Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(8):2189-2201. doi:10.7150/thno.22800 This issue Cite
Research Paper
Glucocorticoids Inhibit Oncogenic RUNX1-ETO in Acute Myeloid Leukemia with Chromosome Translocation t(8;21)
Lianghao Lu1,†, Yefei Wen1,†, Yuan Yao1, Fengju Chen2, Guohui Wang1, Fangrui Wu1, Jingyu Wu1, Padmini Narayanan3,4, Michele Redell3,4, Qianxing Mo2,5, Yongcheng Song1,2, ![]()
1. Department of Pharmacology and Chemical Biology, Baylor College of Medicine, 1 Baylor Plaza, Houston, TX 77030, USA.
2. Dan L. Duncan Cancer Center, Baylor College of Medicine, 1 Baylor Plaza, Houston, TX 77030, USA.
3. Department of Pediatrics, Baylor College of Medicine, 1 Baylor Plaza, Houston, TX 77030, USA.
4. Texas Children's Cancer and Hematology Centers, 1102 Bates Street, Houston, TX 77030, USA.
5. Department of Medicine, Baylor College of Medicine, 1 Baylor Plaza, Houston, TX 77030, USA.
† These authors contributed equally.
Received 2017-9-12; Accepted 2017-11-20; Published 2018-3-8
Abstract

Acute myeloid leukemia (AML) is a major blood cancer with poor prognosis. New therapies are needed to target oncogene-driven leukemia stem cells, which account for relapse and resistance. Chromosome translocation t(8;21), which produces RUNX1-ETO (R-E) fusion oncoprotein, is found in ~13% AML. R-E dominance negatively inhibits global gene expression regulated by RUNX1, a master transcription factor for hematopoiesis, causing increased self-renewal and blocked cell differentiation of hematopoietic progenitor cells, and eventually leukemia initiation.
Methods: Connectivity-Map followed by biological activity testing were used to identify candidate compounds that can inhibit R-E-mediated gene transcription. Molecular mechanistic studies were also performed.
Results: Glucocorticoid drugs, such as betamethasone and dexamethasone, were found to exhibit potent and selective in vitro and in vivo activities against R-E leukemia, as well as strong synergy when combined with chemotherapeutics. Microarray analysis showed that treatment with glucocorticoids significantly inhibited R-E's activity and reactivated that of RUNX1. Such gene expression changes caused differentiation and apoptosis of R-E leukemia cells. Our studies also show a possible molecular mechanism for the targeted therapy. Upon treatment with a glucocorticoid drug, more glucocorticoid receptor (GR) was translocated into the nucleus and bound to DNA, including promoters of RUNX1 target genes. GR was found to associate with RUNX1, but not R-E. This interaction increased binding of RUNX1 to DNA and reduced that of R-E, shifting to a RUNX1 dominance.
Conclusion: Glucocorticoid drugs represent a targeted therapy for AML with chromosome translocation t(8:21). Given their high activity, favorable human pharmacokinetics as well as synergy with chemotherapeutics, glucocorticoids could be clinically useful to treat R-E AML.
Keywords: Acute myeloid leukemia, chromosome translocation t(8, 21), RUNX1-ETO, Glucocorticoid, Glucocorticoid receptor, Targeted therapy
Introduction
Hematopoiesis requires exquisite regulation of gene expression for daily production of ~1011-12 differentiated blood cells from hematopoietic stem cells (HSC) in the bone marrow. Transcription factor RUNX1 (also known as AML1) is a master regulator for hematopoiesis (1, 2). RUNX1 contains an N-terminal RUNT domain (50-177), which is highly conserved from yeast to mammals, a C-terminal transactivation domain (TAD) and an inhibitory domain (ID) (3, 4). The RUNT domain forms a heterodimeric complex with CBFβ and binds to DNA. While CBFβ does not interact with DNA directly, it can significantly increase the DNA binding affinity of RUNT through an allosteric regulation (5). The biological function of RUNX1 is that its RUNT domain recognizes and binds to a range of gene promoters, while TAD recruits other proteins, which can either activate or repress transcription of its target genes, in a promoter- or cell type-specific manner (1, 6-9).
Translocations and mutations of RUNX1 or its partner CBFβ account for the largest portion (~25%) of leukemia in humans (1, 10, 11). Chromosome translocation t(8;21), producing a fusion oncogene/protein RUNX1-ETO (R-E), is found in ~13% of acute myeloid leukemia (AML) (1, 10). R-E contains the N-terminal 1-177 portion of RUNX1 fused with ETO. ETO is a transcription repressor, able to recruit several transcription cofactors to repress or occasionally activate the RUNX1 target genes (12-14). Expression of R-E can block cell differentiation and enhance self-renewal of hematopoietic progenitor cells (15, 16). Although expressing R-E alone does not cause immediate initiation of acute leukemia, a second proto-oncogene (e.g., N-Ras or Kit), which provides a proliferative or survival advantage, can lead to a rapid onset of leukemia (17-20). RUNX1-ETO9a (R-E9a) is an alternative splicing isoform, which was found in most t(8;21) AML patients and is significantly more leukemogenic (21).
Dominant-negative inhibition of RUNX1 by R-E has been found to be the mechanism of the leukemia (1, 2). R-E (or R-E9a) competes with RUNX1 to bind to the same DNA sequences, with ETO regulating gene expression that is otherwise controlled by RUNX1 (22, 23). Chromatin immunoprecipitation (ChIP) followed by sequencing (ChIP-seq) showed that RUNX1 and R-E compete for the same DNA binding loci, with R-E being dominant (24, 25). R-E forms a stable transcription complex with other transcription factors, including HEB/E2A, LYL1 and LMO2, to inhibit hematopoietic differentiation and activate gene expression for stem cell maintenance. Selective knockdown of R-E shifted the balance to a RUNX1 dominance. This event caused a broad range of gene expression changes, inhibited cell proliferation, and induced differentiation and apoptosis. Of interest is that the wild-type RUNX1 in the other allele has also been found to be essential for t(8;21) AML (26). Knockdown of RUNX1 caused apoptosis of these leukemia cells. Both R-E and RUNX1 activities are therefore required to maintain oncogenic gene expression in the leukemia.
It is clear that inhibition of R-E's activity is key to curing leukemia. However, R-E is an onco-transcription factor, which is considered undruggable because it is very difficult to find small molecules that disrupt protein-DNA interactions. Moreover, because R-E and RUNX1 have the same DNA-binding domain, such compounds could also disrupt the binding of RUNX1 to DNA. This could be toxic given RUNX1's critical roles in normal hematopoiesis. An alternative approach is to find compounds that can inhibit the oncogenic gene expression controlled by R-E. Here, using a bioinformatics method followed by biological activity testing, we show several FDA-approved drugs can significantly inhibit R-E-mediated gene expression in t(8;21) AML. These compounds potently inhibited cell proliferation in vitro and in vivo, impaired self-renewal of R-E containing leukemia initiating cells (LICs, also known as leukemia stem cells), and induced differentiation and apoptosis. Our studies also show a possible molecular mechanism for the potent and selective activity against R-E leukemia.
Materials and Methods
Human primary cells and cell lines. Human cell lines Kasumi-1, Jurkat, MCF-7 and WI-38 were obtained from ATCC and cultured in RPMI1640 or DMEM supplemented with 10% fetal bovine serum. NB4 and MV4-11 cells were described in our previous publication. SKNO-1 cells were a gift from Dr. Kimberly Stegmaier.
The primary leukemia cells were obtained from a child diagnosed with AML with t(8;21) who was treated at Texas Children's Cancer Center and whose parents consented to storage of leukemia cells for future research, in accordance with the IRB-approved protocol H-3342.
Chemicals. All compounds were purchased from Sigma-Aldrich, Alfa Aesar, or TCI.
Antiproliferation assay. Antiproliferation assay for leukemia cells was performed using an XTT assay kit (Roche) and that for attachment cell lines MCF-7 and WI-38 was done using an MTT assay, using our previous methods (27-29). EC50 values were determined by Prism (GraphPad) from 3 independent experiments.
Flow cytometry, Western blot and quantitative real-time PCR (qPCR). 105 cells/well were incubated with increasing concentrations of a compound for 3 days. FACS assays to determine differentiation and apoptosis were performed on a FACS Calibur (Applied Biosystems) with cells labeled with fluorochrome-conjugated monoclonal antibodies against human CD14 and CD11b or using the FITC Annexin V Apoptosis Detection Kit I (BD Bioscience) according to the manufacturer's protocol. Data were processed using the program Flowjo (version7.6.5).
Cells were harvested, washed with PBS and lysed with a hypotonic buffer (25 mM Tris pH 7.4, 1 mM MgCl2, 5 mM KCl, 1% NP-40) for 5 min on ice. Supernatant containing cytoplasmic proteins was collected by centrifugation for 8 min at 800 g at 4°C. The nuclei pellet was washed with the hypotonic buffer and lysed with a lysis buffer (25 mM Tris pH 8.0, 150 mM NaCl, 0.5% NP-40, 0.5% sodium deoxycholate and 0.2% SDS). Nuclear proteins in the supernatant were collected by centrifugation. Cytoplasmic proteins and nuclear proteins were used for WB and immunoprecipitation. For Western blot, equal amounts of proteins (2 µg) were separated on SDS-PAGE and transferred to PVDF membranes. The blots were probed with antibodies of GR (cell signaling), ETO (Santa Cruz) and RUNX1 (Santa Cruz), followed by anti-rabbit IgG (Thermo Scientific) secondary antibodies.
For qPCR, RNA was extracted using RNeasy mini kit (Qiagen). 100-1000 ng of total RNA was reverse transcribed using iScript™ Reverse Transcription Supermix (Bio-Rad) using the manufacturer's protocol. Quantitative real-time PCR was carried out using Fast SYBR Green Master Mix (Applied Biosystems) according to the manufacturer's instructions. Measurements were performed in triplicate, using GAPDH as the reference gene. Real-time PCR was performed using Biosystems Step One Plus detection system. Primers used for qPCR are shown in Table S1.
Colony-forming assay. Retrovirus transduction and transformation assays were performed using R-E or R-E9a containing MSCV plasmids (from Addgene), according to methods as described previously. Treatment with various concentrations of a drug was applied in the 3rd round of replating. The colony number of each well was counted, imported into Prism 5.0 and EC50 values were determined using the sigmoidal dose response curve fitting in the software. Colony-forming assay with primary leukemia cells was performed according to our previously published method (30, 31).
In vivo activity studies. All of the mouse studies were conducted in strict compliance with an IRB-approved protocol. NOD-SCID mice (4 to 6 weeks old, from Jackson lab) were obtained and maintained under specific pathogen-free conditions. 2x107 SKNO-1 cells in PBS and Matrigel (1:1, 0.1 mL) were inoculated subcutaneously and palpable tumors (2-3 mm in diameter) developed in ~2 weeks. Mice were treated with a drug (10 or 20 mg/kg/day) in PBS (0.1 mL) administered intraperitoneally for 10x in 2 weeks. Tumors were measured every 2 days and estimated by using the formula a×b2/2.
RNA Amplification and Microarray Data Analysis. 105 cells/well were incubated with Bet (50 nM) for 2 days. Cells were treated with Trizol then frozen in liquid nitrogen. RNA from these samples was isolated, amplified, and hybridized to Agilent Human Gene Expression v3 8x60k microarray according to the manufacturer's protocol. Microarray data were log2 transformed and normalized to have the same median for all the arrays. Moderate t-statistics were used to find genes that were differentially expressed between the samples of interest. Benjamini and Hochberg method was used to correct for multiple comparisons. R and Bioconductor packages were applied for all the statistical analyses (see http://cran.us.r-project.org/, http://www.bioconductor.org/). GSEA analysis was performed using GSEA software from Broad Institute (Boston, MA).
Chromatin immunoprecipitation. Kasumi-1 cells were cross-linked with 1% formaldehyde at room temperature for 10 min, followed by the addition of 125 mM glycine. Cells were lysed with nuclear lysis buffer and sonicated to ~200-500 bp fragments (Diagenode), which were incubated at 4°C overnight with a ChIP-grade antibody. Protein A magnetic Dynabeads (10 µL, Invitrogen) were added and incubated for 2 h. The beads were washed 3x with RIPA buffer and 2x with TE buffer. DNA on the beads was eluted for 2 h at 68°C in 100 μL of an elution buffer (20 mM Tris pH 7.5, 5 mM EDTA, 50 mM NaCl, 1% SDS, 50 μg/mL proteinase K) (2x), and purified using a ChIP DNA Clean & Concentrator kit (Zymo Research).
Results
Bioinformatics search identified compounds having selective activity against R-E leukemia. Connectivity Map (C-map) (32, 33) was used to search potential compounds that can inhibit R-E mediated gene expression. C-map consists of a large collection of whole genome expression data (>7,000 profiles) from cultured human cells treated with 1,309 bioactive, commercially available small molecules, most of which are FDA-approved drugs. Simple pattern-matching algorithms are used to discover connections between compounds, genes and diseases by comparing common gene expression changes.
Previous studies have identified up- or down-regulated genes upon R-E knockdown in Kasumi-1 cells with t(8;21) chromosome translocation (34, 35). Among these genes, we chose a gene signature consisting of 7 upregulated genes (RUNX3, CD11a, CD11b, IL3, BCL2, M-CSF and GM-CSF) as well as 4 downregulated genes (ID1, M-CSFR, p21 and EGR1), because they are known R-E target genes and are closely related to hematopoiesis, differentiation or self-renewal. The C-map search using these genes as a signature yielded 61 compounds showing positive C-scores with statistical significance (p < 0.05), suggesting treatment with these compounds could mimic R-E knockdown (Table S2). In order to find more potential R-E deactivators, we performed a second C-map search by expanding the gene signature to 15 up- and 7 down-regulated genes (Table S3), which yielded additional 28 compounds as well as 14 duplicate compounds for both searches. We next purchased 78 compounds (out of 89 identified by C-map searches) that were commercially available. Activities of these compounds at 1-20 µM (same as that used for the C-map profiling) were tested against proliferation of Kasumi-1 as well as Jurkat leukemia cells that do not have R-E. As shown in Figure 1A, these compounds showed an average of 28.1% anti-proliferative activity against Kasumi-1, while they did not significantly affect the growth of Jurkat cells. The differential activities (p < 0.0001) of these compounds between Kasumi-1 and Jurkat suggest these compounds might target R-E-mediated gene expression.
In vitro activity against R-E leukemia. (A) 78 compounds showed an average of 28.1% inhibition of Kasumi-1 cell growth, while they on average caused 0.2% growth inhibition against Jurkat cells (p<0.0001). (B) Dose-response curves for treatment of Kasumi-1 cells with Dex-P (in blue), Bet (in red) and Hydrocortisone (in green) and those for treatment of Jurkat (in black) and MCF-7 (in brown) cells with Bet. (C) Treatment of primary cells from a t(8;21) AML patient with Bet and hydrocortisone-inhibited colony-forming ability (EC50: ~50 nM and 500 nM). (D) Treatment with Bet and Dex-P potently inhibited the colony-forming ability of R-E or R-E9a transformed LICs (EC50: ~8 nM and 2 nM). (E) Treatment with Bet and Dex-P did not significantly affect colony-forming ability of murine hematopoietic stem/progenitor cells (HSPC).
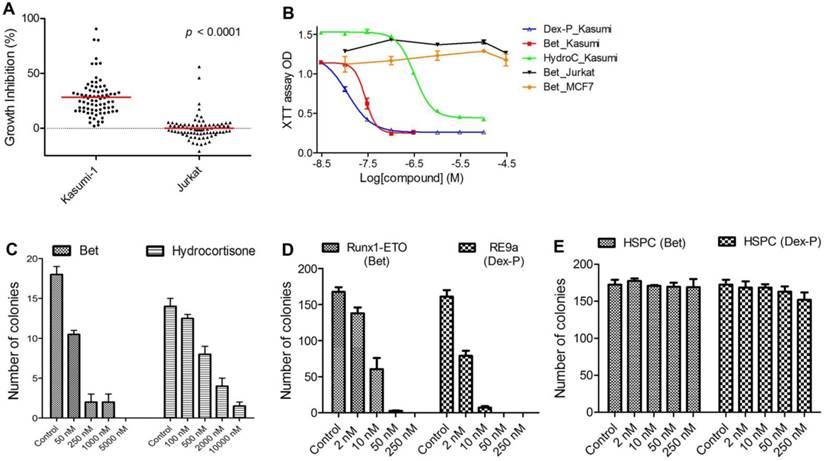
EC50 values for the most active compounds were measured and shown in Table 1. Anti-inflammatory drug betamethasone exhibited very potent activity against Kasumi-1 cells with an EC50 of 28 nM, while it did not affect growth of Jurkat cells even at 50 µM. Hsp90 inhibitors alvespimycin and tanespimycin had strong activities (EC50 = 0.048 µM and 1.1 µM) against Kasumi-1, while they were considerably less active against Jurkat. Nabumetone and Fluninxin, belonging to non-steroid anti-inflammatory drugs (NSAID), also exhibited ~15 µM EC50 values against Kasumi-1, while they were inactive against Jurkat at 50 µM. Several other drugs or natural products (dacarbazine, acepromazine, crotamiton, propafenone and scopoletin) also showed good and selective activity against Kasumi-1 cells.
Antiproliferative activity EC50 (µM) with % inhibition at 50 µM in parenthesis (if EC50 >50 µM).
| Kasumi-1 | Jurkat | |
|---|---|---|
| Betamethasone | 0.028 | >50 (6.9%) |
| Alvespimycin | 0.048 | 1.6 |
| Tanespimycin | 1.1 | ~50 |
| Nabumetone | 18.5 | >50 (-3.5%) |
| Fluninxin | 14.7 | ~50 |
| Dacarbazine | 2.9 | 15.2 |
| Acepromazine | 15.1 | >50 (21%) |
| Crotamiton | 12.5 | >50 (11.2%) |
| Propafenone | 17.2 | >50 (2.5%) |
| Scopoletin | 13.0 | >50 (29.4%) |
Literature survey indicated that 3 classes of the above compounds, i.e., steroid and non-steroid anti-inflammatory drugs and Hsp90 inhibitors, have been reported to have activity against R-E leukemia (36-39). However, to our surprise, in Ref. (37) dexamethasone (a close analog of betamethasone) did not reduce in vivo tumor burden in a mouse model of SKNO-1 leukemia despite its strong in vitro activity. Given the excellent in vitro activity and selectivity, we decided to reevaluate glucocorticoid drugs against R-E leukemia. In addition, Hsp90 inhibitors alvespimycin and tanespimycin were included, while NSAID Nabumetone and Fluninxin were not evaluated further because of their relatively weak activity against Kasumi-1 cells.
Glucocorticoids showed potent and selective cell activity against R-E leukemia. First, 4 glucocorticoid drugs, including betamethasone (Bet), dexamethasone (Dex), hydrocortisone and prednisolone, commonly used in the clinic, were evaluated against an expanded panel of leukemia cell lines, including Kasumi-1 and SKNO-1 having R-E as well as leukemia cells Jurkat, NB4 and MV4-11 without R-E. Also tested were solid tumor MCF-7 (breast cancer) and normal fibroblast WI-38 cells. Two prodrugs, betamethasone-21-acetate (Bet-Ac) and dexamethasone-21-phosphate (Dex-P), were also included with a rationale that prodrugs of these highly hydrophobic drugs can provide enhanced water solubility and/or longer plasma half-life. In vitro antitumor activities of these compounds are summarized in Table 2 and selected dose-response curves from which EC50 values were calculated are shown in Figure 1B. Bet and Dex showed comparable activity against both Kasumi-1 and SKNO-1 with EC50s of 24-59 nM, but they were inactive against all other leukemia, solid tumor or normal cells, showing a high selectivity of >2,000-fold. Hydrocortisone and prednisolone were also strong and selective against R-E leukemia cells (EC50: 110-410 nM), while these two drugs were less active than Bet and Dex. In addition, compared to Bet, Bet-Ac exhibited ~2-fold less activity. However, Dex-P was found to be the most potent compound with EC50s of 9 nM and 17 nM against Kasumi-1 and SKNO-1 cells, being ~2-fold more active than its parent compound.
Antiproliferative activity EC50 values (µM).
| Kasumi-1 | SKNO-1 | Jurkat | NB4 | MV4-11 | MCF-7 | WI-38 | |
|---|---|---|---|---|---|---|---|
| Betamethasone | 0.028 | 0.059 | >50 | >50 | >50 | >50 | >50 |
| Dexamethasone | 0.024 | 0.029 | >50 | >50 | >50 | >50 | >50 |
| Hydrocortisone | 0.32 | 0.41 | >50 | NTa | NT | NT | NT |
| Prednisolone | 0.11 | 0.15 | >50 | NT | NT | NT | NT |
| Bet-Ac | 0.050 | 0.12 | >50 | >50 | >50 | >50 | >50 |
| Dex-P | 0.009 | 0.017 | >50 | >50 | >50 | >50 | >50 |
| Alvespimycin | 0.048 | NT | 1.6 | 0.055 | 0.090 | 5.2 | 27 |
| Tanespimycin | 1.1 | NT | ~50 | 0.42 | 0.52 | >50 | >50 |
Alvespimycin and tanespimycin were less selective (Table 2). These two compounds were comparably active against NB4 and MV4-11 leukemia cells having RML-RARα and MLL-AF4 oncogenes, respectively. This is presumably because Hsp90, a chaperone protein, could be important for the folding and stability of these onco-proteins. For example, inhibition of Hsp90 was found to cause degradation of R-E, causing inhibition of its mediated gene expression (39).
Glucocorticoids potently inhibit R-E leukemia initiating cells. We next evaluated activity of glucocorticoid drugs against R-E containing leukemia initiating cells (LIC). LICs represent a small fraction of leukemia cells that have stem cell-like traits (e.g., self-renewal) and are responsible for initiating leukemia when transplanted into mice (40, 41). The clinical significance of LICs is that since they proliferate relatively slowly, they are intrinsically resistant to chemotherapeutic drugs and thus believed to be responsible for relapse and resistance. First, we tested anti-LIC activity of Bet and hydrocortisone in a colony-forming assay using primary cells from an AML patient with t(8;21) chromosome translocation. As shown in Figure 1C, Bet can potently inhibit colony-forming ability of R-E AML patient samples with an EC50 of ~50 nM. The less potent drug hydrocortisone can also inhibit colony-formation of these cells with an EC50 of ~500 nM.
Retrovirus transduction and transformation assay was used to produce R-E driven LICs, following previously published protocols (21, 42), and these R-E or R-E9a containing LICs have been well characterized. Murine stem cell retrovirus (MSCV) plasmids containing R-E and more leukemogenic R-E9a were used to transfect Phoenix packaging cells. The viral supernatant was used to infect c-kit+ hematopoietic stem/progenitor cells freshly separated from mouse bone marrow cells, after which 10,000 transduced cells/well were plated in 1% semi-solid methylcellulose stem cell culture medium supplemented with growth factors stem cell factor (SCF), IL-3, IL-6 and GM-CSF in the presence of antibiotic G418 in 24-well plates. G418 resistant colonies were replated in the same conditions every 7 days. Non-transformed cells exhausted their colony-forming capability in the second round of experiments, while transformed cells containing R-E or R-E9a had enhanced self-renewal ability, which were able to form secondary and further generations of colonies. Compound treatment was started in the third round of replating. As shown in Figure 1D, Bet and Dex-P potently inhibited colony-formation of R-E and R-E9a transformed LICs with EC50 of ~8 nM and 2 nM, respectively. However, treatment with these two drugs did not significantly affect the colony-forming ability of murine c-kit+ hematopoietic stem/progenitor cells (HSPC) even at 250 nM (Figure 1E).
Glucocorticoids inhibited R-E mediated transcriptome. Next, we performed microarray experiments to analyze global transcriptome changes caused by glucocorticoids in t(8;21) AML. Triplicate samples of Kasumi-1 cells were treated with Bet (50 nM) for 3 days. RNAs from control and treated cells were isolated, amplified and hybridized to Agilent human gene expression microarrays. Data were log2-transformed and normalized to have the same median values for comparative analysis. Moderate t-test was used to search for genes that were differentially transcribed between the control and drug treated samples, using the filter thresholds of p-values < 0.05 and fold changes >4. Gene set enrichment analysis (GSEA) was performed to find whether treatment with Bet caused significant expression alterations in R-E and RUNX1 related gene sets. As shown in Figure 2A and B, the drug treatment led to significant up- and down-regulation of gene sets that correspond to siRNA-mediated R-E knockdown in Kasumi-1 cells (34), showing treatment with Bet can mimic R-E knockdown. These results indicate that Bet can significantly inhibit R-E-mediated gene expression and act as a R-E deactivator. Additional GSEA showed that treatment with Bet can significantly upregulate the RUNX1-upregulated gene set (43) (Figure 2C), while it did not significantly downregulate the RUNX1-downregulated gene set (Figure S1). These results suggest treatment with the glucocorticoid drug can, at least partially, stimulate RUNX1's activity in gene transcription. These results were also confirmed by qPCR (Figure 2D), showing treatment with Bet caused significant downregulation of ID1, M-CSFR, p21, EGR1 and CD34 as well as upregulation of CD11a, CD11b, SLA and BAALC.
Treatment of Kasumi-1 cells with Bet (50 nM) affected R-E/RUNX1-mediated gene expression and induced differentiation and apoptosis. (A-C) GSEA plots show the drug treatment caused significant (A) up- and (B) down-regulation of the R-E up- and down-regulated gene sets, as well as significant (C) upregulation of the RUNX1-upregulated gene set. (D) qPCR results show Bet caused significant expression changes of selected R-E/RUNX1 target genes. (D) The drug treatment caused significantly increased cell populations expressing high levels of CD11b and CD14. (E) Treatment with 30 nM and 100 nM of Bet caused 6.9% and 21.8% apoptosis (as compared to the control).
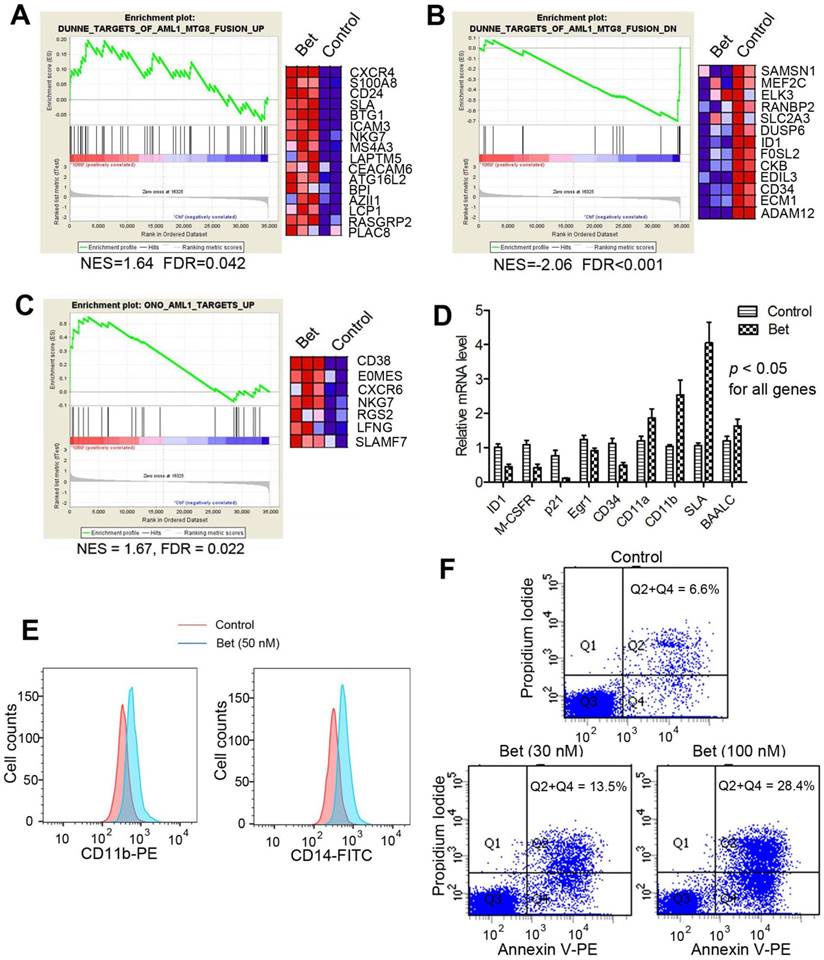
Glucocorticoids induced cell differentiation and apoptosis. Moreover, treatment with Bet was found to induce significant cell differentiation and apoptosis of R-E leukemia cells. As shown in Figure 2E, treatment of Kasumi-1 cells with Bet caused significant expansions of cell populations expressing high levels of CD14 or CD11b, which are two cell surface markers characteristic of differentiated macrophages/monocytes. In addition, treatment with Bet at 30 nM and 100 nM caused 6.9% and 21.8% apoptosis of Kasumi-1 cells (Figure 2F). These results show that activity of glucocorticoid drugs is due to inhibition of R-E-mediated gene expression as well as stimulation of that regulated by RUNX1, which caused inhibited self-renewal of R-E containing LICs (Figure 1C-D) and significant differentiation and apoptosis of R-E leukemia cells.
Dex exhibited potent in vivo antitumor activity against R-E leukemia. With the promising in vitro activities against proliferation and self-renewal of R-E leukemia cells, we evaluated the in vivo antitumor activity of these drugs. Although Dex was reported to have no in vivo activity in a mouse model of SKNO-1 leukemia, only one dosage (15 mg/kg/day i.p. injection for 4 days) was applied (37). Because Dex is a highly lipophilic compound with very low water solubility, lack of activity might be due to inappropriate dosing or formulation. Dex-P (as a disodium salt) is a water-soluble prodrug and can quickly release Dex in plasma or cells. Our in vitro testing also showed Dex-P is ~2x more active than Dex against Kasumi-1 and SKNO-1 cells (Table 2). Dex-P was therefore chosen for in vivo activity testing. In addition, Dex-P was found to have low toxicity, as it can be safely injected intraperitoneally (i.p.) at a high dose of 100 mg/kg/day for 10 injections over a period of 14 days without overt toxicities to mice. A subcutaneous (s.c.) xenograft mouse model of SKNO-1 leukemia was used for antileukemia activity assessment. 2 x 107 SKNO-1 cells/mouse in 0.1 mL of PBS and Matrigel (1:1) were injected s.c. into NOD-SCID mice, which developed palpable tumors (~2-3 mm in diameter) in ~2 weeks. In vivo antitumor experiments of Dex-P were conducted using i.p. doses of 10 and 20 mg/kg/day for 10 injections in 14 days. As shown in Figure 3A, such treatment caused significant tumor growth inhibition as well as prolonged survivals, showing Dex-P exhibited potent in vivo antitumor activity against R-E leukemia. That the 20 mg/kg dosage did not show significantly improved activity over the lower dosage might be due to a short plasma half-life (t1/2) in mice, which have a much faster metabolism as compared to humans. Nevertheless, previous studies showed Dex-P has a good pharmacokinetic profile in humans, with a t1/2 of 4.7 h (44, 45). Moreover, using a non-toxic dose of 2.5 mg/kg (i.v. infusion), the plasma concentrations of Dex were 2.3 µM and 0.75 µM after 4 h and 16 h in humans (45), respectively, which are considerably higher than Dex's in vitro EC50s (24-50 nM) against R-E leukemia cells. These human pharmacokinetic data suggest that Dex-P has the potential to be quickly used in the clinic for R-E AML, given its potent activity and good PK profile.
(A) Treatment with Dex-P caused significant tumor growth inhibition (upper panel) and prolonged survivals (lower). (B) Dose-response inhibitory activities for combination treatment of Kasumi-1 cells with Dex and Cytarabine (upper), together with calculated combination indices (CI) of 0.13-0.75 showing strong synergy (lower).
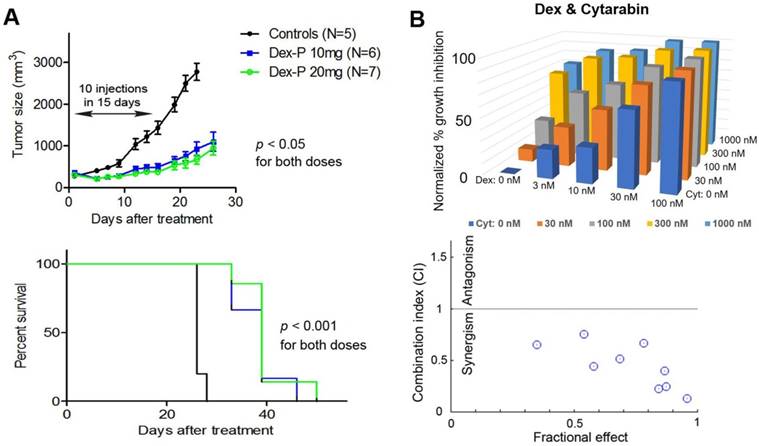
Combination therapy with Dex exhibited strong synergism. We next investigated combination therapies of Dex with cytarabine and doxorubicin, two commonly used chemotherapeutic drugs for AML. The rationale is that combination therapy is essential for modern cancer treatment because of increased clinical efficacy as well as reduced likelihood of developing drug resistance. This strategy could be of particular interest for glucocorticoids as these drugs have adverse side effects in high doses or prolonged treatment. Effective combination therapies could lower the dosage and alleviate these side effects, while retaining antitumor activity. Thus, Kasumi-1 cells were treated with a matrix of increasing concentrations of 0 to ~0.1×, 0.32×, 1×, 3× and 10×EC50 for each individual drug. Cell viability for each drug combination was determined and shown in Figure 3B. Data were analyzed by the program CompuSyn (46), which calculates the combination index (CI) for each drug combination. Combinations with CI < 1 show synergism for the two drugs, while those with CI = 1 exhibit additive effects and those with CI > 1 indicate antagonism. Dex was found to exhibit strong synergism in combination with cytarabine, with CI values ranging from 0.13 to 0.75. Similarly, combinations of Dex with doxorubicin also showed synergistic effects with CI values of 0.42-0.90 (Figure S2). These results indicate combination therapies of Dex with cytarabine or doxorubicin could be useful.
GR associates with RUNX1, but not R-E. Mechanistic investigation was performed in an effort to find the molecular basis underlying the high potency and selectivity of glucocorticoid drugs against R-E leukemia. A previous study (37) showed that glucocorticoids caused a proteasome-dependent degradation of R-E. However, it is still unclear why the drug treatment results in such R-E degradation. Glucocorticoids are ligands of glucocorticoid receptor (GR, also known as nuclear receptor 3C1 or NR3C1), an important transcription factor regulating a wide range of physiological processes including development, metabolism, and immunity (47). Without a ligand, GR is located in the cytoplasm and in complex with several proteins such as Hsp90 (48, 49). Upon binding a glucocorticoid compound such as a natural ligand hydrocortisone or synthetic drug Bet or Dex, GR undergoes considerable conformational changes, homodimerization, and translocation into the nucleus, where it binds to specific DNA responsive elements or attaches to another transcription factor to regulate gene transcription (50, 51).
Kasumi-1 is a good model for the mechanistic studies, as these cells express both R-E and RUNX1, but do not express wild-type ETO (24, 52). Endogenous R-E can be probed by an ETO antibody, while RUNX1 can be independently detected by a RUNX1 antibody that recognizes its C-terminal peptide. First, protein level changes of GR, R-E and RUNX1 were determined. Because GR plays different roles in the cytoplasm and nucleus, cytoplasmic and nuclear proteins were separated using hypertonic lysis buffer followed by nuclear lysis buffer. As shown in Figure 4A, upon treatment with Bet, GR was significantly reduced in the cytoplasm and translocated into the nucleus, causing an increased amount of GR in the nucleus. While negligible amounts of RUNX1 and R-E could be detected in the cytoplasm, Bet treatment caused an increase of RUNX1 in the nucleus as well as a reduction of R-E, the latter of which was also observed in the previous study (37).
Western blot and immunoprecipitation results. (A) Treatment of Kasumi-1 cells with Bet caused significant changes of GR, RUNX1 and R-E in the cytoplasm and nucleus. (B) Immunoprecipitation showed GR is associated with RUNX1, but not R-E in Kasumi-1. (C) Upon transfection of Flag-RUNX1 or Flag-R-E, immunoprecipitation using a Flag antibody also show that GR is associated with RUNX1, but not R-E in 293T cells. (D) Illustrations showing different constructs of RUNX1 used for transfection. (E) Immunoprecipitation results show that WT-, Δ429- and Δ391-RUNX1 is associated with GR, while Δ321-RUNX1 does not interact with GR.
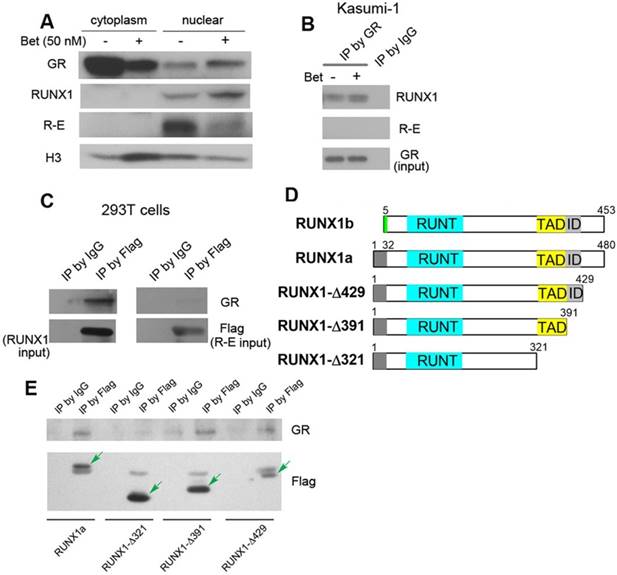
Next, immunoprecipitation (IP) was used to find whether there are physical interactions between GR, RUNX1 and R-E in the nucleus. Kasumi-1 cells were treated with Bet and the nuclear proteins from the control and treatment cells were pulled down with GR antibody-conjugated beads. As shown in Figure 4B and Figure S3, GR was found, for the first time, to be associated with RUNX1, but not R-E, using two RUNX1 antibodies. To confirm these results, pSG5 plasmids containing Flag-tagged RUNX1 or R-E were transfected into 293T cells and cultured for 2 days. The nuclear proteins were immunoprecipitated with FLAG antibody-conjugated beads. The exogenous Flag-RUNX1 was again found to be associated with endogenous GR, while there was no interaction between Flag-R-E and GR (Figure 4C).
It is of interest to know which domain of RUNX1 is essential for the binding to GR. Since R-E does not interact with GR, it is conceivable that the N-terminal 1-177 of RUNX1 is not involved in binding. Thus, three FLAG-tagged pSG5 plasmids containing truncated proteins RUNX1a-Δ429, -Δ391 and -Δ321 were constructed, in which the C-terminal, inhibitory domain (ID), and transactivation domain (TAD) were successively removed. Two isoforms of human RUNX1 (RUNX1a and b) as well as the three truncated RUNX1a are illustrated in Figure 4D. Upon transfection of these plasmids into 293T cells followed by treatment with Bet, the nuclear proteins were immunoprecipitated with FLAG antibody beads and probed with a GR antibody. The results show that interaction between GR and RUNX1 requires the TAD domain (321-391) (Figure 4E), because RUNX1-Δ321 was not immunoprecipitated with GR, while the other two longer proteins (as well as full-length RUNX1) associated with GR.
Treatment with glucocorticoids increased DNA binding of GR and RUNX1, but reduced that of R-E. Next, chromatin immunoprecipitation (ChIP) followed by qPCR was used to investigate how Bet treatment affects the binding of GR, RUNX1 and R-E to CD14, p14ARF and TRKA, which are known RUNX1 target genes. Promoter analysis showed that there are several potential RUNX1 and GR binding sites in the promoter region of these genes (Figure S4). Kasumi-1 cells were treated with Bet for 2 days, harvested, crosslinked with formaldehyde, and lysed. Upon sonication, which sheared the genome into ~200-500 bp DNA fragments, DNA/protein complexes were immunoprecipitated with ChIP-grade antibodies of GR, RUNX1 and ETO that were immobilized on magnetic Dynabeads. Upon thorough washing followed by DNA elution, qPCR was used to quantitate promoter DNAs that bound to GR, RUNX1 or R-E. A non-transcribed DNA region was included in the qPCR assays to characterize non-specific binding. As shown in Figure 5, compared to the non-transcribed region, significant amounts of GR, RUNX1 and R-E were found to bind to the promoters of these three RUNX1-target genes. As compared to controls, treatment with Bet in general significantly increased the binding of GR and RUNX1 onto these gene promoters, while R-E's binding was reduced.
ChIP-qPCR results showing amounts of DNA bound to (A) GR, (B) RUNX1, and (C) ETO in the promoters of CD14, p14ARF and TRKA as well as in a non-transcribed region as non-specific binding controls (*p < 0.05).
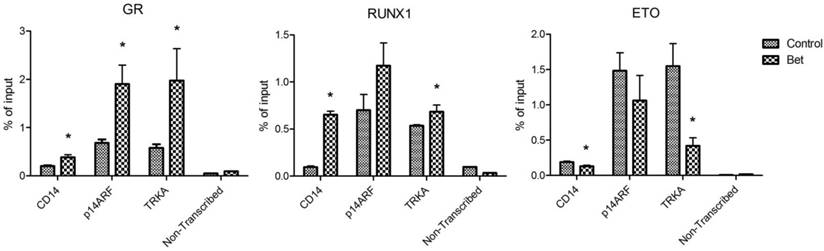
Overall, our results show that treatment of R-E containing leukemia cells with a glucocorticoid drug caused a series of events that eventually led to cell differentiation and apoptosis. First, the drug treatment increased the amount of activated (i.e., ligand-bound) GR in the nucleus (Figure 4A) as well as its binding to DNA including promoters of RUNX1-target genes. Second, the interaction between GR and RUNX1 (but not R-E) increased the binding capability and capacity of RUNX1 to its target genes and decreased those of R-E (Figure 5). The unbound R-E could be subjected to a proteasome-mediated degradation (37), resulting in a reduced level of R-E in the nucleus. Third, the shifted RUNX1/R-E occupancies in DNA towards a RUNX1 dominance altered the global gene expression in R-E-containing AML cells, showing a gene expression pattern mimicking R-E knockdown (Figure 2A-B), as well as stimulated RUNX1's activity (Figure 2C). Fourth, the global gene expression changes promoted RUNX1-mediated hematopoietic differentiation and inhibited R-E-mediated stem cell maintenance, leading to significant differentiation and apoptosis.
Discussion
AML is a major blood cancer and carries a poor prognosis, with 5-year survival rates being <40% for patients younger than 65 years and only 5.2% for older patients (53). With a few exceptions, current treatments are conventional chemotherapeutics, which non-selectively kill all rapidly proliferating cells including normal progenitor cells in the bone marrow and other organs. This causes severe toxicities and side effects and limits the efficacy of these drugs. In addition, LICs can enter quiescence, in which they do not divide, are resistant to chemotherapeutics, and are therefore responsible for relapse. Discovery of novel compounds targeting oncogene/protein-driven LICs is therefore of importance.
Chromosome translocation t(8;21) found in ~13% AML causes leukemia initiation. The resulting fusion oncoprotein R-E outcompetes RUNX1 and predominantly occupies DNA-binding loci of RUNX1 target genes. Such R-E dominance inhibits RUNX1-mediated gene expression for hematopoietic differentiation and promotes gene transcription that maintains a stem cell-like state. A possible targeted therapy is to selectively inhibit R-E's activity, as proven effective at the cellular level by siRNA-mediated R-E knockdown (24). However, the strategy seemed to be undruggable in the perspective of drug discovery, as R-E is a transcription factor and has the identical DNA-binding RUNT domain to RUNX1.
C-map was used to find candidate compounds that can alter gene expression patterns as R-E knockdown does in t(8;21) AML. The bioinformatics search turned out to be effective, and yielded 78 compounds showing selective activity against Kasumi-1 cells. The most active compounds included 3 classes of drugs, i.e., glucocorticoids, NSAID and Hsp90 inhibitors, that were identified previously using different methods. In addition, several other drugs (dacarbazine, acepromazine, crotamiton, propafenone and scopoletin (Table 1)) were also found to have strong and selective activity against R-E leukemia. Further pursuing of these compounds could be worthwhile.
Glucocorticoid drugs such as Dex and Bet attracted most of our attention, because of their low nM activity against R-E leukemia as well as >1000-fold selectivity (Table 2). AML is generally considered to be insensitive to glucocorticoids. Previous clinical studies using short course (3-7 days) treatment with high doses (20-30 mg/kg/day) of glucocorticoid drug methylprednisolone were found to induce differentiation of myeloid leukemia cells in children with different subtypes of AML (54). We decided to reevaluate the in vivo antileukemia activity of these drugs. Although the previous study reported Dex had no in vivo activity (37), this might be due to improper dosing or formulation. We show that Dex-P possessed potent antitumor activity in a mouse model of R-E leukemia. Further investigation revealed that glucocorticoids are non-cytotoxic (Table 2). Rather, treatment with these drugs inhibited the R-E-mediated gene expression and reactivated that of RUNX1. These global gene expression changes cause significant differentiation as well as apoptosis of R-E containing leukemia cells. Particularly noted are potent activities against self-renewal of R-E containing LICs including primary cells from t(8;21) AML patients, as well as strong synergy when combined with cytarabine and doxorubicin, two commonly used drugs for AML (Figure 3B).
Dex-P, a water-soluble prodrug of Dex, showed potent in vivo antileukemia activity in the mouse model of SKNO-1 leukemia. Because pharmacokinetics, pharmacodynamics and other drug behaviors largely differ between mice and humans, we did not perform extensive mouse model studies. Another reason is that glucocorticoids have been widely used in the clinic with well-documented pharmacokinetics, pharmacodynamics and regimes, for anti-inflammation and other indications, e.g., for certain acute lymphoid leukemia (55, 56). Favorable human PK of Dex-P (44, 45) as well as its highly potent and selective activity against R-E leukemia including patient samples strongly support that Dex-P or Dex could be used in the clinic to treat this cancer. Possible drawbacks of using glucocorticoid drugs are immune-suppression and other side-effects. However, given the strong synergy with chemotherapeutic drugs, lower doses could be used in a combination therapy regime for more effective treatment with less pronounced side effects. Moreover, combination therapies including Dex have the potential to eliminate LICs of t(8;21) AML to prevent or reduce likelihoods of relapse and resistance.
In addition to potential clinical applications, this study is of interest because it also reveals the molecular basis for glucocorticoids' potent and selective activity against R-E leukemia. The malignancy is caused by inhibited RUNX1-mediated hematopoietic differentiation, due to R-E's predominant occupancy in RUNX1 target genes. For the first time, our results show that GR interacts with RUNX1's TAD domain. Such interaction increased RUNX1's binding affinity and capacity to DNA and switched to a RUNX1 dominance in R-E containing leukemia cells. This caused inhibition of R-E-mediated gene expression and its associated stem cell maintenance. It also resulted in reactivation of RUNX1, leading to cell differentiation and apoptosis of these cells. Glucocorticoids are therefore a targeted therapy because these compounds exert their activities through interactions with RUNX1/R-E. This mechanism can also elucidate the high selectivity of these drugs for this subtype of leukemia.
It is noted that treatment with a glucocorticoid drug did not significantly affect expression levels of both RUNX1 and R-E, according to our microarray data (not shown). Therefore, the observed increased amount of RUNX1 could be due to increased protein stability or decreased degradation. Association with GR as well as enhanced binding to DNA could be possible reasons. A similar explanation could be applicable to the decreased amount of R-E. The reduced binding of R-E to DNA might promote proteasome-mediated degradation of R-E, which was observed in a previous study (37).
Moreover, linking GR to RUNX1 could be of importance in the study of hematopoiesis. Glucocorticoids and GR are known to affect hematopoiesis: treatment with Dex stimulated erythropoiesis (57, 58), while it inhibited granulopoiesis (59). In addition, GR is required for sustained proliferation of erythroid progenitor cells in vitro as well as in vivo under stress conditions such as hypoxia (58). Our finding that GR associates with RUNX1, a master transcription regulator for hematopoiesis, could be a possible mechanism for the activities of glucocorticoid drugs. Further studies are therefore worthwhile to reveal the relationships between GR, RUNX1 and hematopoiesis.
Abbreviations
AML: acute myeloid leukemia; Bet: betamethasone; CI: combination index; ChIP: chromatin immunoprecipitation; ChIP-seq: ChIP followed by sequencing; C-map: connectivity Map; Dex: dexamethasone; Dex-P: dexamethasone-21-phosphate; GSEA: gene set enrichment analysis; GR: glucocorticoid receptor; ID: inhibitory domain; LIC: leukemia initiating cells; R-E: RUNX1-ETO; R-E9a: RUNX1-ETO9a; TAD: transactivation domain.
Supplementary Material
Supplementary tables (Table S1-S3) and supplementary figures (Figure S1-S3).
Acknowledgements
This work was supported by grants RP150129 and RP140469 from Cancer Prevention and Research Institute of Texas (CPRIT). Flow cytometry was done in the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding support from the NIH (AI036211, CA125123, and RR024574).
Competing interests
The authors declare that they have no competing interests.
References
1. Speck NA, Gilliland DG. Core-binding factors in haematopoiesis and leukaemia. Nat Rev Cancer. 2002;2(7):502-13
2. Ito Y, Bae SC, Chuang LS. The RUNX family: developmental regulators in cancer. Nat Rev Cancer. 2015;15(2):81-95
3. Daga A, Tighe JE, Calabi F. Leukaemia/Drosophila homology. Nature. 1992;356(6369):484
4. Kagoshima H, Shigesada K, Satake M, Ito Y, Miyoshi H, Ohki M. et al. The Runt domain identifies a new family of heteromeric transcriptional regulators. Trends Genet. 1993;9(10):338-41
5. Tahirov TH, Inoue-Bungo T, Morii H, Fujikawa A, Sasaki M, Kimura K. et al. Structural analyses of DNA recognition by the AML1/Runx-1 Runt domain and its allosteric control by CBFbeta. Cell. 2001;104(5):755-67
6. Zhang DE, Hetherington CJ, Meyers S, Rhoades KL, Larson CJ, Chen HM. et al. CCAAT enhancer-binding protein (C/EBP) and AML1 (CBF alpha2) synergistically activate the macrophage colony-stimulating factor receptor promoter. Mol Cell Biol. 1996;16(3):1231-40
7. Takahashi A, Satake M, Yamaguchi-Iwai Y, Bae SC, Lu J, Maruyama M. et al. Positive and negative regulation of granulocyte-macrophage colony-stimulating factor promoter activity by AML1-related transcription factor, PEBP2. Blood. 1995;86(2):607-16
8. Aronson BD, Fisher AL, Blechman K, Caudy M, Gergen JP. Groucho-dependent and -independent repression activities of Runt domain proteins. Mol Cell Biol. 1997;17(9):5581-7
9. Lutterbach B, Westendorf JJ, Linggi B, Isaac S, Seto E, Hiebert SW. A mechanism of repression by acute myeloid leukemia-1, the target of multiple chromosomal translocations in acute leukemia. J Biol Chem. 2000;275(1):651-6
10. Goyama S, Mulloy JC. Molecular pathogenesis of core binding factor leukemia: current knowledge and future prospects. Int J Hematol. 2011;94(2):126-33
11. Sangle NA, Perkins SL. Core-binding factor acute myeloid leukemia. Arch Pathol Lab Med. 2011;135(11):1504-9
12. Peterson LF, Zhang DE. The 8;21 translocation in leukemogenesis. Oncogene. 2004;23(24):4255-62
13. Wang J, Hoshino T, Redner RL, Kajigaya S, Liu JM. ETO, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3/HDAC1 complex. Proc Natl Acad Sci U S A. 1998;95(18):10860-5
14. Amann JM, Nip J, Strom DK, Lutterbach B, Harada H, Lenny N. et al. ETO, a target of t(8;21) in acute leukemia, makes distinct contacts with multiple histone deacetylases and binds mSin3A through its oligomerization domain. Mol Cell Biol. 2001;21(19):6470-83
15. Mulloy JC, Cammenga J, Berguido FJ, Wu K, Zhou P, Comenzo RL. et al. Maintaining the self-renewal and differentiation potential of human CD34+ hematopoietic cells using a single genetic element. Blood. 2003;102(13):4369-76
16. Mulloy JC, Cammenga J, MacKenzie KL, Berguido FJ, Moore MA, Nimer SD. The AML1-ETO fusion protein promotes the expansion of human hematopoietic stem cells. Blood. 2002;99(1):15-23
17. Higuchi M, O'Brien D, Kumaravelu P, Lenny N, Yeoh EJ, Downing JR. Expression of a conditional AML1-ETO oncogene bypasses embryonic lethality and establishes a murine model of human t(8;21) acute myeloid leukemia. Cancer cell. 2002;1(1):63-74
18. Yuan Y, Zhou L, Miyamoto T, Iwasaki H, Harakawa N, Hetherington CJ. et al. AML1-ETO expression is directly involved in the development of acute myeloid leukemia in the presence of additional mutations. Proc Natl Acad Sci U S A. 2001;98(18):10398-403
19. Wang YY, Zhao LJ, Wu CF, Liu P, Shi L, Liang Y. et al. C-KIT mutation cooperates with full-length AML1-ETO to induce acute myeloid leukemia in mice. Proc Natl Acad Sci U S A. 2011;108(6):2450-5
20. Chou FS, Wunderlich M, Griesinger A, Mulloy JC. N-Ras(G12D) induces features of stepwise transformation in preleukemic human umbilical cord blood cultures expressing the AML1-ETO fusion gene. Blood. 2011;117(7):2237-40
21. Yan M, Kanbe E, Peterson LF, Boyapati A, Miao Y, Wang Y. et al. A previously unidentified alternatively spliced isoform of t(8;21) transcript promotes leukemogenesis. Nat Med. 2006;12(8):945-9
22. Yergeau DA, Hetherington CJ, Wang Q, Zhang P, Sharpe AH, Binder M. et al. Embryonic lethality and impairment of haematopoiesis in mice heterozygous for an AML1-ETO fusion gene. Nat Genet. 1997;15(3):303-6
23. Okuda T, Cai Z, Yang S, Lenny N, Lyu CJ, van Deursen JM. et al. Expression of a knocked-in AML1-ETO leukemia gene inhibits the establishment of normal definitive hematopoiesis and directly generates dysplastic hematopoietic progenitors. Blood. 1998;91(9):3134-43
24. Ptasinska A, Assi SA, Mannari D, James SR, Williamson D, Dunne J. et al. Depletion of RUNX1/ETO in t(8;21) AML cells leads to genome-wide changes in chromatin structure and transcription factor binding. Leukemia. 2012;26(8):1829-41
25. Ptasinska A, Assi SA, Martinez-Soria N, Imperato MR, Piper J, Cauchy P. et al. Identification of a dynamic core transcriptional network in t(8;21) AML that regulates differentiation block and self-renewal. Cell Rep. 2014;8(6):1974-88
26. Ben-Ami O, Friedman D, Leshkowitz D, Goldenberg D, Orlovsky K, Pencovich N. et al. Addiction of t(8;21) and inv(16) acute myeloid leukemia to native RUNX1. Cell Rep. 2013;4(6):1131-43
27. Wu F, Zhou C, Yao Y, Wei L, Feng Z, Deng L. et al. 3-(Piperidin-4-ylmethoxy)pyridine Containing Compounds Are Potent Inhibitors of Lysine Specific Demethylase 1. J Med Chem. 2016;59(1):253-63
28. Zhou C, Wu F, Lu L, Wei L, Pai E, Yao Y. et al. Structure activity relationship and modeling studies of inhibitors of lysine specific demethylase 1. PLoS One. 2017;12(2):e0170301
29. Wu F, Jiang H, Zheng B, Kogiso M, Yao Y, Zhou C. et al. Inhibition of Cancer-Associated Mutant Isocitrate Dehydrogenases by 2-Thiohydantoin Compounds. J Med Chem. 2015;58(17):6899-908
30. Feng Z, Yao Y, Zhou C, Chen F, Wu F, Wei L. et al. Pharmacological inhibition of LSD1 for the treatment of MLL-rearranged leukemia. J Hematol Oncol. 2016;9:24
31. Liu W, Deng L, Song Y, Redell M. DOT1L inhibition sensitizes MLL-rearranged AML to chemotherapy. PLoS One. 2014;9(5):e98270
32. Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ. et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929-35
33. Lamb J. The Connectivity Map: a new tool for biomedical research. Nat Rev Cancer. 2007;7(1):54-60
34. Dunne J, Cullmann C, Ritter M, Soria NM, Drescher B, Debernardi S. et al. siRNA-mediated AML1/MTG8 depletion affects differentiation and proliferation-associated gene expression in t(8;21)-positive cell lines and primary AML blasts. Oncogene. 2006;25(45):6067-78
35. Wang L, Gural A, Sun XJ, Zhao X, Perna F, Huang G. et al. The leukemogenicity of AML1-ETO is dependent on site-specific lysine acetylation. Science. 2011;333(6043):765-9
36. Miyoshi H, Ohki M, Nakagawa T, Honma Y. Glucocorticoids induce apoptosis in acute myeloid leukemia cell lines with A t(8;21) chromosome translocation. Leuk Res. 1997;21(1):45-50
37. Corsello SM, Roti G, Ross KN, Chow KT, Galinsky I, DeAngelo DJ. et al. Identification of AML1-ETO modulators by chemical genomics. Blood. 2009;113(24):6193-205
38. Zhang Y, Wang J, Wheat J, Chen X, Jin S, Sadrzadeh H. et al. AML1-ETO mediates hematopoietic self-renewal and leukemogenesis through a COX/beta-catenin signaling pathway. Blood. 2013;121(24):4906-16
39. Yang G, Thompson MA, Brandt SJ, Hiebert SW. Histone deacetylase inhibitors induce the degradation of the t(8;21) fusion oncoprotein. Oncogene. 2007;26(1):91-101
40. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J. et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645-8
41. Misaghian N, Ligresti G, Steelman LS, Bertrand FE, Basecke J, Libra M. et al. Targeting the leukemic stem cell: the Holy Grail of leukemia therapy. Leukemia. 2009;23(1):25-42
42. Nishida S, Hosen N, Shirakata T, Kanato K, Yanagihara M, Nakatsuka S. et al. AML1-ETO rapidly induces acute myeloblastic leukemia in cooperation with the Wilms tumor gene, WT1. Blood. 2006;107(8):3303-12
43. Ono M, Yaguchi H, Ohkura N, Kitabayashi I, Nagamura Y, Nomura T. et al. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature. 2007;446(7136):685-9
44. Miyabo S, Nakamura T, Kuwazima S, Kishida S. A comparison of the bioavailability and potency of dexamethasone phosphate and sulphate in man. Eur J Clin Pharmacol. 1981;20(4):277-82
45. Brady ME, Sartiano GP, Rosenblum SL, Zaglama NE, Bauguess CT. The pharmacokinetics of single high doses of dexamethasone in cancer patients. Eur J Clin Pharmacol. 1987;32(6):593-6
46. Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58(3):621-81
47. Ramamoorthy S, Cidlowski JA. Corticosteroids: Mechanisms of Action in Health and Disease. Rheum Dis Clin North Am. 2016;42(1):15-31 vii
48. Grad I, Picard D. The glucocorticoid responses are shaped by molecular chaperones. Mol Cell Endocrinol. 2007;275(1-2):2-12
49. Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18(3):306-60
50. Beato M. Gene regulation by steroid hormones. Cell. 1989;56(3):335-44
51. Freedman LP. Anatomy of the steroid receptor zinc finger region. Endocr Rev. 1992;13(2):129-45
52. Sun XJ, Wang Z, Wang L, Jiang Y, Kost N, Soong TD. et al. A stable transcription factor complex nucleated by oligomeric AML1-ETO controls leukaemogenesis. Nature. 2013;500(7460):93-7
53. Howlader N, Noone AM, Krapcho M, Miller D, Bishop K, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA (eds). SEER Cancer Statistics Review, 1975-2014, National Cancer Institute. Bethesda, MD, https://seer.cancer.gov/csr/1975_2014/, based on November 2016 SEER data submission, posted to the SEER web site, April. 2017
54. Hicsonmez G. The effect of steroid on myeloid leukemic cells: the potential of short-course high-dose methylprednisolone treatment in inducing differentiation, apoptosis and in stimulating myelopoiesis. Leuk Res. 2006;30(1):60-8
55. Groden BM, Dunnigan MG. Steroid Therapy in Lymphoid Tumours. Br J Cancer. 1963;17:579-82
56. Burningham RA, Restrepo A, Pugh RP, Brown EB, Schlossman SF, Khuri PD. et al. Weekly High-Dosage Glucocorticosteroid Treatment of Lymphocytic Leukemias and Lymphomas. N Engl J Med. 1964;270:1160-6
57. Golde DW, Bersch N, Cline MJ. Potentiation of erythropoiesis in vitro by dexamethasone. J Clin Invest. 1976;57(1):57-62
58. Bauer A, Tronche F, Wessely O, Kellendonk C, Reichardt HM, Steinlein P. et al. The glucocorticoid receptor is required for stress erythropoiesis. Genes Dev. 1999;13(22):2996-3002
59. Golde DW, Bersch N, Quan SG, Cline MJ. Inhibition of murine granulopoiesis in vitro by dexamethasone. Am J Hematol. 1976;1(4):369-73
Author contact
![]() Corresponding author: Address: Department of Pharmacology and Chemical Biology, Baylor College of Medicine, 1 Baylor Plaza, Houston, TX 77030. Tel: 713-798-7415. E-mail: ysongedu.
Corresponding author: Address: Department of Pharmacology and Chemical Biology, Baylor College of Medicine, 1 Baylor Plaza, Houston, TX 77030. Tel: 713-798-7415. E-mail: ysongedu.