Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2012; 2(9):880-888. doi:10.7150/thno.3899 This issue Cite
Research Paper
Involvement of Reactive Oxygen Species in Sonodynamically Induced Apoptosis Using a Novel Porphyrin Derivative
Nagahiko Yumita1 ![]() , Yumiko Iwase1, Koji Nishi1, Hajime Komatsu1, Kazuyoshi Takeda1, Kenji Onodera1, Toshio Fukai1, Toshihiko Ikeda1, Shin-ichiro Umemura2, Kazuho Okudaira3, Yasunori Momose3
, Yumiko Iwase1, Koji Nishi1, Hajime Komatsu1, Kazuyoshi Takeda1, Kenji Onodera1, Toshio Fukai1, Toshihiko Ikeda1, Shin-ichiro Umemura2, Kazuho Okudaira3, Yasunori Momose3
1. School of Pharmacy, Yokohama College of Pharmacy, 601, Matano-cho, Totsuka-ku, Yokohama, Kanagawa 245-0066, Japan;
2. Department of Electrical and Communication Engineering, Tohoku University, Aoba 6-6-05, Aramaki, Aoba-ku, Sendai 980-8579, Japan;
3. School of Pharmaceutical Sciences, Toho University, 2-2-1 Miyama, Funabashi, Chiba 274-8510, Japan.
Received 2011-12-1; Accepted 2012-2-26; Published 2012-9-29
Abstract

In this study, we investigated the induction of apoptosis by ultrasound in the presence of the novel porphyrin derivative DCPH-P-Na(I). HL-60 cells were exposed to ultrasound for up to 3 min in the presence and absence of DCPH-P-Na(I), and the induction of apoptosis was examined by analyzing cell morphology, DNA fragmentation, and caspase-3 activity. Reactive oxygen species were measured by means of ESR and spin trapping technique. Cells treated with 8 μM DCPH-P-Na(I) and ultrasound clearly showed membrane blebbing and cell shrinkage, whereas significant morphologic changes were not observed in cells exposed to either ultrasound or DCPH-P-Na(I) alone. Also, DNA ladder formation and caspase-3 activation were observed in cells treated with both ultrasound and DCPH-P-Na(I) but not in cells treated with ultrasound or DCPH-P-Na(I) alone. In addition, the combination of DCPH-P-Na(I) and the same acoustical arrangement of ultrasound substantially enhanced nitroxide generation by the cells. Sonodynamically induced apoptosis, caspase-3 activation, and nitroxide generation were significantly suppressed by histidine. These results indicate that the combination of ultrasound and DCPH-P-Na(I) induced apoptosis in HL-60 cells. The significant reduction in sonodynamically induced apoptosis, nitroxide generation, and caspase-3 activation by histidine suggests active species such as singlet oxygen are important in the sonodynamic induction of apoptosis. These experimental results support the possibility of sonodynamic treatment for cancer using the induction of apoptosis.
Keywords: Apoptosis, Sonodynamic therapy, Ultrasound, DCPH-P-Na(I), HL-60 cells, Reactive Oxygen, Caspase-3.
Introduction
Ultrasound has a tissue attenuation coefficient that allows it to penetrate intervening tissues and reach internal targets without losing the ability to focus energy into small volumes. This is a unique advantage over electromagnetic modalities such as laser light and microwaves for the noninvasive treatment of internal tumors. Although the use of ultrasound for tumor treatment has been fairly well investigated with respect to the thermal effects of ultrasound absorption [1,2], only a few groups have reported its nonthermal effects, such as potential sonochemical effects [3-7].
Recently, we found that photochemically active porphyrins such as hematoporphyrin, Porfimer Sodium (PF) and a gallium porphyrin complex, 7,12-bis(1-decyloxyethyl)-Ga(III)-3,8,13,17-tetramethyl-porphyrin 2,18-dipropionyl diaspartic acid (ATX-70) can induce significant cell damage when activated by ultrasound [8,9]. When implanted murine tumors are treated after the administration of such chemicals, tumor growth is significantly inhibited at an intensity where ultrasound alone shows only a slight inhibitory effect [10-12]. Therefore, photochemically active porphyrins may be useful for sensitizing tumors to ultrasound. We have proposed that this potential modality be called "sonodynamic therapy" [13,14].
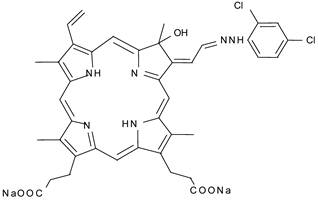
Although this feature of ultrasound is expected to result in an improvement of the tumoricidal effects of SDT, the skin sensitivity caused by photosensitizers still remains to be solved. Recent study reported that 13,17-bis(1-carboxyethyl)-8-[2-(2,4-dichlorophenyl-hydrazono)ethylidene]-3-ethenyl-7-hydroxy-2,7,12,18-tetramethylchlorin, disodium salt(DCPH-P-Na(I)) whose chemical structure is shown in Fig. 1, is much less phototoxic than ATX-70. This weak phototoxicity can be an advantage for avoiding the adverse effect in sonodynamic therapy [15]. In the previous paper, we reported DCPH-P-NA(I) is ultrasonically activated and shows antitumor effect on isolated tumor cells [16].
Chemical structure of DCPH-P-Na(I)
Apoptosis can be initiated by a wide variety of intracellular and extracellular stimuli and is a mechanism for the removal of unnecessary, aged, or damaged cells. Cells undergoing apoptosis exhibit characteristic morphological changes, including initial shrinkage, followed by widespread membrane blebbing, chromatin condensation, and DNA fragmentation. The cell further disassembles into membrane-enclosed vesicles called apoptotic bodies that are rapidly taken up and digested by neighboring cells and phagocytes [17-19].
Recently, ultrasonic exposure has been shown to trigger apoptosis in both normal and malignant cells. Ultrasound-induced apoptotic cell death has been confirmed in K562, HL-60, and U937 leukemia cells [20-24]. In addition, contrast agents are reported to enhance ultrasonically induced apoptosis [25], but only a few report on the effects of sonochemically active agents on ultrasonically induced apoptosis have been published [26]. Therefore, in this study, we examined whether the sonochemically active porphyrin DCPH-P-NA(I) can enhance ultrasonically induced apoptosis in HL60 cells.
Materials and Methods
Chemicals
13,17-bis(1-carboxyethyl)-8-[2-(2,4-dichlorophenyl-hydrazono)ethylidene]-3-ethenyl-7-hydroxy-2,7,12,18-tetramethylchlorin, disodium salt (DCPH-P-Na(I)) was a generous gift from Toyo Hakka Kogyo (Okayama, Japan). Trypan blue, agarose, RNase A, and proteinase K were purchased from Wako (Tokyo, Japan). Histidine, superoxide dismutase (SOD), mannitol, ethidium bromide, 2,2,6,6-Tetramethyl-4-piperidone (TMPone), 2,2,6,6-tetramethyl-4-piperidone-N-oxyl, and 2,2,6,6-tetramethyl-4-piperidol-N-oxyl were purchased from Sigma (St Louis, MO, USA). All the other reagents were of analytical grade.
Cell Culture
Human promyelocytic leukemia HL-60 cells were obtained from the Riken Gene Bank (Tokyo, Japan). Cells were maintained in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (GIBCO BRL, Tokyo, Japan), 100 U/ml penicillin G, 100 μg/ml streptomycin, and 2 mM glutamine (Sigma-Aldrich) in an atmosphere of 5% CO2 in humidified air at 37°C.
Ultrasound Apparatus
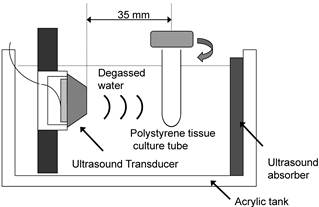
The apparatus for ultrasonic exposure is shown schematically in Figure 2. The ultrasound transducer uses a piezoelectric ceramic disk 24 mm in diameter and was driven at its resonance frequency (1.93 MHz). Low frequency ultrasound waves have greater depth of penetration but are less focused. On the other hands, ultrasound at a frequency of more than 1 MHz is absorbed primarily by tissues but it can be focused into small volume. In consideration of the absorption and the penetration, we employed the frequency of 1.93 MHz for exposure in this experiment [27]. Before exposure, the cells were harvested and washed twice in phosphate-buffered saline (PBS, pH 7.4). HL-60 cells were washed, resuspended at a concentration of 1x106 cells/ml in 2.5 ml of RPMI 1640 (serum free), and transferred into a cylindrical 16×125 mm polystyrene tissue culture tube (Corning, Corning, NY, USA) for exposure to ultrasound. A polystyrene tissue culture tube was suspended 35 mm away from the surface of the plane-wave transducer in degassed water, and insonated at 37°C. Immediately before exposure, DCPH-P-Na(I) was added to the cell suspension. During exposure, the tube was rotated at 60 rpm by a synchronous motor to improve mixing and to provide a uniform exposure [28]. HL-60 cells were exposed to ultrasound for up to 3 min in the presence and absence of PF. The samples treated with PF alone were kept in the same position for the same period as the ultrasonic exposure. Ultrasonic output from the transducer was evaluated in degassed water by placing the axis of the transducer horizontally. The output acoustic power was calibrated by measuring the radiation force on a 2-mm-thick hollow aluminum plate with an area of 20 x 28 mm, suspended at an angle of 45° to the axis. Its horizontal projection was therefore 20 mm x 20 mm. The ultrasonic intensity was calculated by dividing the measured acoustic power by the projected area. The temperature rise in 2.5 ml RPMI 1640 in the container during a 5-min exposure at the highest ultrasonic intensity used in the experiments was less than 1°C. After the treatment procedure, the medium was replaced with fresh RPMI 1640 with 10% FBS and the cells was incubated in an atmosphere humidified with 5% CO2 at 37 °C for 6 h before apoptosis induction was evaluated
Diagram of the ultrasonic exposure apparatus.
Evaluation of Apoptosis and Cell Damage
Apoptosis is characterized by morphological changes such as membrane blebbing, cell shrinkage, and disassembly of the cell into apoptotic bodies. During or after the ultrasound treatment, HL-60 cells were examined using a phase contrast inverted microscope (Olympus, Tokyo, Japan) at 400× magnification. The viability of the treated cells was determined by staining of the cells with trypan blue immediately after the treatment. The fraction of apoptotic cells was determined by counting the number of unstained cells showing morphological changes on a hemocytometer glass plate. The integrity was checked right before each series of treatment, and cell suspensions with integrity above 99 % were used. This number of intact cells before treatment was regarded as the standard for the integrity determination after each treatment.
Electrophoretic Analysis of DNA Fragmentation
After experimental treatments, the cells were harvested, washed in PBS (pH 7.4), and lysed in 100 μl of 0.1 M phosphate-citrate buffer. Following lysis, the samples were centrifuged at 16000 × g for 5 min. Then, supernatants were treated with 200 μg/ml DNase-free RNase at 37°C for 1 h, followed by 1 μg/ml proteinase K at 50°C for 1 h. The samples were separated by electrophoresis on 1.5% (w/v) agarose gels containing 1 μg/ml ethidium bromide. Separated DNA fragments (DNA ladders) were visualized using a UV transilluminator. The size of the DNA fragments was determined by comparison with DNA molecular weight markers (100 bp DNA ladder; Invitrogen, Carlsbad, CA, USA).
Measurement of Caspase-3 Activity
Caspase-3 activity was assayed by a using the specific fluorogenic substrate Ac-DEVD-AFC (MBL, Tokyo, Japan). Treated cells were washed with 50 mM PBS (pH 7.4) and then resuspended in buffer containing 50 mM Tris-HCL (pH 7.4), 1 mM EDTA, and 10 mM EGTA. The cell lysates were then centrifuged at 800× g for 5 min, and the supernatant was incubated with 50 μM of peptide substrate at 37°C for 2 h. The formation of 7-amino-4-trifrluoromethylcoumarin was measured using a fluorescence spectrophotometer (F-3000, Hitachi, Japan) with excitation at 400 nm and emission at 505 nm. The enzyme activity measured just prior to each experiment was used as the control. The caspase activity was expressed as the ratio of released 7-amino-4-trifrluoromethylcoumarin for the experimental condition compared to the untreated control.
Electron Spin Resonance (ESR) Measurements
Ultrasonically induced nitroxide production in the presence and absence of DCPH-P-Na(I) was measured by ESR spectroscopy in an air-saturated aqueous solution of 50 mM TMPone in the presence and absence of oxygen scavengers. The pH of the solutions was adjusted to 9.0 with 50 mM PBS. At appropriate time points, samples were taken for ESR measurement and place in glass capillary tubes with an inner diameter of 1.1-1.2 mm, a wall thickness of 0.2 mm, and a length of 75 mm (Allied Corporation Fisher Scientific, Pittsburgh, PA, USA). The ESR spectra were recorded using a JEOL JES-FE3XG X-band spectrometer (JEOL Ltd., Tokyo, Japan) operating at a 100 kHz modulation frequency and at a 9.26 GHz microwave frequency. The modulation amplitude of the magnetic field was set at 1.0 x10-4 T, and the microwave power was 10 mW. The concentration of the produced nitroxide was determined by comparison with the peak-to-peak ESR signal amplitude of a 1 mM 2,2,6,6-tetramethyl-4-piperidone-N-oxyl solution. The ratio of the amplitude to the nitroxide concentration was also verified by comparison with the peak-to-peak ESR signal amplitude of a 1 mM 2,2,6,6-tetramethyl-4-piperidol-N-oxyl solution.
Statistical Analysis
The results were expressed as the mean ± standard deviation (S.D.). The values were compared by 1 way analysis of variance with 0.05 as the minimum level of significance.
Results
Cell Damage
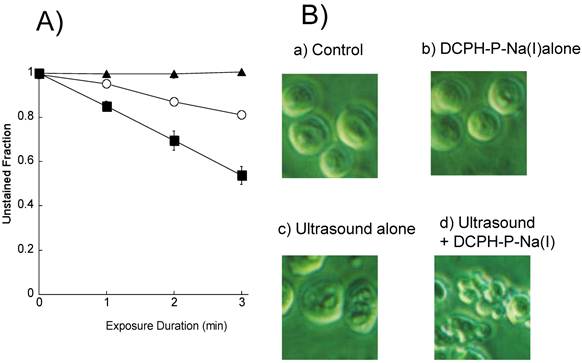
We first exposed HL-60 cells to an ultrasound (6 W/cm2) in the presence and absence of 8 μM DCPH-P-Na(I) and examined their integrity by staining with Trypan blue. Figure 3A shows the fraction of unstained HL-60 cells vs. the duration of exposure. The results show that the unstained (intact) fraction decreased exponentially with the duration of exposure. Following exposure to ultrasound for 3 min in the presence and absence of DCPH-P-Na(I), the fraction of unstained cells was 80% and 56%, respectively. DCPH-P-Na(I) alone did not cause cell damage.
A) Fraction of HL60 cells not stained with Trypan blue following ultrasonic exposure in the presence and absence of DCPH-P-Na(I). ▲, 8 μM DCPH-P-Na(I) alone; ○, ultrasound alone; ■, 8 μM DCPH-P-Na(I) + ultrasound. Values represent the means ± S.D. of four independent experiments. B) Analysis of cellular morphology by phase contrast microscopy after a 4-h incubation under the following conditions: (a) No treated (b) 8 μM DCPH-P-Na(I) alone, (c) ultrasound alone, and (d) 8 μM DCPH-P-Na(I) + ultrasound. C) Fraction of apoptotic HL-60 cells after a 3-min exposure to ultrasound in the presence and absence of DCPH-P-Na(I).□, control; ■, 8 μM DCPH-P-Na(I) alone; ○, ultrasound alone; ●, 8 μM DCPH-P-Na(I) + ultrasound. Values represent the means ± S.D. of four independent experiments. The asterisk symbol indicates significant difference from untreated control at P < 0.05. D) Apoptotic fraction of HL-60 cells as a function of ultrasonic intensity.○, ultrasound alone; ●, 8 μM DCPH-P-Na(I) + ultrasound. Values represent the means ± S.D. of four independent experiments.
Morphological Changes
We next assessed the induction of apoptosis by examining the morphology of the cells with a phase contrast microscope after a 4-h incubation with no addition, 8 μM DCPH-P-Na(I) alone, ultrasound alone (6 W/cm2), and ultrasound in the presence of DCPH-P-Na(I) (Figure 3B). There was no significant morphological change in the cells treated with either DCPH-P-Na(I) alone (b) or ultrasound alone (c). In contrast, the combination of ultrasound and DCPH-P-Na(I) clearly caused membrane blebbing and cell shrinkage (d).
Induction of Apoptosis
From the phase contrast microscope images, we calculated the fractions of apoptotic cells and plotted them vs. time (Figure 3C). Under all conditions, the fraction of apoptotic cells was less than 2% immediately following initiation of the treatment. In cells exposed to ultrasound in the presence of DCPH-P-Na(I), there was a significant increase in the fraction of apoptotic cells that increased with time. The fraction of apoptotic cells reached a maximum after 4 h and then decreased. A significant increase in the apoptotic fraction was not observed in cells exposed to ultrasound or DCPH-P-Na(I) alone.
The apoptotic cell fractions are plotted against the ultrasonic intensity in Figure 3D for the presence or absence of DCPH-P-Na(I). No significant apoptosis induction was observed up to the ultrasonic intensity of 2.5 W/cm2. In the presence of 8 μM DCPH-P-Na(I), the apoptotic fraction increased as the intensity increased from 2.5 to 6 W/cm2 and decreased above 6 W/cm2. On the basis of this result, the ultrasonic intensity was set to 6 W/cm2 in the other ultrasonic exposure experiments.”
DNA Fragmentation
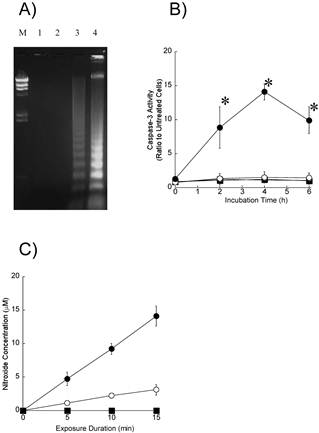
To further explore the induction of apoptosis, we performed agarose gel electrophoresis of DNA samples from the HL60 cells (Figure 4A). The unclear DNA ladder was observed in cells exposed to ultrasound alone (lane 3). A characteristic DNA ladder was observed 4 h after the exposure to ultrasound in the presence of 8 μM DCPH-P-Na(I) (lane 4). An obvious DNA ladder was not observed in cells exposed to DCPH-P-Na(I) alone (lane 3).
A) DNA ladder formation in HL-60 cells 4 h after exposure to DCPH-P-Na(I)and/or ultrasound. Lane M, DNA size markers; Lane 1, No treated; Lane 2, 8 μM DCPH-P-Na(I) alone; lane 3, ultrasound alone; Lane 4, 8 μM DCPH-P-Na(I) + ultrasound. B) Caspase-3 activities in HL-60 cells after a 3-min exposure to ultrasound in the presence and absence of DCPH-P-Na(I). .□, control; ■, 8 μM DCPH-P-Na(I) alone; ○, ultrasound alone; ●, 8 μM DCPH-P-Na(I) + ultrasound. Values represent the means ± S.D. of four independent experiments. The asterisk symbol indicates significant difference from untreated cells at P < 0.05. C) Nitroxide generation in an air-saturated solution of 50 mM TMPone during exposure to ultrasound in the presence and absence of ATX-70. .□, control; ■, 8 μM DCPH-P-Na(I) alone; ○, ultrasound alone; ●, 8 μM DCPH-P-Na(I) + ultrasound. Values represent the means ± S.D. of four independent experiments.
Caspase-3 Activation
To investigate whether caspases are activated by sonodynamic treatment in HL-60 cells, we measured the enzymatic activity of caspase-3 using a fluorescent peptide substrate (Figure 4B). We found that the activity of caspase-3 increased, reached a maximum after 4 h, and then decreased in cells treated with ultrasound in the presence of 8 μM DCPH-P-Na(I). An increase in caspase-3 activity was not observed in cells treated with ultrasound or DCPH-P-Na(I) alone.
Nitroxide generation
The reactive oxygen species by ESR and spin trapping technique to determine whether reactive oxygen species including singlet oxygen and hydroxyl radicals participate in the induction of apoptosis by ultrasound. Figure 4C shows the amounts of nitroxide ultrasonically generated in air-saturated aqueous solutions of 50 mM TMPone with and without DCPH-P-Na(I) under the same acoustic conditions as employed in the cellular experiments. The nitroxide levels were determined from the ESR signal amplitudes and plotted vs. the insonation time. The amount of ultrasonically generated nitroxide increased linearly with the insonation time. DCPH-P-Na(I) at a concentration of 8 μM enhanced the rate approximately three-fold. Nitroxide generation was not observed with DCPH-P-Na(I) alone.
Effect of Reactive Oxygen Scavengers
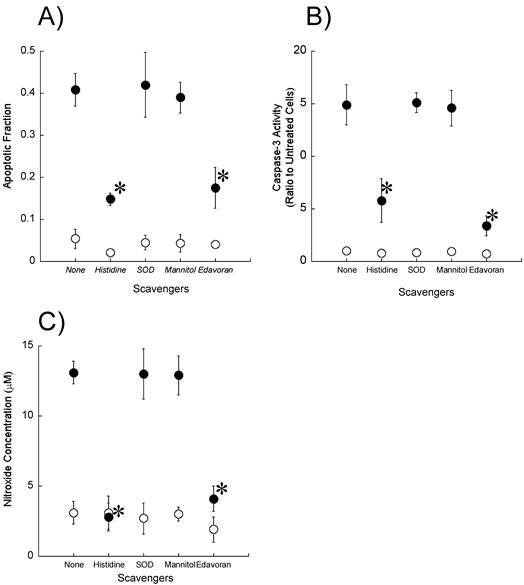
To determine whether reactive oxygen species including singlet oxygen and hydroxyl radicals participate in the induction of apoptosis by ultrasound, we examined the effect of reactive oxygen scavengers (10 mM histidine, 100 μg/ml SOD, and 100 mM mannitol) on the fraction of cells showing morphological changes associated with apoptosis as well as on the caspase-3 activity and the production of nitroxide (Figures 5A-C). Histidine significantly reduced the apoptosis induction, caspase-3 activation, and nitroxide generation caused by exposure to ultrasound in the presence of DCPH-P-Na(I). In contrast, SOD and mannitol did not affect these measurements.
Effect of active oxygen scavengers on A) ultrasonically induced apoptosis, B) caspase-3 activation and C) ultrasonic nitroxide generation in the presence (●) and absence (○) of 8 μM DCPH-P-Na(I). Values represent the means ± S.D. of four independent experiments. The asterisk symbol indicates significant difference from no scavenger treatment at P < 0.05.
Discussion
In our initial morphological studies, we found that ultrasonically induced apoptosis is greatly enhanced by DCPH-P-Na(I). Specifically, we clearly observed membrane blebbing and cell shrinkage when cells were treated with a combination of ultrasound and DCPH-P-Na(I), whereas significant morphologic changes were not observed in the cells exposed to either ultrasound alone or DCPH-P-Na(I) alone. The apoptotic fraction in the cells after the combination treatment was higher than both treatments with ultrasound alone and with DCPH-P-Na(I) alone by more than an order of magnitude. These results clearly demonstrated the synergistic effect between DCPH-P-Na(I) and ultrasound, namely the sonodynamic effect with PF, on the apoptosis. As shown in Fig.3D, the sonodynamically induced apoptosis was significant only when the ultrasonic intensity was above a threshold around 2.5 W/cm2. This is typical for a phenomenon resulting from acoustic cavitation. The increase in ultrasound intensity above 6 W/cm2 leads to a decrease in the incidence of apoptosis. These results show that HL-60 cells are likely to undergo necrosis instead of apoptosis during ultrasonic exposure at an intensity above 6 W/cm2.
The fragmentation of DNA at linker regions between nucleosomes into fragments that are multiples of 180-200 base pairs in length is a hallmark of apoptosis [17]. Using agarose gel electrophoresis, we found that ultrasonic exposure in the presence of DCPH-P-Na(I) resulted in the formation of a characteristic DNA ladder. This was not observed immediately after exposure (data not shown) but clear after the 4 h incubation, indicating that DNA fragmentation was caused by an enzymatic process rather than by direct sonochemical damage to the HL-60 cells.
Caspase-3 is an important enzyme required for the execution of the final phase of apoptosis, and it is active in cells undergoing apoptotic death [19]. We observed a significant activation of caspase-3 after treatment with a combination of ultrasound and DCPH-P-Na(I). This activation of caspase-3 was associated with the induction of apoptosis. Both the apoptotic fraction and the activity of caspase-3 gradually increased to a maximum after 4 h and then decreased, suggesting that caspase-3 acts as the executor caspase responsible for the induction of apoptosis by sonodynamic treatment, but how caspase-3 was activated by sonodynamic treatment remains to be determined.
Like photodynamic reactions, sonodynamic reactions are classified as either type I or type II [30, 31]. In type I reactions, the exited state of the sensitizer reacts with the substrate, whereas in type II reactions, it reacts with oxygen, yielding radicals, radical ions, or singlet oxygen. We observed a substantial enhancement of nitroxide generation in the presence of DCPH-P-Na(I) and under sonodynamic conditions used in the cellular experiments. Studies on the effects of reactive oxygen scavengers and D2O substitution on ultrasonically induced cell damage and on ultrasonic generation of reactive oxygen species suggest that singlet oxygen is the most important reactive oxygen species for the sensitizer effect of porphyrins [13]. We therefore examined the inhibitory effect of reactive oxygen scavengers on the fraction of sonodynamically induced apoptotic cells, caspase-3 activation, and nitroxide generation.
Hiraoka et al. hypothesized that sonodynamically induced cell killing was more likely due to certain mechanical stress such as augmentation of physical disruption of cellular membrane by sensitizers [31]. However, the significant reduction by histidine in both ultrasonically induced apoptosis and caspase-3 activation clearly suggests that some ultrasonically generated active species, which can be deactivated by histidine, stimulated the apoptotic signaling pathways through caspase-3 activation.
The tested scavengers included histidine, which is known to scavenge singlet oxygen and possibly hydroxyl radicals, 100 mM mannitol, a concentration that should scavenge both photodynamically and sonodynamically induced hydroxyl radicals and SOD, which catalyzes the elimination of superoxide radicals [13, 32]. Histidine caused a significant reduction in ultrasonically induced apoptosis, caspase-3 activation, and nitroxide generation, suggesting that singlet oxygen is more important than hydroxyl radicals or superoxide in the induction of apoptosis by ultrasound and DCPH-P-Na(I). These results also suggest that reactive oxygen species stimulate apoptotic signaling pathways via caspase-3 activation.
Misik and Riesz suggest another possible mechanism of sonosensitizer-derived free radicals as mediators of sonodynamically induced cell damage [33]. They proposed that sonosensitization is due to the chemical activation of sonosensitizers in the close vicinity of hot collapsing cavitation bubbles to form sensitizer-derived free radicals. These carbon-centered free radicals react with oxygen to form peroxyl and alkoxyl radicals.
There is no conclusive evidence that cavitation can also occur intracellularly, and the idea that formation of intracellular cavitation is qualitatively different from the inertial cavitation outside the cells because of higher intracellular viscosity has been suggested [34]. On the other hands, Misík et al. reported that the free radical scavenger cysteamine which penetrates the cell wall, has a protective effect in cells exposed to ultrasound, in contrast to the absence of protection by its oxidized cell-nonpermeable form, cystamine. They suggested that Intracellular cavitation can explain how short-lived free radicals reachinternal components of cells. Their results also support the possibility that the cavitation occurring inside the cells can activate sonosensitizer and intracellularly generate reactive oxygen species was also suggested [35].
Histidine can potentially deactivate the exited state of DCPH-P-Na(I) while either mannitol or superoxide dismutase cannot. Therefore, the effectiveness of histidine in contrast with the ineffectiveness of the other two can also be explained by the hypothesis that the observed sonodynamic enhancement of apoptosis by DCPH-P-Na(I) was induced by reactive oxygen species such as singlet oxygen, and sensitizer-derived free radicals. Singlet oxygen could be generated because the cell susupension was air-saturated in the ultrasonic exposure, at least when it was started. These results also suggest that reactive oxygen species such as singlet oxygen and sensitizer-derived free radicals stimulate apoptotic signaling pathways via caspase-3 activation [26].
In conclusion, we demonstrated sonodynamically induced apoptosis in HL-60 cells, as evidenced by morphological changes, DNA ladder formation, and caspase-3 activation. The significant reduction by histidine in the number of sonodynamically induced apoptotic cells, caspase-3 activation, and nitroxide generation suggests that ultrasonically generated singlet oxygen is an important mediator of sonodynamically induced apoptosis. Further studies on the mechanism of sonodynamically induced apoptosis should provide useful information for the improvement of sonodynamic tumor treatment. The present series of experiments was carried out using a single cell line. The investigation of other cell lines is necessary to examine the the possibility of in vivo induction of apoptosis. The results reported in this paper may be preliminary in the senses above, but they significantly support the possibility of in vivo induction of apoptosis by sonodynamic treatment using DCPH-P-Na(I). To expand our results, an in vivo experiment using solid tumor is under a way to examine more physiological effects following sonodynamic treatment, which may provide important information in sonodynamically induced apoptosis.
Acknowledgements
This work was supported, in part, by a Grant-in-Aid for Scientific Research (C) from Japan Society for the Promotion of Science to Nagahiko Yumita (No 22500444), Yumiko Iwase (No 23500569) and Toshio Fukai (No 23500570).
Conflict of Interest
The Authors have declared that no conflict of Interest exists.
References
1. Kremkau FW. Cancer therapy with ultrasound. J Clin Ultrasound. 1979;7:287-300
2. Lele PP. Local hyperthermia by ultrasound. In: Nussbaum GH, ed. Physical aspect of hyperthermia. New York: Am Inst Phys. 1982;39:3
3. Rosenthal I, Sostaric ZJ, Riesz P. Sonodynamic therapy -a review of the synergistic effects of drugs and ultrasound. Ultrason Sonochem. 2004;6:349-63
4. Yu T, Wang Z, Mason TJ. A review of research into the uses of low level ultrasound in cancer therapy. Ultrason Sonochem. 2004;2:95-103
5. Umemura S, Kawabata K, Sasaki K. et al. Recent advances in sonodynamic approach to cancer therapy. Ultrason Sonochem. 1996;3:S187-91
6. Tachibana K, Uchida T, Hisano S. et al. Eliminating adult T-cell leukaemia cells with ultrasound. Lancet. 1997;349:325
7. Abe H, Kuroki M, Tachibana K. et al. Targeted sonodynamic therapy of cancer using a photosensitizer conjugated with antibody against carcinoembryonic antigen. Anticancer Res. 2002;3:1575-80
8. Umemura S, Yumita N, Nishigaki R. et al. Enhancement of ultrasonically induced cell damage by a gallium-porphyrin complex, ATX70. Jpn J Cancer Res. 1993;84:582-8
9. Yumita N, Umemura S, Nishigaki R. Ultrasonically induced cell damage enhanced by photofrin II: mechanism of sonodynamic activation. In Vivo. 2000;14:425-9
10. Yumita N, Nishigaki R, Umemura S. Sonodynamically induced antitumor effect of photofrin II on colon 26 carcinoma. J Cancer Res Clin Oncol. 2000;126:601-6
11. Yumita N, Sasaki K, Umemura S. et al. Sonodynamically induced antitumor effect of a gallium-porphyrin complex, ATX-70. Jpn J Cancer Res. 1996;87:310-6
12. Yumita N, Umemura S. Sonodynamic therapy with photofrin ll on AH130 solid tumor. Pharmacokinetics, tissue distribution and sonodynamic antitumoral efficacy of photofrin ll. Cancer Chemother Pharmacol. 2003;51:174-8
13. Umemura S, Yumita N, Nishigaki R. et al. Mechanism of cell damage by ultrasound in combination with hematoporphyrin. Jpn J Cancer Res. 1990;81:962-6
14. Kuroki M, Hachimine K, Abe H. et al. Sonodynamic therapy of cancer using novel sonosensitizers. Anticancer Res. 2007;27:3673-7
15. Hachimine K, Shibaguchi H, Kuroki M. et al. Sonodynamic therapy of cancer using a novel porphyrin derivative, DCPH-P-Na(I), which is devoid of photosensitivity. Cancer Sci. 2007;98:916-20
16. Yumita N, Iwase Y, Nishi K. et al. Sonodynamically induced cell damage and membrane lipid peroxidation by novel porphyrin derivative, DCPH-P-Na(I). Anticancer Res. 2010;30:2241-6
17. Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456-62
18. Schemp CM, Simon-Haarhaus B, Termeer CC. et al. Hypericin photo-induced apoptosis involves the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and activation of caspase-8. FEBS Lett. 2001;493:26-30
19. Vantieghem A, Assefa Z, Vandenabeele P. et al. Hypericin-induced photosensitization of HeLa cells leads to apoptosis or necrosis. Involvement of cytochrome c and procaspase-3 activation in the mechanism of apoptosis. FEBS Lett. 1998;440:19-24
20. Feril LB Jr, Kondo T, Takaya K. et al. Enhanced ultrasound-induced apoptosis and cell lysis by a hypotonic medium. Int J Radiat Biol. 2004;80:165-75
21. Feril LB Jr, Tsuda Y, Kondo T. et al. Ultrasound-induced killing of monocytic U937 cells enhanced by 2,2'-azobis(2-amidinopropane) dihydrochloride. Cancer Sci. 2004;95:181-5
22. Feril LB Jr, Kondo T, Ogawa R. et al. Dose-dependent inhibition of ultrasound-induced cell killing and free radical production by carbon dioxide. Ultrason Sonochem. 2003;10:81-4
23. Ashush H, Rozenszajn LA, Blass M. et al. Apoptosis induction of human myeloid leukemic cells by ultrasound exposure. Cancer Res. 2000;60:1014-20
24. Lagneaux L, de Meulenaer EC, Delforge A. et al. Ultrasonic low-energy treatment: a novel approach to induce apoptosis in human leukemic cells. Exp Hematol. 2002;30:1293-301
25. Feril LB Jr, Kondo T, Zhao QL. et al. Enhancement of ultrasound-induced apoptosis and cell lysis by echo-contrast agents. Ultrasound Med Biol. 2003;29:331-7
26. Yumita N, Okudaira K, Momose Y. et al. Sonodynamically induced apoptosis and reactive oxygen generation by gallium-porphyrin complex, ATX-70. Cancer Chemother Pharmacol. 2010;66:1071-8
27. Johns LD. Nonthermal effects of therapeutic ultrasound: the frequency resonance hypothesis. J Athl Train. 2002;37:293-9
28. Yumita N, Han QS, Kitazumi I, Umemura S. Sonodynamically-induced apoptosis,necrosis, and active oxygen generation by mono-l-aspartyl chlorin e6. Cancer Sci. 2008Jan;99(1):166-72
29. Coleman AJ, Saunders JE, Crum LA, Dyson M. Acoustic cavitation generated by an extracorporeal shockwave lithotripter. Ultrasound Med Biol. 1987;13:69-76
30. Foote CS. Definition of type I and type II photosensitized oxidation. Photochem Photobiol. 1991;54:659
31. Hiraoka W, Honda H, Feril LB Jr. et al. Comparison between sonodynamic effect and photodynamic effect with photosensitizers on free radical formation and cell killing. Ultrasonic Sonochemistry. 2006;13:535-42
32. Goldstein S, Czapski G. Mannitol as OH scavenger in aqueous solutions and biological systems. Int J Radiat Biol. 1984;46:725-9
33. V. Misik, P. Riesz. Free radical intermediates in sonodynamic therapy. Ann N Y Acad Sci. 2000;899:335-48
34. Rosenthal I, Sostaric JZ, Riesz P. Sonodynamic therapy-a review of the synergistic effects of drugs and ultrasound. Ultrason Sonochem. 2004;11:349-63
35. Misík V, Miyoshi N, Riesz P. Effects of cysteamine and cystamine on the sonochemical accumulation of hydrogen peroxide--implications for their mechanisms of action in ultrasound-exposed cells. Free Radic Biol Med. 1999;26:961-7
Author contact
![]() Corresponding author: Nagahiko Yumita, PhD, School of Pharmacy, Yokohama College of Pharmacy, 601, Matano-cho, Totsuka-ku, Yokohama, Kanagawa 245-0066, Japan, Tel: +81-45-859-1300, Fax: +81-45-859-1301.
Corresponding author: Nagahiko Yumita, PhD, School of Pharmacy, Yokohama College of Pharmacy, 601, Matano-cho, Totsuka-ku, Yokohama, Kanagawa 245-0066, Japan, Tel: +81-45-859-1300, Fax: +81-45-859-1301.