Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(5):1401-1416. doi:10.7150/thno.30701 This issue Cite
Research Paper
Transcriptional Downregulation of miR-4306 serves as a New Therapeutic Target for Triple Negative Breast Cancer
Zitong Zhao1, Lin Li4, Peina Du4, Liying Ma1, Weimin Zhang 6, Leilei Zheng 1, Bo Lan3, Bailin Zhang5, Fei Ma3, Bo Xu2, Qimin Zhan 1,6 ![]() , Yongmei Song1
, Yongmei Song1 ![]()
1. State Key Laboratory of Molecular Oncology, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100021, China
2. Breast Cancer Center and the Key Laboratory of Breast Cancer Prevention and Therapy, Tianjin Medical University Cancer Institute and Hospital, National Clinical Research Center for Cancer, Key Laboratory of Cancer Prevention and Therapy, Tianjin 300060, China
3. Department of Medical Oncology, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100021, China
4. BGI-Shenzhen, Shenzhen, Guangdong 518083, China
5. Department of Breast Surgery, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100021, China
6. Laboratory of Molecular Oncology, Peking University Cancer Hospital, Beijing 100142, China
Received 2018-10-15; Accepted 2018-12-23; Published 2019-2-20
Abstract

Rationale: Triple-negative breast cancer (TNBC) is characterized by the absence of estrogen receptor alpha (ER-α), human epidermal growth factor receptor 2 (HER2) and progesterone receptor (PR) expression, but the effect of lacking the three factors on TNBC is unclear. Whether loss of the three factors contributes to deregulate genes that participate in the progress of TNBC remains unknown.
Methods: We performed microRNA arrays and comprehensive analysis to screen for miRNAs that are transcriptionally regulated by ER-α, HER2 and PR. Functional assays and molecular mechanism studies were used to investigate the role of miR-4306 in TNBC. An orthotopic mouse model of TNBC was used to evaluate the therapeutic potential of a cholesterol-conjugated miR-4306 mimic.
Results: We found that miR-4306 is transcriptionally regulated by ER-α, HER2 and PR, and the downregulation of miR-4306 in TNBC is caused by the loss of ER-α, HER2 and PR. Clinically, low miR-4306 expression is strongly associated with lymph node metastasis and poor survival for TNBC. Upregulation of miR-4306 greatly suppresses TNBC cell proliferation, migration and invasion and abrogates angiogenesis and lymphangiogenesis in vitro. According to in vivo models, miR-4306 overexpression considerably inhibits TNBC growth, lung metastasis, angiogenesis and lymph node metastasis. Mechanistic analyses indicate that miR-4306 directly targets SIX1/Cdc42/VEGFA to inactivate the signaling pathways mediated by SIX1/Cdc42/VEGFA. Finally, the orthotopic mouse model of TNBC reveals that miR-4306 mimic can be used for TNBC treatment in combination with cisplatin.
Conclusions: Our findings suggest that miR-4306 acts as a tumor suppressor in TNBC and is a potential therapeutic target for TNBC treatment.
Keywords: triple-negative breast cancer, transcriptional regulation, microRNA, targeted therapy
Introduction
Triple-negative breast cancer (TNBC) is characterized by the absence of estrogen receptor α (ER-α), human epidermal growth factor receptor 2 (HER2) and progesterone receptor (PR) expression [1]. TNBC accounts for 10% to 15% of all diagnosed breast cancers and is prone to recurrence, distant metastasis and poor clinical prognosis; its five-year survival rate is less than 15% [1]. Currently, chemotherapy is the primary treatment modality for TNBC [2]. Traditional first-line drugs, such as paclitaxel, that are used to treat breast cancer do not yield an ideal therapeutic response when used to treat TNBC. Cisplatin, which is a second-line drug for treating breast cancer, appears to be more effective for treating TNBC. However, approximately one-third of patients develop recurrence within 3 years of adjuvant therapy [3]. The development and metastasis mechanisms of TNBC are not clear, and this fact has hampered the progress of targeted therapy for TNBC.
miRNAs are involved in cancer initiation and progression via their regulation of target genes [4, 5]. A growing body of evidence has demonstrated the practicality of using miRNA mimics or inhibitors as drugs or targets for treating cancer, and miRNA technology is available for tumor treatment in vivo [6, 7].
ER-α and PR are classic nuclear transcription factors [8, 9]. HER2 is a member of the EGF receptor tyrosine kinase family and has been rigorously studied as a signaling molecule of the cell membrane [10]. However, recent studies have reported that HER2 is also expressed in the nucleus and could form a complex at a specific nucleotide sequence of the gene promoter to stimulate its transcription [11, 12]. ER-α, PR and HER2 play key roles in breast cancer; however, whether loss of the three factors contributes to deregulate genes that participate in the progress of TNBC remains unknown.
In this work, we demonstrate that miR-4306 is epigenetically regulated by ER-α, HER2 and PR. miR-4306 is downregulated in TNBC compared with other subtypes of breast cancer, and the downregulation of miR-4306 in TNBC is caused by the loss of ER-α, HER2 and PR. Increased miR-4306 expression levels suppress TNBC metastasis, particularly lymph node metastasis, predominately through targeting SIX1, Cdc42 and VEGFA in vitro and in vivo. Further studies show that a miR-4306 mimic in combination with cisplatin can be used for TNBC treatment. Overall, our study reveals the critical role and underlying mechanism of miR-4306 in suppressing TNBC metastasis and provides a new target for TNBC treatment.
Methods
Cell culture
ZR-75-1, MCF-7, T47D, SK-BR-3, HCC1937, MDA-MB-468, MDA-MB-231, CAL-51, and HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics.
E2 deprivation: the ER-α-positive cell lines were cultured in phenol red-free DMEM in the absence of exogenous E2 and supplemented with 10% dextran charcoal-stripped bovine serum for 48 h.
Human breast cancer tissue samples
A total of 325 paired samples of human breast cancer tissues and their matched adjacent normal tissues were collected at National Cancer Center/ National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College.
RNA extraction, RT-PCR and quantitative real-time PCR
Total RNA was extracted from frozen fresh tissue and cell lines with TRIzol reagent (Invitrogen). cDNAs were synthesized with Superscript II reverse transcriptase (Invitrogen). Quantitative real-time PCR (qPCR) was performed with a SYBR Premix Ex TaqTM II kit (TaKaRa). The qPCR primers used are listed in Table S13.
MicroRNA Array and mRNA Array
The Agilent/Affymetrix microarray was used for miRNA/mRNA expression profiles (CapitalBio).
Cell proliferation assay
The proliferation ability of different cancer cells was determined using the xCELLigence Real-Time Cell Analyzer (RTCA)-MP system (Acea Biosciences/ Roche Applied Science) as reported previously [13].
Transwell migration/invasion assays
Migration and invasion assays were performed as previously described [13].
Oligonucleotide transfection
A miR-4306 mimic and inhibitor and siRNA for Cdc42, SIX1, VEGFA, ER-α, HER2, and PR were purchased from RiboBio (Guangzhou, China). Cells were transfected with siRNA, miRNA mimic and miRNA inhibitor using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's protocol.
Plasmid construction
The Cdc42/SIX1 3'UTR (WT) or mutant (MUT) with a predicted miR-4306 responsive element was inserted downstream of the firefly luciferase gene in the GV272 vector.
The VEGFA 3'UTR (WT) or mutant (MUT) with a predicted miR-4306 responsive element was inserted downstream of the firefly luciferase gene in the pmirGLO plasmid.
The miR-4306 promoter (-2000 bp ~ 0 bp) was inserted upstream of the firefly luciferase gene in the pGL3.0 basic vector.
A Cdc42/SIX1 overexpression plasmid was constructed in the GV230 vector.
Luciferase reporter assay
The Luciferase reporter assay was performed with the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's protocol.
Western blotting analysis
The assays were performed as previously described [13], using anti-Cdc42, anti-P21, anti-vWF, anti- VEGFC (Proteintech), anti-β-actin, anti-E-cadherin, anti-SIX1 anti-cyclinD1(Santa Cruz Biotechnology), anti-ERK, anti-p-ERK, anti-AKT, anti-p-AKT, anti- PLC-γ, anti-p-PLC-γ, anti-STAT3, anti-p-STAT3, anti- EGFR, anti-MMP-9, anti-MMP-11, and anti-PCNA (Cell Signaling Technology) antibodies.
HUVEC/HDLEC tube formation assay
Cell-conditioned media were collected and stored at -80°C. HUVECs/HDLEC (2×105) were suspended in a mixture of conditioned medium (500 μL) and DMEM (500 μL) with 10% FBS and seeded on a 24-well plate coated with 50% Matrigel (300μL/ well). Tube formation was observed after incubation for 3 h at 37 °C. The number of tubular structures was counted in each field [14].
Retroviral infection
The lentivirus for miR-4306 was purchased from GeneChem and infected cells according to the manufacturer's protocol.
ChIP assay
Chromatin immunoprecipitation assays were performed using a Pierce™ Magnetic ChIP Kit (Thermo Scientific™). Immunoprecipitation was performed with anti-ER-α, anti-ERBB2, and anti-PR antibodies (Cell Signaling Technology). Specific regions were quantified via qRT-PCR using the primers listed in Table S13.
RNA immunoprecipitation (RIP) assay
The EZ-Magna RIP Kit (Millipore, USA) was applied to conduct the RIP assay according to the manufacturer's protocol. Immunoprecipitation wasperformed with an anti-Ago2 antibody (Abcam, USA).
Analysis of in vivo tumorigenicity
Four-week-old female BALB/c nude mice were provided by Vital River for the in vivo tumorigenicity study. The mice were injected subcutaneously in their top backs with 1×106 cells in 0.1 mL. In addition, the tail veins and the footpads of the mice were each injected with 1×106 cells in 0.1 mL.
Chicken embryo chorioallantoic membrane (CAM) assay
CAM assay were performed with five-to-seven- day-old chicken embryos (Vital River Co., Beijing, China) as described previously [14].
Study approval
This work was approved by the Cancer Hospital, the Chinese Academy of Medical Sciences, the National GCP Center for Anticancer Drugs, and The Independent Ethics Committee. The collection of all of the human samples used in the experiments was approved by the hospital. Informed consent to participate in this study was obtained from all patients. All experiments involving mice were approved by the Cancer Hospital, the Chinese Academy of Medical Sciences, and the Experimental Animal Ethics Committee and followed the Institutional Animal Welfare Guidelines.
Statistical analysis
The data are presented as means ± SEM. The differences were assessed using 2-tailed Student's t test for group comparisons using the GraphPad Prism 5 software package. Pearson correlation was used to calculate r and P-values. In all experiments, differences were considered significant when P was less than 0.05. *P < 0.05, **P < 0.01, ***P < 0.001. All in vitro experiments were repeated at least 3 times unless stated otherwise. The data were analyzed with the SPSS 17.0 software package.
Results
miR-4306 is a transcriptional downstream target of ER-α, HER2 and PR
To determine the miRNAs regulated by ER-α, HER2 and PR, we profiled the global miRNA expression levels of 2 highly aggressive TNBC cell lines and compared them to those of 2 non-aggressive luminal-type breast cancer cell lines (GEO accession number: GSE110204; for a comparison of the aggressive phenotypes of four breast cancer cell lines, see Figure S1A and B). TNBC is characterized by a lack of ER-α, HER2 and PR, so the miRNAs regulated by the three molecules should be expressed at lower levels in TNBC than in other ER-α, HER2 or PR-positive subtypes. We found five miRNAs that were significantly downregulated in both TNBC cell lines compared with their expression in the 2 luminal lines (using a 4-fold cutoff; all four cell lines expressed these miRNAs according to the miRNA array; Figure 1A). We searched for the putative ER-α-binding element (ERE, GGTCAnnnTGACC) [8], HER2-binding element (TCAAATTTC) [12], and PR-binding element (PRE, GGTACAnnnTGTTCT) [9] in the five downregulated miRNA promoters, and only the promoter of miR-4306 has all three binding elements (Figure 1B). Therefore, in this study, we chose to study miR-4306 and elucidate its targets that may have a function in TNBC tumorigenesis.
ER-α, HER2, and PR transcriptionally regulate miR-4306 promoter status. (A) Heat map of the miRNA profile showing 5 deregulated miRNAs in TNBC cells. (B) Analysis of possible physical associations of regions in the miR-4306 promoter with ER-α, HER2, PR. (C) ChIP assays were performed with ER-α/HER2/PR antibody. IgG served as the negative control. (D) Relative luciferase activity of reporter constructs containing the miR-4306 promoter in cells. Luciferase reporter activity was normalized to Renilla luciferase activity. (E-H) qPCR analysis of miR-4306 levels in cells. (E) MCF-7 cells after treatment with different concentrations of E2 (0 μM, 10 μM, 50 μM, and 100 μM) for 8 h or with 100 μM E2 for different times (0 h, 2 h, 4 h, and 8 h). (F) SK-BR-3 cells were treated with different concentrations of HER2 siRNA for 48 h. (G) MCF-7 cells were treated with different concentrations of PR siRNA for 48 h. (H) SK-BR-3/ZR-75-1 cells were treated with different concentrations of neratinib or trametinib for 24 h. (I) After simultaneous ER-α, HER2, and PR knockdown in ZR-75-1 cells, qPCR analysis of miR-4306 levels and luciferase reporter activity of reporter constructs miR-4306 promoter. The data are representative of three independent experiments. The error bars represent the SEM. *P < 0.05, **P < 0.01, ***P < 0.001; two-tailed unpaired Student's t-test.
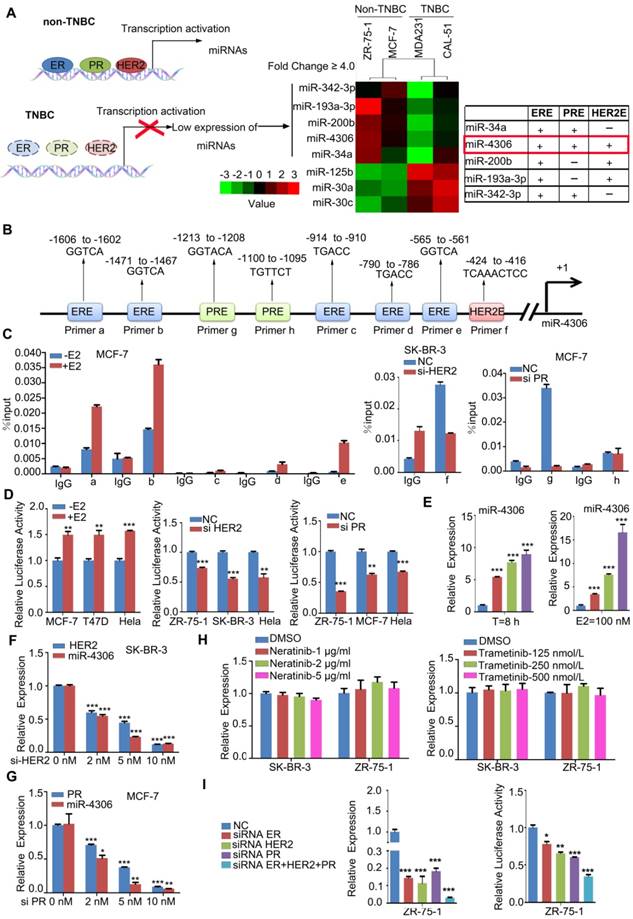
ChIP assays revealed that ER-α, HER2 and PR effectively bound to the corresponding binding elements in the miR-4306 promoter (Figure 1C and Figure S1C). To further examine the transcriptional activity of ER-α/HER2/PR on miR-4306, we constructed a luciferase reporter plasmid containing a -2000 bp ~ 0 bp site within the miR-4306 promoter sequence. The luciferase reporter assays revealed that luciferase activity driven by the miR-4306 promoter sequence was increased in E2-treated cells but decreased in ER-α-silenced cells (Figure 1D and Figure S1D), whereas knocking down HER2 or PR induced the downregulation of the luciferase activity driven by the miR-4306 promoter sequence (Figure 1D). After E2 deprivation for 48 h, miR-4306 expression was increased in MCF-7 cells treated with E2 at different concentrations and for different times (Figure 1E). Similar consequences were obtained in T47D cells (Figure S1E). Conversely, knocking down ER-α/PR/HER2 by siRNA led to a marked decrease in miR-4306 expression levels (Figure S1F-H, Figure 1F and Figure 1G). We used two small molecules to exclude tyrosine kinase function of membrane receptor HER2 on miR-4306 expression. Neratinib (HKI-272) is a highly selective inhibitor of HER2/ EGFR tyrosine kinase activity. Signaling pathways activated by membrane HER2 include the Ras-Raf- MEK-MAPKs pathway. Trametinib (GSK1120212) is an effective inhibitor of MEK1/2. We found these two small molecules had no apparent effect on miR-4306 expression (Figure 1H). These data suggest that ER-α/PR/HER2 transcriptionally regulates miR-4306 by directly targeting the miR-4306 promoter.
In addition to the above data showing that ER-α, HER2 and PR could individually regulate miR-4306, we found that simultaneously knocking down ER-α, HER2 and PR in ZR-75-1 cells with siRNAs led to a considerable decrease in the expression levels of miR-4306 and a significant decrease in the luciferase activity of the miR-4306 promoter sequence (Figure 1I). These experimental results of simultaneously knocking down ER-α, HER2 and PR indicated a more profound effect than knocking down ER-α, HER2 or PR siRNAs alone. These results indicated that the transcriptional deregulation of miR-4306 in TNBC was caused by the loss of ER-α, HER2 and PR.
miR-4306 is downregulated in TNBC
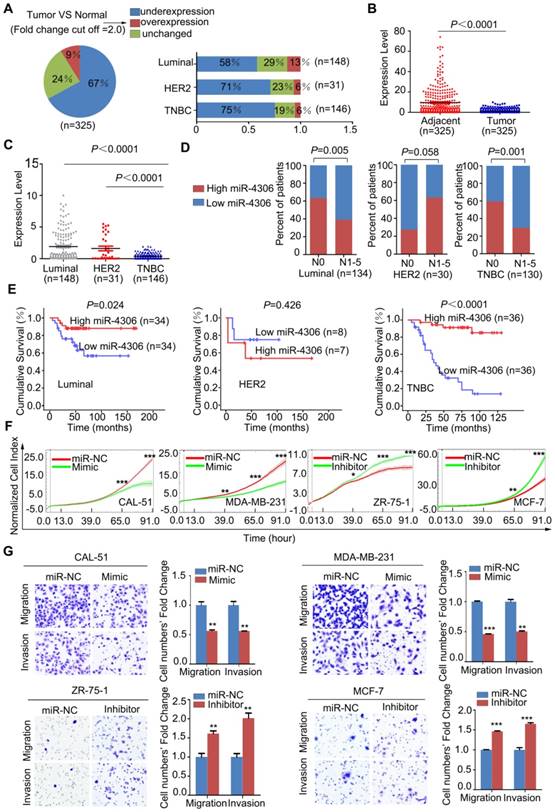
To investigate the clinical value of miR-4306 in breast cancer, qPCR was used to measure miR-4306 expression in 325 tumor tissues and their pair-matched adjacent normal tissues. These samples were taken from 148 patients with luminal-type breast cancer, 31 patients with HER2 type breast cancer and 146 patients with TNBC. The qPCR results revealed that miR-4306 expression was significantly lower (P<0.0001) in tumor tissues than in paired normal tissues (Figure 2A-2B), and miR-4306 expression was significantly lower in patients with TNBC than in patients with other-subtype breast cancer (Figure 2C). The correlation analysis of miR-4306 expression and clinical data revealed a negative correlation between miR-4306 and lymph node metastasis and poor survival in patients with TNBC and luminal-type breast cancer, and the correlation was more significant in patients with TNBC (Figure 2D-2E, Table S1-S3).
Low expression of miR-4306 predicted lymph node metastasis and poor survival in TNBC patients. (A and B) qPCR was used to measure the relative expression levels of miR-4306 in 325 pairs of breast cancer samples and their corresponding noncancerous samples. The results were normalized to the endogenous U6 RNA control. (C) miR-4306 expression levels are lower in TNBC tissues than in luminal-type and HER2 type tissues. (D) Percentages of patients with high miR-4306 expression levels and low miR-4306 expression levels according to lymph node metastasis in different breast cancer subtypes; the patients were classified into tumor lymph node metastasis-negative (N0) and tumor lymph node metastasis-positive (N1-N5) groups. (E) Kaplan-Meier curves for breast cancer patients with low versus high miR-4306 expression levels; log-rank test. (F-G) MDA-MB-231/CAL-51 cells transfected with miR-4306 mimic (Mimic) or miRNA mimic control (miR-NC) and ZR-75-1/MCF-7 cells transfected with miR-4306 inhibitor (Inhibitor) or miRNA inhibitor control (miR-NC). (F) After transfection for 24 h, the xCELLigence Real-Time Cell Analyzer (RTCA)-MP system was used to analyze the effects of miR-4306 on cell proliferation. (G) After transfection for 48 h, transwell assays were performed to analyze the effects of miR-4306 on cell migration and invasion. The quantitative data are presented in the histograms, and the photographs are representative of the migrated/invaded cells. Original magnification, ×100. The data are representative of three independent experiments. The error bars represent the SEM. *P < 0.05, **P < 0.01, ***P < 0.001; two-tailed unpaired Student's t-test.
miR-4306 upregulation suppresses the proliferation, invasion and migration of TNBC cells in vitro
To explore the function of miR-4306 in breast cancer, we transfected the breast cancer cell lines with hsa-miR-4306 mimic oligonucleotides (Mimic) or negative control mimic miRNA (miR-NC) and with hsa-miR-4306 inhibitor oligonucleotides (Inhibitor) or negative control inhibitor miRNA (miR-NC). For the transfection efficiencies, see Figure S2A. Then, we examined the effects of miR-4306 on cellular proliferation, invasion and migration. The growth curves showed that cells transfected with the miR-4306 mimic had significantly lower levels of cell proliferation than those transfected with the miR-NC. In addition, compared to miR-NC, the miR-4306 inhibitor considerably promoted cell proliferation (Figure 2F and Figure S2B). Transwell assays revealed that compared to the miR-NC, the miR-4306 mimic significantly inhibited cellular motility. Similarly, compared to the miR-NC, the miR-4306 inhibitor considerably increased cellular motility (Figure 2G and Figure S2C). These results, along with the above findings of lower miR-4306 expression levels in breast cancer tissues (particularly in TNBC tissues), suggested a tumor-suppressing role for miR-4306 in TNBC cells.
miR-4306 directly targets the 3'UTR of SIX1, Cdc42 and VEGFA
To understand the mechanism of how miR-4306 hampers TNBC progression, we used Affymetrix mRNA microarray analysis to evaluate changes in mRNAs in TNBC cells treated with the miR-4306 mimic. As shown in the cluster heat map, different expression profiles were obtained (Figure 3A left panel; GEO accession number: GSE110202, GSE110203). The right panel of Figure 3A shows the seven overlapping pathways of the top 25 highest ranked pathways in MDA-MB-231 and CAL-51 cells. To search for putative targets of miR-4306, we used the TargetScan, miRDB, and miRanda computational tools and identified a common set of 79 candidate genes whose 3'UTRs contain at least one putative miR-4306 binding sequence. We identified the 79 candidate genes from the microarray results and found that only Cdc42, SIX1 and VEGFA were downregulated in the miR-4306 mimic group compared with their expression in the miR-NC group (Figure 3B). Because miR-4306 is a tumor suppressor, its target genes are expected to be oncogenes for breast cancer. Cdc42, SIX1 and VEGFA have been reported as oncogenes in breast cancer and are involved in the miR-4306-mediated pathways shown in the right panel of Figure 3A. Therefore, we focused on these three genes for our subsequent experiments.
SIX1, Cdc42 and VEGFA are miR-4306 targets. (A) Left panel: Cluster analysis of deregulated genes in cells treated with miR-4306 mimic. Right panel: Pathway enrichment using PANTHER pathways. (B) Venn diagram of putative miR-4306 target genes predicted by computational algorithms from the MicroRNA Target Prediction software package and the microarray results in (A). (C) qPCR analysis of SIX1, Cdc42 and VEGFA levels in MDA-MB-231 and CAL-51 cells treated with miR-4306 mimic. (D) Western blot analysis of SIX1, Cdc42 and VEGFA levels in MDA-MB-231 and CAL-51 cells treated with miR-4306 mimic. (E) The diagram shows the region containing the miR-4306-binding site of the luciferase reporter construct containing the 3' UTR of SIX1, Cdc42 and VEGFA. MDA-MB-231 and CAL-51 cells were transfected with the WT or MUT luciferase vector and miR-4306 mimic or miR-NC. Luciferase reporter activity was normalized to Renilla luciferase activity. The data are representative of three independent experiments. The error bars represent the SEM. **P < 0.01, ***P < 0.001; two-tailed unpaired Student's t-test.
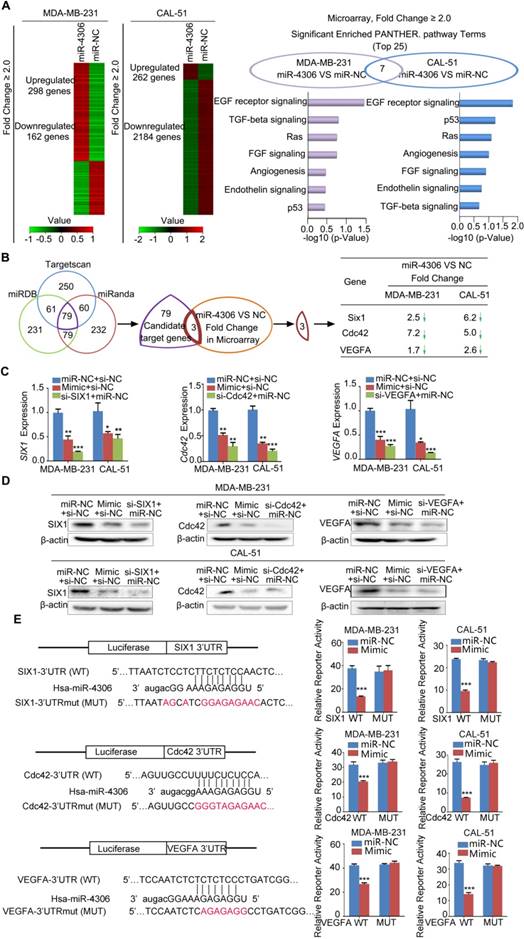
Furthermore, increased miR-4306 expression levels in TNBC cells markedly reduced SIX1, Cdc42, and VEGFA mRNA and protein expression levels (Figure 3C-3D). We performed 3'UTR luciferase reporter assays using pMIR-REPORT constructs: one contained the wild type miR-4306-binding site (WT), and the other contained a mutated miR-4306-binding site (MUT). The results showed that ectopic miR-4306 was able to suppress the luciferase activity of the WT construct but not that of the MUT construct; these data support the direct interaction of miR-4306 with the 3'UTR of SIX1, Cdc42 and VEGFA (Figure 3E). Moreover, it has been well proven that miRNAs exert its function through binding to Ago2. Ago2 is the core component of the RNA-induced silencing complex, and potential targets of miRNAs can be isolated from this complex after Ago2 coimmunoprecipitation [15]. Consistently, the results of RIP assays confirmed that SIX1/Cdc42/VEGFA was a target of miR-4306 in CAL-51/ MDA-MB-231 cells (Figure S2D-2E). Taken together, these results indicate that SIX1, Cdc42 and VEGFA are direct targets of miR-4306.
SIX1, Cdc42 and VEGFA mRNA expression is inversely correlated with miR-4306 expression in TNBC patients
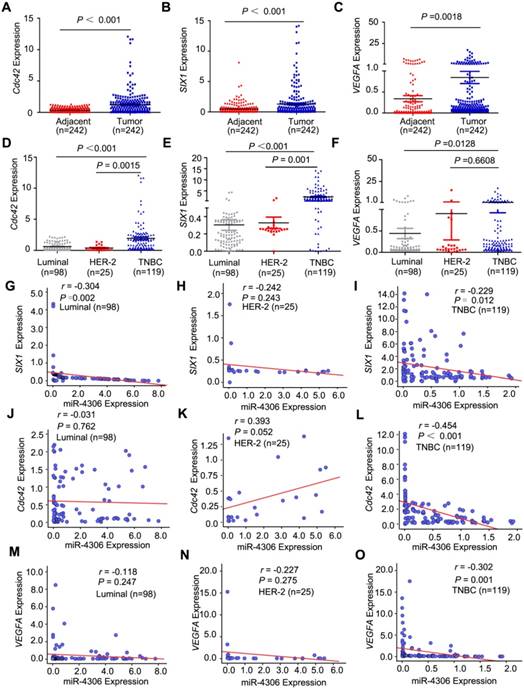
To determine whether SIX1/Cdc42/VEGFA expression is negatively correlated with miR-4306 expression in breast tumors, qPCR was used to measure the mRNA expression levels in the same samples in which we had previously measured miR-4306 expression levels in Figure 2 (242 tumors and their pair-matched adjacent normal tissues from 98 patients with luminal-type breast cancer, 25 patients with HER2-type breast cancer and 119 patients with TNBC). The qPCR results showed that SIX1/ Cdc42/VEGFA were overexpressed in breast cancer (Figure 4A-4C) and more highly expressed in TNBC tissues than in luminal-type and HER2-type tissues (Figure 4D-4F). For the correlation analysis of Cdc42/ SIX1/VEGFA mRNA expression and the clinical data, please see Figure S2F-S2K and Table S4-12. Moreover, there was an inverse correlation between miR-4306 and SIX1/Cdc42/VEGFA expression in the TNBC specimens but not in the luminal- type and HER2 type specimens (Figure 4G-4O). These data suggested that aberrant SIX1/Cdc42/VEGFA expression might be caused by ectopic miR-4306 expression in TNBC. However, aberrant SIX1/Cdc42/ VEGFA expression in other subtypes of breast cancer might be independent of miR-4306 and have other mechanisms.
miR-4306 is negatively correlated with SIX1, Cdc42 and VEGFA expression. (A-C) qPCR was used to measure the relative expression levels of SIX1, Cdc42 and VEGFA in 242 paired tissues. (D-F) SIX1, Cdc42 and VEGFA expression levels in different breast cancer subtype tissues. (G-I) Pearson correlation analysis of miR-4306 and SIX1 mRNA levels in breast cancer. (J-L) Pearson correlation analysis of miR-4306 and Cdc42 mRNA levels in breast cancer. (M-O) Pearson correlation analysis of miR-4306 and VEGFA mRNA levels in breast cancer. The statistical analysis was performed using two-tailed unpaired Student's t-test (A-F) and Pearson's Χ2-test (G-O). *P < 0.05, **P < 0.01, ***P < 0.001.
miR-4306 negatively regulated aggressive TNBC cell phenotypes by suppressing SIX1, Cdc42 and VEGFA expression
The functions of SIX1/Cdc42 focus on cell proliferation, migration and invasion, so we used RTCA-MP system to analyze the effects of miR-4306- SIX1/Cdc42 on cell proliferation, and transwell to study the effect of miR-4306-SIX1/Cdc42 on cell motility. We used human dermal lymphatic endothelial cells (HDLECs) tube formation and migration assays in vitro to study miR-4306-SIX1 on lymphatic metastasis. VEGFA is a kind of potent angiogenic cytokine. The main function of VEGFA is inducing angiogenesis. We conducted the human umbilical vein endothelial cells (HUVEC) tube formation assay in vitro.
Inhibiting SIX1/Cdc42/VEGFA expression by miR-4306 or SIX1/Cdc42/VEGFA siRNAs impaired the development of the TNBC aggressive phenotype. These effects were rescued significantly by the re-expression of SIX1/Cdc42/VEGFA in the miR-4306 mimic-treated cells. Figure 5A-5B and Figure S3B-3C revealed that miR-4306 inhibited the proliferation, migration and invasion of TNBC cells by targeting the 3'UTR of SIX1/Cdc42. Flow cytometry was performed to analyze the cell cycle. Compared to the percentages of cells transfected with the negative control (NC), the percentages of TNBC cells transfected with miR-4306 mimic or siRNA SIX1/Cdc42 were significantly increased in the G1 phase and significantly decreased in the S phase and G2 phase (Figure S3D).
SIX1, Cdc42 and VEGFA mediate the effects of miR-4306 on the aggressive TNBC cell phenotype. (A and B) CAL-51 cells were transfected with miR-4306 mimic or SIX1/Cdc42 siRNA or cotransfected miR-4306 mimic and the Cdc42/SIX1 plasmid. (A) Growth ability analysis using the xCELLigence Real-Time Cell Analyzer (RTCA)-MP system after transfection for 24 h. (B) Migration and invasion ability analysis using transwell assays after transfection for 48 h. The quantitative data are shown in the histograms, and the photographs are representative of the migrated/invaded cells. Original magnification, ×100 (C) Representative images of HDLECs cultured with conditioned medium from miR-4306-overexpressing cells, SIX1-silenced cells, miR-4306-SIX1-overexpressing and control cells. Matrigel tube formation assay (scale bar: 20μm) and migration assay (original magnification, ×100). (D) Representative images of HUVECs cultured with conditioned medium derived from miR-4306-overexpressing cells, VEGFA-silenced cells, miR-4306-overexpressing cells with added VEGFA and control cells. Scale bar: 20 μm. In (A-D), the data are representative of three independent experiments. The error bars represent the SEM. ***P < 0.001, two-tailed unpaired Student's t-test. (E) Western blot analysis showing SIX1 and Cdc42 expression in eight breast cancer cell lines. (F) Western blot analysis showing SIX1 and other related protein expression levels in miR-4306 mimic-treated or SIX1 siRNA-treated cells. (G) Western blot analysis showing Cdc42 and other related protein expression levels in miR-4306 mimic-treated or Cdc42 siRNA-treated cells.
To further confirm the correlation between miR- 4306 and tumor lymphangiogenesis/ angiogenesis, miR-4306 mimic/ miR-4306 inhbitor/ miR-NC-conditioned media (CM) were assayed for lymphangiogenic/angiogenic activity. Tube formation and migration assays revealed that overexpressing miR-4306 or silencing SIX1 strongly abrogated the ability of TNBC cells to induce tube formation (with much lower tube density) and migration in HDLECs, whereas silencing miR-4306 in non-TNBC cells or rescuing SIX1 expression in TNBC cells transfected with miR-4306 mimic promoted tube formation (with much higher tube density) and migration; these findings suggest that miR-4306 suppresses TNBC lymphangiogenesis by targeting the 3'UTR of SIX1 in vitro (Figure 5C, Figure S4A-4C). Likewise, overexpressing miR-4306 or silencing VEGFA obviously suppressed the ability of TNBC cells to induce tube formation (with much lower tube density) in HUVECs, whereas silencing miR-4306 in non-TNBC cells or rescuing VEGFA expression in TNBC cells transfected with miR-4306 mimic promoted tube formation (with much higher tube density); these findings suggest that miR-4306 suppresses TNBC angiogenesis by targeting the 3'UTR of VEGFA in vitro (Figure 5D, Figure S4D-4E).
As shown in Figure 5E, SIX1 and Cdc42 protein levels were markedly higher in the TNBC cell lines than in the non-TNBC cell lines, and their expression levels were almost negatively correlated with miR-4306. To further investigate the molecular mechanism of miR-4306, western blots were used to detect the known SIX1/Cdc42 downstream signaling pathways. As shown in Figure 5F and 5G, we measured the expression levels of cyclin A, cyclin D1 and P21, which are involved in cell cycle regulation and cell proliferation. We found that miR-4306 positively regulated P21 expression but negatively regulated cyclin D1 and cyclin A expression. Moreover, miR-4306 or siRNA SIX1/Cdc42 positively regulated E-cadherin expression but negatively regulated the activation of extracellular signal-regulated kinase (ERK) and its downstream genes matrix metalloprotease (MMP)-2, MMP-9, and MMP-11. These data indicate that miR-4306 might regulate cell metastasis through ERK-MMPs. miR-4306/siRNA SIX1 negatively regulated VEGFC expression, indicating that miR-4306 might regulate TNBC lymph node metastasis through SIX1-VEGFC. After silencing of miR-4306 expression in ZR-75-1/MCF-7 cells, E-cadherin expression was downregulated compared to that in the control (Figure S4F).
Role of miR-4306 in TNBC in vivo
To assess the effects of miR-4306 on tumor suppression in vivo, 2 pooled sets of CAL-51 cells were stably infected with negative control (Control) or miR-4306 lentiviral vectors. As expected, compared to the control-transfected cells, lenti-miR-4306-infected CAL-51 cells expressed much higher levels of miR-4306 (Figure S5A); furthermore, the aggressive phenotype was reduced in these cells (Figure S5B-S5D), the Cdc42, SIX1 and VEGFA mRNA and protein levels were reduced, and a series of changes in downstream pathway proteins were observed (Figure S5E and Figure S5F).
Both pools of cells were implanted subcutaneously into 8 nude mice in each group, and tumor growth was monitored weekly. Tumors in the lenti-miR-4306 group grew significantly slower, which resulted in smaller tumor masses (Figure 6A-6C). As shown in Figure 6D, the tumors formed by CAL-51-lenti-miR-4306 cells exhibited lower levels of SIX1, Cdc42, PCNA, MMP-9, VEGFA, VEGFC and vWF and higher levels of P21 than the control tumors according to IHC analyses.
miR-4306 inhibits TNBC cell growth, lung metastasis, angiogenesis and lymphatic metastasis in vivo. (A-D) The mice were injected subcutaneously in their top backs with 1×106 cells in 0.1 mL. Growth of both control and lenti-miR-4306 cell-induced tumors in nude mice was measured weekly (n=8). (A) Representative tumor mass. (B) Tumor growth curve analysis of mice bearing lenti-miR-4306 cells compared to mice bearing control cells. (C) Tumor mass analysis. (D) Representative IHC staining in CAL-51 xenografts with the indicated treatments. (E) Angiogenic response according to chicken embryo CAM assays after 3 days. Autoclaved filter papers (5 mm in diameter) were applied to CAMs and loaded with 200 μl of medium conditioned with CAL-51-control or CAL-51-miR-4306 two times a day. Neovasculature number/field. The error bars represent the SEM. n=5. * P < 0.05. (F) Rate of lung metastasis after tail injection. Lung metastasis of representative tumors from the CAL-51 control group according to HE analysis. (G) Volumes of popliteal lymph nodes and rates of lymphatic metastasis after foot pad injections. Lymphatic metastasis of representative tumors from the CAL-51 control group according to HE analysis. The statistical analysis in B-E and G-H was performed using the two-tailed Student's t-test. *P < 0.05, **P < 0.01, ***P < 0.001.
To further confirm the correlation between miR-4306 and TNBC angiogenesis, lenti-control- and lenti-miR-4306-conditioned media (CM) were assayed for angiogenic activity, as shown in Figure 6E. In vivo experiments showed that lentiviral-miR-4306 CM induced a weaker angiogenic response in a chick chorioallantoic membrane (CAM) assay than its counterpart.
Both pools of cells were separately injected into the tail veins of nude mice. After two months, the lung metastases were analyzed. Six of ten control mice suffered from lung metastases; in contrast, one of nine lenti-miR-4306 mice suffered from lung metastases (Figure 6F).
The effect of miR-4306 on lymphatic metastasis in TNBC was investigated in vivo using a popliteal lymph node metastasis model. Both pools of cells were inoculated into the footpads of nude mice. The resulting foot pad tumors and popliteal lymph nodes were enucleated and analyzed after 8 weeks. As shown in Figure 6G, the lymph node tumors formed from miR-4306 mimic cells had smaller volumes than those formed from vector control tumors. Strikingly, the ratios of metastatic to total dissected popliteal lymph nodes were markedly lower in the CAL-51-mimic group (14.2% (1/7)) than in the vector control group (100% (7/7)). Taken together, these results indicated that miR-4306 inhibits lymph node metastasis in TNBC in vivo.
miR-4306 combined with cisplatin treatment inhibits TNBC tumor growth in vitro and in vivo
Cisplatin has been used for the treatment of numerous human cancers, including breast cancer. Its mechanism of action has been linked to its ability to crosslink with purine bases in DNA; this action interferes with DNA repair mechanisms to cause DNA damage and subsequently induce apoptosis in cancer cells [16]. After TNBC cells were transfected with miR-4306-mimic, SIX1/Cdc42 siRNA or control, we treated the cells with cisplatin. Then, the cells were stained with FITC-labeled Annexin V and propidium iodide (PI). Flow cytometry analyses indicated that after cisplatin treatment, the number of apoptotic cells transfected with miR-4306 mimic or SIX1/Cdc42 siRNA was substantially greater than the number of apoptotic control cells (Figure S6A). As shown in Figure S6B, higher levels of cleaved-caspase3, cleaved-caspase8, cleaved-caspase9 and cleaved- PARP were detected in miR-4306 mimic-transfected and cisplatin-treated TNBC cells than in cisplatin- only-treated TNBC cells. These results suggested that miR-4306 could promote apoptosis induced by cisplatin in vitro.
We next evaluated the therapeutic potential of a cholesterol-conjugated miR-4306 mimic in an orthotopic mouse model of TNBC (Figure 7A). After six weeks of drug delivery, the tumor sizes were significantly smaller in the control agomir-treated animals receiving cisplatin injections than in those receiving intraperitoneal PBS injections; however, the tumor sizes were much smaller in the miR-4306 agomir-treated mice than in the other two groups (Figure 7B-7C). Consistent with the in vitro data, miR-4306 agomir-treated tumors had lower SIX1, Cdc42, and VEGFA expression levels and higher P21 expression levels than control-treated tumors (Figure 7D-7E). Furthermore, we found that miR-4306 agomir-treated tumors had fewer PCNA-positive cells, higher cleaved caspase 3 expression levels and more apoptosis than control tumors (Figure 7E). These results not only confirmed the tumor suppressor activity of miR-4306 in vivo but also suggested that miR-4306 overexpression combined with cisplatin may represent a novel approach for treating TNBC. Figure 7F presents the proposed mechanism of ectopic miR-4306 expression in TNBC.
Enhanced miR-4306 expression by cholesterol-conjugated delivery promotes TNBC apoptosis induced by cisplatin in vivo. (A) CAL-51 cells were injected into the fat pads of female nude mice. Three weeks after cell injection (tumor volume: 5 mm × 5 mm × 5 mm), the mice were randomly assigned to one of six groups to receive intraperitoneal injections of PBS, intraperitoneal injections of cisplatin (10 mg/kg), intratumoral injections of agomir control (10 nM) or miR-4306 agomir (10 nM), intraperitoneal injections of cisplatin (10 mg/kg), and intratumoral injections of agomir control (10 nM) or miR-4306 agomir (10 nM) every three days (each group n=10). (B) Illustration of the treatment timeline. (C) Representative tumor mass after the indicated treatments. (D) Western blot analysis showing apoptosis-related protein levels in the harvested tumors after different treatments. (E) Representative IHC staining in CAL-51 xenografts with the indicated treatments. ***P < 0.001; two-tailed unpaired Student's t-test. (F) Model of miR-4306-Cdc42/SIX1/VEGFA pathways in regulating TNBC metastasis and progression.
Discussion
ER-α, PR and HER2 play key roles in breast cancer initiation and progress. However, the effect of missing expression of the three factors on TNBC is not yet clear. In this study, we demonstrated for the first time that loss of the three factors contributes to the downregulation of miR-4306 in TNBC. The systematic studies reveal the mechanism and clinical relevance of the aberrant expression of miR-4306. Ultimately, miR-4306 could be classified as a metastasis- suppressing gene and a therapeutic target for TNBC.
There are two major mechanisms that are often considered critical for silencing miRNA expression: genetic (CNV deletion) and epigenetic (aberrant DNA methylation, histone modifications and transcription factor deregulation) [17]. To explore the other mechanisms of ectopic miR-4306 downregulation in TNBC, we acquired somatic copy number alterations from TCGA and analyzed the somatic copy number aberrations (SCNAs) in breast cancer. A total of 978 breast cancer tumor specimens were analyzed. The data revealed that the frequency of miR-4306 copy number loss was remarkably high (21.9%) in breast cancer in both non-TNBC (25%) and TNBC (21.05%) specimens (Figure S1J-S1K). However, there was no significant difference between non-TNBC and TNBC specimens in terms of the CNV level. Moreover, the promoter region of miR-4306 is not located in a typical CpG island, suggesting little possible involvement of DNA methylation in miR-4306 regulation. The TCGA data and the miR-4306 promoter indicate copy number loss and aberrant DNA methylation are not the dominant upstream regulatory mechanism of miR-4306 in TNBC.
ER-α and PR are nuclear receptors whose physiological effects are mediated by hormone interactions to bind to promoters of the target genes to increase gene transcription [18]. Here, we found that ERE, PRE in the miR-4306 promoter, ER-α and PR could transcriptionally activate miR-4306 expression. HER2 is a member of the EGF receptor tyrosine kinase family and has been rigorously studied as a signaling molecule of the cell membrane. We used two small molecules exclude tyrosine kinase function of membrane receptor HER2 on miR-4306 expression. Recent studies have reported that HER2 is also expressed in the nucleus and could form a complex at a specific nucleotide sequence of the gene promoter to stimulate its transcription [19, 20]. Therefore, it is not unexpected that HER2 could directly bind to the promoter of miR-4306 gene and elevate miR-4306 expression. In addition, STAT3 is a transcriptional factor that is activated by nuclear HER2 [21, 22]. We did not find putative STAT3-binding sites in the miR-4306 promoter, so the possibility that miR-4306 is transcriptionally regulated by STAT3 is low.
Furthermore, we simultaneously knocked down ER-α, PR and HER2 in ZR-75-1 cells to mimic the condition in TNBC. The results revealed that miR-4306 transcription and expression levels were changed more obviously with simultaneous knockdown than with individual knockdown of the three transcription factors. These data indicated that miR-4306 downregulation results from the loss of ER-α, HER2 and PR in TNBC. However, due to the sophisticated molecular typing of breast cancer, it is hard to be sure whether these three factors work on the regulation of miR-4306 simultaneously, rather than one of them or two of them in other subtypes except TNBC.
In this study, we demonstrated that Cdc42, SIX1 and VEGFA are downstream targets and effectors of miR-4306. However, the re-expression of Cdc42/SIX1/VEGFA could significantly but not completely rescue the miR-4306-mediated inhibition of the aggressive phenotype. This finding suggests that other potential targets of miR-4306 may exist in TNBC.
SIX1 is a transcription factor for cyclin D1 and cyclin A and is commonly deregulated in multiple types of malignancies [23, 24]. Cdc42 functions as an oncogene in many cancers [25, 26] and plays a key role in regulating multiple cellular processes, such as cell cycle progression, migration, cell cytoskeleton organization, cell fate determination and differentiation [27, 28]. The cell cycle is mainly regulated by a series of cyclins, cyclin-dependent kinases (CDKs) and cyclin-dependent kinase inhibitors (CDKIs) [29]. P21 is one of the best-characterized CDKIs and inhibits the activity of the cyclin D1-CDK4 complex; this action leads to cell cycle arrest at the G1-S phase [30]. The Cdc42/Rac1 signaling pathway activates the proteasomal degradation of P21 (CIP1) [31]. In our study, we found that miR-4306 overexpression in TNBC cells increased P21 levels but reduced cyclin D1 and cyclin A levels. These data are consistent with the results of Cdc42/SIX1 knockdown. Our results demonstrated that miR-4306 mediated cell cycle arrest (at the G1-S phase) and inhibited proliferation via modulating Cdc42/SIX1.
Previous studies have reported that SIX1 facilitates tumor metastasis through a variety of molecular mechanisms [32-34]. SIX1 can activate ERK phosphorylation [35, 36] and suppress CDH1 (encodes E-cadherin) expression by targeting the transcriptional repressors of CDH1 [37]. SIX1 overexpression induces lymphangiogenesis via the transcriptional upregulation of VEGFC [38]. Cdc42 activation inhibits the ubiquitination and degradation of EGFR; these actions result in cellular transformation due to EGFR accumulation and sustained EGF-stimulated ERK activation [39]. ERK activation causes cancer metastasis by inducing MMPs [40]. Cdc42 activation contributes to the mesenchyme-like phenotype by targeting E-cadherin for lysosomal degradation [41]. Upregulation of VEGFA is involved in tumor angiogenesis and metastasis [42]. In our study, the downstream effectors of the miR-4306-Cdc42/SIX1 signaling axis revealed that cdc42 downregulation by the miR-4306 mimic weakened cell metastasis by upregulating E-cadherin and blocking EGFR-ERK-MMPs in TNBC cells. SIX1 downregulation by miR-4306 mimic treatment impairs lymphangiogenesis through decreasing VEGFC levels in TNBC cells. The miR-4306-VEGFA signaling axis indicates that miR-4306 suppresses metastasis by inhibiting angiogenesis.
miRNA-based therapeutics represent a promising approach for cancer therapy [43]. Recent years, numerous preclinical studies have been conducted with miRNA therapeutics. The miRNAs therapeutics has some changlleges for miRNA delivery vehicles, tissue-specific targeting, off-target effects and potential toxicities [44, 45]. MRX34 is a liposome-formulated double-stranded mimic of tumor suppressor miR-34, which is lost or expressed at reduced levels in many solid and hematologic malignancies. A multicenter phase I clinical trial of MRX34 is being conducted in patients with unresectable solid tumors. MRX34 treatment showed evidence of antitumor activity in a subset of patients with refractory advanced solid tumors. However, owing to immune-related adverse events involving patient deaths, the trial was terminated [45, 46].
Estrogen is of paramount importance in breast cancer. As a result, hormone therapy (inhibiting estrogen signaling or estrogen production) is a common clinical treatment for ER-α-positive breast cancer [47]. In addition, Herceptin is a humanized antibody to HER2 and has been shown to have activity as a single agent against HER2-positive breast cancer [48]. However, patients with TNBC do not have the opportunity to receive hormone therapy or Herceptin targeted therapy. Thus, there is no preferred standard form of chemotherapy for TNBC. Initial findings suggest that the neoadjuvant use of cisplatin results in high complete pathological response rates in patients with breast cancer who have BRCA1 mutations, and patients with TNBC frequently have BRCA1 mutations [49, 50]. Excitingly, miR-4306 could indeed promote apoptosis induced by cisplatin in vivo and in vitro. For these reasons, a miR-4306 mimic combined with cisplatin treatment in TNBC may represent a promising targeted therapy with high specificity and limited toxicity.
Abbreviations
CAM: chicken embryo chorioallantoic membrane; CDKs: cyclin-dependent kinases; ChIP: chromatin immunoprecipitation; DMEM: dulbecco's modified eagle's medium; ERK: extracellular signal-regulated kinase; ER-α: estrogen receptor alpha; FBS: fetal bovine serum; HER2: human epidermal growth factor receptor 2; HDLECs: dermal lymphatic endothelial cells; HUVEC: human umbilical vein endothelial cells; MMP: matrix metalloprotease; PI: propidium iodide; PR: progesterone receptor; qPCR: quantitative real-time PCR; RIP: rna immunoprecipitation; TNBC: triple-negative breast cancer.
Acknowledgements
This work was supported by funding from the National Key Research and Development Program of China (no. 2016YFA0500303), the National Natural Science Foundation of China (no. 21335007, 81472661, 81490753, 81230047 and 81672743), the CAMS Innovation Fund for Medical Sciences (CIFMS; grant no. 2016-I2M-1-001), a grant from the Medical Epigenetics Research Center, Chinese Medical Sciences (2018PT31015), and the fund (2016YFC0904600) from the Chinese Ministry of Science and Technology.
Author contributions
Conception and design: Yongmei Song and Qimin Zhan; Development of methodology: Zitong Zhao, Liying Ma, Leilei Zheng and Weimin Zhang; Acquisition of data (provision of animals, acquisition and management of patients, provision of facilities, etc.): Bo Lan, Bailin Zhang, Fei Ma; Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): Lin Li and Peina Du; Writing, review, and/or revision of the manuscript: Yongmei Song and Bo Xu; Study supervision: Yongmei Song and Qimin Zhan.
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363:1938-48
2. Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13:674-90
3. Perez EA, Moreno-Aspitia A, Aubrey Thompson E, Andorfer CA. Adjuvant therapy of triple negative breast cancer. Breast Cancer Res Treat. 2010;120:285-91
4. Yates LA, Norbury CJ, Gilbert RJ. The long and short of microRNA. Cell. 2013;153:516-9
5. Edmonds MD, Boyd KL, Moyo T, Mitra R, Duszynski R, Arrate MP. et al. MicroRNA-31 initiates lung tumorigenesis and promotes mutant KRAS-driven lung cancer. J Clin Invest. 2016;126:349-64
6. Hou J, Lin L, Zhou W, Wang Z, Ding G, Dong Q. et al. Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR-199a/b-3p as therapeutic target for hepatocellular carcinoma. Cancer cell. 2011;19:232-43
7. Van Roosbroeck K, Fanini F, Setoyama T, Ivan C, Rodriguez-Aguayo C, Fuentes-Mattei E. et al. Combining Anti-Mir-155 with Chemotherapy for the Treatment of Lung Cancers. Clin Cancer Res. 2017;23:2891-904
8. Kastrati I, Canestrari E, Frasor J. PHLDA1 expression is controlled by an estrogen receptor-NFkappaB-miR-181 regulatory loop and is essential for formation of ER+ mammospheres. Oncogene. 2015;34:2309-16
9. Wang H, Lee EW, Zhou L, Leung PC, Ross DD, Unadkat JD. et al. Progesterone receptor (PR) isoforms PRA and PRB differentially regulate expression of the breast cancer resistance protein in human placental choriocarcinoma BeWo cells. Mol Pharmacol. 2008;73:845-54
10. Gianni L, Eiermann W, Semiglazov V, Lluch A, Tjulandin S, Zambetti M. et al. Neoadjuvant and adjuvant trastuzumab in patients with HER2-positive locally advanced breast cancer (NOAH): follow-up of a randomised controlled superiority trial with a parallel HER2-negative cohort. Lancet Oncol. 2014;15:640-7
11. Cordo Russo RI, Beguelin W, Diaz Flaque MC, Proietti CJ, Venturutti L, Galigniana N. et al. Targeting ErbB-2 nuclear localization and function inhibits breast cancer growth and overcomes trastuzumab resistance. Oncogene. 2015;34:3413-28
12. Wang SC, Lien HC, Xia W, Chen IF, Lo HW, Wang Z. et al. Binding at and transactivation of the COX-2 promoter by nuclear tyrosine kinase receptor ErbB-2. Cancer cell. 2004;6:251-61
13. Song Y, Li L, Ou Y, Gao Z, Li E, Li X. et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature. 2014;509:91-5
14. Yang F, Zhang W, Li D, Zhan Q. Gadd45a suppresses tumor angiogenesis via inhibition of the mTOR/STAT3 protein pathway. J Biol Chem. 2013;288:6552-60
15. Wang Y, Liu Z, Yao B, Li Q, Wang L, Wang C. et al. Long non-coding RNA CASC2 suppresses epithelial-mesenchymal transition of hepatocellular carcinoma cells through CASC2/miR-367/FBXW7 axis. Mol Cancer. 2017;16:123
16. Hu XC, Zhang J, Xu BH, Cai L, Ragaz J, Wang ZH. et al. Cisplatin plus gemcitabine versus paclitaxel plus gemcitabine as first-line therapy for metastatic triple-negative breast cancer (CBCSG006): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2015;16:436-46
17. Cao J, Song Y, Bi N, Shen J, Liu W, Fan J. et al. DNA methylation-mediated repression of miR-886-3p predicts poor outcome of human small cell lung cancer. Cancer Res. 2013;73:3326-35
18. Badve SS, Baehner FL, Gray RP, Childs BH, Maddala T, Liu ML. et al. Estrogen- and progesterone-receptor status in ECOG 2197: comparison of immunohistochemistry by local and central laboratories and quantitative reverse transcription polymerase chain reaction by central laboratory. J Clin Oncol. 2008;26:2473-81
19. Giri DK, Ali-Seyed M, Li LY, Lee DF, Ling P, Bartholomeusz G. et al. Endosomal transport of ErbB-2: mechanism for nuclear entry of the cell surface receptor. Mol Cell Biol. 2005;25:11005-18
20. Beguelin W, Diaz Flaque MC, Proietti CJ, Cayrol F, Rivas MA, Tkach M. et al. Progesterone receptor induces ErbB-2 nuclear translocation to promote breast cancer growth via a novel transcriptional effect: ErbB-2 function as a coactivator of Stat3. Mol Cell Biol. 2010;30:5456-72
21. Proietti CJ, Rosemblit C, Beguelin W, Rivas MA, Diaz Flaque MC, Charreau EH. et al. Activation of Stat3 by heregulin/ErbB-2 through the co-option of progesterone receptor signaling drives breast cancer growth. Mol Cell Biol. 2009;29:1249-65
22. Venturutti L, Romero LV, Urtreger AJ, Chervo MF, Cordo Russo RI, Mercogliano MF. et al. Stat3 regulates ErbB-2 expression and co-opts ErbB-2 nuclear function to induce miR-21 expression, PDCD4 downregulation and breast cancer metastasis. Oncogene. 2016;35:2208-22
23. Li Z, Tian T, Lv F, Chang Y, Wang X, Zhang L. et al. Six1 promotes proliferation of pancreatic cancer cells via upregulation of cyclin D1 expression. PLoS One. 2013;8:e59203
24. Coletta RD, Christensen K, Reichenberger KJ, Lamb J, Micomonaco D, Huang L. et al. The Six1 homeoprotein stimulates tumorigenesis by reactivation of cyclin A1. Proc Natl Acad Sci U S A. 2004;101:6478-83
25. He Y, Northey JJ, Pelletier A, Kos Z, Meunier L, Haibe-Kains B. et al. The Cdc42/Rac1 regulator CdGAP is a novel E-cadherin transcriptional co-repressor with Zeb2 in breast cancer. Oncogene. 2017;36:3490-503
26. Masuda-Robens JM, Kutney SN, Qi H, Chou MM. The TRE17 oncogene encodes a component of a novel effector pathway for Rho GTPases Cdc42 and Rac1 and stimulates actin remodeling. Mol Cell Biol. 2003;23:2151-61
27. Chrysanthou E, Gorringe KL, Joseph C, Craze M, Nolan CC, Diez-Rodriguez M. et al. Phenotypic characterisation of breast cancer: the role of CDC42. Breast Cancer Res Treat. 2017;164:317-25
28. Lin CW, Sun MS, Liao MY, Chung CH, Chi YH, Chiou LT. et al. Podocalyxin-like 1 promotes invadopodia formation and metastasis through activation of Rac1/Cdc42/cortactin signaling in breast cancer cells. Carcinogenesis. 2014;35:2425-35
29. Shi X, Ma M, Lin S. Cell Cycle-Dependent Expression Dynamics of G1/S Specific Cyclin, Cellulose Synthase and Cellulase in the Dinoflagellate Prorocentrum donghaiense. Front Microbiol. 2017;8:1118
30. Huang GL, Zhang W, Ren HY, Zhou P, Chen Y, Chen QX. et al. Oncogenic retinoic acid receptor alpha promotes human colorectal cancer growth through simultaneously regulating p21 transcription and GSK3beta/beta-catenin signaling. Cancer Lett. 2017;388:118-29
31. Kreis P, Thevenot E, Rousseau V, Boda B, Muller D, Barnier JV. The p21-activated kinase 3 implicated in mental retardation regulates spine morphogenesis through a Cdc42-dependent pathway. J Biol Chem. 2007;282:21497-506
32. Xu H, Zhang Y, Pena MM, Pirisi L, Creek KE. Six1 promotes colorectal cancer growth and metastasis by stimulating angiogenesis and recruiting tumor-associated macrophages. Carcinogenesis. 2017;38:281-92
33. Patrick AN, Cabrera JH, Smith AL, Chen XS, Ford HL, Zhao R. Structure-function analyses of the human SIX1-EYA2 complex reveal insights into metastasis and BOR syndrome. Nat Struct Mol Biol. 2013;20:447-53
34. McCarthy N. Metastasis: SIX1 of the best. Nat Rev Cancer. 2012;12:316
35. Xin X, Li Y, Yang X. SIX1 is overexpressed in endometrial carcinoma and promotes the malignant behavior of cancer cells through ERK and AKT signaling. Oncol Lett. 2016;12:3435-40
36. Yang J, Li G, Zhang K. Pro-survival effects by NF-kappaB, Akt and ERK(1/2) and anti-apoptosis actions by Six1 disrupt apoptotic functions of TRAIL-Dr4/5 pathway in ovarian cancer. Biomed Pharmacother. 2016;84:1078-87
37. Xu H, Zhang Y, Altomare D, Pena MM, Wan F, Pirisi L. et al. Six1 promotes epithelial-mesenchymal transition and malignant conversion in human papillomavirus type 16-immortalized human keratinocytes. Carcinogenesis. 2014;35:1379-88
38. Wang CA, Jedlicka P, Patrick AN, Micalizzi DS, Lemmer KC, Deitsch E. et al. SIX1 induces lymphangiogenesis and metastasis via upregulation of VEGF-C in mouse models of breast cancer. J Clin Invest. 2012;122:1895-906
39. Schmidt MH, Husnjak K, Szymkiewicz I, Haglund K, Dikic I. Cbl escapes Cdc42-mediated inhibition by downregulation of the adaptor molecule betaPix. Oncogene. 2006;25:3071-8
40. Lagares-Tena L, Garcia-Monclus S, Lopez-Alemany R, Almacellas-Rabaiget O, Huertas-Martinez J, Sainz-Jaspeado M. et al. Caveolin-1 promotes Ewing sarcoma metastasis regulating MMP-9 expression through MAPK/ERK pathway. Oncotarget. 2016;7:56889-903
41. Shen Y, Hirsch DS, Sasiela CA, Wu WJ. Cdc42 regulates E-cadherin ubiquitination and degradation through an epidermal growth factor receptor to Src-mediated pathway. J Biol Chem. 2008;283:5127-37
42. English WR, Lunt SJ, Fisher M, Lefley DV, Dhingra M, Lee YC. et al. Differential Expression of VEGFA Isoforms Regulates Metastasis and Response to Anti-VEGFA Therapy in Sarcoma. Cancer Res. 2017;77:2633-46
43. Sicard F, Gayral M, Lulka H, Buscail L, Cordelier P. Targeting miR-21 for the therapy of pancreatic cancer. Mol Ther. 2013;21:986-94
44. Li Z, Rana TM. Therapeutic targeting of microRNAs: current status and future challenges. Nat Rev Drug Discov. 2014;13:622-38
45. Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203-22
46. Beg MS, Brenner AJ, Sachdev J, Borad M, Kang YK, Stoudemire J. et al. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest New Drugs. 2017;35:180-8
47. Fournier A, Mesrine S, Boutron-Ruault MC, Clavel-Chapelon F. Estrogen-progestagen menopausal hormone therapy and breast cancer: does delay from menopause onset to treatment initiation influence risks? J Clin Oncol. 2009;27:5138-43
48. Arpino G, Gutierrez C, Weiss H, Rimawi M, Massarweh S, Bharwani L. et al. Treatment of human epidermal growth factor receptor 2-overexpressing breast cancer xenografts with multiagent HER-targeted therapy. J Natl Cancer Inst. 2007;99:694-705
49. Telli ML, Jensen KC, Vinayak S, Kurian AW, Lipson JA, Flaherty PJ. et al. Phase II Study of Gemcitabine, Carboplatin, and Iniparib As Neoadjuvant Therapy for Triple-Negative and BRCA1/2 Mutation-Associated Breast Cancer With Assessment of a Tumor-Based Measure of Genomic Instability: PrECOG 0105. J Clin Oncol. 2015;33:1895-901
50. Andres R, Pajares I, Balmana J, Llort G, Ramon YCT, Chirivella I. et al. Association of BRCA1 germline mutations in young onset triple-negative breast cancer (TNBC). Clin Transl Oncol. 2014;16:280-4
Author contact
![]() Corresponding authors: Yongmei Song, email: symlh2006com or songymac.cn and Qimin Zhan, email: qiminzhan163.com or zhanqiminedu.cn
Corresponding authors: Yongmei Song, email: symlh2006com or songymac.cn and Qimin Zhan, email: qiminzhan163.com or zhanqiminedu.cn