, Suzhou, 215123, China. Tel/Fax: +86-0512-65883658. E-mail: caocongedu.cn.
Corresponding authors: Prof. Ke-ran Li, The Fourth School of Clinical Medicine, The Affiliated Eye Hospital, Nanjing Medical University,. 138 Han-zhong Road, Nanjing, China, 210029. Tel/Fax: +86-025-86677699. E-mail: kathykeran860327com. Prof. Fang Liu, Department of Neurosurgery, Nanjing Medical University Affiliated Changzhou NO.2 People's Hospital, Changzhou, 213003, Jiangsu, China. Tel/Fax:+86-0519-86557524. Email: czdoctorliucom. Dr. Gang Chen, Professor. Department of Neurosurgery, The First Affiliated Hospital of Soochow University, Suzhou 215006, China. Tel:+86 512 65223637; Fax: +86-512-65223638. E-mail address: nju_neurosurgerycom. Prof. Cong Cao (primary contact), Jiangsu Key Laboratory of Neuropsychiatric Diseases and Institute of Neuroscience, Soochow University, 199 Ren-ai Road, Suzhou, 215123, China. Tel/Fax: +86-0512-65883658. E-mail: caocongedu.cn.