Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2015; 5(10):1068-1082. doi:10.7150/thno.11517 This issue Cite
Research Paper
Photoluminescent Mesoporous Silicon Nanoparticles with siCCR2 Improve the Effects of Mesenchymal Stromal Cell Transplantation after Acute Myocardial Infarction
Wenbin Lu1, 2* ![]() , ZhuoYing Xie3*, Yong Tang1, 2, Ling Bai3, Yuyu Yao1, Cong Fu1, Genshan Ma1
, ZhuoYing Xie3*, Yong Tang1, 2, Ling Bai3, Yuyu Yao1, Cong Fu1, Genshan Ma1 ![]()
1. Department of Cardiology, ZhongDa Hospital affiliated with Southeast University, China;
2. Department of Cardiology, the Second Hospital affiliated with Southeast University, China;
3. State Key Laboratory of Bioelectronics, School of Biological Science and Medical Engineering, Southeast University, China.
*These authors contributed equally to this work.
Received 2015-1-7; Accepted 2015-5-7; Published 2015-6-25
Abstract

Background: Despite the benefits of mesenchymal stromal cell (MSC) transplantation in cardiac tissue, detailed in vivo observations have shown that MSCs only survive for a brief period after transplantation due to harsh microenvironmental conditions, including ischemia, inflammation and anoikis, in the infarcted myocardium. Thus, new strategies are needed to enhance MSC survival and inhibit cardiac remodeling. Studies have now demonstrated that chemokine [C-C motif] ligand 2 (CCL2) and its cognate receptor C-C chemokine receptor 2 (CCR2) promote excessive Ly6Chigh inflammatory monocyte infiltration at the infarct in response to ischemic myocardial injury. Therefore, decreasing the activities of these monocytes immediately after acute myocardial infarction (AMI) could be beneficial for AMI patients. Objectives: This study tested the hypothesis that therapeutic siRNA-loaded photoluminescent mesoporous silicon nanoparticles (PMSNs) targeting CCR2 expression in Ly6Chigh inflammatory monocytes decrease the accumulation of these cells in the infarct, improve the efficacy of MSC transplantation and attenuate myocardial remodeling.
Methods: PMSNs carrying therapeutic siCCR2 were first synthesized without the inclusion of fluorescent materials or dyes. After AMI BALB/c mice were established, 105 5-ethynyl-2'- deoxyuridine (EdU)-labeled MSCs suspended in 100 µl of phosphate buffered saline (PBS) were injected into the border zone of the infarct of each mouse. PMSNs-siCCR2 (25 µg/g) were also intravenously injected via the tail vein immediately following AMI induction. Control mice were injected with an equal amount of PMSNs without siCCR2. PMSNs-siCCR2 were examined in vivo using near-infrared imaging technology. The therapeutic effects of PMSNs-siCCR2 for MSC transplantation were determined at the mRNA, protein and functional levels.
Results: PMSNs-siCCR2 circulated freely in vivo and were cleared in a relatively short period of time (t½=37 min) with no evidence of toxicity. The therapeutic PMSNs-siCCR2 showed higher levels of cellular accumulation in Ly6Chigh monocytes in the spleen and more efficient degradation of CCR2 compared with the control (8.04%±2.17% vs. 20.02%±4.55%, p<0.001). Subsequently, the PMSNs-siCCR2 decreased the accumulation of CD11b-positive monocytes at the infarct (49.3%±17.34% vs. 61.32%±22.43%, p<0.001) on day 1. Increased survival of transplanted MSCs (13±3/mm2 vs. 4±1/mm2, p<0.001) and significantly decreased TdT-mediated dUTP nick end labeling (TUNEL)+ cardiac myocytes (17.44%±6.26% vs. 39.49%±13.28%, p<0.001) were then identified in the infarct zone three days after AMI induction in the PMSNs-siCCR2 group. Three weeks after MSC injection, significant increases were observed in the vascular density (235.5±39.6/mm2 vs. 147.4±20.3/mm2, p<0.001) and the cardiac myosin-positive area (21.7%±8.4% vs. 13.2%±4.4%, p<0.001) of the infarct border zone. In addition, significant amelioration of left ventricular (LV) remodeling (thickness of the LV posterior walls) (0.84±0.11 mm vs. 0.61±0.08 mm, p<0.001) was also observed at the same time compared with the control group.
Conclusions: PMSNs-siCCR2-mediated CCR2 gene silencing in Ly6Chigh monocytes improved the effectiveness of MSC transplantation and selectively ameliorated myocardial remodeling after AMI. These results suggest that PMSNs-siCCR2 could potentially be used to develop an anti-inflammatory therapy for post-AMI MSC transplantation.
Keywords: AMI, PMSNs, siCCR2, MSCs, Ly6Chigh monocytes
Introduction
Acute myocardial infarction (AMI) is the most frequent cause of heart failure, which is an incapacitating disease with a high prevalence and broad socioeconomic impact [1]. The need for an effective treatment for myocardial remodeling after AMI is rapidly increasing. Therapeutic strategies to prevent heart failure in the first 2 weeks after AMI are pivotal because the infarct is still biologically active during this period. Infarct expansion mainly depends on the extent and the quality of the inflammatory response after AMI. Recent recognition of physiological and pathological Ly6Chigh/low monocyte deployment from the spleens of AMI mice provided a fundamental basis for the treatment of inflammation. Proinflammatory Ly6Chigh monocytes are predominant on days 1 to 4 after AMI and promote digestion of the infarcted tissue and removal of the necrotic debris, whereas reparative Ly-6Clow monocytes are predominant during the resolution of inflammation over the next few days [2]. Spleen-derived monocytes, particularly the Ly6Chigh subsets, mainly oppose wound healing and contribute to left ventricular dilatation. In the setting of AMI, hematopoietic tissues in the bone marrow and spleen produce excessive numbers of these cells, which then enter the circulation [3]. Once these monocytes are recruited to the infarcted myocardium, they will release many inflammatory mediators, such as TNF-α, IL-1/6, INF-γ, inducible nitrous oxide synthase (iNOS) and matrix metalloproteinases (MMPs) [4]. Subsequently, the excess inflammatory monocytes and the potent inflammatory microenvironment in the infarct cause a decrease in microvessel generation and collagen deposition, thereby contributing to heart failure.
Monocytes and macrophages are key cells in the initiation and progression of AMI. Mature Ly6Chigh monocytes reside in the subcapsular red pulp of the spleen, from which they can be rapidly deployed in response to inflammatory signals when AMI occurs. The excessive infiltration or prolonged accumulation of Ly6Chigh monocytes impairs the resolution of inflammation and leads to infarct expansion [5]. Ly6Chigh monocytes are known to express the CCR2 receptor, the adhesion molecule L-selectin, and low levels of the chemokine receptor CX3CR1 [6-8]. In addition, the immobilized chemokine responsible for Ly6Chigh monocyte recruitment from the spleen and bone marrow mainly depends on CCL2/CCR2. Many previous studies have demonstrated the beneficial effects of CCR2 deficiency on left ventricular remodeling at 7 and 28 days after AMI in CCR2-/- mice generated by gene targeting [9-11]. Therefore, therapeutic targeting of CCR2 in monocytes could prevent the exaggerated activation and mobilization of the cells and the subsequent severe inflammation in response to AMI, thus leaving the impaired cardiomyocytes, myofibroblasts, and endothelial cells unaffected. Therefore, decreasing the availability of the Ly6Chigh monocytes via CCR2 silencing immediately after AMI could be favorable for these AMI patients.
Nanomaterials that can circulate in the body hold great potential for the diagnosis and treatment of diseases. In the past decade, mesoporous silica nanoparticles (MSNs) have attracted increasing attention due to their potential biomedical applications [12]. Additionally, RNA interference (RNAi) is widely regarded as a promising technology for disease treatment. However, one major obstacle to its clinical application is the lack of efficient siRNA delivery vehicles. Here, we used newly synthesized photoluminescent mesoporous silicon nanoparticles (PMSNs) [13] as a delivery material for small interfering RNA (siCCR2) [14] to decrease Ly6Chigh monocyte recruitment during AMI and subsequent MSC transplantation to aid in the healing of the infarcted myocardium. Near-infrared fluorescence imaging revealed that PMSN-encapsulated siCCR2 rapidly redistributed from the blood pool to the spleen. Highly efficient CCR2 knockdown in Ly6Chigh monocytes was confirmed at the mRNA, protein and functional levels.
Methods
Synthesis of PMSNs
PMSNs were synthesized and prepared as previously described [13]. Briefly, meso-HSiO1.5 was first synthesized in a series of processes using Pluronic-123 surfactant (P123 triblock copolymer, BASF Corporation), NaCl, HCl (aq, 0.1 M), n-butanol (99.9%, ACP), triethoxysilane (HSi (OEt) 3, 95%, Sigma Aldrich). Solid meso-HSiO1.5 was then heated to 1000°C at 20°C/min in a slightly reducing atmosphere (5% H2/95% Ar), and the temperature was maintained for 1 h. After cooling to room temperature, 100-200 nm PMSNs containing 5-10 nm periodic mesopores were obtained.
Preparation, delivery and monitoring of PMSNs-siRNA-PEI
The capacity of siRNA adsorption by the PMSNs was first determined. PMSNs (1 mg) were placed in 125 µl ethanol and incubated under sonication (100 w) for 1 hour. Then, 75 µl siRNA dissolved in water (40 µmol/l) [15] and 40 µl guanidine hydrochloride solution (4 mol/l) were added to a 0.5 ml centrifuge tube with the PMSNs. The mixture was well dispersed by vortexing for 2 minutes and then was continuously shaken at 100 × g at room temperature for an additional 1 hour. The final solution was centrifuged at 1000 × g for 10 min to completely separate PMSNs from the supernatant. The siRNA entrapment efficiency was determined by calculating the differences in the siRNA concentration in solutions before and after the adsorption process using a NanoDrop 2000 BioSpectrometer (Thermo Scientific NanoDrop).
The siRNA-loaded PMSNs isolated above were then dispersed in 250 µl ethanol. After sonication in water for 30 minutes, an equal volume of an ethanol solution of polyethylenimine (PEI 1.5 µg/ul) [16, 17] was added. The mixture was then oscillated lightly for 2 minutes, incubated at room temperature for 30 min, and centrifuged at 1000 × g for 10 min. After removing the supernatant, the PMSN-siRNA-PEI aggregates were placed in water (pH 4.5) and sonicated again for 30 minutes. Finally, the particles were separated out by centrifugation once more and finally dispersed in 200 µl DMEM containing 10% serum to prepare them for use. The final Nano-siRNA particle size and zeta potential were determined using a Malvern Zetasizer NanoZS (Malvern, UK). The loading amount of siRNA in the delivery formulation was approximately 1.77 nmol siRNA/mg PMSNs. For each AMI mouse, the dose of PMSNs-siRNA-PEI delivered was 25 mg/kg. The PMSNs-siRNA-PEI was infused via tail vein injection immediately following AMI. The control was injected with an equal amount of PMSNs without siRNA. The mice were imaged using near-infrared technology (Maestro In Vivo Imaging Systems, Thermo Fisher) every 10 minutes for 2 hours and then at 4 hours, 6 hours, 12 hours, and 24 hours post-injection.
Cytotoxicity evaluation
The toxicity of PMSNs-siCCR2-PEI to CD11b monocytes in vitro was determined using a modified 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay kit. Briefly, monocytes were seeded in 96-well plates at a density of 104 cells/well in complete growth medium and incubated with PMSNs-siCCR2-PEI for 48 hours. The cells were washed with PBS, and 10 μl MTT was added to each well at a concentration of 0.5 mg/ml. Then, the medium in each well was discarded and replaced with dimethyl sulfoxide (DMSO, 150 μl) to dissolve the formazan crystals. The absorbance was read at 490 nm in a microplate reader. The experiment was repeated at least three times. The cell viability was determined in comparison to control cultures that did not include PMSNs-siCCR2-PEI. In vivo investigations to determine the hepatic and renal function of mice treated with or without PMSNs-siCCR2-PEI at 24 hours were also proposed.
Migration assay
Migration assays of cultured Ly-6Chigh monocytes were performed in 24-well transwell plates (8.0 µm pore size) (Millipore). After incubation with PMSNs-siCCR2-PEI for 48 hours, the cells were seeded into the upper chamber of the transwell system at a concentration of 104 cells/well in 100 µl medium, and the lower chamber was filled with CCL2 (10 ng/ml, Peprotech) in 500 µl medium. After 6 hours of incubation at 37°C with 5% CO2, the upper sides of the filters were carefully washed with PBS, and the remaining cells were removed with a cotton wool swab. The cells that migrated to the bottom side of the filter were fixed with 4% paraformaldehyde and stained with DAPI. The migrated cells were manually counted in three random fields per filter at ×200 magnification using a phase contrast microscope.
AMI mouse model
BALB/c male mice (6-8-weeks-old) were used. An anterior wall MI was induced by direct ligation of the left anterior descending (LAD) artery, as previously described [18]. After being anesthetized with pentobarbital sodium (50 mg/kg), the mice were endotracheally intubated and ventilated with room air at 110 breaths per minute using a rodent ventilator (Harvard Apparatus). A left thoracotomy was performed to expose the LV of the heart. The LAD coronary artery was visualized and ligated just below the left auricular level with an 8-0 nylon suture. Blanching and dysfunction of the anterior wall verified the LAD ligation. The animal procedures were carried out according to a protocol approved by the Institutional Animal Care and Use Committee (IACUC) at Southeast University.
MSC Preparation, labeling and delivery
The MSCs were isolated and cultured following standard protocols [19]. The femurs and tibias of 7- to 10-week-old mice were flushed with standard PBS (HyClone) to collect the bone marrow cells. FACSTM Lysing Solution (BD Biosciences) was used to deplete the erythrocytes. After centrifugation at 1000 rpm for 5 minutes, the cells were washed 3 times and then resuspended in minimal essential medium supplemented with 10% FBS (DMEM, Gibco) at a density of 5×106 cells/cm2. Non-adherent cells were removed at day 3, and the cells reached confluence 2 weeks later. Trypsin (0.25%) with EDTA (HyClone) was used for the subsequent passaging. Monoclonal antibodies (Sca-1, CD105, CD90, CD31, CD34, CD45) conjugated to FITC were used to label the cells for flow cytometry (Becton Dickinson Inc., USA). MSCs from passages 5 to 6 were used for the experiments. Two days before infusion, the cells were freshly plated at a ratio of 1:3 and incubated in complete medium with 10 μM/l [20-21] EdU (5-ethynyl-2-deoxyuridine, Invitrogen) for 48 hours to label the cells. Incorporation of EdU was detected by the reaction with azide-conjugated Alexa Fluor 488. The AMI mice, prepared as described above, received 105 MSCs suspended in 100 µl PBS by intramyocardial injection.
Real-time PCR
Three days after AMI, the hearts of the mice were perfused with saline, and the infarcted left ventricles were cut at a level just below the ligation. Total RNA was extracted from the infarct using TRIzol reagent according to the manufacturer's instructions (Invitrogen). The QuantiTect Reverse Transcription Kit (Fermentas) was used for reverse transcription. PCR was performed using Platinum SYBR Green qPCR SuperMix UDG (Invitrogen) in a BIO-RAD MJ Mini Opticon Real-Time PCR System. The primer sequences were as follows: SDF-1 (NM_021704), 5'tgcccttcagattgttgcac3' and 5'ccacggatgtcagccttcc3'; Angiopoietin-1 (NM_001286062.1), 5'cattcttcgctgccattctg3' and 5'gcacattgcccatgttgaatc3'.
Immunostaining
The animals were sacrificed 3 days or 3 weeks after AMI. The tissues were fixed in formalin and embedded in paraffin blocks according to established protocols. The fixed hearts were serially cut at 8 µm from the apex to the level just below the coronary artery ligation site. After antigen retrieval, the specimens were incubated with 1% normal blocking serum in PBS for 60 min to suppress the nonspecific binding of IgG. The slides were then incubated for 30-60 min with each mouse antibody or fluorescent reagent.
Three days after the AMI, the infarcted tissue was stained with EdU (Invitrogen) to identify the live MSCs, and the TUNEL kit (R&D Systems) was used to identify the nuclei of the apoptotic cardiac myocytes in the infarct border zone. Three weeks after the AMI, the infarct border zone was stained with Texas Red-X-conjugated wheat germ agglutinin (WGA, Invitrogen) and FITC-conjugated CD31 (BD Biosciences) to measure the vessel density. The tissue was further stained with anti-myosin heavy chain eFluor 660 (eBioscience) and 4, 6-diamidino-2-phenylindole (DAPI, Roche) to measure the cardiac myosin-positive area in the infarct zone with Image Pro Plus software. Fluorescence microscopy (Olympus BX61) or a confocal laser scanning (CLS) microscopy system (Thorlabs, Inc.) was used as necessary to obtain images of the immunostaining results.
Flow cytometric analysis, Western blots and ELISA
One and three days after the AMI, mouse blood and spleens were obtained, and monocytes were isolated by gradient density centrifugation following erythrocyte lysis. After washing twice with cold PBS, the cells were resuspended at a concentration of 106 cells/ml. Single-cell suspensions were then incubated with FITC-rat anti-mouse CD11b, PE-rat anti-mouse Ly6C (BD Biosciences) or PE-anti-mouse CCR2 (R&D Systems). CD11b+Ly6Chigh monocytes or CCR2-expressing CD11b+ monocytes were further identified using a FACS Calibur flow cytometer (Becton Dickinson). Briefly, CD11b monocytes were gated in a side scatter; subsequently, subpopulations of Ly6Chigh and Ly6Clow monocytes were distinguished by their surface expression pattern of Ly6C The representative process for determining the distribution of the subpopulations of the two subtypes of monocytes in the peripheral blood and spleens of the AMI mice is shown (Supplementary Figure 4).
The infarcted myocardia were washed twice with PBS, lysed in lysis buffer [10 mmol/l Tris- HCl, pH 7.4, containing 1% Triton X-100, 100 mmol/l sodium chloride, 20 mmol/l sodium pyrophosphate, 2 mmol/l sodium orthovanadate, 2 mmol/l EDTA, and 1% protease inhibitor cocktail (Sigma)] and centrifuged at 12,000 g for 30 min at 4°C. The protein concentration was determined using a protein assay kit (Thermo Scientific, USA), and the samples were mixed with SDS-denaturing sample buffer and separated on 10% SDS-PAGE gels. The proteins were transferred to a PVDF membrane by electrophoresis. The membrane was blocked and incubated overnight on a rocking platform at 4°C with antibodies against CCR2 (Santa Cruz Biotechnology) and GAPDH (1:1000; Kangchen Biology Inc., China). The membranes were then incubated with HRP-conjugated secondary antibodies for 1 h and exposed using a Molecular Imager ChemiDoc XRS System (Bio RAD). The relative intensities of the protein bands were analyzed using Image-pro plus 6.0 (Media Cybernetics, Silver Spring, MD, USA).
The concentrations of the inflammatory cytokines IL-1, IL-6, TNF-α and IFN-γ in the serum of the AMI mice were measured using a mouse ELISA kit (R&D Systems Inc.). The absorbance measurements of the test samples were compared with a standard curve. The concentrations were determined in duplicate, and the process was performed according to the manufacturer's instructions.
Echocardiography
At 1 and 21 days after AMI, 2-dimensional echocardiography was performed on mice using the Vevo 770R high-resolution imaging system (Visual Sonic, Canada) and Vevo analysis software (Vevo 2.2.3), as previously described [22-23]. The LV dimensions and the LV posterior wall thickness were quantified from digitally recorded 2-dimensional clips and M-mode images in a short axis view from the mid-LV just below the papillary muscles to allow for consistent measurements from the same anatomical location in each mouse. Each measurement was repeated in each mouse five to six times and taken from 3 randomly chosen m-Mode clips out of the 5 that were recorded by the observer, who was blinded to the experimental group. Fractional shortening (FS) was calculated as (LvEDD-LvESD)/LvEDD×100%.
Processing of the experiments
Our experiments were performed based on the hypothesis that silencing of CCR2 in Ly6Chigh monocytes (Supplementary Figure 1) would decrease the accumulation of the cells in the infarct and improve the results of MSC transplantation. The entire in vivo experimental procedure is shown in an illustration (Supplementary Figures 2 and 3). Briefly, EdU-labeled MSCs were injected into the infarct border zone immediately after AMI was induced. Additionally, PMSNs-siCCR2 (25 µg/g) were also intravenously injected via the tail vein. The therapeutic effects of MSCs were further identified at day 1, day 3 and day 21 using the methods described above.
Statistical analysis
Data management and statistical analysis were performed with Statistical Package for the Social Sciences software (SPSS 18.0 for Windows, SPSS Inc.). The data are expressed as the mean ± standard deviation. Categorical variables were compared using the χ2 test or Fisher's exact test. For continuous data, group comparisons were performed using an unpaired t test or the Mann-Whitney U test. The results were considered statistically significant if the two-sided P-value was ≤0.05.
Results
Characterization of PMSNs-siCCR2-PEI
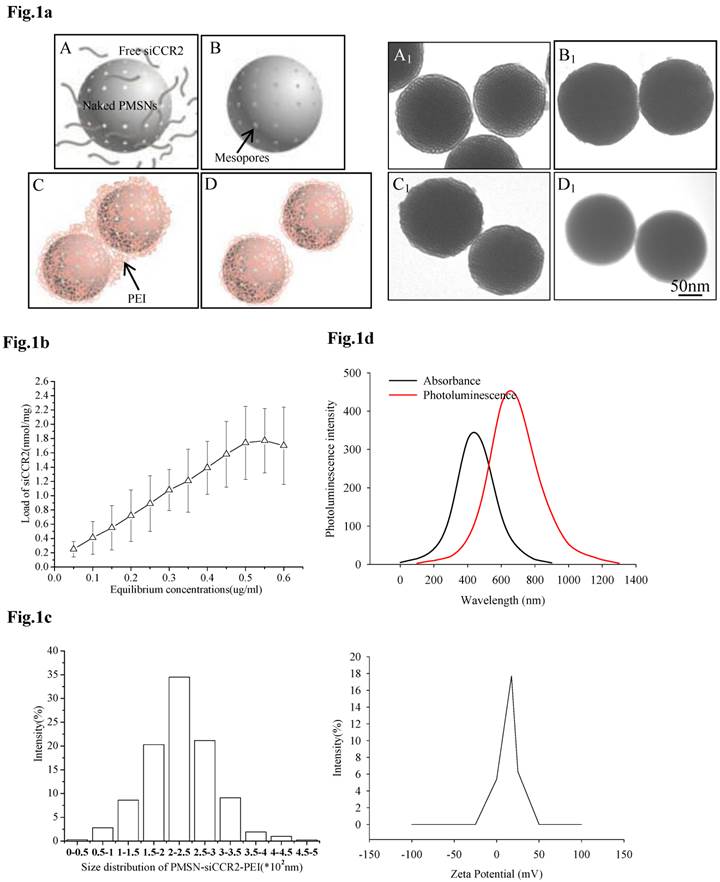
For the efficient loading of siRNA, siCCR2 was first adsorbed onto PMSNs under a strongly dehydrated solution and then mixed with an oppositely charged polyelectrolyte (PEI, 10 kDa) solution to form a polymer layer on the surface. After sonication under acidic conditions, monodispersed PMSNs-siCCR2-PEI were obtained (Fig. 1a). The amount of siCCR2 loaded in the delivery formulation reached a maximum of 1.77 nmol siRNA/mg PMSNs, with an equilibrium concentration of 0.55 µg/ul (Fig. 1b). Consistent with the transmission electron microscopy (TEM) measurements, the mean size of the PMSNs-siCCR2-PEI was 246 nm, as measured by a Nanoparticle Sizer, with a zeta potential of 17.5±2.2 mV (Fig. 1c). The intrinsic photoluminescence of the particle appears at wavelengths between 650 and 800 nm under excitation at 350-450 nm (Fig. 1d), which is suitable for in vivo imaging using near-infrared technology.
Characterization of PMSNs-siCCR2-PEI. (a), Flowchart illustrating the preparation of PMSNs-siCCR2-PEI; A, naked PMSNs and free siCCR2 molecules; B, PMSNs loaded with siCCR2 in the mesopores; C, aggregated PEI-coated PMSNs-siCCR2; D, disaggregated PMSNs-siCCR2-PEI; A1∼D1 are scanning electron microscopic images corresponding to A∼D. (b), The siCCR2 adsorption capacity of PMSNs with increasing siRNA concentrations in solution. (c), Hydrodynamic diameter distribution and zeta potential of the final PMSNs-siCCR2-PEI used in the experiments. (d), The absorption and size-dependent emission spectra of PMSN-siCCR2-PEI compound particles at room temperature.
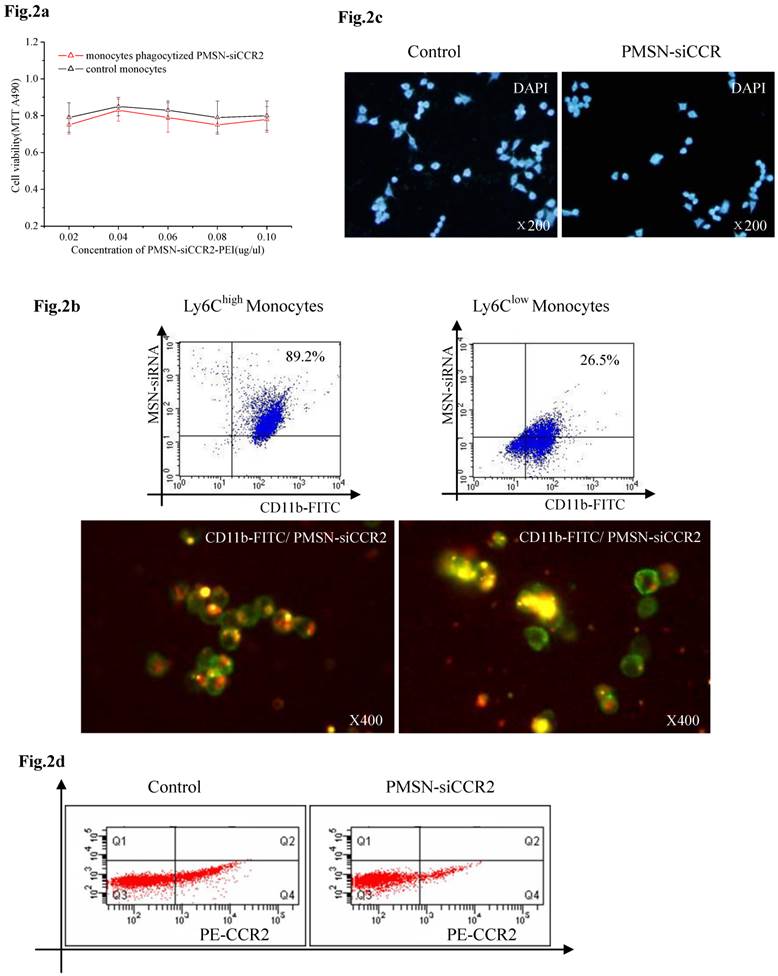
Biocompatibility of PMSNs-siCCR2-PEI and monocytes
We first tested the biocompatibility of PMSNs-siCCR2-PEI in vitro. CD11b-positive monocytes obtained from spleens and cultured in vitro were incubated with increasing concentrations of the particles for 48 h, and the cell viability was quantified using an MTT assay. The results showed that the delivery formulation is relatively non-toxic (p=NS) to CD11b-positive monocytes (Fig. 2a). We next analyzed the toxicity of the particles to mice in vivo. Consistent with the results obtained in vitro, the hepatic and renal function of the AMI mice treated with or without PMSNs-siCCR2-PEI were not significantly different at 24 hours (Supplementary Table 1). The phagocytic capacity of Ly6Chigh/low monocytes for the particle was subsequently determined by fluorescence microscopy and flow cytometry. As shown in Fig. 2b, Ly6Chigh monocytes exhibited greater phagocytosis of the particles compared with Ly6Clow monocytes (89.2%±17.5% vs. 26.5%±6.4%, p<0.001). After the cells phagocytized the PMSNs-siCCR2-PEI, a transwell migration assay was introduced to test the migratory response of the cells to CCL2 (10 ng/ml). We then observed a significant decrease in the migratory response of the monocytes compared with the control, as shown in Fig. 2c (13±4/mm2 vs. 35±11/mm2, p<0.001). Finally, the expression of the chemokine receptor CCR2 on the monocytes was examined. Consistent with the results of the migratory assays, a significant decrease in CCR2 expression was observed in the PMSNs-siCCR2 group (Fig. 2d) compared with the control (19.6%±8.4% vs. 37.1%±13.5%, p<0.001).
Biocompatibility of PMSNs-siCCR2-PEI and monocytes. (a), Determination of the cell viability of monocytes by the MTT assay after incubation with PMSNs-siCCR2-PEI at a concentration of 0.02-0.1 µg/ul; no significant difference was shown at any of the concentrations tested (p=NS). (b), Comparison of the phagocytic capacity of Ly6Chigh/low monocytes for PMSNs-siCCR2-PEI, determined by immunofluorescence staining and flow cytometry. (c), Migration assay of cultured Ly-6Chigh monocytes using CCL2 (10 ng/ml) as a chemoattractant. Cells were harvested after incubation with PMSNs-siCCR2-PEI for 48 h. (d), Analysis of CCR2 expression on Ly-6Chigh monocytes by flow cytometry (n=12).
PMSNs-siCCR2-PEI distribution
The specific siRNA sequence used in this study (5′-uGcuAAAcGucucuGcAAAdTsdT-3′, 5′- UUUGcAGAGACGUUuAGcAdTsdT-3′) to target CCR2 (siCCR2/fluorescent dye-labeled siCCR2, synthesized by Gene Pharma, China) was previously reported by Matthias Nahrendorf et al [24]. After the siRNA was scaled up and encapsulated in the particles, the PMSNs-siCCR2-PEI were intravenously injected into AMI mice. In vivo data were acquired serially for up to 24 hours using near-infrared technology. Ten minutes after injection, a strong fluorescent signal was observed in the cardiac region and the circulation (0.0144±0.00214 scaled counts/s), reflecting the presence of PMSNs-siRNA-PEI in the blood. These nanoparticles accumulate and degrade quickly in the circulation, with a short half-life of 37 minutes (Fig. 3a).
Dynamic fluorescence imaging of the distribution and metabolism of PMSNs-siCCR2-PEI in vivo determined using a near-infrared imaging system (n=12). (a), In vivo images of PMSNs-siCCR2-PEI. The mice were imaged at multiple time points after intravenous injection of the compounds (25 mg/kg). The white and red arrowheads indicate the liver and spleen, respectively. Imaging the dynamic biodistribution of siCCR2 shows major uptake in the spleen and liver. (b), Fluorescence images showing the in vivo biodistribution of PMSNs-siCCR2 in the heart, liver, spleen, kidney and lung over time; the compounds are rapidly cleared from the blood in 12 hours and were excreted via the hepatobiliary and renal routes during this time. (c), Confocal laser scanning microscopy of the spleen red pulp shows colocalization of PMSNs-siCCR2 (red) and CD11b-expressing monocytes (green). (d), Immunofluorescence microscopy of spleen sections reveals detached siCCR2 and PMSNs with green fluorescence and red fluorescence, respectively.
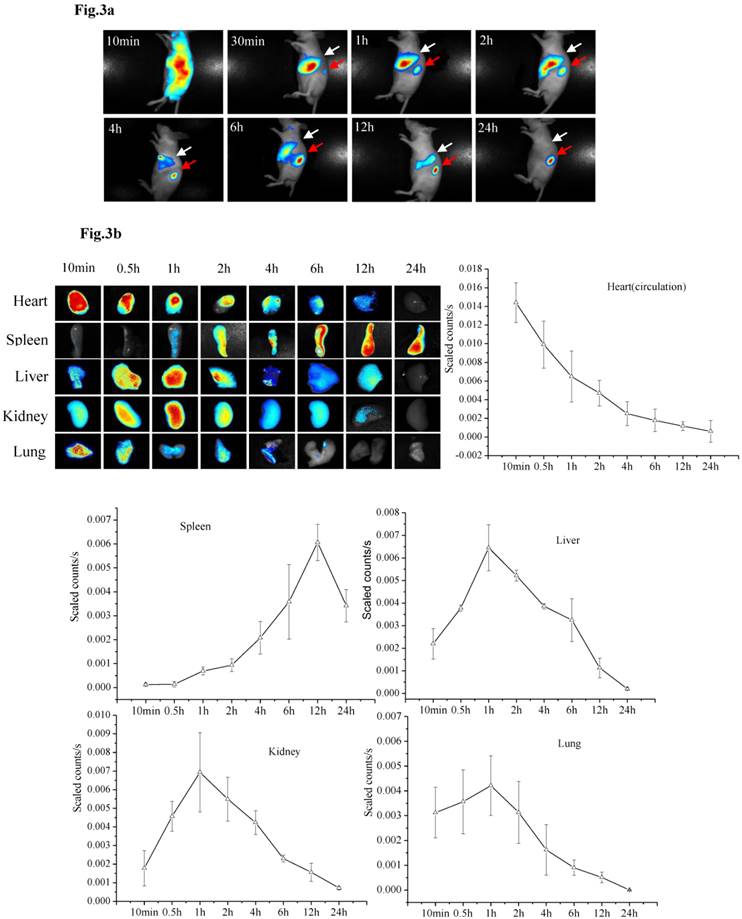
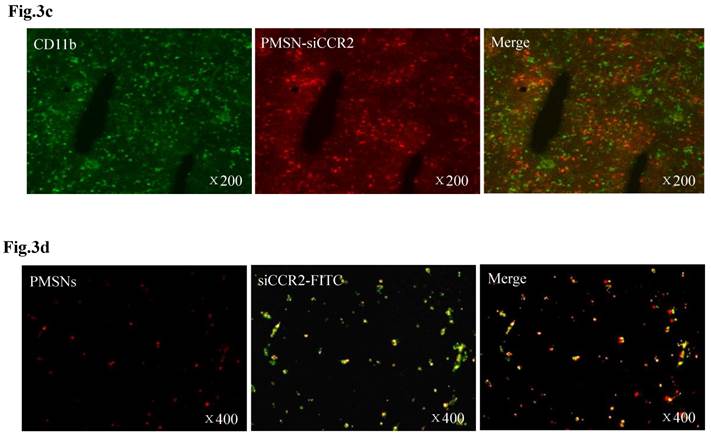
Interestingly, the biodistribution pattern of the PMSNs-siCCR2-PEI was different in the organs. Compared with the liver (0.00113±0.000437 scaled counts/s) and kidney (0.00156±0.00049 scaled counts/s), the spleen contained many more particles, with a peak fluorescence of 0.00606±0.000758 scaled counts/s at 12 hours (p<0.001) (Fig. 3b). An extremely weak fluorescent signal was observed in the lungs (0.000509±0.000217 scaled counts/s) at 12 hours. Dynamic in vivo imaging showed that the particles were removed via hepatobiliary and renal clearance. The distribution of PMSNs-siCCR2-PEI in the spleen was further detected by confocal laser scanning microscopy. We found that most of the particles resided in CD11b-monocytes and accumulated in the subcapsular red pulp (Fig. 3c). Importantly, independent green or red dots in the cytoplasm, labeling the naked PMSNs (red) and free siCCR2 (FAM-labeled, green), respectively, were observed, which implies the successful release of siCCR2 into the cell cytoplasm from the delivery particles (Fig. 3d).
Silencing of CCR2 ameliorates inflammation after AMI
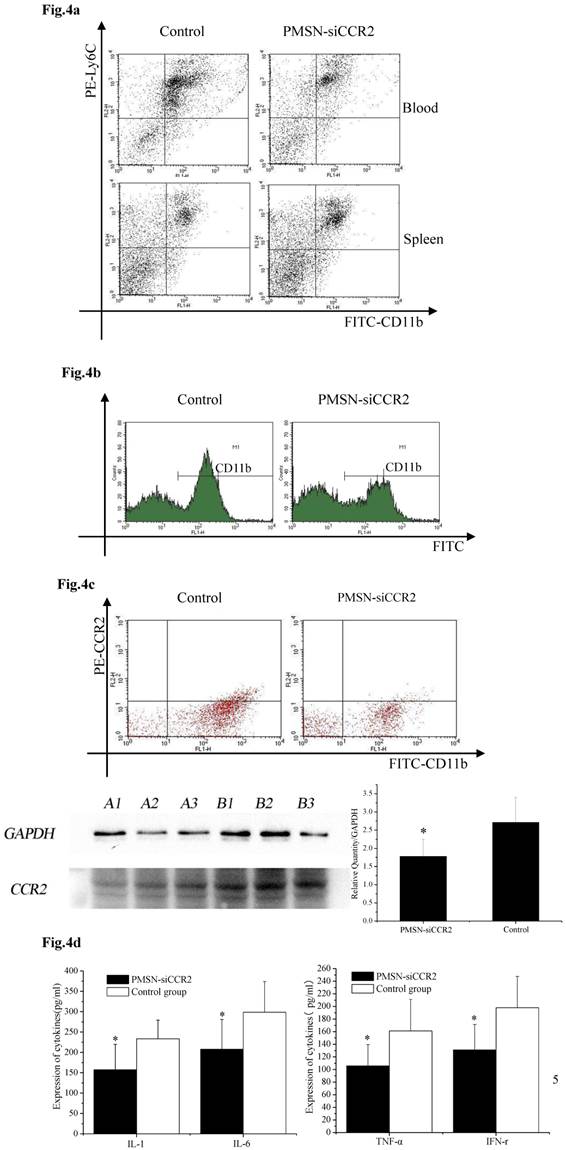
Ly6Chigh monocytes are potent inflammatory mediators and are believed to be the dominant source of inflammation after AMI; therefore, we tested the deployment of Ly6Chigh monocytes after AMI and PMSN-siCCR2 administration. Flow cytometric analysis showed a significant reduction in the number of inflammatory Ly6Chigh monocytes (43.62%±11.32% vs. 67.95%±17.04%, p<0.001) in the circulation one day after AMI in mice treated with PMSNs-siCCR2-PEI compared with the control group. The number of cells present in the spleen also changed significantly between the groups (29.78%±4.38% vs. 20.06%± 4.25%, p<0.001) (Fig. 4a). CD11b-positive monocytes in the infarcts were further examined at the same time, and the results revealed that PMSNs-siCCR2-PEI could significantly reduce the levels of the cells compared with the groups not given siCCR2 (49.3%±17.34% vs. 61.32%± 22.43%, p<0.001) (Fig. 4b). These findings were further confirmed by the decreased levels of CCR2 protein expression on spleen monocytes, as examined by flow cytometry (8.04%±2.07% vs. 20.02%±4.55%, p<0.001) and Western blot (1.78±0.47 vs. 2.71±0.69, p<0.001) (Fig. 4c), and the decreased expression of inflammatory cytokines (pg/ml), including IL-1 (157.49±62.36 vs. 233.37±45.78, p<0.001), IL-6 (207.88±73.3 vs. 298.62±75.5, p<0.001), TNF-α (105.74±33.87 vs. 161.09±50.24, p<0.001) and IFN-γ (131.07±40.34 vs. 197.98±49.77, p<0.001), in the serum, as detected by ELISA kits (Fig. 4d).
The effect of PMSNs-siCCR2 on inflammatory infiltration post-AMI (n=12). (a), Flow cytometry reveals a different pattern of CD11b+Ly6Chigh in the circulation and the spleen of AMI mice one day after treatment with PMSNs-siCCR2. Significantly reduced counts of the cells were shown in the siCCR2 group. (b), Flow cytometry revealed reduced CD11b-positive cells in the infarct zones of AMI mice one day after treatment with PMSNs-siCCR2. The expression of CCR2 in the infarction was further determined by Western-blot (A1~3 in the PMSNs-siCCR2 group, B1~3 in the control). (c), FACS analysis of the CCR2 protein levels on the spleen CD11b+Ly6Chigh monocytes of the AMI mice confirmed the decreased levels of CCR2 in the PMSN-siCCR2 group. (d), Levels of the inflammatory cytokines IL-1, IL-6, TNF-α and IFN-γ in the serum of AMI mice after treatment with PMSNs-siCCR2 were determined using immunosorbent assays (ELISAs), *p<0.05 vs. the corresponding control group.
Silencing of CCR2 benefits MSC transplantation
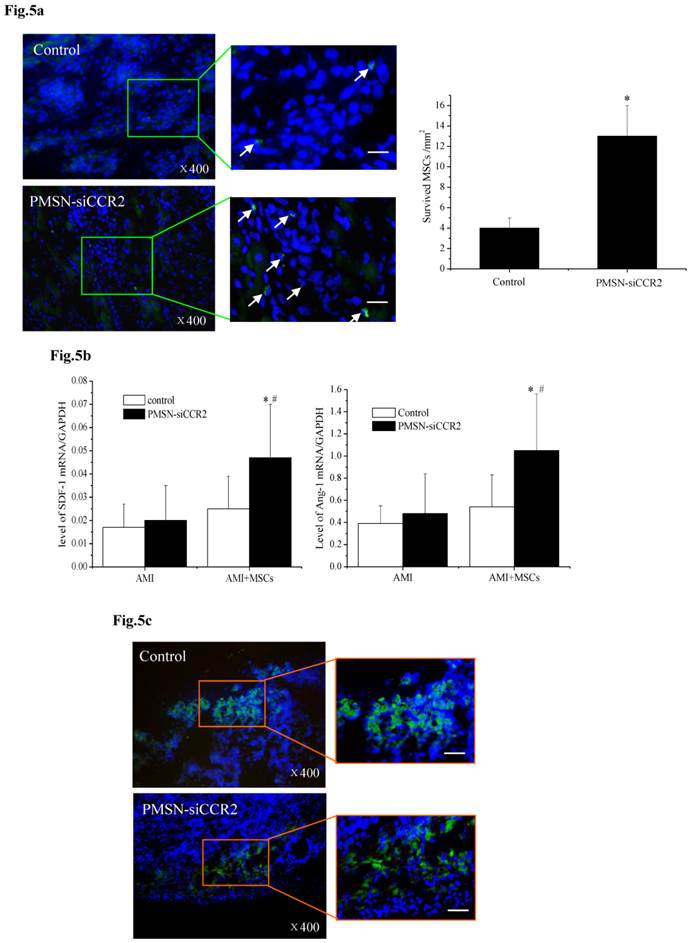
A current limitation of MSC therapy is the impact of the inflammatory microenvironment on cell behavior [25]. We next tested the therapeutic potential of MSCs in the context of PMSN-siCCR2 treatment. After infarction, the hearts were harvested and embedded in paraffin, and the sections were incubated with azide-conjugated Alexa Fluor 488. We then observed a significant difference in the number of EdU-positive cells (Fig. 5a) in the infarction (13±3/mm2 vs. 4±1/mm2, p<0.001) between the two groups three days after MSC transplantation. In addition, increased levels of SDF-1 (0.047±0.023 vs. 0.025±0.014, p<0.001) and Ang-1 (1.05±0.51 vs. 0.54±0.29, p<0.001) mRNA were identified in the infarct zone of the siCCR2 group (Fig. 5b).
PMSNs-siCCR2-PEI enhanced the efficiency of MSC transplantation and attenuated myocardial remodeling (n=12). (a), The surviving MSCs in the infarct zone following cell therapy are shown by representative immunofluorescent staining for EdU (5-Ethynyl-2'-deoxyuridine, green) and visualization of the nuclei (DAPI, blue) within the infarct zone 3 days after cell transplantation. Scale bar=50 μm. (b), SDF-1 and Ang-1 mRNA levels in the infarct zone after MSC transplantation. The data represent the mean±SD, *p<0.05 vs. the corresponding group at 1 day post-AMI; #p<0.05 vs. the control group at 3 days after cell transplantation. (c), Representative image of immunofluorescent staining for TUNEL (Alexa Fluor 488, green) and DAPI in the infarct border zone 3 days after AMI; the number of TUNEL-positive cardiomyocytes was calculated. Scale bar=50 μm. (d), The vascular density was determined by immunofluorescent staining for wheat germ agglutinin (WGA, red), endothelial cells (staining of CD31, green) and DAPI (blue) in the infarct zone at 21 days after MSC transplantation. (e), Confocal image of representative immunofluorescent staining for cardiac myosin heavy chain (eFluor® 660, red) and DAPI (blue) staining of the nuclei within the infarct zone at 21 days after MSC transplantation. (f), Infarct size at baseline and at 21 days after MSC transplantation, Arrowheads (blue) indicate the diastolic thickness of the LV posterior walls (0.84±0.11 mm vs. 0.61±0.08 mm, p<0.001) over time in the two groups. *p<0.05 vs. the corresponding 1-day-post-AMI group; #p<0.05 vs. the control group at 21 days after cell transplantation.
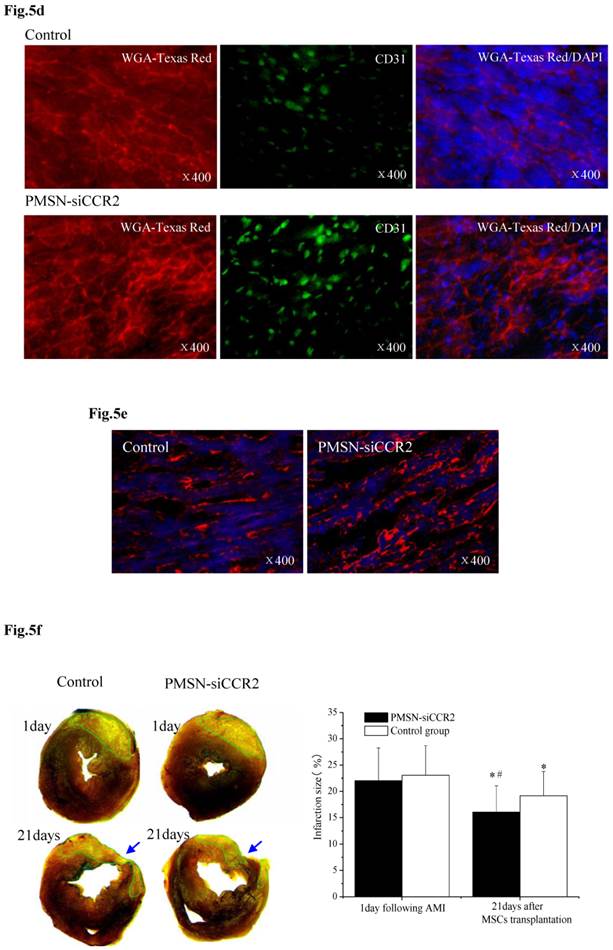
These results further suggested that the presence of PMSNs-siCCR2 decreased the inflammation in AMI mice and restored the MSCs in the heart. Consistent with the restored MSCs, we also observed a significant reduction in the number of TUNEL+ cardiac myocytes (17.44%±6.26% vs. 39.49%±13.28%, p<0.001) within the infarct zone compared with the control at three days post-AMI (Fig. 5c).
Three weeks after MSC infusion, the vascular density in the border zone of the infarction was examined using WGA and CD31 (Fig. 5d). A significant increase in the number of capillaries and small arterioles was observed in the siCCR2 group (235.5±39.6/mm2 vs. 147.4±20.3/mm2, p<0.001). Consistent with the increased vascular density, a significant increase in the myosin-positive cardiac area within the infarct zone in the siCCR2 group (21.7%±8.4% vs. 13.2%±4.4%, p<0.001) was shown (Fig. 5e). Finally, the infarction size (%) (16.04±5.01 vs. 19.17±4.6, p<0.001) and LV remodeling (thickness of the LV posterior wall) (0.84±0.11 mm vs. 0.61±0.08 mm, p<0.001) were measured three weeks after MSC infusion; both were greatly ameliorated in the siCCR2 group compared with the control (Fig. 5f, Supplementary Table 1). Taken together, these data demonstrated that silencing CCR2 in Ly6Chigh monocytes played a critical role in MSC transplantation post-AMI.
Discussion
Numerous studies have attempted to utilize MSCs or other stem cells as a cell therapy to treat AMI; however, only modest improvements in patient outcomes have been reported in meta-analyses, and none of the therapies have become a standard of care [26-27]. In fact, increasing evidence suggests that acute inflammation caused by AMI impairs the function of the transplanted marrow-derived cells [28-29]. Thus, a method to reduce the rate of apoptosis of the transplanted cells is needed to allow the use of these cells as a myocardial reparative therapy. Thus, additional work is required to improve our understanding of the different patterns of inflammation after AMI that promote the decreased survival of transplanted cells.
The involvement of inflammation in the pathophysiology of AMI and subsequent MSC transplantation has been described in recent years. Ly6Chigh monocytes have been shown to be key players in this process [30], as they aggravate inflammation and expand in AMI. MSC transplantation has resulted in improved wound healing, but it is limited by sub-optimal cell survival and engraftment due to the harsh microenvironment of the cells. Therefore, controlling the deployment of Ly6Chigh monocytes from the spleen is essential during AMI and stem cell therapy. Researchers are now working to apply methods that modulate inflammation during AMI. The inflammatory effects of Ly6Chigh monocytes on MSC transplantation have not been previously investigated. Here, using an siCCR2 delivery formulation, we investigated the effects of silencing CCR2 in Ly6Chigh monocytes on MSC transplantation after AMI. The present study demonstrated that (1) the PMSNs used as a delivery vehicle can be loaded with siCCR2 with high efficiency, and the siRNA delivery system was phagocytized by Ly6Chigh monocytes in the spleen and showed little cytotoxicity to the cells; (2) AMI induced a local or systemic inflammatory response that could be inhibited by the formulation of PMSNs-siCCR2-PEI by silencing the receptor CCR2 on Ly6Chigh monocytes in the spleen; (3) Marked improvements in the outcome of MSC transplantation were observed after administration of PMSNs-siCCR2-PEI, as evidenced by the increased survival of EdU-positive cells, the reduced apoptosis of cardiomyocytes at three days, the increased regeneration of capillaries and the amelioration of LV remodeling at three weeks post-AMI.
Previous reports have shown that spleen Ly6Chigh monocytes infiltrate the ischemic heart and express proteases in response to AMI [31-33], thereby promoting adverse myocardial remodeling and cardiac rupture. Inflammatory CCL2/CCR2 signaling is known to be a key regulator of the recruitment and infiltration of Ly6Chigh monocytes in the ischemic heart. This emphasizes the need to control pro-inflammatory monocytes by targeting CCL2/CCR2. RNA interference through siCCR2 was introduced in this study, and the effects of siCCR2 treatment on splenic reservoir monocytes were particularly intriguing in AMI mice for at least three reasons. First, CCR2 expression in Ly6Chigh monocytes was shown to be decreased at the protein level, which implies that siCCR2 provided cellular specificity for the therapeutic target in this study. Second, consistent with a previous report in CCL2/CCR-2-double-deficient mice [31], acute inflammation caused by Ly6Chigh monocytes derived from the spleen was decreased greatly, as evidenced by reduced numbers of CD11b-positive monocytes infiltrating the infarct and decreased inflammatory cytokines present in the serum. Third, the therapeutic effects of MSC transplantation were extremely encouraging, as shown by increases in the SDF-1 and Ang-1 mRNA levels and the cardiac myosin-positive area. These results further confirmed previous reports that MSC engraftment leads to a significant increase in cardiac myocyte preservation in the context of gene or protein modification[34-35]. Taken together, these data directly demonstrated the efficiency of MSC transplantation when CCR2 was silenced in Ly6Chigh monocytes by PMSNs-siCCR2-PEI.
Notably, the therapeutic effects of siCCR2 in this study were based on delivery by PMSNs. The current challenge for the application of RNA interference in vivo is the development of an efficient and nontoxic delivery vector. For efficient loading of siCCR2 in the mesoporous PMSNs, we used strongly dehydrated conditions and increased siRNA equilibrium concentrations, as described in other studies [36-37], and we then determined the siRNA adsorption capacity using a BioSpectrometer. Near-infrared imaging technology and confocal microscopy imaging revealed that intravenously injected PMSNs distributed siCCR2 to monocytes in the spleen, resulting in efficient and specific CCR2 knockdown. In addition, after the PMSNs-siCCR2-PEI were injected intravenously, the particles accumulated mainly in monocyte-related organs (such as the spleen); furthermore, they were removed from these organs in vivo via renal and hepatobiliary clearance. In summary, for use in vivo, PMSNs-siCCR2-PEI exhibit a high siRNA loading efficiency and provide an attractive chemical alternative to traditional nanoparticles, particularly because they have been shown to be nontoxic in biological environments by MTT assays.
Lipid nanoparticles, carbon nanotubes and gold nanoparticles have all been used previously as siRNA vectors by researchers [38-39]; in contrast to these traditional nanoparticles, PMSN-based materials provide more opportunities for on-demand biological therapy and pave the way for the development of simple treatments for challenging diseases [13,40]. PMSNs offer a high surface area and pore volume as well as the capacity for surface charge control. The versatility, non-toxicity, biocompatibility, biodegradability, and homogenous distribution of siCCR2 into the porous space and free dispersal throughout the particle are also beneficial features. In addition, the acceptable stability of PMSNs ensures resistance to mechanical stress and harsh pH conditions in vivo. Furthermore, the intrinsic brightly photoluminescent properties of PMSNs enable the non-invasive monitoring of their biodistribution and degradation in vivo due to their near-infrared emission (650-800 nm) when excited at 350-450 nm.
The PMSNs used in this study ranged in size from 100 to 200 nm, which may not only prevent their rapid clearance from circulation but may also allow for an increased siRNA loading capacity. In addition, the intrinsic luminescence of PMSNs at 100-200 nm enabled the non-invasive monitoring of their biodistribution and degradation in mice. Here, we present the first example of biodegradable photoluminescent mesoporous silicon nanoparticles for biological applications after AMI. This system exhibited considerable potential for MSC transplantation. It should be noted that the PEI coating on the external surface of the siRNA-loaded PMSNs is critical; this coating prevents the detachment of siCCR2 from the particles during biological applications. In addition, PEI has a high buffering capacity in the endosomal pH range and can efficiently deliver nucleic acids by overcoming lysosomal sequestration via the proton sponge effect [41-42]. However, recently, much more efficient polycations and fusogenic peptides have been reported for the coating of nanocarriers [43-44]; thus, the PMSNs used in the study as delivery vehicles can still be optimized for greater efficiency in the future.
In conclusion, we showed that photoluminescent mesoporous silicon nanoparticles represented a suitable delivery vehicle for siCCR2, which can then be applied to target inflammatory monocytes during AMI and MSC transplantation. Further, we confirmed the therapeutic effect of the particles in the context of MSC transplantation in AMI mice at the gene, protein and functional levels. We believe these findings using PMSNs-siCCR2-PEI provide a promising pathway for clinical translation in AMI patients and subsequent MSC transplantation. In view of the findings in this paper, we hope that this strategy will contribute to the development of new treatment methods or drugs for AMI patients.
Supplementary Material
Supplementary Figures 1-4, Supplementary Tables 1-2.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81300160, 81470400, 51102043) and Qing Lan Project of Jiangsu Province and The Scientific Research Foundation of Graduate School of Southeast University.
Competing Interests
The authors hereby declare that no competing interests exist.
References
1. Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010;121:2437-45
2. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P. et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612-6
3. Robbins CS, Chudnovskiy A, Rauch PJ, Figueiredo JL, Iwamoto Y, Gorbatov R. et al. Extramedullary hematopoiesis generates Ly-6C (high) monocytes that infiltrate atherosclerotic lesions. Circulation. 2012;125:364-74
4. Ley K, Miller YI, Hedrick CC. Monocyte and macrophage dynamics during atherogenesis. Arterioscler Thromb Vasc Biol. 2011;31:1506-16
5. Gui T, Shimokado A, Sun Y, Akasaka T, Muragaki Y. Diverse roles of macrophages in atherosclerosis:from inflammatory biology to biomarker discovery. Mediators Inflamm. 2012:693083 1-14
6. van der Laan AM, Ter Horst EN, Delewi R, Begieneman MP, Krijnen PA, Hirsch A. et al. Monocyte subset accumulation in the human heart following acute myocardial infarction and the role of thespleen as monocyte reservoir. Eur Heart J. 2014;35:376-85
7. Mestas J, Ley K. Monocyte-Endothelial Cell Interactions in the Development of Atherosclerosis. Trends Cardiovasc Med. 2008;18:228-32
8. Ruparelia N, Digby JE, Jefferson A, Medway DJ, Neubauer S, Lygate CA. et al. Myocardial infarction causes inflammation and leukocyte recruitment at remote sites in the myocardium and in the renal glomerulus. Inflamm Res. 2013;62:515-25
9. Jerath MR, Liu P, Struthers M, Demartino JA, Peng R, Peterson LB. et al. Dual targeting of CCR2 and CX3CR1 in an arterial injury model of vascular inflammation. Thromb J. 2010;8:14
10. Kaikita K, Hayasaki T, Okuma T Kuziel WA, Ogawa H, Takeya M. et al. Targeted Deletion of CC Chemokine Receptor 2 Attenuates Left Ventricular Remodeling after Experimental Myocardial Infarction. Am J Pathol. 2004;165:439-47
11. Osterholzer JJ, Olszewski MA, Murdock BJ, Chen GH, Erb-Downward JR, Subbotina N. et al. Implicating exudate macrophages andLy-6C(high) monocytes in CCR2-dependent lung fibrosis following gene-targeted alveolar injury. J Immunol. 2013;190:3447-57
12. Tang F, Li L, Chen D. Mesoporous Silica Nanoparticles: Synthesis, Biocompatibility and Drug Delivery. Adv Mater. 2012;24:1504-34
13. Xie Z, Henderson EJ, Dag Ö, Wang W, Lofgreen JE, Kübel C. et al. Periodic Mesoporous Hydridosilica-Synthesis of an “Impossible” Material and Its Thermal Transformation into Brightly Photoluminescent Periodic Mesoporous Nanocrystal Silicon-Silica Composite. J Am Chem Soc. 2011;133:5094-102
14. Majmudar MD, Keliher EJ, Heidt T, Leuschner F, Truelove J, Sena BF. et al. Monocyte-directed RNAi targeting CCR2 improves infarct healing in atherosclerosis-prone mice. Circulation. 2013;127:2038-46
15. Gouda N, Miyata K, Christie RJ, Suma T, Kishimura A, Fukushima S. et al. Silica nanogelling of environment-responsive PEGylated polyplexes for enhanced stability and intracellular delivery of siRNA. Biomaterials. 2013;34:562-70
16. Li X, Chen Y, Wang M, Ma Y, Xia W, Gu H. A mesoporous silica nanoparticle-PEI -Fusogenic peptide system for siRNA delivery in cancer therapy. Biomaterials. 2013;34:1391-401
17. Li TS, Yawata T, Honke K. Efficient siRNA delivery and tumor accumulation mediated by ionically cross-linked folic acid-poly (ethylene glycol)-chitosan oligosaccharide lactate nanoparticles: for the potential targeted ovarian cancer gene therapy. Eur J Pharm Sci. 2014;52:48-61
18. Cochain C, Auvynet C, Poupel L, Vilar J, Dumeau E, Richart A. et al. The Chemokine Decoy Receptor D6 Prevents Excessive Inflammation and Adverse Ventricular Remodeling After Myocardial Infarction. Arterioscler Thromb Vasc Biol. 2012;32:2206-13
19. Pedersen TO, Blois AL, Xue Y, Xing Z, Sun Y, Finne-Wistrand A. et al. Mesenchymal stem cells induce endothelial cell quiescence and promote capillary formation. Stem Cell Res Ther. 2014;5:23
20. Ning H, Albersen M, Lin G, Lue TF, Lin CS. Effects of EdU Labeling on Mesenchymal Stem Cells. Cytotherapy. 2013;15(1):57-63
21. Lin G, Huang YC, Shindel AW, Banie L, Wang G, Lue TF. et al. Labeling and Tracking of Mesenchymal Stem Cells with EdU. Cytotherapy. 2009;11:864-73
22. Sheng Z, Yao Y, Li Y. et al. Bradykinin Preconditioning Improves Therapeutic Potential of Human Endothelial Progenitor Cells in Infarcted Myocardium. PLoS One. 2013;8:e81505
23. Yan F, Yao Y, Chen L, Li Y, Sheng Z, Ma G. Hypoxic Preconditioning Improves Survival of Cardiac Progenitor Cells: Role of Stromal Cell Derived Factor-1a-CXCR4 Axis. PLoS One. 2012;7:e37948
24. Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G. et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol. 2011;29:1005-10
25. Ennis WJ, Sui A, Bartholomew A. Stem Cells and Healing: Impact on Inflammation. Adv Wound Care (New Rochelle). 2013;2:369-378
26. Ale A, Siebenhaar F, Kosanke K, Aichler M, Radrich K, Heydrich S. et al. Cardioprotective C-kit+ Bone Marrow Cells Attenuate Apoptosis after Acute Myocardial Infarction in Mice - In-vivo Assessment with Fluorescence Molecular Imaging. Theranostics. 2013;3:903-913
27. Ieda M. Heart regeneration using reprogramming technology. Proc Jpn Acad Ser B Phys Biol Sci. 2013;89:118-28
28. Raicevic G, Najar M, Najimi M, El Taghdouini A, van Grunsven LA, Sokal E. et al. Influence of inflammation on the immunological profile of adult-derived human liver mesenchymal stromal cells and stellate cells. Cytotherapy. 2015;17:174-85
29. Sesia SB, Duhr R, da Cunha CM, Todorov A, Schaeren S, Padovan E. et al. Anti-inflammatory/tissue repair macrophages enhance the cartilage-forming capacity of human bone marrow-derived mesenchymal stromal cells. J Cell Physiol. 2015;230:1258-69
30. Charo IF. Blinding the monocytes to protect the heart. Circulation. 2013;127:2006-8
31. Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS. et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487:325-9
32. Jacquelin S, Licata F, Dorgham K, Hermand P, Poupel L, Guyon E. et al. CX3CR1 reduces Ly6Chigh-monocyte motility within and release from the bone marrow after chemotherapy in mice. Blood. 2013;122:674-83
33. Schuette-Nuetgen K, Strecker JK, Minnerup J, Ringelstein EB, Schilling M. MCP-1/CCR-2-double-deficiency severely impairs the migration of hematogenous inflammatory cells following transient cerebral ischemia in mice. Exp Neurol. 2012;233:849-58
34. Cho J, Zhai P, Maejima Y, Sadoshima J. Myocardial injection with GSK-3β-overexpressing bone marrow-derived mesenchymal stem cells attenuates cardiac dysfunction after myocardial infarction. Circ Res. 2011;108:478-89
35. McGinley LM, McMahon J, Stocca A, Duffy A, Flynn A, O'Toole D. et al. Mesenchymal stem cell survival in the infarcted heart is enhanced by lent virus vector-mediated heat shock protein 27 expression. Hum Gene Ther. 2013;24:840-51
36. Chen AM, Taratula O, Wei D, Yen HI, Thomas T, Thomas TJ. et al. Labile catalytic packaging of DNA/siRNA: control of gold nanoparticles "out" of DNA/siRNA complexes. ACS Nano. 2010;4:3679-88
37. Li X, Xie QR, Zhang J, Xia W, Gu H. The packaging of siRNA within the mesoporous structure of silica nanoparticles. Biomaterials. 2011;32:9546-56
38. Park JH, Gu L, von Maltzahn G, Ruoslahti E, Bhatia SN, Sailor MJ. et al. Biodegradable luminescent porous silicon nanoparticles for in vivo applications. Nat Mater. 2009;8:331-6
39. Lee HU, Park SY, Park ES, Son B, Lee SC, Lee JW. et al. Photoluminescent carbon nanotags from harmful cyanobacteria for drug delivery and imaging in cancer cells. Sci Rep. 2014;4:4665
40. Prabhakar N, Näreoja T, von Haartman E, Karaman DŞ, Jiang H, Koho S. et al. Core-shell designs of photoluminescent nanodiamonds with porous silica coatings for bioimaging and drug delivery II: application. Nanoscale. 2013;5:3713-22
41. Richard I, Thibault M, De Crescenzo G, Buschmann MD, Lavertu M. Ionization behavior of chitosan and chitosan-DNA polyplexes indicate that chitosan has a similar capability to induce a proton-sponge effect as PEI. Biomacromolecules. 2013;14(6):1732-40
42. Yaqiong D, Jun Y, Hongmei L, Tianyou W, Suoqin T, Jinchao Z. et al. Site-Specific Drug-Releasing Polypeptide Nanocarriers Based on Dual-pH Response for Enhanced Therapeutic Efficacy against Drug-Resistant Tumors. Theranostics. 2015;5:890-904
43. Freeman EC, Weiland LM, Meng WS. Modeling the proton sponge hypothesis: examining proton sponge effectiveness for enhancing intracellular gene delivery through multiscale modeling. J Biomater Sci Polym Ed. 2013;24:398-416
44. Bienvenu C, Martínez Á, Jiménez Blanco JL, Di Giorgio C, Vierling P, Ortiz Mellet C. et al. Polycationic amphiphilic cyclodextrins as gene vectors: effect of the macrocyclic ring size on the DNA complexing and delivery properties. Org Biomol Chem. 2012;10:5570-81
Author contact
![]() Corresponding authors: Genshan Ma MD. PhD. 89#, Dingjiaqiao Road, Nanjing 210009, China. Email: magenshancom / magenshanseucom or Wenbin Lu MD. 87#, Dingjiaqiao Road, Nanjing 210009, China. Email: 230139545edu.cn / luwenbinseucom
Corresponding authors: Genshan Ma MD. PhD. 89#, Dingjiaqiao Road, Nanjing 210009, China. Email: magenshancom / magenshanseucom or Wenbin Lu MD. 87#, Dingjiaqiao Road, Nanjing 210009, China. Email: 230139545edu.cn / luwenbinseucom