Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2021; 11(16):7685-7699. doi:10.7150/thno.60190 This issue Cite
Research Paper
Attenuation of tonic inhibition prevents chronic neurovascular impairments in a Thy1-ChR2 mouse model of repeated, mild traumatic brain injury
James R Mester1,2 ![]() , Paolo Bazzigaluppi2,3, Adrienne Dorr2, Tina Beckett2,3, Matthew Burke5,6, JoAnne McLaurin3,7, John G Sled1,4, Bojana Stefanovic1,2
, Paolo Bazzigaluppi2,3, Adrienne Dorr2, Tina Beckett2,3, Matthew Burke5,6, JoAnne McLaurin3,7, John G Sled1,4, Bojana Stefanovic1,2
1. University of Toronto, Department of Medical Biophysics, Toronto, Ontario, Canada.
2. Physical Sciences, Sunnybrook Research Institute, Toronto, Ontario, Canada.
3. Biological Sciences, Sunnybrook Research Institute, Toronto, Ontario, Canada.
4. Mouse Imaging Centre, The Hospital for Sick Children, Toronto, Ontario, Canada.
5. Neuropsychiatry Program, Department of Psychiatry and Division of Neurology, Department of Medicine, Sunnybrook Health Sciences Centre, University of Toronto, Toronto, ON, Canada.
6. Division of Cognitive Neurology, Department of Neurology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, USA.
7. University of Toronto, Department of Laboratory Medicine and Pathology, Toronto, Ontario, Canada.
Received 2021-3-7; Accepted 2021-6-4; Published 2021-6-16
Abstract

Rationale: Mild traumatic brain injury (mTBI), the most common type of brain trauma, frequently leads to chronic cognitive and neurobehavioral deficits. Intervening effectively is impeded by our poor understanding of its pathophysiological sequelae.
Methods: To elucidate the long-term neurovascular sequelae of mTBI, we combined optogenetics, two-photon fluorescence microscopy, and intracortical electrophysiological recordings in mice to selectively stimulate peri-contusional neurons weeks following repeated closed-head injury and probe individual vessel's function and local neuronal reactivity.
Results: Compared to sham-operated animals, mTBI mice showed doubled cortical venular speeds (115 ± 25%) and strongly elevated cortical venular reactivity (53 ± 17%). Concomitantly, the pericontusional neurons exhibited attenuated spontaneous activity (-57 ± 79%) and decreased reactivity (-47 ± 28%). Post-mortem immunofluorescence revealed signs of peri-contusional senescence and DNA damage, in the absence of neuronal loss or gliosis. Alteration of neuronal and vascular functioning was largely prevented by chronic, low dose, systemic administration of a GABA-A receptor inverse agonist (L-655,708), commencing 3 days following the third impact.
Conclusions: Our findings indicate that repeated mTBI leads to dramatic changes in the neurovascular unit function and that attenuation of tonic inhibition can prevent these alterations. The sustained disruption of the neurovascular function may underlie the concussed brain's long-term susceptibility to injury, and calls for development of better functional assays as well as of neurovascularly targeted interventions.
Keywords: Traumatic brain injury, Two-photon fluorescence microscopy, Optogenetics, Neurovascular coupling, L-655, 708
Introduction
Mild traumatic brain injury (mTBI) is the most common type of traumatic brain injury globally, with an incidence of 369 in 100,000 [1]. Ninety percent of all TBI are mild [2, 3] and many cases of mTBI are thought to go undiagnosed [3]. In addition to short-term cognitive dysfunction such as fatigue, mental clouding, and confusion, mTBI can lead to long-term neuropsychiatric and neurological deficits [4-9], with mTBI raising the probability of dementia up to 6-fold [6]. Adverse effects are compounded when mTBI occurs repeatedly [10, 11]. Many organizations have consequently implemented a three-strike rule in which individuals suffering mTBI for the third time are isolated from situations that previously posed risk of injury irrespectively of any symptoms [12]. Clinical assessment of mTBI is confounded by the absence of contrast in the peri-contusional (proximal to injury focus) region on structural neuroimaging [13-15]. Hitherto, the majority of the preclinical mTBI research effort has focused on acute neurological changes and diagnosis, leaving major gaps in our understanding of the lasting deficits after repeated mTBI. The absence of detectable neuroimaging contrast and the gaps in the understanding of chronic pathology have encumbered development of effective interventions: currently, pharmacological treatments are prescribed in the subacute and chronic stage of TBI to treat symptoms and not the root causes of dysfunction [16].
Particularly significant to understanding the lasting deficits post TBI is evaluation of the neurovascular unit, given its importance for normal brain functioning [17]; and its high dynamicity in the subacute stage post injury [18]. To date, studies of the neurovascular unit at the individual vessel level post mTBI have been limited to recordings at rest or ex vivo [19-22], leaving injury-induced changes to functional hyperemia at the single vessel level unclear. At the same time, dysregulation of functional hyperemia has been suggested as a promising therapeutic target post TBI [23]. In parallel, neuronal evaluations post-mTBI have shown reduced peri-contusional firing and attenuated evoked potentials [8, 9, 24-29], and neuronal loss [20, 25, 27, 30, 31]. Attenuated neuronal activity may result from differential susceptibility of excitatory vs. inhibitory neurons to injury [32], and in turn lead to further neurovascular unit disruptions [33]. Addressing post-injury excitatory/inhibitory imbalances [8, 9, 27] has recently been proposed a means of correcting both neuronal and cerebrovascular disruptions [9]. Two recent studies accordingly investigated GABA-A receptor (GABAAR) inverse agonism [26, 27]. In ischemia and chronic stress, beneficial effects on functional recovery have been shown [34-36] by GABAAR antagonism via sustained, low dose administration of L-655,708, a GABAAR inverse agonist with dose-dependent specificity [37]. Its applications in mTBI models have resulted in partial behavioural recovery and alleviation of post-injury increases in GABAergic tone in slice preparations [26, 27]; however, the effects of GABAA receptor inverse agonism on in situ neurophysiological state are not known. These gaps are particularly significant given the critical role of the re-instatement of neurovascular coupling for functional recovery [17]. Finally, quantitative functional assays are key to enable reliable cross-study comparisons and synthesis of results [13, 38].
Here we use simultaneous two-photon fluorescence microscopy and intracerebral electrophysiology all recordings in an optogenetic murine model of repeated mTBI to quantify the sustained effects on the neurovascular unit with low invasiveness and high spatio-temporal precision, and assess the potential for neurovascular function normalization by a disinhibitory intervention. For this study, mTBI was modeled by three closed head injuries, an intervention that has previously been shown to produce no contrast on conventional neuroimaging [39]. Neurovascular readouts were quantified in the impacted tissue region (henceforth peri-contusional tissue). Focused photostimulation of channelrhodopsin-2 was used to probe the neurovascular unit function in light of our earlier work showing this paradigm provides more robust neuronal and vascular responses than do physiological stimuli and offers greater spatiotemporal control over the stimulation, while still providing a correlate of responses to physiological stimuli [40]. We found sustained neurovascular function aberrations that parallel earlier mesoscopic scale observations in clinical populations but that have not been thus far observed in preclinical models of mTBI. The observed mTBI-induced neurovascular deficits were largely prevented via a disinhibitory treatment with L-655,708, an inverse GABAAR agonist.
Methods
Animals
All experimental procedures in this study followed the ARRIVE guidelines and were approved by the Animal Care Committee of the Sunnybrook Research Institute, which adheres to the Policies and Guidelines of the Canadian Council on Animal Care and meets all the requirements of the Provincial Statute of Ontario, Animals for Research Act as well as those of the Canadian Federal Health of Animals Act. Ninety-four adult (51 male and 43 female) Thy1-ChR2-YFP mice (wild-type Channelrhodopsin-2, ChR2, in pyramidal neurons [41], Jackson labs 007612, line 18), 3-6 months of age, were used in the study. Cohort sizes and sex ratios are summarized in Table 1.
Summary of animals and vessel recordings across cohorts.
Group | 2PFM Cohort | 2PFM + EP Cohort | ||
|---|---|---|---|---|
| Mice | Vessels | Mice | Vessels | |
| Sham/DMSO vehicle | 6 | 34 arterioles 61 venules | 5 | 50 arterioles 44 venules |
| Sham/L-655,708 | 6 | 49 arterioles 63 venules | 5 | 43 arterioles 41 venules |
| TBI/DMSO vehicle | 10 | 50 arterioles 82 venules | 5 | 42 arterioles 46 venules |
| TBI/L-655,708 | 7 | 56 arterioles 79 venules | 5 | 28 arterioles 35 venules |
Closed head injury surgery
The induction of closed head injury was performed by adapting a previously published protocol [25]. Mice were anesthetized in an induction chamber with 5% isoflurane in oxygen enriched medical air (30% O2, balance N2) and transferred to a stereotaxic frame, where they were maintained under 1.5-2% isoflurane via a nose cone. Mice were subcutaneously administered Baytril (0.02 mg/mL), Marcaine (0.1 mL), and Ringer's lactate solution (0.5-1 mL of solution with 130 mM Na, 4 mM K, 1.5 mM Ca, 109 mM Cl, 28 mM Lactate, Hospira, Canada). The scalp was shaved and following sterile surgical procedures, an incision was made in the scalp to expose the skull surrounding the impact site at +1mm ML, +0.5 mm AP. Connective tissue was removed and an impact delivered at the aforementioned location via a stereotaxic impactor (Leica Cortical Impactor, Leica, US) using the following parameters: 1.5 mm tip diameter, 2 m/s impact speed, 1 mm impact depth and 200 ms impact dwell time (corresponding to 359 kPa). Following the impact, the scalp was sutured, Ringer's lactate solution re-administered, and Betadine reapplied over the incision site. Mice were then monitored and allowed to recover in a recovery cage on a warming pad. After approximately 30 min, fully awake and alert animals were returned to their home cage in the colony. Each mouse was subjected to three cortical impacts, delivered with an inter-impact interval of 72 h. A 9% mortality rate was observed in the 47 mice that underwent impact procedures and in vivo experiments, due to skulls cracking in three mice as well as one incident of poor recovery from surgery. Sham animals underwent the same surgical procedures but with no impact delivery.
Osmotic pump implant surgery
Osmotic pump implant surgeries were performed 3 days following the third closed head injury surgery. The 3-day delay to start of L-655,708 administration was chosen based on previous reports of the timing of hyper-/hypoexcitability shifts occurring in the days following CNS damage [8,9,34,35] Osmotic pumps (Alzet micro-osmotic pump Model 1002, Durect, USA) were prepared one day prior to implantation following established procedures [35]. Pumps were loaded with 5 mM L-655,708 (Bio-techne, USA) in 50% DMSO/50% saline to be delivered at a dose of 200 μg/kg/day over 12 days, previously described as a dose level for GABAAR ɑ5-subunit specificity with significant effects counteracting increased tonic inhibition [35, 37, 42] in the days following injury [8, 9]. The dosing was set to both significantly affect the ɑ5-subunit containing GABAARs and avoid convulsive behaviour [35, 37]. Vehicle pumps were loaded with 50% DMSO/50% saline. Loaded pumps were incubated at 37°C overnight prior to surgery. On the day of the pump implantation surgery, mice were anesthetized in an induction chamber under 5% isoflurane and transferred to a stereotaxic frame with auxiliary ear bars for head fixation where they were maintained under 1.5-2% isoflurane delivered via the nose cone. Mice were given subcutaneous Baytril (0.05 mg/mL), Marcaine (0.1 mL), and Ringer's lactate solution (1 mL). The skin behind the right shoulder was shaved and cleaned with alcohol wipes and Betadine and ocular gel applied to the eyes. Following sterile surgical procedures, an incision was made behind the right shoulder and subcutaneous space was gently cleared through the incision with blunt surgical clamps. Pumps were then placed subcutaneously through the incision such that they were fully enclosed under the skin. The incision was then sutured, Betadine reapplied and another 1 mL of Ringer's lactate solution administered. Mice were allowed to recover on a warming pad in a recovery cage. When mice were awake and alert, they were returned to their home cage. Over the subsequent 48 h, the mice were monitored closely for any signs of complications due to the implant surgery.
Surgical procedures in preparation for imaging
Mice were anesthetized with 5% isoflurane and maintained at 1.5-2-2.5% isoflurane while on a feedback-controlled temperature pad (CWE Inc., Ardmore, PA) set to 37°C. A tracheostomy was performed to enable mechanical ventilation (SAR 830/P, CWE Inc.) and a tail vein catheter placed to allow for fluorophore and anesthetic infusion. Breath rate, heart rate, arterial oxygen saturation, and pulse/breath distention were measured throughout experiments via a pulse oximeter (MouseOx, STARR Life Sciences) and recorded via an MP150 acquisition system (Biopac Systems, Canada). Representative physiological data acquisition is shown in Figure S2 and the recorded values are summarized in Table 2.
Summary of physiological readouts of injury/treatment groups. No significant difference in any physiological parameters was seen across groups.
| Sham/DMSO vehicle | Sham/ L-655,708 | TBI/DMSO vehicle | TBI/L-655,708 | |
|---|---|---|---|---|
| End tidal CO2 (mmHg) | 15 ± 3 | 18 ± 5 | 19 ± 3 | 16 ± 3 |
| Body temperature (°C) | 37.1 ± 0.1 | 37.1 ± 0.6 | 37.1 ± 0.1 | 37.0 ± 0.2 |
| O2 saturation (%) | 97 ± 2 | 97 ± 3 | 98 ± 1 | 97.3 ± 0.6 |
| Heart rate (bpm) | 353 ± 61 | 338 ± 42 | 397 ± 102 | 376 ± 39 |
Cranial window implantation was performed following established protocols [43]. Isoflurane was lowered to 1.25-1.5% and mice were head-fixed on a stereotactic frame (Narishige, Japan). Subcutaneous Ringer's lactate solution and Xylocaine (10 mg/mL, 50-100 μL volume, AstraZeneca Canada) were administered for hydration and local anesthesia, respectively. The scalp was removed and a dental drill used to create a 3-4mm craniotomy centered over the impact location, at +1 mm ML, +0.5 mm AP. The skull cap was removed; the dura was left intact unless it remained attached to the removed skull cap, and 1% agarose in phosphate-buffered saline (PBS) (Sigma-Aldrich, Canada) applied to the brain surface. A 5 mm glass coverslip for imaging only experiments or a small section of ~1 mm thick silicone-based polydimethylsiloxane (PDMS) [44] for combined imaging and electrophysiology experiments was placed over the agarose and secured with cyanoacrylate glue. Dental cement (Land Dental, USA) was then used to create an immersion well surrounding the window. Prior to imaging, a 70 kDa Texas Red dextran (25 mg/kg body weight, Invitrogen, USA) dissolved in PBS (8.33 mg/mL) was injected via a tail vein catheter.
Two-photon fluorescence microscopy (2PFM) and focused optogenetic stimulation
Two-photon fluorescence microscopy was performed following established methods [40]. 2PFM was performed on an FVMPE-RS multiphoton microscope (Olympus, Japan) with 25x/1.05 NA or 10x/0.6 NA objective lenses (Olympus, Japan) for imaging and imaging/electrophysiology experiments, respectively. The mouse was positioned under the microscope and an Insight Ti:Sapphire laser (SpectraPhysics, USA) was used to excite YFP-labelled ChR2-expressing neurons and Texas Red-labelled vasculature at 900 nm. Emitted signals were acquired by photomultiplier tubes (PMTs) preceded by 485-540 nm or 575-630 nm barrier filters, separated by a 570 nm dichroic mirror (Chroma Technology, USA). Images down to cortical depths of 500-600 μm were captured prior to functional imaging (Figure 2). Thereafter, isofluorane was discontinued and anesthesia switched to alpha-chloralose (75 mg/kg induction, 40 mg/kg/hr maintenance) via an infusion pump (Harvard Apparatus, USA). The objective was positioned over the injury focus, and cortical penetrating vessels within that 2PFM imaging FOV (508 x 508 μm for the 25x objective, 1272 x 1272 μm for the 10x objective) were interrogated. The red blood cell (RBC) speed in these vessels was recorded through galvanometer-driven line scans (1.1-1.3 ms/line, 2 μs/pixel) along the longitudinal axes of these vessels, at the level of pre-diving point segments parallel to the cortical surface. The 458 nm (0.5 mW/mm2) photostimulation was raster scanned with a separate galvanometer over a 120 μm-diameter circular ROI centered over each penetrating vessel with 144-502 ms repetition period, 4 μs/pixel, 2-2.1 s duration, so as to stimulate neuronal soma and processes proximal to the imaged vessel while maintaining >70% of the photostimulated area extravascular. Photostimulation power was measured via a power meter (Model 842-PE, Newport, USA). Power density conversion was performed using the area of the stimulation ROI (120 μm diameter circular ROI).
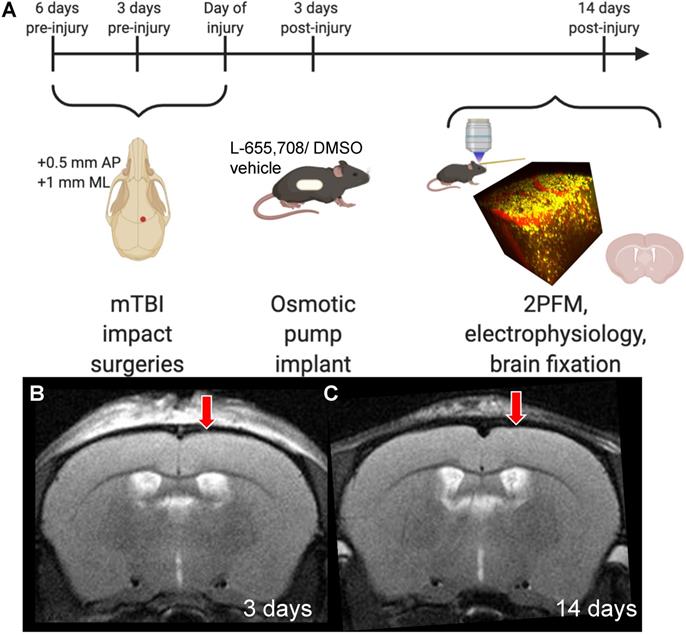
Experimental timeline. A. Mice received 3 mild impacts over the skull at the location indicated by the red dot (+0.5 mm AP, +1 mm ML) with a 3 day inter-impact interval. Impacts were delivered with a 1.5 mm impactor tip diameter, at 2 m/s, with a 200-ms dwell time. 3 days post-injury (DPI), mice were subcutaneously implanted with an osmotic pump containing either L-655,708 in saline/DMSO solution or vehicle alone. At 14 DPI, mice underwent 2PFM imaging and a subset of mice underwent concurrent local field potential recordings via intracortical tungsten microelectrode. A subset of mice brains did not undergo imaging, but were instead fixed for immunohistochemistry. B. Representative T2-weighted MR image in an injured mouse at 3 DPI. Red arrow indicates impact location. C. Representative T2-weighted MR image in an injured mouse at 14 DPI. Red arrow indicates impact location. Images in panel A were generated via BioRender.com.
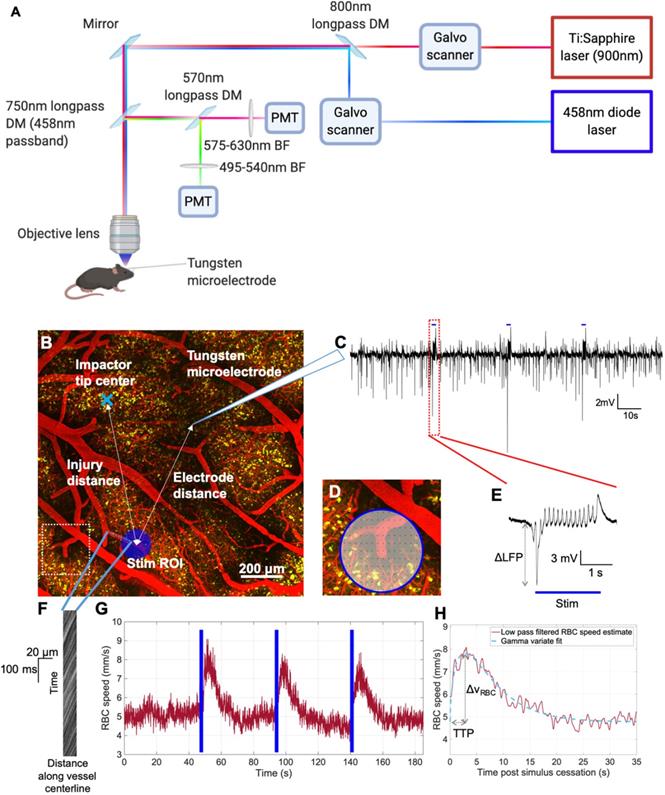
In vivo experimental paradigm for focused photostimulation and combined 2PFM/electrophysiological recording. A. Lightpath diagram of imaging/stimulation configuration in the FVMPE-RS system (Olympus, Japan). DM: dichroic mirror. BF: barrier filter. PMT: photomultiplier tube. B. Maximum intensity projection of a 2PFM image stack over 500 μm of cortical depth, with the image acquired using 900 nm excitation wavelength with a 10x/0.5 NA water immersion objective (Olympus, Japan) at 1024x1024 matrix size (1.243 μm/pixel). Red: intravenous Texas Red (Invitrogen, USA). Yellow: ChR2-eYFP in pyramidal neurons. Light blue “x” denotes impactor tip centre, while dark blue circle indicates raster-scanned focused photostimulation ROI, with light blue dots representing the imaging linescan trajectory inside the vessel. The inset in D corresponds to the dotted white square in B. C. Representative trace of LFP recordings. Blue bars indicate focused photostimulation periods. D. Representative pixel trajectory for focused photostimulation presentation. E. LFP response to focused photostimulation. Blue bar indicates focused photostimulation period. F. Example XT image collected in a penetrating venule. G. vRBC estimation via LSPIV method [46]. Blue bars denote photostimulation periods. H. Low pass-filtered vRBC response to focused photostimulation (red) and the corresponding gamma variate fit (light blue). Images in panel A generated via BioRender.com.
Local field potential recordings and data analysis
In a subset of experiments, local field potentials (LFPs) were recorded simultaneously with RBC speed. Mice were prepared as described above, but the cranial window was covered using PDMS instead of glass so as to allow for tungsten electrodes to be positioned in the cortical tissue below the cranial window [44], at a cortical depth of ca. 50 μm. LFP activity was captured via tungsten electrode mounted on a multi-manipulator (Scientifica, UK) and recorded through a patch-clamp amplifier operating in current clamp mode (MultiClamp 700B, Molecular Devices, USA) with an analog 20 Hz low-pass filter used to decrease background noise and enable real-time response visualization; and subsequently digitized for computer acquisition (Digidata 1440A, Molecular Devices, USA). Recordings were low pass-filtered post-hoc and LFP response parameters were extracted aparametrically. Extracted response parameters were peak LFP magnitude (ΔLFP) and area under the curve (AUC) during stimulus; as well as peak LFP magnitude and AUC following stimulus offset. To characterize neuronal activity at rest, power spectra of 20 s pre-stimulus baseline segments were acquired via FFT and divided into frequency bands as previously described [45]. ΔLFP increased with decreasing distance between the region of the photostimulation and the location of the recording electrode tip. This effect lead us to scale ΔLFP, on a subject-wise basis, to the level observed at the mean electrode-to-photostimulation ROI distance of all mice (Figure 2B): all LFP readouts depicted were linearly corrected in this way, as shown in Figure S1.
RBC speed data analysis
RBC speed was estimated from line scans as described earlier [46], allowing for robust speed estimation of fast-moving RBCs commonly seen in cortical penetrating arterioles. After image normalization and DC offset correction, sequential line scans were Fourier transformed, one FFT was conjugated and multiplied by the FFT of the other, and the inverse FFT was performed on the result to compute their cross-correlation. Gaussian peak-fitting was performed to calculate the spatial shift between adjacent line scans. RBC speed was calculated from this spatial shift while taking into account the line scan acquisition rate. RBC speed traces were then low-pass filtered and a 10-point moving average was applied to minimize noise and effects of respiratory/cardiac fluctuations. RBC traces following each stimulus presentation were fitted to a gamma function to estimate response magnitudes (ΔvRBC), time to peak (TTP), full-width-at-half-maximum (FWHM), and area under the curve (AUC).
Paired neurovascular measurements and neurovascular coupling ratio
In mice that underwent combined electrophysiology and 2PFM experiments, simultaneous neuronal and vascular recordings were made at rest and in response to photostimulation. At rest, Θ band power, acquired from spectral characterization of LFP baseline, and baseline RBC speed (vbase) were linearly regressed. In the evoked state, the neurovascular coupling (NVC) ratio was calculated as the quotient of venular ΔvRBC AUC to ΔLFP AUC.
Histological characterization
At experimental endpoint peripheral blood was flushed with phosphate buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4; pH 7.4) containing 0.1% heparin (1 i.u./mL; LEO Pharma Inc, Thornhill, ON) followed by 4% paraformaldehyde (PFA) (w/v) in PBS. Brains were post-fixed in 4% PFA overnight followed by cryoprotection in 30% sucrose (w/v) in PBS at 4°C. Brains were sectioned at a thickness of 40 µm via sliding freezing microtome (Leica Biosystems, Wetzlar, Germany). Brain slices were imaged on a Zeiss Observer Z1 microscope and images were analyzed via custom analysis in ImageJ. LaminB1 fluorescence was normalized on a slicewise basis to DAPI co-stain fluorescence to account for intersubject variance in staining. (Figure 7C). ɣH2AX expression was calculated from the fluorescence within neuronal nuclei, as determined by co-expression of DAPI and NeuN, and expressed as a percentage of area of those nuclei (Figure 7F). GFAP fluorescence was summed across images and normalized to the number of slices per image.
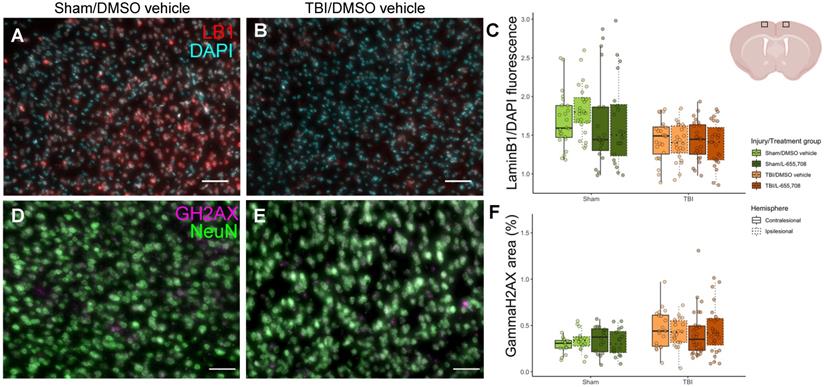
Histological characterization of the mTBI model. A. DAPI (cyan) /LaminB1 (red) staining of a brain slice maximum intensity projection along the A-P dimension in the ipsi-contusional hemisphere of a vehicle-treated sham mouse at the sham impact coordinates. B. DAPI/LaminB1 staining of a brain slice maximum intensity projection along the A-P dimension in the right hemisphere of an mTBI mouse at the impact coordinates. Scale bar = 50 μm. C. Total LaminB1 fluorescence normalized by total DAPI fluorescence in each image slice across injury/treatment groups. LaminB1 fluorescence is significantly reduced in mTBI mice (ΔLaminB1/DAPI = -17 ± 8%, pinjury = 0.047). Nmice = 23 (6 sham/vehicle, 5 sham/L-655,708, 6 TBI/vehicle, 6 TBI/L-655,708), Nimages = 184 (4 slices/subject, 2 contra/ipsi images/slice). Representation of imaging locations on the brain slices. Image from biorender.com. D. NeuN (green) /ɣH2AX (magenta) staining of a brain slice maximum intensity projection along the A-P dimension in the right hemisphere of a vehicle-treated sham mouse at the sham impact coordinates. E. NeuN/ɣH2AX staining of a brain slice maximum intensity projection along the A-P dimension in the right hemisphere of an mTBI mouse at the impact coordinates. Scale bar = 50 μm. F. Fractional area of ɣH2AX expression within neuronal nuclei in each image slice across injury/treatment groups. ɣH2AX expression is significantly increased in mTBI mice (ΔɣH2AX = 43 ± 13%, pinjury = 0.011). Nmice = 20 (4 sham/vehicle, 4 sham/L-655,708, 5 TBI/vehicle, 7 TBI/L-655,708), Nimages = 160 (4 slices/subject, 2 contra/ipsi images/slice).
Glial fibrillary acidic protein (GFAP) immunofluorescent staining
Forty-micron coronal sections were collected with a sliding microtome. 2 sections per animal at 160 µm apart were selected around the injury site (Bregma + 0.5 mm) for immunofluorescent staining. Tissue sections were washed with PBS (3 x 10 min) followed by an incubation in a blocking buffer (PBS with 0.2% triton-X-100, 0.2% bovine serum albumin (BSA) and 2% donkey serum) for 1 h at room temperature. Sections were incubated with rabbit polyclonal antibody against GFAP (Z0334, 1:500; DAKO) in blocking buffer overnight at 4°C. Alexa Fluor 647 Donkey anti-Rabbit secondary antibody (A31573, 1:200; Invitrogen) was incubated with DAPI (NucBlue Fixed Cell ReadyProbes Reagent, 2 drops/mL; Thermo Fisher) in PBS with 0.2% triton-X-100, 0.2% BSA for 2 h at RT. Sections were washed with PBS (3 x 10 min) and then mounted on VistaVision HistoBond slides (VWR) with polyvinyl-alcohol mounting medium with DABCO (PVA-DABCO, Sigma) and sealed with a coverslip.
Lamin-B1 immunofluorescent staining
Free-floating tissue sections underwent antigen retrieval (10 mM Sodium Citrate buffer @ 81°C, 20 min) followed by incubation in a blocking buffer (PBS with 0.3% triton-X-100 and 5% serum) for 1 h at room temperature. Sections were incubated with rabbit polyclonal antibody against Lamin B1 (ab16048, 1:1000; abcam, Cambridge, MA) in blocking buffer overnight at 4°C. AlexaFluor Gt anti-Rb secondary antibody (A-11037, 1:200; Thermo Fisher, Waltham, MA) was incubated with DAPI (NucBlue Fixed Cell ReadyProbes Reagent, 2 drops/mL as per manufacturer's instructions; Thermo Fisher) in PBS with 0.1% triton and 0.5% bovine serum albumin (BSA) for 2 h at RT. Tissue sections were washed with PBS between each step (3 x 10 min). Tissue sections were then mounted on VistaVision HistoBond slides (VWR, Mississauga, ON) with a polyvinyl-alcohol mounting medium with DABCO (PVA-DABCO, Sigma, Oakville, ON) and sealed with a coverslip. Nmice = 23 (6 sham/vehicle, 5 sham/L-655,708, 6 TBI/vehicle, 6 TBI/L-655,708), Nimages = 184 (4 slices/subject, 2 contra/ipsi images/slice).
ɣH2AX/NeuN immunofluorescent staining
Free-floating tissue sections underwent antigen retrieval (0.25 mM EDTA at 95°C, 40 min), were allowed to cool (10 min, benchtop), and were then incubated in blocking buffer (Tris Buffered Saline (TBS) with 4% BSA, 0.02% tween-20, and 2% serum) for 1 h at RT. Sections were incubated with primary antibodies against phospho-Histone H2A.X and NeuN (mouse anti-H2A.X (05-636, 1:200), guinea pig anti-NeuN (ABN90, 1:500); both Sigma-Aldrich) in blocking buffer overnight at 4°C. AlexaFluor-labeled secondary antibodies (Dk-anti-Mo-594, A21203, Thermo Fisher; Dk-anti-GP-647, AP193SA6, Sigma) were diluted at 1:200 in TBS with 4% BSA and 0.02% tween-20 and incubated for 2 h at RT. Tissue sections were washed with TBS between each step (3 x 10 min). Tissue sections were then mounted on VistaVision HistoBond slides (VWR) with PVA-DABCO (Sigma) and sealed with a coverslip. Nmice = 20 (4 sham/vehicle, 4 sham/L-655,708, 5 TBI/vehicle, 7 TBI/L-655,708), Nimages = 160 (4 slices/subject, 2 contra/ipsi images/slice).
Statistical analysis
The effects of injury and treatment were assessed by generalised linear modelling using the lme4, lmerTest, and nlme packages in R, with subjects treated as a random effect. This modelling accounts for variation within and between subjects to yield robust estimates in the presence of unbalanced group sizes. Fixed effects in linear mixed effect models were condition (injury/sham), treatment (L-655,708/vehicle), and, for histological data, hemisphere (contra-/ipsilateral to injury). Interaction terms between fixed effects were included in the modelling. The contrast across condition and treatment groups of the different readouts was thus modeled as:
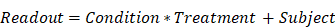
where multiplication in this notation denotes both the main effects and interaction between the two terms. Since the LFP magnitude estimates were not normal, they were log transformed prior to linear modelling. Sex was included as a fixed effect in preliminary analysis but was not found to exert a significant effect on any readout and was thus not made a part of the model in the final analysis.
Results
Resting venular RBC speed and neuronal Θ power are altered in mTBI mice
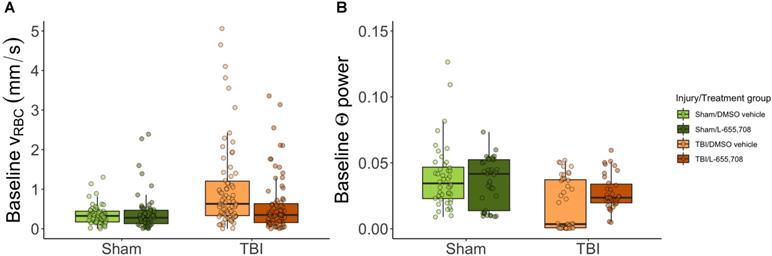
We first examined the resting hemodynamics by comparing pre-stimulus vRBC (vbase) across groups. At rest, arterioles did not exhibit any baseline RBC speed contrast. In contrast, RBC speed in penetrating venules of mTBI mice was twice that of sham animals (Δvbase injury = 115 ± 25%, pinjury = 0.000005, ptreatment = 0.78). Such increased venular baseline RBC speeds were not present in mTBI mice treated with L-655,708 (Δvbase = -88 ± 35%, pinjury x treatment = 0.01) as displayed in Figure 3A. Resting vessel diameters were not found to be significantly different across groups aside from a trend toward venular dilation due to mTBI (Δdvenule = 14.5 ± 9.1%, pvenule injury = 0.1). The standard deviation of venular baseline vRBC (σbase) was increased in mTBI (Δσbase= 60 ± 39%, pinjury = 0.0002, ptreatment = 0.54, pinjury x treatment = 0.07, Figure S3D). There was, however, no difference in the corresponding coefficient of variation (COV = σbase/vbase), indicating that the contrast in standard deviation resulted from the proportionate increase in vbase (Figure 3B). Analysis of power spectra of the baseline LFP recordings indicated that the theta band (4-8 Hz [45]) neuronal power was reduced in mTBI mice (Figure 3B, ΔΘinjury = -57 ± 79%, pinjury = 0.01). We also observed a trend toward a reduced effect of injury on neuronal theta band power in L-655,708 treated mTBI mice (ΔΘinjury x treatment = 27 ± 59%, pinjury x treatment = 0.08).
Baseline RBC speeds in penetrating venules and baseline neuronal Θ power are elevated in mTBI mice but normalized to sham levels in L-655,708 treated mTBI animals. A-B. Points indicate the mean of pre-stimulus baseline vRBC across 3 photostimulation presentations on a single vessel. A. Pre-stimulus baseline vRBC in penetrating venules. Baseline venular speeds are elevated in vehicle-treated mTBI mice, but indistinguishable from sham levels in L-655,708-treated mTBI mice. Δvbase injury = 115 ± 25%, pinjury = 0.000005, ptreatment = 0.78, Δvbase injury x treatment = -88 ± 35%, pinjury x treatment = 0.01. NSham/DMSO vehicle = 6, NSham/L-655,708 = 6, NTBI/DMSO vehicle = 10, NTBI/L-655,708 = 7. B. Neuronal Θ (4-8 Hz) power recorded during venular linescanning shown in A. mTBI induced an attenuation in Θ power, with a trend toward amelioration of this effect in L-655,708 treated cohort. ΔΘinjury = -57 ± 79% pinjury = 0.01, ptreatment = 0.93, ΔΘinjury x treatment = 27 ± 59%, pinjury x treatment = 0.08. NSham/DMSO vehicle = 5, NSham/L-655,708 = 5, NTBI/DMSO vehicle = 5, NTBI/L-655,708 = 5.
Reduced neuronal reactivity in mTBI mice
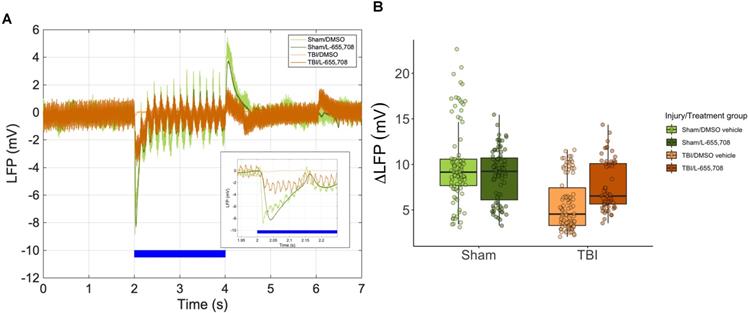
To investigate changes in local neuronal activity and neurovascular unit function post-injury, 2PFM measurements of vascular reactivity were paired with local field potential recordings via a tungsten microelectrode [44]. ΔLFP was attenuated in the mTBI group (ΔLFP = -47 ± 28%, p = 0.018), as shown in Figure 4. A simplified linear model further showed that there was no statistically significant difference between sham and mTBI L-655,708-treated mice (p = 0.59). Of note, as shown in Figure 2B, the Euclidean distance between photostimulation ROI/vessel diving point and the injury focus was recorded: however, no significant correlation between this distance and any neuronal measures were observed within the 1272 x 1272 μm imaging FOV used here. These findings show that mTBI may elicit sustained reduction in neuronal reactivity and that low-dose L-655,708 intervention can prevent this effect.
Neuronal hyporeactivity in mTBI mice is partially normalized in L-655,708 treated mTBI animals. A. Representative LFP traces during photostimulation. Inset depicts the early response. Blue bar indicates the photostimulation period. B. Peak magnitude of electrode distance-corrected LFP response to focused photostimulation. Lower responses were seen in vehicle-treated mTBI mice (ΔLFP = -47 ± 28%, p= 0.018), and a simplified linear model indicated that Sham/L-655,708 and TBI/L-655,708 groups were indistinguishable (p = 0.59). Points indicate the mean LFP peak across 3 stimulus presentations on each vessel. NSham/DMSO vehicle = 5, NSham/L-655,708 = 5, NTBI/DMSO vehicle = 5, NTBI/L-655,708 = 5.
Augmented venular reactivity in mTBI mice
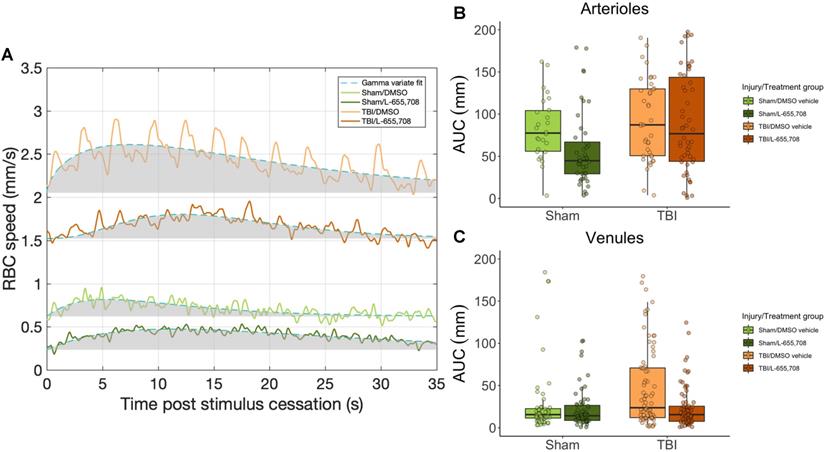
Vascular responsivity was evaluated in the chronic stage of repeated mTBI by measuring single cortical penetrating vessel responses to focused photostimulation (Figure 5). Area under the curve (AUC) of arteriolar responses to focused photostimulation was found to be indistinguishable between cohorts (Figure 5B), whereas venular reactivity was elevated in mTBI mice (Figure 5C, ΔAUC = 53 ± 17%, p = 0.001). This venular hyperreactivity was prevented by L-655,708 treatment (ΔAUC = -66 ± 24%, p = 0.005). Our findings here indicated sustained venular hyperreactivity post mTBI and a potential for its prevention by an early, low dose L-655,708 intervention. Similarly to neuronal recordings, no significant correlations between distance from injury focus and any vascular measures were observed within our 508 x 508 μm imaging FOV.
Venular hyperreactivity in mTBI mice is normalized to sham levels following L-655,708 administration. A. Venular vRBC responses to focused photostimulation in representative animals from each cohort. Shaded grey area represents the AUC plotted across all animals in B. B. AUC of arteriolar responses to focused photostimulation. Each point represents the mean response AUC across 3 stimulus presentations on each vessel. C. AUC of venular responses to photostimulation was significantly elevated in vehicle-treated mTBI mice. Each point represents the mean response AUC across 3 stimulus presentations on each vessel. L-655,708 treatment prevented this elevation. ΔAUCinjury = 53 ± 17%, pinjury = 0.001, ΔAUCinjury x treatment = -66 ± 24%, pinjury x treatment = 0.005. NSham/DMSO vehicle = 6, NSham/L-655,708 = 6, NTBI/DMSO vehicle = 10, NTBI/L-655,708 = 7.
Neurovascular coupling is altered in mTBI mice
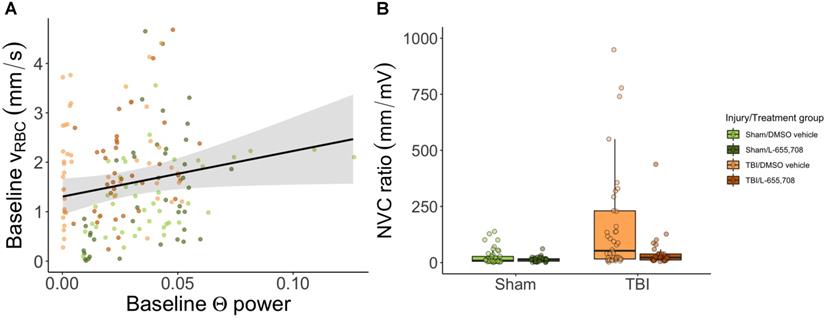
Lastly, we examined the relationship between neuronal and vascular reactivity to test for changes to neurovascular coupling. Paired neuronal and venular function measurements at rest were linearly regressed to assess the effects of mTBI on resting state (Figure 6). We observed a significant dependence (p = 0.009, slope = 9 ± 4) of the baseline venular vRBC on neuronal theta power at baseline (Figure 6A), with no effect of either injury or treatment. For responses to photostimulation, the neurovascular coupling (NVC) ratio was calculated as the ratio of vascular to neuronal AUC following photostimulation. We observed a significant increase in NVC ratio due to mTBI as shown in Figure 6B (ΔNVC = 674 ± 304%, p = 0.039) and further that mTBI mice treated with L-655,708 were indistinguishable from their uninjured counterparts (p = 0.88), implying an increased venular NVC component in the injured state that may be prevented through GABAAR inverse agonism.
Neurovascular coupling. A. Linear regression of baseline vRBC of all penetrating venules versus baseline theta power based on electrophysiological data acquired via the electrode in the imaging field of view. There was a significant positive linear dependence of baseline vRBC on baseline Θ power (p = 0.009, slope = 9 ± 4), but no dependence on injury (p = 0.89) or treatment (p = 0.44). Solid line represents a linear mixed effects model fit of all groups. B. Neurovascular coupling (NVC) ratio was calculated as the ratio of vascular response AUC to paired neuronal response AUC. Shown here are venular NVC ratios depicting an increased NVC in mTBI vehicle mice (ΔNVC = 674 ± 304%, p = 0.039), with L-655,708-treated sham and L-655,708-treated mTBI groups being indistinguishable (p = 0.88). NSham/DMSO vehicle = 5, NSham/L-655,708 = 5, NTBI/DMSO vehicle = 5, NTBI/L-655,708 = 5.
Reduced LaminB1 expression and increased ɣH2AX expression in mTBI mice
To independently assess the mTBI sequelae, we evaluated several histological markers in the pericontusional tissue. Neuronal survival (on NeuN) and glial activation (on GFAP) were indistinguishable between mTBI and sham cohorts. We next evaluated laminB1 and ɣH2AX as prior clinical and preclinical studies reported on TBI-elicited senescence, manifested as nuclear membrane loss (and thus reduced cerebral laminB1 expression) [47, 48], and DNA damage (and hence increased ɣH2AX expression in neuronal nuclei) [47]. LaminB1 expression was visibly reduced in mTBI vs. sham mice as shown in Figure 7C (ΔLaminB1/DAPI = -17 ± 8%, pinjury = 0.047). ɣH2AX expression was increased in mTBI cohort, as demonstrated in Figure 7F (ΔɣH2AX = 43 ± 13%, pinjury = 0.011). YFP expression in brain slices was also quantified to test whether ChR2 expression were altered following mTBI; however, no such changes were observed (Figure S4). No significant differences were observed between ipsi- and contracontusional hemispheres in mTBI mice in any histological readout (pLaminB1 hemisphere = 0.25, pɣH2AX hemisphere = 0.84).
Discussion
The cellular level changes elicited by mild traumatic brain injury are not well understood [49-51]. Previous preclinical mTBI studies have largely focused on structural changes and/or mesoscopic scale functional measures [21, 24, 25, 31, 52-56]. Here, for the first time, we paired single vessel measurements with intracortical recordings and applied focal photostimulation to elucidate neurovascular unit dysfunction post mTBI. We demonstrated beneficial effects of delayed, chronic, low dose disinhibition, via GABAAR inverse agonism, on the neurovascular function. Given the importance of neurovascular physiology for brain health [17], our findings motivate the development of post-concussion interventions that target the neurovascular unit to promote long term resilience of the concussed brain. Successful application of such interventions in heterogeneous clinical populations will be predicated on the development of translational biomarkers of neurovascular dysfunction post mTBI, thereby allowing optimization of treatment to individual patients' needs.
The impact parameters in this model were set within previously-described guidelines for inducing mild injury with the controlled cortical impact [57, 58]. The resulting injury was designated as 'mild' given lack of contrast on structural MRI, the criterion commonly applied in clinical practice [13-15]. That the injury produced was mild was further supported by the lack of group contrast on both NeuN and GFAP staining. While microhemorrhages, dural damage, and vessel rupture are common occurrences in the acute stage of mTBI [59-62], they are expected to resolve rapidly and were indeed not presently observed at two weeks post final insult. Prior preclinical studies have characterized TBI pathophysiology via morphological, functional, and behavioural assays [7-9, 19, 20, 22, 24-26, 30, 31, 50-52, 54-56, 63-71]. The presence and extent of these changes has varied widely across studies depending on the age of subject [30, 65, 72], time post-injury [19, 31, 54, 63, 64, 70], number of injuries [30, 31], and injury model [65, 67]. In the case of mild TBI, it has been shown that long-term changes may arise in both patients and in rodent models from either single or repeated injuries [11, 49]. Consistent with this, we observed neurovascular impairments two weeks post third impact. The current study used laminB1 and ɣH2AX expression as markers of senescence in the absence of cohort-wise differences with respect to either NeuN or GFAP expression. Dramatic laminB1 expression reductions, which are associated with nuclear membrane ruptures and cell death [73], have been observed in TBI and in preclinical models of TBI [47, 72]. In the present mild TBI model, a limited decrease of laminB1 expression was observed as well. Furthermore, heightened ɣH2AX expression, indicative of DNA damage, has been reported post mortem in TBI brains [47], a feature recapitulated by our model. Of note, these effects were not specific to the impacted hemisphere, indicating that closed head impacts elicit bilateral damage and/or that the injury is relayed by the two-week time point. Acute bilateral damage is likely a result of contrecoup injury [61], with the impactor causing deformation of the impacted cortex and contusion of the contralateral cortex. This is in line with cortical impact models being described as “mainly focal” [57]. Neuronal survival and astrogliosis, as measured by NeuN and GFAP expression, were indistinguishable among groups. In a similar model, we have previously reported no NeuN contrast alongside ipsicontusional GFAP contrast [25], as with earlier work [31, 74]; other studies, in contrast, did find reductions in neuronal survival [75-77]. Despite the lack of group contrast, gliosis was visible in some of our mTBI mice.
The cerebrovascular changes observed in mTBI patients in the chronic stage post trauma exhibit high variance with respect to presence, directionality, and magnitude of cerebral blood flow (CBF) signal changes [7, 13, 20, 25, 50-54, 68, 78]. The majority of studies have reported a reduction in baseline arterial blood flow [13, 52, 78] and an attenuation of stimulation-elicited cerebrovascular responses measured 6+ months post mTBI [13, 52]. Several studies have reported decreases [79-82] in CBF in the weeks to months following mTBI via ASL fMRI, whereas others have shown increases [78, 83-85], no change [86-89], or a combination of spatially-dependent changes [90-93] in this timeframe. In preclinical models of mTBI, hypoperfusion has been observed at 1-4 weeks post TBI [25, 70] and within 1 hour to 1 day of injury [19, 22, 24]. Similar to the clinical data, findings in preclinical models are highly heterogeneous but widely report CBF and cerebrovascular reactivity reductions post-injury [38, 94], whereas we presently report venular hyperreactivity, analogous to previous clinical findings [13, 78] and the first preclinical study to report this to our knowledge. While conventional readouts may be valuable for establishing the presence and severity of injury, quantitative functional measurements at the single-unit level are necessary for an in-depth understanding of the neurovascular sequelae of TBI pathophysiology [8, 9, 49, 50, 56] at a single-unit level [49, 50], and for establishing the absolute magnitude of these changes [13, 95]. Here, we observed doubling of venular baseline RBC velocities, a trend toward venular dilatation of about 15%, as well as a ~50% elevation in venular reactivity. Analogously, earlier work in mTBI patients reported increased CBF using BOLD and ASL fMRI 5-6 weeks [83] and 6+ months [52, 56, 68, 78, 87] post injury. Evaluation of BOLD fMRI responses to working memory task in mTBI patients 4+ months post-injury has revealed attenuated responses of peri-contusional vasculature [87], whereas other studies using hypercapnic challenge with BOLD fMRI have shown increased reactivity 2+ weeks following injury [89] and with ASL 1 week following injury [88]. The presently-observed venular specificity of the treatment effect on the brain vasculature may be due to venular measures reflecting an integration of all upstream changes in the microvascular network [96] capillaries are likely affected by trauma as well [21, 97], acquiring a representative sample size of these numerous microvessels would have taken prohibitively long. Additionally, the intrinsically slow nature of laser scanning (vs. relatively high speed of RBCs in cortical penetrating vessels) prevented us from making robust measurements of photostimulation-induced changes in the vessel diameter concomitant to the vessel's vRBC estimation.
Neurovascular coupling was interrogated in this study through ratios of paired neuronal and venular measures. While resting coupling between neuronal and vascular function remain unaltered, the NVC ratio was increased by mTBI. This finding suggests an alteration of neurovascular signaling that may be an adverse sequela of mTBI or a compensatory effect to potentiate the support of the injured tissue. At rest, venular baseline vRBC was linearly dependent on baseline neuronal theta power; this dependence was not affected by either injury or treatment. While gamma band neuronal power would have been interesting to evaluate due to its role in neurovascular coupling [98], such analysis was presently precluded by the 20 Hz low-pass analog filtering used to mitigate the noise in the electrophysiological recordings introduced by the concurrent microscopy and by the use of a tungsten electrode (to accommodate a bulky objective of concurrent 2PFM and electrophysiology recordings). On the whole, our observations support the notion of evoked response assays being more sensitive measures of brain (dys)function than the baseline state recordings [13].
Interneuron-mediated tonic inhibition has been previously implicated in preclinical mTBI pathophysiology [8, 9, 26, 27]. L-655,708 distribution in the brain following i.p. administration of 3 mg/kg (vs. 2.4 mg delivered subcutaneously, over 12 days, in the current study) has previously been shown to result, within 1-2 h following injection, in widespread distribution across the neocortical tissue [99]. In contrast to the neocortex, little L-655,708 was observed in the cerebellum and the subcortical regions. Excitatory/inhibitory signaling balance is disrupted in mTBI as well as in severe injuries [8, 9] and this imbalance is thought to cause hyperreactivity in the immediate aftermath of injury followed by hyporeactivity in the subacute phase of injury [9]. A delayed interventional window may thus be present in the hyporeactive phase post-injury. In months following mTBI, it has been shown that functional changes mediated by GABAA receptor currents occur in absence of an increase in receptor count [27]. Additionally, one week following mTBI, impaired behaviour and synaptic plasticity were previously prevented via administration of L-655,708 intraperitoneally [26]. Here we demonstrate the neurophysiological effects of such treatment at the single-unit level in vivo: specifically, a prevention of venular deficits and a normalization of neuronal responses in the L-655,708 treated vs. vehicle administered mTBI animals. The venular normalization observed in mTBI/L-655,708 mice may have been secondary to the normalization of neuronal tone and/or induced by the decreased availability of GABA, which is itself vasoactive [100, 101]. This indicates that a well-timed (subacute phase), sustained, low-dose GABAA receptor inverse agonism may have preventative effects on the neurovascular aberrations elicited by mTBI and identifies the excitation/inhibition signaling imbalance as an important mechanism of injury and hence treatment target. Furthermore, this study was performed with functional recordings in vivo, providing a greater translational potential than the previously reported ex vivo measures [8, 9, 26, 27]. Of note, L-655,708 administration in this study was systemic (via a subcutaneous osmotic pump) and is thus easily translatable. The resulting widespread effect on the neocortex [99] may have been beneficial considering the aforementioned spatial propagation of injury.
In summary, the present study describes sustained neurovascular impairments in a preclinical model of repeated closed head injury. We showed that in addition to neuronal hyporeactivity (-47 ± 28%) and reduced activity at rest (-57 ± 79%), the pericontusional venular function is abnormally elevated at rest (115 ± 25%) and upon stimulation (53 ± 17%). Increased resting cerebral blood flow has been observed at a regional level via MRI in mTBI patients [13, 56, 68, 78] but has not been shown at the level of individual vessels. Elevated venular responses to neuronal activation is a novel finding in preclinical mTBI, with analogous pathology described clinically [13, 88, 89]. Delayed, low dose GABAA receptor inverse agonism via L-655,708 [26, 27, 34, 35] to alleviate hypothesized tonic inhibition post TBI [8, 9, 26, 27, 34, 35] prevented the mTBI-induced venular impairment and normalized the neuronal reactivity. Our study provides new insight into the neurophysiological sequelae of mTBI, constitutes a first step in the development of translational markers of neurovascular pathology in mild TBI that is silent on neuroimaging, and suggests benefits of attenuating tonic inhibition in the subacute to chronic stage of injury.
Supplementary Material
Supplementary figures.
Acknowledgements
The authors would like to acknowledge BioRender (BioRender.com) for figure assembly.
Funding
Funding for this study was provided by the Natural Sciences and Engineering Research Council (NSERC RGPIN-2020-06590) and the Canadian Institutes of Health Research (CIHR PJT156179 and PJT376309).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Global Burden of Disease [Internet]. 2019 [cited 26 October 2020]. Available at: https://www.thelancet.com/gbd.
2. Cassidy JD, Carroll LJ, Peloso PM. et al. Incidence, risk factors and prevention of mild traumatic brain injury: results of the WHO Collaborating Centre Task Force on Mild Traumatic Brain Injury. J Rehabil Med. 2004:28-60
3. Prince C, Bruhns ME. Evaluation and Treatment of Mild Traumatic Brain Injury: The Role of Neuropsychology. Brain Sci [Internet]. 2017 7. Available at: http://dx.doi.org/10.3390/brainsci7080105
4. Bryant R. Post-traumatic stress disorder vs traumatic brain injury. Dialogues Clin Neurosci. 2011;13:251-62
5. Van Den Heuvel C, Thornton E, Vink R. Traumatic brain injury and Alzheimer's disease: a review. Prog Brain Res. 2007;161:303-16
6. Lee Y-K, Hou S-W, Lee C-C, Hsu C-Y, Huang Y-S, Su Y-C. Increased risk of dementia in patients with mild traumatic brain injury: a nationwide cohort study. PLoS One. 2013;8:e62422
7. Logsdon AF, Lucke-Wold BP, Turner RC, Huber JD, Rosen CL, Simpkins JW. Role of Microvascular Disruption in Brain Damage from Traumatic Brain Injury. Compr Physiol. 2015;5:1147-60
8. McGuire JL, Ngwenya LB, McCullumsmith RE. Neurotransmitter changes after traumatic brain injury: an update for new treatment strategies. Mol Psychiatry. 2019;24:995-1012
9. Guerriero RM, Giza CC, Rotenberg A. Glutamate and GABA imbalance following traumatic brain injury. Curr Neurol Neurosci Rep. 2015;15:27
10. Mouzon B, Chaytow H, Crynen G. et al. Repetitive mild traumatic brain injury in a mouse model produces learning and memory deficits accompanied by histological changes. J Neurotrauma. 2012;29:2761-73
11. Bailes JE, Dashnaw ML, Petraglia AL, Turner RC. Cumulative effects of repetitive mild traumatic brain injury. Prog Neurol Surg. 2014;28:50-62
12. US Department of Defense. DoD Policy Guidance for Management of Mild Traumatic Brain Injury [Internet]. 2012. Available at: https://www.hsdl.org/?view&did=722999.
13. Wang Y, Bartels HM, Nelson LD. A Systematic Review of ASL Perfusion MRI in Mild TBI. Neuropsychol Rev [Internet]. 2020 Available at: http://dx.doi.org/10.1007/s11065-020-09451-7
14. Son BC, Park CK, Choi BG. et al. Metabolic changes in pericontusional oedematous areas in mild head injury evaluated by 1H MRS. Acta Neurochir Suppl. 2000;76:13-6
15. Wu H-M, Huang S-C, Vespa P, Hovda DA, Bergsneider M. Redefining the pericontusional penumbra following traumatic brain injury: evidence of deteriorating metabolic derangements based on positron emission tomography. J Neurotrauma. 2013;30:352-60
16. Galgano M, Toshkezi G, Qiu X, Russell T, Chin L, Zhao L-R. Traumatic Brain Injury: Current Treatment Strategies and Future Endeavors. Cell Transplant. 2017;26:1118-30
17. Iadecola C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron. 2017;96:17-42
18. Joy MT, Carmichael ST. Encouraging an excitable brain state: mechanisms of brain repair in stroke. Nat Rev Neurosci. 2021;22:38-53
19. Han X, Chai Z, Ping X. et al. In vivo Two-Photon Imaging Reveals Acute Cerebral Vascular Spasm and Microthrombosis After Mild Traumatic Brain Injury in Mice. Front Neurosci. 2020;14:210
20. Ichkova A, Rodriguez-Grande B, Zub E. et al. Early cerebrovascular and long-term neurological modifications ensue following juvenile mild traumatic brain injury in male mice. Neurobiol Dis. 2020 104952
21. Østergaard L, Engedal TS, Aamand R. et al. Capillary transit time heterogeneity and flow-metabolism coupling after traumatic brain injury. J Cereb Blood Flow Metab. 2014;34:1585-98
22. Sword J, Masuda T, Croom D, Kirov SA. Evolution of neuronal and astroglial disruption in the peri-contusional cortex of mice revealed by in vivo two-photon imaging. Brain. 2013;136:1446-61
23. Toth P, Szarka N, Farkas E. et al. Traumatic brain injury-induced autoregulatory dysfunction and spreading depression-related neurovascular uncoupling: Pathomechanisms, perspectives, and therapeutic implications. Am J Physiol Heart Circ Physiol. 2016;311:H1118-31
24. Jang H, Huang S, Hammer DX. et al. Alterations in neurovascular coupling following acute traumatic brain injury. Neurophotonics. 2017;4:045007
25. Adams C, Bazzigaluppi P, Beckett TL. et al. Neurogliovascular dysfunction in a model of repeated traumatic brain injury. Theranostics. 2018;8:4824-36
26. Khodaei S, Avramescu S, Wang D-S. et al. Inhibiting α5 Subunit-Containing γ-Aminobutyric Acid Type A Receptors Attenuates Cognitive Deficits After Traumatic Brain Injury. Crit Care Med. 2020;48:533-44
27. Boychuk JA, Butler CR, Halmos KC, Smith BN. Enduring changes in tonic GABAA receptor signaling in dentate granule cells after controlled cortical impact brain injury in mice. Exp Neurol. 2016;277:178-89
28. Allitt BJ, Iva P, Yan EB, Rajan R. Hypo-excitation across all cortical laminae defines intermediate stages of cortical neuronal dysfunction in diffuse traumatic brain injury. Neuroscience. 2016;334:290-308
29. Carron SF, Alwis DS, Rajan R. Traumatic Brain Injury and Neuronal Functionality Changes in Sensory Cortex. Front Syst Neurosci. 2016;10:47
30. McColl TJ, Brady RD, Shultz SR. et al. Mild Traumatic Brain Injury in Adolescent Mice Alters Skull Bone Properties to Influence a Subsequent Brain Impact at Adulthood: A Pilot Study. Front Neurol. 2018;9:372
31. Gold EM, Vasilevko V, Hasselmann J. et al. Repeated Mild Closed Head Injuries Induce Long-Term White Matter Pathology and Neuronal Loss That Are Correlated With Behavioral Deficits. ASN Neuro. 2018;10:1759091418781921
32. Fairless R, Williams SK, Diem R. Calcium-Binding Proteins as Determinants of Central Nervous System Neuronal Vulnerability to Disease. Int J Mol Sci [Internet]. 2019 20. Available at: http://dx.doi.org/10.3390/ijms20092146
33. Cauli B, Hamel E. Revisiting the role of neurons in neurovascular coupling. Front Neuroenergetics. 2010;2:9
34. Lake EMR, Chaudhuri J, Thomason L. et al. The effects of delayed reduction of tonic inhibition on ischemic lesion and sensorimotor function. J Cereb Blood Flow Metab. 2015;35:1601-9
35. Clarkson AN, Huang BS, Macisaac SE, Mody I, Carmichael T. Reducing excessive GABAergic tonic inhibition promotes post- stroke functional recovery. Nature. 2011;468:305-9
36. Fischell J, Van Dyke AM, Kvarta MD, LeGates TA, Thompson SM. Rapid Antidepressant Action and Restoration of Excitatory Synaptic Strength After Chronic Stress by Negative Modulators of Alpha5-Containing GABAA Receptors. Neuropsychopharmacology. 2015;40:2499-509
37. Atack JR, Bayley PJ, Seabrook GR, Wafford KA, McKernan RM, Dawson GR. L-655,708 enhances cognition in rats but is not proconvulsant at a dose selective for alpha5-containing GABAA receptors. Neuropharmacology. 2006;51:1023-9
38. Bodnar CN, Roberts KN, Higgins EK, Bachstetter AD. A Systematic Review of Closed Head Injury Models of Mild Traumatic Brain Injury in Mice and Rats. J Neurotrauma [Internet]. 2019; Available at: http://dx.doi.org/10.1089/neu. 2018 6127
39. Koerte IK, Hufschmidt J, Muehlmann M, Lin AP, Shenton ME. Advanced Neuroimaging of Mild Traumatic Brain Injury. In: Laskowitz D, Grant G, Eds. Translational Research in Traumatic Brain Injury. Boca Raton (FL): CRC Press/Taylor and Francis Group. 2015
40. Mester JR, Bazzigaluppi P, Weisspapir I. et al. In vivo neurovascular response to focused photoactivation of Channelrhodopsin-2. Neuroimage. 2019;192:135-44
41. Arenkiel BR, Peca J, Davison IG. et al. In vivo light-induced activation of neural circuitry in transgenic mice expressing channelrhodopsin-2. Neuron. 2007;54:205-18
42. Atack JR, Pike A, Clarke A. et al. Rat pharmacokinetics and pharmacodynamics of a sustained release formulation of the GABAA alpha5-selective compound L-655,708. Drug Metab Dispos. 2006;34:887-93
43. Dorr A, Sahota B, Chinta LV. et al. Amyloid-β-dependent compromise of microvascular structure and function in a model of Alzheimer's disease. Brain. 2012;135:3039-50
44. Heo C, Park H, Kim Y-T. et al. A soft, transparent, freely accessible cranial window for chronic imaging and electrophysiology. Sci Rep. 2016;6:27818
45. Groppe DM, Bickel S, Keller CJ. et al. Dominant frequencies of resting human brain activity as measured by the electrocorticogram. Neuroimage. 2013;79:223-33
46. Kim TN, Goodwill PW, Chen Y. et al. Line-Scanning Particle Image Velocimetry: An Optical Approach for Quantifying a Wide Range of Blood Flow Speeds in Live Animals. Secomb TW, Ed. PLoS One. 2012;7:e38590
47. Schwab N, Grenier K, Hazrati L-N. DNA repair deficiency and senescence in concussed professional athletes involved in contact sports. Acta Neuropathol Commun. 2019;7:182
48. Henderson M, Rice B, Sebastian A. et al. Neuroproteomic study of nitrated proteins in moderate traumatic brain injured rats treated with gamma glutamyl cysteine ethyl ester administration post injury: Insight into the role of glutathione elevation in nitrosative stress. Proteomics Clin Appl. 2016;10:1218-24
49. Fehily B, Fitzgerald M. Repeated Mild Traumatic Brain Injury: Potential Mechanisms of Damage. Cell Transplant. 2017;26:1131-55
50. Salehi A, Zhang JH, Obenaus A. Response of the cerebral vasculature following traumatic brain injury. J Cereb Blood Flow Metab. 2017;37:2320-39
51. Chen S, Chen Y, Xu L. et al. Venous system in acute brain injury: Mechanisms of pathophysiological change and function. Exp Neurol. 2015;272:4-10
52. Ding K, Tarumi T, Tomoto T. et al. Impaired cerebral blood flow regulation in chronic traumatic brain injury. Brain Res. 2020 146924
53. Kim J, Whyte J, Patel S. et al. A perfusion fMRI study of the neural correlates of sustained-attention and working-memory deficits in chronic traumatic brain injury. Neurorehabil Neural Repair. 2012;26:870-80
54. Lyons DN, Vekaria H, Macheda T. et al. A Mild Traumatic Brain Injury in Mice Produces Lasting Deficits in Brain Metabolism. J Neurotrauma. 2018;35:2435-47
55. Raizman R, Tavor I, Biegon A. et al. Traumatic Brain Injury Severity in a Network Perspective: A Diffusion MRI Based Connectome Study. Sci Rep. 2020;10:9121
56. Pomschar A, Koerte I, Lee S. et al. MRI evidence for altered venous drainage and intracranial compliance in mild traumatic brain injury. PLoS One. 2013;8:e55447
57. Ma X, Aravind A, Pfister BJ, Chandra N, Haorah J. Animal Models of Traumatic Brain Injury and Assessment of Injury Severity. Mol Neurobiol. 2019;56:5332-45
58. Siebold L, Obenaus A, Goyal R. Criteria to define mild, moderate, and severe traumatic brain injury in the mouse controlled cortical impact model. Exp Neurol. 2018;310:48-57
59. Irimia A, Van Horn JD, Vespa PM. Cerebral microhemorrhages due to traumatic brain injury and their effects on the aging human brain. Neurobiol Aging. 2018;66:158-64
60. Hellal F, Bonnefont-Rousselot D, Croci N, Palmier B, Plotkine M, Marchand-Verrecchia C. Pattern of cerebral edema and hemorrhage in a mice model of diffuse brain injury. Neurosci Lett. 2004;357:21-4
61. Johnson VE, Meaney DF, Cullen DK, Smith DH. Animal models of traumatic brain injury. Handb Clin Neurol. 2015;127:115-28
62. Kallakuri S, Bandaru S, Zakaria N. et al. Traumatic Brain Injury by a Closed Head Injury Device Induces Cerebral Blood Flow Changes and Microhemorrhages. J Clin Imaging Sci. 2015;5:52
63. Angoa-Pérez M, Zagorac B, Anneken JH. et al. Repetitive, mild traumatic brain injury results in a progressive white matter pathology, cognitive deterioration, and a transient gut microbiota dysbiosis. Sci Rep. 2020;10:8949
64. Jullienne A, Salehi A, Affeldt B. et al. Male and Female Mice Exhibit Divergent Responses of the Cortical Vasculature to Traumatic Brain Injury. J Neurotrauma. 2018;35:1646-58
65. Brickler TR, Hazy A, Guilhaume Correa F. et al. Angiopoietin/Tie2 Axis Regulates the Age-at-Injury Cerebrovascular Response to Traumatic Brain Injury. J Neurosci. 2018;38:9618-34
66. Pernici CD, Rowe RK, Doughty PT, Madadi M, Lifshitz J, Murray TA. Longitudinal optical imaging technique to visualize progressive axonal damage after brain injury in mice reveals responses to different minocycline treatments. Sci Rep. 2020;10:7815
67. Tagge CA, Fisher AM, Minaeva OV. et al. Concussion, microvascular injury, and early tauopathy in young athletes after impact head injury and an impact concussion mouse model. Brain. 2018;141:422-58
68. Champagne AA, Coverdale NS, Ross A. et al. Multi-modal normalization of resting-state using local physiology reduces changes in functional connectivity patterns observed in mTBI patients. Neuroimage Clin. 2020;26:102204
69. Pacheco JM, Hines-Lanham A, Stratton C. et al. Spreading Depolarizations Occur in Mild Traumatic Brain Injuries and Are Associated with Postinjury Behavior. eNeuro [Internet]. 2019; 6. Available at: http://dx.doi.org/10.1523/ENEURO.0070-19. 2019
70. Steinman J, Cahill LS, Koletar MM, Stefanovic B, Sled JG. Acute and chronic stage adaptations of vascular architecture and cerebral blood flow in a mouse model of TBI. Neuroimage. 2019;202:116101
71. Steinman J, Cahill LS, Stortz G, Macgowan CK, Stefanovic B, Sled JG. Non-Invasive Ultrasound Detection of Cerebrovascular Changes in a Mouse Model of Traumatic Brain Injury. J Neurotrauma. 2020;37:2157-68
72. Ritzel RM, Doran SJ, Glaser EP. et al. Old age increases microglial senescence, exacerbates secondary neuroinflammation, and worsens neurological outcomes after acute traumatic brain injury in mice. Neurobiol Aging. 2019;77:194-206
73. Chen NY, Yang Y, Weston TA. et al. An absence of lamin B1 in migrating neurons causes nuclear membrane ruptures and cell death. Proc Natl Acad Sci U S A. 2019;116:25870-9
74. Rachmany L, Tweedie D, Rubovitch V. et al. Cognitive impairments accompanying rodent mild traumatic brain injury involve p53-dependent neuronal cell death and are ameliorated by the tetrahydrobenzothiazole PFT-α. PLoS One. 2013;8:e79837
75. Gao X, Chen J. Mild traumatic brain injury results in extensive neuronal degeneration in the cerebral cortex. J Neuropathol Exp Neurol. 2011;70:183-91
76. Bu W, Ren H, Deng Y. et al. Mild Traumatic Brain Injury Produces Neuron Loss That Can Be Rescued by Modulating Microglial Activation Using a CB2 Receptor Inverse Agonist. Front Neurosci. 2016;10:449
77. Ritzel RM, Li Y, He J. et al. Sustained neuronal and microglial alterations are associated with diverse neurobehavioral dysfunction long after experimental brain injury. Neurobiol Dis. 2020;136:104713
78. Stephens JA, Liu P, Lu H, Suskauer SJ. Cerebral Blood Flow after Mild Traumatic Brain Injury: Associations between Symptoms and Post-Injury Perfusion. J Neurotrauma. 2018;35:241-8
79. Ge Y, Patel MB, Chen Q. et al. Assessment of thalamic perfusion in patients with mild traumatic brain injury by true FISP arterial spin labelling MR imaging at 3T. Brain Inj. 2009;23:666-74
80. Hamer J, Churchill NW, Hutchison MG, Graham SJ, Schweizer TA. Sex Differences in Cerebral Blood Flow Associated with a History of Concussion. J Neurotrauma. 2020;37:1197-203
81. Lin C-M, Tseng Y-C, Hsu H-L. et al. Arterial Spin Labeling Perfusion Study in the Patients with Subacute Mild Traumatic Brain Injury. PLoS One. 2016;11:e0149109
82. Meier TB, Bellgowan PSF, Singh R, Kuplicki R, Polanski DW, Mayer AR. Recovery of cerebral blood flow following sports-related concussion. JAMA Neurol. 2015;72:530-8
83. Barlow KM, Marcil LD, Dewey D. et al. Cerebral Perfusion Changes in Post-Concussion Syndrome: A Prospective Controlled Cohort Study. J Neurotrauma. 2017;34:996-1004
84. Churchill NW, Hutchison MG, Graham SJ, Schweizer TA. Mapping brain recovery after concussion: From acute injury to 1 year after medical clearance. Neurology. 2019;93:e1980-92
85. Doshi H, Wiseman N, Liu J. et al. Cerebral hemodynamic changes of mild traumatic brain injury at the acute stage. PLoS One. 2015;10:e0118061
86. Churchill NW, Hutchison MG, Graham SJ, Schweizer TA. Evaluating Cerebrovascular Reactivity during the Early Symptomatic Phase of Sport Concussion. J Neurotrauma. 2019;36:1518-25
87. Coverdale NS, Fernandez-Ruiz J, Champagne AA, Mark CI, Cook DJ. Co-localized impaired regional cerebrovascular reactivity in chronic concussion is associated with BOLD activation differences during a working memory task. Brain Imaging Behav. 2020;14:2438-49
88. Militana AR, Donahue MJ, Sills AK. et al. Alterations in default-mode network connectivity may be influenced by cerebrovascular changes within 1 week of sports related concussion in college varsity athletes: a pilot study. Brain Imaging Behav. 2016;10:559-68
89. Mutch WAC, Ellis MJ, Ryner LN. et al. Patient-Specific Alterations in CO2 Cerebrovascular Responsiveness in Acute and Sub-Acute Sports-Related Concussion. Front Neurol. 2018;9:23
90. Brooks BL, Low TA, Plourde V. et al. Cerebral blood flow in children and adolescents several years after concussion. Brain Inj. 2019;33:233-41
91. Churchill NW, Hutchison MG, Graham SJ, Schweizer TA. Symptom correlates of cerebral blood flow following acute concussion. Neuroimage Clin. 2017;16:234-9
92. Liu K, Li B, Qian S. et al. Mental fatigue after mild traumatic brain injury: a 3D-ASL perfusion study. Brain Imaging Behav. 2016;10:857-68
93. Möller MC, Nordin LE, Bartfai A, Julin P, Li T-Q. Fatigue and Cognitive Fatigability in Mild Traumatic Brain Injury are Correlated with Altered Neural Activity during Vigilance Test Performance. Front Neurol. 2017;8:496
94. Hajiaghamemar M, Seidi M, Oeur RA, Margulies SS. Toward development of clinically translatable diagnostic and prognostic metrics of traumatic brain injury using animal models: A review and a look forward. Exp Neurol. 2019;318:101-23
95. Borogovac A, Asllani I. Arterial Spin Labeling (ASL) fMRI: advantages, theoretical constrains, and experimental challenges in neurosciences. Int J Biomed Imaging. 2012;2012:818456
96. Nishimura N, Schaffer CB, Friedman B, Lyden PD, Kleinfeld D. Penetrating arterioles are a bottleneck in the perfusion of neocortex. Proc Natl Acad Sci U S A. 2007;104:365-70
97. Bragina OA, Lara DA, Nemoto EM, Shuttleworth CW, Semyachkina-Glushkovskaya OV, Bragin DE. Increases in Microvascular Perfusion and Tissue Oxygenation via Vasodilatation After Anodal Transcranial Direct Current Stimulation in the Healthy and Traumatized Mouse Brain. Adv Exp Med Biol. 2018;1072:27-31
98. Han K, Min J, Lee M. et al. Neurovascular Coupling under Chronic Stress Is Modified by Altered GABAergic Interneuron Activity. J Neurosci. 2019;39:10081-95
99. Atack JR, Alder L, Cook SM, Smith AJ, McKernan RM. In vivo labelling of alpha5 subunit-containing GABA(A) receptors using the selective radioligand [(3)H]L-655,708. Neuropharmacology. 2005;49:220-9
100. Kocharyan A, Fernandes P, Tong X-K, Vaucher E, Hamel E. Specific subtypes of cortical GABA interneurons contribute to the neurovascular coupling response to basal forebrain stimulation. J Cereb Blood Flow Metab. 2008;28:221-31
101. Hinds K, Monaghan KP, Frølund B, McGeown JG, Curtis TM. GABAergic control of arteriolar diameter in the rat retina. Invest Ophthalmol Vis Sci. 2013;54:6798-805
Author contact
![]() Corresponding author: james.mesterutoronto.ca
Corresponding author: james.mesterutoronto.ca