Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2021; 11(9):4483-4501. doi:10.7150/thno.54473 This issue Cite
Research Paper
The subcellular redistribution of NLRC5 promotes angiogenesis via interacting with STAT3 in endothelial cells
Xu Xu1*, Yefei Shi1*, Peipei Luan1, Wenxin Kou1, Bo Li1, Ming Zhai1, Shuangjie You1, Qing Yu1, Jianhui Zhuang1, Weixia Jian2 ![]() , Mark W. Feinberg3
, Mark W. Feinberg3 ![]() , Wenhui Peng1
, Wenhui Peng1 ![]()
1. Department of Cardiology, Shanghai Tenth People's Hospital, Tongji University School of Medicine, Shanghai, China.
2. Department of Endocrinology, Xinhua Hospital, Shanghai Jiaotong University, School of Medicine, Shanghai, China.
3. Cardiovascular Division, Department of Medicine, Brigham and Women's Hospital, Harvard Medical School, Boston, Massachusetts, USA.
*Co-first authors.
Received 2020-10-14; Accepted 2021-2-9; Published 2021-3-4
Abstract

Angiogenesis is a critical step in repair of tissue injury. The pattern recognition receptors (PRRs) recognize pathogen and damage associated molecular patterns (DAMPs) during injury and achieve host defense directly. However, the role of NLR family CARD domain containing 5 (NLRC5), an important member of PPRs, beyond host defense in angiogenesis during tissue repair remains unknown.
Methods: In vitro, western blot and real-time PCR (RT-PCR) were used to detect the expression of NLRC5 in endothelial cells (ECs). Immunofluorescence microscopy was used to reveal the subcellular location of NLRC5 in ECs. Cell proliferation, wound healing, tube formation assays of ECs were performed to study the role of NLRC5 in angiogenesis. By using Tie2Cre-NLRC5flox/flox mice and bone marrow transplantation studies, we defined an EC-specific role for NLRC5 in angiogenesis. Mechanistically, co-immunoprecipitation studies and RNA sequencing indicated that signal transducer and activator of transcription 3 (STAT3) was the target of NLRC5 in the nucleus. And Co-IP was used to verify the specific domain of NLRC5 binding with STAT3. ChIP assay determined the genes regulated by interaction of STAT3 and NLRC5.
Results: Knockdown of NLRC5 in vitro or in vivo inhibited pathological angiogenesis, but had no effect on physiological angiogenesis. NLRC5 was also identified to bind to STAT3 in the nucleus required the integrated death-domain and nucleotide-binding domain (DD+NACHT domain) of NLRC5. And the interaction of STAT3 and NLRC5 could enhance the transcription of angiopoietin-2 (Ang2) and cyclin D1 (CCND1) to participate in angiogenesis.
Conclusions: In the ischemic microenvironment, NLRC5 protein accumulates in the nucleus of ECs and enhances STAT3 transcriptional activity for angiogenesis. These findings establish NLRC5 as a novel modulator of VEGFA signaling, providing a new target for angiogenic therapy to foster tissue regeneration.
Keywords: angiogenesis, NLRC5, STAT3, signal transduction, endothelial cell
Introduction
Pathogen products termed pathogen associated molecular patterns (PAMPs e.g., lipopolysaccharide, proteoglycan, viral RNA) and nuclear or cytosolic molecules termed damage associated molecular patterns (DAMPs e.g., HMGB-1, ATP, DNA) are the two main triggers of inflammation [1, 2]. The pattern recognition receptors (PRRs) in host cells recognize the PAMPs or DAMPs and facilitate host defense directly. PPRs are widely studied for their roles in inflammation; yet, their true roles in other disease processes remain poorly defined [2-4]. Recently, several PRRs have been found to directly regulate angiogenesis [5, 6]. As PRRs are extensively expressed in ischemic and inflammatory microenvironments, it is reasonable to hypothesize that PRRs would not only be involved in tissue injury, but also tissue repair, a process that involves pathological neovascularization.
PPRs have several subfamilies including Toll like receptors (TLRs), Retinoic acid-inducible gene-I-like receptors (RLRs), C-type lectin receptors (CLRs), and the NOD-like receptors (NLRs). NLR family CARD domain containing 5 (NLRC5), is the longest member of the NLRs family [7]. It contains an atypical caspase recruitment domain (CARD domain), a central nucleotide-binding domain (NBD domain) and leucine-rich repeats domain (LRRs domain) [8]. The small nuclear localization signal ahead of the NBD domain determines the subcellular distribution of NLRC5. The structure characteristics of NLRC5 make its function similar to major histocompatibility class II (MHC II) transactivator (CIITA), another member of NLRs [7, 9]. As CIITA has been recognized to regulate MHC II related genes [10-12], NLRC5 regulates the MHC I gene in an analogous manner by forming a complex in the SXY (containing of W/S, X1, X2 and Y boxes) module of the MHC I promoter. Therefore, NLRC5 is also called major histocompatibility class I transactivator (CITA) [13, 14]. Early studies demonstrated that NLRC5 inhibits the NF-κB signaling pathway [15-17] and regulates the secretion pro-inflammatory cytokines such as interleukin-6 (IL-6), tumor necrosis factor α (TNFα), and interleukin-1β (IL-1β) both in vitro and in vivo [7, 16, 17]. Moreover, NLRC5 impacts the formation of the NACHT, LRR and PYD domains-containing protein 3 inflammasome [18, 19]. Although the inflammatory role of NLRC5 is correlated with its location in the cytoplasm, there remains uncertainty about the nuclear function of NLRC5 beyond classical MHC I regulation. Our group reported that NLRC5 not only modulated the TGFβ signaling pathway, but also regulated the phenotype of vascular smooth muscle cells by directly binding to peroxisome proliferator activated receptor γ (PPARγ) in the nucleus [20, 21]. In addition to vascular remodeling [21], we also found that the expression of NLRC5 was regulated in the vascular endothelium. Based on these observations, we hypothesize that NLRC5 may also play an essential role in endothelial cells (ECs).
Angiogenesis is the process that sprouting or intussusceptive ECs form new blood vessels from preexistent ones [22, 23]. It exists as early as the embryonic development stage in response to proangiogenic gradients. Such angiogenesis during development is defined as physiological angiogenesis [24]. In contrast, pathological angiogenesis, such as angiogenesis involved in ischemia or cancer, is a distinct type due to its microenvironment being infiltrated with PAMPs, DAMPs, and inflammatory cells [25-28]. In general, pathological angiogenesis, a rapid, partial, and restricted form of angiogenesis [29], assists injured tissues in cellular clearance and tissue regeneration [27, 30, 31]. However, the initiating events enabling heterogeneous ECs to participate in pathological angiogenesis remains poorly defined.
In this study, we identify that NLRC5 translocation to the nucleus of ECs will promote pathological angiogenesis, serving as an activator for STAT3 and STAT3-related genes involved in angiogenesis.
Methods
Human sample collection and ethical approval
Human lung tissue sample was obtained from patients undergoing trauma surgery. Human saphenous vein and human internal mammary artery were obtained from patients undergoing coronary artery bypass grafting. The study received approval by the Ethical Committee of Shanghai Tenth People's Hospital and experiments were conducted in compliance with all relevant ethical regulations. The written informed consent was collected from each patient and/or their relatives.
Cell culture and stimulation
Human umbilical vein endothelial cells (HUVECs) were purchased from Sciencell Research Laboratories (Cat# 8000, Sciencell) and passage 2 to 8 were used for experiments. Cells were cultured in endothelial cells medium (Cat# 1001, Sciencell) with 5% fetal calf serum (FBS), 1% endothelial cells growth supplement (ECGS), 1% penicillin and streptomycin. HUVECs were starved for 4 h, then the medium was supplied with VEGFA-165 (50 ng/mL, Cat# 100-20, PeproTech) or lipopolysaccharide (LPS, 100 ng/mL, Cat# L3024, Sigma-Aldrich) for another 6, 12, 24 h. After stimulations, HUVECs were harvested for other experiments. Human aortic smooth muscle cells (HASMCs) were purchased from Sciencell Research Laboratories (Cat# 6110, Sciencell), and passage 3 to 8 were used for the experiments. Cells were cultured in the smooth muscle cells medium (Cat# 1101, Sciencell) containing 2% FBS, 1% smooth muscle cells growth supplement and 1% penicillin and streptomycin. Human mononuclear cells (THP-1) and human embryonic kidney 293T (HEK293T) cells were obtained from Fudan University Institutes of Biomedical Sciences Cell Center (Shanghai, China) and EA.hy926 cells were purchased from Shanghai Zhongqiaoxinzhou Biotechnology (Cat# ZQ0079). They were cultured in DMEM supplemented with 10% FBS and 1% penicillin and 1% streptomycin.
Mouse lung endothelial cells isolation and culture
7-10 days old newborn mice were anesthetized with 2% isoflurane for the isolation of lung endothelial cells. Lungs were quickly dissected and cut into small pieces (1 mm or smaller) and digested with Hank's balanced salt solution (HBSS) containing 1 mg/mL collagenase I (Cat# 1148089, Sigma-Aldrich) for 40 min at 37 °C. The mixture of tissues and cells were filtered using sterile 0.45 µm nylon mesh and washed in HBSS containing 5% FBS. The filtered cell suspension was then incubated with Dynabeads (Cat# 557355, Invitrogen) at 4 °C for 30 min to capture ECs. The beads were first coated with anti-mouse CD31 monoclonal antibody (Cat# 557355, BD biosciences) overnight at 4 °C and then washed with PBS. After isolation, mouse lung endothelial cells were cultured in endothelial cells medium supplemented with 10% FBS, 2% ECGS, 1% penicillin and streptomycin, and TGFβ inhibitor 1:1000 (Cat# SB431542, Medchemexpress).
siRNA knockdown experiments
HUVECs and EA.hy926 were transfected using Lipofectamine 2000 (Cat# 11668027, Invitrogen) with duplex small interfering RNA (siRNA) against human NLRC5 (SASI_Hs02_00359503, Sigma, 50 nM) or human STAT3 (50 nM, GenePharma) and control siRNA (HSS 12935-300, Sigma, 50 nM/20 nM) for 48 h.
In vitro adenovirus preparation and infection
HUVECs were seeded in 6-well-plates over 70% confluence and used for in vitro adenovirus infection. The NLRC5 and control adenovirus, which were constructed and purchased from Shanghai Genechem Co, Ltd, were added into the serum-free medium at the concentration of 50 mM for 24 h. The medium was replaced by the complete medium for another 24 h and used for further experiments.
Construction and transfection of plasmid
STAT3-Flag, ΔNTD domain-Flag (2-120 aa delated), ΔCCD domain-Flag (141-313 aa delated), ΔDBD domain-Flag (325-464 aa delated), ΔSH2 domain-Flag (584-647 aa delated);myc-NLRC5 NACHT (222-539 aa), myc-NLRC5 DD+NACHT (1-539 aa), myc-NLRC5 DD (1-221 aa) were constructed and purchased from Shanghai Genechem Co, Ltd. The following plasmids were generated from pGV219 vector with myc tag including myc-NLRC5 full length (Cat# 37509, Addgene), myc-NLRC ΔDD (Cat# 37511, Addgene) and obtained from Addgene Institution.
Immunofluorescence Staining and Fluorescence Confocal Microscopy
We injected 10 mL of PBS/Heparin (Cat# H3149, Sigma-Aldrich, 10 U/mL) and 10 mL of 4% paraformaldehyde solution consecutively at a rate of 2 mL/min into left ventricle of the mice heart. Gastrocnemius muscles were collected after perfusion fixation. Tissue samples were transferred into 30% sucrose solution at 4 °C overnight and embedded in optimal cutting temperature compound and frozen in liquid nitrogen, followed by sectioning at 8 mm thickness. Sections were incubated in primary antibodies (Table S1.1). DAPI 1:5000 (Cat# ab228549, Abcam) was used as a nuclear counterstain. Tissue was visualized using objective on Nikon Eclipse TE-2000U immunofluorescent microscope (Nikon, Japan). The area and percentage of staining was quantified using NIS-elements v3.0 (Nikon) and expressed as a percentage of the total surface area of the tissue section. Necrotic area in the gastrocnemius muscle was analyzed by hematoxylin and eosin staining. Necrotic cells displayed a glassy homogeneous appearance in the cytoplasm with increased neutrophils.
RNA Isolation and RT-qPCR Analysis
Total RNA was isolated using Trizol (Cat# 15596026, Thermo Fisher) and extracted by chloroform and isopropanol. The yield and purity of the total RNA was assessed by Nanodrop 2000 (Thermo Scientific). The total mass of 1000 ng RNA was taken for subsequent reverse transcription using PrimerScript RT Reagent Kit (Cat# RR047A, Takara, Japan). Then, qPCR was performed by KAPA SYBR FAST kit (Cat# KM4101, KAPA Biosystem) and using the Roche LightCycler96 (Roche). Each sample was run in triplicate. Data was normalized by using GAPDH as the control. The expression of target genes was compared with fold-change between reference control and experimental groups. The primers were listed in the Table S2.
Extraction of Cytoplasmic and Nuclear Proteins
HUVECs were prepared in 6 cm dishes for extraction of cytoplasmic and nuclear proteins. In short, after infection of adenovirus for 48 h, HUVECs were starved for 4 h and IL-6 (20 ng/mL, Cat# 200-06, PeproTech) was added into the medium for 0, 15, 30, 60 min. HUVECs were then digested by 0.025% trypsin with EDTA and suspended in completed medium. The cytoplasmic and nuclear proteins were extracted following the protocol of NE-PERTM Nuclear and Cytoplasmic Extraction Reagent (Cat# 7883, Invitrogen). HUVECs were also stimulated with VEGFA-165 (50 ng/mL, 12 h) or LPS (100 ng/mL, 24 h), and then were subjected to the same procedures mentioned above.
Western Blot Analysis
Whole cell lysis of HUVECs were harvested for western blot using 1× cell lysis (Cat# 9803, Cell Signaling Technologies) with protease inhibitors (Cat# 04693159001, Roche Molecular Biochemicals, USA). Lysates were centrifuged at the speed of 12,000 g at 4 °C for 10 min. The supernatants were quantified using bicinchoninic acid kit (Cat# 20201ES76, Yeasen) and subjected to SDS-polyacrylamide gel electrophoresis and electrotransferred onto PVDF membranes. Primary antibodies were listed on Table S1.1. The PVDF membranes were washed in PBST and then incubated in PBST containing a 1:5,000 dilution of indicated secondary antibodies listed on Table S1.2. HRP was detected using the Super Signal chemiluminescence reagent substrate (Cat# 32134, Thermo Fish Scientific) and signal was visualized on the Biorad Chemdoc system. All experiments were performed at least three times.
Co-immunoprecipitation
HUVECs or HEK293T cells were harvested in 1× cell lysis buffer (Cat# 9803, Cell Signaling Technologies) with protease inhibitors (Cat# 04693159001, Roche). After centrifugation, 1000 µg of cell lysate was incubated with the indicated primary antibodies (Table S1.1) at 4 °C overnight. The lysate immunoprecipitated with anti-Immunoglobulin G antibody served as negative control. The immune complexes were then purified by 20 µl of protein A/G agarose (Cat# sc-2003, Santa Cruz) at 4 °C for 5 h and washed by precooled cell lysis buffer. The immunoprecipitated protein were then used for Western Blot.
Edu Assay
After starvation, VEGFA-165 was used to stimulate HUVECs for 12 h. Edu solution was incubated with the cells for 8 h in cell incubator, and subsequently fixed in 4% paraformaldehyde solution. The images of Edu-positive cells were taken by Leica DMI6000 microscopy. Three or four different fields of each section were taken for each group. The rate of proliferation was calculated as Edu-positive cells/4',6-diamidino-2-phenylindole (DAPI). All the reagents were included in Click-iT™ EdU Cell Proliferation Kit for Imaging, Alexa Fluor™ 594 dye (Cat# 10339, Invitrogen) or Alexa Fluor™ 488 dye (Cat# C10637, Invitrogen).
TUNEL Assay
HUVECs were treated with siNLRC5 for 48 h. Then phosphate buffered saline (PBS) containing 2% hydrogen peroxide was added into cells for 5 min and washed with PBS 3 times for 5 min. TdT enzyme buffer were added for 5 min. Next, TdT enzyme reaction liquid was added to the cells and incubated for 1 h at 37 °C. HUVECs were washed with PBS, and peroxidase labeled anti-digoxin antibody were added to the cells for 30 min at room temperature. The results were obtained by measuring the TUNEL positive cells divided by the number of DAPI-positive cells per scope. All the reagents were included in the In situ Cell Death Detection Kit (Cat# 12156792910, Roche). The results were observed by Leica DMI6000 microscopy.
Tube Formation Assay
The Matrigel (Cat# 354230, Corning Matrigel) was thawed on ice overnight. Then the liquid Matrigel was injected into precooled 96-well-plate (50 µl/well) and plates were placed into the cell incubator. 100 µl with 20,000 HUVECs were seeded into the well and incubated for 8 h. Each group was taken 4 replications. The pictures were also taken by Leica DMI6000 microscopy. The total length and branch points was calculated to determining the ability of angiogenesis.
Migration assay
HUVECs in 6-well-plate (pretreated) at 100% confluence were scratched by tip. VEGFA-165 was then added into the medium to enhance the migration of HUVECs. 4 pictures (n = 4) were captured by Leica DMI6000 microscopy in 0 h, 12 h, 24 h after scratching. The area of migration was calculated as the difference between the initial area (S0) and the area measured at each time (St). The migration rate was defined as the migrated area divided by the initial area: Migration rate = (S0 - St)/S0 * 100%.
Luciferase assays
pGL4.45 [luc2p/ISRE/Hygro] vector (REF# E414, Promega) and pGL4.47 [luc2p/SIE/Hygro] vector (REF# E4041, Promega) in combination with various vectors for luciferase assays were carried out using FuGene reagent (Cat# E2311, Promega) according to the manufacturer's protocol. Briefly, transfections contained 100 ng renilla expression vector, and 1 µg of sis induce element (SIE) and interferon sensitive response element (ISRE) luciferase reporter plasmid or control empty vectors were conducted for 24 h and IL-6 (20 ng/mL) was added for another 18 h. For siRNA knockdown in HEK293T cells, NLRC5 was knocked down for 24 h and subjected to the transfection of vectors and stimulation. Cytosolic fractions were prepared at 24 h post-transfection using the Promega kit (Cat# E1910, Promega). Samples were analyzed with a luminometer and normalized to renilla activity to calculate the transfection efficiency.
ChIP assay
HUVECs cultured in 10 cm dishes were transfected with vector or NLRC5 for 48 h (8 dishes per group). 1% formaldehyde was used to treat cells to cross-link the DNA and protein for 10 min at room temperature. Glycine was added to stop fixation. Next, cells were harvested with cold PBS. Cell nucleus was centrifugated at 3000 rpm for 5 min at 4 °C and resuspended in nuclei lysis buffer. Nuclear lysates were treated with 0.15ul Micrococcal Nuclease for 30 min at 37 °C. The lysate was then sonicated to shear DNA into fragments ranging 150~ 900 bp and equal amounts of chromatin were incubated overnight with anti-STAT3 and anti-Flag antibodies as listed in Table S1.1. 30 ul ChIP-grade protein G magnetic beads were used to absorb the chromatin. The DNA was purified by adsorption column. All of the regents were included in the SimpleChIP® Enzymatic Chromatin IP Kit (Cat# 9003, Cell Signaling Technologies). The Primers used for ChIP assay were listed in Table S2.
RNA sequencing
Sample preparation, sequencing, and alignment: human umbilical vein endothelial cells were transfected with siRNA (siNLRC5 or negative control, 50 nM) for 48 h. After starvation for 12 h, VEGFA-165 (50 ng/mL) was used to stimulate HUVECs for another 12 h. RNA sequencing was conducted with the help of Genergy Bio-technology (China). Briefly, RNA was harvested using Trizol reagent, Illumina TruSeq RNA Sample Prep Kit (Cat# FC-122-1001) was used with 1 µg of total RNA for the construction of sequencing libraries. RNA libraries were prepared for sequencing using standard Illumina protocols, and RNA sequencing was subsequently performed using the Illumina novaseq6000 platform. Finally, differentially expressed genes were screened using DESeq (version 1.22.1). Heatmaps of selected genes were created by using conditional formatting tool in Microsoft Excel with the highest and lowest expression for each gene displayed as red and blue (row min and row max). Raw and processed data were deposited in GEO (accession GSE159377).
GSEA analysis
Gene Set Enrichment Analysis (GSEA, version 4.1.0, the broad institute of MIT and Harvard) was used to discuss whether a genetically defined genome is statistically significant between the two groups [32]. We used R studio to construct RNK file according to each gene's log2FoldChange and -log10P.value compared between two groups based on our RNA-seq data. Then the profile was uploaded to the GSEA software. GSEA was performed with default algorithm as 1000 permutations, minimum term size of 15, and maximum term size of 500. HALLMARK was served as our annotated gene sets, collected from the Molecular Signature Database 3.0 [33]. Enriched gene sets were assigned based on normal P-value < 0.05 and FDR q-value < 0.25.
Generation of NLRC5 global mice and EC specific KO mice
All experimental procedures involving animals were performed in accordance with the guidelines of the National Institutes of Health for the care and use of laboratory animals (NIH Publication, 8th Edition, 2011) and approved by the Animal Care and Use Committees of Shanghai Tenth People's Hospital.
Global NLRC5-KO mice were obtained from Shanghai Biomodel Organism and bred as previously described [34]. To generate the EC-specific NLRC5-KO mice, 5'loxP sites were inserted in the upstream of exons 1 where the promoter of EGE-LJL-121 was located. The 3'loxP was inserted into intron1 that was big and loxP element would not interfere mRNA splicing. To minimize the possibility of disruption of EGE-LJL-12 expression, two loxP sites were inserted into non-conserved regions. Southern blot and PCR were used by amplifying the region with the specific primers to identify the existence of loxP (details shown in Figure S5). Then, the NLRC5flox/flox mice were crossed with Tie2Cre mice (purchased from Biocytogen) to obtain Tie2Cre-NLRC5flox/flox mice. The study received approval by the Animal Care and Use Committees of Shanghai Tenth People's Hospital for animal welfare.
Femoral artery ligation
Animals (8-10 weeks old) were anesthetized with 2% isoflurane. The left superficial femoral artery was ligated proximal to the deep femoral artery and proximal to the branching of the tibial arteries, leaving nerve and vein intact. The skin was sutured with 6-0 monofilament sutures. Then, mice were anesthetized by 2% isoflurane and were put on the platform to scan the blood flow of functional recovery (moorLDI Laser Doppler Perfusion Imager, England). The recovery rate of blood flow was measured as blood flow of ligated leg/sham leg. The score of necrosis in mice could be evaluated 28 days after ligation. Scores were calculated as following, 0 = none necrosis; 1 = 1-3 fingertips peeling; 2 = 4-5 fingertips peeling; 3 = necrosis of 1-3 toes; 4 = necrosis of 4-5 toes; 5 = necrosis of 1/3 soles; 6 = necrosis of 2/3 soles; 8 = necrosis of the full foot; 10 = necrosis of 1/3 legs; 12 = necrosis of 2/3 legs; 14 = necrosis of the whole leg.
Bone marrow transplantation
NLRC5flox/flox and Tie2Cre-NLRC5flox/flox (male, 6-8 weeks) were used as recipients and WT mice were used as donors (indicated in Figure 4A). Recipient mice were irradiated with 9 Gy of radiation before injection. After sacrificing the donors, clean femur and tibia were collected in cold and sterilized PBS. Bone marrow was flushed from the cut bone as intact as possible using 1 mL syringe needle filled with RPMI 1640 medium. Then, cells were centrifuged and suspended with 1 mL RPMI 1640 after filtration of 0.45 µm strainer. Cells were transplanted to recipients (1×107, 400 µl/recipient) through tail vein injection.
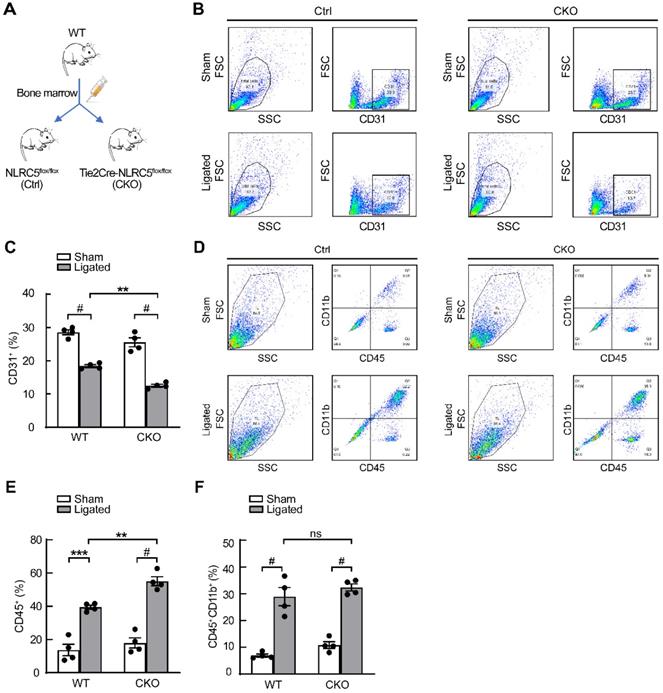
Tie2Cre-NLRC5flox/flox mice exhibited severe inflammation and decreased angiogenesis without the contribution of myeloid cells. (A) Schematic of bone marrow transplantation studies. WT mice were the donors. NLRC5flox/flox (Ctrl) mice and Tie2Cre-NLRC5flox/flox (CKO) mice were the recipients. (B) The CD31+ cells were measured by fluorescence activated cell sorter (FACS). (C) Quantification of B. n = 4 mice/group. Data are mean ± SEM, two-way ANOVA with Bonferroni post-test, ** P < 0.01, # P < 0.001. (D) The CD45+CD11b+ cells were measured by fluorescence activated cell sorter (FACS). (E-F) Quantification of D. n = 4 mice/group. Data are mean ± SEM, two-way ANOVA with Bonferroni post-test, ** P < 0.01, *** P < 0.005, # P < 0.001.
Retinal angiogenesis assay
The P5 pups were sacrificed and eyes were fixed in 4% paraformaldehyde for 2 h and dissected, flatted, and permeabilized in 0.1% Triton X-100 in PBS for 30 min. 10% normal goat serum was used to block the retina at 4 °C overnight. They were incubated in biotin conjugated isolectin B4 (Bandeiraea simplicifolia, Cat# L-2140; Sigma-Aldrich) 20 µg/mL in 1% BSA at 4 °C overnight. The streptavidin-fluorescein isothiocyanate conjugate (Cat# SA10002, Invitrogen™) was added to the retina for 30 min in the second day. After being washed in PBS, the retinas were flat mounted.
Tumor transplant
The B16F10 murine melanoma cell line was a gift from Prof. Ping Wang of Tongji University. In brief, cells were digested and suspended (1×107 cells/mL, 100 µl/C57BL/6 mice) in cold PBS, and then injected subcutaneously into the abdominal area of 6 to 8-weeks-old male NLRC5flox/flox mice and Tie2Cre-NLRC5flox/flox mice. When tumors started to form, tumor growth was measured every two days by measuring the length and width and the tumor of volume (mm3) was calculated as 4π/3×(width/2)2×(length/2). Tumor was harvested (tumor size smaller than 1,000 mm3) and weighted at 14 days after injection. The vessel density was detected as the CD31-positive cells/scope (4 scopes per mice, 6 mice per group).
En-face staining
According to Kyung Ae Ko et al. [35], the aorta was exposed and incised after the mice were sacrificed. The aorta was then fixed in 4% paraformaldehyde in PBS for 5 min. After removal of fat and connective tissues, the endothelium inside the vessel was exposed longitudinally and permeabilized by 0.1% Triton X-100 in PBS 30 min and blocked in 10% goat normal serum overnight. The antibodies (Table S1.1) diluted with 0.5% normal serum were incubated with vessels overnight. The specific secondary antibody (Table S1.2) and DAPI were used to incubate with the aorta for 30 min. Last, photos of aorta were taken by Nikon Eclipse TE-2000U microscope (Nikon) with HeNe laser and driven by EZ-C1 Viewer v3.5 software (Nikon).
Acute lung injury model
Male 8-10 weeks C57BL/6 mice were used for the acute lung injury model. In short, the mice were intraperitoneal injected with LPS (10 mg/kg). 6 h later, the mice were sacrificed and perfused with 10 mL 4% paraformaldehyde in PBS. Then, the left lung was fixed in optimal cutting temperature compound.
Flow cytometry analysis
As described before [36], anesthetized mice (8-10 weeks, male) were used to collect the leg skeletal muscles. The muscles without fat and connective tissue were cut into small pieces. Tissue was then suspended and digested in 5 mL HBSS containing 1 mL (100 U/mL) collagenase II (Cat# 1148089, Sigma-Aldrich) and 2 mL (1 U/mL) dispase (Cat# 354235, Corning) for 1 h at 37 °C. The mixture was filtered using sterile 0.45 µm nylon mesh and suspended in 1 U/mL dispase in HSBB. After digestion for another 30 min, the cells were filtered again. Centrifuged cells (1×106, 100 µl) were blocked by 10% FBS for 30 min. The fluorescence labeled rat antibody (Table S1.2) was incubated with the cells in 1% FBS for 30 min. Finally, cells were fixed in 4% paraformaldehyde to perform flow cytometry analysis.
Statistics
Student's t-test (two-sided), one-way ANOVA or two-way ANOVA followed by Bonferroni's pos-hoc test were used to calculate P value accordingly. Error bars represented standard errors, and numbers of experiments (n) were as indicated. P < 0.05 was regarded as significant.
Results
VEGFA-165 regulates the expression and redistribution of NLRC5 in ECs
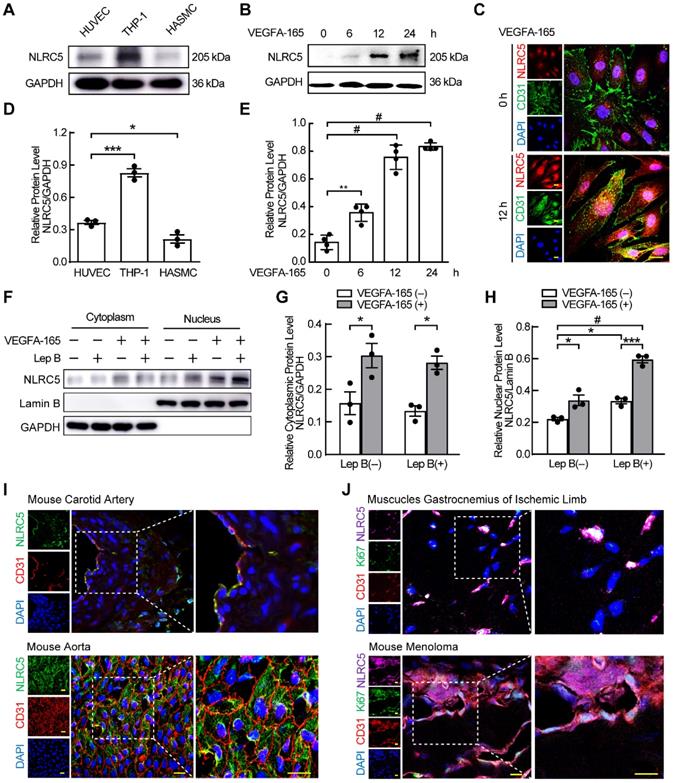
We previously found that NLRC5 was expressed in the endothelium of carotid arteries [21]. To understand the relative expression of NLRC5 in the vasculature, we measured and compared NLRC5 protein expression in HUVECs, HASMCs, and THP-1 cells. NLRC5 was expressed higher in HUVECs compared with HASMCs (Figure 1A and D), and significantly increased in HUVECs after VEGFA-165 stimulation for 12 h (0.14 ± 0.03 folds vs 0.75 ± 0.04 folds, P < 0.001, Figure 1B and E). In contrast, CIITA, another member of the NLRs family with similar structure and function of NLRC5, could not be detected in the static aortic endothelium, pulmonary endothelium or endothelial cells in the ischemic limb of mice (Figure S1A-C). In vitro, CIITA was still almost undetectable in HUVECs with or without VEGFA-165 stimulation (Figure S1D-E). Notably, the increased NLRC5 expression in ECs was mainly located in the nucleus of HUVECs after treated with VEGFA-165 as shown by immunofluorescence (Figure 1C). Leptomycin B has been reported to inhibit the export of NLRC5 in the nucleus as NLRC5 is shuttled from cytoplasm to nucleus continually [37]. After HUVECs were pretreated with leptomycin B, they were stimulated with VEGFA-165 for another 12 h followed by cell separation for nuclear and cytoplasmic protein fractions. NLRC5 expression was blocked in the nucleus. In addition, the expression of NLRC5 in the nucleus could also be enhanced after VEGFA-165 stimulation (Figure 1F-H). In contrast, in vivo experiments demonstrated that NLRC5 expression was located in the cytoplasm of quiescent endothelium of mice or human vessels (Figure 1I and Figure S2A). VEGFA is known to be highly concentrated in ischemic tissues or tumors with high demand of oxygen and nutrition [38]. Indeed, NLRC5 expression was detected in the nucleus of some ECs in the vessels of both ischemic limbs and melanoma tumors in mice (Figure 1J). Finally, these NLRC5 nuclear positive ECs were in a proliferative state as they were also Ki67 positive, a marker of cell proliferation [39] (Figure 1J).
NLR family CARD domain containing 5 (NLRC5) was increased by VEGFA-165 in human umbilical vein endothelial cells (HUVECs) and translocated into the nucleus. (A) NLRC5 expression was moderately expressed in HUVECs compared to human myeloid leukemia mononuclear cells (THP-1) and human aortic smooth muscle cells (HASMCs). The protein level of NLRC5 was detected by western blot analysis. (B) HUVECs were stimulated with VEGFA-165 (50 ng/mL) for different time points. The protein level of NLRC5 was detected by western blot analysis. (C) HUVECs were treated with VEGFA-165 (50 ng/mL) for 0 and 12 h. Representative confocal microscopy images of immunofluorescence staining for NLRC5 (green), CD31 (red), 4'-6-diamidino-2 -phenylindole (DAPI, blue). Scar bar, 20 µm. (D) Quantification of A. Data are mean ± SEM, n = 3 independent experiments. One-way ANOVA with Bonferroni post-test, * P < 0.05, *** P < 0.005. (E) Quantification of B. Data are mean ± SEM, n = 4 independent experiments. One-way ANOVA with Bonferroni post-test, ** P < 0.01, # P < 0.001. (F) HUVECs were treated with VEGFA-165 (50 ng/mL) for 12 h with or without leptomycin B (100 µM) pretreated for 6 h. Nuclear and cytoplasmic fractions were extracted from HUVECs. The protein level of NLRC5 was detected by western blot analysis. (G) Quantification of NLRC5 expression in the cytoplasmic fraction. (H) Quantification of NLRC5 expression in the nuclear fraction. Data are mean ± SEM, n = 3 independent experiments. Two-way ANOVA with Bonferroni post-test, * P < 0.05, *** P < 0.005, # P < 0.001. (I) NLRC5 was localized in the cytoplasm of vascular endothelial cells in static mouse vessels. Representative confocal microscopy images of immunofluorescence staining for NLRC5 (green), CD31 (red), Ki67 (magenta), DAPI (blue). Scar bar, 50 µm. (J) NLRC5 was localized in the nucleus of proliferative vascular endothelial cells in ischemic legs and melanoma tumors of mice. Representative confocal microscopy images of immunofluorescence staining for NLRC5 (green), CD31 (red), Ki67 (magenta), DAPI (blue). Scar bar, 50 µm.
Since LPS could enhance the expression of NLRC5, we stimulated HUVECs with LPS to further investigate the expression and subcellular distribution of NLRC5. As expected, LPS slightly enhanced the expression of NLRC5 after 24 h (Figure S2C). And the increased NLRC5 was mainly located in cytoplasm (Figure S2E-F). Nonetheless, LPS-induced NLRC5 expression was dominantly located in the cytoplasm of ECs both in vivo using an acute lung injury model and in vitro after LPS stimulation of HUVECs (Figure S2B-D). Taken together, these results suggest that NLRC5 may serve as an important functional role in the nucleus of VEGFA-165 treated ECs.
Deficiency of NLRC5 decreases angiogenesis in vitro
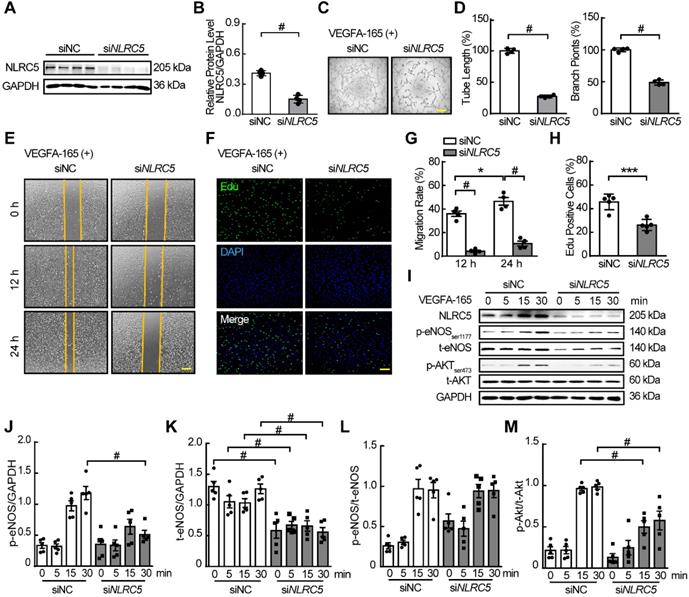
As VEGFA was a strong proangiogenic cytokine by promoting EC migration, survival, proliferation, and permeability in ischemic or neoplastic tissues [38], we hypothesized that the function of NLRC5 in the EC nucleus stimulated by VEGFA was to regulate angiogenesis. To prove this hypothesis, we first used siRNA to knockdown the expression of NLRC5 in HUVECs (siNC 0.41 ± 0.03 folds vs siNLRC5 0.15 ± 0.02 folds, P < 0.001) (Figure 2A-B). In response to NLRC5 knockdown, tube formation was significantly decreased as quantified by total tube length or the number of branch points (Figure 2C-D). After VEGFA stimulation, the siNLRC5 knockdown groups exhibited lower migration rates at 12 and 24 h as quantified by scratching (Figure 2E and G), and proliferation by Edu staining (Figure 2F and H). On the other hand, TUNEL assay revealed that the decreased expression of NLRC5 had no significant influence on apoptosis (Figure S3). Moreover, the deficiency of NLRC5 in ECs led to decreased phosphorylation of Akt and phosphorylation of endothelial nitric oxide synthase (eNOS), two important signal pathways for angiogenesis [40] (Figure 2I-M).
siRNA knockdown of NLRC5 in HUVECs inhibited tube formation, migration, and proliferation and markedly decreased the phosphorylation of eNOS and AKT. (A) siRNA knockdown decreased NLRC5 protein level in HUVECs. The expression of NLRC5 was detected by western blot analysis. (B) Quantification of A. Data are mean ± SEM, n = 4 independent experiments. Unpaired Student's t-test, # P < 0.001. (C) Tube formation of NLRC5 decreased HUVECs. Scar bar, 100 µm. (D) Quantification of total tube length and branch points. Data are mean ± SEM, n = 4 each group. Unpaired Student's t-test, # P < 0.001. (E) Migration of NLRC5 decreased HUVECs. Scar bar, 100 µm. (F) Proliferation of NLRC5 decreased HUVECs, 5-ethynyl-2'-deoxyuridine (Edu, green), DAPI (blue). Bar, 100 µm. (G) Quantification of C. Data are mean ± SEM, n = 4 each group. Two-way ANOVA with Bonferroni post-test, * P < 0.05, # P < 0.001. (H) Quantification of F. Data are mean ± SEM, n = 5 each group. Unpaired Student's t-test, *** P < 0.005. (I) p-AKT, p-eNOS decreased in NLRC5 knockdown HUVECs. (J-M), Quantification of I. Data are mean ± SEM, n = 5, independent experiments. Two-way ANOVA with Bonferroni post-test, # P < 0.001.
The function of NLRC5 on endothelial cells was further investigated by primary mouse lung endothelial cells (MLECs) isolated from global NLRC5 KO and WT mice. AdNLRC5 was also used to rescue the expression of NLRC5 in MLECs isolated from KO mice (Figure S4A and B). The overexpressed NLRC5 would translocate into nucleus in VEGFA stimulated MLECs as well (Figure S4C). Certainly, MLECs isolated from KO mice exhibited decreased tube formation, which could be improved by the overexpression of NLRC5 (Figure S4D-F). Moreover, the decreased migration (Figure S4G-H) and proliferation (Figure S4I-J) of NLRC5 knockout MLECs were significantly rescued in the presence of NLRC5 overexpression. Collectively, these data indicate that NLRC5 expression is critical for EC angiogenic properties such as proliferation and migration, and suggest that it may play a role in pathological angiogenesis.
Downregulation of NLRC5 inhibits pathological angiogenesis in vivo
To investigate the function of NLRC5 in angiogenesis in vivo, the NLRC5flox/flox mice were crossed with Tie2Cre mice to generate Tie2Cre-NLRC5flox/flox mice in which NLRC5 was knocked out specifically in ECs and myeloid/microglia [41] (Figure S5A-C). The deletion of NLRC5 was confirmed by qPCR in mouse lung tissues of the Tie2Cre-NLRC5flox/flox mice (Figure S5D). Since angiogenesis took place in early embryo development, defined as physiological angiogenesis, we used the postnatal retina to investigate the role of NLRC5 in physiological angiogenesis. No significant changes of outgrowth length, superficial plexus, and sprout filopodia were observed in the retina of NLRC5flox/flox mice compared with Tie2Cre-NLRC5flox/flox mice (Figure S6A-F). Consistent with these findings, the expression of NLRC5 in the retina of mouse pups was fairly low in the early postnatal days, and increased after completion of angiogenesis in the superficial retina (Figure S6G-H). This might be the main reason for the negligible contribution of NLRC5 in retina angiogenesis.
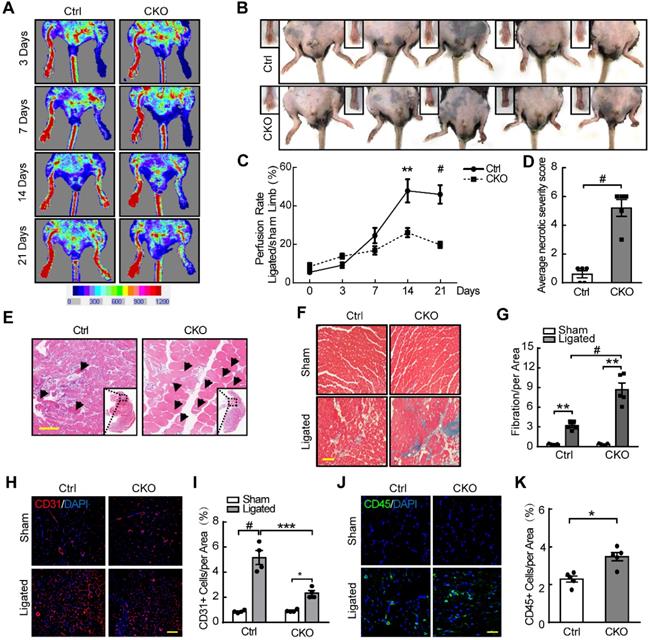
We further investigated the role of NLRC5 in pathological angiogenesis using femoral artery ligation as a limb ischemia mouse model. The blood flow recovery in the ischemic limbs of Tie2Cre-NLRC5flox/flox mice was significantly lower compared with control at day 14 (Tie2Cre-NLRC5flox/flox 26.11 ± 2.38% vs NLRC5flox/flox 47.86 ± 6.0%, P < 0.01) and after 21 days (Tie2Cre-NLRC5flox/flox 19.75 ± 1.67% vs NLRC5flox/flox 46.0 ± 0.7%, P < 0.001) (Figure 3A and C). More severe myocyte necrosis was observed on the early time point (3 days) after femoral artery ligation (Figure 3B, D-E), while a greater degree of fibrosis was observed later at 28 days (Tie2Cre-NLRC5flox/flox 8.21 ± 2.11% vs NLRC5flox/flox 2.08 ± 0.41%, P < 0.001) (Figure 3F-G). Using immunofluorescence, we found that CD31-positive cells in the ischemic area was significantly reduced in knockout mice compared with the controls (Figure 3H-I). Moreover, CD45-positive cells in the ischemic area were more increased in knockout mice (Figure 3J-K), supporting that inflammation was involved in angiogenesis post-ischemia. In contrast, the enlargement of arteries measured by artery diameter remained unchanged between groups (Figure S7A-B). These findings suggested that NLRC5 mainly influenced angiogenesis, but not arteriogenesis.
Decreased angiogenesis of Tie2Cre-NLRC5flox/flox mice resulted in severe necrosis and fibrosis of ischemic legs. (A) The blood flow recovery in Tie2Cre-NLRC5flox/flox (CKO) and NLRC5flox/flox (Ctrl) mice. Blood flow was measured by tissue Doppler analysis. (B) The necrosis was quantified in Tie2Cre-NLRC5flox/flox mice and NLRC5flox/flox mice (28 days). (C) Quantification of A. n = 5 mice/group. Two-way ANOVA with Bonferroni post-test, ** P < 0.01, # P < 0.001. (D) Quantification of B. The average necrotic severity score. n = 5 mice/group. Unpaired Student's t-test, # P < 0.001. (E) Hematoxylin-eosin (HE) staining for necrotic cells in the cross section of ligated musculus gastrocnemius muscle (3 days). Scar bar, 50 µm. (F) Masson staining for collagen deposition in the cross section of musculus gastrocnemius muscle after femoral artery ligation (28 days). Scar bar, 100 µm. (G) Quantification of F. n = 5 mice /group, 4 scopes/mice. Data are mean ± SEM, two-way ANOVA with Bonferroni post-test, ** P < 0.01, # P < 0.001. (H) CD31-positive cells in the cross section of musculus gastrocnemius muscles after femoral artery ligation (14 days). Representative confocal microscopy images of immunofluorescence staining for CD31 (red), DAPI (blue). Scar bar, 100 µm. (I) Quantification of H. n = 4 mice/group, 3 scopes/mice. Two-way ANOVA with Bonferroni post-test, * P < 0.05, *** P < 0.005, # P < 0.001. (J) CD45-positive cells in the cross section of musculus gastrocnemius muscles after femoral artery ligation (14 days). Representative confocal microscopy images of immunofluorescence staining for CD45 (green), DAPI (blue). Scar bar, 100 µm. (K) Quantification of J. n = 5 mice/group, 3 scopes/mice. Two-way ANOVA with Bonferroni post-test, * P < 0.05.
As NLRC5 was also known to be expressed in myeloid cells [42], bone marrow transplantation studies were conducted to exclude the effect of myeloid cells-derived NLRC5 on angiogenesis (Figure 4A). The data from flow cytometry analyses demonstrated that Tie2Cre-NLRC5flox/flox mice still had less CD31-positive cells compared with NLRC5flox/flox mice after they were transplanted with WT bone marrow (Tie2Cre-NLRC5flox/flox 12.5 ± 0.43% vs NLRC5flox/flox 18.4 ± 0.42%, P < 0.01, Figure 4B-C). Despite the greater level of CD45+ inflammatory cells that accumulated in the local injured tissues there was no difference of bone marrow-derived CD45+CD11b+ myeloid cell recruitment in the injured legs between groups (Figure 4D-F). Taken together, these findings indicate that there is a limited role of bone marrow-derived cells, particularly myeloid cell-derived NLRC5-deficient cells, contributing to the decreased angiogenesis observed in Tie2Cre-NLRC5flox/flox mice.
Finally, as angiogenesis was essential in tumor growth through which tumor tissues gain its nutrient supply. Melanoma is a cutaneous neoplasia with rapid angiogenesis, which might dramatically increase the risk of lethality [43]. And the expression of NLRC5 was correlated with survival rate in patients with melanoma which suggested that NLRC5 might have effect on the procession of melanoma [44]. As a result, we used melanoma as another model that mimics a complicated microenvironment and further explored the contributing role of NLRC5 in angiogenesis. The melanoma cells (B16F10) were injected into mice subcutaneously. Remarkably, tumors in Tie2Cre-NLRC5flox/flox mice grew slower than the NLRC5flox/flox mice as quantified by final tumor size (Tie2Cre-NLRC5flox/flox 56.95 ± 23.27 mm3 vs NLRC5flox/flox 525.5 ± 112.8 mm3, P < 0.001) and tumor weight (Tie2Cre-NLRC5flox/flox 19.5 ± 7.6 mg vs NLRC5flox/flox mice, 249.5 ± 24.0 mg, P < 0.001) without much difference in body weight (Figure S8A-D). In addition, Tie2Cre-NLRC5flox/flox mice had less vessel density (CD31+ cells number) (Figure S8E-F).
NLRC5 binds with STAT3 but not STAT1 in VEGFA treated ECs
To investigate the potential mechanism of pro-angiogenesis effects of NLRC5, we conducted GSEA analysis based on our RNA-seq results. Several angiogenesis related pathways were found to be suppressed in the NLRC5 knock down group, the mainly enriched pathway and related genes' profile of GSEA results (Figure 5A-B). Of note, down regulation of NLRC5 was positively related to IL-6-JAK-STAT3 signaling pathway's suppression according to the GSEA analysis results (Figure 5C). This analysis gives us a clue to further explore the possible rationale for the interaction of NLRC5 and STAT3.
Signal transducer and activator of transcription 3 (STAT3) bound with NLRC5 in VEGFA-165 stimulated ECs. (A) Flow chart of sample preparation for RNA sequencing. (B) Heat maps of related genes based on RNA-seq. (C) Gene set enrichment analysis (GSEA) was performed using the gene in the IL-6-JAK-STAT3 pathway generated from RNA-seq. (D) Identification of STAT3 as a binding partner of NLRC5 in VEGFA-165 treated HUVECs. HUVECs were stimulated with VEGFA-165 for 0 or 12 h. The lysate was immunoprecipitated with anti-STAT3 antibody and then immunoblotted with the indicated antibodies. (E) Co-immunoprecipitation of myc-NLRC5 overexpressed HEK293T cells. The lysate was immunoprecipitated with anti-myc antibody and then immunoblotted with the indicated antibodies. The protein expression was detected by western blot analysis. (F) Co-immunoprecipitation of myc-NLRC5 in combination with Flag-STAT3 in HEK293T cells. The lysate was immunoprecipitated with anti-Flag antibody and then immunoblotted with the indicated antibodies. The protein expression was detected by western blot analysis. (G) Identification of STAT1 as a binding partner of NLRC5 in IFN-γ-treated HUVECs, but not in VEGFA-165-treated HUVECs. HUVECs were stimulated with IFN-γ and VEGFA-165 for 12 h. The lysate was immunoprecipitated with anti-STAT1 antibody and then immunoblotted with the indicated antibodies. (H) Co-immunoprecipitation of myc-NLRC5 overexpressed HEK293T cells. The lysate was immunoprecipitated with anti-myc antibody and then immunoblotted with the indicated antibodies. The protein level was detected by western blot analysis.
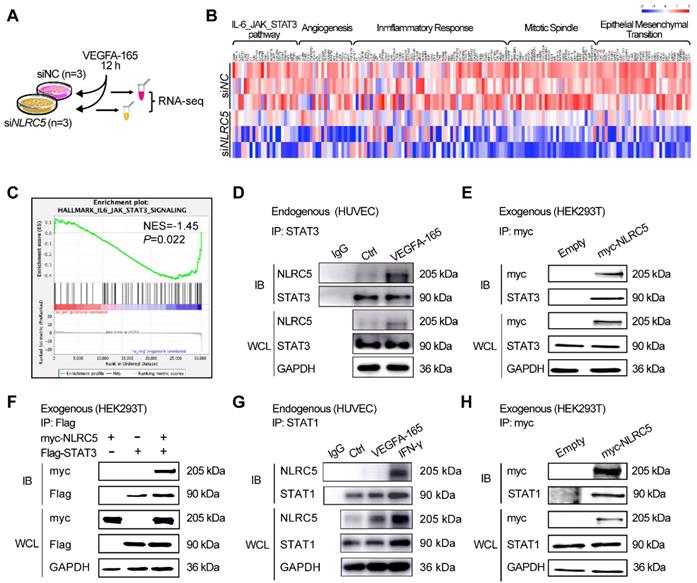
Based on previous studies, NLRC5 regulates gene expression through forming interactions with several transcription factors [21, 45]. Our presented data indicated that NLRC5 might serve as a transcriptional regulator or coactivator in the nucleus of ECs. STAT3 is a transcription factor that has been clarified its function in angiogenesis beyond classical role in regulating tumorigenesis or inflammation [46]. Lysates of HUVECs treated with VEGFA-165 were immunoprecipitated with anti-STAT3 antibodies. As shown in Figure 5D, the binding of STAT3 to NLRC5 was enhanced after VEGFA-165 stimulation. This finding was in line with the results of co-immunoprecipitation studies of NLRC5-myc and STAT3-Flag overexpressed in HEK293T cells (Figure 5E-F). Together, these results showed that STAT3 was enhanced in VEGFA-165 stimulated HUVECs and banded with NLRC5.
Because STAT1 and STAT3 shared more than 75% of their homology sequence [47], so it urged us to suppose that NLRC5 would also combine with STAT1. The results showed that STAT1 was a binding partner of NLRC5 in IFN-γ-treated HUVECs, but not in VEGFA-165 treated HUVECs (Figure 5G). And the result from co-immunoprecipitation of myc-NLRC5 overexpressed HEK293T cells revealed the binding of Flag-STAT1 and myc-NLRC5 as well (Figure 5H).
DD+NACHT domain is a key domain for NLRC5 binding to STAT3 in the nucleus
NLRC5 is an atypical member of the NLRs family because of its unusual caspase activation and recruitment domain (CARD domain or DD domain) and its longest leucine rich repeats (LRRs domain) [7]. As our data suggested above, NLRC5 interacted with STAT3 in ECs. To investigate which specific domain of NLRC5 mediated this interaction, several plasmids containing different NLRC5 domains (myc-tagged) including the DD domain (1-221aa), NACHT domain (222-539 aa), DD+NACHT domain (1-539 aa), ΔDD domain (135-1855 aa) and the NLRC5 full-length plasmid were transfected together with STAT3-Flag in HEK293T cells. Co-immunoprecipitation showed that while neither DD nor NACHT domains interacted with STAT3, the DD+NACHT domain did bind STAT3 (Figure 6A). Moreover, the mutant ΔDD domain, 1-134 amino acids deleted from the N-terminal of NLRC5, also bound to STAT3 as well (Figure 6A). Yet, this ΔDD domain only localized to the cytoplasm while DD+NACHT domain as well as the NLRC5 full-length plasmid could shuttle into nucleus besides interacting with STAT3 (Figure 6B). Further, domain deleted STAT3 plasmids were transfected into HEK293T in combination with NLRC5 respectively. Co-IP was conducted to determine which main domain of STAT3 (Flag-tagged) interacted with NLRC5 (myc-tagged). As shown in Figure S9A, ΔNTD domain (2-120 aa deleted), ΔCCD domain (141-313 aa deleted), ΔDBD domain (325-464 aa deleted) and ΔSH2 domain (584-647 aa deleted) were constructed. And the result of Co-IP also has clarified CCD domain was the main domain for the interaction of NLRC5 and STAT3 (Figure S9B). In brief, these results indicated that the structural integrity of DD+NACHT domain was essential for the interaction of NLRC5 and STAT3 in the nucleus.
The integrity of the DD+NACHT domain of NLRC5 was critical for NLRC5 transcriptional activity. (A) Flag-STAT3 was co-transfected with myc-DD, myc-NACHT, myc-DD+NACHT, myc-ΔDD, and myc-NLRC5 full length plasmids, respectively, in HEK293T cells. The lysates were immunoprecipitated and then immunoblotted with antibodies against the indicated proteins. (B) Representative confocal microscopy images of immunofluorescence staining for myc (green), Flag (red), DAPI (blue). Scar bar, 20 µm. (C) myc-NLRC5 was transfected in HEK293T cells and the efficiency was detected by western blot analysis. (D) Endogenous NLRC5 was knockdown by siRNA. (E) Quantification of C. Data are mean ± SEM, n = 3 independent experiments. One-way ANOVA with Bonferroni post-test, ** P < 0.01. (F) Quantification of D. Data are mean ± SEM, n = 3 independent experiments. Unpaired Student's t-test, *** P < 0.005. (G) Sis induce element (SIE) promoter luciferase reporter plasmid was respectively transfected with myc-NLRC5, myc-DD, or myc-ΔDD into HEK293T cells for 24 h. Cells were treated with PBS or IL-6 (20 ng/mL) for another 18 h. Promoter activities were normalized to renilla luciferase. The results were expressed as relative luciferase activity. Data are mean ± SEM, n = 3 independent experiments. One-way ANOVA with Bonferroni post-test, ** P < 0.01, *** P < 0.005. (H) After transfection, the cells were stimulated with IL-6 (20 ng/mL) for 18 h. Promoter activities were normalized to renilla luciferase. Data are mean ± SEM, n = 3 independent experiments. Two-way ANOVA with Bonferroni post-test, # P < 0.001.
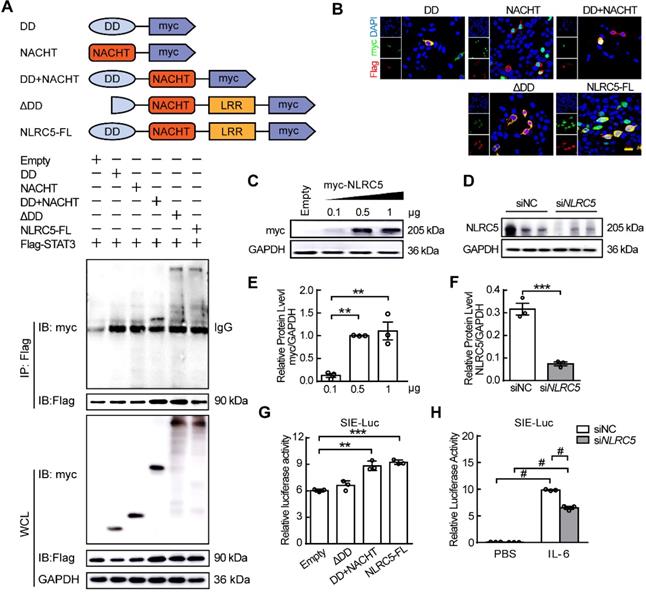
To explore the effects of the various domains on the expression of the downstream STAT-responsive elements, the SIE luciferase reporter was transfected along with the DD+NACHT domain, ΔDD domain, or the full length NLRC5 into HEK293T cells and stimulated with IL-6. Although the DD+NACHT domain and full length NLRC5 could enhance the relative luciferase activity, the ΔDD domain remained unchanged (Figure 6C, E and G). In addition, knockdown of NLRC5 in HEK293T cells inhibited the relative luciferase activity significantly (Figure 6D, F and H). Taken together, these data indicated that the DD+NACHT domain is necessary for the ability of NLRC5 to maintain its transcriptional activity.
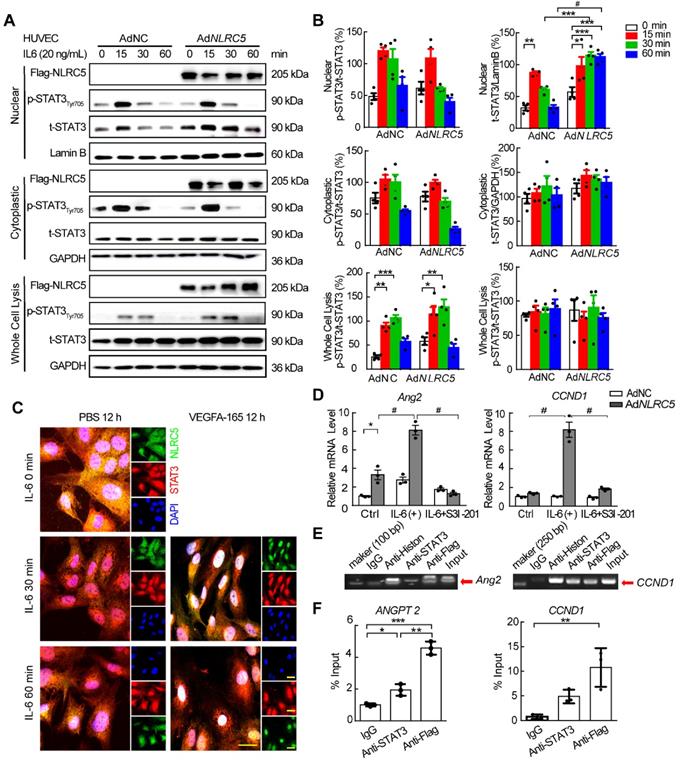
NLRC5 promotes STAT3 transcriptional activity by enhancing the accumulation of unphosphorylated STAT3 in the nucleus
As it had been reported that the unphosphorylated form of STAT3 accumulated in the nucleus to regulate gene expression [48, 49], we further hypothesized that NLRC5 might regulate the STAT3 function by binding and trapping unphosphorylated STAT3 in the nucleus. To verify this hypothesis, NLRC5-Flag tagged adenovirus was constructed and used to infect HUVECs stimulated with interleukin-6 (IL-6). The overexpressed NLRC5 delayed the translocation of unphosphorylated STAT3 out of the nucleus by 30 min after IL-6 stimulation in HUVECs and was sustained until 60 min (AdNC 32.63 ± 4.18 folds vs AdNLRC5 114.74 ± 5.19 folds, P < 0.001) (Figure 7A-B). Similar results were further confirmed by immunofluorescence staining (Figure 7C). Overexpression of NLRC5 in HUVECs also promoted some STAT3-regulated genes expression such as angiopoietin-2 (Ang2) and cyclin D1 (CCND1) (Figure 7D and Figure S10), suggesting the subsequent transcriptional activity brought about by the trapped unphosphorylated STAT3. Furthermore, such enhanced transcriptional activity could be blocked by S3I-201, an inhibitor of STAT3 [50] (Figure 7D). ChIP assay of the Ang2 and CCND1 promoter provided further evidence that the binding of NLRC5 and STAT3 in the nucleus increased the expression of Ang2 or cyclin D1 targeted to the promoter region (Figure 7E-F).
Overexpressed NLRC5 prolonged the accumulation of STAT3 in the nucleus after interleukin 6 (IL-6) stimulation in HUVECs. (A) HUVECs were transfected with AdNC or AdNLRC5 for 48 h and then treated with IL-6 (20 ng/mL) for the indicated time points over 60 min. Nuclear and cytoplasmic fractions were extracted from HUVECs. The protein expression of Flag-NLRC5, t-STAT3, p-STAT3(Tyr705), Lamin B, and GAPDH were measured by western blot analysis. (B) Quantification of A. Data are mean ± SEM. n = 4 independent experiments, two-way ANOVA with Bonferroni post-test, * P < 0.05, ** P < 0.01, *** P < 0.005, # P < 0.001. (C) Representative confocal microscopy images of immunofluorescence staining for Flag-NLRC5 (green), STAT3 (red) and DAPI (blue). Scar bar, 20 µm. (D) NLRC5 overexpression increased mRNA expression of angiopoietin-2 and cyclin D1 in IL-6 induced HUVECs, and the enhancement was inhibited by the STAT3 specific inhibitor S3I-201. Data are mean ± SEM, n = 3 independent experiments. Two-way ANOVA with Bonferroni post-test, * P < 0.05, # P < 0.001. (E-F) ChIP assay for the promoter of angiopoietin-2 and cyclin D1. HUVECs were transfected with AdNLRC5 and the sonicated nuclear lysates were incubated with anti-STAT3 or anti-Flag antibodies. The purified DNA was amplificated by qPCR and confirmed by southern blot. Data are mean ± SEM, n = 3 independent experiments. Two-way ANOVA with Bonferroni post-test, * P < 0.05, ** P < 0.01, *** P < 0.005.
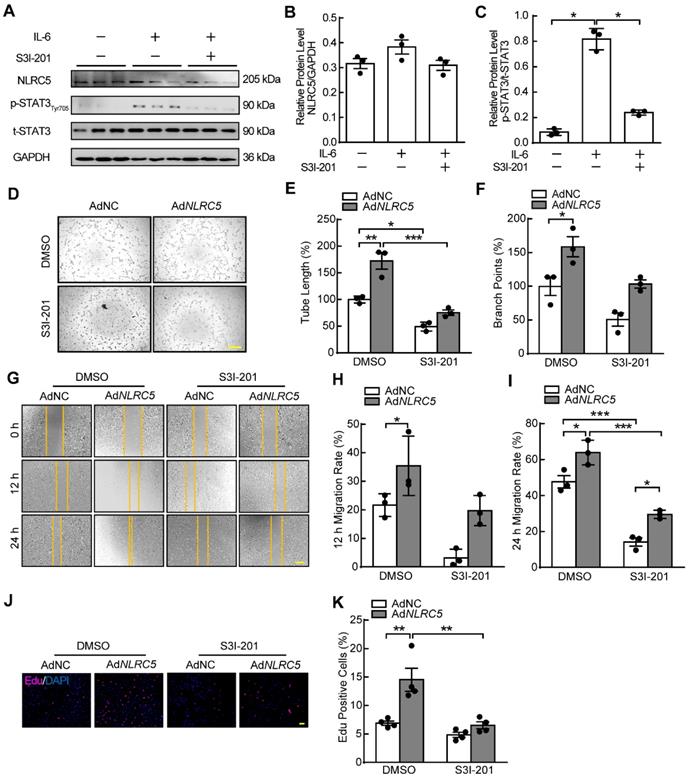
Inhibiting STAT3 suppresses the proangiogenic properties of NLRC5 in ECs
STAT3 inhibitor S3I-201 and siRNA were used to block the activity of STAT3. The phosphorylation of STAT3 induced by IL-6 stimulation was significantly inhibited without altering expression of NLRC5 when HUVECs were treated with S3I-201 (Figure 8A-C). At the same time, the protein level of STAT3 was suppressed after siSTAT3 knockdown in HUVECs. S3I-201and siSTAT3 reversed the proangiogenic properties of overexpressed NLRC5 on EC tube formation (Figure 8D-F & Figure S11A-C), migration (Figure 8G-I & Figure S11D-E), and proliferation (Figure 8J-K & Figure S11F-G). Moreover, overexpression of STAT3 (WT) rather than STAT3 (Y705F) (Figure S12A-C) has rescued the decreased angiogenesis in NLRC5 defected endothelial cell line. Taken together, inhibition of STAT3 activity significantly reduced NLRC5-mediated pro-angiogenic properties in ECs.
STAT3 specific inhibitor S3I-201 impaired the induced enhancement of EC tube formation, migration, and proliferation mediated by NLRC5 overexpression. (A) The efficiency of S3I-201 to bind with STAT3 and to decrease the phosphorylation of STAT3. (B-C) Quantification of A. Data are mean ± SEM, n = 3 independent experiments. Two-way ANOVA with Bonferroni post-test, * P < 0.05. (D) S3I-201 inhibited the tube formation in NLRC5 overexpressed in HUVECs. Scar bar, 100 µm. (E-F) Quantification of D. Data are mean ± SEM, n = 3 independent experiments. Two-way ANOVA with Bonferroni post-test, * P < 0.05, ** P < 0.01, *** P < 0.005. (G) S3I-201 inhibited the migration in NLRC5 overexpressed in HUVECs. Scar bar, 100 µm. (H-I) Quantification of G. Data are mean ± SEM, n = 3 independent experiments. Two-way ANOVA with Bonferroni post-test, * P < 0.05, *** P < 0.005. (J) S3I-201 inhibited the proliferation in NLRC5 overexpressed in HUVECs. 5-ethynyl-2'-deoxyuridine (Edu, red), DAPI (blue). Scar bar, 100 µm. (K) Quantification of J. Data are mean ± SEM, n = 4 independent experiments. Two-way ANOVA with Bonferroni post-test, ** P < 0.01.
Discussion
In this study, we demonstrate the existence of an intertwined regulation between the VEGF and IL-6/STAT3 signaling pathways and pathological angiogenesis. NLRC5 promotes pathological angiogenesis through its interaction with STAT3 and subsequent upregulation of angiogenesis-related genes.
Prior studies of NLRC5 mainly focused on its role in the inflammatory response as it was first reported in the cytoplasm of hemocytes [21]. Consistent with these studies, we found that LPS could slightly enhance the cytoplasmic expression of NLRC5 in ECs in vitro or in MLECs from an acute lung injury model induced by LPS in vivo. Such effect was attributed to cytoplasmic NLRC5 as the LPS did not change the subcellular distribution of NLRC5 protein. NLRC5 could also regulate the expression of ICAM-1 and VCAM-1 in ECs (Figure S2G), raising the possibility that NLRC5 in the cytoplasm might suppress NF-κB activities and decrease the expression of ICAM-1 and VCAM-1.
Data from our experiments further suggested that NLRC5 in ECs served as a transactivator of STAT3 and stimulated angiogenic gene expression. Induction of angiogenesis by NLRC5 stemmed largely by its interaction with STAT3 since we did not find the interaction of STAT1. Phosphorylated STAT1 homodimers induced by IFN-γ are known to enhance ISRE, which eventually increased NLRC5 expression [51, 52]. Although STAT1 could bind to NLRC5 following IFN-γ stimulation in HUVECs, STAT1 did not interact with NLRC5 in VEGFA-165-stimulated HUVECs. In line with these data, our results also demonstrated that STAT1 bound with NLRC5 in HEK293T cells and enhanced ISRE luciferase activities (Figure S13), meaning that the feedback mechanism of NLRC5 and STAT1 was more intricate.
STAT1 and STAT3 share more than 75% of their homology sequence [47] and both can recognize similar DNA consensus sequences based on a TTCNNN(T, G)AA motif [53]. However, STAT1 often has the opposite function to STAT3 in a range of cellular paradigms such as inflammation or angiogenesis [47, 54]. Accumulating studies demonstrate that STAT3 regulates angiogenesis by increasing the proliferation or migration of ECs [55-57]. Traditionally, the phosphorylated STAT3 dimer evoked by IL-6 or other factors will translocate to the nucleus and transactivate target genes [58-60]. In fact, STAT3 could activates transcription in the form of unphosphorylated STAT3 as well as phosphorylated STAT3. A recent study has demonstrated that unphosphorylated STAT3 in the nucleus interacted with yes-associated protein and enhances angiogenesis of ECs [61]. In addition to this, STAT3 could regulate transcription of related genes by acetylation, methylation and even palmitoylation[62-64]. It is hard to exclude from the interaction with other forms of STAT3 in the present experiments since they could be concluded as unphosphorylated STAT3. Meanwhile, our results showed that NLRC5 preferentially bound to STAT3 rather than STAT1 in VEGFA-165-treated ECs in vitro. Although IL-6 is generally thought to be a pro-angiogenic cytokine [65, 66], its role in angiogenesis is not fully elucidated. Some studies indicate that the pro-angiogenic ability of IL-6 depends on the presence of VEGF in vivo [67-69]. Our study provides a clue that NLRC5 is not only an essential sensor for inflammation since NLRC5 expression increases in the complicated microenvironment of tissue injury, but also a signal transducer of injury to promote tissue repair, through neovascularization by coordinating with the VEGF and IL-6/STAT3 pathways. In short, the functional variability of NLRC5 in multiple microenvironments is likely attributed to its interactions with various transcription factors in response to specific ligands in different cell-types. The proangiogenic characteristics of NLRC5 may be attributed to its unique structure to form a loop to integrate STAT3. The other NLRs that lack the α-helix-riched caspase domain may have more difficult to form a loop. Hence, the DD+NACHT domain of NLRC5 could be a druggable target for clinical therapy of ischemic disease states.
Interestingly, tumor growth and angiogenesis were also markedly reduced in the melanoma tumor transplant model of Tie2Ce-NLRC5flox/flox mice. Since NLRC5 regulates MHC class I antigen presentation and T cell responses, the deficient mice are potentially more susceptible to pathogen infection [66, 70]. In our hand, we observed that some MHCI related genes such as HLA-B, HLA-F and HLA-G were suppressed in siNLRC5 knockdown HUVECs (Figure S14). In theory, the NLRC5-deficient mice could also aggravate tumor progression as NLRC5/CITA plays a crucial role in human cancer immunity through the recruitment and activation of tumor killing CD8+ T cells [71]. However, the strong inhibition of tumor growth that we observed suggests that the STAT3 function in vivo is largely depended on NLRC5. These results provide evidence that the multifunctional role of NLRC5 in the nucleus and the specific binding partner of NLRC5 and STAT3, determined the fate of cells.
There are several limitations in this study. First of all, we used Tie2Cre mice to generate knockout mice. Since Tie2 is expressed in both ECs and myeloid cells, the phenotype observed in Tie2Cre mice is not exclusively of ECs origin. To eliminate the effect of myeloid cells where NLRC5 is also expressed, we perform bone marrow transplantation. The data of cytometry analyses have indicated there was no significant difference of CD45+CD11b+ (myeloid cell) between ligated legs of CKO and Ctrl mice. The data suggest the role of NLRC5 from ECs rather than hematopoietic cell-origin myeloid cells on angiogenesis in hindlimb ischemia studies. Meanwhile, the increase of CD45+ cells suggested the enhanced accumulation of CD45+CD11b- cells in CKO ligated limbs. Therefore, it is needed for us to explore the subset of these cells and their contribution to angiogenesis in further study. Secondly, HUVECs were used to study the mechanism of NLRC5 in angiogenesis, but not the primary neonatal endothelial cells. HUVECs are isolated from umbilical vein of neonate with great potential of proliferation and are regarded as an easy tool to investigate endothelium in vitro [72, 73]. ECs isolated from neonatal KO and WT mice might have better performance in comparing their potential on angiogenesis. But they are easily to differentiate into fibroblast-like cells (mesenchymal cells) as reported [74, 75]. Hence, they are not stable to be used to investigate the mechanism of NLRC5 on angiogenesis. Thirdly, we cannot exclude a participatory role for other NLRC5 interacting factors in the nucleus beyond STAT3, although our data strongly suggests that STAT3 is at least sufficient to mediate NLRC5's angiogenic properties in vitro. Finally, we cannot rule out various combinations of domains of NLRC5 mediating the STAT3 interaction in specific cells types or in response to divergent pro-angiogenic stimuli. Future studies will be of interest to clarify these points further.
In summary, we presented new insights for NLRC5 in pathological angiogenesis in this study. Endothelial NLRC5 deficiency in vitro or in vivo inhibited pathological angiogenesis, but had no effect on physiological angiogenesis. The accumulation of NLRC5 sustained the expression of unphosphorylated STAT3 in the nucleus, which in turn increased the STAT3-regulated gene Ang2 and proangiogenic properties. Finally, we identified that the DD+NACHT domain of NLRC5 was required for binding to STAT3. Collectively, these findings reveal an unanticipated role for NLRC5 as a key molecular switch in response to tissue injury to facilitate neovascularization during tissue regeneration.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank patients who participated in this study. This study is supported by grants No. 9193910, No. 81670746, and No. 81800424 from the Chinese National Natural Science Foundation and 2016YFC1301202 from National Key Research and Development Program of China. We are grateful to Ping Wang from Tongji University for providing the B16F10.
Author Contributions
Xu Xu performed the animal experiments, cell function, and conducted western blot, immunofluorescence, and analyzed data. Yefei Shi performed the cell culture, animal experiments and western blot. Peipei Luan conducted co-immunoprecipitation assays and immunoblots. Wenxi Kou and Bo Li performed the animal experiments. Qing Yu performed mice lung endothelial cells isolation and culture. Ming Zhai and Shuangjie You performed the bioinformation analysis. Mark W. Feinberg helped interpret the data and wrote the manuscript. Jianhui Zhuang performed the animal experiments, conducted luciferase and Ch-IP assays, interpreted results. Weixia Jian and Wenhui Peng conceived the project, designed experiments, analyzed data, interpreted results and wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209-23
2. Akira S, Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett. 2003;85:85-95
3. Sadeghi K, Wessner B, Laggner U, Ploder M, Tamandl D, Friedl J. et al. Vitamin D3 down-regulates monocyte TLR expression and triggers hyporesponsiveness to pathogen-associated molecular patterns. Eur J Immunol. 2006;36:361-70
4. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805-20
5. Grote K, Schütt H, Schieffer B. Toll-like receptors in angiogenesis. ScientificWorldJournal. 2011;11:981-91
6. Daniels BP, Holman DW, Cruz-Orengo L, Jujjavarapu H, Durrant DM, Klein RS. Viral pathogen-associated molecular patterns regulate blood-brain barrier integrity via competing innate cytokine signals. mBio. 2014;5:e01476-14
7. Benko S, Magalhaes JG, Philpott DJ, Girardin SE. NLRC5 limits the activation of inflammatory pathways. J Immunol. 2010;185:1681-91
8. Martinon F, Tschopp J. NLRs join TLRs as innate sensors of pathogens. Trends Immunol. 2005;26:447-54
9. Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E. et al. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc Natl Acad Sci U S A. 2010;107:13794-9
10. Cressman DE, Chin KC, Taxman DJ, Ting JP. A defect in the nuclear translocation of CIITA causes a form of type II bare lymphocyte syndrome. Immunity. 1999;10:163-71
11. Spilianakis C, Papamatheakis J, Kretsovali A. Acetylation by PCAF enhances CIITA nuclear accumulation and transactivation of major histocompatibility complex class II genes. Mol Cell Biol. 2000;20:8489-98
12. Cressman DE, O'Connor WJ, Greer SF, Zhu XS, Ting JP. Mechanisms of nuclear import and export that control the subcellular localization of class II transactivator. J Immunol. 2001;167:3626-34
13. Meissner TB, Liu YJ, Lee KH, Li A, Biswas A, van Eggermond MC. et al. NLRC5 cooperates with the RFX transcription factor complex to induce MHC class I gene expression. J Immunol. 2012;188:4951-8
14. Neerincx A, Rodriguez GM, Steimle V, Kufer TA. NLRC5 controls basal MHC class I gene expression in an MHC enhanceosome-dependent manner. J Immunol. 2012;188:4940-50
15. Meng Q, Cai C, Sun T, Wang Q, Xie W, Wang R. et al. Reversible ubiquitination shapes NLRC5 function and modulates NF-κB activation switch. J Cell Biol. 2015;211:1025-40
16. Cui J, Zhu L, Xia X, Wang HY, Legras X, Hong J. et al. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell. 2010;141:483-96
17. Tong Y, Cui J, Li Q, Zou J, Wang HY, Wang RF. Enhanced TLR-induced NF-κB signaling and type I interferon responses in NLRC5 deficient mice. Cell Res. 2012;22:822-35
18. Davis BK, Roberts RA, Huang MT, Willingham SB, Conti BJ, Brickey WJ. et al. Cutting edge: NLRC5-dependent activation of the inflammasome. J Immunol. 2011;186:1333-37
19. Triantafilou K, Kar S, van Kuppeveld FJ, Triantafilou M. Rhinovirus-induced calcium flux triggers NLRP3 and NLRC5 activation in bronchial cells. Am J Respir Cell Mol Biol. 2013;49:923-34
20. Luan P, Zhuang J, Zou J, Li H, Shuai P, Xu X. et al. NLRC5 deficiency ameliorates diabetic nephropathy through alleviating inflammation. FASEB J. 2018;32:1070-84
21. Luan P, Jian W, Xu X, Kou W, Yu Q, Hu H. et al. NLRC5 inhibits neointima formation following vascular injury and directly interacts with PPARγ. Nat Commun. 2019;10:2882
22. Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873-87
23. Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6:389-95
24. Adams RH, Eichmann A. Axon guidance molecules in vascular patterning. Cold Spring Harb Perspect Biol. 2010;2:a001875
25. Stahl A, Connor KM, Sapieha P, Chen J, Dennison RJ, Krah NM. et al. The Mouse Retina as an Angiogenesis Model. Invest Ophthalmol Vis Sci. 2010;51:2813-26
26. Staton CA, Reed MW, Brown NJ. A critical analysis of current in vitro and in vivo angiogenesis assays. Int J Exp Pathol. 2009;90:195-221
27. Boerckel JD, Uhrig BA, Willett NJ, Huebsch N, Guldberg RE. Mechanical regulation of vascular growth and tissue regeneration in vivo. Proc Natl Acad Sci U S A. 2011;108:E674-80
28. Kilarski WW, Samolov B, Petersson L, Kvanta A, Gerwins P. Biomechanical regulation of blood vessel growth during tissue vascularization. Nat Med. 2009;15:657-64
29. Kreuger J, Phillipson M. Targeting vascular and leukocyte communication in angiogenesis, inflammation and fibrosis. Nat Rev Drug Discov. 2016;15:125-42
30. Vaeyens MM, Jorge-Peñas A, Barrasa-Fano J, Steuwe C, Heck T, Carmeliet P. et al. Matrix deformations around angiogenic sprouts correlate to sprout dynamics and suggest pulling activity. Angiogenesis. 2020;23:315-24
31. Zhou T, Zheng Y, Sun L, Badea SR, Jin Y, Liu Y. et al. Microvascular endothelial cells engulf myelin debris and promote macrophage recruitment and fibrosis after neural injury. Nat Neurosci. 2019;22:421-35
32. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545-50
33. Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739-40
34. Hehlert P, Hofferek V, Heier C, Eichmann TO, Riedel D, Rosenberg J. et al. The α/β-hydrolase domain-containing 4- and 5-related phospholipase Pummelig controls energy storage in Drosophila. J Lipid Res. 2019;60:1365-78
35. Ko KA, Fujiwara K, Krishnan S, Abe JI. En Face Preparation of Mouse Blood Vessels. J Vis Exp. 2017;123:e55460
36. Liu L, Cheung TH, Charville GW, Rando TA. Isolation of skeletal muscle stem cells by fluorescence-activated cell sorting. Nat Protoc. 2015;10:1612-24
37. Meissner TB, Li A, Liu YJ, Gagnon E, Kobayashi KS. The nucleotide-binding domain of NLRC5 is critical for nuclear import and transactivation activity. Biochem Biophys Res Commun. 2012;418:786-91
38. Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT, De Bruijn EA. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev. 2004;56:549-80
39. Weigel MT, Dowsett M. Current and emerging biomarkers in breast cancer: prognosis and prediction. Endocr Relat Cancer. 2010;17:R245-62
40. Lee MY, Gamez-Mendez A, Zhang J, Zhuang Z, Vinyard DJ, Kraehling J. et al. Endothelial Cell Autonomous Role of Akt1: Regulation of Vascular Tone and Ischemia-Induced Arteriogenesis. Arterioscler Thromb Vasc Biol. 2018;38:870-9
41. Tang Y, Harrington A, Yang X, Friesel RE, Liaw L. The contribution of the Tie2+ lineage to primitive and definitive hematopoietic cells. Genesis. 2010;48:563-7
42. Yu Q, Kou W, Xu X, Zhou S, Luan P, Xu X. et al. FNDC5/Irisin inhibits pathological cardiac hypertrophy. Clin Sci (Lond). 2019;133:611-27
43. Mahabeleshwar GH, Byzova TV. Angiogenesis in melanoma. Semin Oncol. 2007;34:555-65
44. Yoshihama S, Roszik J, Downs I, Meissner TB, Vijayan S, Chapuy B. et al. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc Natl Acad Sci U S A. 2016;113:5999-6004
45. Kobayashi KS, van den Elsen PJ. NLRC5: a key regulator of MHC class I-dependent immune responses. Nat Rev Immunol. 2012;12:813-20
46. Chen Z, Han ZC. STAT3: a critical transcription activator in angiogenesis. Med Res Rev. 2008;28:185-200
47. Fagard R, Metelev V, Souissi I, Baran-Marszak F. STAT3 inhibitors for cancer therapy: Have all roads been explored? JAKSTAT. 2013;2:e22882
48. Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 2007;21:1396-408
49. Carpenter RL, Lo HW. STAT3 Target Genes Relevant to Human Cancers. Cancers (Basel). 2014;6:897-925
50. Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR. et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci U S A. 2007;104:7391-6
51. Funovics P, Brostjan C, Nigisch A, Fila A, Grochot A, Mleczko K. et al. Effects of 15d-PGJ(2) on VEGF-induced angiogenic activities and expression of VEGF receptors in endothelial cells. Prostaglandins Other Lipid Mediat. 2006;79:230-44
52. Huang W, Eum SY, András IE, Hennig B, Toborek M. PPARalpha and PPARgamma attenuate HIV-induced dysregulation of tight junction proteins by modulations of matrix metalloproteinase and proteasome activities. FASEB J. 2009;23:1596-606
53. Staehli F, Ludigs K, Heinz LX, Seguín-Estévez Q, Ferrero I, Braun M. et al. NLRC5 deficiency selectively impairs MHC class I- dependent lymphocyte killing by cytotoxic T cells. J Immunol. 2012;188:3820-28
54. Darnell JE Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415-21
55. Levy DE, Lee CK. What does Stat3 do? J Clin Invest. 2002;109:1143-8
56. Huang S, Bucana CD, Van Arsdall M, Fidler IJ. Stat1 negatively regulates angiogenesis, tumorigenicity and metastasis of tumor cells. Oncogene. 2002;21:2504-12
57. Krishnamurthy P, Rajasingh J, Lambers E, Qin G, Losordo DW, Kishore R. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ Res. 2009;104:e9-18
58. Hoffmann CJ, Harms U, Rex A, Szulzewsky F, Wolf SA, Grittner U. et al. Vascular signal transducer and activator of transcription-3 promotes angiogenesis and neuroplasticity long-term after stroke. Circulation. 2015;131:1772-82
59. Miyazaki T, Taketomi Y, Saito Y, Hosono T, Lei XF, Kim-Kaneyama JR. et al. Calpastatin counteracts pathological angiogenesis by inhibiting suppressor of cytokine signaling 3 degradation in vascular endothelial cells. Circ Res. 2015;116:1170-81
60. Reich NC, Liu L. Tracking STAT nuclear traffic. Nat Rev Immunol. 2006;6:602-12
61. Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol. 2003;3:900-11
62. Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005;307:269-73
63. Yang J, Huang J, Dasgupta M, Sears N, Miyagi M, Wang B. et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc Natl Acad Sci U S A. 2010;107:21499-504
64. Zhang M, Zhou L, Xu Y, Yang M, Xu Y, Komaniecki GP. et al. A STAT3 palmitoylation cycle promotes T(H)17 differentiation and colitis. Nature. 2020;586:434-9
65. Liu L, McBride KM, Reich NC. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc Natl Acad Sci U S A. 2005;102:8150-5
66. He J, Bao Q, Zhang Y, Liu M, Lv H, Liu Y. et al. Yes-Associated Protein Promotes Angiogenesis via Signal Transducer and Activator of Transcription 3 in Endothelial Cells. Circ Res. 2018;122:591-605
67. Gopinathan G, Milagre C, Pearce OM, Reynolds LE, Hodivala-Dilke K, Leinster DA. et al. Interleukin-6 Stimulates Defective Angiogenesis. Cancer Res. 2015;75:3098-107
68. Gertz K, Kronenberg G, Kälin RE, Baldinger T, Werner C, Balkaya M. et al. Essential role of interleukin-6 in post-stroke angiogenesis. Brain. 2012;135:1964-80
69. Wei LH, Kuo ML, Chen CA, Chou CH, Lai KB, Lee CN. et al. Interleukin-6 promotes cervical tumor growth by VEGF-dependent angiogenesis via a STAT3 pathway. Oncogene. 2003;22:1517-27
70. Nagasaki T, Hara M, Nakanishi H, Takahashi H, Sato M, Takeyama H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. Br J Cancer. 2014;110:469-78
71. Yao Y, Wang Y, Chen F, Huang Y, Zhu S, Leng Q. et al. NLRC5 regulates MHC class I antigen presentation in host defense against intracellular pathogens. Cell Res. 2012;22:836-47
72. Lamalice L, Boeuf FL, Huot J. Endothelial Cell Migration During Angiogenesis. Circ Res. 2007;100:782-94
73. Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359-71
74. Wang J, Niu N, Xu S, Jin ZG. A simple protocol for isolating mouse lung endothelial cells. Sci Rep. 2019;9:1458
75. Ni CW, Kumar S, Ankeny CJ, Jo H. Development of immortalized mouse aortic endothelial cell lines. Vasc Cell. 2014;6:7
Author contact
![]() Corresponding authors: Wenhui Peng, Department of Cardiology, Shanghai Tenth People's Hospital, Tongji University School of Medicine, 301 Middle Yanchang Road, Shanghai, 200072, China. E-mail: pwenhuiedu.cn; or Mark W. Feinberg, Division of Cardiovascular Medicine, Brigham and Women's Hospital, Harvard Medical School, Boston, MA, 02115, USA. Phone: 617-525-4381, E-mail: mfeinbergharvard.edu; or Weixia Jian, Department of Endocrinology, Xinhua Hospital, Shanghai Jiaotong University School of Medicine, 1665 Kongjiang Road, Shanghai, 200092, China. E-mail: jianweixiacom.cn.
Corresponding authors: Wenhui Peng, Department of Cardiology, Shanghai Tenth People's Hospital, Tongji University School of Medicine, 301 Middle Yanchang Road, Shanghai, 200072, China. E-mail: pwenhuiedu.cn; or Mark W. Feinberg, Division of Cardiovascular Medicine, Brigham and Women's Hospital, Harvard Medical School, Boston, MA, 02115, USA. Phone: 617-525-4381, E-mail: mfeinbergharvard.edu; or Weixia Jian, Department of Endocrinology, Xinhua Hospital, Shanghai Jiaotong University School of Medicine, 1665 Kongjiang Road, Shanghai, 200092, China. E-mail: jianweixiacom.cn.