Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Basic components and...
3. Mechanism of NETs formation
4. NETs and primary cancer...
5. Role of NETs in cancer...
6. NETs and cancer-related...
7. NET interaction with immune...
8. Target NETs for cancer...
9. Conclusion and perspectives
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2025; 15(12):5846-5869. doi:10.7150/thno.111096 This issue Cite
Review
Targeting neutrophil extracellular traps in cancer progression and metastasis
Ji Zhang1,2,3, Changhong Miao1,2,3 ![]() , Hao Zhang1,2,3
, Hao Zhang1,2,3 ![]()
1. Department of Anesthesiology, Zhongshan Hospital, Fudan University, Shanghai, China.
2. Shanghai Key Laboratory of Perioperative Stress and Protection, Shanghai, China.
3. Department of Anesthesiology, Shanghai Medical College, Fudan University, China.
Received 2025-1-25; Accepted 2025-4-7; Published 2025-4-22
Abstract

Neutrophils serve as pivotal effectors and regulators of the intricate immune system. Their contributions are indispensable, encompassing the obliteration of pathogens and a significant role in both cancer initiation and progression. Conversely, malignancies profoundly affect neutrophil activity, maturation, and lifespans. Cancer cells manipulate their biology to enhance or suppress the key functions of neutrophils. This manipulation is one of the most remarkable defensive mechanisms used by neutrophils, including the formation of neutrophil extracellular traps (NETs). NETs are filamentous structures comprising DNA, histones, and proteins derived from cytotoxic granules. In this review, we discuss the bidirectional interplay in which cancer elicits NET formation, and NETs concurrently facilitate cancer progression. Here, we discuss how vascular dysfunction and thrombosis induced by neutrophils and NETs contribute to an elevated risk of mortality from cardiovascular complications in patients with cancer. Ultimately, we propose a series of therapeutic strategies that hold promise for effectively targeting NETs in clinical settings.
Keywords: neutrophil extracellular traps, cancer progression, metastasis, tumor microenvironment, therapeutic targets
1. Introduction
Neutrophils represent a pivotal category of white blood cells found within the circulatory system, comprising 50%-70% of the total leukocyte population, and serve as frontline defenders of the immune response [1]. These formidable cells primarily perform their roles by orchestrating the recruitment of additional immune entities through phagocytosis, inciting the generation of reactive oxygen species (ROS), modulating the degradation and release of proteases or other cytotoxic agents, and activating fellow immune components [2, 3]. Moreover, they engage in sophisticated defense mechanisms, known as neutrophil extracellular traps (NETs), among various functions aimed at combating invading pathogens [4]. The process by which neutrophils generate NETs is termed NETosis, and can be divided into two categories: suicidal and active. The former denotes a condition in which neutrophils succumb during NET formation, whereas active NETosis allows the retention of viable neutrophilic functionalities [5]. As discovered by Brinkmann et al., NETs comprise a novel defensive strategy used by neutrophils; their chemical composition predominantly comprises chromatin, deoxyribonucleic acid (DNA), and protein [6, 7]. NETs function not only as instruments through which neutrophils ensnare and eradicate pathogens, but also as pathological indicators associated with various diseases due to their excessive release [8]. Notable examples include preeclampsia, chronic inflammatory disorders, and myocardial ischemia-reperfusion injuries, which culminate in the overwhelming discharge of NETs.
An essential biomarker of tumorigenesis is a heightened inflammatory milieu, exemplified by the augmented release of NETs [9]. Excessive secretion of NETs results in chronic inflammation throughout the body. An abundance of NETs fosters tumor angiogenesis through mechanisms such as degradation of the extracellular matrix, entrapment of circulating tumor cells, compromise of vascular integrity, and induction of epithelial-mesenchymal transition (EMT), thereby creating an opportune "premetastatic microenvironment" conducive to both tumor initiation and metastatic spread [10]. Nonetheless, neoplastic cells adeptly elude their primary site, infiltrate the bloodstream, evade the immune system facilitated by NETs, and subsequently establish colonies at distal sites [11]. This surge in NETs production engenders a permissive microenvironment that supports tumor cell proliferation and expedites metastasis [12]. Given this understanding, modulating the expression levels of NETs may inhibit tumor cell growth while simultaneously dismantling the metastatic landscape, which has emerged as a novel avenue for cancer therapy [11]. Recent investigations have revealed that NETs can potentiate the immunosuppressive cell functions while undermining the functions of CD8+ T and natural killer (NK) cells. Consequently, this dual effect compromises anti-tumor immunity and accentuates oncogenic processes [12]. Given the intricate nature of the Tumor Microenvironment (TME), our knowledge of the interplay between NETs and immune responses remains limited, and existing strategies aimed at inhibiting NET formation are usually simple. Such constraints have significantly curtailed the broader application of targeting NETs in contemporary cancer immunotherapy (Figure 1).
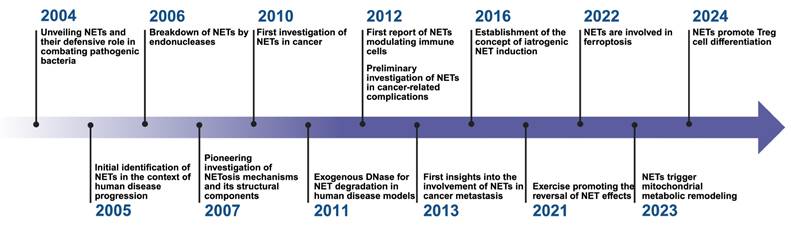
Research history of NETs. This timeline highlights significant milestones in the research and understanding of Neutrophil Extracellular Traps (NETs) from 2004 to 2024. The timeline illustrates the evolution of key discoveries and their impact on various domains, particularly in the field of oncology and immunology. *Created with BioRender.com.
Herein, we elucidate the potential triggers and associated signaling cascades that precipitate NET formation, particularly focusing on their implications within the tumor microenvironment. Subsequent evidence has increasingly underscored the critical role of NETs as crucial mediators of various aspects of tumor progression, including awakening of dormant cancer cells, extravasation of circulating tumor cells (CTCs), and recurrence following metastasis. Furthermore, NETs are intricately involved in inflammation-induced disturbances that culminate in the establishment of an immunosuppressive milieu, facilitating immune evasion and promoting tumor survival and proliferation. Considering the multifaceted roles attributed to NETs, it is plausible to consider them not only as prospective prognostic biomarkers but also as promising therapeutic targets in oncology.
2. Basic components and functions of NETs
In 2004, Brinkmann et al. published a groundbreaking article in Science that elucidated the fundamental architecture and function of NETs for the first time. This pioneering study revealed that NETs represent an intricate extracellular network composed of chromatin intertwined with a diverse array of protein constituents, designed to restrain and obliterate invading pathogens [6]. The myriad functions of NETs are associated with their constituent components. Over 200 proteins have been identified within these structures [13], underscoring the complexity of their function. For instance, NETs have been implicated in disease severity, such as bronchiectasis severity index, quality of life, future risk of hospital admission, and mortality in patients with bronchiectasis [14]. NF-κB-dependent autoimmunity induced by NETs-DNA via cGAS/TLR9 in chronic obstructive pulmonary disease [15]. Furthermore, they play a pivotal role in the pathogenesis of rheumatoid arthritis by inducing carbamylation and histone-mediated osteoclast formation [16]. Besides chronic inflammation and autoimmune disorders, emerging evidence suggests that NETs contribute to tumorigenesis. Zhan et al. found HBV-induced S100A9 activates RAGE/TLR4-ROS signaling, leading to abundant NETs formation, which subsequently promoted the growth and metastasis of hepatocellular carcinoma (HCC) cells [17]. In addition, HMGB1 present within NETs has been found to accelerate the progression of diffuse large B-cell lymphoma and colorectal cancer via the TLR9-MAPK signaling pathways [18, 19]. Beyond their protein constituents, DNA elements contained within NETs can initiate the β-parvin-RAC1-CDC42 signaling pathway through the membrane receptor Coiled-coil domain containing 25(CCDC25), mediating metastasis in MDA-MB-231 cells [20]. Collectively, these investigations highlight that NETs can influence tumor advancement from a multifaceted perspective; however, their complex composition poses significant challenges to related research. Future studies should adopt a holistic approach to examine the roles of various NET components in tumor progression. Moreover, it is essential to further delineate how these structures impact tumor immunity, potentially positioning this line of inquiry among upcoming priorities (Figure 2).
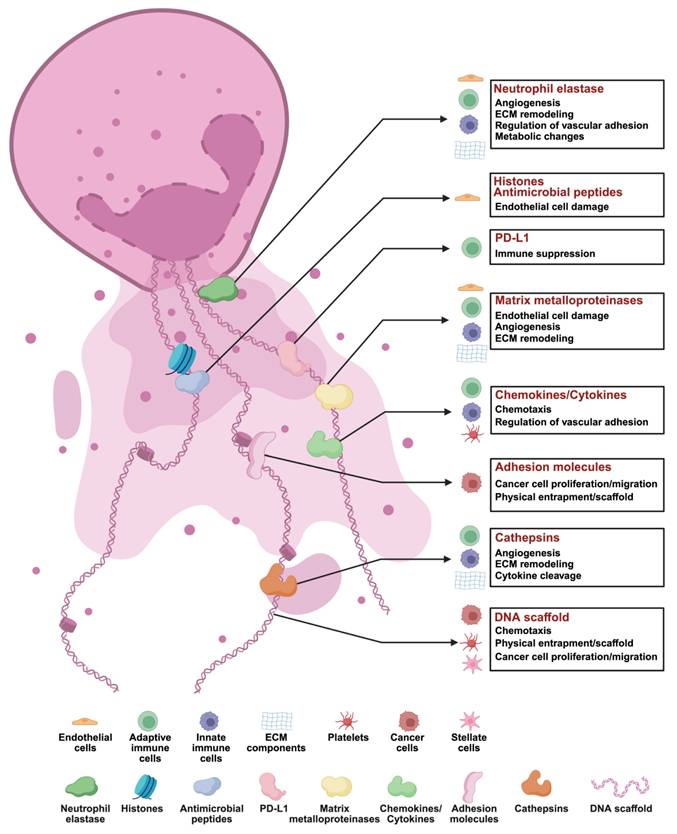
Composition of NETs. Neutrophil extracellular traps (NETs) are composed of a DNA scaffold released by neutrophils and a variety of functional proteins that play critical roles in tumor biology, immune modulation, and microenvironment remodeling. The core components and their functions include: (1) Neutrophil Elastase: Facilitates angiogenesis by degrading the extracellular matrix (ECM) and modulates endothelial adhesion, inducing metabolic changes that impact immune cells, ECM, and endothelial cell functions. (2) Histones and Antimicrobial Peptides: Directly cause endothelial cell damage and exhibit antimicrobial activity against pathogens. (3) Programmed Death-Ligand 1 (PD-L1): Suppresses immune responses by modulating adaptive immune cells. (4) Matrix Metalloproteinases (MMPs): Promote endothelial damage, angiogenesis, and ECM remodeling through degradation, influencing immune cells, ECM, and endothelial cell dynamics. (5) Chemokines and Cytokines: Recruit immune cells and platelets by inducing chemotaxis and regulating vascular adhesion, thereby orchestrating inflammation and tumor progression. (6) Adhesion Molecules: Act as scaffolds or physical barriers for immune cells, promoting cancer cell proliferation, migration, and immune evasion. (7) Cathepsins: Contribute to angiogenesis, ECM remodeling, and cytokine cleavage, modulating immune responses and ECM architecture. (8) DNA Scaffold: Provides structural support and physical entrapment for immune and tumor cells, while generating chemotactic signals that influence tumor migration and differentiation. *Created with BioRender.com.
3. Mechanism of NETs formation
As early as 1996, Takei et al. made significant observations, revealing that phorbol-12-myristate-13-acetate (PMA) could induce distinct morphological alterations in neutrophils that are divergent from the processes of apoptosis and necrosis. This pioneering study serves as a foundation for future studies related to NETs [21]. Currently, NET formation involves two principal pathways: NETosis and Non-lytic NETosis. Regarding morphology and functionality, neutrophils undergoing NETosis exhibit marked loss of their characteristic cellular structure and activity upon NET formation. Conversely, cells undergoing Non-lytic NETosis retain their transient phagocytic capabilities following NET production. Regarding the stimuli and mechanisms underpinning these processes, PMA, antibodies, and cholesterol crystallization can facilitate the progression of NETosis by inducing ROS, which instigates chromatin depolymerization. In contrast, Staphylococcus aureus and Escherichia coli promote Non-lytic NETosis through vesicular release of protein-modified chromatin via TLR2 and TLR4 along a non-reactive oxygen species-dependent pathway [22-24]. Nevertheless, whether functional disparities exist between differentially formed NETs remains an area that warrants further investigation. Chromatin depolymerization constitutes a critical prerequisite for NET formation, and studies have identified peptidyl arginine deiminase 4 (PAD4) as an integral part of this process. The disruption of NET formation observed in PAD4 knockout mice underscores its indispensable role in facilitating connections within pathways, leading to net assembly [25, 26]. PAD4 catalyzes the citrullination of histone H3's arginine residues while simultaneously inhibiting their binding affinity to negatively charged DNA backbones. This promotes chromatin densification followed by depolymerization, which is crucial for effective NET development [27]. Moreover, intracellular ROS levels have been shown to influence neurogenesis. NADPH oxidase-derived or mitochondria-associated ROS accelerates chromatin depolymerization by synergistically activating myeloperoxidase (MPO) alongside neutrophil elastase (NE), propelling these factors into the nucleus [28-30].
Besides directly facilitating NET formation, neutrophil elastase possesses the capacity to activate Gasdermin D (GSDMD), further enhancing NET production [31]. Moreover, DEK functions analogously to MPO, inducing NET formation after binding to DNA [32]. Collectively, these studies showed that the intricate process of NETosis in neutrophils is orchestrated by a plethora of crucial proteins, among which PAD4 has emerged as a pivotal target and has been extensively studied for its role in promoting NET formation. Numerous contemporary investigations have corroborated that various PAD4 inhibitors can markedly disrupt NET synthesis in vivo, while concurrently impeding tumor progression [33-35]. Furthermore, the precise mechanisms that govern neutrophils within the tumor microenvironment remain unclear. Consequently, probing whether functional variances exist between NETs formed via disparate pathways could prove advantageous for targeted inhibition strategies aimed at mitigating the undesired off-target effects associated with inhibitors. In addition, determining whether tumor heterogeneity influences the dynamics of NET formation is a principal focus for guiding future research on NETs (Figure 3).
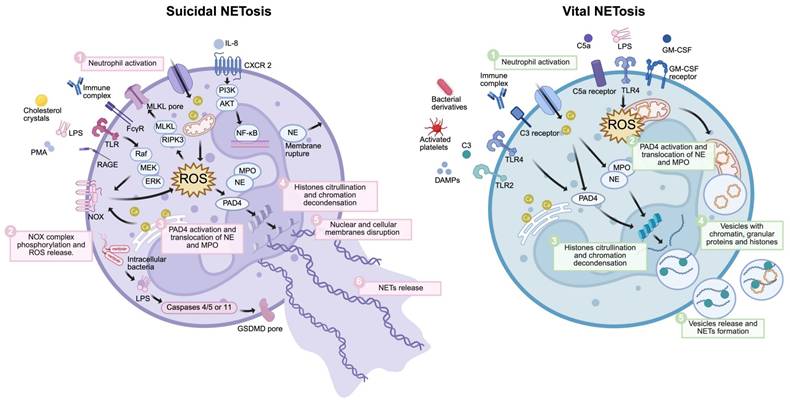
Formation of NETs. The formation of NETs includes suicide NETosis and vital NETosis. Suicidal NETosis (left) is initiated by stimuli such as PMA, LPS, immune complexes, cholesterol crystals, or IL-8. These extracellular signals mainly activate the NOX complex via several pathways, such as the Raf-MEK-ERK signaling pathways. IL-8 also activates NF-κB via PI3K-AKT pathways. The influx of extracellular calcium ions can activate mitochondria. These signaling pathways subsequently generate ROS in the cytoplasm, resulting in the release of NE and MPO from azurophilic granules, activation of PAD4, and their translocation into the cell nucleus. Subsequently, activated PAD4 catalyzes the citrullination of histones and chromatin decondensation with the aid of NE and MPO. Additionally, ROS can both activate RIPK3-MLKL, leading to membrane perforation, and cause membrane rupture through NE release. Moreover, intracellular bacterial LPS forms GSDMD pores via caspases 4/5 or 11. When the nuclear membrane breaks down, the decondensed chromatin enters the cytoplasm, mixes with granular proteins, and forms NETs. Finally, NETs are released following the membrane and the mechanical action of swollen chromatin. Vital NET formation (right) is initiated by stimuli such as S. aureus, DAMPs, LPS, activated platelets, and bacterial derivatives. One process is mainly mediated by Ca²⁺ but is independent of the NOX complex. Activated PAD4, NE, and MPO also translocate into the nucleus to promote chromatin decondensation. Mitochondria participate in another pathway by releasing mtDNA and generating mtROS. Lastly, NETs, which may include nuclear DNA and mtDNA, are stored within vesicles budding from nuclei and released by neutrophils without membrane rupture. Abbreviations: LPS, lipopolysaccharide; PMA, phorbol-12-myristate-13-acetate; RAGE, Receptor for Advanced Glycation End-products; FcR, Fc receptor; IL-8, interleukin-8; DAMPs, damage-associated molecular patterns; TLR, toll-like receptor; MPO, myeloperoxidase; NE, neutrophil elastase; NOX, NADPH oxidase; PAD4, protein-arginine deiminase 4; ROS, reactive oxygen species; GSDMD, gasdermin D; mtDNA, mitochondrial DNA; MLKL, mixed lineage kinase domain-like protein.*Created with BioRender.com.
3.1. Suicidal NETosis
Pathogens, including bacteria, fungi, viruses, neutrophil cytoplasmic antibodies, and nutate and bacterial lipopolysaccharides, along with calcium ionophoresis and other stimuli, can induce neutrophils to release NETs [36]. The intricate process of NETs release is initiated by the engagement of cellular receptors that prompt the efflux of calcium ions from the endoplasmic reticulum. This cascade subsequently activates protein kinase C and NADPH oxidase complex, helping to generate ROS [27]. ROS then stimulates PAD4, which catalyzes the conversion of arginine residues on histones into citrulline. Citrullinated histones facilitate chromatin depolymerization and compaction, orchestrating NET formation [37]. Moreover, proteins such as neutrophil granuloprotein and MPO play pivotal roles in directing NE toward nuclear translocation [38]. This translocation results in the concomitant release of condensed chromatin and granular materials into the extracellular milieu, ultimately leading to NET extrusion via the disruption of the plasma membrane. This process culminates in neutrophil demise post-NET formation. Within the tumor microenvironment, endothelial cells that produce interleukin-8 (IL-8) and tumor cell-derived granulocyte colony-stimulating factor (G-CSF) mediate NETs formation. Research conducted by Park et al. and Gupta et al. revealed that co-culturing activated endothelial cells with neutrophils precipitated notable NETs production, a phenomenon partially driven by IL-8 secreted from these stimulated endothelial cells [39, 40]. Conversely, G-CSF is often overexpressed within malignant domains, which leads to increased peripheral neutrophilia coupled with enhanced ROS production and subsequent induction of NETosis [41, 42].
3.2. Vital NETosis
NET genesis independent of NADPH oxidase occurs because of a lack of ROS production and cell death following stimulation, a phenomenon called non-soluble NETosis [43, 44]. In this alternative pathway for NET formation, which bypasses NADPH oxidase activation, neutrophils are stimulated by various factors, including bacteria, bacterial derivatives, activated platelets, and complement proteins. PAD4 plays a pivotal role in promoting chromatin condensation [26, 37]. Subsequently, NETs associate with granule proteins and cytoplasmic components before being extruded from the cell via exocytosis while preserving membrane integrity. Remarkably, upon NET release, neutrophils retain their viability and continue to exhibit phagocytic activity and chemotactic responsiveness [45, 46].
3.3 Crosstalk between NETs and cancer cells
Recent evidence suggests bidirectional interactions between NETs and cancer cells [39, 47-49]. Various cancer-derived factors contribute to NET formation [50], which, in turn, promotes both hypercoagulability and tumor progression [51, 52]. Among these factors, cytokines play a crucial role in modulating NET function. For example, chemotherapy-exposed cancer cells secrete IL-1β, which induces NET formation, subsequently driving cancer cells to undergo EMT [53, 54]. IL-8 is another key cytokine involved in this process. In diffuse large B-cell lymphoma (DLBCL), IL-8 secreted by tumor cells binds to its receptor, CXCR2, on neutrophils, promoting NET formation. The newly formed NETs activate the TLR9 pathway in DLBCL cells, thereby facilitating tumor progression [18]. Cytokine IL-8, derived from CC cells, triggers NETs in an NADPH oxidase-dependent manner, while NET-associated CG promotes cancer metastasis. Clinically, elevated CG protein expression in tumor tissues is closely associated with poor prognosis in HCC patients [55, 56]. Additionally, IL-17 [57] and TGF-β [11], secreted by cancer cells, have been reported to exert similar effects. In contrast, Chi3l1 facilitates neutrophil recruitment and NET formation, ultimately enhancing anti-tumor immunity [58].
In addition to cytokines, cancer cell metabolites play a pivotal role in NET formation. In gastric cancer, NETs are driven by a hypoxic tumor microenvironment, thereby promoting tumor growth [59]. Moreover, lactate, a key product of the Warburg effect, induces NET formation by regulating gene expression, potentially contributing to tumor progression within the TME [60]. Notably, lysine (K)-specific demethylase 6A (KDM6A), a frequently mutated tumor suppressor gene in pancreatic ductal adenocarcinoma (PDAC), has been implicated in NET regulation. Yang et al. [61] demonstrated that KDM6A loss correlates with increased tumor-associated neutrophils (TANs) and NETs, both of which are known to promote PDAC progression. Additionally, extracellular RNAs from lung cancer cells activate epithelial cells and may indirectly induce NET formation, contributing to lung cancer oncogenesis [62]. Overall, the complex interplay and feedback loops between NETs and cancer cells present numerous potential therapeutic targets, warranting further investigation.
4. NETs and primary cancer progression
Extensive research has demonstrated a strong correlation between NETs and chronic inflammation in the human body, elucidating their capacity to facilitate tumorigenesis by inducing persistent tissue damage and eliciting DNA disruption [63]. Following tumor initiation, NETs not only catalyze the proliferation of neoplastic cells through the activation of pivotal signaling pathways [18], providing crucial dynamic support for tumor expansion, but also invigorate mitochondrial biogenesis in cancerous cells and amplify ATP production, providing essential energy reserves for tumor growth [64]. Concurrently, NETs may engage in tumor progression via an array of pro-tumor mechanisms. They have been implicated in the induction of EMT [65], endowing malignant cells with invasive and migratory capabilities. NETs facilitate the dissemination of malignancies [66]. Their ability to compromise vascular integrity expedites access of neoplastic cells into the circulatory system [67]. In addition, NETs enhance the metastatic potential of CTCs by ensnaring them and mediating immune evasion mechanisms [68, 69]. Furthermore, studies have shown that NET formation is intricately linked to recurrence in various forms of cancers [70]. Specifically, NET-associated NE and Matrix Metalloproteinase 9 (MMP9) possess the capacity to cleave laminin proteins; this action can reawaken dormant breast cancer cell populations from both human origins and murine models, fostering lung metastasis through activation of the Integrinα3β1-FAK-ERK-MLC2 signaling cascade [71]. This finding not only uncovered a novel mechanistic pathway whereby NETs contribute to oncological relapse but also provides innovative perspectives toward devising targeted therapeutic strategies aimed at quelling dormant malignant populations. In summary, we assert that NETs play an instrumental role in all phases, encompassing the emergence, evolution, and recurrence of tumors. Targeted interventions against these traps, including strategies geared toward inhibiting their formation, dismantling established NETs structures, or obstructing their pro-oncogenic functionalities, could represent a pioneering approach for curtailing tumor proliferation and thwarting recurrence (Figure 4).
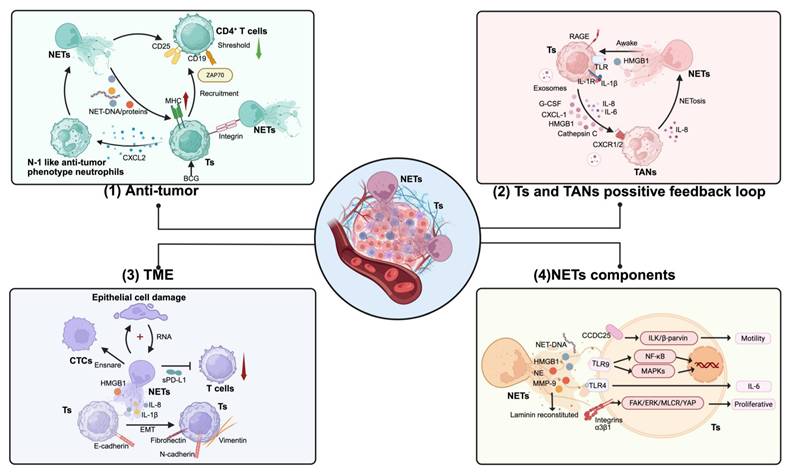
Role of NETs in the initial progression of cancer. The impact of Neutrophil Extracellular Traps (NETs) on the initial progression of cancer can be divided into four categories: (1) Anti-tumor Effect: NETs release DNA and associated proteins that are processed and presented by MHC molecules on tumor cells. Tumor cells recruit CD4+ T cells and downregulate their activation threshold through ZAP70. NETs interact with tumor cells and T cells through integrin-mediated adhesion, promoting proximity for effective immune cell-tumor cell interactions. Bacillus Calmette-Guérin (BCG), as an immunotherapeutic agent, augments this response by enhancing the anti-tumor properties of neutrophils. (2) Tumors and Tumor-Associated Neutrophils (TANs) Positive Feedback Loop: Tumors stimulate TANs to secrete IL-8 through exosomes and cellular molecules (G-CSF, CXCL-1, HMGB1, and Cathepsin C), which in turn leads to the formation of NETs. NETs activate tumors via substances such as HMGB1, promoting their differentiation and metastasis. This positive feedback loop forms a malignant cycle, facilitated by molecules such as HMGB1 that activate TANs via TLR and RAGE signaling pathways, inducing further NETosis and secretion of proinflammatory cytokines (IL-1β, IL-6, IL-8), enhancing the recruitment and activation of additional TANs. (3) NETs' Impact on the Tumor Microenvironment: NETs damage epithelial cells and capture circulating tumor cells (CTCs), providing potential support for tumor colonization. They promote tumor migration through the secretion of HMGB1 and cytokines via epithelial-mesenchymal transition (EMT). Additionally, NETs inhibit the cytotoxic activity of T cells through soluble PD-L1 (sPD-L1). (4) Components and Signaling Pathways of NETs in Tumor Cells: NET-derived components, including NET-DNA, HMGB1, neutrophil elastase (NE), and matrix metalloproteinase-9 (MMP-9), interact with tumor cells (Ts) through multiple pathways. CCDC25 and TLR9 recognize NET-DNA, activating the ILK/β-parvin pathway to promote tumor cell motility. TLR4 engagement triggers NF-κB and MAPK signaling cascades. Additionally, reconstituted laminin binds to integrins α3β1, activating the FAK/ERK/MLCR/YAP signaling axis to enhance tumor cell proliferation and IL-6 production. Abbreviations: NETs, Neutrophil Extracellular Traps; Ts, Tumor cells; BCG, Bacillus Calmette-Guérin; MHC, Major Histocompatibility Complex; TANs, Tumor-Associated Neutrophils; G-CSF, Granulocyte Colony-Stimulating Factor; CXCL-1, C-X-C Motif Chemokine Ligand 1; HMGB1, High Mobility Group Box 1; RAGE, Receptor for Advanced Glycation End-products; IL-8, Interleukin 8; TLR, Toll-Like Receptor; CTCs, Circulating Tumor Cells; NE, Neutrophil Elastase; MMP-9, Matrix Metallopeptidase 9; CCDC25, Coiled-Coil Domain Containing 25. *Created with BioRender.com.
4.1. Role of NETs in cancer progression
4.1.1. Anti-tumor effects
The role of NETs in tumor progression is highly context-dependent, shaped by tumor type, microenvironmental factors, and immune interactions. Their anti-tumor effects primarily arise from direct cytotoxicity against malignant cells and immune activation [72]. In a comprehensive melanoma biopsy analysis, Schedel et al. detected NETs in all 27 ulcerative melanomas but none in seven non-ulcerative cases, with no correlation to disease stage. In vitro studies demonstrate that NETs inhibit melanoma cell migration via integrin-mediated adhesion and induce necrosis, though their abundance does not correlate with tumor progression [73]. In a pancreatic cancer model, Chan et al. [74] found that melatonin induces tumor cells to secrete CXCL2, recruiting TANs, which adopt an N1-like anti-tumor phenotype, release NETs, and promote tumor apoptosis via ROS-dependent NETosis. This suggests that the anti-tumor function of NETs is influenced by neutrophil subtypes and NET formation mechanisms. In addition to neutrophils, immune interactions are critical. CD16⁺ neutrophils promote colorectal cancer by suppressing NK cell activity through NET-mediated NKp46 cleavage [75]. Similarly, pancreatic cancer-induced MMP-9 and IDO contribute to NK cell dysfunction [76], highlighting the immunomodulatory role of NET components. Therapeutic interventions targeting NETs have shown promise. An injectable hemostatic gel formulated with a tumor acidity neutralizer and NET lyase has been shown to enhance adoptive NK cell therapy, effectively mitigating post-resection recurrence of hepatocellular carcinoma [77]. Interestingly, the role of genes in influencing the effects of NETs on tumor progression is noteworthy. Shen et al. [78] utilized gene set enrichment analysis of NET-related gene signatures to evaluate NET levels across various cancer types. Their analysis revealed two distinct survival patterns: in cancers such as prostate, esophageal, breast, and colon cancer, higher NET scores were associated with better survival outcomes, whereas in cancers like pancreatic cancer, lung squamous cell carcinoma, low-grade glioma, ovarian cancer, gastric cancer, and bladder cancer, higher NET scores correlated with poorer survival.
Overall, the effects of NETs on tumor progression are complex and multifactorial. While their role remains debated, increasing evidence suggests they facilitate malignancy under certain conditions. Further elucidation of NET-driven molecular mechanisms may inform novel therapeutic strategies in cancer immunotherapy.
4.1.2. Pro-tumor effects
In the tumor microenvironment, neutrophils are readily activated by various stimuli to generate NETs, which in turn promote tumor proliferation, invasion, and metastasis, creating a deleterious vicious cycle [39, 79]. Clinically, elevated plasma levels of NETs have been observed in patients with a range of malignancies, including lung, pancreatic, and bladder cancers, suggesting a direct role in oncogenesis [80]. In colorectal cancer, NET presence is independent of tumor location or stage, yet their excessive accumulation is associated with poor patient outcomes [62]. NETs also facilitate the reactivation of dormant tumor cells, thus promoting subsequent metastasis [81], as evidenced in models of chronic pulmonary inflammation [82]. NETs serve as potent attractants for additional neutrophils that accumulate at the tumor locus while simultaneously ensnaring CTCs. This adhesion allows these cells to attach to vascular walls and subsequently undergo exosmosis. Furthermore, NETs interact with adhesion molecules, such as fibronectin, through proteolytic activity and enlist platelets that envelop CTCs, enhancing immune evasion mechanisms and fostering metastatic spread to distant sites (e.g., the lungs). Ultimately, copious amounts of NETs robustly adhere to blood vessels, providing scaffolding conducive to platelet attachment, activation, and thrombin generation, a process that precipitates thrombosis.
In addition to promoting tumor progression, NETs are implicated in therapeutic resistance and immune suppression. Chemotherapy-induced IL-1β secretion stimulates NET formation, thereby activating TGF-β pathways that mediate chemoresistance [53]. Concurrently, NETs upregulate programmed death-ligand 1 (PD-L1), contributing to T cell dysfunction and the establishment of an immunosuppressive tumor microenvironment [83]. In ischemia-reperfusion models, blockade of PD-L1 led to significant tumor regression and T cell functional recovery, underscoring the immunosuppressive role of NETs.
4.2. NETs components affecting cancer
NETs comprise a diverse array of constituents, including MMP-9, NE, cathepsin G (CG), chemokines, antimicrobial peptides, and histone antibodies. These components are intricately linked to the pathophysiology of endothelial cell injury, angiogenesis, and modulation of vascular adhesion and extracellular matrix (ECM) [84]. The factors implicated in cancer cell proliferation and tumor progression include NE, MMP-9, CG, histones, and DNA. In this section, we discuss pivotal elements of NETs and explore their roles in advancing cancer progression.
4.2.1. NE
NE is a serine protease secreted by neutrophils that plays a pivotal role in enhancing phagocytosis and exhibits bactericidal properties, thus serving as an acute-phase protein that is integral to the body's inflammatory response. Remarkably, Cui et al. discovered that NE can liberate proteolytic fragments encapsulating death domains by cleaving recombinant CD95 proteins, which subsequently exhibit selective cytotoxicity toward cancer cells upon interaction with the high-abundance histone H1 subtypes present within these malignant cells, underscoring NE's inhibitory influence on tumor cell proliferation [84]. Conversely, other studies have revealed that NE may ostensibly facilitate tumor cell proliferation. In studies involving murine models of lung cancer conducted by Houghton et al., it was observed that NE could directly penetrate the cellular endosomal spaces, diminishing insulin receptor substrate expression and encouraging the expansion of lung carcinoma cells [85]. Research has linked elevated NE levels with breast cancer metastasis. Consequently, monitoring NE levels may provide prognostic insights in patients with this malignancy [86]. In summary, NE has a dual effect on the metastatic processes associated with cancer. It fosters metastasis through intricate remodeling of the TME by stimulating tumor cell growth directly [87], cleaving membrane ligands [88], fostering angiogenesis [88], and synergizing with other components released by the neutrophils [89]. Given the complex nature of TME dynamics that influence NE functionality, further research is warranted to ascertain its viability as a clinical prognostic marker [90].
4.2.2. MMP-9
MMP plays pivotal roles in an array of physiological processes, including tissue development, wound healing, tissue remodeling, organ morphogenesis, and angiogenesis. Its expression remains relatively low in vivo, and it is subject to stringent regulatory mechanisms. Extensive research has shown that MMP-9 is markedly upregulated in various human malignancies and plays a crucial role in the proliferation and metastasis of cancer cells [91]. MMP-9 facilitates the degradation of non-ECM molecules, such as IL-1β and TNF-α. Several studies have substantiated MMP-9's involvement in catalyzing both the onset and metastasis across a spectrum of cancers, including gastric cancer [92], breast cancer [93, 94], colon cancer [95], lung cancer [96, 97], among others.
4.2.3. CG
CG can be synthesized within cells and possesses a remarkable ability to degrade a multitude of intracellular matrix precursor proteins, including collagen, elastin, and laminin. It plays a pivotal role in modulating the secretion and deposition of matrix precursors under normal physiological conditions and in the context of malignancies [98]. The hydrolytic activity of CG is significantly influenced by neutrophil-derived DNA, which facilitates its hydrolysis by obstructing the protective effects exerted by endogenous protease inhibitors present in the tissues. Where CG operates in a non-hydrolytic fashion, it exhibits the capacity to activate platelets [99], which subsequently envelops tumor cells and transfers their major histocompatibility complex (MHC) into the same neoplastic cells, resulting in elevated levels of MHC on tumor cell surfaces. This process further activates NK cells, thwarting potential immune evasion strategies used by tumor cells [100] while simultaneously exerting inhibitory effects on them. Additional research suggests that CG may function as a tumor suppressor gene in various malignancies, including breast cancer [101], bladder cancer [102], and colorectal cancer [103]. Given that arginine residues embedded within CG sequences dictate whether they act in a hydrolyzed or non-hydrolyzed manner, their propensity to either foster or impede malignant metastasis is contingent on their mode of action. Therefore, the effects of CG on cancer metastasis warrant further investigation.
4.2.4. Histones and DNA
Because histones within NETs possess inherent cytotoxic properties that can directly inflict damage to endothelial cells, they have been shown to elicit proinflammatory signals at certain toxic concentrations, which exceed those induced by DNA alone [104, 105]. Furthermore, NET-derived DNA, as a chemokine, induces cancer cell metastasis. In their investigation using a murine sepsis model, Najmh et al. [106] discovered that NET-DNA can capture cancerous cells through integrins, facilitating their navigation across the vascular barrier and enabling the colonization of secondary tissues such as the lungs and liver, which helps to promote metastatic spread. In addition, the precursor protein CCDC25 functions as a signal transducer; upon recognizing NET-DNA, it activates the ILK-β-parvin pathway, which subsequently fosters oncogenic metastasis [20]. However, the precise mechanisms underpinning how CCDC25 regulates tumor cell behavior remain enigmatic. Tang et al. [107] demonstrated that cholesterol enhances CCDC25 expression and revealed an intriguing correlation in which both CCDC25 and fovein-1 were observed to increase and colocalize within 4T1 cells subjected to ASPP2 knockdown. Their findings also indicate that cholesterol inhibitors concurrently diminished the expression of both CCDC25 and fovein-1. Currently, literature pertaining to histones and DNA associated with NETs remains sparse. Existing studies have implied that interactions between histones and DNA may facilitate cancer metastasis. However, thorough elucidation of these specific mechanisms warrants further exploration in future studies.
5. Role of NETs in cancer metastasis
Single-cell technology has a remarkable capacity to reveal the profound heterogeneity of neutrophils at the cellular level, which may correlate with variations in their migratory capabilities and NET formation at distinct stages throughout their life cycle, as highlighted in several studies [108]. Research indicates that NETosis can assume both pro-tumorigenic and anti-tumorigenic roles contingent on TME [109]. Given that NETs predominantly facilitate metastasis in various types of cancer, this section elucidates the contribution of these structures to cancer metastasis. Furthermore, NETs play a pivotal role in facilitating the distal metastasis of tumors [110]. Although surgical resection remains a fundamental approach to tumor treatment, adverse prognosis is often exacerbated by metastatic progression (Figure 5).
NET-mediated metastasis in various tumor types. NETs facilitate tumor metastasis through distinct organ-specific pathways: In the nervous system, GBM metastasis is promoted via HMGB1/RAGE and IL-8/PI3K/AKT/ROS signaling. In OSCC, NETs facilitate cancer progression through the induction of epithelial-mesenchymal transition (EMT), enhancing tumor cell motility and invasiveness. NETs contribute to lung metastasis by supporting polarization of neutrophils and EMT, influenced by cytokines such as IL-4 and IL-13, and pathways including STAT3 and NAMPT/SIRT1. Additionally, lung cancer cells can promote NET formation through the release of mitochondrial DNA. Colorectal cancer metastasis features mutant KRAS-driven NET formation and mast cell interactions. Omental metastasis occurs through NET-mediated CTC entrapment. Breast cancer metastasis involves NET-induced P53/IL-1β/NF-κB signaling, promoting CD44high/CD24low phenotype transition, EMT, and angiogenesis. Liver metastasis is facilitated through multiple NET-dependent pathways: DNA webs activating RAGE/TLR9, cholesterol-mediated mechanisms, and integrin α3β1-dependent ECM remodeling. Gastric cancer metastasis is driven by TGF-β/ANGPT2/Tie2 axis activation. In pancreatic cancer, NETs promote metastasis through inflammatory pathways (IL-1β/IL-17) and collagen-mediated EMT. *Created with BioRender.com.
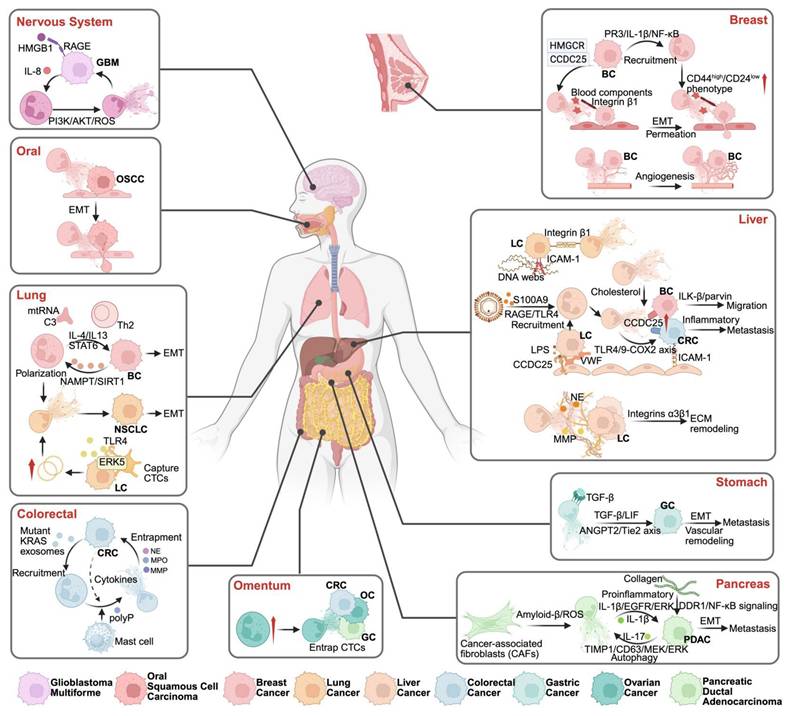
5.1. NETs and inflammation
In an in vivo model in which mouse LEWIS lung cancer cells were administered intravenously following cecal puncture and ligation, instigating a state of systemic inflammation, it was observed that the entrapment of NETs within the hepatic sinuses, encompassing cancer cells, was significantly correlated with metastatic progression. The introduction of DNases to disintegrate NETs abolished this correlation, supporting the hypothesis that inflammation may be intricately entwined with distant metastasis induced by NETs [111]. Previous research has similarly articulated that systemic inflammation augments the adherence of CTCs in peripheral circulation to the hepatic sinuses, consequently facilitating the distant micrometastatic spread of malignancies [112]. The integrin β2 present on neutrophils engages intercellular adhesion molecule-1 (ICAM-1) expressed by CTCs, orchestrating neutrophilic modulation that fosters CTC adhesion and obstructs hepatic sinuses, a pivotal element in the metastatic cascade of cancerous cells [113]. A study conducted by Yang et al. [12] elucidated how an elevated frequency of NETs in patients with hepatocellular carcinoma further propels tumor cell metastasis via the blockade of TLR4/9. In contrast, impairment due to prior inflammatory responses ameliorates NET-mediated metastatic potential. In conclusion, the mechanism by which NETs facilitate tumor cell metastasis appears to be closely associated with inflammatory responses. Nuclear factor-kappa B(NF-κB), an essential transcription factor governing IL-8 expression, is a proinflammatory chemokine that elevates IL-8 secretion within the tumor microenvironment. Zha et al. [114] further elucidated that IL-8 expression was elevated in patients with high-grade gliomas. Moreover, HMGB1, a pivotal constituent of NETs, engages with the receptor of advanced glycation end-products to activate the nuclear factor kappa-light-chain-enhancer of NF-κB. This interaction substantiates the notion that NETs facilitate the proliferation, migration, and invasion of tumor cells. Tumor cells reciprocally induce the secretion of IL-8, triggering inflammation and augmenting the infiltration of neutrophils into the tumor tissue, helping to form additional NETs. Although surgical resection remains the most prevalent strategy for oncological intervention, it often carries inherent risks associated with postoperative infections.
In addition, surgical procedures may induce stress responses that alter the procoagulant and inflammatory profiles, which promote tumor metastasis. They documented an alarming increase in NET formation post-surgery in a cohort of patients undergoing hepatectomy for metastatic colorectal cancer. Conversely, those achieving disease-free survival exhibited a striking reduction in NET levels to merely one-quarter of the baseline values [115]. The postoperative inflammatory milieu can precipitate systemic immunosuppression [116], engendering the upregulation of various pro-tumorigenic factors and culminating in expedited tumor recurrence. This provokes neutrophils to congregate at sites of trauma, where they release NETs that ensnare CTCs. Najmeh et al. adeptly simulated a postoperative inflammatory environment using mouse abdominal sepsis models and found that integrin β1 expression was markedly upregulated under these conditions in vivo. β1 integrin mediates CTC adhesion to NETs, further reinforcing the assertion that NETs facilitate tumor metastasis [106]. Moreover, Spicer et al. made intriguing observations wherein NETs were deposited and seized circulating lung cancer cells in mouse models subjected to cecal ligation and puncture, with liver metastases emerging just 48 h following lung cancer cell injection, with an increase in metastatic load evident 2 weeks later [69]. Tohme et al. further substantiated the premise that NET formation exacerbates tumor metastasis after surgical stress [115]. Ren et al. [117] provided compelling evidence that platelets enhance NET-mediated capture of CTCs and consequently propel metastatic progression. NETs play a critical role in promoting cancer metastasis by regulating inflammatory responses.
5.2. NETs and pre-metastasis niche
Laminin, a key ECM component, undergoes degradation that disrupts the basement membrane, initiating tumor invasion and metastasis [66]. NET-derived proteases, such as NE and MMP-9, remodel laminin and activate integrin α3β1 signaling, promoting cancer cell proliferation [82]. Notably, integrin β1 signaling drives the transition from dormancy to metastatic growth [118]. Different NET components contribute to metastasis through distinct mechanisms. NE promotes invasion by directly degrading ECM proteins and activating protease cascades [119], while MMP-9 disrupts ECM integrity by degrading type IV collagen in the basement membrane, facilitating tumor cell dissemination [120]. Additionally, CG enhances metastasis by activating MMPs and hydrolyzing ECM components, as demonstrated in liver cancer models [55].
NETs may capture CTCs, exposing them to a protein-rich microenvironment that promotes proliferation and metastasis. This interaction could further enhance the ability of CTCs to colonize distant organs [121]. Before the actual metastatic process, the primary neoplasm cultivates a microenvironment within a remote organ that fosters cancer dissemination—this phenomenon is termed a “premetastatic niche (PMN)” [122]." Castano et al. [123] elucidated that in early-stage ovarian cancer, factors derived from ovarian malignancies initially catalyze the formation of NETs and establish PMNs, forging an optimal milieu for subsequent metastasis. Subsequently, NETs act as mediators, through which CTCs amplify the spread of OCs. Wculek et al. [124] noted that neutrophils residing within these PMNs enhance subpopulations of malignant cells by releasing leukotrienes, promoting breast cancer dissemination. Owing to the existence of PMNs, carcinoma cells originating from primary tumors usually remain quiescent upon the invasion of other tissues. However, NETs can rouse dormant carcinogenic cells back into active proliferation. Albrengues et al. [82] discovered that lipopolysaccharides can awaken dormant cancer cells; however, depletion of neutrophils can negate this effect. The utilization of PAD4 inhibitors or DNases has been shown to diminish both LPS-induced formation of NETs and awakening of cancer cells. They proposed a nuanced mechanism through which NETs rouse these quiescent cells: NE and MMP-9 derived from NETs cleave and remodel laminin, activating the integrin α3β1 signaling pathway and fostering cancer cell proliferation.
Metastasis remains a pivotal factor contributing to poor survival rates in patients with cancer. Dormant cancer cells can remain concealed within PMNs until favorable sites for colonization are identified; this phenomenon is prevalent in breast cancer, prostate carcinoma, and melanoma. Consequently, investigating the intricate relationship between NETs and PMNs is important and offers valuable insights into strategies aimed at curtailing cancer metastasis.
5.3. NETs and angiogenesis
To sustain tumor growth, an adequate supply of blood is required [125]. Aldabbous et al. [126] demonstrated that NETs directly promote angiogenesis. Tumor cells traverse the vascular endothelial barrier, infiltrate the circulation, and colonize distant tissues, facilitating metastasis [127, 128]. This process depends on two key factors: cancer cell plasticity and vascular integrity [129]. Jiang et al. [67] pioneered an in vitro co-culture system using human umbilical vein endothelial cells (HUVECs) to investigate the impact of NETs on oncological fluidity. Their findings revealed that NETs downregulated vascular endothelial cadherin, compromised endothelial integrity, and facilitated extravascular infiltration by HCC, augmenting the metastatic potential. Given the tortuous and highly permeable nature of tumor vasculature, it is particularly vulnerable to NET-mediated remodeling. NETs have been shown to enhance endothelial sprouting, tube formation, and vascular permeability [126]. McDowell et al. [120] discovered that NET formation significantly affects endothelial cell-cell and contacts and enhances vascular permeability, which catalyzed cancer metastasis. The NET-DNA receptor CCDC25, expressed in HUVECs, mediates NET-driven endothelial proliferation, migration, and tubulation. In an in vitro rat aortic explant model, NETs promoted endothelial survival and chemotaxis, effects comparable to VEGF stimulation [130]. NETs play a role in pathological vascular remodeling in other disease contexts. In proliferative diabetic retinopathy and ischemic retinopathy models, senescent vasculature releases factors that attract neutrophils, triggering NET formation. This process clears dysfunctional endothelial cells and facilitates vessel remodeling, further supporting the role of NET-associated proteases in shaping the tumor vasculature [131]. Moreover, MMP-9—an integral component of NETs—has been implicated in promoting angiogenesis, reinforcing the concept that NETs contribute to the vascular adaptations necessary for tumor progression.
5.4. NETs and metastasis cascade
In a mouse in situ breast cancer model constructed by Park et al. [39] without systemic inflammation, tumors induced by metastatic cell lines recruited a higher proportion of NETs to the primary tumor than those induced by non-metastatic cell lines, and intravenous injection of metastatic cell lines resulted in the deposition of NETs in the lung, which was conducive to the formation of a metastatic microenvironment. One mechanism by which NETs stimulate cancer cell migration is through chemotaxis of NET-DNA to DNA sensors, such as CCDC25. Yang et al. [20] found that neutrophils, NETs markers MPO, and CitH3 exist in primary tumors and liver metastatic tumors of patients with breast cancer, and proved that the DNA components of NETs in the liver interact with the transmembrane protein CCDC25, and DNA-CCC25 triggers a cascade of intracellular signaling. The promotion of anticancer antibodies against CCDC25 significantly reduced NET-mediated liver metastasis in breast cancer. Tang et al. [107] further expanded upon these findings by investigating 4T1 cells and discovered that ASPP2-induced promotion of cholesterol biosynthesis culminated in NET formation, indicating that de novo cholesterol synthesis plays a crucial role in neutrophil recruitment and the subsequent development of NETs, whereas high levels of cholesterol stimulated CCDC25 expression, a regulatory mechanism requiring deeper exploration.
NETs can also catalyze cancer metastasis through mechanisms distinct from protein interactions, such as eliciting endothelial damage and facilitating EMT. EMT induction augmented the aggressiveness of epithelial carcinoma cells. Co-culturing NETs with cancer cells within a breast cancer milieu revealed downregulation of E-cadherin coupled with upregulation of mesenchymal markers [65], substantiating the notion that NEThe induces gastric cancer cell metastasis via EMT activation, specifically targeting NETs. Zhu et al. [132] observed a marked decrease in the levels of E-cadherin in gastric adenocarcinoma cells, concomitant with an elevation in mesenchymal markers. In a comprehensive analysis conducted by Martins-Cardoso et al. [65], the influence of NETs on the phenotypic characteristics of human breast cancer cells was examined using a sophisticated breast cancer model. Their findings revealed that exposure to NETs effectively transformed the epithelial morphology of MCF-7 cells into a mesenchymal phenotype, accompanied by significant alterations in the EMT attributes. There was an upregulation of N-cadherin and fibronectin expression, whereas E-cadherin levels were inhibited. These EMT modifications may not only correlate with morphological changes at the cellular level, but can also augment cellular migratory capabilities. Such investigations highlight that NETs possess the potential to facilitate cancer metastasis through intricate protein interactions or by orchestrating metastatic cascades such as tumor-derived EMT processes.
6. NETs and cancer-related thrombosis
Demers et al. meticulously examined the intricate relationship between NETs and tumor-associated thrombosis, ultimately concluding that after chemotherapy for tumors, the liberation of NETs alongside the thrombin-antithrombin complex precipitates a process known as NETosis. This cascade exacerbates both inflammation and coagulation processes; these inflammatory responses activate neutrophils, further promoting the generation of additional NETs. Hence, NETs are believed to function as potent enhancers of coagulation in malignancy-related thrombosis [133]. Jung et al. investigated whether NET formation catalyzes endogenous plasma thrombin production. Their experiments revealed that pancreatic cancer cells and their conditioned media can induce NETs formation, significantly augmenting their ability to produce normal endogenous plasma thrombin [52]. Boone et al., through their findings, corroborated that thrombosis associated with pancreatic carcinoma is intrinsically linked to NET formation. In an experimental cohort involving mice burdened with tumors, but genetically modified to lack PAD4, an essential enzyme for NETs creation, it was observed that these subjects generated no NETs. Moreover, hydroxychloroquine (an inhibitor of NETs formation) has demonstrated efficacy in diminishing platelet aggregation, curtailing circulating tissue factors, and mitigating blood hypercoagulability. Clinical data have revealed a striking reduction in venous thromboembolism rates among patients with pancreatic cancer treated with hydroxychloroquine after chemotherapy, from 30% to 9.1% [134]. Seo et al. elucidated the mechanism underlying portal vein thrombosis in patients with liver cancer through clinical evaluation involving 177 hepatitis carrier cases, including 77 afflicted individuals with portal vein thrombosis. They compared them with 48 healthy controls, in which markers indicative of NETs formation (such as DNA-histone complexes, double-stranded DNA fragments, and NE levels) exhibited significantly elevated concentrations relative to those found in healthy subjects. This finding substantiates contact system activation as a novel mechanistic pathway that contributes to thrombus development [135]. NETs represent a pivotal mechanism of the innate immune response within the body and play an integral role in the intricate process of thrombosis. The distinct reticular architecture of NETs serves as a scaffold for thrombus formation. NETs facilitate thrombosis through dynamic interactions with coagulation factors, complement proteins, platelets, endothelial cells, and erythrocytes (Figure 6).
NETs promote coagulation and cancer-associated thrombosis. Platelets increase intracellular calcium due to histone activation, which triggers neutrophil activation through P-selectin signaling. Tumor cells (Ts) recruit tumor-associated neutrophils (TANs) and induce their transformation into NETs. Activated endothelial cells secrete IL-8, promoting NET formation. Depending on the microenvironment, NET components can activate or induce apoptosis in endothelial cells. Erythrocytes infected with Phasmodium species and free heme participate directly or indirectly in the conversion of neutrophils into NETs. NETs contribute to thrombosis through several mechanisms. Initially, they promote platelet adhesion, activation, and aggregation through interactions with von Willebrand factor (VWF), fibrinogen, and intermediate filament 1(IF1). Both exogenous and endogenous pathways involving histones, oxidative stress markers, phosphatidylserine, and tissue factor (TF) pathway inhibitors facilitate NET-mediated fibrin formation. Additionally, a positive feedback loop involving neutrophils exists: complement activation through C3a receptor (C3aR), signal STAT4/ROS signaling promotes NET formation. Concurrently, C3b and C5a mediate further neutrophil recruitment and activation. These processes collectively contribute to erythrocyte lysis and aggregation, crucial for thrombus formation in cancer-related contexts. Abbreviations: TANs, tumor-associated neutrophils; Ts, Tumor cells; VWF, von Willebrand factor; IF1, intermediate filament 1; TF, tissue factor; C3aR, C3a receptor; ROS, reactive oxygen species. *Created with BioRender.com.
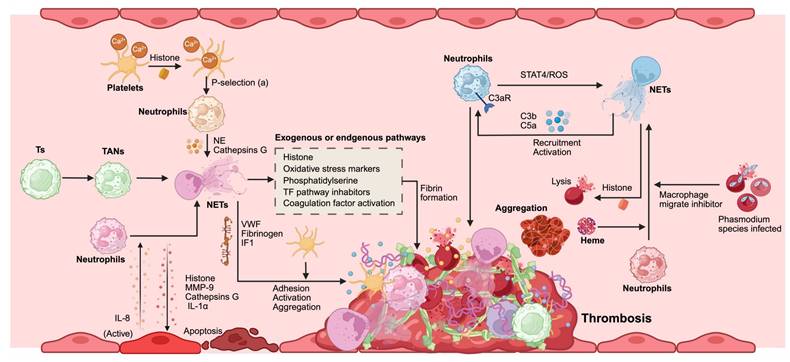
6.1. NETs and coagulation cascade
NETs possess the intriguing capability to initiate the coagulation cascade directly. DNA embedded within NETs enhances the activity of serine proteases, whereas histones are implicated in augmenting thrombin production. Paradoxically, intact NETs cannot activate coagulation in vitro [136]. This phenomenon may be because of the selective exposure of tissue factors present within NET structures [137]. Furthermore, negatively charged NETs can engage directly with and activate endogenous promoters of the coagulation pathway, notably the coagulation factor XII (FⅫ). NET-induced thrombin generation was markedly diminished in plasma derived from mice deficient in either F-XI or F-XII [138]. Wang et al. [139] also showed that neutrophil microparticles can attach to NETs through interactions between histones and phosphatidylserine, instigating thrombin production via endogenous mechanisms of coagulation. In addition, NE and cathepsin G within NETs play a significant role in facilitating fibrin formation by degrading tissue factor pathway inhibitors [9]. The processes of fibrin formation and deposition transpire upon activation by either exogenous or endogenous coagulation pathways on NETs structures; however, fibrin clots intertwined with NETs exhibit enhanced resistance to plasminase. This characteristic may constitute a pivotal challenge for thromboembolic diseases [137].
6.2. NETs and component systems
The initial indication of the impact of the complement system on NETs emerged from observations that neutrophils derived from complement 3 (C3) or complement C3a receptor(C3aR) knockout mice exhibited a marked inability to form NETs [140]. Guglietta et al. [141] elucidated that the release of LPS-induced NETs is contingent on the activation of the complement cascade and the subsequent upregulation of C3aR on neutrophil surfaces, which fosters coagulation. The complement component C5a plays a pivotal role in recruiting and activating neutrophils, and serves as a frequently used irritant in prompting NETosis. Chen et al. [142] demonstrated that C5a amplified ROS production by inhibiting STAT4 activity during NETosis, stimulating NETosis, and facilitating arterial thrombosis formation. The released NETs create an environment that is conducive to complement activation. The complement component C1q deposited onto these structures stabilizes their integrity by impeding the action of deoxyribonuclease I [143]. In contrast, it enhances macrophage activation through the high expression of the C1q receptor induced by NETs [144]. Furthermore, complement factor H has been shown to suppress PMA-induced NET formation while diminishing the deposition of complement fragment C3b on these traps [145, 146]. In addition, proteins such as MPO residing within NETs actively participate in complement activation processes, cleaving complement component C5 into its active fragments, and cathepsin G and neutrophil elastase target C3 [137]. This intricate interplay further highlights the correlation between thrombosis onset, NET release dynamics, and the activation of complementary pathways.
6.3. NETs and platelets
After perfusion of whole blood with NETs, one can observe that NETs serve as scaffolding structures, facilitating the adsorption of platelet aggregation [147]. Activated platelets express P-selectin, which interacts with P-selectin glycoprotein ligand 1 present on the surface of neutrophils, promoting the formation of NETs [148, 149]. Furthermore, platelets release the HMGB1 protein, a crucial factor that enhances NET formation. HMGB1 elicits the release of NETs by activating neutrophils via multiple receptors, such as TLR2 and TLR4 [150]. Experiments have demonstrated that the interaction between platelets and NETs is contingent on histones; purified histones stimulate calcium influx into platelets in vitro, inducing their activation [9]. In addition, NE and cathepsin G within NETs can promote platelet aggregation by activating platelet surface receptors [151]. The interplay between NETs and platelets may also be mediated by various adhesion molecules, including von Willebrand factor (VWF) and fibrinogen [151]. In summary, a significant interaction exists between activated platelets and NETs. Activated platelets trigger the release of NETs, which further amplify platelet adhesion, activation, and aggregation, ultimately engaging in procoagulant activity that creates a positive feedback loop that enhances coagulation.
6.4. NETs and endothelial cells
Preservation of the integrity of endothelial architecture and its multifaceted functions is paramount for sustaining vascular homeostasis; however, endothelial injury is a pivotal contributor to thrombosis. Co-culture of endothelial cells and neutrophils demonstrated that activated endothelial cells can precipitate the release of NETs, a mechanism intricately reliant on IL-8 secreted during the activation of these cells [40]. Concurrently, components associated with NETs can induce both the activation and apoptosis of endothelial cells, and this reciprocal feedback loop significantly exacerbates thrombotic processes. Histone proteins exhibit pronounced affinity for phospholipids, facilitating their binding to cellular membranes and inducing calcium ion influx. This cascade further amplifies endothelial cell activation while promoting exocytosis in Weibel-Palade bodies [152]. MMP-9 present within NETs may induce damage to endothelial cells through an activation pathway involving MMP-2, participating in pathological phenomena such as thrombosis [153, 154]. Folco et al. [155] reported that NETs activate endothelial cells to express adhesion molecules via synergistic interactions between IL-1α and cathepsin G, culminating in localized vascular inflammation accompanied by thrombosis. In parallel with the research conducted by Blanco et al. [156], it was revealed that small RNAs encapsulated within NETs could be internalized by endothelial cells, triggering a type I interferon response, an event believed to mediate vascular injury and advance atherosclerotic processes. VWF, a glycoprotein produced by activated platelets or directly from endothelium-derived sources, plays an essential role in hemostasis. The release of NETs from neutrophils has been shown to elevate VWF concentrations while fostering polymer formation through mechanisms such as oxidation, citrullination, hydrolysis mediated by ADAMTS13, or competitive interactions with VWF itself [157]. Henceforth, we observed how NETs serve not merely as agents, but help to perpetuate dysfunction within the endothelium associated with thrombus formation.
6.5. NETs and red blood cells
Red blood cells are intrinsically linked to thrombosis and can facilitate coagulation through a myriad of mechanisms, including alterations in blood viscosity, exposure to phosphatidylserine, and adhesion to platelets [158]. Immunohistochemical staining of murine models of deep vein thrombosis and human coronary artery thrombi revealed pronounced accumulation of red blood cells surrounding NETs [159, 160]. This observation implies that the interplay between NETs and blood cells may play a pivotal role in thrombosis progression. Kordbacheh et al. [161] discovered that extracellular histones can provoke erythrocyte aggregation and lysis; moreover, heme liberated from ruptured erythrocytes has been shown to instigate NETs formation [162]. Red blood cells infected with Plasmodium species can stimulate neutrophils to release NETs by discharging macrophage migration inhibitors and heme substances [162, 163]. However, the exact mechanisms underlying the interaction between blood cells and NETs require further investigation.
7. NET interaction with immune cells in cancer
The immunosuppressive TME is a key driver of tumor immune evasion [164]. Persistent inflammatory stimuli, chemokines, and metabolic factors drive neutrophil chemotaxis toward tumor tissues, where they actively reshape the TME. This remodeling fosters the infiltration of immunosuppressive cells, particularly myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs), thereby reinforcing mechanisms of tumor recurrence and metastasis [165, 166]. As a key player in this process, NETs not only sustain an immunosuppressive niche but also disrupt anti-tumor immune responses, positioning them as critical regulators of immune escape (Figure 7). The emergence of immune checkpoint inhibitors, such as anti-PD-1/PD-L1 therapies, has transformed cancer treatment. However, accumulating evidence suggests that NETs may limit their efficacy. Studies indicate that targeting NETs can enhance the therapeutic effects of anti-PD-1 therapy, as demonstrated in MC-38 colon cancer models [167], while NET-based prognostic models show promise in predicting immunotherapy responses [168]. Furthermore, inhibiting NET formation has been linked to improved anti-PD-1 treatment outcomes in liver, pancreatic, and breast cancers [58, 169]. These findings highlight NETs as key mediators of tumor progression through immune suppression, warranting further investigation into their role as therapeutic targets.
NETs regulate the functions of immune cells in the tumor microenvironment. NETs modulate multiple immune cell populations through distinct mechanisms: In CD4+ T cells, NETs trigger TLR4-mediated oxidative phosphorylation gene regulation, affecting both regulatory T cells (Treg) and Th17 differentiation. CD8+ T cells respond to NETs through multiple pathways: forming physical barriers that inhibit infiltration, suppressing cytokine production (IFN-γ, TNF-α, IL-2), inducing functional proteins (Tim-3, LAG3, PD-1), and causing T cell depletion via TMO6/TCR signaling. NETs promote Th17 cell differentiation through TLR2/STAT pathway activation. In the NK cell compartment, NETs create physical barriers preventing infiltration, while inducing apoptosis and NK cell dysfunction through Angiopoietin-2 and inflammatory cytokines (IL-2, IL-15). NETs-derived cathepsin G and MMP-9 regulate NK cell receptor expression (e.g., NKp46) affecting IFN-γ production and function. Dendritic cells (DCs) respond to NETs through two opposite pathways: activation of DCs and mitochondrial damage-induced apoptosis. Macrophages exposed to NETs undergo polarization toward an M2 phenotype. Abbreviations: Treg; regulatory T cells; NK, natural killer cells; DC, dendritic cells. *Created with BioRender.com.
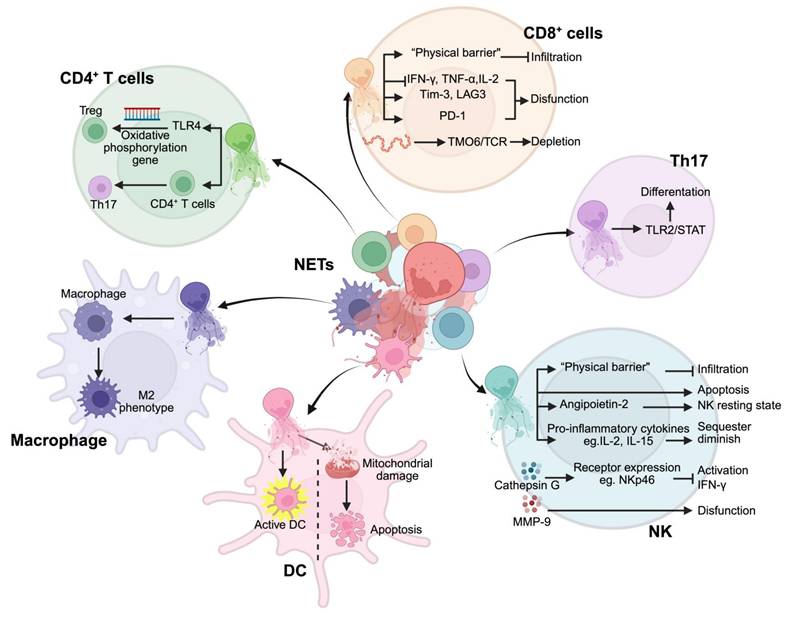
Despite these insights, the mechanistic underpinnings of NET-driven immune resistance remain poorly defined. How NETs modulate T cell exhaustion, suppress antigen presentation, or interact with metabolic constraints within the TME is not fully understood. This gap underscores the need for refined therapeutic strategies that selectively dismantle NET-mediated immunosuppression while preserving their potential role in host defense. Future research should aim to dissect these molecular pathways and explore the synergistic potential of NET-targeting approaches in combination with immunotherapy, ultimately paving the way for more effective and durable cancer treatments.
7.1. NETs and immunosuppressive cells
A multitude of immunosuppressive cells exists within the TME, including MDSCs, Tregs, TAMs, and Th17 cells. NETs can modulate the expression of genes linked to oxidative phosphorylation in naïve CD4+ T cells via TLR4 signaling, inducing their differentiation into Tregs and facilitating the progression from nonalcoholic steatohepatitis to hepatocellular carcinoma. In addition, NETs have been shown to enhance the infiltration of Tregs and support the local advancement and metastasis of breast cancer [170]. According to research conducted by Yu et al. [171], NETs can mediate macrophage polarization toward an M2 phenotype, further advancing gastric cancer development. Combined with this phenomenon, they collectively promote invasion and migration of the A549 cell lines [172]. While there is currently a lack of reports detailing the interplay between NETs and Th17 cells within the tumoral context, recent investigations suggest that NETs may stimulate Th17 cell differentiation through activation of the TLR2-STAT signaling pathway [173]. Furthermore, these traps have been implicated in driving CD4+ T cell transformation into the Th17 phenotype, specifically in acute lung injury [174]. Taken together, these results demonstrate that NETs critically contribute to tumor advancement through enhancing the activity of diverse immunosuppressive cell populations.
7.2. NETs and dendritic cells
Dendritic cells (DCs) are the principal antigen-presenting entities within the body and play a pivotal role in curbing tumor progression by eliciting an antigen-specific adaptive immune response [175]. Numerous studies have shown that NETs possess the capability to activate DCs while simultaneously delivering pertinent antigens that foster specific immunity, impeding the advancement of NPM-mutant myeloid leukemia. Conversely, NETs induce DC apoptosis by inducing mitochondrial damage [176, 177]. In collection, these investigations underscore the dualistic nature of NETs' influence on DCs; they not only facilitate antigen presentation and prime the body's immune response, but may also precipitate apoptotic pathways within DC, ultimately hindering adaptive immunity. Factors such as NET composition and the tumor microenvironment likely dictate whether NETs enhance antigen presentation or induce DC apoptosis. This complexity underscores the need for targeted strategies that mitigate NET-induced immunosuppression while preserving their immunostimulatory potential.
7.3. NETs and CD8+ T cells
Dysfunction of CD8+ T cells is intricately linked to tumorigenesis, progression, and evasion of the disease [178]. NETs are strategically positioned around tumor cells, acting as a formidable "physical barrier" that facilitates tumor advancement by constraining the infiltration of CD8+ T cells and NK cells. Beyond merely inhibiting cellular infiltration, NETs profoundly induce CD8+ T cells in various tumor models. For instance, these structures diminish the secretion of critical effector cytokines from CD8+ T cells, such as interferon-gamma (IFN-γ), TNF-α, and IL-2, while simultaneously upregulating immune checkpoint molecules, such as Tim-3 and LAG3. This indicates that NETs play a pivotal role in lung cancer progression by inducing CD8+ T cell dysfunction [179]. Moreover, components such as PD-L1 present within NETs have been shown to inhibit the functionality of CD8+ T cells by engaging PD-1 receptors on the surface membranes of these lymphocytes [83]. In liver cancer, DNA fragments derived from NETs obstruct both T cell receptor (TCR) signaling and activation pathways, including NF-kB, by binding to TMCO6 on the membranes of CD8+ T cells. This interaction precipitates a depletion effect on these crucial immune players, while concurrently promoting malignant growth [180]. Research focusing on breast cancer has revealed that targeting NETs can effectively curtail T cell infiltration and significantly enhance the therapeutic efficacy of anti-PD-1 monoclonal antibodies [58]. Besides breast carcinoma, numerous studies have independently demonstrated that disrupting NET formation augments the effectiveness of anti-PD-1 therapy in various malignancies, including colorectal cancer, lung cancer, and hepatocellular carcinoma [181, 182]. Collectively, these investigations underscore how NETs compromise CD8+ T cell function and hinder the effective action of immune checkpoint inhibitors in tumors. Therefore, strategies aimed at targeting NETs hold promise for revitalizing CD8+ T cell efficacy and improving immunotherapeutic outcomes.
7.4. NETs and NK cells
NETs can impede the anti-tumor efficacy of NK cells via a multitude of mechanisms. First, as previously indicated, NK cells are hindered from effectively engaging tumor cells because of the obstruction posed by NETs. Furthermore, NETs can degrade proinflammatory cytokines that are imperative for the activation, proliferation, and functional performance of NK cells, such as IL-2 and IL-15 [183, 184]. Consequently, NETs may indirectly suppress the anti-tumor properties of NK cells by sequestering and diminishing critical proinflammatory signals. In addition, NETs have been shown to directly induce apoptosis in NK cells [75]. CG is a vital component of NETs; it can undermine NK cell activation as well as IFN-γ release and degranulation processes by downregulating the expression of activated receptors, such as NKp46 [185]. Moreover, MMP-9 abundance within NETs may further contribute to the dysfunctional behavior of NK cells, facilitating immune evasion by malignant tumor cells [76]. NETs interact with various elements within the TME to modulate NK cell function. For instance, in gastric cancer models, NETs have been observed to elevate angiopoietin-2 levels within the TME, which is intricately linked to the maintenance of intact resting states among NK cell populations [186], suggesting that NETs might indirectly influence the anti-tumor activity of these immune effector cells via the regulation of TME constituents. Cheng et al. [77] discovered that inhibiting NET formation significantly enhances both functionally active capacities in liver cancer-associated NK cells while simultaneously impeding disease progression, underscoring the potential for future exploration of interactions between NET profiles and NK cell dynamics to optimize therapeutic strategies focused on enhancing cellular immunotherapy against tumors.
In conclusion, the presence of NETs significantly amplified the infiltration and functionality of immunosuppressive cells while markedly undermining the activities of NK and CD8+ T cells. This disruption leads to the breakdown of immune homeostasis within the body, promoting immune evasion by tumor cells and facilitating disease progression. These findings highlight the potential of NETs as novel therapeutic targets in tumor immunotherapy. Within the rapidly advancing landscape of cancer immunotherapy, strategic targeting of NETs may greatly enhance the efficacy of immune checkpoint inhibitors in various malignancies. This approach offers innovative strategies to address the challenges posed by suboptimal treatment response rates and emerging resistance mechanisms. We posit that a thorough investigation of the specific mechanisms used by NETs within the tumor microenvironment, along with the development of effective and targeted interventions against NETs, will be crucial for evaluating their synergistic effects with other immunotherapeutic modalities. This trajectory promises to propel continued innovation and advancement in the field of tumor immunotherapy.
8. Target NETs for cancer treatment
8.1. DNA inhibitor
Recombinant human DNase I is a multifaceted therapeutic agent used to manage bronchiectasis and lung abscesses. Its myriad functions include the absorption of nucleotide nutrients, modulation of biofilm formation, facilitation of pathogen invasion, degradation of DNA matrices, and regulation of immune responses, among others [187]. However, their clinical application is hindered by the short half-life of the endonuclease enzymes. While the U.S. The Food and Drug Administration has sanctioned an inhalable formulation, pulmozyme, indicated for cystic fibrosis treatment, but the ability of such aerosolized preparations to penetrate the systemic circulation remains limited. This results in suboptimal efficacy when addressing NETs beyond the pulmonary or vascular confines. Moreover, this approach exhibits insufficient targeting capabilities and poses certain risks to overall health [188]. In an innovative stride forward, Xia et al. [189] engineered an adeno-associated virus (AAV) gene therapy vector specifically designed for the hepatic expression of DNase I. Their research demonstrated that AAV-mediated delivery of DNase I significantly inhibited liver metastasis from colorectal cancer in murine models following a single intravenous administration. These findings suggested that AAV-mediated DNase I treatment is a safe and effective therapeutic strategy. Consequently, DNase I inhibitors exhibit substantial potential as targeted therapies for NETs and warrant further investigation to elucidate their clinical applications.
8.2. PAD4 inhibitors
PAD4 is a pivotal molecule in the intricate formation of NETs and exerts regulatory control over the citrullination of histone proteins. This process culminates in the disruption of chromatin DNA within neutrophil nuclei, facilitating the subsequent generation of NETs [190]. Inhibition of PAD4 has demonstrated efficacy in diminishing circulating levels of NETs and abolishing cancer-induced NET formation. Studies involving PAD4-deficient murine models have revealed pronounced inhibition of NETs formation, which concurrently impedes tumor progression and prolongs the survival span of these subjects [191]. Research indicates that targeting PAD4 through its inhibition is an exceptional strategy for curtailing NET activity; among the potential agents are small-molecule inhibitors such as chloramidine, an irreversible inhibitor, and GSK484, which offers reversible inhibitory action against PAD4 [192]. Nevertheless, challenges remain owing to the rapid metabolic degradation and suboptimal oral bioavailability of the existing PAD4 inhibitors. Considering these drawbacks, recent studies have focused on innovative nanodelivery systems for PAD4 inhibitor development. These include the exploration of AuNPs used as nanomaterials [193], covalent linkages with chitosan to fabricate oxidative stress-responsive nanosystems [194], and liposomal carriers engineered for targeted delivery [195].
8.3. Inhibiting inflammatory factors
Another approach for inhibiting NET formation involves targeting upstream mediators, a domain that has garnered considerable attention in studies of inflammation-related factors. CXCR1/2, a G-protein-coupled transmembrane receptor on the surface of neutrophils, is a critical mediator of nuclear chemotactic recruitment of neutrophils [196]. IL-8 is a pivotal and efficacious agent that facilitates chemotaxis by interacting with the aforementioned receptor. Recent reports have indicated that the synergistic application of IL-8 monoclonal antibodies with CXCR1/2 inhibitors demonstrates promising efficacy and has the potential to be an innovative therapeutic strategy for targeting NETs [197]. Several pharmacological agents are under development for repurposing. For instance, anti-interleukin-17 antibodies used in psoriasis treatment may possess the capacity to modulate neutrophil recruitment while simultaneously inhibiting NET formation [57]. In addition, complement C5a receptor 1 inhibitors have received approval for their use in combating antineutrophil-associated small vessel vasculitis [198].
8.4. Traditional Chinese medicine
Tang et al. [39] elucidated that lipid-lowering agents, such as simvastatin, possess the capacity to modulate cholesterol synthesis and indirectly influence the expression of CCDC25, curbing the formation of NETs. Berberine, a naturally occurring compound prevalent in traditional Chinese medicine, is recognized for its lipid-lowering properties and its regulatory effects on NET formation. Dihydrotanshinone I (DHT), the principal active constituent of Salvia miltiorrhiza, inhibits the proliferation of breast cancer cells. Zhao et al. [199] posited that DHT could diminish the expression of tissue inhibitor of metalloproteinase-1 (TIMP1), which is generated during NET formation, consequently mitigating NET production. Pan et al. [200] discovered that Huangqin Decoction effectively reduced the levels of IL-1 and MMP-9 in a murine model of colorectal cancer while simultaneously inhibiting PAD4 expression, which impeded the progression of colorectal carcinoma by hindering NETs generation. Haute et al. [201] demonstrated the ability of octyl gallate to suppress the release of ROS and regulate lipopolysaccharide-induced neutrophil apoptosis. However, the intricate mechanisms underlying its inhibitory effects on NETs production remain unclear. The bioactive constituents derived from single herbs and compounds in traditional Chinese medicine demonstrate the potential to regulate NETs by modulating factors such as ROS, PAD4, and TIMP1. Nonetheless, current research on traditional Chinese medicine remains nascent, with insufficient clinical trials. There is an imperative need for further investigation of the precise mechanistic pathways associated with the components found in traditional Chinese medicines to more effectively underscore their therapeutic value against targeted NETs.
8.5. Antibiotics
Antibiotics represent a distinct class of secondary metabolites synthesized by microorganisms, higher plants, and animals that confer resistance against pathogens. Bystrzycka et al. [202] investigated neutrophils treated with chloramphenicol and azithromycin and found a significant reduction in NET formation. Furthermore, azithromycin administration appears to diminish the likelihood of respiratory disease outbreaks. Although agents such as gentamicin have similar inhibitory effects on NET formation, cefotaxime demonstrates no therapeutic efficacy. Consequently, it is imperative to conduct more in-depth research on the judicious application of antibiotics targeting NETs to better understand their implications and potential benefits.
8.6. Nanodrugs for targeting NETs
As mentioned, various strategies have been explored to inhibit NETs and suppress tumor metastasis (Supplementary Table 1), yet clinical translation faces major challenges. Many inhibitors have failed due to low oral bioavailability and poor targeting. Moreover, NET inhibition may impair neutrophil-mediated pathogen defense, increasing infection and carcinogenesis risks, as seen with PAD4 inhibitors and histone citrullination blockade [203]. NET degradation may also release DNA-bound proteins like CitH3 and MPO, triggering systemic inflammation [204]. Thus, minimizing the adverse effects of NET-targeted therapy remains crucial. Combining NET inhibitors with antibiotics or antivirals may enhance efficacy [192], but optimizing their potency and specificity remains a key challenge.
Nanotechnology has addressed major limitations of non-specific chemotherapy by enhancing drug circulation, enabling precise tumor targeting, and minimizing toxicity to healthy cells [205] (Supplementary Table 1). For example, Yin et al. [206] developed a smart nanocarrier (mP-NPs-DNase/PTX) targeting tumor-associated NETs, comprising a paclitaxel (PTX) prodrug nanoparticle core and a PLL-based DNase I shell, cleavable by MMP-9. Upon tumor accumulation, MMP-9 triggers DNase I release to degrade NETs, exposing a cell-penetrating peptide that facilitates tumor cell uptake. Intracellularly, high glutathione levels induce PTX release, exerting cytotoxic effects. Beyond its role as a therapeutic agent, nanomedicine can also enhance adoptive NK cell therapy. An injectable hydrogel rapidly forms an adhesive gel, inhibits immunosuppressive cell infiltration by neutralizing tumor acidity, and degrades NETs by releasing pH-responsive DNase I [77]. This approach enhances NK cell infiltration and reduces post-surgical HCC recurrence with minimal systemic toxicity [77].
However, TME, characterized by features such as hypoxia, acidity, and elevated interstitial fluid pressure, presents significant challenges that impede the delivery and efficacy of nanomedicines [207, 208]. Nanomedicines can interact with neutrophils and potentially induce NETosis, a process that may have dual effects: either promoting anti-tumor immune responses [209] or, if uncontrolled, exacerbating tumor progression [210]. Therefore, a comprehensive understanding of the molecular mechanisms, transcription factors, and signaling pathways involved in NETosis—and how these can be modulated by nanomedicines—is crucial [209]. Despite notable progress, several gaps remain in our understanding of nanomedicine, particularly regarding its interaction with the TME and immune system. Further research is required to establish foundational principles for nanomedicine design, develop advanced material platforms, and create relevant animal models that closely mirror human tumor biology [211]. Additionally, in-depth studies on tumor biology, including intratumor heterogeneity and nano-bio interactions, are essential for designing nanomedicines capable of effectively navigating the TME and delivering therapeutic payloads.
9. Conclusion and perspectives
NETs have attracted increasing attention in cancer research due to their role in tumor progression. Evidence suggests that NETs facilitate metastasis by modulating the tumor immune microenvironment, vascular system, and premetastatic niche formation. However, some studies indicate that NETs may exhibit tumor-suppressive effects under certain conditions. The functional outcomes of NET-tumor interactions are influenced by neutrophil subtypes, NET composition and formation mechanisms, tumor microenvironment heterogeneity, and experimental conditions. Thus, further elucidation of the determinants shaping NET activity in cancer is of critical importance.
Despite growing research efforts, the precise mechanisms underlying NET-mediated tumor progression remain unclear, limiting their clinical translation. Inhibitors such as chloramidine, recombinant human DNase I, antibiotics, and certain traditional Chinese medicine formulations have demonstrated efficacy in suppressing NET formation, offering potential therapeutic benefits. Additionally, the combination of NET inhibitors with immunosuppressants may represent a novel anti-tumor strategy. Advances in nanotechnology further enable the development of smart drug delivery systems, enhancing targeted therapy while mitigating adverse effects.
Several key knowledge gaps warrant further investigation. First, the extent to which NET formation mechanisms influence their structural composition and biological function remains unresolved. Second, despite the increasing volume of research on NETs since their discovery 18 years ago, the absence of a standardized detection method complicates cross-study comparisons and reproducibility. Third, while substantial progress has been made in understanding NETs in tumor progression, their effects are highly tissue- and context-dependent. Distinct polymorphonuclear neutrophil subpopulations may generate functionally diverse NETs, and variations in the immune microenvironment may modulate their impact. Lastly, interdisciplinary efforts are essential for the development of spatiotemporally controlled drug delivery systems targeting NET-associated cancers, necessitating further clinical validation. In conclusion, a mechanistic dissection of NET-cancer interactions holds significant promise and warrants rigorous investigation to inform future clinical applications.
Supplementary Material
Supplementary table.
Acknowledgements
Funding
This review was supported by the National Natural Science Foundation of China (No. 82472183), Shanghai Pujiang Talents Program (No. 21PJD013), and Shanghai Raising Star Program (24QA2707800).
Author contributions
Ji Zhang contributed to the literature review and research, writing, formatting, and editing of the manuscripts. Hao Zhang and Changhong Miao assisted with the review and polishing of the manuscripts.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, Mantovani A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer. 2020;20:485-503
2. Burn GL, Foti A, Marsman G, Patel DF, Zychlinsky A. The Neutrophil. Immunity. 2021;54:1377-91
3. Zhang F, Xia Y, Su J, Quan F, Zhou H, Li Q. et al. Neutrophil diversity and function in health and disease. Signal Transduct Target Ther. 2024;9:343
4. Hidalgo A, Libby P, Soehnlein O, Aramburu IV, Papayannopoulos V, Silvestre-Roig C. Neutrophil extracellular traps: from physiology to pathology. Cardiovasc Res. 2022;118:2737-53
5. Poli V, Zanoni I. Neutrophil intrinsic and extrinsic regulation of NETosis in health and disease. Trends Microbiol. 2023;31:280-93
6. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS. et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532-5
7. Hickey MJ, Kubes P. Intravascular immunity: the host-pathogen encounter in blood vessels. Nat Rev Immunol. 2009;9:364-75
8. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18:134-47
9. Wang H, Kim SJ, Lei Y, Wang S, Wang H, Huang H. et al. Neutrophil extracellular traps in homeostasis and disease. Signal Transduct Target Ther. 2024;9:235
10. Ma Y, Wei J, He W, Ren J. Neutrophil extracellular traps in cancer. MedComm (2020). 2024;5:e647
11. Zhang F, Yan Y, Cao X, Guo C, Wang K, Lv S. TGF-β-driven LIF expression influences neutrophil extracellular traps (NETs) and contributes to peritoneal metastasis in gastric cancer. Cell Death Dis. 2024;15:218
12. Yang LY, Luo Q, Lu L, Zhu WW, Sun HT, Wei R. et al. Increased neutrophil extracellular traps promote metastasis potential of hepatocellular carcinoma via provoking tumorous inflammatory response. J Hematol Oncol. 2020;13:3
13. Petretto A, Bruschi M, Pratesi F, Croia C, Candiano G, Ghiggeri G. et al. Neutrophil extracellular traps (NET) induced by different stimuli: A comparative proteomic analysis. PLoS One. 2019;14:e0218946
14. Keir HR, Shoemark A, Dicker AJ, Perea L, Pollock J, Giam YH. et al. Neutrophil extracellular traps, disease severity, and antibiotic response in bronchiectasis: an international, observational, multicohort study. Lancet Respir Med. 2021;9:873-84
15. Chen J, Wang T, Li X, Gao L, Wang K, Cheng M. et al. DNA of neutrophil extracellular traps promote NF-κB-dependent autoimmunity via cGAS/TLR9 in chronic obstructive pulmonary disease. Signal Transduct Target Ther. 2024;9:163
16. O'Neil LJ, Oliveira CB, Wang X, Navarrete M, Barrera-Vargas A, Merayo-Chalico J. et al. Neutrophil extracellular trap-associated carbamylation and histones trigger osteoclast formation in rheumatoid arthritis. Ann Rheum Dis. 2023;82:630-8
17. Zhan X, Wu R, Kong XH, You Y, He K, Sun XY. et al. Elevated neutrophil extracellular traps by HBV-mediated S100A9-TLR4/RAGE-ROS cascade facilitate the growth and metastasis of hepatocellular carcinoma. Cancer Commun (Lond). 2023;43:225-45
18. Nie M, Yang L, Bi X, Wang Y, Sun P, Yang H. et al. Neutrophil Extracellular Traps Induced by IL8 Promote Diffuse Large B-cell Lymphoma Progression via the TLR9 Signaling. Clin Cancer Res. 2019;25:1867-79
19. Wang WW, Wu L, Lu W, Chen W, Yan W, Qi C. et al. Lipopolysaccharides increase the risk of colorectal cancer recurrence and metastasis due to the induction of neutrophil extracellular traps after curative resection. J Cancer Res Clin Oncol. 2021;147:2609-19
20. Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J. et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature. 2020;583:133-8
21. Takei H, Araki A, Watanabe H, Ichinose A, Sendo F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J Leukoc Biol. 1996;59:229-40
22. Khan U, Chowdhury S, Billah MM, Islam KMD, Thorlacius H, Rahman M. Neutrophil Extracellular Traps in Colorectal Cancer Progression and Metastasis. Int J Mol Sci. 2021;22:7260
23. Baz AA, Hao H, Lan S, Li Z, Liu S, Chen S. et al. Neutrophil extracellular traps in bacterial infections and evasion strategies. Front Immunol. 2024;15:1357967
24. Speziale P, Pietrocola G. Staphylococcus aureus induces neutrophil extracellular traps (NETs) and neutralizes their bactericidal potential. Comput Struct Biotechnol J. 2021;19:3451-7
25. Thiam HR, Wong SL, Qiu R, Kittisopikul M, Vahabikashi A, Goldman AE. et al. NETosis proceeds by cytoskeleton and endomembrane disassembly and PAD4-mediated chromatin decondensation and nuclear envelope rupture. Proc Natl Acad Sci U S A. 2020;117:7326-37
26. Zhu YP, Speir M, Tan Z, Lee JC, Nowell CJ, Chen AA. et al. NET formation is a default epigenetic program controlled by PAD4 in apoptotic neutrophils. Sci Adv. 2023;9:eadj1397
27. Thiam HR, Wong SL, Wagner DD, Waterman CM. Cellular Mechanisms of NETosis. Annu Rev Cell Dev Biol. 2020;36:191-218
28. Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A. et al. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol. 2011;7:75-7
29. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191:677-91
30. Douda DN, Khan MA, Grasemann H, Palaniyar N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci U S A. 2015;112:2817-22
31. Zhao Q, Chen DP, Chen HD, Wang YZ, Shi W, Lu YT. et al. NK-cell-elicited gasdermin-D-dependent hepatocyte pyroptosis induces neutrophil extracellular traps that facilitate HBV-related acute-on-chronic liver failure. Hepatology. 2025;81:917-31
32. Mor-Vaknin N, Saha A, Legendre M, Carmona-Rivera C, Amin MA, Rabquer BJ. et al. DEK-targeting DNA aptamers as therapeutics for inflammatory arthritis. Nat Commun. 2017;8:14252
33. Jia Y, Jia R, Taledaohan A, Wang Y, Wang Y. Structure-Activity Relationship of PAD4 Inhibitors and Their Role in Tumor Immunotherapy. Pharmaceutics. 2024;16:335
34. Wu X, Chan L, Zhu D, Pang Y, Jin M, Wang Y. et al. Mitosis targeting in non-small lung cancer cells by inhibition of PAD4. Heliyon. 2024;10:e27313
35. Wang B, Su X, Zhang B, Pan S. GSK484, an inhibitor of peptidyl arginine deiminase 4, increases the radiosensitivity of colorectal cancer and inhibits neutrophil extracellular traps. J Gene Med. 2023;25:e3530
36. Erpenbeck L, Schön MP. Neutrophil extracellular traps: protagonists of cancer progression? Oncogene. 2017;36:2483-90
37. Guo W, Gong Q, Zong X, Wu D, Li Y, Xiao H. et al. GPR109A controls neutrophil extracellular traps formation and improve early sepsis by regulating ROS/PAD4/Cit-H3 signal axis. Exp Hematol Oncol. 2023;12:15
38. Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A, Papayannopoulos V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014;8:883-96
39. Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J. et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med. 2016;8:361ra138
40. Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S. et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010;584:3193-7
41. Bun M, Kawano M, Yamamoto G, Sakata M, Shimura K, Toda A. et al. G-CSF induces neutrophil extracellular traps formation and promotes ovarian cancer peritoneal dissemination. J Leukoc Biol. 2024;116:1157-68
42. Cho Y, Bukong TN, Tornai D, Babuta M, Vlachos IS, Kanata E. et al. Neutrophil extracellular traps contribute to liver damage and increase defective low-density neutrophils in alcohol-associated hepatitis. J Hepatol. 2023;78:28-44
43. Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. 2017;23:279-87
44. Honda M, Kubes P. Neutrophils and neutrophil extracellular traps in the liver and gastrointestinal system. Nat Rev Gastroenterol Hepatol. 2018;15:206-21
45. Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD. et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. 2012;18:1386-93
46. Rochael NC, Guimarães-Costa AB, Nascimento MT, DeSouza-Vieira TS, Oliveira MP, Garcia e Souza LF. et al. Classical ROS-dependent and early/rapid ROS-independent release of Neutrophil Extracellular Traps triggered by Leishmania parasites. Sci Rep. 2015;5:18302
47. Rayes RF, Mouhanna JG, Nicolau I, Bourdeau F, Giannias B, Rousseau S. et al. Primary tumors induce neutrophil extracellular traps with targetable metastasis promoting effects. JCI Insight. 2019;5:e128008
48. Wang Y, Liu F, Chen L, Fang C, Li S, Yuan S. et al. Neutrophil Extracellular Traps (NETs) Promote Non-Small Cell Lung Cancer Metastasis by Suppressing lncRNA MIR503HG to Activate the NF-κB/NLRP3 Inflammasome Pathway. Front Immunol. 2022;13:867516
49. Zeng J, Cheng Y, Xie W, Lin X, Ding C, Xu H. et al. Calcium-sensing receptor and NF-κB pathways in TN breast cancer contribute to cancer-induced cardiomyocyte damage via activating neutrophil extracellular traps formation. Cell Mol Life Sci. 2024;81:19
50. Miller-Ocuin JL, Liang X, Boone BA, Doerfler WR, Singhi AD, Tang D. et al. DNA released from neutrophil extracellular traps (NETs) activates pancreatic stellate cells and enhances pancreatic tumor growth. Oncoimmunology. 2019;8:e1605822
51. Leal AC, Mizurini DM, Gomes T, Rochael NC, Saraiva EM, Dias MS. et al. Tumor-Derived Exosomes Induce the Formation of Neutrophil Extracellular Traps: Implications For The Establishment of Cancer-Associated Thrombosis. Sci Rep. 2017;7:6438
52. Jung HS, Gu J, Kim JE, Nam Y, Song JW, Kim HK. Cancer cell-induced neutrophil extracellular traps promote both hypercoagulability and cancer progression. PLoS One. 2019;14:e0216055
53. Mousset A, Lecorgne E, Bourget I, Lopez P, Jenovai K, Cherfils-Vicini J. et al. Neutrophil extracellular traps formed during chemotherapy confer treatment resistance via TGF-β activation. Cancer Cell. 2023;41:757-75.e10
54. Jin W, Yin H, Li H, Yu XJ, Xu HX, Liu L. Neutrophil extracellular DNA traps promote pancreatic cancer cells migration and invasion by activating EGFR/ERK pathway. J Cell Mol Med. 2021;25:5443-56
55. Guan X, Lu Y, Zhu H, Yu S, Zhao W, Chi X. et al. The Crosstalk Between Cancer Cells and Neutrophils Enhances Hepatocellular Carcinoma Metastasis via Neutrophil Extracellular Traps-Associated Cathepsin G Component: A Potential Therapeutic Target. J Hepatocell Carcinoma. 2021;8:451-65
56. Xiao Y, Cong M, Li J, He D, Wu Q, Tian P. et al. Cathepsin C promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell. 2021;39:423-37.e7
57. Zhang Y, Chandra V, Riquelme Sanchez E, Dutta P, Quesada PR, Rakoski A. et al. Interleukin-17-induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J Exp Med. 2020;217:e20190354
58. Taifour T, Attalla SS, Zuo D, Gu Y, Sanguin-Gendreau V, Proud H. et al. The tumor-derived cytokine Chi3l1 induces neutrophil extracellular traps that promote T cell exclusion in triple-negative breast cancer. Immunity. 2023;56:2755-72.e8
59. Li J, Xia Y, Sun B, Zheng N, Li Y, Pang X. et al. Neutrophil extracellular traps induced by the hypoxic microenvironment in gastric cancer augment tumour growth. Cell Commun Signal. 2023;21:86
60. Sun X, Gui Y, Yang T, Chen L, Zhang Y, Yan L. et al. PD-L1(+) neutrophils induced NETs in malignant ascites is a potential biomarker in HCC. Cancer Immunol Immunother. 2024;73:254
61. Yang J, Jin L, Kim HS, Tian F, Yi Z, Bedi K. et al. KDM6A Loss Recruits Tumor-Associated Neutrophils and Promotes Neutrophil Extracellular Trap Formation in Pancreatic Cancer. Cancer Res. 2022;82:4247-60
62. Li Y, Yang Y, Gan T, Zhou J, Hu F, Hao N. et al. Extracellular RNAs from lung cancer cells activate epithelial cells and induce neutrophil extracellular traps. Int J Oncol. 2019;55:69-80
63. Hu Y, Wang H, Liu Y. NETosis: Sculpting tumor metastasis and immunotherapy. Immunol Rev. 2024;321:263-79
64. Yazdani HO, Roy E, Comerci AJ, van der Windt DJ, Zhang H, Huang H. et al. Neutrophil Extracellular Traps Drive Mitochondrial Homeostasis in Tumors to Augment Growth. Cancer Res. 2019;79:5626-39
65. Martins-Cardoso K, Almeida VH, Bagri KM, Rossi MID, Mermelstein CS, König S. et al. Neutrophil Extracellular Traps (NETs) Promote Pro-Metastatic Phenotype in Human Breast Cancer Cells through Epithelial-Mesenchymal Transition. Cancers (Basel). 2020;12:1542
66. Chen Q, Zhang L, Li X, Zhuo W. Neutrophil Extracellular Traps in Tumor Metastasis: Pathological Functions and Clinical Applications. Cancers (Basel). 2021;13:2832
67. Jiang ZZ, Peng ZP, Liu XC, Guo HF, Zhou MM, Jiang D. et al. Neutrophil extracellular traps induce tumor metastasis through dual effects on cancer and endothelial cells. Oncoimmunology. 2022;11:2052418
68. Xiong G, Chen Z, Liu Q, Peng F, Zhang C, Cheng M. et al. CD276 regulates the immune escape of esophageal squamous cell carcinoma through CXCL1-CXCR2 induced NETs. J Immunother Cancer. 2024;12:e008662
69. Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B. et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. 2013;123:3446-58
70. Adrover JM, McDowell SAC, He XY, Quail DF, Egeblad M. NETworking with cancer: The bidirectional interplay between cancer and neutrophil extracellular traps. Cancer Cell. 2023;41:505-26
71. Berger-Achituv S, Brinkmann V, Abed UA, Kühn LI, Ben-Ezra J, Elhasid R. et al. A proposed role for neutrophil extracellular traps in cancer immunoediting. Front Immunol. 2013;4:48
72. Liu Y, Liu L. The pro-tumor effect and the anti-tumor effect of neutrophils extracellular traps. Biosci Trends. 2020;13:469-75
73. Schedel F, Mayer-Hain S, Pappelbaum KI, Metze D, Stock M, Goerge T. et al. Evidence and impact of neutrophil extracellular traps in malignant melanoma. Pigment Cell Melanoma Res. 2020;33:63-73
74. Chan YT, Tan HY, Lu Y, Zhang C, Cheng CS, Wu J. et al. Pancreatic melatonin enhances anti-tumor immunity in pancreatic adenocarcinoma through regulating tumor-associated neutrophils infiltration and NETosis. Acta Pharm Sin B. 2023;13:1554-67
75. Zhang Y, Wang Z, Lu Y, Sanchez DJ, Li J, Wang L. et al. Region-Specific CD16(+) Neutrophils Promote Colorectal Cancer Progression by Inhibiting Natural Killer Cells. Adv Sci (Weinh). 2024;11:e2403414
76. Peng YP, Zhang JJ, Liang WB, Tu M, Lu ZP, Wei JS. et al. Elevation of MMP-9 and IDO induced by pancreatic cancer cells mediates natural killer cell dysfunction. BMC Cancer. 2014;14:738
77. Cheng Y, Gong Y, Chen X, Zhang Q, Zhang X, He Y. et al. Injectable adhesive hemostatic gel with tumor acidity neutralizer and neutrophil extracellular traps lyase for enhancing adoptive NK cell therapy prevents post-resection recurrence of hepatocellular carcinoma. Biomaterials. 2022;284:121506
78. Shen XT, Xie SZ, Xu J, Yang LY, Qin LX. Pan-Cancer Analysis Reveals a Distinct Neutrophil Extracellular Trap-Associated Regulatory Pattern. Front Immunol. 2022;13:798022
79. Li Y, Liu F, Cai Q, Deng L, Ouyang Q, Zhang XH. et al. Invasion and metastasis in cancer: molecular insights and therapeutic targets. Signal Transduct Target Ther. 2025;10:57
80. Zeng F, Shao Y, Wu J, Luo J, Yue Y, Shen Y. et al. Tumor metastasis and recurrence: The role of perioperative NETosis. Cancer Lett. 2024;611:217413
81. Zhou H, Zhu C, Zhao Q, Ni J, Zhang H, Yang G. et al. Wrecking neutrophil extracellular traps and antagonizing cancer-associated neurotransmitters by interpenetrating network hydrogels prevent postsurgical cancer relapse and metastases. Bioact Mater. 2024;39:14-24
82. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME. et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. 2018;361:eaao4227
83. Kaltenmeier C, Yazdani HO, Morder K, Geller DA, Simmons RL, Tohme S. Neutrophil Extracellular Traps Promote T Cell Exhaustion in the Tumor Microenvironment. Front Immunol. 2021;12:785222
84. Cui C, Chakraborty K, Tang XA, Zhou G, Schoenfelt KQ, Becker KM. et al. Neutrophil elastase selectively kills cancer cells and attenuates tumorigenesis. Cell. 2021;184:3163-77.e21
85. Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE. et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med. 2010;16:219-23
86. Huang H, Zhang H, Onuma AE, Tsung A. Neutrophil Elastase and Neutrophil Extracellular Traps in the Tumor Microenvironment. Adv Exp Med Biol. 2020;1263:13-23
87. Lerman I, Garcia-Hernandez ML, Rangel-Moreno J, Chiriboga L, Pan C, Nastiuk KL. et al. Infiltrating Myeloid Cells Exert Protumorigenic Actions via Neutrophil Elastase. Mol Cancer Res. 2017;15:1138-52
88. Wada Y, Yoshida K, Tsutani Y, Shigematsu H, Oeda M, Sanada Y. et al. Neutrophil elastase induces cell proliferation and migration by the release of TGF-alpha, PDGF and VEGF in esophageal cell lines. Oncol Rep. 2007;17:161-7
89. Bekes EM, Schweighofer B, Kupriyanova TA, Zajac E, Ardi VC, Quigley JP. et al. Tumor-recruited neutrophils and neutrophil TIMP-free MMP-9 regulate coordinately the levels of tumor angiogenesis and efficiency of malignant cell intravasation. Am J Pathol. 2011;179:1455-70
90. Liu Z, Chen J, Ren Y, Liu S, Ba Y, Zuo A. et al. Multi-stage mechanisms of tumor metastasis and therapeutic strategies. Signal Transduct Target Ther. 2024;9:270
91. Nguyen TT, Ding D, Wolter WR, Pérez RL, Champion MM, Mahasenan KV. et al. Validation of Matrix Metalloproteinase-9 (MMP-9) as a Novel Target for Treatment of Diabetic Foot Ulcers in Humans and Discovery of a Potent and Selective Small-Molecule MMP-9 Inhibitor That Accelerates Healing. J Med Chem. 2018;61:8825-37
92. Yoshikawa AK, Yamaguchi K, Muro K, Takashima A, Ichimura T, Sakai D. et al. Safety and tolerability of andecaliximab as monotherapy and in combination with an anti-PD-1 antibody in Japanese patients with gastric or gastroesophageal junction adenocarcinoma: a phase 1b study. J Immunother Cancer. 2022;10:e003518
93. Yang Y, Song S, Li S, Kang J, Li Y, Zhao N. et al. GATA4 regulates the transcription of MMP9 to suppress the invasion and migration of breast cancer cells via HDAC1-mediated p65 deacetylation. Cell Death Dis. 2024;15:289
94. Kochumon S, Al-Sayyar A, Jacob T, Bahman F, Akhter N, Wilson A. et al. TGF-β and TNF-α interaction promotes the expression of MMP-9 through H3K36 dimethylation: implications in breast cancer metastasis. Front Immunol. 2024;15:1430187
95. Shin Y, Kim S, Liang G, An W. MMP-9-dependent proteolysis of the histone H3 N-terminal tail: a critical epigenetic step in driving oncogenic transcription and colon tumorigenesis. Mol Oncol. 2024;18:2001-19
96. Kowalczyk A, Nisiewicz MK, Bamburowicz-Klimkowska M, Kasprzak A, Ruzycka-Ayoush M, Koszytkowska-Stawińska M. et al. Effective voltammetric tool for simultaneous detection of MMP-1, MMP-2, and MMP-9; important non-small cell lung cancer biomarkers. Biosens Bioelectron. 2023;229:115212
97. Gou S, Wang G, Zou Y, Geng W, He T, Qin Z. et al. Non-Pore Dependent and MMP-9 Responsive Gelatin/Silk Fibroin Composite Microparticles as Universal Delivery Platform for Inhaled Treatment of Lung Cancer. Adv Mater. 2023;35:e2303718
98. Cox TR. The matrix in cancer. Nat Rev Cancer. 2021;21:217-38
99. Fu Z, Akula S, Thorpe M, Hellman L. Potent and Broad but not Unselective Cleavage of Cytokines and Chemokines by Human Neutrophil Elastase and Proteinase 3. Int J Mol Sci. 2020;21:651
100. Burster T, Knippschild U, Molnár F, Zhanapiya A. Cathepsin G and its Dichotomous Role in Modulating Levels of MHC Class I Molecules. Arch Immunol Ther Exp (Warsz). 2020;68:25
101. Morimoto-Kamata R, Yui S. Insulin-like growth factor-1 signaling is responsible for cathepsin G-induced aggregation of breast cancer MCF-7 cells. Cancer Sci. 2017;108:1574-83
102. Song Y, Jin D, Chen J, Luo Z, Chen G, Yang Y. et al. Identification of an immune-related long non-coding RNA signature and nomogram as prognostic target for muscle-invasive bladder cancer. Aging (Albany NY). 2020;12:12051-73
103. Chan S, Wang X, Wang Z, Du Y, Zuo X, Chen J. et al. CTSG Suppresses Colorectal Cancer Progression through Negative Regulation of Akt/mTOR/Bcl2 Signaling Pathway. Int J Biol Sci. 2023;19:2220-33
104. Tsourouktsoglou TD, Warnatsch A, Ioannou M, Hoving D, Wang Q, Papayannopoulos V. Histones, DNA, and Citrullination Promote Neutrophil Extracellular Trap Inflammation by Regulating the Localization and Activation of TLR4. Cell Rep. 2020;31:107602
105. Ioannou M, Hoving D, Aramburu IV, Temkin MI, De Vasconcelos NM, Tsourouktsoglou TD. et al. Microbe capture by splenic macrophages triggers sepsis via T cell-death-dependent neutrophil lifespan shortening. Nat Commun. 2022;13:4658
106. Najmeh S, Cools-Lartigue J, Rayes RF, Gowing S, Vourtzoumis P, Bourdeau F. et al. Neutrophil extracellular traps sequester circulating tumor cells via β1-integrin mediated interactions. Int J Cancer. 2017;140:2321-30
107. Tang Q, Liang B, Zhang L, Li X, Li H, Jing W. et al. Enhanced CHOLESTEROL biosynthesis promotes breast cancer metastasis via modulating CCDC25 expression and neutrophil extracellular traps formation. Sci Rep. 2022;12:17350
108. Hedrick CC, Malanchi I. Neutrophils in cancer: heterogeneous and multifaceted. Nat Rev Immunol. 2022;22:173-87
109. Fang Q, Stehr AM, Naschberger E, Knopf J, Herrmann M, Stürzl M. No NETs no TIME: Crosstalk between neutrophil extracellular traps and the tumor immune microenvironment. Front Immunol. 2022;13:1075260
110. Shi X, Wang X, Yao W, Shi D, Shao X, Lu Z. et al. Mechanism insights and therapeutic intervention of tumor metastasis: latest developments and perspectives. Signal Transduct Target Ther. 2024;9:192
111. Xie Y, Liu F, Wu Y, Zhu Y, Jiang Y, Wu Q. et al. Inflammation in cancer: therapeutic opportunities from new insights. Mol Cancer. 2025;24:51
112. Wang Y, Jia J, Wang F, Fang Y, Yang Y, Zhou Q. et al. Pre-metastatic niche: formation, characteristics and therapeutic implication. Signal Transduct Target Ther. 2024;9:236
113. Di Russo S, Liberati FR, Riva A, Di Fonzo F, Macone A, Giardina G. et al. Beyond the barrier: the immune-inspired pathways of tumor extravasation. Cell Commun Signal. 2024;22:104
114. Zha C, Meng X, Li L, Mi S, Qian D, Li Z. et al. Neutrophil extracellular traps mediate the crosstalk between glioma progression and the tumor microenvironment via the HMGB1/RAGE/IL-8 axis. Cancer Biol Med. 2020;17:154-68
115. Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, Mowen K. et al. Neutrophil Extracellular Traps Promote the Development and Progression of Liver Metastases after Surgical Stress. Cancer Res. 2016;76:1367-80
116. Onuma AE, Zhang H, Gil L, Huang H, Tsung A. Surgical Stress Promotes Tumor Progression: A Focus on the Impact of the Immune Response. J Clin Med. 2020;9:4096
117. Ren J, He J, Zhang H, Xia Y, Hu Z, Loughran P. et al. Platelet TLR4-ERK5 Axis Facilitates NET-Mediated Capturing of Circulating Tumor Cells and Distant Metastasis after Surgical Stress. Cancer Res. 2021;81:2373-85
118. Shibue T, Brooks MW, Weinberg RA. An integrin-linked machinery of cytoskeletal regulation that enables experimental tumor initiation and metastatic colonization. Cancer Cell. 2013;24:481-98
119. Radisky ES. Extracellular proteolysis in cancer: Proteases, substrates, and mechanisms in tumor progression and metastasis. J Biol Chem. 2024;300:107347
120. McDowell SAC, Luo RBE, Arabzadeh A, Doré S, Bennett NC, Breton V. et al. Neutrophil oxidative stress mediates obesity-associated vascular dysfunction and metastatic transmigration. Nat Cancer. 2021;2:545-62
121. Lei G, Zhuang L, Gan B. The roles of ferroptosis in cancer: Tumor suppression, tumor microenvironment, and therapeutic interventions. Cancer Cell. 2024;42:513-34
122. Wu J, Dong W, Pan Y, Wang J, Wu M, Yu Y. Crosstalk between gut microbiota and metastasis in colorectal cancer: implication of neutrophil extracellular traps. Front Immunol. 2023;14:1296783
123. Castaño M, Tomás-Pérez S, González-Cantó E, Aghababyan C, Mascarós-Martínez A, Santonja N. et al. Neutrophil Extracellular Traps and Cancer: Trapping Our Attention with Their Involvement in Ovarian Cancer. Int J Mol Sci. 2023;24:5995
124. Wculek SK, Malanchi I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature. 2015;528:413-7
125. Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022;12:31-46
126. Aldabbous L, Abdul-Salam V, McKinnon T, Duluc L, Pepke-Zaba J, Southwood M. et al. Neutrophil Extracellular Traps Promote Angiogenesis: Evidence From Vascular Pathology in Pulmonary Hypertension. Arterioscler Thromb Vasc Biol. 2016;36:2078-87
127. Dosch AR, Martos MP, Singh S, Kodia K, Merchant NB, Nagathihalli NS. The Role of Myeloid Cells on the Development of Hepatic Metastases in Gastrointestinal Cancer. Gastro Hep Adv. 2025;4:100538
128. Liao CY, Li G, Kang FP, Lin CF, Xie CK, Wu YD. et al. Necroptosis enhances 'don't eat me' signal and induces macrophage extracellular traps to promote pancreatic cancer liver metastasis. Nat Commun. 2024;15:6043
129. Wieland E, Rodriguez-Vita J, Liebler SS, Mogler C, Moll I, Herberich SE. et al. Endothelial Notch1 Activity Facilitates Metastasis. Cancer Cell. 2017;31:355-67
130. Yang S, Sun B, Li J, Li N, Zhang A, Zhang X. et al. Neutrophil extracellular traps promote angiogenesis in gastric cancer. Cell Commun Signal. 2023;21:176
131. Binet F, Cagnone G, Crespo-Garcia S, Hata M, Neault M, Dejda A. et al. Neutrophil extracellular traps target senescent vasculature for tissue remodeling in retinopathy. Science. 2020;369:eaay5356
132. Zhu T, Zou X, Yang C, Li L, Wang B, Li R. et al. Neutrophil extracellular traps promote gastric cancer metastasis by inducing epithelial-mesenchymal transition. Int J Mol Med. 2021;48:127
133. Herre M, Cedervall J, Mackman N, Olsson AK. Neutrophil extracellular traps in the pathology of cancer and other inflammatory diseases. Physiol Rev. 2023;103:277-312
134. Boone BA, Murthy P, Miller-Ocuin J, Doerfler WR, Ellis JT, Liang X. et al. Chloroquine reduces hypercoagulability in pancreatic cancer through inhibition of neutrophil extracellular traps. BMC Cancer. 2018;18:678
135. Seo JD, Gu JY, Jung HS, Kim YJ, Kim HK. Contact System Activation and Neutrophil Extracellular Trap Markers: Risk Factors for Portal Vein Thrombosis in Patients With Hepatocellular Carcinoma. Clin Appl Thromb Hemost. 2019;25:1076029618825310
136. Wienkamp AK, Erpenbeck L, Rossaint J. Platelets in the NETworks interweaving inflammation and thrombosis. Front Immunol. 2022;13:953129
137. de Bont CM, Boelens WC, Pruijn GJM. NETosis, complement, and coagulation: a triangular relationship. Cell Mol Immunol. 2019;16:19-27
138. Ngo AT, Skidmore A, Oberg J, Yarovoi I, Sarkar A, Levine N. et al. Platelet factor 4 limits neutrophil extracellular trap- and cell-free DNA-induced thrombogenicity and endothelial injury. JCI Insight. 2023;8:e171054
139. Wang Y, Luo L, Braun O, Westman J, Madhi R, Herwald H. et al. Neutrophil extracellular trap-microparticle complexes enhance thrombin generation via the intrinsic pathway of coagulation in mice. Sci Rep. 2018;8:4020
140. Wu X, You D, Pan M, Weng M, Xie Q, Guan Y. et al. Knockout of the C3a receptor protects against renal ischemia reperfusion injury by reduction of NETs formation. Cell Mol Life Sci. 2023;80:322
141. Guglietta S, Chiavelli A, Zagato E, Krieg C, Gandini S, Ravenda PS. et al. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat Commun. 2016;7:11037
142. Chen Y, Li X, Lin X, Liang H, Liu X, Zhang X. et al. Complement C5a induces the generation of neutrophil extracellular traps by inhibiting mitochondrial STAT3 to promote the development of arterial thrombosis. Thromb J. 2022;20:24
143. Leffler J, Martin M, Gullstrand B, Tydén H, Lood C, Truedsson L. et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol. 2012;188:3522-31
144. Ribon M, Seninet S, Mussard J, Sebbag M, Clavel C, Serre G. et al. Neutrophil extracellular traps exert both pro- and anti-inflammatory actions in rheumatoid arthritis that are modulated by C1q and LL-37. J Autoimmun. 2019;98:122-31
145. Schneider AE, Sándor N, Kárpáti É, Józsi M. Complement factor H modulates the activation of human neutrophil granulocytes and the generation of neutrophil extracellular traps. Mol Immunol. 2016;72:37-48
146. Kárpáti É, Kremlitzka M, Sándor N, Hajnal D, Schneider AE, Józsi M. Complement Factor H Family Proteins Modulate Monocyte and Neutrophil Granulocyte Functions. Front Immunol. 2021;12:660852
147. Burkard P, Schonhart C, Vögtle T, Köhler D, Tang L, Johnson D. et al. A key role for platelet GPVI in neutrophil recruitment, migration, and NETosis in the early stages of acute lung injury. Blood. 2023;142:1463-77
148. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood. 2015;126:242-6
149. Manfredi AA, Ramirez GA, Godino C, Capobianco A, Monno A, Franchini S. et al. Platelet Phagocytosis via P-selectin Glycoprotein Ligand 1 and Accumulation of Microparticles in Systemic Sclerosis. Arthritis Rheumatol. 2022;74:318-28
150. Stark K, Philippi V, Stockhausen S, Busse J, Antonelli A, Miller M. et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood. 2016;128:2435-49
151. Zucoloto AZ, Jenne CN. Platelet-Neutrophil Interplay: Insights Into Neutrophil Extracellular Trap (NET)-Driven Coagulation in Infection. Front Cardiovasc Med. 2019;6:85
152. Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP. et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One. 2012;7:e32366
153. Carmona-Rivera C, Zhao W, Yalavarthi S, Kaplan MJ. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann Rheum Dis. 2015;74:1417-24
154. Meteva D, Vinci R, Seppelt C, Abdelwahed YS, Pedicino D, Nelles G. et al. Toll-like receptor 2, hyaluronan, and neutrophils play a key role in plaque erosion: the OPTICO-ACS study. Eur Heart J. 2023;44:3892-907
155. Folco EJ, Mawson TL, Vromman A, Bernardes-Souza B, Franck G, Persson O. et al. Neutrophil Extracellular Traps Induce Endothelial Cell Activation and Tissue Factor Production Through Interleukin-1α and Cathepsin G. Arterioscler Thromb Vasc Biol. 2018;38:1901-12
156. Blanco LP, Wang X, Carlucci PM, Torres-Ruiz JJ, Romo-Tena J, Sun HW. et al. RNA Externalized by Neutrophil Extracellular Traps Promotes Inflammatory Pathways in Endothelial Cells. Arthritis Rheumatol. 2021;73:2282-92
157. Yang J, Wu Z, Long Q, Huang J, Hong T, Liu W. et al. Insights Into Immunothrombosis: The Interplay Among Neutrophil Extracellular Trap, von Willebrand Factor, and ADAMTS13. Front Immunol. 2020;11:610696
158. Byrnes JR, Wolberg AS. Red blood cells in thrombosis. Blood. 2017;130:1795-9
159. Chilingaryan Z, Deshmukh T, Leung HHL, Perdomo J, Emerson P, Kurup R. et al. Erythrocyte interaction with neutrophil extracellular traps in coronary artery thrombosis following myocardial infarction. Pathology. 2022;54:87-94
160. Brill A, Fuchs TA, Savchenko AS, Thomas GM, Martinod K, De Meyer SF. et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost. 2012;10:136-44
161. Kordbacheh F, O'Meara CH, Coupland LA, Lelliott PM, Parish CR. Extracellular histones induce erythrocyte fragility and anemia. Blood. 2017;130:2884-8
162. Knackstedt SL, Georgiadou A, Apel F, Abu-Abed U, Moxon CA, Cunnington AJ. et al. Neutrophil extracellular traps drive inflammatory pathogenesis in malaria. Sci Immunol. 2019;4:eaaw0336
163. Rodrigues DAS, Prestes EB, Gama AMS, Silva LS, Pinheiro AAS, Ribeiro JMC. et al. CXCR4 and MIF are required for neutrophil extracellular trap release triggered by Plasmodium-infected erythrocytes. PLoS Pathog. 2020;16:e1008230
164. Chen C, Wang Z, Ding Y, Qin Y. Tumor microenvironment-mediated immune evasion in hepatocellular carcinoma. Front Immunol. 2023;14:1133308
165. Cheng X, Zhang H, Hamad A, Huang H, Tsung A. Surgery-mediated tumor-promoting effects on the immune microenvironment. Semin Cancer Biol. 2022;86:408-19
166. Shi Y, Wu D, Wang Y, Shao Y, Zeng F, Zhou D. et al. Treg and neutrophil extracellular trap interaction contributes to the development of immunosuppression in sepsis. JCI Insight. 2024;9:e180132
167. Zhang H, Wang Y, Onuma A, He J, Wang H, Xia Y. et al. Neutrophils Extracellular Traps Inhibition Improves PD-1 Blockade Immunotherapy in Colorectal Cancer. Cancers (Basel). 2021;13:5333
168. Feng C, Li Y, Tai Y, Zhang W, Wang H, Lian S. et al. A neutrophil extracellular traps-related classification predicts prognosis and response to immunotherapy in colon cancer. Sci Rep. 2023;13:19297
169. Shen XT, Xie SZ, Zheng X, Zou TT, Hu BY, Xu J. et al. Cirrhotic-extracellular matrix attenuates aPD-1 treatment response by initiating immunosuppressive neutrophil extracellular traps formation in hepatocellular carcinoma. Exp Hematol Oncol. 2024;13:20
170. Li H, Li J, Bai Z, Yan S, Li J. Collagen-induced DDR1 upregulates CXCL5 to promote neutrophil extracellular traps formation and Treg infiltration in breast cancer. Int Immunopharmacol. 2023;120:110235
171. Yu C, Zhou G, Shi Z, Yu L, Zhou X. TREM1 facilitates the development of gastric cancer through regulating neutrophil extracellular traps-mediated macrophage polarization. Dig Liver Dis. 2024;56:1237-47
172. Zhang L, Yi H, Chen J, Li H, Luo Y, Cheng T. et al. Neutrophil Extracellular Traps Facilitate A549 Cell Invasion and Migration in a Macrophage-Maintained Inflammatory Microenvironment. Biomed Res Int. 2022;2022:8316525
173. Wilson AS, Randall KL, Pettitt JA, Ellyard JI, Blumenthal A, Enders A. et al. Neutrophil extracellular traps and their histones promote Th17 cell differentiation directly via TLR2. Nat Commun. 2022;13:528
174. Chen W, Chen H, Yang ZT, Mao EQ, Chen Y, Chen EZ. Free fatty acids-induced neutrophil extracellular traps lead to dendritic cells activation and T cell differentiation in acute lung injury. Aging (Albany NY). 2021;13:26148-60
175. Heras-Murillo I, Adán-Barrientos I, Galán M, Wculek SK, Sancho D. Dendritic cells as orchestrators of anticancer immunity and immunotherapy. Nat Rev Clin Oncol. 2024;21:257-77
176. Tripodo C, Bassani B, Jachetti E, Cancila V, Chiodoni C, Portararo P. et al. Neutrophil extracellular traps arm DC vaccination against NPM-mutant myeloproliferation. Elife. 2022;11:e69257
177. Donis-Maturano L, Sánchez-Torres LE, Cerbulo-Vázquez A, Chacón-Salinas R, García-Romo GS, Orozco-Uribe MC. et al. Prolonged exposure to neutrophil extracellular traps can induce mitochondrial damage in macrophages and dendritic cells. Springerplus. 2015;4:161
178. Kyrysyuk O, Wucherpfennig KW. Designing Cancer Immunotherapies That Engage T Cells and NK Cells. Annu Rev Immunol. 2023;41:17-38
179. Lei Q, Zhen S, Zhang L, Zhao Q, Yang L, Zhang Y. A2AR-mediated CXCL5 upregulation on macrophages promotes NSCLC progression via NETosis. Cancer Immunol Immunother. 2024;73:108
180. Song M, Zhang C, Cheng S, Ouyang D, Ping Y, Yang J. et al. DNA of Neutrophil Extracellular Traps Binds TMCO6 to Impair CD8+ T-cell Immunity in Hepatocellular Carcinoma. Cancer Res. 2024;84:1613-29
181. Chen D, Li Q, Liang H, Huang L, Zhou H, Zheng X. et al. Exenatide enhanced the antitumor efficacy on PD-1 blockade by the attenuation of neutrophil extracellular traps. Biochem Biophys Res Commun. 2022;619:97-103
182. Chen D, Liang H, Huang L, Zhou H, Wang Z. Liraglutide enhances the effect of checkpoint blockade in lung and liver cancers through the inhibition of neutrophil extracellular traps. FEBS Open Bio. 2024;14:1365-77
183. Abel AM, Yang C, Thakar MS, Malarkannan S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front Immunol. 2018;9:1869
184. Zhang H, Wang Y, Qu M, Li W, Wu D, Cata JP. et al. Neutrophil, neutrophil extracellular traps and endothelial cell dysfunction in sepsis. Clin Transl Med. 2023;13:e1170
185. Valayer A, Brea D, Lajoie L, Avezard L, Combes-Soia L, Labas V. et al. Neutrophils can disarm NK cell response through cleavage of NKp46. J Leukoc Biol. 2017;101:253-9
186. Yang S, Zou X, Li J, Yang H, Zhang A, Zhu Y. et al. Immunoregulation and clinical significance of neutrophils/NETs-ANGPT2 in tumor microenvironment of gastric cancer. Front Immunol. 2022;13:1010434
187. Sharma P, Garg N, Sharma A, Capalash N, Singh R. Nucleases of bacterial pathogens as virulence factors, therapeutic targets and diagnostic markers. Int J Med Microbiol. 2019;309:151354
188. Papayannopoulos V, Staab D, Zychlinsky A. Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving DNase therapy. PLoS One. 2011;6:e28526
189. Xia Y, He J, Zhang H, Wang H, Tetz G, Maguire CA. et al. AAV-mediated gene transfer of DNase I in the liver of mice with colorectal cancer reduces liver metastasis and restores local innate and adaptive immune response. Mol Oncol. 2020;14:2920-35
190. Ansari J, Vital SA, Yadav S, Gavins FNE. Regulating Neutrophil PAD4/NOX-Dependent Cerebrovasular Thromboinflammation. Int J Biol Sci. 2023;19:852-64
191. Li M, Lin C, Deng H, Strnad J, Bernabei L, Vogl DT. et al. A Novel Peptidylarginine Deiminase 4 (PAD4) Inhibitor BMS-P5 Blocks Formation of Neutrophil Extracellular Traps and Delays Progression of Multiple Myeloma. Mol Cancer Ther. 2020;19:1530-8
192. Mutua V, Gershwin LJ. A Review of Neutrophil Extracellular Traps (NETs) in Disease: Potential Anti-NETs Therapeutics. Clin Rev Allergy Immunol. 2021;61:194-211
193. Song S, Gui L, Feng Q, Taledaohan A, Li Y, Wang W. et al. TAT-Modified Gold Nanoparticles Enhance the Antitumor Activity of PAD4 Inhibitors. Int J Nanomedicine. 2020;15:6659-71
194. Jia Y, Taledaohan A, Jia R, Wang X, Jia Y, Liu J. et al. Chitosan nanomedicine containing RGD peptide and PAD4 inhibitor based on phenyl boronate coupling inhibition of primary tumor growth and lung metastasis. Biomed Pharmacother. 2023;168:115826
195. Sun S, Lv W, Li S, Zhang Q, He W, Min Z. et al. Smart Liposomal Nanocarrier Enhanced the Treatment of Ischemic Stroke through Neutrophil Extracellular Traps and Cyclic Guanosine Monophosphate-Adenosine Monophosphate Synthase-Stimulator of Interferon Genes (cGAS-STING) Pathway Inhibition of Ischemic Penumbra. ACS Nano. 2023;17:17845-57
196. Teijeira Á, Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A. et al. CXCR1 and CXCR2 Chemokine Receptor Agonists Produced by Tumors Induce Neutrophil Extracellular Traps that Interfere with Immune Cytotoxicity. Immunity. 2020;52:856-71.e8
197. Teijeira A, Garasa S, Ochoa MC, Villalba M, Olivera I, Cirella A. et al. IL8, Neutrophils, and NETs in a Collusion against Cancer Immunity and Immunotherapy. Clin Cancer Res. 2021;27:2383-93
198. O'Sullivan KM, Holdsworth SR. Neutrophil Extracellular Traps: A Potential Therapeutic Target in MPO-ANCA Associated Vasculitis? Front Immunol. 2021;12:635188
199. Zhao H, Liang Y, Sun C, Zhai Y, Li X, Jiang M. et al. Dihydrotanshinone I Inhibits the Lung Metastasis of Breast Cancer by Suppressing Neutrophil Extracellular Traps Formation. Int J Mol Sci. 2022;23:15180
200. Pan Z, Xie X, Chen Y, Pan S, Wu Z, Yang C. et al. Huang Qin Decoction inhibits the initiation of experimental colitis associated carcinogenesis by controlling the PAD4 dependent NETs. Phytomedicine. 2022;107:154454
201. Haute GV, Luft C, Pedrazza L, Donadio MVF, de Oliveira JR. Octyl gallate decrease lymphocyte activation and regulates neutrophil extracellular traps release. Mol Biol Rep. 2022;49:1593-9
202. Bystrzycka W, Manda-Handzlik A, Sieczkowska S, Moskalik A, Demkow U, Ciepiela O. Azithromycin and Chloramphenicol Diminish Neutrophil Extracellular Traps (NETs) Release. Int J Mol Sci. 2017;18:2666
203. Zhao Z, Pan Z, Zhang S, Ma G, Zhang W, Song J. et al. Neutrophil extracellular traps: A novel target for the treatment of stroke. Pharmacol Ther. 2023;241:108328
204. Tan C, Aziz M, Wang P. The vitals of NETs. J Leukoc Biol. 2021;110:797-808
205. Wang B, Hu S, Teng Y, Chen J, Wang H, Xu Y. et al. Current advance of nanotechnology in diagnosis and treatment for malignant tumors. Signal Transduct Target Ther. 2024;9:200
206. Yin H, Lu H, Xiong Y, Ye L, Teng C, Cao X. et al. Tumor-Associated Neutrophil Extracellular Traps Regulating Nanocarrier-Enhanced Inhibition of Malignant Tumor Growth and Distant Metastasis. ACS Appl Mater Interfaces. 2021;13:59683-94
207. Li J, Burgess DJ. Nanomedicine-based drug delivery towards tumor biological and immunological microenvironment. Acta Pharm Sin B. 2020;10:2110-24
208. Peng S, Xiao F, Chen M, Gao H. Tumor-Microenvironment-Responsive Nanomedicine for Enhanced Cancer Immunotherapy. Adv Sci (Weinh). 2022;9:e2103836
209. Wang Y, Liu C, Fang C, Peng Q, Qin W, Yan X. et al. Engineered Cancer Nanovaccines: A New Frontier in Cancer Therapy. Nanomicro Lett. 2024;17:30
210. Muñoz LE, Bilyy R, Biermann MH, Kienhöfer D, Maueröder C, Hahn J. et al. Nanoparticles size-dependently initiate self-limiting NETosis-driven inflammation. Proc Natl Acad Sci U S A. 2016;113:E5856-e65
211. Lin X, Jiao R, Cui H, Yan X, Zhang K. Physiochemically and Genetically Engineered Bacteria: Instructive Design Principles and Diverse Applications. Adv Sci (Weinh). 2024;11:e2403156
Author contact
![]() Corresponding authors: Hao Zhang, M.D., Ph.D.: Department of Anesthesiology, Zhongshan Hospital, Fudan University, 180# Feng-Lin Road, Shanghai, 200032, China; E-mail: zhang.haosh.cn. Changhong Miao, M.D., Ph.D.: Department of Anesthesiology, Zhongshan Hospital, Fudan University, 180# Feng-Lin Road, Shanghai, China; E-mail: miaochanghong_zscom.
Corresponding authors: Hao Zhang, M.D., Ph.D.: Department of Anesthesiology, Zhongshan Hospital, Fudan University, 180# Feng-Lin Road, Shanghai, 200032, China; E-mail: zhang.haosh.cn. Changhong Miao, M.D., Ph.D.: Department of Anesthesiology, Zhongshan Hospital, Fudan University, 180# Feng-Lin Road, Shanghai, China; E-mail: miaochanghong_zscom.