Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Summary and conclusions
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(11):1753-1767. doi:10.7150/thno.15616 This issue Cite
Research Paper
PET Imaging with [18F]FSPG Evidences the Role of System xc- on Brain Inflammation Following Cerebral Ischemia in Rats
Maria Domercq1, Boguslaw Szczupak2, Jon Gejo1, Vanessa Gómez-Vallejo2,3, Daniel Padro2,4, Kiran Babu Gona2,3, Frédéric Dollé5, Makoto Higuchi6, Carlos Matute1, Jordi Llop2,3, Abraham Martín2 ![]()
1. Department of Neurosciences, University of the Basque Country, Barrio Sarriena s/n, 48940 Leioa, Spain, Achucarro Basque Center for Neuroscience-UPV/EHU, 48170 Zamudio, Spain and Instituto de Salud Carlos III, Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED), 48940 Leioa, Spain.
2. Molecular Imaging Unit, CIC biomaGUNE, Pº Miramon 182, San Sebastian, Spain.
3. Radiochemistry and Nuclear Imaging, CIC biomaGUNE, Pº Miramon 182, San Sebastian, Spain.
4. Magnetic Resonance Imaging. CIC biomaGUNE, Pº Miramon 182, San Sebastian, Spain.
5. CEA, Institut d'imagerie biomédicale, Service Hospitalier Frédéric Joliot, Orsay, France.
6. Molecular Imaging Center, National Institute of Radiological Sciences, Chiba, Japan.
Received 2016-3-22; Accepted 2016-5-18; Published 2016-7-9
Abstract

In vivo Positron Emission Tomography (PET) imaging of the cystine-glutamate antiporter (system xc-) activity with [18F]FSPG is meant to be an attractive tool for the diagnosis and therapy evaluation of brain diseases. However, the role of system xc- in cerebral ischemia and its involvement in inflammatory reaction has been scarcely explored. In this work, we report the longitudinal investigation of the neuroinflammatory process following transient middle cerebral artery occlusion (MCAO) in rats using PET with [18F]FSPG and the translocator protein (TSPO) ligand [18F]DPA-714. In the ischemic territory, [18F]FSPG showed a progressive binding increase that peaked at days 3 to 7 and was followed by a progressive decrease from days 14 to 28 after reperfusion. In contrast, [18F]DPA-714 evidenced maximum binding uptake values over day 7 after reperfusion. Ex vivo immnunohistochemistry confirmed the up-regulation of system xc- in microglial cells and marginally in astrocytes. Inhibition of system xc- with sulfasalazine and S-4-CPG resulted in increased arginase (anti-inflammatory M2 marker) expression at day 7 after ischemia, together with a decrease in TSPO and microglial M1 proinflammatory markers (CCL2, TNF and iNOS) expression. Taken together, these results suggest that system xc- plays a key role in the inflammatory reaction underlying experimental stroke.
Keywords: T2W-MRI, [18F]FSPG, [18F]DPA-714, PET, cerebral ischemia.
Introduction
Cystine-glutamate antiporter (system xc-) is a heterodimer composed by 4F2hc and xCT that mediates the cellular import of cystine used in glutathione production and oxidative protection in exchange for export of glutamate [1-3]. Indeed, the need for oxidative protection in the pathologic brain induces extrasynaptic glutamate release through system xc- contributing to neuronal damage [4]. System xc- is expressed in a wide variety of cells both outside and inside the central nervous system (CNS) including immature cortical neurons [5], astrocytes [6] and microglia [7-9]. The activation of microglia from a resting state and migration into injured brain areas has not only beneficial neuroprotective and neurothrophic effects, but also a detrimental action through excitotoxic glutamate release [7, 10-14]. Likewise, the involvement of microglia on excitotoxic stress has been linked to a wide variety of neurodegerative diseases such as Alzheimer's disease [14], amyotrophic lateral sclerosis [8], glioma-derived epileptic seizures [15] and multiple sclerosis [9, 16, 17]. Therefore, in vivo imaging of system xc- with positron emission tomography (PET) technique might be crucial to further understanding the role of these receptors on inflammation underlying brain diseases. BAY 94-9392 ((4S)-4-(3-[18F]fluoropropyl)-L-glutamate (herein referred to as [18F]FSPG), a fluorine-18-labeled L-glutamate derivative, is taken up by system xc- due to the lack of discrimination between its natural substrate cystine and glutamate for the inward transport [18, 19]. [18F]FSPG has shown to be a promising radiotracer to image the amino acid antiporter system xc- in patients with cancer [20, 21] and infectious lesions [22]. In this sense, the PET imaging of amino acid transporters with [18F]BAAs (boramino acids) have also showed high tumor-specific accumulation, suggesting that PET imaging of amino acid transporters are promising targeted radiotracers for molecular imaging in oncology and mental disorders [23]. Within the CNS, [18F]FSPG has been used to explore cystine-glutamate antiporter activity after early acute cerebral ischemia [4] and multiple sclerosis [24] in rats. Recently, we have demonstrated with PET imaging that the system xc- was overexpressed in microglial cells in those areas more severely affected in experimental autoimmune encephalomyelitis (EAE) in rats [24], suggesting the potential of this radioligand to monitor neuroinflammatory damage. Despite this, the use of [18F]FSPG to evaluate the contribution of system xc- on the neuroinflammatory response following cerebral ischemia has not been explored before. The purpose of the present study was to investigate the levels of system xc- in the rat brain after cerebral ischemia using [18F]FSPG-PET and immunohistochemistry. In particular, we were interested in clarifying the relationship of the system xc- activity with the activation of glial cells after cerebral ischemia in rats. For this reason, ischemic rats were subjected to PET studies with [18F]DPA-714, a specific radioligand for the translocator protein (18kDa) (TSPO), to image brain inflammation [25, 26]. Likewise, [18F]DPA-714 binding and RT-PCR were used to evaluate the effect of pharmacological inhibition of system xc- (SAS and S-4-CPG) on the neuroinflammatory reaction after ischemic stroke in rats. The results reported here provide novel information about the activity of cystine/glutamate antiporter in microglial cells after cerebral ischemia and might ultimately contribute to a better design of anti-inflammatory strategies for the treatment of ischemic stroke.
Materials and Methods
Cerebral ischemia and treatment
Adult male Sprague-Dawley rats (300 g body weight; Janvier, France) (n=52) were used. Animal studies were approved by the animal ethics committee of CIC biomaGUNE and local authorities and were conducted in accordance with the Directives of the European Union on animal ethics and welfare. Transient focal ischemia was produced by a 90 minutes intraluminal occlusion of the middle cerebral artery (MCA) followed by reperfusion as described elsewhere [27]. Briefly, rats were anaesthetized with 4% isofluorane in 100 % O2 and a 2.6-cm length of 4-0 monofilament nylon suture was introduced into the right external carotid artery up to the level where the MCA branches out. Animals were then sutured and placed in their cages with free access to water and food. After 90 minutes, the animals were re-anesthetized and the filament was removed to allow reperfusion. Twelve rats were repeatedly examined before (day 0) and at 1, 3, 7, 14, 21 and 28 days after ischemia to evaluate the temporal PET binding of both xc- system and TSPO. The animals studied at day 0 have been considered as the baseline control group.
Two groups of seven and eight rats, respectively, were inoculated daily for a total of seven days from one hour following MCAO with 0.1 mL Sulfasalazine (SAS) (8 mg/Kg, i.p.) or 0.1 mL (S)-4-Carboxyphenylglycine (S-4-CPG) (1 mg/Kg, i.p.). A control ischemic group of five rats received daily the same volume of vehicle (normal saline). At day seven, treated and control rats were imaged with PET to determine the effect of both SAS and S-4-CPG on the TSPO expression. Sixteen rats were used to perform ex vivo studies (immunohistochemistry) for xc- system at 0, 3, 7 and 28 days after cerebral ischemia. Rats used in imaging studies for the assessment of the effect of the treatments were used to perform immunohistochemistry for TSPO (n=20). Therefore, MCAO was induced to forty-four rats.
Magnetic resonance imaging
T2-weighted (T2W) MRI scans were performed in ischemic animals at 24 hours after reperfusion to select the rats (n=12) presenting cortico-striatal lesions to be included in the PET studies. Before the scans, anesthesia was induced with 4% isofluorane and maintained by 2-2.5% of isofluorane in 100% O2 during the scan. Animals were placed into a rat holder compatible with the MRI acquisition systems and maintained normothermic using a water-based heating blanket at 37ºC. MRI experiments were performed on a 7 Tesla Bruker Biospec 70/30 MRI system (Bruker Biospin GmbH, Ettlingen, Germany), interfaced to an AVANCE III console. The BGA12-S imaging gradient (maximum gradient strength 400 mT/m switchable within 80 µs), an 82 mm inner diameter quadrature volume resonator for transition and surface rat brain coil for reception were used. T2W images were acquired with a RARE sequence with the following parameters: RARE factor 2, TR/TE = 4400/40 ms, FOV = 25 mm x 25 mm, ACQ Matrix =256 x 256, Slice Thickness = 1 mm, 2 averages and 24 contiguous slices. Contiguous slices covering all the infarcted volume were acquired and fat suppression was used.
Magnetic resonance imaging image analysis
MRI (T2W) images were used to calculate the lesion volume. Regions of Interest (ROIs) were manually defined using the Open Source software 3D Slicer image analysis software (Version 3.6.3 www.slicer.org) for each rat on the region of increased signal in the ipsilateral hemisphere. The total lesion volume was calculated by summing the area of the infarcted regions of all slices affected by the lesion.
Radiochemistry
Fluorine-18 production: Fluorine-18 (T1/2 = 109.8 min) as [18F]fluoride was cyclotron-produced (Cyclone-18/9, IBA, Louvain-la-Neuve, Belgium) by proton irradiation of an 18O-enriched (98%) water target (2.9 mL) via the 18O(p,n)18F nuclear reaction. [18F]FSPG preparation: The synthesis of (4S)-4-[3-[18F]fluoropropyl]-L-glutamate ([18F]FSPG) was performed using a TRACERlab FXFN synthesis module (GE Healthcare), based on a two-step procedure, as previously described [18] with minor modification: i) 18F-fluorination of the appropriately protected precursor for labeling (di-tert-butyl (2S,4S)-2-tert-butoxycarbonylamino-4-(3-nitrophenylsulfonyloxy-propyl)-pentanedioate), followed by ii) removal of the protective groups. Briefly, once transferred into a dedicated (ventilated and lead-shielded) hot-cell, [18F]fluoride was first trapped on a pre-conditioned Sep-Pak® Accell Plus QMA Light cartridge (Waters, Milford, MA, USA), then eluted from the cartridge with a solution of Kryptofix K2.2.2/K2CO3 in a mixture of water and acetonitrile. A solution of the precursor for labeling (5 mg) in acetonitrile (1 mL) was then added and the mixture heated at 80ºC for 10 minutes. The crude reaction mixture was then diluted with a solution of acetonitrile and water (1/1, 2 mL) and purified by high performance liquid chromatography (HPLC) using a Nucleosil 100-7 C18 column (Macherey-Nagel, Düren, Germany) as stationary phase and water/acetonitrile (30/70) as the mobile phase at a flow rate of 8 mL/min. The desired fraction (11-13 min) was collected and diluted with water (50 mL). The labeled specie was retained on a C-18 cartridge (Sep-Pak® Light, Waters, Milford, MA, USA) and further eluted with ethanol (1 mL). This solution was treated with 0.5 ml of 4M HCl (500µL); after hydrolysis, the solution was neutralized with 2N NaOH (650 µL) and phosphate buffer solution (PBS, pH=7.4, 3 mL). After filtration through a 0.22 μm filter, the radiotracer was ready for injection. Radiochemical yields (decay-corrected) were in the range 30-35% and radiochemical purity was always >93% at the time of injection.
[18F]DPA-714 preparation: The synthesis of N,N-diethyl-2-(2-(4-(2-fluoroethoxy)phenyl)-5,7-dimethylpyrazolo[1,5-a]pyrimidin-3-yl)acetamide ([18F]DPA-714) was performed using a TRACERlab FXFN synthesis module (GE Healthcare), based on a one-step procedure, as previously described [28]. Briefly, once transferred into a dedicated (ventilated and lead-shielded) hot-cell, [18F]fluoride was first trapped on a pre-conditioned Sep-Pak® Accell Plus QMA Light cartridge (Waters, Milford, MA, USA), then eluted from the cartridge with a solution of Kryptofix K2.2.2/K2CO3 in a mixture of water and acetonitrile. A solution containing the appropriate precursor for labeling (N,N-diethyl-2-(2-(4-(2-toluenesulfonyloxyethoxy)phenyl)-5,7-dimethylpyrazolo[1,5-a]pyrimidin-3-yl)acetamide, 4 mg) in dimethylsulfoxide (0.7 mL) was added and the mixture heated at 165°C for 5 minutes. The reactor was then cooled at room temperature, the reaction crude diluted with a mixture of acetonitrile and water (2/1, 3 mL), and purified by HPLC using a Nucleosil 100-7 C18 column (Macherey-Nagel, Düren, Germany) as stationary phase and 0.1M aqueous ammonium formate solution (pH = 3.9)/acetonitrile (30/70) as the mobile phase at a flow rate of 7 mL/min. The desired fraction (10-11 min) was collected, diluted with water (20 mL), and the radiotracer was retained on a C-18 cartridge (Sep-Pak® Light, Waters, Milford, MA, USA) and further eluted with ethanol (1 mL). The ethanol solution was finally reconstituted with saline solution (9 mL). Filtration through a 0.22 μm filter yielded the final solution, ready for injection. Radiochemical yields (non-decay corrected) were in the range 8-13% and radiochemical purity was always >95% at the time of injection.
Positron emission tomography scans and data acquisition
PET scans were repeatedly performed before (day 0) and at 1, 3, 7, 14, 21 and 28 days after reperfusion using a General Electric eXplore Vista CT camera (GE Healthcare). Scans were performed in rats anaesthetized with 4% isofluorane and maintained by 2-2.5% of isofluorane in 100% O2. The tail vein was catheterized with a 24-gauge catheter for intravenous administration of the radiotracer. Animals were placed into a rat holder compatible with the PET acquisition system and maintained normothermic using a water-based heating blanket. Animals were subjected to PET scans to assess system xc- ([18F]FSPG) and TSPO binding ([18F]DPA-714) at each time point before and after ischemia onset. The appropriate radiotracer ([18F]FSPG or [18F]DPA-714, ~70 MBq) was injected concomitantly with the start of the PET acquisition and dynamic brain images were acquired (24 frames: 1x5, 1x15, 3x30, 5x60, 5x120, 3x180, 6x300 seconds) in the 400-700 keV energetic window, with a total acquisition time of 57.5 minutes. For evaluation of SAS and S-4-CPG treatment efficacy after ischemia, ~70 MBq of [18F]DPA-714 were injected concomitantly with the start of the PET acquisition and brain dynamic images were acquired for a total of 30 minutes (23 frames 3x5, 3x15, 4x30, 4x60, 4x120, 5x180 seconds). After each PET scan, CT acquisitions were also performed (140 µA intensity, 40 kV voltage), providing anatomical information of each animal as well as the attenuation map for the later image reconstruction. Dynamic acquisitions were reconstructed (decay and CT-based attenuation corrected) with filtered back projection (FBP) using a Ramp filter with a cutoff frequency of 0.5 mm-1.
Positron emission tomography image analysis
PET images were analyzed using PMOD image analysis software (PMOD Technologies Ltd, Zürich, Switzerland). To verify the anatomical location of the signal, PET images were co-registered to the anatomical data of a MRI rat brain template. Two type of Volumes of Interest (VOIs) were established as follows: (i) A first set of VOIs was defined to study the whole brain [18F]FSPG and [18F]DPA-714 PET signal over time. Whole brain VOIs were manually drawn in both the entire ipsilateral and contralateral hemispheres containing the territory irrigated by the middle cerebral artery on slices of a MRI (T2W) rat brain template from the PMOD software. (ii) A second set of VOIs was automatically generated in the cortex and the striatum by using the regions proposed by the PMOD rat brain template, to study the evolution of PET signal in these specific regions in both the ipsilateral and contralateral cerebral hemispheres. The last four frames in steady state were used to calculate the summed PET binding uptake during the last 15 minutes of acquisition for both radiotracers. PET signal uptake was averaged in each ROI and expressed as percentage of injected dose per cubic centimetre (%ID/cc).
Immunohistochemistry and cell counts
Immunohistochemistry staining was performed at (i) control (day 0), day 3, day 7 and day 28 after reperfusion and (ii) at day 7 after ischemia in animals treated with SAS and S-4-CPG and non-treated rats. Animals were terminally anesthetized and killed by decapitation. The brain was removed, frozen and cut in 5-μm-thick sections in a cryostat. Sections were fixed in acetone (-20°C) during 2minutes, washed with phosphate-buffered saline (PBS) and saturated with a solution of bovine serum albumine (BSA) 5%/Tween 0.5% in PBS during 15 minutes at room temperature, and incubated during 1 hour at room temperature with primary antibodies BSA (5%)/Tween (0.5%) in PBS. The first set of sections were stained for xCT with rabbit anti-rat xCT (1:100, AbCam, Cambridge, UK), for CD11b with mouse anti-rat CD11b (1:300; Serotec, Raleigh, NC, USA) and for the glial fibrillary acidic protein (GFAP) with chicken anti-rat GFAP (1:500; AbCam, Cambridge, UK). The second set of sections was stained for TSPO with a rabbit anti-rat TSPO (NP155, 1:1000), for CD11b and for GFAP. Sections were washed (3 × 10minutes) in PBS and incubated for 1hour at room temperature with secondary antibodies Alexa Fluor 350 goat anti-rabbit IgG, Alexa Fluor 594 goat anti-mouse IgG and Alexa Fluor 488 goat anti-chicken IgG (Molecular Probes, Life Technologies, Madrid, Spain, 1:1,000) in BSA 5%/Tween 0.5% in PBS, washed again (3 × 10minutes) in PBS, and mounted with a prolong antifade kit in slices (Molecular Probes Life Technologies, Madrid). Standardized images acquisition was performed with an Axio Observer Z1 (Zeiss, Le Pecq, France) equipped with a motorized stage. The number of microglial and astrocytic cells immunopositive for xCT and TSPO within the ischemic area was assessed at 0, 3, 7 and 28 days after ischemia (xCT+/CD11b+ and xCT+/GFAP+) and at day 7 to evaluate the inflammatory effect of xc- system inhibition (TSPO+/CD11b+ and TSPO+/GFAP+). Cells were manually counted in ten representative and different fields at 100x magnification by using Image J (NIH) software.
Neurological assessment
The assessment of neurological outcome induced by cerebral ischemia was based on a previously reported 9-neuroscore test [29]. Four consecutive tests were performed on ischemic animals submitted to longitudinal imaging studies at 0, 1, 3, 7, 14, 21 and 28 days, and before (day 0) and at day 7 after ischemia in treated rats as follows: (a) spontaneous activity (moving and exploring = 0, moving without exploring = 1, no moving = 2); (b) left drifting during displacement (none = 0, drifting only when elevated by the tail and pushed or pulled = 1, spontaneous drifting = 2, circling without displacement or spinning = 3), (c) parachute reflex (symmetrical = 0, asymmetrical = 1, contralateral forelimb retracted = 2), and (d) resistance to left forepaw stretching (stretching not allowed = 0, stretching allowed after some attempts = 1, no resistance = 2). Total score could range from 0 (normal) to a 9 (highest handicap) point-scale.
Real-time quantitative PCR (qPCR)
Frozen tissue was cut into 40 µm slices on a cryostat and a pool of 3-4 different slices of ipsilateral or contralateral hemispheres was used per each condition. Total RNA was isolated from tissue using Trizol (Invitrogen) according to the manufacturer's instructions. Subsequently, cDNA synthesis was conducted using SuperScript III retrotranscriptase (200 U/µL; Invitrogen) and random hexamers as primers (Promega). Primers specific for the rat M1 and M2 markers were designed using Primer Express software (Applied Biosystems) at exon junctions to avoid genomic DNA amplification (see Table 1). Data was normalized to HPRT gene (best M coefficient using GeNorm Software v3.5; Vandesompele et al., 2002). Real-time quantitative PCR reactions were performed with SYBR-Green using a BioRad CFX96 Real-time PCR detection system as described previously [30].
Primers used to analyze M1 and M2 gene expression.
| Gen | Forward primer sequence | Reverse primer Sequence |
|---|---|---|
| CCL2 | GTGCTGTCTCAGCCAGATGCA | GCTGCTGGTGATTCTCTTGTAGTTC |
| TNF α | GGTCCCAACAAGGAGGAGAAG | GTCTGGGCCATGGAACTGA |
| iNOS | ATGGCTCCTTCAAAGAGGCA | CTATTTCCTTTGTTACGGCTTCCA |
| Arginase | GTGAAGAACCCACGGTCTGTG | GAGATGCTTCCAATTGCCATACTG |
| Mannose receptor | TGGTACGCAGACTGCACCTC | CTCTTGCTGCCCTCAAACTG |
| TGF β | CTGAGTGGCTGTCTTTTGACGTC | AAGCCCTGTATTCCGTCTCCTTG |
Statistical analyses
For PET signal values, the statistical analysis was performed as follows: The percentage of injected dose per cubic centimeter (%ID/cc) for each animal, brain hemisphere (ipsilateral and contralateral) and region (cortex and striatum) were calculated at each time point after cerebral ischemia. Values of %ID/cc within each region and time point following cerebral ischemia and after treatment were averaged and compared with the averaged baseline control values (day 0) and non-treated ischemic rats respectively using one-way ANOVA followed by Dunnet's multiple-comparison tests for post-hoc analysis. Likewise, cellular expression of both microglial/xCT and astrocytic/xCT receptors before (day 0) and at days 3, 7 and 28 after ischemia and microglial/TSPO and astrocytic/TSPO after treatment were compared using the same statistical analysis than that for PET imaging. Neurological outcome comparisons were performed as follows: animals were subjected to the 9-neuroscore test before MCAO and at 1, 3, 7, 14, 21 and 28 days after cerebral ischemia. The results within each time point and after treatment were averaged and compared with baseline average values using Mann-Whitney U-tests. The effect of treatments in the mRNA levels for M1/M2 microglial phenotypes in sham, treated and non-treated ischemic rats were compared using a one-way ANOVA followed by Bonferroni's multiple-comparison tests for post hoc analysis. Finally, the level of arginase mRNA in treated ischemic rats was compared with non-treated ischemic rats using an unpaired t-test. The level of significance was regularly set at P<0.05. Statistical analyses were performed with GraphPad Prism version 6 software.
Results
Hyperintensities of T2W images showed similar infarct extents as well as locations affected. All ischemic rats subjected to nuclear studies showed cortical and striatal MRI alterations (mean ± s.d. 344.49 ± 59.44 mm3, n=12) (Figure 1).
Magnetic resonance imaging (MRI) (T2-weighting (T2W)) and positron emission tomography (PET) images of [18F]FSPG at control (day 0), day, 3, day 7, day 14 and day 28 after middle cerebral artery occlusion (MCAO). Serial MRI (T2W) (A) and xc- system PET binding (B) images of coronal planes at the level of the lesion. PET images are co-registered with the MRI (T2W) of the same animal at different times to localize the PET signal. Images correspond to the lesion evolution of the same animal over time.
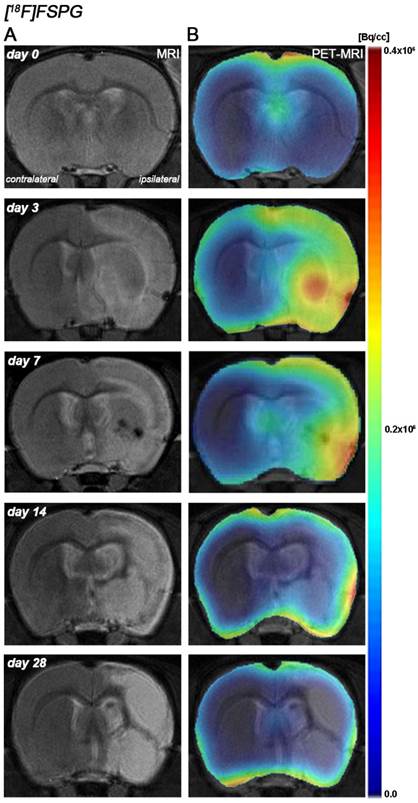
[18F]FSPG PET after cerebral ischemia
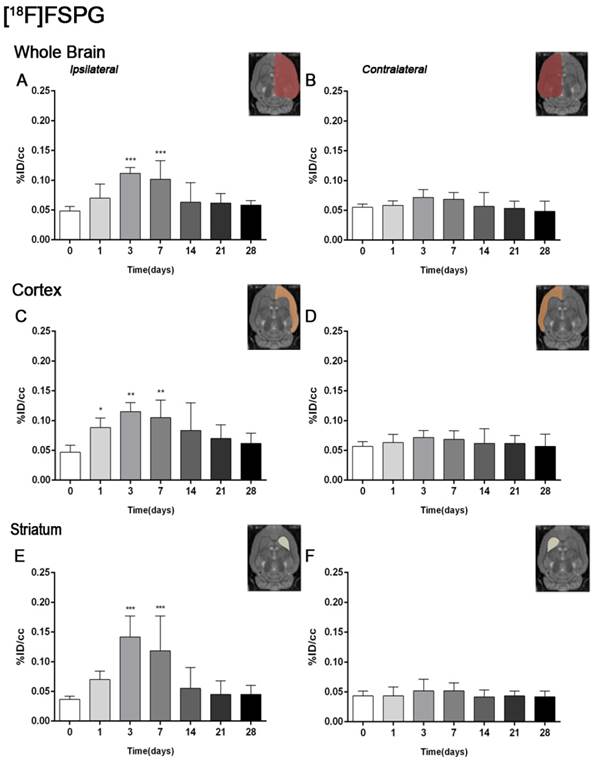
The coronal brain images with normalized color scale shown in Figure 1 illustrate the evolution of the [18F]FSPG-PET signals in ischemic animals at day 0 (control) and at 3, 7, 14 and 28 days after reperfusion. Quantification of the images provided information related to the time-course activity of system xc- in both the ipsilateral and contralateral cortex, in the striatum and the whole brain at days 0 (control) and 1, 3, 7, 14, 21 and 28 days after MCAO (Figure 2, n=6). All brain regions considered evidenced a similar [18F]FSPG binding evolution after long-term focal cerebral ischemia. In the ipsilateral whole brain (cerebrum), the PET signal for [18F]FSPG showed a non-significant increase at day 1 followed by a significant uptake increase from days 3 to 7 after ischemia in comparison to control (day 0) values (P<0.001, Figure 2A). In fact, the highest binding value was reached at day 3 after MCAO. Subsequently, the PET signal evidenced a progressive decrease from day 14 to day 28 compared to days 3 and 7. In the contralateral whole brain, [18F]FSPG PET signal at day 1 showed similar values to those at day 0 (control) followed by a non-statistically mild increase from days 3 to 7 compared to control values. This was followed by a progressive decline from days 14 to 28 after cerebral ischemia (Figure 2B). The cerebral cortex in the ipsilateral hemisphere evidenced a statistically [18F]FSPG PET signal progressive increase from day 1 to day 3 and 7 followed by a progressive decrease at days 14 and onwards (P<0.05; P<0.01, with respect to control animals, Figure 2C). In contrast, non-statistically significant differences were observed in the contralateral hemisphere despite the weak increase of PET signal observed from day 3 to day 7 following reperfusion (Figure 2D). Likewise, the non-ischemic cortex showed higher PET signal values than those observed in the non-infarcted striatum, however, this situation was reverted after cerebral ischemia where striatum displayed higher [18F]FSPG binding levels at days 3 and 7 after reperfusion than those observed in the cerebral cortex (P<0.001 with respect to control animals, Figure 2E).
Time course of the progression of the [18F]FSPG PET signal before and after cerebral ischemia. %ID/cc (mean ±SD) of [18F]FSPG was quantified in six VOIs. The entire ipsilateral cerebral hemisphere (A), contralateral hemisphere (B), ipsilateral cortex (C), contralateral cortex (D), ipsilateral striatum (E) and contralateral striatum (F) are shown. The upper right panels of each figure show the selected brain ROIs for the quantification defined on a slice of a MRI (T2W) template. Rats (n=6) were repeatedly examined by PET before (day 0) and at 1, 3, 7, 14, 21 and 28 after ischemia. *p<0.05, **p<0.01 and ***p<0.001 compared with control.
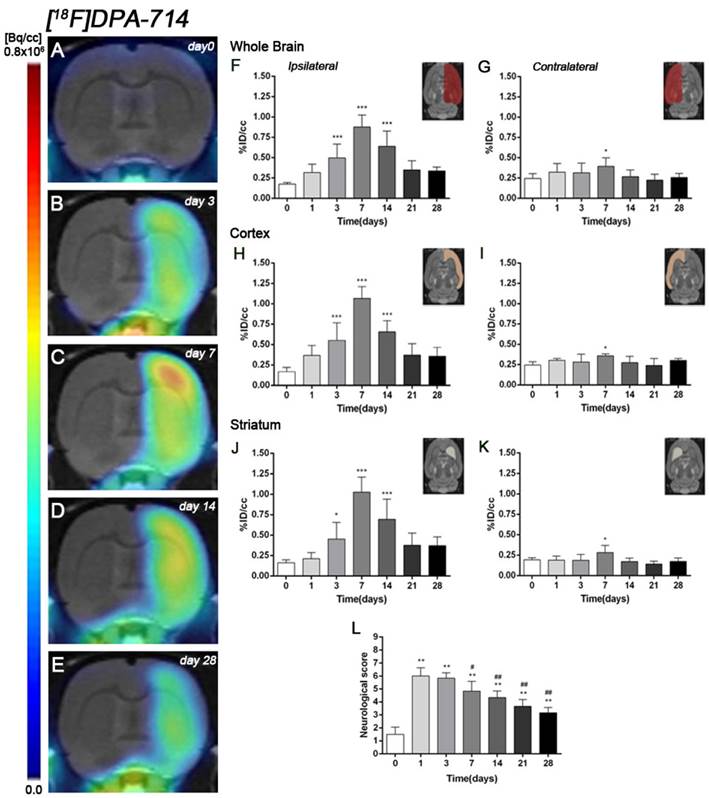
[18F]DPA-714 PET after cerebral ischemia
The coronal brain images with normalized color scale shown in Figure 3A-E illustrate the evolution of the [18F]DPA-714 PET signals in the ischemic lesion at 0, 3, 7, 14 and 28 days after reperfusion. Quantification of the images provided information related to the time-course activity of TSPO in both the ipsilateral and contralateral cortex, in the striatum and the whole brain at days 0 (control) and 1, 3, 7, 14, 21 and 28 days after MCAO (Figure 3, n=6). Quantification of the images at day 0 (control) and 1, 3, 7, 14, 21 and 28 days after MCAO show low TSPO expression in healthy cerebral tissue, followed by a dramatic up-regulation due to the inflammatory reaction, evidencing the usefulness of this radiotracer to evaluate neuroinflammation by PET imaging [31]. All brain regions considered evidenced a similar [18F]DPA-714 binding profile during the following month to cerebral ischemia. In the ischemic territory, [18F]DPA-714 uptake displayed a progressive increase during the first week after MCAO, from circa 0.20-0.30 %ID/cc at days 0 and 1, to 0.50 at day 3, peaking to ca. 1 at day 7. Uptake of [18F]DPA-714 then decreased from 0.70 at day 14 to 0.30 at days 21-28 (P<0.001, with respect to control animals, Figure 3F). In the contralateral hemisphere, [18F]DPA-714 PET uptake showed a significant increase at day 7 after reperfusion in relation to day 0 (P<0.05, Figure 3G). The cerebral cortex in the ipsilateral hemisphere evidenced a statistically [18F]FSPG PET signal progressive increase from day 1 to day 3 and 7 followed by a progressive decrease at days 14 and onwards (P<0.001, with respect to control animals, Figure 3H). Likewise, the contralateral cortex showed a significant binding increase at day 7 after reperfusion (P<0.05, Figure 3I). The ipsilateral and contralateral striatums evidenced a similar TSPO expression than that observed in the cerebral cortex after cerebral ischemia; (i) a progressive increase from day 3 to day 7 and followed by a progressive decrease from day 14 to 21 (P<0.05; P<0.001, with respect to control animals, Figure 3J) in the ischemic striatum and (ii) a significant TSPO over-expression at day 7 after ischemia in the contralateral striatum (P<0.05, with respect to control animals, Figure 3K).
Serial images of [18F]DPA-714 at control (day 0), day 3, day 7, day 14 and day 28 after MCAO. Normalized coronal PET images of [18F]DPA-714 signal after cerebral ischemia are co-registered with a MRI (T2W) rat template to localize anatomically the PET signal (A-E). Images correspond to the same representative animal for each time point. [18F]DPA-714 uptake was quantified as %ID/cc (mean ±SD) in the entire ipsilateral hemisphere (F), contralateral hemisphere (G), ipsilateral cortex (H), contralateral cortex (I), ipsilateral striatum (J) and contralateral striatum (K). The upper right panels of each figure show the selected brain ROIs for the quantification defined on a slice of a MRI (T2W) template. Rats (n=6) were repeatedly examined by PET before (day 0) and at 1, 3, 7, 14, 21 and 28 after ischemia. The neurologic score shows a neurofunctional improvement over time (L). *p<0.05 and ***p<0.001 compared with control; #p<0.05 and ##p<0.01 compared with day 1.
Time course of neurologic score after cerebral ischemia
Ischemic animals subjected to [18F]DPA-714 PET studies (n=6) presented a major impairment from days 1 to day 7 after MCAO in relation to control (day 0). The neurologic impairment showed a significant increase with respect to control at days 1, 3, 7, 14, 21 and 28 (P<0.01). After day 3, rats exhibited a trend to progressive functional recovery over time at days 7, 14, 21 and 28 in relation to day 1 after ischemia (P<0.05; P<0.01, Figure 3L).
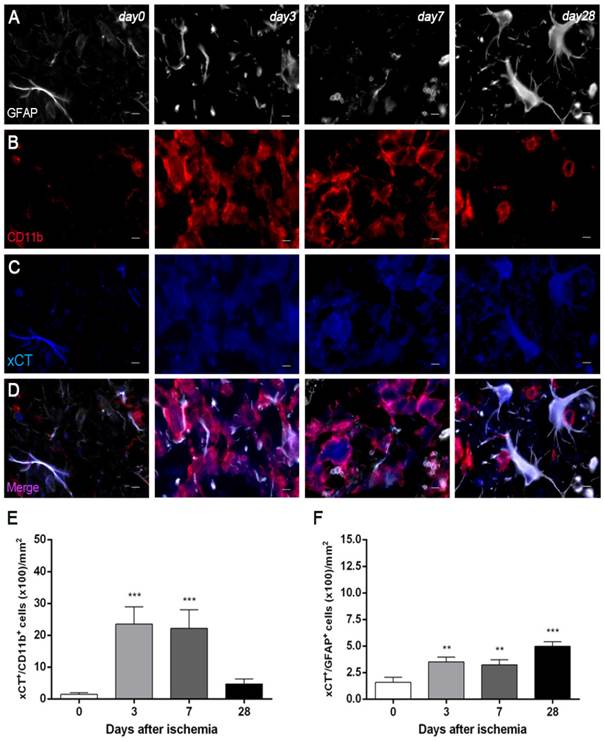
Expression of system xc- in microglia and astrocytes after MCAO
Immunofluorescence staining exhibited xCT expression in both microglia/macrophages and astrocytes after ischemia (Figure 4). At days 3 and 7, cells with the morphology of amoeboid reactive microglia/macrophages showed intense CD11b immunoreactivity in the lesion (in red; Figure 4B) followed by a decrease at day 28 (in red; Figure 4B). The over-reactivity of microglia co-localized with the cellular expression of xCT (in blue and red; Figure 4D) at days 3, 7 and 28 after ischemia. The number of xCT+/CD11b+ cells displayed a significant increase at days 3 and 7 in relation to day 0 followed by a sharp decrease at day 28 after reperfusion (P<0.001, Figure 4E). Likewise, astrocytes displayed an increase of the GFAP immunoreactivity from days 3-7 to 28 (in white; Figure 4A) after ischemia, forming a thin astrocytic rim in the vicinity of the lesion. In addition, reactive astrocytes showed co-localization with xCT expression (in blue and white; Figure 4D) after cerebral ischemia (3, 7 and 28 days). The number of xCT+/GFAP+ cells showed a significant progressive increase from days 3-7 to day 28 after MCAO (P<0.01; P<0.001, Figure 3F).
Immunofluorescent labeling of GFAP (white), CD11b (red) and xCT (blue) in the ischemic area, shown as three channels. The data show temporal evolution of xCT in microglial and astrocytic cells at day 0 (control) (left column, n=4), day 3 (middle left column, n=4), day 7 (middle right column, n=4) and day 28 (right column, n=4) after ischemia. GFAP-positive astrocytes increase in the ischemic area over time (A). CD11b-reactive microglia/macrophages increase at days 3 to 7 (B) corresponding to the temporal xCT immunoreactivity (C). (D) Merged images of three immunofluorescent antibodies at different time points. The number of CD11b-reactive microglia/macrophages expressing xCT increase at days 3 and 7 after cerebral ischemia (E). The number of GFAP-reactive astrocytes expressing xCT increase in the ischemic area following reperfusion (F). **p<0.01 and ***p<0.001 compared with control. Scale bars, 5 μm.
Effect of system xc- inhibition on neuroinflammation following cerebral ischemia
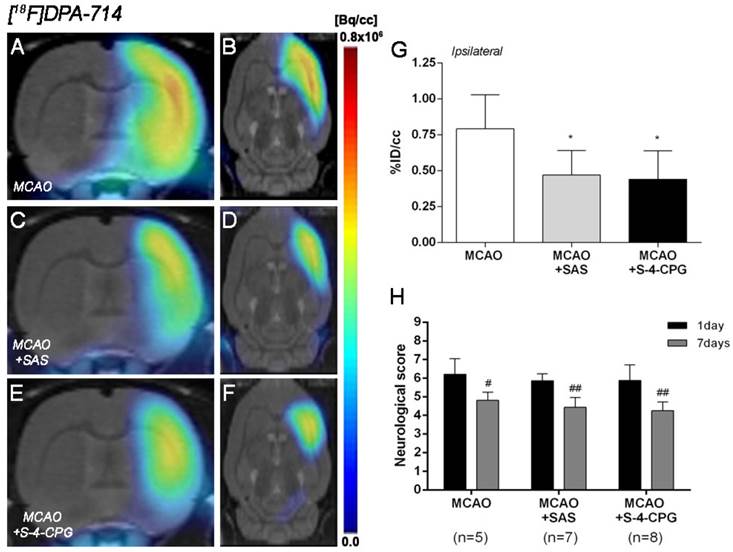
The effect of system xc- inhibition with SAS and S-4-CPG on neuroinflammation was explored by PET imaging of TSPO with [18F]DPA-714 at day 7 following MCAO (Figure 5). All the images were quantified in standard units, i.e., %ID/cc. Coronal and axial images with normalized color scale illustrate the [18F]DPA-714 PET uptake in vehicle (MCAO) (Figure 5A and B), SAS (MCAO+SAS) (Figure 5C and D) and S-4-CPG (MCAO+S-4-CPG) (Figure 5E and F) ischemic rats. Both SAS and S-4-CPG treatments evidenced a significant decrease of [18F]DPA-714 binding in the ischemic cerebral hemisphere in relation to non-treated ischemic rats (P<0.05, Figure 5G). Neurological score at day 1 after ischemia showed similar values reflecting that the groups presented similar neurological impairment before the start of the treatments (Figure 5H). At day 7, treated ischemic rats with SAS and S-4-CPG displayed a superior neurofunctional improvement in relation to that showed by non-treated ischemic rats (P<0.05; P<0.01, Figure 5H).
Normalized coronal (A, C, E) and axial (B, D, F) PET images of [18F]DPA-714 at day 7 after middle cerebral artery occlusion (MCAO) in vehicle (A, B), SAS (C, D) and S-4-CPG-treated (E, F) rats at day 7 after cerebral ischemia. PET images are co-registered with a MRI (T2W) rat template to localize anatomically the PET signal. [18F]DPA-714 uptake was quantified as %ID/cc (mean ± SD) in the entire ipsilateral cerebral hemisphere. Vehicle (n=5), SAS (n=7) and S-4-CPG-treated rats (n=8) were examined by PET 7 days after ischemia (G). The neurologic score show similar neurologic outcome at day 1 after ischemia (before the start of treatments) followed by a higher neurological outcome improvement at day 7 after SAS and S-4-CPG treatments than vehicle treated rats (H). *p<0.05 compared with vehicle; #p<0.05 and ##p<0.01 compared with day 1.
Expression of TSPO receptors in microglia and astrocytes after treatments in ischemic rats
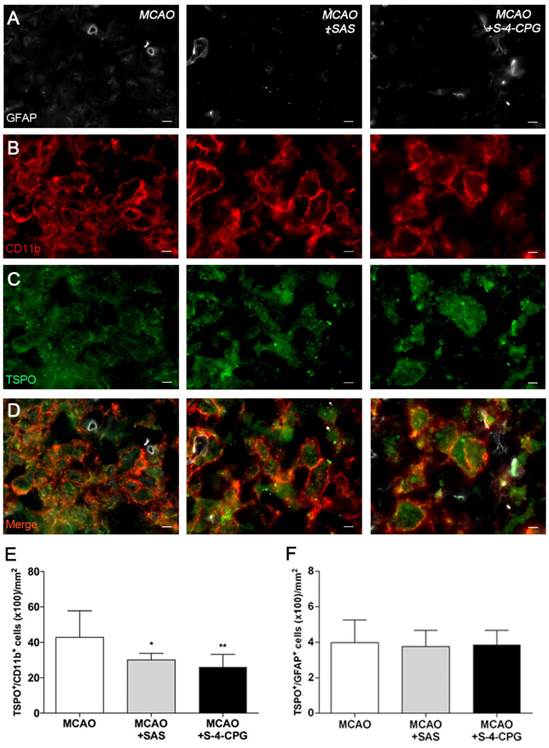
Immunofluorescence staining exhibited TSPO expression in both microglia/macrophages and astrocytes after ischemia in vehicle and treated-rats (Figure 6). At day 7, cells with the morphology of amoeboid reactive microglia/macrophages showed intense CD11b immunoreactivity (in red; Figure 6B) in the lesion that co-localized with TSPO receptor expression (in green and red; Figure 6D). The number of TSPO+/CD11b+ cells displayed a significant decrease in treated ischemic rats with both SAS and S-4-CPG at day 7 after ischemia in comparison with control ischemic rats (P<0.05; P<0.01, Figure 6E). Likewise, rats treated with S-4-CPG showed a non-significant decrease of cerebral TSPO+/CD11b+ cells in comparison to SAS treated ischemic rats. In contrast, equivalent number of TSPO+/GFAP+ cells was observed for non-treated and treated ischemic rats at 7 days after ischemia (Figure 6F).
Immunofluorescent labeling of GFAP (white), CD11b (red) and TSPO (green) in the ischemic area, shown as three channels. The data show temporal evolution of TSPO in microglial and astrocytic cells at day 7 after MCAO in vehicle (left column, n=5), SAS (middle column, n=7) and S-4-CPG-treated rats (right column, n=8). GFAP-positive astrocytes do not change after treatments (A). CD11b-reactive microglia/macrophages (B) and TSPO receptor (C) decrease after MCAO in SAS and S-4-CPG-treated rats. (D) Merged images of three immunofluorescent antibodies. The number of CD11b-reactive microglia/macrophages expressing TSPO decrease at day 7 after daily treatment with SAS and S-4-CPG (E). The number of GFAP-reactive astrocytes expressing TSPO shows similar values following treatment (F). *p<0.05 and **p<0.01 compared with vehicle. Scale bars, 5 μm.
Effect of system xc- inhibition on the expression of M1 and M2 microglia/macrophages
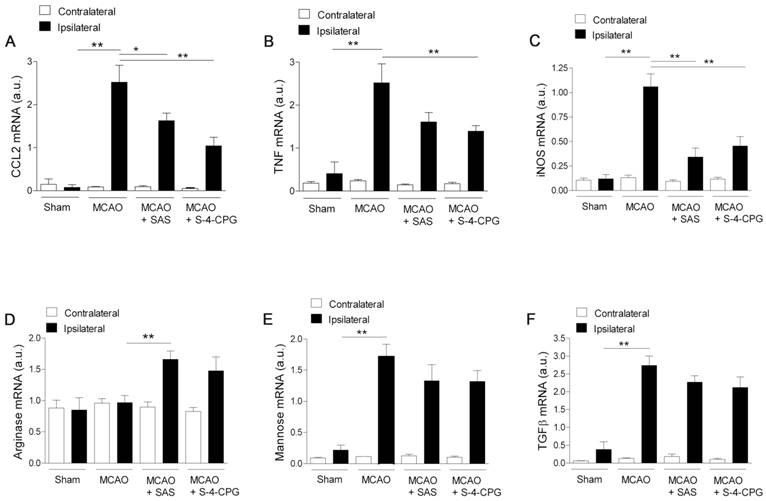
We next investigated the impact of system xc- inhibition on microglia phenotype after stroke. Polarized microglia/macrophages after cerebral ischemia are commonly distinguished by their expression of signature genes for surface markers and cytokines/chemokines [32]. Reverse-transcription polymerase chain reaction (RT-PCR) was used to measure the effect of system xc- inhibition in the expression of mRNA for M1 (CCL2, TNF and iNOS) and M2 (arginase, mannose receptor and TGFβ) markers in the region of the infarction and the in contralateral brain hemisphere at sham control and at day 7 after MCAO in vehicle-, SAS- and S-4-CPG-treated rats (Figure 7). M1 gene markers (CCL2, TNF and iNOS) evidenced a significant overexpression following cerebral ischemia in vehicle-treated rats (MCAO) (P<0.01, Figure 7A to C). M1 markers expression displayed a significant decrease in ischemic rats after system xc- inhibition with SAS and S-4-CPG (P<0.05; P<0.01, Figure 7A to C) with respect to non-treated rats. In contrast, M2 markers showed more complex patterns of expression after stroke. Firstly, arginase expression displayed no expression changes following cerebral ischemia and was significantly increased in ischemic rats treated with SAS but not-significantly with S-4-CPG (Figure 7D). In contrast mannose receptor and TGFβ mRNA expression evidenced a significant increase after MCAO that was slightly decreased in rats treated with SAS and S-4-CPG (P<0.01, Figure 7E and F). Altogether, data showed here demonstrated that system xc- modulates microglial inflammatory reaction.
Cystine/glutamate antiporter inhibition induces changes in mRNA expression of M1 and M2 microglia polarization markers. Reverse-transcription polymerase chain reaction (RT-PCR) was performed using RNA extracted from the region of the infarction at sham (control) (n=5) and at day 7 after MCAO in vehicle (n=5), SAS (n=7) and S-4-CPG-treated rats (n=8). Expression of mRNA for M1 markers CCL2 (A), TNF (B) and iNOS (C) and for M2 markers Arginase (D), Mannose receptor (E) and TGFβ (F) were performed after ischemia. *p<0.05 and **p<0.01 compared with vehicle.
Discussion
Following brain injury, system xc- has not only a detrimental function via glutamate release but also a beneficial neuroprotective and neurothrophic effect through the import of L-cysteine for glutathione production and oxidative protection [33]. Indeed, Piani and Fontana showed that microglial cells overexpressed system xc- as a response to the oxidative stress underlying CNS disease [7] and the conversion to excitotoxic stress was related to the exacerbation of neurodegeneration [14, 16]. Therefore, the identification of new up-regulated transporters in reactive glial cells, such as system xc- is of great interest to the in vivo imaging research of cerebral ischemia. Because of this, we have assessed in parallel the in vivo expression of the system xc- and TSPO in reactive glial cells using PET imaging procedures, in combination with immunohistochemistry, RT-PCR and neurofunctional evaluation after experimental stroke in rats.
PET imaging of system xc- with [18F]FSPG
Recent preclinical and clinical PET studies performed with [18F]FSPG have shown a low background uptake in the normal brain [34] that improves the visualization of the system xc- function increase following pathological situations [4, 35]. These findings are consistent with the low [18F]FSPG binding uptake in the rat brain before the induction of the cerebral ischemia. In the present study, the PET [18F]FSPG binding uptake in the healthy rat brain was mainly concentrated in the leptomeninges and ventricles (Figure 1) which are in agreement with the well-known distribution of system xc- in the rat healthy brain [36, 37].
In vivo and ex vivo evidence of system xc- overexpression after cerebral ischemia
Previous in vivo PET imaging of system xc- with [18F]FSPG evidenced a significant progressive increased uptake at early phase following MCAO in rats. Indeed, these results evidenced the role of system xc- on excitotoxic damage after reperfusion [4]. In the present study, [18F]FSPG binding was evaluated from day 1 to day 28 after MCAO in rats, to show its relationship to the neuroinflammatory reaction after cerebral ischemia. In the infarcted brain hemisphere, [18F]FSPG binding experienced a non-significant increase at day 1 followed by a maximum binding at days 3 to 7 that was followed by a progressive decline to pseudo-control values at days 21 to 28 after reperfusion (Figure 1 and 2). Nevertheless, these results stand in contrast with the in vivo PET imaging distribution of TSPO, a well-known marker for neuroinflammation, following cerebral ischemia [26, 38] (Figure 3). In our study, the ischemic brain hemisphere experienced a gradual [18F]DPA-714 binding increase from days 1 to 3 which peaked at day 7, followed by a slight decline afterwards. These results are in agreement with Martin and collaborators who, after monitoring the in vivo post-ischemic TSPO receptor expression with [18F]DPA-714 PET, observed a progressive binding increase that peaked at day 11 followed by a progressive decline later on [26]. Likewise, the peak of the inflammatory reaction observed by PET imaging during the first week after ischemia runs in parallel with the gradual recovery of neurological handicap (Figure 3L). Thus, animals experienced a quasi-recovery of functional outcome one month after stroke. Likewise, such capacity to undergo motor and cognitive recovery has been monitored in adult human brain following stroke [39].
Altogether, these results evidence an earlier overexpression of system xc- compared to TSPO that could suggest the existence of a different involvement of both receptors on the neuroinflammatory response after stroke. The first step to confirm this hypothesis was the characterization of system xc- by ex vivo immunohistochemistry at days 3, 7 and 28 after reperfusion (Figure 4). These results verified the expression of system xc- in both microglia/macrophages and astrocytes following cerebral ischemia. Moreover, immunohistochemistry showed an over-increase of microglial cells expressing system xc-(xCT+/CD11b+) at days 3 and 7 in relation to day 0 followed by a dramatic decrease at day 28 after cerebral ischemia. Likewise, astrocytes expressing system xc- (xCT+/GFAP+) evidenced a mild progressive increase from day 3 to day 28 after cerebral ischemia (Figure 4). Hence, the present results supported the previous PET imaging results obtained with [18F]FSPG evidencing that the system xc- over-expression at days 3 to 7 was mainly promoted by the system xc- expression increase by microglial cells. Indeed, these findings are in concordance with the role of system xc- on microglial reactivity following amyotrophic lateral sclerosis [8] and multiple sclerosis [24] in rodents. Despite this, little is known concerning the role of system xc- on inflammation underlying experimental ischemic stroke.
System xc- inhibition attenuates neuroinflammatory after cerebral ischemia
Pharmacological inhibition of system xc- with sulfasalazine depletes the inflammatory response following EAE [17] and amyothrophic lateral sclerosis [8]. Likewise, since sulfasalazine is an inhibitor of the antiinflammatory NF-ΚB factor [40], other blockers of system xc- that do not have any effect on NF-ΚB such as S-4-CPG have also been tested to reduce the inflammatory reaction after EAE in mice [17]. Additionally, the depletion of microglial population using clodronate induced a reduction on system xc- expression following experimental multiple sclerosis in rats [24]. Altogether these studies confirmed the role of system xc- on the inflammatory reaction underlying neurodegenerative diseases. In our study, we explored the effect of SAS and S-4-CPG on neuroinflammation by using in vivo [18F]DPA-714 PET binding following cerebral ischemia (Figure 5). The inhibition of system xc- with both SAS and S-4-CPG induced a significant decrease of the TSPO expression following seven days after reperfusion (Figure 5G). Besides, treatment with system xc- inhibitors evidenced a major neurological handicap improvement at day 7 after reperfusion in relation to the non-treated rats. These findings support those showed by Evonuk and collaborators who observed a clinical improvement after system xc- inhibition in EAE mice [17]. Besides, rats treated with SAS and S-4-CPG displayed a significant decrease of microglial population expressing TSPO (Figure 6), confirming the results obtained with [18F]DPA-714 PET and the role of the system xc- on microglial reactivity after cerebral ischemia in rats.
Effect of system xc- inhibition on M1/M2 microglial state
Microglial activation can be influenced by both extracellular glutamate and intracellular glutathione, by acting through microglial expressed glutamate receptors [41, 42]. Thus, system xc- inhibition might have a direct influence on the microglial function through the control of microglial M1/M2 polarization state following stroke. For this reason, we evaluated the mRNA expression of typical M1 and M2 markers after inhibition of system xc- with both sulfasalazine and S-4-CPG in ischemic rats. The expression of known M1 proinflammatory/neurotoxic markers; CCL2, TNF and iNOS at day 7 after cerebral ischemia showed a significant increase in relation to sham-operated rats (Figure 7). These findings stand in agreement with the expression profile experienced by proinflammatory markers after MCAO in mice [32]. The inhibition of system xc- showed a significant decrease of CCL2, TNF and iNOS (Figure 7). On the contrary, the antiinflammatory M2 mRNA markers arginase, mannose receptor and TGFβ showed an asymmetrical temporal expression after cerebral ischemia in mice. While mannose receptor and TGFβ mRNA showed high expression at day 7 after reperfusion, arginase evidenced a peak at day 3 followed by a sharp decline from day 5 to day 7 afterwards [32]. Our results support these previous findings since arginase showed similar mRNA values at day 7 after ischemia to those obtained for sham-operated rats, and both mannose receptor and TGFβ mRNA levels displayed a significant increase in ischemic animals in relation to non-ischemic rats (Figure 7). Therefore, this may be a possible explanation why only arginase experienced an increase in mRNA expression following treatment with both sulfasalazine and S-4-CPG after cerebral ischemia in rats. Hence, these results suggest that system xc- inhibition in stroke leads to a change in microglial phenotype markers. Interestingly, similar results have also been described in ALS in mice, indicating the regulation of microglial functions by system xc- [8].
Summary and conclusions
We report here [18F]FSPG PET imaging to assess system xc- activity and its relationship with neuroinflammatory response after cerebral ischemia in rats. Our results confirmed the [18F]FSPG binding increase in the ipsilateral hemisphere during the first week after cerebral ischemia followed by a progressive decrease later on. In addition, our findings displayed an earlier expression of system xc- compared to TSPO with [18F]DPA-714 imaging, evidencing its novel role on inflammation after cerebral ischemia. Besides, the overexpression of system xc- was identified in microglial cells by ex vivo immunohistochemistry and the inhibition of system xc- was able to promote a decrease of TSPO expression and a shift of pro-inflammatory microglia state towards an anti-inflammatory phenotype after MCAO in rats. These findings provide novel insights on the inflammatory reaction underlying cerebral ischemia that might contribute to the identification of a novel target at improving both diagnosis and therapy evaluation of stroke.
Acknowledgements
The authors would like to thank M González, A Leukona, M Errasti, A Arrieta and A Cano for technical support in the radiosynthesis and technical assistance in the PET studies, and the Department of Industry of the Basque Government and Spanish Ministry of Economy and Competitiveness through grant SAF2014-54070-JIN and SAF2013-45084-R for financial support.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bannai S, Kitamura E. Transport interaction of L-cystine and L-glutamate in human diploid fibroblasts in culture. J Biol Chem. 1980;255:2372-2376
2. Lo M, Wang YZ, Gout PW. The x(c)- cystine/glutamate antiporter: a potential target for therapy of cancer and other diseases. J Cell Physiol. 2008;215:593-602
3. Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-): cystine supplier and beyond. Amino Acids. 2012;42:231-246
4. Soria FN, Perez-Samartin A, Martin A, Gona KB, Llop J, Szczupak B. et al. Extrasynaptic glutamate release through cystine/glutamate antiporter contributes to ischemic damage. J Clin Invest. 2014;124:3645-3655
5. Murphy TH, Schnaar RL, Coyle JT. Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. Faseb J. 1990;4:1624-1633
6. Cho Y, Bannai S. Uptake of glutamate and cysteine in C-6 glioma cells and in cultured astrocytes. J Neurochem. 1990;55:2091-2097
7. Piani D, Fontana A. Involvement of the cystine transport system xc- in the macrophage-induced glutamate-dependent cytotoxicity to neurons. J Immunol. 1994;152:3578-3585
8. Mesci P, Zaidi S, Lobsiger CS, Millecamps S, Escartin C, Seilhean D. et al. System xC- is a mediator of microglial function and its deletion slows symptoms in amyotrophic lateral sclerosis mice. Brain. 2015;138:53-68
9. Pampliega O, Domercq M, Soria FN, Villoslada P, Rodriguez-Antiguedad A, Matute C. Increased expression of cystine/glutamate antiporter in multiple sclerosis. J Neuroinflammation. 2011;8:1742-2094
10. Espey MG, Kustova Y, Sei Y, Basile AS. Extracellular glutamate levels are chronically elevated in the brains of LP-BM5-infected mice: a mechanism of retrovirus-induced encephalopathy. J Neurochem. 1998;71:2079-2087
11. Qin S, Colin C, Hinners I, Gervais A, Cheret C, Mallat M. System Xc- and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid-beta peptide 1-40. J Neurosci. 2006;26:3345-3356
12. Shih AY, Erb H, Sun X, Toda S, Kalivas PW, Murphy TH. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J Neurosci. 2006;26:10514-10523
13. Barger SW, Goodwin ME, Porter MM, Beggs ML. Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J Neurochem. 2007;101:1205-1213
14. Barger SW, Basile AS. Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J Neurochem. 2001;76:846-854
15. Buckingham SC, Campbell SL, Haas BR, Montana V, Robel S, Ogunrinu T. et al. Glutamate release by primary brain tumors induces epileptic activity. Nat Med. 2011;17:1269-1274
16. Domercq M, Sanchez-Gomez MV, Sherwin C, Etxebarria E, Fern R, Matute C. System xc- and glutamate transporter inhibition mediates microglial toxicity to oligodendrocytes. J Immunol. 2007;178:6549-6556
17. Evonuk KS, Baker BJ, Doyle RE, Moseley CE, Sestero CM, Johnston BP. et al. Inhibition of System Xc(-) Transporter Attenuates Autoimmune Inflammatory Demyelination. J Immunol. 2015;195:450-463
18. Koglin N, Mueller A, Berndt M, Schmitt-Willich H, Toschi L, Stephens AW. et al. Specific PET imaging of xC- transporter activity using a (1)(8)F-labeled glutamate derivative reveals a dominant pathway in tumor metabolism. Clin Cancer Res. 2011;17(18):6000-6011
19. Patel SA, Warren BA, Rhoderick JF, Bridges RJ. Differentiation of substrate and non-substrate inhibitors of transport system xc(-): an obligate exchanger of L-glutamate and L-cystine. Neuropharmacology. 2004;46(2):273-284
20. Baek S, Mueller A, Lim YS, Lee HC, Lee YJ, Gong G. et al. (4S)-4-(3-18F-fluoropropyl)-L-glutamate for imaging of xC transporter activity in hepatocellular carcinoma using PET: preclinical and exploratory clinical studies. J Nucl Med. 2013;54:117-123
21. Baek S, Choi CM, Ahn SH, Lee JW, Gong G, Ryu JS. et al. Exploratory clinical trial of (4S)-4-(3-[18F]fluoropropyl)-L-glutamate for imaging xC- transporter using positron emission tomography in patients with non-small cell lung or breast cancer. Clin Cancer Res. 2012;18:5427-5437
22. Chae SY, Choi CM, Shim TS, Park Y, Park CS, Lee HS. et al. Exploratory Clinical Investigation of (4S)-4-(3-18F-Fluoropropyl)-l-Glutamate PET of Inflammatory and Infectious Lesions. J Nucl Med. 2016;57:67-69
23. Liu Z, Chen H, Chen K, Shao Y, Kiesewetter DO, Niu G. et al. Boramino acid as a marker for amino acid transporters. Sci Adv. 2015:1 (8)
24. Martin A, Vazquez-Villoldo N, Gomez-Vallejo V, Padro D, Soria FN, Szczupak B. et al. In vivo imaging of system xc- as a novel approach to monitor multiple sclerosis. Eur J Nucl Med Mol Imaging. 2016;43:1124-38
25. Rojas S, Martin A, Arranz MJ, Pareto D, Purroy J, Verdaguer E. et al. Imaging brain inflammation with [(11)C]PK11195 by PET and induction of the peripheral-type benzodiazepine receptor after transient focal ischemia in rats. J Cereb Blood Flow Metab. 2007;27:1975-1986
26. Martin A, Boisgard R, Theze B, Van Camp N, Kuhnast B, Damont A. et al. Evaluation of the PBR/TSPO radioligand [(18)F]DPA-714 in a rat model of focal cerebral ischemia. J Cereb Blood Flow Metab. 2010;30:230-241
27. Justicia C, Perez-Asensio FJ, Burguete MC, Salom JB, Planas AM. Administration of transforming growth factor-alpha reduces infarct volume after transient focal cerebral ischemia in the rat. J Cereb Blood Flow Metab. 2001;21:1097-1104
28. Damont A, Hinnen F, Kuhnast B, Schöllhorn-Peyronneau M-A, James M, Luus C. et al. Radiosynthesis of [18F]DPA-714, a selective radioligand for imaging the translocator protein (18 kDa) with PET. Journal of Labelled Compounds and Radiopharmaceuticals. 2008;51:286-292
29. Menzies SA, Hoff JT, Betz AL. Middle cerebral artery occlusion in rats: a neurological and pathological evaluation of a reproducible model. Neurosurgery. 1992;31:100-106
30. Vallejo-Illarramendi A, Domercq M, Perez-Cerda F, Ravid R, Matute C. Increased expression and function of glutamate transporters in multiple sclerosis. Neurobiol Dis. 2006;21:154-164
31. Chen MK, Guilarte TR. Translocator protein 18 kDa (TSPO): molecular sensor of brain injury and repair. Pharmacol Ther. 2008;118:1-17
32. Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S. et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43:3063-3070
33. Bridges RJ, Natale NR, Patel SA. System xc(-) cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol. 2012;165:20-34
34. Mosci C, Kumar M, Smolarz K, Koglin N, Stephens AW, Schwaiger M. et al. Characterization of Physiologic F FSPG Uptake in Healthy Volunteers. Radiology. 2016;18:142000
35. Mittra ES, Koglin N, Mosci C, Kumar M, Hoehne A, Keu KV. et al. Pilot Preclinical and Clinical Evaluation of (4S)-4-(3-[18F]Fluoropropyl)-L-Glutamate (18F-FSPG) for PET/CT Imaging of Intracranial Malignancies. PLoS One. 2016:11 (2)
36. Sato H, Tamba M, Okuno S, Sato K, Keino-Masu K, Masu M. et al. Distribution of cystine/glutamate exchange transporter, system x(c)-, in the mouse brain. J Neurosci. 2002;22:8028-8033
37. Sasaki H, Sato H, Kuriyama-Matsumura K, Sato K, Maebara K, Wang H. et al. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J Biol Chem. 2002;277:44765-44771
38. Martin A, Szczupak B, Gomez-Vallejo V, Domercq M, Cano A, Padro D. et al. In vivo PET imaging of the alpha4beta2 nicotinic acetylcholine receptor as a marker for brain inflammation after cerebral ischemia. J Neurosci. 2015;35:5998-6009
39. Rossini PM, Calautti C, Pauri F, Baron JC. Post-stroke plastic reorganisation in the adult brain. Lancet Neurol. 2003;2:493-502
40. Chung WJ, Sontheimer H. Sulfasalazine inhibits the growth of primary brain tumors independent of nuclear factor-kappaB. J Neurochem. 2009;110:182-193
41. Kaindl AM, Degos V, Peineau S, Gouadon E, Chhor V, Loron G. et al. Activation of microglial N-methyl-D-aspartate receptors triggers inflammation and neuronal cell death in the developing and mature brain. Ann Neurol. 2012;72(4):536-549
42. Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW. et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal. 2013;18:522-555
Author contact
![]() Corresponding author: Abraham Martín, Molecular Imaging Unit, CIC biomaGUNE, Pº Miramon 182, San Sebastian, Spain. E-mail address: amartines.
Corresponding author: Abraham Martín, Molecular Imaging Unit, CIC biomaGUNE, Pº Miramon 182, San Sebastian, Spain. E-mail address: amartines.