Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Structure and function of SIRT3
Endogenous regulators of SIRT3
SIRT3 and human disease
Targeting SIRT3 for potential...
Conclusions
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(18):8315-8342. doi:10.7150/thno.45922 This issue Cite
Review
Mitochondrial Sirtuin 3: New emerging biological function and therapeutic target
Jin Zhang1, Honggang Xiang1, Jie Liu1, Yi Chen1 ![]() , Rong-Rong He2
, Rong-Rong He2 ![]() , Bo Liu1
, Bo Liu1 ![]()
1. State Key Laboratory of Biotherapy and Cancer Center and Department of Gastrointestinal Surgery, West China Hospital, Sichuan University, and Collaborative Innovation Center of Biotherapy, Chengdu 610041, China.
2. Anti-Stress and Health Research Center, College of Pharmacy, Jinan University, Guangzhou, Guangdong 510632, China.
Received 2020-3-12; Accepted 2020-6-8; Published 2020-7-9
Abstract

Sirtuin 3 (SIRT3) is one of the most prominent deacetylases that can regulate acetylation levels in mitochondria, which are essential for eukaryotic life and inextricably linked to the metabolism of multiple organs. Hitherto, SIRT3 has been substantiated to be involved in almost all aspects of mitochondrial metabolism and homeostasis, protecting mitochondria from a variety of damage. Accumulating evidence has recently documented that SIRT3 is associated with many types of human diseases, including age-related diseases, cancer, heart disease and metabolic diseases, indicating that SIRT3 can be a potential therapeutic target. Here we focus on summarizing the intricate mechanisms of SIRT3 in human diseases, and recent notable advances in the field of small-molecule activators or inhibitors targeting SIRT3 as well as their potential therapeutic applications for future drug discovery.
Keywords: SIRT3, Mitochondrial homeostasis, Age-related disease, Cancer, SIRT3 activator, SIRT3 inhibitor
Introduction
Sirtuins, a family of NAD+-dependent protein deacetylases, contain seven members (SIRT1-7) in mammals while bacteria and archaea possess only one or two members [1]. Sirtuins are divided into five subclasses based on the conserved catalytic core domain. Class I is comprised of SIRT1, 2, and 3, which exhibit robust deacetylase activity. SIRT4, a class II Sirtuin, functions predominantly as an ADP-ribosyltransferase in mitochondria. SIRT5 belongs to Class III, while SIRT6 and 7 are assigned to Class IV. The U class sirtuins have only been observed in bacteria. Although subcellular localization and functions are different, mammalian sirtuins have different divisions of labor from regulating genome stability and energy metabolism to responding to cellular stress, working together to regulate the fate of cells as well as participating in various disease processes (Table 1) [2-4]. Among them, SIRT3 localizes mainly to the mitochondrial matrix and plays an important role in regulating mitochondrial metabolism, including the tricarboxylic acid (TCA) cycle, the urea cycle, amino acid metabolism, fatty acid oxidation, ETC/oxidative phosphorylation (OXPHOS), ROS detoxification, mitochondrial dynamics and the mitochondrial unfolded protein response (UPR) [5, 6].
Classification, function and characteristics of mammal sirtuins
| Sirtuin | Class | Enzymatic activity | Subcellular localization | Function | Disease | Reference |
|---|---|---|---|---|---|---|
| SIRT1 | I | Deacetylase | Cytoplasm & Nuclear | a. Metabolism regulation b. Regulation of chromatin and transcription c. DNA repair d. Inflammation suppression | a. Aging b. Neurodegeneration c. Metabolic disease d. Cardiovascular disease e. Cancer | [1, 7] |
| SIRT2 | I | Deacetylase | Cytoplasmic & Nuclear | a. Cell differentiation b. Metabolism regulation c. Cell cycle regulation d. Microtubule dynamics e. Inflammation regulation | a. Cancer b Neurodegeneration | [1, 7, 8] |
| SIRT3 | I | Deacetylase | Mitochondria & Nuclear | a. Metabolism regulation b. Inflammation suppression c. Inhibition of oxidative stress d. Apoptosis regulation e. Autophagy regulation | a. Aging b. Neurodegeneration c. Metabolic disease d. Cardiovascular disease e. Cancer | [1, 7] |
| SIRT4 | II | a. ADP-ribosyltransferase b. Lipoamidase c. Deacetylase | Mitochondria | Metabolism regulation | a. Metabolic disease b. Cancer c. Neurodegeneration | [1, 7] |
| SIRT5 | III | a. Deacetylase b. Desuccinylase c. Demalonylase d. Deglutarylase | Mitochondria | a. Metabolism regulation b. Immune response modulation | a. Metabolic disease b. Cancer | [1, 7, 9] |
| SIRT6 | IV | a. ADP-ribosyltransferase b. Deacylase c. Deacetylase | Nuclear | a. Chromatin and DNA repair b. Metabolism regulation | a. Aging b. Cancer c. Metabolic disease d. Cardiovascular disease | [1, 4, 7, 10] |
| SIRT7 | IV | Deacetylase | Nuclear | a. Transcription regulation b. Chromatin remodeling c. Metabolism regulation | a. Cancer b. Metabolic disease c. Cardiovascular disease | [1, 7, 11] |
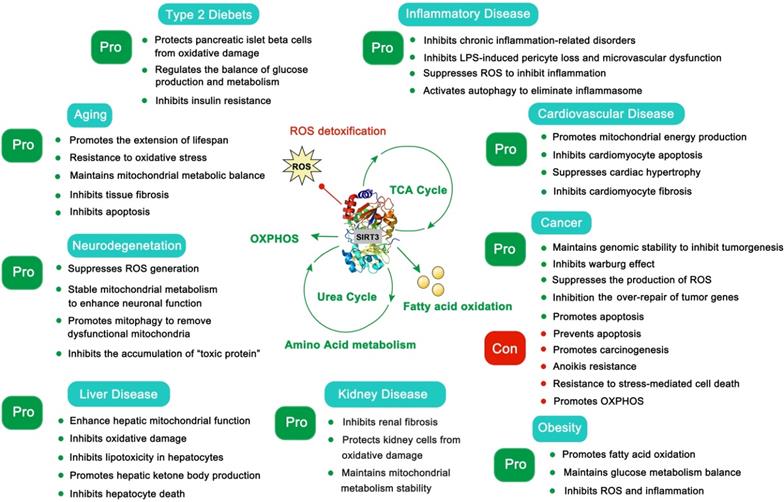
The SIRT3 protein is widely expressed in mitochondria-rich tissues, such as kidney, heart, brain and liver tissue [12]. The acetylation modifications that are regulated by SIRT3 are essential for maintaining mitochondrial function in these tissues. Given the crucial role of mitochondria in energy generation, metabolism, apoptosis and intracellular signaling [13], these highly metabolic tissues are more sensitive to mitochondrial dysfunction. Not surprisingly, SIRT3 has been verified to regulate aging, neurodegeneration, liver disease, kidney disease, heart disease and other metabolic diseases (Figure 1) [13]. Moreover, the dual role SIRT3 plays in cancer development is intriguing [14]. Therefore, SIRT3 has been proposed to be a promising therapeutic target for multiple human diseases, and a series of small-molecule compounds targeting SIRT3 have displayed favorable therapeutic effects [15, 16]. In this review, we focus on summarizing the intricate mechanisms of SIRT3 in human diseases, the development of small molecule compounds targeting SIRT3 as well as their potential applications, and the potential superiorities and shortcomings regarding future drug discoveries with SIRT3 as a potential druggable target.
The pros and cons of SIRT3 in type 2 diabetes, aging, neurodegeneration, liver disease, inflammatory disease, cardiovascular disease, cancer, kidney disease and obesity.
Structure and function of SIRT3
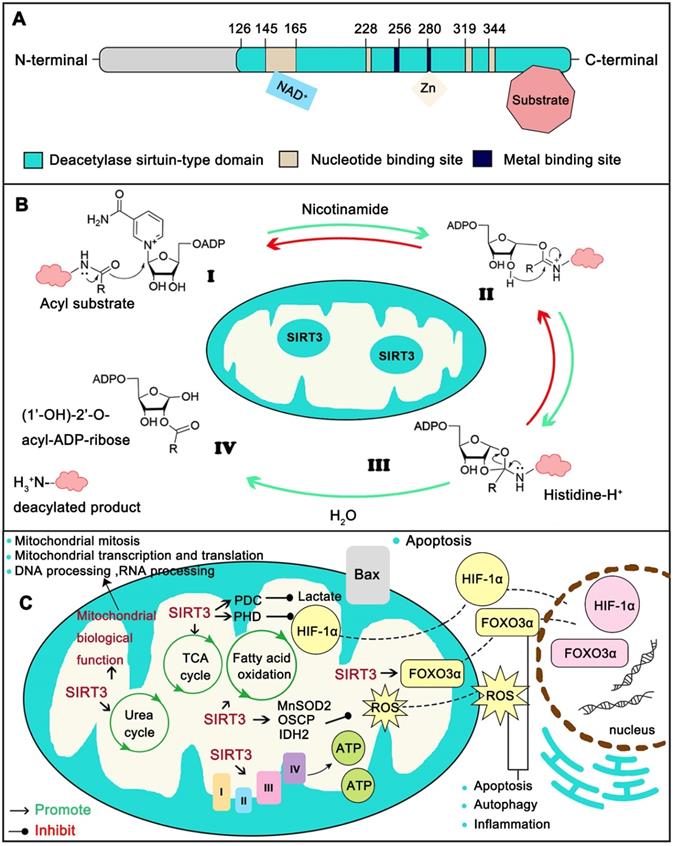
As a typical sirtuin, SIRT3 has a conserved enzymatic core (aa126-399) undertaking the deacetylation function and acts in an NAD+ dependent manner. The core region contains a large Rossmann fold domain for NAD+ binding and a smaller domain comprising of a helical bundle and a zinc-binding motif which is formed by two loops extending from the large domain. The remainder of the enzymatic core is composed of the binding sites for SIRT3 substrates (Figure 2A) [14]. Strictly speaking, the NAD+-dependent SIRT3 deacetylation reaction process includes four steps. First, the acetylated substrate and the NAD+ co-substrate bind in a cleft between the Rossmann-fold and zinc-binding domains, thereby inducing closure of the active site and stabilization of the NAD+ binding loop. Then the nicotinamide moiety of NAD+ is induced and buried in the nearby highly conserved hydrophobic pocket in a productive conformation. Subsequently, this conformation orients the α-face of NAD+ toward the acyl substrate, exposing the C1 atom of the ribose ring for direct nucleophilic attack from the carbonyl oxygen of acyl substrate. The acetyl group consequently transfers from the substrate to the ADP-ribose moiety of NAD+ coupled with cleavage of the nicotinamide from NAD+. Next, the C1'-O-alkylamidate intermediate converts to the bicyclic intermediate with the aid of the conserved His 224, which induces a nucleophilic attack of the 2'-OH group of the ribose onto the iminium carbon of the O-alkylamidate intermediate. Finally, the bicyclic intermediate is disrupted by an activated water molecule to produce the deacetylated protein and 2'-O-acetyl-ADP-ribose (Figure 2B). SIRT3 not only removes acetyl groups, but also crotonyl and myristoyl groups. However lysine acetylation is the most extensively studied modification [17]. Mitochondrial proteins are relatively highly acetylated (nearly 20%) [18] and acetylation modification is involved in all aspects of mitochondria function. Therefore, acetylation modification is pivotal to mitochondrial destiny. In a recent report, the most important mitochondria sirtuin, SIRT3, can directly interact with at least 84 mitochondria proteins. These proteins are involved in all aspects of mitochondria biological function, including mitochondrial mitosis (CHCHD3, IMMT), transcription (TFAM, MTIF2, etc.), translation (MRPL11, MRPS34, etc.), DNA processing (POLB, POLDIP2,etc.), RNA processing (PNPT1, RNMTL1, etc.), lipid metabolism (ACADM, AGK , etc.), ETC/OXPHOS (NDUFA5, ATP5B and etc.), the TCA cycle (OGDH, DLST and etc.), as well as amino acid metabolism (GLUD1, OAT, etc.) [19]. Together, SIRT3 supports the maintenance of mitochondrial homeostasis by regulating the acetylation levels of its substrates (Figure 2C).
Structure and function of SIRT3. (A) The conserved enzymatic core of SIRT3 contains a NAD+ binding domain, a zinc binding motif and the binding sites of SIRT3 substrates. (B) The modification by SIRT3 is deacetylate its substrate with a NAD+ dependent manner. (C) Typical SIRT3 regulated biological function. SIRT3 assists mitochondria to maintain metabolic stability including the homeostasis of TCA cycle, Urea cycle, Amino acid metabolism, Fatty acid oxidation, ETC/OXPHOS, ROS detoxification and mitochondrial dynamics. Moreover, SIRT3 is closely related with oxidative stress, apoptosis, autophagy and inflammation.
Endogenous regulators of SIRT3
SIRT3 is important in mitochondria, and therefore it is regulated in various ways. There are several studies on endogenous regulators of its expression, as well as transcriptional and post-translational modifications on this protein (Table 2). SIRT3 is a sensor of mitochondrial energy, thus levels of the metabolic co-factor (NAD+) and the byproduct nicotinamide are direct regulators of its activity. As the co-factor of SIRT3, NAD+ promotes the deacetylation process [20] while nicotinamide inhibits this process through accelerating the reverse reaction by binding to the reaction product [17, 21]. In addition, caloric restriction (CR) is another crucial factor that can obviously stimulate the expression of SIRT3, which is also a biological self-protection strategy. In response to CR, the up-regulated SIRT3 activates mitochondrial isocitrate dehydrogenase 2 (IDH2) by deacetylation, thus increasing the NADPH level to reduce oxidative damage and enhance the mitochondrial antioxidant defense system [20]. SIRT3 is transcribed in the nucleus and full-length SIRT3 (FLSIRT3) has no enzyme activity. FLSIRT3 is imported into the mitochondria, then its NH2 terminus is proteolytically cleaved in the mitochondrial matrix to become the active form, SIRT3 (an approximately 28kDa product). During this process, matrix processing peptidase (MPP) acts as “scissors” to accomplish this post-translational modification [22]. SUMOylation is another post-translational modification on SIRT3, which further inhibits its activity. SUMO specific protease SENP1 can de-SUMOylate and activate SIRT3 to promote mitochondria metabolism [23]. SIRT3 can also be regulated by covalent modification. 4-Hydroxynonenal (4-HNE) is an endogenous product of lipid peroxidation which inhibits SIRT3 activity by occupying its zinc-binding residue (Cys 280) [24]. In addition, transcriptional modification is another major regulation of SIRT3. NF-κB, which is a pleiotropic transcription factor, was recently identified to bind to the SIRT3 promoter to enhance its expression [25]. Peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1 (PGC-1α) could bind to the SIRT3 promoter to stimulate SIRT3 transcription [26, 27], while SNAI1 and Zinc finger E-box-binding homeobox 1 (ZEB1) negatively regulates the SIRT3 promoter activity to inhibit its expression [28, 29]. Of note, microRNAs (miRNAs) are mainly post-transcriptional regulators that affect mRNA stability and protein levels which can also regulate SIRT3 activity. As a class of non-coding RNA molecules, miRNAs bind to complementary target mRNAs, resulting in mRNA translational inhibition or degradation [30]. Studies have proven that miR-195 [31], miR-421 [32], miR-494-3p [33], miR-708-5p [34], miR-31 [35], miR-145 [36], miR-298 [37] could directly target the 3'UTR of SIRT3, thus inhibiting its gene expression and protein levels. In addition, miR-210 targets and represses the iron-sulfur cluster assembly protein (ISCU), which changes the NAD+/NADH ratio, indirectly influencing SIRT3 [38]. Long non-coding RNA (LncRNAs) play a similar role to miRNAs as well. LncRNA TUG1 suppresses the mRNA expression of miR-145, which can further positively regulate SIRT3 [36], while LncRNA DYNLRB2-2 inhibits miR-298 to activate the transcription of SIRT3 [37]. The protein-protein network is another primary method of regulating SIRT3 activity. Profilin1, an actin-associated protein, can interact with SIRT3 and promote its expression [39]. β-catenin, a key downstream effector in the Wnt signaling pathway, can suppress SIRT3 promoter activity thus inhibiting its expression [40]. As mentioned above, these endogenous regulators modulate the expression and activities of SIRT3 in response to cellular stresses, thus maintaining homeostasis.
Endogenous direct regulators of SIRT3
| Name | Classification | Regulatory Mechanism | References |
|---|---|---|---|
| NAD+ | cofactor | Promotes the deacetylation process of SIRT3 | [20] |
| Nicotinamide | Deacetylation product | Nicotinamide inhibits SIRT3 through rebinding of the reaction product to the enzyme accelerates the reverse reaction | [17, 21] |
| Caloric Restriction | - | Increases SIRT3 expression and activity | [20] |
| MPP | Peptidase | Proteolytic processing of FLSIRT3 to active SIRT3 | [22] |
| SENP1 | SUMOspecific protease | SENP1 can de-SUMOylates and activates SIRT3 | [23] |
| 4-Hydroxynonenal | Endogenous product | 4-Hydroxynonenal inhibits SIRT3 activity by occupy its zinc-binding residue Cys(280). | [24] |
| NF-κB | Transcription factor | NF-κB binds to the SIRT3 promoter to enhance its expression | [25] |
| PGC-1α | Transcriptional coactivator | PGC-1α bounds to the SIRT3 promoter as its transcription factor to regulate SIRT3 expression | [26, 27] |
| SNAI1 | Transcriptional repressor | SNAI1 inhibits SIRT3 promoter activity | [28] |
| ZEB1 | Transcriptional repressor | ZEB1 inhibits SIRT3 promoter activity. | [29] |
| miR-195 | MicroRNA | miR-195 down-regulates SIRT3 expression through direct 3'-untranslated region targeting | [31] |
| miR-421 | MicroRNA | miR-421 targets the 3'UTR of SIRT3 and decreases SIRT3 protein level | [32] |
| miR-494-3p | MicroRNA | miR-494-3p targets the 3'UTR of SIRT3 and inhibits SIRT3 expression at mRNA and protein levels | [33] |
| miR -708-5p | MicroRNA | MiRNA-708-5p targets the 3'UTR of SIRT3 and decreases SIRT3 protein level | [34] |
| miR-31 | MicroRNA | miR-31 directly targets SIRT3 to repress its expression | [35] |
| miR-145 | MicroRNA | miR-31 directly targets SIRT3 to reduce its expression | [36] |
| miR-298 | MicroRNA | miR-298 directly targets SIRT3 to inhibit its expression | [37] |
| miR-210 | MicroRNA | miR-210 targets and represses ISCU to change the NAD+/NADH ratio thus indirectly negative regulate SIRT3 | [38] |
| TUG1 | Long non-coding RNA | TUG1 negatively regulates the expression of miR-145 thus indirectly positively regulate SIRT3 | [36] |
| DYNLRB2-2 | Long non-coding RNA | DYNLRB2-2 suppresses the mRNA expression of miR-298 thus indirectly activate SIRT3 | [37] |
| Profilin-1 | Protein | Profilin-1 interacts with SIRT3 and promotes its expression | [41] |
| β-catenin | Protein | β-catenin suppresses SIRT3 promotor activity to negative regulate its expression | [40] |
SIRT3 and human disease
“Small” modification, “big” function
The biological function of SIRT3, which is just deacetylation of its substrate, seems to be relatively simple and limited, but it does participate in numerous biological processes and the development of various diseases [41, 42]. The most fascinating role of SIRT3 is in longevity. It was first reported in 2003 that SIRT3 is positively associated with longevity [43], and much subsequent work has confirmed and expanded this result [44-46]. More delightfully, studies have proven that SIRT3 protects human heart, brain, muscle, liver, kidney and other tissues from dysfunction and disease [46]. SIRT3 is also involved in the overall progression of cancer. It may function differently in various conditions, but it mostly inhibits the development of cancer [14]. Here, we will discuss how SIRT3 fights against human diseases by regulating specific substrate proteins and their corresponding signaling pathways.
SIRT3 in age-related disease
Age-related disease usually occurs with high frequency after a certain point, and these diseases are always accompanied by the gradual decline of function in various organs. In this section, we mainly focus on aging, Alzheimer's disease (AD) [47], Parkinson's disease (PD) [48], Huntington's disease (HD) [49], amyotrophic lateral sclerosis (ALS) [50] and age-related hearing loss (Figure 3). Aging is a complex and irreversible process. Although the pathological features of aging vary considerably, they all bear the common characteristic that the smallest components of lesions, such as neuronal or muscle cells, have decreased or imbalanced mitochondrial function and increased oxidative damage [51]. Under normal circumstances, SIRT3 is highly expressed in the brain and nervous system, but decreased SIRT3 expression is always observed in age-related diseases and this age-dependent defect of SIRT3 is a major risk factor of pathogenesis [52-54].
SIRT3 in age-related disease. Age-related diseases are always accompanied with a decline in mitochondrial function, high oxidative stress and accumulation of toxic proteins. SIRT3 activates a range of substrates by deacetylation to promote mitochondrial function, enhance ATP production, accelerate ROS clearance, and maintain mitochondrial metabolic homeostasis. In addition, SIRT3 can activate mitophagy to accelerate mitochondrial renewal. It is worth noting that SIRT3 also inhibits the production of misfolded proteins and accelerates their clearance. The pink proteins represent the substrates of SIRT3. Gray circles represent acetylation modifications.
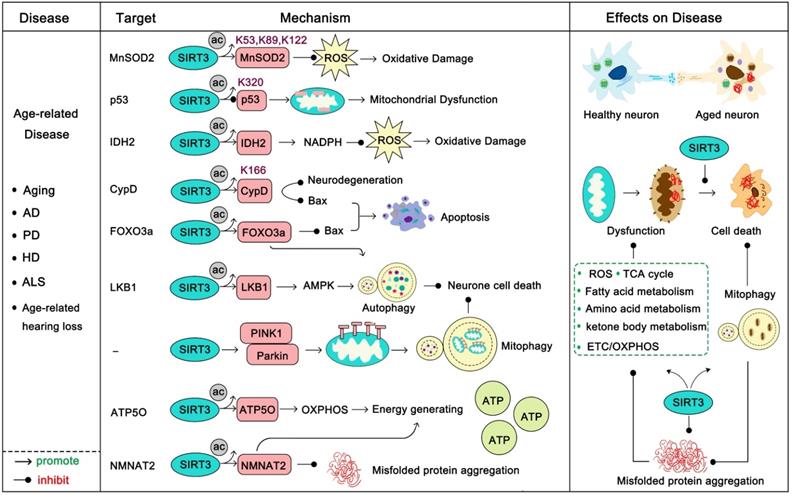
Another well-studied role of SIRT3 in age-related disease is that it can act to resist apoptosis. Progressive loss and death of neuronal cells is a feature of these diseases, and apoptosis plays an important role in this process. P53, the most famous tumor suppressor gene, is highly acetylated in neurodegenerative diseases. Ac-p53K320 targets mitochondria and directly reduces its function, inducing mitochondrial dysfunction which leads to mitochondrial-dependent apoptosis. SIRT3 deacetylates p53 to prevent its localization-induced releasing of cytochrome c from the mitochondria to the cytoplasm [53]. Moreover, SIRT3 activates FOXO3α, a transcription factor of the FOXO family which is involved in metabolism-regulation and stress response, to inhibit Bax-regulated apoptosis [57]. SIRT3 can deacetylate CypD, a regulatory component of the mitochondrial permeability transition pore (mPTP), to inhibit the mitochondrial permeability transition to prevent apoptosis and mitochondrial dysfunction [58]. In response to age-dependent mitochondrial capacity reduction, SIRT3 binds to ATP5O, a mitochondrial ATP synthase, strengthening the ETC/OXPHOS process to enhance cellular ATP production [19]. Recently, studies have shown that the activation of SIRT3 and the inhibition of apoptosis play an important role in the improvement of age-related diseases. For example, celastrol can ameliorate age-related macular degeneration via SIRT3 activation and apoptosis inhibition [59]. Melatonin decelerated age-induced ovarian aging via activating SIRT3-regulated mitochondrial function enhancement together with apoptosis inhibition [60]. Not surprisingly, daily melatonin supplementation protects vascular endothelium from aging via SIRT3-regulated oxidative stress suppression and apoptosis inhibition [61]. Accordingly, up-regulation of SIRT3 plus apoptosis inhibitors might provide new directions for age-related disease treatment. SIRT3 can activate autophagy, a notable cellular self-protective mechanism. SIRT3 can deacetylate LKB1 to activate the AMPK-mTOR autophagy pathway [62]. More interesting, deacetylated FOXO3α can activate expression of various autophagy genes (ULK1, ATG5, ATG7, et al) to protect cells from apoptosis [63]. Mitophagy is indispensable for maintaining mitochondrial homeostasis and is a specific form of autophagy that selectively removes dysfunctional mitochondria. When an organ is unable to repair dysfunctional mitochondria, mitophagy helps to provide for metabolic needs and contributes to mitochondrial renewal [64]. SIRT3 induces the initiation and activation of the PINK1-Parkin mitophagy pathway to inhibit cell death [65]. Induction of autophagy is a new hotspot in the treatment of neurodegenerative diseases [66, 67]. Therefore, SIRT3 activation along with autophagy activators (like ULK1 activators [68, 69], rapamycin [70]) will be a new candidate method for treatment of these diseases. Of note, another major pathogenesis of age-related disease is the aggregation of pathological proteins (such as Aβ, tau in AD, and α-synuclein in PD) [47-50]. Recently, nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2), a substrate of SIRT3, was found to be a neuron protector by interacting with heat shock protein 90 (HSP90) to refold aggregated protein substrates [71]. SIRT3 can interact with NMNAT2 and activate it to improve mitochondrial function and inhibit apoptosis [72]. More interestingly, NMNAT2 is a neuron-specific and effective protector which inhibits axonal degeneration and dysfunction, and it is very likely that SIRT3 can promote this function [73]. It is noteworthy that SIRT3-induced autophagy might also be an effective method to eliminate these pathological proteins, thus SIRT3 activation together with “toxic protein” scavenging drugs might be beneficial. Taken together, SIRT3 is the nemesis of age-related diseases and it fights against almost all the aspects of their development. Properly targeting SIRT3 will be promising in these diseases.
SIRT3 in cancer
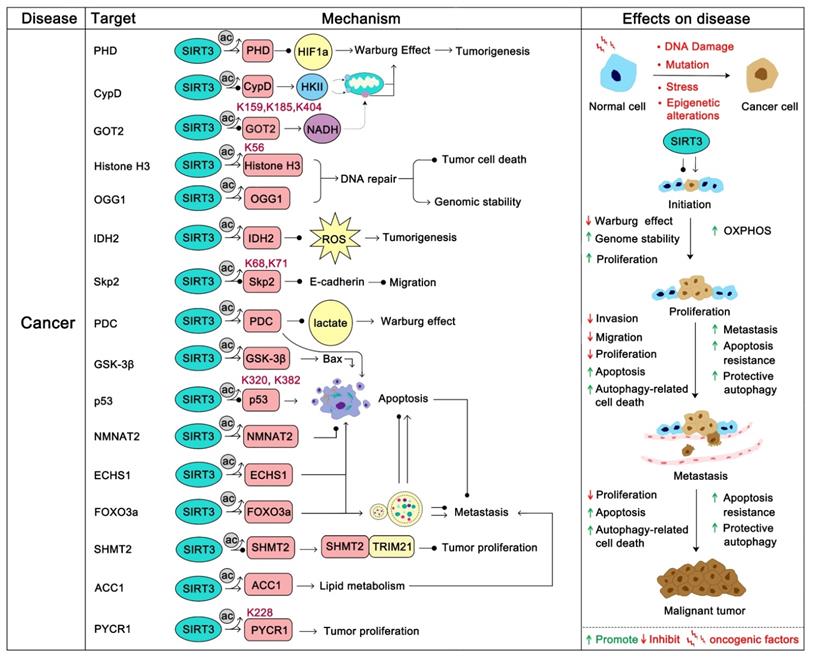
Cancer cells exhibit metabolic patterns that are distinct from normal cells. The feature of mitochondria metabolic reprogramming is the “Warburg effect” [74], which is that most cancer cells are more dependent on aerobic glycolysis than oxidative phosphorylation, regardless of the fact that glycolysis produces ATP inefficiently. Glycolysis not only provides a rapid energy supply, but also provides a number of favorable factors for the occurrence and development of tumor microenvironments, such as accelerating the instability of genome and the positive response to multiple cell proliferation signals (PI3K/AKT, c-Myc, etc.) [75, 76]. Of course, in some specific cancers or specific circumstances (such as brain cancer and acute myeloid leukemia), OXPHOS is still their more suitable means of energy supply, and the survival of these cancers is more dependent on the continued activity of OXPHOS [77-79]. The tumor microenvironment of different cancers varies considerably, and the pressure of survival makes them evolve to choose the most suitable metabolic pathway. In short, SIRT3 promotes oxidative phosphorylation and inhibits glycolysis in the overall regulation of mitochondrial metabolism [80, 81]. Intriguingly, a recent study found that SIRT3 changes to an oncogene to promote HFD-induced tumorigenesis in mice [82]. Therefore, the role of SIRT3 in cancer appears context-dependent [83]. In most cases, it is carcinogenic in some cancers that are addicted to OXPHOS [77]. However, it is also a tumor suppressor in glycolysis-dependent cancers [80, 81, 84]. Since the role of SIRT3 in cancer has been widely reported and is well summarized, in this part we mainly focus on how SIRT3 regulates its substrates to play a double-sided role in cancer.
Tumor suppressor role of SIRT3
The most important tumor suppression role of SIRT3 is that it hinders cancer metabolism changes. The inhibition of hypoxia-inducible factor-1a (HIF1α) by SIRT3 is the most studied pathway. HIF1α is a key factor that activates a series of glycolysis genes which contribute to the “Warburg phenotype”. SIRT3 can destabilize HIF1α to prevent its deleterious role of “Warburg effect” promotion. Interestingly, HIF1α is not the direct substrate of SIRT3. SIRT3 regulates the activity of HIF1α by direct deacetylation of prolyl hydroxylase (PHD). Activated PHDs then hydroxylate HIF1α to affect its stability and decrease its tumor promoting effect [81]. The pyruvate dehydrogenase complex (PDCs) is another SIRT3 substrate related to glycolysis. In the process of hypoxia and tumor growth, lysine acetylation of PDC plays an important role in promoting glycolysis and subsequent cancer cell proliferation. SIRT3 is the upstream deacetylase of PDC that deacetylates and activates PDC to inhibit glycolysis and promote apoptosis in cancer cells [85]. In addition, SIRT3 could stabilize p53 to inhibit glycolysis in wt-p53 cancer cells. During this process, full-length SIRT3 can interact with PTEN in the nucleus to increase PTEN activity thus inhibiting MDM2 transcription, which is responsible for p53 degradation [86]. The stabilization of p53 further impedes glycolysis via inhibition of the transcription of numerous key glycolytic enzymes [87]. Moreover, SIRT3 can inhibit breast carcinoma glycolysis through deacetylation and inactivation of cyclophilin D. This further inhibits the binding of the lactate metabolism enzyme hexokinase II (HK II) to mitochondria to obstruct glycolysis [88]. Glutamate oxaloacetate transaminase 2 (GOT2), which is a limiting enzyme in regulating glycolysis processing, is deacetylated at Lys 159, 185, and 404 by SIRT3 thus inhibiting GOT2 activity and hindering pancreatic tumor growth [89]. Accordingly, in most glycolysis-addiction cancers, SIRT3 activation will be beneficial and SIRT3 activators might be candidate adjuvants. In addition, SIRT3 directly or indirectly inhibits ROS-regulated tumorigenesis and metastasis [90]. Of which, SIRT3 represses ROS dependent Src/FAK signaling and promotes the proliferation, migration and metastasis of cancer cells [91]. ROS elimination by SIRT3 was also proven to contribute to the growth inhibition of lung adenocarcinoma cells [92]. In chronic lymphocytic leukemia (CLL), SIRT3 activates MnSOD2 to eliminate ROS, thus inhibiting CLL progression [93]. Additionally, programmed cell death induction is another strategy SIRT3 uses to inhibit tumor progression. For instance, SIRT3 can activate glycogen synthase kinase-3β (GSK-3β) to promote the Bax-regulated apoptosis pathway [94]. SIRT3 up-regulation of MnSOD2 and p53 activity further induced Bax- and Fas-regulated apoptosis in HCC [95]. Moreover, SIRT3 plays a role in monitoring the stability of tumor genomes. Histone H3, which participates in the reaction to DNA damage, can be deacetylated by SIRT3 at K56 to enhance DNA nonhomologous end joining repair [96]. 8-oxoguanine-DNA glycosylase 1 (OGG1) is a DNA repair enzyme that is important in the inhibition of genome damage. SIRT3 binds and blocks the degradation of OGG1 to inhibit tumorigenesis [97].
Of note, SIRT3 can also inhibit cancer progression by deacetylation of its substrate to modulate proliferation and migration. The F-box protein S-phase kinase associated protein 2 (Skp2) is a proto-oncogene which inhibits the activity of numerous tumor suppressor proteins (such as p21, p27 and E-cadherin) via ubiquitination and destruction to positively regulate cell cycle progression and migration, and negatively regulate apoptosis [98, 99]. Acetylation of Skp2 by histone acetyltransferase p300 at Lys 68 and 71 will increase its stability and cytoplasmic localization, which promotes the degradation of E-cadherin. SIRT3 can directly deacetylate Skp2 to prevent this process [100]. Similarly, Enoyl-CoA hydratase-1 (ECHS1) is also highly acetylated in cancer cells. Acetylation blocks the activity of ECHS1 and activates the mTOR-regulated proliferation pathway. SIRT3 inhibits this hyperacetylation to restore mitochondrial translocation and ECHS1 activity [101]. Hyperacetylation of Glutamate oxaloacetate transaminases (GOT) promotes tumor growth while SIRT3 reverses this process [89]. It has been shown in recent studies that in some highly malignant tumors, the activation of SIRT3 might be a possible treatment method, especially for some drug-resistant cancers. For example, sorafenib is a well-known drug approved for clinical use in hepatocellular carcinoma (HCC), but is very prone to drug resistance, which makes its treatment of liver cancer less satisfactory. Interestingly, studies revealed that sorafenib could decrease the expression of SIRT3, which contributes to its reduced drug sensitivity, while up-regulation of SIRT3 can re-sensitize HCC to sorafenib treatment [102-104]. In addition, SIRT3 activation by ABT737 contributes to ameliorating cisplatin resistance in ovarian cancer [105].
Oncogenic role of SIRT3
The tumor promoting effect of SIRT3 has also been well studied, especially in some hematological malignancies that exhibit oxidative phosphorylation addiction [77, 106]. IDH2 is a key enzyme acting in the forward Krebs cycle and was identified as a hallmark of hematological malignancies. SIRT3 can deacetylate IDH2 to increase its activity in promoting carcinogenesis [107]. In diffuse large B cell lymphomas (DLBCLs), SIRT3 was proven to accelerate TCA cycle metabolism via enhancing GDH activity to promote lymphomagenesis [108]. Not surprisingly, SIRT3 has also been found to promote tumor progression in some other cancers. In non-small cell lung cancer, SIRT3 promotes the oncogenic role of NMNAT2 to stimulate cancer cell proliferation [72]. Similarly, SIRT3 deacetylates p53 at Lys 320 and 382 to promote its degradation, thus hindering its tumor inhibition role in PTEN-deficient non-small cell lung cancer [109]. Therefore, in PTEN-deficient tumors, SIRT3 inhibition might be a better treatment strategy. Recently, another study demonstrated that SIRT3 could promote colorectal carcinogenesis. SIRT3 deacetylates serine hydroxymethyltransferase 2 (SHMT2) at Lys 95 and inhibits its lysosome-dependent degradation. Acetylated SHMT2 exhibits deficient enzymatic activity, which inhibits carcinogenesis, while SIRT3 activates SHMT2 to promote colorectal cancer cell proliferation [39]. In addition, SIRT3 can increase SOD2 activity to properly regulate ROS production to prevent apoptosis [110]. In ovarian cancer cells, SIRT3 fine-tunes SOD2 activity to adapt to cellular stress and anoikis resistance in order to ensure cell survival [111]. Interestingly, the activation of SOD2 mediated by SIRT3 can promote epithelial-mesenchymal transition (EMT) in triple negative breast cancer (TNBC) cells [25]. In cervical cancer cells, SIRT3 deacetylates Acetyl-CoA carboxylase (ACC1) to promote lipid metabolism. This fatty acid metabolism reprogramming promotes cancer migration and invasion [112]. Pyrroline-5-carboxylate reductase 1 (PYCR1) is another substrate of SIRT3. Acetylation at Lys 228 suppresses tumor proliferation, while deacetylation by SIRT3 promotes breast cancer cell and lung cancer cell survival [113]. Accordingly, we should be thoughtful in trying to regulate SIRT3 for cancer therapy.
Overall, SIRT3 is capable of metabolic reprogramming and contributes greatly in the fate of cancers (Figure 4). Highly acetylated modifications frequently occur in cancer, which is conducive to the survival of most tumors. SIRT3 regulates tumor progression by changing this excessive modification back to a normal condition. The role of SIRT3 in cancer is a double-edged sword which to some extent increases the confusion and risk of SIRT3 as a target for cancer treatment. More regrettably, there are no satisfactory SIRT3 activators or inhibitors that have been developed successfully for cancer therapy, which makes recognition of SIRT3 as a druggable target in cancer more difficult and questionable. Even so, we believe it is a promising drug target in cancer if we can intelligently regulate it in personalized therapy.
SIRT3 in cancer. SIRT3 plays a two-sided role in cancer. In most cancers, SIRT3 plays a tumor suppressor role. On the one hand, SIRT3 can maintain the stability of the cancer genome and inhibit carcinogenesis. On the other hand, SIRT3 inhibits the Warburg effect of cancer to inhibit the development of tumors. In addition, SIRT3 can inhibit tumor proliferation and metastasis. SIRT3 induced apoptosis and autophagy also involved in this progress. However, in some colorectal cancers and lung cancers, SIRT3 plays an oncogenic role by promoting proliferation and metastasis via deacetylation of specific substrates. The pink protein represents the substrate for SIRT3. Gray circles represent acetylation modifications.
SIRT3 in heart disease
Heart disease is one of the most common diseases and one of the biggest killers in the field of human health [97]. The role of the heart is to encourage blood flow, provide sufficient blood to organs and tissues to supply oxygen and various nutrients, and remove metabolic products to maintain metabolism and homeostasis. The normal work of the heart requires huge amounts of energy production and consumption, which is mainly provided by the mitochondria [114]. The normal function of mitochondria as energy-producing engines is the basis of the normal function of the heart. Conversely, the dysfunction of mitochondria directly or indirectly contributes to the progression of a series of heart diseases, among which are heart failure, cardiac hypertrophy, atherosclerosis, and dilated cardiomyopathy [115, 116]. As an eminent guardian of mitochondrial homeostasis, SIRT3 also plays an irreplaceable role in heart disease. Loss of SIRT3 impairs the contractile function of the heart [117], and in a later study SIRT3 was found to be down-regulated in heart failure. In this process, miR-195 targets SIRT3 to inhibit its expression thus disturbing oxidative phosphorylation, resulting in the myocardial energy metabolism imbalance [31]. Another study reported that rarefaction of cardiac microvessels and functional hypoxia are more likely to occur in SIRT3-KO mice than WT mice. And SIRT3-KO mice exhibit distinct mitochondrial dysfunction and enhanced cardiac fibrosis marker expression [118]. Moreover, SIRT3-KO mice exhibited diastolic dysfunction and decreased angiogenesis [119]. Remarkably, SIRT3 deletion mice are more susceptible to a high-fat diet and exhibit higher ROS production and lower cardiac function [120]. In addition, SIRT3 can inhibit arterial thrombosis by suppressing neutrophil extracellular traps and decreasing plasma tissue factor activity [121]. More interestingly, a study found that acetylation of mitochondrial proteins mostly occurs nonenzymatically and SIRT3 just modifies the acetylation back to the normal level. A more prominent role of SIRT3 is to remove acetylation lesions on its substrates, such as at Lys 127 on Glycine N-acyltransferase (GLYAT) and at Lys 48 on hydroxymethylglutaryl-CoA lyase (HMGCL), to protect metabolic fidelity [122]. In this section we will mainly discuss how SIRT3 regulates its substrates to carry out its heart protector role (Figure 5).
SIRT3 in heart disease. Heart disease often manifests as dysfunction of cardiomyocytes, such as local hypoxia, death of cardiomyocytes, fibrosis, and the like. Dysfunction of cardiomyocytes ultimately leads to myocardial ischemia, cardiac hypertrophy, heart failure. SIRT3 can increase the mitochondrial function of cardiomyocytes and increase energy production by deacetylating its substrate. In addition, SIRT3 can deacetylate its substrates to inhibit AKT-mTOR/ERK1/2/TGF-β-smad3-induced myocardial fibrosis. During this process, SIRT3 can also activate GSK-3β to contribute to myocardial fibrosis inhibition. SIRT3 also directly inhibits cardiomyocytes apoptosis. Last but not the least, SIRT3 can eliminate ROS and activate mitophagy to inhibit cardiac remodeling. The pink protein represents the substrate for SIRT3. Gray circles represent acetylation modifications.
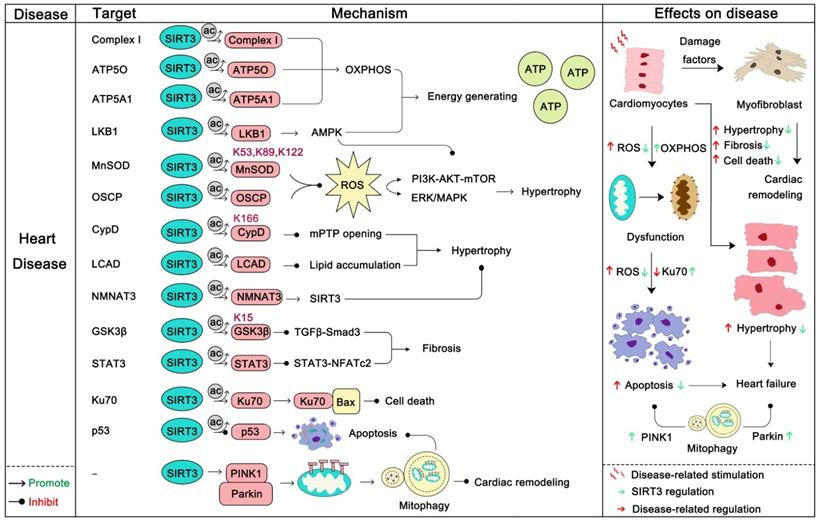
The first protective effect of SIRT3 on the heart is to increase the energy production of mitochondria. SIRT3 can activate mitochondrial complex I and enhance the ETC function to maintain ATP homeostasis [123]. ATP5O and ATP5A1, two mitochondrial ATP synthases, can be deacetylated by SIRT3 to enhance ATP production [19, 124]. In addition, SIRT3 actives the LKB1-AMPK pathway to generate ATP [125]. Optic atrophy 1 (OPA1), another target of SIRT3, can be triggered by SIRT3 to improve cardiac mitochondrial bioenergetics [126]. In addition, SIRT3 can regulate a series of substrates to block cardiac hypertrophy. Cardiac hypertrophy is a stress adaptive response to a range of heart conditions, but it also increases mortality and the risk of heart disease. Thus, inhibiting hypertrophy may be beneficial to the heart. Of note, Rho/Rho kinase signaling, mTOR signaling, ROS signaling are three key villains of cardiac hypertrophy [127]. Of note, the LKB1-AMPK pathway is the upstream negative regulator of mTOR signaling, and SIRT3 can active LKB1-AMPK signaling to inhibit mTOR regulated protein synthesis, which inhibits cardiac hypertrophy [125]. More interestingly, the SIRT3 substrate FOXO3α not only functions well in regulating apoptosis and autophagy, but also it regulates mTOR and Rho/Rho kinase signaling [128, 129]. SIRT3 can active the activity and nuclear translocation of FOXO3α, thereby inhibiting mTOR and Rho/Rho kinase signaling and preventing cardiac hypertrophy. To inhibit ROS regulated signaling, SIRT3 can directly deacetylate, activate MnSOD2 and oligomycin-sensitivity conferring protein (OSCP), inhibiting the synthesis and aggregation of ROS, thus preventing cardiac hypertrophy [16]. Cyclophilin D (CypD), which is an integral part of the mitochondrial permeability transition pore (mPTP), can be deacetylated by SIRT3 at lysine 166 to prevent the opening of mPTP, thus inhibiting stress-induced cardiac hypertrophy and apoptosis [58]. Hypertrophy-related lipid accumulation is another problem of cardiac hypertrophy, which will further lead to heart failure. SIRT3 could downregulate the acetylation of long-chain acyl CoA dehydrogenase (LCAD) to restore lipid metabolism homeostasis [130]. In addition, Nicotinamide mononucleotide adenylyltransferase 3 (NMNAT3), can also bind to SIRT3 and be deacetylated. Deacetylation of NMNAT3 enhances its enzyme activity and in-turn promotes the anti-hypertrophic effects of SIRT3 [131].
Cardiac fibrosis is another pathological manifestation of cardiac remodeling in heart disease. SIRT3 can ameliorate cardiac fibrosis through blocking the TGF-β/Smad3 pathway [132]. SIRT3 can deacetylate glycogen synthase kinase 3β (GSK3β) at residue Lys 15 to promote GSK3β activity. The activation of GSK3β thereby resists TGF-β/Smad3 regulated fibrotic genes expression [133]. Moreover, signal transducer and activator of transcription 3 (STAT3) can be deacetylated by SIRT3 to inhibit STAT3-NFATc2 regulated fibrosis [134]. Also the role of SIRT3 in eliminating ROS also helps the SIRT3/ROS/ERK1/2 cascade inhibit cardiac remodeling [135]. In addition, the role of SIRT3-induced autophagy and apoptosis inhibition in heart disease should not be ignored. Parkin-dependent mitosis induced by SIRT3 can clear damaged mitochondria and prevent the remodeling of hypertensive heart and the death of myocardial cells [65]. Ku70, another substrate of SIRT3, can be deacetylated and activated by SIRT3. After deacetylation, Ku70 interacts with Bax to inhibit the apoptosis of cardiomyocytes [136]. SIRT3 can protect cardiomyocytes by inhibiting apoptosis through deacetylating p53. As mentioned above, SIRT3 has a wide protective role in heart disease, and targeting SIRT3 can be a new strategy for the treatment of heart disease. It is gratifying that the SIRT3 activator honokiol has been proven to have cardioprotective effects [16, 137], which also provides evidence for SIRT3 as a druggable target for improving heart disease.
SIRT3 in metabolic disease
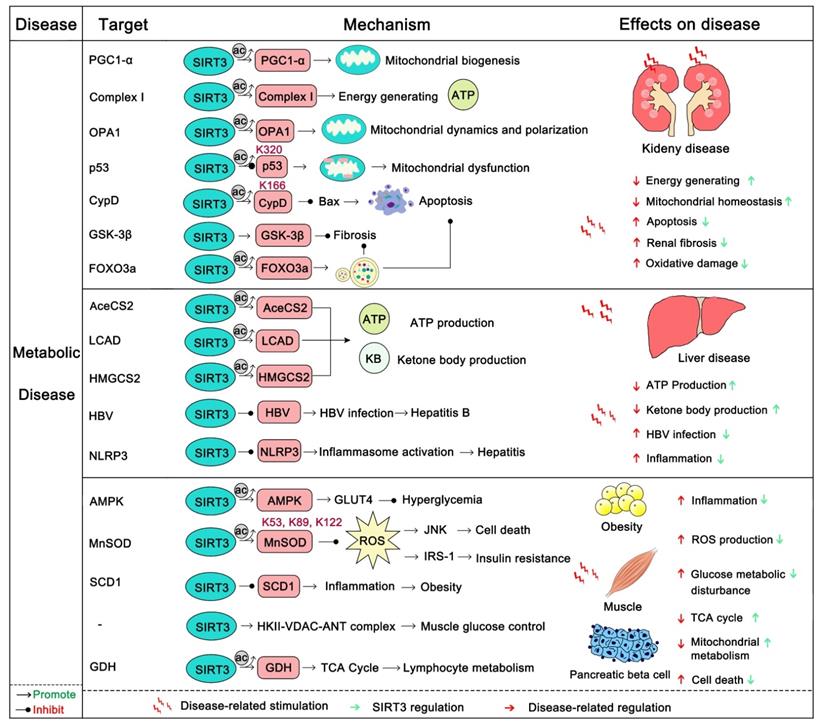
Metabolic diseases generally have a long course over which energy metabolism, glycometabolism, fatty acid metabolism, and amino acid metabolism gradually become out of balance. Eventually this leads to obesity, diabetes, liver disease, and kidney disease [138]. The kidney is a highly energy-consuming organ because it not only removes metabolites from the body by generating urine, but also maintains the balance between water and electrolytes. Additionally, it secretes some active substances, such as erythropoietin and active vitamin D3 to promote the growth and development of the body [139]. The kidney is rich in mitochondria in order to satisfy its energy needs, and the mitochondrion is a highly mobile organelle that can change its position and numbers as needed [140]. SIRT3, as a key regulator of mitochondrial dynamics, plays an important role in the energy supply of renal cells [141]. The overexpression of SIRT3 can improve kidney function, attenuate oxidative injury, and suppress the inflammatory damage and apoptosis of renal tubular epithelial cells [142]. The absence of SIRT3 expression will aggravate acute renal injury (AKI), and increase ROS levels and apoptosis. Activation of SIRT3 decreases the acetylation of CypD, thereby inhibiting mitochondrial damage of AKI, and thus protecting the kidneys [143, 144]. Activation of SIRT3 also inhibits the acetylation of p53 which blocks apoptosis in AKI [145]. In addition, SIRT3 deacetylates PGC1-α and mitochondrial complex I to enhance mitochondrial biogenesis and energy generating for resisting AKI [139]. Nephrolithiasis is a form of kidney metabolic disease and its main cause is damage to renal epithelial cells from calcium oxalate. A recent study found that nephrolithiasis-afflicted mice always exhibit a significant reduction in SIRT3 expression. SIRT3 could deacetylate FOXO3a thus activating it. After deacetylation, FOXO3a binds with the promoter of LC3 to induce autophagy and suppress renal tubular epithelial cell injury [146]. Additionally, the SIRT3 regulated NRF2/HO-1 pathway may also contribute to inhibiting the formation of kidney stones [147]. Furthermore, SIRT3 can inhibit renal fibrosis. Endothelial-to-mesenchymal transition (EndoMT) has emerged as an important contributor to renal fibrosis. Mice with SIRT3 loss more easily develop renal dysfunction with increased ROS production and EndoMT. Interestingly, SIRT3 can activate FOXO3a to inhibit this progress [148]. SIRT3 can also inhibit renal fibrosis by deacetylation and activation of GSK3 β to inhibit the expression of fibrosis genes [133]. Liraglutide can protect the kidney from diabetic nephropathy, while deletion of SIRT3 abrogated its kidney protection effect [31]. Optic atrophy 1 (OPA1), a regulator of mitochondrial fusion, can be up-regulated by SIRT3, which subsequently enhances the fusion of renal mitochondria and improve the production of kidney energy [142]. In short, SIRT3 can protect the kidney from metabolic disease.
Liver is the main metabolic organ of the human body. It governs the metabolism of glucose, protein, fat and carbohydrates. Also, it is also the largest detoxification organ in the human body. Abnormal liver metabolism will induce a series of hepatic metabolic diseases, of which fatty liver is the most common. SIRT3 is always down-regulated in patients with fatty liver [149-151]. SIRT3 overexpression can restore liver function, inhibit inflammation and apoptosis, and alleviate liver fibrosis. SIRT3 activates ERK-CREB signaling to promote BNIP3 activity, thus inducing BNIP3 regulated mitotic resistance to nonalcoholic fatty liver disease [31]. SIRT3-induced autophagy can also protect the liver from alcohol-induced injury [152]. However, there are often two sides to complex issues. Another study demonstrated that, in contrast, SIRT3 could be a negative regulator of autophagy, which can lead to NAFLD induced by lipotoxicity [153]. More interestingly, SIRT3 can repress hepatitis caused by infection with the Hepatitis B virus (HBV). In a recent study, SIRT3 was found to aggregate in the covalently closed circular DNA (cccDNA) of HBV and deacetylate Lys 9 of Histone H3, inhibiting HBV transcription and replication [154]. As it does in other organs, SIRT3 can enhance ROS clearance to protect liver from oxidative damage by activating MnSOD2 and FOXO3a. Also SIRT3 protects hepatocytes by inhibiting mitochondrial damage and apoptosis through deacetylation of Ku70 [155, 156]. In addition, SIRT3 can alleviate liver fibrosis. The deacetylation and activation of GSK3β at Lys 15 by SIRT3 inhibit the expression of fibrosis genes in the liver [133]. ROS promotes liver fibrosis through AKT-mTOR and ERK1/2 signaling, while SIRT3 resists liver fibrosis by eliminating harmful ROS. SIRT3 is definitely a liver protector by safeguarding metabolic stability.
Obesity and diabetes are becoming ever more common around the world and are now trending with younger people. Although a recent study suggests that SIRT3 is dispensable in adipocyte metabolism and obesity-induced metabolic complications [157], SIRT3 in fact is important in obesity and diabetes. Of note, insulin resistance and vascular dysfunction are common in obese patients, which are exacerbated by SIRT3 deficiency [158, 159]. SIRT3 can regulate endothelial cell glycolytic metabolism, and mice lacking the expression of SIRT3 will increase insulin resistance because of the dysfunction of glucose uptake and mitochondrial function [119, 160]. In addition, SIRT3 can protect endothelial cells from mitochondrial ROS damage and increase NO release to benefit vasodilatation [159]. Moreover, SIRT3-mediated SOD2 deacetylation also contributes to maintaining the escape of endothelial progenitor cells (EPCs) from dysfunction and injury as well as decreased vascular inflammation [161, 162]. Maternal obesity is another key problem in human beings. The resulting high oxidative stress and meiotic defects of oocytes will damage reproductive health. Activation 7of SIRT3 can attenuate oxidative damage and improve the oocyte quality in obese woman [163]. Of note, the deficiency of SIRT3 causes pancreatic beta cells to be more sensitive to cellular stress and oxidative damage, impairs their function and promotes the development of diabetes [164]. Activation of SIRT3 can also regulate skeletal muscle metabolism and activate insulin signaling to improve diabetes. In this process, SIRT3 removes ROS to inhibit the activation of JNK and ISR-1 [165]. Interestingly, activation of pro-inflammatory macrophages is fateful in the pathogenesis of insulin resistance in diabetes. Loss of SIRT3 leads to the increasing expression of inflammatory cytokines and high risk of diabetes [166]. Pyruvate dehydrogenase (PDH) is a key regulator in the TCA cycle and glucose oxidation. SIRT3 can deacetylate its E1α subunit to increase its enzymatic activity [167]. SIRT3 also promotes HKII-VDAC-ANT complex formation to improve glucose control [168]. As it does in other diseases, SIRT3 activates MnSOD2 and IDH2 to remove harmful ROS and activates FOXO3a to induce cell-protective autophagy to improve obesity and diabetes. Thus, SIRT3 protects human from metabolic diseases (Figure 6), and SIRT3 modulators will shine in the prevention and early treatment of metabolic diseases.
SIRT3 in metabolic disease. The body's energy metabolism, glycometabolism, fatty acid metabolism, and amino acid metabolism are generally imbalanced in metabolic diseases. SIRT3 can regulate a series of substrates to maintain the metabolic balance and stability of different organisms, and inhibit the occurrence and development of metabolic diseases. In addition, SIRT3 can inhibit the fibrosis of each organ and protect its normal function. It is worth noting that SIRT3 is also involved in the fight against viral infections and inflammatory responses. The pink protein represents the substrate for SIRT3. Gray circles represent acetylation modifications.
SIRT3 in other diseases
In addition to several major diseases, SIRT3 offers protection from other common diseases and improves the quality of human life. SIRT3 can maintain bone metabolism by enhancing the AMPK-PGC-1β axis [169]. SIRT3 can also protect microvasculature from LPS-induced damage. LPS upregulates Ang-2, leading to vascular leakage, while SIRT3 inhibits the expression of Ang-2 to maintain vascular integrity [170]. Up-regulation of SIRT3 can attenuate endothelial cellular senescence to decrease the risk of atherosclerosis [171]. In addition, activation of SIRT3 can suppress osteoarthritis by maintaining mitochondrial metabolism stability [172]. Overall, SIRT3 is very important in maintaining human health and proper regulation of SIRT3 will be beneficial to human health.
Targeting SIRT3 for potential therapies
As a result of the importance of SIRT3 in various diseases, several SIRT3 regulatory compounds have been discovered or designed synthetically. These compounds can deacetylate SIRT3 or regulate its expression level through different mechanisms. Based on their influence on SIRT3, these compounds were divided into two broad categories, SIRT3 activators (strictly speaking, “positive modulators”) and inhibitors. In this section we will discuss these compounds, the related mechanisms and potential therapeutic implications.
Positive modulators of SIRT3
SIRT3 dysfunction is closely related to the occurrence and development of various diseases, and activation of SIRT3 appears to be an effective strategy for the treatment of many diseases. Unfortunately, to date SIRT3 agonist has ever been reported to date. Several positive modulators of SIRT3 were reported, which can stimulate SIRT3 by elevating SIRT3 expression. By upregulating SIRT3, they have displayed promising therapeutic effects in some diseases such as cardiac hypertrophy, acute kidney injury, and others (Table 3). Of note, most positive regulators of SIRT3 are derived from natural products. For instance, Honokiol is one of the most studied SIRT3 activators and is a natural lignan derived from the bark of Magnolia. Honokiol could increase SIRT3 expression and deacetylation activity, which have a favorable effect on heart disease [16, 137], renal disease [173], surgery/anesthesia-induced cognitive decline [174] and Vitiligo [175]. For heart disease, Honokiol activation of SIRT3 further decreases the acetylation levels of MnSOD2 and OSCP, resulting in improved mitochondrial rate of oxygen consumption and inhibition of ROS synthesis. In addition, Honokiol suppresses cardiac hypertrophy and fibrosis via SIRT3-regulated AKT and ERK1/2 inhibition in mice with cardiac hypertrophy [16]. Furthermore, in mice with doxorubicin-induced cardiomyopathy, Honokiol activated SIRT3 to promote mitochondrial fusion and inhibit apoptosis. More intriguingly, activation of SIRT3 by Honokiol can protect against doxorubicin-induced cardiotoxicity in tumor-xenograft mice without affecting the anti-tumor effect of doxorubicin. This makes it possible for Honokiol to contribute to adjuvant therapy of chemotherapy [137]. In renal disease, Honokiol stimulates SIRT3 activity to block the NF-κB-TGF- β1/Smad regulated inflammation and fibrosis signaling in renal fibrosis mice model [173]. Surprisingly, Honokiol can ameliorate surgery-induced or anesthesia-induced cognitive decline in mice via SIRT3 activation mediating ROS elimination and apoptosis inhibition [174]. Interestingly, honokiol could improve vitiligo via melanocyte apoptosis inhibition through SIRT3-OPA1 axis activation [175]. These studies suggest that Honokiol may be helpful in the treatment of various diseases, whether used alone or as an adjunct therapy. Silybin is another natural plant-derived SIRT3 activator. It is isolated from the seeds of blessed milkthistle (Silybum marianum) and is used as a hepatoprotectant in traditional Chinese medicine. It was reported that Silybin can improve kidney mitochondrial function via activating SIRT3 in a mouse model of cisplatin-induced acute kidney injury. Up-regulation of SIRT3 eliminates ROS and inhibits apoptosis to protect kidney cells from death. It is noteworthy that this protective effect can decrease cisplatin-induced renal toxicity and may contribute to clinical adjuvant treatment in cisplatin chemotherapy [176]. Resveratrol, a famous SIRT1 activator (although resveratrol-mediated activation of sirtuins has been repeatedly shown to be an experimental artifact, it does have biological activity in vivo), can also increase the expression of SIRT3 to attenuate acute kidney injury [177]. Fascinatingly, Dihydromyricetin has a similar chemical structure to Resveratrol and it can elevate the expression of SIRT3 through SIRT3 mediated cytoprotection and inflammatory resistance, thus treating osteoarthritis [172]. Polydatin, a polyphenolic compound isolated from Polygonum cuspidatum, can initiate SIRT3-regulated mitochondrial autophagy to protect cardiomyocytes from myocardial infarction [178]. Moreover, in a mouse model of sulfur mustard-induced hepatic injury, this natural product exerts its hepatoprotective effects via SIRT3 [179]. Pyrroloquinoline quinone, another natural product, can improve liver metabolic diseases through increasing the expression of SIRT3 [180].
Positive modulators of SIRT3
| Compound | Chemical Structure | Target/Pathways | Disease/cell | Biological Activity | Reference |
|---|---|---|---|---|---|
| Honokiol | 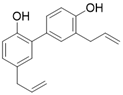 | Honokiol increases SIRT3 expression and activity | a. Cardiac Hypertrophy b. Renal disease c. Surgery/anesthesia-induced cognitive decline d. Vitiligo | / | [16, 137, 173-175] |
| Silybin | 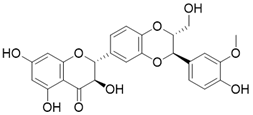 | Silybin Increases SIRT3 expression | Acute Kidney Injury | / | [176] |
| Resveratrol | 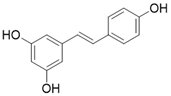 | Resveratrol increases SIRT3 expression | Acute Kidney Injury | / | [177] |
| Polydatin | 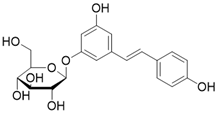 | Polydatin increases SIRT3 activity | a. Myocardial infarction b. Ulfur mustard-induced hepatic injury | / | [178, 179] |
| Dihydromyricetin | 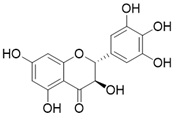 | Dihydromyricetin increases the expression and activity of SIRT3 via activation of PGC-1α | Osteoarthritis | / | [172] |
| Pyrroloquinoline quinone | 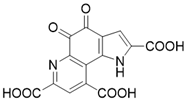 | Pyrroloquinoline quinone increases the expression and activity of SIRT3 | Liver metabolic diseases | / | [180] |
| Metformin | 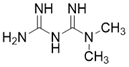 | Metformin increases SIRT3 expression | Atherosclerosis | / | [181] |
| Adjudin | 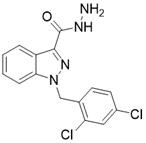 | Adjudin increase the expression of SIRT3 | Hearing loss | / | [182] |
| Melatonin | 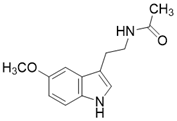 | Melatonin activates SIRT3 signaling pathway | a. myocardial ischemia reperfusion injury b. liver injury c. atherosclerosis | / | [183-185] |
| 7-hydroxy-3-(4'-methoxyphenyl) coumarin (C12) | 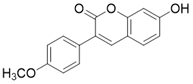 | C12 binds to the MnSODK68AcK-SIRT3 complex and promotes the deacetylation and activation of MnSOD | unclear | (SIRT3)Kd=3.9μM; IC50(MnSODK68AcK)= 75.78 μM | [15] |
Importantly, a growing number of reports show that some “old drugs” have a capacity of activating SIRT3 with a clear action mechanism. Metformin is a known AMPK activator which applied as the first-line drug for type 2 diabetes. Metformin can improve atherosclerosis aroused by type 2 diabetes via up-regulating SIRT3 [181]. Hearing loss is a common age-related disease with cell degeneration. The Cl- channel blocker, Adjudin, prevents gentamicin-induced hair cell loss via SIRT3 [182]. Interestingly, a hormone named Melatonin, which targets the melatonin receptor to play a role in sleep, now has been found to be a SIRT3 activator that plays a protective role in heart disease [183], liver injury [184] and atherosclerosis [185]. In a mouse model of myocardial ischemia/reperfusion (MI/R) injury, melatonin also increases SIRT3 expression and activity to inhibit apoptosis of cardiomyocytes and maintain the stability of mitochondrial metabolism [183]. In addition, in cadmium-induced hepatotoxicity in vitro and in vivo models, melatonin enhances the activity of SIRT3 to inhibit both ROS production and cadmium-triggered autophagic cell death [184]. Additionally, in a mouse model of atherosclerosis, melatonin activates SIRT3-FOXO3a-Parkin regulated mitophagy to prevent inflammation and atherosclerotic progression [185].
Another type of SIRT3 activator is a substrate-dependent activator. 7-hydroxy-3-(4'-methoxyphenyl) coumarin (C12) is a compound that promotes the deacetylation of MnSODK68AcK (MnSOD acetylated at Lys 68) through SIRT3. C12 was identified based on the crystal structure of MnSODK68AcK. C12 binds to the MnSODK68AcK-SIRT3 complex and promotes the deacetylation and activation of MnSOD. The Kd value of C12 binds to SIRT3 is 3.9 μM and the 50% activation concentration is 75.78 μM towards MnSODK68AcK [15]. Of note, among all these SIRT3 activators, C12 is closest to the real SIRT3 activator.
As a fascinating target for disease treatment, SIRT3 has always been a research hotspot. Although these positive modulators of SIRT3 have certain therapeutic potentials for disease, the design of targeted small-molecule activators of SIRT3 still faces significant challenges.
The design of SIRT3 activators, which control the SIRT3 catalytic activity level by some exact mechanism, lacks a theoretical and structural basis. At present, allosteric activators have attracted much attention in the design of targeted activators of SIRTs (such as SIRT1 and SIRT6). Further elucidation of the structure and biological function of SIRT3 will promote the development of small molecule activators targeting SIRT3.
SIRT3 inhibitors
Compared with the discovery of SIRT3 activators, the development of SIRT3 inhibitors has been much easier. The deacetylation process of SIRT3 (Figure 7) logically suggests a series of methods to inhibit this chemical reaction. In addition, as the crystal structure of the SIRT3 protein has been identified, another strategy for the discovery of SIRT3 inhibitors is structure-based design. Last but not least, chemical library screening is another common way to discover SIRT3 inhibitors [17]. Of note, another type of SIRT3 inhibitor has been found fortuitously, which can inhibit SIRT3 expression or activity [186, 187]. Through these strategies, a variety of SIRT3 inhibitors have been developed and they have shown good viability in a variety of diseases (Table 4), especially in cancer.
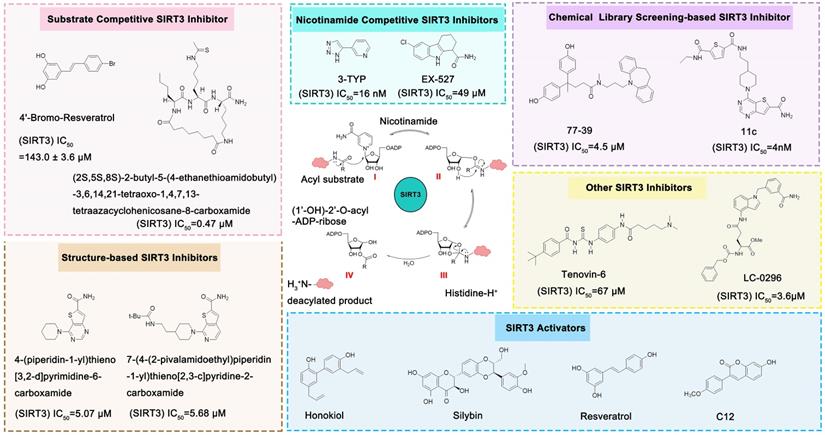
The NAD+-dependent SIRT3 deacetylation reaction process, SIRT3 activators and SIRT3 inhibitors. The NAD+-dependent SIRT3 deacetylation reaction process is roughly divided into four steps. I, The acetylated substrate and the NAD+ co-substrate binding to SIRT3. II, the acetyl group consequently transfer from substrate to ADP-ribose moiety of NAD+. III, Generation of bicyclic intermediates. IV, Produce the deacetylated protein. SIRT3 inhibitors are divided into five types. Substrate competitive SIRT3 inhibitors, Nicotinamide competitive SIRT3 inhibitors, chemical library screening-based SIRT3 inhibitors, structure-based SIRT3 inhibitors and other SIRT3 inhibitors. The chemical structures of representative SIRT3 inhibitors and activators are displayed in the figure.
SIRT3 Inhibitors
| Compound | Chemical Structure | Type | Disease/cell | Biological Activity | Reference |
|---|---|---|---|---|---|
| 4'-Bromo-Resveratrol | 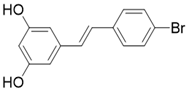 | Substrate competitive SIRT3 inhibitor | Melanoma | SIRT3 IC50=143.0 ± 3.6 μM | [188, 189] |
| /(4- [(2-Hydroxy-6-phenylnaphthalen-1-yl) methyl]-5- (4-methylphenyl) -2,3-dihydro-1H-pyrazol-3-one) | 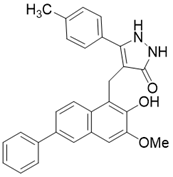 | Substrate competitive SIRT3 inhibitor | Cancer | SIRT3 IC50=6 μM | [190] |
| (2S,5S,8S)-5-(4-ethanethioamidobutyl)-2-(naphthalen-2-ylmethyl)-3,6,13,20-tetraoxo-1,4,7,12-tetraazacycloicosane-8-carboxamide | 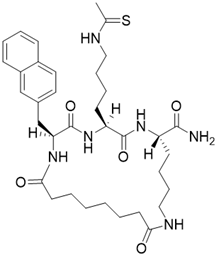 | Substrate competitive SIRT3 inhibitor | Cancer | SIRT3 IC50=1.94 μM | [190] |
| (3S,6S,9S)-9-butyl-6-(4-ethanethioamidobutyl)-5,8,11,18-tetraoxo-1,4,7,10-tetraazacyclooctadecane-3-carboxamide | 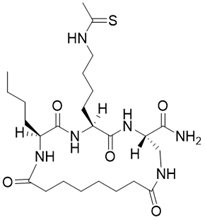 | Substrate competitive SIRT3 inhibitor | Cancer | SIRT3 IC50=1.06 μM | [191] |
| (2S,5S,8S)-2-butyl-5-(4-ethanethioamidobutyl)-3,6,12,19-tetraoxo-1,4,7,11-tetraazacyclononadecane-8-carboxamide | 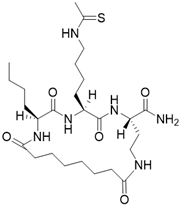 | Substrate competitive SIRT3 inhibitor | Cancer | SIRT3 IC50=1.48 μM | [191] |
| (2S,5S,8S)-2-butyl-5-(4-ethanethioamidobutyl)-3,6,13,20-tetraoxo-1,4,7,12-tetraazacycloicosane-8-carboxamide | 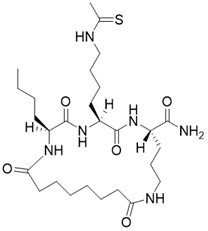 | Substrate competitive SIRT3 inhibitor | Cancer | SIRT3 IC50=1.82 μM | [191] |
| (2S,5S,8S)-2-butyl-5-(4-ethanethioamidobutyl)-3,6,14,21-tetraoxo-1,4,7,13-tetraazacyclohenicosane-8-carboxamide | 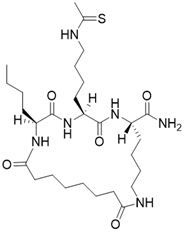 | Substrate competitive SIRT3 inhibitor | Cancer | SIRT3 IC50=0.47 μM | [191] |
| (S)-2-((S)-4-([1,1'-biphenyl]-4-yl)-2-acetamidobutanamido)-N-((S)-6-acetamido-1-amino-1-oxohexan-2-yl)-6-ethanethioamidohexanamide | 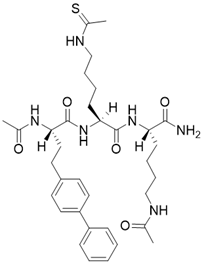 | Substrate competitive SIRT3 inhibitor | / | SIRT3 IC50= 0.48 μM | [192] |
| N,N'-((S)-6-(((S)-1-(((S)-4-acetamido-1-amino-1-oxobutan-2-yl)amino)-6-ethanethioamido-1-oxohexan-2-yl)amino)-6-oxohexane-1,5-diyl)diacetamide | 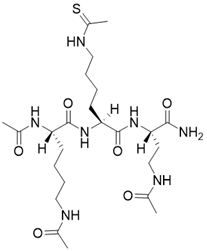 | Substrate competitive SIRT3 inhibitor | / | SIRT3 IC50= 0.36 μM | [192] |
| N,N'-((S)-6-(((S)-1-(((S)-1-amino-1-oxohexan-2-yl)amino)-6-ethanethioamido-1-oxohexan-2-yl)amino)-6-oxohexane-1,5-diyl)diacetamide | 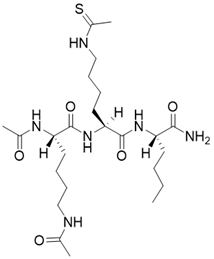 | Substrate competitive SIRT3 inhibitor | / | SIRT3 IC50=0.48 μM | [192] |
| N,N'-((S)-6-(((S)-1-(((S)-1-amino-4-(naphthalen-2-yl)-1-oxobutan-2-yl)amino)-6-ethanethioamido-1-oxohexan-2-yl)amino)-6-oxohexane-1,5-diyl)diacetamide | 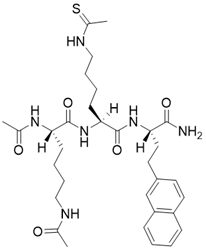 | Substrate competitive SIRT3 inhibitor | SIRT3 IC50=2.1 μM | [192] | |
| YC8-02 | 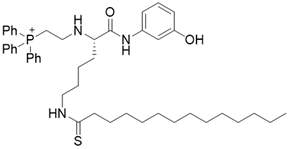 | Substrate competitive SIRT3 inhibitor | Lymphoma | SIRT3 IC50=0.53 μM | [108] |
| JH-T4 | 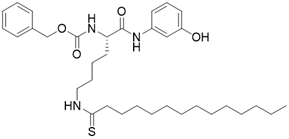 | Substrate competitive SIRT3 inhibitor | Lymphoma | SIRT3 IC50=2.5 μM | [108] |
| 3-TYP | 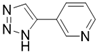 | Nicotinamide competitive SIRT3 inhibitors | Tool medicine | SIRT3 IC50=16 nM | [193] |
| EX-527 | 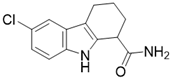 | Nicotinamide competitive SIRT3 inhibitors | Cancer | SIRT3 IC50=49 μM | [194] |
| 4-(4-(acetamidomethyl)piperidin-1-yl)thieno[3,2-d]pyrimidine-6-carboxamide | 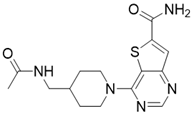 | Structure-based SIRT3 inhibitors | / | SIRT3 IC50=5.36 μM | [195] |
| 4-(4-(2-pivalamidoethyl)piperidin-1-yl)furo[3,2-d]pyrimidine-6-carboxamide | 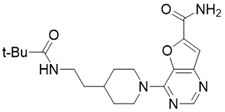 | Structure-based SIRT3 inhibitors | / | SIRT3 IC50=5.89 μM | [195] |
| 7-(4-(2-pivalamidoethyl)piperidin-1-yl)thieno[2,3-c]pyridine-2-carboxamide | 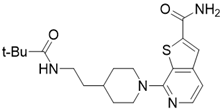 | Structure-based SIRT3 inhibitors | / | SIRT3 IC50=5.68 μM | [195] |
| 4-(piperidin-1-yl)thieno[3,2-d]pyrimidine-6-carboxamide | 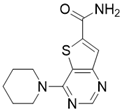 | Structure-based SIRT3 inhibitors | / | SIRT3 IC50=5.07 μM | [195] |
| 77-39 | 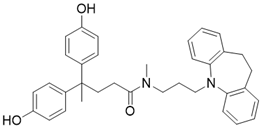 | Chemical library screening-based SIRT3 inhibitor | / | SIRT3 IC50=4.5 μM; Kd =2.14 µM | [196] |
| 11c (N2-(2-(1-(6-carbamoylthieno[3,2-d]pyrimidin-4-yl)piperidin-4-yl)ethyl)-N5-ethylthiophene-2,5-dicarboxamide) | 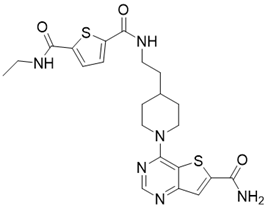 | Chemical library screening-based SIRT3 inhibitors | / | SIRT3 IC50=4nM | [197] |
| Tenovin-6 | 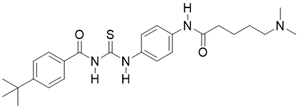 | other | Cancer | SIRT3 IC50=67 μM | [186] |
| LC-0296 | 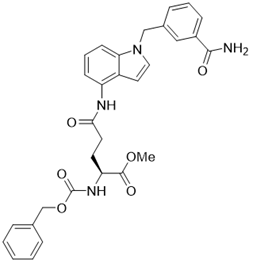 | other | Head and Neck Cancer | SIRT3 IC50=3.6μM | [198] |
| Trimethylamine-N-oxide (TMAO) | 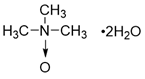 | other | Vascular Inflammation | / | [199] |
| Albendazole | 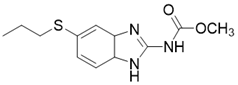 | other | leukemia U937 and HL60 cells | / | [187] |
| 2-methoxyestradiol | 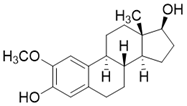 | binding to both the canonical and allosteric inhibitor binding sites | Osteosarcoma Cancer | / | [200] |
Catalytic Mechanism-based SIRT3 Inhibitors
The deacetylation of SIRT3 is a multi-step, continuous and complex process involving the coenzyme NAD+ and the acetylated substrate. Targeting the dynamic process of deacetylation is an effective strategy for the design of SIRT3 inhibitors. On one hand, structural analogues of endogenous acetylated substrates can effectively and competitively inhibit the deacetylation activity of SIRT3. On the other hand, NAD+ coenzyme competitive inhibitors can accelerate the reverse reaction of deacetylation to inhibit the progress of the deacetylate reaction. Notably, it is easy to achieve considerable satisfactory inhibition efficiency and selectivity with the catalytic mechanism-based inhibitors. This is the fascinating characteristic of this type of SIRT3 inhibitor and, of course, why it is one of the most studied.
Substrate Competitive SIRT3 Inhibitors
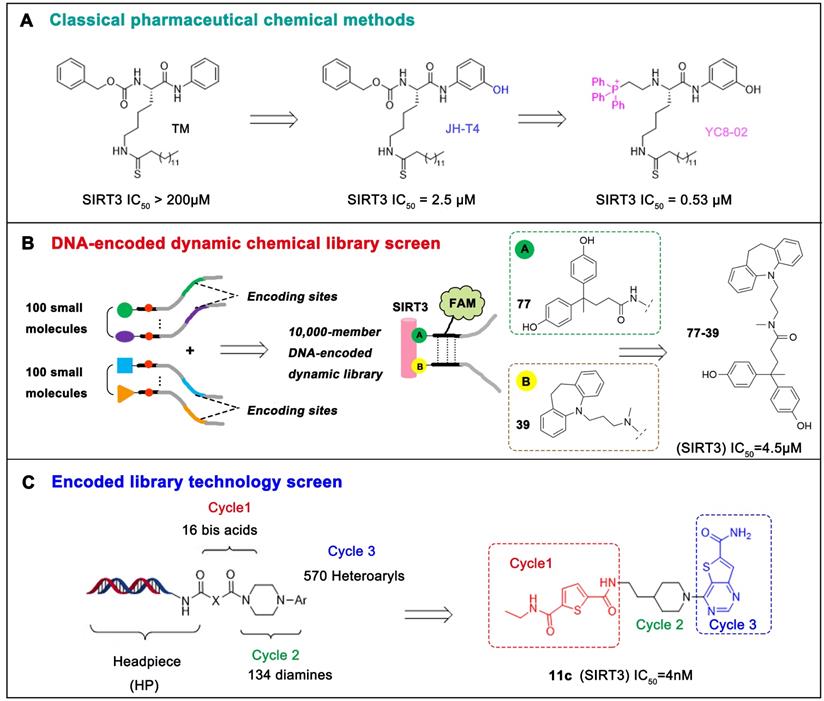
The most common and effective inhibitor design strategies use substrate competitive inhibitors. 4'-bromo-resveratrol is an ACS2 peptide substrate competitive inhibitor discovered in 2013 [188]. Recently, 4'-bromo-resveratrol was found to inhibit melanoma progression via SIRT3 mediated mitochondrial metabolic reprogramming. In this process, 4'-bromo-resveratrol was also confirmed to induce apoptosis and G0/G1 cell cycle arrest [189]. Simon et al. discovered a peptide substrate competitive SIRT1/2 inhibitor, cambinol, which has a potential effect on cancer treatment. After that, they designed a series of cambinol analogues and discovered the SIRT3 selective inhibitor 4- [(2-Hydroxy-6-phenylnaphthalen-1-yl) methyl]-5-(4-methylphenyl) -2, 3-dihydro-1H-pyrazol-3-one (SIRT3 IC50 = 6 μM) that has good anti-cancer potential [190]. Analogs of NƐ-acyl-lysine are important substrate competitive SIRT3 inhibitors. Peptide-based SIRT3 inhibitors are one of the analogs of NƐ-acyl-lysine with excellent proteolytic stability and cell permeability. Chen et al. designed a series of peptides containing NƐ-thioacetyl-lysine that exhibit SIRT1/2/3 inhibition effects. Their effects vary from 0.47 μM to 10.8 μM and the most potent one can efficiently inhibit SIRTs activity in HCT116 human colon cancer cells. Unfortunately, despite its acceptable SIRT3 inhibitory activity, this one is not a selective SIRT3 inhibitor (SIRT3 IC50 = 0.22 μM; SIRT3 IC50 = 0.24 μM; SIRT3 IC50 = 0.47 μM) [191]. This also suggests that if we want to develop specific substrate-competitive SIRT3 inhibitors, we may need to select SIRT3-specific substrate action sites for targeted design. Zheng et al. also designed a variety of potent SIRT3 tripeptidic inhibitors containing NƐ-thioacetyl-lysine. However, their selectivity was generally not strong [192]. SIRT3-selective inhibitors can be obtained from other SIRT inhibitors by classical medicinal chemistry methods [108]. One case in point is the thioacyl lysine compound TM reported by Hui Jing et al. It was identified as targeting SIRT2 with little SIRT3 inhibition. 3-hydroxy substitution (generating JH-T4) remarkably enhances SIRT3 inhibition with an IC50 of 2.5 μM. Considering the fact that SIRT3 is abundant in mitochondria, researchers modified JH-T4 by replacing the benzyl carbamoyl group with a triphenylphosphonium (TPP) mitochondrial targeting moiety to obtain the compound YC8-02, which achieved superior penetration into mitochondria. Accordingly, the SIRT3 IC50 was further increased to 0.53 μM. YC8-02 not only exhibits strong SIRT3 inhibition effects, but also inhibits lymphomagenesis by selectively inhibiting SIRT3 (Figure 8A) [108].
Potential therapeutic strategies of representative SIRT3 inhibitors. (A) SIRT3 inhibitor discovered by classical pharmaceutical chemical method. (B) SIRT3 inhibitor discovered by DNA-encoded dynamic chemical library screen strategy. (C) SIRT3 inhibitor discovered by encoded library technology screen method.
Nicotinamide Competitive SIRT3 Inhibitors
Nicotinamide is an endogenous inhibitor of SIRTs, but the one major drawback is that it has no selectivity for SIRTs. However, it is undeniable that nicotinamide analogues are an important component of SIRT3 inhibitors. 3-TYP is essentially a nicotinamide analogue which is a widely used tool in medicine with high selective SIRT3 inhibition (SIRT3 IC50=16 nM) [193]. EX-527 is a selective SIRTs inhibitor that functions by occupying the nicotinamide site and neighboring pocket contacting NAD+. EX-527 always been defied as a SIRT1 inhibitor, but it can also inhibit SIRT3 with weak activity (SIRT3 IC50=49 μM) [194]. Interestingly, the discovery of EX-527 revealed a new SIRT inhibition mechanism, namely, the formation of trimer Sirtuin complex with a NAD+-derived coproduct.
Structure-based SIRT3 Inhibitors
Docking and binding free energy calculations are methods to discover novel SIRT3 inhibitors based on its reported structure. In this way, Berin et al. discovered a fair number of compounds that exhibit fine SIRT3 inhibition effects. Unfortunately, their biological activity has not been detected, so their application may require more work to support [195].
Chemical Library Screening-based SIRT3 Inhibitors
Chemical library screening is an effective way to discover novel SIRT3 inhibitors. Recently, the novel SIRT3 inhibitor 77-39 (SIRT3 IC50 = 4.5 μM) was discovered through the DNA-encoded Dynamic Chemical Library (Figure 8B). 77-39 also exhibits excellent cellular SIRT3 inhibition activity, which increases the global mitochondrial acetylation level of HeLa cells. Moreover, 77-39 showed only little cytotoxicity [194]. Encoded Library Technology screening is another efficient strategy to identify novel SIRT3 inhibitors. Through this strategy, the potent SIRT3 inhibitor 11c ((N2-(2-(1-(6-carbamoylthieno [3, 2-d] pyrimidin-4-yl) piperidin-4-yl) ethyl)-N5-ethylthiophene-2, 5-dicarboxamide)) (SIRT3 IC50 = 4.0 nM) was discovered (Figure 8C) [197].
Other SIRT3 Inhibitors
Tenovin-6 is a p53 activator with biological activity that later was also found to be a SIRT3 inhibitor (SIRT3 IC50 = 67 μM) with anti-tumor activity. The inhibitory effect on SIRT3 was unclear, but it has been proven to act as a non-competitive inhibitor [186]. LC-0296 is a synthetic SIRT3 inhibitor with good inhibitory effect (SIRT3 IC50 = 3.6 μM) and its mechanism is also unclear. Judging by its structure, LC-0296 might be a NAD+ competitive inhibitor. It has good activity against squamous cell carcinoma of the head and neck through inhibiting proliferation and promoting apoptosis [198]. Trimethylamine-N-oxide (TMAO) is a choline metabolite that can promote vascular inflammation through SIRT3 inhibition-induced ROS-NLRP3 activation [199]. Albendazole is an anthelmintic drug with microtubule-targeting ability. Recently, albendazole was found to induce SIRT3 degradation to inhibit leukemia cell survival [187]. 2-methoxyestradiol (2-ME) is an anti-cancer drug, which has been found to bind to the typical and allosteric inhibitor binding sites on SIRT3 to inhibit its activity. By inhibiting SIRT3, 2-ME can disturb normal mitochondrial functions and kill osteosarcoma cells [200].
Although the design and discovery of SIRT3 inhibitors are easier than activators, the development of proprietary and highly efficient SIRT3 inhibitors remains a challenge. The application of SIRT3 inhibitors is something that requires more attention. Given the cytoprotective role of SIRT3 in multiple organs, inhibition of SIRT3 in certain cancers should be carefully evaluated and discussed. Therefore, how to develop a highly efficient SIRT3 inhibitor without organ toxicity is widely expected. In addition, the introduction of new drug delivery methods, such as target organ delivery, may open a new door for the application of SIRT3 inhibitors.
Conclusions
SIRT3, the major deacetylase in mitochondria, has been well-established to be involved in all aspects of mitochondrial metabolism as well as the synthesis and movement of mitochondria. Mitochondria play a very important role in cells, and their disorders induce a variety of diseases. The dysregulation of SIRT3 has been confirmed in many mitochondria-related diseases.
An accumulation of current evidence has gradually revealed the mysteries of the inherent biological functions of SIRT3 and medicinal applications in human diseases. SIRT3 has been attracting extensive attention from its original role as the longevity gene to being a potential “superstar” target in many diseases. Several studies have demonstrated that the improved role of SIRT3 in many diseases is mainly due to its scavenging effect on ROS. Excessive accumulation of ROS is often an important factor in the development of age-related diseases, heart disease, cancer and metabolic diseases. SIRT3 activates its substrates such as MnSOD2, IDH2 and PHD by deacetylation and then removes excess ROS. On the other hand, SIRT3 regulates TCA, OXPHOS, fatty acid and amino acid metabolism to maintain mitochondria homeostasis in normal or slightly damaged mitochondria to influence cell fitness and survival. However, when the mitochondria are too damaged to be repaired, SIRT3 can deacetylate and activate FOXO3a to induce mitophagy, thus eliminating and circulating mitochondria. Interestingly, regarding to its inherent complexity, SIRT3 has been demonstrated to play the Janus role in cancer, indicating that SIRT3 activators or inhibitors would be utilized as anti-tumor agents in specific types of cancers, respectively. For instance, it cannot be ignored that the application of SIRT3 inhibitors in some OXPHOS-addiction cancers seems very promising, but in regarding to the protective effects of SIRT3 on various human organs, inhibition of SIRT3 might partially bring multiple organ toxicities. On the other hand, the excessive pursuit of SIRT3 activation might also lead to unpredictable carcinogenesis under some circumstances. Thus, considering either up-regulation or down-regulation expression of SIRT3 seems to be so limited for the discovery of new small-molecule inhibitor or activator in the treatment of different types of diseases. Maybe, the combination of SIRT3 activator/inhibitor and the other drug in a new delivery system targeting specific human organ would be an alternative potential therapeutic strategy.
Hitherto, no specific SIRT3 activators or potent inhibitors have been successfully discovered and thus being utilized for potential therapeutics. But still, several small-molecule compounds with therapeutic potential have been reported. For example, the most famous SIRT3 activator, Honokiol, exhibits some therapeutic effects in heart disease, inflammation-related diseases, cancer and metabolic diseases. However, its beneficial effects are not entirely attributed to the activation of SIRT3. The structural characteristics of its polyphenols determine its outstanding antioxidant capacity, which is also the key to its therapeutic activity. Similarly, although the highly selective SIRT3 inhibitor 3-TYP has good properties, it is only used as a probe with almost no therapeutic applications.
Therefore, how to develop an effective SIRT3 activator or inhibitor for therapeutic purposes remains to be an enigma. In this regard, scientific drug design strategy is particularly important. For the design of SIRT3 activators, there is still a lack of effective and scientific design methods at present. Because the mechanism of the interaction between small molecule agonists and SIRT3 is still a black box, there is no basis for structure-based drug design. At present, allosteric agonists are one of the most convincing methods for the design of SIRT3 agonists. Recently, artificial intelligence has been applied in the field of drug design, which has opened a new era of revolutionary drug design. However, much effort still needs to be made to elucidate the molecular mechanism of SIRT3 allosteric activation.
As a plan for SIRT3 inhibitor design, structure-based drug design is a mature and effective strategy, including receptor- and ligand-based technology. Among them, basing this on the binding characteristics of the substrate and SIRT3 complex is a better choice for selective SIRT3 inhibitors. Co-enzyme structure-based design is more likely to lose selectivity, resulting in serious side effects. Additionally, PROTAC technology is a new strategy to design potent targeted inhibitors. Taken together, SIRT3, from a mitochondrial metabolic regulator to a promising therapeutic target, would shed new light on exploiting more candidate drugs for fighting human diseases in the near future.
Abbreviations
SIRT3: Sirtuin 3; TCA: tricarboxylic acid; UPR: unfolded protein response; OXPHOS: oxidative phosphorylation; CR: Caloric Restriction; IDH2: isocitrate dehydrogenase 2; FLSIRT3: full length SIRT3; MPP: Matrix processing peptidase; 4-HNE: 4-Hydroxynonenal; PPARγ: Peroxisome proliferator-activated receptor γ; PGC-1α: PPARγ coactivator 1; miRNA: MicroRNA; ISCU: iron-sulfur cluster assembly protein; LncRNAs: Long non-coding RNA; AD: Alzheimer's disease; PD: Parkinson's disease; HD: Huntington's disease; AML: Amyotrophic lateral sclerosis; MnSOD2: manganous superoxide dismutase 2; ROS: reactive oxygen species; mPTP: mitochondrial permeability transition pore; NMNAT2: Nicotinamide mononucleotide adenylyltransferase 2; HSP90: heat shock protein 90; HIF1α: hypoxia-inducible factor-1a; PHD: prolyl hydroxylase; PDC: pyruvate dehydrogenase complex; Skp2: the F-box protein S-phase kinase associated protein 2; GSK-3β: glycogen synthase kinase-3β; ECHS1: Enoyl-CoA hydratase-1; GOT: Glutamate oxaloacetate transaminases; OGG1: 8-oxoguanine-DNA glycosylase 1; SHMT2: serine hydroxymethyltransferase 2; PYCR1: Pyrroline-5-carboxylate reductase 1; OPA1: Optic atrophy 1; OSCP: oligomycin-sensitivity conferring protein; CypD: cyclophilin D; LCAD: long-chain acyl CoA dehydrogenase; NMNAT3: Nicotinamide mononucleotide adenylyltransferase 3; STAT3: signal transducer and activator of transcription 3; AKI: acute kidney injury; EndoMT: Endothelial-to-mesenchymal transition; NAFLD: nonalcoholic fatty liver disease; HBV: Hepatitis B virus; cccDNA: covalently closed circular DNA; PDH: Pyruvate dehydrogenase; ZEB1: Zinc finger E-box-binding homeobox 1; GOT2: glutamate oxaloacetate transaminases 2; CLL: chronic lymphocytic leukemia; HCC: hepatocellular carcinoma; DLBCLs: diffuse large B cell lymphomas; EMT: epithelial-mesenchymal transition; TNBC: triple negative breast cancer; ACC1: acetyl-coA carboxylase 1; GLYAT: Glycine N-acyltransferase; HMGCL: hydroxymethylglutaryl-CoA lyase; EPCs: endothelial progenitor cells.
Acknowledgements
We are grateful to the assistance with the proper usage of scientific English provided by Dr. L.J. Sparvero (University of Pittsburgh). We also thank Dr. Xin Wen (University of Michigan, Ann Arbor), Prof. Shengyong Yang and Prof. Canhua Huang (Sichuan University) for their good discussions on this manuscript. This work was supported in part by National Natural Science Foundation of China (Grant No. 81673455, Grant No. 81803365, Grant No. 81573290, and Grant No. U1603123), Post-Doctor Research Project (Grant No. 2018M643510), Post-Doctor Research Project of West China Hospital, Sichuan University (Grant No. 2018HXBH065), and the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (Grant No. 2017BT01Y036) and GDUPS (2019), National Science and Technology Major Project of the Ministry of Science and Technology of China (No. 2018ZX09735005). Some picture materials in the graphical abstract and figures are produced by Servier Medical Art (https://smart.servier.com) under CC BY 3.0 license, we are grateful for their generosity and kindness.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Schwer B, Verdin E. Conserved metabolic regulatory functions of sirtuins. Cell Metab. 2008;7:104-12
2. Frye RA. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem Biophys Res Commun. 1999;260:273-9
3. Hirschey MD. Old enzymes, new tricks: sirtuins are NAD(+)-dependent de-acylases. Cell Metab. 2011;14:718-9
4. Zhang Y, Nie L, Xu K, Fu Y, Zhong J, Gu K. et al. SIRT6, a novel direct transcriptional target of FoxO3a, mediates colon cancer therapy. Theranostics. 2019;9:2380-94
5. Samant SA, Zhang HJ, Hong Z, Pillai VB, Sundaresan NR, Wolfgeher D. et al. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol Cell Biol. 2014;34:807-19
6. Papa L, Germain D. Correction for Papa and Germain, "SirT3 regulates a novel arm of the mitochondrial unfolded protein response". Mol Cell Biol. 2017 37
7. Anderson KA, Green MF, Huynh FK, Wagner GR, Hirschey MD. Snapshot: mammalian sirtuins. Cell. 2014;159:956-e1
8. Wang Y, Yang J, Hong T, Chen X, Cui L. SIRT2: controversy and multiple roles in disease and physiology. Ageing Res Rev. 2019;55:100961
9. Liu X, Zhu C, Zha H, Tang J, Rong F, Chen X. et al. SIRT5 impairs aggregation and activation of the signaling adaptor MAVS through catalyzing lysine desuccinylation. EMBO J. 2020: e103285.
10. D'Onofrio N, Servillo L, Balestrieri ML. SIRT1 and SIRT6 signaling pathways in cardiovascular disease protection. Antioxid Redox Signal. 2018;28:711-32
11. Tang M, Li Z, Zhang C, Lu X, Tu B, Cao Z. et al. SIRT7-mediated ATM deacetylation is essential for its deactivation and DNA damage repair. Sci Adv. 2019;5:eaav1118
12. Jin L, Galonek H, Israelian K, Choy W, Morrison M, Xia Y. et al. Biochemical characterization, localization, and tissue distribution of the longer form of mouse SIRT3. Protein Sci. 2009;18:514-25
13. Verdin E, Hirschey MD, Finley LW, Haigis MC. Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci. 2010;35:669-75
14. Chen Y, Fu LL, Wen X, Wang XY, Liu J, Cheng Y. et al. Sirtuin-3 (SIRT3), a therapeutic target with oncogenic and tumor-suppressive function in cancer. Cell Death Dis. 2014;5:e1047
15. Lu J, Zhang H, Chen X, Zou Y, Li J, Wang L. et al. A small molecule activator of SIRT3 promotes deacetylation and activation of manganese superoxide dismutase. Free Radic Biol Med. 2017;112:287-97
16. Pillai VB, Samant S, Sundaresan NR, Raghuraman H, Kim G, Bonner MY. et al. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat Commun. 2015;6:6656
17. Jiang Y, Liu J, Chen D, Yan L, Zheng W. Sirtuin inhibition: strategies, inhibitors, and therapeutic potential. Trends Pharmacol Sci. 2017;38:459-72
18. Finley LW, Haigis MC. Metabolic regulation by SIRT3: implications for tumorigenesis. Trends Mol Med. 2012;18:516-23
19. Yang W, Nagasawa K, Münch C, Xu Y, Satterstrom K, Jeong S. et al. Mitochondrial sirtuin network reveals dynamic SIRT3-dependent deacetylation in response to membrane depolarization. Cell. 2016;167:985-1000.e21
20. Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C. et al. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802-12
21. Guan X, Lin P, Knoll E, Chakrabarti R. Mechanism of inhibition of the human sirtuin enzyme SIRT3 by nicotinamide: computational and experimental studies. PLoS One. 2014;9:e107729
22. Schwer B, North BJ, Frye RA, Ott M, Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J Cell Biol. 2002;158:647-57
23. Wang T, Cao Y, Zheng Q, Tu J, Zhou W, He J. et al. SENP1-Sirt3 signaling controls mitochondrial protein acetylation and metabolism. Mol Cell. 2019;75:823-34.e5
24. Fritz KS, Galligan JJ, Smathers RL, Roede JR, Shearn CT, Reigan P. et al. 4-Hydroxynonenal inhibits SIRT3 via thiol-specific modification. Chem Res Toxicol. 2011;24:651-62
25. Neeli PK, Gollavilli PN, Mallappa S, Hari SG, Kotamraju S. A novel metadherinΔ7 splice variant enhances triple negative breast cancer aggressiveness by modulating mitochondrial function via NFĸB-SIRT3 axis. Oncogene. 2020;39:2088-102
26. Giralt A, Hondares E, Villena JA, Ribas F, Díaz-Delfín J, Giralt M. et al. Peroxisome proliferator-activated receptor-gamma coactivator-1alpha controls transcription of the Sirt3 gene, an essential component of the thermogenic brown adipocyte phenotype. J Biol Chem. 2011;286:16958-66
27. Nogueiras R, Habegger KM, Chaudhary N, Finan B, Banks AS, Dietrich MO. et al. Sirtuin 1 and sirtuin 3: physiological modulators of metabolism. Physiol Rev. 2012;92:1479-514
28. Zhang J, Wang G, Zhou Y, Chen Y, Ouyang L, Liu B. Mechanisms of autophagy and relevant small-molecule compounds for targeted cancer therapy. Cell Mol Life Sci. 2018;75:1803-26
29. Xu WY, Hu QS, Qin Y, Zhang B, Liu WS, Ni QX. et al. Zinc finger E-box-binding homeobox 1 mediates aerobic glycolysis via suppression of sirtuin 3 in pancreatic cancer. World J Gastroenterol. 2018;24:4893-905
30. Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203-22
31. Zhang X, Ji R, Liao X, Castillero E, Kennel PJ, Brunjes DL. et al. MicroRNA-195 regulates metabolism in failing myocardium via alterations in sirtuin 3 expression and mitochondrial protein acetylation. Circulation. 2018;137:2052-67
32. Cheng Y, Mai J, Hou T, Ping J. MicroRNA-421 induces hepatic mitochondrial dysfunction in non-alcoholic fatty liver disease mice by inhibiting sirtuin 3. Biochem Biophys Res Commun. 2016;474:57-63
33. Geng L, Zhang T, Liu W, Chen Y. MiR-494-3p modulates the progression of in vitro and in vivo Parkinson's disease models by targeting SIRT3. Neurosci Lett. 2018;675:23-30
34. Huang S, Guo H, Cao Y, Xiong J. MiR-708-5p inhibits the progression of pancreatic ductal adenocarcinoma by targeting Sirt3. Pathol Res Pract. 2019;215:794-800
35. Kao YY, Chou CH, Yeh LY, Chen YF, Chang KW, Liu CJ. et al. MicroRNA miR-31 targets SIRT3 to disrupt mitochondrial activity and increase oxidative stress in oral carcinoma. Cancer Lett. 2019;456:40-8
36. Zeng B, Ye H, Chen J, Cheng D, Cai C, Chen G. et al. LncRNA TUG1 sponges miR-145 to promote cancer progression and regulate glutamine metabolism via Sirt3/GDH axis. Oncotarget. 2017;8:113650-61
37. Li Y, Sun T, Shen S, Wang L, Yan J. LncRNA DYNLRB2-2 inhibits THP-1 macrophage foam cell formation by enhancing autophagy. Biol Chem. 2019
38. Sun W, Zhao L, Song X, Zhang J, Xing Y, Liu N. et al. MicroRNA-210 modulates the cellular energy metabolism shift during H2O2-induced oxidative stress by repressing ISCU in H9c2 cardiomyocytes. Cell Physiol Biochem. 2017;43:383-94
39. Wei Z, Song J, Wang G, Cui X, Zheng J, Tang Y. et al. Deacetylation of serine hydroxymethyl-transferase 2 by SIRT3 promotes colorectal carcinogenesis. Nat Commun. 2018;9:4468
40. Jiang W, He T, Liu S, Zheng Y, Xiang L, Pei X. et al. The PIK3CA E542K and E545K mutations promote glycolysis and proliferation via induction of the β-catenin/SIRT3 signaling pathway in cervical cancer. J Hematol Oncol. 2018;11:139
41. Yao W, Ji S, Qin Y, Yang J, Xu J, Zhang B. et al. Profilin-1 suppresses tumorigenicity in pancreatic cancer through regulation of the SIRT3-HIF1α axis. Mol Cancer. 2014;13:187
42. Baur JA, Ungvari Z, Minor RK, Le Couteur DG, de Cabo R. Are sirtuins viable targets for improving healthspan and lifespan? Nat Rev Drug Discov. 2012;11:443-61
43. Rose G, Dato S, Altomare K, Bellizzi D, Garasto S, Greco V. et al. Variability of the SIRT3 gene, human silent information regulator Sir2 homologue, and survivorship in the elderly. Exp Gerontol. 2003;38:1065-70
44. Brown K, Xie S, Qiu X, Mohrin M, Shin J, Liu Y. et al. SIRT3 reverses aging-associated degeneration. Cell Rep. 2013;3:319-27
45. Denu RA. SIRT3 enhances mesenchymal stem cell longevity and differentiation. Oxid Med Cell Longev. 2017;2017:5841716
46. McDonnell E, Peterson BS, Bomze HM, Hirschey MD. SIRT3 regulates progression and development of diseases of aging. Trends Endocrinol Metab. 2015;26:486-92
47. Turner RS. Alzheimer's disease. Semin Neurol. 2006;26:499-506
48. Kalia LV, Lang AE. Parkinson's disease. Lancet. 2015;386:896-912
49. Caron NS, Dorsey ER, Hayden MR. Therapeutic approaches to Huntington disease: from the bench to the clinic. Nat Rev Drug Discov. 2018;17:729-50
50. Brown RH Jr, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377:1602
51. van de Ven RAH, Santos D, Haigis MC. Mitochondrial sirtuins and molecular mechanisms of aging. Trends Mol Med. 2017;23:320-31
52. Fu J, Jin J, Cichewicz RH, Hageman SA, Ellis TK, Xiang L. et al. trans-(-)-ε-Viniferin increases mitochondrial sirtuin 3 (SIRT3), activates AMP-activated protein kinase (AMPK), and protects cells in models of Huntington disease. J Biol Chem. 2012;287:24460-72
53. Lee J, Kim Y, Liu T, Hwang YJ, Hyeon SJ, Im H. et al. SIRT3 deregulation is linked to mitochondrial dysfunction in Alzheimer's disease. Aging Cell. 2018 17
54. Shi H, Deng HX, Gius D, Schumacker PT, Surmeier DJ, Ma YC. Sirt3 protects dopaminergic neurons from mitochondrial oxidative stress. Hum Mol Genet. 2017;26:1915-26
55. Ansari A, Rahman MS, Saha SK, Saikot FK, Deep A, Kim KH. Function of the SIRT3 mitochondrial deacetylase in cellular physiology, cancer, and neurodegenerative disease. Aging Cell. 2017;16:4-16
56. Wong GH, Elwell JH, Oberley LW, Goeddel DV. Manganous superoxide dismutase is essential for cellular resistance to cytotoxicity of tumor necrosis factor. Cell. 1989;58:923-31
57. Jacobs KM, Pennington JD, Bisht KS, Aykin-Burns N, Kim HS, Mishra M. et al. SIRT3 interacts with the daf-16 homolog FOXO3a in the mitochondria, as well as increases FOXO3a dependent gene expression. Int J Biol Sci. 2008;4:291-9
58. Ronkainen PH, Pöllänen E, Alén M, Pitkänen R, Puolakka J, Kujala UM. et al. Global gene expression profiles in skeletal muscle of monozygotic female twins discordant for hormone replacement therapy. Aging Cell. 2010;9:1098-110
59. Du Z, Zhang W, Wang S, Zhang J, He J, Wang Y. et al. Celastrol protects human retinal pigment epithelial cells against hydrogen peroxide mediated oxidative stress, autophagy, and apoptosis through sirtuin 3 signal pathway. J Cell Biochem. 2019;120:10413-20
60. Song C, Peng W, Yin S, Zhao J, Fu B, Zhang J. et al. Melatonin improves age-induced fertility decline and attenuates ovarian mitochondrial oxidative stress in mice. Sci Rep. 2016;6:35165
61. Lee FY, Sun CK, Sung PH, Chen KH, Chua S, Sheu JJ. et al. Daily melatonin protects the endothelial lineage and functional integrity against the aging process, oxidative stress, and toxic environment and restores blood flow in critical limb ischemia area in mice. J Pineal Res. 2018;65:e12489
62. Zhang M, Deng YN, Zhang JY, Liu J, Li YB, Su H. et al. SIRT3 protects Rotenone-induced injury in SH-SY5Y cells by promoting autophagy through the LKB1-AMPK-mTOR pathway. Aging Dis. 2018;9:273-86
63. Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R. et al. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature. 2013;494:323-7
64. Harper JW, Ordureau A, Heo JM. Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol. 2018;19:93-108
65. Li Y, Ma Y, Song L, Yu L, Zhang L, Zhang Y. et al. SIRT3 deficiency exacerbates p53/Parkin-mediated mitophagy inhibition and promotes mitochondrial dysfunction: implication for aged hearts. Int J Mol Med. 2018;41:3517-26
66. Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176:11-42
67. Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov. 2017;16:487-511
68. Ouyang L, Zhang L, Zhang S, Yao D, Zhao Y, Wang G. et al. Small-molecule activator of UNC-51-like knase 1 (ULK1) that induces cytoprotective autophagy for Parkinson's disease treatment. J Med Chem. 2018;61:2776-92
69. Ouyang L, Zhang L, Fu L, Liu B. A small-molecule activator induces ULK1-modulating autophagy-associated cell death in triple negative breast cancer. Autophagy. 2017;13:777-8
70. Bové J, Martínez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci. 2011;12:437-52
71. Ali YO, Allen HM, Yu L, Li-Kroeger D, Bakhshizadehmahmoudi D, Hatcher A. et al. NMNAT2:HSP90 complex mediates proteostasis in proteinopathies. PLoS Biol. 2016;14:e1002472
72. Li H, Feng Z, Wu W, Li J, Zhang J, Xia T. SIRT3 regulates cell proliferation and apoptosis related to energy metabolism in non-small cell lung cancer cells through deacetylation of NMNAT2. Int J Oncol. 2013;43:1420-30
73. Di Stefano M, Loreto A, Orsomando G, Mori V, Zamporlini F, Hulse RP. et al. NMN deamidase delays wallerian degeneration and rescues axonal defects caused by NMNAT2 deficiency in vivo. Curr Biol. 2017;27:784-94
74. Warburg O. On the origin of cancer cells. Science. 1956;123:309-14
75. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029-33
76. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27-47
77. Baccelli I, Gareau Y, Lehnertz B, Gingras S, Spinella JF, Corneau S. et al. Mubritinib targets the electron transport chain complex I and reveals the landscape of OXPHOS dependency in acute myeloid leukemia. Cancer Cell. 2019;36:84-99.e8
78. Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C. et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med. 2018;24:1036-46
79. Pollyea DA, Stevens BM, Jones CL, Winters A, Pei S, Minhajuddin M. et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med. 2018;24:1859-66
80. Bell EL, Emerling BM, Ricoult SJ, Guarente L. SirT3 suppresses hypoxia inducible factor 1α and tumor growth by inhibiting mitochondrial ROS production. Oncogene. 2011;30:2986-96
81. Finley LW, Carracedo A, Lee J, Souza A, Egia A, Zhang J. et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization. Cancer Cell. 2011;19:416-28
82. Ahmed MA, O'Callaghan C, Chang ED, Jiang H, Vassilopoulos A. Context-dependent roles for SIRT2 and SIRT3 in tumor development upon calorie restriction or high fat diet. Front Oncol. 2019;9:1462
83. Torrens-Mas M, Oliver J, Roca P, Sastre-Serra J. SIRT3: oncogene and tumor suppressor in cancer. Cancers (Basel). 2017 9
84. Bell EL, Guarente L. The SirT3 divining rod points to oxidative stress. Mol Cell. 2011;42:561-8
85. Fan J, Shan C, Kang HB, Elf S, Xie J, Tucker M. et al. Tyr phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3 to regulate the pyruvate dehydrogenase complex. Mol Cell. 2014;53:534-48
86. Zhao K, Zhou Y, Qiao C, Ni T, Li Z, Wang X. et al. Oroxylin A promotes PTEN-mediated negative regulation of MDM2 transcription via SIRT3-mediated deacetylation to stabilize p53 and inhibit glycolysis in wt-p53 cancer cells. J Hematol Oncol. 2015;8:41
87. Puzio-Kuter AM. The role of p53 in metabolic regulation. Genes Cancer. 2011;2:385-91
88. Wei L, Zhou Y, Dai Q, Qiao C, Zhao L, Hui H. et al. Oroxylin A induces dissociation of hexokinase II from the mitochondria and inhibits glycolysis by SIRT3-mediated deacetylation of cyclophilin D in breast carcinoma. Cell Death Dis. 2013;4:e601
89. Yang H, Zhou L, Shi Q, Zhao Y, Lin H, Zhang M. et al. SIRT3-dependent GOT2 acetylation status affects the malate-aspartate NADH shuttle activity and pancreatic tumor growth. EMBO J. 2015;34:1110-25
90. Xiong Y, Wang M, Zhao J, Han Y, Jia L. Sirtuin 3: a Janus face in cancer (review). Int J Oncol. 2016;49:2227-35
91. Lee JJ, van de Ven RAH, Zaganjor E, Ng MR, Barakat A, Demmers J. et al. Inhibition of epithelial cell migration and Src/FAK signaling by SIRT3. Proc Natl Acad Sci U S A. 2018;115:7057-62
92. Xiao K, Jiang J, Wang W, Cao S, Zhu L, Zeng H. et al. Sirt3 is a tumor suppressor in lung adenocarcinoma cells. Oncol Rep. 2013;30:1323-8
93. Yu W, Denu RA, Krautkramer KA, Grindle KM, Yang DT, Asimakopoulos F. et al. Loss of SIRT3 provides growth advantage for B cell malignancies. J Biol Chem. 2016;291:3268-79
94. Song CL, Tang H, Ran LK, Ko BC, Zhang ZZ, Chen X. et al. Sirtuin 3 inhibits hepatocellular carcinoma growth through the glycogen synthase kinase-3β/BCL2-associated X protein-dependent apoptotic pathway. Oncogene. 2016;35:631-41
95. Liu Y, Liu YL, Cheng W, Yin XM, Jiang B. The expression of SIRT3 in primary hepatocellular carcinoma and the mechanism of its tumor suppressing effects. Eur Rev Med Pharmacol Sci. 2017;21:978-98
96. Sengupta A, Haldar D. Human sirtuin 3 (SIRT3) deacetylates histone H3 lysine 56 to promote nonhomologous end joining repair. DNA Repair (Amst). 2018;61:1-16
97. Cheng Y, Ren X, Gowda AS, Shan Y, Zhang L, Yuan YS. et al. Interaction of Sirt3 with OGG1 contributes to repair of mitochondrial DNA and protects from apoptotic cell death under oxidative stress. Cell Death Dis. 2013;4:e731
98. Lu Z, Bauzon F, Fu H, Cui J, Zhao H, Nakayama K. et al. Skp2 suppresses apoptosis in Rb1-deficient tumours by limiting E2F1 activity. Nat Commun. 2014;5:3463
99. Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, Kaelin WG Jr. Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature. 2004;428:194-8
100. Inuzuka H, Gao D, Finley LW, Yang W, Wan L, Fukushima H. et al. Acetylation-dependent regulation of Skp2 function. Cell. 2012;150:179-93
101. Zhang YK, Qu YY, Lin Y, Wu XH, Chen HZ, Wang X. et al. Enoyl-CoA hydratase-1 regulates mTOR signaling and apoptosis by sensing nutrients. Nat Commun. 2017;8:464
102. Jo H, Park Y, Kim T, Kim J, Lee JS, Kim SY. et al. Modulation of SIRT3 expression through CDK4/6 enhances the anti-cancer effect of sorafenib in hepatocellular carcinoma cells. BMC Cancer. 2020;20:332
103. Casadei Gardini A, Faloppi L, De Matteis S, Foschi FG, Silvestris N, Tovoli F. et al. Metformin and insulin impact on clinical outcome in patients with advanced hepatocellular carcinoma receiving sorafenib: validation study and biological rationale. Eur J Cancer. 2017;86:106-14
104. Tao NN, Zhou HZ, Tang H, Cai XF, Zhang WL, Ren JH. et al. Sirtuin 3 enhanced drug sensitivity of human hepatoma cells through glutathione S-transferase pi 1/JNK signaling pathway. Oncotarget. 2016;7:50117-30
105. Hou L, Wang R, Wei H, Li S, Liu L, Lu X. et al. ABT737 enhances ovarian cancer cells sensitivity to cisplatin through regulation of mitochondrial fission via Sirt3 activation. Life Sci. 2019;232:116561
106. Ma J, Liu B, Yu D, Zuo Y, Cai R, Yang J. et al. SIRT3 deacetylase activity confers chemoresistance in AML via regulation of mitochondrial oxidative phosphorylation. Br J Haematol. 2019;187:49-64
107. Bergaggio E, Riganti C, Garaffo G, Vitale N, Mereu E, Bandini C. et al. IDH2 inhibition enhances proteasome inhibitor responsiveness in hematological malignancies. Blood. 2019;133:156-67
108. Li M, Chiang YL, Lyssiotis CA, Teater MR, Hong JY, Shen H. et al. Non-oncogene addiction to SIRT3 plays a critical role in lymphomagenesis. Cancer Cell. 2019;35:916-31.e9
109. Xiong Y, Wang L, Wang S, Wang M, Zhao J, Zhang Z. et al. SIRT3 deacetylates and promotes degradation of P53 in PTEN-defective non-small cell lung cancer. J Cancer Res Clin Oncol. 2018;144:189-98
110. Wang Q, Ye S, Chen X, Xu P, Li K, Zeng S. et al. Mitochondrial NOS1 suppresses apoptosis in colon cancer cells through increasing SIRT3 activity. Biochem Biophys Res Commun. 2019;515:517-23
111. Kim YS, Gupta Vallur P, Jones VM, Worley BL, Shimko S, Shin DH. et al. Context-dependent activation of SIRT3 is necessary for anchorage-independent survival and metastasis of ovarian cancer cells. Oncogene. 2020;39:1619-33
112. Xu LX, Hao LJ, Ma JQ, Liu JK, Hasim A. SIRT3 promotes the invasion and metastasis of cervical cancer cells by regulating fatty acid synthase. Mol Cell Biochem. 2020;464:11-20
113. Chen S, Yang X, Yu M, Wang Z, Liu B, Liu M. et al. SIRT3 regulates cancer cell proliferation through deacetylation of PYCR1 in proline metabolism. Neoplasia. 2019;21:665-75
114. Ventura-Clapier R, Garnier A, Veksler V. Energy metabolism in heart failure. J Physiol. 2004;555:1-13
115. Murphy MP, Hartley RC. Mitochondria as a therapeutic target for common pathologies. Nat Rev Drug Discov. 2018;17:865-86
116. Dominic EA, Ramezani A, Anker SD, Verma M, Mehta N, Rao M. Mitochondrial cytopathies and cardiovascular disease. Heart. 2014;100:611-8
117. Koentges C, Pfeil K, Schnick T, Wiese S, Dahlbock R, Cimolai MC. et al. SIRT3 deficiency impairs mitochondrial and contractile function in the heart. Basic Res Cardiol. 2015;110:36
118. Wei T, Huang G, Gao J, Huang C, Sun M, Wu J. et al. Sirtuin 3 deficiency accelerates hypertensive cardiac remodeling by impairing angiogenesis. J Am Heart Assoc. 2017 6
119. He X, Zeng H, Chen ST, Roman RJ, Aschner JL, Didion S. et al. Endothelial specific SIRT3 deletion impairs glycolysis and angiogenesis and causes diastolic dysfunction. J Mol Cell Cardiol. 2017;112:104-13
120. Zeng H, Vaka VR, He X, Booz GW, Chen JX. High-fat diet induces cardiac remodelling and dysfunction: assessment of the role played by SIRT3 loss. J Cell Mol Med. 2015;19:1847-56
121. Gaul DS, Weber J, van Tits LJ, Sluka S, Pasterk L, Reiner MF. et al. Loss of Sirt3 accelerates arterial thrombosis by increasing formation of neutrophil extracellular traps and plasma tissue factor activity. Cardiovasc Res. 2018;114:1178-88
122. Weinert BT, Moustafa T, Iesmantavicius V, Zechner R, Choudhary C. Analysis of acetylation stoichiometry suggests that SIRT3 repairs nonenzymatic acetylation lesions. EMBO J. 2015;34:2620-32
123. Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A. et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A. 2008;105:14447-52
124. Koentges C, Cimolai MC, Pfeil K, Wolf D, Marchini T, Tarkhnishvili A. et al. Impaired SIRT3 activity mediates cardiac dysfunction in endotoxemia by calpain-dependent disruption of ATP synthesis. J Mol Cell Cardiol. 2019;133:138-47
125. Pillai VB, Sundaresan NR, Kim G, Gupta M, Rajamohan SB, Pillai JB. et al. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J Biol Chem. 2010;285:3133-44
126. Benigni A, Cassis P, Conti S, Perico L, Corna D, Cerullo D. et al. Sirt3 deficiency shortens life span and impairs cardiac mitochondrial function rescued by Opa1 gene transfer. Antioxid Redox Signal. 2019;31:1255-71
127. McKinsey TA, Kass DA. Small-molecule therapies for cardiac hypertrophy: moving beneath the cell surface. Nat Rev Drug Discov. 2007;6:617-35
128. Skurk C, Izumiya Y, Maatz H, Razeghi P, Shiojima I, Sandri M. et al. The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signaling. J Biol Chem. 2005;280:20814-23
129. Joseph J, Ametepe ES, Haribabu N, Agbayani G, Krishnan L, Blais A. et al. Inhibition of ROS and upregulation of inflammatory cytokines by FoxO3a promotes survival against Salmonella typhimurium. Nat Commun. 2016;7:12748
130. Chen T, Liu J, Li N, Wang S, Liu H, Li J. et al. Mouse SIRT3 attenuates hypertrophy-related lipid accumulation in the heart through the deacetylation of LCAD. PLoS One. 2015;10:e0118909
131. Yue Z, Ma Y, You J, Li Z, Ding Y, He P. et al. NMNAT3 is involved in the protective effect of SIRT3 in Ang II-induced cardiac hypertrophy. Exp Cell Res. 2016;347:261-73
132. Chen T, Li J, Liu J, Li N, Wang S, Liu H. et al. Activation of SIRT3 by resveratrol ameliorates cardiac fibrosis and improves cardiac function via the TGF-β/Smad3 pathway. Am J Physiol Heart Circ Physiol. 2015;308:H424-34
133. Sundaresan NR, Bindu S, Pillai VB, Samant S, Pan Y, Huang JY. et al. SIRT3 blocks aging-associated tissue fibrosis in mice by deacetylating and activating glycogen synthase kinase 3β. Mol Cell Biol. 2015;36:678-92
134. Guo X, Yan F, Li J, Zhang C, Bu P. SIRT3 attenuates AngII-induced cardiac fibrosis by inhibiting myofibroblasts transdifferentiation via STAT3-NFATc2 pathway. Am J Transl Res. 2017;9:3258-69
135. Fan D, Yang Z, Liu FY, Jin YG, Zhang N, Ni J. et al. Sesamin protects against cardiac remodeling via Sirt3/ROS pathway. Cell Physiol Biochem. 2017;44:2212-27
136. Sundaresan NR, Samant SA, Pillai VB, Rajamohan SB, Gupta MP. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol Cell Biol. 2008;28:6384-401
137. Pillai VB, Kanwal A, Fang YH, Sharp WW, Samant S, Arbiser J. et al. Honokiol, an activator of Sirtuin-3 (SIRT3) preserves mitochondria and protects the heart from doxorubicin-induced cardiomyopathy in mice. Oncotarget. 2017;8:34082-98
138. Chalkiadaki A, Guarente L. Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat Rev Endocrinol. 2012;8:287-96
139. Morigi M, Perico L, Benigni A. Sirtuins in renal health and disease. J Am Soc Nephrol. 2018;29:1799-809
140. Zhan M, Brooks C, Liu F, Sun L, Dong Z. Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013;83:568-81
141. Morigi M, Perico L, Rota C, Longaretti L, Conti S, Rottoli D. et al. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J Clin Invest. 2015;125:715-26
142. Wang Q, Xu J, Li X, Liu Z, Han Y, Xu X. et al. Sirt3 modulate renal ischemia-reperfusion injury through enhancing mitochondrial fusion and activating the ERK-OPA1 signaling pathway. J Cell Physiol. 2019;234:23495-506
143. Zhang Q, Liu X, Li N, Zhang J, Yang J, Bu P. Sirtuin 3 deficiency aggravates contrast-induced acute kidney injury. J Transl Med. 2018;16:313
144. Si Y, Bao H, Han L, Chen L, Zeng L, Jing L. et al. Dexmedetomidine attenuation of renal ischaemia-reperfusion injury requires sirtuin 3 activation. Br J Anaesth. 2018;121:1260-71
145. Ouyang J, Zeng Z, Fang H, Li F, Zhang X, Tan W. SIRT3 inactivation promotes acute kidney injury through elevated acetylation of SOD2 and p53. J Surg Res. 2019;233:221-30
146. Peng Y, Yang C, Shi X, Li L, Dong H, Liu C. et al. Sirt3 suppresses calcium oxalate-induced renal tubular epithelial cell injury via modification of FoxO3a-mediated autophagy. Cell Death Dis. 2019;10:34
147. Xi J, Jing J, Zhang Y, Liang C, Hao Z, Zhang L. et al. SIRT3 inhibited the formation of calcium oxalate-induced kidney stones through regulating NRF2/HO-1 signaling pathway. J Cell Biochem. 2018
148. Lin JR, Zheng YJ, Zhang ZB, Shen WL, Li XD, Wei T. et al. Suppression of endothelial-to-mesenchymal transition by SIRT (Sirtuin) 3 alleviated the development of hypertensive renal injury. Hypertension. 2018;72:350-60
149. Komatsu M, Kanda T, Urai H, Kurokochi A, Kitahama R, Shigaki S. et al. NNMT activation can contribute to the development of fatty liver disease by modulating the NAD (+) metabolism. Sci Rep. 2018;8:8637
150. Kendrick AA, Choudhury M, Rahman SM, McCurdy CE, Friederich M, Van Hove JL. et al. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem J. 2011;433:505-14
151. Li R, Xin T, Li D, Wang C, Zhu H, Zhou H. Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: the role of the ERK-CREB pathway and bnip3-mediated mitophagy. Redox Biol. 2018;18:229-43
152. Ma Y, Chai H, Ding Q, Qian Q, Yan Z, Ding B. et al. Hepatic SIRT3 upregulation in response to chronic alcohol consumption contributes to alcoholic liver disease in mice. Front Physiol. 2019;10:1042
153. Li S, Dou X, Ning H, Song Q, Wei W, Zhang X. et al. Sirtuin 3 acts as a negative regulator of autophagy dictating hepatocyte susceptibility to lipotoxicity. Hepatology. 2017;66:936-52
154. Ren JH, Hu JL, Cheng ST, Yu HB, Wong VKW, Law BYK. et al. SIRT3 restricts hepatitis B virus transcription and replication through epigenetic regulation of covalently closed circular DNA involving suppressor of variegation 3-9 homolog 1 and SET domain containing 1A histone methyltransferases. Hepatology. 2018;68:1260-76
155. Liu J, Li D, Zhang T, Tong Q, Ye RD, Lin L. SIRT3 protects hepatocytes from oxidative injury by enhancing ROS scavenging and mitochondrial integrity. Cell Death Dis. 2017;8:e3158
156. Song C, Zhao J, Fu B, Li D, Mao T, Peng W. et al. Melatonin-mediated upregulation of Sirt3 attenuates sodium fluoride-induced hepatotoxicity by activating the MT1-PI3K/AKT-PGC-1α signaling pathway. Free Radic Biol Med. 2017;112:616-30
157. Porter LC, Franczyk MP, Pietka T, Yamaguchi S, Lin JB, Sasaki Y. et al. NAD(+)-dependent deacetylase SIRT3 in adipocytes is dispensable for maintaining normal adipose tissue mitochondrial function and whole body metabolism. Am J Physiol Endocrinol Metab. 2018;315:E520-e30
158. Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B. et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell. 2011;44:177-90
159. Yang L, Zhang J, Xing W, Zhang X, Xu J, Zhang H. et al. SIRT3 deficiency induces endothelial insulin resistance and blunts endothelial-dependent vasorelaxation in mice and human with obesity. Sci Rep. 2016;6:23366
160. Lantier L, Williams AS, Williams IM, Yang KK, Bracy DP, Goelzer M. et al. SIRT3 is crucial for maintaining skeletal muscle insulin action and protects against severe insulin resistance in high-fat-fed mice. Diabetes. 2015;64:3081-92
161. Solman RT, Dain SJ, Keech SL. Color-mediated contrast sensitivity in disabled readers. Optom Vis Sci. 1991;68:331-7
162. He J, Liu X, Su C, Wu F, Sun J, Zhang J. et al. Inhibition of mitochondrial oxidative damage improves reendothelialization capacity of endothelial progenitor cells via SIRT3 (Sirtuin 3)-enhanced SOD2 (Superoxide Dismutase 2) deacetylation in hypertension. Arterioscler Thromb Vasc Biol. 2019;39:1682-98
163. Han L, Wang H, Li L, Li X, Ge J, Reiter RJ. et al. Melatonin protects against maternal obesity-associated oxidative stress and meiotic defects in oocytes via the SIRT3-SOD2-dependent pathway. J Pineal Res. 2017 63
164. Zhou Y, Chung ACK, Fan R, Lee HM, Xu G, Tomlinson B. et al. Sirt3 deficiency increased the vulnerability of pancreatic beta cells to oxidative stress-induced dysfunction. Antioxid Redox Signal. 2017;27:962-76
165. Jing E, Emanuelli B, Hirschey MD, Boucher J, Lee KY, Lombard D. et al. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc Natl Acad Sci U S A. 2011;108:14608-13
166. Xu H, Hertzel AV, Steen KA, Bernlohr DA. Loss of fatty acid binding protein 4/aP2 reduces macrophage inflammation through activation of SIRT3. Mol Endocrinol. 2016;30:325-34
167. Jing E, O'Neill BT, Rardin MJ, Kleinridders A, Ilkeyeva OR, Ussar S. et al. Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes. 2013;62:3404-17
168. Huynh FK, Muoio DM, Hirschey MD. SIRT3 directs carbon traffic in muscle to promote glucose control. Diabetes. 2015;64:3058-60
169. Huh JE, Shin JH, Jang ES, Park SJ, Park DR, Ko R. et al. Sirtuin 3 (SIRT3) maintains bone homeostasis by regulating AMPK-PGC-1β axis in mice. Sci Rep. 2016;6:22511
170. Zeng H, He X, Tuo QH, Liao DF, Zhang GQ, Chen JX. LPS causes pericyte loss and microvascular dysfunction via disruption of Sirt3/angiopoietins/Tie-2 and HIF-2α/Notch3 pathways. Sci Rep. 2016;6:20931
171. Xing SS, Li J, Chen L, Yang YF, He PL, Li J. et al. Salidroside attenuates endothelial cellular senescence via decreasing the expression of inflammatory cytokines and increasing the expression of SIRT3. Mech Ageing Dev. 2018;175:1-6
172. Wang J, Wang K, Huang C, Lin D, Zhou Y, Wu Y. et al. SIRT3 activation by dihydromyricetin suppresses chondrocytes degeneration via maintaining mitochondrial homeostasis. Int J Biol Sci. 2018;14:1873-82
173. Quan Y, Park W, Jin J, Kim W, Park SK, Kang KP. Sirtuin 3 activation by honokiol decreases unilateral ureteral obstruction-induced renal inflammation and fibrosis via regulation of mitochondrial dynamics and the renal NF-κBTGF-β1/Smad signaling pathway. Int J Mol Sci. 2020 21
174. Ye JS, Chen L, Lu YY, Lei SQ, Peng M, Xia ZY. SIRT3 activator honokiol ameliorates surgery/anesthesia-induced cognitive decline in mice through anti-oxidative stress and anti-inflammatory in hippocampus. CNS Neurosci Ther. 2019;25:355-66
175. Yi X, Guo W, Shi Q, Yang Y, Zhang W, Chen X. et al. SIRT3-dependent mitochondrial dynamics remodeling contributes to oxidative stress-induced melanocyte degeneration in vitiligo. Theranostics. 2019;9:1614-33
176. Li Y, Ye Z, Lai W, Rao J, Huang W, Zhang X. et al. Activation of Sirtuin 3 by silybin attenuates mitochondrial dysfunction in cisplatin-induced acute kidney injury. Front Pharmacol. 2017;8:178
177. Xu S, Gao Y, Zhang Q, Wei S, Chen Z, Dai X. et al. SIRT1/3 activation by resveratrol attenuates acute kidney injury in a septic rat model. Oxid Med Cell Longev. 2016;2016:7296092
178. Zhang M, Zhao Z, Shen M, Zhang Y, Duan J, Guo Y. et al. Polydatin protects cardiomyocytes against myocardial infarction injury by activating Sirt3. Biochim Biophys Acta Mol Basis Dis. 2017;1863:1962-72
179. Zhang H, Chen Y, Pei Z, Gao H, Shi W, Sun M. et al. Protective effects of polydatin against sulfur mustard-induced hepatic injury. Toxicol Appl Pharmacol. 2019;367:1-11
180. Zhang J, Meruvu S, Bedi YS, Chau J, Arguelles A, Rucker R. et al. Pyrroloquinoline quinone increases the expression and activity of Sirt1 and -3 genes in HepG2 cells. Nutr Res. 2015;35:844-9
181. Diaz-Morales N, Rovira-Llopis S, Bañuls C, Lopez-Domenech S, Escribano-Lopez I, Veses S. et al. Does metformin protect diabetic patients from oxidative stress and leukocyte-endothelium interactions? Antioxid Redox Signal. 2017;27:1439-45
182. Quan Y, Xia L, Shao J, Yin S, Cheng CY, Xia W. et al. Adjudin protects rodent cochlear hair cells against gentamicin ototoxicity via the SIRT3-ROS pathway. Sci Rep. 2015;5:8181
183. Zhai M, Li B, Duan W, Jing L, Zhang B, Zhang M. et al. Melatonin ameliorates myocardial ischemia reperfusion injury through SIRT3-dependent regulation of oxidative stress and apoptosis. J Pineal Res. 2017 63
184. Pi H, Xu S, Reiter RJ, Guo P, Zhang L, Li Y. et al. SIRT3-SOD2-mROS-dependent autophagy in cadmium-induced hepatotoxicity and salvage by melatonin. Autophagy. 2015;11:1037-51
185. Ma S, Chen J, Feng J, Zhang R, Fan M, Han D. et al. Melatonin ameliorates the progression of atherosclerosis via mitophagy activation and NLRP3 inflammasome inhibition. Oxid Med Cell Longev. 2018;2018:9286458
186. Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M. et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell. 2008;13:454-63
187. Wang LJ, Lee YC, Huang CH, Shi YJ, Chen YJ, Pei SN. et al. Non-mitotic effect of albendazole triggers apoptosis of human leukemia cells via SIRT3/ROS/p38 MAPK/TTP axis-mediated TNF-α upregulation. Biochem Pharmacol. 2019;162:154-68
188. Nguyen GT, Gertz M, Steegborn C. Crystal structures of Sirt3 complexes with 4'-bromo-resveratrol reveal binding sites and inhibition mechanism. Chem Biol. 2013;20:1375-85
189. George J, Nihal M, Singh CK, Ahmad N. 4'-Bromo-resveratrol, a dual Sirtuin-1 and Sirtuin-3 inhibitor, inhibits melanoma cell growth through mitochondrial metabolic reprogramming. Mol Carcinog. 2019;58:1876-85
190. Mahajan SS, Scian M, Sripathy S, Posakony J, Lao U, Loe TK. et al. Development of pyrazolone and isoxazol-5-one cambinol analogues as sirtuin inhibitors. J Med Chem. 2014;57:3283-94
191. Chen D, Zheng W. Cyclic peptide-based potent and selective SIRT1/2 dual inhibitors harboring N(ε)-thioacetyl-lysine. Bioorg Med Chem Lett. 2016;26:5234-9
192. Chen B, Wang J, Huang Y, Zheng W. Human SIRT3 tripeptidic inhibitors containing N(ε)-thioacetyl-lysine. Bioorg Med Chem Lett. 2015;25:3481-7
193. Galli U, Mesenzani O, Coppo C, Sorba G, Canonico PL, Tron GC. et al. Identification of a sirtuin 3 inhibitor that displays selectivity over sirtuin 1 and 2. Eur J Med Chem. 2012;55:58-66
194. Gertz M, Fischer F, Nguyen GT, Lakshminarasimhan M, Schutkowski M, Weyand M. et al. Ex-527 inhibits Sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc Natl Acad Sci U S A. 2013;110:E2772-81
195. Karaman B, Sippl W. Docking and binding free energy calculations of sirtuin inhibitors. Eur J Med Chem. 2015;93:584-98
196. Zhou Y, Li C, Peng J, Xie L, Meng L, Li Q. et al. DNA-encoded dynamic chemical library and its applications in ligand discovery. J Am Chem Soc. 2018;140:15859-67
197. Disch JS, Evindar G, Chiu CH, Blum CA, Dai H, Jin L. et al. Discovery of thieno[3,2-d]pyrimidine-6-carboxamides as potent inhibitors of SIRT1, SIRT2, and SIRT3. J Med Chem. 2013;56:3666-79
198. Alhazzazi TY, Kamarajan P, Xu Y, Ai T, Chen L, Verdin E. et al. A novel sirtuin-3 inhibitor, LC-0296, inhibits cell survival and proliferation, and promotes apoptosis of head and neck cancer cells. Anticancer Res. 2016;36:49-60
199. Chen ML, Zhu XH, Ran L, Lang HD, Yi L, Mi MT. Trimethylamine-N-Oxide induces vascular inflammation by activating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS signaling pathway. J Am Heart Assoc. 2017 6
200. Gorska-Ponikowska M, Kuban-Jankowska A, Eisler SA, Perricone U, Lo Bosco G, Barone G. et al. 2-Methoxyestradiol affects mitochondrial biogenesis pathway and succinate dehydrogenase complex flavoprotein subunit a in osteosarcoma cancer cells. Cancer Genomics Proteomics. 2018;15:73-89
Author contact
![]() Corresponding authors: Bo Liu (B.L.), E-mail: liubo2400com; Phone: (+86)28-85164063. Rong-Rong He (R.H.) E-mail: rongrongheedu.cn; Phone: (+86)20-85221559. Yi Chen (Y.C.), E-mail: toddychancom; Phone: (+86)28-85164063.
Corresponding authors: Bo Liu (B.L.), E-mail: liubo2400com; Phone: (+86)28-85164063. Rong-Rong He (R.H.) E-mail: rongrongheedu.cn; Phone: (+86)20-85221559. Yi Chen (Y.C.), E-mail: toddychancom; Phone: (+86)28-85164063.