Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(20):5575-5592. doi:10.7150/thno.27372 This issue Cite
Research Paper
Alkaline Phosphatase Controls Lineage Switching of Mesenchymal Stem Cells by Regulating the LRP6/GSK3β Complex in Hypophosphatasia
Wenjia Liu1,2 *, Liqiang Zhang1,2 *, Kun Xuan1*, Chenghu Hu2, Liya Li2, Yongjie Zhang2, Fang Jin1 ![]() , Yan Jin1,2
, Yan Jin1,2 ![]()
1. MS-State Key Laboratory & National Clinical Research Center for Oral Diseases & Shaanxi International Joint Research Center for Oral Diseases, Center for Tissue Engineering, School of Stomatology, Fourth Military Medical University, Xi'an, 710032, China.
2. Xi'an Institute of Tissue Engineering and Regenerative Medicine, Xi'an, 710032, China.
*W.J.L, L.Q.Z and K.X. made equal contribution.
Received 2018-5-20; Accepted 2018-9-18; Published 2018-11-9
Abstract

Lineage differentiation of bone marrow mesenchymal stem cells (BMMSCs) is the key to bone-fat reciprocity in bone marrow. To date, the regulators of BMMSC lineage switching have all been identified to be transcription factors, and researchers have not determined whether other genes control this process. This study aims to reveal a previously unknown role of tissue-nonspecific alkaline phosphatase (TNSALP) in controlling BMMSC lineage selection.
Methods: We compared the characteristics of cultured BMMSCs from patients with hypophosphatasia (HPP), which is caused by mutations in the liver/bone/kidney alkaline phosphatase (ALPL) gene, and an ALPL knockout (ko) mouse model. We performed ALPL downregulation and overexpression experiments to investigate the regulatory role of ALPL in BMMSC lineage switching. Using the PathScan array, coimmunoprecipitation experiments and pathway-guided small molecule treatments, we explored the possible mechanism underlying the regulatory effects of ALPL on cell differentiation and evaluated its therapeutic effect on ALPL ko mice.
Results: BMMSCs from both patients with HPP and ALPL ko mice exhibited defective lineage differentiation, including a decrease in osteogenic differentiation and a parallel increase in adipogenic differentiation. Mechanistically, TNSALP directly interacted with LRP6 and regulated the phosphorylation of GSK3β, subsequently resulting in lineage switching of BMMSCs. Re-phosphorylation of GSK3β induced by LiCl treatment restored differentiation of BMMSCs and attenuated skeletal deformities in Alpl+/- mice.
Conclusion: Based on our findings, TNSALP acts as a signal regulator to control lineage switching of BMMSCs by regulating the LRP6/GSK3β cascade.
Keywords: TNSALP, MSC lineage switch, LRP6/GSK3β complex, HPP
Introduction
The balance of bone-fat reciprocity in bone marrow (BM) is generally linked to skeletal diseases [1], particularly osteopenia and osteoporosis [2]. At the cellular level, bone marrow mesenchymal stem cells (BMMSCs) are the common progenitors of osteoblasts and adipocytes [3, 4]. Importantly, long-term maintenance of bone homeostasis is mediated by BMMSCs rather than mature osteoblasts, which undergo rapid turnover in vivo [5]. According to studies from our group and other researchers, the impaired lineage differentiation of BMMSCs is probably a major cause of osteopenia/osteoporotic disorders [2, 6, 7].
Several crucial transcription factors have been implicated in controlling the lineage selection of mesenchymal stem cells (MSCs), including peroxisome proliferator activated receptor gamma (PPARγ) and CCAAT/enhancer binding protein, alpha/beta/ delta (CEBPα/β/δ) [8, 9], critical regulators of adipocyte formation, whereas runt-related transcription factor 2 (Runx2) and osterix are required for osteoblast differentiation [10]. Recently, another transcription factor, forkhead box P1 (FOXP1), has been shown to regulate the lineage selection of BMMSCs and attenuate their senescence [7]. Currently, the well-known regulators of osteo-/ adipogenic reciprocity have been identified as transcription factors, and researchers have not determined whether other genes control lineage switching of BMMSCs.
Tissue-nonspecific alkaline phosphatases (TNSALP), which is encoded by the liver/bone/kidney alkaline phosphatase (ALPL) gene [11, 12], is necessary for bone and tooth development and mineralization [13, 14, 15]. Clinically, loss-of-function mutations in the ALPL gene lead to hypophosphatasia (HPP), which manifests as the most broad-ranging severity of all skeletal diseases [16], from stillbirth without mineralized bone to pathological fractures developing later in adulthood as a function of the degree of the TNSALP deficiency [17, 18]. Recently, a clinical trial including 101 patients with HPP patients showed that although the mean Z-score for bone mineral density (BMD) was nearly normal, a considerable percentage of patients had lower spine and hip BMD values (Z-scores ≤ -2) compared to the normal group. Specifically, extremely low BMD values are observed in severe childhood or infantile cases of HPP [16]. Supplementation with bone-targeted TNSALP successfully increases bone mass in homozygous Alpl knockout (Alpl-/-) mice [19] and children with life-threatening HPP, and even extends the lives of affected individuals [20]. Based on the results from these clinical studies, the TNSALP level may affect the bone mass and structure, but evidence showing how TNSALP regulates the skeletal phenotype is lacking.
The dysfunction of osteoblasts caused by a TNSALP deficiency was the main cause of demineralization in subjects with HPP [21]. However, according to a recent study, targeted deletion of Alpl in osteoblasts (Col1a1-Cre) and mesenchymal cells (Prx1-Cre, used to delete genes from mesenchymal progenitor cells) both phenocopy the skeletal and dental deformities of HPP [22]. Notably, BMMSCs isolated from patients with HPP exhibit extremely low ALP activity and diminished osteogenic differentiation [23]. Since TNSALP is expressed at high levels on the BMMSC membrane [4, 24], we hypothesize that TNSALP may regulate the function of BMMSCs and subsequently affect bone characteristics [4, 24].
We revealed that TNSALP controlled bone-fat reciprocity mainly by regulating lineage switching of BMMSCs from patients with HPP and Alpl+/- knockout mice. Specifically, TNSALP regulated the phosphorylation of GSK3β by binding to LRP6, which subsequently affected the canonical Wnt/β-catenin pathway and governed BMMSC lineage selection. Reactivation of the phosphorylation of GSK3β by the small molecule LiCl not only rescued the function of BMMSCs from patients with HPP but also ameliorated skeletal deformities in Alpl+/- mice.
Methods
Human subjects
Two patients with HPP (male), aged 8 and 13 years, respectively, were treated at the Affiliated Hospital of the Fourth Military Medical University for osteodynia and tooth loss. Healthy human BM samples were collected from five teenagers aged 8 to 17 years who were undergoing alveolar bone cleft repair through an auto-ilium transplantation. All clinical data are presented in Table 1 and Table S1.
Characteristics of the HPP patients.
| Characteristic | Patient 1 | Patient 2 |
|---|---|---|
| Sex | Male | Male |
| Age (year) | 8 | 13 |
| ALPL mutation | c.551G>A, p.R184Q | c.407G>A, p.R136H; c.551 G>A, p.R184Q |
| Exon | 6 | 5&6 |
| Serum ALP (Normal range) (U/L) | 76 (79-267) | 12 (40-150) |
| Plasma PLP | + | ++ |
| T value (Osteopenia, -1<T<-2.5) | -1.82 | -2.05 |
The clinical study was approved by the Ethics Committee of the Affiliated Hospital of the Fourth Military Medical University, and written informed consent was obtained from all participants prior to blood collection.
DNA sequencing
The total blood genomic DNA was extracted from peripheral blood samples from the patients using a QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA, USA). Primer sequences targeting ALPL exons (1-12) are listed in Table S2. PCR was performed using a PCR Amplification Analysis Instrument (BBI, Grand Forks, ND, USA). The PCR fragments of 12 exons were gel purified with an E.Z.N.A.® Gel Extraction Kit (Omega, Bethesda, MD, USA) according to the manufacturer's protocol. DNA sequencing analyses were performed using an ABI Prism 3730 DNA analyzer sequencer (ABI, Framingham, MA, USA).
Alkaline phosphatase activity assay
Serum and intracellular alkaline phosphatase (ALP) activities were estimated using a kit from NJJCBIO company (NJJCBIO, Nanjing, China) according to the manufacturer's instructions. ALP activity was ultimately reported as micromoles of p-nitrophenol release.
Serum ALP activity was calculated using the formula: ALP activity (U/L) = A / V / T, where A is the amount of pNP generated by samples (in μmol), V is the volume of sample added to the assay well [59], and T is the reaction time (in min).
ALP activity in cells was calculated using the formula: ALP activity (U/g) = A / V / T, where A is the amount of pNP generated by samples (in μmol), V is volume of sample added to the assay well (in g), and T is the reaction time (in min).
Bone alkaline phosphatase activity assay
Bone-specific ALP (BALP) activity in serum was examined using an ELISA kit (FSSYBIO, Shanghai, China) according to the manufacturer's instructions. Briefly, the human serum samples were incubated with a purified human/mouse BALP antibody to coat wells of the microtiter plate for 0.5 h at 37 °C; then, an HRP-labeled goat anti-human/mouse BALP antibody was added and incubated for 0.5 h at 37 °C to form the antibody-antigen-enzyme antibody complex. Next, the TMB substrate was added and incubated for 10 min at 37 °C, and the absorbance was then measured at 450 nm using a spectrophotometer. The concentration of human BALP in serum was then determined by comparing the OD value of the samples to the standard curve.
Enzyme-linked immunosorbent assay (ELISA)
We analyzed serum samples using human PINP and CTX-1 ELISA kits (BOSTER, Wuhan, China). All ELISA assays were performed according to the manufacturer's instructions.
Isolation of human BMMSCs
Cells were purified from the BM using the Percoll density gradient centrifugation method and cultured in α-MEM supplemented with 10% FBS (Gibco BRL), 2 mM L-glutamine (Invitrogen), 100 U/mL penicillin, and 100 mg/mL streptomycin (Invitrogen) at 37 °C in 5% CO2. Only third-passage BMMSCs were subjected to induction of osteogenic and adipogenic differentiation. The results of the surface markers are shown in Figure S1.
Immunofluorescence staining
Bone slices and BMMSCs that had been seeded in a 24-well plate for 24 h were collected for staining. After washes with PBS and fixation with 4% paraformaldehyde for 15 min, bone samples were incubated with a phospho-GSK-3β (Ser9) antibody (Cell Signaling Technology, Boston, MA, USA, 9323). BMMSC samples were incubated with TNSALP (Abcam, ab65834; R&D Systems, AF2910), CD146 (Abcam, ab24577) and active β-catenin (anti-ABC) (Millipore, Billerica, MA, USA, 05-665) antibodies for 2 h and subsequently incubated with the appropriate secondary antibodies. The positively stained cells were examined under a laser scanning confocal microscope (Olympus FluoView FV 1000, Tokyo, Japan).
Flow cytometry analysis
Approximately 5×105 human BMMSCs were incubated with TNSALP (R&D Systems, FAB1448P), STRO-1-Phycoerythrin (Abcam, ab190282), CD105-PE (Biolegend, 323206), CD90/Thy1-PE (eBioscience, 12-0909), CD34-PE (Biolegend, 343506), CD45-PE (eBioscience, 304008), or CD 146-PE (eBioscience, 12-1469) antibodies for 30 min on ice. Samples were analyzed using a fluorescence-activated cell sorting (FACS) Aria flow cytometer (BD Bioscience) and FACS DIVE software version 6.1.3 (BD Biosciences).
Osteogenic and adipogenic differentiation assays
Human and mouse BMMSCs were incubated with osteogenic medium (human cells: 100 nM dexamethasone, 50 μg/mL ascorbic acid, and 5 mM β-glycerophosphate; mouse cells: 100 nM dexamethasone, 50 μg/mL ascorbic acid, and 1 mM β-glycerophosphate) (Sigma) for 3 and 4 weeks, respectively, according to the manufacturer's instructions. Cells were fixed with 60% isopropanol and stained with 1% alizarin red (Sigma) to assess osteogenic differentiation. Runx2 and OCN levels were determined by Western blotting.
Human and mouse BMMSCs were cultured with adipogenic medium (0.5 mM methylisobutylxanthine, 0.5 mM hydrocortisone, and 60 mM indomethacin; Sigma) for 2 weeks. Intracellular lipid accumulation was detected by staining with an Oil Red O solution. PPARγ levels were examined by Western blotting.
In vivo bone formation assay
After 3 days of culture, approximately 5×106 BMMSCs were mixed with 40 mg of hydroxyapatite/ tricalcium phosphate (HA/TCP) ceramic particles (40 mg, Sigma) and implanted into subcutaneous pockets on the backs of 8-week-old immunocompromised mice. The control group was implanted into the other side of the same host. The implants were removed 8 weeks after transplantation, fixed with 4% paraformaldehyde, and decalcified with buffered 10% EDTA (pH 7.0). For the histological analysis, sections were stained with hematoxylin and eosin (HE) or Masson's trichrome (BaSO Diagnostic, Inc., Guangdong, China) according to the manufacturer's instructions. Sections were analyzed using Image-Pro Plus software. Five fields were selected and the area of newly formed mineralized tissue in each field was calculated and reported as a percentage of the total tissue area.
Western blot analysis
BMMSCs were harvested in RIPA lysis buffer (Beyotime Co., Shanghai, China, P0013B). After the concentrations of whole cell protein extracts were quantified using the BCA assay, extracts were separated on NuPAGE 10-12% polyacrylamide gels, transferred to PVDF membranes (Millipore, Billerica, MA, USA), and blocked with 5% BSA in TBST for 1 h before an overnight incubation with primary antibodies diluted in blocking solution. The primary antibodies included β-actin (Abcam, ab179467), TNSALP (Abcam, ab65834), Runx2 (Cell Signaling Technology, 12556), OCN (Abcam, ab93876), PPAR-γ (Abcam, ab19481), OPG (Santa Cruz Biotechnology, sc-11383), RANKL (Santa Cruz Biotechnology, sc-52950), ERK1/2 (Cell Signaling, 9102S), phospho-ERK1/2 (Thr202/Tyr204) (Cell Signaling Technology, 9101S), AMPKα (Cell Signaling Technology, 2532), phospho-AMPKα (Thr172) (Cell Signaling Technology, 2531), GSK-3β (Abcam, ab93926), phospho-GSK-3β (Ser9) (Cell Signaling Technology, 9323), β-catenin (Abcam, ab32572), active β-catenin (anti-ABC) (Millipore, 05-665), LRP5 (Abcam, ab38311), LRP6 (Abcam, ab134146), p-LRP6 (Cell Signaling Technology, 2568), FZD1 (R&D Systems, MAB1120-SP), FZD2 (Proteintech, 24272-1-AP), FZD3 (GeneTex, GTX100182), FZD4 (R&D Systems, MAB194-SP), FZD5 (Proteintech, 21519-1-AP), FZD6 (R&D Systems, MAB1120-SP), FZD7 (Proteintech, 16974-1-AP), FZD8 (Proteintech, 55093-1-AP), FZD9 (Proteintech, 13865-1-AP), and FZD10 (Proteintech, 18175-1-AP). β-actin was used as a loading control. Signals were revealed after incubation with a peroxidase-conjugated secondary antibody (Cwbiobiotech, Beijing, China) using ECL.
Lentiviral vector construction and transduction
ALPL was amplified from human genomic DNA (293T cells) by PCR to construct a lentiviral vector expressing human ALPL. The PCR product was digested with the restriction enzymes Age I and EcoR I and inserted into the pLko.1 vector (Invitrogen) or digested with Kpn I and EcoR V and inserted into the pLenti 6.3/v5-DEST vector (Invitrogen). The inserted fragments were verified by Sanger sequencing. A lentiviral construct containing the scrambled ALPL sequence was used as a negative control. The lentivirus was produced by co-transfecting 293T cells with the transfer vector and two packaging vectors (i.e., psPAX2 and pMD2.G). The virus was subsequently purified by ultracentrifugation. MSCs were plated in 6-well plates and transduced with lentiviral constructs and 5 µg/mL polybrene (Sigma, 107689). Primers used to construct the ALPL lentiviral vector are listed in Table S3.
The GSK3β shRNA plasmid for downregulating the expression of GSK3β was purchased from Santa Cruz Biotechnology (sc-35527-SH). The GSK3β shRNA lentiviral particles were collected from the culture medium of 293T cells 48 h after they had been transfected with GSK3β shRNA plasmid, psPax plasmid and pMD2G plasmid. The virus was subsequently purified by ultracentrifugation. For the downregulation of GSK3β expression in BMMSCs, the GSK3β shRNA lentiviral constructs and 5 µg/mL polybrene (Sigma, 107689) were added to the cell culture medium and replaced with normal cell culture medium after 24 h.
Transfection assay
Small interfering RNA (siRNA) duplex oligonucleotides targeting human LRP6 were also obtained from Santa Cruz Biotechnology. Non-targeting control siRNAs (Santa Cruz Biotechnology) were used as negative controls. These siRNAs were transfected into cells at a final concentration of 50 nM using siPORT NeoFX (Invitrogen). The medium was replaced 8 h after transfection.
Analysis of the phosphorylation of intracellular signaling intermediates
We examined the phosphorylation of intracellular signaling intermediates using a PathScan® array kit (Cell Signaling Technology, Danvers, MA, USA, #7323), which detects the levels of the indicated cellular proteins and signaling nodes only when they are phosphorylated or cleaved at the specified residues. We prepared cell lysates of human MSCs and then performed all assays according to the manufacturer's instructions.
RNA isolation and transcription-polymerase chain reaction
Total RNA was isolated from cells using RNAiso plus (TaKaRa, Tokyo, Japan, 9109) according to the manufacturer's instructions. The mRNA samples were reverse transcribed into cDNAs and real-time RT-PCR was performed using PrimescriptTM RT master mix (TaKaRa, RR036A) and SYBR Premix Ex TaqTMⅡ (TaKaRa, RR820A), respectively. A CFX96 Trademark teal-time PCR detection system (Bio-Rad, Richmond, CA, USA) was used for RT-PCR. Expression levels of ERK, AMPK, GSK3β, Axin2 and CCND1 (TaKaRa) were examined.
Co-immunoprecipitation (Co-IP)
Cells were lysed in RIPA lysis buffer supplemented with a cocktail of protease inhibitors (Roche, Switzerland, 04693132001) and PMSF. Briefly, the proteins were incubated with primary antibodies against TNSALP (1 μg) (Abcam, ab108337) or mIgG (1 μg) at 4 °C for at least 12 h to form antibody-antigen complexes, followed by incubation with Protein G magnetic beads (Millipore) at 4 °C for 3 h. Immune complexes were washed at least 4 times with PBS containing 0.1% Tween 20 and the precipitated proteins were eluted with ice-cold lysis buffer for Western blotting. The beads were removed from the lysis buffer mixture and antibodies against the predicted target proteins Lrp6, GSK3β (Cell Signaling Technology, 12456) and β-catenin (Abcam, ab32572) overnight. The target proteins were detected by SDS-PAGE followed by Western blotting, as described above.
Luciferase reporter assays
A TOPFlash luciferase reporter plasmid (Millipore, USA) containing the TCF/LEF consensus sequence was used to assay β-catenin-dependent promoter activity, and a FOPFlash reporter plasmid (Millipore, USA) containing mutated TCF binding sites was used as a control. Cells in six-well plates were transfected with TOPFlash and FOPFlash plasmids with Fugene transfection reagent (Roche, IN, USA). Cells were cotransfected with an internal control reporter Renilla luciferase plasmid (pRL-TK; Promega, WI, USA) to normalize the transfection efficiency. The luciferase assay was performed after 24 h using the Dual Luciferase Assay System kit, according to the manufacturer's protocol. The relative luciferase activities of TOPFlash and FOPFlash are presented as fold induction after normalization for transfection efficiency.
Inorganic pyrophosphate (PPi) measurements
Extracellular and intracellular PPi concentrations were measured with a spectrophotometric method using EnzChek® Pyrophosphate Detection Kit (Molecular Probes, Madison, USA, E-6645), according to the manufacturer's protocol. Briefly, we collected the culture medium or cell lysates, then added 10 μL samples to a final 100 μL reaction mixture in 96-well plates. The reaction mixture was incubated for another 30-60 min at 22 °C, and the absorbance was recorded at 360 nm. A Pi-free laboratory must be used when collecting samples.
TNSALP mimic treatment
We treated BMMSCs from patients with HPP with 1 U/mL TNSALP mimics from porcine kidney (Sigma, P4439) to examine the effect of TNSALP on the lineage selection of BMMSCs. Then, we cultured cells in osteogenic or adipogenic induction medium for 2 or 4 weeks, respectively, and collected cells for staining and Western blot analyses.
Wnt3a and LiCl treatments in vitro
A total of 3×105 BMMSCs were plated in each well of a 6-well plate and cultured. After the cells reached 70-80% confluence, they were cultured in osteogenic/adipogenic differentiation medium containing 25 ng/mL recombinant Wnt3a (R&D Systems, 5036) or 10 mM LiCl (Sigma, 213233) as described above, and the medium was changed every 2 days. On days 7 and 14, we harvested the cells and subjected them to in vitro osteogenic/adipogenic differentiation assays.
Mice
Female Alpl+/+ and Alpl+/- mice (B6.129S7-Akp2tm1Sor/J, pure C57BL/6J genetic background) were purchased from Jackson labs (Bar Harbor, ME, USA). Female immunocompromised nude mice (CAnN.Cg-Foxn1nu/CrlVr) were purchased from Vital River Laboratory Animal Technology Co. Ltd. (Beijing, China). All of the procedures that involved animals were approved by the animal use and care committee of the Fourth Military Medical University (license number: SYXK 2012-0023).
Primers used for genotyping include: oIMR0137 - CCG TGC ATC TGC CAG TTT GAG GGG A - Mutant, oIMR0138 - CTG GCA CAA AAG AGT TGG TAA GGC AG - Wild type, oIMR0139 - GAT CGG AAC GTC AAT TAA CGT CAA T - Common.
Microcomputer tomography (μCT) scanning and analysis
Mouse tibias/femora were scanned with the Inveon μCT system (Siemens AG, Germany). Cross-sectional volumetric bone mineral density (BMD) was measured at right tibia mid-diaphysis. Using two-dimensional images, a region of interest in secondary spongiosa was manually drawn near the endocortical surface, and cancellous bone morphometric parameters, including bone mineral density (BMD, mg/cc) and bone volume relative to tissue volume (BV/TV, %) were assessed.
Oil Red O section staining
To assess the fat tissue in trabecular areas, femurs were fixed in 4% paraformaldehyde and decalcified with 5% EDTA (pH 7.4), followed by cryosectioning. 10 μm thick sections were stained with Oil Red O for 10-20 min at room temperature, and positive areas were quantified by microscopy and shown as a percentage of the total area. Then, sections were washed with 60% isopropanol and twice with PBS and observed under a microscope. All reagents for Oil Red O staining were purchased from Sigma-Aldrich.
Colony forming unit (CFU) assay
To assess the CFU efficiency of mouse BMMSCs, primary cultured BMMSCs 1×103 cells/mL were seeded in 5 cm culture dishes (Corning, Lowell, MA, USA). After culturing in proliferation or osteogenic differentiation medium for 2 weeks, the newly formed colonies were visualized with 0.1% crystal violet or Alizarin Red/ALP staining following 4% paraformaldehyde fixation. Aggregates of 50 or more cells were scored as colonies under the microscope. CFU efficiency was determined by the number of colonies relative to the total number of seeded cells in each plate.
Bone histomorphometric analysis
To examine new bone formation rate, mice received a double injection of calcein intraperitoneally (10 mg/kg body weight; Sigma, St. Louis, MO, USA) 10 and 2 days prior to euthanasia. The tibiae and femora were isolated, fixed in 95% ethanol, and embedded in methyl methacrylate. A microtome was used to prepare 50 μm longitudinal sections. Bone dynamic histomorphometric analyses for BFR/BS were performed using Image J image analysis software of fluorescence images (Lecia, Wetzlar, Germany, DMI6000B).
TNSALP/TRAP staining
TNSALP and tartrate-resistant acid phosphatase (TRAP) are known as marker enzymes for osteoblasts and osteoclasts respectively. TNSALP/TNAP staining was performed using the TNSALP/TNAP Stain Kit (Wako, Japan, 294-67001) following the manufacturer's instruction. Sections were washed with distilled water, dried, and observed under a microscope (Leica Microsystems, Heerbrugg, Switzerland).
Isolation of mouse BMMSCs
Mouse BM cells (2-3×107) were flushed out from long bones with 3% FBS in PBS. A single-cell suspension of all nuclear cells was obtained by passing all BM cells through a 70 μm cell strainer (Bioscience, Dümmer, Germany). Then, 2.5×105 cells/cm2 were seeded on 10 cm culture dishes (Corning, Lowell, MA, USA) and incubated at 37 ℃ in 5% CO2. After 48 h, the cultures were washed with PBS to eliminate non-adherent cells. The attached cells were cultured for 10 to 15 days with α-modified essential medium (α-MEM; Gibco BRL, Gaithersburg, MA, USA) supplemented with 20% fetal bovine serum (Gibco BRL), 2 mM L-glutamine, 100 U/mL penicillin, and 100 mg/mL streptomycin (Invitrogen, Gaithersburg, MD, USA). The cell culture protocol and surface markers identification are described in our previous studies [2, 6].
LiCl injection in vivo
LiCl (100 mg/kg body weight dissolved in 15 μL NaCl, Sigma, 213233) was injected into the femur BM cavity of 4-month-old Alpl+/- mice twice per month for one month. The control mice received a comparable volume of NaCl (n = 8 per group).
4-month-old Alpl+/- mice were injected with 100 mg/kg body weight of LiCl by intraperitoneal injection three times per week for 4 weeks. The control mice received a comparable volume of NaCl (n = 8 per group). All mice were harvested at day 30 after injection for analysis.
Statistics
Data are presented as mean ± s.d. Data were assessed for normal distributions and similar variances between groups before further analysis. We used unpaired, two-tailed Student's t-tests for comparisons between two groups and one-way analysis of variance (ANOVA) with Bonferroni's correction for multiple comparisons. All experiments were repeated more than three times, and representative experiments are shown. P values less than 0.05 were considered significant. The sizes of all experimental groups were chosen to ensure adequate statistical power, despite the highly variable nature of the studies performed. No animals or samples were excluded from the analysis, and animals were randomly assigned to groups for the studies.
Results
Patients with HPP present with a loss of bone mass, which is linked to the degree of TNSALP deficiency
According to a previous study, patients with severe cases of childhood and infantile HPP have lower spine and hip BMD Z-scores than normal controls [16]. We examined two patients with osteodynia and tooth loss in the clinic to further investigate the relationship between bone mass and TNSALP. DNA sequencing confirmed that patient 1 carried a p.R184Q (c.551G>A) mutation and patient 2 carried p.R136H (c.407G>A) and p.R184Q (c.551G>A) two mutations (Figure 1A and Table 1) (http://www.sesep.uvsq.fr/database_hypo/Mutation.html). The serum ALP and bone-specific ALP (BALP) activities in these patients were lower than the normal range, and serum pyridoxal 5'-phosphate (PLP) was positive in the two patients with HPP (Figure 1B and Table 1). According to the age of onset, clinical examinations and DNA sequencing, both patients were diagnosed with childhood HPP.
TNSALP deficiency reflecting ALPL leads to bone mass loss in humans. (A) DNA sequence analysis for two HPP patients. (B) Serum BALP activity in normal controls (Normal) and HPP patients (HPP1,2). (C) Radiographs of long bone of normal control and two patients. (D) Enzyme-linked immunosorbent assay (ELISA) analysis revealed PINP and CTX-1 expression in the serum of control and HPP patients. Normal n = 5, HPP n = 2. The data are presented as mean ± standard deviation (s.d.) for triplicate samples from a representative experiment. *P < 0.05, **P < 0.01. One-way analysis of variance (ANOVA).
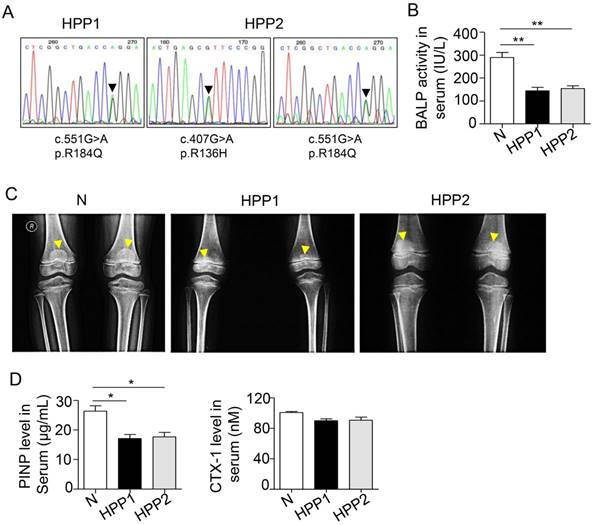
Childhood HPP cases show areas of decreased transradiancy and thin cortical bone, but the whole-body BMD is at the lower end of the normal range according to the Z-score [16, 25]. Another clinical study including 101 patients with HPP showed that although the mean BMD Z-score was nearly normal, a considerable percentage of patients had lower spine and hip BMD values than the normal group [16], particularly patients with severe childhood and infantile HPP. Here, the two patients with HPP both had rachitic changes in the metaphysis (yellow arrows in Figure 1C) and widespread bone mass loss. Additionally, their T scores for the L1-L4 lumbar spine, which are widely used in the clinic to determine BMD, were lower than the normal range (the T scores of patients with HPP were -2.05 and -1.82; -1 < T < -2.5 is diagnosed as osteopenia) (Table 1). Because bone mass is maintained by both bone formation by osteoblasts and bone resorption by osteoclasts, we next evaluated the changes in bone formation and resorption capacities in patients with HPP. A reduced serum PINP level was observed in patients with HPP, indicating the bone formation capacity was reduced due to TNSALP deficiency. In contrast, the serum level of the osteoclast-related marker CTX-1 was not different from the WT controls (Figure 1D), suggesting that osteoclast activity may not be affected in the two patients with HPP. Based on these data, the TNSALP deficiency induces bone mass loss in patients with HPP, thus reflecting the reduced bone formation capacity independent of osteoclast activity.
BMMSCs from patients with HPP show impaired lineage differentiation due to TNSALP deficiency
We isolated BMMSCs from both patients with HPP and normal controls and used them in subsequent experiments designed to examine whether the osteopenia phenotype of patients with HPP is cell autonomous (Figure S1). Both HPP and normal BMMSCs co-expressed TNSALP with CD146 (Figure 2A), and reduced ALP activity and decreased TNSALP expression were observed in HPP BMMSCs compared with normal cells, as evidenced by the ALP activity assay, Western blotting and flow cytometry analyses (Figure 2B-C).
BMMSCs of HPP patients exhibit diminished osteogenic differentiation but preferential adipogenic differentiation. (A) Double immunostaining showed that CD146+ (red) human BMMSCs co-expressed TNSALP (green). Scale bars, 50 μm. (B) TNSALP expression was analyzed by flow cytometry analysis (FCM). (C) The ALP activity and TNSALP expression in normal and HPP BMMSCs were examined using the ALP activity assay and western blotting. (D) Alizarin red staining and quantification of mineralized nodules were performed at day 28 after osteogenic induction (OS). Runx2 and OCN expression levels were examined at day 7 after induction by western blotting. (E) HE/Masson's trichrome staining and quantitative analysis showed formation of bone (B), bone marrow (BM), and collagen fiber (CF) around HA/TCP (HA) carrier when BMMSCs were implanted into nude mice. Scale bars, 200 μm. (F) Oil Red O staining and quantification of fat depots were performed at day 14 after adipogenic induction (AD). PPAR-γ expression was examined at day 7 after induction by western blotting. Scale bars, 100 μm. (G) OPG and RANKL expression levels in BMMSCs were examined by western blotting. Normal control n = 5, HPP n = 2. The data are presented as mean ± s.d. for triplicate samples from a representative experiment. *P < 0.05, **P < 0.01. One-way analysis of variance (ANOVA).
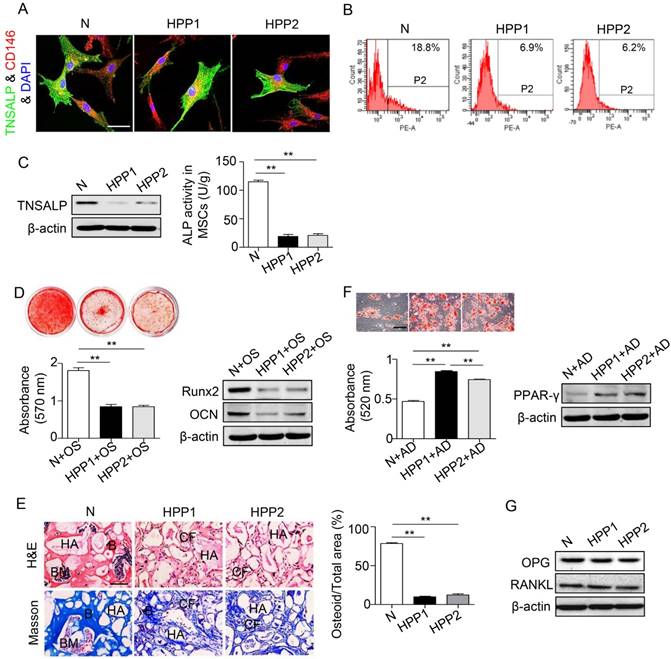
When cultured in induction medium, the BMMSCs from the two patients showed inhibited osteogenesis, including a reduced number of mineralization nodules, decreased expression of Runx2 and OCN in vitro and a reduction in new bone formation capacity in vivo (Figure 2D-E). In contrast, these BMMSCs exhibited preferential adipogenic differentiation, as indicated by Oil Red O staining and PPARγ expression (Figure 2F). In contrast to osteo-/adipogenesis, the expressions of osteoprotegerin (OPG) and nuclear factor kappa B ligand (RANKL) were similar in BMMSCs from patients with HPP and normal controls, as shown by Western blotting assays (Figure 2G). Thus, the TNSALP deficiency impairs lineage differentiation of BMMSCs, but does not affect osteoclastogenesis.
TNSALP controls lineage switching of BMMSCs
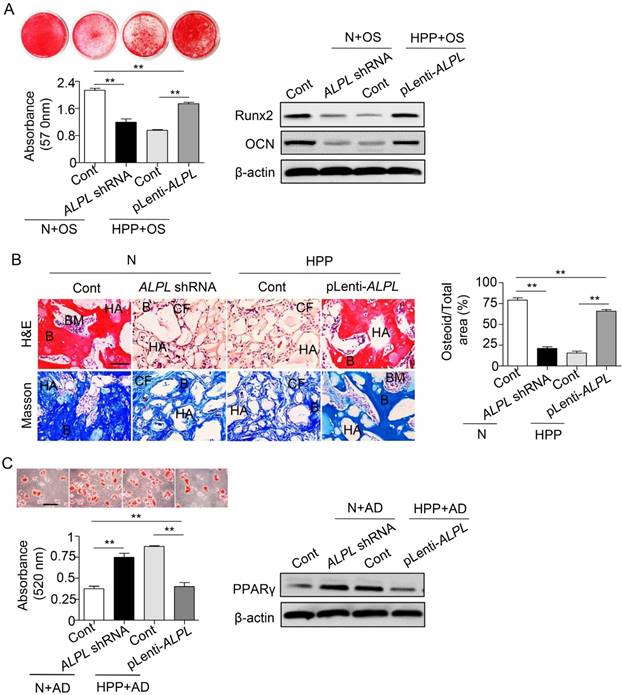
Because the HPP BMMSCs exhibited impaired differentiation due to TNSALP deficiency, we next evaluated the regulatory role of TNSALP in BMMSC differentiation. Downregulation of ALPL in normal BMMSCs impaired their osteogenic differentiation but enhanced adipogenic differentiation, mimicking the characteristics of HPP BMMSCs. However, ALPL overexpression in HPP BMMSCs rescued the lineage differentiation of these cells, as confirmed by alizarin red and Oil Red O staining, Runx2, OCN and PPARγ expression in vitro, and regeneration of new bone in vivo (Figure S2 and Figure 3). Collectively, these data reveal a previously unrecognized role for TNSALP in BMMSC lineage selection that subsequently affects bone-fat reciprocity in postnatal bone. The next question we asked is how TNSALP regulates the lineage differentiation of BMMSCs.
TNSALP controls the lineage differentiation of BMMSCs. (A) Downregulated ALPL expression in normal BMMSCs and upregulated expression in HPP BMMSCs through lentiviral vectors. Alizarin red staining and quantification of mineralized nodules were performed at day 28 after osteogenic induction (OS). Expression of Runx2 and OCN was examined through western blotting at day 7 after induction. (B) HE/Masson's trichrome staining and quantitative analysis revealed formation of bone (B), bone marrow (BM), and collagen fiber (CF) around HA/TCP (HA) carrier when BMMSCs were implanted into nude mice. Scale bars, 200 μm. (C) Oil Red O staining and quantification of fat depots were performed at day 14 after adipogenic induction (AD). PPAR-γ expression was examined at day 7 after induction by western blotting. Scale bars, 100 μm. Normal control n = 5, HPP n = 2. Data are shown as mean ± s.d. of independent experiments performed in triplicate. *P < 0.05, **P < 0.01. One-way analysis of variance (ANOVA).
TNSALP deficiency-induced inactivation of GSK3β/β-catenin signaling contributes to the altered lineage specification of BMMSCs
Because TNSALP is a phosphatase [12], we next explored the signaling mechanisms of TNSALP in determining the lineage differentiation of BMMSCs by examining the phosphorylation of intracellular signaling intermediates in BMMSCs. According to the array data, intermediates in most pathways were hypo-phosphorylated in HPP BMMSCs (Figure 4A), particularly extracellular signal-regulated kinase1/2 (ERK1/2), AMP-activated protein kinase α (AMPKα) and glycogen synthase kinase 3β (GSK3β), which are all closely associated with BMMSC function [26-28]. However, the total protein levels of these enzymes, except for ERK1/2, did not change in HPP BMMSCs (Figure 4B). Furthermore, the mRNA levels of intermediates in the three signaling pathways were not decreased in HPP BMMSCs (Figure 4C).
TNSALP regulates the canonical Wnt pathway via dephosphorylating GSK3β. (A) The phosphorylation of intracellular signaling in normal and HPP BMMSCs was examined by intracellular protein assay. (B) The expression levels of total ERK1/2, p-ERK1/2, total AMPKα, p-AMPKα, total GSK3β and p-GSK3β were analyzed by western blotting. (C) The expression levels of ERK1/2, AMPK and GSK3β were analyzed by qRT-PCR. (D-E) Western blotting (D) and immunostaining (E) for total and active β-catenin in normal and HPP BMMSCs. Scale bars, 50 μm. (F) Downstream genes of canonical Wnt pathway, CCND1 and Axin2 mRNA levels were analyzed by qRT-PCR. (G) Western blotting of p-GSK3β and active β-catenin in normal BMMSCs, normal control shRNA and ALPL shRNA, HPP BMMSCs, HPP pLenti-control and pLenti-ALPL. (H) Activation of the canonical Wnt pathway was examined 48 h after transfection by luciferase assay. (I) Upregulated ALPL expression in HPP BMMSCs and downregulation of GSK3β through lentiviral vectors and siRNA. The expressions of p-GSK3β and active β-catenin were examined by western blotting. (J) Upregulated ALPL expression in HPP BMMSCs and downregulation of GSK3β through lentiviral vectors. Expression of Runx2, OCN and PPARγ was examined through western blotting at day 7 after induction. Normal control n = 5, HPP n = 2. The data are presented as mean ± s.d. for triplicate samples from a representative experiment. *P < 0.05, **P < 0.01. (C, F) One-way analysis of variance (ANOVA). (H) Unpaired two-tailed Student's t-test.
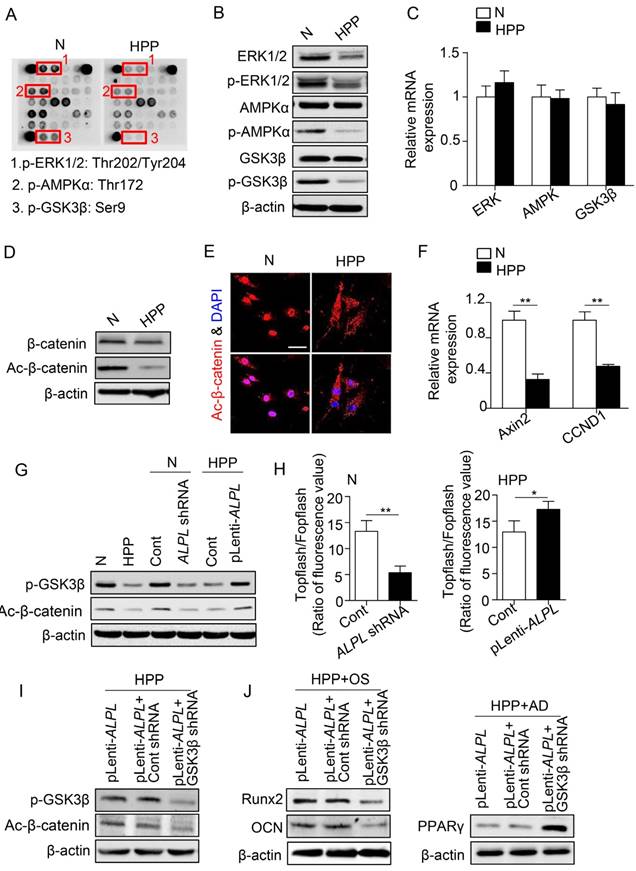
Among the three signaling pathways, GSK3β prevents proteasomal degradation of the transcriptional coactivator β-catenin and thereby inhibits the Wnt/β-catenin pathway [29], which has emerged as a critical regulator of stem cell lineage specification [27, 28]. As expected, lower levels of active β-catenin were detected in HPP BMMSCs, whereas the total β-catenin level was almost the same as that of normal BMMSCs. In addition, active β-catenin mainly accumulated in the nucleus of normal BMMSCs, whereas it was dispersed in the cytoplasm of HPP BMMSCs, as shown by immunostaining. Furthermore, the levels of CCND1 and Axin2 mRNAs, downstream intermediates in the Wnt/β-catenin pathway, were much lower in HPP BMMSCs, suggesting that the Wnt/β-catenin pathway was inactivated in HPP BMMSCs (Figure 4D-F). Furthermore, the GSK3β/β-catenin cascade was activated or inactivated, depending on the level of TNSALP, as indicated by Western blotting and the Topflash assay (Figure 4G-H). More strikingly, upon GSK3β knockdown with a lentiviral vector, ALPL overexpression did not activate the GSK3β/β-catenin cascade and rescue the impaired differentiation of HPP MSCs (Figure 4I-J). Based on these data, TNSALP, which functions as a signal regulator, affects the phosphorylation of GSK3β and promotes β-catenin translocation to the nucleus, thus controlling lineage switching of postnatal BMMSCs.
Direct binding of TNSALP and LRP6 regulates the GSK3β-mediated Wnt/β-catenin pathway
Next, we investigated the mechanism by which TNSALP regulates the phosphorylation of GSK3β. Notably, the immunoprecipitation assay showed that TNSALP did not directly interact with GSK3β or β-catenin (Figure 5A). We therefore inferred that TNSALP may not directly regulate the phosphorylation of intracellular signaling intermediates. Because TNSALP has been reported to hydrolyze PPi to promote osteoblast mineralization [21], we therefore determined the levels of PPi and their effect on the regulation of intracellular proteins. Although the extracellular PPi concentrations varied according to the ALPL level, the intracellular PPi levels were not affected (Figure 5B-C). More importantly, an exogenous PPi treatment did not inhibit the phosphorylation of intermediates in the GSK3β/ β-catenin cascade, suggesting that PPi is not the major cause of hypo-phosphorylation of intracellular proteins caused by TNSALP deficiency (Figure 5D).
Direct binding of TNSALP and LRP6 consequently affects the phosphorylation of GSK3β. (A) Co-immunoprecipitation showed GSK3β-, β-catenin- and TNSALP-binding protein in normal BMMSCs. (B-C) Extracellular and intracellular PPi concentrations were assayed after 48 h of transducing different lentiviral vectors. (D) The expression levels of p-GSK3β and active-β-catenin were analyzed by western blotting after treatment with 10 μM PPi at different time points. (E) The expression levels of frizzled class receptor 1-10 in normal and HPP BMMSCs were analyzed by western blotting. (F) The expression levels of LRP5/6 in normal and HPP BMMSCs were also analyzed by western blotting. (G) Expression levels of FZD2, FZD9, LRP6 and p-LRP6 were assayed after 48 h of transducing different lentiviral vectors by western blotting. (H) Expression levels of p-LRP6, p-GSK3β and active-β-catenin were analyzed by western blotting after treatment with 25 ng/mL Wnt3a. (I) TNSALP immunoprecipitated LRP6 in normal BMMSCs. Left panel shows the expression of LRP6, and right panel shows the level of LRP6 following immunoprecipitation with an anti-TNSALP antibody. Normal n = 5, HPP n = 2 for all groups. The data are presented as mean ± s.d. of independent experiments performed in triplicate. *P < 0.05, **P < 0.01. (B-C) One-way analysis of variance (ANOVA).
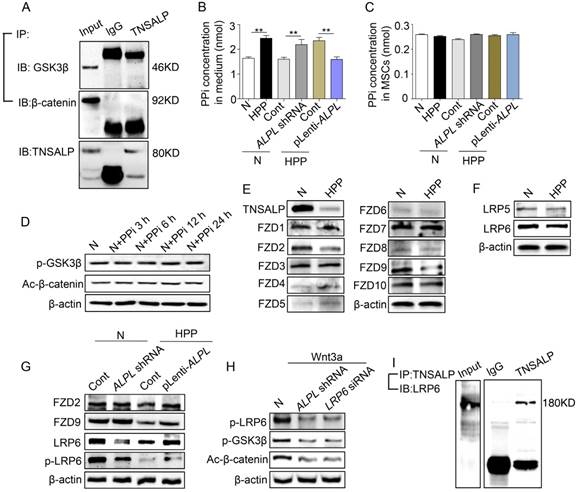
Activation of the canonical Wnt/β-catenin signaling pathway is initiated by the binding of Wnt proteins to two distinct cell surface receptors. One is a member of the Frizzled protein family (FZD), and the other are low-density lipoprotein receptor-related protein (LRP) 5 and LRP6 [30, 31]. The Wnt-FZD-LRP5/6 trimeric complex promotes translocation of β-catenin into the nucleus via phosphorylation of GSK3β [32]. In our study, the levels of FZD2, FZD9 and LRP6 were all decreased in HPP BMMSCs, as indicated by Western blotting (Figure 5E-F). However, only LRP6 expression was proportional to the level of ALPL, as shown by Western blotting (Figure 5G). Since Wnt induces LRP6 phosphorylation [33], we therefore added 25 ng/mL Wnt3a to the culture medium of normal BMMSCs. Wnt3a successfully increased the levels of p-LRP6 and p-GSK3β, and subsequently induced translocation of β-catenin into the nucleus of normal BMMSCs. However, this effect of Wnt3a was partially blocked after the downregulation of ALPL and LRP6 (Figure 5H).
We then further explored the mechanism underlying the membrane localization of TNSALP and LRP6 in normal BMMSCs. Coimmunoprecipitation showed that a control antibody did not precipitate either protein, but an anti-TNSALP antibody coimmunoprecipitated LRP6 (Figure 5I and Figure S4). Therefore, TNSALP directly interacts with LRP6 and subsequently affects the activation of GSK3β-mediated Wnt/β-catenin pathway.
Phosphorylation of GSK3β by LiCl exerts a better therapeutic effect on rescuing lineage differentiation of HPP BMMSCs
As expected, knockdown of GSK3β in normal BMMSCs inhibited the Wnt/β-catenin pathway and then inhibited osteogenic differentiation and enhanced adipogenic differentiation (Figure S3). Next, we evaluated the role of the Wnt/β-catenin pathway in regulating the differentiation of HPP BMMSCs. Wnt3a was chosen as a positive control because it is widely used to activate the Wnt/β-catenin pathway [34]. The small molecule lithium chloride (LiCl), which induces the phosphorylation of GSK3β at Ser9 [35, 36], was also employed. Although both treatments restored the lineage differentiation of HPP BMMSCs, LiCl exerted a much stronger effect, including increased osteogenic differentiation and decreased adipogenic differentiation (Figure 6A-B). Based on these results, inactivation of the GSK3β/β-catenin cascade by TNSALP deficiency may contribute to BMMSC impairments, and LiCl successfully restored their function in vitro.
Phosphorylation of GSK3β by LiCl manifests a stronger effect than TNSALP mimic on rescuing the impaired function of HPP BMMSCs. (A) Alizarin red staining and quantification of mineralized nodules performed at day 28 after osteogenic induction (OS) in Wnt3a-treated (25 ng/mL) and LiCl-treated (10 mM) HPP BMMSCs. Expression levels of Runx2 and OCN were examined by western blotting. (B) Oil Red O staining and quantification of fat depots were performed at day 14 after adipogenic induction. PPAR-γ expression was examined by western blotting. Scale bars, 100 μm. (C) Alizarin red staining and quantification of mineralized nodules performed at day 28 after OS in TNSALP-treated (10 U/mL) and LiCl-treated (10 mM) HPP BMMSCs. Expression levels of Runx2 and OCN were examined through western blotting at day 7 after induction. (D) Oil Red O staining and quantification of fat depots were performed at day 14 after adipogenic induction (AD). PPAR-γ expression was examined at day 7 after induction by western blotting. Scale bars, 100 μm. HPP n = 2 per group. Data shown as mean ± s.d. for triplicate samples from a representative experiment. *P < 0.05. One-way analysis of variance (ANOVA).
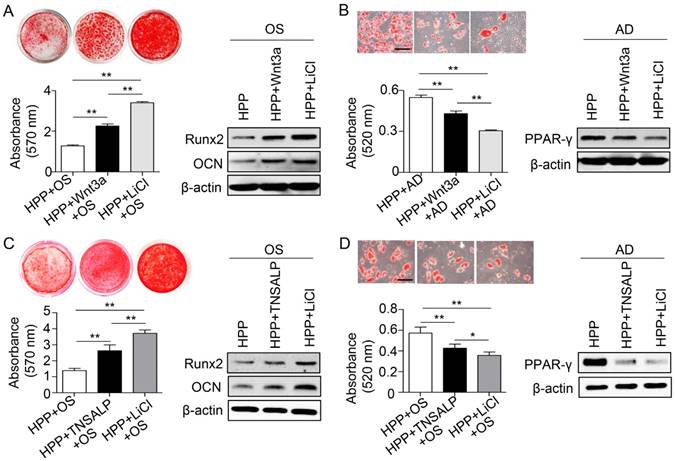
Because enzyme replacement therapy with mineral-targeting TNSAP (sALP-FcD10, also known as ENB-0040 or asfotase alfa) has shown promising results in life-threatening cases of HPP [20], we next compared the therapeutic effects of TNSALP and LiCl on the functional recovery of HPP BMMSCs. Interestingly, LiCl also exhibited a stronger capacity to rescue the function of HPP BMMSCs compared with TNSALP mimics, including improved osteogenic differentiation and diminished adipogenic differentiation (Figure 6C-D). Collectively, the LiCl treatment successfully rescued the impaired differentiation of BMMSCs caused by TNSALP deficiency.
LiCl rescues endogenous BMMSC function and alleviates skeletal deformities in Alpl+/- mice
We used Alpl knockout (C57BL/6J-Alpl+/-) mice to verify whether LiCl exerts a therapeutic effect in vivo. Homozygous Alpl-/- mice exhibit a global deficiency in plasma TNSALP activity and severe skeletal hypomineralization due to impaired and delayed mineralization of osteoblasts [37-41]; however, these mice die at 20 days of age in most cases [42]. Therefore, they are not appropriate for studying the relationship between bone-fat reciprocity and TNSALP. In contrast to homozygous Alpl-/- mice, heterozygous Alpl+/- mice exhibit a normal lifespan, and a nearly 50% reduction in serum ALP activity was observed in Alpl+/- mice compared with their WT counterparts (Figure S5A). According to previous studies, carrier (Alpl+/-) mice do not present significant radiographic evidence of skeletal disease until 3-4 months after birth [38]. In our study, μCT and histological analyses showed that the BMD, bone volume/total volume (BV/TV), and distal femoral trabecular bone structure of 4-month-old Alpl+/- mice were significantly decreased compared with those of WT mice (Figure S5B-C). In contrast, the bone marrow fat content in the femur of Alpl+/- mice was 7-fold higher than that of WT controls (Figure S5D). Moreover, Alpl+/- MSCs had proliferation capacities similar to WT cells, but exhibited a reduced osteogenic colony formation capability (CFU-ALP and CFU-Ob), as shown by colony formation unit analyses (Figure S5E-F). Consistent with the findings obtained from HPP BMMSCs, the Alpl+/- BMMSCs also showed a decrease in osteogenic differentiation and a parallel increase in adipogenic differentiation (data not shown). Furthermore, osteogenesis was significantly decreased in Alpl+/- mice, but osteoclastogenesis was not affected, as indicated by TNSALP/TRAP staining (Figure S5G). Based on these observations, TNSALP deficiency decreases bone mass and concomitantly increases BM adiposity in Alpl+/- mice.
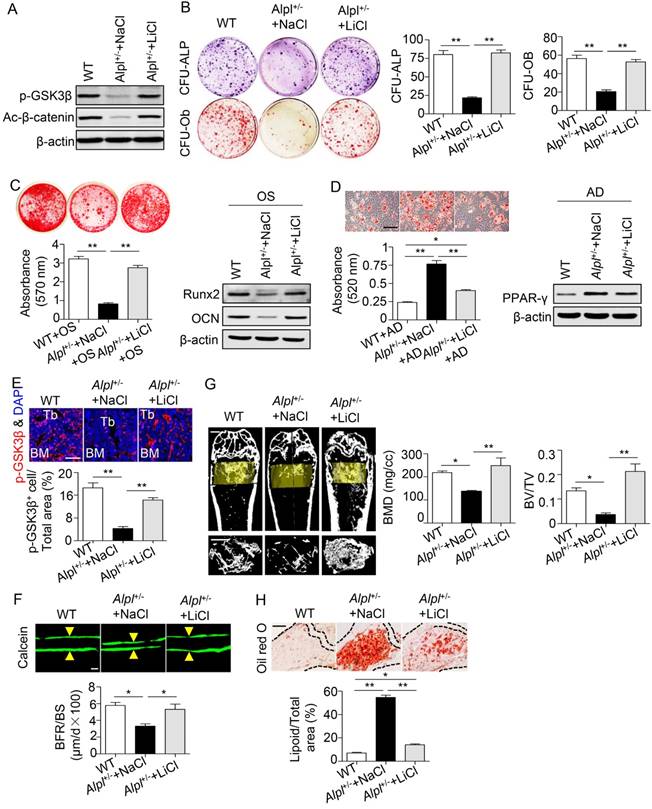
We next injected 20 mg/kg LiCl into the femoral BM cavity of 4-month-old Alpl+/- mice twice per month for one month to confirm the therapeutic effect of LiCl in vivo. The phosphorylation of GSK3β and β-catenin was restored in BMMSCs after the LiCl injection (Figure 7A). Moreover, the osteogenic colony formation of Alpl+/- BMMSCs was elevated approximately 3-fold, similar to that of WT cells (Figure 7B). Moreover, the impaired lineage differentiation of Alpl+/- BMMSCs was also rescued by LiCl, as indicated by enhanced mineralized nodule formation, increased Runx2 and OCN expression, and decreased adipogenic differentiation (Figure 7C-D). Importantly, LiCl also restored the phosphorylation of GSK3β in the BM of Alpl+/- mice, thereby promoting new bone formation, as assessed by bone formation rate/bone surface (BFR/BS) using double calcein labeling (Figure 7E-F). Thus, the skeletal deformities of Alpl+/- mice were rescued, including a 1.8-fold increase in BMD and a 5-fold increase in BV/TV compared with the NaCl group (Figure 7G). Furthermore, the bone marrow fat content of Alpl+/- mice was reduced approximately 74% after the LiCl injection (Figure 7H).
Recovery of GSK3β phosphorylation rescues impaired BMMSC function and skeletal deformities in Alpl+/- mice. We injected LiCl into the femoral BM cavity of 4-month-old Alpl+/- mice every two weeks at 20 mg/kg for 1 month (total 2 injections) and NaCl was used as a control. (A) Expression levels of p-GSK3β and active-β-catenin in BMMSCs were analyzed by western blotting. (B) CFU-ALP (top panels) and CFU-Ob (bottom panels) assays of harvested mice BMMSCs; quantitative analyses, right panel. (C) Alizarin red staining and quantification of mineralized nodules were performed at day 21 after osteogenic induction in BMMSCs of WT and Alpl+/- mice injected with NaCl and LiCl. Expression levels of Runx2 and OCN were examined by western blotting at day 7 after induction. (D) Oil Red O staining and quantification of fat depots were performed at day 14 after adipogenic induction. Scale bars, 100 μm. PPAR-γ expression was examined by western blotting. (E) Immunostaining analysis of p-GSK3β (red) and nuclear staining (blue, DAPI) of femoral diaphysis. Quantification of GSK3β+ cells is indicated in the bottom panel. Scale bars, 200 μm. (F) Images of calcein double labeling of trabecular (TB) with quantification of BFR/BS. Scale bars, 50 μm. (G) μCT images and quantification of BMD and BV/TV. Scale bars, 1 mm. (H) Oil Red O staining images and quantitative analysis of the area of adipose tissue over the total area of the proximal femoral diaphysis. Scale bars, 500 μm. n = 8 for all groups. All experiments were performed in three independent experiments. The data are presented as mean ± s.d. *P < 0.05, **P < 0.01. One-way analysis of variance (ANOVA).
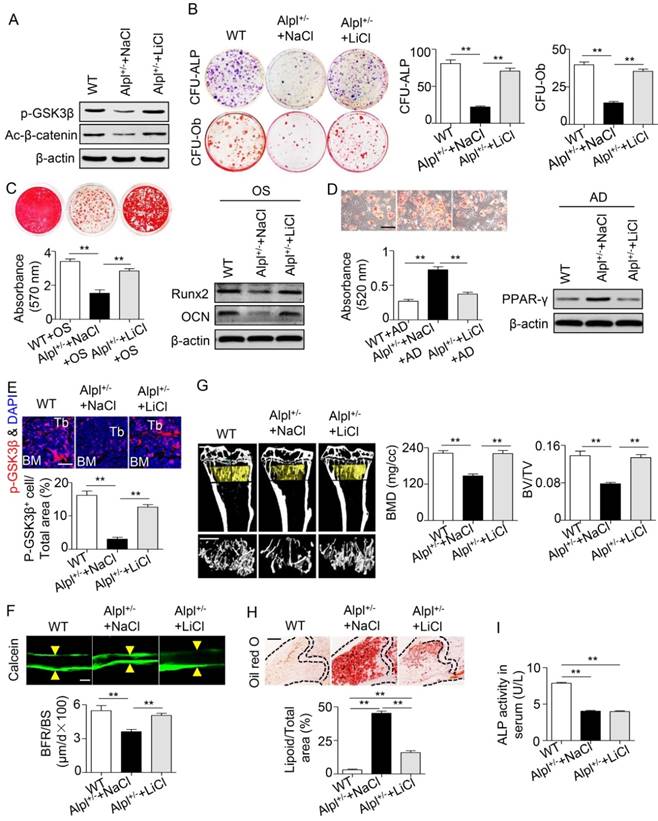
Based on the results of the intra-BM injection, we intraperitoneally injected 100 mg/kg LiCl into Alpl+/- mice every other day for 28 days (a total of 14 injections), and NaCl was used as a control. Systemic injections also rescued BMMSC function and skeletal deformities in Alpl+/- mice, similar to local injections (Figure 8A-H). The most interesting finding is that although the skeletal deformities were reversed after LiCl administration, the serum ALP activity remained unchanged (Figure 8I). Thus, TNSALP deficiency lead to dephosphorylation of intermediates in the GSK3β/β-catenin cascade, which subsequently caused defects in BMMSC differentiation. Either intra-BM injections or systemic administration of LiCl successfully alleviated the skeletal deformities by restoring the lineage differentiation of BMMSCs.
Systemic LiCl injection also rescues skeletal deformities in Alpl+/- mice. We intraperitoneally injected LiCl into 4-month-old Alpl+/- mice every other day at 100 mg/kg for 28 days (total 14 injections) and NaCl was used as control. (A) Expression levels of p-GSK3β and active-β-catenin in BMMSCs were analyzed by western blotting. (B) CFU-ALP (top panels) and CFU-Ob (bottom panels) assays of harvested BMMSCs; quantitative analyses, right panel. (C) Alizarin red staining and quantification of mineralized nodules were performed at day 21 after osteogenic induction in MSCs of Alpl+/+ and Alpl+/- mice injected NaCl and LiCl. Expression levels of Runx2 and OCN were examined by western blotting at day 7 after induction. (D) Oil Red O staining and quantification of fat depots were performed at day 14 after adipogenic induction. PPAR-γ expression was examined by western blotting. Scale bars, 100 μm. (E) Immunostaining analysis of p-GSK3β (red) and nuclear staining (blue, DAPI) of femoral diaphysis. Quantification of GSK3β+ cells is indicated in the bottom panel. Scale bars, 200 μm. (F) Images of calcein double labeling of trabecular (TB) with quantification of bone formation rate (BFR/BS). Scale bars, 50 μm. (G) μCT images and quantification of bone mineral density (BMD) and trabecular bone fraction (BV/TV). Scale bars, 1 mm. (H) Oil Red O staining images and quantitative analysis of the area of adipose tissue over the total area of the proximal femoral diaphysis. Scale bars, 500 μm. n = 8 for all groups. All experiments were performed in three independent experiments. The data are presented as mean ± s.d. *P < 0.05, **P < 0.01. One-way analysis of variance (ANOVA).
Discussion
Since its first description by Suzuki and colleagues in 1907 [43], ALP has been investigated continuously and extensively. We propose here that in addition to the traditional transcription factors, the phosphohydrolase TNSALP controls lineage switching of BMMSCs and subsequently regulates bone-fat reciprocity in BM. Specifically, TNSALP deficiency causes dephosphorylation of GSK3β, prevents β-catenin translocation to the nucleus and inactivates the canonical Wnt pathway, which is the main factor responsible for the lineage specification of BMMSCs. Pharmacological findings reveal that the restoration of GSK3β phosphorylation induced by LiCl treatment restores the functions of impaired resident BMMSCs and then alleviates skeletal deformities in Alpl+/- mice. Collectively, we have revealed previously unrecognized roles for TNSALP in the BMMSC lineage switch and the maintenance of bone homeostasis and provided experimental evidence that LiCl treatment may be an effective therapy for aberrant bone-fat reciprocity due to TNSALP deficiency (Figure S6).
TNSALP deficiency has been shown to induce substantial extracellular accumulation of PPi in osteoblasts and subsequent inhibition of matrix mineralization [21]. However, long-term maintenance of the skeletal phenotype is mediated by BMMSCs rather than mature osteoblasts [5]. BMMSCs isolated from patients with HPP exhibit extremely low ALP activity and do not produce a mineralized bone matrix, even under osteogenic culture conditions [23], indicating that the osteogenic differentiation of BMMSCs was impaired due to the TNSALP deficiency. In the present study, TNSALP-deficient BMMSCs not only displayed decreased osteogenic differentiation but also showed a preference for adipogenic differentiation. Currently, the regulators of lineage reciprocity of BMMSCs have all been identified as transcription factors [7-9], and researchers have not determined whether other genes control the lineage differentiation of BMMSCs. TNSALP has already been proposed to ensure that ES cells are maintained in an undifferentiated state [44, 45], suggesting that TNSALP plays one or more functions in these undifferentiated cells and/or during their multipotential differentiation [46]. In the present study, downregulation of ALPL in normal BMMSCs decreased osteogenic differentiation but increased adipogenic differentiation, whereas the lineage differentiation of HPP BMMSCs was rescued by ALPL overexpression, indicating that the differentiation properties of BMMSCs were regulated by the TNSALP level. Because TNSALP is located in the membrane of BMMSCs, we revealed that TNSALP controls lineage switching of postnatal BMMSCs, and does not merely function as an osteoblastic marker [2, 47].
Currently, four ALP isozymes have been identified, including germ cell ALP (GCALP) encoded by the ALPP2 gene, placental ALP (PALP) encoded by the ALPP gene, intestinal ALP (IALP) encoded by the ALPI gene, and TNSALP encoded by the ALPL gene [48, 49]. All four isozymes are plasma membrane-bound glycoproteins [50, 51]. To date, TNSALP has been implicated in the extracellular hydrolysis of substrates, including phosphoethanolamine, pyridoxal 5ʹ-phosphate (PLP) and PPi, as well as the dephosphorylation of extracellular proteins [52]. However, researchers have not clearly determined whether TNSALP induces a series of intracellular changes. ALP is a downstream intermediate in the canonical Wnt pathway [53-56]. In the present study, TNSALP deficiency directly induced dephosphorylation of GSK3β and prevented β-catenin translocation to the nucleus and its interactions with members of the T-cell factor (Tcf)/lymphoid enhancer factor (Lef) family of transcription factors, which subsequently inhibited the canonical Wnt pathway. Furthermore, re-phosphorylation of GSK3β by LiCl not only rescued the function of HPP BMMSCs but also alleviated skeletal deformities in Alpl+/- mice, probably by restoring lineage differentiation of endogenous BMMSCs. Thus, we have preliminarily shown that TNSALP functions to regulate the phosphorylation of intermediates in the GSK3β/ β-catenin cascade, thereby promoting lineage switching of BMMSCs.
Bone-targeted TNSALP (asfotase alfa) successfully increases bone mass in children with life-threatening HPP, particularly by decreasing the plasma PPi and PLP levels and increasing serum levels of PTH and dietary calcium supplements [20]. Here, re-phosphorylation of intermediates in the GSK3β/β-catenin cascade via mechanism-guided LiCl treatments, which have been proven to increase bone formation [57] and decrease the risk of potential fractures [58] in both mice and humans, exhibited a stronger capacity to rescue the function of HPP BMMSCs than TNSALP mimics. We postulate that the differences may be because the TNSALP mimics used here were extracted from the porcine kidney and were not the fusion protein containing the catalytic domain of human TNSALP, like asfotase alfa. Furthermore, although the extracellular substrate PPi was hydrolyzed by TNSALP, the impaired signaling pathways in BMMSCs were still incompletely restored, which resulted in a weaker therapeutic effect on the recovery of BMMSCs function in the present study. Based on our results, either local administration or systemic injection of LiCl rescued the impairments of resident BMMSCs and subsequently alleviated skeletal deformities in Alpl+/- mice. The intra-BM injection might exert a stronger effect on restoring bone mass, as shown by the elevated BMD and BV/TV in the LiCl-injected group compared with the WT group. Collectively, we propose that the pathway-guided LiCl treatment may represent an effective therapeutic treatment for the aberrant bone-fat reciprocity caused by TNSALP deficiency.
Although the present study focused on alterations in the lineage specification of BMMSCs, we were not able to exclude the possibility that local or systemic LiCl injections also affected other types of cells in Alpl+/- mice. Indeed, the ratio of Treg/Th17 cells was low, and the levels of some pro-inflammatory cytokines, such as interleukin (IL)-1β, IL-6, and IL-17, were elevated in patients with HPP and Alpl+/- mice (data not shown). Additional experiments might be required to examine whether LiCl exerts therapeutic benefits on immune cells. However, bone-fat reciprocity is primarily maintained by BMMSCs. As shown in our previous study, the recovery of resident BMMSC function is an effective therapeutic method for rescuing bone mass loss in subjects with lupus [6]. Thus, the effect of LiCl on attenuating the skeletal deformities in Alpl+/- mice likely primarily reflects the functional recovery of resident BMMSCs. Based on these results, TNSALP controls lineage switching of BMMSCs by regulating the GSK3β/β-catenin cascade. Finally, a pathway-guided LiCl treatment is a potential therapeutic treatment for abnormal bone-fat reciprocity.
Abbreviations
BMMSCs: bone marrow mesenchymal stem cells; TNSALP: tissue-nonspecific alkaline phosphatase; HPP: hypophosphatasia; ALPL: liver/bone/ kidney alkaline phosphatase gene; BM: bone marrow; PPAR-γ: peroxisome proliferator-activated receptor gamma; CEBPα/β/δ: CCAAT/enhancer binding protein, alpha/beta/delta; Runx2: runt-related transcription factor 2; FOXP1: forkhead box P1; ALP: alkaline phosphatase; BALP: bone-specific ALP; PLP: pyridoxal 5'-phosphate; PINP: N-terminal propeptide of type I procollagen; CTX-I: C-terminal telopeptide of type I collagen; OPG: osteoprotegerin; RANKL: nuclear factor kappa B ligand; ERK1/2: extracellular signal-regulated kinase1/2; AMPKα: AMP-activated protein kinase α; GSK3β: glycogen synthase kinase 3β; PPi: inorganic pyrophosphate; FZD: Frizzled protein family; LRP 5/6: low-density lipoprotein receptor-related protein 5/6; H&E: hematoxylin and eosin; FBS: fetal bovine serum; BMD: bone mineral density; BV/TV: bone volume/total volume; BFR/BS: bone formation rate/bone surface.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was financially supported through grants from the Nature Science Foundation of China (81620108007), the National key Research and Development Program of China (2016YFC1101400), and the Nature Science Foundation of China (31571532, 31601099 and 81670947).
Author contributions
JWL conceived the ideas for experimental designs, conducted most of the experiments, analyzed the data and prepared the manuscript. QLZ and KX collected the HPP patients and normal controls, maintained mice, collected tissue samples, performed microcomputation tomography analyses, conducted immunohistochemical and immunofluorescence analyses, cell culture and induction analysis, western blot experiments, and assisted with data analysis. HCH and LYL assisted with cell culture and flow cytometry and manuscript preparation. YJZ contributed to the experimental design and assisted in drafting the manuscript. FJ and YJ developed the concept, supervised the project, conceived the experiments and revised the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Singh L, Brennan TA, Russell E, Kim JH, Chen QJ, Johnson FB. et al. Aging alters bone-fat reciprocity by shifting in vivo mesenchymal precursor cell fate towards an adipogenic lineage. Bone. 2016;85:29-36
2. Liu Y, Yang R, Liu X, Zhou Y, Qu C, Kikuiri T. et al. Hydrogen sulfide maintains mesenchymal stem cell function and bone homeostasis via regulation of Ca(2+) channel sulfhydration. Cell Stem Cell. 2014;15:66-78
3. Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD. et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143-7
4. Bianco P, Cao X, Frenette PS, Mao JJ, Robey PG, Simmons PJ. et al. The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat Med. 2013;19:35-42
5. Park D, Spencer JA, Koh BI, Kobayashi T, Fujisaki J, Clemens TL. et al. Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell. 2012;10:259-72
6. Liu S, Liu D, Chen C, Hamamura K, Moshaverinia A, Yang R. et al. MSC Transplantation Improves Osteopenia via Epigenetic Regulation of Notch Signaling in Lupus. Cell Metab. 2015;22:606-18
7. Li HJ, Liu P, Xu SQ, Li YH, Dekker JD, Li BJ. et al. FOXP1 controls mesenchymal stem cell commitment and senescence during skeletal aging. J Clin Invest. 2017;127:1241-53
8. Farmer SR. Transcriptional control of adipocyte formation. Cell metabolism. 2006;4:263-73
9. Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747-54
10. Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR. et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17-29
11. Van Hoof VO, De Broe ME. Interpretation and clinical significance of alkaline phosphatase isoenzyme patterns. Crit Rev Clin Lab Sci. 1994;31:197-293
12. Coleman JE, Gettins P. Alkaline phosphatase, solution structure, and mechanism. Adv Enzymol Relat Areas Mol Biol. 1983;55:381-452
13. Hahnel AC, Rappolee DA, Millan JL, Manes T, Ziomek CA, Theodosiou NG. et al. Two alkaline phosphatase genes are expressed during early development in the mouse embryo. Development. 1990;110:555-64
14. MacGregor GR, Zambrowicz BP, Soriano P. Tissue non-specific alkaline phosphatase is expressed in both embryonic and extraembryonic lineages during mouse embryogenesis but is not required for migration of primordial germ cells. Development. 1995;121:1487-96
15. Buchet R, Millan JL, Magne D. Multisystemic functions of alkaline phosphatases. Methods Mol Biol. 2013;1053:27-51
16. Whyte MP, Wenkert D, Zhang F. Hypophosphatasia: Natural history study of 101 affected children investigated at one research center. Bone. 2016;93:125-38
17. Whyte MP. Hypophosphatasia and the role of alkaline phosphatase in skeletal mineralization. Endocr Rev. 1994;15:439-61
18. Mornet E. Hypophosphatasia. Best Pract Res Clin Rheumatol. 2008;22:113-27
19. Yadav MC, Lemire I, Leonard P, Boileau G, Blond L, Beliveau M. et al. Dose response of bone-targeted enzyme replacement for murine hypophosphatasia. Bone. 2011;49:250-6
20. Whyte MP, Greenberg CR, Salman NJ, Bober MB, McAlister WH, Wenkert D. et al. Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med. 2012;366:904-13
21. Wennberg C, Hessle L, Lundberg P, Mauro S, Narisawa S, Lerner UH. et al. Functional characterization of osteoblasts and osteoclasts from alkaline phosphatase knockout mice. J Bone Miner Res. 2000;15:1879-88
22. Foster BL, Kuss P, Yadav MC, Kolli TN, Narisawa S, Lukashova L. et al. Conditional alpl ablation phenocopies dental defects of hypophosphatasia. J Dent Res. 2017;96:81-91
23. Katsube Y, Kotobuki N, Tadokoro M, Kanai R, Taketani T, Yamaguchi S. et al. Restoration of cellular function of mesenchymal stem cells from a hypophosphatasia patient. Gene Ther. 2010;17:494-502
24. Sacchetti B, Funari A, Michienzi S, Di Cesare S, Piersanti S, Saggio I. et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. 2007;131:324-36
25. Girschick HJ, Schneider P, Kruse K, Huppertz HI. Bone metabolism and bone mineral density in childhood hypophosphatasia. Bone. 1999;25:361-7
26. Chen H, Liu X, Chen H, Cao J, Zhang L, Hu X. et al. Role of SIRT1 and AMPK in mesenchymal stem cells differentiation. Ageing Res Rev. 2014;13:55-64
27. Krishnan V, Bryant HU, Macdougald OA. Regulation of bone mass by Wnt signaling. J Clin Invest. 2006;116:1202-9
28. Ross SE, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL. et al. Inhibition of adipogenesis by Wnt signaling. Science. 2000;289:950-3
29. Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y. et al. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837-47
30. Wehrli M, Dougan ST, Caldwell K, O'Keefe L, Schwartz S, Vaizel-Ohayon D. et al. arrow encodes an LDL-receptor-related protein essential for Wingless signalling. Nature. 2000;407:527-30
31. Tamai K, Semenov M, Kato Y, Spokony R, Liu C, Katsuyama Y. et al. LDL-receptor-related proteins in Wnt signal transduction. Nature. 2000;407:530-5
32. Bodine PV. Wnt signaling control of bone cell apoptosis. Cell Res. 2008;18:248-53
33. Tamai K, Zeng X, Liu C, Zhang X, Harada Y, Chang Z. et al. A mechanism for Wnt coreceptor activation. Mol Cell. 2004;13:149-56
34. Ling L, Dombrowski C, Foong KM, Haupt LM, Stein GS, Nurcombe V. et al. Synergism between Wnt3a and heparin enhances osteogenesis via a phosphoinositide 3-kinase/Akt/RUNX2 pathway. J Biol Chem. 2010;285:26233-44
35. Hedgepeth CM, Conrad LJ, Zhang J, Huang HC, Lee VM, Klein PS. Activation of the Wnt signaling pathway: a molecular mechanism for lithium action. Dev Biol. 1997;185:82-91
36. Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA. 1996;93:8455-9
37. Waymire KG, Mahuren JD, Jaje JM, Guilarte TR, Coburn SP, MacGregor GR. Mice lacking tissue non-specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B-6. Nat Genet. 1995;11:45-51
38. Fedde KN, Blair L, Silverstein J, Coburn SP, Ryan LM, Weinstein RS. et al. Alkaline phosphatase knock-out mice recapitulate the metabolic and skeletal defects of infantile hypophosphatasia. J Bone Miner Res. 1999;14:2015-26
39. Harmey D, Johnson KA, Zelken J, Camacho NP, Hoylaerts MF, Noda M. et al. Elevated skeletal osteopontin levels contribute to the hypophosphatasia phenotype in Akp2(-/-) mice. J Bone Miner Res. 2006;21:1377-86
40. Narisawa S, Frohlander N, Millan JL. Inactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasia. Dev Dyn. 1997;208:432-46
41. Yamamoto S, Orimo H, Matsumoto T, Iijima O, Narisawa S, Maeda T. et al. Prolonged survival and phenotypic correction of Akp2(-/-) hypophosphatasia mice by lentiviral gene therapy. J Bone Miner Res. 2011;26:135-42
42. Matsumoto T, Miyake K, Yamamoto S, Orimo H, Miyake N, Odagaki Y. et al. Rescue of severe infantile hypophosphatasia mice by AAV-mediated sustained expression of soluble alkaline phosphatase. Hum Gene Ther. 2011;22:1355-64
43. Suzuki U, Yoshimura K, Takaishi M. Uberein enzyme-phytasek das anhydro-oxy-methylen-diphosphorsaure spaltet. Bull Coll Agric. 1907;7:503-12
44. Merchant-Larios H, Mendlovic F, Alvarez-Buylla A. Characterization of alkaline phosphatase from primordial germ cells and ontogenesis of this enzyme in the mouse. Differentiation. 1985;29:145-51
45. Millan JL. The in vivo role of TNAP. Millan JL. Mammalian alkaline phosphatases: from biology to applications in medicine and biotechnology. 1st ed. New York: Wiley-Blackwell. 2006:105-129
46. Esteve D, Galitzky J, Bouloumie A, Fonta C, Buchet R, Magne D. Multiple functions of MSCA-1/TNAP in adult mesenchymal progenitor/stromal cells. Stem Cells Inter. 2016;2016:1815982
47. Liu W, Liu Y, Guo T, Hu C, Luo H, Zhang L. et al. TCF3, a novel positive regulator of osteogenesis, plays a crucial role in miR-17 modulating the diverse effect of canonical Wnt signaling in different microenvironments. Cell Death Dis. 2013;4:e539
48. Van Hoof VO, De Broe ME. Interpretation and clinical significance of alkaline phosphatase isoenzyme patterns. Crit Rev Clin Lab Sci. 1994;31:197-293
49. Millan JL. Alkaline phosphatases: Structure, substrate specificity and functional relatedness to other members of a large superfamily of enzymes. Purinergic Signal. 2006;2:335-41
50. McComb RB, Bowers GN, Posen S. Alkaline phosphatase. New York: Plenum Press. 1979
51. Tsai LC, Hung MW, Chen YH, Su WC, Chang GG, Chang TC. Expression and regulation of alkaline phosphatases in human breast cancer MCF-7 cells. Eur J Biochem. 2000;267:1330-9
52. Whyte MP. Hypophosphatasia - aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2016;12:233-46
53. Bennett CN, Longo KA, Wright WS, Suva LJ, Lane TF, Hankenson KD. et al. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc Natl Acad Sci USA. 2005;102:3324-9
54. Gaur T, Lengner CJ, Hovhannisyan H, Bhat RA, Bodine PV, Komm BS. et al. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J Biol Chem. 2005;280:33132-40
55. Bain G, Muller T, Wang X, Papkoff J. Activated beta-catenin induces osteoblast differentiation of C3H10T1/2 cells and participates in BMP2 mediated signal transduction. Biochem Biophys Res Commun. 2003;301:84-91
56. Rawadi G, Vayssiere B, Dunn F, Baron R, Roman-Roman S. BMP-2 controls alkaline phosphatase expression and osteoblast mineralization by a Wnt autocrine loop. J Bone Miner Res. 2003;18:1842-53
57. Clement-Lacroix P, Ai M, Morvan F, Roman-Roman S, Vayssiere B, Belleville C. et al. Lrp5-independent activation of Wnt signaling by lithium chloride increases bone formation and bone mass in mice. Proc Natl Acad Sci USA. 2005;102:17406-11
58. Vestergaard P, Rejnmark L, Mosekilde L. Reduced relative risk of fractures among users of lithium. Calcif Tissue Int. 2005;77:1-8
59. Fabian Molina G, Cabral RJ, Mazzola I, Brain Lascano L, Frencken JE. Biaxial flexural strength of high-viscosity glass-ionomer cements heat-cured with an LED lamp during setting. Biomed Res Int. 2013;2013:838460
Author contact
![]() Corresponding authors: Yan Jin, School of Stomatology, Fourth Military Medical University, 145 West Chang Le Road, Xin cheng District, Xi'an, China, 710032; e-mail: yanjinedu.cn; Fang Jin, School of Stomatology, Fourth Military Medical University, 145 West Chang Le Road, Xin cheng District, Xi'an, China, 710032; e-mail: jinfangedu.cn.
Corresponding authors: Yan Jin, School of Stomatology, Fourth Military Medical University, 145 West Chang Le Road, Xin cheng District, Xi'an, China, 710032; e-mail: yanjinedu.cn; Fang Jin, School of Stomatology, Fourth Military Medical University, 145 West Chang Le Road, Xin cheng District, Xi'an, China, 710032; e-mail: jinfangedu.cn.