Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Bioengineering...
3. Microphysiological...
4. Current challenges and future...
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2026; 16(12):6463-6484. doi:10.7150/thno.130030 This issue Cite
Review
Human Multi-Organ-on-a-Chip Platforms for Next-Generation Drug Delivery Strategies
Cosmo S. Mitchell1*, Hong-Il Yoo2,3*, Carolina Gracia Diaz3,4,5, Jordan J. Green3,5,6,7,10, Deok-Ho Kim3,7,8,9,10 ![]()
1. Chemistry-Biology Interface Program, Johns Hopkins University, Baltimore, MD 21218, USA.
2. Department of Anatomy and Neurosciences, Eulji University School of Medicine, Daejeon 34824, Republic of Korea.
3. Department of Biomedical Engineering, Johns Hopkins University, Baltimore, MD 21205, USA.
4. GSK, Collegeville, PA 19426, USA.
5. Translational ImmunoEngineering Center, Johns Hopkins University, Baltimore, MD 21205, USA.
6. Departments of Materials Science & Engineering, Chemical & Biomolecular Engineering, Neurosurgery, Oncology, and Ophthalmology, Johns Hopkins University, Baltimore, MD 21205, USA.
7. Center for Microphysiological Systems, Johns Hopkins University, Baltimore, MD 21205, USA.
8. Department of Medicine, Johns Hopkins University, Baltimore, MD 21205, USA.
9. Departments of Mechanical Engineering and Neurology, Johns Hopkins University, Baltimore, MD 21205, USA.
10. Institute of NanoBioTechnology, Johns Hopkins University, Baltimore, MD 21205, USA.
*, Equally contributed.
Received 2025-12-16; Accepted 2026-4-8; Published 2026-4-23
Abstract

Human multi-organ-on-a-chip (MOoC) platforms have rapidly emerged as next-generation preclinical models for evaluating therapeutic delivery and efficacy. By integrating dynamic perfusion, physiologically relevant microenvironments, and systemic tissue-tissue crosstalk, MOoCs provide unprecedented opportunities to study drug absorption, distribution, metabolism, and toxicity under human-relevant conditions. In this review, we summarize recent progress in MOoC technologies and their application to diverse drug delivery strategies, including stem cell-based carriers, gene and nucleic acid therapies, nanoparticles, extracellular vesicles, and biomaterials. We highlight how these systems overcome key limitations of conventional in vitro and animal models, offering more predictive insight into therapeutic distribution and pharmacodynamics. Key technical and biological considerations, including material limitations, physiological scaling, and incorporation of patient-specific components, are discussed in the context of ongoing engineering developments. These platforms enable assessment of organ-specific accumulation, metabolic transformation, and off-target effects that are poorly captured by traditional methods. Finally, we outline future perspectives for MOoCs as translational platforms poised to accelerate drug discovery, reduce clinical attrition, and support the development of precision medicines.
Keywords: multi-organ-on-a-chip (MOoC), microphysiological systems (MPS), drug delivery, new approach methodologies (NAMs)
1. Introduction
Preclinical drug testing provides valuable information early into the drug development process. In vitro and animal models provide insight into pharmacokinetics (PK), pharmacodynamics (PD), and off-target effects of a potential therapeutic. However, despite encouraging results in cellular or animal experiments, translation to human clinical trials remains a critical bottleneck. A vast majority of drug candidates that enter clinical testing fail due to a lack of efficacy or unacceptable toxicity, resulting in only a small fraction of compounds ultimately reaching patients [1,2]. This high attrition rate highlights the limitations of traditional preclinical systems in capturing human-specific responses.
The concept of an organ-on-a-chip (OoC) emerged in the 2000s as a promising advancement in in vitro modeling to better emulate human physiology [3]. Soon after, the concept of multi-organ-on-a-chip (MOoC) was introduced as a more complex and accurate model that includes the dynamic crosstalk and systemic circulation between different organs [4]. Traditional in vitro methods can provide some insight into the cellular responses and local toxicities of drugs, while animal models provide further detail into the pharmacokinetics and immune or metabolic interactions of a whole organism. However, while these both serve as a baseline for preclinical evaluation, organ-on-chip and multi-organ-on-chip technologies offer models more relevant to humans with the potential to reduce cost and attrition at this early stage [5,6]. Major avenues through which drugs fail in testing include poor systemic distribution, limited absorption, or unexpected organ-specific toxicity. As such, the appeal of MOoCs lies in their ability to predict these failures earlier in the drug development process and serve as a human-relevant alternative for preclinical testing and screening.
We will review recent developments in MOoC platforms, focusing on cutting-edge advances including: iPSC-derived platforms for patient-specific disease modeling, physiologically interconnected organ systems spanning neurological (gut-brain axis, neurovascular unit, neuromuscular junction), cardiovascular, hepatic, and renal compartments, advanced biosensing and real-time analytical monitoring, 3D bioprinted tissue constructs with biomimetic extracellular matrices, and scalable high-throughput screening formats. We will examine how MOoCs have been applied to study diverse drug delivery strategies, including viral vectors, nanoparticles (NPs), extracellular vesicles (EVs), cell-based therapies, and biomaterials. By evaluating representative examples across these modalities, we aim to provide a comprehensive perspective on how MOoCs contribute to overcoming delivery barriers and modeling therapeutic responses in physiologically relevant contexts. Finally, we will discuss the outlook of MOoCs as transformative platforms for future drug delivery and translational medicine, emphasizing their potential to bridge the gap between preclinical evaluation and clinical success.
To fully appreciate the capabilities of multi-organ systems, it is essential to understand the foundational principles of organ-on-a-chip technology. Single-organ systems and their underlying engineering approaches have been extensively reviewed elsewhere [3,6-11]. These reviews provide comprehensive coverage of microfluidic device design, cell culture techniques, material selection, and validation methods that underpin all OoC platforms.
Key literature we discussed in this review was identified using search databases, including PubMed, Web of Science, and Google Scholar. Key search terms included a combination “multi-organ”, “multi-organ-on-a-chip”, “interconnected MPS”, “drug delivery”, “ADME”, with a focus on identifying field-defining reviews, and an emphasis on key studies published within the last 10 years.
2. Bioengineering multi-organ-on-a-chip platforms
While single-organ-on-a-chip systems have demonstrated remarkable capabilities in replicating organ-level functions, multi-organ integration addresses a critical limitation: the absence of intersystem crosstalk essential for predicting whole-body drug behavior and disease progression. Pharmacokinetic processes, including absorption, metabolism, distribution, and excretion, inherently span multiple tissues. Modeling these organs in isolation provides incomplete insight, whereas integrating targeted organs with metabolic and clearance pathways offers a holistic view of drug delivery and systemic exposure.
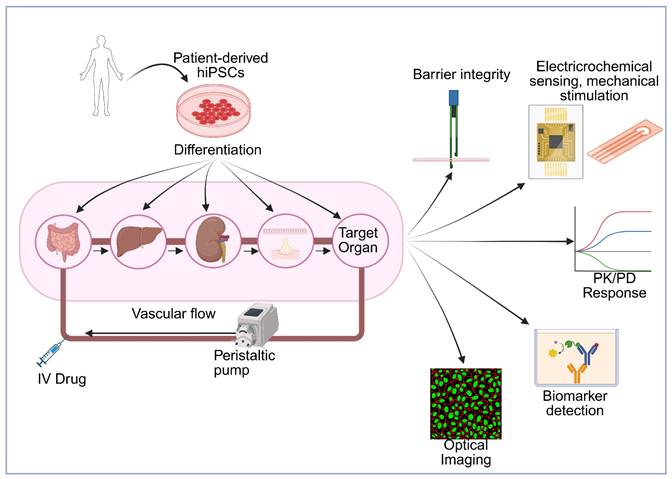
The Multi-Organ-on-a-Chip, as it is often defined, is a platform of multiple tissues connected through microfluidic circulatory systems, allowing continuous perfusion, soluble factor exchange, and systemic exposure to drugs or metabolites (Figure 1). One of the first designs for a MOoC model used for absorption, distribution, metabolism, excretion (ADME) profiling included a 4-organ chip with a co-culture of intestine, liver, skin, and kidney cells [4]. Through a shared recirculating medium, these organs shared a circulating fluid, mimicking blood flow. Dynamic perfusion and organ-organ communication was demonstrated for 28 days, establishing one of the first long-term, physiologically integrated human tissue platforms for drug testing. They were also capable of measuring organ-specific functionality and systemic drug metabolism, including cytochrome P activity, barrier integrity, and metabolite distribution across compartments.
Schematic overview of patient-specific multi-organ-on-a-chip platform opportunities for drug evaluation. Patient-derived human induced pluripotent stem cells (hiPSCs) are differentiated into multiple organ-specific cell types and integrated into a microfluidic multi-organ system connected via vascular flow with peristaltic pump control. The platform incorporates key organs involved in drug metabolism, clearance, and barriers (gut, liver, kidney, NVU, NMJ) along with a target organ, enabling modeling of drug absorption, distribution, metabolism, and excretion (ADME). Drugs can be administered via intravenous (IV) injection or other routes. The system can integrate multiple analytical modalities including: (1) barrier integrity monitoring through transepithelial/transendothelial electrical resistance (TEER) measurements, (2) electrochemical sensing and mechanical stimulation for physiological relevance, (3) optical imaging for real-time cellular visualization, (4) biomarker detection for assessing therapeutic and toxic responses, and (5) pharmacokinetic/pharmacodynamic (PK/PD) response profiling to generate dose-response curves across different organs. This integrated platform enables prediction of human-relevant drug responses, organ-specific toxicity, and inter-organ communication under controlled physiological conditions, supporting personalized medicine approaches and reducing reliance on animal testing. Created in BioRender (https://BioRender.com/64au3m2).
Recent engineering advances have enabled the sophistication of contemporary MOoC platforms (Table 1). Progress toward physiological relevance has been achieved through synthetic hydrogels supporting 3D tissue constructs that mimic native extracellular matrix properties, with bioprinting enabling tailor-made ECMs for specific organs to enhance cell viability [12]. Advances in barrier function and tissue-tissue interfaces allow for the recreation of complex native barriers such as the blood-brain barrier, incorporating tight junctions and dynamic shear stress to replicate in vivo permeability. Platform designs supporting long-term culture have also emerged, including pumpless circulation systems using inter-well pressure gradients [13] and closed-loop circulation that minimizes tubing and external media reservoirs to decrease media loss and contamination risk [14]. Methods such as dual-flow systems enable tissue polarization, exemplified by a 96-well microfluidic platform allowing separate perfusion of apical and basolateral components in renal barrier models [15]. Scaling to high-throughput formats has been achieved through plate-formatted systems, including 96-well chip arrays with pneumatic pressure control that enable independent yet synchronized operation of multiple organ units with automated liquid handling, imaging, and real-time monitoring [15,16].
Engineering advances enabling MOoC development
| Engineering Feature | Technology/Approach | Advantages | Key Applications | Representative Systems | References |
|---|---|---|---|---|---|
| Perfusion Systems | Pumpless gravity-driven flow | No external pumps, reduced complexity, inter-well pressure gradients | Long-term culture (14+ days), reduced contamination | Multi-organ PK-PD platform | [13] |
| Closed-loop circulation | Minimal media loss, reduced tubing | 28-day liver-skin co-culture | Dynamic MOoC | [14] | |
| Pneumatic pressure control | Programmable flow, centralized control | High-throughput screening | 96-well pneumatic platform | [16] | |
| Scaling & Throughput | 96-well plate format | Standard laboratory equipment, automated handling | Drug screening, toxicology testing | Dual perfusion bioreactor | [15] |
| Modular compartments | Independent organ control, flexible configuration | Custom organ pairing, disease modeling | Various gut-brain designs | [17,19,20] | |
| Systemic multi-organ integration | Holistic inter-organ communication modeling | Disease pathways, drug metabolism | Multi-organ systemic platforms | [9] | |
| Barrier Engineering | Permeable membranes with tight junctions | Physiological permeability, selective transport | BBB, gut epithelium, kidney tubules | NVU chips, renal barriers | [31] |
| Dual-flow systems | Separate apical/basolateral perfusion | Polarized epithelial function | 96-well renal platform | [15] | |
| 3D self-organized barriers | Physiological microvessel architecture | BBB modeling with realistic structure | Microvascular BBB models | [25] | |
| Biomaterials | Synthetic hydrogels | 3D tissue constructs, tunable ECM properties | Tissue maturation, cell viability | 3-tissue organ platform | [12,153] |
| Bioprinting | Organ-specific ECM, spatial control | Custom tissue architecture | Various applications | [12,153,154] | |
| Biomaterial-drug depots | Spatiotemporal drug release control | Localized therapeutic action | Hydrogel mesh for glioblastoma, shape memory polymers | [156,157,160] | |
| Tumor modeling biomaterials | Biomimetic tumor microenvironment | Cancer drug testing, metastasis studies | Tumor-on-chip platforms | [155] | |
| Monitoring & Sensing | Integrated biosensors | Real-time readouts, continuous monitoring | TEER, oxygen, metabolites | Gut-brain donepezil study, BBB-on-chip | [20,38] |
| Multi-electrode arrays (MEA) | Electrophysiological monitoring, functional assessment | Cardiac and neural activity, drug-induced effects | Heart-on-chip, neuromuscular systems | [84,86,87] | |
| Microplate readers | Automated imaging, high-content analysis | Throughput screening | 96-well systems | [15,16] | |
| Integrated bioelectronics | Extra- and intracellular monitoring | Cardiac electrophysiology under stress | Heart-on-chip with hypoxia | [86] | |
| Advanced Integration | Optogenetic control | Dynamic barrier modulation, temporal control | Endothelial barrier studies, neuromuscular actuation | OptoBarrier platform, optogenetic neurons | [60,159] |
| AI integration | Enhanced predictive modeling, data analysis | Future drug screening platforms | Cancer modeling | [161] | |
| Patient-specific iPSC systems | Personalized disease modeling, precision medicine | Patient-specific drug screening | BBB chips, cardiac models | [33,75] | |
| Validation & Maturity | Standardization approaches | Reproducibility, quality control | Regulatory acceptance, industrial adoption | MPS as reliable tools | [10,131] |
| Biological product assessment | Testing of biologics, new modalities | Therapeutic antibodies, cell therapies | Advanced MPS platforms | [126] | |
| New drug modality testing | Toxicity assessment beyond small molecules | Gene therapies, biologics, complex formulations | Comprehensive MPS systems | [11,169] |
Building upon these foundational multi-organ systems and engineering capabilities, contemporary MOoC platforms have evolved to address specific physiological axes and therapeutic challenges (Table 2). The following sections highlight representative systems organized by their biological focus, demonstrating how targeted multi-organ designs enable mechanistic investigation of disease pathology and drug delivery in physiologically relevant contexts.
Comparison of representative MOoC platform designs
| Platform Type | Organs/Tissues Included | Perfusion Method | Culture Duration | Key Readouts | Reference |
|---|---|---|---|---|---|
| 4-Organ Chip | Intestine, liver, skin, kidney | Recirculating medium | 28 days | CYP activity, barrier integrity, metabolite distribution | [4] |
| 3-Tissue Platform | Liver, cardiac, skeletal muscle | Integrated microfluidics | 14+ days | Multi-tissue interactions, drug metabolism | [12] |
| Gut-Brain Axis | Gut epithelium (Caco-2/iPSC), BBB (hBMECs, astrocytes, pericytes) | Dual compartment with independent flow | 7-14 days | TEER, permeability, cytokine panels, neurotransmitter levels, EV-mediated communication | [19,20,149] |
| Neurovascular Unit | Endothelial cells, pericytes, astrocytes, neurons | Dynamic perfusion with shear stress | 14-21 days | Tight junction integrity, transporter expression, electrophysiology, stem cell therapy evaluation | [32] |
| Liver-Kidney | Hepatocytes, proximal tubule epithelium | Microfluidic circulation | 14-28 days | Transporter activity (OAT, OCT), metabolite clearance, albumin production, EV biodistribution | [97,98] |
| Lung-Liver | Alveolar epithelium, hepatocytes | Pneumatic microfluidics | 7-14 days | Cytokine production, barrier function, metabolic conversion, inflammatory crosstalk | [94] |
| Heart-Liver | Cardiac tissue, hepatocytes | Microfluidic perfusion | 14+ days | Hepatic metabolism effects on cardiotoxicity, metabolite-induced cardiac effects | [92] |
| Heart-Liver-Skin | Cardiac, hepatic, skin | Microphysiological perfusion | 7-14 days | Topical drug delivery, systemic distribution, organ-specific responses | [79] |
| Multi-organ (Heart-Liver-Tumor-Lung) | Cardiac, hepatic, tumor spheroids, lung | Pumpless gravity-driven | 14 days | Drug efficacy, off-target toxicity, PK/PD modeling | [95] |
| Cardiovascular-Kidney-Metabolic | Heart, kidney, metabolic tissues | Integrated circulation | Variable | Interorgan crosstalk, CKM syndrome modeling | [93] |
| Heart-Brain Codevelopoid | Cardiac and neural tissues via codevelopment | Trans-germ-layer integration | Extended culture | Heart-brain axis interactions, codevelopment signaling | [80] |
| iPSC-derived Personalized BBB | Patient-specific endothelial, pericytes, astrocytes | Perfused microfluidic | 7-14 days | Disease modeling (AD, PD), personalized drug screening | [33] |
2.1 Gut-brain-axis-on-a-chip
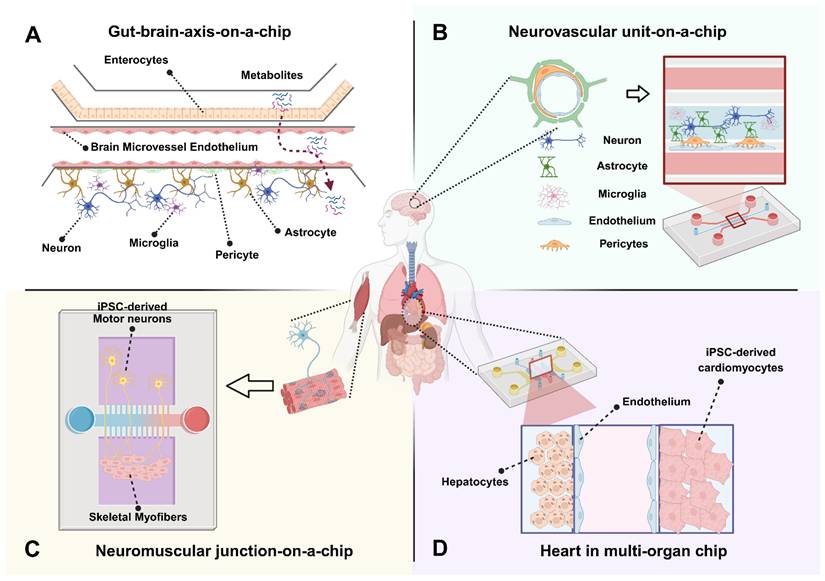
The gut-brain axis is a system that has gained a lot of attention in recent years due to its relevance in neurodegeneration and inflammatory diseases. This system is particularly relevant for understanding drug delivery that utilizes an oral delivery method, as the gut is often the first barrier for absorption. In addition, with the brain having one of the most selective barriers, this model provides a substantial perspective on a full absorption to target pathway. Most gut-brain chip designs utilize modular compartments separated by permeable membranes. An important component of these designs is the creation of distinct microenvironments tailored to each organ, meeting specific needs of oxygenation, media, and flow dynamics (Figure 2A). These systems utilize distinct compartments that allow for co-culture of gut and brain cell types (typically Caco-2 or induced pluripotent stem cell (iPSC)-derived enterocytes for the gut; hBMECs, astrocytes, and pericytes for the BBB) and allow for maintenance of physiological flow rates and shear stress for both compartments [17,18].
Schematic overview of multi-organ-on-a-chip platforms for targeted drug-delivery evaluation. A human body silhouette highlights key organs involved in systemic drug absorption, distribution, metabolism, and response. A dual-compartment microfluidic platform integrates an intestinal epithelial barrier with a BBB module. The gut chamber incorporates microvilli-bearing enterocytes that enable assessment of oral drug absorption, microbial metabolites, and epithelial transport. The downstream BBB module includes endothelial cells, astrocyte endfeet, and pericytes, enabling evaluation of molecular permeability, TEER dynamics, cytokine-mediated inflammation, and gut-derived metabolites transported to the brain (A). A perfusable endothelial microvessel is surrounded by astrocytes, neurons, microglia, and pericytes to reconstruct the structural and functional components of the human NVU. This platform allows measurement of BBB integrity, neuroinflammatory signaling, neurovascular coupling, electrophysiological activity, and drug or nanoparticle transport under controlled shear stress. The model enables patient-specific and disease-relevant investigation of ischemia, neurodegeneration, and barrier dysfunction (B). A compartmentalized microfluidic device links iPSC-derived motor neurons to aligned skeletal myofibers through directed axonal projection. This system facilitates real-time assessment of neuromuscular synaptogenesis, neurotransmission, calcium dynamics, optogenetically induced contraction, and synaptic deficits associated with neuromuscular disorders such as ALS. Functional readouts include MEA-based electrophysiology, force generation profiles, and acetylcholine receptor clustering (C). A coupled hepatocyte-cardiac platform models drug metabolism and metabolism-dependent cardiotoxicity. The liver compartment supports phase I/II metabolic processing, while the connected cardiac module contains beating cardiomyocytes with electrophysiological readouts. This integrated system enables evaluation of parent compounds, hepatic metabolites, biomarker release (BNP, troponin), and PK/PD responses under dynamic perfusion (D). Created in BioRender (https://BioRender.com/0gptvri).
The use of iPSC-derived cells has become of particular interest because they allow for patient-specific modeling and the generation of mature neuronal or gut epithelial subtypes, which improves disease relevance and expands the potential for personalized medicine. However, these studies also highlight limitations including the challenges of maintaining functional maturation and long-term viability in co-culture systems, as well as variability between iPSC lines.
Important analytical readouts include confirming barrier integrity for both the gut and brain compartments, typically measured using TEER and permeability assays. Additionally, in the context of inflammation, cytokine release panels (e.g., IL-6, TNF-α) can track immune responses to microbial or drug stimuli across the interface. Metabolite and neurotransmitter quantification can elucidate chemical communication across the axis, including serotonin, short-chain fatty acids, or drug metabolites. Also, live imaging and reporter assays can provide important information on the status of tight junctions, oxidative stress, or inflammatory signaling within each tissue compartment.
Few reliable gut-brain multi-organ platforms have been made at this point, as the majority of brain-related OoC studies have been focused on creating an accurate model of the blood-brain barrier. Some current gut-brain chip designs attempt to link gut models with BBB models and include confirmation of formation of both barriers and transport observations of known microbial products and gut-derived exosomes [19]. Additionally, gut-liver-brain models have been examined in the context of Parkinson's disease, as short-chain fatty acids are thought to induce changes in neurodegenerative pathology. These studies suggest systemic metabolite transfer and immune activation may influence CNS phenotypes. Additionally, a gut-brain-immune axis model has been proposed to better capture bidirectional signaling involving circulating immune cells, though few models have successfully integrated all three components in dynamic co-culture [17].
In a recent example of functional multi-organ modeling, a gut-brain chip was constructed and challenged with donepezil, an Alzheimer's medication [20]. In this study, they demonstrated the ability to monitor gut and brain barriers in real time using permeability assays and integrated biosensors, and they were able to monitor drug absorption from the gut to the brain under dynamic perfusion conditions. They also observed pharmacodynamic effects including acetylcholine modulation and downstream neuroinflammatory markers.
Despite these advances, there remain significant limitations with current models. As mentioned, current work on brain-on-a-chip units is still mostly focused on simulating a complete and accurate blood-brain barrier, which is a necessary advancement in the path of producing a fully functional neurovascular unit (NVU) before integrating into a multi-organ model. Another roadblock is establishing a complete gut microbiome, mostly due to the difficulties in establishing a unique anaerobic coculture for the gut microbiome while also allowing for aerobic brain tissue viability and media recirculation. Furthermore, iPSC-based approaches offer promise for disease models and personalized testing, but there are still challenges with cost and creating reproducible cell populations and barrier properties [21]. In terms of future directions, efforts are focused on integrating immune cells, simulating microbiomes, and developing hormonal cues to better reflect the bidirectional complexity of the gut-brain axis.
2.2 Neurovascular unit (NVU)-on-a-chip
The neurovascular unit (NVU) represents the fundamental structural and functional interface linking the cerebral vasculature with neural and glial networks [22,23]. NVU-on-a-chip platforms recapitulate this multicellular microenvironment by integrating brain microvascular endothelial cells, pericytes, astrocytes, neurons, oligodendrocytes and often microglia within microfluidic architectures (Figure 2B). These platforms enable simulation of complex in vivo cellular interactions that govern blood-brain barrier (BBB) integrity, cerebral metabolic processes, and neuroimmune interactions [24]. Unlike conventional BBB-on-chip models that primarily focus on endothelial barrier properties, NVU chips incorporate neuronal and glial components to reproduce bidirectional signaling between vascular and parenchymal compartments, thereby bridging vascular physiology with neural function and pathology [25,26].
Recent engineering advances have enabled three-dimensional (3D) co-culture configurations in which perfusable endothelial tubes enveloped by astrocytes and neurons embedded within extracellular matrix hydrogels, allowing astrocytic endfeet to contact the vascular wall and neuronal processes to extend through the matrix, facilitating spontaneous synaptic activity and physiologically relevant signaling [27,28]. Controlled microfluidic perfusion generates shear stress that promotes endothelial tight-junction assembly and transporter expression, thus improving barrier fidelity and enabling quantitative assessment of molecular permeability, neuroinflammation, and neurovascular coupling under defined hemodynamic conditions [29-31].
NVU-on-a-chip models have been leveraged to study a range of central-nervous-system (CNS) disorders including ischemic stroke, Alzheimer's disease (AD), and amyotrophic lateral sclerosis (ALS). Transient ischemia can be reproduced by regulating flow or oxygen tension, allowing evaluation of barrier breakdown, excitotoxicity, and neurorestorative interventions such as stem-cell-based repair [32]. iPSC-derived human NVU chips carrying familial AD mutations exhibit hallmark phenotypes such as β-amyloid accumulation, pericyte dysfunction, and impaired barrier integrity, mirroring patient-specific pathophysiology [33-35]. These personalized microphysiological systems enable mechanistic dissection of endothelial-glial signaling and preclinical screening of BBB-penetrant therapeutics, neuroprotective compounds, and nanoparticle-based delivery vehicles [36-38].
Integration of biosensing and real-time analytics further enhances the translational potential of NVU chips [39-41]. Embedded microelectrodes and impedance sensors permit continuous monitoring of trans-endothelial electrical resistance (TEER), neuronal firing, and cytokine dynamics, facilitating kinetic profiling of drug transport and immune-vascular interactions in defined microenvironments [42-44]. Exposure to inflammatory cytokines or amyloid-β oligomers induces quantifiable barrier leakage and endothelial toxicity, reproducing early pathological events of neurodegenerative disease progression [45].
Despite these advances, achieving standardized, long-term NVU-on-a-chip systems remains a major challenge. Maintaining co-culture stability, incorporating microglia without excessive inflammatory drift, and establishing reproducible manufacturing pipelines are ongoing priorities. Future designs are expected to integrate metabolic coupling with peripheral organ modules, such as gut or liver, to capture systemic influences on the brain and expand applicability for CNS drug-delivery testing [21,46]. Furthermore, incorporating glymphatic-like fluid dynamics to replicate cerebrospinal-interstitial exchange and metabolic waste clearance, together with the use of patient-derived iPSCs to reflect interindividual variability in neurovascular function, represents a crucial direction for future translational research [47-49].
2.3 Neuromuscular junction (NMJ)-on-a-chip
Neuromuscular junction (NMJ)-on-a-chip systems emulate the synaptic interface between motor neurons and skeletal muscle fibers, creating a microphysiologically relevant environment for studying signal transmission, excitation-contraction coupling, and neuromuscular disorders. Microfluidic compartmentalization allows directed axonal projection from neuronal chambers toward muscle compartments, enabling compartment-specific control of chemical gradients and trophic cues (Figure 2C) [50,51]. The integration of 3D hydrogel matrices and aligned myofiber scaffolds contributes to structural fidelity, physiological contractility, and mechanical feedback [52,53]. These engineered systems combined with microelectrode arrays (MEAs) or optogenetic stimulation tools provide precise quantitative readouts of neurotransmission, muscle contractility, and synaptic function under defined perfusion conditions [54,55].
The application of iPSC technology has advanced NMJ modeling by enabling patient-specific neuromuscular disease reconstruction [56-58]. Recently, rapid iPSC-based NMJ-on-a-chip system was introduced wherein cryopreserved human motor neurons and skeletal myotubes formed mature synaptic junctions within 12 days, recapitulating SOD1 mutation-related degeneration observed in amyotrophic lateral sclerosis (ALS) [59]. This system demonstrated functional NMJ coupling validated through α-bungarotoxin labeling and electrophysiological recordings. Similarly, Osaki et al presented an ALS-on-a-chip integrating iPSC-derived motor neurons with optogenetically controlled synaptic inputs to model glutamate excitotoxic injury and its impact on NMJ integrity, thereby linking neurodegeneration to synaptic dysfunction [60]. More recently, geometrically engineered assembloid-on-a-chip systems have expanded this framework by incorporating three-dimensional, self-organizing co-cultures of motor neurons, Schwann cells, and muscle fibers, achieving synchronized optogenetically evoked contractions that mimic in vivo neuromuscular circuitry [61]. Comparative analyses across these models highlight that while dense neuronal populations enhance motor neuron viability, optimal NMJ formation often relies on lower neuronal density to promote selective neuron-to-muscle connectivity.
NMJ-on-a-chip systems employ diverse analytical modalities to quantify pre- and postsynaptic functionality. MEA-based recordings provide electrophysiological insights into presynaptic burst patterns and muscle depolarization, whereas calcium imaging enables real-time visualization of excitation-contraction coupling kinetics [62,63]. Molecular assays, such as acetylcholine receptor clustering, agrin and MuSK signaling assessment, and postsynaptic membrane stability tests, serve as biomarkers of synaptic maturation and responsiveness [64]. Beyond disease modeling, emerging bioactuated applications integrate NMJ tissues into soft robotic and neuromorphic frameworks, enabling biologically controlled mechanical outputs [65]. The integration of machine learning algorithms and biosensor feedback loops further refines automatic detection of synaptic fatigue, transmission failure, and plasticity, positioning NMJ-on-chip models as valuable testbeds for neurotoxicology and regenerative drug discovery [66,67].
Although substantial progress has been made, several challenges remain in sustaining long-term synaptic integrity, coordinated cell survival, and functional adaptability during prolonged stimulation. Future directions involve incorporating myelinating Schwann cells and fibroblasts to restore trophic support and synaptic repair pathways, as well as coupling NMJ units with vascular or hepatic modules in multi-organ circuits to model metabolic and inflammatory influences on neuromuscular health [68,69]. Simultaneously, efforts are ongoing to enhance throughput via automated optical stimulation and AI-assisted image analysis, enabling large-scale screening of therapeutic compounds targeting neuromuscular transmission pathways.
2.4 Heart in multi-organ platforms
The heart plays a central role in multi-organ physiology and pharmacokinetics, acting as both a target and mediator of systemic drug circulation. Integrating a heart-on-a-chip (HoC) into MOoC systems is particularly critical for evaluating drug efficacy and cardiotoxicity under physiologically relevant flow and mechanical conditions [70,71]. HoC devices replicate the myocardium's electrophysiological and contractile properties using microfluidic and tissue-engineering approaches, which enable real-time evaluation of cardiac function, rhythm, and metabolic coupling under controlled perfusion [72,73]. By embedding cardiomyocytes, often derived from iPSCs, within mechanically active microenvironments, these systems recapitulate the beating and electromechanical synchronization characteristic of native cardiac tissue, surpassing traditional 2D culture limitations (Figure 2D) [74,75].
In multi-organ contexts, the HoC often interfaces with key metabolic and regulatory organs such as the liver, kidney, or brain to evaluate systemic pharmacokinetic behavior and multi-organ drug responses [76,77]. For example, heart-liver chip models have been successfully used to assess cardiotoxic effects of hepatic drug metabolites. Co-cultured liver-heart chip could simulate clomipramine-induced cardiotoxicity dependent on hepatic metabolic processing, thereby illustrating the predictive power of inter-organ coupling. Such systems allow evaluation of both primary cardiac drug effects and secondary, metabolism-driven toxicity, which are otherwise challenging to predict using single-organ assays [78,79].
More advanced integrations include neuro-cardiac-on-a-chip constructs, which investigate bidirectional signaling between neurons and cardiomyocytes [80,81]. These hybrid systems recreate neuro-cardiac junctions essential for understanding arrhythmias and stress-induced heart dysfunctions [82,83]. The combination of human iPSC-derived cardiomyocytes and neurons within microfluidic co-culture platforms has enabled dynamic, patient-specific modeling of autonomic regulation in cardiac pathophysiology [84,85].
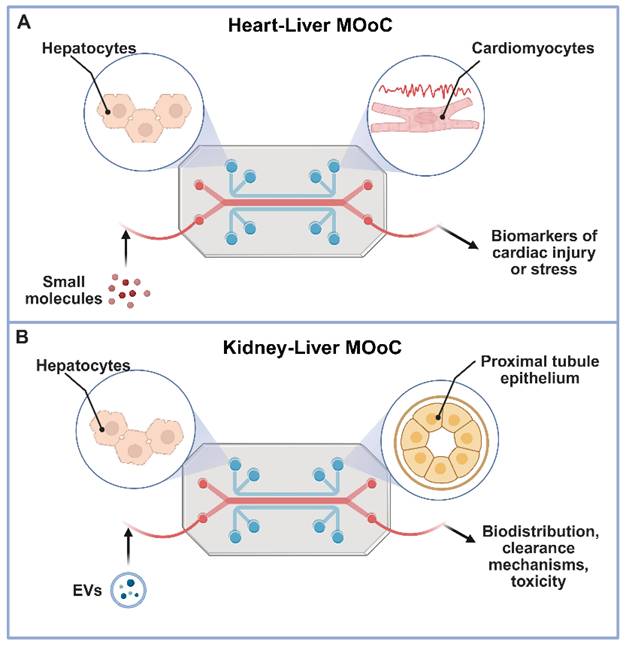
Analytical endpoints in heart-integrated MOoC setups combine electrical, biochemical, and mechanical measurements. Typical readouts include contractile amplitude and frequency, field potential duration, and calcium transients to assess tissue excitability and drug response [86-88]. Simultaneous measurement of biomarkers such as troponin I or brain natriuretic peptide (BNP) provides indicators of cardiac injury or stress [89,90]. Integration of biosensors directly within these systems enables real-time monitoring of beating patterns and metabolic fluxes under cyclical perfusion. Additionally, liver-heart coupled systems have adopted metabolomics and electrophysiological metrics to distinguish between reversible and irreversible toxicity profiles, improving preclinical evaluation accuracy [9,91]. Multi-organ circuits containing hepatic, cardiac, and vascular modules have also been developed to replicate systemic circulation and realistic drug distribution profiles, moving closer to whole-body pharmacokinetic simulation (Figure 3A) [92,93].
Multi-organ-on-a-chip systems for comprehensive drug delivery evaluation. This figure illustrates the power of multi-organ-on-a-chip (MOoC) platforms in evaluating advanced drug delivery strategies by modeling inter-organ communication and systemic effects under physiologically relevant conditions. Panel A depicts a Heart-Liver MOoC, demonstrating its utility in assessing metabolism-driven cardiotoxicity, where a parent drug is metabolized in the liver, and the resulting metabolites circulate to induce potential adverse effects in the heart [92]. Panel B showcases a Kidney-Liver MOoC, highlighting its role in tracking the biodistribution and renal impact of agents like extracellular vesicles (EVs) or natural compounds, which are processed by the liver before circulating to the kidney, allowing for the evaluation of clearance, nephrotoxicity, or therapeutic outcomes [98]. Together, these examples underscore how MOoC systems provide crucial, human-relevant insights into the complex systemic fate and organ-specific responses to diverse advanced therapeutics. Created in BioRender (https://biorender.com/qe5ibkt).
2.5 Liver in multi-organ platforms
The liver is extremely relevant in drug metabolism, specifically with Phase I and II reactions. It is responsible for the biotransformation of a wide range of compounds, including oxidation, reduction, conjugation, and hydrolysis reactions that can either activate or inactivate therapeutic agents. Drugs taken orally, after absorption through the intestine, encounter the “first-pass” effect when passing through the liver, where a large portion of the drug is metabolized and removed from the bloodstream. Since this organ can have significant impact on the bioavailability of a drug, it is crucial to understand how hepatic metabolism alters the active form, concentration, and half-life of administered compounds, and it demonstrates the need to link this system to upstream (such as gut and skin) and downstream (such as kidney and tumor) modules in order to accurately predict metabolic effects.
We return to the early four-organ-chip model to understand how the inclusion of liver contributes to systemic drug processing. This system linked intestine, liver, kidney, and skin tissues via dynamic flow, and demonstrated metabolic activity sustained for over four weeks. This liver model showed some enzymatic activity and albumin production comparable to in vitro monocultures and demonstrates that some liver functionality is capable in liver-on-chip models [4].
Experiments that explore the liver-on-chip module as a metabolic gatekeeper observed controlled changes in concentration of parent drugs and metabolites at downstream heart, tumor, and lung modules, in the context of chemotherapeutics and targeted small molecules [94,95]. These studies demonstrated that hepatic metabolism could either reduce drug efficacy at the tumor site or amplify off-target toxicity at cardiac tissues, depending on the metabolic fate of the compound. The lung-liver platform additionally shows that cytokines produced in one tissue would affect barrier integrity and response in the other, indicating inflammatory crosstalk between organ modules in response to metabolic products or stress [94].
A key consideration in this system is the need for long-term maintenance of hepatic function and viability under continuous flow. Since chronic exposure is relevant in many toxicological and pharmacokinetic studies, it is necessary to ensure that these systems can maintain function for extended time periods. Use of dynamic perfusion systems such as peristaltic micropumps or pumpless gravity-driven flow setups can assist with the required viability and function. In terms of future directions, there is growing relevance in integrating iPSC-derived hepatocytes, modeling disease-specific liver phenotypes, and mimicking spatial heterogeneity in liver function [6].
2.6 Kidney in multi-organ platforms
The kidney plays an essential role in eliminating hydrophilic drugs from the bloodstream, and as such can have a significant effect on systemic clearance and drug half-life. Drug-relevant processes that occur within the kidney, specifically in the nephrons, include glomerular filtration and phase III transporter-mediated secretion and reabsorption, all processes which can influence drug efficacy and toxicity.
One of the key components of getting accurate pharmacokinetic information from a kidney model is the presence and detection of key transporter activity. An accurate kidney model requires polarized proximal tubule epithelium expressing active transporters such as OATs, OCTs, and MATE family transporter proteins [96]. In the work done by Chang et al., they demonstrate that maintaining dynamic fluid flow is necessary in this system to simulate shear stress and produce functional differentiation, resulting in transporter expression. They further emphasize that fluidic shear is essential for sustaining this expression over time and preventing de-differentiation of epithelial cells [97].
In an example of multi-organ interaction, Nguyen et al. showed a functional kidney-liver on-a-chip model altering the biodistribution of extracellular vesicles and natural compounds such as silymarin and berberine. They demonstrated crosstalk between the liver and kidney model and the effect of liver dysfunction on the kidney model, which mirrored clinically relevant hepatorenal effects observed in diseases like MAFLD. They also exposed the system to natural compounds, which were metabolized by the liver and then received by the kidney, allowing them to assess nephrotoxicity, clearance, and the downstream impact of liver-derived metabolites on renal function (Figure 3B) [98].
Many current MOoC designs rely on computational or other artificial modes of elimination that may serve as a rudimentary stand-in for renal clearance but lack physiologically relevant transporter expression and inter-organ feedback. Future directions should integrate functional kidney modules with glomerular and tubular components, real-time readouts of renal transport, and disease-relevant models that can better emulate impaired renal clearance.
2.7 Organoids in multi-organ platforms
Three-dimensional organoids have emerged as powerful in vitro models that recapitulate native tissue cytoarchitecture, multicellular organization, and developmental patterning in ways that monolayer cultures or simplified co-culture systems cannot achieve. Derived from human iPSCs or primary patient biopsies, organoids self-organize into physiologically relevant structures that recapitulate spatial gradients, stem cell niches, and functionally distinct cellular subtypes [99]. While traditional organoid systems lack vascular perfusion and often exhibit diffusion-limited nutrient and oxygen transport, integrating organoids within microfluidic platforms has enabled a new class of organoid-on-a-chip technologies that merge the physiological strengths of 3D tissues with the controlled perfusion and barrier dynamics of microfluidics [100].
Microengineered organoid-on-chip platforms have been reported for a range of tissues, including gut, liver, kidney, brain, and tumor microenvironments, each offering enhancements over 2D or planar engineered constructs [100]. For example, the incorporation of primary or iPSC-derived intestinal organoids into microfluidic channels has been shown to promote crypt-villus architecture, mucus layer formation, and regionalized epithelial maturation under physiologically relevant flow and shear stress [101,102]. Similarly, cerebral and neurovascular organoid-on-chip systems support increased neuronal maturation, functional network activity, and improved modeling of neurodevelopmental or neurodegenerative processes by combining organoid self-organization with perfusable vascular-like structures [103-105]. These constructs represent important complements to the BBB- and NVU-on-chip technologies described earlier and serve as promising tools for investigating neuronal-vascular coupling, inflammatory signaling, and patient-specific neurological phenotypes.
Organoid models also provide substantial advantages for metabolic and clearance organs, where 3D architecture, ECM-rich microenvironments, and zonated functional domains are key drivers of physiological function. Multi-organ systems incorporating liver or kidney organoids have demonstrated improved long-term viability and metabolic fidelity compared to 2D or scaffold-seeded tissues. A notable example includes a kidney-liver organoid-based MOoC used to study extracellular vesicle biodistribution and hepatorenal crosstalk, revealing disease-relevant alterations in clearance and toxicity pathways under dynamic perfusion [98]. Tumor organoid-on-chip systems have similarly advanced the study of cancer drug responses by enabling vascularized, stromalized 3D tumor architecture with improved prediction of drug penetration, therapeutic efficacy, and off-target toxicity [106-109]; capabilities that are difficult to achieve in static organoid cultures alone.
The integration of organoids into multi-organ microphysiological circuits represents an important step toward achieving physiologically scalable multi-tissue interactions. Organoid-on-chip models have been applied to study gut-brain axis communication, NVU dysfunction, metabolic disease, and tumor-immune dynamics, offering enhanced biological fidelity for drug absorption, metabolism, and toxicity evaluation. Their patient-specific nature also positions them as valuable platforms for personalized medicine and precision drug screening, particularly for modalities such as biologics, nanoparticles, gene therapies, and extracellular vesicles that interact with complex 3D tissue structures and restrictive biological barriers.
Despite these advantages, several challenges remain. Organoids exhibit batch-to-batch variability, and their self-organized geometry can complicate standardization and reproducibility. Achieving robust vascular integration, scaling organoid size for perfusion compatibility, and incorporating immune or stromal elements remain active areas of research. Nevertheless, ongoing engineering advances, including organoid vascularization strategies, guided morphogenesis, and controlled spatial seeding within microfluidic compartments, continue to improve consistency and functional maturation. As these techniques are refined, organoid-on-a-chip platforms are expected to play a central role in next-generation multi-organ systems by bridging the gap between developmental biology, engineered microenvironments, and human-relevant drug evaluation.
2.8 Pharmacokinetic validation and computational approaches in multi-organ platforms
MOoC platforms are increasingly positioned as human-relevant tools for improving prediction of ADME. However, their translational utility critically depends on the extent to which chip-derived data can quantitatively reproduce PK parameters observed in vivo or in clinical settings. In recent years, the field has progressed beyond qualitative demonstrations toward more rigorous, quantitative validation through integration with physiologically based pharmacokinetic (PBPK) modeling frameworks [110,111].
A landmark study by Herland et al. demonstrated that fluidically coupled, vascularized human organ chips could quantitatively predict human plasma concentration-time profiles for multiple drugs [112]. By linking liver, intestine, kidney, and other organ modules under shared circulation and coupling experimentally measured transport and metabolism parameters with PBPK models, the authors achieved close agreement with clinical PK data across different compounds. This work provided one of the first demonstrations that MOoC-derived parameters, such as intrinsic clearance, permeability, and organ-specific extraction, can be directly translated into whole-body PK predictions.
Subsequent studies have further clarified the role of MOoC platforms as experimental parameter generators for PBPK modeling rather than standalone predictors [113,114]. MOoC systems can provide human-specific inputs including metabolic turnover rates, barrier permeability coefficients, transporter activity, and time-resolved concentration profiles. When incorporated into PBPK frameworks, these parameters improve mechanistic interpretability and reduce uncertainty associated with interspecies scaling, particularly for drugs with complex metabolism or barrier-limited distribution [115].
Despite these advances, important limitations remain. Quantitative concordance is strongest for small-molecule therapeutics, while validation for biologics, nanoparticles, and gene-based modalities is still emerging. Challenges related to physiological scaling of organ size and flow, material-dependent drug adsorption, simplified renal and immune clearance pathways, and limited culture duration continue to constrain absolute PK accuracy. As emphasized by Ingber [116], regulatory agencies currently view organ-chip technologies not as replacements for animal models, but as complementary, decision-support tools that can strengthen mechanistic confidence and inform translational decision-making when used within defined contexts.
In recent years, computational approaches have progressed beyond conceptual demonstrations and are now being practically coupled with MOoC-derived data to inform physiologically based pharmacokinetic (PBPK) models. For example, human liver-on-chip systems have been used to generate intrinsic clearance and metabolic turnover rates that accurately parameterize PBPK models, yielding plasma concentration-time profiles concordant with clinical observations across multiple small-molecule drugs [115,117]. Similarly, intestine-liver two-organ chips have provided experimentally derived permeability, efflux ratios, and first-pass metabolism constants that, when incorporated into mechanistic absorption models, improve the prediction of oral bioavailability and hepatic extraction [117,118]. BBB-on-chip platforms have generated human-relevant drug permeability coefficients and TEER values that integrate directly into CNS PBPK frameworks, enabling quantitative prediction of brain partitioning and exposure. Chip-based hepatic models have also produced transporter and enzyme inhibition constants suitable for drug-drug interaction modeling, with PBPK simulations accurately reproducing known clinical interactions [115]. Together, these examples highlight that MOoC platforms can now supply validated, human-specific parameters for computational models, including clearance, permeability, metabolism, partitioning, and transport kinetics. Despite this progress, several limitations remain. Current efforts often rely on simplified chip geometries, incomplete representation of systemic clearance pathways, or short culture durations that limit long-term PK/PD prediction [118], and scaling non-physiological flow rates, surface-area-to-volume ratios, and organoid sizes into whole-body PBPK models remains challenging [115]. Moreover, integration of large biologics, nanoparticles, gene therapies, and cell-based modalities into PBPK frameworks is only beginning to emerge, given the limited availability of quantitative chip-derived ADME data. Continued development of computational models informed by rigorously standardized MOoC parameters will be essential for enabling predictive human-relevant PK/PD estimations in the future.
3. Microphysiological systems-driven advances in drug delivery strategies
In this section, we focus on how microphysiological systems (MPS), which are engineered in vitro platforms that recapitulate key structural, mechanical and functional features of human tissues using microfluidics and advanced biomaterials, can be leveraged to evaluate next generation drug delivery strategies. Within this broad category, MOoC platforms represent a subset of MPS in which multiple organ tissue or organ modules are interconnected by a shared perfusion circuit to model pharmacokinetics and inter-organ crosstalk. Unless otherwise noted we use “MPS” to refer to both single-organ and multi-organ platforms, and “MOoC” specifically for interconnected multi-organ systems.
3.1 Stem cell-based delivery in MPS
Stem cells have emerged as promising therapeutic carriers owing to their intrinsic regenerative capacity, tissue-homing properties, and immunomodulatory potential [119]. Initially, many strategies focused on using whole cells as delivery vehicles engineered to secrete therapeutic proteins, cytokines, and growth factors at disease sites [120]. However, the intrinsic complexity and heterogeneity of live cells pose substantial challenges for manufacturing, quality control and long-term safety. As a result, the primary use of stem cells in drug delivery applications shifted towards harnessing stem-cell derived extracellular vesicles (EVs), which we review in Section 3.4. Regardless, here we briefly describe whole-cell drug delivery approaches in the literature and highlight potential MPS applications.
Neural stem cells (NSCs), for example, demonstrate translational relevance for central nervous system (CNS) disorders, where they can migrate across the blood-brain barrier (BBB) and deliver therapeutic payloads to sites of pathology [121]. Mesenchymal stem cells (MSCs) have also been studied as delivery vehicles in oncology due to their ability to home to tumors [122]. Taking advantage of this feature, researchers have investigated genetically modified MSCs as targeted delivery systems, engineering them to secrete therapeutic proteins such as interferon (IFN)-β or TRAIL within the tumor microenvironment to suppress tumor growth [122,123]. In another example, polymer-based gene delivery systems, such as those using poly(beta-amino ester) (PBAE) nanoparticles, can be used to ex vivo non-virally engineer MSCs to secrete VEGF to induce angiogenesis and tissue regeneration [124] or secrete BMP4 to target and turn off brain tumor initiating cells, putative brain cancer stem cells, to extend brain cancer survival in mice [123].
Nevertheless, critical hurdles remain beyond manufacturing, including immune rejection, uncontrolled differentiation, and tumorigenic risk. These limitations have motivated a shift toward acellular stem cell-derived products, particularly EVs, but also highlight the need for human-relevant platforms to systematically evaluate whole-stem cell therapies where they remain of interest. Indeed, MPS and MOoC platforms offer controlled microenvironments in which stem cell-based therapies can be evaluated for homing, engraftment, and functional activity, while simultaneously providing an opportunity to assess biodistribution, safety, and therapeutic efficacy across interconnected organ models under physiologically relevant conditions [125].
3.2 Gene and nucleic acid delivery in MPS
Gene delivery technologies have rapidly progressed through viral and non-viral approaches. Viral vectors such as adeno-associated viruses (AAVs) and lentiviruses remain the most widely employed owing to their efficiency and tissue tropism, with next-generation capsids engineered to minimize immunogenicity and enhance payload specificity [126]. Non-viral carriers, including lipid nanoparticles (LNPs) and polymeric nanoparticles (PNPs), as well as physical methods of delivery, such as electroporation, are increasingly being investigated due to their advantages for safety, manufacturing scalability with a low cost of goods, large cargo capacity, flexible targeting capability, and tunable gene expression durability [121,127]. More recently, CRISPR/Cas9 and other precision editing platforms have been incorporated into delivery systems, improving on-target activity for durable changes in gene expression while reducing off-target events [128].
For instance, an iPSC-derived retina-on-a-chip system was recently used to characterize AAVs based on their transduction efficacy and cell tropism [129]. While many studies have interrogated AAV-driven gene delivery in human organoid models, this study was the first to use MPS to test AAV transduction efficacy (Table 3) [129,130]. This work illustrates the great potential of MPS technologies as gene delivery screening platforms, allowing for the evaluation of vector biodistribution, organ-specific uptake, and immunological responses, which are factors that are often insufficiently captured in static culture or animal models [131].
Drug delivery modalities tested in MOoC systems
| Delivery Strategy | Specific Examples | MOoC Platform Used | Key Findings | Current Limitations | References |
|---|---|---|---|---|---|
| Stem Cell-Based | MSCs for immunomodulation, NSCs for CNS delivery, engineered stem cells | NVU-on-chip, ischemic stroke model, various organ chips | MSCs restored BBB integrity and neuronal survival post-ischemia; stem cells as delivery vehicles demonstrated | Immune rejection risk, differentiation control, limited multi-organ studies, scalability | [31,113,116-118,173] |
| Gene Therapy | AAV vectors for retinal delivery, CRISPR/Cas9 delivery, gene transfer nanoparticles | Retina-on-chip (single organ), various MPS platforms | Capsid variant screening, long-term viability, improved targeting and efficiency | Lacks immune components in most models; needs multi-organ integration for systemic delivery; off-target effects | [121,128-130] |
| Nanoparticles | Polymeric NPs, lipid NPs, metal NPs, functionalized nanorods, nanoshuttles | Tumor-on-chip, BBB-on-chip, liver-kidney systems, tumor microenvironment chips | Biodistribution tracking, clearance pathways, toxicity assessment, BBB penetration strategies, tumor-specific delivery | Limited systematic multi-organ PK/PD studies; need for better predictive in vivo correlation | [7,127,133-135,138-140,142,143] |
| Extracellular Vesicles | MSC-derived EVs, engineered exosomes, iPSC-organoid-derived EVs | Liver-kidney organoid MOoC, lung injury chip, gut-brain axis, feto-maternal interface, cardiac regeneration models | Biodistribution altered by liver dysfunction; therapeutic effects in acute lung injury; gut-brain communication; pregnancy pathology modeling | Scale-up production, standardization challenges, loading efficiency, targeting specificity | [98,144,147-151] |
| Biomaterials | Hydrogels, scaffold-based depots, shape memory polymers, drug-eluting meshes, microcapsules, hybrid systems | 3-tissue platform, wound repair models, glioblastoma models, tumor models | Sustained release, spatiotemporal control, tissue integration, localized drug delivery, controlled release mechanisms | Limited representation in current MOoC literature; need for more multi-organ validation | [12,153-158,160] |
| Small Molecules | Chemotherapeutics, targeted drugs, donepezil, antidepressants, topical agents | Heart-liver-tumor-lung, gut-brain axis, multi-organoid systems | Metabolic conversion, organ-specific toxicity, PK/PD correlation, off-target effects, safety assessment | Most common modality; well-studied but still gaps in complex multi-organ interactions | [20,78,79,94,95] |
Indeed, MPS/MOoC platforms could provide a marked advantage for quantifying the transduction efficiency, cell-type specificity, and durability of expression of gene and nucleic acid therapeutics in human-relevant tissues under controlled flow and barrier conditions [132]. When extended to MOoC configurations that link metabolic and clearance organs with target tissues, these systems can reveal patterns of off-target transduction in a single experiment, helping to de-risk gene delivery vectors and refine dosing strategies before clinical studies.
3.3 Nanoparticle and nanocarrier delivery in MPS
Nanoparticles (NP) are among the most versatile and widely investigated drug delivery systems. Polymeric, lipid-based, metallic, silica, and hybrid nanoparticles have been engineered to encapsulate diverse therapeutic cargos, regulate release kinetics, and prolong circulation [133]. Surface functionalization with antibodies, peptides, or aptamers enables active targeting, while stimuli-responsive designs provide spatiotemporal control over cargo release [134]. Despite these advances, challenges such as immune clearance and systemic instability persist. Biomimetic nanoparticles, such as red blood cell membrane-coated systems, are being developed to circumvent immune recognition [135]. Biomimetic particles can mimic particle size, shape, and surfaces of naturally occurring biological cells or particulates to have improved biodistribution and targeting properties [136].
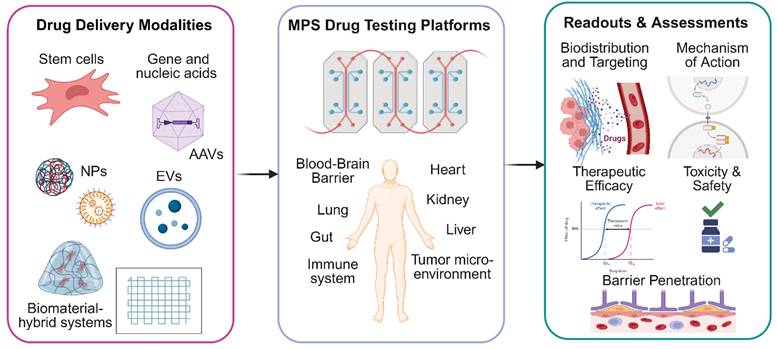
MPS technologies offer advanced platforms for preclinical assessment of engineered nanoparticles by mimicking the complex structure-function of human organs, and by investigating how properties like size, shape and surface charge affect their targeted delivery and therapeutic efficacy (Table 3) [133,137]. Studies have used tumor-on-a-chip models to show that smaller nanoparticles penetrate tumor tissue more effectively [138]. Moreover, the ability to replicate the microenvironment, such as fluid shear stress and 3D tissue architecture allows for more accurate prediction of how nanoparticles will behave in vivo compared to 2D cultures [139]. MPS models are key for evaluating the transport properties of new medicines, including when the medicines have unique sustained release profiles or unique extracellular interactions. For example, vascularized tumor-on-a-chip systems can best quantify drug flux through stroma and tumor in a way that is predictive of patient responses, especially if patient-specific cells are utilized [140]. Beyond efficacy, MPS models have been used to test potential nanoparticle toxicity and safety. For example, lung-on-a-chip systems demonstrated that mechanical forces from breathing can worsen the adverse effects of silica nanoparticles, which are toxic to the alveolar-capillary barrier [141]. Indeed, MPS platforms enable detailed assessment of NP biodistribution, cellular uptake, clearance pathways, and toxicity under physiologically relevant fluid dynamics, thereby improving translational accuracy compared to traditional in vitro or in vivo testing [142,143].
Compared with 2D cultures, MPS platforms incorporate flow conditions, vascular architecture tissue barriers, all of which influence nanoparticle margination, extravasation and tissue penetration. For example, tumor-on-a-chip systems with perfused microvasculature enable systematic tuning of nanoparticle size, shape, and surface chemistry while directly measuring intratumoral transport and therapeutic response [138-140]. MPS platforms are also powerful systems for interrogating nanoparticle biodistribution, metabolism, clearance, and off-target toxicity, and therefore represent valuable tools for de-risking nanoparticle-based therapeutics prior to clinical translation (Figure 4).
Microphysiological systems (MPS) for evaluating next generation drug delivery strategies. This schematic summarizes the application of multi-organ-on-a-chip (MOoC) platforms as advanced preclinical models for the evaluation of novel drug delivery modalities. Diverse therapeutic carriers, encompassing stem cell-based system, gene and nucleic acid therapies, engineered nanoparticles (NPs), extracellular vesicles (EVs) and biomaterial-hybrid systems are introduced into these integrated microphysiological platforms. The MOoC systems incorporate human-relevant organ and organ-system models, such as tumor, blood-brain barrier (BBB), liver, kidney, lung, gut, and cardiac tissues, interconnected through dynamic microfluidic circulation. This integrated approach facilitated the assessment of crucial parameters: (1) Biodistribution and Organ-Specific Accumulation of the therapeutic agent; (2) Barrier Penetration across physiological interfaces like the BBB; (3) Therapeutic Efficacy at target tissues (e.g., tumors); (4) Off-Target Toxicity and (5) Elucidation of Mechanisms of Action. Created in BioRender (https://BioRender.com/xdj22lo).
3.4 Extracellular vesicle-mediated delivery in MPS
Extracellular vesicles (EVs), including exosomes and microvesicles, are increasingly recognized as natural nanocarriers due to their intrinsic biocompatibility, ability to cross biological barriers, and suitability for precision drug delivery [144-146]. EVs can be derived from diverse cell types, including MSCs and engineered producer lines, and can be loaded with therapeutic RNAs, proteins, or genome-editing cargos. Current strategies focus on enhancing EV targeting specificity via ligand engineering, scaling up production, and implementing standardized purification protocols [147].
MPS enables the creation of physiologically relevant disease models where EV efficacy can be evaluated. For instance, Nguyen et al. developed a human kidney-on-a-chip to model acute kidney injury, demonstrating that MSC-derived EVs can recover the renal epithelium after injury [98]. Similarly, Saferzadeh et al. used a complex fetal-maternal interface organ-on-a-chip to model infection-induced inflammation and showed that delivery of IL-10 via engineered EVs reduced inflammation associated with preterm birth [148]. In addition to single-organ applications, MOoC platforms are well-suited for studying extracellular vesicle (EV) biodistribution and organ tropism. Kim et al. proposed a modular MOoC system to investigate EV-mediated interactions within the gut-brain-axis (GBA) [149]. This system features a main body chip that houses multiple GBA organ modules, and its integrated circulation route connects each element, allowing for the observation of labeled EV delivery [149].
Because EVs interact with multiple cell types and barriers, MOoC platforms are particularly powerful for mapping their organ tropism and systemic effects. By integrating relevant organ modules under shared circulation, these systems can reveal how EV dosing and tissue-specific uptake can translate into therapeutic benefit. Critically important in the field of drug development, MPS systems also allow for investigation into EV drug delivered mechanisms of action. For example, Safarzadeh et al. leveraged their platform to show that their IL-10 therapy worked by activating IL-10 signaling pathways while inhibiting pro-inflammatory NF-kB activation [148]. These systems also serve as robust preclinical platforms for assessing pharmacokinetics and safety, as demonstrated by the same study, which tracked EV propagation across a multi-layer biological barrier and confirmed the therapy's lack of cytotoxicity. Collectively, these studies highlight the utility of MPSs to accelerate the translation of EV therapeutics by providing real-time evaluation of biodistribution, immune modulation, and therapeutic outcomes across multiple tissue compartments, thereby accelerating clinical translation (Table 3) [150-152].
3.5 Biomaterial and hybrid delivery strategies in MPS
Biomaterials such as hydrogels, scaffolds, and synthetic extracellular matrix analogues provide spatiotemporal control of therapeutic release while supporting cell viability and tissue integration [153]. These materials can be dynamically tuned to mimic native biochemical and mechanical cues, thereby enabling more predictive preclinical testing [154,155]. Integration of biomaterial-based drug depots into MPS architectures further allows simulation of physiologically relevant pharmacokinetics and localized therapeutic action (Table 3) [156]. Drug delivery systems can also achieve spatiotemporal control of payload release external triggers such as light or ultrasound. For example, thermoplastic polymeric particles can be constructed to release drugs upon a light trigger [157] and elastic eccentric microcapsules can have triggered release following an ultrasound trigger [158]. These approaches can penetrate through the length scales of many MPS constructs. Another intriguing method of spatiotemporal control of biomolecules in a MPS device can be enabled using genetic cellular design combined with an external trigger. For example, Gordana Vunjak-Novakovic and coworkers recently demonstrated the ability to tune endothelial barrier permeability via light-stimulated RhoA signaling in an optogenetic organ-on-a-chip platform [159].
Compelling examples of spatiotemporal control of drug delivery in MPSs include the development of hybrid systems for localized cancer therapy. Chehri et al utilized a 'GlioMesh,' which combined a 3D-printed alginate hydrogel scaffold with drug-loaded PLGA microparticles [160]. This hybrid biomaterial provided spatiotemporal control over the release of two different chemotherapeutics. Crucially, its efficacy was evaluated within a bespoke MPS containing 3D glioblastoma tumoroids embedded in a collagen matrix. This integrated 'tumoroid-on-a-plate' platform enabled the researchers to simultaneously evaluate the therapeutic efficacy and anti-invasive effects of the localized drug depot in a context that mimics the native tumor microenvironment, paving the way for more predictive, personalized therapeutic design [160]. Hybrid systems that combine nanoparticles, EVs, and stem cell carriers with biomaterial scaffolds are emerging as cutting-edge approaches. In multi-organ MPS platforms, such hybrid strategies enable simultaneous evaluation of efficacy, toxicity, and off-target effects in interconnected organ systems, paving the way for personalized therapeutic design [11,161].
Biomaterial depots and hybrid constructs often exhibit complex, spatiotemporal controlled release profiles that interact with local tissue architecture. Embedding these biomaterial systems into MPS and MOoC models allows for measurement of release kinetics, gradient formation, and therapeutic impact into 3D human-relevant microenvironments. In addition, they can help capture how locally released drugs distribute to distal organs and whether degradation of biomaterials can induce off-target toxicity. Indeed, MPS systems can help provide a human-centric comprehensive view of safety and efficacy difficult to achieve with traditional models.
3.6 Immune system and lymphatic transport modeling in MPS
MOoC platforms that simultaneously integrate immune components, such as the mononuclear phagocyte system cells, and lymphatic structures to evaluate next-generation drug delivery systems under immune-mediated clearance and lymphatic transport conditions are not yet well established. Existing studies instead address isolated aspects of these questions, often using murine rather than human cells and tissues, typically in single-organ or lymph node focused MPS. As a result, there is currently no chip-based model that comprehensively links NP or EV design to immune activation, clearance and lymphatic routing within a unified MOoC framework.
Despite this limitation, recent single-organ lymphatic models have begun to elucidate how NPs interact with lymphatic endothelial cells (LECs). Lu et al. developed a lymphatics-on-a-chip model incorporating an engineered lymphatic vessel that drains interstitial fluid containing PLGA-b-PEG NPs of defined sizes. In this system, smaller particles (30 and 50 nm) diffused more rapidly through the interstitial matrix and were preferentially internalized and retained by LECs, paradoxically slowing their appearance in the lymphatic lumen compared with larger 70 nm particles [162]. Through pharmacologic inhibition, Lu et al. further showed that dynamin-, caveolin-, and macropinocytosis-mediated endocytosis, together with regulation of endothelial junctions, are critical determinants of this size-dependent lymphatic transport [162].
Lymph-node-on-a-chip (LNOC) models have primarily been used to probe immune cell behavior and tissue-like architecture, with only limited, non-systematic exploration of carrier design. German et al. introduced a polydimethylsiloxane (PDMS)-based LNOC incorporating a freeze-cast collagen sponge supporting a 3D 4T1 tumor spheroid and examined penetration of fluorescent bovine serum albumin (BSA)-tannic-acid (TA) capsules (0.3 - 4 µm) delivered with lymphocytes, finding the smallest capsules penetrated more effectively into the spheroid [163]. In addition, Cook et al. developed a 3D-printed, multi-compartment-on-a-chip platform that connects an upstream agarose “mock injection site” to a downstream slice of murine skin-draining lymph node tissue under recirculating flow [164]. After introducing a model vaccine (Rho-OVA/R848) into the mock injection site, antigen drainage and spatial accumulation within the mouse LN slice was observed. The resulting patterns of antigen distribution and early activation marker expression resembled those seen after vaccination in mice [164]. Complementing these murine tissue-based systems, Hallfors et al. developed a multi-compartment human LNOC supporting co-cultured Jurkat T cells and Raji B cells under continuous flow, allowing real-time analysis of lymphocyte motility and viability [165].
Together, these studies highlight that current MOoC platforms are still far from capturing the complexity of how innate and adaptive immune mechanisms, lymphatic transport, and clearance physiology influence the pharmacokinetics, biodistribution, and efficacy of next-generation drug modalities. The integration of human lymphatic endothelium, lymph node-like structures, and MPS organs into multi-organ, immune-competent systems thus remains an important and largely unmet goal for the field and represents a key opportunity to make MOoC platforms more predictive for immune-sensitive delivery strategies.
4. Current challenges and future perspectives
Multi-organ-on-a-chip platforms represent a transformative convergence of tissue engineering, microfluidics, and pharmaceutical sciences, offering unprecedented opportunities to bridge the translational gap between preclinical models and clinical outcomes. By recapitulating organ-level functionality, inter-organ crosstalk, and systemic pharmacokinetic behavior within physiologically scaled microenvironments, MOoCs address critical limitations of conventional in vitro assays and animal models [166]. As demonstrated throughout this review, these systems have already enabled mechanistic insights into diverse drug delivery modalities, from viral vectors and nanoparticles to extracellular vesicles and cell-based therapies, while revealing delivery barriers and toxicity profiles that would be difficult to predict using traditional approaches. Looking forward, a variety of biological and technical challenges, along with regulatory advances define the landscape for future progress within the field. Here we highlight several of these challenges.
Biological limitations include cell sourcing; as iPSCs, though desirable for the ability to create patient-specific and diverse cell cultures, face challenges with reproducibility across donors and experiments and often exhibit incomplete maturation during extended culture [17,21,167]. Emerging use of isogenic and genome-edited iPSC lines offers partial solutions by improving batch consistency and enabling controlled genotype-phenotype comparisons [33,34]. The lack of immune cell components also poses a significant gap in understanding of a major part of clinical evaluation that current MOoC models fail to capture [168].
Pharmacological limitations highlight PK/PD modeling difficulties, due to the issue of scaling chip models and organ-to-organ ratios to physiological values. Also, there is a significant bias towards small-molecule studies with MOoC platforms. While much progress has been made in terms of MOoC models for examining small-molecule drug delivery, many other modalities have yet to be thoroughly examined using this new technology. Larger biologics and peptides, gene therapies, and nanoparticles have all been tested on single-organ chip models, but a significant gap lies in holistically examining their pharmacokinetic and pharmacodynamic properties across multiple relevant tissues under shared, physiologically relevant flow conditions [169]. As stated in section 2.8 of this review, computational modeling and digital-twin frameworks are beginning to emerge as tools to bridge this gap, linking experimental chip data with in silico PK/PD scaling for whole-body predictions [9].
As opposed to most small molecules, biologics and peptides are often too large to undergo passive diffusion and therefore rely on mechanisms such as receptor-mediated endocytosis, particularly at restrictive barriers such as the blood-brain barrier [36]. In this context, the inclusion of sequential organ modules such as gut, liver, and BBB within a single MOoC could capture an entire sequence of regions of loss along the path to CNS exposure. The development of endothelial barriers in multi-organ models offers a mode of tracking how these larger molecules pass through each barrier in a sequential manner.
Gene therapies and nucleic acid-based therapeutics face a different major hurdle, namely reaching specific disease-relevant cells while avoiding degradation and expression in off-target sites. Traditional in vitro cultures and animal modeling often lack the reproduction of human-specific biodistribution, clearance, and immune responses needed for a holistic understanding of vector fate. A paper by Achberger et al. has shown the use of a retina-on-chip model for validating adeno-associated virus (AAV) retinal gene therapy vectors. They demonstrated that their model accurately distinguished capsid variants under controlled microfluidic perfusion and retained retinal tissue viability for long-term monitoring [129]. Further development into a multi-organ model could enable mimicking vector delivery through systemic circulation. This model, with further tissue complexity, would be important because these interactions can significantly affect efficacy in patients. Perhaps most importantly, the authors identify that a major shortcoming of their single-organ model is the lack of immune components in their system, resulting in an inability to assess immune responses to AAV vectors. This could be enabled through a multi-organ model incorporating immune cell populations. Future directions may also include coupling vector biodistribution assays with liver, spleen, and kidney modules to capture clearance and innate-immune recognition dynamics [126,130].
For nanoparticles, future MOoCs could be designed to focus on understanding biodistribution, passing through clearance organs such as the liver, before reaching their target. Studies have emphasized that the development of nanomaterials, while becoming quite popular in clinical applications, is significantly delayed due to the inability to test efficacy [7]. The integration of multiple tissue types together within the same MPS, including especially macrophages and immune system phagocytosis, kidney filtration and clearance, and potential hepatotoxicity of degradation products, are all key to modeling the many delivery challenges faced by next-generation therapeutics. With the many possibilities offered by MOoCs, these platforms can help the field move away from animal testing toward improved, clinically relevant evaluation of particle-based therapeutics at lower cost and higher throughput.
A fundamental limitation of current MOoC platforms is the inability to fully recapitulate functional circulatory networks that are essential for physiologically accurate drug distribution and excretion. Although vascularized organ-on-chip models have advanced considerably, many existing systems rely on simplified endothelial-lined channels or non-perfusable capillary networks that lack the hierarchical branching, flow dynamics, and barrier properties of native vasculature [170,171]. True perfusable microvascular networks that approximate physiological vessel diameters, branching architecture, and appropriate shear stress profiles remain technically challenging to engineer and maintain long-term, particularly when integrated across multiple organ modules [171,172]. The lymphatic system, which plays critical roles in fluid homeostasis, immune cell trafficking, and drug clearance, is even more underrepresented in MOoC designs. Recent proof-of-concept studies have demonstrated lymphatic endothelial cell integration into single-organ chips and shown differential biomarker expression under lymphatic versus blood flow condition [173], however developed lymphatic drainage systems connected to blood vasculature within multi-organ platforms are virtually nonexistent. This absence prevents accurate modeling of the role of lymphatic clearance in drug pharmacokinetics, particularly for biologics and nanoparticles that preferentially drain through lymphatics.
There are still several technical limitations being considered in the development of MOoCs. PDMS, a polymer often utilized in chip fabrication, often causes significant, non-specific absorption and adsorption of small, hydrophobic drug molecules. This phenomenon causes substantial loss of drug concentration, sometimes over 90%, leading to inaccurate dose-response results, impaired translatability, and incorrect pharmacological data [174]. Quantitative studies have established that absorption is strongly correlated with a compound's partition coefficient, with molecules exceeding a logP threshold of approximately 2.5 being particularly susceptible, though parameters such as topological polar surface area and exposure time further complicate predictions. Finite element modeling approaches have since been employed to simulate drug concentration dynamics within PDMS devices, enabling more accurate dose-response corrections without requiring full material substitution [175,176]. This has prompted exploration of alternative polymers or surface coatings [5,8]. Recent efforts have focused on replacing PDMS with cyclic olefin copolymers (COC/COP), thiol-ene polymers, and high-resolution 3D-printed photopolymers that minimize drug sorption and improve optical and mechanical stability [6,16]. A recent study by Mair et al. shows that utilizing a PDMS-PEG copolymer, and pretreatment of the polymer with the drug prior to experimental testing can significantly reduce drug absorption [177]. Additionally, new modes of perfusion are being developed to attain more physiologically accurate flow systems for long-term culture, with the goal of maintaining stable shear stress profiles and minimizing media loss or contamination over extended studies [14,16]. Parallel advances in automated plate-based and pneumatic circulation systems have begun to improve throughput, yet reproducibility and cross-site standardization remain major obstacles [16].
From a fabrication standpoint, producing MOoC systems presents substantially greater challenges compared to single-organ platforms. While soft lithography remains a predominant technique, MOoC manufacturing requires intricate multi-step lithographic processes, precise alignment of multiple layers, and integration of distinct organ compartments with controlled geometries and volume ratios [178]. These alignment and bonding steps become increasingly labor-intensive and error-prone as system complexity increases, with device yields often compromised by fabrication defects or fluidic leakage. Fluidic interconnection is also a key challenge, as connecting multiple organ modules via tubing, pumps, and reservoirs introduces dead volumes, increases contamination risk, and requires robust long-term sealing strategies [179,180]. Bubble formation within microfluidic channels can occlude flow and cause experimental failure, an issue exacerbated in multi-compartment systems [179]. Bioprinting has emerged as a promising alternative for constructing tissue-laden MOoC platforms, but still presents limitations, including limited resolution compared to photolithography, challenges in post-printing device sealing and assembly, difficulties in achieving vascularization within printed constructs, and the need for bioinks that maintain cell viability across all printing stages. Additionally, integrating bioprinted tissues into closed-loop microfluidic systems remains technically demanding, as printed constructs must withstand shear stress induced by perfusion, while maintaining structural integrity over culture duration [181].
In parallel with technological advances, the regulatory and policy landscape surrounding MPS and MOoC platforms has evolved rapidly. The passage of the U.S. FDA Modernization Act 2.0 formally acknowledged New Approach Methodologies (NAMs), including organ-on-chip technologies, as potential contributors to drug development and regulatory decision-making [182]. Also, the European Medicines Agency (EMA) have emphasized the potential value of MPS platforms for evaluating human-specific metabolism, toxicity, and complex modalities such as biologics, gene therapies, and nanoparticle-based therapeutics, while underscoring the importance of context-specific validation [183]. This shift reflects increasing recognition that human-relevant in vitro systems can complement traditional animal models, particularly in areas where animal-to-human translation is limited [184]. Importantly, regulatory agencies do not currently position MOoC platforms as direct replacements for animal testing, but rather as complementary, decision-support tools that can improve mechanistic understanding, enable early de-risking, and inform candidate selection and study design [185].
Concurrently, significant efforts are underway to establish technical standards and best practices for MPS and MOoC development [186,187]. International organizations and consortia, including the OECD, ASTM International, and the IQ MPS Affiliate, are actively developing guidelines related to device characterization, reproducibility, quality control, and data reporting. These standardization initiatives aim to enhance cross-platform comparability and experimental robustness, which are essential prerequisites for broader industrial adoption and regulatory confidence. Together, evolving regulatory engagement and standardization efforts signal a transition of MOoC platforms from exploratory research tools toward increasingly translational technologies. Continued dialogue among technology developers, end users, and regulatory agencies will be critical for defining appropriate contexts of use, validation benchmarks, and pathways for integrating MOoC-derived data into pharmaceutical development pipelines.
Acknowledgements
This work was supported by the Global Industrial Technology Cooperation Center (GITCC) through a grant agreement with the Korea Institute for Advancement of Technology (KIAT; project number P0028364), and also supported by the Ministry of Health & Welfare of the Republic of Korea (RS-2024-00438628), and the National Institutes of Health (R01NS133965, R01CA279948, UH3TR003271, R01HL164936, R21AI168886, P41EB028239, P41EB024495).
Language editing was assisted by an AI language model (ChatGPT-5.1). The AI tool was used to improve clarity, grammar, and flow of portions of the text. All scientific content, data interpretation, and references were conceived, selected, and evaluated by the authors.
Competing Interests
D.-H.K. is a scientific founder and shareholder of Curi Bio. Additionally, D.-H.K. serves on the advisory boards of Samsung Biologics and Samsung Bioepis. The authors declare no other conflicts of interest.
References
1. Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotechnol. 2014;32:40-51
2. Van Norman GA. Limitations of animal studies for predicting toxicity in clinical trials. JACC Basic Transl Sci. 2019;4:845-54
3. Bhatia SN, Ingber DE. Microfluidic organs-on-chips. Nat Biotechnol. 2014;32:760-72
4. Maschmeyer I, Lorenz AK, Schimek K, Hasenberg T, Ramme AP, Hübner J. et al. A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents. Lab Chip. 2015;15:2688-99
5. Sung JH. Multi-organ-on-a-chip for pharmacokinetics and toxicokinetic study of drugs. Expert Opin Drug Metab Toxicol. 2021;17:969-86
6. Zhang B, Korolj A, Lai BFL, Radisic M. Advances in organ-on-a-chip engineering. Nat Rev Mater. 2018;3:257-78
7. Ashammakhi N, Darabi MA, Çelebi-Saltik B, Tutar R, Hartel MC, Lee J. et al. Microphysiological systems: next generation systems for assessing toxicity and therapeutic effects of nanomaterials. Small Methods. 2020;4:1900589
8. Cecen B, Karavasili C, Nazir M, Bhusal A, Dogan E, Shahriyari F. et al. Multi-organs-on-chips for testing small-molecule drugs: challenges and perspectives. Pharmaceutics. 2021;13:1657
9. Picollet-D'hahan N, Zuchowska A, Lemeunier I, Le Gac S. Multiorgan-on-a-chip: a systemic approach to model and decipher inter-organ communication. Trends Biotechnol. 2021;39:788-810
10. Ma C, Peng Y, Li H, Chen W. Organ-on-a-chip: a new paradigm for drug development. Trends Pharmacol Sci. 2021;42:119-33
11. Maulana TI, Wevers NR, Kristoforus T, Chandler M, Lanz HL, Joore J. et al. Opportunities for microphysiological systems in toxicity testing of new drug modalities. Annu Rev Pharmacol Toxicol. 2025;65:47-69
12. Skardal A, Murphy SV, Devarasetty M, Mead I, Kang H-W, Seol Y-J. et al. Multi-tissue interactions in an integrated three-tissue organ-on-a-chip platform. Sci Rep. 2017;7:8837
13. Lee H, Kim DS, Ha SK, Choi I, Lee JM, Sung JH. A pumpless multi-organ-on-a-chip (MOC) combined with a pharmacokinetic-pharmacodynamic (PK-PD) model. Biotechnol Bioeng. 2017;114:432-43
14. Wagner I, Materne E-M, Brincker S, Süßbier U, Frädrich C, Busek M. et al. A dynamic multi-organ-chip for long-term cultivation and substance testing proven by 3D human liver and skin tissue co-culture. Lab Chip. 2013;13:3538-3547
15. Parrish J, Lim KS, Baer K, Hooper GJ, Woodfield TBF. A 96-well microplate bioreactor platform supporting individual dual perfusion and high-throughput assessment of simple or biofabricated 3D tissue models. Lab Chip. 2018;18:2757-75
16. Satoh T, Sugiura S, Shin K, Onuki-Nagasaki R, Ishida S, Kikuchi K. et al. A multi-throughput multi-organ-on-a-chip system on a plate formatted pneumatic pressure-driven medium circulation platform. Lab Chip. 2017;18:115-25
17. Kim R, Sung JH. Recent advances in gut- and gut-organ-axis-on-a-chip models. Adv Healthc Mater. 2024;13:2302777
18. Steinway SN, Saleh J, Koo B-K, Delacour D, Kim D-H. Human microphysiological models of intestinal tissue and gut microbiome. Front Bioeng Biotechnol. 2020;8:725
19. Kim M-H, Kim D, Sung JH. A gut-brain axis-on-a-chip for studying transport across epithelial and endothelial barriers. J Ind Eng Chem. 2021;101:126-34
20. Fanizza F, Perottoni S, Boeri L, Donnaloja F, Negro F, Pugli F. et al. A gut-brain axis on-a-chip platform for drug testing challenged with donepezil. Lab Chip. 2025;25:1854-74
21. Hall V, Bendtsen KMS. Getting closer to modeling the gut-brain axis using induced pluripotent stem cells. Front Cell Dev Biol. 2023;11:1146062
22. Kaplan L, Chow BW, Gu C. Neuronal regulation of the blood-brain barrier and neurovascular coupling. Nat Rev Neurosci. 2020;21:416-32
23. Terstappen GC, Meyer AH, Bell RD, Zhang W. Strategies for delivering therapeutics across the blood-brain barrier. Nat Rev Drug Discov. 2021;20:362-83
24. Mulay AR, Hwang J, Kim D-H. Microphysiological blood-brain barrier systems for disease modeling and drug development. Adv Healthc Mater. 2024;13:e2303180
25. Campisi M, Shin Y, Osaki T, Hajal C, Chiono V, Kamm RD. 3D self-organized microvascular model of the human blood-brain barrier with endothelial cells, pericytes and astrocytes. Biomaterials. 2018;180:117-29
26. Yan L, Moriarty RA, Stroka KM. Recent progress and new challenges in modeling of human pluripotent stem cell-derived blood-brain barrier. Theranostics. 2021;11:10148-70
27. Bang S, Lee S-R, Ko J, Son K, Tahk D, Ahn J. et al. A low permeability microfluidic blood-brain barrier platform with direct contact between perfusable vascular network and astrocytes. Sci Rep. 2017;7:8083
28. de Rus Jacquet A, Alpaugh M, Denis HL, Tancredi JL, Boutin M, Decaestecker J. et al. The contribution of inflammatory astrocytes to BBB impairments in a brain-chip model of Parkinson's disease. Nat Commun. 2023;14:3651
29. Brown JA, Codreanu SG, Shi M, Sherrod SD, Markov DA, Neely MD. et al. Metabolic consequences of inflammatory disruption of the blood-brain barrier in an organ-on-chip model of the human neurovascular unit. J Neuroinflammation. 2016;13:306
30. Jeong S, Seo J-H, Garud KS, Park SW, Lee M-Y. Numerical approach-based simulation to predict cerebrovascular shear stress in a blood-brain barrier organ-on-a-chip. Biosens Bioelectron. 2021;183:113197
31. Hajal C, Offeddu GS, Shin Y, Zhang S, Morozova O, Hickman D. et al. Engineered human blood-brain barrier microfluidic model for vascular permeability analyses. Nat Protoc. 2022;17:95-128
32. Lyu Z, Park J, Kim K-M, Jin H-J, Wu H, Rajadas J. et al. A neurovascular-unit-on-a-chip for the evaluation of the restorative potential of stem cell therapies for ischaemic stroke. Nat Biomed Eng. 2021;5:847-63
33. Vatine GD, Barrile R, Workman MJ, Sances S, Barriga BK, Rahnama M. et al. Human iPSC-derived blood-brain barrier chips enable disease modeling and personalized medicine applications. Cell Stem Cell. 2019;24:995-1005.e6
34. Balestri W, Sharma R, da Silva VA, Bobotis BC, Curle AJ, Kothakota V. et al. Modeling the neuroimmune system in Alzheimer's and Parkinson's diseases. J Neuroinflammation. 2024;21:32
35. Gu L, Mao X, Tian C, Yang Y, Yang K, Canfield SG. et al. Engineering blood-brain barrier microphysiological systems to model Alzheimer's disease monocyte penetration and infiltration. Biomater Sci. 2025;13:3650-61
36. Dong X. Current strategies for brain drug delivery. Theranostics. 2018;8:1481-93
37. Zhang W, Mehta A, Tong Z, Esser L, Voelcker NH. Development of polymeric nanoparticles for blood-brain barrier transfer—strategies and challenges. Adv Sci (Weinh). 2021;8:2003937
38. Palma-Florez S, López-Canosa A, Moralez-Zavala F, Castaño O, Kogan MJ, Samitier J. et al. BBB-on-a-chip with integrated micro-TEER for permeability evaluation of multi-functionalized gold nanorods against Alzheimer's disease. J Nanobiotechnology. 2023;21:115
39. Wevers NR, Kasi DG, Gray T, Wilschut KJ, Smith B, van Vught R. et al. A perfused human blood-brain barrier on-a-chip for high-throughput assessment of barrier function and antibody transport. Fluids Barriers CNS. 2018;15:23
40. Choi J-W, Kim K, Mukhambetiyar K, Lee NK, Sabaté del Río J, Joo J. et al. Organ-on-a-chip approach for accelerating blood-brain barrier nanoshuttle discovery. ACS Nano. 2024;18:14388-402
41. Chang Y, Chen T, Geng S, Wang Y, Zhang W, Hu Q. et al. A scenario-adaptive microfluidic chip for constructing in vitro models of biological barriers. Anal Chem. 2025;97:3816-21
42. Booth R, Kim H. Characterization of a microfluidic in vitro model of the blood-brain barrier (μBBB). Lab Chip. 2012;12:1784-92
43. Zeng Y-L, Du Y, Xu X-X, Wang Y-J, Yu S-X, Liu T. et al. On-chip modeling of physiological and pathological blood-brain barrier microenvironment for studying glial responses to neuroinflammation. Nano Today. 2023;52:101947
44. Servais B, Mahmoudi N, Gautam V, Tong W, Ibbotson MR, Nisbet DR. et al. Engineering brain-on-a-chip platforms. Nat Rev Bioeng. 2024;2:691-709
45. Raimondi I, Izzo L, Tunesi M, Comar M, Albani D, Giordano C. Organ-on-a-chip in vitro models of the brain and the blood-brain barrier and their value to study the microbiota-gut-brain axis in neurodegeneration. Front Bioeng Biotechnol. 2020;7:435
46. Kawakita S, Mandal K, Mou L, Mecwan MM, Zhu Y, Li S. et al. Organ-on-a-chip models of the blood-brain barrier: Recent advances and future prospects. Small. 2022;18:e2201401
47. Koh I, Hagiwara M. Modular tissue-in-a-CUBE platform to model blood-brain barrier (BBB) and brain interaction. Commun Biol. 2024;7:177
48. Ma Y, Han Y. Targeting the brain's glymphatic pathway: A novel therapeutic approach for cerebral small vessel disease. Neural Regen Res. 2024;21:433-42
49. Park R, Peng Y, Yslas AR, Lee E. Astrocyte-driven vasoconstriction impairs glymphatic clearance in a human tauopathy-on-chip model. APL Bioeng. 2025;9:026126
50. Ionescu A, Zahavi EE, Gradus T, Ben-Yaakov K, Perlson E. Compartmental microfluidic system for studying muscle-neuron communication and neuromuscular junction maintenance. Eur J Cell Biol. 2016;95:69-88
51. Bellmann J, Goswami RY, Girardo S, Rein N, Hosseinzadeh Z, Hicks MR. et al. A customizable microfluidic platform for medium-throughput modeling of neuromuscular circuits. Biomaterials. 2019;225:119537
52. Osaki T, Uzel SGM, Kamm RD. On-chip 3D neuromuscular model for drug screening and precision medicine in neuromuscular disease. Nat Protoc. 2020;15:421-49
53. Raffa P, Easler M, Urciuolo A. Three-dimensional in vitro models of neuromuscular tissue. Neural Regen Res. 2022;17:759-766
54. Leng Y, Li X, Zheng F, Liu H, Wang C, Wang X. et al. Advances in in vitro models of neuromuscular junction: Focusing on organ-on-a-chip, organoids, and biohybrid robotics. Adv Mater. 2023;35:2211059
55. Duc P, Vignes M, Hugon G, Sebban A, Carnac G, Malyshev E. et al. Human neuromuscular junction on micro-structured microfluidic devices implemented with a custom micro electrode array (MEA). Lab Chip. 2021;21:4223-36
56. Lall D, Workman MJ, Sances S, Ondatje BN, Bell S, Lawless G. et al. An organ-chip model of sporadic ALS using iPSC-derived spinal cord motor neurons and an integrated blood-brain-like barrier. Cell Stem Cell. 2025;32:1139-1153.e7
57. Scherrer C, Loret C, Védrenne N, Buckley C, Lia A-S, Kermene V. et al. From in vivo models to in vitro bioengineered neuromuscular junctions for the study of Charcot-Marie-Tooth disease. J Tissue Eng. 2025;16:20417314241310508
58. Shin M, Ha T, Lim J, An J, Beak G, Choi J-H. et al. Human motor system-based biohybrid robot-on-a-chip for drug evaluation of neurodegenerative disease. Adv Sci (Weinh). 2024;11:2305371
59. Ting H-C, Guo Y-T, Su H-L, Chen Y-S, Lin S-Z, Harn H-J. et al. Rapid iPSC-derived neuromuscular junction model uncovers motor neuron dominance in amyotrophic lateral sclerosis cytopathy. Cell Death Discov. 2025;11:23
60. Osaki T, Uzel SGM, Kamm RD. Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Sci Adv. 2018;4:eaat5847
61. Zhang W, Yu L, Pan J, Deng J, Tong X, Zhao B. et al. Geometrically-engineered human motor assembloids-on-a-chip for neuromuscular interaction readout and hypoxia-driven disease modeling. Nat Commun. 2025;16:8693
62. Tong Z, Seira O, Casas C, Reginensi D, Homs-Corbera A, Samitier J. et al. Engineering a functional neuro-muscular junction model in a chip. RSC Adv. 2014;4:54788-97
63. Yamaoka N, Shimizu K, Imaizumi Y, Ito T, Okada Y, Honda H. Open-chamber co-culture microdevices for single-cell analysis of skeletal muscle myotubes and motor neurons with neuromuscular junctions. Biochip J. 2019;13:127-32
64. Fornetti E, Testa S, De Paolis F, Fuoco C, Bernardini S, Pozo Devoto V. et al. Dystrophic muscle affects motoneuron axon outgrowth and nmj assembly. Adv Mater Technol. 2022;7:2101216
65. Tetsuka H, Gobbi S, Hatanaka T, Pirrami L, Shin SR. Wirelessly steerable bioelectronic neuromuscular robots adapting neurocardiac junctions. Sci Robot. 2024;9:eado0051
66. Johns AE, Maragakis NJ. Exploring motor neuron diseases using iPSC platforms. Stem Cells. 2022;40:2-13
67. Park M, Yang JY, Yeom MJ, Bae B, Baek Y, Yoo G. et al. An artificial neuromuscular junction for enhanced reflexes and oculomotor dynamics based on a ferroelectric CuInP2S6/GaN HEMT. Sci Adv. 2023;9:eadh9889
68. Faustino Martins J-M, Fischer C, Urzi A, Vidal R, Kunz S, Ruffault P-L. et al. Self-organizing 3D human trunk neuromuscular organoids. Cell Stem Cell. 2020;26:172-186.e6
69. Auletta B, Chiolerio P, Cecconi G, Rossi L, Sartore L, Cecchinato F. et al. Tissue-engineered neuromuscular organoids. Commun Biol. 2025;8:1074
70. Criscione J, Rezaei Z, Hernandez Cantu CM, Murphy S, Shin SR, Kim D-H. Heart-on-a-chip platforms and biosensor integration for disease modeling and phenotypic drug screening. Biosens Bioelectron. 2023;220:114840
71. Mourad O, Yee R, Li M, Nunes SS. Modeling heart diseases on a chip: advantages and future opportunities. Circ Res. 2023;132:483-97
72. Liu B, Wang S, Ma H, Deng Y, Du J, Zhao Y. et al. Heart-on-a-chip: a revolutionary organ-on-chip platform for cardiovascular disease modeling. J Transl Med. 2025;23:132
73. Tang Y, Tian F, Miao X, Wu D, Wang Y, Wang H. et al. Heart-on-a-chip using human iPSC-derived cardiomyocytes with an integrated vascular endothelial layer based on a culture patch as a potential platform for drug evaluation. Biofabrication. 2022;15:015010
74. Butler D, Reyes DR. Heart-on-a-chip systems: disease modeling and drug screening applications. Lab Chip. 2024;24:1494-528
75. Ewoldt JK, DePalma SJ, Jewett ME, Karakan MÇ, Lin Y-M, Mir Hashemian P. et al. Induced pluripotent stem cell-derived cardiomyocyte in vitro models: benchmarking progress and ongoing challenges. Nat Methods. 2025;22:24-40
76. Mladěnka P, Applová L, Patočka J, Costa VM, Remiao F, Pourová J. et al. Comprehensive review of cardiovascular toxicity of drugs and related agents. Med Res Rev. 2018;38:1332-403
77. Xu H, Liu G, Gong J, Zhang Y, Gu S, Wan Z. et al. Investigating and resolving cardiotoxicity induced by COVID-19 treatments using human pluripotent stem cell-derived cardiomyocytes and engineered heart tissues. Adv Sci (Weinh). 2022;9:2203388
78. Yin F, Zhang X, Wang L, Wang Y, Zhu Y, Li Z. et al. HiPSC-derived multi-organoids-on-chip system for safety assessment of antidepressant drugs. Lab Chip. 2021;21:571-81
79. de Mello CPP, Carmona-Moran C, McAleer CW, Perez J, Coln EA, Long CJ. et al. Microphysiological heart-liver body-on-a-chip system with skin mimic for evaluating topical drug delivery. Lab Chip. 2020;20:749-59
80. Huang X, Zhao W, Wang Y, Wang Y, Chen T, Zhu L. et al. Bioengineering of heart-brain codevelopoid model via trans-germ-layer codevelopment organoid chip. Engineering. 2025 In press
81. Choi SJ, Liu Z, Yang F, Wang H, George D, Gracias DH. et al. 3D spatiotemporal electrophysiology of cardiac organoids using shell microelectrode arrays. Adv Mater. 2026;38:e06793
82. Silva AC, Matthys OB, Joy DA, Kauss MA, Natarajan V, Lai MH. et al. Co-emergence of cardiac and gut tissues promotes cardiomyocyte maturation within human iPSC-derived organoids. Cell Stem Cell. 2021;28:2137-2152.e6
83. Ziegler KA, Ahles A, Dueck A, Esfandyari D, Pichler P, Weber K. et al. Immune-mediated denervation of the pineal gland underlies sleep disturbance in cardiac disease. Science. 2023;381:285-90
84. Spira ME, Hai A. Multi-electrode array technologies for neuroscience and cardiology. Nature Nanotech. 2013;8:83-94
85. Simats A, Sager HB, Liesz A. Heart-brain axis in health and disease: role of innate and adaptive immunity. Cardiovasc Res. 2025;120:2325-35
86. Liu H, Bolonduro OA, Hu N, Ju J, Rao AA, Duffy BM. et al. Heart-on-a-chip model with integrated extra- and intracellular bioelectronics for monitoring cardiac electrophysiology under acute hypoxia. Nano Lett. 2020;20:2585-93
87. Kim B, Choi JS, Zhu Y, Kim J, Kim YS, Parra A. et al. Effect of electrochemical topology on detection sensitivity in MEA assay for drug-induced cardiotoxicity screening. Biosens Bioelectron. 2025;272:117082
88. Choi JS, Lee HJ, Rajaraman S, Kim D-H. Recent advances in three-dimensional microelectrode array technologies for in vitro and in vivo cardiac and neuronal interfaces. Biosens Bioelectron. 2021;171:112687
89. Goetze JP, Christoffersen C, Perko M, Arendrup H, Rehfeld JF, Kastrup J. et al. Increased cardiac BNP expression associated with myocardial ischemia. FASEB J. 2003;17:1105-7
90. Wereski R, Kimenai DM, Taggart C, Doudesis D, Lee KK, Lowry MTH. et al. Cardiac troponin thresholds and kinetics to differentiate myocardial injury and myocardial infarction. Circulation. 2021;144:528-38
91. Deir S, Mozhdehbakhsh Mofrad Y, Mashayekhan S, Shamloo A, Mansoori-Kermani A. Step-by-step fabrication of heart-on-chip systems as models for cardiac disease modeling and drug screening. Talanta. 2024;266:124901
92. Oleaga C, Riu A, Rothemund S, Lavado A, McAleer CW, Long CJ. et al. Investigation of the effect of hepatic metabolism on off-target cardiotoxicity in a multi-organ human-on-a-chip system. Biomaterials. 2018;182:176-90
93. Juguilon C, Khosravi R, Radisic M, Wu JC. In vitro modeling of interorgan crosstalk: multi-organ-on-a-chip for studying cardiovascular-kidney-metabolic syndrome. Circ Res. 2025;136:1476-93
94. Bovard D, Sandoz A, Luettich K, Frentzel S, Iskandar A, Marescotti D. et al. A lung/liver-on-a-chip platform for acute and chronic toxicity studies. Lab Chip. 2018;18:3814-29
95. McAleer CW, Long CJ, Elbrecht D, Sasserath T, Bridges LR, Rumsey JW. et al. Multi-organ system for the evaluation of efficacy and off-target toxicity of anticancer therapeutics. Sci Transl Med. 2019;11:eaav1386
96. Shin J, Tabatabaei Rezaei N, Choi S, Li Z, Kim D, Kim K. Photocrosslinkable Kidney Decellularized Extracellular Matrix-Based Bioink for 3D Bioprinting. Adv Healthc Mater. 2025;14:2501616
97. Chang S, Weber E, Ness KV, Eaton D, Kelly E. Liver and kidney on chips: microphysiological models to understand transporter function. Clin Pharmacol Ther. 2016;100:464-78
98. Nguyen VVT, Ye S, Gkouzioti V, van Wolferen ME, Yengej FY, Melkert D. et al. A human kidney and liver organoid-based multi-organ-on-a-chip model to study the therapeutic effects and biodistribution of mesenchymal stromal cell-derived extracellular vesicles. J Extracell Vesicles. 2022;11:12280
99. Yang F, Pavon N, Matsubara T, Sebastian R, Martinez-Martin B, Pak C. et al. Distinct spatial patterning and transcriptomic landscapes of human neural organoids by localized delivery of morphogens. Cell Stem Cell. 2026;33:438-453.e7
100. Park SE, Georgescu A, Huh D. Organoids-on-a-chip. Science. 2019;364:960-5
101. Kasendra M, Tovaglieri A, Sontheimer-Phelps A, Jalili-Firoozinezhad S, Bein A, Chalkiadaki A. et al. Development of a primary human small intestine-on-a-chip using biopsy-derived organoids. Sci Rep. 2018;8:2871
102. Nikolaev M, Mitrofanova O, Broguiere N, Geraldo S, Dutta D, Tabata Y. et al. Homeostatic mini-intestines through scaffold-guided organoid morphogenesis. Nature. 2020;585:574-8
103. Salmon I, Grebenyuk S, Fattah ARA, Rustandi G, Pilkington T, Verfaillie C. et al. Engineering neurovascular organoids with 3D printed microfluidic chips. Lab Chip. 2022;22:1615-29
104. Shi Y, Sun L, Wang M, Liu J, Zhong S, Li R. et al. Vascularized human cortical organoids (vOrganoids) model cortical development in vivo. PLoS Biol. 2020;18:e3000705
105. Cakir B, Xiang Y, Tanaka Y, Kural MH, Parent M, Kang Y-J. et al. Engineering of human brain organoids with a functional vascular-like system. Nat Methods. 2019;16:1169-75
106. Sobrino A, Phan DTT, Datta R, Wang X, Hachey SJ, Romero-López M. et al. 3D microtumors in vitro supported by perfused vascular networks. Sci Rep. 2016;6:31589
107. Hachey SJ, Movsesyan S, Nguyen QH, Burton-Sojo G, Tankazyan A, Wu J. et al. An in vitro vascularized micro-tumor model of human colorectal cancer recapitulates in vivo responses to standard-of-care therapy. Lab Chip. 2021;21:1333-51
108. Lim J, Ching H, Yoon J-K, Jeon NL, Kim Y. Microvascularized tumor organoids-on-chips: advancing preclinical drug screening with pathophysiological relevance. Nano Converg. 2021;8:12
109. Kim HN, Habbit NL, Su C-Y, Choi N, Ahn EH, Lipke EA. et al. Microphysiological systems as enabling tools for modeling complexity in the tumor microenvironment and accelerating cancer drug development. Adv Funct Mater. 2019;29:1807553
110. Glieberman AL, Pope BD, Zimmerman JF, Liu Q, Ferrier JP, Kenty JHR. et al. Synchronized stimulation and continuous insulin sensing in a microfluidic human Islet on a Chip designed for scalable manufacturing. Lab Chip. 2019;19:2993-3010
111. Li B, Tang Y, Huang Z, Ma L, Song J, Xue L. Synergistic innovation in organ-on-a-chip and organoid technologies: reshaping the future of disease modeling, drug development, and precision medicine. Protein Cell. 2025 pwaf058
112. Herland A, Maoz BM, Das D, Somayaji MR, Prantil-Baun R, Novak R. et al. Quantitative prediction of human pharmacokinetic responses to drugs via fluidically coupled vascularized organ chips. Nat Biomed Eng. 2020;4:421-36
113. Nawroth JC, Petropolis DB, Manatakis DV, Maulana TI, Burchett G, Schlünder K. et al. Modeling alcohol-associated liver disease in a human liver-chip. Cell Rep. 2021;36:109393
114. Chakrabarty S, Quiros-Solano WF, Kuijten MMP, Haspels B, Mallya S, Lo CSY. et al. A microfluidic cancer-on-chip platform predicts drug response using organotypic tumor slice culture. Cancer Res. 2022;82:510-20
115. Yang Y, Chen Y, Wang L, Xu S, Fang G, Guo X. et al. PBPK modeling on organs-on-chips: an overview of recent advancements. Front Bioeng Biotechnol. 2022;10:900481
116. Ingber DE. Challenges and opportunities for human organ chips in FDA assessments and pharma pipelines. Cell Stem Cell. 2026;33:176-83
117. Choi S, Lee J, Kim O, Jung Y, Ryu T, Kim SJ. et al. Integrating a microphysiological system and physiologically based pharmacokinetic modeling to predict human responses to diclofenac. Biochip J. 2025;19:350-66
118. Sung JH, Srinivasan B, Esch MB, McLamb WT, Bernabini C, Shuler ML. et al. Using PBPK guided “body-on-a-chip” systems to predict mammalian response to drug and chemical exposure. Exp Biol Med (Maywood). 2014;239:1225-39
119. Bush LM, Healy CP, Javdan SB, Emmons JC, Deans TL. Biological cells as therapeutic delivery vehicles. Trends Pharmacol Sci. 2021;42:106-18
120. Labusca L, Herea DD, Mashayekhi K. Stem cells as delivery vehicles for regenerative medicine-challenges and perspectives. World J Stem Cells. 2018;10:43-56
121. Kavanagh EW, Green JJ. Toward gene transfer nanoparticles as therapeutics. Adv Healthc Mater. 2022;11:2102145
122. Zhang Q, Xiang W, Yi D, Xue B, Wen W, Abdelmaksoud A. et al. Current status and potential challenges of mesenchymal stem cell-based therapy for malignant gliomas. Stem Cell Res Ther. 2018;9:228
123. Mangraviti A, Tzeng SY, Gullotti D, Kozielski KL, Kim JE, Seng M. et al. Non-virally engineered human adipose mesenchymal stem cells produce bmp4, target brain tumors, and extend survival. Biomaterials. 2016;100:53-66
124. Yang F, Cho S-W, Son SM, Bogatyrev SR, Singh D, Green JJ. et al. Genetic engineering of human stem cells for enhanced angiogenesis using biodegradable polymeric nanoparticles. Proc Natl Acad Sci U S A. 2010;107:3317-22
125. Gupta K, Kim D-H, Ellison D, Smith C, Kundu A, Tuan J. et al. Lab-on-a-chip devices as an emerging platform for stem cell biology. Lab Chip. 2010;10:2019-31
126. Mansouri M, Lam J, E Sung K. Progress in developing microphysiological systems for biological product assessment. Lab Chip. 2024;24:1293-306
127. Chen X, Zhang YS, Zhang X, Liu C. Organ-on-a-chip platforms for accelerating the evaluation of nanomedicine. Bioact Mater. 2021;6:1012-27
128. De Masi C, Spitalieri P, Murdocca M, Novelli G, Sangiuolo F. Application of CRISPR/Cas9 to human-induced pluripotent stem cells: from gene editing to drug discovery. Hum Genomics. 2020;14:25
129. Achberger K, Cipriano M, Düchs MJ, Schön C, Michelfelder S, Stierstorfer B. et al. Human stem cell-based retina on chip as new translational model for validation of AAV retinal gene therapy vectors. Stem Cell Reports. 2021;16:2242-56
130. Ramamurthy RM, Atala A, Porada CD, Almeida-Porada G. Organoids and microphysiological systems: Promising models for accelerating AAV gene therapy studies. Front Immunol. 2022;13:3389
131. Moon H-R, Surianarayanan N, Singh T, Han B. Microphysiological systems as reliable drug discovery and evaluation tools: Evolution from innovation to maturity. Biomicrofluidics. 2023;17:061504
132. Kim J, Hwang I, Britain D, Chung TD, Sun Y, Kim D-H. Microfluidic approaches for gene delivery and gene therapy. Lab Chip. 2011;11:3941-8
133. Kang S, Park SE, Huh DD. Organ-on-a-chip technology for nanoparticle research. Nano Converg. 2021;8:20
134. Lu RXZ, Radisic M. Organ-on-a-chip platforms for evaluation of environmental nanoparticle toxicity. Bioact Mater. 2021;6:2801-19
135. Sampaio AR, Maia RF, Ciardulli MC, Santos HA, Sarmento B. Organ-on-chip platforms for nanoparticle toxicity and efficacy assessment: Advancing beyond traditional in vitro and in vivo models. Mater Today Bio. 2025;33:102053
136. Green JJ, Elisseeff JH. Mimicking biological functionality with polymers for biomedical applications. Nature. 2016;540:386-94
137. Tsui JH, Lee W, Pun SH, Kim J, Kim D-H. Microfluidics-assisted in vitro drug screening and carrier production. Adv Drug Deliv Rev. 2013;65:1575-88
138. Kwak B, Ozcelikkale A, Shin CS, Park K, Han B. Simulation of complex transport of nanoparticles around a tumor using tumor-microenvironment-on-chip. J Control Release. 2014;194:157-67
139. Albanese A, Lam AK, Sykes EA, Rocheleau JV, Chan WCW. Tumour-on-a-chip provides an optical window into nanoparticle tissue transport. Nat Commun. 2013;4:2718
140. Kim H, Cho S, Kim HN. Vascularized tumor-on-a-chip model as a platform for studying tumor-microenvironment-drug interaction. Macromol Biosci. 2025;25:e00240
141. Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Yuan Hsin H, Ingber DE. Reconstituting organ-level lung functions on a chip. Science. 2010;328:1662-8
142. Rodrigues RO, Sousa PC, Gaspar J, Bañobre-López M, Lima R, Minas G. Organ-on-a-chip: a preclinical microfluidic platform for the progress of nanomedicine. Small. 2020;16:2003517
143. de Roode KE, Hashemi K, Verdurmen WPR, Brock R. Tumor-on-a-chip models for predicting in vivo nanoparticle behavior. Small. 2024;20:2402311
144. Fusco C, De Rosa G, Spatocco I, Vitiello E, Procaccini C, Frigè C. et al. Extracellular vesicles as human therapeutics: A scoping review of the literature. J Extracell Vesicles. 2024;13:e12433
145. Abdal Dayem A, Yan E, Do M, Kim Y, Lee Y, Cho S-G. et al. Engineering extracellular vesicles for ROS scavenging and tissue regeneration. Nano Converg. 2024;11:24
146. Kwon S, Shin S, Do M, Oh BH, Song Y, Bui VD. et al. Engineering approaches for effective therapeutic applications based on extracellular vesicles. J Controlled Release. 2021;330:15-30
147. Chen W, Zhu Y, Liu R, Kong B, Xia N, Zhao Y. et al. Screening therapeutic effects of msc-evs to acute lung injury model on a chip. Adv Healthc Mater. 2024;13:2303123
148. Safarzadeh M, Richardson LS, Kammala AK, Mosebarger A, Bettayeb M, Kim S. et al. A multi-organ, feto-maternal interface organ-on-chip, models pregnancy pathology and is a useful preclinical extracellular vesicle drug trial platform. Extracell Vesicle. 2024;3:100035
149. Kim M-H, van Noort D, Sung JH, Park S. Organ-on-a-chip for studying gut-brain interaction mediated by extracellular vesicles in the gut microenvironment. Int J Mol Sci. 2021;22:13513
150. Kwak S, Song CL, Lee J, Kim S, Nam S, Park Y-J. et al. Development of pluripotent stem cell-derived epidermal organoids that generate effective extracellular vesicles in skin regeneration. Biomaterials. 2024;307:122522
151. Wagner KT, Nash TR, Liu B, Vunjak-Novakovic G, Radisic M. Extracellular vesicles in cardiac regeneration: potential applications for tissues-on-a-chip. Trends Biotechnol. 2021;39:755-73
152. Chun C, Smith AST, Kim H, Kamenz DS, Lee JH, Lee JB. et al. Astrocyte-derived extracellular vesicles enhance the survival and electrophysiological function of human cortical neurons in vitro. Biomaterials. 2021;271:120700
153. Sun L, Bian F, Xu D, Luo Y, Wang Y, Zhao Y. Tailoring biomaterials for biomimetic organs-on-chips. Mater Horiz. 2023;10:4724-45
154. Guttenplan APM, Tahmasebi Birgani Z, Giselbrecht S, Truckenmüller RK, Habibović P. Chips for biomaterials and biomaterials for chips: recent advances at the interface between microfabrication and biomaterials research. Adv Healthc Mater. 2021;10:2100371
155. Monteiro CF, Deus IA, Custódio CA, Mano JF. Biomaterials meet organ-on-chips - a perspective on tumor modeling. Int Mater Rev. 2025;70:31-68
156. Leoni G, Neumann P-A, Kamaly N, Quiros M, Nishio H, Jones HR. et al. Annexin A1-containing extracellular vesicles and polymeric nanoparticles promote epithelial wound repair. J Clin Invest. 2015;125:1215-27
157. Guo Q, Bishop CJ, Meyer RA, Wilson DR, Olasov L, Schlesinger DE. et al. Entanglement-based thermoplastic shape memory polymeric particles with photothermal actuation for biomedical applications. ACS Appl Mater Interfaces. 2018;10:13333-41
158. Tang J, Mi J, Huang W, Zhong H, Li Y, Zhou J. et al. Controlled drug release from ultrasound-visualized elastic eccentric microcapsules using different resonant modes. J Mater Chem B. 2018;6:1920-9
159. Fleischer S, Liberman M, Summers M, Li V, Nash TR, Vunjak-Novakovic G. OptoBarrier: An optogenetic platform for modulating endothelial barriers in vitro. ACS Biomater Sci Eng. 2025;11:5542-53
160. Chehri B, Liu K, Vaseghi G, Seyfoori A, Akbari M. In Vitro glioblastoma model on a plate for localized drug release study from a 3D-printed drug-eluted hydrogel mesh. Cells. 2024;13:363
161. Fetah KL, DiPardo BJ, Kongadzem E-M, Tomlinson JS, Elzgheid A, Elmusrati M. et al. Cancer modeling-on-a-chip with future artificial intelligence integration. Small. 2019;15:e1901985
162. Lu R, Lee BJ, Lee E. Three-dimensional lymphatics-on-a-chip reveals distinct, size-dependent nanoparticle transport mechanisms in lymphatic drug delivery. ACS Biomater Sci Eng. 2024;10:5752-63
163. German SV, Abalymov AA, Kurochkin MA, Kan Y, Gorin DA, Novoselova MV. Plug-and-play lymph node-on-chip: secondary tumor modeling by the combination of cell spheroid, collagen sponge and T-cells. Int J Mol Sci. 2023;24:3183
164. Cook SR, Ball AG, Mohammad A, Pompano RR. A 3D-printed multi-compartment organ-on-chip platform with a tubing-free pump models communication with the lymph node. Lab Chip. 2025;25:155-74
165. Hallfors N, Shanti A, Sapudom J, Teo J, Petroianu G, Lee S. et al. Multi-compartment lymph-node-on-a-chip enables measurement of immune cell motility in response to drugs. Bioengineering (Basel). 2021;8:19
166. Harriot AD, Ward CW, Kim D-H. Microphysiological systems to advance human pathophysiology and translational medicine. J Appl Physiol (1985). 2024;137:1494-501
167. Park S, Laskow TC, Chen J, Guha P, Dawn B, Kim D. Microphysiological systems for human aging research. Aging Cell. 2024;23:e14070
168. Miller CP, Shin W, Ahn EH, Kim HJ, Kim D-H. Engineering microphysiological immune system responses on chips. Trends Biotechnol. 2020;38:857-72
169. Vasconez Martinez MG, Frauenlob M, Rothbauer M. An update on microfluidic multi-organ-on-a-chip systems for reproducing drug pharmacokinetics: the current state-of-the-art. Expert Opin Drug Metab Toxicol. 2024;20:459-71
170. Lim J, Fang H-W, Bupphathong S, Sung P-C, Yeh C-E, Huang W. et al. The edifice of vasculature-on-chips: a focused review on the key elements and assembly of angiogenesis models. ACS Biomater Sci Eng. 2024;10:3548-67
171. Yin H, Wang Y, Liu N, Zhong S, Li L, Zhang Q. et al. Advances in the model structure of in vitro vascularized organ-on-a-chip. Cyborg Bionic Syst. 2024;5:0107
172. Wang R, Zhang C, Li D, Yao Y. Tumor-on-a-chip: Perfusable vascular incorporation brings new approach to tumor metastasis research and drug development. Front Bioeng Biotechnol. 2022;10:1057913
173. Morrison AI, Jäger J, de Winde CM, Konijn T, Roest HP, van der Laan LJW. et al. Integration of lymphatic vasculature to a human lymph node-on-chip enhances physiological immune properties. Mater Today Bio. 2025;35:102326
174. Kemas AM, Zandi Shafagh R, Taebnia N, Michel M, Preiss L, Hofmann U. et al. Compound absorption in polymer devices impairs the translatability of preclinical safety assessments. Adv Healthc Mater. 2024;13:2303561
175. Shirure VS, George SC. Design considerations to minimize the impact of drug absorption in polymer-based organ-on-a-chip platforms. Lab Chip. 2017;17:681-90
176. Grant J, Ӧzkan A, Oh C, Mahajan G, Prantil-Baun R, Ingber DE. Simulating drug concentrations in PDMS microfluidic organ chips. Lab Chip. 2021;21:3509-19
177. Mair DB, Williams MAC, Chen JF, Goldstein A, Wu A, Lee PHU. et al. PDMS-PEG block copolymer and pretreatment for arresting drug absorption in microphysiological devices. ACS Appl Mater Interfaces. 2022;14:38541-9
178. Ferreira DA, Rothbauer M, Conde JP, Ertl P, Oliveira C, Granja PL. A fast alternative to soft lithography for the fabrication of organ-on-a-chip elastomeric-based devices and microactuators. Adv Sci (Weinh). 2021;8:2003273
179. Novak R, Ingram M, Marquez S, Das D, Delahanty A, Herland A. et al. Robotic fluidic coupling and interrogation of multiple vascularized organ chips. Nat Biomed Eng. 2020;4:407-20
180. Graf H, Gaier M, Culp C, Loskill P. Plug and play multi-organ chips: Integrated µgaskets for the facile and reversible connection of individual organ-on-chip modules. Micromachines (Basel). 2025;16:1251
181. Chliara MA, Elezoglou S, Zergioti I. Bioprinting on organ-on-chip: development and applications. Biosensors. 2022;12:1135
182. FDA announces plan to phase out animal testing requirement for monoclonal antibodies and other drugs. FDA; 2025
183. Non-clinical and new approach methodologies european specialised expert community | European Medicines Agency (EMA); 2023
184. Ahmed SM, Shivnaraine RV, Wu JC. FDA Modernization Act 2.0 paves the way to computational biology and clinical trials in a dish. Circulation. 2023;148:309-11
185. Zushin P-JH, Mukherjee S, Wu JC. FDA Modernization Act 2.0: transitioning beyond animal models with human cells, organoids, and AI/ML-based approaches. J Clin Invest. 2023;133:e175824
186. Reyes DR, Esch MB, Ewart L, Nasiri R, Herland A, Sung K. et al. From animal testing to in vitro systems: advancing standardization in microphysiological systems. Lab Chip. 2024;24:1076-87
187. Wang J, Chen X, Li R, Wang S, Geng Z, Shi Z. et al. Standardization and consensus in the development and application of bone organoids. Theranostics. 2025;15:682-706
Author contact
![]() Corresponding author: dhkimedu.
Corresponding author: dhkimedu.