Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(17):4649-4663. doi:10.7150/thno.26789 This issue Cite
Research Paper
Hypoxia-induced up-regulation of VASP promotes invasiveness and metastasis of hepatocellular carcinoma
Zhikui Liu#, Yufeng Wang#, Changwei Dou, Meng Xu, Liankang Sun, Liang Wang, Bowen Yao, Qing Li, Wei Yang, Kangsheng Tu ![]() , Qingguang Liu
, Qingguang Liu ![]()
Department of Hepatobiliary Surgery, the First Affiliated Hospital of Xi'an Jiaotong University, 277 Yanta West Road, Xi'an 710061, China
# Contributed equally
Received 2018-4-20; Accepted 2018-8-10; Published 2018-9-9
Abstract

Rational: Patients with hepatocellular carcinoma (HCC) have a poor prognosis mostly due to intrahepatic as well as distal metastasis. Vasodilator-stimulated phosphoprotein (VASP), a regulator of actin cytoskeleton and cell migration, is overexpressed in HCC and correlated with its malignant features and poor prognosis. Very little is known about its function in HCC.
Methods: qRT-PCR, Western blot and IHC were used to detect the VASP expression in tissues and cells. Transwell and wound healing assays were used to measure the migration and invasion of HCC cells. Immunoblotting and immunofluorescence were used for detection of epithelial-to-mesenchymal transition (EMT) progression in HCC cells. A lung metastasis mouse model was used to evaluate metastasis of HCC in vivo. The putative targets of miR-204 were disclosed by public databases and a dual-luciferase reporter assay. IP was used to show the interaction between VASP and CRKL. ChIP was used to analyze the binding of HIF-1α to VASP promoter region.
Results: Our data involving both gain- and loss-of-function studies revealed that VASP activated AKT and ERK signaling and promoted HCC migration and invasion in vitro and in vivo by altering the EMT phenotype and expression of MMPs. We investigated the positive correlation between VASP and an adapter protein, CRKL. VASP dynamically co-localized at the SH3N domain of CRKL and mediated its function. Mechanistically, VASP overexpression at the transcriptional level was mediated by HIF-1α through direct binding to two hypoxia response elements (HRE) in the VASP promoter region. Furthermore, we identified hypoxia-induced down-regulation of miR-204, which functioned as the regulator of VASP overexpression at the post-transcriptional level. Also, hypoxia-activated p-Smad3 dependent TGF-β signaling indirectly promoted VASP expression.
Conclusion: A variety of hypoxia-induced molecular mechanisms contributed to the upregulation of VASP at transcriptional and post-transcriptional levels. These mechanisms involved CRKL, HIF-1α, miR-204, and TGF-β activating the AKT and ERK signaling to promote EMT and expression of MMPs. Taken together, our results defined VASP as an oncogene of HCC pathogenesis and metastasis with the potential to serve as a prognostic biomarker.
Keywords: VASP, hypoxia microenvironment, hepatocellular carcinoma, metastasis, CRKL
Introduction
Hepatocellular carcinoma (HCC) is one of the most common, aggressive malignancies and the third leading cause of cancer-related mortality worldwide [1]. Despite recent remarkable advancements and progress in the diagnosis and therapeutic strategies for HCC, the long-term prognosis remains poor due to high occurrence of local invasion and distant metastasis [2, 3]. However, the molecular mechanisms controlling HCC metastasis remain poorly understood.
Hypoxia is a critical tumor microenvironment parameter attributed to excessive oxygen consumption and insufficient vascular supply in rapidly growing tumors [4, 5]. Hypoxia microenvironment facilitates cell proliferation, glucose metabolism, neovascularization, phenotype conversion, and distant metastasis through hypoxia-inducible factor (HIF) family [6-8]. HIF-1α is highly expressed and significantly associated with advanced stage and aggressive phenotype in HCC and is indicative of poor prognosis [9, 10]. Noticeably, hypoxia triggers overexpression of HIF-1α, which has previously been shown to induce epithelial-mesenchymal transition (EMT) of cancer cells in HCC [11, 12]. This shift from an epithelial to mesenchymal phenotype can facilitate the transition of the primary tumor to a metastatic and invasive type [13]. However, the exact mechanisms involved in hypoxia-driven HCC metastasis have not been well investigated so far.
Vasodilator-stimulated phosphoprotein (VASP), which belongs to Ena/VASP family, links the cytoskeletal system to signal transduction pathways and plays a critical role in cytoskeletal dynamics, cell migration, cell cycle, and cell adhesion [14, 15]. VASP governs cell migration and spreading by regulating the formation and stability of protrusive membrane structures driven by actin polymerization [16, 17]. Recently, conflicting studies have confused the VASP field, pointing to its role both in the stimulation and inhibition of cell migration [18-20]. VASP was involved in migfilin-mediated cell-matrix adhesions and migration; however, VASP exerted its positive modulation of migration and invasion via Rac1 in human breast cancer cells [21-23]. Thus, the molecular details of the functional role of VASP in cell motility and migration are still controversial. Previous studies have also confirmed that HIF-1α could regulate VASP expression in breast cancer cells or during acute lung injury [24, 25]. This suggests that hypoxia microenvironment may play an essential role in regulating VASP; however, until now, the potential role of VASP in HCC metastasis and invasion and its relationship with hypoxia remain mostly unknown.
In this study, we showed, for the first time, that VASP was overexpressed in HCC, and its overexpression promoted HCC cell migration and invasion in vitro and metastasis in vivo. HIF-1α, by directly binding to and transactivating the VASP promoter, mediated the overexpression of VASP in HCC. Hypoxia-induced down-regulation of miR-204 expression in human HCC also contributed to VASP up-regulation by relieving its post-transcriptional regulation. Furthermore, hypoxia-driven TGF-β secretion indirectly promoted VASP expression. Functional analyses revealed that the kinase CRK-like adapter protein (CRKL), due to its effects on the activation of ERK and AKT pathways, was involved in VASP-mediated cell migration. In cells with high expression of VASP, we detected increased EMT and translocation of Twist to the nucleus. These observations warrant a comprehensive investigation into the functional and pathological roles of VASP in HCC.
Methods
Clinical specimens and cell culture
Clinical tissues and matched adjacent non-tumor tissues were obtained from 126 patients in the First Affiliated Hospital of Xi'an Jiaotong University from January 2006 to December 2009. No patients received preoperative chemo- or radiotherapy before surgery. Written informed consent was obtained from each patient. The human immortalized normal hepatic cell lines (LO2, HL-7702) and HCC cell lines (MHCC-97H, Hep3B, SMMC-7721, Huh7, MHCC-97L, and HCCLM3) were obtained from the Chinese Academy of Sciences (Shanghai, China). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Gibco, GrandIsland, NY, USA), 100 units/mL ampicillin, and 100 μg/mL streptomycin at 37 °C with 5% CO2 considered as the normoxic condition. To evaluate the effects of hypoxia, cells were cultured under the normoxic condition to 60-70% confluence and subsequently cultured in a consistent 1% O2 hypoxic condition for 48 h.
RNA extraction and quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from tissues or cultured cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer's instructions. Quantitative real-time PCR was performed using SYBR Premix Ex Taq II (TaKaRa). TaqMan microRNA assays (Applied Biosystems, Foster City, California, USA) were used to quantify the expression levels. The amount of each target gene was quantitated by the comparative C (T) method using GAPDH as the normalization control. qPCR primers were purchased from Genecopoeia (Guangzhou, China).
Immunohistochemical staining (IHC)
Immunohistochemistry was performed on paraformaldehyde-fixed paraffin sections. VASP, E-cadherin, N-cadherin, Vimentin and MMP9 primary antibodies were used for immunohistochemistry using a streptavidin peroxidase-conjugated (SP-IHC) method. The immunohistochemistry procedure was performed as previously reported [26, 27]. Staining intensity was divided into four grades: 0, none; 1, weak; 2, moderate; 3, strong. The percentage of specifically positive staining of tumor cells was classified with the following grades: 0 (<5%), 1 (6%-25%), 2 (26%-50%), 3 (51%-75%), and 4 (>75%). The final score was expressed by multiplying the staining intensity and the percentage of specifically positive staining tumor cells.
Immunofluorescence (IF)
HCC cells were fixed with 4% paraformaldehyde and permeabilized using 0.2% Triton X-100. Subsequently, the fixed cells were incubated with the β-catenin, E-cadherin, N-cadherin, VASP, CRKL, and F-actin primary antibodies. The secondary antibody was an Alexa Fluor-conjugated IgG (Invitrogen, Carlsbad, CA, USA). Fluorescence confocal images were captured using an LSM 5 Pascal Laser Scanning Microscope (Zeiss Germany, Oberkochen, Germany) using a 40X lens and Laser Scanning Microscope LSM PASCAL software (version 4.2 SP1).
Western blot analysis
Total protein was extracted from HCC cells following which 20 μg of isolated protein was separated by 10% SDS-PAGE and transferred onto a PVDF membrane (Bio-Rad Laboratories, Hercules, CA, USA). The membranes were probed with the respective primary antibodies (Table S6) overnight. Subsequently, the membranes were incubated with the HRP-conjugated goat anti-mouse or anti-rabbit IgG antibody (ZSGB-BIO, China). Protein bands were visualized using an enhanced chemiluminescence kit (Amersham, Little Chalfont, UK). Experimental details were as previously described [28, 29].
miRNA array
To identify hypoxia-regulated miRNAs, Huh7 cells were cultured in normoxic (20% O2) and hypoxic incubators (1% O2) for 24 h. RNA extraction for these cells was performed using Trizol. RNA quality and quantity were assessed by ThremoNanodrop 2000 and Agilent 2100 Bioanalyzer along with the Agilent RNA 6000 Nano Kit (Agilent Technologies, Shanghai, China). RNA samples with A260/A280 in the range of 1.7 to 2.2, RIN>7.0 and 28S/18S>0.7 were subjected to further analysis. These RNA samples were analyzed using an Affymetrix GeneChip miRNA Array v. 4.0 (Affymetrix, Santa Clara, CA, U.S.). RNAs were labeled with biotin using a FlashTag Biotin HSR RNA Labeling Kit (Genisphere, Hatfield, PA, U.S.). The GeneChip® miRNA 4.0 arrays were washed and stained using the GeneChip Hybridization Wash and Stain Kit and were then scanned with the Affymetrix GeneChip Scanner 3000 7G (Affymetrix, Santa Clara, CA, U.S.). To identify microRNAs that were differentially expressed between Huh7 cells cultured in normoxic and hypoxic condition, the raw reads for each miRNA were normalized and subjected to statistical analysis. MiRNAs with fold change ≥ 2 were considered to be differentially expressed between Huh7 cells cultured in normoxic and hypoxic conditions.
RNA interference
The specific small interfering RNA (siRNA) for HIF-1α and a negative control siRNA were obtained from GenePharm (Shanghai, China). Cells (2×105 per well) were cultured in a 6-well plate and transfected with 100 nM siRNA using lipofectamine 2000 (Invitrogen, CA, USA), according to the manufacturers' instructions. Forty-eight hours after transfection, the cells were harvested for further experiments.
Transwell migration and invasion assay
The migration and invasion assays were performed using Transwell chamber (Millipore, Billerica, USA). For migration assay, the transfected cells were seeded into the upper chamber with serum-free medium (2.5×104 cells), and the bottom of the chamber contained the DMEM medium with 10% FBS. For the invasion assay, the chamber was coated with Matrigel (BD Biosciences, Franklin Lakes, NJ, USA), and the subsequent steps were similar to the migration assay. After the cells migrated or invaded for 24 h, they were fixed and stained with crystal violet. Migrated and invaded HCC cells were counted under an inverted light microscope. The number of migrated or invaded cells was quantified by counting the number of cells from 10 random fields at ×100 magnification.
Wound healing assay
The cells were seeded in 6-well plates at a high density and allowed to form cell monolayers overnight. A 200 μL sterile plastic tip was used to create a wound line across the surface of plates, and the suspension cells were removed with PBS. Cells were cultured in reduced serum DMEM medium in a humidified 5% CO2 incubator at 37 °C for 48 h, and then images were taken with a phase-contrast microscope. Each assay was replicated three times.
In vivo metastasis assay
4-6-week-old male BALB/c nude mice (Centre of Laboratory Animals, The Medical College of Xi'an Jiaotong University, Xi'an, China) were randomized into two groups (n=5). Transfected cells (1×106) were injected into the tail veins for the establishment of the pulmonary metastatic model. Mice were sacrificed 3 weeks post-injection and examined microscopically by H&E staining for the development of lung metastatic foci. Animals were housed in cages under standard conditions. All in vivo protocols were approved by the Institutional Animal Care and Use Committee of Xi'an Jiaotong University.
Coimmunoprecipitation assay
For coimmunoprecipitation (Co-IP) assay, cells were lysed with lysis buffer. Cell lysates or control immunoglobulin (IgG). IP 2-5 µg of primary antibody was used for each antibody pull down Antibodies used for IP were anti-HA (12CA5) (Roche Diagnostics) and anti-VASP (610447, BD Transduction Laboratory). After extensive washing, precipitates were analyzed by western blotting. Western blotting analysis was performed as previously described.
Chromatin immunoprecipitation assay (ChIP)
ChIP assay was performed using a commercial kit (Upstate Biotechnology) according to the manufacturer's instructions. The PCR primers are indicated in Table S3 and Table S5.
Statistical analysis
Data are presented as the mean ± SD from at least three independent replicates. SPSS software, 16.0 (SPSS, Inc, Chicago, IL, USA) was used to conduct the analysis, and a two-tailed Student t-test was employed to analyze the differences between two groups. Pearson's correlation analysis was used to analyze the correlation between two indices. Differences were considered statistically significant at P < 0.05.
Results
VASP is frequently over-expressed in HCC tissues and is positively associated with metastatic potential
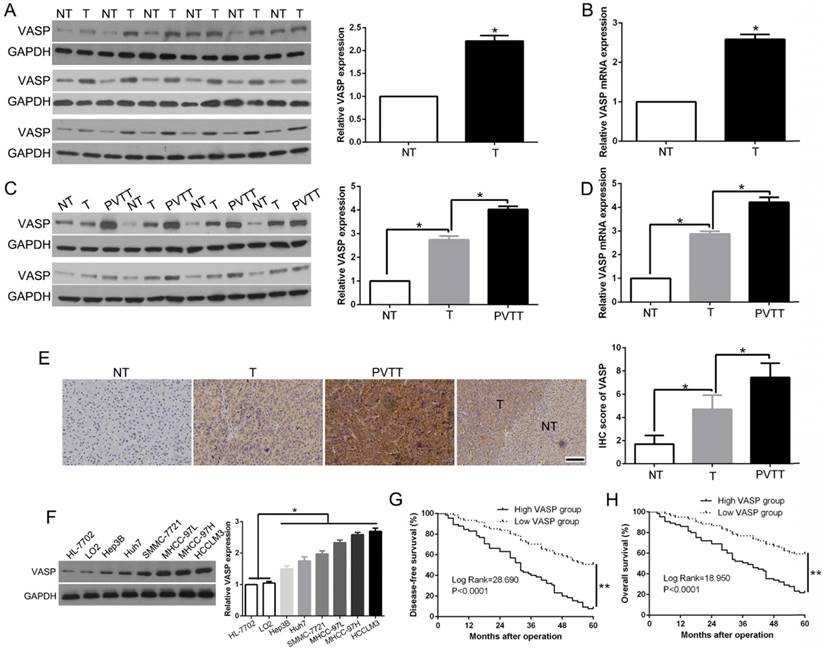
To validate the role of VASP in HCC progression, we examined VASP levels in tumors and paired adjacent nontumor liver tissues. VASP protein (Figure 1A) and mRNA (Figure 1B) levels were remarkably up-regulated in HCC tissues compared to corresponding normal liver tissues suggesting that VASP was activated at the transcriptional level during HCC progression. Portal vein tumor thrombus (PVTT), identified as an invasive tissue, serves as a poor prognostic factor for predicting frequent recurrence and intrahepatic metastasis [30]. We detected increased VASP mRNA and protein in PVTT samples (Figure 1C-D, P<0.05), suggesting its potential role not only in original tissues but also in the invasive progression of HCC. We performed immunohistochemical staining to validate low levels of VASP protein in normal liver tissues; however, it was expressed at a relatively higher level in primary HCC and further increased in PVTT (Figure 1E, P<0.05). We also examined the VASP protein in serval HCC cell lines with varying metastatic abilities. Its levels increased progressively from normal liver cells (HL-7702 and LO2) to low metastatic Hep3B, Huh7, SMMC-7721 and MHCC-97L and to highly metastatic MHCC-97H and HCCLM3 cells (Figure 1F, P<0.05). Taken together, these data indicated that VASP may play a promotive role in the metastasis of HCC.
VASP is frequently overexpressed in HCC tissues and correlates with HCC progression and survival. (A) Left: Western blot analysis of VASP levels in paired human HCC and matched adjacent non-tumorous tissues. Right: The chart shows VASP protein expression levels in HCC specimens versus paired adjacent normal liver tissues. NT: non-tumor; T: tumor. (B) qRT-PCR for VASP was performed in 126 pairs of HCC and matched adjacent NT liver tissues. *, P<0.05 by t-test. (C) Western blot analysis of VASP levels in paired human HCC and matched adjacent NT, portal vein tumor thrombus (PVTT). (D) qRT-PCR for VASP was performed in paired HCC and matched adjacent NT, PVTT. (E) Representative images of IHC staining of VASP in differentiated HCC, NT, and PVTT. Comparison of VASP expression in HCC. 95% CI = 1.892-7.853. (F) Expression of VASP protein in normal hepatic cells (LO2 and HL-7702) and a panel of HCC cells was assessed by Western blotting. (G) Disease-free and (H) overall survival rates of 126 patients with HCC were compared between the low-VASP and high-VASP groups.
Elevated expression of VASP predicts poor prognosis in HCC patients
To determine the significance of VASP in the clinical features and outcome of HCC patients, we divided the HCC patients into two groups according to the VASP expression. As shown in Table S1 (Pearson chi-square test), we found that overexpression of VASP was significantly associated with tumor-node-metastasis (TNM) stage (III+IV, P=0.004), venous invasion (P=0.004) and the presence of multiple tumor nodes (P=0.006). Intriguingly, Kaplan-Meier analysis showed that patients with high VASP had poor disease-free survival (DFS, log-rank, 28.690; P<0.0001, Figure 1G) and overall survival rates (OS, log-rank, 18.95; P<0.0001, Figure 1H). Cox proportional hazard regression analysis showed that overexpression of VASP was an independent prognostic factor for the OS of HCC patients (P=0.001, Table S2). Furthermore, we explored the publicly available database compiled in The Human Protein Atlas and found similar results of association of higher expression of VASP with a worse OS (P<0.05, Figure S11A). These findings suggested that up-regulation of VASP has significant pathological implications in HCC development.
Ectopic expression of VASP promotes HCC migration and invasion in vitro and in vivo
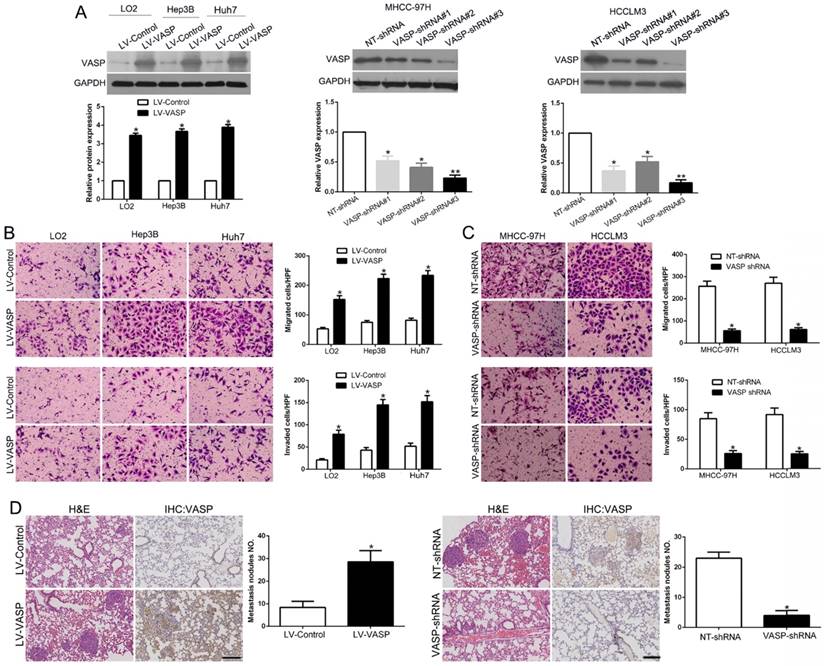
Metastasis is considered a critical factor for poor prognosis of HCC [31]. Clinical data showed that VASP overexpression was significantly associated with HCC metastasis. To investigate the functional role of VASP in HCC metastasis, we used lentiviral transduction to establish stable cell lines with VASP overexpression or knockdown (Figure 2A). Of the three pairs of shRNAs, LV-shVASP#3 most efficiently knocked down VASP expression and was used in the subsequent functional studies. Transwell migration and invasion assays revealed that VASP overexpression in Hep3B and Huh7 HCC cells increased cell migration and invasion compared with normal hepatic LO2 cells (Figure 2B, P<0.05). Similarly, compared to control cells, the migratory activity in wound-healing assays was enhanced by VASP overexpression (Figure S1A, P<0.05). However, knockdown of VASP exhibited opposite effects in MHCC-97H and HCCLM3 cells (Figure 2C and Figure S1B, P<0.05). Immunofluorescence analysis confirmed regional co-localization of VASP and F-actin (Figure S1C). VASP overexpression caused a spindle-shaped, more elongated morphology, while VASP-knockdown cells displayed a rounded shape. To further validate the role of VASP in HCC metastasis, in vivo experiments were performed. Hep3B cells overexpressing VASP and MHCC-97H cells with VASP knockdown were administered into mice via tail vein injections. As expected, Hep3B cells promoted lung metastasis while MHCC-97H cells reduced lung metastasis as observed by microscopic evaluation (P<0.05) (Figure 2D). Metastasis to the liver and abdominal organs caused by VASP-overexpressing Hep3B cells was visually evident (Figure S5A). To control for off-target effects of shRNA, we used shRNA#1 to knock down VASP in HCCLM3 cells and it also showed similar effects (Figure S2). Collectively, these results indicated that VASP could stimulate the aggressive and metastatic phenotype of HCC both in vitro and in vivo.
VASP promotes HCC cell migration and invasion. (A) Retrovirus encoding VASP vector was transduced into LO2, Hep3B, and Huh7 cells to establish stably overexpressing VASP cells. Lentivirus encoding VASP shRNA was transduced into MHCC-97H and HCCLM3 cells to establish VASP knockdown cells. (B) Comparison of the migration and invasion potential of LO2, Hep3B, and Huh7 cells transfected with LV-VASP using Transwell. (C) Comparison of migration and invasion potential of MHCC-97H and HCCLM3 cells transfected with VASP shRNA using the Transwell assay. (D) Representative hematoxylin and eosin (H&E) images of metastatic nodules from the mouse lung tissue sections of the Hep3B-VASP group (upper) and MHCC-97H shRNA group (bottom). n = six independent experiments. *P<0.05, **P<0.01.
VASP induces EMT and MMPs expression
EMT has been recognized as a critical regulator of metastasis in HCC [32]. To explore the molecular mechanisms underlying the role of VASP in HCC metastasis, EMT markers and related transcription factors were measured. As shown in Figure 3A, VASP overexpression in Hep3B and Huh7 cells led to a decrease in the epithelial markers E-cadherin and α-E-catenin but increased mesenchymal markers including fibronectin, vimentin, and N-cadherin as well as transcriptional factors Snail and Twist. However, no significant difference was observed in total β-catenin. Conversely, VASP knockdown in MHCC-97H and HCCLM3 cells induced a reverse trend (P<0.05) (Figure S3A). MMPs are proteolytic enzymes that can degrade basement membrane extracellular matrix proteins to establish metastatic foci at distant sites. Matrix metalloproteinases (MMP)-2 and 9 are the key MMPs responsible for cell migration and invasion. Increased expression of MMP-2 and MMP-9 was detected in VASP-overexpressing cells (P<0.05) (Figure 3A and Figure S3A), but no changes were observed in the expression levels of tissue inhibitors of metalloproteases (TIMPs). The MMP activity also showed a similar change after VASP alteration (P<0.05) (Figure S4). Opposite expression patterns of these markers were observed in VASP knockdown cells (Figure 3A). The data from R2: Genomics Analysis and Visualization Platform database showed a positive correlation between the expression of VASP and MMP2, MMP9, Snail, Twist, Vimentin, and S100A4 expression (Figure S11C-H), confirming the effects of VASP on EMT and MMPs expression. We then tested the sub-cellular distribution of β-catenin and found that VASP overexpression promoted translocation of β-catenin from cytosolic to nuclear fractions (P<0.05) (Figure 3B and Figure S3B). IF analysis also confirmed increased accumulation of β-catenin in the nucleus in VASP-overexpressing cells (Figure 3C and Figure S3D).
VASP induces EMT and increases nuclear translocation of β-catenin. (A) Western blot analysis was used to compare expression of epithelial and mesenchymal markers between VASP-transfected cells and shVASP-transfected cells. GAPDH was used as the loading control. (B) Level of β-catenin in nuclear and cytosolic fraction was detected by Western blot analysis. Histone H3 and GAPDH were used as loading controls. (C) LO2 and Hep3B cells were transfected with VASP expression vector and MHCC-97H cells were transfected with VASP shRNA. 72 h after transfection, the expression of cytosolic and nuclear levels of β-catenin (red) in the cells were analyzed by triple IF staining. (D) Immunofluorescence assay was performed to compare the expression patterns of E-cadherin (green) and N-cadherin (red). The nuclei were stained with DAPI in blue color. (E) Immunoblots were performed to detect expression of indicated molecules in Hep3B LV-VASP or Huh7 LV-VASP cells transfected with siRNA targeting Twist1 (Twist1 siRNA) or scrambled siRNA (Control siRNA). (F) Representative pictures of immunochemical staining of serial lung sections for VASP, E-cadherin, N-cadherin, Vimentin, and MMP9 in the Hep3B-LV-control group and the Hep3B-LV-VASP group.
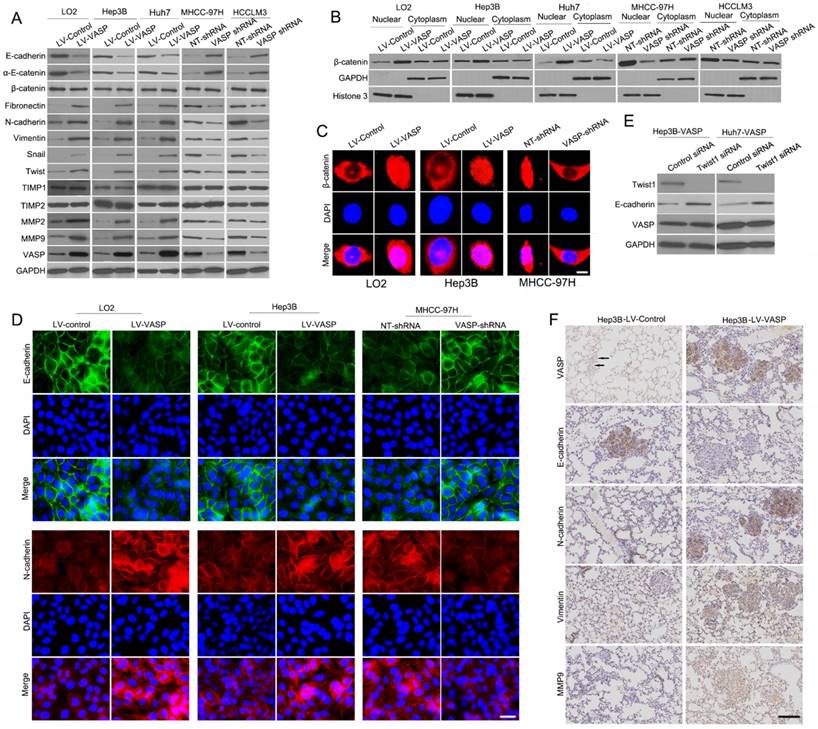
To address if VASP affected the transcriptional activity of β-catenin, we performed TOP-Flash promoter activity and found that VASP overexpression significantly augmented the reporter activity (P<0.05) (Figure S3C). However, the FOP-flash promoter reporter did not show any response supporting that VASP acted explicitly on β-catenin. Furthermore, IF validated the effects of VASP on EMT (Figure 3D). We also explored whether Twist1 and Snail were involved in VASP-induced down-regulation of E-cadherin. The expression of E-cadherin could be rescued by silencing Twist1 or Snail in Hep3B-VASP or Huh7-VASP cells (Figure 3E and Figure S3E-F), suggesting that Twist1 and Snail were required for VASP-driven EMT. We next investigated the presence of EMT in vivo by staining the EMT markers in lung sections. There was increased N-cadherin and vimentin expression but decreased E-cadherin expression in lung sections with overexpressed VASP (Figure 3F). We further explored the correlation between VASP expression and EMT markers in HCC tissues. We found that the E-cadherin expression in the high VASP group was lower than that in the low VASP group. Conversely, the expression level of N-cadherin and vimentin in the high VASP group was markedly higher than that in the low VASP group (P<0.05) (Figure S5B). Collectively, these results indicated that VASP is capable of regulating EMT phenotype of HCC both in vitro and in vivo.
VASP exerts oncogenic effects via ERK and AKT signaling pathways in HCC cells
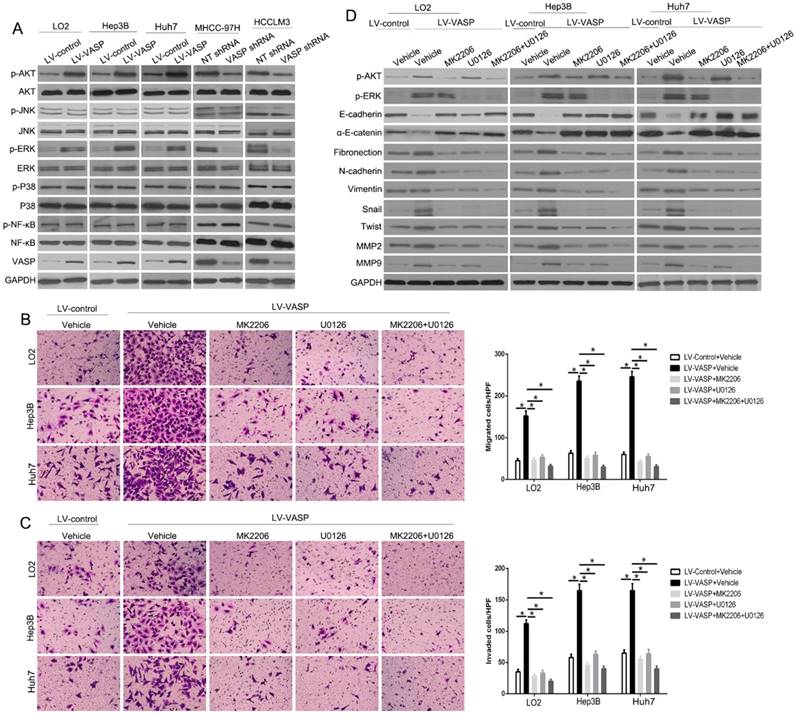
To determine how VASP regulates EMT and MMPs expression, we explored the phosphorylation levels of the upstream signaling pathways by Western blot analysis after altering VASP expression. Only p-AKT and p-ERK had changed with altered VASP expression (P<0.05) (Figure 4A and Figure S6). To confirm whether AKT and ERK signaling pathways were necessary for VASP-mediated increased HCC metastasis, we used AKT-specific inhibitor MK2206 or ERK-specific inhibitor U0126 to block the respective signaling pathways. As displayed in Figure 4B-C, the migration and invasion of both MHCC-97H-VASP and HCCLM3-VASP cells were remarkably attenuated upon treatment with AKT or ERK inhibitors. Furthermore, an inhibitory effect of blocking AKT or ERK signaling on EMT and MMPs expression was detected by VASP overexpression (Figure 4D). Together, these data suggested that AKT- and ERK-mediated signaling plays a critical role in the modulation of VASP-induced HCC migration and invasion.
VASP exerts oncogenic effects on HCC cells by activating AKT and ERK pathways. (A) Western blot was performed to investigate the influence of VASP on AKT, ERK, JNK, MAPK, and NF-κB pathways in indicated cells. GAPDH was used as an internal control. (B-D) LO2, Hep3B, and Huh7 cells overexpressing VASP and corresponding cells in the control group were treated with MK2206 (AKT inhibitor) and U0126 (p-ERK inhibitor) for 24 h and subjected to (B) migration, (C) invasion, and (D) Western blotting.
The N-terminal SH3 domain of CRKL dynamically interacts with VASP and mediates its functional effects
In the public database (http://www.cbioportal.org), the co-expression of VASP and CRKL in HCC series was significant (P<0.05). Furthermore, a previous study reported an oncogenic role of CRKL in HCC. Thus, we chose CRKL, an oncogenic kinase, to understand the mechanisms of regulation of AKT and ERK by VASP. First, we observed that CRKL protein was significantly up-regulated in HCC compared to non-tumor tissues (P<0.05) (Figure 5A) and that VASP had a positive association with CRKL in HCC samples (P<0.05) (Figure 5B). To determine whether VASP could interact with CRKL, Flag-tagged VASP and HA-tagged CRKL were co-expressed in Hep3B and MHCC-97H cells following which co-IP assays were performed. Anti-Flag antibodies, but not control IgG, immunoprecipitated CRKL from cell lysates (Figure 5C, left panel). Reciprocally, anti-HA antibodies could immunoprecipitate VASP from cell lysates (Figure 5C, right panel). Similar results were observed in MHCC-97H and HCCLM3 cells, confirming the endogenous nature of protein-protein interactions (Figure S7A). To further evaluate whether the interaction between VASP and CRKL occurred in HCC tissues, we performed Co-IP in three HCC samples with a high expression of both VASP and CRKL. Anti-VASP antibodies, but not control IgG, could coimmunoprecipitate CRKL protein in these samples (Figure 5D). Reciprocally, anti-CRKL antibodies could coimmunoprecipitate VASP protein. We also used corresponding adjacent non-tumor tissues as negative controls (Figure S7B). VASP-CRKL interaction was further analyzed by confocal immunofluorescence microscopy in HCC cells (Figure 5E). The adapter CRKL was composed of an N-terminal SH2 domain and two SH3 domains, SH3N and SH3C. To address which domain was involved in direct interaction with VASP, we expressed a series of GST-CRKL fusion proteins (Figure 5F). We found that GST-FL-CRKL and GST-SH3N-CRKL but not GST-SH3C-CRKL efficiently precipitated VASP (Figure 5G). Thus, we demonstrated that VASP interacted directly with CRKL, suggesting that the SH3N domain of CRKL was involved in binding to VASP. To explore the functional role of CRKL in VASP-induced migration and invasion, we knocked down CRKL by specific siRNA and found that it almost reversed VASP overexpression-induced promotion of cell migration and invasion (Figure 5H and Figure S7C) as well as Rac1 activation in Huh7 cells (P<0.05) (Figure S7D). These data indicated that the migration-promotive function of VASP is CRKL dependent and that CRKL mediated the role of Rac1 activation by VASP. Notably, AKT, ERK phosphorylation and EMT and MMPs markers were also altered (P<0.05) (Figure 5I and Figure S7E). This suggested that the AKT and ERK signaling induced by VASP was also dependent on CRK. In conclusion, these data demonstrated that VASP interacted directly with the SH3N domain of CRKL, which then mediated the function of VASP.
The N-terminal SH3 domain of CRKL dynamically interacts with VASP and mediates its functional effects. (A) Expression of CRKL in paired human HCC and matched adjacent non-tumorous tissues. (B) Correlation between VASP and CRKL in human HCC tissues. (C) Co-IP analysis of the interaction between VASP protein and CRKL protein in HCC cells. (D) Co-IP analysis of the interaction between VASP protein and CRKL protein in HCC tissues. (E) Confocal images of LO2, Hep3B, and MHCC-97H cells stained for CRKL (red) and VASP (green) and 4′,6-diamidino-2-phenylindole (DAPI; blue). Colocalization was indicated by the merged images showing yellow immunofluorescence. (F) Schematic diagram of the CRKL domain organization and the different GST-CRKL fusion proteins used in this study. (G) GST pull-down assay with lysates of human cells. Equal amounts of lysates were incubated with equimolar amounts of the depicted, immobilized GST fusion proteins or GST alone. After extensive washing, precipitated material was analyzed by Western blotting with anti-VASP antibodies. Cells overexpressing VASP and corresponding cells in the control group were transfected with CRKL siRNA or control siRNA. 48 h after transfection, cells were subjected to the Transwell assay for migration and invasion (H) and Western blotting for EMT markers (I). n = six independent experiments.
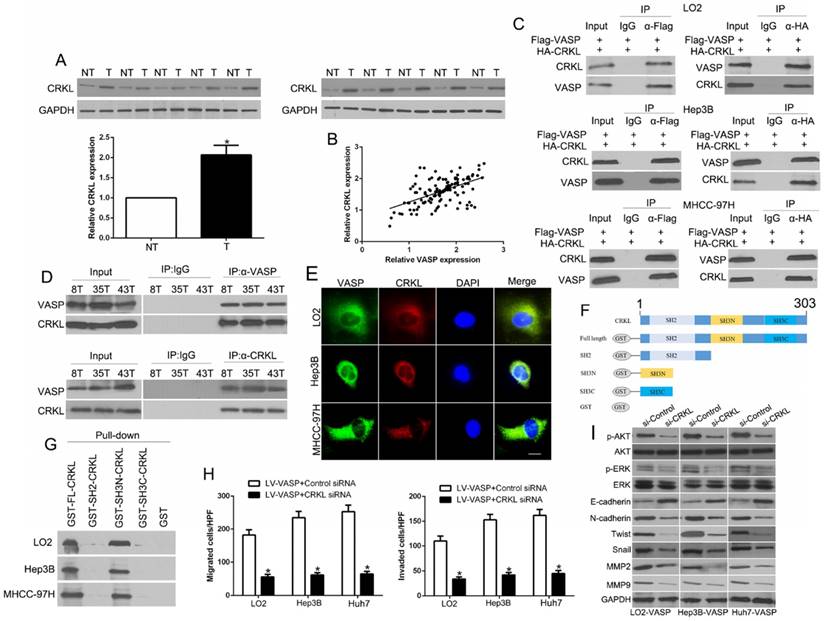
HIF-1α mediates hypoxia-induced VASP overexpression by binding to HRE in the VASP gene promoter
The microenvironment of solid tumors is often hypoxic, and our study identified HIF-1α as a critical transcriptional factor in the migration and invasion of HCC [33]. To confirm whether hypoxia promotes the expression of VASP in HCC, we performed qRT-PCR to determine the VASP expression in HCC cells cultured under normoxia (21% O2) or hypoxia (1% O2) for 24 h. Our data showed that VASP mRNA was significantly increased under hypoxia compared with normoxia. VEGF expression was used as a positive control and was found to be increased (P<0.05) (Figure 6A). Western blot analysis showed significant induction of VASP with the upregulation of HIF-1α under hypoxia (Figure 6B). To investigate whether hypoxia increased VASP in a HIF-1α-dependent manner, we used specific siRNA to knockdown HIF-1α and found it dramatically decreased VASP expression whereas silencing HIF-2α, another subunit of the HIF protein, did not affect VASP expression (Figure 6C). Moreover, deferoxamine mesylate (DFO), a known HIF-1α activator, induced HIF-1α expression in a concentration-dependent manner and up-regulated VASP in HCC cells (Figure 6D). To better understand the correlation between HIF-1α and VASP in HCC tissues, immunohistochemical staining was performed. VASP protein levels in HCC tissues positively correlated with HIF-1α expression (R=0.661, P<0.001) (Figure 6E). Also, the data from R2: Genomics Analysis and Visualization Platform database showed a positive correlation between HIF-1α and VASP expression (Figure S11B). To understand the underlying mechanism of VASP overexpression in hypoxia, we surveyed the promoter region of the VASP gene and identified 7 hypoxia response elements (HREs) (Figure 6F). To investigate whether HIF-1α directly binds to VASP promoter, we performed the ChIP assay in Hep3B cells under hypoxia. In chromatin fractions pulled down by anti- HIF-1α antibody, only the HRE site1 and site 3 of VASP promoter were detected (Figure 6F and Table S3). To determine whether the binding of HIF-1α activates VASP promoter, we constructed a full-length VASP luciferase promoter vector construct (containing sites 1, 3) and co-transfected it with or without HIF-1α cDNA into Hep3B cells. Luciferase analysis showed that HIF-1α overexpression (pcDNA- HIF-1α) significantly increased VASP promoter activity in Hep3B cells (P<0.05) (Figure 6G). To determine whether the HRE site 1 or 3 was required for HIF-1α to transactivate VASP promoter, these binding sites were mutated. As shown in Figure 6G, the mutation of HRE site 1 and 3 almost abolished the transactivation of VASP promoter by HIF-1α; VEGF promoter was used as a positive control by HIF-1α activation. To explore the role of VASP in hypoxia-induced HIF-1α-mediated migration, we ectopically expressed VASP in HIF-1α knockdown cells. VASP overexpression at least partially rescued the inhibitory effect of HIF-1α knockdown in HCC migration and invasion assay (P<0.05 (Figure 6H and Figure S8A) and EMT progression (P<0.05) (Figure 6I and Figure S8B), suggesting that VASP was involved in HIF-1α-mediated migration and EMT.
HIF-1α mediates hypoxia-induced VASP overexpression by binding to the HRE in the VASP gene promoter. (A) qRT-PCR and (B) Western blot analysis in LO2, Hep3B, and Huh7 cells cultured under normoxia (21% O2) or hypoxia (1% O2). *, P < 0.05 versus control. (C) Western blot analysis in LO2, He3B, and Huh7 cells after knockdown of si-HIF-1α (50 nM) under hypoxia. GAPDH was used as a normalization control. (D) Western blotting of HIF-1α and VASP after treatment with DFO in Hep3B and Huh7 cells. (E) Positive association between HIF-1α and VASP expression in HCC tissues. (F) Upper: Chromatin immunoprecipitation (ChIP) analysis of HIF-1α binding to VASP promoter in Hep3B cells. Lower: ChIP assay. Anti-IgG and anti- HIF-1α antibodies were used in the ChIP assay. (G) Luciferase analysis in Hep3B cells. Hep3B cells overexpressing HIF-1α expression plasmid (pcDNA-HIF1α) or control vector (pcDNA3.1) were transfected with the pGL3-VASP promoter, pGL3-VASP-promoter mutation, or pGL3-VEGF promoter. 48 h after transfection, cells were subjected to dual luciferase analysis. Results were expressed as a fold induction relative to the cells transfected with the control vector (pcDNA3.1) after normalization to Renilla activity. The experiments were performed independently three times. (H-I) HIF-1α siRNA was transfected into LO2, Hep3B, and Huh7 cells to knock down HIF-1α under hypoxia. Transwell assay for migration and invasion (H) and Western blotting (I) for EMT markers were performed.
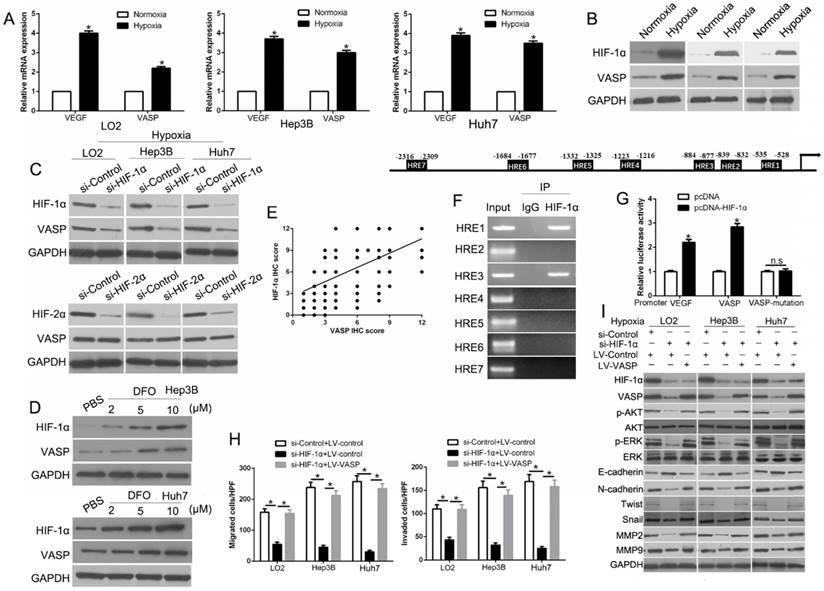
Hypoxia-induced loss of miR-204 contributes to VASP up-regulation in HCC
Hypoxia has been shown to regulate the role of microRNAs as post-transcriptional modulators, which prompted us to investigate whether deregulation of miRNA(s) contributes to VASP up-regulation in human HCC. We performed miRNA arrays of Huh7 cells cultured under normoxic and hypoxic conditions. The data showed that hypoxia led to 64 up-regulated and 25 down-regulated miRNAs (Figure 7A and Table S4). We used the TargetScan and Miranda databases to predict which of the differentially expressed miRNAs identified in the microarray analysis mediated hypoxia-induced VASP up-regulation by interacting with VASP 3'-UTR. We identified the down-regulated hypoxia-responsive miR-204 as the potential mediator of the elevated VASP expression. Subsequently, we performed qRT-PCR and confirmed that hypoxia led to significant down-regulation of miR-204 in HCC cells (P<0.05) (Figure 7B). However, suppression of miR-204 was significantly abolished upon knockdown of HIF-1α but not HIF-2α, indicating that suppression of miR-204 by hypoxia was HIF-1α dependent (P<0.05) (Figure 7C). We performed luciferase assay for VASP 3'-UTR to clarify whether miR-204 was involved in the up-regulation of VASP induced by hypoxia/ HIF-1α signaling and interacted with VASP 3'-UTR as predicted (Figure S9A). We found that miR-204 overexpression significantly inhibited the luciferase activity of wild-type (wt) VASP 3'UTR in Hep3B cells, while miR-204 knockdown increased the luciferase activity of wt VASP 3'UTR but not mutant VASP 3'-UTR (P<0.05) (Figure 7D and Figure S9B). Furthermore, miR-204 overexpression markedly reduced, while miR-204 knockdown increased the mRNA and protein levels of VASP in HCC cells (P<0.05) (Figure 7E). To confirm the relationship between miR-204 and VASP, we measured the miR-204 level in HCC tissues with different levels of VASP expression. As expected, our data showed that the levels of miR-204 and VASP mRNAs were inversely correlated (P<0.05) (Figure 7F-G). Importantly, miR-204 overexpression partially abolished the increase of hypoxia-induced VASP-mediated migration and invasion and EMT (P<0.05) (Figure 7H-I and Figure S9C-D). In summary, these data demonstrated that hypoxia/ HIF-1α signaling-induced miR-204 down-regulation could indirectly contribute to VASP up-regulation at the post-transcriptional level.
Hypoxia-induced loss of miR-204 contributes to VASP up-regulation in HCC. (A) miRNA array was used to identify the differentially expressed miRNAs between Huh7 cells cultured in normoxic and hypoxic conditions. (B) qRT-PCR was performed for miR-204 in HCC cells cultured under normoxia and hypoxia. (C) qRT-PCR was performed for miR-204 in HIF-1α-knockdown LO2, Hep3B, and Huh7 cells in hypoxic condition. (D) miR-204 overexpression significantly suppressed, while miR-204 loss increased, the luciferase activity that carried wild-type (wt) but not mutant (mt) 3'-UTR of VASP. (E) LO2, Hep3B, and Huh7 cells were transfected with miR-204 inhibitors (anti-miR-204) and MHCC-97H and HCCLM3 cells were transfected with precursor miR-204 and were subjected to qRT-PCR for VASP mRNA expression. (F) The expression of miR-204 in VASP high-expressing tumors was significantly lower than that in VASP low-expressing tumors, as determined by qRT-PCR. (G) An inverse correlation between the levels of miR-204 and VASP mRNA was observed in HCC tissues. (H-I) LO2, Hep3B, and Huh7 cells cultured in normoxic or hypoxic condition were transduced with miR-204 vector and were subjected to Transwell assay for migration and invasion (H), and Western blotting (I) for EMT markers.
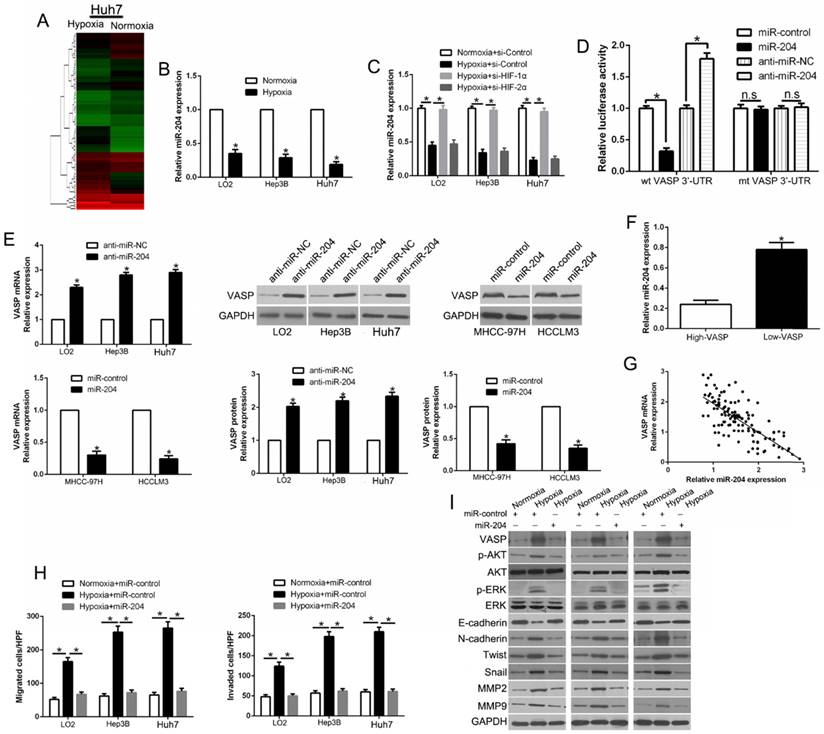
Hypoxia-induced TGF-β/Smad signaling activation contributes to VASP up-regulation in HCC
Hypoxia and TGF-β are central upstream regulators that drive HCC metastasis. However, how hypoxia influences TGF-β signaling and whether it plays a role in the upregulation of VASP is unknown. To investigate this, we performed ELISA assay and found TGF-β1 expression was time-dependent under the hypoxic condition, which did not affect the expression of TGF-β2 and TGF-β3 (Figure 8A). Also, the data from R2: Genomics Analysis and Visualization Platform database showed a positive correlation between TGF-β1 and VASP expression (Figure S11I). We also observed increased expression of p-Smad2 and p-Smad3 (Figure 8B), which was indicative of the activation of TGF-β/Smad signaling in hypoxia-exposed HCC cells. We, therefore, explored the effects of activation of TGF-β signaling. Treatment of HCC cells with TGF-β increased VASP expression (Figure 8C). In contrast, VASP expression in HCC cells was inhibited in hypoxia upon treatment with specific p-Smad3 inhibitor SIS3 or Smad3 shRNA (Figure 8D). These data showed p-Smad3 was important in the hypoxia-induced increased expression of VASP. By bioinformatic software prediction, we detected two binding sites for p-Smad3 in the VASP promoter. To confirm whether p-Smad3 directly binds to VASP promoter, ChIP assay was performed following TGF-β treatment. In chromatin fractions pulled down by an anti-p-Smad3 antibody, both sites were not detected (Figure 8E and Table S5). This indicated that the promoting effect of TGF-β signaling was not the direct effect of the binding of p-Smad3 to VASP promoter. In functional assays, p-Smad3 inhibitor could abolish the effects on migration, invasion (Figure 8F and Figure S10A), and EMT (Figure 8G). Moreover, we determined the effect of p-Smad3 inhibitor, SB431542, on in vivo metastasis. The data showed that SB431542 significantly inhibited the number of lung metastases (P<0.05) (Figure S10B). Taken together, we demonstrated that hypoxia promoted the expression of VASP by activating TGF-β signaling.
Hypoxia-induced TGF-β/Smad signaling activation contributes to VASP up-regulation in HCC. (A) Elisa assay was performed to detect the expression of TGF-β1, 2, and 3 in conditioned media of Hep3B cells exposed to hypoxia. (B) Western blot determined the TGF-β signaling-related protein expression in LO2, Hep3B, and Huh7 cells under normoxia or hypoxia. (C) Western blotting of the TGF-β signaling-related protein expression in LO2, Hep3B, and Huh7 cells following TGF-β treatment. (D) Western blot analysis of VASP expression of Hep3B cells under hypoxia after transfection with Smad3 shRNA or specific p-Smad3 inhibitor SIS3. (E) Upper: ChIP analysis of p-Smad3 binding to VASP promoter in Hep3B cells. Lower: ChIP assay using anti-IgG and anti-p-Smad3 antibodies. (F-G) LO2, Hep3B, and Huh7 cells cultured in normoxic or hypoxic condition were treated with p-Smad3 inhibitor, SIS3, and were subjected to Transwell assay for migration and invasion (F) and Western blot analysis (G) for EMT markers.
Discussion
Metastasis and recurrence are the most important prognostic factors for HCC patients. Therefore, there is a critical need to explore the molecular mechanisms governing the pathogenesis of HCC metastasis [34]. In this study, we investigated the role of VASP in HCC progression and metastasis. Our data showed that VASP was expressed at a higher level in HCC than in adjacent non-tumor tissues. Remarkably, VASP overexpression was also observed in metastatic HCC PVTT tissues. The increased expression of VASP was correlated with clinicopathological features, including TNM stage, venous invasion, and the presence of multiple tumor nodes. Furthermore, VASP was an independent prognostic factor in predicting survival of HCC patients. Mechanistically, in terms of downstream signaling, VASP interacted with the SH3N domain of CRKL, enhancing AKT and ERK signaling by increased phosphorylation, which, in turn, promoted the twist1-dependent EMT and expression of MMPs leading to increased migration and invasion of HCC cells. Our study also elucidated several molecular events upstream of VASP overexpression in HCC that were mainly mediated directly or indirectly by HIF-1α. First, HIF-1α directly binds to a hypoxia response element on VASP promoter. It has been reported that HIF-1α acts downstream of TNF-α to inhibit VASP expression and modulates the adhesion and proliferation of breast cancer cells [24]. Importantly, HIF-1α inhibition drastically reduced the expression level of VASP protein. Second, hypoxia-induced miR-204 downregulation indirectly promoted VASP overexpression at the post-transcriptional level. Third, hypoxia-induced activation of TGF-β signaling caused p-Smad3-dependent VASP up-regulation. Taken together, hypoxia induced multiple mechanisms in HCC-mediated VASP overexpression, thus promoting cancer invasiveness and metastasis (Figure S12).
VASP was shown to be associated with microfilaments by promoting the assembly and polymerization of actin [35, 36]. Given the crucial function of VASP in the regulation of adherent junctions in epithelial cells, we focused on its role in HCC and the underlying mechanisms that contributed to frequent up-regulation of VASP. In HCC, VASP expression had a positive correlation with CRKL, which encodes a protein kinase and is vital in malignant transformation of multiple cancers, including HCC [37]. The immunoprecipitation assay showed a direct interaction between VASP and CRKL involving the SH3N domain of CRKL. The IF assay showed co-location of VASP and CRKL in HCC cells. CRKL has been shown to recruit tyrosine kinases. Consistent with this notion, we found that CRKL mediated its function by activating p-AKT and p-ERK.
Solid tumors often have a hypoxic environment resulting from an imbalance of the high proliferation rate of tumor cells and abnormal vascularization. It is well established that hypoxia contributes to aggressive progression and poor prognosis. Constitutive HIF-1α expression in solid tumors was also previously reported. Our data indicated that elevated HIF-1α in HCC transactivated VASP expression at both transcription and protein levels, which in turn dysregulated the actin cytoskeleton in HCC to promote invasion and metastasis. It is of note that HIF-1α also transactivated transcription of the pro-metastatic actin-bundling proteins fascin-1 and LASP-1. In addition to HIF-1α/VASP signaling, other signaling pathways such as CXCL12/CXCR4 also influence HCC metastasis. Understanding the interaction among these pathways may provide new clues for inhibiting metastasis of HCC.
In summary, we demonstrated that VASP was up-regulated, especially in metastatic HCC and was indicative of poor clinical prognosis. VASP promoted an aggressive phenotype and metastasis both in vitro and in vivo. VASP-CRKL complex activated AKT and ERK signaling to promote EMT and expression of MMPs. Importantly, hypoxia-induced HIF-1α regulated VASP expression at the transcriptional level by binding to the HRE in the promoter region of VASP. Also, hypoxia-induced down-regulation of miR-204 contributed to VASP up-regulation at the post-transcriptional level. Furthermore, hypoxia-induced activation of TGF-β signaling led to VASP overexpression. Thus, it appears that VASP is an oncogene that is functionally important to HCC metastasis, and inhibiting VASP expression may constitute an effective strategy for the treatment of metastatic HCC.
Abbreviations
ChIP: chromatin immunoprecipitation; CRKL: CRK-like adapter protein; EMT: epithelial-mesenchymal transition; HCC: hepatocellular carcinoma; IF: immunofluorescence; IHC: immunohistochemistry; qRT-PCR: quantitative reverse transcription polymerase chain reaction; shRNA: short hairpin RNA; VASP: vasodilator-stimulated phosphoprotein; WB: Western blot.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (81572847, 81773123), the Natural Science Basic Research Plan in Shaanxi Province of China (2016JQ8047), Innovation Capacity Support Plan in Shaanxi Province of China (2018KJXX-045), the Fundamental Research Funds for the Central Universities (7N010011015) and Beijing Medical Reward Foundation (YJHYXKYJJ-211).
Competing Interests
The authors have declared that no competing interest exists.
References
1. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterol. 2007;132:2557-76
2. Bruix J, Sherman M, American Association for the Study of Liver D. Management of hepatocellular carcinoma: an update. Hepatol. 2011;53:1020-2
3. Maluccio M, Covey A. Recent progress in understanding, diagnosing, and treating hepatocellular carcinoma. CA Cancer J Clin. 2012;62:394-9
4. Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nat. 2006;441:437-43
5. Carnero A, Lleonart M. The hypoxic microenvironment: A determinant of cancer stem cell evolution. BioEssays. 2016;38(Suppl 1):S65-74
6. Harris AL. Hypoxia-a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38-47
7. Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Sci. 2016;352:175-80
8. Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer. 2008;8:180-92
9. Soni S, Padwad YS. HIF-1 in cancer therapy: two decade long story of a transcription factor. Acta Oncol. 2017;56:503-15
10. Wu XZ, Xie GR, Chen D. Hypoxia and hepatocellular carcinoma: The therapeutic target for hepatocellular carcinoma. J Gastroenterol Hepatol. 2007;22:1178-82
11. Zhang J, Zhang Q, Lou Y, Fu Q, Chen Q, Wei T. et al. HIF-1alpha/IL-1beta signaling enhances hepatoma epithelial-mesenchymal transition via macrophages in a hypoxic-inflammatory microenvironment. Hepatol. 2018;67:1872-89
12. Yoo YG, Na TY, Seo HW, Seong JK, Park CK, Shin YK. et al. Hepatitis B virus X protein induces the expression of MTA1 and HDAC1, which enhances hypoxia signaling in hepatocellular carcinoma cells. Oncogene. 2008;27:3405-13
13. Hu F, Deng X, Yang X, Jin H, Gu D, Lv X. et al. Hypoxia upregulates Rab11-family interacting protein 4 through HIF-1alpha to promote the metastasis of hepatocellular carcinoma. Oncogene. 2015;34:6007-17
14. Kwiatkowski AV, Gertler FB, Loureiro JJ. Function and regulation of Ena/VASP proteins. Trends Cell Biol. 2003;13:386-92
15. Krause M, Dent EW, Bear JE, Loureiro JJ, Gertler FB. Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol. 2003;19:541-64
16. Bear JE, Svitkina TM, Krause M, Schafer DA, Loureiro JJ, Strasser GA. et al. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 2002;109:509-21
17. Geese M, Loureiro JJ, Bear JE, Wehland J, Gertler FB, Sechi AS. Contribution of Ena/VASP proteins to intracellular motility of listeria requires phosphorylation and proline-rich core but not F-actin binding or multimerization. Mol Biol Cell. 2002;13:2383-96
18. Wang J, Zhang J, Wu J, Luo D, Su K, Shi W. et al. MicroRNA-610 inhibits the migration and invasion of gastric cancer cells by suppressing the expression of vasodilator-stimulated phosphoprotein. Eur J Cancer. 2012;48:1904-13
19. Gan L, Li L. Interleukin-1 Receptor-Associated Kinase-1 (IRAK-1) functionally associates with PKCepsilon and VASP in the regulation of macrophage migration. Mol Immunol. 2010;47:1278-82
20. Galler AB, Garcia Arguinzonis MI, Baumgartner W, Kuhn M, Smolenski A, Simm A. et al. VASP-dependent regulation of actin cytoskeleton rigidity, cell adhesion, and detachment. Histochem Cell Biol. 2006;125:457-74
21. Zhang Y, Tu Y, Gkretsi V, Wu C. Migfilin interacts with vasodilator-stimulated phosphoprotein (VASP) and regulates VASP localization to cell-matrix adhesions and migration. J Biol Chem. 2006;281:12397-407
22. Bae YH, Ding Z, Zou L, Wells A, Gertler F, Roy P. Loss of profilin-1 expression enhances breast cancer cell motility by Ena/VASP proteins. J Cell Physiol. 2009;219:354-64
23. Han G, Fan B, Zhang Y, Zhou X, Wang Y, Dong H. et al. Positive regulation of migration and invasion by vasodilator-stimulated phosphoprotein via Rac1 pathway in human breast cancer cells. Oncol Rep. 2008;20:929-39
24. Su K, Tian Y, Wang J, Shi W, Luo D, Liu J. et al. HIF-1alpha acts downstream of TNF-alpha to inhibit vasodilator-stimulated phosphoprotein expression and modulates the adhesion and proliferation of breast cancer cells. DNA Cell Biol. 2012;31:1078-87
25. Tang M, Tian Y, Li D, Lv J, Li Q, Kuang C. et al. TNF-alpha mediated increase of HIF-1alpha inhibits VASP expression, which reduces alveolar-capillary barrier function during acute lung injury (ALI). PloS One. 2014;9:e102967
26. Liu Z, Dou C, Yao B, Xu M, Ding L, Wang Y. et al. Methylation-mediated repression of microRNA-129-2 suppresses cell aggressiveness by inhibiting high mobility group box 1 in human hepatocellular carcinoma. Oncotarget. 2016;7:36909-23
27. Liu Z, Dou C, Yao B, Xu M, Ding L, Wang Y. et al. Ftx non coding RNA-derived miR-545 promotes cell proliferation by targeting RIG-I in hepatocellular carcinoma. Oncotarget. 2016;7:25350-65
28. Liu Z, Dou C, Jia Y, Li Q, Zheng X, Yao Y. et al. RIG-I suppresses the migration and invasion of hepatocellular carcinoma cells by regulating MMP9. Int J Oncol. 2015;46:1710-20
29. Liu Z, Dou C, Wang Y, Jia Y, Li Q, Zheng X. et al. Highmobility group box 1 has a prognostic role and contributes to epithelial mesenchymal transition in human hepatocellular carcinoma. Mol Med Rep. 2015;12:5997-6004
30. Sun J, Shi J, Huang B, Cheng F, Guo W, Lau WY. et al. The degree of hepatic arterial blood supply of portal vein tumor thrombus in patients with hepatocellular carcinoma and its impact on overall survival after transarterial chemoembolization. Oncotarget. 2017;8:79816-24
31. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245-55
32. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442-54
33. Chiu DK, Tse AP, Xu IM, Di Cui J, Lai RK, Li LL. et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun. 2017;8:517
34. Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563-72
35. Wu Y, Gunst SJ. Vasodilator-stimulated phosphoprotein (VASP) regulates actin polymerization and contraction in airway smooth muscle by a vinculin-dependent mechanism. J Biol Chem. 2015;290:11403-16
36. Thom SR, Bhopale VM, Yang M, Bogush M, Huang S, Milovanova TN. Neutrophil beta2 integrin inhibition by enhanced interactions of vasodilator-stimulated phosphoprotein with S-nitrosylated actin. J Biol Chem. 2011;286:32854-65
37. Ren Y, Shang J, Li J, Liu W, Zhang Z, Yuan J. et al. The long noncoding RNA PCAT-1 links the microRNA miR-215 to oncogene CRKL-mediated signaling in hepatocellular carcinoma. J Biol Chem. 2017;292:17939-49
Author contact
![]() Corresponding authors: Dr. Kangsheng Tu and Qingguang Liu, Department of Hepatobiliary Surgery, the First Affiliated Hospital of Xi'an Jiaotong University, 277 Yanta West Road, Xi'an 710061, China. E-mail: tks0912com or liuzk0319com; Phone: +086-029-85323905; Fax: +086-029-85323209
Corresponding authors: Dr. Kangsheng Tu and Qingguang Liu, Department of Hepatobiliary Surgery, the First Affiliated Hospital of Xi'an Jiaotong University, 277 Yanta West Road, Xi'an 710061, China. E-mail: tks0912com or liuzk0319com; Phone: +086-029-85323905; Fax: +086-029-85323209