Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
The Structure of STK17A and...
STK17 and Its Evolution in...
Regulation of STK17A Expression
Molecular Interactions of STK17A
Physiological role of STK17A...
STK17A in Cancers
STK17A in Other Diseases
Comparison STK17A / STK17B
STK17A/B Inhibitors
Conclusion
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2026; 16(11):5830-5855. doi:10.7150/thno.129910 This issue Cite
Review
STK17A (DRAK1) at the crossroads of apoptosis, immunity, and cancer: Emerging roles and therapeutic opportunities
Leticia Christina Pires Gonçalves1‡, Olivia Rastoin1‡, Vira Morozova2,3, Clément Buzet1,2, Audrey Bennetot4, Gilles Pagès1,4, Cyril Ronco1,2,5 ![]() , Maeva Dufies1,4
, Maeva Dufies1,4 ![]()
1. Roca Therapeutics, Nice, France.
2. University Côte d'Azur, Institut de Chimie de Nice (ICN), CNRS UMR 7272, Nice, France.
3. V. N. Karazin Kharkiv National University, School of Chemistry, Kharkiv, Ukraine.
4. University Côte d'Azur, Institute for Research on Cancer and Aging of Nice (IRCAN), CNRS UMR 7284, INSERM U1081, Nice, France.
5. Institut Universitaire de France (IUF), Paris, France.
‡L.C.P.G. and O.R. contributed equally to this work.
Received 2025-12-13; Accepted 2026-2-11; Published 2026-3-30
Abstract

Death-associated protein kinase-Related Apoptosis-inducing protein Kinase 1 (DRAK1/STK17A) is a serine/threonine kinase of the Death Associated Protein Kinase (DAPK) family. STK17A is widely expressed and enriched in immune tissues, and is primarily localized in the nucleus, though it can translocate to the cytoplasm in response to specific stimuli. STK17A stimulates apoptosis and cytoskeletal dynamics, but its physiological roles remain incompletely defined, in part due to limited availability of potent/selective chemical probes and the absence of STK17A in commonly used rodent models.
In this review, we summarize current knowledge on STK17A, including its structure, evolution, expression patterns, molecular interactions, and roles in cancer as well as in autoimmune, cardiovascular, infectious, and neurological disorders. We also compare STK17A with its closest homolog, STK17B, highlighting both shared features and functional distinctions.
The review further examines recent medicinal chemistry efforts that have yielded the first small-molecule modulators of STK17A (DRAK1) and STK17B (DRAK2), including dual inhibitors and emerging selective scaffolds. These compounds can serve as valuable chemical probes and hold promising therapeutic potential. Nonetheless, challenges of selectivity and functional validation remain, emphasizing the need for continued medicinal chemistry efforts to unlock the full potential of STK17A as a therapeutic target across cancer, autoimmune, and neurodegenerative diseases.
Introduction
Protein kinases play a fundamental role in regulating cellular signaling and metabolic pathways. Their activity is essential for processes such as cell proliferation, differentiation, and apoptosis. Dysregulation of kinase function has been implicated in a wide range of diseases, including cancer, inflammatory conditions and neurodegenerative disorders. Understanding kinase structure and function, therefore, remains a central focus in biomedical research, and modulation of their activity offers significant therapeutic potential for addressing diverse pathologies. Within the broad family of kinases, the calcium/calmodulin-dependent protein kinases (CAMKs), and particularly their subfamilies, occupy a central role in apoptosis and cytoskeletal regulation. The CAMKs are composed of three main subfamilies: the Death-Associated Protein Kinases (DAPKs), the Myosin Light chain-related Kinases (MLKs), and the TRIple functional dOmain protein-related kinases (TRIOs), all of which are implicated in apoptotic processes, cytoskeletal dynamics, and cellular plasticity. Although classified within the CAMK superfamily, only about half of the kinases in the subfamilies possess an additional C-terminal autoinhibitory domain capable of binding calcium/calmodulin to regulate kinase activity. Instead, many are regulated through alternative mechanisms, including phosphorylation or interaction with other protein ligands [1].
DAPKs are a subset of calcium/calmodulin (CaM)-dependent serine/threonine kinases localized in the cytoskeleton, characterized by the presence of a death domain, and promote apoptotic signaling. This family includes five kinases: DAPK1, DAPK2, DAPK3 (also known as ZIPK), and the less-characterized STK17A (DRAK1) and STK17B (DRAK2). Importantly, overexpression of DAPK family members induces direct cell death in multiple cell lines in a kinase activity-dependent manner [2]. Their kinase domain is situated near the N-terminus, and DAPK1, DAPK2, and DAPK3 share sequence motifs within the upper lobe of their catalytic domains. DAPK1 and DAPK2 are more closely related to DAPK3 and possess a calcium/calmodulin-binding autoinhibitory domain, unlike DAPK3, which instead contains three serine/threonine phosphorylation sites within its catalytic domain [1].
The DAPK family contributes to apoptosis notably via tumor necrosis factor-alpha (TNF-α) and Fas-induced cell death pathways [3]. Additionally, DAPK activity impairs cell adhesion by inhibiting integrin functions and survival signaling pathways, thereby activating p53. Integrins mediate the connection between the extracellular matrix and the cytoskeleton and promote survival pathways such as focal adhesion kinase (FAK) activation [4]. DAPK activity also exerts anti-metastatic effects by impeding the survival of anchorage-dependent cells [5].
The catalytic domain homology and regulatory mechanisms of DAPK family members are sufficiently conserved to classify them as a distinct serine/threonine kinase subfamily. Within this group, STK17A, STK17B and DAPK3, lack the CaM domain and are predominantly localized in the nucleus [6]. Although several functions have been described, this protein family remains poorly characterized and likely carries out additional roles that are not yet fully understood.
Within this context, STK17A emerges as one of the least studied members of the DAPK family.
Unlike other members of the death-associated protein kinase family, STK17A exhibits several distinctive features that complicate its biological and translational interpretation. STK17A lacks the death domain and calcium/calmodulin regulatory motifs characteristic of DAPK1-3, and its gene is absent in murid genomes, precluding conventional genetic interrogation in widely used mouse models. As a result, much of the current literature relies on overexpression or knockdown approaches in transformed cell lines, raising important questions regarding physiological relevance. Additional bottlenecks include limited validation of endogenous kinase activity and substrates, frequent reliance on transcriptomic or network-based associations, and apparent context-dependent functional outputs that vary across cellular and disease settings. Together, these features distinguish STK17A from other death-associated kinases and underscore the need for cautious interpretation of existing data, as well as for improved experimental and chemical tools to resolve its precise biological roles.
Accordingly, this review focuses on STK17A, with brief comparisons to STK17B, describing their structure and distribution, summarizing their molecular interactions, and highlighting their roles in cancer and other human diseases. In addition, we provide a comprehensive and critical analysis of all known or designed small molecules that modulate their activity.
The Structure of STK17A and STK17B
Death-associated protein kinase-Related Apoptosis-inducing protein Kinase 1 and 2 (DRAK1/STK17A and DRAK2/STK17B, respectively) are two serine/threonine kinases belonging to the DAPK family, first discovered in 1998 [7]. Also known respectively as serine/threonine kinase 17A (STK17A) and serine/threonine kinase 17B (STK17B), they are ubiquitously expressed across tissues.
Structurally, STK17A and STK17B possess catalytic domains that are highly homologous to those of DAPK1 and DAPK3. However, they lack several regulatory domains present in DAPK1, notably the death domain and the calcium/calmodulin-binding regulatory domain, which are critical for canonical DAPK1 regulation [6].
STK17A is encoded by the STK17A gene, located on the chromosome 7p13,spanning 44,622 base pairs and comprising seven exons and seven introns [8]. STK17B is encoded by the STK17B gene, located on the chromosome 2q32.3. The corresponding open reading frames encode a protein of 414 amino acids for STK17A and 372 amino acids for STK17B. Both kinases contain N-terminal kinase domains, sharing approximately 67% sequence homology, and encompassing 11 conserved subdomains typical of serine/threonine kinases which are found across the DAPK family, as well as other CAMK-related kinases such as CaMKII and MLCK.
At the sequence level, the catalytic domains of STK17A and STK17B harbor all canonical motifs required for kinase activity, although their absolute positions differ due to variations in N-terminal length. In STK17A, the glycine-rich loop (subdomain I) is defined by the motif GRGKFA at amino acids 68-73, marking the functional onset of the kinase domain, whereas the equivalent motif in STK17B is located at amino acids 40-45. The catalytic lysine within subdomain II is positioned at residues 80-81 in STK17A and 65-66 in STK17B, enabling ATP coordination. The conserved catalytic loop motif HLD (subdomain VI) is present at residues 184-186 in STK17A and 156-158 in STK17B, followed by the activation loop DFG motif (subdomain VII) at residues 207-209 and 179-181, respectively. The activation segment terminates with the conserved APE motif (subdomain VIII) at residues 231-233 in STK17A and 203-205 in STK17B, collectively defining a single, conserved kinase fold in both proteins.
Although STK17A contains an additional glycine-rich sequence (GSGRAG, residues 19-24) upstream of its catalytic domain, this motif is not embedded within a complete kinase fold and lacks accompanying catalytic elements and therefore does not constitute a second functional ATP-binding kinase domain.
Outside the catalytic domain, STK17A and STK17B share ~20-24% sequence homology within their conserved C-terminal region outside the kinase domain (C-terminal tail region; see full-length alignment in Figure 1A) [9]. This region is comparatively divergent and is not shared with other serine/threonine kinases, suggesting potential roles in localization and protein-protein interactions.
Structure of STK17A and STK17B proteins. (A) Amino acids sequence alignment of STK17A and STK17B. MUSCLE algorithm was used to align the full sequences of STK17A (1-414) and STK17B (1-372), followed by pairwise alignment to determine the percentage of identity between STK17A and STK17B. The important residues for the catalytic activity are colored in blue. (B) Schematic representation of the kinase and NLS (nuclear localization signals) domains. (C) 3D structure superimposition of STK17A (blue - PDB 7QUF) and STK17B (magenta - PDB 6ZJF). The catalytic pocket is represented by a red circle. (D) and (E) 3D structure of the structural divergent region and key amino acids of STK17A (blue, D) and STK17B (magenta, E).
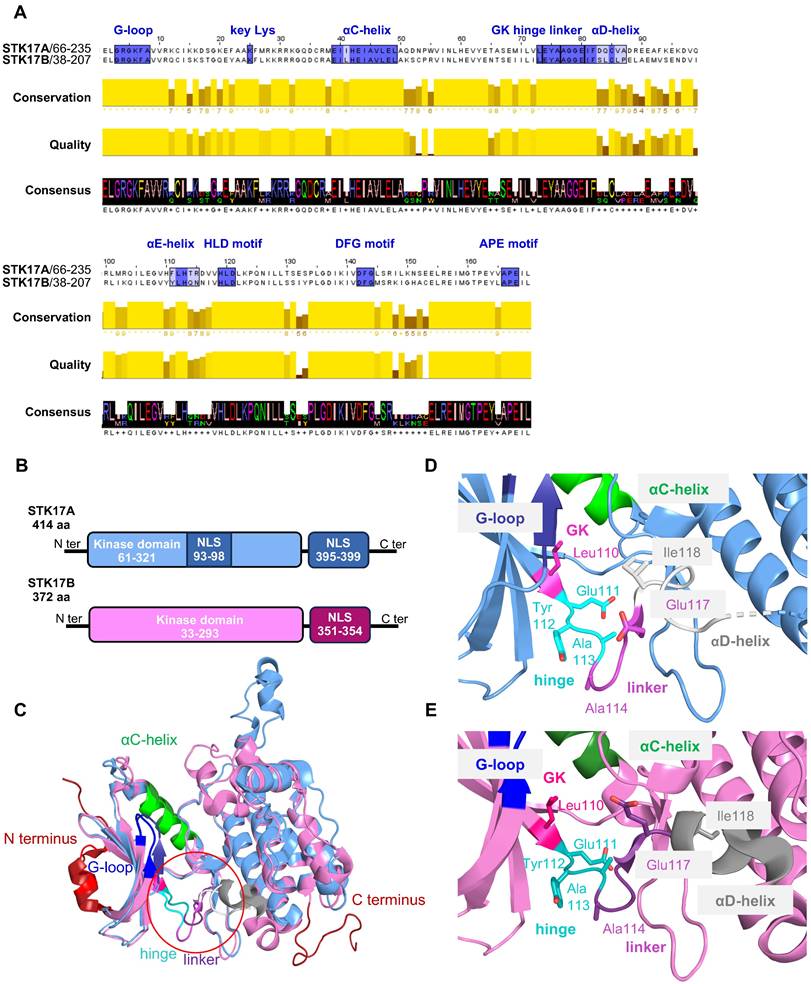
A notable feature of STK17A is the presence of two predicted nuclear localization signals (NLS): one proximal to the kinase domain (amino acids 93-97, sequence RKRRK) and a second within the C-terminal region (amino acids 396-399, sequence KRFK). In contrast, STK17B contains a single C-terminal NLS (amino acids 351-354, sequence KRFR). The presence of these NLS motifs enables predominant nuclear localization of both kinases, although protein kinase C (PKC) activation has been reported to promote their context-dependent translocation to the cytoplasm [1,7,10].
The ATP-binding sites of STK17A and STK17B are highly conserved (Figure 1A), not only between the two kinases but also across the broader DAPK family [11]. Nevertheless, sequence alignment reveals notable differences in the αD-helix, αE-helix, and xDGF+1 subdomains, with the αD-helix showing the most pronounced divergence (Figure 1A). Functionally, this helix is critical for stabilizing the catalytic pocket and orienting residues that interact with ATP. These structural differences in STK17A and STK17B result in substantial changes in the architecture and volume of the catalytic pocket (∼349 ų for STK17A vs. ∼708 ų for STK17) and appear to influence the conformations of the linker and hinge regions, albeit to a lesser extent (Figure 1D-E). Despite these differences, the residues directly involved in catalysis are conserved, underscoring the αD-helix as the primary structural divergence between the two kinases. These distinct structural features may be exploited for the rational design of selective inhibitors targeting STK17A or STK17B, by specifically interacting with residues of the αD-helix.
STK17 and Its Evolution in Eukaryotes
STK17A is absent in two of the most commonly used animal research models, the mouse (Mus musculus) and the rat (Rattus norvegicus) [12], which severely limits the availability of genetic in vivo validation. Although STK17B is present in these species, there is currently no direct experimental evidence demonstrating functional compensation for STK17A loss. It has been proposed that the STK17A gene was lost in the evolutionary lineage of murids (rats and mice), a phenomenon known as gene loss or pseudogenization, which is relatively common in evolution. This loss may result from genomic rearrangements involving a synteny break: a 50.4 kb genomic deletion between two chromosomal blocks that are conserved in other species, such as chimpanzees, pigs, and cows [13].
Despite its absence in mice and rat, STK17A is widely conserved across more than 600 animal species (UniProt database). It is present in numerous invertebrate species as well as key vertebrate models, including zebrafish (Danio rerio). In zebrafish, STK17A is located on chromosome 24 and contains seven exons, mirroring the human gene structure [14]. To date, however, functional studies of STK17A in zebrafish are lacking.
In contrast, STK17A has been studied in several other organisms. In the Japanese flounder (Paralichthys olivaceus), STK17A is dominantly expressed in the liver and exhibits pro-apoptotic activity, particularly in response to bacterial infection and inflammatory stimuli [15].
In rabbits, STK17A shares approximately 88% sequence identity with its human ortholog, retains the conserved DAPK catalytic motifs, and localizes to the nucleus. Functionally, it promotes apoptosis when overexpressed in osteoclasts [16]. Similar pro-apoptotic roles have been reported in pigs, where STK17A expression is associated with malignant melanoma progression [17], and in chickens, where STK17A polymorphisms correlated with anxiety-like behaviors [18].
In some invertebrates, the DAPK repertoire is reduced to a single family member. For example, in fruit flies (Drosophila melanogaster) and silkworms (Bombyx mori), only one DAPK family protein, orthologous to STK17, is present [19]. In Drosophila, this STK17 ortholog plays a crucial role in cytoskeletal regulation by phosphorylating Spaghetti Squash (Sqh), the fly homolog of the myosin regulatory light chain (MRLC) [20]. STK17A-mediated phosphorylation of MRLC is essential for smooth muscle contraction and for regulating cytoskeletal tension [21-23].
Together, evolutionary analyses reveal an unexpectedly high degree of conservation of STK17A's apoptotic and cytoskeletal regulatory functions across diverse species. At the same time, species-specific adaptations gene losses and functional redundancies highlight the evolutionary plasticity of the DAPK family.
Regulation of STK17A Expression
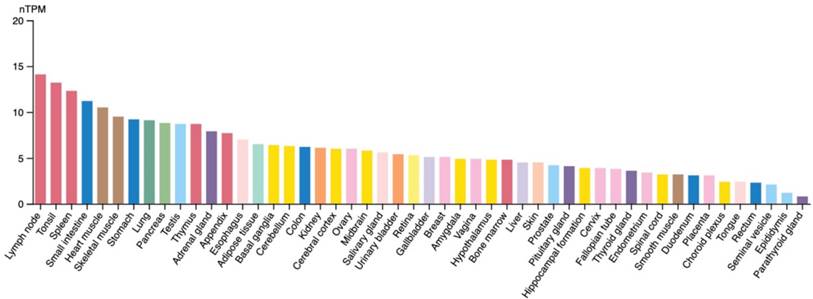
The expression of STK17A is governed by complex regulatory networks involving transcriptional, post-transcriptional, and signaling-dependent mechanisms. Expression profiling (Figure 2) indicates that STK17A and STK17B are broadly expressed but show higher transcript abundance in immune-related tissues (e.g., lymph nodes, thymus, spleen).
STK17A mRNA expression in different organs (Protein Atlas). RNA expression shows RNA-data from two different sources: generated from Human Protein Atlas (HPA) RNA-seq data and RNA seq data from the Genotype-Tissue Expression (GTEx) project. nTPM: relatively comparing RNA level, transcripts per million.
The expression of STK17A is regulated by multiple upstream signals, including transcription factors, cytokines-triggered pathways, and microRNAs (miRNAs). Throughout this section, we distinguish direct regulation (e.g., promoter binding, response elements) from indirect associations inferred from stress signaling or pharmacologic perturbation.
Regulation by Transcription Factors
One of the key regulators of STK17A expression is the tumor suppressor p53, a guardian of genomic stability with central roles in DNA damage response, reactive oxygen species (ROS) regulation, senescence, and apoptosis. Upon cellular stress, particularly DNA damage, p53 activates a network of downstream genes that mediate cell-cycle arrest and programmed cell death.
STK17A gene has been identified as a direct transcriptional target of p53, regulated through a p53 response element (p53RE) located near its transcription start site. This direct regulation has been confirmed by promoter analyses and chromatin immunoprecipitation demonstrating p53 binding to the response element [24]. p53-dependent upregulation of STK17A has been observed across multiple cell types following exposure to DNA-damaging agents such as cisplatin. This activation may be cell-type dependent, as STK17A does not consistently participate in feedback loop with p53 in testicular cancer cells [25].
This transcriptional axis should be distinguished from reports suggesting that STK17A phosphorylates p53, which—so far—derive from in vitro or ectopic-expression. STK17A can phosphorylate p53 at Ser20 and Thr18. The phosphorylation of p53 is known to stimulate the p53 transcriptional response and plays an important role in controlling its function; thus, making it an interesting target to study [26].
Regulation by Cytokines
STK17A expression is also induced by cytokine-mediated signaling pathways, notably the type I interferon (IFN-I)/ RNase L pathway. Interferons are critical mediators of antiviral defense and can initiate apoptosis in infected cells. Activation of RNase L down-stream of IFN-I leads to RNA degradation and apoptotic signaling through the c-Jun N-terminal kinase (JNK) pathway.
Studies have shown that IFN-I stimulation, particularly in the presence of double-stranded RNA (dsRNA), results in robust upregulation of STK17A mRNA levels. RNase L-deficient cells exhibit reduced STK17A induction, confirming the importance of this pathway [27]. In cell-line models, ectopic STK17A expression has been reported to increase JNK phosphorylation and markers of mitochondrial apoptosis (activation of caspase-3 and cleavage of PARP, and mitochondrial membrane permeabilization through Bax translocation to mitochondria). However, these findings should be interpreted as demonstrating the capacity of STK17A to engage apoptotic signaling under artificial expression conditions, rather than establishing an endogenous requirement.
Collectively, these findings suggested that STK17A overexpression is sufficient to promote intrinsic apoptosis.
Regulation by microRNAs
At the post-transcriptional level, microRNAs (miRNAs) further modulate STK17A expression. For example, miR-182-5p directly targets STK17A by binding to two distinct sites within its 3' untranslated region (3'UTR), thereby reducing mRNA stability and translation efficiency [12].
Thus, STK17A expression is precisely regulated through an integrated network of transcriptional, cytokine-mediated, and post-transcriptional mechanisms. This multilayered regulation positions STK17A as a central node in the cellular response to stress, DNA damage, and viral infection.
Molecular Interactions of STK17A
Once expressed and/or relocalized, STK17A has been reported to interact with multiple proteins implicated in apoptosis, inflammation, and cytoskeletal regulation (Figures 3 and 4). Importantly, most interactions have been identified in overexpression or co-immunoprecipitation settings, and direct kinase-substrate relationships remain largely unvalidated at endogenous levels.
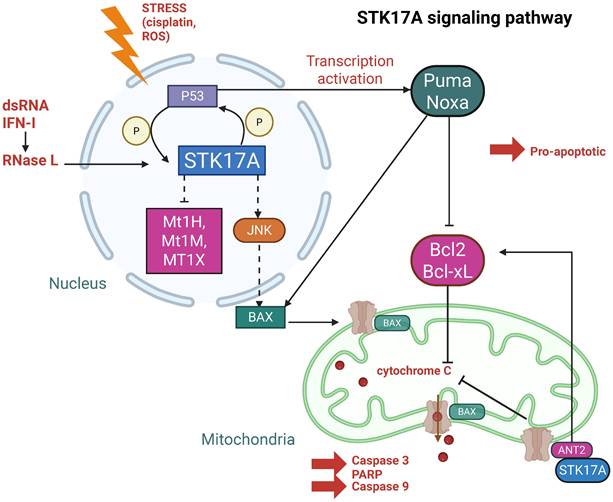
STK17A and its interactions in the nucleus. Created with BioRender.com. Solid lines represent reported direct protein-protein interactions, whereas dashed lines indicate indirect regulatory relationships inferred from functional or correlative evidence.
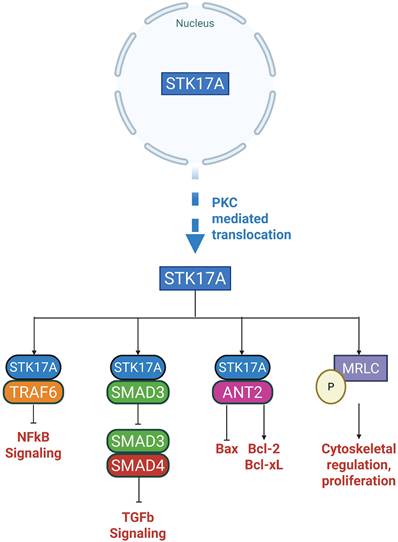
STK17A and its binding partners in the cytoplasm. Created with BioRender.com. STK17A (STK17A) reported cytoplasmic interactors and putative functional outputs.
Nuclear Targets
In the nucleus (Figure 3), STK17A exerts its kinase activity, although its endogenous substrates remain largely unidentified. Nuclear STK17A is essential for the activation of apoptotic pathways, particularly through activation of the JNK signaling cascade. Overexpression of STK17A indirectly activates JNK, which subsequently phosphorylates Bax facilitating its translocation. This translocation promotes cytochrome C release and leads to the cleavage of caspase-3 and PARP [27].
Moreover, knockdown of STK17A results in elevated of antioxidation response genes such as Mt1H, Mt1M and Mt1X, suggesting that STK17A contributes to the enhancement of intracellular ROS by repressing these genes [28].
Cytoplasmic Targets
While predominantly nuclear, STK17A can translocate to the cytoplasm under specific conditions (Figure 4), such as activation of Protein Kinase C (PKC). Upon cytoplasmic translocation, STK17A interacts with several important regulatory proteins:
Smad3
STK17A binds to Smad3, a key mediator of TGF-β1 signaling. Notably, STK17A binding does not result in Smad3 phosphorylation. Instead, STK17A inhibits the formation of the Smad3/Smad4 complex, thereby blocking the nuclear translocation of Smad3 and the subsequent activation of TGF-β1 target genes. This interference impairs TGF-β1's signaling [29].
TRAF6
STK17A also interacts with TNF receptor-associated factor 6 (TRAF6), an E3 ubiquitin ligase critical for the activation of NF-kB signaling and the inflammatory response. STK17A binds to TRAF6 through its kinase domain (residues 112-120), engaging the TRAF domain (residues 358-364) of TRAF6. Although kinase-inactive STK17A mutants can bind TRAF6, binding of STK17A inhibits the auto-ubiquitination of TRAF6, thereby suppressing NF-kB-mediated inflammatory signaling [30].
Mitochondrial ANT2
Another important cytoplasmic binding partner is adenine nucleotide translocase 2 (ANT2), a mitochondrial inner membrane protein with well-established anti-apoptotic functions in cancer. ANT2 regulates mitochondrial permeability and catalyzes the exchange of ADP/ATP across the inner membrane. It promotes cell survival by inhibiting the pro-apoptotic Bax and by regulating the anti-apoptotic Bcl-2 and Bcl-xL. Upon cytoplasmic translocation to mitochondria, the catalytic domain of STK17A binds ANT2. Although it remains unclear whether STK17A directly phosphorylates ANT2, this interaction suggests that STK17A may modulate apoptosis by inhibiting ANT2. STK17A mutants that are constitutively retained in the cytoplasm display an even higher affinity for ANT2 [10].
Myosin Light Chain (MLC)
STK17A has also been shown to phosphorylate myosin light chain (MLC), an exogenous substrate essential for cytoskeletal organization. This interaction was demonstrated in fibroblast-like monkey kidney cells (CO7 cells), where a kinase-defective mutant of STK17A failed to phosphorylate MLC. In mice fibroblasts (NIH3T3), overexpression of wild-type STK17A resulted in increased apoptosis and morphological changes [7]. A similar role has been observed in Drosophila, where STK17 (DRAK) phosphorylates Spaghetti Squash (Sqh), the MLC ortholog, regulating cellularization and cytoskeletal dynamics [21].
Physiological role of STK17A under non-pathological conditions
Despite the growing body of literature describing STK17A in disease contexts, its physiological function under normal conditions remains poorly defined. Unlike STK17B, whose roles are supported by genetic and in vivo models, current knowledge of STK17A is largely derived from in vitro studies and stress-induced systems. Available evidence suggests that STK17A may function as a stress-responsive kinase involved in cell fate regulation, including modulation of apoptosis, redox balance, and signaling homeostasis. However, whether STK17A plays an essential role in normal tissue maintenance or acts primarily as a context-dependent modulator activated under stress remains unclear. This knowledge gap represents an important limitation of the field and highlights the need for future studies using physiological and in vivo models to define the basal functions of STK17A.
STK17A in Cancers
STK17A Positive Outcomes in Cancers
STK17A has emerged as a significant regulator of apoptosis in a variety of cancers, where it often contributes to tumor suppression by promoting tumor cell death. Apoptosis in cancer cells can be initiated through intrinsic pathways (e.g., mitochondrial membrane permeabilization) and extrinsic pathways (e.g., death receptor activation by Fas ligand or TNF-α) [31]. The extrinsic signaling pathway involves receptors from the tumor necrosis factor (TNF) receptor superfamily and are best characterized by the FasL/FasR and TNF-α/TNFR1 ligand models. The intrinsic signaling pathway is triggered by non-receptor stimuli, including growth factors deprivation, oxidative stress or radiation. These stimuli result in the release of pro-apoptotic factors from the mitochondria into the cytosol.
Among intrinsic mechanisms, the p53 signaling pathway is particularly significant. TP53 gene is mutated in most human cancers, contributing to tumor development. In healthy cells, p53 is targeted by the E3 ubiquitin ligase MDM2 for degradation. In response to stress such as DNA damage, p53 promotes G1 cell cycle arrest or mitochondrial permeabilization and subsequent apoptosis. Its downstream effectors include p21, which inhibits cyclin-dependent kinases to induce cell-cycle arrest; Noxa, which neutralizes the anti-apoptotic Bcl-2 proteins to promote cytochrome c release and caspase activation; and puma, which similarly promotes mitochondrial outer membrane permeabilization. Pro-apoptotic p53 targets such as Bax and Bad can also regulate the activity of mitochondrial ANT isoforms, thereby influencing mitochondrial apoptosis [10].
STK17A participates in multiple apoptotic pathways, but its role within the p53-dependent apoptotic cascade is particularly well established.
Testicular Cancer and Osteosarcoma
In testicular cancer, treatment with cisplatin — a DNA-damaging chemotherapeutic — upregulates STK17A expression in a dose-dependent manner, independently of STK17B. Similar observations have been made in osteosarcoma cell lines, where cisplatin induces both STK17A expression and p53 phosphorylation at Ser392, enhancing p53's transcriptional activity [10]. Silencing p53 or STK17A markedly reduces sensitivity to cisplatin, while STK17A overexpression increases ROS production and promotes apoptosis [25].
Moreover, STK17A upregulation is not limited to cisplatin: vinblastine, doxorubicin, and etoposide also induce STK17A expression, indicating that STK17A responds broadly to genotoxic stress.
Prostate and Adenocarcinoma Cells
STK17A expression can be induced in the context of dsRNA and type I interferon (IFN) signaling, consistent with engagement of antiviral stress pathways. Current data support association with IFN/RNase L-linked apoptosis in specific cell-line models, but do not establish a causal role in virus-driven tumorigenesis. In prostate cancer and adenocarcinoma cell lines, the combination of dsRNA and IFN induces STK17A expression, which correlates with enhanced apoptosis via JNK activation and mitochondrial apoptotic pathways [27].
Cervical Cancer
In cervical cancers, STK17A exhibits anti-inflammatory properties. By binding to and destabilizing TRAF6, STK17A impairs the NF-kB pathway and reduces the expression of pro-inflammatory cytokines such as IL-1β, IL-8, and CXCL1 and CXCL2. Functionally, STK17A silencing enhances proliferation and invasiveness in HeLa and CaSki cervical cancer cell lines, further confirming its tumor-suppressive role [30].
Colon and Ovarian Cancers
In colon carcinoma, oxaliplatin-resistant cell lines exhibit downregulation of STK17A, while anti-apoptotic genes are upregulated [32]. Similarly, in ovarian cancer (OVCAR3 cell line), STK17A overexpression does not trigger apoptosis per se but significantly sensitizes cells to chemotherapeutic agents such as carboplatin and paclitaxel. Conversely, STK17A knockdown enhances chemoresistance [33].
STK17A Negative Outcomes in Cancers
Although STK17A is traditionally linked to apoptosis and tumor suppression, it paradoxically drives tumor progression and therapy resistance in certain cancers.
Glioblastoma
In glioblastoma, STK17A exhibits a pro-tumorigenic effect with expression levels increasing in a grade-dependent manner and correlating with malignancy. Silencing STK17A in glioblastoma cell lines (U87 and U251) reduces proliferation, migration, and invasion. Conversely, STK17A overexpression enhances cell survival and resistance to genotoxic stress, but does not significantly increase proliferation or migration, suggesting that while STK17A contributes to cell motility, it is not the primary driver [28].
In glioblastoma and low-grade of glioma datasets, STK17A expression has been reported to correlate with cytoskeletal regulators (e.g., MRLC, anillin) and with EGFR expression. In glial tumorigenesis, EGFR is overexpressed and PI3K mutated (active constitutive). STK17A could be a downstream of EGFR/PI3K and its co-overexpression with EGFR can promote cell proliferation and glial EGFR-PI3K-driven neoplasia, by directly phosphorylating MRLC at Thr18 and Ser19 which regulates changes to the cytoskeleton by promoting aniline binding to Zipper proteins [21].
Head and Neck Squamous Cell Carcinoma (HNSCC)
In HNSCC, STK17A is overexpressed at both the mRNA and protein levels and exerts a pro-tumoral role. It binds competitively to Smad3 in the cytoplasm, preventing its association with Smad4 and subsequent nuclear translocation. This inhibits TGF-β1 tumor suppressor signaling, which normally induces cell cycle arrest and restricts epithelial tumor progression. STK17A overexpression decreases TGF-β1 signaling and promotes tumor cell proliferation, whereas STK17A silencing restores TGF-β1 activity and reduces tumor cell proliferation [29].
Gastric Cancer
In advanced gastric cancer, STK17A expression is significantly upregulated and is associated with poor prognosis. Overexpression of STK17A in gastric cancer cells (MKN45 and SNU1) promotes proliferation, migration, and colony formation. It also induces epithelial-mesenchymal transition (EMT) markers, increasing N-cadherin and vimentin levels while decreasing E-cadherin expression — key features that facilitate metastasis [34].
In silico Analysis
We analyzed STK17A mRNA expression across various cancer types using data from The Cancer Genome Atlas (TCGA) and compared tumor versus normal tissues usings GEPIA software (http://gepia.cancer-pku.cn). These analyses describe transcript-level associations and are limited by the absence of direct evidence for mechanistic causality or kinase activity.
STK17A is overexpressed in several cancer types, including cervical cancer, colorectal cancer, diffuse large B-cell lymphoma, glioblastoma, head and neck squamous cell carcinoma, glioma, and pancreatic adenocarcinoma (Figure 5).
Cancers exhibiting STK17A mRNA overexpression (GEPIA). Red: STK17A mRNA level in tumor. Grey: STK17A mRNA level in non-tumoral tissue.
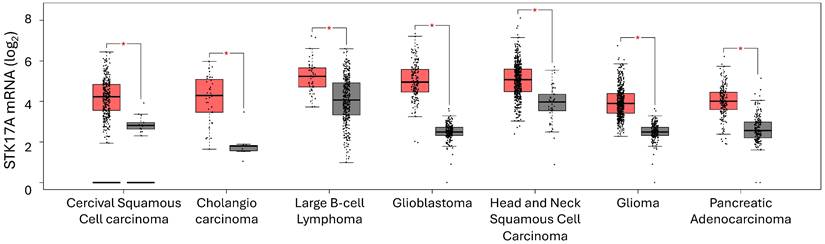
We subsequently investigated the association between STK17A overexpression and overall survival (OS) across all cancer types in the TCGA dataset using the cBioPortal software https://www.cbioportal.org/. High STK17A expression correlates with longer overall survival (OS) in melanoma and showed a favorable trend in cervical and colorectal cancers. Previous studies have reported an anti-tumor role for STK17A in cervical and colorectal cancers, although its function in melanoma remains to be elucidated.
Conversely, STK17A overexpression was associated with a shorter OS in glioma, head and neck squamous cell carcinoma, and gastric cancer. These findings are consistent with existing literature supporting a pro-tumor role of STK17A in these malignancies.
Possible Explanations for the Dual Role of STK17A
The divergent roles of STK17A across cancer types are likely highly context-dependent (Table 1) and reflect differences in (i) baseline expression/addiction, (ii) dominant upstream signaling networks, and (iii) subcellular localization and interactome. Basal abundance may set the stage for whether STK17A is primarily engaged as a stress-effector (low baseline, inducible) versus a pathway “node” co-opted by oncogenic signaling (high baseline, constitutive). STK17A can be induced by genotoxic stress (cisplatin, vinblastine, doxorubicin, etoposide) and acts within a p53-linked apoptotic program, where STK17A supports ROS production and apoptotic sensitivity; in such settings, p53/STK17A silencing reduces chemotherapy responsiveness, consistent with a tumor-suppressive function. Conversely, in tumors where growth-factor signaling dominates, STK17A may be integrated downstream of oncogenic pathways: in glioblastoma/glioma, STK17A is described as pro-tumorigenic, associated with grade, and functionally connected to EGFR/PI3K circuitry and cytoskeletal control via MRLC phosphorylation, supporting proliferation programs and resistance to stress. Similarly, in HNSCC, STK17A can blunt tumor-suppressive TGF-β signaling by sequestering Smad3 in the cytoplasm and preventing Smad3/Smad4 complex formation, thereby favoring proliferation. In gastric cancer, STK17A overexpression is linked to poor prognosis and induction of EMT-like changes, consistent with a pro-invasive role. Mechanistically, STK17A's localization switch (nuclear vs cytoplasmic/mitochondrial) and partner availability may further bias outcomes: STK17A can interact with TRAF6 to dampen NF-kB inflammatory outputs (tumor-suppressive context in cervical cancer) or engage mitochondrial/cytoskeletal partners such as ANT2 and MRLC, which could differentially tune apoptosis versus motility depending on cellular state. Finally, microenvironment-derived signals (e.g., the IFN/RNase axis) together with post-transcriptional regulatory mechanisms may rewire STK17A-associated signaling networks, helping to explain how the same kinase can function as a tumor suppressor in some lineages yet promote tumor progression in others.
STK17A expression and effects in various cancer types
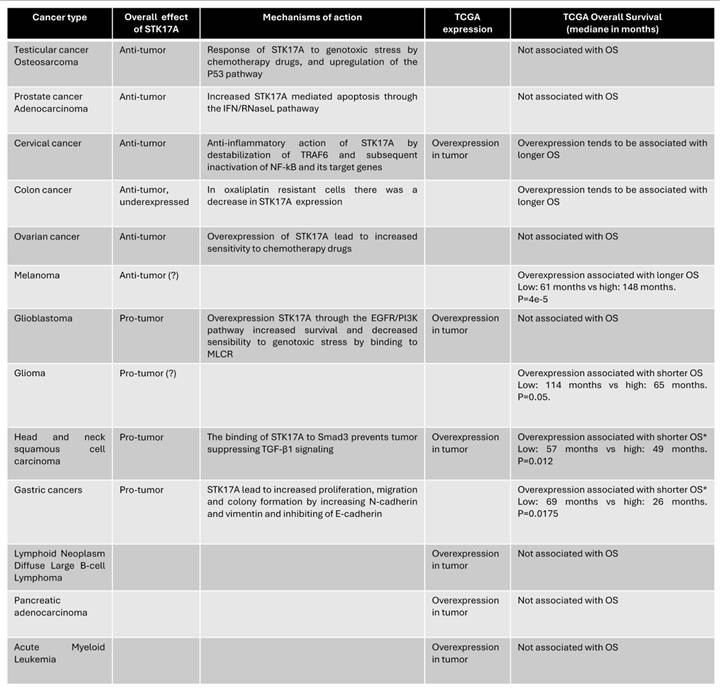
TCGA: The Cancer Genome Atlas; OS: overall survival.
In conclusion, STK17A can be conceptualized as a context-dependent signaling node rather than a classical oncogene or tumor suppressor. In apoptosis-competent settings with intact stress-response machinery, STK17A acts as an apoptosis amplifier or chemosensitizer. In contrast, in tumors dominated by oncogenic signaling and defective apoptotic checkpoints, STK17A may function as a non-oncogene dependency supporting survival, migration, or therapy resistance. These divergent outcomes likely reflect differences in baseline expression, localization, interactome, and signaling context.
STK17A in Other Diseases
Beyond its roles in cancer, STK17A is involved in a wide range of non-oncological diseases, including cardiovascular, autoimmune, infectious, and neurological disorders.
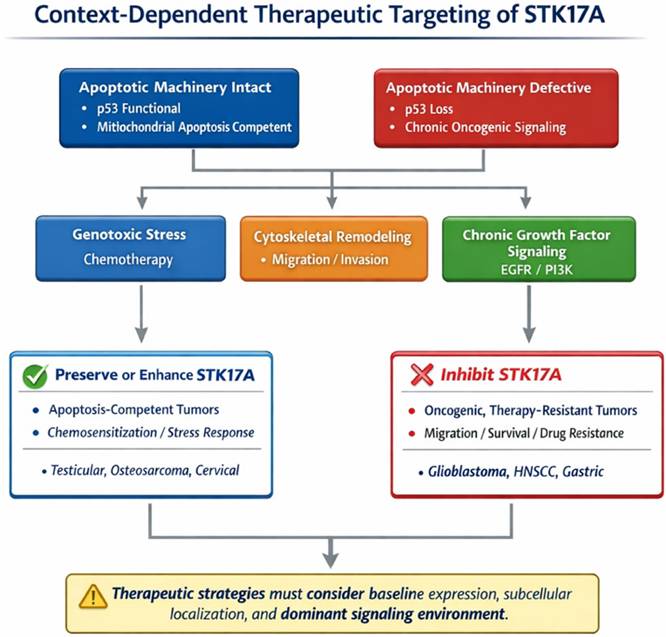
Context-dependent therapeutic targeting of STK17A in cancer. Decision framework scheme for context-dependent therapeutic targeting of STK17A in cancer. In apoptosis-competent tumors with intact p53 function and preserved mitochondrial apoptotic machinery, STK17A appears to act as an apoptosis amplifier and stress-response effector, supporting chemo sensitization under genotoxic stress. In contrast, in tumors characterized by defective apoptotic pathways, chronic oncogenic signaling, and cytoskeletal remodeling, STK17A may function as a non-oncogene dependency that promotes survival, migration, and therapy resistance. Accordingly, therapeutic strategies may favor preservation or enhancement of STK17A activity in apoptosis-competent contexts, whereas pharmacological inhibition may be advantageous in oncogenic, therapy-resistant settings. The scheme emphasizes that optimal targeting of STK17A requires consideration of baseline expression levels, subcellular localization, and the dominant signaling environment.
Organ Transplantation
Evidence linking STK17A to organ transplantation outcomes is limited to observational human tissue studies. In liver transplantation cohorts (human samples), STK17A protein expression has been described in bile canaliculi of healthy grafts, with reduced or altered staining patterns observed during chronic rejection. STK17A has been proposed as a potential early biomarker for liver graft rejection [35].
These findings are correlative and do not establish a functional role for STK17A in graft rejection or tolerance. No mechanistic studies or genetic models have yet validated STK17A as a driver of biliary injury or immune-mediated rejection, and its potential utility should therefore be considered exploratory and biomarker-oriented rather than causal.
Autoimmune Diseases
In autoimmune conditions such as systemic lupus erythematosus (SLE), STK17A expression is altered. SLE is an autoimmune disease in which about 50% of patients develop nephritis and cerebral vasculitis. During active phases of SLE, STK17A expression is repressed, which may contribute to impaired DNA damage repair, a hallmark of SLE pathology [8]. Genetic polymorphisms in STK17A are associated with increased susceptibility to the disease.
Nevertheless, associations between STK17A and autoimmune diseases are primarily supported by human genetic and transcriptomic studies. These observations are restricted to human cohorts and lack functional validation. Importantly, causal involvement of STK17A in autoimmunity has not been demonstrated in vivo, in contrast to STK17B, whose immune regulatory functions are supported by genetic mouse models. Sex-specific effects have not been systematically addressed and remain an important unresolved variable.
Neurodegenerative Diseases
Alzheimer's disease is characterized by progressive neuronal loss and the accumulation of insoluble protein aggregates, such as amyloid beta, accompanied by secondary inflammatory and immune responses. In Drosophila models, STK17A orthologs have been implicated in the regulation of amyloid precursor protein (APP) metabolism, suggesting a possible protective role of STK17A against amyloid beta accumulation [36]. These findings suggest evolutionary conservation of stress-related functions but cannot be directly extrapolated to human disease. No direct evidence currently demonstrates a role for STK17A in mammalian neurodegeneration, and reported associations should be interpreted as hypothesis-generating rather than mechanistic.
Neurological and Psychiatric Disorders
Emerging evidence links STK17A to neurological and psychiatric conditions. STK17A is abnormally expressed in the brains of individuals with major depressive disorder, suggesting a potential involvement in psychiatric diseases [37]. In chickens, STK17A polymorphisms have been correlated with anxiety-like behaviors, indicating a possible conserved role in stress response regulation across species [18]. Nevertheless, as in neurodegenerative disease, there is no experimental evidence clearly demonstrating a direct link between STK17A and psychiatric conditions.
Infectious Diseases
STK17A plays also a role in host defense against bacterial infections. In infections caused by Mycobacterium tuberculosis, Mycobacterium avium, or Mycobacterium abscessus, STK17A expression is upregulated in macrophages, promoting apoptosis and limiting pathogen replication [38,39]. Genetic studies have shown that mutations on chromosome 7p13, where STK17A is located, reduce STK17A expression and increase susceptibility to non-tuberculous mycobacterial pulmonary disease [40]. However, direct genetic or functional evidence demonstrated that STK17A is required for host defense, pathogen clearance, or apoptosis in vivo is currently lacking.
Age/sex-related pathologies
Current evidence linking STK17A to age/sex-related pathologies remains limited and largely indirect. Transcript-level analyses of human bone marrow samples reported sex-associated increases in STK17A expression, particularly in females, with correlations to markers of osteoclast genesis and apoptotic regulation in age-related disorders such as osteoarthritis and rheumatoid arthritis, without demonstrating mechanistic causality. It also reported higher STK17A expression in osteoarthritis and rheumatoid arthritis patients compared with normal bone marrow cells [41]. More recently, genome-wide association studies identified STK17A among shared genetic loci associated with age-related cataract and hearing difficulties [42]. Collectively, these findings support a potential association between STK17A and aging-related tissue stress responses, while a direct causal role in age-related disease pathogenesis has yet to be established.
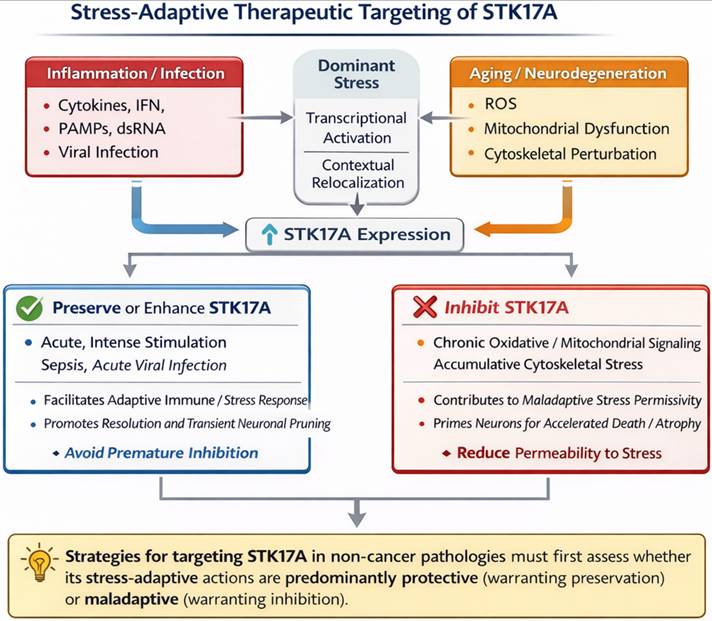
Stress-adaptive therapeutic targeting of STK17A in non-cancer pathologies. This schematic proposes a stress-adaptive framework for therapeutic modulation of STK17A in non-cancer pathologies. Diverse stressors, including inflammation or infection (cytokines, interferons, PAMPs, dsRNA) and aging- or neurodegeneration-associated stress (oxidative stress, mitochondrial dysfunction, cytoskeletal perturbation), converge on transcriptional induction and context-dependent relocalization of STK17A. In acute or intense stress settings, preservation or enhancement of STK17A activity may facilitate adaptive immune or stress responses and support resolution processes, suggesting that premature inhibition could be detrimental. Conversely, under chronic or cumulative stress conditions, sustained STK17A signaling may contribute to maladaptive stress permissiveness, neuronal vulnerability, or tissue degeneration, thereby providing a rationale for pharmacological inhibition.
Metabolic disorders (Diabetes mellitus)
Diabetes mellitus is a disorder characterized by metabolic anomalies marked by insulin resistance, relative insulin deficiency, and persistent hyperglycemia. During the progression of diabetes, STK17A may contribute to the advancement of the disease by regulating oxidative stress, programmed cell death pathways, and critical signaling pathways such as p53 and MAPK, thereby promoting the death of islet cells [43]. Moreover, STK17A has been also discovered as a gene that can differentiate between individuals with obstructive sleep apnea and diabetes mellitus based on four machine learning methods. Indeed, chronic intermittent hypoxemia increases oxidative stress by enhancing relative oxygen production and oxidative/antioxidative imbalance in obstructive sleep apnea patients and oxidative stress permeates the development of diabetes and its complications [44]. Although STK17A has been proposed to influence β-cell survival through stress-associated pathways, no direct functional or genetic evidence confirms its involvement in diabetes onset or progression.
Comparison STK17A / STK17B
STK17B functions are supported by genetic and in vivo models—particularly in immune regulation—whereas STK17A evidence relies predominantly on cell-line perturbation and correlative analyses. This asymmetry has important implications for therapeutic prioritization: confidence in STK17A mechanisms and safety liabilities is necessarily lower until validated by improved models and selective chemical probes (Table 2 and Figure 8).
Comparative biological and therapeutic features of STK17A and STK17B
| Feature | STK17A | STK17B |
|---|---|---|
| Genetic in vivo models | Absent (murine loss) | Robust knockout models |
| Dominant Biology | Stress/apoptosis, cytoskeleton | Immune tolerance, T-cell signaling |
| Cancer targeting logic | Context-dependent | Limited evidence |
| Autoimmune relevance | Indirect | Strong |
| Drug-development complexity | Higher (model limitation) | Lower |
Distinct biological roles and therapeutic implications of STK17A and STK17B.
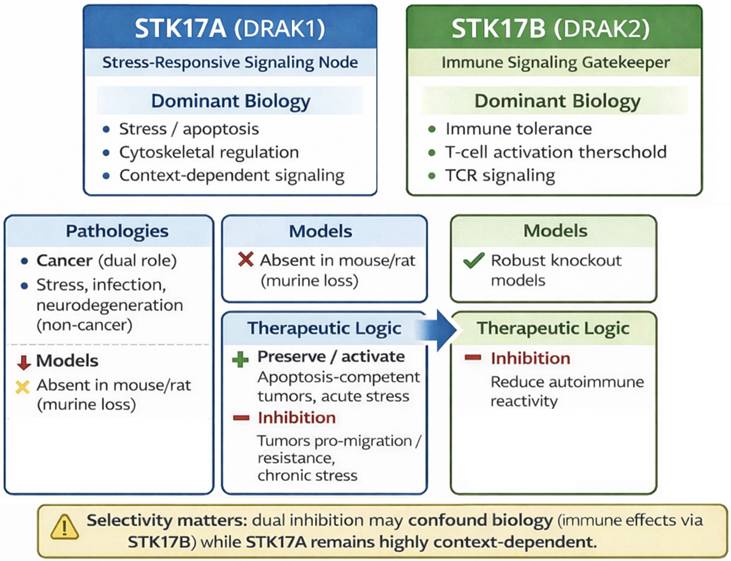
STK17A and STK17B share a high degree of sequence similarity within their catalytic serine/threonine kinase domains, reflecting their common origin as members of the DAPK subfamily. Both proteins lack the CaM-binding domain characteristic of other DAPK family members and are primarily localized in the nucleus, where they contribute to the regulation of cell death pathways. Despite these structural and functional similarities, several differences distinguish between the two kinases. STK17B is predominantly expressed in lymphoid tissues and has been extensively implicated in immune cell homeostasis and tolerance. By contrast, STK17A shows a broader but generally lower expression profile and remains comparatively less studied. Emerging evidence suggests that STK17A may play a role in apoptosis and tumorigenesis, whereas STK17B has been more clearly associated with T cell signaling and autoimmune regulation. These observations highlight that, while structurally related, STK17A and STK17B may fulfill non-redundant, context-dependent biological functions.
Role in T Cells and Immune Regulation
STK17B is predominantly expressed in lymphoid tissues and is highly enriched in B cells and T cells. It acts as a negative regulator of T cell activation, fine-tuning immune responses. During T cell development, STK17B expression fluctuates and is tightly regulated. STK17B deficiency results in T cell hypersensitivity to TCR (T cell receptor) stimulation, requiring less co-stimulatory signaling for activation [45]. In STK17B knockout mice, T cells are hyperresponsive but surprisingly exhibit resistance to autoimmune diseases such as experimental autoimmune encephalomyelitis (EAE), a model for multiple sclerosis, and type 1 diabetes in NOD mice [46].
Conversely, overexpression of STK17B in transgenic mice enhances T cell apoptosis in an IL-2-dependent manner and impairs memory T cell development [47]. Thus, STK17B plays a critical role in balancing T cell activation, survival, and memory formation. In comparison, STK17A is expressed in several tissues, but its role in T lymphocytes is less well characterized than that of STK17B [47].
Interaction with TGF-β Signaling
Unlike STK17A, which binds to Smad3, STK17B inhibits TGF-β signaling through a different mechanism. It binds directly to the TGF-β type I receptor (TβRI), blocking the phosphorylation and activation of Smad2/3 [48]. Notably, STK17B can bind to both active and inactive forms of TβRI, distinguishing it from other negative regulators such as Smad7.
This interaction has been demonstrated in breast cancer, hepatocellular carcinoma, and cervical cancer cell lines, where STK17B overexpression suppresses TGF-β-induced tumor-suppressive signaling. Interestingly, despite its nuclear localization in many cancer cells, STK17B also localizes to the cytoplasm and membrane where it interacts with TβRI.
However, in T cells, STK17B does not inhibit TGF-β signaling, as Smad3/4 activation remains intact in wild-type and STK17B knockout T cells [49].
Tumorigenic and Tumor Suppressor Roles
STK17B displays context-dependent effects in cancers. In breast cancer, STK17B overexpression promotes tumor progression whereas in myeloid leukemia and in colorectal cancer, STK17B acts as a tumor suppressor, enhancing apoptosis when overexpressed. In myeloid leukemia models, oncogene silencing de-repressing STK17B leads to increased apoptosis [50].
Interestingly, despite these in vitro effects, STK17B knockout mice show no difference in spontaneous tumor development, even when crossed with p53-deficient mice or exposed to carcinogens, suggesting that STK17B alone may not play a dominant role in tumor surveillance in vivo [50].
Compared to STK17A, the role of STK17B in cancer is less extensively characterized; however, both kinases exhibit distinct and context-dependent functions across different tumor types.
Role in Neuronal Development
STK17B also phosphorylates myosin-II regulatory light chain (MRLC), like Rho kinases, influencing cytoskeletal dynamics. In neurons, STK17B inhibits axon outgrowth, elongation, and branching. Pharmacological inhibition of STK17B with compounds such as SC82510 enhances axonal regeneration, suggesting that STK17B acts as a negative regulator of neuronal regeneration [51].
Conclusion
The biological complexity and context-dependent roles of STK17A and STK17B underscore their relevance as signaling nodes in apoptosis, immunity, cytoskeletal control and tumorigenesis.
However, despite growing insights into their molecular interactions and disease associations, the precise mechanisms governing their activity remain incompletely understood. Indeed, major limitations constrain current interpretation for STK17A: (i) reliance on overexpression/knockdown systems; (ii) limited validation of endogenous substrates and kinase activity; (iii) absence of murine genetic models; and (iv) frequent overinterpretation of transcriptomic or network associations as mechanistic causality. Future work should prioritize (a) development and broad dissemination of selective, cell-active STK17 chemical probes; (b) quantitative phosphoproteomics and orthogonal substrate-validation pipelines and (c) alternative in vivo models (e.g., zebrafish or humanized systems) that enable genetic interrogation.
Particularly, the development of small-molecule inhibitors would not only provide essential probes to clarify the distinct and overlapping roles of STK17A and STK17B, but also offer opportunities for therapeutic intervention in cancers, autoimmune, and neurodegenerative diseases. Recent medicinal chemistry efforts have, therefore, focused on designing scaffolds with improved potency and selectivity on each STK17 kinase, paving the way for both mechanistic studies and potential clinical applications.
STK17A/B Inhibitors
Despite the therapeutic promise of STK17A, only a few STK17A inhibitors have been reported to date [9]. Conversely, inhibitors of STK17B have attracted more attention, particularly for the treatment of autoimmune diseases and the prevention of transplant rejection [52]. Although this review primarily focuses on STK17A, the identification of inhibitors targeting both STK17A and STK17B is of significant interest for advancing our understanding of their biological functions and therapeutic implications, as well as for the development of novel pharmacological agents. The discovery of new, potent, and truly selective chemical scaffolds could serve as a foundation for further optimization in drug discovery and provide valuable insights into the distinct and overlapping roles of these kinases in biological systems.
Notably, the development of STK17A-selective inhibitors remains a critical unmet need. Such compounds would enable precise dissection of STK17A-specific signaling pathways and functions while minimizing unintended interference with STK17B-mediated immune regulation. Beyond their utility as mechanistic probes, selective STK17A inhibitors could offer safer and more effective therapeutic strategies in cancers and non-oncological diseases in which STK17A plays a pivotal role.
The first STK17 inhibitor report is a STK17B inhibitor (SC82510, small molecule with undisclosed structure) described only in 2014 [51]. It exhibits a low micromolar IC50 yet promotes in cellulo activity at 1-5 nM, suggesting that significant off-target effects may be expected. SC82510 is an improved derivative of 2-(2-(trifluoromethoxy)benzamido)-N-(6-(trifluoromethyl)-1H-benzo[d]imidazol-2-yl)thiazole-4-carboxamide, another kinase (CK1δ/ε) inhibitor [53]. SC82510 had a much cleaner profile in the KINOMEscan™ assay, inhibiting only STK17A, STK17B and RPSK2, although less active than the other analogues tested. Interestingly, inactive compounds resulted from a benzothiazole core versus a benzimidazole one. Furthermore, the inhibition of STK17B in cells was associated to axon outgrowth, proposing a pro-regenerative activity with potential therapeutic applications following axonal lesions [51].
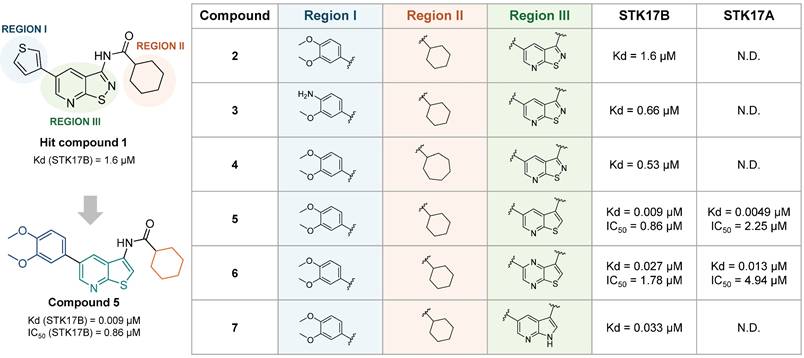
In the same year, Gao et al. reported an isothiazolo[5,4-b]pyridine backbone (1, Figure 9) as novel STK17B ligand (Kd = 1.6 µM) discovered from a 150 compounds library screening in the DiscoverX binding assays [52]. The optimization of this structure towards affinity enhancement led to thieno[2,3-b] pyridine derivatives, revealing compound 5 (Figure 9) as a dual STK17A (Kd = 5 nM, IC50 = 2.25 µM) and STK17B (Kd = 9 nM, IC50 = 0.86 µM) inhibitor. Modifications of the two regions, namely 5-aryl (region I, Figure 9) and cyclohexyl (region II, Figure 9), did not lead to affinity improvement, with the best compounds as 2, 3, and 4 (Figure 9). Besides, the replacement of cycloalkyl amides by non-cylic and aromatic amides led to complete loss of STK17B binding. Scaffold hopping on the isothiazolopyridine scaffold led to compound 5 with the highest affinity for STK17B (Kd = 9 nM) and compound 6 with Kd = 27 nM (Figure 9). Nevertheless, both compounds were not selective to STK17B, showing also strong STK17A binding (Kd = 4.9 nM and 13 nM, respectively). Functional activity against both STK17A and STK17B enzymes confirmed the dual inhibitory properties of compounds 5 and 6, albeit with IC₅₀ values in the micromolar range. The difference in affinity and activity values remains unclear, despite the suggestion that these compounds act as ATP competitive inhibitors might partially account for it. Interestingly, compound 7 shows strong affinity for STK17B but lacks completely functional inhibitory activity.
Discovery of thieno[2,3-b]pyridine derivatives as potent STK17A/B ligands [52]. Representative compounds with corresponding binding constants (Kd) and enzymatic activity IC50 values.
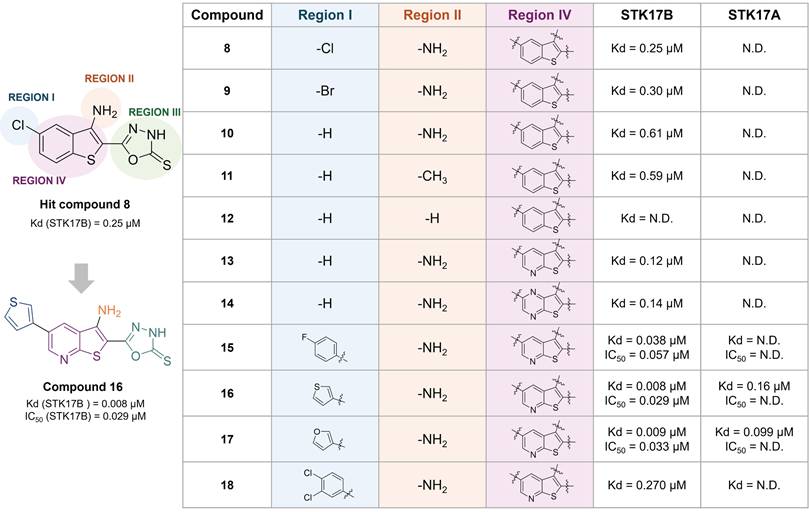
The same research group reported a benzothiophene hit (8) with high affinity to STK17B (Kd = 0.25 µM), optimized towards compound 16 (Figure 10) [54]. This optimized inhibitor still shows low selectivity within the DAPK family but shows improved inhibitory activity for STK17B (Kd = 8 nM, IC50 = 29 nM, Figure 10) than the previous developed compound 5 (Kd = 9 nM, IC50 = 860 nM, Figure 9) [52]. SAR was conducted based on the modifications of 4 regions (Figure 10). The substitution of the phenyl ring with electron-withdrawing and electron-donating substituents did not afford better analogues than the hit compound 8. The presence of the halogen (compound 8, 9 vs 10) in position 6 and of the amino in position 3 (compound 10 vs 11) only showed a slight beneficial effect (Figure 10). In contrast, the thioxo group in the oxadiazole moiety in region III was revealed to be essential for improving activity. The modulation of the benzothiophene (region IV) by introduction of nitrogen atoms in the 6-membered ring (compound 13, 14) resulted in compounds with 5-fold tighter binding than the hit compound 8. Therefore, the thieno[2,3-b]pyridine scaffold was selected for further optimization of region I. Introduction of aryl substituents in position 6 resulted in drastic binding improvements with low nanomolar Kd values. In addition, the 3-thiophenyl and 3-furanyl derivatives 16 and 17 demonstrated high enzymatic activity (IC50 = 29 nM and 33 nM, respectively, Figure 10). However, the selectivity of these compounds remained limited, with compound 18 showing the highest selectivity although with lower activity.
Optimization of thieno[2,3-b]pyridine scaffold as potent non-selective STK17B ligand [54]. Representative compounds with corresponding binding constants (Kd) and enzymatic activity IC50 values.
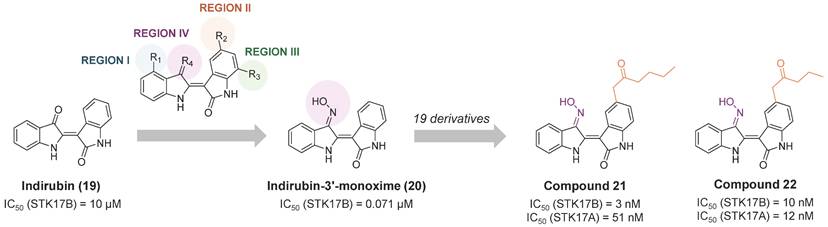
In 2016, indirubin derivatives were identified as a promising scaffold for STK17B inhibition on a high throughput screening [55]. Optimization of the hit compound indirubin (compound 19, Figure 11) —the active component of a traditional Chinese medicinal formulation, Danggui Longhui Wan—revealed that the oxime group at R4 position and small aliphatic groups at R5 are critical for enhancing STK17B inhibitory activity. Derivatives of indirubin-3'-monoxime (compound 20) led to the discovery of compound 21 as the most potent derivative with an inhibitory value for STK17B IC50 = 3 nM and Ki = 0.25 nM. Interestingly, compound 22, bearing a propylketone (region II) instead of the butylketone (compound 21), exhibited high activity against both STK17B and STK17A (Figure 11). Enzyme kinetics and molecular docking studies indicated that compound 21 is an ATP-competitive inhibitor. Although all analogues exhibited moderate inhibitory activity against DAPK1-3 and strong inhibition for STK17A, compound 21 demonstrated the highest selectivity. Docking studies were consistent with the higher binding affinity observed for STK17B compared to other DAPK family members, attributing this selectivity to the interaction of the inhibitor with the active site residue Tyr112.
Optimization of indirubin toward potent indirubin-3'-monoxime non-selective STK17A/B ligands [36]. Representative compounds with corresponding enzymatic IC50 values.
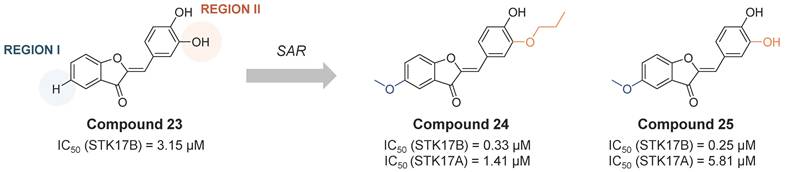
Another class of STK17B inhibitors presenting a benzofuran-3(2H)-one scaffold was discovered on a high throughput screening [56]. From the hit compound 23 (IC50 STK17B = 3.15 µM, Figure 12), the SAR study let to the obtention of compounds 24 and 25 (Figure 12) with IC50 values around 10-fold lower than the initial hit (IC50 STK17B = 0.33 µM and 0.25 µM, respectively). Although both compounds are nonselective STK17B inhibitors with residual activities against other members of the DAPK family, including STK17A, compound 24 showed a more favorable selectivity profile than compound 25 against other kinases of the DAPK family. Both compounds were found to protect islet b-cells from apoptosis in dose dependent manners, supporting the hypothesis that STK17B inhibitors might be a promising approach for the treatment of diabetes.
Optimization of benzofuran-3(2H)-one non-selective STK17B inhibitors [56]. Representative compounds with corresponding enzymatic IC50 values.
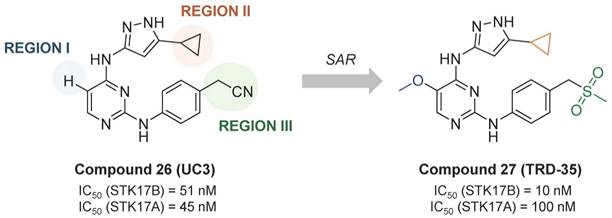
From the promiscuous kinase inhibitor (compound 26, UC-3, IC50 STK17B = 51 nM), Ali et. al. developed a promising quite selective 3-aminopyrazoylpyrimidine STK17B inhibitor (compound 27, TRD-35, IC50 STK17B = 10 nM), though no cellular activity is reported (Figure 13) [57]. Interestingly, the replacement of the cyclopropyl by tert-butyl led to total loss of activity (IC50 > 10000 nM).
Optimization of promiscuous 3-aminopyrazoylpyrimidine (compound 26, UC-3) toward potent and selective STK17B inhibitor compound 27 (TRD-35) [57]. Representative compounds with corresponding enzymatic IC50 values.
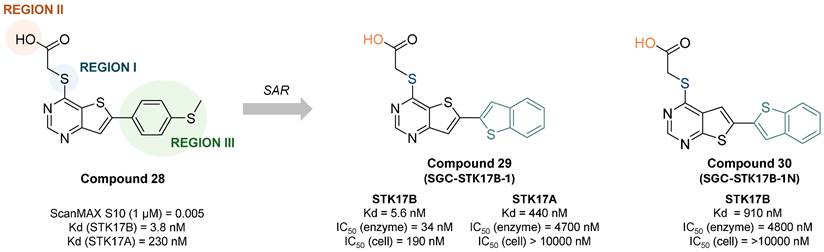
The most selective and in cellulo active STK17B inhibitor reported to date was described by Picado et al. in 2020 [58]. Thieno[3,2d] pyrimidines were screened in the DiscoverX scanMAX panel screen against 403 wild-type human kinases leading to the discovery of PFE-PKIS 43 (compound 28, Figure 14). They also revealed by MD simulations that a unique P-loop conformation appears to be the molecular basis of their remarkable potency and selectivity. SAR studies demonstrated the critical need of the sulfur atom in region I as well as the carboxylic acid in region II for activity. Extensive SAR study on region III led to optimized compound 29 (SGC-STK17B-1, IC50 STK17B = 34 nM) with high selectivity and potency contrary to its isomer 30 (SGC-STK17B-1N, IC50 STK17B = 4800 nM). The drastic loss in activity for compound 30 was attributed to the reduced basicity of the pyrimidine N1, which does not support a similar strength of molecular recognition.
Discovery of thieno[3,2d] pyrimidines as selective and potent STK17B inhibitors. Representative compounds with corresponding binding constants (Kd) and activity IC50 values. ScanMAX, S10 (1 μM) = number of kinases with 10% or less of control activity remaining at 1 μM / number of wild type kinases tested (403), n = 1. Kd determined by dose response, n = 2. IC50 for enzyme inhibition at Km ATP determined at Eurofins, n = 2. Target engagement IC50 determined by NanoBRET assay in HEK293 cells [58].
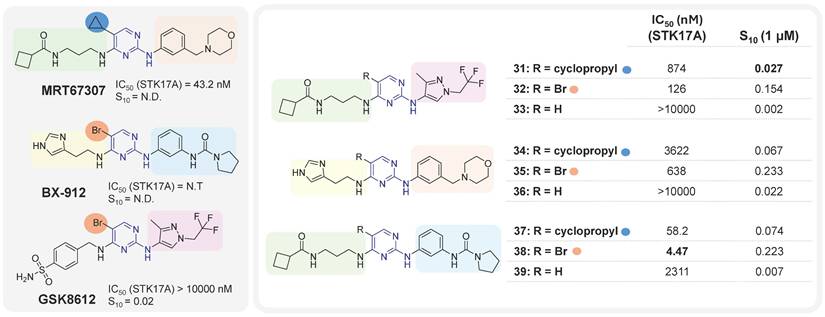
It was only from 2021 onwards that studies focusing on the development of selective STK17A inhibitors began to emerge. In that year, Drewry at al. identified six 2,4-diaminopyrimidine-based compounds as non-selective submicromolar STK17A inhibitors, in the search of ligands for understudied kinases implicated in driving neurodegeneration [59]. These pyrimidine-based derivatives were developed by diversely substituting both amino groups. This strategy can lead to an additional hydrogen bond with the kinase conserved hinge region found in nearly all human kinases along with the key hydrogen bond associated with nitrogen of the pyrimidine scaffold, extensively used in the design of kinase inhibitors. The derivatives were designed based on the structure of three previously studied TBK1 inhibitors MRT67307, BX-912 and GSK8612 (Figure 15). The mixing and matching of the side chains and cores from these three inhibitors led to 21 derivatives [59]. A first kinase panel screening (14 kinases, Eurofins) demonstrated the enzymatic activity of some compounds towards STK17A, which was subsequently confirmed through single-dose and dose-response assays using NanoBRET. Compounds 31, 32, 35, 38, 37 and MRT67307 (Figure 15) were shown to have submicromolar activity in the NanoBRET STK17A assay. Interestingly, substitution in position 5 of the pyrimidine (-R, Figure 15) appeared critical for activity, with bromine as the best substituent. Compounds 35, 34, 38, and 37 also showed submicromolar inhibitory activities against MARK4 kinase in NanoBRET, whereas compounds 35 and 38 showed submicromolar activities against MARK3. Overall, compound 31 may represent the most promising molecule of this series, despite its moderate activity (S10 = 0.027, IC50 = 874 nM, Figure 15).
Aminopyrimidines derivatives and corresponding STK17A IC50 values (NanoBRET cell assay) and selectivity indexes S10 [59]. N.D.: not determined. S10 (selectivity index at 1μM): percentage of screened kinases with percentage of control values < 10% at 1μM. The lower the S10 value, the higher the selectivity.
In 2022, Kurz et. al. reported the first selective STK17A inhibitor (CK156, compound 40, Figure 16), exhibiting high in vitro potency (Kd = 21 nM, IC50 = 49 nM) and remarkable selectivity in kinome widescreens (S35 = 0.005 at 100 nM and S35 = 0.02 at 1 mM; S35: selectivity score) [60]. This achievement was permitted by structure-guided optimization of a pyrazolo[1,5-a]pyrimidine-based macrocyclic scaffold (Figure 16), previously found as a RIPK2 inhibitor [61]. Macrocyclic kinase inhibitors typically contain an ATP-mimetic pharmacophore, which is constrained by a linker of 12 or more atoms, connecting flexible moieties of acyclic conventional inhibitors [62].
Macrocyclic kinase inhibitors and their activities against STK17A determined using various analytical techniques [60]. DSF: differential scanning fluorimetry; ITC: isothermal titration calorimetry; 33PanQuinase (Reaction Biology): enzymatic activity assay; NanoBRET (Promega): target engagement assays; KINOMEscan (Eurofins Discovery): selectivity profile. S35: selectivity index score. The S-score quantifies the selectivity at the measured concentration; threshold of %Ctrl = 35% [S35 = (number of non-mutant kinases with %Ctrl <35)/ (number of non-mutant kinases tested)]. The lower the S35 value, the higher the selectivity.
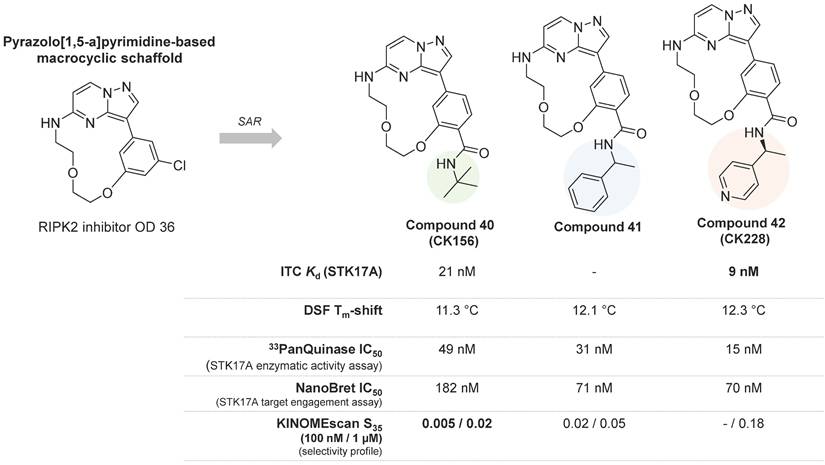
The optimization of the pyrazolo[1,5-a] pyrimidine-based macrocyclic scaffold resulted in better potent inhibitors of STK17A, compounds 41 and 42 (Figure 16), with in vitro activity values of 31 nM and 15 nM, respectively [60]. Nevertheless, compounds 41 and 42 showed lower selectivity in the KINOMEscan (high throughput site-directed competition affinity binding assay over 500 kinase domain-containing human wildtype and clinically relevant mutant targets) in comparison to compound 40 (Figure 16), which showed exclusive affinity to STK17A, with an IC50 value of 182 nM. Indeed, affinities for other potential off-target kinases were 30-fold lower compared to STK17A. The first crystal structure of STK17A was obtained through co-crystallization with compound 40, revealing a classical type I inhibitor binding mode. The tert-butyl group of compounds 40 was observed to point to the back pocket of STK17A and does not disturb key H-bonds with the hinge region and Lys90. In addition, cell viability studies showed no toxicity of compound 40 in HEK293T, U2OS, and MRC-9 cells (1 μM concentration). Despite the unknown in vivo activity and bioavailability of the pyrazolo[1,5-a] pyrimidine-based STK17A inhibitors, compound 40 is a very promising chemical probe and provides valuable insights for the development of selective STK17A inhibitors.
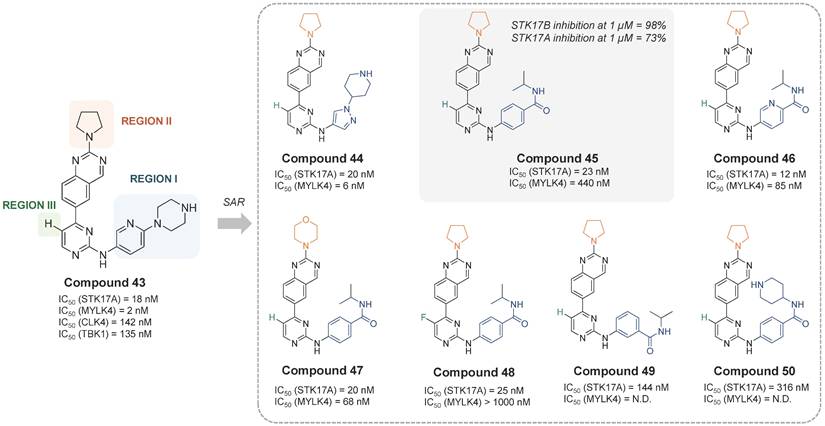
In the most recent work, Chaudhry, S. presented a quinazoline-based STK17A/B dual inhibitors with good drug-like profile and oral bioavailability [63]. The developed inhibitors selectively target STK17A and STK17B and were shown to be effective in viability assays against cancer cell lines, suggesting a role of STK17A as a therapeutic kinase target in various cancers. Compound 43 was the first hit in a profiling study against a panel of 375 kinases. It showed strong inhibition of MYLK4 (IC50 = 2 nM) and STK17A (IC50 = 18 nM), and moderate potency against TBK1 (IC50 = 135 nM) and CLK4 (IC50 = 142 nM). The SAR study over 19 analogues revealed that the modifications in the region I with an N-piperidine pyrazole moiety (compound 44, IC50 = 20 nM) or a 4-yl-isopropylbenzamide moiety (compound 45, IC50 = 23 nM) led to derivatives with STK17A inhibitory potency comparable to that of compound 40 (Figure 16). Yet, better potency was also observed when the aromatic ring in Region-I was a pyridine instead of a benzene moiety (compound 46, IC50 =12 nM). Nevertheless, neither the modification of region II by a morpholine ring (compound 47, IC50 = 20 nM) nor the modification of region III by a F-substitution (compound 48, IC50 = 25 nM) showed significant improvement (Figure 17). Besides, reduction on the STK17A inhibition was observed with the 3-yl-isopropylbenzamide moiety (compound 49, IC50 = 144 nM) and further with the 4-yl-piperidinebenzamide moiety (compound 50, IC50 = 316 nM), probably due to the additional NH that could form H-bonds with surrounding protein residues. The selectivity against MYLK4 and CLK4 kinases was evaluated, revealing that compounds 45 and 48 were the most selective (Figure 17). Both compounds also displayed favorable in vitro DMPK properties, including good human/mouse microsomal stability and low inhibition of cytochrome P450 isoforms. Compound 45 was selected as the lead candidate, and in silico studies suggested a Type-I kinase inhibition mode. However, final selectivity assessment of compound 45 toward STK17A showed 98% inhibition of STK17B versus 73% inhibition of STK17A at 1 µM, indicating that compound 45 acts as a dual STK17A/B inhibitor with greater potency against STK17B. Biological evaluation of compound 45 across three acute myeloid leukemia (AML) cell lines (MOLM-13, HEL, and Kasumi-1) revealed EC₅₀ values in the low micromolar range, while demonstrating good selectivity over the immortalized cell line HEK-293T (IC50 ≈ 100 μM). However, direct target-engagement evidence supporting this AML activity remains to be established. Pharmacokinetics studies in C57BL/6 mice after oral administration (vehicle: 2% Tween80 / 10% DMSO / 88% PBS) of compound 45 (dose: 10 mg/kg) indicated a reasonable exposure (t1/2 = 2.7 h, Tmax = 1h, Cmax = 1 μM, AUC = 4.2 μM·h). Compound 45 has shown activity on tumor progression in an isogenic K562 xenograft mice experiment (TGI around 46%), being patented for treatment of proliferative disorders, including myelodysplastic syndrome and leukemia (WO2023212181).
Quinazoline-based STK17A/B inhibitors and their activities [63]. IC50 values were based on enzyme activity assays with an ATP concentration of 10 μM (Reaction Biology Corporation). STK17B inhibition was tested only with compound 45 (1 µM). CLK4 IC50 is not included in the figure because it is either > 10,000 nM or not determined.
Overall, research on STK17 inhibitors remains very recent. Initial efforts were primarily directed toward STK17B inhibitors, with the first reports of selective STK17A inhibitors emerging only from 2021 onward. To date, the chemical space explored for both kinases remains highly limited, with only five distinct scaffolds described for STK17B inhibition and three for STK17A inhibition (Table 3). This narrow diversity highlights rather the novelty of the field than identified significant challenges associated with identifying drug-like and selective inhibitors.
Summary of main small molecule STK17A and STK17B inhibitors.
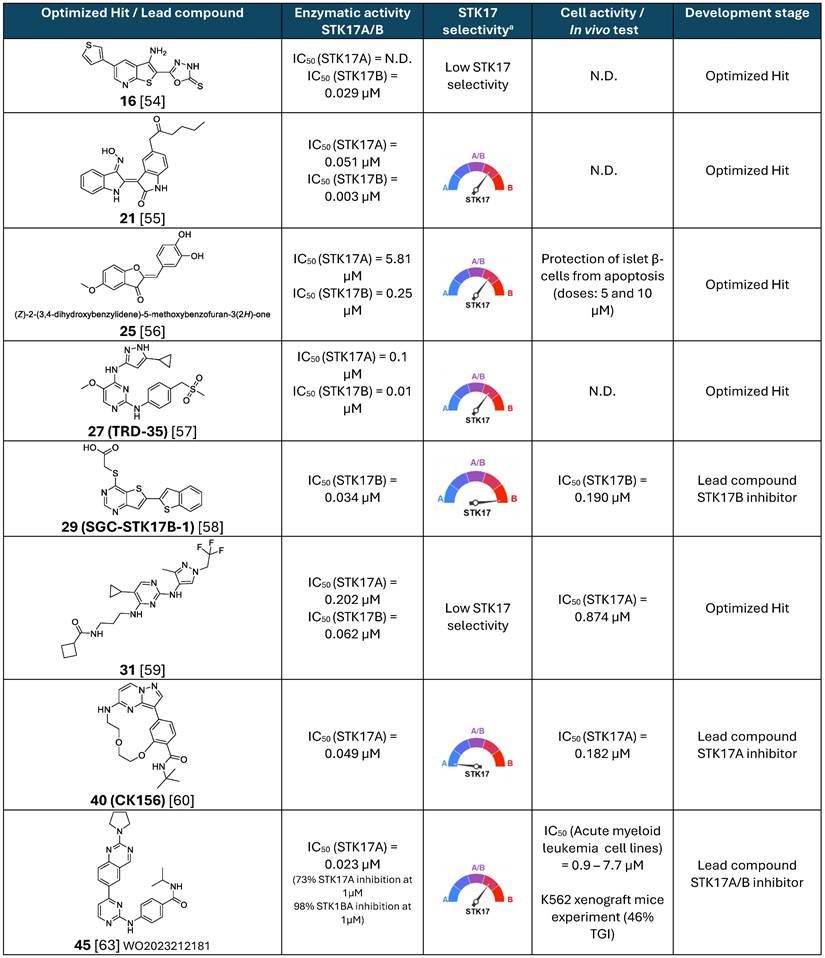
N.D.: not determined. a Low STK17 selectivity: IC50 difference between STK17 and other kinases < 5-fold.
STK17A/B selectivity:

Among STK17B inhibitors, the thieno[2,3-b] pyridine scaffold demonstrates promising two-digit nanomolar enzymatic potency; but exhibits limited selectivity over other DAPK kinase. In addition, physicochemical liabilities may constrain their use in biochemical or cellular studies. For example, the lead compound 16 displays a very low predicted solubility (cLogS = -6.5), likely due to its rigid structure and limited number of rotatable bonds. Experimental validation of solubility will be important to determine its suitability as a chemical probe. Future optimization could focus on the introduction of polar or solubilizing substituents, e.g. through further exploration of region II, which to date exhibits relatively flat structure-activity relationships.
The indirubin scaffold shows high inhibitory activity against STK17 kinases but suffers from poor selectivity. Moreover, structural features such as oxime moiety and overall molecular rigidity raise concerns regarding suitability in biological systems. These characteristics may negatively impact solubility and metabolic stability and could be associated with toxicity liabilities. Comprehensive evaluation of aqueous solubility, plasma stability, and potential oxime-related toxicity would therefore be essential before considering indirubin derivatives as viable chemical probes or therapeutic leads.
The benzofuran-3(2H)-one scaffold displays only modest submicromolar activity, with suboptimal selectivity even for the most advanced compound (25). Furthermore, the presence of a Michael acceptor motif, combined with phenol- or catechol-enriched substituents, raises concerns about chemical reactivity and stability under physiological conditions. Such features may lead to off-target reactivity or rapid degradation, limiting their applicability in cellular or in vivo studies.
Conversely, the 3-aminopyrazolylpyrimidine scaffold, and particularly its lead compound 27 (TRD-35), displays more favorable physicochemical properties. Nevertheless, both potency and selectivity remain suboptimal and would require further optimization to support its development as a robust chemical probe.
Among the STK17B inhibitor series reported to date, the thieno[3,2-d] pyrimidine scaffold appears to be the most promising. Compounds from this series combine high two-digit nanomolar enzymatic potency with excellent selectivity over a broad kinase panel, including STK17A, and demonstrate sub micromolar cellular activity supported by direct target engagement using NanoBRET assays. In addition, the predicted physicochemical properties of the lead compound 29 are encouraging. The next steps toward validating this scaffold as a selective STK17B chemical probe will be the comprehensive characterization of its pharmacological profile via in vitro ADMET assays.
With respect to STK17A inhibitors, an initial aminopyrimidine scaffold has been described; however, the lead compound 31 still lacks sufficient potency and selectivity to qualify as a reliable chemical tool.
In contrast, the macrocyclic series — particularly the lead compound 40 (CK156) — represents a highly promising advancement. This scaffold exhibits two-digit nanomolar enzymatic potency that is largely preserved in cellular settings, with three-digit nanomolar activity observed in cellulo. Selectivity over other kinases also appears favorable, although direct comparisons across series remain challenging due to differences in selectivity metrics. Importantly, this series enabled the first co-crystal structures of STK17A, providing valuable structural insights into ligand binding within the catalytic pocket. It should be noted, however, that ligand-protein interactions observed for a given chemotype are not necessarily generalizable, and alternative scaffolds may engage STK17A through distinct binding modes. Additional structural and comparative studies across different chemotypes would therefore be highly valuable to the field.
Finally, the quinazoline-based scaffold represents a dual STK17A/B inhibitor series with a preference for STK17B. Although strong enzymatic inhibition has been reported for STK17B, residual activity against MYLK4 and STK17A limits the suitability of these compounds as selective chemical probes. Nonetheless, their demonstrated cellular activity and oral efficacy in vivo, particularly in acute myeloid leukemia models, support their further evaluation as preclinical oncology candidates. Notably, direct evidence of target engagement underlying the observed cellular and in vivo effects is still lacking and will be essential to establish the mechanistic basis of their pharmacological activity.
Overall, only a limited number of STK17 inhibitors have been reported, and substantial optimization is still required to develop truly ideal chemical probes. SGC-STK17B-1 (compound 29) currently represents the most advanced candidate for studying STK17B inhibition, pending full pharmacological validation. However, there remains a clear unmet need for a highly selective STK17A probe. CK156 (compound 40) is the most promising STK17A-directed compound to date, but further optimization is needed to achieve fully satisfactory potency, selectivity, and drug-like properties. An ideal STK17A chemical probe would combine single-digit nanomolar enzymatic potency with ≥50-fold selectivity over other kinases and possess physicochemical characteristics suitable for biological studies, including adequate solubility, favorable plasma and metabolic stability, and absence of obvious toxicophores. Crucially, robust evidence of target engagement in cellulo will be essential to directly link enzymatic or binding activity to observed cellular effects. This requirement is particularly important given that the biological consequences of selective STK17A inhibition remain largely unexplored. In this context, the use of selective chemical probes in genetically modified cell systems, such as STK17A knockout or siRNA-mediated knockdown cells, will be of highest interest for confirming known STK17A-specific functions, elucidating novel ones, and validating therapeutic hypothesis.
Conclusion
STK17A, a member of the DAPK family within the CAMK superfamily, has emerged as a pivotal regulator at the intersection of apoptosis, cytoskeletal organization, immunity, and tumor biology. Its functions are highly context-dependent, acting either as a tumor suppressor or promoter depending on cellular environment and cancer type, and extending beyond oncology to influence autoimmune, infectious, cardiovascular, and neurodegenerative processes. Despite its physiological importance, progress in understanding STK17A has been hindered by the absence of the kinase in commonly used rodent models, as well as the reliance on in vitro overexpression or knockdown systems, which may not fully reflect physiological expression or localization. Mechanistic insights remain limited, with many reported associations lacking definitive evidence of kinase activity, substrate specificity, or pathway dependency.
STK17B, while structurally related, plays distinct biological roles, particularly in immune regulation, and transplant rejection. Dual STK17A/B inhibitors therefore carry the risk of producing confounding effects: inhibition of STK17B can lower T cell activation thresholds, enhance immune responsiveness, and disrupt homeostasis, potentially exacerbating autoimmunity or altering transplant outcomes. This highlights the necessity of selective chemical tools to interrogate STK17A independently and to avoid unintended immunological consequences.
Encouragingly, recent medicinal chemistry efforts have provided the first small-molecule inhibitors targeting STK17A and STK17B, including several dual inhibitors and a very limited number of selective compounds. The thieno[3,2-d]pyrimidine scaffold shows promise as a selective STK17B inhibitor, while the macrocyclic pyrazolopyrimidine compound 40 (CK156) has emerged as potential selective STK17A inhibitor. Nevertheless, the field still lacks optimized chemical probes that combine high potency, clear selectivity, and drug-like properties. Particularly, the development of new highly potent and selective STK17A inhibitors, sparing STK17B, is a key priority, as such compounds would enable precise functional studies, validate STK17A as a therapeutic target, and pave the way for novel interventions in cancer, autoimmune disorders, and neurodegenerative diseases. Beyond their use as chemical probes to dissect kinase-specific pathways, STK17A/B inhibitors, such as compound 45, also hold potential as therapeutic leads for drug development, provided they display suitable pharmacological and safety profiles.
Complementarily, research should focus on generating physiological and tissue-specific STK17A models, including genetic loss- and gain-of-function systems, to delineate basal and stress-induced roles. Systematic identification of bona fide STK17A substrates and careful distinction between kinase-dependent and kinase-independent functions will be critical. Integrating these biological advances with the continued development of selective, well-characterized chemical probes will be essential for assessing STK17A's true translational potential.
In summary, STK17A represents both a promising and challenging kinase target. Fully realizing its therapeutic potential will require a synergistic approach that integrates structural biology, chemical biology, and translational studies, supported by potent, selective chemical tools. Such efforts have the potential to illuminate STK17A's multifaceted roles and unlock novel therapeutic opportunities across oncology, immunology, and neurodegeneration.
Acknowledgements
This work was financially supported by Université Côte d'Azur, CNRS and Roca Therapeutics.
Funding
We extend our sincere thanks to the Région Sud Provence-Alpes-Côte d'Azur, the FEDER, the European Union, BPI France, the Ministère de l'Enseignement Supérieur and INSERM.
This work received financial support from Roca Therapeutics, CNRS, Université Côte d'Azur, the Canceropôle PACA, ANR, INCA, the H2020 TheraLymph project (Grant ID: 874708), La Ligue Nationale Contre le Cancer (Equipe Labellisée 2019), Fondation ARC pour la Recherche sur le Cancer (Programme Labellisé 2022), and the ARCAGEING2023020006332 program.
Author contributions
All authors wrote and discussed the draft.
Maeva Dufies and Cyril Ronco: Writing-Original draft preparation, Conceptualization, Visualization, Supervision, Writing-Reviewing and Editing and Funding acquisition; Leticia Christina Pires Gonçalves, Olivia Rastoin: Writing-Original draft preparation, Conceptualization, Visualization. Gilles Pagès: Writing- Reviewing and Editing; Vira Morozova, Clément Buzet, Audrey Bennetot: Visualization, Conceptualization.
Language editing
AI-assisted tools were used to support language editing and text refinement to improve readability and linguistic quality (e.g.: InstaText, ChatGPT). The manuscript has also undergone professional linguistic revision and editing by the Office of International Scientific Visibility at Université Côte d'Azur to ensure the highest standards of English clarity and presentation.
All scientific content, interpretation, and conclusions were developed and validated by the authors.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Temmerman K, Simon B, Wilmanns M. Structural and functional diversity in the activity and regulation of DAPK -related protein kinases. The FEBS Journal. 2013;280:5533-50
2. Makgoo L, Mosebi S, Mbita Z. The Role of Death-Associated Protein Kinase-1 in Cell Homeostasis-Related Processes. Genes. 2023;14:1274
3. Cohen O, Inbal B, Kissil JL, Raveh T, Berissi H. et al. DAP-kinase Participates in TNF-a- and Fas-induced Apoptosis and Its Function Requires the Death Domain. J Cell Biol. 1999;146:141-8
4. Wang W-J, Kuo J-C, Yao C-C, Chen R-H. DAP-kinase induces apoptosis by suppressing integrin activity and disrupting matrix survival signals. The Journal of Cell Biology. 2002;159:169-79
5. Raveh T, Kimchi A. DAP Kinase—A Proapoptotic Gene That Functions as a Tumor Suppressor. Experimental Cell Research. 2001;264:185-92
6. Inbal B, Shani G, Cohen O, Kissil JL, Kimchi A. Death-Associated Protein Kinase-Related Protein 1, a Novel Serine/Threonine Kinase Involved in Apoptosis. Molecular and Cellular Biology. 2000;20:1044-54
7. Sanjo H, Kawai T, Akira S. DRAKs, Novel Serine/Threonine Kinases Related to Death-associated Protein Kinase That Trigger Apoptosis. Journal of Biological Chemistry. 1998;273:29066-71
8. Da Silva Fonseca AM, De Azevedo Silva J, Pancotto JAT, Donadi EA, Segat L, Crovella S. et al. Polymorphisms in STK17A gene are associated with systemic lupus erythematosus and its clinical manifestations. Gene. 2013;527:435-9
9. Zhang L, Luo B, Lu Y, Chen Y. Targeting Death-Associated Protein Kinases for Treatment of Human Diseases: Recent Advances and Future Directions. J Med Chem. 2023;66:1112-36
10. Oue Y, Murakami S, Isshiki K, Tsuji A, Yuasa K. Intracellular localization and binding partners of death associated protein kinase-related apoptosis-inducing protein kinase 1. Biochemical and Biophysical Research Communications. 2018;496:1222-8
11. Liu C, Zhang Y, Zhang Y, Liu Z, Mao F, Chai Z. Mechanistic Insights into the Mechanism of Inhibitor Selectivity toward the Dark Kinase STK17B against Its High Homology STK17A. Molecules. 2022;27:4655
12. Li X, Jin Y. Inhibition of miR-182-5p attenuates ROS and protects against myocardial ischemia-reperfusion injury by targeting STK17A. Cell Cycle. 2022;21:1639-50
13. Fitzgerald J, Bateman JF. Why mice have lost genes for COL21A1, STK17A, GPR145 and AHRI: evidence for gene deletion at evolutionary breakpoints in the rodent lineage. Trends in Genetics. 2004;20:408-12
14. Pasquier J, Cabau C, Nguyen T, Jouanno E, Severac D, Braasch I. et al. Gene evolution and gene expression after whole genome duplication in fish: the PhyloFish database. BMC Genomics. 2016;17:368
15. Xu Y, Feng Y, Li S, Sun J. Identification and characterization of apoptosis-related gene serine/threonine kinase 17A (STK17A) from Japanese flounder Paralichthys olivaceus. Fish & Shellfish Immunology. 2020;98:1008-16
16. Kojima H, Nemoto A, Uemura T, Honma R, Ogura M, Liu Y. rDRAK1, a Novel Kinase Related to Apoptosis, Is Strongly Expressed in Active Osteoclasts and Induces Apoptosis. Journal of Biological Chemistry. 2001;276:19238-43
17. Okomo-Adhiambo M, Rink A, Rauw WM, Gomez-Raya L. Gene expression in Sinclair swine with malignant melanoma. Animal. 2012;6:179-92
18. Johnsson M, Williams MJ, Jensen P, Wright D. Genetical Genomics of Behavior: A Novel Chicken Genomic Model for Anxiety Behavior. Genetics. 2016;202:327-40
19. Chuang M, Chisholm AD. Insights into the functions of the death associated protein kinases from C. elegans and other invertebrates. Apoptosis. 2014;19:392-7
20. Sitbon YH, Yadav S, Kazmierczak K, Szczesna-Cordary D. Insights into myosin regulatory and essential light chains: a focus on their roles in cardiac and skeletal muscle function, development and disease. J Muscle Res Cell Motil. 2020;41:313-27
21. Chen AS, Wardwell-Ozgo J, Shah NN, Wright D, Appin CL, Vigneswaran K. et al. Drak/STK17A Drives Neoplastic Glial Proliferation through Modulation of MRLC Signaling. Cancer Research. 2019;79:1085-97
22. Chougule AB, Hastert MC, Thomas JH. Drak Is Required for Actomyosin Organization During Drosophila Cellularization. G3 Genes|Genomes|Genetics. 2016;6:819-28
23. Neubueser D, Hipfner DR. Overlapping Roles of Drosophila Drak and Rok Kinases in Epithelial Tissue Morphogenesis. Margolis B, Ed. MBoC. 2010;21:2869-79
24. Wang B, Xiao Z, Ko HL, Ren E-C. The p53 response element and transcriptional repression. Cell Cycle. 2010;9:870-9
25. Mao P, Hever MP, Niemaszyk LM, Hhaghkerdar JM, Yanco EG, Desai D. et al. Serine/Threonine Kinase 17A Is a Novel p53 Target Gene and Modulator of Cisplatin Toxicity and Reactive Oxygen Species in Testicular Cancer Cells. Journal of Biological Chemistry. 2011;286:19381-91
26. Craig AL, Chrystal JA, Fraser JA, Sphyris N, Lin Y, Harrison BJ. et al. The MDM2 Ubiquitination Signal in the DNA-Binding Domain of p53 Forms a Docking Site for Calcium Calmodulin Kinase Superfamily Members. Molecular and Cellular Biology. 2007;27:3542-55
27. Manivannan P, Reddy V, Mukherjee S, Clark KN, Malathi K. RNase L Induces Expression of A Novel Serine/Threonine Protein Kinase, DRAK1, to Promote Apoptosis. IJMS. 2019;20:3535
28. Mao P, Hever-Jardine MP, Rahme GJ, Yang E, Tam J, Kodali A. et al. Serine/Threonine Kinase 17A Is a Novel Candidate for Therapeutic Targeting in Glioblastoma. Fillmore H, Ed. PLoS ONE. 2013;8:e81803
29. Park Y, Kim W, Lee J-M, Park J, Cho JK, Lee J. et al. Cytoplasmic DRAK1 overexpressed in head and neck cancers inhibits TGF-β1 tumor suppressor activity by binding to Smad3 to interrupt its complex formation with Smad4. Oncogene. 2015;34:5037-45
30. Park Y, Pang K, Park J, Hong E, Lee J, Ooshima A. et al. Destablilization of TRAF6 by DRAK1 Suppresses Tumor Growth and Metastasis in Cervical Cancer Cells. Cancer Research. 2020;80:2537-49
31. Elmore S. Apoptosis: A Review of Programmed Cell Death. Toxicol Pathol. 2007;35:495-516
32. Tang H, Liu Y-J, Liu M, Li X. Establishment and gene analysis of an oxaliplatin-resistant colon cancer cell line THC8307/L-OHP. Anti-Cancer Drugs. 2007;18:633-9
33. Gao J, Liu D, Li J, Song Q, Wang Q. Effect of STK17A on the sensitivity of ovarian cancer cells to paclitaxel and carboplatin. Oncology Letters. 2016;12:1107-12
34. Wang Z, Wang C, Jiang B-H. et al. Predictive significance of STK17A in patients with gastric cancer and association with gastric cancer cell proliferation and migration. Oncol Rep. 2021;45:119
35. Ozeki M, Salah A, Aini W, Tamaki K, Haga H, Miyagawa-Hayashino A. Abnormal Localization of STK17A in Bile Canaliculi in Liver Allografts: An Early Sign of Chronic Rejection. Alpini G, Ed. PLoS ONE. 2015;10:e0136381
36. Huichalaf CH, Al-Ramahi I, Park K-W, Grunke SD, Lu N, De Haro M. et al. Cross-species genetic screens to identify kinase targets for APP reduction in Alzheimer's disease. Human Molecular Genetics. 2019;28:2014-29
37. Lee S-A. Construction and analysis of the protein-protein interaction networks for schizophrenia, bipolar disorder, and major depression. 2011.
38. Kabara E, Kloss CC, Wilson M. et al. A large-scale study of differential gene expression in monocyte-derived macrophages infected with several strains of Mycobacterium avium subspecies paratuberculosis. Briefings in Functional Genomics. 2010;9:220-37
39. Silver RF, Walrath J, Lee H, Jacobson BA, Horton H, Bowman MR. et al. Human Alveolar Macrophage Gene Responses to Mycobacterium tuberculosis Strains H37Ra and H37Rv. Am J Respir Cell Mol Biol. 2009;40:491-504
40. Cho J, Park K, Choi SM, Lee J, Lee C-H, Lee J-K. et al. Genome-wide association study of non-tuberculous mycobacterial pulmonary disease. Thorax. 2021;76:169-177
41. Jiang Y, Mishima H, Sakai S, Liu Y, Ohyabu Y, Uemura T. Gene expression analysis of major lineage-defining factors in human bone marrow cells: Effect of aging, gender, and age-related disorders. Journal Orthopaedic Research. 2008;26:910-7
42. Zhang X, Wang S, Liu S, Du Z, Wu G, Liang Y. et al. Epidemiologic association and shared genetic architecture between cataract and hearing difficulties among middle-aged and older adults. Hum Genomics. 2024;18:39
43. Luo D, Gao X, Zhu X, Xu J, Gao P, Zou J. et al. Biomarker screening using integrated bioinformatics for the development of “normal—impaired glucose intolerance—type 2 diabetes mellitus”. Sci Rep. 2024;14:4558
44. Yang J, Han Y, Diao X, Yuan B, Gu J. Screening of obstructive sleep apnea and diabetes mellitus -related biomarkers based on integrated bioinformatics analysis and machine learning. Sleep Breath. 2025;29:74
45. McGargill MA, Wen BG, Walsh CM, Hedrick SM. A Deficiency in Drak2 Results in a T Cell Hypersensitivity and an Unexpected Resistance to Autoimmunity. Immunity. 2004;21:781-91
46. McGargill MA, Choy C, Wen BG, Hedrick SM. Drak2 Regulates the Survival of Activated T Cells and Is Required for Organ-Specific Autoimmune Disease. The Journal of Immunology. 2008;181:7593-605
47. Mao J, Qiao X, Luo H, Wu J. Transgenic Drak2 Overexpression in Mice Leads to Increased T Cell Apoptosis and Compromised Memory T Cell Development. Journal of Biological Chemistry. 2006;281:12587-95
48. Yang K-M, Kim W, Bae E, Gim J, Weist BM, Jung Y. et al. DRAK2 Participates in a Negative Feedback Loop to Control TGF-β/Smads Signaling by Binding to Type I TGF-β Receptor. Cell Reports. 2012;2:1286-99
49. Harris TL, McGargill MA. Drak2 Does Not Regulate TGF-β Signaling in T Cells. Wiendl H, Ed. PLoS ONE. 2015;10:e0123650
50. Edwards BA, Harris TL, Floersh H, Lukens JR, Zaki MH, Vogel P. et al. Drak2 is not required for tumor surveillance and suppression. International Immunology. 2015;27:161-6
51. Marvaldi L, Hausott B, Auer M, Leban J, Klimaschewski L. A Novel DRAK Inhibitor, SC82510, Promotes Axon Branching of Adult Sensory Neurons In vitro. Neurochem Res. 2014;39:403-7
52. Gao L-J, Kovackova S, Šála M, Ramadori AT, De Jonghe S, Herdewijn P. Discovery of Dual Death-Associated Protein Related Apoptosis Inducing Protein Kinase 1 and 2 Inhibitors by a Scaffold Hopping Approach. J Med Chem. 2014;57:7624-43
53. Bischof J, Leban J, Zaja M, Grothey A, Radunsky B, Othersen O. et al. 2-Benzamido-N-(1H-benzo[d]imidazol-2-yl)thiazole-4-carboxamide derivatives as potent inhibitors of CK1δ/ε. Amino Acids. 2012;43:1577-91
54. Leonczak P, Gao L, Ramadori AT, Lescrinier E, Rozenski J, De Jonghe S. et al. Synthesis and Structure-Activity Relationship Studies of 2-(1,3,4-Oxadiazole-2(3 H )-thione)-3-amino-5-arylthieno[2,3- b ]pyridines as Inhibitors of DRAK2. ChemMedChem. 2014;9:2587-601
55. Jung ME, Byun BJ, Kim H-M, Lee JY, Park J-H, Lee N. et al. Discovery of indirubin derivatives as new class of DRAK2 inhibitors from high throughput screening. Bioorganic & Medicinal Chemistry Letters. 2016;26:2719-23
56. Wang S, Xu L, Lu Y-T, Lui Y-F, Han B, Liu T. et al. Discovery of benzofuran-3(2 H )-one derivatives as novel DRAK2 inhibitors that protect islet β-cells from apoptosis. European Journal of Medicinal Chemistry. 2017;130:195-208
57. Ali I, Park S, Jung ME, Lee N, Bibi M, Chae CH. et al. Identification of TRD-35 as Potent and Selective DRAK2 Inhibitor. Bulletin Korean Chem Soc. 2020;41:567-9
58. Picado A, Chaikuad A, Wells CI, Shrestha S, Zuercher WJ, Pickett JE. et al. A Chemical Probe for Dark Kinase STK17B Derives Its Potency and High Selectivity through a Unique P-Loop Conformation. J Med Chem. 2020;63:14626-46
59. Drewry DH, Annor-Gyamfi JK, Wells CI, Pickett JE, Dederer V, Preuss F. et al. Identification of Pyrimidine-Based Lead Compounds for Understudied Kinases Implicated in Driving Neurodegeneration. J Med Chem. 2022;65:1313-28
60. Kurz CG, Preuss F, Tjaden A, Cusak M, Amrhein JA, Chatterjess D. et al. Illuminating the Dark: Highly Selective Inhibition of Serine/Threonine Kinase 17A with Pyrazolo[1,5- a ]pyrimidine-Based Macrocycles. J Med Chem. 2022;65:7799-817
61. Krämer A, Kurz CG, Berger B-T, Celik IE, Tjaden A, Greco FA. et al. Optimization of pyrazolo[1,5-a]pyrimidines lead to the identification of a highly selective casein kinase 2 inhibitor. European Journal of Medicinal Chemistry. 2020;208:112770
62. Marsault E, Peterson ML. Macrocycles Are Great Cycles: Applications, Opportunities, and Challenges of Synthetic Macrocycles in Drug Discovery. J Med Chem. 2011;54:1961-2004
63. Chaudhry S, Castro JR, Totiger TM, Afaghani J, Khurshid R, Nicholls M. et al. Potent, Selective, and Orally Bioavailable Quinazoline-Based STK17A/B Dual Inhibitors. ACS Med Chem Lett. 2024;15:945-9
Author contact
![]() Corresponding authors: Cyril Ronco, cyril.roncofr; Maeva Dufies. maeva.dufiesfr.
Corresponding authors: Cyril Ronco, cyril.roncofr; Maeva Dufies. maeva.dufiesfr.