Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Design and Development of PFC...
3. Manufacturing of PFC NEs for...
4. Potential Challenges in the...
5. What's next? Targeted...
6. Conclusions and Future Outlook
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2025; 15(7):3013-3034. doi:10.7150/thno.103820 This issue Cite
Review
Perfluorocarbon nanoemulsions in drug delivery: design, development, and manufacturing
Riddhi Vichare, Jelena Janjic ![]()
School of Pharmacy, Graduate School of Pharmaceutical Sciences, Duquesne University, Pittsburgh, PA 15282, USA.
Received 2024-9-18; Accepted 2025-1-30; Published 2025-2-10
Abstract

Perfluorocarbons (PFCs) are formulated into kinetically stable nanoemulsions (NEs) having a droplet diameter less than 500 nm for biomedical applications. PFC NEs are developed for various applications, such as 19F magnetic resonance imaging tracers, ultrasound contrast agents, and oxygen carriers. They are an attractive platform for theranostic development, as PFC NEs can be designed to deliver a multitude of therapeutics, from small molecules to biologics. Although many reviews have been published on designing PFC NEs as imaging agents or oxygen carriers, there is no comprehensive review of the formulation strategies and manufacturing of PFC NEs as drug delivery platform. From a “formulator's perspective,” the presented review addresses this gap as it describes the selection of formulation components such as the PFCs, surfactants, and hydrocarbon oils and provides an overview of manufacturing techniques. In this review, we will also identify potential shortcomings in the published literature on the manufacturing of PFC NEs. We will discuss the need for the implementing Quality by Design (QbD) approach in the manufacturing of PFC NEs to achieve scalability and necessary quality control for supporting both preclinical and clinical applications. Finally, we will review different surface-conjugation strategies for developing targeted PFC NEs. Overall, this article aims to increase understanding of PFC NEs designed for delivering drugs, including both small molecules and biologics.
Keywords: Perfluorocarbons, Nanoemulsions, Drug delivery, Theranostic, Targeted drug delivery, Manufacturing, Quality by Design
1. Introduction
1.1 Brief Overview of Perfluorocarbon Nanoemulsions (PFC NEs)
Perfluorocarbons (PFCs) were initially synthesized as inert solvents for separating uranium isotopes [1]. It was not until 1966, when Clark and Gollan, in their 'liquid breathing' experiment, delineated the use of oxygen-saturated PFC liquids as an alternative respiratory medium [2]. PFCs are colorless and odorless synthetic compounds in which all the hydrogen atoms on a saturated linear or cyclic carbon backbone are substituted with fluorine atoms, with a few exceptions where heteroatoms are present in the molecular structure [3]. PFCs are chemically inert due to their electronic structure and the spatial arrangement of fluorine atoms on the C-C backbone. Fluorine substitution changes the physicochemical properties of the hydrocarbon bond due to its high electronegativity, high ionization potential, and low polarizability compared to the hydrogen atom. The C-F bond is a stronger covalent single bond compared to the C-H bond, requiring roughly 530 kJmol-1 of energy to break the bond. Additionally, fluorine substitutions force the C-C backbone to adopt a helical configuration, deviating from the planar zigzag configuration observed in the presence of hydrogen atoms [3, 4]. The large trans/gauche conversion energy barrier (4.6 kJmol-1) imparts rigidity to the structure, sterically shielding the C-C bonds from chemical attacks [5]. Early on, the scientific community recognized the need to emulsify PFCs for clinical applications involving parenteral administration to prevent fatal vascular embolism [2, 6]. In 1967, Solviter was the first to report the emulsification of PFCs in water using bovine serum albumin through sonication (droplet diameter: 2-3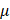 m) for perfusing isolated rat brains [7]. Around the 1980s, the HIV-virus-contaminated donor blood epidemic further intensified research on PFCs as artificial blood substitutes owing to their high oxygen-carrying capacity [8]. Although termed blood substitutes, PFC emulsions did not perform all the functions of blood, like coagulation and nutrient transport. In 1989, Fluosol-DA® (droplet diameter < 200 nm) became the first FDA-approved oxygen-carrying PFC emulsion for perfusing coronary arteries, which was manufactured using the high-pressure homogenization technique [9]. Unfortunately, Fluosol-DA® was withdrawn from the market mainly due to its poor stability and low oxygen-carrying capacity. In parallel, a paper discussing PFCs as tracer agents for 19F magnetic resonance imaging (MRI) was published [10]. The 19F atom has a negligible biological abundance, and therefore fluorinated materials can be non-invasively detected in vivo using 19F MRI without background [11]. Indeed, the late 20th century was a “golden period” for PFCs, officially marking their entry into the biomedical field.
m) for perfusing isolated rat brains [7]. Around the 1980s, the HIV-virus-contaminated donor blood epidemic further intensified research on PFCs as artificial blood substitutes owing to their high oxygen-carrying capacity [8]. Although termed blood substitutes, PFC emulsions did not perform all the functions of blood, like coagulation and nutrient transport. In 1989, Fluosol-DA® (droplet diameter < 200 nm) became the first FDA-approved oxygen-carrying PFC emulsion for perfusing coronary arteries, which was manufactured using the high-pressure homogenization technique [9]. Unfortunately, Fluosol-DA® was withdrawn from the market mainly due to its poor stability and low oxygen-carrying capacity. In parallel, a paper discussing PFCs as tracer agents for 19F magnetic resonance imaging (MRI) was published [10]. The 19F atom has a negligible biological abundance, and therefore fluorinated materials can be non-invasively detected in vivo using 19F MRI without background [11]. Indeed, the late 20th century was a “golden period” for PFCs, officially marking their entry into the biomedical field.
During the same timeline, emulsification technology extended from the food industry to the pharmaceutical sector. Consequently, a metric prefix terminology, nanoemulsions (NEs), was introduced in 1996 for describing droplets having a diameter in the nanometer range [12, 13]. NEs are kinetically stable colloidal dispersions of two immiscible liquids (oil and water), where a high oil fraction is stabilized with a low surfactant concentration (5-10% w/v). This is distinct from thermodynamically stable microemulsions, which are formulated at a higher surfactant-to-oil ratio (SOR) [14]. There is no consensus within the field over the upper limit of droplet size considered acceptable for NEs. The droplet size of NEs, particularly PFC NEs, formulated for clinical applications ranges between 100 nm and 500 nm [15-19]. Through continued research efforts, PFC NEs are being designed and formulated to serve as 19F MRI tracers, ultrasound contrast agents, ex vivo cell labeling agents, probes for intracellular pH measurements, and as oxygen carriers in regenerative medicine and oncology to reverse tissue hypoxia, which are comprehensively covered elsewhere [20-26].
Recently, research interest has shifted towards designing PFC NEs as drug delivery platform, with their imaging or oxygen-carrying capabilities serving as added functionalities. This interest is primarily because of the biologically inert nature of PFCs. Following intravenous (i.v) administration, PFC NE droplets are removed from the bloodstream by the reticuloendothelial system (RES), with the liver and spleen playing a major role in this process. In RES organs, PFC NEs are first demulsified, and then PFC droplets are transported across cell membranes to blood vessels at a rate contingent on the lipophilicity of the PFC. Subsequently, PFC droplets are taken up by circulating lipoproteins and transported to the lung, where they are eventually excreted unmetabolized through exhalation [27, 28]. Typically, PFC NEs for oxygen delivery (OxygentTM and Oxycyte®) are formulated at a higher PFC concentration (~60% w/v) [29, 30]. Whereas for formulating PFC NEs for successful drug delivery and cell labeling applications, PFC concentrations up to 30% w/v are reported. Unlike the existing literature reviews, which primarily focus on the diagnostic or oxygen delivery aspect of PFC NEs, this review will summarize the selection of different components used while formulating PFC NEs as delivery vehicles for small molecules and biologics. We will discuss two distinct processing techniques, microfluidization and ultrasonication, commonly reported for manufacturing PFC NEs. Furthermore, we will enlist a few surface conjugation strategies used for developing targeted PFC NEs. A differentiating factor of this review is that we will identify key problems in the published literature on PFC NE manufacturing and introduce application of Quality by Design (QbD) for developing PFC NEs for drug delivery.
1.2 Types of Perfluorocarbon Nanoemulsions
Perfluorocarbon nanoemulsions for drug delivery applications can be formulated as biphasic PFC NEs, which consist of PFC as the dispersed phase, water as the continuous phase, and surfactant to stabilize the interface [31]. Exceptions to this commonly reported type are triphasic PFC NEs and W1/PFC/W2 double nanoemulsions. In triphasic PFC NEs, hydrocarbon oil is incorporated as an additional dispersed phase along with PFC, water, and surfactants [32-34]. In the presence of oil, PFCs form a third “fluorous” phase owing to their low propensity to participate in induced dipole-induced dipole interactions with the hydrocarbon oil and induced dipole-dipole interactions with the water [3]. In order to formulate W1/PFC/W2 double nanoemulsions, Lee Y-H and coworkers used a combination of surfactants and followed the two-step emulsification technique. In the first step, a water-in-PFC (W1/PFC) emulsion was developed by emulsifying PFC in the water phase using polyethoxylated fluorosurfactant. This primary emulsion was later added to a solution of triblock copolymer and subjected to sonication to formulate a W1/PFC/W2 double nanoemulsion [35-37]. Compositional differences between the three types of PFC NEs are depicted in Figure 1A-D.
Compositional differences between PFC NEs. A. Illustrates the types of PFC NEs used for drug delivery applications. Created with BioRender.com. B. The image of Cryo-Transmission Electron Microscopy (TEM) for heat sterilized PFOB NE emulsified with phospholipids was observed using a Leo 912 Ω-mega, Carl Zeiss instrument operated at 120 kV. Figure adapted from [31] under the Creative Commons Attribution License (CC BY 4.0), PLOS One. C. Cryo-TEM image for PFC NE with PFOB, Tributyl O-acetyl Citrate oil, and fluorinated surfactant was observed using a MET Hitachi instrument operated at 80 kV. Figure adapted from [34] under the Creative Commons Attribution License (CC BY 4.0), MDPI. D. Cryo-TEM image of W1/PFOB/W2 double nanoemulsions stabilized by polyethoxylated fluorosurfactant and carboxylic Pluronic F-68 copolymer was observed using a JEM-1400. Figure adapted from [37] under the Creative Commons Attribution License (CC BY 4.0), MDPI.
2. Design and Development of PFC NEs as a Drug Delivery Platform
Perfluorocarbon nanoemulsions are a versatile drug delivery platform, reflected by their ability to deliver different therapeutic payloads ranging from small molecules [16, 17, 35, 37-43] to biologics such as small interfering RNAs (siRNAs) [18, 44, 45], proteins [19, 46], peptides and enzymes [47-50], and oligonucleotides [51] (Table 1).
Summary of different therapeutic payloads that are delivered through PFC NEs, indication, PFC type, targeting mechanism, manufacturing method, Z-average size analyzed by dynamic light scattering (DLS), and route of delivery.
| Drugs | Indication | Type of PFC | Targeting mechanism | Manufacturing conditions | Size (nm) | Route | Ref. | |
|---|---|---|---|---|---|---|---|---|
| Small molecules | Rapamycin | Muscular dystrophy | PFOB | - | MF, M110S- 4 minutes 20,000 psi | ~185 | Intra-venous | [16] |
| Celecoxib* | Neuro-inflammation | PFCE | - | MF on ice M110S- 6 passes 17,500 psi | ~140 | Intra-venous | [17] | |
| Osimertinib (EGFR-TK1) | Non-small cell lung cancer | PFCE | Folic acid | M110P- 4 minutes 20,000 psi | 100.9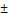 8.0 8.0 | Intra-venous | [38] | |
| Resveratrol* | Anti-inflammatory | PFPE | - | MF on ice M110S- 6 passes 17,500 psi | 155.1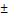 3.1 3.1 | - | [39] | |
| Paclitaxel | Ovarian cancer | PFP | US-TDD | UE on ice Power-500 W | ~250 | Intra-venous | [40] | |
| Sn2 lipase labile-Fumagillin prodrug | Angiogenesis | PFOB | αvβ3-integrin | MF, M110S- 4 minutes 20,000 psi | ~230 | Intra-venous | [42] | |
| Thymoquinone | Cancer | PFP | as1411 aptamer | UE on ice Amplitude-60% Pulse mode | ~195 | - | [43] | |
| Rifampicin# | Antimicrobial | PFOB | - | UE on ice 5-10 minutes | 238.6 ± 7.51 | Intra-venous | [37] | |
| Doxorubicin# | Breast Cancer | PFOB | Anti-HER2 | UE on ice | ~340 | - | [35] | |
| Isoflurane | General Anesthesia | PFTBA | MF on ice, M110Y- 8 minutes 8-10,000 psi | ~125 | Intra-venous | [41] | ||
| siRNA | siSTAT3 | Idiopathic Pulmonary Fibrosis | PFOB | CXCR4 | UE on ice Amplitude-30% Pulse mode | 175 2.0 2.0 | Intra-tracheal | [18] |
| siNGF | Pancreatic cancer | PFOB | CXCR4 | UE on ice Power-200 W Amplitude-80% Pulse mode | ~147 | Intra-peritoneal | [44] | |
| siSTAT3 | Lung metastatic osteosarcoma | PFOB | CXCR4 | UE on ice Amplitude-30% Pulse mode | ~170 | Intra-tracheal | [45] | |
| Proteins | Model protein-GFP | - | PFH/ PFP | US-TDD and Folic acid | Vigorous vortexing | ~300 | Intra-vascular | [19] |
| Pigment epithelium-derived factor | Acute myocardial injury | PFOB | - | Miniextruder | 140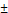 4.7 4.7 | Intra-tracheal | [46] | |
| Peptides and enzymes | PPACK | Atherosclerosis | PFOB | - | MF, M110S- 4 minutes 20,000 psi | 160.5 0 ± 2.6 | Intra-venous | [48] |
| Melittin | Cancer | PFOB | αvβ3-integrin | ~7 | Intra-venous | [49] | ||
| Lactate oxidase | Breast Cancer | PFTBA | PD-1-expressing cell membrane | UE 260 W 8 minutes | 133.1 | Intra-venous | [50] | |
| pDNA | Model oligonucleotide-eGFP | - | PFOB | - | UE on ice Power-125 W Amplitude-35% Continuous mode | ~170 | - | [51] |
Abbreviations: EGFR-TKI: Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitor, siSTAT3: Signal transducer and activator of transcription 3, siNGF: Nerve growth factor, PPACK: Phe-Pro-Arg-Chloromethylketone, US-TDD: Ultrasound-Targeted drug delivery, CXCR4: C-X-C chemokine receptor type 4, as1411: G-quadruplex aptamer, MF: Microfluidization, UE: Ultrasound Emulsification. * Triphasic PFC NEs, # W1/PFC/W2 double nanoemulsion.
In biphasic PFC NEs, poorly soluble drugs or biologics are incorporated into the surfactant layer. In triphasic PFC NEs, small molecules are loaded in the oil phase. As NEs are formulated using low surfactant concentrations, the amount of therapeutic drug that can be loaded into the surfactant layer is limited. By incorporating an oil phase, it is feasible to improve the capacity for loading poorly soluble drugs or lipophilic dyes [33]. In recent research, an alternative strategy has been investigated where the therapeutic payloads are dispersed within the PFC core of biphasic PFC NEs. This approach is particularly applied for delivering macromolecules, as the fluorous PFC core can efficiently shield the biological cargo from an external degradative environment [19, 51]. Sloand and coworkers [19] screened a small library of fluoroalkanes, carboxylic acid fluoroalkanes, and fluorobenzenes to select optimal fluorous tags (FTags). Mechanistically, FTags can “fluorous mask” the protein of interest to facilitate its dispersion in the PFC core. This is in contrast of loading the macromolecule in the surfactant layer. The research group demonstrated that the selected FTag (perfluorononanoic acid) noncovalently complexes with the model protein via hydrogen bonds that are formed between the carboxylic end of the FTag and the polar side chains of the protein. Importantly, the chemical modification with the FTag did not compromise the native conformational state and retained the biological function of the protein. In another study based on the same concept, Estabrook and coworkers demonstrated that cationic tags (ammonium with two C6F13 chains) can associate with the anionic backbone of pDNA through electrostatic interactions, which allows the fluorous-tagged pDNA to be loaded into the PFC core of the NE. The fluorous-tagged eGFP pDNA was loaded in PFC NE and was successfully delivered to promote in cellulo eGFP expression (Figure 2A and B) [51].
Strategy for loading pDNA into PFC core. A. Fluorous amine tag complexes with eGFP pDNA (model pDNA) to facilitate its solubilization in the PFC liquid. In the next step, the fluorous-tagged eGFP pDNA was sonicated in the presence of a reduction-sensitive surfactant, PFC, and water to form eGFP-loaded PFC NE. B. Schematic of in cellulo eGFP pDNA delivery. The cell uptake of eGFP pDNA-loaded PFC NE via endocytosis (i). This is followed by endosomal escape in cytosol (ii), and release of eGFP pDNA due to reduction of disulfide-linked surfactant in presence of cytosol glutathione (iii). Nuclear entry and expression of eGFP (iv). Reprinted with permission from [51]. Copyright (2021) from John Wiley and Sons, Inc.
The success of PFC NEs as a drug delivery platform relies on the selection of formulation components like PFC, surfactant, and hydrocarbon oil. As described below, through this careful selection, formulators can design PFC NEs that not only deliver therapeutic payloads but also offer additional benefits such as diagnostic capability or provide spatiotemporal control over drug release.
2.1 Selection of PFCs: Building Blocks of PFC NEs
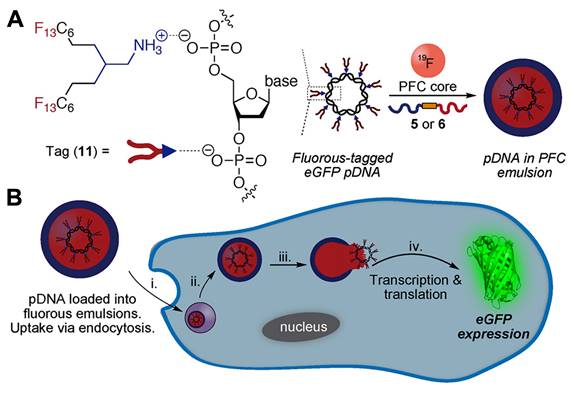
PFCs used for drug delivery applications can be broadly categorized into two groups: (1) Perfluorinated compounds with a molecular weight less than 1000 g/mol. This group includes compounds like perfluorooctyl bromide (PFOB), perfluoro-15-crown-5-ether (PFCE), perfluorodecalin (PFD), perfluorotributyl amine (PFTBA), and others. (2) Perfluorinated polymers with a molecular weight greater than 1000 g/mol. An example in this category is perfluoropolyether (PFPE) (for all chemical structures and molecular weights, refer to Figure 3). When selecting PFCs for drug delivery applications, organ clearance, which is inversely proportional to the molecular weight of the PFC, has been given precedence primarily to avoid long-term organ retention. Although for most PFCs, molecular weight is a reliable predictor of organ clearance, there are a few exceptions to this rule. For example, PFOB shows faster organ clearance (~4 days) compared to perfluorotripropylamine (~65 days), despite a comparable molecular weight [4]. This is attributed to the presence of a polarizable bromine atom at the terminal position of PFOB, which increases its molecular lipophilicity. As a result, circulating lipoproteins can easily take up PFOB and transport it for pulmonary removal [27, 52]. Bérard et al. [34] recently investigated the in vivo biodistribution of PFOB NE in major organs at different time points post i.v injection. The authors' data corroborated the value reported in the literature, as PFOB was completely excreted from major organs as early as 3 days without any signs of organ toxicity. PFOB is commonly selected by researchers for developing drug-delivering PFC NEs [18, 42, 49] based on its favorable organ clearance time and proven safety up to 1.35 g of PFOB dose/kg body weight in humans [29].
Structures of the PFCs and surfactants commonly used for formulating PFC NEs for drug delivery. Created with Biorender.com. All the chemical structures were drawn using ChemDraw 21.0.0.
A molecularly symmetric PFCE is selected when the goal is to develop theranostic PFC NEs [17, 38, 53, 54]. Theranostic nanomedicines represent a recent advancement in the field of drug delivery, designed to simultaneously perform therapeutic and diagnostic functions. The capability to image the therapeutic during or after administration presents a prospect for developing personalized medicines in the future [55]. Despite the favorable organ clearance time, PFOB is not ideal for imaging purposes. The 19F NMR spectra of PFOB consist of eight resonance peaks. Therefore, special frequency-selective MRI pulse sequences and extended scan times are required to improve the spatial resolution of images by minimizing chemical shift artifacts. Jacoby et al. [27] implemented chemical shift imaging to provide signal contributions from all fluorine nuclei in PFOB to generate artifact-free 19F MR images. However, the in vivo detection sensitivity of 50.5% w/w PFOB NE was only 37% compared to 10% w/w PFCE NE. Yang et al. [38] selected PFCE as the objective was not only to deliver the Osimertinib drug but also to conduct real-time monitoring of PFCE NE bioaccumulation in the tumor tissue by the 19F MRI technique. PFCE has 20 magnetically equivalent fluorine atoms that result in a single peak on NMR spectra. This increases the signal-to-noise ratio and results in high-contrast 19F “hot spots” during clinical MRI, which can be placed within the anatomical context using 1H MRI. One concern raised by Flögel and colleagues regarding the biomedical use of PFCE was the occurrence of transient spleen enlargement after injecting a high i.v dose [27]. However, in their study, a single i.v dose of PFCE NE resulted in a high PFC load (14 g of PFC/kg in mice). For drug delivery applications, a low i.v dose of PFCE NE is injected, which results in minimal PFC load in major organs [38, 54]. For example, a single i.v injection of Osimertinib-loaded PFCE NE corresponded to ~2 g of PFCE/kg in mice. Further, no signs of tissue necrosis or inflammation were observed even after two weeks, as confirmed by hematoxylin and eosin staining of major organs. Nevertheless, long-term studies observing for signs of splenomegaly or monitoring the changes in aspartate transaminase and alanine transaminase levels (markers of liver damage) are required, especially when planning multi-dosing experiments with PFCE NEs.
Historically, PFC NEs were used to deliver oxygen to the lungs through liquid-assisted ventilation. This has inspired the use of PFC NEs for pulmonary drug delivery applications. Systemic drug delivery to the injured lung fails to achieve the desired drug concentration as the blood flow is redirected away from the site of injury. Pulmonary drug delivery overcomes this challenge and helps to achieve increased local concentrations of drugs without any global systemic exposure [18, 46]. Ding et al. [18] selected PFOB, a low vapor pressure (10.4 torr at 37 °C) PFC, to formulate a NE for delivering siRNA targeting STAT3 through the intratracheal route. As per the literature, PFCs with low vapor pressure (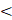 20 torr) do not increase pulmonary residual volume or compromise lung compliance [56]. However, in addition to low vapor pressure, the spreading coefficient of the PFCs is also a critical parameter that needs to be taken into consideration for achieving wide intrapulmonary distribution of the therapeutic payload. For example, even though PFD has a low vapor pressure (13.6 torr at 37 °C) it is not a suitable candidate for pulmonary delivery as it has a low spreading coefficient of -1.5 dyne/cm [57]. This would limit PFD from penetrating mucus-filled, collapsed, and under-ventilated areas of the lungs. Thus, the success of pulmonary drug delivery relies on identifying PFCs with a high spreading coefficient that can deliver payloads to lower lung lobes without triggering transient respiratory depression.
20 torr) do not increase pulmonary residual volume or compromise lung compliance [56]. However, in addition to low vapor pressure, the spreading coefficient of the PFCs is also a critical parameter that needs to be taken into consideration for achieving wide intrapulmonary distribution of the therapeutic payload. For example, even though PFD has a low vapor pressure (13.6 torr at 37 °C) it is not a suitable candidate for pulmonary delivery as it has a low spreading coefficient of -1.5 dyne/cm [57]. This would limit PFD from penetrating mucus-filled, collapsed, and under-ventilated areas of the lungs. Thus, the success of pulmonary drug delivery relies on identifying PFCs with a high spreading coefficient that can deliver payloads to lower lung lobes without triggering transient respiratory depression.
PFCs can be selected for specific applications based on their suitability. Ultrasound-triggered drug release is investigated as a non-invasive approach for achieving spatiotemporal control over drug delivery by using ultrasound waves as an external trigger. An ultrasonic wave is a longitudinal pressure wave with a frequency above the human audible range [58]. PFC NEs are developed as “phase-shift microbubble precursors” to overcome limitations of PFC gas-filled microbubbles like poor drug loading and short circulation time (minutes) [15, 59]. However, the developed PFC NE should be ultrasound-responsive, remain stable in the blood circulation, and release the therapeutic payload at sub-ablative ultrasound energy levels to prevent tissue damage [59]. Rapoport et al. [40] used a low-boiling-point (29 °C) PFC such as Perfluoropentane (PFP), as it requires low levels of ultrasound energy to trigger vaporization. Although PFP NE droplets might be expected to vaporize prematurely at physiological temperature (37 °C) before reaching the intended site, the authors theoretically showed an inverse relationship between the temperature required for PFP NE vaporization and the droplet diameter. As the droplet diameter decreases, the Laplace pressure on the PFC core increases, consequently increasing the boiling point of PFP. The group further supported their theoretical findings with in vivo experimental data, showing that PFP NE undergoes a droplet-to-bubble transition, also known as acoustic droplet vaporization (AVD), under ultrasound exposure. This facilitates an increase in surface area, causing the “ripping off” and release of Paclitaxel in ovarian carcinoma tumors without any unwanted bioeffects. Although the authors selected PFP based on its low boiling point, they did not comment on the vaporization efficiency of PFP (i.e., how many microbubbles are formed from nanodroplets under the given ultrasound parameters and time), which has been identified as a critical parameter for low boiling point PFCs determining the amount of drug release [60].
PFCs are also selected to increase the stability of NEs, which is primarily compromised by a destabilization mechanism known as Ostwald ripening. Ostwald ripening is a molecular diffusion phenomenon in which the large droplets grow at the expense of the small droplets, leading to an irreversible increase in droplet diameter and ultimately phase separation. According to Lifshitz, Slyzov, and Wagner (LSW) theory, the thermodynamic driving force for Ostwald ripening is the increased water solubility of the dispersed phase as the droplet radius decreases. PFCs having high molecular weight and low continuous-phase solubility are reported to decrease Ostwald ripening rates in PFC NEs [61]. Patel et al. [62] selected perfluorinated polymers like PFPE to formulate 7.24% w/v PFPE NE with celecoxib and monitored changes in the droplet diameter using dynamic light scattering (DLS) over a period of three months as an indicator of Ostwald ripening. Their data showed no changes in the droplet diameter at 4 °C (refrigeration temperature) and 25 °C (room temperature), supporting the use of PFPE to formulate PFC NEs with long-term colloidal stability. In another example, Ashrafi et al. [41] systematically screened twenty-seven PFCs to show a direct correlation between the observed evaporation rate of PFC and the colloidal stability of PFC NE when the Pluronic surfactant system remained constant. The authors selected PFTBA with an evaporation rate less than 0.01 g/h to formulate volatile anesthetics delivering PFC NE. PFTBA/isoflurane NE batches stored at room temperature over a period of 360 days showed 4% decrease in droplet diameter with no significant loss in isoflurane content. It is, however, important to emphasize that the stability of PFC NEs is not solely determined by the properties of PFCs. Other variables, such as surfactant properties, surfactant concentration, and the manufacturing process, also need to be optimized to achieve PFC NEs with long-term stability. Taken together, the selection of PFC core is application-specific, and a formulator has to evaluate several factors, including biological half-life [16], ability to develop artifact-free 19F MRI images [17], desired route of administration [46], intended application [19, 40], and potential to improve the stability of PFC NE [62, 63] during selection.
2.2 Selection of Surfactants
A surfactant is an amphiphilic molecule that reduces the interfacial tension to facilitate droplet break-up and subsequently stabilizes the newly formed interfaces to prevent recoalescence [14, 64]. Older literature emphasized that the colloidal stability of PFC NEs is governed by the properties of PFCs rather than the emulsifier [61]. However, recent literature contradicts this, as PFC NEs emulsified with surfactants varying in their hydrophilic-lipophilic balance (HLB) values at the same PFC-to-surfactant ratio and manufacturing conditions showed different colloidal stabilities over time [65, 66]. For formulating PFC NEs as drug delivery carriers, researchers have selected surfactants that were initially used to develop FDA-approved PFC NEs for oxygen delivery. Consequently, the selected surfactants can be categorized into (1) non-ionic surfactants [51, 62, 67] and (2) lipid-based surfactants [46]. This preference may be attributed to the available empirical data demonstrating their effectiveness to stabilize PFC in water, their proven biocompatibility in preclinical studies, and their hydrophilic nature, as indicated by their high HLB values (> 8). Surfactants with high HLB values favor the formation of PFC-in-water NEs rather than water-in-PFC NEs. However, a few recent studies have explored a new class of fluorinated polymeric surfactants [34, 68], synthesized to be compatible with the PFC core (for chemical structures, refer to Figure 3).
(a) Non-ionic surfactants
The first generation of FDA-approved PFC NEs for oxygen delivery were stabilized using water-soluble triblock copolymers of poly(ethylene oxide) (PEO) and poly(propylene oxide) (PPO). Pluronic F-68 (PEO76PPO29PEO76) was the first non-ionic emulsifier used in Fluosol® [9]. Pluronics consist of a hydrophobic PPO block that anchors at the droplet surface and two hydrophilic PEO blocks that extend into the aqueous phase for steric stabilization. These stabilizing PEO chains extend to form shells of nanometer thickness (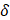 ). When two droplets approach at a separation distance (h) smaller than two times the thickness (h
). When two droplets approach at a separation distance (h) smaller than two times the thickness (h 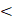 2δ), the chains experience strong repulsive forces to avoid loss in configurational entropy at the overlapping region [69]. However, repeated administration of Fluosol® triggered mild flu-like symptoms, attributed to the F-68 dose-dependent immune activation [70]. Therefore, research efforts are directed towards investigating other biocompatible polymeric surfactants that can emulsify PFC in water. Sletten and her team published a polymer library of amphiphilic diblock and triblock poly(2-oxazoline) (POx) copolymers designed to mimic F-68 based on their structure-property relationships [65, 66]. For triblock polymers, the hydrophilic PEO block in F-68 was replaced with either a poly(2-methyl-2-oxazoline) [P(MeOx)] block or a poly(2-ethyl-2-oxazoline) [P(EtOx)] block, and the hydrophobic PEO block was replaced with a poly(2-nonyl-2-oxazoline) block [P(NonOx)]. The hydrophilic and hydrophobic block lengths of POx polymer were controlled to be comparable to the ratio in F-68. The authors systematically showed that the droplet size stability of PFC NEs stabilized with triblock POx surfactants increases with an increase in hydrophilic block length, which favors steric stabilization. Note that in this study, the total PFC content (7:3 PFD/PFTPA) was 10 vol% and required 2.8 wt% of triblock POx emulsifier to formulate colloidally stable PFC NE. The total PFC content in Fluosol® was 20 vol% (7:3 PFD/PFTPA) and was emulsified with 2.72 wt% of F-68 [9]. This means that, compared to F-68, a higher POx surfactant concentration might be required to emulsify the same amount of PFC in the formulation. Further, the authors compared the relative surface protein adsorption on PFC NEs emulsified with POx polymers to that with a polymer having poly(ethylene oxide) (PEG) as a hydrophilic block. Their Bradford assay data showed no significant differences in total protein adsorption, indicating POx polymers can effectively shield non-specific protein interactions to offer a stealth effect [66]. Thus, POx polymers can be selected to formulate antifouling PFC NEs with prolonged circulation times. One avenue to explore is whether PFC NEs stabilized with POx polymers could be used as a safe alternative for delivering drugs to human populations positive for anti-PEG antibodies. Non-ionic polymeric surfactants can also be designed to leverage unique microenvironments within cells. The same research group showed the development of POx amphiphiles with disulfide linkers (P(MeOx)27-SS-P(NonOx)8). This reduction-sensitive surfactant was designed to respond to the increased concentrations of glutathione (GSH) within cells (2-10 mM) compared to the extracellular fluid (0.02-0.1 mM). The presence of high GSH led to the irreversible cleavage of disulfide bonds at the liquid-liquid interface. Consequently, demulsifying PFC NEs and releasing the therapeutic payload intracellularly [51]. In another example, PFCE NE was formulated using poly(ethylene oxide)-co-poly(d,l-lactide) (PEG-PDLA), a biodegradable and pH-sensitive surfactant, to promote the endo-lysosomal escape of paclitaxel-loaded PFCE NE in tumor cells [67].
2δ), the chains experience strong repulsive forces to avoid loss in configurational entropy at the overlapping region [69]. However, repeated administration of Fluosol® triggered mild flu-like symptoms, attributed to the F-68 dose-dependent immune activation [70]. Therefore, research efforts are directed towards investigating other biocompatible polymeric surfactants that can emulsify PFC in water. Sletten and her team published a polymer library of amphiphilic diblock and triblock poly(2-oxazoline) (POx) copolymers designed to mimic F-68 based on their structure-property relationships [65, 66]. For triblock polymers, the hydrophilic PEO block in F-68 was replaced with either a poly(2-methyl-2-oxazoline) [P(MeOx)] block or a poly(2-ethyl-2-oxazoline) [P(EtOx)] block, and the hydrophobic PEO block was replaced with a poly(2-nonyl-2-oxazoline) block [P(NonOx)]. The hydrophilic and hydrophobic block lengths of POx polymer were controlled to be comparable to the ratio in F-68. The authors systematically showed that the droplet size stability of PFC NEs stabilized with triblock POx surfactants increases with an increase in hydrophilic block length, which favors steric stabilization. Note that in this study, the total PFC content (7:3 PFD/PFTPA) was 10 vol% and required 2.8 wt% of triblock POx emulsifier to formulate colloidally stable PFC NE. The total PFC content in Fluosol® was 20 vol% (7:3 PFD/PFTPA) and was emulsified with 2.72 wt% of F-68 [9]. This means that, compared to F-68, a higher POx surfactant concentration might be required to emulsify the same amount of PFC in the formulation. Further, the authors compared the relative surface protein adsorption on PFC NEs emulsified with POx polymers to that with a polymer having poly(ethylene oxide) (PEG) as a hydrophilic block. Their Bradford assay data showed no significant differences in total protein adsorption, indicating POx polymers can effectively shield non-specific protein interactions to offer a stealth effect [66]. Thus, POx polymers can be selected to formulate antifouling PFC NEs with prolonged circulation times. One avenue to explore is whether PFC NEs stabilized with POx polymers could be used as a safe alternative for delivering drugs to human populations positive for anti-PEG antibodies. Non-ionic polymeric surfactants can also be designed to leverage unique microenvironments within cells. The same research group showed the development of POx amphiphiles with disulfide linkers (P(MeOx)27-SS-P(NonOx)8). This reduction-sensitive surfactant was designed to respond to the increased concentrations of glutathione (GSH) within cells (2-10 mM) compared to the extracellular fluid (0.02-0.1 mM). The presence of high GSH led to the irreversible cleavage of disulfide bonds at the liquid-liquid interface. Consequently, demulsifying PFC NEs and releasing the therapeutic payload intracellularly [51]. In another example, PFCE NE was formulated using poly(ethylene oxide)-co-poly(d,l-lactide) (PEG-PDLA), a biodegradable and pH-sensitive surfactant, to promote the endo-lysosomal escape of paclitaxel-loaded PFCE NE in tumor cells [67].
Another direction in the field is to use PluronicsTM triblock copolymers with lower HLB values as compared to F-68 [71]. This selection criteria is particularly applied for stabilizing triphasic PFC NEs with hydrocarbon oil. The HLB scale provides a numerical value to characterize the balance between the hydrophilic and lipophilic groups in a surfactant and indicates the solubility of the surfactant in either the oil or water phase. On the other hand, hydrocarbon oils have required HLB values typically in the 10-15 range, which should be matched by the selected emulsifiers to develop colloidally stable NEs with water as an external phase [72]. F-68 is primarily a hydrophilic surfactant with a high HLB value of 29 and is not an ideal candidate for emulsifying hydrocarbon oils [71]. Therefore, pluronics with relatively low HLB values, such as pluronic P105 (HLB = 15), are selected to match the required HLB values of the selected oil. Additionally, as the HLB value of an individual surfactant may not always directly correspond with the required HLB value for oil, different non-ionic surfactants with varying HLB values are selected and blended in different ratios such that the HLB of the surfactant blend matches the targeted HLB requirement of oil [55]. While the HLB value of non-ionic surfactants serves as a good metric to narrow down the selection list of emulsifiers, this semi-empirical approach is limited to triphasic PFC NEs. This is because, unlike hydrocarbon oils, PFC liquids do not have the required HLB values. Consequently, the selection of non-ionic surfactants for biphasic PFC NEs depends on conducting extensive screening experiments with different surfactants. Furthermore, PFC NEs stabilized with triblock copolymers are reported to maintain their colloidal stability in the presence of serum proteins [55, 73, 74]. Recently, Zhalimov et al. [75] demonstrated that the amount of plasma protein adsorbed on the surface of the PFC NE droplets stabilized with triblock copolymers was directly proportional to the 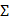 PEO/
PEO/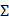 PPO ratio, rather than other parameters like molecular weight and length of the PEO and PPO block lengths.
PPO ratio, rather than other parameters like molecular weight and length of the PEO and PPO block lengths.
(b) Lipid-Based Surfactants
The second generation of FDA-approved PFC NEs were sterically stabilized using lipid-based surfactants such as egg yolk phospholipids (EYP) (HLB = 10) and hydrogenated soybean phospholipid (HLB = 9) [42]. Phospholipids are amphiphilic molecules composed of a phosphate group (polar head) and fatty acid chains (nonpolar tails) connected by a glycerol backbone [76]. Based on the literature, phospholipids are preferred when the aim is to effectively deliver biologics intracellularly without exposing them to the acidic pH of endosomes [46, 49, 77]. For PFC NEs, the selected surfactant dictates the cell uptake mechanism [66, 77]. Mechanisms of cell uptake can be investigated using different inhibitors as shown in Figure 4A. Estabrook et al. [65] showed PFC NEs stabilized with POx or Pluronic F-68 surfactants are primarily internalized through endocytosis, regardless of droplet diameter (150-300 nm), in both phagocytic (RAW 264.7 macrophages) and non-phagocytic (A375) cell lines. Day et al. [66] corroborated these findings by showing colocalization of the rhodamine-labeled PFC NE droplets with Lysotracker green dye, which selectively labels acidic cell organelles like endosomes and lysosomes. Further, clathrin-dependent endocytosis was the dominant route of internalization for PFC NEs emulsified with non-ionic surfactants, as a  40% decrease was observed in the presence of chlorpromazine, an inhibitor of clathrin-mediated endocytosis (Figure 4B and C). In contrast, Soman et al. [78] showed that PFC NEs emulsified with EYP (droplet diameter: ~240 nm) avoid endosomal uptake and deliver therapeutic cargo directly to the cytoplasm through 'contact-facilitated drug delivery'. The authors performed deep-etch platinum replica electron microscopy to investigate a mechanism that depends on lipid mixing between the surfactant monolayer and the bilayer membrane to form a hemifusion complex (Figure 4D) [49]. Importantly, molecular dynamics simulations supported the role of the PFC core in the reorientation and protrusion of the hydrophobic tails of the surfactant [79]. The formation of the hemifusion complex enabled direct shuttling of the cargo to the cell membrane, which was further trafficked intracellularly via an energy-dependent process known as caveolae/lipid raft-mediated internalization [49, 78, 80]. Lipid rafts are dynamin-dependent, cholesterol-sensitive membrane domains that are intracellularly trafficked to caveolin-1-positive endocytic compartments, which are at a neutral pH, unlike acidic early endosomes [81]. The research group used a panel of cell uptake inhibitors and showed that cell uptake was five-fold lower in the presence of filipin, which specifically inhibits lipid raft formation by binding to membrane cholesterol (Figure 4E) [80]. Recently, Qin et al. [46] showed minimal overlap of the fluorescence signal between the fluorescently labeled Cou-6-PFOB NE emulsified with phospholipid (~150 nm) and the LysoTracker Red DND-99 dye (Figure 4F). Furthermore, the group developed protein-phospholipid complexes (PPCs) between HSPC and pigment epithelium-derived factor (PEDF), a glycoprotein that protects against cellular oxidative stress. PPCs increase the lipophilicity of the therapeutic proteins, thereby increasing protein loading in the PFOB NE scaffold.
40% decrease was observed in the presence of chlorpromazine, an inhibitor of clathrin-mediated endocytosis (Figure 4B and C). In contrast, Soman et al. [78] showed that PFC NEs emulsified with EYP (droplet diameter: ~240 nm) avoid endosomal uptake and deliver therapeutic cargo directly to the cytoplasm through 'contact-facilitated drug delivery'. The authors performed deep-etch platinum replica electron microscopy to investigate a mechanism that depends on lipid mixing between the surfactant monolayer and the bilayer membrane to form a hemifusion complex (Figure 4D) [49]. Importantly, molecular dynamics simulations supported the role of the PFC core in the reorientation and protrusion of the hydrophobic tails of the surfactant [79]. The formation of the hemifusion complex enabled direct shuttling of the cargo to the cell membrane, which was further trafficked intracellularly via an energy-dependent process known as caveolae/lipid raft-mediated internalization [49, 78, 80]. Lipid rafts are dynamin-dependent, cholesterol-sensitive membrane domains that are intracellularly trafficked to caveolin-1-positive endocytic compartments, which are at a neutral pH, unlike acidic early endosomes [81]. The research group used a panel of cell uptake inhibitors and showed that cell uptake was five-fold lower in the presence of filipin, which specifically inhibits lipid raft formation by binding to membrane cholesterol (Figure 4E) [80]. Recently, Qin et al. [46] showed minimal overlap of the fluorescence signal between the fluorescently labeled Cou-6-PFOB NE emulsified with phospholipid (~150 nm) and the LysoTracker Red DND-99 dye (Figure 4F). Furthermore, the group developed protein-phospholipid complexes (PPCs) between HSPC and pigment epithelium-derived factor (PEDF), a glycoprotein that protects against cellular oxidative stress. PPCs increase the lipophilicity of the therapeutic proteins, thereby increasing protein loading in the PFOB NE scaffold.
Influence of surfactants on the uptake of PFC NEs. A. Schematic representation of cellular uptake and commonly selected inhibitors for inhibiting each uptake mechanism. Created with Biorender.com. B. Quantified FACS analysis of the uptake of rhodamine-labeled PFC NEs emulsified with non-ionic surfactants. RAW264.7 macrophages were treated with the listed inhibitors. C. Confocal microscopy of RAW264.7 macrophages showing the uptake of PFC NEs emulsified with POx polymer. Rhodamine labeled PFC NE (red, Ex 532nm), LysoTracker (green, Ex 488nm), and nuclei stained with Hoescht (blue, Ex 405nm). Scale bar = 7.5 μm. Figures B and C adapted with permission from [66]. Copyright (2020), American Chemical Society. D. Deep-etch platinum electron microscopy showing a hemifusion complex between the EYP stabilized PFC NE droplet and liposome (model bilayer membrane). Reprinted with permission from [78]. Copyright (2008), American Chemical Society. E. Analysis of pixel intensities within the C32 melanoma cell cytosol in the presence or absence of inhibitors. Adapted from [80] with permission. Copyright (2008), Elsevier. F. Confocal microscopy in H9c2 cells shows the uptake of PFC NEs emulsified with phospholipids. Coumarin-6-labeled PFC NE (green) and LysoTracker dye (Red) visualized on a 60 × objective lens; scale bar = 10 μm. Adapted from [46] with permission. Copyright (2021), Elsevier.
One factor to consider during formulation is that lipid-based surfactants are susceptible to oxidative degradation due to the presence of unsaturated fatty acid chains within phospholipids. This compromises their functionality to stabilize interfaces [82], which negatively impacts the storage stability of PFC NEs. To address this, Qin et al. [46] incorporated a low concentration of  -tocopherol in the PFC NE formulation to prevent oxidative degradation of phospholipids. Tocopherol is an effective interfacial antioxidant that prevents the oxidation of phospholipids due to its free radical scavenging activity at interfaces [83]. However, the group did not present data demonstrating the long-term stability of the formulated PFC NE or the effectiveness of tocopherol at PFC/water, which requires further investigations.
-tocopherol in the PFC NE formulation to prevent oxidative degradation of phospholipids. Tocopherol is an effective interfacial antioxidant that prevents the oxidation of phospholipids due to its free radical scavenging activity at interfaces [83]. However, the group did not present data demonstrating the long-term stability of the formulated PFC NE or the effectiveness of tocopherol at PFC/water, which requires further investigations.
(c) Semifluorinated Polymeric Surfactants
Semifluorinated polymeric surfactants represent a new class of amphiphilic polymers composed of hydrophilic head groups and fluoroalkyl tails. Initially, these surfactants were used to formulate PFC NEs for i.v delivery of fluorinated anesthetics to induce rapid anesthesia compared to inhalation. However, the presence of large fluorophilic block triggered allergic responses due to histamine release in large animal studies, resulting in failures of these PFC NEs [84]. However, recent advances in synthetic polymer chemistry have supported the development of fluorinated polymers with different chemical structures for biomedical applications [85]. Decato et al. [86] synthesized a series of semifluorinated polymers and showed that the total fluorine content of the surfactant determines the initial droplet diameter and long-term stability of PFOB NEs. Polymers composed of three perfluoro-tert-butyl (PFtBTRI) groups as the fluorophilic tail resulted in PFOB NE with a smaller droplet diameter and a decreased Ostwald ripening rate as opposed to the polymers with a single PFtB group (PFtBMONO), irrespective of the chain length of the hydrophilic monomethyl poly(ethylene glycol) (mPEG) head group. This difference in PFOB NE stability was explained by determining the critical micelle concentration (CMC) based on surface tension measurements. The mPEGx-(PFtBMONO) polymers did not drive the self-assembly to form micelles. In contrast, mPEGx-(PFtBTRI) polymers showed acceptable CMC values below 1 mM, driving self-assembly due to the increased interaction between the fluorous tails on the polymer. Nevertheless, the scope of using semifluorinated amphiphiles is limited, as volatile fluorinated anesthetics that solubilize in PFC cores can be delivered through these systems. To broaden their applicability for loading hydrophobic drugs, further work should explore whether an intermediate hydrocarbon segment can be incorporated into the surfactant design and the implications of this strategic installation on surfactant properties such as CMC values.
Another notable example in this category is fluorinated polyethyleneimine (F-PEI) surfactants, specially synthesized to deliver siRNA for achieving sequence-specific gene silencing. Polycationic polymers like PEI condense siRNA through electrostatic interactions to form polyplexes. Polyplexes provide superior transfection efficiency by facilitating the endosomal escape of siRNA through the proton-sponge mechanism, which is an osmosis-driven process triggered by the proton buffering capacity of PEI. However, the use of PEI is limited due to its cytotoxicity [87]. Chen et al. [68] proposed that formulating PFD NE stabilized with F-PEI would minimize the cytotoxicity of PEI. To test their hypothesis, the research group first synthesized F-PEI by grafting the fluoroalkyl chains from heptafluorobutyric anhydride onto the primary and secondary amines present in PEI. In the second step, F-PEI was used to emulsify PFD (FPEI@ PFD NE). Following this, the process of siRNA condensation on F-PEI@PFD NE was driven by low-speed vortexing. The cell viability data supported their hypothesis, as F-PEI@PFD NE was significantly less toxic compared to naïve PEI even at high wPEI/wsiRNA ratios owing to the decreased PEI mobility [87]. Another important aspect of their work was that the F-PEI@PFD NE (~150 nm) had the same buffering capacity as naïve PEI and condensed siRNA at the same wPEI/wsiRNA ratio, indicating that covalent modification of amine groups on PEI with fluoroalkyl tails did not compromise polycation functionality. However, the colloidal stability of the formulated F-PEI@PFD NE stored at 4 °C was monitored for 7 days. In a comparable study, Lv, Jia et al. [88] used the same synthetic chemistry to develop F-PEI as Chen et al. and showed that under aqueous conditions, the amide bond linking the fluoroalkyl tails to PEI undergoes hydrolysis within 10 days. Therefore, the study duration for monitoring the stability of F-PEI@PFD NE should be extended beyond 10 days to observe if the stability of F-PEI@PFD NE is compromised due to F-PEI hydrolysis or whether the presence of a hydrophobic PFD core protects the embedded amide bond from hydrolytic degradation. In another example, Gao et al. [89] synthesized a fluorinated cationic polymer (C11F17-PBLA-DET) by performing an aminolysis reaction of C11F17-poly(β-benzyl-l-aspartate) (C11F17-PBLA) with diethylenetriamine (DET). PFP was emulsified with C11F17-PBLA-DET, followed by coating with PGA-g-mPEG to form nanodroplets capable of delivering nucleic acids to the cancer cells. Although semi-fluorinated surfactants open new avenues in drug delivery science, several critical aspects need further investigation, including surfactant characterization, the ability to scale up synthesis, and proving preclinical efficacy and safety.
After surfactant selection, the next step is to determine the optimal surfactant concentration required for formulating PFC NEs. This relies on optimization experiments, regardless of the surfactant type [34, 66]. The required surfactant concentration is selected based on achieving the predefined goals, such as targeted droplet size, low dispersity, long-term droplet size stability of PFC NEs under the set processing conditions.
2.3 Selection of Hydrocarbon oil: A Key Component in Triphasic PFC NEs
The developers of OxyfluorTM (HemaGen/PFC Ltd., MO, USA) first reported increased storage stability of PFC NEs formulated with 2% w/v soybean oil compared to PFC NE formulated without oil. The increased stability was attributed to the long-chain triglyceride, which covers the dispersed fluorous phase to improve the adsorption of hydrophobic surfactant tails [70]. In 1992, the company patented this three-liquid-phase system innovation [90]. Later studies have reported similar findings, as increased stability of PFC NEs was observed in the presence of hydrocarbon oil [71, 91]. However, recent efforts are directed towards repurposing this small percentage oil (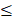 5% w/v) phase to incorporate hydrophobic small molecules (~1000 Da) and lipophilic dyes (e.g., carbocyanine dyes) as a second imaging modality [17, 34, 55]. Consequently, the selection of hydrocarbon oil primarily depends on the solubility of the hydrophobic drug in the oil phase, as it directly affects the amount of drug loaded in triphasic PFC NEs. For instance, Herneisey et al. [39] performed solubility studies to determine the solubility of Resveratrol, a natural antioxidant, in Miglyol 812, propylene glycol, and olive oil. The authors selected propylene glycol based on the maximum solubility of resveratrol, an anti-oxidant drug, compared to the other two tested oils. Additionally, the presence of the oil phase allows the formulators to incorporate FDA-approved solubilizers (
5% w/v) phase to incorporate hydrophobic small molecules (~1000 Da) and lipophilic dyes (e.g., carbocyanine dyes) as a second imaging modality [17, 34, 55]. Consequently, the selection of hydrocarbon oil primarily depends on the solubility of the hydrophobic drug in the oil phase, as it directly affects the amount of drug loaded in triphasic PFC NEs. For instance, Herneisey et al. [39] performed solubility studies to determine the solubility of Resveratrol, a natural antioxidant, in Miglyol 812, propylene glycol, and olive oil. The authors selected propylene glycol based on the maximum solubility of resveratrol, an anti-oxidant drug, compared to the other two tested oils. Additionally, the presence of the oil phase allows the formulators to incorporate FDA-approved solubilizers (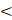 1% w/v) if required to increase the solubility of the hydrophobic drugs. For example, in another study, diethylene glycol-monoethyl ether (Transcutol® HP) was incorporated in a small percentage along with Miglyol 812 to increase the amount of celecoxib, a non-steroidal anti-inflammatory drug, that can be loaded into the PFC NE [74].
1% w/v) if required to increase the solubility of the hydrophobic drugs. For example, in another study, diethylene glycol-monoethyl ether (Transcutol® HP) was incorporated in a small percentage along with Miglyol 812 to increase the amount of celecoxib, a non-steroidal anti-inflammatory drug, that can be loaded into the PFC NE [74].
In addition to the rational selection of formulation components and determining the required concentration of each component, an emulsion scientist must carefully select the manufacturing technique and optimize processing parameters, as they influence the physicochemical properties of PFC NEs [74]. Therefore, the following sections will focus on the manufacturing aspect of PFC NEs.
3. Manufacturing of PFC NEs for Drug Delivery Applications
The process of manufacturing PFC NEs can be divided into two steps. The first step is the formation of a coarse emulsion, typically achieved by low-speed vortexing or magnetic stirring of the components. The second step is focused on droplet size reduction, either by microfluidization (MF) or ultrasonication emulsification (UE). For instrumentation, mechanism of droplet break-up, and processing parameters refer to Figure 5. Although both the homogenization techniques are categorized under high-energy methods (input energy density: ~108-1010 W kg-1) [92], the mechanism of droplet breakup differs between the two techniques [93]. In general, NEs can also be manufactured using low-energy techniques (input energy density: ~103 W kg-1). However, the popularity of selecting high-energy methods is because of the high interfacial tension at the PFC/water interface (50-60 mNm-1) that opposes the dispersion of PFC in the aqueous phase as compared to the HC oil/water interface (30-40 mNm-1) [4]. Therefore, in this section, we will focus on MF and UE, two commonly reported techniques for manufacturing PFC NEs.
Manufacturing techniques for developing PFC NEs. Illustrative figure showing instrumentation, mechanism of droplet breakup and processing parameters for A. Microfluidization (MF) and B. Ultrasonication emulsification (UE). Created with Biorender.com.
3.1 Microfluidization
In microfluidizers, the coarse emulsion enters the system through an inlet reservoir and is accelerated by using a high-pressure pneumatic pump capable of generating operating pressure as high as 40,000 psi. The coarse emulsion is then directed through a microchannel with an axially varying geometry, either Y-type or Z-type, often referred to as an interaction chamber. The enclosure of this interaction chamber is made of stainless steel, while the interior is either aluminum oxide ceramic or polycrystalline diamond, which offers chamber wear resistance. For liquid-liquid dispersions, such as NEs, a Y-type interaction chamber with microchannel diameters ranging from 75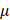 m to 125
m to 125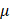 m is generally preferred to achieve a narrow-size distribution. Furthermore, due to the high level of turbulence, an auxiliary processing module (APM; also known as a return pressure chamber) can be used in conjunction with the interaction chamber. For a Y-type interaction chamber, a Z-type AMP is installed downstream to add backpressure. This prevents interaction chamber wear-off and stabilizes flow rate [94, 95]. In the interaction chamber, the process of droplet breakup occurs due to turbulent viscous (TV) and turbulent inertial (TI) forces. As the fluid is accelerated, the droplets experience turbulent flow fields and interact with short-lived eddies. The nature of the interaction between the droplet and the eddies depends on the droplet diameter (d) and the length scale of the eddies (
m is generally preferred to achieve a narrow-size distribution. Furthermore, due to the high level of turbulence, an auxiliary processing module (APM; also known as a return pressure chamber) can be used in conjunction with the interaction chamber. For a Y-type interaction chamber, a Z-type AMP is installed downstream to add backpressure. This prevents interaction chamber wear-off and stabilizes flow rate [94, 95]. In the interaction chamber, the process of droplet breakup occurs due to turbulent viscous (TV) and turbulent inertial (TI) forces. As the fluid is accelerated, the droplets experience turbulent flow fields and interact with short-lived eddies. The nature of the interaction between the droplet and the eddies depends on the droplet diameter (d) and the length scale of the eddies (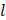 ). If the droplet diameter is smaller than the length scale of the eddy (d
). If the droplet diameter is smaller than the length scale of the eddy (d 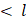 ), the droplet experiences viscous shear at the interface because of the velocity gradient created by the eddy. Depending on the strength of the viscous gradient and the time scale of the droplet-eddy interaction, the droplet undergoes elongation and eventually breaks up. This process is referred to as the TV droplet breakup. In contrast, for droplet breakup by TI forces, short-length scale eddies (d
), the droplet experiences viscous shear at the interface because of the velocity gradient created by the eddy. Depending on the strength of the viscous gradient and the time scale of the droplet-eddy interaction, the droplet undergoes elongation and eventually breaks up. This process is referred to as the TV droplet breakup. In contrast, for droplet breakup by TI forces, short-length scale eddies (d 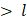 ) with low kinetic energies participate in generating pressure fluctuations at the interface [96]. Following a fixed number of microfluidization passes through the interaction chamber, the sample exits through an outlet, where heat exchangers or cooling coils can be installed to prevent overheating of PFC NEs. The microfluidization process established on a lab scale can be linearly scaled to pilot or commercial scale by using interaction chambers consisting of parallel arrangements of multiple fixed-geometry microchannels. This design approach helps to achieve shear and pressure profiles similar to those obtained during lab-scale manufacturing [97]. A few studies have shown implementation of MF technology to scale up (
) with low kinetic energies participate in generating pressure fluctuations at the interface [96]. Following a fixed number of microfluidization passes through the interaction chamber, the sample exits through an outlet, where heat exchangers or cooling coils can be installed to prevent overheating of PFC NEs. The microfluidization process established on a lab scale can be linearly scaled to pilot or commercial scale by using interaction chambers consisting of parallel arrangements of multiple fixed-geometry microchannels. This design approach helps to achieve shear and pressure profiles similar to those obtained during lab-scale manufacturing [97]. A few studies have shown implementation of MF technology to scale up (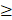 50 mL) drug-loaded PFC NEs [41, 55, 98].
50 mL) drug-loaded PFC NEs [41, 55, 98].
In microfluidizers, the operating pump pressure and number of passes are the critical processing parameters (CPPs) primarily governing the measurable critical quality attributes (CQAs) of PFC NEs, such as droplet diameter, dispersity index, and long-term droplet size stability [74]. However, there is an interplay between the processing parameters and the composition of PFC NE. For example, Herneisey et al. [74] reported that changing formulation compositions of triphasic PFC NEs resulted in varying CQAs despite being processed under the same homogenizing conditions. Although a systematic study investigating the influence of different operating pump pressures on the CQAs of PFC NEs has not yet been reported in the field, the reported operating pressure for manufacturing biphasic or triphasic PFC NEs on the lab scale and pilot scale models (M110S, M110P, and M110EH) lies between 10,000 psi and 20,000 psi. Liu et al. [98] formulated PFC NEs with a fixed composition and demonstrated that the droplet diameter of PFC NEs decreases with an increase in the number of microfluidization passes at a constant operating pressure. It is, however, important to note that no significant decrease in droplet diameter was observed after increasing the number of passes beyond a critical number. This implies that increasing the number of passes to decrease the droplet diameter of PFC NEs applies until a size threshold is achieved. After that, the rate of surfactant adsorption can become a limiting factor, increasing the risk of overprocessing PFC NEs. The phenomenon of overprocessing occurs when the time scale of collision between the droplets is smaller than the time scale for surfactant adsorption, leading to an irreversible increase in droplet diameter [99]. In general, another concern associated with high-energy homogenization techniques is the increased product temperature, which can lead to drug degradation. Mao et al. [100] in their work on M110EH, recorded an increase in NE temperature as the operating pressure was increased from 40 MPa to 120 MPa. However, their data showed that even at a high operating pressure of 120 MPa (~17,500 psi), the final product temperature was ~32 °C, owing to the high emulsification efficiency of microfluidizers compared to other high-energy methods. Although the NEs formulated by Mao et al. do not contain PFCs, studies conducted by other researchers on triphasic PFC NEs corroborated the findings [62, 98]. Liu et al. recorded ~25 °C as the final product temperature when processed at an operating pressure of 15,000 psi for 6 passes on M110EH [98]. This observation was primarily attributed to the design of microfluidizers, which allows the interaction chamber to be ice-cooled prior to manufacturing.
3.2 Ultrasonication Emulsification
The droplet breakup in UE is achieved due to sound waves with frequencies above 20 kHz. An ultrasonicator consists of a metal probe, an electric generator, and a piezoelectric transducer, which converts the electrical voltages into mechanical vibrations of the same frequency. The mechanical vibrations are amplified and directed through the metal probe to the tip submerged in the sample. This generates sinusoidal pressure fluctuations in the liquid and forms cavitation bubbles near the tip. The bubbles undergo a series of compressions and expansions before experiencing a catastrophic collapse to generate high shear forces that cause droplet breakup. This mechanism is also referred to as cavitation-induced emulsification. The generated acoustic pressure depends on CPPs like maximum amplitude, time of sonication, and frequency [101].
Bérard et al. [34] varied output amplitude settings from 20% to 80% on a 750 W power commercial ultrasonicator. Their results indicated that as the percent amplitude increases, the droplet diameter decreases. This can be explained as increasing amplitude proportionally increases the power emitted into the PFC NEs. However, this observation holds true only at lower surfactant concentrations, as at high surfactant concentrations the authors reported no changes in the droplet diameter, irrespective of percent amplitude. The result suggested that at high surfactant concentrations, the droplet diameter becomes insensitive to ultrasonication amplitude, similar to findings reported by other researchers in the NE literature [92, 102]. The time of ultrasonication can be controlled by performing UE either in continuous mode or pulse mode. Throughout the PFC NE literature, researchers have preferred using pulse mode, perhaps to avoid overheating the final product. For example, Li et al. [103] performed ultrasonication with a 1.5 s power-on and 2 s power-off cycle for 30 min at a 30% output amplitude setting in an iced water bath to avoid overheating of PFOB NE. Although ultrasonication generally is performed by placing the vessel in a cold water bath [15, 45], researchers have used a sequential manufacturing approach to avoid subjecting thermosensitive payloads, especially biologics, to high temperatures during the initial stages [18]. For example, Chen et al. [68] first emulsified PFD NE with a fluorinated polycationic polymer (F-PEI) through UE at 45% amplitude in pulse mode for 30 min on an ice bath. In the later step, the pre-formulated PFD NE with F-PEI as a chemical handle was vortexed at a low speed with siRNA to develop functional polyplexes demonstrating gene silencing activity.
Further investigations are required to determine how apparatus-specific parameters like the geometry of the vessel containing the coarse emulsion, probe diameter, position of the tip within the sample, and fluctuations in temperature of the water bath have any impact on the CQAs of PFC NEs. Although slight variations in these parameters may not significantly contribute to CQA changes during small processing volumes, they will significantly alter the CQAs during processing large-scale volumes (liters). Despite the limited knowledge in the PFC NE field on UE, a cited reason for selecting UE is its suitability for low processing volumes (1 to 5 mL) as opposed to MF (~25 mL). This is particularly beneficial during optimization experiments where limited monetary resources constrain the research project.
4. Potential Challenges in the Reported Literature on PFC NE
The first challenge is that information on the processing parameters is often inadequately reported in research articles. This compromises the reproducibility and creates barriers for integrating scientific findings in the field. For example, researchers report on the amplitude or the time of ultrasonication [68], but the details on the power of the ultrasonicator, diameter of the probe, vessel geometry, or name of the specific ultrasonicator model are often omitted. For MF, a few studies [16, 38] report the total time required for MF, while the other studies report the total number of MF passes or stokes required to achieve a targeted droplet diameter on a particular microfluidizer model [17, 55]. Reporting on the total time for MF can provide comprehensive information, provided supporting details, such as the total volume processed during manufacturing or the time required per MF pass at a set operating pressure, are specified by the researchers.
The second challenge is that in current publications, emphasis is placed on conducting empirical assessments to validate the therapeutic efficacy of PFC NEs, while limited focus is given to understanding the manufacturing aspect of PFC NEs. However, during early development, it is recommended to simultaneously address questions such as: Does the selected manufacturing technique support the scale-up of the product? Which material attributes and processing parameters would influence the CQAs if the product was scaled up in the future? Recently, product development approaches like Quality by Design (QbD) have gained popularity for developing various nanoparticles. Nevertheless, the application of QbD for optimizing PFC NE composition and manufacturing parameters is limited (Figure 6A). According to the ICH Q8 guidelines, the US FDA defines QbD as “a systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control based on sound science and quality risk management” [104]. For example, after demonstrating the therapeutic effectiveness of celecoxib-loaded theranostic PFC NE in an inflammatory pain rodent model [17, 73], Herneisey et. al. [74, 105] implemented the Quality by Design (QbD) approach as a next step to gain a thorough understanding of the manufacturing process and to define a design space for formulating PFC NEs with improved quality. The authors performed the first-ever risk assessment on triphasic PFC NEs to narrow down the high-risk material and processing parameters that would significantly impact the desired CQAs. This guided the authors to identify factors such as the number of microfluidization passes, type of PFC, and concentration of oil as a few of the critical processing parameters (CPPs) and critical material attributes (CMAs) that needed to be systematically investigated by performing design of experiments (DOE). DOE serves as a statistical tool that helps to systematically study and optimize the values of CMAs and CPPs such that specified CQAs are achieved within the design space. The authors concluded these studies by developing predictive multivariate linear regression (MLR) models to establish a relationship between the changes in the CPPs and CMAs and their effect on CQAs, such as droplet diameter, drug loading, and long-term stability (Figure 6B). Overall, implementation of QbD principles helps to increase process understanding, guides researchers to develop PFC NEs with improved colloidal stability and drug loading and establishes control over the manufacturing process [74, 105]. For an in-depth understanding of the basic elements of QbD and the need for QbD application in academic nanomedicine research, we direct the readers to read an excellent review [106]. Currently, there are no approved PFC NEs for drug delivery on the market, but we speculate that following FDA-recommended pharmaceutical manufacturing approaches at the academic level to develop products that consistently meet target specifications can take the field one step closer to clinical translation. Additionally, in the future, this can prevent situations where manufacturing flaws of unidentified nature led to supply stoppages and shortages of nanomedicines like Doxil™ in the USA.
Application of QbD for developing PFC NEs. A. Academic landscape showing proportion of publication counts on therapeutic NEs and PFC NEs administered parenterally. Less than 1% of publication employed QbD approach for manufacturing therapeutic NEs or PFC NEs. Literature counts were obtained from Scopus® using Keywords: Nanoemulsions; Perflurocarbon AND Nanoemulsions; Drug delivery; QbD. NEs delivered topically, intraocular, or orally were omitted. PFC NEs include both biphasic and triphasic NEs. B. Simplistic representation of QbD in PFC NE manufacturing. Created with Biorender.com.
The third challenge is the need for performing robust characterization of PFC NEs developed for drug delivery applications. In majority of the publications, droplet diameter measured using DLS. Grapentine et al. [31] recommended using cryo-TEM as an orthogonal sizing technique for monitoring the long-term size stability of heat-sterilized PFC NEs stabilized with phospholipids. In contrast to DLS, which only provides averaged hydrodynamic diameter, cryo-TEM distinguished between PFC NE droplets and liposomes (PFC-free nanovesicles) formed during long-term storage. Thus, using techniques like cryo-TEM along with DLS can provide in-depth information on the droplet diameter changes without any bias. Recently, Janjic et al. [107] published a list of routine in vitro quality assessments for characterizing PFC NEs, which can be performed before in vivo testing. These tests include assessing droplet size stability after sterile filtration (0.22 μm filters), high-speed centrifugation (mechanical stress), thermal cycling, and post-exposure to high serum-containing biological media (biological stress). Few reports have also published accelerated stability testing conditions, where PFC NEs were subjected to extreme conditions such as elevated temperatures [41, 105]. As compared to assessing longitudinal stability, the development of accelerated stability testing protocols for PFC NEs can save time and resources for manufacturers. Herneisey et al. in parallel developed an accelerated stability testing model and a shelf-life stability testing model to evaluate whether the accelerated stability model (7 days) can accurately predict changes in size, polydispersity index, and fluorescence signal loss of theranostic PFC NEs upon long-term storage (7 months). The authors showed that the accelerated stability testing model accurately predicted changes in CQAs in a time-efficient manner, speeding the identification of stable PFC NEs [105].
5. What's next? Targeted Perfluorocarbon Nanoemulsions for Drug Delivery
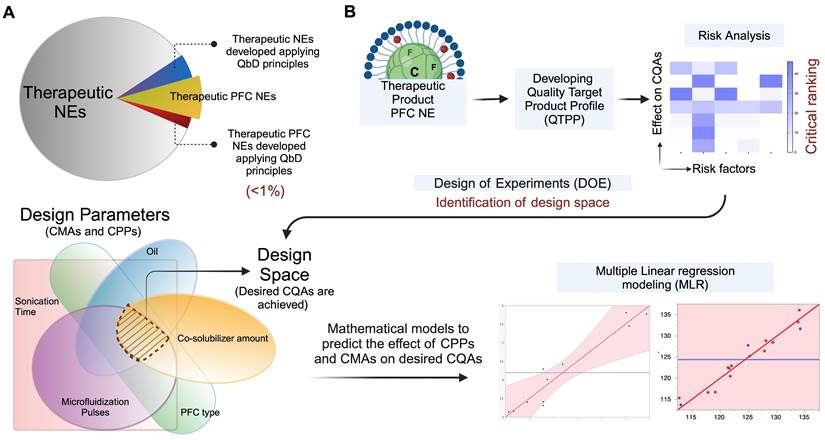
The next leap in the field is to design PFC NEs with targeting ligands for achieving precise spatial control over drug delivery. Currently, no therapeutic nanomedicines with an active targeting ligand are commercially available [108]. However, relying on passive targeting mechanisms, such as the EPR effect or RES-mediated uptake, produces variable results between preclinical models and clinical trials [109]. From a clinical perspective, the impetus for developing targeted nanoparticles as an innovative therapeutic intervention lies in their potential to alter biodistribution and pharmacokinetic profiles, reduce unwanted systemic exposure, and improve therapeutic efficacy [109]. A variety of ligands, such as small molecules, antibodies, peptides, and aptamers, have been conjugated to the surface of PFC NEs to support active targeting.
The targeting ligand enables ligand-mediated receptor interactions, which facilitates the binding of PFC NE droplets to the cell surface and intracellular transport via endocytosis. Several studies have demonstrated that increased intracellular drug delivery translates into improved biological outcomes compared to non-targeted PFC NEs [43, 55]. For example, Vichare et al. [55] showed that the uptake of folic-acid-conjugated celecoxib-loaded PFC NE was significantly higher in LPS-activated macrophages (M1-like phenotype) overexpressing folate receptors (FR) compared to non-targeted celecoxib-loaded PFC NE. This increased uptake correlated with improved therapeutic efficacy, as increased suppression of proinflammatory cytokines (TNF-α and IL-6) was observed in these macrophages. Bae et al. [110] developed folate-PFC/rhodamine NEs to specifically target FR-positive tumors. The authors showed that folate-targeted PFC NE specifically accumulated in the FR-positive xenograft tumors in mice post-intravenous administration. In contrast, minimal accumulation was observed in the tumor tissue with non-targeted PFC NE. While this work did not incorporate a therapeutic agent, it shows that targeting can significantly improve the biodistribution of the drugs to the desired site with minimal off-target effects. In another example, Hingorani et al. [111] developed PFC NE displaying cell penetrating peptides (CPPs) derived from the transactivator of transcription (TAT) component of the human immunodeficiency virus type-1 to facilitate ex vivo labeling of engineered chimeric antigen receptor lymphocytes (CAR T) cells (Figure 7A). CARs are synthetic receptors that redirect lymphocytes (T cells) to recognize and eliminate cells expressing a specific target protein or antigen. One of the limitations of CAR T cell therapy is its inability to track CAR T cell trafficking [112]. Ex vivo labeling of CAR T cells can (1) allow for real-time monitoring of CAR T biodistribution, (2) help quantify the number of survival therapeutic cells, (3) guide optimization of the required dose, and (4) provide insights into off-target toxicities. However, two major technical challenges during ex vivo labeling of CAR T cells are their small cytoplasmic volume and their poor ability to perform phagocytosis, which limits the uptake of imaging probes by the CAR T cells. The group showed that engineering PFC NE droplets to display CPPs can overcome this challenge, as an 8.2-fold higher uptake of the imaging probe was observed in the therapeutic CAR T cells with TAT surface-conjugated PFC NE droplets compared to the unmodified PFCE NE droplets (Figure 7B and C). Moreover, PFCE labeling of therapeutic CAR-T cells did not affect the phenotype or their functionality.
A. Schematic representation of TAT peptide surface-conjugated to PFCE NE droplet (TATP-F68-PFC) for increasing uptake in CAR T cells. In vivo 19F MRI signal enhancement in TATP-F68-PFC labeled human CAR T cells. B. A histogram of the 19F signal-to-noise ratio for each image voxel in the tumors is displayed and shows sensitivity improvement of the TATP-containing PFC NE compared to control. C. Comparison of apparent 19F atoms per tumor, measured in vivo for mice (n=4). * indicates p<0.001 for TAT-F68-PFC nanoemulsions compared to control. Figures adapted with permission from [111]. Copyright (2020) John Wiley and Sons, Inc.
Recently, hybrid cell membrane-coated PFC NEs have been developed as a successful strategy for site-specific accumulation of payloads [50, 113]. For instance, Zhang et al. [50] formulated PFTBA NE loaded with lactate oxidase (LOX), which catalyzes the conversion of lactic acid to pyruvate and generates toxic hydrogen peroxide (H2O2) in cancer cells. The oxygen required for this reaction was provided by the PFTBA, as PFCs have excellent oxygen-carrying capacity. Next, the authors camouflaged PFTBA nanoemulsion using hybrid membrane coatings (PSHM) composed of outer membrane vesicles of Salmonella typhimurium (SM) and the membrane of PD-1-expressing HEK293T cells (PM) (Figure 8). The group showed a three-pronged, synergistic therapeutic approach to inhibit tumor growth. First, by upregulating the expression of PD-L1 on the surface of tumor cells due to SM, thereby improving response to immune checkpoint inhibition. Second, by the targeted blocking of PD-L1 of tumor cells due to PM. Third, by boosting specific anti-tumor responses using photothermal therapy and generation of H2O2. The tumor growth inhibition rate of biomimetic PHL@PSHM plus laser radiation was as high as 92.8% compared to the PBS treatment in 4T1 breast tumor-bearing mice after a single i.v dose administration.
The preparation process of PHL@PSHM. HEK 293T cell line expressing a fusion protein PD-1/DsRed were amplified and homogenized to obtain the PD-1-expressed cell membranes (PM). Bacterial outer membrane vesicles (OMVs) were obtained from strain (VNP20009) of Salmonella typhimurium (SM). Hybrid membrane vesicles (PSHM NVs) were formed by co-extrusion. PFTBA@HSA/Lox nanoemulsion (PHL) was prepared by sonication of PFTBA and the aqueous solution of HSA and LOX. The final construct, PHL@PSHM was prepared by co-extruding PHL with PSHM. Reprinted with permission from [50]. Copyright (2023) Elsevier.
5.1 Approaches for manufacturing targeted PFC NEs
The successful development of targeted PFC NEs for drug delivery applications is dependent on the choice of manufacturing strategy. Researchers must analyze the type of ligand (biomolecules or synthetic compounds) and the physicochemical properties of the selected ligand (e.g., pH and heat sensitivity) before deciding at which step the selected ligand will be introduced in the manufacturing process. There are three general manufacturing strategies for developing surface-functionalized PFC NEs for active targeting (Figure 9). The first strategy involves the covalent conjugation of the targeting ligand to a lipid tail anchor, such as dipalmitoyl phosphatidylethanolamine (DPPE) [38] or dioctadecylamine [114]. Subsequently, a lower concentration (0.05-5 mol%) of the targeting ligand is mixed with surfactants and PFC. The formed coarse emulsions are then subjected to high-pressure homogenization techniques (pre-insertion technique, approach 1). The pre-insertion technique is limited to robust synthetic ligands that are not sensitive to high sheer stress or localized heat. One example of such a ligand is folic acid [38, 55], which binds with high affinity (Kd∼ 0.1-1 nM) to tissue-specific folate receptors (FRs). For instance, overexpressed FR-α isoform in epithelial malignancies or upregulated FR-β isoform on proinflammatory macrophages. Yang et al. [38] manufactured FR-α targeting PFCE NE for delivering Osimertinib in a non-small cell lung cancer model. Folic acid was covalently conjugated to the headgroups of DPPE lipid tails and was added to phospholipids and PFCE oil to form a coarse emulsion prior to processing at 20,000 psi on a microfluidizer. The targeting ability of folic acid was preserved despite undergoing MF, resulting in a higher uptake of PFCE NEs within the FR-α-positive tumors.
Summary of surface-functionalized PFC NEs used for drug delivery applications. Created with Biorender.com. All the chemical structures were drawn using ChemDraw 21.0.0.
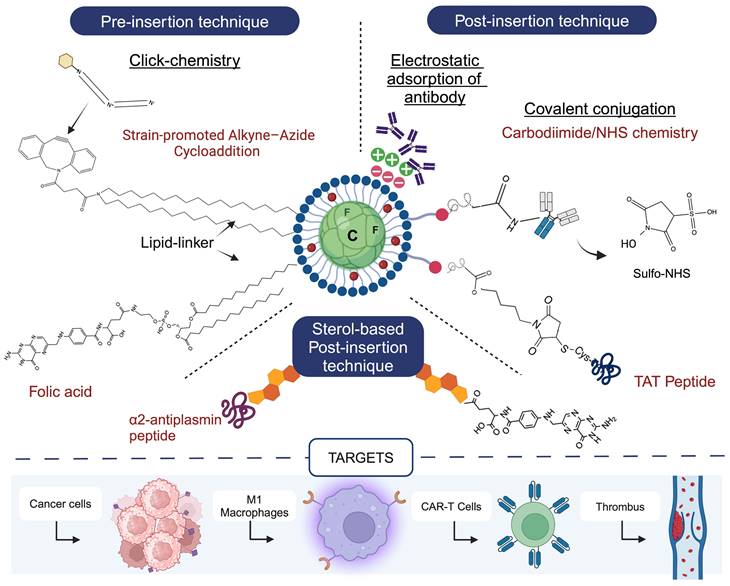
In the second strategy, PFC NEs are first manufactured with chemically modified surfactants. The pre-formed PFC NEs can display reactive groups such as maleimide (-SH), N-hydroxysuccinimide (-NHS) esters, or carboxyl (-COOH) on the surface. In the next step, the targeting ligand is chemically conjugated on the surface of the nanodroplet (post-insertion technique, approach 2). This two-step process minimizes the exposure of sensitive ligands to harsh conditions and provides control over ligand conjugation. Nevertheless, careful optimization of manufacturing parameters is required to avoid the degradation of reactive groups or chemical handles. Macromolecules like antibodies [35, 36, 115] or peptides [111] are surface-functionalized on PFC NEs through approach 2. Lee YH et al. [35] immobilized an anti-human epidermal growth factor receptor 2 (HER2) monoclonal antibody on the surface of the doxorubicin-loaded PFC NE. First, PFOB was emulsified with carboxyl-terminated pluronic F-68 block copolymer (CT-F68), which allowed the polycationic PEI polymer to react with the negative surface charge of carboxyl moieties. Later, the HER2 antibody was immobilized on the surface through ionic adsorption. Surface conjugation with HER2 resulted in a three-fold higher uptake in the HER2+ breast cancer cell line. The same group also functionalized PFOB NE with an anti-human epidermal growth factor receptor (anti-EGFR) antibody and encapsulated two payloads (ICG dye and Mitomycin C) to specifically target overexpressed EGFR in metastatic bladder cancer [36]. In another example, Bae et al. [115] proposed covalent coupling based on the carbodiimide/NHS chemistry that offers higher stability due to covalent linking between the primary amines present in the anti-HER2 antibody and the PEGylated phospholipid NSH-ester based surfactant. Both groups successfully demonstrated preservation of bioactivity following conjugation of the antibody to the surface of the PFC NE droplet. The spatial orientation or density of the antibody molecules on the PFOB NE surface are the critical parameters that directly influence the antigen-binding capacity. To prevent random immobilization of antibodies on the surface of nanoemulsions, site-selective antibody conjugation approaches like maleimide chemistry have been studied. Such chemistries can ensure “end-on” antibody immobilization on PFC NEs scaffolds. For instance, Murphy et al. [43] used maleimide-conjugated lipids to attach as1411 aptamer via the thiol groups at the 3' end.
The third strategy is based on using cholesterol anchors, where the cholesterol concentration is several-fold higher than the selected ligand. The developed cholesterol-ligand conjugates are inserted spontaneously in the phospholipid surfactant monolayer of pre-formed PFC NE (sterol-based post-insertion technique (SPIT), approach 3). The popularity of the SPIT is driven by its advantages, including mild processing conditions for ligand insertion and the feasibility of using a small volume of PFC NE. This can allow for routine ligand screening experiments. Temme et al. [116] developed first-in-class α2-antiplasmin peptide (α2AP)-labeled thrombin-inhibiting PFCE NEs using SPIT. The α2AP-PFCE NE specifically targeted factor XIIIa (FXIIIa) in developing thrombi (diameter <1 mm). SPIT provides flexibility to researchers for manufacturing PFC NEs using high sheer techniques, as the spontaneous insertion of cholesterol-ligand conjugates into the surfactant layer occurs under mild post-insertion conditions, such as shaking at room temperature.
6. Conclusions and Future Outlook
The first step in the preclinical development of PFC NEs for drug delivery applications involves screening and selection of formulation components. The presented review highlighted different studies that showed PFC NEs can be developed as smart nanomedicines by carefully selecting formulation components. For example, through rational selection of PFCs, formulators can design PFC NEs that can respond to exogenous stimuli, like ultrasound energy or pH alterations, to enable site-specific drug delivery. PFC NEs can also be designed to co-deliver therapeutic and diagnostic payloads or to facilitate targeted drug delivery. Based on the literature review, the interdisciplinary nature of formulation science for PFC NEs was evident. Material and formulation scientists are actively synthesizing novel polymeric surfactants for emulsifying PFC in water. However, it is important to address questions regarding the in vivo safety, biodistribution, and batch-to-batch consistency of these synthetic polymers. This review highlighted the potential limitations in the published academic literature on PFC NEs. It is recommended to provide detailed information on the selected critical processing parameters. Such collective efforts would help to integrate scientific findings in the field and facilitate a smooth transition of technology from academia to industry. Additionally, with advances in artificial intelligence and the development of machine learning algorithms to predict formulation parameters of nanomedicines, comprehensive data must be published for developing reliable mathematical models in the future.
Research efforts are also required to develop analytical methods or devices that would enable industries to perform in-line monitoring of the physicochemical characteristics of PFC NEs during manufacturing. Conventional DLS has several limitations, such as it relies on end-point size measurements and cannot be applied to turbid samples or in liquid flow conditions. Using advanced techniques like spatially resolved DLS (SR-DLS), which is based on the Fourier Domain Low Coherence Interferometry, can allow fast (in sec) size measurements of undiluted samples without interrupting the liquid flow. Recently, Bardsley et al. [117] designed optical sensors measuring turbidity that can be placed in the fluid path during the manufacturing of PFC NEs. These sensors have an ability to inform on the particle size changes in real time. The information obtained from such sensors can provide immediate feedback and guide manufacturers to optimize processing parameters like duration of homogenization in order to ensure product consistency and process reproducibility, especially during scale-up of PFC NEs.
It is also important that the formulation design of drug-delivering PFC NEs should not be overcomplicated. While this may result in publication in scientific journals, the clinical translational potential of the formulation in the future would become highly questionable. Perhaps a lesson to be learned from the discrepancy between the number of published articles on nanoparticles and commercially available nanomedicines is that during the early stages of development, researchers should think from a translational perspective when designing nanomedicines like PFC NEs. Although the progress of developing PFC NEs for drug delivery has been more like a marathon than a sprint, the field has made tremendous progress in recent years, expanding the applicability of PFC NEs to not only deliver small molecules but also biologics.
Acknowledgements
The authors wish to thank the Pharmaceutics Discipline Faculty of the Graduate School of Pharmaceutical Sciences, Dr. Devika Manikam, Dr. Wilson Meng, Dr. Peter Wildfong, Dr. Carl Anderson, and Dr. Ira Buckner, who critically reviewed this manuscript and provided invaluable feedback.
Funding
This work was supported by the following CDMRP awards: HT9425-24-1-0828, W81XWH-20-1-0730, W81XWH-20-1-0276, W81XWH-20-1-0854 and W81XWH-19-1-0828 awarded to JMJ.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Simons J. Production of fluorocarbons: I. The generalized procedure and its use with nitrogen compounds. J Electrochem Soc. 1949;95:47
2. Clark LC Jr, Gollan F. Survival of mammals breathing organic liquids equilibrated with oxygen at atmospheric pressure. Science. 1966;152:1755-6
3. Miller MA, Sletten EM. Perfluorocarbons in Chemical Biology. Chembiochem. 2020;21:3451-62
4. Riess JG. Understanding the fundamentals of perfluorocarbons and perfluorocarbon emulsions relevant to in vivo oxygen delivery. Artif Cells Blood Substit Immobil Biotechnol. 2005;33:47-63
5. Dalvi VH, Rossky PJ. Molecular origins of fluorocarbon hydrophobicity. Proc Natl Acad Sci U S A. 2010;107:13603-7
6. Clark LC Jr, Kaplan S, Becattini F, Benzing G 3rd. Perfusion of whole animals with perfluorinated liquid emulsions using the Clark bubble-defoam heart-lung machine. Fed Proc. 1970;29:1764-70
7. Sloviter HA, Kamimoto T. Erythrocyte substitute for perfusion of brain. Nature. 1967;216:458-60
8. Winslow RM. Blood substitutes. Adv Drug Deliv Rev. 2000;40:131-42
9. Lowe KC. Chapter 25 - Fluosol®: The First Commercial Injectable Perfluorocarbon Oxygen Carrier. In: Winslow RM, editor. Blood Substitutes. Oxford: Academic Press. 2006 p. 276-87
10. Holland G, Bottomley PA, Hinshaw W. 19F magnetic resonance imaging. J Magn Reson (1969). 1977;28:133-6
11. Ahrens ET, Flores R, Xu H, Morel PA. In vivo imaging platform for tracking immunotherapeutic cells. Nat Biotechnol. 2005;23:983-7
12. Calvo P, Vila-Jato JL, Alonso MJ. Comparative in vitro evaluation of several colloidal systems, nanoparticles, nanocapsules, and nanoemulsions, as ocular drug carriers. J Pharm Sci. 1996;85:530-6
13. Mason TG. Emulsiogenesis and the Emergence of Nanoemulsions. Matter. 2019;1:542-6
14. McClements DJ. Nanoemulsions versus microemulsions: terminology, differences, and similarities. Soft Matter. 2012;8:1719-29
15. Rapoport N, Payne A, Dillon C, Shea J, Scaife C, Gupta R. Focused ultrasound-mediated drug delivery to pancreatic cancer in a mouse model. J Ther Ultrasound. 2013;1:11
16. Bibee KP, Cheng YJ, Ching JK, Marsh JN, Li AJ, Keeling RM. et al. Rapamycin nanoparticles target defective autophagy in muscular dystrophy to enhance both strength and cardiac function. Faseb j. 2014;28:2047-61
17. Janjic JM, Vasudeva K, Saleem M, Stevens A, Liu L, Patel S. et al. Low-dose NSAIDs reduce pain via macrophage targeted nanoemulsion delivery to neuroinflammation of the sciatic nerve in rat. J Neuroimmunol. 2018;318:72-9
18. Ding L, Tang S, Tang W, Mosley DD, Yu A, Sil D. et al. Perfluorocarbon Nanoemulsions Enhance Therapeutic siRNA Delivery in the Treatment of Pulmonary Fibrosis. Adv Sci (Weinh). 2022;9:e2103676
19. Sloand JN, Nguyen TT, Zinck SA, Cook EC, Zimudzi TJ, Showalter SA. et al. Ultrasound-Guided Cytosolic Protein Delivery via Transient Fluorous Masks. ACS Nano. 2020;14:4061-73
20. Wu L, Liu F, Liu S, Xu X, Liu Z, Sun X. Perfluorocarbons-Based (19)F Magnetic Resonance Imaging in Biomedicine. Int J Nanomedicine. 2020;15:7377-95
21. Fernandes DA, Kolios MC. Near-infrared absorbing nanoemulsions as nonlinear ultrasound contrast agents for cancer theranostics. J Mol Liq. 2019;287:110848
22. Lambert E, Gorantla VS, Janjic JM. Pharmaceutical design and development of perfluorocarbon nanocolloids for oxygen delivery in regenerative medicine. Nanomedicine (Lond). 2019;14:2697-712
23. Janjic JM, Ahrens ET. Fluorine-containing nanoemulsions for MRI cell tracking. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2009;1:492-501
24. Patrick MJ, Janjic JM, Teng H, O'Hear MR, Brown CW, Stokum JA. et al. Intracellular pH measurements using perfluorocarbon nanoemulsions. J Am Chem Soc. 2013;135:18445-57
25. Yang Y, Liu Y, Jiang Y. Recent Advances in Perfluorocarbon-Based Delivery Systems for Cancer Theranostics. Mol Pharm. 2023;20:3254-77
26. Holman R, Lorton O, Guillemin PC, Desgranges S, Contino-Pépin C, Salomir R. Perfluorocarbon Emulsion Contrast Agents: A Mini Review. Front Chem. 2021;9:810029
27. Jacoby C, Temme S, Mayenfels F, Benoit N, Krafft MP, Schubert R. et al. Probing different perfluorocarbons for in vivo inflammation imaging by 19F MRI: image reconstruction, biological half-lives and sensitivity. NMR Biomed. 2014;27:261-71
28. Tsuda Y, Yamanouchi K, Yokoyama K, Suyama T, Watanabe M, Ohyanagi H. et al. Discussion and considerations for the excretion mechanism of perfluorochemical emulsion. Biomater Artif Cells Artif Organs. 1988;16:473-83
29. Keipert PE. Use of Oxygent, a perfluorochemical-based oxygen carrier, as an alternative to intraoperative blood transfusion. Artif Cells Blood Substit Immobil Biotechnol. 1995;23:381-94
30. Haque A, Scultetus AH, Arnaud F, Dickson LJ, Chun S, McNamee G. et al. The Emulsified PFC Oxycyte(®) Improved Oxygen Content and Lung Injury Score in a Swine Model of Oleic Acid Lung Injury (OALI). Lung. 2016;194:945-57
31. Grapentin C, Barnert S, Schubert R. Monitoring the Stability of Perfluorocarbon Nanoemulsions by Cryo-TEM Image Analysis and Dynamic Light Scattering. PLoS One. 2015;10:e0130674
32. Patel SK, Janjic JM. Macrophage targeted theranostics as personalized nanomedicine strategies for inflammatory diseases. Theranostics. 2015;5:150-72
33. Janjic JM, Shao P, Zhang S, Yang X, Patel SK, Bai M. Perfluorocarbon nanoemulsions with fluorescent, colloidal and magnetic properties. Biomaterials. 2014;35:4958-68
34. Bérard C, Desgranges S, Dumas N, Novell A, Larrat B, Hamimed M. et al. Perfluorocarbon Nanodroplets as Potential Nanocarriers for Brain Delivery Assisted by Focused Ultrasound-Mediated Blood-Brain Barrier Disruption. Pharmaceutics. 2022;14(7):1498
35. Lee YH, Ma YT. Synthesis, characterization, and biological verification of anti-HER2 indocyanine green-doxorubicin-loaded polyethyleneimine-coated perfluorocarbon double nanoemulsions for targeted photochemotherapy of breast cancer cells. J Nanobiotechnology. 2017;15:41
36. Lee YH, Lin YC. Anti-EGFR Indocyanine Green-Mitomycin C-Loaded Perfluorocarbon Double Nanoemulsion: A Novel Nanostructure for Targeted Photochemotherapy of Bladder Cancer Cells. Nanomaterials (Basel). 2018Apr26;8(5):283
37. Hsiao KH, Huang CM, Lee YH. Novel Rifampicin and Indocyanine Green Co-Loaded Perfluorocarbon Nanodroplets Provide Effective In Vivo Photo-Chemo-Probiotic Antimicrobility against Pathogen of Acne Vulgaris Cutibacterium acnes. Nanomaterials (Basel). 2020Jun1;10(6):1095
38. Yang J, Li Y, Sun J, Zou H, Sun Y, Luo J. et al. An Osimertinib-Perfluorocarbon Nanoemulsion with Excellent Targeted Therapeutic Efficacy in Non-small Cell Lung Cancer: Achieving Intratracheal and Intravenous Administration. ACS Nano. 2022;16:12590-605
39. Herneisey M, Williams J, Mirtic J, Liu L, Potdar S, Bagia C. et al. Development and characterization of resveratrol nanoemulsions carrying dual-imaging agents. Ther Deliv. 2016;7:795-808
40. Rapoport NY, Kennedy AM, Shea JE, Scaife CL, Nam KH. Controlled and targeted tumor chemotherapy by ultrasound-activated nanoemulsions/microbubbles. J Control Release. 2009;138:268-76
41. Ashrafi B, Tootoonchi MH, Bardsley R, Molano RD, Ruiz P, Pretto EA Jr. et al. Stable perfluorocarbon emulsions for the delivery of halogenated ether anesthetics. Colloids Surf B Biointerfaces. 2018;172:797-805
42. Esser AK, Schmieder AH, Ross MH, Xiang J, Su X, Cui G. et al. Dual-therapy with αvβ3-targeted Sn2 lipase-labile fumagillin-prodrug nanoparticles and zoledronic acid in the Vx2 rabbit tumor model. Nanomedicine. 2016;12:201-11
43. Murphy EM, Centner CS, Bates PJ, Malik MT, Kopechek JA. Delivery of thymoquinone to cancer cells with as1411-conjugated nanodroplets. PLoS One. 2020;15:e0233466
44. Ding L, Tang S, Yu A, Wang A, Tang W, Jia H. et al. Nanoemulsion-Assisted siRNA Delivery to Modulate the Nervous Tumor Microenvironment in the Treatment of Pancreatic Cancer. ACS Appl Mater Interfaces. 2022;14:10015-29
45. Li Z, Shen Y, Wang Y, Zhu L, Zhu C, Qian C. et al. Perfluorocarbon nanoemulsions for combined pulmonary siRNA treatment of lung metastatic osteosarcoma. Adv Ther (Weinh). 2019;2:1900039
46. Qin X, Zhou Y, Wang Y, Wang Z, Wang Y, Chen J. et al. Preparation and Characterization of Protein-loaded PFC Nanoemulsions for the Treatment of Heart Diseases by Pulmonary Administration. Eur J Pharm Sci. 2021;158:105690
47. Pan H, Soman NR, Schlesinger PH, Lanza GM, Wickline SA. Cytolytic peptide nanoparticles ('NanoBees') for cancer therapy. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2011;3:318-27
48. Palekar RU, Jallouk AP, Myerson JW, Pan H, Wickline SA. Inhibition of Thrombin With PPACK-Nanoparticles Restores Disrupted Endothelial Barriers and Attenuates Thrombotic Risk in Experimental Atherosclerosis. Arterioscler Thromb Vasc Biol. 2016;36:446-55
49. Soman NR, Baldwin SL, Hu G, Marsh JN, Lanza GM, Heuser JE. et al. Molecularly targeted nanocarriers deliver the cytolytic peptide melittin specifically to tumor cells in mice, reducing tumor growth. J Clin Invest. 2009;119:2830-42
50. Zhang Y, Liao Y, Bai F, Xing S, Wang M, Dang Z. et al. Genetically engineered eukaryocyte-bacteria hybrid membrane-camouflaged nanoemulsion for three-pronged synergistic cancer therapy. Nano Today. 2024;54:102076
51. Estabrook DA, Day RA, Sletten EM. Redox-Responsive Gene Delivery from Perfluorocarbon Nanoemulsions through Cleavable Poly(2-oxazoline) Surfactants. Angew Chem Int Ed Engl. 2021;60:17362-7
52. Priimagi A, Cavallo G, Metrangolo P, Resnati G. The halogen bond in the design of functional supramolecular materials: recent advances. Acc Chem Res. 2013;46:2686-95
53. Saleem M, Deal B, Nehl E, Janjic JM, Pollock JA. Nanomedicine-driven neuropathic pain relief in a rat model is associated with macrophage polarity and mast cell activation. Acta Neuropathol Commun. 2019;7:108
54. Liu L, Karagoz H, Herneisey M, Zor F, Komatsu T, Loftus S. et al. Sex Differences Revealed in a Mouse CFA Inflammation Model with Macrophage Targeted Nanotheranostics. Theranostics. 2020;10:1694-707
55. Vichare R, Crelli C, Liu L, McCallin R, Cowan A, Stratimirovic S. et al. Folate-conjugated near-infrared fluorescent perfluorocarbon nanoemulsions as theranostics for activated macrophage COX-2 inhibition. Sci Rep. 2023;13:15229
56. Riess JG. Oxygen carriers ("blood substitutes")-raison d'etre, chemistry, and some physiology. Chem Rev. 2001;101:2797-920
57. Chenoune M, De Rochefort L, Bruneval P, Lidouren F, Kohlhauer M, Seemann A. et al. Evaluation of lung recovery after static administration of three different perfluorocarbons in pigs. BMC Pharmacol Toxicol. 2014;15:53
58. Delaney LJ, Isguven S, Eisenbrey JR, Hickok NJ, Forsberg F. Making waves: how ultrasound-targeted drug delivery is changing pharmaceutical approaches. Mater Adv. 2022;3:3023-40
59. Rapoport N. Drug-Loaded Perfluorocarbon Nanodroplets for Ultrasound-Mediated Drug Delivery. Adv Exp Med Biol. 2016;880:221-41
60. Wu SY, Fix SM, Arena CB, Chen CC, Zheng W, Olumolade OO. et al. Focused ultrasound-facilitated brain drug delivery using optimized nanodroplets: vaporization efficiency dictates large molecular delivery. Phys Med Biol. 2018;63:035002
61. Kabalnov AS, Shchukin ED. Ostwald ripening theory: applications to fluorocarbon emulsion stability. Adv Colloid Interface Sci. 1992;38:69-97
62. Patel SK, Zhang Y, Pollock JA, Janjic JM. Cyclooxgenase-2 inhibiting perfluoropoly (ethylene glycol) ether theranostic nanoemulsions-in vitro study. PLoS One. 2013;8:e55802
63. Kosugi I, Yamaguchi Y, Kitagaki T, Kawashima Y, Mizota H. [Effects of perfluorodecalin (Fluosol-DC) on tissue oxygenation]. Masui. 1976;25:293-9
64. Gupta A, Eral HB, Hatton TA, Doyle PS. Nanoemulsions: formation, properties and applications. Soft Matter. 2016;12:2826-41
65. Estabrook DA, Ennis AF, Day RA, Sletten EM. Controlling nanoemulsion surface chemistry with poly(2-oxazoline) amphiphiles. Chem Sci. 2019;10:3994-4003
66. Day RA, Estabrook DA, Wu C, Chapman JO, Togle AJ, Sletten EM. Systematic Study of Perfluorocarbon Nanoemulsions Stabilized by Polymer Amphiphiles. ACS Appl Mater Interfaces. 2020;12:38887-98
67. Gupta R, Shea J, Scafe C, Shurlygina A, Rapoport N. Polymeric micelles and nanoemulsions as drug carriers: Therapeutic efficacy, toxicity, and drug resistance. J Control Release. 2015;212:70-7
68. Chen G, Wang K, Wu P, Wang Y, Zhou Z, Yin L. et al. Development of fluorinated polyplex nanoemulsions for improved small interfering RNA delivery and cancer therapy. Nano Res. 2018;11:3746-61
69. Tadros TF, Vandamme A, Levecke B, Booten K, Stevens CV. Stabilization of emulsions using polymeric surfactants based on inulin. Adv Colloid Interface Sci. 2004;108-109:207-26
70. Shaw RF, Richard TJ. Chapter 27 - Rational Development of Oxyfluor™. In: Winslow RM, editor. Blood Substitutes. Oxford: Academic Press. 2006 p. 298-311
71. O'Hanlon CE, Amede KG, O'Hear MR, Janjic JM. NIR-labeled perfluoropolyether nanoemulsions for drug delivery and imaging. J Fluor Chem. 2012;137:27-33
72. Kaustav B. Chapter 6 - Importance of Surface Energy in Nanoemulsion. In: Kai Seng K, Voon Loong W, editors. Nanoemulsions. Rijeka: IntechOpen. 2019
73. Patel SK, Beaino W, Anderson CJ, Janjic JM. Theranostic nanoemulsions for macrophage COX-2 inhibition in a murine inflammation model. Clin Immunol. 2015;160:59-70
74. Herneisey M, Liu L, Lambert E, Schmitz N, Loftus S, Janjic JM. Development of Theranostic Perfluorocarbon Nanoemulsions as a Model Non-Opioid Pain Nanomedicine Using a Quality by Design (QbD) Approach. AAPS PharmSciTech. 2019;20:65
75. Zhalimov V, Sklifas A, Kaptsov V, Penkov N, Temnov A, Kukushkin N. The chemical structure of triblock copolymers and the adsorption capacity of perfluorocarbon—core nanoparticles stabilized by them. Colloid Polym Sci. 2018;296:251-7
76. Li J, He Y, Anankanbil S, Guo Z. Chapter 7 - Phospholipid-Based Surfactants. In: Hayes DG, Solaiman DKY, Ashby RD, editors. Biobased Surfactants (Second Edition). AOCS Press. 2019 p. 243-86
77. Kaneda MM, Caruthers S, Lanza GM, Wickline SA. Perfluorocarbon nanoemulsions for quantitative molecular imaging and targeted therapeutics. Ann Biomed Eng. 2009;37:1922-33
78. Soman NR, Lanza GM, Heuser JM, Schlesinger PH, Wickline SA. Synthesis and characterization of stable fluorocarbon nanostructures as drug delivery vehicles for cytolytic peptides. Nano Lett. 2008;8:1131-6
79. Lee SJ, Schlesinger PH, Wickline SA, Lanza GM, Baker NA. Simulation of fusion-mediated nanoemulsion interactions with model lipid bilayers. Soft Matter. 2012;8:3024-35
80. Partlow KC, Lanza GM, Wickline SA. Exploiting lipid raft transport with membrane targeted nanoparticles: a strategy for cytosolic drug delivery. Biomaterials. 2008;29:3367-75
81. Nabi IR, Le PU. Caveolae/raft-dependent endocytosis. J Cell Biol. 2003;161:673-7
82. Pichot R, Watson RL, Norton IT. Phospholipids at the interface: current trends and challenges. Int J Mol Sci. 2013;14:11767-94
83. McClements DJ, Decker E. Interfacial Antioxidants: A Review of Natural and Synthetic Emulsifiers and Coemulsifiers That Can Inhibit Lipid Oxidation. J Agric Food Chem. 2018;66:20-35
84. Johnson RA, Simmons KT, Fast JP, Schroeder CA, Pearce RA, Albrecht RM. et al. Histamine release associated with intravenous delivery of a fluorocarbon-based sevoflurane emulsion in canines. J Pharm Sci. 2011;100:2685-92
85. Cardoso VF, Correia DM, Ribeiro C, Fernandes MM, Lanceros-Méndez S. Fluorinated polymers as smart materials for advanced biomedical applications. Polymers. 2018;10:161
86. Decato S, Bemis T, Madsen E, Mecozzi S. Synthesis and characterization of perfluoro-tert-butyl semifluorinated amphiphilic polymers and their potential application in hydrophobic drug delivery. Polym Chem. 2014;5:6461-71
87. Wang H, Zhang S, Lv J, Cheng Y. Design of polymers for siRNA delivery: Recent progress and challenges. View. 2021;2:20200026
88. Lv J, Chang H, Wang Y, Wang M, Xiao J, Zhang Q. et al. Fluorination on polyethylenimine allows efficient 2D and 3D cell culture gene delivery. J Mater Chem B. 2015;3:642-50
89. Gao D, Xu M, Cao Z, Gao J, Chen Y, Li Y. et al. Ultrasound-Triggered Phase-Transition Cationic Nanodroplets for Enhanced Gene Delivery. ACS Appl Mater Interfaces. 2015;7:13524-37
90. Kaufman RJ, Richard TJ, inventors; Hemagen PFC LP, assignee; Emulsions of highly fluorinated organic compounds. United State patents. US-5171755-A. 1992 December 15; Available from:. https://pubchem.ncbi.nlm.nih.gov/patent/US-5171755-A
91. Lambert E, Janjic JM. Multiple linear regression applied to predicting droplet size of complex perfluorocarbon nanoemulsions for biomedical applications. Pharm Dev Technol. 2019;24:700-10
92. Gupta A, Eral HB, Hatton TA, Doyle PS. Controlling and predicting droplet size of nanoemulsions: scaling relations with experimental validation. Soft Matter. 2016;12:1452-8
93. Jafari SM, McClements DJ. Nanoemulsions: formulation, applications, and characterization. Academic Press. 2018
94. Microfluidics. Microfluidizer Processor User Guide [Internet]. MicrofluidicsTM; 2014 December. Available from: https://www.alfatest.it/keyportal/uploads/2017-microfluidics-chamber-user-guide.pdf
95. Villalobos-Castillejos F, Granillo-Guerrero VG, Leyva-Daniel DE, Alamilla-Beltrán L, Gutiérrez-López GF, Monroy-Villagrana A. et al. Chapter 8 - Fabrication of Nanoemulsions by Microfluidization. In: Jafari SM, McClements DJ, editors. Nanoemulsions. Cambridge, MA, USA: Academic Press. 2018 p. 207-32
96. Vankova N, Tcholakova S, Denkov ND, Ivanov IB, Vulchev VD, Danner T. Emulsification in turbulent flow 1. Mean and maximum drop diameters in inertial and viscous regimes. J Colloid Interface Sci. 2007;312:363-80
97. Kavinila S, Nimbkar S, Moses J, Anandharamakrishnan C. Emerging applications of microfluidization in the food industry. J Agric Food Res. 2023;12:100537
98. Liu L, Bagia C, Janjic JM. The First Scale-Up Production of Theranostic Nanoemulsions. Biores Open Access. 2015;4:218-28
99. Qian C, McClements DJ. Formation of nanoemulsions stabilized by model food-grade emulsifiers using high-pressure homogenization: Factors affecting particle size. Food Hydrocoll. 2011;25:1000-8
100. Mao L, Yang J, Xu D, Yuan F, Gao Y. Effects of homogenization models and emulsifiers on the physicochemical properties of β-carotene nanoemulsions. J Dispers Sci Technol. 2010;31:986-93
101. Leong TS, Martin GJ, Ashokkumar M. Ultrasonic encapsulation - A review. Ultrason Sonochem. 2017;35:605-14
102. Tang SY, Shridharan P, Sivakumar M. Impact of process parameters in the generation of novel aspirin nanoemulsions-comparative studies between ultrasound cavitation and microfluidizer. Ultrason Sonochem. 2013;20:485-97
103. Li Z, Chen G, Ding L, Wang Y, Zhu C, Wang K. et al. Increased Survival by Pulmonary Treatment of Established Lung Metastases with Dual STAT3/CXCR4 Inhibition by siRNA Nanoemulsions. Mol Ther. 2019;27:2100-10
104. Testas M, da Cunha Sais T, Medinilha LP, Niwa KNI, de Carvalho LS, Maia SD. et al. An industrial case study: QbD to accelerate time-to-market of a drug product. AAPS Open. 2021;7:12
105. Herneisey M, Janjic JM. Multiple Linear Regression Predictive Modeling of Colloidal and Fluorescence Stability of Theranostic Perfluorocarbon Nanoemulsions. Pharmaceutics. 2023;15:1103
106. Dailey LA. Pharmaceutical quality by design in academic nanomedicine research: stifling innovation or creativity through constraint? J Interdiscip Nanomed. 2018;3:175-82
107. Janjic JM, McCallin R, Liu L, Crelli C, Das AC, Troidle A. In vitro Quality Assessments of Perfluorocarbon Nanoemulsions for Near-infrared Fluorescence Imaging of Inflammation in Preclinical Models. Bio Protoc. 2023;13:e4842
108. Thapa RK, Kim JO. Nanomedicine-based commercial formulations: current developments and future prospects. J Pharm Investig. 2023;53:19-33
109. Rosenblum D, Joshi N, Tao W, Karp JM, Peer D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun. 2018;9:1410
110. Bae PK, Jung J, Lim SJ, Kim D, Kim SK, Chung BH. Bimodal perfluorocarbon nanoemulsions for nasopharyngeal carcinoma targeting. Mol Imaging Biol. 2013;15:401-10
111. Hingorani DV, Chapelin F, Stares E, Adams SR, Okada H, Ahrens ET. Cell penetrating peptide functionalized perfluorocarbon nanoemulsions for targeted cell labeling and enhanced fluorine-19 MRI detection. Magn Reson Med. 2020;83:974-87
112. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11:69
113. Zhang Y, Liao Y, Tang Q, Lin J, Huang P. Biomimetic Nanoemulsion for Synergistic Photodynamic-Immunotherapy Against Hypoxic Breast Tumor. Angew Chem Int Ed Engl. 2021;60:10647-53
114. Perez AS, Zhou J, Leach B, Xu H, Lister D, Adams SR. et al. Click-Ready Perfluorocarbon Nanoemulsion for (19)F MRI and Multimodal Cellular Detection. ACS Nanosci Au. 2022;2:102-10
115. Bae PK, Chung BH. Multiplexed detection of various breast cancer cells by perfluorocarbon/quantum dot nanoemulsions conjugated with antibodies. Nano Converg. 2014;1:23
116. Temme S, Grapentin C, Quast C, Jacoby C, Grandoch M, Ding Z. et al. Noninvasive Imaging of Early Venous Thrombosis by 19F Magnetic Resonance Imaging With Targeted Perfluorocarbon Nanoemulsions. Circulation. 2015;131:1405-14
117. Bardsley R, Gardner G, Tse HM, Fraker CA. Optical sensor arrays designed for guided manufacture of perfluorocarbon nanoemulsions with a non-synthetic stabilizer. Acta Biomater. 2021;136:558-69
Author contact
![]() Corresponding author: janjicjedu.
Corresponding author: janjicjedu.